Embed Size (px)
Citation preview

Polyhedron 20 (2001) 3091–3099
The synthesis of three novel pendant armed macrocyclic ligands.The X-ray crystal structure of a cadmium complex derived fromthe first oxaazamacrocycle bearing two alkylated aromatic amine
functions
Laura Valencia a, Harry Adams b, Rufina Bastida a,*, David E. Fenton b,Alejandro Macıas a, Jose Mahıa c, Sharon E. Spey b
a Departamento de Quimica Inorganica, Uni�ersidad de Santiago de Compostela, A�da. de las Ciencias s/n 15706,Santiago de Compostela, Spain
b Department of Chemistry, The Uni�ersity of Sheffield, Sheffield S3 7HF, UKc Ser�icios Xerais de Apoio a In�estigacion SXAI, Uni�ersidade de A Coruna, La Coruna, Spain
Received 8 June 2001; accepted 15 August 2001
Abstract
Three new macrocyclic ligands derived from 2,6-pyridinedicarbaldehyde have been prepared by N-alkylation of a secondaryaliphatic amine and two secondary phenylic amines of the oxaazamacrocycle precursor (L) with p-nitrobenzylbromide andbromoacetonitrile. The trialkylated ligand 2 is the first example of a macrocyclic ligand bearing pendant arms on secondaryaromatic amine nitrogens. Complexation reactions of macrocycles 2 and 3 with different hydrated metal salts were carried out inorder to investigate the complexation capability of these new pendant arm macrocyclic ligands. The X-ray crystal structures ofligands 1 and 3 and of the cadmium complex of 2 have been obtained. © 2001 Elsevier Science Ltd. All rights reserved.
Keywords: Macrocycle; Pendant arm; Metal complexes; Crystal structures
www.elsevier.com/locate/poly
1. Introduction
In recent years stable metal complexes of macrocyclicligands incorporating pendant arms have attracted con-siderable interest mainly for radiopharmaceutical andbiomedical applications [1,2]. The introduction of pen-dant-arms into the macrocyclic framework can lead toimportant changes in the complexation capability of theligands, and can enhance the metal-ion selectivity andthe stability of metal complexes depending on the co-ordination properties of the pendant arms. Although awide variety of polyaza- or oxaaza-macrocyclic ligandsincorporating pendant arms attached to secondaryaliphatic amines have been reported [3–5] to date, no
examples of oxaazamacrocyclic ligands bearing pendantarms on secondary aromatic amines have appeared.
The ligands used in this work have been synthesisedfrom the ligand L, derived from 2,6-pyridinedicarbalde-hyde [6,7] and N,N-bis-[2-(2-aminophenoxy)ethyl]amine[8,9]. The proximity of aromatic rings to the aminefunctions in L make introduction of pendant groupsinto the macrocyclic framework difficult due to elec-tronic effects and steric hindrance. A range of alkylat-ing agents have been used in different reactionsattempting to prepare trialkylated pendant arm macro-cyclic ligands, however, in most cases, this led only tomonoalkylation of the aliphatic amine function of L.
Previously, we have reported the coordination capa-bility of the precursor ligand L as well as that of thecorresponding Schiff base macrocyclic ligand [9]. Con-tinuing with these studies we have synthesised threenew pendant-armed macrocyclic ligands 1–3 (Scheme1) derived from L, and some metal complexes with 2
* Corresponding author. Tel.: +34-981-591-076; fax: +34-981-597-525.
E-mail address: [email protected] (R. Bastida).
0277-5387/01/$ - see front matter © 2001 Elsevier Science Ltd. All rights reserved.PII: S 0 2 7 7 -5387 (01 )00909 -3

L. Valencia et al. / Polyhedron 20 (2001) 3091–30993092
and 3 have been prepared in order to compare thedifferent coordination properties of these ligands withrespect the precursor L.
2. Experimental
2.1. Measurements
Elemental analysis were performed in a Carlo-ErbaEA microanalyser. Infrared spectra were recorded asKBr discs in a Bruker IFS-66V spectrophotometer.FAB mass spectra were recorded using a Kratos-MS-50T spectrometer connected to a DS90 data systemusing 3-nitrobenzyl alcohol as the matrix. Conductivitymeasurements were carried out in 10−3 mol dm−3 ace-tonitrile solutions at 20 °C using a WTW LF3 conduc-tivitymeter. 1H and 13C NMR spectra were recorded ina Brucker AMX 300 MHz instrument against TMS asan internal standard and DEPT 135 and HMQC 1H–13C in a Brucker 500 MHz, CD3CN was used as adeuterated solvent in all cases.
2.2. Chemicals and starting materials
p-Nitrobenzylbromide, bromoacetonitrile and metalsalts were commercial products (from Alfa and Aldrich)and were used without further purification. Solventswere of reagent grade and were purified by the usualmethods. The ligand L was prepared according to aliterature method [9]. Caution : perchlorate salts arepotentially explosive!
2.3. X-ray crystallography
All the crystals were obtained by slow recrystallisa-tion of the compound from acetonitrile. The details ofthe X-ray crystal data, and the structure solution andrefinement for 1, 3 and [Cd2(NO3)][Cd(NO3)3]·3.5CH3CN are given in Table 2. Measurements weremade in a Siemens SMART CCD area detector for[Cd2(NO3)][Cd(NO3)3]·3.5CH3CN, in a BrukerSMART CCD area diffractometer for the free macro-cycle 1 and in a Siemens P4 diffractometer for 3. Alldata were corrected for Lorentz and polarisation ef-fects. Absorption corrections were also applied for 1(semiempirical) and [Cd2(NO3)][Cd(NO3)3]·3.5CH3CN(empirical) [10]. Complex scattering factors were takenfrom the program package SHELXTL [11] for 1 and 3.[Cd2(NO3)][Cd(NO3)3]·3.5CH3CN was solved usingSHELX-97 [12]. The structures were solved by directmethods which revealed the position of all non-hydro-gen atoms. All the structures were refined on F2 by afull-matrix least-squares procedure using anisotropicdisplacement parameters for all non-hydrogen atoms.The hydrogen atoms were located in their calculatedpositions and refined using a riding model.
