Embed Size (px)
Citation preview

Thermosetting Polymers from Renewable Resources
by
Bernal Sibaja Hernandez
A dissertation submitted to the Graduate Faculty of Auburn University
in partial fulfillment of the requirements for the Degree of
Doctor of Philosophy
Auburn, Alabama August 06, 2016
Keywords: renewable resources, polymers, thermosets, biorefinery, chemical modification, thermo-mechanical properties
Copyright 2016 by Bernal Sibaja Hernandez
Approved by
Maria L. Auad, Chair, Professor of Chemical Engineering Mario Eden, Professor of Chemical Engineering
Sushil Adhikari, Professor of Department of Biosystems Engineering Brian Via, Professor of School of Forestry and Wildlife Sciences
Mahesh Hosur, Materials Science Engineering Department

ii
Abstract
Polymer science was born as an answer to the need to produce and to understand materials such
as plastics, rubber, adhesives, fibers, and paints. It has been recognize as an interdisciplinary
field, combining theoretical and experimental knowledge from areas such as chemistry, physics,
biology, materials and chemical engineering, etc. It has also been an industry of high levels of
innovation, leading the major investments in research and development programs. The
continued use of finite fossil fuel resources has shifted thinking towards the future energy
scenario of the world, and the polymer science has not escaped to the impact of this trend. The
interest in renewable resources is constantly increasing, backed up by new environmental
regulations and economic considerations. Biomass is abundant, and polymeric materials based
on renewable biomass feedstocks represent a viable alternative to fossil resources. The present
work aims to synthesize new, potentially bio-based thermosetting materials from biomass-
derived compounds, use them to prepare bio-based thermosetting resins, and investigate the
structure-property relations of the resulting polymers. In the first Chapter, background
information is provided about the current status of industrial biomass, emphasizing on numbers
as well as on new governmental regulations aiming to impulse the use and study of biomass as
an alternative chemical feedstock. In the next Chapter, the cationic copolymerization of tung
oil, employing limonene and myrcene as comonomers, is presented and discussed.
Characterization revealed that the resulting materials behave as elastomeric materials, with the
glass-rubber transition occurring at room temperature, and modulus values in the order of MPa.

iii
Chapter III investigates the chemical modification of two different functional groups of biomass
derived materials; the carbon-carbon double bonds of triglycerides, and the hydroxyl groups of
phenolic compounds. These chemical modifications were addressed to introduce more reactive
functional groups able to polymerize and to produce materials with a better thermo-mechanical
performance. Testing revealed the deep impact that the chemical modification brings up to the
thermo-mechanical performance of these materials, generating resins with properties similar to
those shown by commercial resins.
Chapter IV considers the preparation of interpenetrating polymer networks or IPNs, a particular
class of hybrid polymer consisting of two (or more) crosslinked polymers. These
multicomponent polymer systems seek to combine the best properties of two or more different
polymer networks in order to achieve a material with better properties compared to their not-
interpenetrated counterparts. Materials with high toughenability were produced and vastly study.
Finally, in certain thermo-mechanically demanding applications, aromatic raw materials are
needed to impart the required properties to the thermoset. The most available source of
phenolics in nature lies in lignocellulosic. Molecular aromatics can be obtained on a large scale
by means of new and arising biorefinery technologies. Chapter V evaluates the viability of
synthesizing a high performance bio-based epoxy polymer using fast pyrolysis bio-oil
(containing lignin fragments) as a source of phenolic compound.

iv
Acknowledgments
I am deeply indebted to my parents and family for their love and support, as well as for having
planted and nurtured the importance of education in my person from a very early age. Without
them this work would not have been possible. I would also like to thank Dr. Maria Auad for her
guide and for giving me the opportunity to pursue a PhD degree. I am also deeply thankful to Dr.
Gisela Buschle-Diller for her kindness and friendship. I want to thank my close friends as well.
I would also like to thank the committee members, Dr. Mario Eden, Dr. Sushil Adhikari, Dr.
Brian Via, and Dr. Mahesh Hosur for their contributions to this project. Thanks to Dr. Allan
David for his willing to be the outside reader for this dissertation.
This work was supported by the NSF under grant # NSF EPS-115882 and by the CREST under
grant # HDR-1137681.

v
Table of Contents
Abstract ......................................................................................................................................... ii
Acknowledgments........................................................................................................................ iv
List of Tables .............................................................................................................................. xii
List of Figures ............................................................................................................................ xiv
Chapter I. Introduction ................................................................................................................. 1
1.1. Overview of the global synthetic polymer industry ............................................................. 1
1.2. Thermosetting plastics and composite materials ................................................................. 3
1.3. Challenges for the future and innovation of the polymeric materials .................................. 5
1.4. Biomass as an alternative to produce green plastics ............................................................ 6
1.5. General pathways towards synthesis of renewable monomers .......................................... 11
1.5.1. Molecular biomass direct from nature ............................................................................ 11
1.5.2. Fermentation or biological transformation ..................................................................... 18
1.5.3. Chemical transformation of natural polymers ................................................................ 20
1.6 Research Objective ............................................................................................................. 26
References ................................................................................................................................. 30
Chapter II. Renewable Thermoset Copolymers from Tung Oil and Natural Terpenes ............ 41
1. Introduction ............................................................................................................................ 44
2. Methods ................................................................................................................................. 44
2.1. Materials ............................................................................................................................ 44

vi
2.2. Polymer preparation via cationic polymerization .............................................................. 44
2.3. Characterization ................................................................................................................. 45
3. Results and Discussion ......................................................................................................... 46
3.1. Cationic Polymerization of vegetable oils ......................................................................... 46
3.2. Characterization of the polymer networks ......................................................................... 48
3.2.1. Thermo-mechanical behavior ......................................................................................... 49
3.3. Theoretical Prediction of Tg based on the Fox-Loshaek Model ........................................ 57
3.4. Conclusions ........................................................................................................................ 61
References ................................................................................................................................. 62
Chapter III. Triglycerides and Phenolic Compounds as Precursors of Bio-Based Thermosetting Epoxy Resins ................................................................................................................ 66
1. Introduction ........................................................................................................................... 66
2. Experimental Section ............................................................................................................ 70
2.1. Materials ............................................................................................................................ 70
2.2. Synthesis Procedures ......................................................................................................... 71
2.2.1. Synthesis of epoxidized triglycerides ............................................................................. 71
2.2.2. Synthesis of polyol .......................................................................................................... 71
2.2.3. Functionalization of phenolic compounds with epoxy groups. Synthesis of triglycidylated
ether of α-resorcylic acid .............................................................................................. 72
2.2.4. Synthesis of bio-based epoxy resins ............................................................................... 72
2.2.5. Preparation of the linseed oil based polyurethane .......................................................... 73

vii
2.3. Characterization of starting and modified materials .......................................................... 74
2.3.1. Fourier transform infrared spectroscopy (FTIR) ............................................................ 74
2.3.2. Epoxide equivalent weight determination ...................................................................... 74
2.3.3. Iodine value determination ............................................................................................. 75
2.3.4. Hydroxyl number determination ..................................................................................... 76
2.4. Characterization of the epoxy networks ............................................................................ 76
2.4.1. Soxhlet extraction ........................................................................................................... 76
2.4.2. Scanning electron microscopy (SEM) ............................................................................ 77
2.4.3. Thermal properties .......................................................................................................... 77
2.4.4. Thermo-mechanical properties of the networks ............................................................. 77
3. Results and Discussion ......................................................................................................... 78
3.1. Synthesis of epoxidized triglycerides ................................................................................ 78
3.2. Synthesis of linseed oil based polyol ................................................................................... 81
3.3. Functionalization of phenolic compounds with epoxy groups .......................................... 84
3.3.1. Synthesis of triglycidylated ether of α-resorcylic acid ................................................... 84
3.3.2. Synthesis of diglycidylated ether of resorcinol ............................................................. 86
3.4. Thermo-mechanical characterization of bio-based epoxy thermosetting resins ................ 87
3.4.1. Polymerization of epoxidized linseed oil through step-growth process ......................... 87
3.5. Preparation of the linseed oil based polyurethane ............................................................. 89
3.6. Polymerization of acrylated soybean oil through free radical mechanism ........................ 89

viii
3.7. Copolymerization of epoxidized linseed oil and epoxidized α-resorcylic acid through chain growth process .............................................................................................................. 92
3.8. Scanning electron microscopy (SEM) .............................................................................. 94
3.9. Thermo-mechanical characterization of the epoxidized linseed oil (ELO) and α-resorcylic acid (ERA) copolymers .................................................................................................. 96
4. Conclusions ......................................................................................................................... 108
References ............................................................................................................................... 110
Chapter IV. Synthesis and Characterization of Interpenetrating Polymer Networks (IPNs) using Biomass Derived Materials .......................................................................................... 116
1. Introduction ......................................................................................................................... 116
2. Experimental Section .......................................................................................................... 121
2.1. Materials .......................................................................................................................... 121
2.2. Synthesis procedures ........................................................................................................ 122
2.2.1. Synthesis of epoxidized triglycerides ........................................................................... 122
2.2.2. General procedure for the glycidylation of α-resorcylic acid ....................................... 122
2.2.3. Synthesis of IPNs ............................................................................................................ 122
2.3. Characterization of materials and networks ....................................................................... 124
2.3.1. Fourier transform infrared spectroscopy (FTIR) ............................................................ 124
2.3.2. Kinetics of gelation ....................................................................................................... 125
2.3.3. Determination and measurement of the extent of the reaction at the gel time ............. 125
2.3.4. Soxhlet extraction ......................................................................................................... 126

ix
2.3.5. Scanning electron microscopy (SEM) .......................................................................... 127
2.3.6. Thermal properties .......................................................................................................... 127
2.3.7. Thermo-mechanical properties of the networks ........................................................... 127
2.3.8. Linear elastic fracture mechanics .................................................................................. 128
3. Results and Discussion ....................................................................................................... 129
3.1. Chemical functionalization of raw materials ................................................................... 129
3.1.1. Synthesis of epoxidized triglycerides ........................................................................... 129
3.1.2. Functionalization of phenolic compounds with epoxy groups. Synthesis of triglycidylated
ether of α-resorcylic acid ........................................................................................... 129
3.2. Characterization of the IPNs ............................................................................................ 129
3.2.1. System A: Epoxidized linseed/acrylated soybean oil IPNs .......................................... 129
3.2.1.1. Scanning electron microscopy ................................................................................... 129
3.2.1.2. Thermo-mechanical characterization of IPNs ........................................................... 132
3.2.2. System B: epoxidized α-resorcylic acid/acrylated oil IPNs ......................................... 138
3.2.2.1. Scanning electron microscopy ................................................................................... 138
3.2.2.2. Kinetics of gelation .................................................................................................... 141
3.2.2.3. Curing behavior of IPNs ............................................................................................ 146
3.2.2.4. Thermo-mechanical characterization ......................................................................... 150
3.2.2.5. Linear elastic fracture mechanics ............................................................................... 151
4. Conclusions ......................................................................................................................... 160
References ............................................................................................................................... 162

x
Chapter V. Fast Pyrolysis Bio-oil as precursor of Thermosetting Epoxy Resins ................... 169
1. Introduction ......................................................................................................................... 169
2. Experimental section ........................................................................................................... 175
2.1. Materials .......................................................................................................................... 175
2.2. Synthesis procedures ........................................................................................................ 176
2.2.1. General method for the glycidylation (functionalization with epoxy groups) of bio-oil........................................................................................................................ 176
2.3. Characterization of starting and modified materials ........................................................ 177
2.3.1. Gas chromatography –Mass spectrometry (GC-MS) ................................................... 177
2.3.2. Hydroxyl group analysis through 31P-NMR spectroscopy ........................................... 177
2.3.3. Fourier Transform Infrared Spectroscopy (FTIR) ........................................................ 178
2.3.4. Epoxide equivalent weight determination .................................................................... 178
2.4. General procedure for the formulation of crosslinked epoxy networks .......................... 179
2.5. Characterization of epoxy networks ................................................................................ 179
2.5.1. Sohxlet extraction ......................................................................................................... 179
2.5.2. Scanning electron microscopy (SEM) .......................................................................... 180
2.5.3. Thermo-mechanical properties of the networks ........................................................... 180
3. Results and Discussion ....................................................................................................... 181
3.1. Gas chromatography – Mass spectroscopy (GC-MS) ....................................................... 181
3.2. Hydroxyl group analysis through 31P-NMR spectroscopy ............................................... 185

