Embed Size (px)
Citation preview

This article was downloaded by: [University Of Pittsburgh]On: 13 November 2014, At: 16:32Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Molecular Physics: An International Journal at theInterface Between Chemistry and PhysicsPublication details, including instructions for authors and subscription information:http://www.tandfonline.com/loi/tmph20
Theoretical study of low-lying excited states ofX2CY (X = F, Cl; Y = O, S) using the equation-of-motioncoupled-cluster theoryHeechol Choi & Kyoung Koo Baecka Department of Chemistry , Kangnung National University , Gangwon-do, 210-702, Koreab Department of Chemistry , Kangnung National University , Gangwon-do, 210-702, Korea E-mail:Published online: 21 Feb 2007.
To cite this article: Heechol Choi & Kyoung Koo Baeck (2005) Theoretical study of low-lying excited states of X2CY (X = F, Cl;Y = O, S) using the equation-of-motion coupled-cluster theory, Molecular Physics: An International Journal at the InterfaceBetween Chemistry and Physics, 103:15-16, 2247-2254, DOI: 10.1080/00268970500083341
To link to this article: http://dx.doi.org/10.1080/00268970500083341
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) containedin the publications on our platform. However, Taylor & Francis, our agents, and our licensors make norepresentations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of theContent. Any opinions and views expressed in this publication are the opinions and views of the authors, andare not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon andshould be independently verified with primary sources of information. Taylor and Francis shall not be liable forany losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoeveror howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use ofthe Content.
This article may be used for research, teaching, and private study purposes. Any substantial or systematicreproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in anyform to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http://www.tandfonline.com/page/terms-and-conditions

Theoretical study of low-lying excited states of X2CY (X¼F, Cl;Y¼O, S) using the equation-of-motion coupled-cluster theory
HEECHOL CHOI and KYOUNG KOO BAECK*
Department of Chemistry, Kangnung National University, Gangwon-do, 210-702, Korea
(Received 1 August 2004; in final form 12 November 2004)
The spectroscopic constants of the ground ( ~XX1A1) and low-lying excited states (~aa3A00, ~AA1A00,~BB1A0) of Cl2CS, F2CS Cl2CO and F2CO were studied using the coupled-cluster andthe equation-of-motion coupled-cluster singles and doubles (CCSD/EOM-CCSD) method.A comparison of the calculated results for Cl2CS with available experimental values showsthat the results from the CCSD/EOM-CCSD method are quite reliable. Many otherspectroscopic constants of the excited states of the four molecules are provided in this work.It is shown that the fluorescence corresponding to the S2!S1/S2! S0 transition is alsoexpected in F2CS and F2CO as well as in Cl2CS, an already known violation of Kasha’s rule.
Keywords: Equation-of-motion coupled-cluster theory; Low-lying excited states
1. Introduction
The chemistry of carbonyl and thiocarbonyl halideshas been the subject of much research because of theirapplications in the chemical industry, such as the use ofphosgene as an intermediate in polyurethane productionand as a reagent in the synthesis of pharmaceuticals [1].The study of these compounds also plays an importantrole in the development of our understanding of thephotochemistry and photophysics of polyatomic mole-cules [2, 3]. The development of better laser methods forpumping and probing target molecules with complexityand short lifetimes, and supersonic expansion techni-ques for synthesizing and characterizing van der Waalsmolecules, clusters, and other unstable species hascontributed greatly to our understanding of the photo-chemistry and photophysics of carbonyl and thiocarbo-nyl systems, as summarized by Maciejewski and Steera decade ago [4]. Among these moleules, thiophosgene,Cl2CS, has attracted special interest because the S2!S0fluorescence violates Kasha’s rule [5].A further detailed understanding of the photo-
chemistry and photophysics of these systems can befacilitated with the help of theoretical calculations onthe structures of the ground and excited electronic statesof the molecules. Although Kapur, Steer, and Mezeycalculated the potential energy surfaces and equilibriumgeometries for the ground and the lowest triplet statesof Cl2CS, F2CS, and some thiocarbonyl halides using
ab initio SCF MO methods [6–8], theoretical studieson the halogen-substituted carbonyl and thiocarbonylmolecules using post-HF methods are quite limitedexcept for a recent CASSCF calculation for Cl2CS [9].The large number of electrons in the molecules seemsto be the main reason for the lack of post-HF results.The difficulties in analysing recent work with increas-ingly sophisticated experimental techniques, however,demands more detailed and reliable results fromtheoretical studies with higher levels of theory [10, 11].
As part of theoretical work on these molecules, thegeometrical, vibrational and energetic properties ofthe ground singlet state, the lowest triplet state, andthe low-lying excited singlet states of X2CY (X¼F, Cl;Y¼O, S) were studied using the equation-of-motioncoupled-cluster singles and doubles (EOM-CCSD)method. The calculated results are compared withavailable experimental values, and several spectroscopicproperties of the low-lying excited states of X2CYwill be given in the present work for the first time. Thesimilarities and differences among the low-lying excitedstates of the molecules are also discussed briefly.