2.4. Synthesis of the macrocycle 1
The ligand L (1 mmol, 0.39 g) was dissolved inacetonitrile (30 ml) under reflux and p-nitrobenzylbro-mide (1 mmol, 0.432 g) and Na2CO3 (1 mmol, 0.106 g)were added. The mixture was refluxed for 6 h. Thesolution was evaporated to dryness and the residueextracted with water–chloroform. The organic layerwas dried over MgSO4 and evaporated to yield a yellowsolid. This product was dissolved in absolute ethanol(60 ml) with 0.025 g of Pd/C; after that 3 ml ofhydrated hydrazine was added. The mixture wasrefluxed for 2 h and filtered while hot. The solution wasconcentrated to dryness and the solid obtained wasrecrystallised in acetonitrile giving a white solid.
Anal. Calc. for C30H33N5O2: C, 72.7; H, 7.7; N, 13.8.Found: C, 72.3; H, 7.5; N, 13.6%. Yield: 60%. IR (KBr,cm−1): 3457m, 3427m [�(NH2)], 3368m [�(NH)], 1601s,1450s [�(C�C)ar, �(C�N)py]. FAB MS; m/z : 496. 1HNMR (CD3CN): �=7.74 (t, 1H, py), 7.31 (d, 2H, py),4.43 (d, 4H, py-CH2), 5.73 (t, 2H, NH), 6.59–6.88 (m,12H, ar), 4.15 (t, 4H, O�CH2), 3.15 (t, 4H,O�CH2�CH2�NH), 3.80 (s, 2H, N�CH2�ar).
2.5. Synthesis of the macrocycle 2
The ligand L (1 mmol, 0.39 g) was dissolved inacetonitrile (30 ml) under reflux and p-nitrobenzylbro-mide (3 mmol, 1.296 g) and Na2CO3 (3 mmol, 0.318 g)were added. The mixture was refluxed for 6 h. Thesolution was evaporated to dryness and the residueScheme 1.

L. Valencia et al. / Polyhedron 20 (2001) 3091–3099 3093
Table 11H and 13C NMR spectra of 2 in acetonitrile
AssignmentAssignment � (ppm)� (ppm)
H1 H21 7.60 (m, 3H)C1 136.5H2121.0 7.20 (d, 2H)C2
159.0C3
59.1, 55.9C4, C13 H4, H13 4.45 (s, 4H)4.55 (s, 4H)
148.0C5
121.4C6 H6 6.72 (d, 2H)H7 H8114.7, 123.3 6.80 (m, 4H)C7, C8
H9C9 6.91 (d, 2H)122.2153.1C10
67.6C11 H11 4.12 (t, 4H)54.4C12 H12 2.90 (t, 4H)
140.1C14, C19
123.4C15 H15 7.53 (d, 4H)H16129.3 7.95 (d, 4H)C16
C17, C22 147.2H1858.1 3.85 (s, 2H)C18
123.6C20 H20 8.11 (d, 2H)129.7C21
Yield: 58%. Analytical data: Found: C, 68.9; H, 6.7;N, 16.6. Calc. for C25H27N5O2: C, 69.9; H, 6.3; N,16.6%; FAB MS (mNBA): 430; IR (KBr disc, cm−1):�(NH) 3392, �(C�C)ar �(C�N)py 1603, 1446. 1H NMR(CD3CN): � (ppm) 7.74 (t, 1H, py), 7.30 (d, 2H, py),4.40 (d, 4H, py�CH2), 5.73 (t, 2H, NH), 6.45–6.95 (m,12H, ar), 4.15 (t, 4H, O�CH2), 3.02 (t, 4H,O�CH2�CH2�NH), 3.55 (s, 2H, N�CH2�CN).
2.7. Synthesis of the metal complexes. Generalprocedure
The appropriate metal salt (0.25 mmol) was dissolvedin acetonitrile (10 ml) and added to a refluxed solutionof the corresponding ligand (0.25 mmol) in acetonitrile(30 ml). The solution was refluxed for 2 h, concentratedin a rotary evaporator until approximately 5–6 ml. Theproduct obtained after 2–3 days was filtered off anddried under vacuum. When the precipitation did notoccur a few drops of ethyl ether were added.
2.8. [Zn2](ClO4)2 ·3H2O
Anal. Calc for C44H47N7O19Cl2Zn: C, 47.4; H, 4.3; N,8.8. Found: C, 47.7, H, 4.5; N, 8.2%. Yield: 44%. IR(KBr, cm−1): 1348s [�(NO2)], 1608s, 1454m [�(C�C)and �(C�N)py], 1106b, 623s, [�(ClO4
−)]. FAB MS; m/z :960 [Zn2(ClO4)]+. �M (�−1cm2 mol−1, CH3CN): 230(2:1). Colour: white.