xi
3.3. General method for the glycidylation (functionalization with epoxy groups) of bio-oil ....................................................................................................................... 187
3.3.1. Glycidylation of the α-resorcylic acid .......................................................................... 187
3.3.2. Glycidylation of bio-oil................................................................................................... 187
3.4. Fourier transform infrared spectroscopy (FTIR) ............................................................. 189
4. Characterization .................................................................................................................. 190
4.1. Soxhlet extraction ............................................................................................................ 190
4.2. Scanning electron microscopy (SEM) ............................................................................. 192
4.3. Thermo-mechanical characterization of bio-oil based epoxy thermosetting resin ............ 194
4. Conclusions ........................................................................................................................... 200
References ............................................................................................................................... 201
General conclusions ................................................................................................................ 206
Future work ............................................................................................................................. 211

xii
List of Tables
Table 1.1 Global plastic consumption for the year 2012 .............................................................. 2
Table 1.2 Annual consumption (2012) of major thermosets (1000 tons and percentage). ........... 3
Table 1.3 Average biomass content of the bio-based polymers currently developed. ................. 7
Table 1.4 Classification of mayor molecular biomass ……………………………...……..…..10
Table 1.5 Names and double bonds positions of common fatty acids. ................................. …..13
Table 1.6 Platform molecules identified by the United States Department of Energy. .............. 19
Table 1.7 Chemical classification of bio-oil components ........................................................... 23
Table 2.1 Properties of the tung oil-limonene copolymers ......................................................... 52
Table 2.2 Properties of the tung oil-myrcene copolymers .......................................................... 54
Table 2.3 Properties of the tung oil-styrene copolymers ............................................................ 56
Table 3.1 Properties of some common oils. ................................................................................ 79
Table 3.2 Properties of the resulting linseed oil based polyol. ................................................... 82
Table 3.3 Properties of the acrylated soybean oil polymers. ...................................................... 90
Table 3.4 Mechanical properties of the triglycidylated ether of α-RA and epoxidized linseed oil epoxy resins. ............................................................................................................. 97
Table 3.5 Mechanical properties of the triglycidylated ether of α-RA and resin EPON 828 epoxy resins. ................................................................................................................ 104
Table 3.6 Thermo-mechanical properties of the triglycidylated ether of α-RA and diglycidylated ether of resorcinol copolymers. ........................................................... 105

xiii
Table 4.1 Evolution of the storage modulus (E’) and glass transition temperature as a function of epoxy/acrylate weight proportion.. .......................................................................... 132
Table 4.2 Extent of the reaction at the gel point for IPNs based on epoxidized α-resorcylic acid and acrylated soybean oil.. ........................................................................................... 145
Table 4.3 Onset and maximum of curing peak temperatures for the IPNs based on epoxidized α- resorcylic acid and acrylated soybean oil.. .............................................................. 146
Table 4.4 Mechanical Properties of the IPNs based on epoxidized α-resorcylic acid and acrylated soybean oil.. ................................................................................................................. 153
Table 4.5 Experimental KIc and GIc values. .............................................................................. 156
Table 5.1 Major components found in pyrolysis oil. ................................................................ 173
Table 5.2 Major pyrolysis decomposition products identified by GS/MS. .............................. 183
Table 5.3 Fractions of phenolic compounds found in bio-oil. .................................................. 184
Table 5.4 Hydroxyl number (OHN) of pyrolysis oil determined by quantitative31P-NMR…………….. ………………………………………............. …186
Table 5.5 Thermo-mechanical properties of epoxidized bio-oil and the triglycidylated ether of α-resorcylic acid (epoxidized α-resorcylic) resins. .................................................. 196
Table 5.6 Mechanical properties of the epoxidized bio-oil, triglycidylated α-RA and Super Sap 100 epoxy. .................................................................................................................... 199

xiv
List of Figures
Figure 1.1 Global production and demand of plastics by geographical area by percentage (2012) ....................................................................................................... 2
Figure 1.2 Plastics classification ................................................................................................... 5
Figure 1.3 Trend of publications for polymers from renewable resources ................................... 9
Figure 1.4 Major pathways to produce monomers (or precursors) from biomass. ..................... 11
Figure 1.5 Structure of a triglyceride. Where R1, R2, and R3 are fatty acid chains .................... 12
Figure 1.6 Chemical transformation of biomass. ........................................................................ 21
Figure 2.1 Cationic polymerization of tung oil with limonene, myrcene, and linalool .............. 47
Figure 2.2 Temperature dependence of the (2.2.a) storage modulus (E’) for the copolymers tung oil-limonene. (2.2.b) Loss factor (tan δ) as a function of the temperature for the tung oil- limonene copolymers. .................................................................................................. 50
Figure 2.3 (2.3.a) Glass transition temperature of tung oil-limonene copolymers as a function of
the composition: (▲) values measured with DMA, (♦) linear copolymer contribution, (■) glass transition temperature of the copolymer. (2.3.b) Glass transition temperature of tung oil-mycene copolymers as a function of the composition: (▲) values measured
with DMA, (♦) linear copolymer contribution, (■) glass transition temperature of the copolymer. .................................................................................................................... 58
Figure 3.1 Epoxidation of triglycerides ...................................................................................... 79
Figure 3.2 FTIR spectrum of epoxidized linseed oil .................................................................. 81
Figure 3.3 Methanolysis of epoxidized linseed oil ..................................................................... 81

xv
Figure 3.4 FTIR of hydroxylated linseed oil .............................................................................. 83
Figure 3.5 Glycidylation reaction of α-resorcylic acid with epichlorohydrin ............................ 84
Figure 3.6 FTIR spectra of α-resorcylic acid and epoxidized α-resorcylic acid ........................ 85
Figure 3.7 Glycidylation reaction of resorcinol acid with epichlorohydrin................................ 86
Figure 3.8 FTIR spectra of resorcinol and epoxidized resorcinol .............................................. 87
Figure 3.9 Storage modulus and tan δ of epoxidized linseed oil as a function epoxy/amine ratio ........................................................................................... 88
Figure 3.10 Anionic polymerization of epoxy monomers. ......................................................... 93
Figure 3.11 SEM micrographs of the fracture surface of 100 % α-RA (3.11.a), 80 % α-RA – 20 %
ELO (3.11.b), 70 % α-RA – 30 % ELO (3.11.c), and 100 % ELO (3.11.d). ..................... 94
Figure 3.12 Temperature dependence of the storage modulus (3.12.a) and loss factor or tan δ (3.12.b) as a function of the composition of the epoxy resins ...................................... 96
Figure 3.13 Relationship between 1/Tg and relative epox. α-resorcylic acid weight fraction .............................................................................................................. 99
Figure 3.14 SEM micrographs of the fracture surface of 100 wt% α-RA (3.14.a), 75 wt%
α-RA – 25 wt% EPON 828 (3.14.b), 50 wt% α-RA – 50 wt% EPON 828 (3.14.c), and
100 wt% EPON 828 (3.14.d). .................................................................................... 102
Figure 3.15 Temperature dependence of the storage modulus (3.15.a) and loss factor or tan δ (3.15.b) as a function of the composition of the resorcinol weight content for the
resorcinol – α-resorcylic acid epoxy resins ............................................................. 106
Figure 4.1 Generic schemes of the IPNs containing epoxy and acrylate networks. ................ 120

xvi
Figure 4.2 Chemical structure of the acrylated soybean oil. .................................................... 122
Figure 4.3 SEM micrographs of the fracture surface of 100 % epoxidized linseed oil (4.3.a), IPN 80% epoxy - 20% acrylate (4.3.b), IPN 60% epoxy - 40% acrylate (4.3.c), IPN 40% epoxy – 60%acrylate (4.3.d), IPN 20% epoxy-80% acrylate (4.3.e), 100% acrylated soybean oil (4.3.f) ............................................................................. 130
Figure 4.4 Crosslinking densities as a function of the epoxy weight fraction. ......................... 134
Figure 4.5. SEM micrographs of the fracture surface of 100 wt% epoxidized α-resorcylic acid (4.5.a), IPN 75 wt% epoxy – 25 wt% acrylate (4.5.b), IPN 50 wt% epoxy – 50 wt% acrylate (4.5.c), IPN 25 wt% epoxy – 75 wt% acrylate (4.5.d), 100 wt% acrylated soybean oil (4.5.e)…………………………………………………………………....139
Figure 4.6. Gel times at 65°C for the studied IPN systems based on epoxidized α-resorcylic acid and acrylated soybean oil.. .................................................................................. 142
Figure 4.7. Extent of conversion of stoichiometric mixture of epoxidized α-resorcylic acid and multifunctional amine (Jeffamine T-403). ................................................................. 144
Figure 4.8 Heats of polymerization (4.8.a) and conversions (4.8.b) of the IPNs based on
epoxidized α-resorcylic acid and acrylated soybean ................................................. 147
Figure 4.9. Dynamic mechanical response of IPNs based on epoxidized α-resorcylic acid and acrylated soybean oil.................................................................................................. 150
Figure 4.10. GIc versus Mc1/2 for the studied IPNs. ................................................................... 159
Figure 5.1. Monomeric phenylpropane unit structures.. ........................................................... 171
Figure 5.2. Total ion GC/MS chromatogram of fast pyrolysis bio-oil.. ................................... 182

xvii
Figure 5.3. Glycidylation of bio-oil with epichlorohydrin in alkaline medium. ....................... 188
Figure 5.4. FTIR spectra of raw bio-oil, modified bio-oil, and epichlorohydrin. ..................... 190
Figure 5.5. Mass loss (wt%) of bio-oil / α-resorcylic acid epoxy resins. ................................. 191
Figure 5.6. SEM micrographs of the 100 wt% epoxidized bio-oil (5.6.a), 50 wt% epoxidized
biooil – 50 wt% epoxidized α-RA (5.6.b), and 100 wt% epoxidized α-RA (5.6.c) resins.. ........................................................................................................................ 193
Figure 5.7. Temperature dependence of the storage modulus (5.7.a) and loss factor or tan δ (5.7.b) as a function of the composition of the epoxy resins. .................................... 194
Figure 5.8. Relationship between 1/Tg and relative epoxy bio-oil weight content (Wbio-oil). ... 198

