Embed Size (px)
Citation preview

Theoretical Study of the Structuresand Properties of Cyclic Nitramines:Tetranitrotetraazadecalin (TNAD)and Its Isomers
LING QIU, HE MING XIAO, XUE HAI JU, XUE DONG GONGDepartment of Chemistry, Nanjing University of Science and Technology,Nanjing 210094, People’s Republic of China
Received 8 November 2004; accepted 16 March 2005Published online 18 May 2005 in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/qua.20674
ABSTRACT: Density Functional Theory (DFT) was employed to study thegeometries, electronic structures, infrared vibrational spectra, and thermodynamicproperties of seven isomeric cyclic nitramines of C6H10N8O8 (i.e., TNAD and its sixisomers) at the B3LYP/6-31G* level of theory. The experimental results available forTNAD were used to determine the reliability of the DFT method for generatingstructural and IR spectroscopic values for these molecular systems. The relativestabilities of the conformers were evaluated from the energy differences of thestructures. Detonation properties of various conformers were evaluated using theKamlet-Jacobs equations, and it was found that all the calculated results are comparableto the available experimental data. In addition, the calculated results demonstrate thatall title compounds can be used as excellent propellant ingredients. © 2005 WileyPeriodicals, Inc. Int J Quantum Chem 105: 48–56, 2005
Key words: cyclic nitramines; density functional theory; structure; properties
Introduction
N itramines have long been important in tech-nological applications. This group is still a
source of explosives or propellants that possesspredominantly high energy contents [1–3]. An im-portant point for selection and exploitation of this
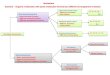
kind of compound is the study of both their phys-ical and chemical properties. At present, the saidtype of attractive nitramines involves a series ofcyclic polynitramines, such as trans-1,4,5,8-tetrani-tro-1,4,5,8-tetraazadecalin (trans-1458TNAD) [4–5],trans-1,3,5,7-tetranitro-1,3,5,7-tetraazadecalin-(trans-1357TNAD) [6], cis-1,3,5,7-tetranitro-1,3,5,7-tetraazadecalin(cis-1357TNAD) [6], and other pos-sible structural isomers (see Fig. 1). Obviously, allthe compounds shown in Figure 1 (1A–4A) areCorrespondence to: H. M. Xiao; e-mail: [email protected]
International Journal of Quantum Chemistry, Vol 105, 48–56 (2005)© 2005 Wiley Periodicals, Inc.

structural isomers of C6H10N8O8. Furthermore,among these compounds 1B, 2B, and 3B in Figure 1are stereoisomers of 1A, 2A, and 3A, respectively.The reported syntheses of TNAD [4–6] are the re-sult of efforts in this direction to develop energeticmaterials with improved crystal density and highstored energy to meet superior performance re-quirements.
To date, there have been many investigations on theseven cyclic nitramines, especially on trans-1458TNAD,such as their syntheses, sensitivities, thermal decompo-sition, lattice energies, heats of fusion and sublimation,IR spectra, densities, and detonation properties, and soon [4–20]. However, to date there are no systematicstudies on these isomeric compounds and limited exper-imental geometries of these compounds are available,especially those in gaseous phase. Because of the impor-tance of the geometrical information for further under-standing of the properties, it is necessary to perform atheoretical investigation on them, for example, using abinitio molecular orbital methods. The calculated resultsfrom ab initio methods are quite accurate and reliable atcertain levels. Therefore, they have been widely em-ployed in investigations. However, high-level ab initiocalculations are not always practicable. Therefore, Den-sity Functional Theory (DFT) [21, 22], an alternative ap-proach which requires relatively fewer computer re-sources and is able to produce reliable geometries,vibrational frequencies, and thermochemical properties,is used more and more widely. Our recent studies [23–26] on nitramines using the hybrid B3LYP [27, 28]
functional with the 6-31G* basis set [29] showedthat the results obtained at the B3LYP/6-31G* levelare satisfactory. Thus, here we have chosen theB3LYP/6-31G* method to study the following com-pounds: (1A) trans-1,4,5,8-tetranitro-1,4,5,8-tet-raazadecalin, (1B)cis-1,4,5,8-tetranitro-1,4,5,8-tetraazadecalin,(2A)trans-1,3,5,7-tetranitro-1,3,5,7-tetraazadecalin,(2B)cis-1,3,5,7-tetranitro-1,3,5,7-tetraazadecalin,(3A)(R*,R*)-1,1�,3,3�-tetranitro-4,4�-biimidazolidine,(3B)(R*,S*)-1,1�,3,3�-tetranitro-4,4�-biimidazolidine,and(4A)1,3,7,9-tetranitro-1,3,7,9-tetraazaspiro[4.5]decane (see Fig. 1). Among theseenergetic compounds, 1B has not been synthesizedyet. This article is a continuation of our computer-aided work on evaluating and searching for highenergy density materials (HEDM).