2. Calculations
The coupled-cluster and equation-of-motion coupled-cluster (CC/EOM-CC) theory is now established as oneof the best theoretical methods for the ground andexcited electronic states [12]. For ground states, coupled-cluster singles and doubles augmented with the non-iterative inclusion of triple excitations (CCSD(T)) [13]*Corresponding author. Email: [email protected]
Molecular Physics, Vol. 103, No. 15–16, 10–20 August 2005, 2247–2254
Molecular PhysicsISSN 0026–8976 print/ISSN 1362–3028 online # 2005 Taylor & Francis Group Ltd
http://www.tandf.co.uk/journalsDOI: 10.1080/00268970500083341
Dow
nloa
ded
by [
Uni
vers
ity O
f Pi
ttsbu
rgh]
at 1
6:32
13
Nov
embe
r 20
14

is well established as a reliable and computationallyfeasible method for small- and medium-sized molecules.Although EOM-CC methods for excited states thatinclude effects of triple excitations non-iteratively havebeen developed [14], these methods have not generallybeen as successful as CCSD(T). The cost of the tripleexcitation part of these methods is at least twice the costof that part of CCSD(T). In addition, the excitationsstudied in this work have predominantly one-electroncharacter, as indicated by the fact that the largestmagnitude of the approximate-excitation level of all thestates is 1.13, so effects of triple excitations are notexpected to be significant. Therefore, the CCSD [15] andEOM-CCSD [16] methods have been employed in thepresent work.The analytic gradient methods [17, 18] for CCSD and
EOM-CCSD energies offer an efficient way to studyspectroscopic properties, but the molecular orbitals(MOs) corresponding to the 1s atomic orbital (AO) ofcarbon, oxygen, and fluorine atoms as well as the 1s, 2s,and 2p AOs of sulfur and chlorine atoms are droppedin the post-HF steps to reduce the computation timefurther without a deterioration in the final results. As aresult, the four, eight, 12, and 16 innermost MOs aredropped in the calculations for F2CO, F2CS, Cl2CO,and Cl2CS, respectively. The effects of omitting theMOs on the spectroscopic constants have already beenstudied for the ground states [19] and excited states[20, 21] of simple molecules. Previous results have shownthat the effects of the dropped MOs on bond lengths,angles, excitation energies, and harmonic frequenciesare about 0.3 pm, 0.5�, 0.03 eV, and 3%, respectively.Effects of similar magnitude are expected in the resultsof the present work on X2CY molecules. Test calcula-tions on the effects for the Cl2CS molecule alsoconfirmed almost the same trend and magnitude forthe effects, but the detailed results are not included herefor brevity.All calculations in this work were performed using the
ACES-II suite of programs [22], and the 6-31þG(d)basis set with six Cartesian d functions. The use of theCCSD method for the spectroscopic properties of theground electronic states as well as the use of the EOM-CCSD for the single-point excitation energies are nowwell established, and need no further description. Theapplication of the EOM-CCSD method for the search ofa stationary structure and for the computation of thevibrational frequencies of an excited state, however,needs special care and experience, especially whenthe second derivatives of the excited state energy arecomputed by numerical differentiation of analytic first-order derivatives. The proper initial guess for the targetroot in excited state calculations is an essential require-ment for the successful application of the EOM-CCSD
method, especially for the harmonic frequency of thenon-totally symmetric vibrational modes of an excitedstate. The results of the EOM-CCSD computation forthe single-point excitation energy provided us with theinformation for the proper initial guess [23]. In thepresent work, all the calculated energies of each excitedstate during the numerical differentiation of the analyticderivatives were carefully examined to eliminate anydoubtful results.
The lowest triplet states of the present work werecalculated using both the UHF and ROHF molecularorbitals. The CCSD results for both types of referenceMOs are identical within the numerical accuracy givenin the tables of this work.
3. Results and discussion
The calculated results for Cl2CS will be discussed firstbecause much more experimental work has beenconducted for this molecule than the others. The moreplentiful experimental data for the excited states ofCl2CS should be compared with our theoretical resultsprior to the predictive production of spectroscopicconstants for the other molecules. A short discussionof the similarities and differences among the excitedstates of the four molecules is also included.
3.1. Cl2CS
The calculated stationary geometries and vibrationalfrequencies of the ground electronic state ( ~XX1A1) as wellas the three lowest excited states (~aa3A00, ~AA1A00, ~BB1A0) ofthiophosgene (Cl2CS) are collected and compared withavailable experimental values in table 1. The optimizedbond lengths and angles of the ground state ~XX1A1 are ingood agreement with the experimental values obtainedby Carpenter et al. [24], which is also supported by thedata of Nakata et al. [25]. The discrepancies in the bondlengths and angles for the ground state are about 1.0 pmand 0.3�, respectively. The calculated harmonic frequen-cies are also in good accordance with the experimentaldata extracted from laser fluorescence excitation spec-troscopy (LFES) by Clouthier and Moule [26], witha maximum deviation of 6% from the correspondingexperimental frequency. The descriptions of the vibra-tional modes are �1 (C–S stretch), �2 (sym. C–Cl stretch),�3 (CCl2 scissor), �4 (out-of-plane bend), �5 (asym. C–Clstretch), and �6 (CCl2 rock).
Clouthier and Moule have also determined all of thevibrational frequencies of the first excited triplet andsinglet states, except for �6 of ~aa3A00 and �5 of ~aa3A00 and~AA1A00 [26]. All of our computed harmonic frequencies ofthe states are in fairly good agreement with the availableexperimental values. The calculated harmonic frequency
2248 H. Choi and K. K. Baeck
Dow
nloa
ded
by [
Uni
vers
ity O
f Pi
ttsbu
rgh]
at 1
6:32
13
Nov
embe
r 20
14
of the out-of-plane bending mode of the excited states,!4, is compared with the 0–2 spacing of the experimental�4 because the potential energy surface along the modehas two symmetric double wells.Both the ~XX ! ~aa and ~XX ! ~AA transitions correspond
to electronic excitation from a non-bonding orbitalnðb2Þ to an anti-bonding orbital p�ðb1Þ. The similarityis reflected in the very similar bond lengths, angles, andvibrational frequencies of the ~aa and ~AA states. As a resultof electronic excitation from a non-bonding MO to ananti-bonding MO, which is localized between the carbonand sulphur atoms, the bond length between the twoatoms is increased, whereas the bond length betweenthe C and Cl atoms is slightly shortened. The overallmolecular structure becomes non-planar. The calculatedout-of-plane angles of the ~aa and ~AA states, 32.4� and26.7�, are also very close to the experimentally suggestedvalues, 32� and 27.2�, respectively. The computed
inversion barrier along the out-of-plane bending mode�4 of the ~aa state (819 cm�1) is also comparable to theexperimentally suggested value (726 cm�1). The com-puted barrier for the ~AA state (377 cm�1) is muchsmaller than that suggested experimentally (620 cm�1).The old experimental analysis seems to be questionablein this case, as is the case for the ~BB1A0 state discussedbelow.