1
CHAPTER I
Introduction
Plastics, or synthetic polymers, are one of the first primary man-made materials and they have
been applied in a variety of applications. Plastics are made of raw chemicals that mainly include
petroleum based products of its refinery process, natural gas, carbon, and other natural
compounds. Their impact in the modern society can be traced to research-related developments,
the discovery of new and innovative materials able to fulfill the needs of the industrial sectors,
and the gradual substitution of heavier and less versatile materials, such as metals, glass,
ceramics, etc. Starting in the 1950s with the development of the macromolecular theory
developed by scientists such as Herman Staudinger, H.W. Carothers, Paul Flory, etc., the
worldwide production of plastic has shown an important increase. In the following section, an
overview of the global plastic industry is given. The goal of the figures and details is to help
clarify the real perspective about the current state of the plastic industry.
1. Overview of the global synthetic polymer industry.
For the year 2012, the worldwide plastic consumption was of the order of 270 million tons, with
a turnover of around $1,100 billion or more (Table 1.1). Over the past years, the global plastic
consumption has grown consistently, showing an average annual rate that is consistent with this
trend. For the next years, this growth is estimated to have an annual increase of approximately
5.5%, reaching an estimate average value of 400 million tons by the year 2020 [1].
2
Other
common-
wealth
3% Japan
5%
Latin
America
5%Middle
East, And
Africa
7%
Rest of
Asia
16%Europe
20%
China
24%
NAFTA
20%
Demand
Table 1.1. Global plastic consumption for the year 2012
Million tons %
Thermoplastics 215 80
Thermosets 30 11
Composites 10 4
Others 15 5
Total 270 100
Source: Biron, M. Thermosets and Composites, 2014
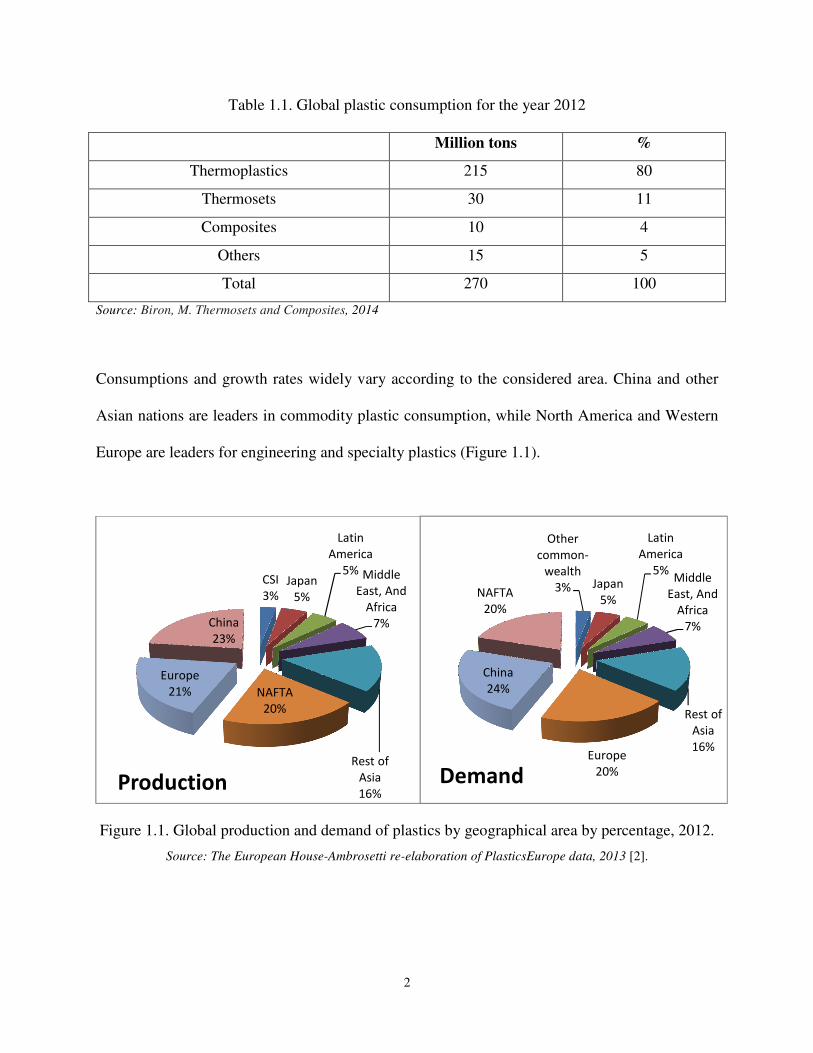
Consumptions and growth rates widely vary according to the considered area. China and other
Asian nations are leaders in commodity plastic consumption, while North America and Western
Europe are leaders for engineering and specialty plastics (Figure 1.1).
Figure 1.1. Global production and demand of plastics by geographical area by percentage, 2012.
Source: The European House-Ambrosetti re-elaboration of PlasticsEurope data, 2013 [2].
CSI
3%
Japan
5%
Latin
America
5% Middle
East, And
Africa
7%
Rest of
Asia
16%
NAFTA
20%
Europe
21%
China
23%
Production
3
1.2. Thermosetting plastics and composite materials
Thermosetting materials are polymeric materials that cure irreversibly. They are generally
stronger than thermoplastic materials due to their three-dimensional network of bonds (cross-
linking), and are also better suited to high-temperature applications up to the decomposition
temperature, however, they are considerably brittle. Since their shape is permanent, they tend not
to be recyclable as a source for newly made plastic. According to the review “Global
Thermosetting Plastics Market - Segmented by Type, Industry and Geography - Trends and
Forecasts (2015-2020)” [3], the global thermosetting plastics production was estimated to be
33.74 million tons in 2014, and is expected to reach 41.96 million tons by 2020; showing a
compound annual growth rate of 3.7%. Asia-Pacific currently dominates the market for
thermosetting plastics followed by North America and Europe. This increase is primarily
supported by the growth in the use of unsaturated polyurethanes, phenol-formaldehyde and
amino/epoxy resins, to supply the needs of an increasing number of markets from construction,
adhesives & coatings, electrical & electronics, automotive & transportation and a number of
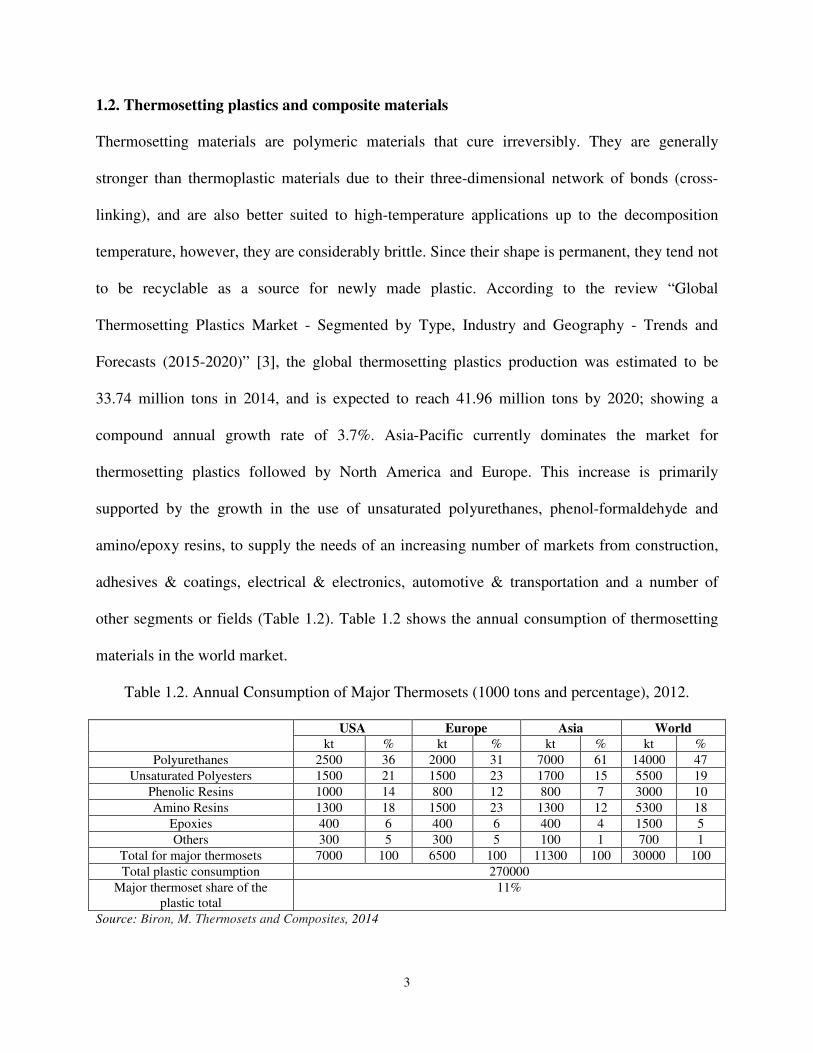
other segments or fields (Table 1.2). Table 1.2 shows the annual consumption of thermosetting
materials in the world market.
Table 1.2. Annual Consumption of Major Thermosets (1000 tons and percentage), 2012.
USA Europe Asia World
kt % kt % kt % kt %
Polyurethanes 2500 36 2000 31 7000 61 14000 47
Unsaturated Polyesters 1500 21 1500 23 1700 15 5500 19
Phenolic Resins 1000 14 800 12 800 7 3000 10
Amino Resins 1300 18 1500 23 1300 12 5300 18
Epoxies 400 6 400 6 400 4 1500 5
Others 300 5 300 5 100 1 700 1
Total for major thermosets 7000 100 6500 100 11300 100 30000 100
Total plastic consumption 270000
Major thermoset share of the plastic total
11%
Source: Biron, M. Thermosets and Composites, 2014

4
The mentioned thermosets represent 11% of global plastic consumption. Likewise, interest in
composite materials has constantly grown in the last decades, striving for obtaining new
composites with superior mechanical strength, better thermal and electrical properties, and high
resistance to external factors while reducing the weight [4]. In most of the structural and
engineering applications, composites generally comprise a bulk phase (known as the matrix) and
a reinforcing phase (the reinforcement). The matrix provides the composite with its shape,
surface appearance, environmental tolerance and durability by integrally binding the
reinforcement; being the last one the component carrying most of the structural loads [5]. In the
case of polymer composites, matrix materials can be either a thermosets or thermoplastics.
Common thermosetting polymers employed in composite materials include polyurethanes, epoxy
resins and polyesters. Generally, their mechanical performance requires them to have modulus
values in the range of 0.025 to 6.0 GPa [6]. It is due to this direct link between the composites
and the polymeric materials that the composites markets are highly influenced by the changes
and innovations in the macromolecular science. Based on these figures and future projections, it
is clear that the polymer industry plays a prominent role and is essential to a variety of industries.
Nowadays, most commercially available polymeric materials are derived from non-renewable
resources and account for approximately 7% of global oil and gas use [7]. It is the combination
of environmental awareness, the depletion of fossil feedstocks and fluctuations in their prices, as
well as financial drivers and global policies that have catapulted the utilization of renewable
resources as raw materials for the production of polymeric materials; and the generation of
biomass-derived materials with properties similar to those shown by their commercial
counterparts remains as a challenge in the academic and industrial sectors.

5
1.3. Challenges for the future and innovation of the polymeric materials.
The polymer industry traditionally has been an area of high level of innovation, leading to major
investment in research and development programs, in response to modern day needs, such as low
cost, high performance, recycling, light weight, and low environmental impact. In the future,
green composite materials and bioplastics will be two further important sectors. Bioplastic is a
term that has been coined as a term for describing plastic derived completely, or partially, from
biological and renewable raw materials instead of fossil fuels (carbon, crude oil and natural gas)
[2]. The term bioplastic depicts a chemical process during which microorganisms that are
available in the environment convert materials into natural substances such as water, carbon
dioxide, and compost. The process of biodegradation is dependent on factors such as
environmental conditions, material, and application. To illustrate the distinction between these
two terms, Figure 1.2 shows the European Bioplastics two-axis model, that encompasses all
plastics types and combinations [8].
Figure 1.2. Plastics classification
Source: http://www.european-bioplastics.org/bioplastics/
Bio-based
Fossil-based
Non
Biodegradable Biodegradable
Bioplastics
e.g. PLA, PHA
PBS, Starch blends
Bioplastics
e.g. PBAT, PCL
Bioplastics
e.g. bio-based PE,
PET, PA, PTT
Conventional
Plastics
e.g. PE, PP, PET