Materials and Methods
Seven cyclic nitramines, i.e., TNAD and its sixstable isomers, obtained from ChemBats 3D soft-ware, were fully optimized at the B3LYP level [27,28] with the 6-31G* basis set [29] using the Bernygradient optimization method. Harmonic vibra-tional analyses were performed subsequently oneach optimized structure at the same level. Accord-ing to the previous studies, the computed frequen-cies were scaled using an average factor 0.96 tocorrect the systematic errors for B3LYP calculations[30]. Based on the principle of statistical thermody-namics [31], the standard thermodynamic functionsof title compounds ranging from 200–800K, includ-ing standard molar heat capacity (Cp,m
o ), entropy(Sm
o ), and enthalpy (Hmo ), were derived from the
scaled frequencies by using a self-compiled pro-gram.
Density, detonation velocity, and detonationpressure are the important characters of energeticmaterials, which would be one of the importantcriteria for evaluating the performances of energeticmaterials. Currently, estimation of detonationproperties for C, H, O, and N containing explosivesrelies on the empirical Kamlet-Jacobs equations[32]:
D � 1.01�NM1/ 2Q1/ 2�1/ 2�1 � 1.30�� (1)
P � 1.558�02NM1/ 2Q1/ 2 (2)
Where each term in Eqs. (1) and (2) is defined asfollows: D, detonation velocity [km/s]; P, detona-
FIGURE 1. Illustration of the molecular structures ofthe seven title compounds.
TNAD AND ITS ISOMERS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 49

tion pressure [GPa]; N, moles of gaseous detonationproducts per gram of explosive; M, average molec-ular weight of gaseous products; Q, chemical en-ergy of detonation [cal/g]; and �, density of explo-sive [g/cm3]. Here, the molecular densities of thetitle compounds were obtained by dividing the av-erage molecular volume by the molecular weight.The average molecular volume of each moleculewas yielded from the statistical average value of 40molar volumes of each molecule. The molar volumeof each molecule was calculated by the Monte-Carlo method in the Gaussian98 program package[33], which is defined as the volume inside a con-tour of 0.001e/Bohr3 density [34]. Detonation energies(Q) of the title compounds were evaluated using theformula derived from the compositional conditiondefined in the Kamlet-Jacobs equations and the cor-responding heats of formation, which were obtaineddirectly from the semiempirical PM3 method.
All of these calculations were carried out withthe Gaussian98 program package on a Pentium-IVpersonal computer in our laboratory using the de-fault Gaussian convergence criteria.
Results and Discussion
This section presents and discusses the results,including geometric features, Mulliken bondingpopulations, energies, IR spectra, thermodynamicproperties, and detonation properties of seven
structural isomers of C6H10N8O8 obtained in thetheoretical study.
GEOMETRIES
According to the harmonic vibrational analyses,each structure of the title compounds is confirmedto be the true minimum on the potential energysurface by the absence of imaginary frequency. Theoptimized geometries and molecular symmetry ofseven title compounds obtained at the B3LYP/6-31G* level are presented in Table I. Of all the titlecompounds, (3A) (R*, R*)-1,1�,3,3�-tetranitro-4,4�-bi-imidazolidine has the highest symmetry and be-longs to the D2 group.