2.9. [Cd2](ClO4)2 ·3H2O
Anal. Calc for C44H47N7O19Cl2Cd: C, 45.5; H, 4.1; N,8.4. Found: C, 45.6, H, 3.8; N, 8.3%. Yield: 67%. IR(KBr, cm−1): 1348s [�(NO2)], 1607s, 1459m [�(C�C)and �(C�N)py], 1108b, 623s, [�(ClO4
−)]. FAB MS; m/z :1107 [Cd2(ClO4)]+. �M (�−1 cm2 mol−1, CH3CN): 226(2:1). Colour: white.
2.10. [Ni2](NO3)2 ·3H2O
Anal. Calc for C44H47N9O17Ni: C, 51.2; H, 4.5; N,12.2. Found: C, 51.7, H, 4.6; N, 12.0%. Yield: 38%. IR(KBr, cm−1): 1348s [�(NO2)], 1606s, 1452m [�(C�C)and �(C�N)py], 704m, 821w, 1016m, 1306s, 1384s,[�(NO3
−)]. FAB MS; m/z : 915 [Ni2(NO3)]+. �M
(�−1 cm2 mol−1, CH3CN): 121 (1:1). Colour: green.
2.11. [Cd2](NO3)2 ·H2O
Anal. Calc for C44H43N9O15Cd: C, 50.32; H, 4.13; N,12.0. Found: C, 50.7, H, 4.0; N, 11.8%. Yield: 75%. IR(KBr, cm−1): 1348s [�(NO2)], 1606s, 1497m [�(C�C)and �(C�N)py], 705m, 823m, 1024s, 1291m, 1384s,1460m [�(NO3
−)]. FAB MS; m/z : 971 [Cd2(NO3)]+. �M
(�−1 cm2 mol−1, CH3CN): 126 (1:1). Colour: white.
extracted with water–chloroform. The organic layerwas dried over MgSO4 and evaporated to yield anorange solid.
Anal. Calc. for C44H41N7O8: C, 65.4; H, 5.2; N, 12.3.Found: C, 65.3; H, 5.5; N, 12.6%. Yield: 83%. IR (KBr,cm−1): 1345s, 1500s [�(NO2)], 1596s, 1453s [�(C�C)ar,�(C�N)py]. FAB MS; m/z : 796. 1H and 13C NMR dataare given in Table 1.
2.6. Synthesis of the macrocycle 3
The ligand L (1 mmol, 0.39 g) was dissolved inacetonitrile (30 ml) under reflux and bromoacetonitrile(1.5 mmol, 0.179 g) and Na2CO3 (1.5 mmol, 0.16 g)were added. The mixture was refluxed for 12 h. Thesolution was filtered off and evaporated to drynessunder vacuum. The residue was extracted with water–chloroform, the organic layer was dried over MgSO4
and evaporated to yield an orange solid.

L. Valencia et al. / Polyhedron 20 (2001) 3091–30993094
2.12. [Zn3](ClO4)2 ·3H2O
Anal. Calc for C25H33N5O13Cl2Zn: C, 40.1; H, 4.4; N,9.4. Found: C, 40.3, H, 4.6; N, 9.0%. Yield: 53%. IR(KBr, cm−1): 1612m, 1445m [�(C�C) and �(C�N)py],1105b, 623s, [�(ClO4
−)]. FAB MS; m/z : 594[Zn3(ClO4)]+, 493 [Zn3]+. �M (�−1 cm2 mol−1,CH3CN): 246 (2:1). Colour: white.
2.13. [Cd3](ClO4)2 ·3H2O
Anal. Calc for C25H33N5O13Cl2Cd: C, 37.8; H, 4.2; N,8.8. Found: C, 37.7, H, 4.6; N, 9.2%. Yield: 83%. IR(KBr, cm−1): 2263w [�(C�N)], 1607m, 1447m [�(C�C)and �(C�N)py], 1105b, 623s, [�(ClO4
−)]. FAB MS; m/z :641 [Cd3(ClO4)]+. �M (�−1 cm2 mol−1, CH3CN): 220(2:1). Colour: white.
2.14. [Co3](ClO4)2 ·2H2O
Anal. Calc for C25H31N5O12Cl2Co: C, 41.5; H, 4.3; N,9.7. Found: C, 42.1, H, 4.6; N, 9.3%. Yield: 35%. IR(KBr, cm−1): 1602m, 1441m [�(C�C) and �(C�N)py],1109b, 626s, [�(ClO4
−)]. FAB MS; m/z : 587[Co3(ClO4)]+, 486 [Co3]+. �M (�−1 cm2 mol−1,CH3CN): 230 (2:1). Colour: red.
2.15. [Cd3](NO3)2 ·H2O
Anal. Calc for C25H29N7O9Cd: C, 43.9; H, 4.2; N,14.3. Found: C, 44.4, H, 4.6; N, 13.8%. Yield: 75%. IR(KBr, cm−1): 1605m, 1448s [�(C�C) and �(C�N)py],752m, 816w, 1025w, 1300s, 1384s [�(NO3
−)]. FAB MS;m/z : 605 [Cd3(NO3)]+. �M (�−1 cm2 mol−1, CH3CN):124 (1:1). Colour: white.
2.16. [Co3](NO3)2 ·2H2O
Anal. Calc for C25H31N7O10Co: C, 46.3; H, 4.8; N,15.1. Found: C, 46.1, H, 4.6; N, 15.3%. Yield: 76%. IR(KBr, cm−1): 1592m, 1447m [�(C�C) and �(C�N)py],751m, 830w, 1384s [�(NO3
−)]. FAB MS; m/z : 548[Co3(NO3)]+, 486 [Co3]+. �M (�−1 cm2 mol−1,CH3CN): 145 (1:1). Colour: red.