The calculated adiabatic excitation energies (Te) of the~aa3A00 and ~AA1A00 states are 1.97 and 2.46 eV, whereasthe corresponding experimental values are 2.17 and2.32 eV, respectively [27, 28]. Considering the size ofthe basis sets used in this work, the deviations of thecalculated adiabatic excitation energies are quite reason-able. The results of calculations on the triplet stateusing the UHF orbitals do not show any sign of spincontamination. To be sure, we carried out all thecalculations for geometry optimization and vibrational
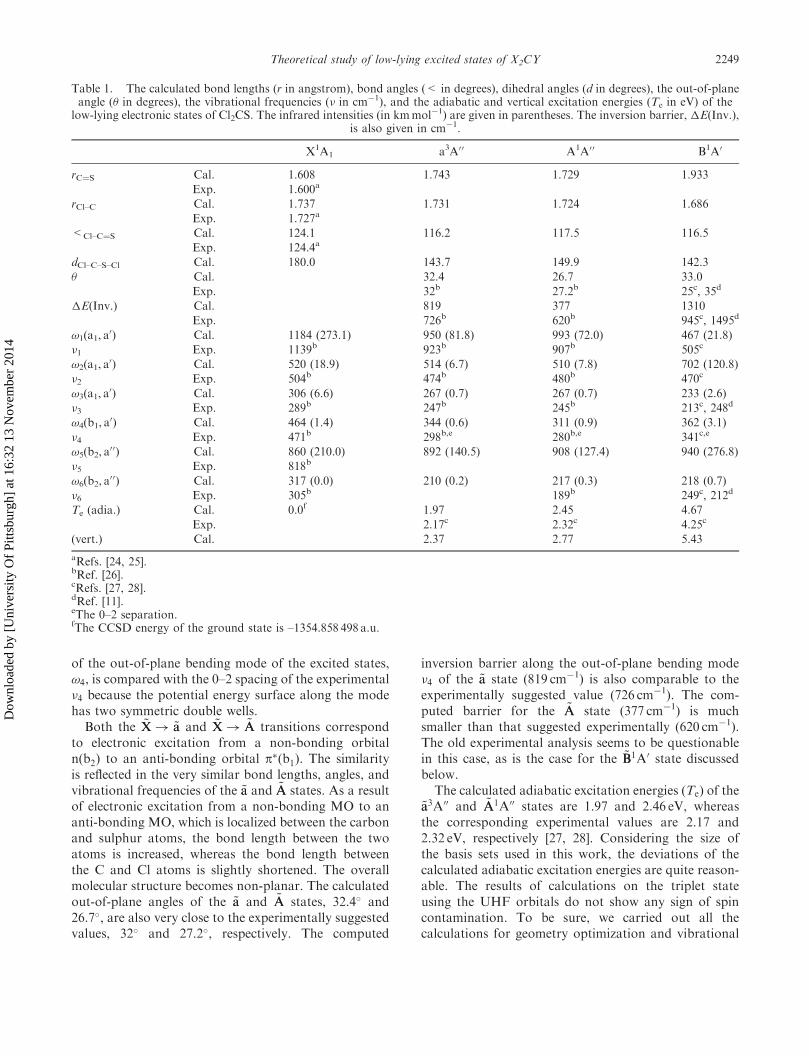
Table 1. The calculated bond lengths (r in angstrom), bond angles (< in degrees), dihedral angles (d in degrees), the out-of-planeangle (� in degrees), the vibrational frequencies (� in cm�1), and the adiabatic and vertical excitation energies (Te in eV) of the
low-lying electronic states of Cl2CS. The infrared intensities (in kmmol�1) are given in parentheses. The inversion barrier, �E(Inv.),is also given in cm�1.
X1A1 a3A0 0 A1A0 0 B1A0
rC¼S Cal. 1.608 1.743 1.729 1.933
Exp. 1.600a
rCl–C Cal. 1.737 1.731 1.724 1.686
Exp. 1.727a
<Cl–C¼S Cal. 124.1 116.2 117.5 116.5
Exp. 124.4a
dCl–C–S–Cl Cal. 180.0 143.7 149.9 142.3
� Cal. 32.4 26.7 33.0
Exp. 32b 27.2b 25c, 35d
�E(Inv.) Cal. 819 377 1310
Exp. 726b 620b 945c, 1495d
!1(a1, a0) Cal. 1184 (273.1) 950 (81.8) 993 (72.0) 467 (21.8)
�1 Exp. 1139b 923b 907b 505c
!2(a1, a0) Cal. 520 (18.9) 514 (6.7) 510 (7.8) 702 (120.8)
�2 Exp. 504b 474b 480b 470c
!3(a1, a0) Cal. 306 (6.6) 267 (0.7) 267 (0.7) 233 (2.6)
�3 Exp. 289b 247b 245b 213c, 248d
!4(b1, a0) Cal. 464 (1.4) 344 (0.6) 311 (0.9) 362 (3.1)
�4 Exp. 471b 298b,e 280b,e 341c,e
!5(b2, a00) Cal. 860 (210.0) 892 (140.5) 908 (127.4) 940 (276.8)
�5 Exp. 818b
!6(b2, a00) Cal. 317 (0.0) 210 (0.2) 217 (0.3) 218 (0.7)
�6 Exp. 305b 189b 249c, 212d
Te (adia.) Cal. 0.0f 1.97 2.45 4.67
Exp. 2.17c 2.32c 4.25c
(vert.) Cal. 2.37 2.77 5.43
aRefs. [24, 25].bRef. [26].cRefs. [27, 28].dRef. [11].eThe 0–2 separation.fThe CCSD energy of the ground state is –1354.858 498 a.u.