Investigating the optimized geometries in TableI, the longest N–N bond in (1A) trans-1458TNAD is1.442 Å, and average N–N bond length is 1.412 Å.This is in a reasonable agreement with the referencedata 1.417 Å and 1.414 Å, respectively [35]. Therelative errors are 1.73% and –0.14%, respectively.Variations (i.e., the differences between the maxi-mum and minimum values) of the calculated re-sults for the C–C, C–N, and N–N bond lengths andN–C–N, C–N–C, and C–N–N angles are muchlarger than those of the other parameters, indicatingthat these geometrical parameters are more sensi-tive to the environment or molecular structures. Forexample, from the present calculations the varia-tions of the bond lengths C–C, C–N, N–N, N–O,and C–H are 0.054, 0.059, 0.108, 0.017, and 0.014 Å,
TABLE I ______________________________________________________________________________________________Geometrical parameters of title compounds obtained at the B3LYP/6-31G* level.a,b
Compd. 1A 1B 2A 2B 3A 3B 4A
Sym.c C1 C2 C1 C1 D2 C1 C1
C–C 1.514–1.568 1.527–1.548 1.531–1.555 1.543–1.555 1.541, 1.544 1.545–1.550 1.546–1.565C–N 1.441–1.493 1.449–1.475 1.439–1.498 1.444–1.471 1.458–1.483 1.456–1.486 1.452–1.497N–N 1.398–1.442 1.398, 1.408 1.408–1.418 1.395–1.420 1.393, 1.429 1.374–1.421 1.382–1.482N–O 1.219–1.230 1.224–1.227 1.220–1.229 1.221–1.228 1.222–1.229 1.221–1.231 1.213–1.230C–H 1.088–1.098 1.088–1.095 1.084–1.098 1.085–1.097 1.087–1.095 1.085–1.095 1.084–1.097H–C–H 106.6–109.2 107.7–107.9 109.0–110.1 107.3–109.7 108.8, 109.7 107.9–109.7 107.4–110.1N–C–N 115.9, 119.6 112.5 111.4, 111.5 109.1–112.5 104.1 102.5, 103.0 102.7, 112.0C–N–C 110.9–122.3 120.0–121.7 112.0–120.6 115.4–119.5 107.7, 111.6 108.1–113.8 104.7–117.3C–N–N 110.2–122.4 115.1–118.4 110.4–118.2 115.9–117.8 115.4–117.1 114.2–119.4 106.4–118.9N–N–O 115.0–118.8 114.9–118.4 116.2–117.7 115.7–117.6 116.1–116.8 115.5–117.4 113.7–119.1O–N–O 126.2–127.3 126.7–126.8 126.1–126.6 126.5–126.8 126.9, 127.0 126.6–127.1 126.6–127.2N–N–O–O 177.0–178.6 177.2–180.0 176.4–179.1 176.3–178.4 174.8, 176.4 174.7–178.6 175.0–180.0
a Bond length in angstroms, angle in degrees.b 1A, 1B, 2A, 2B, 3A, 3B, and 4A represent the compounds in Figure 1.c Sym. denotes the molecular symmetry.
QIU ET AL.
50 VOL. 105, NO. 1

respectively; and those of the angles H–C–H,N–C–N, C–N–C, C–N–N, N–N–O, O–N–O, andN–N–O–O are 3.5, 17.1, 17.6, 16.0, 5.4, 1.2, and 5.3°,respectively. Comparison of the calculated resultsof title compounds shows that the placements of thenitramino groups far away from each other havelarger influences on the geometries than the effectof the stereochemistry at the ring junction.
MULLIKEN BONDING POPULATIONS
Mulliken bonding populations of all title com-pounds are shown in Table II. Although the Mul-liken populations fail to give reliable characteriza-tion of bond strength, it obviously demonstratesthat the N–NO2 bonds are the weakest since theyhave much smaller populations than all the otherbonds at an identical computational level. Thismeans that the N–NO2 bond may be the initial bondin pyrolysis and explosion according to the princi-ple of the smallest bond order (PSBO) [3], which isin good agreement with the conclusion drawn fromour previous studies on the pyrolysis mechanismsand impact sensitivities of nitramines [3, 23–26]. Inaddition, the calculated results also show that Mul-liken populations of some C–N bonds in 1A aresmaller than those of N–NO2 bonds, which seemsto imply that it is possible for the title compoundsto be pyrolyzed by the C–N bond breaking in thering. This also indicates that 1A might have largerstrain in rings than other isomers.