6
The horizontal axis shows the biodegradability of the plastics, whereas the vertical axis depicts
the source from which the plastics are produced. The plastics are therefore divided into four
different groups, which include plastics which are not biodegradable and made from
petrochemical resources, biodegradable plastics from renewable resources, biodegradable
plastics from fossil resources, and non-biodegradable plastics from renewable resources [9].
In 2011, the global production of bioplastics was 1.2 million tons, and it is expected to increase
up to 5.8 million tons in 2016. Asia (34.6%), Latin America (32.8%), and Europe (18.5%) are
the worldwide leaders of bioplastics. It is clear that fossil sources are still required for the
production of major part of the polymers, as only 5% of chemicals are currently produced from
renewable feedstocks [10]. However, the application of natural chemicals as feedstock for the
manufacture of polymers is steadily increasing. Taking into account the great diversity of
biomass; consisting of polysaccharides, aromatic compounds, triglycerides, proteins, etc., a great
number of alternative monomers and novel polymer are potentially accessible [10].
1.4. Biomass as an alternative to produce “green materials”
Biomass is considered the most abundant natural resource with an annual production of 1.3
billion dry tons in the U.S.A. [11]. Biomass is any organic matter that is renewable over time,
and in the context of energy it is often used to mean plant-based materials such as agricultural
residues, forestry wastes, and energy crops [12]. The world energy crisis has led to the
development of programs that have provoked extensive research programs on biomass
conversion. The U.S. Department of Energy (DOE) and the U.S. Department of Agriculture
(USDA) have prioritized the development of bioenergy and bioproducts, and they have a goal to
7
produce 18% of the current U.S. chemical commodities from biomass by 2020, and 25% by 2030
[11,13]. Increased research on the utilization of lignocellulosic biomass to produce bio-based
resins has also been the subject of industrial entrepreneurships. On 2012, the nova-Institute in
Germany released the results of a market study named “Bio-based polymers in the world -
production, capacities, and applications: status quo and trends towards 2020” [14]. This market
report is continually updated, and it is considered the most comprehensive study in this field ever
made. It is the first time that a study has looked at every kind of bio-based polymer produced by
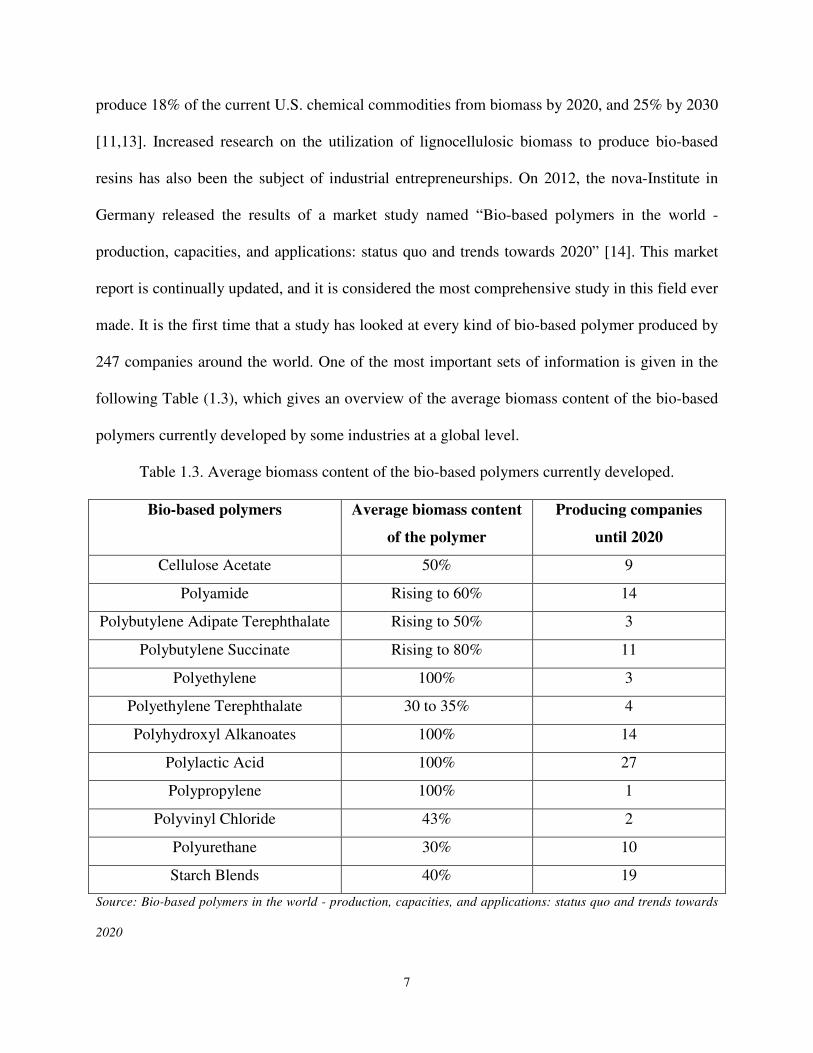
247 companies around the world. One of the most important sets of information is given in the
following Table (1.3), which gives an overview of the average biomass content of the bio-based
polymers currently developed by some industries at a global level.
Table 1.3. Average biomass content of the bio-based polymers currently developed.
Bio-based polymers Average biomass content
of the polymer
Producing companies
until 2020
Cellulose Acetate 50% 9
Polyamide Rising to 60% 14
Polybutylene Adipate Terephthalate Rising to 50% 3
Polybutylene Succinate Rising to 80% 11
Polyethylene 100% 3
Polyethylene Terephthalate 30 to 35% 4
Polyhydroxyl Alkanoates 100% 14
Polylactic Acid 100% 27
Polypropylene 100% 1
Polyvinyl Chloride 43% 2
Polyurethane 30% 10
Starch Blends 40% 19
Source: Bio-based polymers in the world - production, capacities, and applications: status quo and trends towards
2020

8
The report shows that the production capacity of bio-based polymers will triple from 3.5 million
tons in 2011 to nearly 12 million tons by 2020. Mayor players in bulk biopolymers production
include companies like Metabolix, Dow Chemicals, Novamont, Cereplast, Teijin, Natureworks,
Hisun, Tianan, Plantic, Innovia, Proter & Gamble, Kaneka, and Arkema [15]. In the case of
bioplastics, some mayor companies include BASF, DuPont and Braskem [16].
According to this results, it is clear that they development of green polymers is growing, and that
it is supported not only by the industrial sectors, but also by governmental entities and agencies.
In the case of the academic field, the worldwide interest in bio-based polymers has increased
progressively in the past years, as the advances in technology have generated an accumulated
experience, allowing the development of new routes to produce bioplastics to reduce the
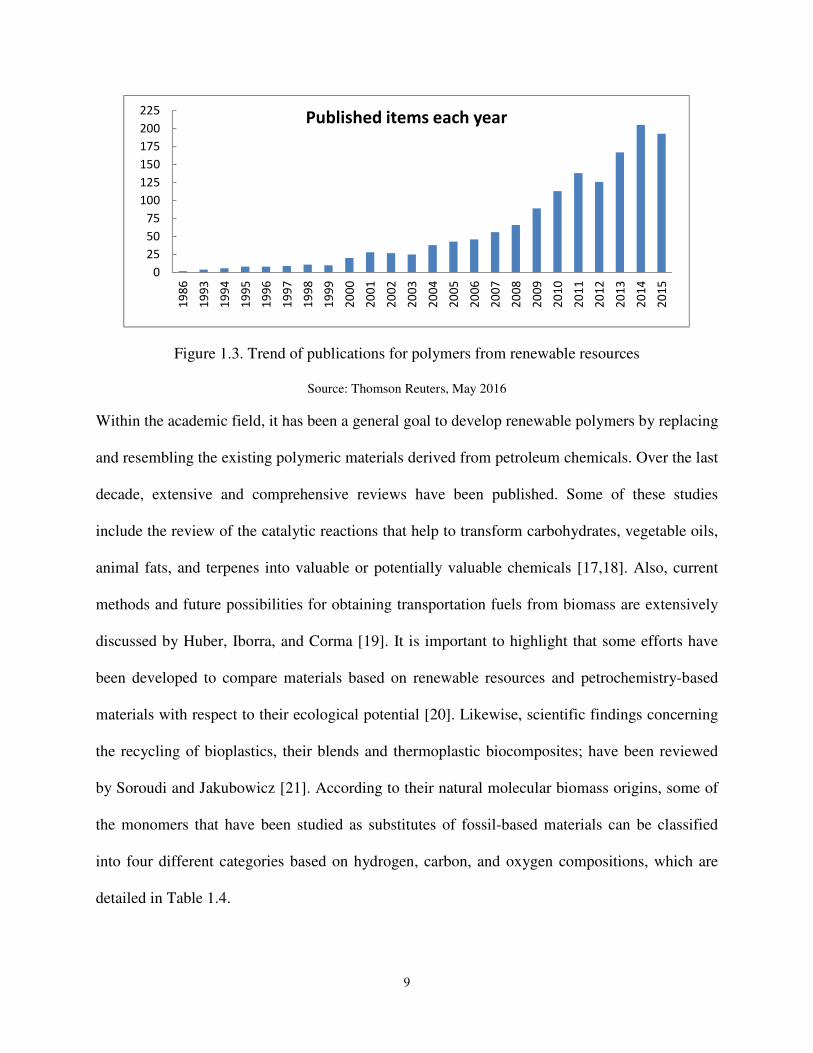
dependency on fossil fuels. As an example of the evolution of this field, a search of the Web of
Science database using the words “polymers from renewable resources” identified a total of
more than 1500 scientific articles currently available on the subject. Figure 1.3 shows that there
has been an exponential growth in the last 20 years, with approximately 86% of them published
in the last decade, clearly evidencing the increasing interest in this promising field. Concerning
the countries, the USA leads the trend in publications in the bioplastic development, ranking first
with approx. 23 % of the more than 1500 papers matching the online search, followed by
countries like Spain, Japan, and China.
9
Figure 1.3. Trend of publications for polymers from renewable resources
Source: Thomson Reuters, May 2016
Within the academic field, it has been a general goal to develop renewable polymers by replacing
and resembling the existing polymeric materials derived from petroleum chemicals. Over the last
decade, extensive and comprehensive reviews have been published. Some of these studies
include the review of the catalytic reactions that help to transform carbohydrates, vegetable oils,
animal fats, and terpenes into valuable or potentially valuable chemicals [17,18]. Also, current
methods and future possibilities for obtaining transportation fuels from biomass are extensively
discussed by Huber, Iborra, and Corma [19]. It is important to highlight that some efforts have
been developed to compare materials based on renewable resources and petrochemistry-based
materials with respect to their ecological potential [20]. Likewise, scientific findings concerning
the recycling of bioplastics, their blends and thermoplastic biocomposites; have been reviewed
by Soroudi and Jakubowicz [21]. According to their natural molecular biomass origins, some of
the monomers that have been studied as substitutes of fossil-based materials can be classified
into four different categories based on hydrogen, carbon, and oxygen compositions, which are
detailed in Table 1.4.
0
25
50
75
100
125
150
175
200
225
19
86
19
93
19
94
19
95
19
96
19
97
19
98
19
99
20
00
20
01
20
02
20
03
20
04
20
05
20
06
20
07
20
08
20
09
20
10
20
11
20
12
20
13
20
14
20
15
Published items each year

10
Table 1.4. Classification of mayor molecular biomass
Natural Molecular Biomass
Oxygen-rich biomass
Hydrocarbon-rich
biomass
Hydrocarbon
biomass
Non-hydrocarbon
biomass
Carboxylic Acids Vegetable Oils Ethene Carbon Dioxide
Polyols Fatty Acids Propene Carbon Monoxide
Dianhydroalditols Terpenes Isoprene
Furans Terpenoids Butene
Resin Acids
Source: K. Yao, C. Tang, 46 (2013) 1689–17
As a general trend, there are three major pathways to produce monomers (or their precursors)
from natural resources, which include and molecular biomass direct from nature, fermentation or
biological transformation, and chemical transformation of natural polymers [22]. Figure 1.4
depicts a scheme of the three approaches to obtain chemicals for the polymer industry. In the
following section, the molecular biomass direct from nature and chemical transformations are
discussed more in depth than fermentation, which is briefly described, due to their significant
role in the production of the monomers and precursors studied in this work.