Theoretical study of low-lying excited states of X2CY 2249
Dow
nloa
ded
by [
Uni
vers
ity O
f Pi
ttsbu
rgh]
at 1
6:32
13
Nov
embe
r 20
14

frequencies again using ROHF orbitals. The bondlengths, angles, and vibrational frequencies remainunchanged within the numerical precision given intable 1.The second excited singlet state, ~BB1A0, has been the
subject of numerous studies since its fluorescenceactivity violates Kasha’s rule [5]. The vertical excitationenergy of the S1 ( ~AA
1A00) state, 2.77 eV, is calculated to bemuch smaller than the adiabatic excitation energy ofthe S2 ( ~BB1A0) state, 4.25 eV. This provides one simpleexplanation of why thiophosgene can violate Kasha’srule. The ~XX ! ~BB spectrum, however, turns out to beextremely difficult to analyse due to the large change ofgeometry in the transition. The assignment of the bandorigin and other vibronic bands has long been thesubject of controversy [29]. After much effort over morethan two decades, the assignment was established by thework of Simard et al., who reconciled the discrepanciesamong previous experimental studies [28]. Very recently,however, Fujiwara et al. [11] have reversed the assign-ment of �3 and �6, as shown in the last column of table 1.The ~XX ! ~BB transition corresponds to the nðb2Þ !
p�ðb1Þ electronic excitation. The formal bond orderbetween C and S atoms changes from two to one, whichis manifested in the large increase of the bond distancebetween the C and S atoms. The change is also reflectedin the noticeable decrease of the harmonic frequency ofthe C–S stretching mode from 900 to 1200 cm�1 in the ~XX,~aa and ~AA states to s500 cm�1 in the ~BB state. On the otherhand, the C–Cl bond length is decreased, and the C–Clstretch frequency is increased by about 200 cm�1.The computed frequencies for �1 (C–S stretch) and �4(CCl2 wagging, out-of-plane) are in good agreementwith the experimental values, and our calculated valuesfor �3 (sym. Cl–C–Cl bend) and �6 (antisym. Cl–C–Clbend) also support the reversed assignment of Fujiwaraet al. [11], as shown in the last column of table 1. Ourcalculated values for the out-of-plane angle and theinversion barrier along the �4 mode (33.0� and1310 cm�1) are also much closer to the recent experi-mental values (35� and 1495 cm�1) [11] than to theprevious values (25� and 945 cm�1) [27, 28].The only noticeable deviation is between the calcu-
lated !2 (702 cm�1) and the experimental �2 (470 cm
�1).The deviation of the calculated adiabatic excitationenergy, Te (4.67 eV), from the recently established experi-mental value, 4.25 eV [28, 29], is also a little larger thanexpected. The 6-31þG(d) basis set may not be largeenough in this case, but our test calculations with largerbasis sets do not change the computed values of !2 andTe much. Although recent work using the CASSCF(6/6)/VDZ method generated vibrational frequenciesvery close to the previous experimental values [28], thecalculation overestimated the excitation energy by
about 0.9 eV. The interpretation and assignment of pre-vious experimental results may need to be re-examined.
The bond lengths and angles as well as the vibrationalfrequencies �5 have never been explicitly reported forthe ~aa3A00, ~AA1A00, and ~BB1A0 states in any previousexperimental and theoretical work, and we expect thevalues first reported here will be useful in futureexperimental and theoretical work.
3.2. F2CS
Experimental work on F2CS, Cl2CO, and F2CO isquite limited. The vibrational properties of the groundstates of the three molecules were determined by Hopperet al. [30, 31], and the geometry of the ground stateof F2CS was reported by Careless et al. [32]. With theexception of these data, the numbers in Herzberg’smonograph [33] are the only available experimentaldata for F2CS, Cl2CO, and F2CO to the best of ourknowledge.
The optimized geometry of the ground state of F2CSgiven in table 2 is in good agreement with theexperimental value of Careless et al. [32], with differ-ences of less than 0.02 for the bond lengths. Thecalculated vibrational frequencies of the ~XX states arealso in good accordance with the experimental resultsof Hopper et al. [30, 31] with an average error of about2%, which is about the same magnitude as in the abovecase of Cl2CS. The absence of experimental valuesfor the exited states of F2CS prevents any further com-parison, but we expect our calculated results to be asreliable as in the case of Cl2CS.
When the values in table 2 are compared with thecorresponding values in table 1, it is easy to see that theexcited states and possible transitions among the statesof F2CS are almost identical to those of Cl2CS. Thecharacter of the electronic transitions leading to excitedstates and the consequent change in geometricalparameters and vibrational frequencies almost parallelthose observed in the case of Cl2CS. The energy gapbetween the S1 ( ~AA1A00) and S2 ( ~BB1A0) states is largeenough and comparable to that in Cl2CS. The verticalTe of S1 is also much smaller than the adiabatic Te of S2.The molecular structures of S1 and S2 are similar exceptfor the C¼S bond length. Although the existence andthe role of a second triplet state, T2, are not exploredhere, all of these facts imply the possibility of theexperimental observation of florescence correspondingto both the S2! S1 and S2! S0 transitions, which havenot yet been studied.
One noticeable change from Cl2CS to F2CS is thedifference between the vertical and the adiabaticexcitation energies of each state, the lowest triplet stateT1 and the first excited singlet state S1. The differences
2250 H. Choi and K. K. Baeck
Dow
nloa
ded
by [
Uni
vers
ity O
f Pi
ttsbu
rgh]
at 1
6:32
13
Nov
embe
r 20
14
for the S1 and T1 states are only 0.3–0.4 eV in Cl2CS,whereas they are 0.9–1.0 eV in F2CS. This reflectsthe fact that the potential wells of the S1 and T1 statesare steeper in F2CS than in Cl2CS.