ENERGY
Table III reports the total energies, zero pointenergies (ZPE), the total energies including ZPEcorrection, the energies of the highest occupied andlowest unoccupied molecular orbitals (EHOMO andELUMO, respectively), and their gap (�E) obtained atthe B3LYP/6-31G* level. From Table III we can seethat the uncorrected and corrected total energiesare both in the order of 1B � 1A, 2B � 2A, 3B � 3A,and 3B � 2B/3A � 2A � 1B � 4A � 1A. Thecalculated total energies of 1A, 1B, 2A, and 2B seemto reflect the amount of steric strain in the mole-cules as estimated by the inspection of molecularmodels of the compounds. The trans-decalin 1Aand 2A are highly strained because of a very unfa-vorable interaction between the nitro groups andthe methylene groups, while these interactions areabsent in the cis-decalin 1B and 2B. Furthermore,since there exists larger hindrance among nitrogroups and larger interactions between the nitro T
AB
LEII
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
____
___
Mul
liken
bo
ndin
gp
op
ulat
ions
of
titl
eco
mp
oun
ds
calc
ulat
edat
the
B3L
YP
/6-3
1G*
leve
l.
1A1B
2A2B
3A3B
4A
C–C
0.23
66–0
.317
30.
2902
–0.3
133
0.29
79–0
.331
30.
2996
–0.3
249
0.31
32,0
.337
70.
2894
–0.3
341
0.28
88–0
.324
1C
–N0.
1710
–0.2
539
0.18
67–0
.253
20.
1961
–0.2
451
0.19
92–0
.245
80.
2096
–0.2
491
0.19
97–0
.250
70.
1870
–0.2
559
N–N
0.17
25–0
.184
10.
1724
,0.1
823
0.16
63–0
.180
60.
1714
–0.1
806
0.17
77,0
.178
30.
1735
–0.1
807
0.17
52–0
.182
7N
–O0.
3078
–0.3
395
0.32
44–0
.338
20.
3081
–0.3
331
0.32
41–0
.334
00.
3231
–0.3
309
0.30
35–0
.333
30.
2920
–0.3
339
C–H
0.35
92–0
.383
60.
3627
–0.3
785
0.35
27–0
.386
30.
3513
–0.3
878
0.35
34–0
.385
00.
3482
–0.3
772
0.35
08–0
.388
1
TNAD AND ITS ISOMERS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 51

and the methylene groups in 1A than in the otherisomers, the energy difference between 1A and 1Bis much bigger than those between 2A and 2B, 3Aand 3B. The total energy of 3B is relatively thesmallest, which implies that 3B may be the moststable conformer in the seven isomers ofC6H10N8O8.
Inspecting the energies of the highest occupiedmolecular orbital (HOMO) and the lowest unoccu-pied molecular orbital (LUMO), that is, EHOMO andELUMO in Table III, shows that the EHOMO andELUMO of structures 1A–4A are close. The energygaps (�E) between the EHOMO and ELUMO in all thestructures are close, and the magnitudes are 0.21–0.22 au.
IR SPECTRUM
The simulated infrared (IR) spectra of title com-pounds (1A–4A) are shown in Figure 2, where theintensity is plotted against the harmonic vibrationalfrequencies (the scale factor is 0.96 [30]). For thecomplexity of vibrational modes, it is difficult toassign all bands, so we only analyzed some mainvibrational modes that would facilitate assignmentof the observed peaks. For compound 1A, it isfound that N–NO2 characteristic asymmetricstretching vibrations at 1607 cm�1 have the highestintensity and the N–NO2 symmetric stretchingmodes at 1260–1283 cm�1 also have strong intensi-ties. The characteristic vibrations in the range of2925–3062 cm�1 are assigned to the C–H asymmet-ric and symmetric stretching vibrational modes.The band below 1246 cm�1 is the fingerprint regionof 1A, which is composed of the stretching of rings,wagging and rocking of C–H, and the out-of-planevibration of nitro groups. This can be used to iden-
tify the isomers. The corresponding experimentalresults of �as(NNO2), �s(NNO2), �(C–H), and thefingerprint region for 1A are 1555, 1190–1290,2950–3040, and below 1120 cm�1, respectively [5,19]. It is obvious that the calculated results agreewell with the experimental data, although there area few discrepancies. The existence of discrepanciesis due to the intermolecular interactions in crystal,but such interactions are not present in the DFTcalculations applied here. Considering the solid-state effects they are rather compatible as a whole.That is to say, the calculated vibrational frequenciesand intensities can only be validated by comparisonwith the corresponding experimental values mea-sured under low-pressure, gas phase conditions.Thus, it is reasonable to believe that the B3LYP/6-31G* method could generate reliable IR spectra forcyclic nitramines.