11
Figure 1.4. Major pathways to produce monomers (or their precursors) from biomass
1.5. General pathways towards synthesis of renewable monomers.
1.5.1. Molecular biomass direct from nature.
Different from the compounds obtained through processes such as fermentation or chemical /
thermal degradation, there is a considerable amount of chemicals or precursors that can be
directly obtained from nature. This pathway already stands as one of the cornerstones for using
renewable resources, both on large (pulp, paper and starch industry) and on small scale
(production of fine chemicals). Within this category, a rich pallet of monomers structures such as
terpenes, rosin, sugars, polycarboxylic acids and aminoacids, furans, and vegetable oils can be
exploited as a source of materials for macromolecules industry. Since a considerable part of the
experimental work described in this manuscript deals with the use of vegetable oils as a platform
for polymeric materials, the next paragraphs review basic information regarding the reasons why
Biomass
Molecular biomass direct from nature
Vegetable oils, carbohydrates, phenolic acids,
terpenes, proteins
Fermentation or biological
transformation
Etanol, sorbitol, glucose, diacids,
glycerol
Chemical Transformation
Thermochemical
degradation
Chemical treatments

12
these compounds have awakened great interest as sustainable and green materials. Also, other
types of chemical precursors obtained from biomass are considered and reviewed.
Oil based bio-products
In the year 2000, the production of oils and fats amounted to 17.4 kg per year per capita and a
growth rate of 3.3 % per year was estimated. The total worldwide yearly production of
renewable materials was over 110 million tons in 2002, and vegetal oils constitutes the 80 % of
this production (the remaining 20 % being animal fats). Only 15.6 million tons (around 15 per
cent) of these raw materials are used as precursors to the synthesis of new chemical commodities
and materials [23]. Nowadays, their main application lies in the field of biodiesel, which can be
used as an alternative engine fuel. However, considerable work on polymers derived from plant
oils have been performed in the last few years [24,25]. Vegetable oils are hydrocarbon rich
compounds that are liquid at room temperature and mostly composed of triglycerides. These
chemical compounds have a three-armed star structure in which three fatty acids are joined at a
glycerol junction (Figure 1.5), and these fatty acids contribute from 94 – 96% of the total weight
of one molecule of triglyceride [25]. The chain length of these fatty acids can vary from 14 to 22
carbons and contain 0 – 5 double bonds situated at different positions along the chain and in
conjugated or unconjugated sequences [23].
O
O
O R1
O
O
R3
O R2
Figure 1.5. Structure of a triglyceride. Where R1, R2, and R3 are fatty acid chains.

13
Table 1.5 collects the 16 most important oils with their specific chemical structures. All of the
mentioned oils do not include conjugated double bonds in their structures (polymers, monomers,
composites).
Table 1.5. Names and double bonds positions of common fatty acids.
Chain length : number of double bonds
Systematic name Trivial name Double bond position
12:0 Dodecanoic Lauric -
14:0 Tetradecanoic Mystiric -
16:0 Hexadecanoic Palmitic -
18:0 Octadecanoic Stearic -
18:1 9-Octadecanoic Oleic 9
18:2 9,12-Octadecanoic Linoleic 9;12
18:3 6,9,12-Octadecanoic γ-linolenic 6,9,12
18:3 9,12,15-Octadecatrienoic α-linolenic 9,12,15
20:0 Eicosanoic Arachidic -
20:1 Eicosanoeic - 9
20:4 Eicosatetraenoic Arachidonic 5,8,11,14
20:5 Eicosapentaenoic EPA 5,8,11,14,17
22:0 Docosanoic - -
22:1 Docosenoic Euric 13
22:5 Docopentanoic DPA 7,10,13,16,19
22:6 Docosahexanoic DHA 4,7,10,13,16,19 Source: A. Gandini, M.N. Belgacem, Monomers, Polymers and Composites, Elsevier, UK, 2008
As shown, some fatty acids are saturated (having no double bonds). On the other hand,
unsaturated fatty acids can have one or more than one double bond in their structure. It is
important to mention that, additionally to the above described oils, some naturally fatty acids
have different organic functional groups attached to the hydrocarbon backbone, such as
hydroxyl, epoxy, and triple bonds. This structural diversity, as well as the number of double
bonds and their position within the aliphatic chain, strongly affects the chemical and physical
properties of the oils [26]. When vegetable oils are used as a source of monomers to produce
polymeric materials, the double bonds, allylic positions, ester groups and other functional groups
are the main targets of chemical reactions [27].

14
During the last decade, a variety of oil-based polymeric systems have been developed and
studied, mainly address to replace or diminish the use of existing petroleum-based resins.
Unmodified plant oils have been used to prepare renewable polymers by thermal [28], and
cationic [29] polymerization methods. Copolymerization is a common practice in polymer
science, and it is used to modify the properties of manufactured plastics to meet specific needs of
the market and final application. Plant oils have not been an exception to this approach, and
several investigations have been published concerning the copolymerization of unmodified oils
with common crosslinking agents such as styrene, divinylbenzene, norbornadiene, and
dicyclopentadiene [30,31]. Less common commercial agents such as 4-vinylphenyl boronic acid
and tris(4-vinylphenyl)boroxine have been also studied [32]. By varying the structure and
stoichiometry of the alkene comonomers and the vegetable oils, polymers ranging from soft to
hard rubbers can be obtained. However, this approach generates partially bio-based resins, and
for obtaining more rigid and crosslinked materials; higher amounts of these expensive
commercial comonomers are required [33].
Due the richness of the hydrocarbon chemistry, several oil-based polymeric materials can be
synthesized by introducing new functional groups through reactions or chemical modifications.
Most of the natural unsaturated oils contain cis-configured alkenes allowing, in principle, the
application of well-known reaction of petrochemical alkenes. Remarkably, only very few
reactions across the double bond of unsaturated oils are currently applied in chemical industry.
The key concept behind chemical modification is to reach a higher level of molecular weight and
crosslinking density, factors that are known to deeply impact stiffness in the polymer network
[34]. This can be accomplished by several pathways. From natural triglycerides, it is possible to

15
attach maleates [35], epoxy [36], acrylate [37], allylic double bonds [38], amine [39,40], and
hydroxyl [41] functionalities. Such transformations make the triglyceride capable of reaction via
ring opening, free radial or polycondensation reactions, which are common mechanisms to
polymerize commercial monomers. These approaches involve the modification of triglycerides.
A different method of chemical modification is to convert the triglyceride into monoglycerides
through glycerolysis or amidation reactions. An extension of this method implies the
functionalization of the unsaturated sites, and then a reduction of the triglyceride into
monoglycerides. These derivatives have found applications as coatings [34].
Recently, relatively more advanced polymerization methods such as ring opening metathesis
(ROMP) of modified oils containing norbornene moiesties have been employed to prepare
macromolecular materials out of vegetable oils [42]. Likewise, in the composites field, glass
fiber bio-based composites have been prepared by the same technique using a vegetable oil
derivative bearing an unsaturated bicyclic moiety and dicyclopentadiene [43]. A thorough and
vast review about possible reactions using oil and fats as substrates is given by Biermann et al.
[44]. From the literature review, it was been very clear that all of the above mentioned
approaches lead to polymeric materials which are not fully, but partially, derived from biomass.
Evidently, there is a need to provide materials with higher contributions of biomass derived
compounds per weight basis.

16
Terpenes
In contrast to common petroleum derived monomers for the plastic industry, such as ethylene
with a molecular weight of 28 g/mole; triglycerides obtained from plant oils can be considered as
organic “macromonomers”, with molecular weights approaching 880 g/mol. As previously
stated, triglycerides are composed of chains with a number of carbon atoms ranging to 14 to 22
attached to a glycerol core, and these chains structurally mimic the behavior of olefins such as
ethylene, propylene, and isoprene. As one of the major classes of petroleum chemicals,
cycloaliphatic, and aromatic compounds such as styrene, divinyl benzene, etc. confer rigidity and
hydrophobicity to the polymers derived from them [45]. Besides from biomass refinery,
compounds structurally resembling cycloaliphatic and aromatic compounds can be directly
obtained from biomass. Within this category, it is of worth to mention the possibility of
exploring terpenes, terpenoids, and rosin as a class of important bearing cyclic and aromatic
structures. A vast review about sources, structure and reactivity of rosin is given by Maiti, Ray,
and Kundu [46].
Terpenes are unsaturated aliphatic structures, predominantly derived from turpentine, the volatile
fraction of resins exuded from conifers. Turpedine production amounts to approx. 350 000
tons/year. Most terpenes have a cycloaliphatic structure with isoprene (C5H8) as a carbon
skeleton building unit, and they can be classified according to the number of isoprene units [23].
Terpenoids can be considered as modified terpenes, wherein methyl groups have been moved o
removed, or oxygen atoms added [45]. Terpenes are a good starting material for the synthesis of
fine chemicals due to their similar carbon skeleton. Monoterpenes such as Pinene and Limonene
(which represent more than 90 % of the orange peel oil) are abundant and inexpensive and have

17
been extensively used as chemicals for fragrances, flavors, pharmaceuticals, and solvents [47].
Concerning their use as starters of polymeric materials, very few of these compounds have been
the subject of studies. In Chapter II, cationic copolymerization of tung oil (a natural vegetable
oil) with three different terpenes; such as limonene, myrcene and linalool as reactive
comonomers is studied and discussed. No other study has been reported on copolymerizing plant
oils with terpenes.
Sugars and starch bioproducts
As previously presented, the diversity and the worldwide production capabilities for renewable
and sustainable biomass are enormous. These two factors, along with governmental legislations
mandating increases in the gross domestic and energy and chemical production from renewable
resources, are contributing to the intensified interest in the development of technology and
processes for biomass valorization. From long ago, the chemical literature on this topic has
mostly focus on the use of biopolymers, primarily on products obtained from cellulose [48]. The
use of lignin, plant oils, terpenes, sugars, polyphenols and other biomass derived compounds as
chemical intermediate platforms to develop polymeric materials with potential structural and
composite applications stands up as a field in which considerable scientific research can be
developed.
Concerning sugar and starch bioproducts, novel bio-based high functionality epoxy resins have
been synthetized by the epoxidation of sucrose esters resins of vegetable oils fatty acids [49].
The same research group extended their work using dipentaerythritol, and tripentaerythrytol as
core polyols that were also substituted with epoxidized soybean oil fatty acids [50]. Relatively

18
recently, Chrysanthos, Galy and Pascault [51], developed a bio-based epoxy network from
isosorbide diglycidyl ether via epichlorohydrin that mimics the structure of the well-known
diglycidyl ether of bisphenol A (DGEBA). An overview of advances in sugar-based polymers is
given by Feng et al. [52]. Starch has also impacted the polymers technology. In starch-based
bioplastics starch is fully utilized with a yield very close to 100 %, whereas in starch-derived
bioplastics (synthesized from monomers resulting from the fermentation of glucose syrup). The
yield is generally less than 45 %. Polymers based on starch have a wide range of final properties,
being as flexible as polyethylene or as rigid as polystyrene [53].
Cellulose derivatives, fibers, and plastics
In the case of cellulose, making pure cellulose bioplastics from bio-sources is still highly
challenging due to the fact that it has a strong and highly structured network that cannot be melt
or dissolved by standard process such as thermoforming or dissolution [54]. Due to this
limitation, cellulose has been used as filler for biodegradable plastics and composites. Goffin et
al. [55] incorporated cellulose nanowhiskers into polylactide based composites. Likewise,
recycled cellulose fibers have been combined with bacterial polyester, PHBV, by melt mixing
technique [56].
1.5.2. Fermentation or Biological Transformation
In these processes biological agents take over the task of facilitating the synthesis of products.
This is the most-developed pathway to transform biomass into monomers via fermentation of
carbohydrates [22]. An extensive review considering the use of carbohydrates as a source of