2.17. [Ni3](NO3)2 ·3H2O
Anal. Calc for C25H31N7O10Ni: C, 45.1; H, 4.9; N,14.7. Found: C, 44.9, H, 5.0; N, 14.5%. Yield: 40%. IR(KBr, cm−1): 1607m, 1447m [�(C�C) and �(C�N)py],751m, 821w, 1384s [�(NO3
−)]. FAB MS; m/z : 549[Ni3(NO3)]+, 487 [Ni3]+. �M (�−1 cm2 mol−1,CH3CN): 146 (1:1). Colour: green.
Attempts to prepare the metal complexes of 1 wereunsuccessful.
3. Results and discussion
The macrocyclic product obtained by using p-ni-trobenzylbromide as the alkylating agent was found tobe dependent on the molar ratio of L and the alkylatingagent. The reaction between L, p-nitrobenzylbromideand Na2CO3 in a 1:1:1 molar ratio in acetonitrile,followed by catalytic reduction of the product obtained,gives rise only to the monoalkyated ligand, 1. The FABmass spectrum indicates the presence of this ligand, asit displays a peak at m/z 496 corresponding to [1+H]+
. The IR spectrum shows bands at 3457 and 3427 cm−1
assignable to the aromatic primary amine and a band at3386 cm−1 due to the secondary aromatic aminestretch. The 1H NMR spectrum run in CD3CN, confi-rms the integrity of the ligand and shows a signal at5.73 ppm (2H) assignable to the unreacted secondaryaromatic amines. This type of functionalised aminopendant-armed macrocycle should provide opportuni-ties for the synthesis of a wide range of derivatives, bya Schiff-base condensation with a corresponding car-bonyl compound, suitable as precursors for polynuclearcomplexation.
When the reaction was carried out with L, p-ni-trobenzylbromide and Na2CO3 in a 1:3:3 molar ratio inacetonitrile trialkylation took place and ligand 2 wasobtained. The FAB mass spectrum displays an intensepeak at m/z 796 corresponding to [2+H]+. The disap-pearance of the band due to N�H stretching in the IRspectrum as well as the presence of two bands due toasymmetric and symmetric stretching of the NO bondat 1500 and 1345 cm−1 confirms the incorporation ofthe pendant groups in the macrocyclic framework [13].The 1H and 13C NMR spectra of 2 were carried out inCD3CN at room temperature (Table 1) and the identityof the macrocycle was confirmed by full assignment ofthe spectra. In the 1H NMR spectrum no signal corre-sponding to a secondary aromatic amine is present,indicating that alkylation had occurred. The reductionof the nitro groups should provide the opportunity toobtain a new amino pendant-armed macrocyclic ligand,which offers exciting possibilities to construct novelsupramolecular moieties.
In contrast, when bromoacetonitrile was used as thealkylating agent the nature of the macrocyclic productwas not dependent on the molar ratio of L and only themonoalkyated ligand, 3 is obtained, alkylation of thesecondary aromatic amines did not take place. Theseresults suggest that the reaction should take placethrough a SN1 mechanism, which also justifies thedifferent reactivity of the alkylating agents. The FABmass spectrum indicates the presence of the ligand, as itdisplays a peak at m/z 430 corresponding to [3+H]+.The IR spectrum shows a band at 3392 cm−1
assignable to the secondary aromatic amine stretch.The 1H NMR spectrum, run in CD3CN, confirms the

L. Valencia et al. / Polyhedron 20 (2001) 3091–3099 3095
integrity of the ligand and shows a signal at 5.73 ppm(2H) assignable to the unreacted secondary aromaticamines.
3.1. Metal complexes of 2 and 3 ligands
Complexation reactions of ligands 2 and 3 withhydrated metal salts (2: M=Ni, Zn and Cd; X=NO3
−,ClO4
−; 3: M=Co, Ni, Zn, Cd; X=NO3−, ClO4
−) werecarried out in order to investigate the coordinationcapability of the ligands. In all cases, the microanalyti-cal data were in accord with the formation of mononu-clear complexes of the type [M2]X2·xH2O or[M3]X2·xH2O. The FAB mass spectra of the complexesshow in all cases a peak corresponding to the fragment[M2X]+ or [M3X]+ and in most spectra a peakassignable to the fragment [M2]+ or [M3]+ is alsopresent.
The presence of the water molecules is confirmed bythe appearance of an intensive broad band centredabout 3400–3300 cm−1 in the IR spectra of the com-plexes. The bands at about 1600 and 1450 cm−1 areassociated with �(C�N) and �(C�C) vibrations from thepyridine and phenyl rings which undergo a shift to-wards high frequencies on complexation, suggestinginteraction between the metal and the pyridine nitrogenatom [14]. In the complexes with the macrocyclic ligand3, the secondary amine stretches in the region 3280–3250 cm−1 which are split and/or shifted relative tothat of the free macrocycle (3392 cm−1) suggest thecoordination of this group [15].
The band due to symmetric stretching of the NObond at 1348 cm−1 is present in the spectra of all thecomplexes with 2. This band appears at the samefrequency as in the free ligand indicating that thependant arm groups are not coordinated to the metal.
For all the nitrate complexes, the presence of severalbands in the region associated with nitrate vibrationssuggests the presence of coordinated nitrate groups [16].The most intense nitrate absorptions, �(N�O) and�a(NO2), appear at 1500 and 1330 cm−1 and the magni-tude of the separation of these two highest frequencybands is indicative of a bidentate interaction with themetal ion [17].