3.3. Cl2CO
The results for Cl2CO are given in table 3, where thesecond excited singlet state, ~BB1A0, is not includedbecause the state turns out to be dissociative, as will bediscussed below. The calculated geometry and theharmonic frequencies of the ground state are also ingood agreement with the experimental geometry [33]and the vibrational frequencies [30, 31], respectively.The changes in geometry and vibrational frequenciesin both the ~XX ! ~aa and ~XX ! ~AA transitions are verysimilar to those observed in the F2CS and Cl2COsystems. The geometry is distorted from the planarstructure and the C¼O bond length is increased due tothe nðb2Þ ! p�ðb1Þ electronic excitation.Two experimental vibrational frequencies, 581 and
430 cm�1, are reported for the ~AA state in Herzberg’smonograph [33], who assigned them to �4 and �6,respectively, from a comparison with the correspondingfrequency of the ground state. The calculated values for�4 and �6 are 458 and 294 cm�1, whereas the computed
frequency of �2 is 458 cm�1. Based on the accuracy and
reliability of our calculated results, as demonstratedin the above case of the ~AA state of Cl2CS, we suggesta re-assignment of the experimental frequencies to �2and �4, as shown in table 3.
The stationary structure of the second excited singletstate, ~BB, was not obtained in spite of repeated attemptsstarting from many different initial guesses of the geom-etry. The geometry optimization results in the simul-taneous dissociation of C–Cl bonds, while the dihedralangle, dCl–C–O–Cl, increases to near 90�. The statecorresponds to the dissociation channel of CO and twochlorine radicals. These facts are in accordance with theexperimental observation that the absorption spectrumof the ~XX ! ~BB transition is a continuum with a peakmaximum at 8.02 eV (64 700 cm�1) [33], which is com-parable to the calculated vertical Te of the state, 6.97 eV.
3.4. F2CO
The optimized geometry and vibrational properties ofthe ground state of F2CO, shown in table 4, also showgood agreement with experimental values, as expected.No experimental values are available for the spectro-scopic constants of the excited states. The changes inbond lengths and vibrational frequencies of the ~XX ! ~aa
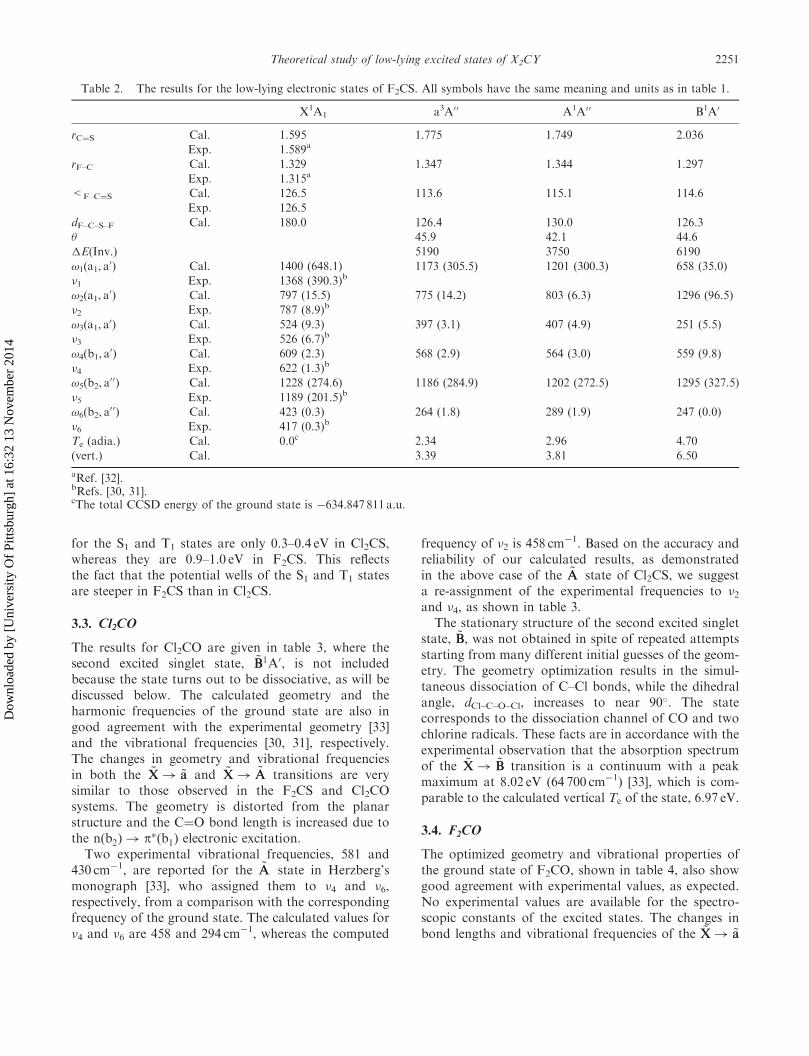
Table 2. The results for the low-lying electronic states of F2CS. All symbols have the same meaning and units as in table 1.
X1A1 a3A00 A1A0 0 B1A0
rC¼S Cal. 1.595 1.775 1.749 2.036
Exp. 1.589a
rF–C Cal. 1.329 1.347 1.344 1.297
Exp. 1.315a
<F–C¼S Cal. 126.5 113.6 115.1 114.6
Exp. 126.5
dF–C–S–F Cal. 180.0 126.4 130.0 126.3
� 45.9 42.1 44.6
�E(Inv.) 5190 3750 6190
!1(a1, a0) Cal. 1400 (648.1) 1173 (305.5) 1201 (300.3) 658 (35.0)
�1 Exp. 1368 (390.3)b
!2(a1, a0) Cal. 797 (15.5) 775 (14.2) 803 (6.3) 1296 (96.5)
�2 Exp. 787 (8.9)b
!3(a1, a0) Cal. 524 (9.3) 397 (3.1) 407 (4.9) 251 (5.5)
�3 Exp. 526 (6.7)b
!4(b1, a0) Cal. 609 (2.3) 568 (2.9) 564 (3.0) 559 (9.8)
�4 Exp. 622 (1.3)b
!5(b2, a00) Cal. 1228 (274.6) 1186 (284.9) 1202 (272.5) 1295 (327.5)
�5 Exp. 1189 (201.5)b
!6(b2, a00) Cal. 423 (0.3) 264 (1.8) 289 (1.9) 247 (0.0)
�6 Exp. 417 (0.3)b
Te (adia.) Cal. 0.0c 2.34 2.96 4.70
(vert.) Cal. 3.39 3.81 6.50
aRef. [32].bRefs. [30, 31].cThe total CCSD energy of the ground state is �634.847 811 a.u.