19
compounds such as lactic acid, acids and alcohols by means of fermentations and
biotechnological conversions is given by Lichtenthaler [57].
According to the biorefinery scheme described in the biomass program of the US Department of
Energy [58], part of the biomass is converted to fuels via thermal treatments (pyrolysis and
gasification), and the other is converted by fermentation or chemo-catalytic routes. Table 1.6
provides the 12 platform chemicals that have been identified by the US Department of Energy as
starting materials to produce chemicals and polymeric materials via catalytic routes [58]. The
fermentation processes employed for the synthesis of some of these chemicals are pair with the
advances in genomics and bioprocess engineering, where new genetically modified bacteria or
yeasts continuously improve their production.
Table 1.6. Platform molecules identified by the United States Department of Energy.
Aspartic acid 1,4-Diacids (succinic, fumaric, malic)
Glutamic acid 3-hydroxypropionic acid
Levulinic acid Glycerol
2-hydroxypropionic acid Sorbitol
2,5-Furan dicarboxylic acid Xylitol / arabitol
Glucaric acid 1,4-Diacids (succinic, fumaric, malic)
Itaconic acid 3-hydroxypropionic acid
Source: P. Gallezot, Catal. Today. 121 (2007) 76–91.
From Table 1.6, glycerol needs to be highlighted as a valuable carbon source for the production
of fuels and chemicals. It is a byproduct generated during both bioethanol and biodiesel
production processes, and the massive growth of these two industrial has led to a decrease in
crude glycerol prices [59]. By means of anaerobic fermentation, the use of glycerol permits the

20
co-production of ethanol and formic acid (or ethanol and hydrogen), 1,3-propanediol, 1,2-
propanediol, succinic acid, propionic acid, ethanol and butanol [60].
1.5.3 Chemical transformation of natural polymers.
This pathway involves degradation and transformation of natural materials by means of two
main approaches, the chemical treatments and the thermochemical degradations, which are
shown in Figure 1.6. The main challenge for biorefinery is the efficient, economic, and
sustainable design of pretreatment processes allowing breaking and opening the complex
lignocellulosic structure to facilitate the obtention of chemicals and fuels [61]. These chemical
treatments have gained attention as promising methods to improve the biodegradability of
cellulose, by removing the lignin and hemicellulose fractions. Common chemical treatments
require agents such as oxidizing agents, alkali, acids, organic solvents, etc. able to break bonds,
and thus lowering the degree of polymerization, or by extracting certain compounds of interest,
such as oligomers, alcohols, glucose, xylose, etc. [62,63].
In spite the progress in the chemical treatments, the thermo-chemical conversion remains
dominant because the technologies applied to the conversion of biomass into energy and fuels
are characterized by a highly efficient and sustainable operation with low environmental impact
[62]. Biomass can be thermally treated by gasification or pyrolysis. With these processes,
complex macromolecules found in biomass are broken into smaller molecules in gaseous or
liquid state (with remaining solid products such as ash and carbon), which can subsequently be
used as building blocks for further chemical synthesis [64].
21
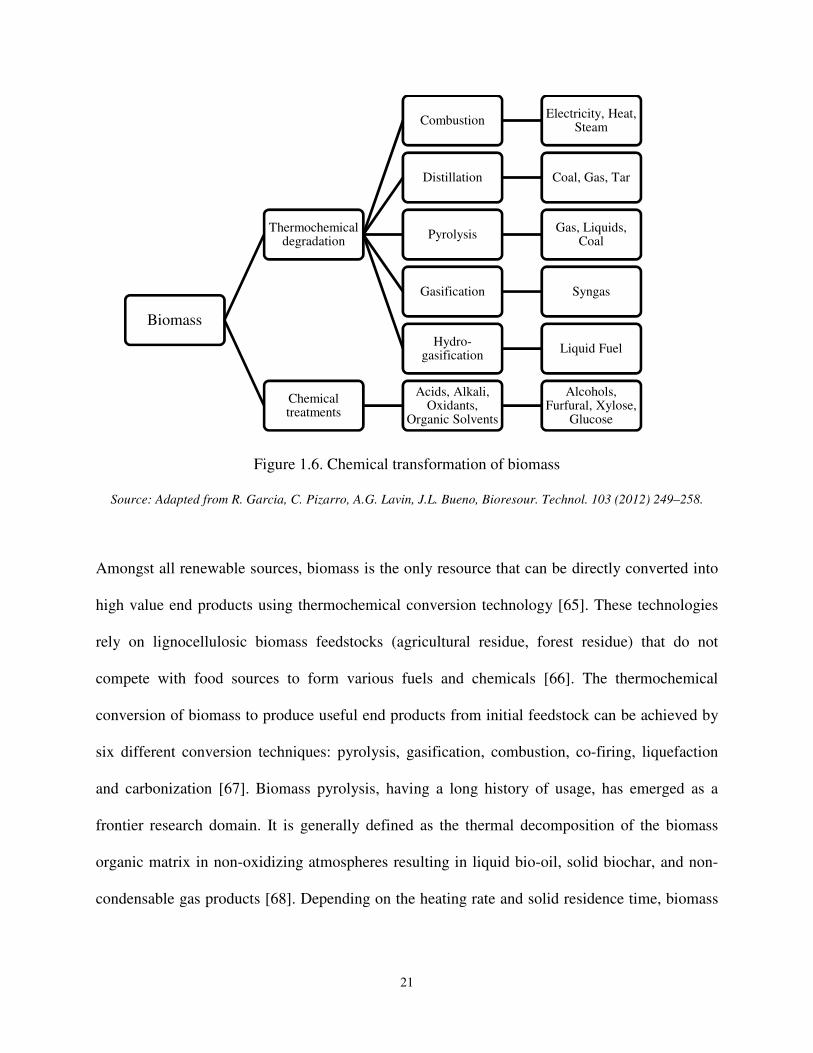
Figure 1.6. Chemical transformation of biomass
Source: Adapted from R. Garcia, C. Pizarro, A.G. Lavin, J.L. Bueno, Bioresour. Technol. 103 (2012) 249–258.
Amongst all renewable sources, biomass is the only resource that can be directly converted into
high value end products using thermochemical conversion technology [65]. These technologies
rely on lignocellulosic biomass feedstocks (agricultural residue, forest residue) that do not
compete with food sources to form various fuels and chemicals [66]. The thermochemical
conversion of biomass to produce useful end products from initial feedstock can be achieved by
six different conversion techniques: pyrolysis, gasification, combustion, co-firing, liquefaction
and carbonization [67]. Biomass pyrolysis, having a long history of usage, has emerged as a
frontier research domain. It is generally defined as the thermal decomposition of the biomass
organic matrix in non-oxidizing atmospheres resulting in liquid bio-oil, solid biochar, and non-
condensable gas products [68]. Depending on the heating rate and solid residence time, biomass
Biomass
Thermochemical degradation
CombustionElectricity, Heat,
Steam
Distillation Coal, Gas, Tar
PyrolysisGas, Liquids,
Coal
Gasification Syngas
Hydro-gasification
Liquid Fuel
Chemical treatments
Acids, Alkali, Oxidants,
Organic Solvents
Alcohols, Furfural, Xylose,
Glucose

22
pyrolysis can be divided into three main types, including slow pyrolysis, fast pyrolysis, and flash
pyrolysis [69].
Bio-oil is a product of fast pyrolysis process, which is a dark brown, free flowing organic liquid
mixture. Bio-oil generally comprises a great amount of water, and hundreds of organic
compounds, such as acids, alcohols, ketones, aldehydes, phenols, ethers, esters, sugars, furans,
alkenes, nitrogen compounds and miscellaneous oxygenates, as well as solid particles [70]. The
abundant output of reviews and research papers on bio-oils in the last years is a glaring
indication of the vigorous growth of this area. Again, a search of the Web of Science database
using the words “pyrolysis bio-oil” identified a total of more than 3000 scientific articles
currently available on the subject that were published in the last decade (Thomson Reuters
2016). Most of these investigations are concerned with the optimization of the pyrolysis
parameters [68], as well as the use of bio-oil as bio-fuels [71,72], as an alternative to heat and
power generation [73], hydrogen production [74], and valued-added chemicals [75]. Due to the
complexity of bio-oil and the multitude of its components (which in turn depend on the source of
biomass, type of pyrolysis, temperature, residence time, etc.), analysis of bio-oil content often
employs instrumental techniques such as gas chromatography (GC), gas chromatography-mass
spectrometry (GC-MS), nuclear magnetic resonance (NMR), etc. The following Table (1.7)
depicts a model chemical group classification for bio-oil derived from wheat-wood sawdust [76].

23
Table 1.7. Chemical classification of bio-oil components
Chemical Classes Amount present in bio-oil (%)
Hydrocarbons 2.6
Low molecular weight fatty acids/esters 13.7
Hexadecanoic acid 23.6
Furanoids 3.2
Pyranoids 1.2
Benzenoid hydrocarbons 2.6
Oxygenated benzenoids 14.6
Low mol. wt. alcohols / aldehydes / ketones 1.7
High molecular weight alcohols 19.5
High molecular weight waxy compounds 2.1
Source: K. Jacobson, K.C. Maheria, A. Kumar Dalai, Renew. Sustain. Energy Rev. 23 (2013) 91–106.
All these components are derived primarily from depolymerization and fragmentation reactions
of the three building blocks of lignocellulosic biomass: cellulose, hemicellulose, and lignin. The
phenols, guaiacols, and syringols are formed from the lignin fraction. Miscellaneous oxygenates,
sugars, and furans are produced from cellulose and hemicellulose. The esters, acids, alcohols,
ketones, and aldehydes probably form as bi-products of the decomposition of oxygenated
compounds, furans, and sugars [19]. As seen, these pyrolysis technologies based on biomass
refinery appear as a good alternative to stand as a sustainable way for chemicals biosourcing.
However, in spite of the potential of the wood chemicals and forest derivatives, the biomass
refinery still facing several challenges that have limited its spread and extensive exploitation.
These factors include the feedstock diversity, biomass collection and transportation logistics,
seasonal variation, lad usage, compatibility with refinery infrastructure and facilities, market and
economic viability, sustainability and consistent R&D investments [77].

24
Concerning the last factor, government, academia, and industry have made significant
contributions in developing feedstocks and technologies to impulse biorefinery. Nevertheless,
many of these technologies remain in early stages of development. This stands as the key reason
why only very few studies considering the use of biorefinery products in the polymer synthesis
have been reported. On-going and consistent support, achieved through research and scientific
understanding, is essential to promote the development of this field and to make biorefinery a
profitable industry. In traditional bio-refinery, ethanol has provided opportunities to produce
hydrocarbon fuels and varieties of chemicals besides its application as fuel or fuels additive.
Ethanol can be converted into diethyl ether, ethylene, higher hydrocarbons or aromatics over
zeolites catalysts [78]. Likewise, efforts have been also focus on bio-butanols as a source of
chemicals and chemicals. At present, butanol has a wide range of market potentials as precursor
of solvents like butyl acetate, dibutyl acetate, butyl glycol ethers and butyl propionate. The
isobutanol has also potential as feedstock for synthetic rubbers, plastics and polyesters; such as
butyl acrylate, butyl methacrylate and butyl vinyl ether [78].
In the case of lignin, only small amounts of this material are presently used for commercial
purposes. Almost all lignin that is currently commercially used in non-energy applications is
ligno-sulfonate produced from spent sulfite pulping liqueurs in cement applications [79]. Issues
related to low reactivity associated to steric hindrance have surrounded the lignin for a long time;
however efforts to use lignin for the production of enhanced composites, carbon fibers, and
nanomaterials are found in literature [80]. Regarding the use of bio-oil as a source of chemicals
for macromolecular synthesis, very few studies have been published so far. Bio-oil chemistry
still in an early stage of development, and considerable work needs to be performed in order to

25
make it a profitable and fruitful source of chemicals. As it will be detailed later in Chapter V, this
fact opens up a huge opportunity to perform scientific research on this field; and the crude bio-oil
could serve as raw materials for the obtention and production of specialty chemicals and resins,
such as epoxy, phenol-formaldehyde, polyurethane, acrylic, etc.
Presently, the use of bio-oil in the polymer science has been addressed to partially replace
commercial resins. Wei et al. (2014) studied the use of bio-oil derived from switchgrass to cure a
commercial epoxy EPON 828 resin. Liquefaction time and temperature, as well as the optimal
epoxy / bio-oil ratio were studied [81]. On the same verge, Celikbag et al (2014) employed bio-
oil derived from loblolly pine to also crosslink EPON 828 [82]. The effect of the liquefaction
temperature and time on the hydroxyl number, as well as its impact of the thermo-mechanical
properties of the obtained materials were assessed.
Recently, vanillin and its derivatives have attracted attention as renewable building blocks for
high performance polymers, mainly because of its aromatic nature. Vanillin, and similar
aromatic compounds, has been identified in pyrolysis bio-oils [83]. Zhang et al. copolymerized
bio-monomer-methacrylated vanillin with acrylated soybean oil at different ratios [84]. The
materials were studied by dynamic-mechanical analysis, thermo-gravimetric and tensile tests.