For the perchlorate complexes absorptions at-tributable to ionic perchlorate were found approxi-mately 1100 and 625 cm−1 [18]. The lack of splitting ofthese bands suggests that the perchlorate anions are notcoordinated. This result is supported by conductancemeasurements of the complexes in acetonitrile at 25 °Cwhere molar conductance values for the perchloratecomplexes lie in the range characteristic of 2:1 [19]. Themolar conductance values for the nitrate complexes,measured under the same conditions, are in the rangecharacteristic of 1:1 electrolytes indicating the strongercoordinating capability of the nitrate anion.
The 1H NMR spectra of the Zn(II) and Cd(II)macrocyclic complexes have been recorded usingdeuterated acetonitrile as solvent. The 1H NMR spectraof the complexes show in general downfield shifts ofsignals on coordination suggesting an interaction of themetal with the available donor atoms. For 2, the Cdcomplexes exhibit well-resolved signals for all the pro-tons of the ligand, whilst the 1H NMR spectrum of[Zn2](ClO4)2·3H2O complex shows a large number ofsignals which could be indicative of the presence ofdifferent conformers in solution. Similar results havebeen obtained for the Zn and Cd complexes with L,and it could be related with ‘structural dislocation’processes between the respective complexes [20–24].
3.2. Crystal structure of 1
Single crystals of 1 suitable for X-ray analysis wereobtained by slow recrystallisation from acetonitrile.Crystal and structure refinement data are given in Table2, and the molecular structure is shown in Fig. 1. Thebond lengths and angles do not show any deviationfrom the expected values. The overall conformation ofthe macrocyclic ligand is quite folded; the aromaticrings are planar (r.m.s. for the pyridinic ring is 0.01 and0.006 and 0.0026 for the phenylic rings which arealmost perpendicular to each other, the dihedral anglebetween them being 89.2°). One of the phenyl ringsforms an angle of 88.87° with the pyridinyl head unit,whilst the dihedral angle between the second phenylring and the pyridinyl ring is 12.46°. The pendant armincorporated at the secondary aliphatic amine radiatesout away from the cavity of the macrocyclic ligand.
3.3. Crystal structure of 3
Single crystals of 3 suitable for X-ray analysis wereobtained by slow recrystallisation from acetonitrile.Crystal and structure refinement data are given in Table2, and the molecular structure is shown in Fig. 2. Thebond lengths and angles do not show any deviationfrom the expected values. The overall conformation ofthe macrocyclic ligand is quite folded; the aromaticrings are planar (r.m.s. for the pyridinic ring is 0.0083and 0.0165 and 0.1510 for the phenylic rings and thedihedral angle between them being 64.85°). One of thephenyl rings forms an angle of 46.35° with the pyridinylhead unit, whilst the dihedral angle between the secondphenyl ring and the pyridinyl ring is 31.42°. As in thecrystal structure of the ligand 1, the pendant armincorporated at the secondary aliphatic amine radiatesout away from the cavity of the macrocyclic ligand.
3.4. Crystal structure of[Cd2(NO3)][Cd(NO3)3] ·3.5CH3CN
Slow recrystallisation of the compound [Cd2](NO3)2·

L. Valencia et al. / Polyhedron 20 (2001) 3091–30993096
H2O from acetonitrile gave crystals suitable for X-raycrystal structure analysis. Crystal and structure refine-ment data are given in Table 2. The molecular structureof the complex is shown in Fig. 3. The structure foundis consistent with the formula [Cd2(NO3)][Cd(NO3)3]·3.5CH3CN, solvation having occurred on recrystallisa-tion. In the cationic complex the metal atom is seven-coordinated, with a distorted capped trigonal prismatic(1:4:2) geometry, by the pyridinic nitrogen, the nitrogenof the aliphatic bridge, one of the aromatic nitrogens,and both ether oxygens of the macrocyclic ligand. Twooxygen atoms from a bidentate nitrate group occupythe two remaining coordination sites. The macrocyclicbackbone is folded, the dihedral angle between thephenylic rings being 78.08°. The three pendant groupsradiate out away from the metal center so none of theoxygen atoms of the three p-nitrobenzyl pendant armsare coordinated to the metal ion. The bond lengthsfound in the structure are within the normal ranges forthis geometry, and the pyridinic nitrogen atoms providethe strongest bond to the cadmium ion (Cd1�N1 2.3257
A� ). A similar disposition at the metal was found in thecadmium nitrate complexes of L and the correspondingdiiminic ligand where the metal is seven coordinatedwith a distorted capped trigonal prismatic (1:4:2 geome-try) arising from coordination by the four nitrogenatoms of the ligand and three oxygen atoms frommonodentate and bidentate nitrate groups [9].
In the anionic complex [Cd(NO3)3]− (Fig. 4) the Cdatom and two of the nitrate ions have a site occupationfactor of 0.5. The cadmium is four coordinated beingbonded to four monodentate nitrate groups having adistorted tetrahedral environment. The distances be-tween the Cd and the other oxygen atoms of the nitrategroups are too long for considering them as bidentate.