Theoretical study of low-lying excited states of X2CY 2251
Dow
nloa
ded
by [
Uni
vers
ity O
f Pi
ttsbu
rgh]
at 1
6:32
13
Nov
embe
r 20
14
and ~XX ! ~AA transitions show that the transitionscorrespond to the same transitions of the previousthree molecules.The second excited singlet state, ~BB1A0, turns out to
have a stationary geometry, in contrast to the Cl2COcase. Although the vertical Te of S1 ( ~AA1A00) is slightlylarger than the adiabatic Te of S2 ( ~BB1A0), the energeticseparation between the S1 and the S2 states, s2.2 eV,is large enough. The molecular structures of S1 and S2are very similar, except for the CO bond length. Thesefacts are very similar to those for the F2CS electronicstates. We therefore expect the possibility of observingfluorescence corresponding to both the S2!S0 andS2! S1 transitions in F2CO.The differences between the vertical and the adia-
batic excitation energies of each state are very largefor the excited states of Cl2CO and F2CO, whichimplies steep potential wells for these states. Largeinversion barriers �E(Inv.) (10 551, 8894, and1092 cm�1) along the out-of-plane bending mode arealso observed. The consequences of these facts for theoverall dynamics of the excited states are openquestions, which will be explored later.
4. Conclusions
The spectroscopic constants of the ground states( ~XX1A1), the lowest triplet states (~aa3A00), and two excitedsinglet states ( ~AA1A00 and ~BB1A0) of Cl2CS, F2CS, Cl2CO,and F2CO have been studied using the CCSD/EOM-CCSD theoretical methods with the 6-31þG(d )basis set. Several new spectroscopic constants areprovided in the present work. On the basis of theaccuracy and reliability demonstrated by the compar-ison of the calculated spectroscopic constants ofthiophosgene with the available corresponding experi-mental values, we expect the other calculated values tobe sufficiently reliable.
Comparisons between the calculated results for F2CSand F2CO with those for Cl2CS, however, suggest thepossibility of observing fluorescence corresponding tothe S2! S1/S2!S0 transition, not only in Cl2CS, butalso in F2CS and F2CO, which will be new casesviolating Kasha’s rule.
Computations using the CCSD/EOMCCSD theorywith the 6-31þG(d) basis sets may not be accurateenough for the study of the ~BB1A0 state. The present
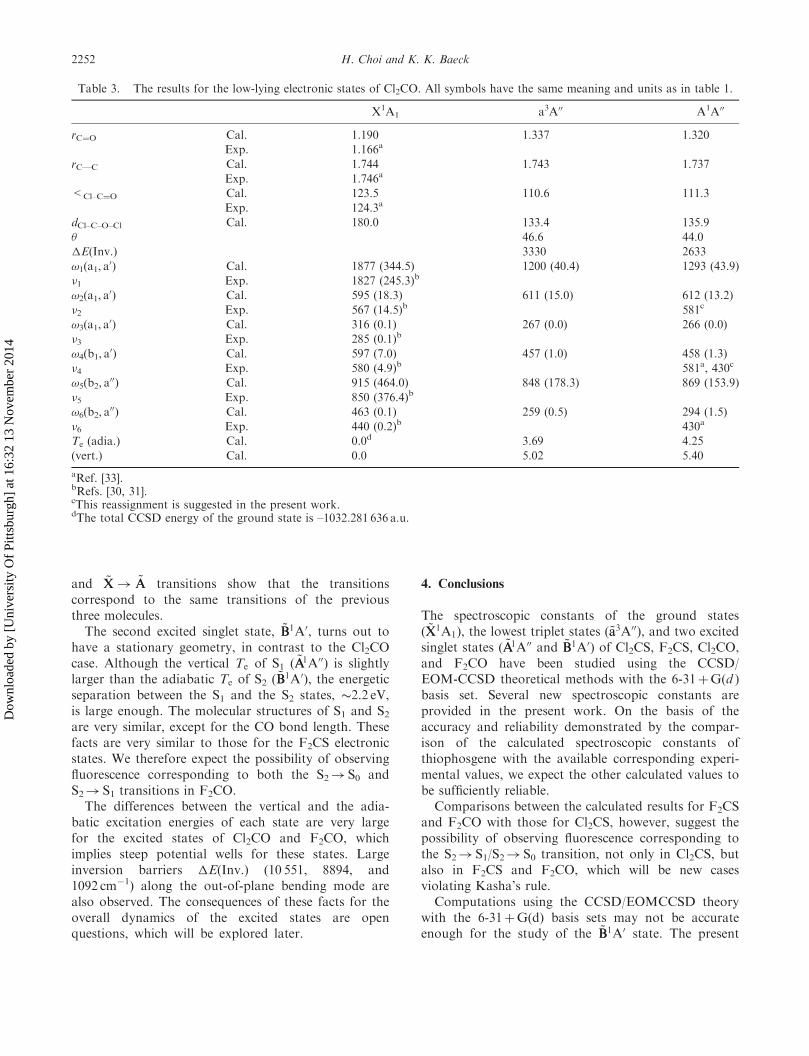
Table 3. The results for the low-lying electronic states of Cl2CO. All symbols have the same meaning and units as in table 1.