26
1.6. Research objective
According to what has been reviewed, it is clear that the development of green polymers is
growing, and that it is supported not only by the industrial sectors, but also by governmental
entities and agencies. In order to approach the exploitation of natural resources as a source of
raw materials for the polymer industry, four different strategies can be defined [85], namely:
(1) The preparation of monomers which are already widely used, as obtained from fossil
resources.
(2) The synthesis of polymers which are intended to simulate the performance of existing
counterparts from non-renewable sources.
(3) Original materials with novel properties and applications.
(4) The chemical or physical modification of natural polymers with the intent of improving or
radically modifying their pristine properties.
The second strategy constitutes the corner stone of this research, and can be used meaningfully in
the construction and understanding of the work that is presented in the following sections; which
deal with the synthesis of novel polymers, intended to simulate the performance of existing
counterparts from non-renewable sources. Therefore, the objective of this project is the use of
monomers or macromonomers derived from the biomass, which bear reactive sites that can be
exploited, as such, or after appropriate modifications, to generate novel macro-molecular
architectures. Many studies have revolved around the development of biomass derived materials;
however, the obtention of polymers with good thermo-mechanical properties remains as a major
challenge, as well as the development of materials with higher contributions of biomass-derived
compounds per weight basis. This research stands as an effort to contribute to overcome these

27
difficulties, by producing biomass-derived thermosetting polymers with properties comparable to
their commercial counterparts. This document is divided into five sections. The first one explains
the overall organization of the work. The rest of them present an introduction, which main
purpose is to provide an insight about the major theoretical and technical aspects of the contents
and experimental results that are subsequently detailed.
The second section discusses the cationic copolymerization of tung oil, limonene and myrcene as
comonomers, initiated by boron trifluoride. Dynamic mechanical analysis (DMA) revealed that
all copolymers behave as thermosets. FTIR spectra for both copolymers, after extraction with
dichloromethane, suggested that the major component of the insoluble fraction was reacted tung
oil (a cross-linked triglyceride network). Likewise, unreacted tung oil was found to be the main
component of the soluble phase. Also, all the copolymers showed only one tan δ peak, indicating
no phase separation. Glass transition temperature (Tg) increased with the myrcene content and
decreased almost linearly as the limonene content increased. Furthermore, the Fox and Loshaek
model showed a relatively good prediction of the Tg values of the polymers. The Young`s
modulus ranged from 33.8 to 4.7 MPa for all tested thermosets.
The third one focuses on the chemical modification of two different functional groups of biomass
derived materials; the carbon-carbon double bonds of triglycerides, and the hydroxyl groups of
phenolic compounds. These chemical modifications were addressed to introduce more reactive
functional groups able to polymerize and to produce materials with a better thermo-mechanical
performance. Efforts are centered on the epoxy functional groups, and the feasibility of using
step-growth or chain-growth polymerizations for their conversion into macromolecular materials

28
is also assessed. Linseed oil was epoxidized using a peracetic acid generated in situ from reaction
between acetic acid and hydrogen peroxide, meanwhile α-resorcylic acid was reacted with
epichlorohydrin in alkaline medium using benzyltriethylammonium chloride as phase transfer
catalyst. The epoxy content of the modified monomers was measured by means of a titration
using HBr in acetic acid solution, and the grafting of epoxy functions onto the monomer`s
structure was studied by FTIR. The epoxidized derivatives of α-resorcylic acid and linseed oil
were cured in epoxy polymer with DMPA. Thermo-mechanical characterization showed that the
obtained materials behave as thermoset amorphous polymers, exhibiting moduli values in the
order of Giga Pascals at room temperature and glass transition temperatures above 100 °C. SEM
images confirmed the glassy brittle nature of the materials and revealed possible thermodynamic
incompatibility of these two compounds. When compared to samples of commercial origin,
testing showed the mechanical properties of the triglycidylated α-RA based resins are similar to
those shown by EPON 828 epoxy resin.
The fourth focus on the synthesis of interpenetrating polymer networks (IPNs) using an epoxy
phase synthesized either from linseed oil or α-resorcylic acid, and an acrylate phase employing
acrylated soybean oil are the main focus of this study. Thermo-mechanical characterization
showed that the obtained materials behave as thermoset amorphous polymers, and that both the
modulus and glass transition are extremely dependent on the epoxy/acrylate weight ratio, as well
as on the chemical structure of the epoxy monomers. In the case of the systems epoxidized
linseed oil / acrylated soybean oil, the mechanical properties of the IPNs were not superior to
those of the constituent polymers as a result of the plasticizing effect of the triglyceride flexible
chains in the macromolecular assembly of the IPNs. However, IPNs based on epoxidized α-

29
resorcylic acid and acrylated soy bean oil provided considerably better mechanical properties
than the system epoxidized linseed oil and acrylated soy bean oil. For the α-resorcylic acid and
soy bean oil system, modulus values ranged from 0.7 to 3.3 GPa at 30 °C, and glass transition
temperatures ranging from around 58 °C to approx. 130 °C. Both parameters were affected by
the epoxy/acrylate ratio, showing an increase as the epoxy proportion was enlarged.
Interpenetrating networks containing equivalent weight proportions of the parent resins showed
the highest fracture toughness of the series, exhibiting a KIc value of around 2.1 MPa·m1/2.
The final section discusses the use of fast pyrolysis bio-oils as precursors of thermosetting epoxy
resins. In this context, with the objective to synthetize a high performance bio-based epoxy
polymer, fast pyrolysis bio-oil (containing lignin fragments) was employed as a source of
phenolic compounds in the production of a bio-based polymeric network. The bio-oil was
reacted with epichlorohydrin and an aqueous solution of sodium hydroxide in the presence of
benzyltriethylammonium chloride as a phase transfer catalyst. The amount of free phenolic
hydroxyl groups before and after modification was quantified through P31-NMR spectroscopy;
and the epoxy content of the bio-oil upon chemical functionalization was measured by means of
a titration using HBr in acetic acid solution. Grafting of epoxy functions onto the monomer`s
structure was studied by FTIR. Likewise, α-resorcylic acid was also modified with reactive
epoxy moieties and used as a model to study the mechanical behavior of phenolic-based epoxy
polymers. The epoxidized derivatives of the bio-oil were cured in epoxy polymer with DMPA.
Thermo-mechanical characterization showed that the obtained materials behave as thermoset
amorphous polymers, exhibiting moduli values in the order of Giga Pascals at room temperature
and glass transition temperatures above 100 °C.

30
References
[1] M. Biron, Thermosets and Composites: Material Selection, Applications, Manufacturing
and Cost Analysis, Elsevier Science, 2013.
https://books.google.com/books?id=IZwlAAAAQBAJ&pgis=1 (accessed May 30, 2016).
[2] Metef & Golf- Executive-Summary-inglese.pdf, (n.d.). http://www.ambrosetti.eu/wp-
content/uploads/Executive-Summary-inglese.pdf (accessed May 30, 2016).
[3] Global Thermosetting Plastics Market - Segmented by Type, Industry and Geography -
Trends and, (n.d.). http://www.prnewswire.com/news-releases/global-thermosetting-
plastics-market---segmented-by-type-industry-and-geography---trends-and-forecasts-
2015-2020---reportlinker-review-300145372.html (accessed May 30, 2016).
[4] U. Szeluga, B. Kumanek, B. Trzebicka, Synergy in hybrid polymer/nanocarbon
composites. A review, Compos. Part A Appl. Sci. Manuf. 73 (2015) 204–231.
doi:10.1016/j.compositesa.2015.02.021.
[5] J. Karger-Kocsis, H. Mahmood, A. Pegoretti, Recent advances in fiber/matrix interphase
engineering for polymer composites, Prog. Mater. Sci. 73 (2015) 1–43.
doi:10.1016/j.pmatsci.2015.02.003.
[6] T. Väisänen, A. Haapala, R. Lappalainen, L. Tomppo, Utilization of agricultural and
forest industry waste and residues in natural fiber-polymer composites: A review, Waste
Manag. 54 (2016) 62–73. doi:10.1016/j.wasman.2016.04.037.
[7] C. Williams, M. Hillmyer, Polymers from Renewable Resources: A Perspective for a
Special Issue of Polymer Reviews, Polym. Rev. 48 (2008) 1–10.
doi:10.1080/15583720701834133.

31
[8] Bioplastics – European Bioplastics e.V., (n.d.). http://www.european-
bioplastics.org/bioplastics/ (accessed May 30, 2016).
[9] Bioplastics - Sustainable Plastics, (n.d.).
https://sites.google.com/site/sustainableplastics/bioplastics (accessed May 30, 2016).
[10] I. Delidovich, P.J.C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose, R. Palkovits,
Alternative Monomers Based on Lignocellulose and Their Use for Polymer Production,
Chem. Rev. 116 (2016) 1540–1599. doi:10.1021/acs.chemrev.5b00354.
[11] Biomass as Feedstock for a Bioenergy and Bioproducts Industry: The Technical
Feasibility of a Billion-Ton Annual Supply - final_billionton_vision_report2.pdf, (n.d.).
https://www1.eere.energy.gov/bioenergy/pdfs/final_billionton_vision_report2.pdf
(accessed May 30, 2016).
[12] C. Li, X. Zhao, A. Wang, G.W. Huber, T. Zhang, Catalytic Transformation of Lignin for
the Production of Chemicals and Fuels, Chem. Rev. 115 (2015) 11559–11624.
doi:10.1021/acs.chemrev.5b00155.
[13] N. Scarlat, J.F. Dallemand, F. Monforti-Ferrario, V. Nita, The role of biomass and
bioenergy in a future bioeconomy: Policies and facts, Environ. Dev. 15 (2015) 3–34.
doi:10.1016/j.envdev.2015.03.006.
[14] NOVA -Market-Study-on-Bio-based-Polymers.pdf, (n.d.).
https://biobs.jrc.ec.europa.eu/sites/default/files/generated/files/stakeholders/NOVA -
Market-Study-on-Bio-based-Polymers.pdf (accessed May 30, 2016).
[15] H.P. Meyer, Sustainability and biotechnology, Org. Process Res. Dev. 15 (2011) 180–188.
doi:10.1021/op100206p.

32
[16] A. Iles, A.N. Martin, Expanding bioplastics production: Sustainable business innovation
in the chemical industry, J. Clean. Prod. 45 (2013) 38–49.
doi:10.1016/j.jclepro.2012.05.008.
[17] A. Corma Canos, S. Iborra, A. Velty, Chemical routes for the transformation of biomass
into chemicals, Chem. Rev. 107 (2007) 2411–2502. doi:10.1021/cr050989d.
[18] R.T. Mathers, How well can renewable resources mimic commodity monomers and
polymers?, J. Polym. Sci. Part A Polym. Chem. 50 (2012) 1–15. doi:10.1002/pola.24939.
[19] G.W. Huber, I. Sara, A. Corma, Synthesis of Transportation Fuels from Biomass, Chem
Rev. 2 (2006) 4044–4098.
[20] S. Mecking, Nature or petrochemistry? - Biologically degradable materials, Angew.
Chemie - Int. Ed. 43 (2004) 1078–1085. doi:10.1002/anie.200301655.
[21] A. Soroudi, I. Jakubowicz, Recycling of bioplastics, their blends and biocomposites: A
review, Eur. Polym. J. 49 (2013) 2839–2858. doi:10.1016/j.eurpolymj.2013.07.025.
[22] K. Yao, C. Tang, Controlled polymerization of next-generation renewable monomers and
beyond, Macromolecules. 46 (2013) 1689–1712. doi:10.1021/ma3019574.
[23] A. Gandini, M.N. Belgacem, Lignins as components of macromolecular materials, in
M.N. Belgacem, A. Gandini (EDs.), Monomers, Polymers and Composites, Elsevier, UK,
2008, pp. 243-272.
[24] L. Montero De Espinosa, M.A.R. Meier, Plant oils: The perfect renewable resource for
polymer science?, Eur. Polym. J. 47 (2011) 837–852.
doi:10.1016/j.eurpolymj.2010.11.020.