For compound 1B, it has characteristic vibrationsof �as(NNO2), �s(NNO2), and �(C–H) in the range1605–1619, 1260–1276, and 2953–3063 cm�1, respec-tively. From the calculated results and Figure 2, wecan see that the IR behavior of compounds 1A and1B is similar. Similarly, the IR characteristics ofcompounds 2A and 2B are also close to each other.For example, 2A has the characteristic vibrations of�as(NNO2), �s(NNO2), and �(C–H) in the range1602–1626, 1254–1284, and 2933–3113 cm�1, and thecorresponding vibrational modes of 2B are locatedin the range 1599–1629, 1255–1290, and 2937–3103cm�1, respectively. The characteristic vibrations of�as(NNO2), �s(NNO2), and �(C–H) for 3A are in therange 1597–1626, 1246–1291, and 2965–3083 cm�1,while for 3B are 1588–1619, 1257–1275, and 2964–3100 cm�1, respectively. The peaks at 1250–1292,1588–1649, and 2938–3109 cm�1 of 4A are assignedto the N–NO2 symmetric and asymmetric stretch-
TABLE III _____________________________________________________________________________________________Energies of seven C6H10N8O8 isomers calculated at the B3LYP/6-31G* level.a
Compd. E ZPE EZPE EHOMO ELUMO �E
1A �3344703.50 599.24 �3344104.26 �0.29271 �0.08542 0.207291B �3344801.74 599.49 �3344202.25 �0.29629 �0.08129 0.215002A �3344803.98 600.70 �3344203.28 �0.29534 �0.09123 0.204112B �3344807.71 599.28 �3344208.43 �0.29710 �0.08714 0.209963A �3344806.14 597.54 �3344208.60 �0.30587 �0.08468 0.221193B �3344809.17 598.32 �3344210.85 �0.29820 �0.08257 0.215634A �3344770.47 598.33 �3344172.14 �0.30118 �0.09474 0.20644
a E, ZPE, and EZPE are the total energy, zero-point energy, and ZPE corrected total energy (kJ/mol), respectively. EHOMO, ELUMO, and�E are the energies of the highest occupied and lowest unoccupied molecular orbitals, and their gap (au), respectively.
QIU ET AL.
52 VOL. 105, NO. 1

FIGURE 2. IR spectra of the seven title compounds.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 53

ing, and C–H stretching vibrational modes, respec-tively.
THERMODYNAMIC PROPERTIES
The computed thermodynamic properties (Cp,mo ,
Smo , and Hm
o ) of the title compounds in the temper-ature range 200–800K are listed in Table IV. Asthere are no corresponding experimental values, wecannot make any comparison. We hope that ourresults may be used as reference data for futureinvestigations.
Table IV shows that the thermodynamic proper-ties of the seven compounds are similar to eachother at a given temperature, due to the fact thatthey are isomers with similar molecular and elec-tronic structures. That is to say, the stereochemistryhas little effect on the title compounds’ thermody-namic properties. Meanwhile, it is evident that allthe thermodynamic functions increase with increas-ing temperature. This phenomenon is due to thefact that the main contributions to the thermody-namic functions are from the translations and rota-tions of molecules at a lower temperature. But at ahigher temperature, the vibrations contribute moreto the thermodynamic functions, which results in
the increase of thermodynamic functions. In addi-tion, the increase extent in both Cp,m
o and Smo de-
crease with increasing temperature, but increasesfor Hm
o .