4. Conclusions
Starting from the precursor ligand L, three newpendant-armed macrocyclic ligands have been obtained.We have found that in some alkylating reactions start-
Table 2Crystal data and structure refinement for 1, 3 and [Cd2(NO3)][Cd(NO3)3]·3.5CH3CN
[Cd2(NO3)][Cd(NO3)3]· 1 33.5CH3CN
Empirical formula C51H51.50Cd2N14.50O20 C30H33N5O2 C25H27N5O2
495.611294.16 429.52Formula weight298(2)Temperature (K) 150(2) 293(2)
0.710730.71073 0.71073Wavelength (A� )orthorhombicmonoclinicCrystal system monoclinic
C2/c Iba2Space group CcUnit cell dimensions
21.369(9)15.567(4)a (A� ) 21.1992(6)17.803(5) 22.880(12)18.4818(5)b (A� )
c (A� ) 9.074(7)11.127(3)30.5581(9)121.914(4)90.1690(10)� (°)
4436(5)11972.6(6) 2617.5(11)V (A� 3)8Z 4 8
Dcalc (Mg m−3) 1.436 1.258 1.286Absorption coefficient (mm−1) 0.0840.0810.615
1056 18245280F(000)0.50×0.35×0.20Crystal size (mm) 0.45×0.45×0.20 0.80×o.50×0.301.33–25.00Theta range for data collection (°) 1.92–28.28 1.30–25.14
Index ranges −25�h�1, −27�k�1,−20�h�20, −17�k�22,−25�h�19, −21�k�21,−13�l�14−31�l�36 −10�l�1
24 223 8478Reflections collected 260510 479 [Rint=0.0668]Independent reflections 5143 [Rint=0.1026] 2344 [Rint=0.0403]
Completeness (%) to theta 99.5 (25.00°) 96.1 (28.28°)empiricalAbsorption correction semi-empirical semi-empirical
Max./min. transmission 1.000, 0.632 0.9840, 0.9646full-matrix least-squares on F2 full-matrix least-squares on F2Refinement method full-matrix least-squares on F2
10 479/0/791 5143/2/334Data/restraints/parameters 2344/1/289Goodness-of-fit on F2 1.066 1.012 1.044Final R indices [I�2�(I)] R1=0.0919, wR2=0.2397 R1=0.0461, wR2=0.1192 R1=0.0838, wR2=0.2033
R1=0.1331, wR2=0.2729R indices (all data) R1=0.0721, wR2=0.1459 R1=0.1513, wR2=0.2763−3.2 (14)Absolute structure parameter −4 (6)
2.000 and −2.833 0.278 and −0.201 0.297 and −0.316Largest difference peak and hole(e A� −3)
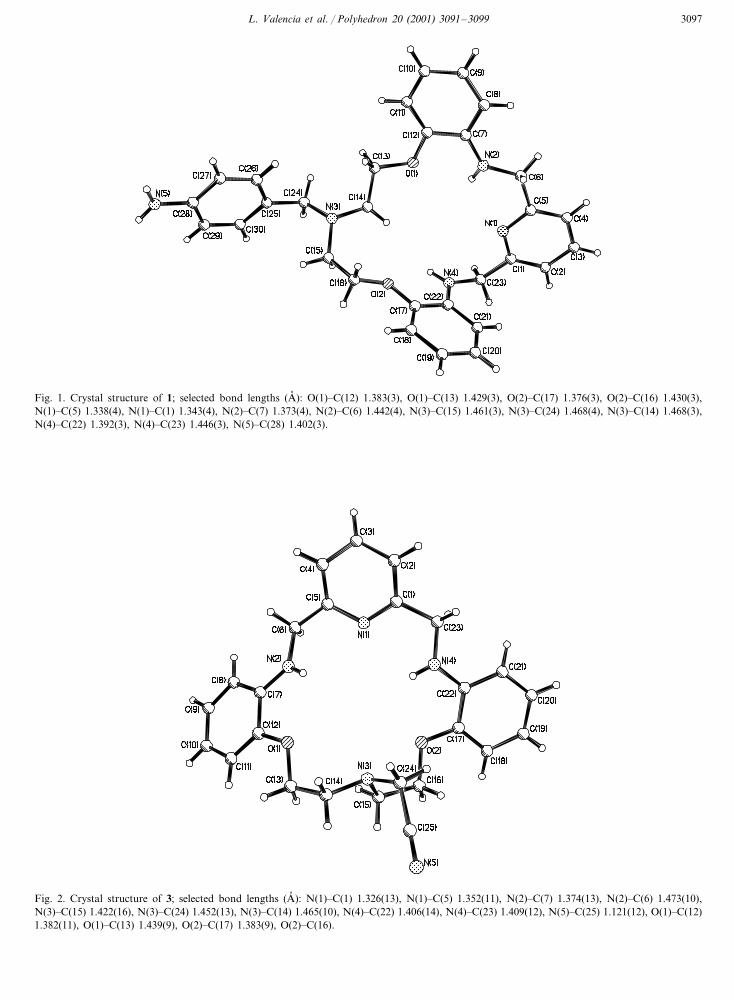
L. Valencia et al. / Polyhedron 20 (2001) 3091–3099 3097
Fig. 1. Crystal structure of 1; selected bond lengths (A� ): O(1)�C(12) 1.383(3), O(1)�C(13) 1.429(3), O(2)�C(17) 1.376(3), O(2)�C(16) 1.430(3),N(1)�C(5) 1.338(4), N(1)�C(1) 1.343(4), N(2)�C(7) 1.373(4), N(2)�C(6) 1.442(4), N(3)�C(15) 1.461(3), N(3)�C(24) 1.468(4), N(3)�C(14) 1.468(3),N(4)�C(22) 1.392(3), N(4)�C(23) 1.446(3), N(5)�C(28) 1.402(3).
Fig. 2. Crystal structure of 3; selected bond lengths (A� ): N(1)�C(1) 1.326(13), N(1)�C(5) 1.352(11), N(2)�C(7) 1.374(13), N(2)�C(6) 1.473(10),N(3)�C(15) 1.422(16), N(3)�C(24) 1.452(13), N(3)�C(14) 1.465(10), N(4)�C(22) 1.406(14), N(4)�C(23) 1.409(12), N(5)�C(25) 1.121(12), O(1)�C(12)1.382(11), O(1)�C(13) 1.439(9), O(2)�C(17) 1.383(9), O(2)�C(16).