X1A1 a3A00 A1A00
rC¼O Cal. 1.190 1.337 1.320
Exp. 1.166a
rC—C Cal. 1.744 1.743 1.737
Exp. 1.746a
<Cl–C¼O Cal. 123.5 110.6 111.3
Exp. 124.3a
dCl–C–O–Cl Cal. 180.0 133.4 135.9
� 46.6 44.0
�E(Inv.) 3330 2633
!1(a1, a0) Cal. 1877 (344.5) 1200 (40.4) 1293 (43.9)
�1 Exp. 1827 (245.3)b
!2(a1, a0) Cal. 595 (18.3) 611 (15.0) 612 (13.2)
�2 Exp. 567 (14.5)b 581c
!3(a1, a0) Cal. 316 (0.1) 267 (0.0) 266 (0.0)
�3 Exp. 285 (0.1)b
!4(b1, a0) Cal. 597 (7.0) 457 (1.0) 458 (1.3)
�4 Exp. 580 (4.9)b 581a, 430c
!5(b2, a00) Cal. 915 (464.0) 848 (178.3) 869 (153.9)
�5 Exp. 850 (376.4)b
!6(b2, a00) Cal. 463 (0.1) 259 (0.5) 294 (1.5)
�6 Exp. 440 (0.2)b 430a
Te (adia.) Cal. 0.0d 3.69 4.25
(vert.) Cal. 0.0 5.02 5.40
aRef. [33].bRefs. [30, 31].cThis reassignment is suggested in the present work.dThe total CCSD energy of the ground state is –1032.281 636 a.u.
2252 H. Choi and K. K. Baeck
Dow
nloa
ded
by [
Uni
vers
ity O
f Pi
ttsbu
rgh]
at 1
6:32
13
Nov
embe
r 20
14
prediction of fluorescence from S2 is also notcertain because the presence and the role of the secondtriplet state were not studied in detail. Other aspects ofthe dynamics of the excited state were also not explored.The current results, however, are expected to besufficiently useful for future theoretical and experi-mental works on these molecules. We expect ourresults will initiate further study of the possibility offluorescence corresponding to the S2!S1/S2! S0transitions and the detailed dynamics of the excitedstates.
Acknowledgements
This paper is dedicated to Professor Rodney J. Bartletton the occasion of his 60th birthday. His pioneering rolein the development of coupled-cluster and equation-of-motion coupled-cluster theories and the contributionof all of his coworkers to the development of theACES-II program are the essential basis of the presentwork. This work was supported by Korea Scienceand Engineering Foundation grant R05-2004-000-10995-0.
References
[1] T.A. Ryan, C. Ryan, E.A. Seddon, K.R. Seddon.Phosgene and Related Carbonyl Halides, Elsevier,Amsterdam (1996).
[2] S.J. Formosinho, L.G. Arnaut. Adv. Photochem., 16, 67(1991).
[3] P. Wagner, B.-S. Park. Org. Photochem., 11, 227 (1991).[4] A. Maciejewski, R.P. Steer. Chem. Rev., 93, 67 (1993).[5] M. Kasha. Discuss. Faraday Soc., 9, 14 (1950).[6] A. Kapur, R.P. Steer, P.G. Mezey. J. chem. Phys., 69, 968
(1978).[7] A. Kapur, R.P. Steer, P.G. Mezey. J. chem. Phys., 70, 745
(1979).[8] S. Simard, A.E. Bruno, P.G. Mezey, R.P. Steer. Chem.
Phys., 103, 75 (1986).[9] B. Strickler, M. Gruebele. Chem. Phys. Lett., 349, 137
(2001).[10] A.M. Warsylewicz, K.J. Falk, R.P. Steer. Chem. Phys.
Lett., 352, 48 (2002).[11] T. Fujiwara, E.C. Lim, D.C. Moule. J. chem. Phys., 119,
7741 (2003).[12] R.J. Bartlett, J.F. Stanton. Rev. Comput. Chem., 5, 65
(1994).[13] K. Raghavachari, G.W. Trucks, J.A. Pople, M. Head-
Gordon. Chem. Phys. Lett., 157, 479 (1989).[14] J.D. Watts, R.J. Bartlett. Chem. Phys. Lett., 258, 581
(1996); O. Christiansen, H. Koch, P. Jørgensen. J. chem.Phys., 105, 1451 (1996).
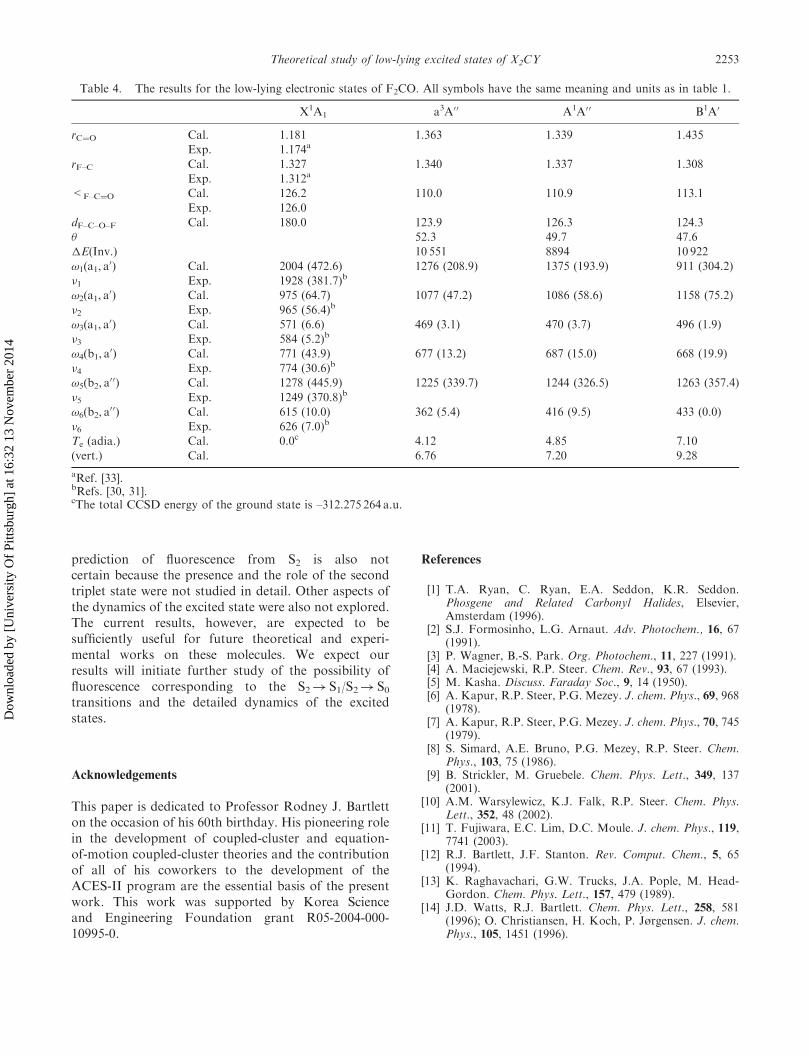
Table 4. The results for the low-lying electronic states of F2CO. All symbols have the same meaning and units as in table 1.