DETONATION PROPERTIES
All the calculated and available experimentaldensities (�), detonation velocities (D), and detona-tion pressures (P) of the seven C6H10N8O8 isomersare presented in Table V. In addition, the availablemeasured melting points (mp) of the title com-pounds [5, 6] are also listed in Table V. We can seethat all the calculated results of the title compoundsin Table V agree reasonably well with the availableexperimental values. The relative errors in the esti-mate of densities for compounds 1A, 2A, 2B, 3A,and 4A are –1.69%, 3.31%, –1.14%, 0.58%, and6.08%, respectively. This suggests that the resultscalculated at the B3LYP/6-31G* level are reliableexcept for 4A, with somewhat larger relative error.The evaluated detonation velocity and detonationpressure of 1A compare favorably with the experi-mental values [5] with relative errors of 1.65% and2.30%, respectively. All these further confirm thereliability of the calculation method. Furthermore,
TABLE IV _____________________________________________________________________________________________Thermodynamic properties of C6H10N8O8 isomers at different temperatures.
T (K) 200.0 298.2 400.0 500.0 600.0 700.0 800.0
1A Cp,mo (J � mol�1 � K�1) 221.07 303.47 383.78 450.07 503.20 545.47 579.41
Smo (J � mol�1 � K�1) 494.91 598.52 699.18 792.19 879.13 959.99 1035.12
Hmo (kJ � mol�1) 26.34 52.08 87.16 128.97 176.74 229.25 285.55
1B Cp,mo (J � mol�1 � K�1) 220.98 303.35 383.92 450.34 503.48 545.69 579.54
Smo (J � mol�1 � K�1) 498.31 601.86 702.52 795.58 882.57 963.47 1038.63
Hmo (kJ � mol�1) 26.58 52.30 87.39 129.22 177.01 229.55 285.87
2A Cp,mo (J � mol�1 � K�1) 217.58 302.17 383.73 450.50 503.71 545.89 579.67
Smo (J � mol�1 � K�1) 500.97 603.62 704.10 797.16 884.19 965.12 1040.30
Hmo (kJ � mol�1) 26.21 51.72 86.74 128.57 176.39 228.95 285.29
2B Cp,mo (J � mol�1 � K�1) 220.03 303.12 384.03 450.55 503.69 545.86 579.66
Smo (J � mol�1 � K�1) 522.59 625.90 726.55 819.64 906.67 987.60 1062.78
Hmo (kJ � mol�1) 27.04 52.71 87.80 129.64 177.46 230.01 286.35
3A Cp,mo (J � mol�1 � K�1) 220.22 303.18 384.65 451.48 504.68 546.78 580.48
Smo (J � mol�1 � K�1) 529.45 632.76 733.51 826.78 913.99 995.07 1070.36
Hmo (kJ � mol�1) 27.59 53.26 88.38 130.30 178.22 230.87 287.29
3B Cp,mo (J � mol�1 � K�1) 220.81 303.01 384.17 450.98 504.24 546.42 580.19
Smo (J � mol�1 � K�1) 510.65 614.04 714.68 807.84 894.96 975.98 1051.23
Hmo (kJ � mol�1) 27.13 52.82 87.90 129.78 177.64 230.26 286.65
4A Cp,mo (J � mol�1 � K�1) 218.80 304.42 386.26 452.81 505.67 547.50 580.99
Smo (J � mol�1 � K�1) 498.89 602.24 703.43 797.04 884.46 965.67 1041.04
Hmo (kJ � mol�1) 26.18 51.86 87.14 129.21 177.24 229.98 286.46
QIU ET AL.
54 VOL. 105, NO. 1

we can see that the calculated values of �, D, and Pfor the seven isomers are close to each other, whichis in accord with the previous study that the calcu-lated crystal density varies slightly between theisomeric structures [36]. This may be due to the factthat they have similar molecular and electronicstructures, and the stereochemistry has little effecton the isomers’ detonation properties.