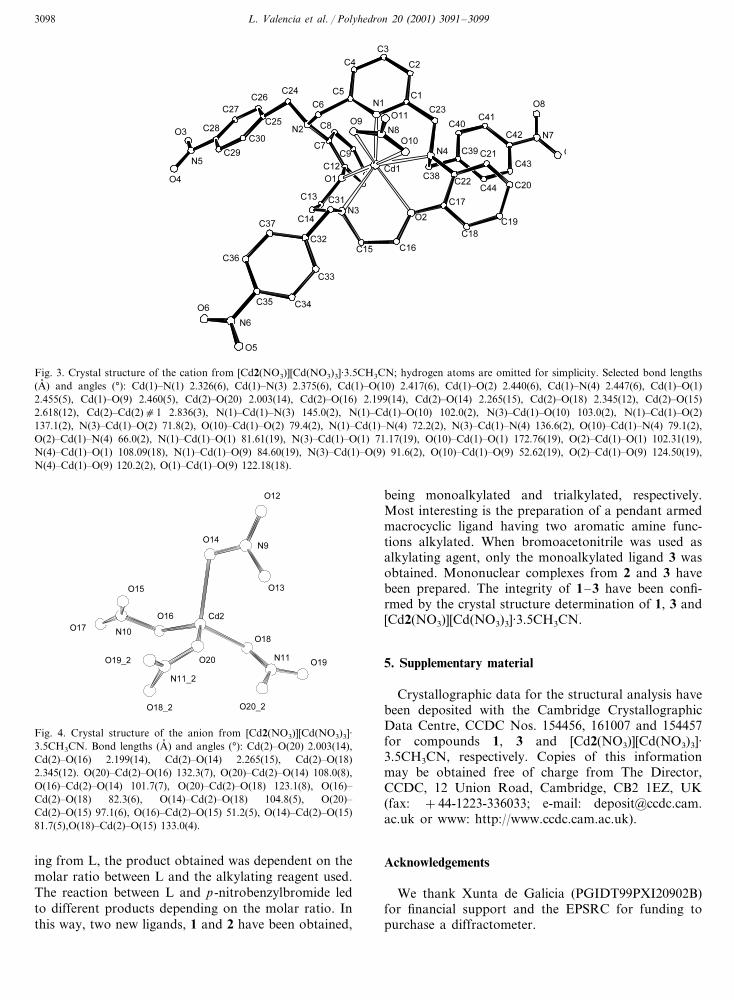
L. Valencia et al. / Polyhedron 20 (2001) 3091–30993098
Fig. 3. Crystal structure of the cation from [Cd2(NO3)][Cd(NO3)3]·3.5CH3CN; hydrogen atoms are omitted for simplicity. Selected bond lengths(A� ) and angles (°): Cd(1)�N(1) 2.326(6), Cd(1)�N(3) 2.375(6), Cd(1)�O(10) 2.417(6), Cd(1)�O(2) 2.440(6), Cd(1)�N(4) 2.447(6), Cd(1)�O(1)2.455(5), Cd(1)�O(9) 2.460(5), Cd(2)�O(20) 2.003(14), Cd(2)�O(16) 2.199(14), Cd(2)�O(14) 2.265(15), Cd(2)�O(18) 2.345(12), Cd(2)�O(15)2.618(12), Cd(2)�Cd(2)c1 2.836(3), N(1)�Cd(1)�N(3) 145.0(2), N(1)�Cd(1)�O(10) 102.0(2), N(3)�Cd(1)�O(10) 103.0(2), N(1)�Cd(1)�O(2)137.1(2), N(3)�Cd(1)�O(2) 71.8(2), O(10)�Cd(1)�O(2) 79.4(2), N(1)�Cd(1)�N(4) 72.2(2), N(3)�Cd(1)�N(4) 136.6(2), O(10)�Cd(1)�N(4) 79.1(2),O(2)�Cd(1)�N(4) 66.0(2), N(1)�Cd(1)�O(1) 81.61(19), N(3)�Cd(1)�O(1) 71.17(19), O(10)�Cd(1)�O(1) 172.76(19), O(2)�Cd(1)�O(1) 102.31(19),N(4)�Cd(1)�O(1) 108.09(18), N(1)�Cd(1)�O(9) 84.60(19), N(3)�Cd(1)�O(9) 91.6(2), O(10)�Cd(1)�O(9) 52.62(19), O(2)�Cd(1)�O(9) 124.50(19),N(4)�Cd(1)�O(9) 120.2(2), O(1)�Cd(1)�O(9) 122.18(18).
Fig. 4. Crystal structure of the anion from [Cd2(NO3)][Cd(NO3)3]·3.5CH3CN. Bond lengths (A� ) and angles (°): Cd(2)�O(20) 2.003(14),Cd(2)�O(16) 2.199(14), Cd(2)�O(14) 2.265(15), Cd(2)�O(18)2.345(12). O(20)�Cd(2)�O(16) 132.3(7), O(20)�Cd(2)�O(14) 108.0(8),O(16)�Cd(2)�O(14) 101.7(7), O(20)�Cd(2)�O(18) 123.1(8), O(16)�Cd(2)�O(18) 82.3(6), O(14)�Cd(2)�O(18) 104.8(5), O(20)�Cd(2)�O(15) 97.1(6), O(16)�Cd(2)�O(15) 51.2(5), O(14)�Cd(2)�O(15)81.7(5),O(18)�Cd(2)�O(15) 133.0(4).
being monoalkylated and trialkylated, respectively.Most interesting is the preparation of a pendant armedmacrocyclic ligand having two aromatic amine func-tions alkylated. When bromoacetonitrile was used asalkylating agent, only the monoalkylated ligand 3 wasobtained. Mononuclear complexes from 2 and 3 havebeen prepared. The integrity of 1–3 have been confi-rmed by the crystal structure determination of 1, 3 and[Cd2(NO3)][Cd(NO3)3]·3.5CH3CN.