X1A1 a3A00 A1A0 0 B1A0
rC¼O Cal. 1.181 1.363 1.339 1.435
Exp. 1.174a
rF–C Cal. 1.327 1.340 1.337 1.308
Exp. 1.312a
<F–C¼O Cal. 126.2 110.0 110.9 113.1
Exp. 126.0
dF–C–O–F Cal. 180.0 123.9 126.3 124.3
� 52.3 49.7 47.6
�E(Inv.) 10 551 8894 10 922
!1(a1, a0) Cal. 2004 (472.6) 1276 (208.9) 1375 (193.9) 911 (304.2)
�1 Exp. 1928 (381.7)b
!2(a1, a0) Cal. 975 (64.7) 1077 (47.2) 1086 (58.6) 1158 (75.2)
�2 Exp. 965 (56.4)b
!3(a1, a0) Cal. 571 (6.6) 469 (3.1) 470 (3.7) 496 (1.9)
�3 Exp. 584 (5.2)b
!4(b1, a0) Cal. 771 (43.9) 677 (13.2) 687 (15.0) 668 (19.9)
�4 Exp. 774 (30.6)b
!5(b2, a00) Cal. 1278 (445.9) 1225 (339.7) 1244 (326.5) 1263 (357.4)
�5 Exp. 1249 (370.8)b
!6(b2, a00) Cal. 615 (10.0) 362 (5.4) 416 (9.5) 433 (0.0)
�6 Exp. 626 (7.0)b
Te (adia.) Cal. 0.0c 4.12 4.85 7.10
(vert.) Cal. 6.76 7.20 9.28
aRef. [33].bRefs. [30, 31].cThe total CCSD energy of the ground state is –312.275 264 a.u.
Theoretical study of low-lying excited states of X2CY 2253
Dow
nloa
ded
by [
Uni
vers
ity O
f Pi
ttsbu
rgh]
at 1
6:32
13
Nov
embe
r 20
14

[15] G.D. Purvis, III, R.J. Bartlett. J. chem. Phys., 76, 1910(1982).
[16] J.F. Stanton, R.J. Bartlett. J. chem. Phys., 98, 7029(1993).
[17] J. Gauss, J.F. Stanton, R.J. Bartlett. J. chem. Phys., 95,2623 (1991).
[18] J.F. Stanton, J. Gauss. J. chem. Phys., 100, 4695 (1994).[19] K.K. Baeck, J.D. Watts, R.J. Bartlett. J. chem. Phys., 107,
3853 (1997).[20] K.K. Baeck. J. chem. Phys., 112, 1 (2000).[21] K.K. Baeck, S.I. Jeon. Bull. Korean Chem. Soc., 21, 720
(2000).[22] ACES-II is a program product of the Quantum Theory
Project, University of Florida. Authors: J.F. Stanton,J. Gauss, J.D. Watts, M. Nooijen, N. Oliphant,S.A. Perera, P.G. Szalay,W.J. Lauderdale, S.R. Gwaltney,S. Beck, A. Balkova, D.E. Berhholdt, K.K. Baeck,P. Rozyczko, H. Sekino, C. Huber, R.J. Bartlett. Integralpackages included are VMOL (J. Almlof, P.R. Taylor),VPROPS (P. Taylor), ABACUS (T. Helgaker,H.J.Aa. Jensen, P. Jørgensen, J. Olsen, P.R. Taylor).
[23] Refer to the ‘InputManual forACES-II’ for further details.
[24] D.R. Carpenter, D.F. Rimmer, J.G. Smith, D.H. Whiffen.J. chem. Soc. Faraday Trans., 271, 1752 (1975).
[25] M. Nakata, T. Fukuyama, K. Kuchitsu. J. molec. Struct.,81, 121 (1982).
[26] D.J. Clouthier, D.C. Moule. J. molec. Spectrosc., 87, 471(1981).
[27] D.C. Moule, C.R. Subramainam. J. molec. Spectrosc., 48,336 (1973).
[28] B. Simard, V.J. MacKenzie, P.A. Hackett. Can. J. chem.,72, 745 (1994).
[29] M. Ludwiczak, D.R. Latimer, R.P. Steer. J. molec.Spectrosc., 147, 414 (1991).
[30] M.J. Hopper, J.W. Russell, J. Overend. J. chem. Phys., 48,3765 (1968).
[31] M.J. Hopper, J.W. Russell, J. Overend. Spectrochim.Acta A, 28, 1215 (1972).
[32] A.J. Careless, H.W. Kroto, B.M. Landsberg. Chem.Phys., 1, 371 (1973).
[33] G. Herzberg. Molecular Spectra and Molecular StructureIII. Electronic Spectra and Electronic Structure ofPolyatomic Molecules, Van Nostrand, Princeton, NJ(1966).
2254 H. Choi and K. K. Baeck
Dow
nloa
ded
by [
Uni
vers
ity O
f Pi
ttsbu
rgh]
at 1
6:32
13
Nov
embe
r 20
14
























![[DPnF] Lying Lips](https://img.pdfslide.net/doc/110x75/577cdae41a28ab9e78a6cf58/dpnf-lying-lips.jpg)