Inspecting the melting point in Table V, it isevident that the title compounds have considerablethermal stability with high melting points. Further-more, the reported impact sensitivities (height fromwhich a falling 2.5 kg weight will cause detonation50% of the time) of 1A, 2A, and 4A are 35, 65, and50 cm, respectively, while the corresponding valuesfor RDX and �-HMX are 25–28 and 18–26 cm, re-spectively [5, 37]. This indicates that the three iso-meric cyclic nitramines are less sensitive to impactthan either RDX or HMX. This property of titlecompounds coupled with their high melting points,high energy, and low cost of the starting materialdemonstrate that they can be used as excellent pro-pellant ingredients.
Conclusions
According to the DFT calculations on the sevenisomeric cyclic polynitramines of C6H10N8O8 at theB3LYP/6-31G* level, we can draw the followingconclusions.
1. The optimized geometries of the seven con-formers (1A, 1B, 2A, 2B, 3A, 3B, and 4A) agreereasonably well with the available reference data.Variations of the C–C, C–N, and N–N bond lengthsand N–C–N, C–N–C, and C–N–N angles in the titlecompounds are much larger than those of the otherparameters, indicating that these geometrical pa-rameters are more sensitive to the environment ormolecular structures. The placements of the nitra-mino groups far away from each other have larger
influences on the geometries than the effect of thestereochemistry at the ring junction.
2. Comparisons of Mulliken bonding popula-tions show that the N–NO2 bond may be the initialbond in pyrolysis and explosion. Meanwhile, it ispossible that the pyrolysis initiation reaction takesplace by the C–N bond breaking in the ring.
3. The differences in the total energies of theseven isomeric structures are small. The overallorder of stability for the seven optimized moleculesis 3B � 2B/3A � 2A � 1B � 4A � 1A according tothe B3LYP calculated results.
4. For each of the studied cyclic nitramines, IRspectra were computed with the B3LYP/6-31G*method, which has been proven to generate reliablefrequencies. The IR behavior of stereoisomers 1Aand 1B, 2A and 2B, 3A and 3B are similar.
5. The thermodynamic properties of the titlecompounds are very similar due to the fact thatthey are isomers with similar molecular and elec-tronic structures. All the thermodynamic functionsincrease with increasing temperature because thevibrations contribute more to the thermodynamicfunctions at higher temperatures than at lower tem-peratures.
6. The calculated detonation properties of thetitle compounds are comparable to the availableexperimental data, and close to each other, for theyhave similar molecular and electronic structures.Their low impact sensitivities coupled with their highmelting points and high energies indicate that theycan be used as excellent propellant ingredients.
References
1. Borman, S. Chem Eng News, 1994, 72, 18.2. Olah, G. A.; Squire, D. R. Chemistry of Energetic Materials;
Academic Press: San Diego, CA, 1991.3. Xiao, H. M. The Molecular Orbital Theory of Nitro Com-
pounds; National Defense Industry Press: Beijing, 1993.
TABLE V ______________________________________________________________________________________________Density (�), detonation velocity (D), and detonation pressure (P) of C6H10N8O8 isomers.a
1A 1B 2A 2B 3A 3B 4A
� (g � cm�3) 1.77 (1.80) 1.79 1.81 (1.75) 1.76 (1.78) 1.72 (1.71) 1.77 1.81 (1.70)D (km � s�1) 8.50 (8.36) 8.51 8.59 8.41 8.24 8.40 8.56P (GPa) 31.73 (31) 32.05 32.05 30.94 29.31 30.99 32.62mp (°C) 232–234 — 251–252 236–237 213–214 198–199 —
a Data in parentheses are the experimental results taken from Refs. [5, 6].
TNAD AND ITS ISOMERS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 55

4. Willer, R. L. Synthesis of a new explosive compound, trans-1,4,5,8-tetranitro-1,4,5,8-tetraaza-decalin. Report, NWC-TP-6303, SBI-AD-E450017; Order No. AD-A116666, 16 pp. Avail.NTIS From: Gov. Rep. Announce. Index (U.S.) 1982, 82(22),4738.