5. Supplementary material
Crystallographic data for the structural analysis havebeen deposited with the Cambridge CrystallographicData Centre, CCDC Nos. 154456, 161007 and 154457for compounds 1, 3 and [Cd2(NO3)][Cd(NO3)3]·3.5CH3CN, respectively. Copies of this informationmay be obtained free of charge from The Director,CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK(fax: +44-1223-336033; e-mail: [email protected] or www: http://www.ccdc.cam.ac.uk).
Acknowledgements
We thank Xunta de Galicia (PGIDT99PXI20902B)for financial support and the EPSRC for funding topurchase a diffractometer.
ing from L, the product obtained was dependent on themolar ratio between L and the alkylating reagent used.The reaction between L and p-nitrobenzylbromide ledto different products depending on the molar ratio. Inthis way, two new ligands, 1 and 2 have been obtained,

L. Valencia et al. / Polyhedron 20 (2001) 3091–3099 3099
References
[1] D. Parker, J.A.G. Williams, J. Chem. Soc., Dalton Trans. (1996)3613.
[2] C.C. Bryden, C.N. Reilley, J.F. Desreux, Anal. Chem. 53 (1981)1418.
[3] V. Alexander, Chem. Rev. 95 (1995) 273.[4] H. Okawa, H. Furutachi, D.E. Fenton, Coord. Chem. Rev. 174
(1998) 51.[5] L.F. Lindoy, Pure Appl. Chem. (1997) 2179.[6] D. Jerchel, J. Heider, H. Wagner, Liebigs Ann. Chem. 613 (1958)
153.[7] E.P. Papadoupoulus, A. Jarrar, C.H. Issidorides, J. Org. Chem.
31 (1966) 615.[8] H. Adams, R. Bastida, D.E. Fenton, B.E. Mann, L. Valencia,
Eur. J. Org. Chem. (1999) 1843.[9] H. Adams, R. Bastida, D.E. Fenton, A. Macıas, S.E. Spey, L.
Valencia, J. Chem. Soc., Dalton Trans. (1999) 4131.[10] G.M. Sheldrick, SADABS, Program for Empirical Absorption
Correction of Area Detector Data, University of Gottingen,Gottingen, Germany, 1996.
[11] SHELXTL version, An Integrated System for Solving and RefiningCrystal Structures from Diffraction Data (revision 5.1), BrukerAXS LTD.
[12] G.M. Sheldrick, SHELX-97, An Integrated System for Solvingand Refining Crystal Structures from Diffraction Data, Univer-sity of Gottingen, Gottingen, Germany, 1997.
[13] K. Nakamoto, Infrared and Raman Spectra of Inorganic andCoordination Compounds, Wiley, New York, 1997, p. 1997.
[14] N.S. Gill, R.H. Nuttall, D.E. Scaife, J. Inorg. Nucl. Chem. 18(1961) 79.
[15] N.A. Bailey, D.E. Fenton, S.J. Kitchen, T.H. Lilley, M.G.Williams, P.A. Tasker, A.J. Leong, L.F. Lindoy, J. Chem. Soc.,Dalton Trans. (1991) 627.
[16] W.T. Carnall, S. Siegel, J.R. Ferraro, B. Tani, E. Gebert, Inorg.Chem. 12 (1973) 560.
[17] P. Guerriero, U. Casellato, S. Tamburini, P.A. Vigato, R. Grazi-ani, Inorg. Chim. Acta 129 (1987) 127.
[18] M.R. Rosenthal, J. Chem. Educ. 50 (1973) 331.[19] W.J. Geary, Coord. Chem. Rev. 7 (1971) 81.[20] K.R. Adam, K.P. Dancey, A.J. Leong, L.F Lindoy, B.J. Mc-
Cool, M. McPartlin, P.A. Tasker, J. Am. Chem. Soc. 110 (1988)8471.
[21] K.R. Adam, K.P. Dancey, B.A. Harrison, A.J. Leong, L.FLindoy, B.J. McCool, M. McPartlin, P.A. Tasker, J. Chem. Soc.,Chem. Commun. (1983) 1351.
[22] K.R. Adam, C.W.G. Ansell, K.P. Dancey, L.A. Drummond,A.J. Leong, L.F Lindoy, P.A. Tasker, J. Chem. Soc., Chem.Commun. (1986) 1011.
[23] K.R. Adam, S. Donnelly, A.J. Leong, L.F Lindoy, B.J. McCool,A. Bashall, M.R. Dent, B.P. Murphy, M. McPartlin, D.E.Fenton, P.A. Tasker, J. Chem. Soc., Dalton Trans. (1990) 1635.
[24] H. Adams, N.A. Bailey, D.E. Fenton, I.G. Ford, S.J. Kitchen,M.G. Williams, P.A. Tasker, A.J. Leong, L.F Lindoy, J. Chem.Soc., Dalton Trans. (1991) 1665.





