5. Willer, R. L. Propell Explos Pyrotech 1983, 8, 65.
6. Willer, R. L. J Org Chem 1984, 49, 5150.
7. Edwards, A.; Webb, G. A. J Chem Soc Perkin Trans I 1977,18, 1989.
8. Willer, R. L. NWC; TM4703, Naval Weapons Center, ChinaLake, CA, January, 1982, 10 pp.
9. Ritter, H.; Licht, H. H.; Michaud, P. DE 19501377 A1 25 July1996, 3 pp.
10. Politzer, P.; Murray, J. S.; Lane, P.; Sjoberg, P.; Adolph, H. G.Chem Phys Lett 1991, 181, 78.
11. Zeman, V.; Koci, J.; Zeman, S. Energ Mater 1999, 7, 172.
12. Zeman, S. Propell Explos Pyrotech 2003, 28, 308.
13. Brill, T. B.; Oyumi, Y. J Phys Chem 1986, 90, 6848.
14. Oyumi, Y.; Brill, T. B. Combust Flame 1987, 68, 209.
15. Zeman, S. Thermochim Acta 1997, 302, 11.
16. Jalovy, Z.; Zeman, S. International Annual Conference ofICT, 1999, 30th, 104/1-104/8.
17. Sorescu, D. C.; Rice, B. M.; Thompson, D. L. J Phys Chem A1998,102, 8386.
18. Zeman, S.; Krupka, M. Energ Mater 2002, 10, 27.
19. Prabhakaran, K. V.; Bhide, N. M.; Kurian, E. M. ThermochimActa 1995, 249, 249.
20. Liu, M. H.; Chen, C.; Hong, Y. S. J Mol Struct (Theochem)2004, 710, 207.
21. Parr, R. G. Density Functional Theory of Atoms and Mole-cules; Oxford University Press: Oxford, 1995.
22. Seminario, J. M.; Politzer, P. Modern Density FunctionalTheory: A Tool for Chemistry; Elsevier Press: New York,1995.
23. Zhang, J.; Xiao, H. M.; Ji, G. F. Acta Chim Sinica 2001, 59,1265.
24. Xiao, J. J.; Ji, G. F.; Yang D.; Xiao H. M. Chin J Struct Chem2002, 21, 437.
25. Xiao, J. J.; Zhang J.; Yang D.; Xiao H. M. Chin J Chem Phys2002, 15, 41.
26. Xiao, J. J.; Gong X. D.; Xiao H. M. Chin J Chem Phys 2002, 15,433.
27. Becke, A. D. J Chem Phys 1993, 98, 5648.28. Lee, C.; Yang, W.; Parr, R. G. Phys Rev 1988, B37, 785.29. Francl, M. M.; Pietro, W. J.; Hehre, W. J.; Binkley, J. S.;
Gordon, M. S.; Defrees, D. J.; Pople, J. A. J Chem Phys 1982,77, 3654.
30. Scott, A. P.; Radom, L. J Phys Chem 1996, 100, 16502.31. Hill, T. L. Introduction to Statistical Thermodynamics; Ad-
dision-Wesley: New York, 1960.32. Kamlet, M. J.; Jacobs, S. J. J Chem Phys 1968, 48, 23.33. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;
Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgom-ery, J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Mil-lam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas,O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci,B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Peters-son, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.;Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski,J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz,P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.;Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.;Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.;Chen, W.; Wong, M. W.; Andres, J. L.; Head-Gordon, M.;Replogle, E. S.; Pople, J. A. Gaussian98, Revision A.7; Gauss-ian Inc.: Pittsburgh, PA, 1998.
34. Wong, M. W.; Wiberg, K. B.; Frisch, M. J. J Comp Chem 1995,16(3), 385.
35. Murray, J. S.; Politzer, P. Computational Studies of EnergeticNitramines; in: Bulusu, S. N. Ed.; Chemistry and Physics ofEnergetic Materials; NATO ASI Series, Vol. 309; Kluwer:Dordrecht, The Netherlands, 1990, 175.
36. Molchanova, M. S.; Pivina, T. S.; Arnautova, E. A.; Zefirov,N. S. J Mol Struct (Theochem) 1999, 465, 11.
37. Lowe-Ma, C. K. Acta Cryst 1990, C46, 1029.
QIU ET AL.
56 VOL. 105, NO. 1












![Isomers [compatibility mode]](https://img.pdfslide.net/doc/110x75/5590bc1e1a28abbf308b46da/isomers-compatibility-mode-5593e8f124020.jpg)