33
[25] F. Seniha Güner, Y. Yaǧci, A. Tuncer Erciyes, Polymers from triglyceride oils, Prog.
Polym. Sci. 31 (2006) 633–670. doi:10.1016/j.progpolymsci.2006.07.001.
[26] A. Gandini, T.M. Lacerda, A.J.F. Carvalho, E. Trovatti, Progress of Polymers from
Renewable Resources: Furans, Vegetable Oils, and Polysaccharides, Chem. Rev. (2015)
150820135704001. doi:10.1021/acs.chemrev.5b00264.
[27] S. Miao, P. Wang, Z. Su, S. Zhang, Vegetable-oil-based polymers as future polymeric
biomaterials, Acta Biomater. 10 (2014) 1692–1704. doi:10.1016/j.actbio.2013.08.040.
[28] F. Li, R.C. Larock, Synthesis, structure and properties of new tung oil-styrene-
divinylbenzene copolymers prepared by thermal polymerization., Biomacromolecules. 4
(2003) 1018–25. doi:10.1021/bm034049j.
[29] F. Li, M.. Hanson, R.. Larock, Soybean oil–divinylbenzene thermosetting polymers:
synthesis, structure, properties and their relationships, Polymer (Guildf). 42 (2001) 1567–
1579. doi:10.1016/S0032-3861(00)00546-2.
[30] F. Li, R.C. Larock, New soybean oil-styrene-divinylbenzene thermosetting copolymers.
V. Shape memory effect, J. Appl. Polym. Sci. 84 (2002) 1533–1543.
doi:10.1002/app.10493.
[31] D.P. Pfister, R.C. Larock, Thermophysical properties of conjugated soybean oil/corn
stover biocomposites, Bioresour. Technol. 101 (2010) 6200–6206.
doi:10.1016/j.biortech.2010.02.070.
[32] M. Sacristan, J.C. Ronda, M. Galias, V. Cadiz, Synthesis and properties of boron-
containing soybean oil based thermosetting copolymers, Polymer (Guildf). 51 (2010)
6099–6106. doi:10.1016/j.polymer.2010.11.003.

34
[33] D.D. Andjelkovic, R.C. Larock, Novel rubbers from cationic copolymerization of soybean
oils and dicyclopentadiene. 1. Synthesis and characterization, Biomacromolecules. 7
(2006) 927–936. doi:10.1021/bm050787r.
[34] S.N. Khot, J.J. Lascala, E. Can, S.S. Morye, G.I. Williams, G.R. Palmese, et al.,
Development and application of triglyceride-based polymers and composites, J. Appl.
Polym. Sci. 82 (2001) 703–723. doi:10.1002/app.1897.
[35] J.M. Ferri, O. Fenollar, R. Balart, The effect of maleinized linseed oil ( MLO ) on
mechanical performance of poly ( lactic acid ) -thermoplastic starch ( PLA-TPS ) blends,
147 (2016) 60–68.
[36] R. Mungroo, V. V. Goud, S.N. Naik, A.K. Dalai, Utilization of green seed canola oil for
in situ epoxidation, Eur. J. Lipid Sci. Technol. 113 (2011) 768–774.
doi:10.1002/ejlt.201000500.
[37] P. Zhang, J. Zhang, One-step acrylation of soybean oil (SO) for the preparation of SO-
based macromonomers, Green Chem. 15 (2013) 641. doi:10.1039/c3gc36961g.
[38] Q. Luo, M. Lui, Y. Xu, M. Ionescu, Z.S. Petrovic, Thermosetting allyl resins derived from
soybean fatty acids, J. Appl. Polym. Sci. 127 (2013) 432–438. doi:10.1002/app.37814.
[39] O. Türünç, M. Firdaus, G. Klein, M. a. R. Meier, Fatty acid derived renewable polyamides
via thiol–ene additions, Green Chem. 14 (2012) 2577. doi:10.1039/c2gc35982k.
[40] M. Stemmelen, F. Pessel, V. Lapinte, S. Caillol, J.P. Habas, J.J. Robin, A fully biobased
epoxy resin from vegetable oils: From the synthesis of the precursors by thiol-ene reaction
to the study of the final material, J. Polym. Sci. Part A Polym. Chem. 49 (2011) 2434–
2444. doi:10.1002/pola.24674.

35
[41] S. Caillol, M. Desroches, G. Boutevin, C. Loubat, R. Auvergne, B. Boutevin, Synthesis of
new polyester polyols from epoxidized vegetable oils and biobased acids, Eur. J. Lipid
Sci. Technol. 114 (2012) 1447–1459. doi:10.1002/ejlt.201200199.
[42] P.H. Henna, R.C. Larock, Rubbery thermosets by ring-opening metathesis polymerization
of a functionalized castor oil and cyclooctene, Macromol. Mater. Eng. 292 (2007) 1201–
1209. doi:10.1002/mame.200700209.
[43] P.H. Henna, M.R. Kessler, R.C. Larock, Fabrication and properties of vegetable-oil-based
glass fiber composites by ring-opening metathesis polymerization, Macromol. Mater. Eng.
293 (2008) 979–990. doi:10.1002/mame.200800202.
[44] U. Biermann, U. Bornscheuer, M.A.R. Meier, J.O. Metzger, H.J. Schaefer, Oils and fats as
renewable raw materials in chemistry, Angew. Chemie - Int. Ed. 50 (2011) 3854–3871.
doi:10.1002/anie.201002767.
[45] P.A. Wilbon, F. Chu, C. Tang, Progress in renewable polymers from natural terpenes,
terpenoids, and rosin, Macromol. Rapid Commun. 34 (2013) 8–37.
doi:10.1002/marc.201200513.
[46] S. Maiti, S.S. Ray, A.K. Kundu, Rosin: a renewable resource for polymers and polymer
chemicals, Prog. Polym. Sci. 14 (1989) 297–338. doi:10.1016/0079-6700(89)90005-1.
[47] J.L. Bicas, A.P. Dionisio, G.M. Pastore, Bio-oxidation of terpenes: An approach for the
flavor Industry, Chem. Rev. 109 (2009) 4518–4531. doi:10.1021/cr800190y.
[48] J. Zakzeski, P.C.A. Bruijnincx, A.L. Jongerius, B.M. Weckhuysen, The Catalytic
Valorization of Ligning for the Production of Renewable Chemicals, Chem. Rev. 110
(2010) 3552–3599. doi:10.1021/cr900354u.

36
[49] X. Pan, P. Sengupta, D.C. Webster, Novel biobased epoxy compounds: epoxidized
sucrose esters of fatty acids, Green Chem. 13 (2011) 965. doi:10.1039/c0gc00882f.
[50] X. Pan, D.C. Webster, Impact of Structure and Functionality of Core Polyol in Highly
Functional Biobased Epoxy Resins., Macromol. Rapid Commun. (2011) 1324–1330.
doi:10.1002/marc.201100215.
[51] M. Chrysanthos, J. Galy, J.-P. Pascault, Preparation and properties of bio-based epoxy
networks derived from isosorbide diglycidyl ether, Polymer (Guildf). 52 (2011) 3611–
3620. doi:10.1016/j.polymer.2011.06.001.
[52] X. Feng, A.J. East, W.B. Hammond, Y. Zhang, M. Jaffe, Overview of advances in sugar-
based polymers, Polym. Adv. Technol. 22 (2011) 139–150. doi:10.1002/pat.1859.
[53] C. Bastioli, P. Magistrali, S. Gesti Garcia, Starch in polymers technology, ACS Symp.
Ser. 1114 (2012) 87–112. doi:10.1021/bk-2012-1114.ch007.
[54] I.S. Bayer, S. Guzman-Puyol, J.A. Heredia-Guerrero, L. Ceseracciu, F. Pignatelli, R.
Ruffilli, et al., Direct transformation of edible vegetable waste into bioplastics,
Macromolecules. 47 (2014) 5135–5143. doi:10.1021/ma5008557.
[55] A.-L. Goffin, J.-M. Raquez, E. Duquesne, G. Siqueira, Y. Habibi, A. Dufresne, et al.,
From Interfacial Ring-Opening Polymerization to Melt Processing of Cellulose
Nanowhisker-Filled Polylactide-Based Nanocomposites, Biomacromolecules. 12 (2011)
2456–2465. doi:10.1021/bm200581h.
[56] R. Bhardwaj, A.K. Mohanty, L.T. Drzal, F. Pourboghrat, M. Misra, Renewable resource-
based green composites from recycled cellulose fiber and poly(3-hydroxybutyrate-co-3-
hydroxyvalerate) bioplastic, Biomacromolecules. 7 (2006) 2044–2051.
doi:10.1021/bm050897y.

37
[57] M. Cadek, O. Vostrowsky, a Hirsch, Carbon, 7. Fullerenes and Carbon Nanomaterials,
Ullmann’s Encycl. Ind. Chem. (2012) 23–44. doi:10.1002/14356007.n05.
[58] P. Gallezot, Catalytic routes from renewables to fine chemicals, Catal. Today. 121 (2007)
76–91. doi:10.1016/j.cattod.2006.11.019.
[59] S.S. Yazdani, R. Gonzalez, Anaerobic fermentation of glycerol: a path to economic
viability for the biofuels industry, Curr. Opin. Biotechnol. 18 (2007) 213–219.
doi:10.1016/j.copbio.2007.05.002.
[60] J.M. Clomburg, R. Gonzalez, Anaerobic fermentation of glycerol: A platform for
renewable fuels and chemicals, Trends Biotechnol. 31 (2013) 20–28.
doi:10.1016/j.tibtech.2012.10.006.
[61] R. Travaini, J. Martin-Juarez, A. Lorenzo-Hernando, S. Bolado-Rodriguez, Ozonolysis:
An advantageous pretreatment for lignocellulosic biomass revisited, Bioresour. Technol.
199 (2016) 2–12. doi:10.1016/j.biortech.2015.08.143.
[62] R. Garcia, C. Pizarro, A.G. Lavin, J.L. Bueno, Characterization of Spanish biomass wastes
for energy use, Bioresour. Technol. 103 (2012) 249–258.
doi:10.1016/j.biortech.2011.10.004.
[63] S. Behera, R. Arora, N. Nandhagopal, S. Kumar, Importance of chemical pretreatment for
bioconversion of lignocellulosic biomass, Renew. Sustain. Energy Rev. 36 (2014) 91–106.
doi:10.1016/j.rser.2014.04.047.
[64] M. Narodoslawsky, A. Niederl-Schmidinger, L. Halasz, Utilising renewable resources
economically: new challenges and chances for process development, J. Clean. Prod. 16
(2008) 164–170. doi:10.1016/j.jclepro.2006.08.023.

38
[65] C.N. Hamelinck, A.P.C. Faaij, Outlook for advanced biofuels, Energy Policy. 34 (2006)
3268–3283. doi:10.1016/j.enpol.2005.06.012.
[66] I.M. Area, Biomass resource facilities and biomass conversion processing for fuels and
chemicals Biomass resource facilities and biomass conversion processing for fuels and
chemicals, 42 (2014) 2–3.
[67] M. Patel, X. Zhang, A. Kumar, Techno-economic and life cycle assessment on
lignocellulosic biomass thermochemical conversion technologies: A review, Renew.
Sustain. Energy Rev. 53 (2016) 1486–1489. doi:10.1016/j.rser.2015.09.070.
[68] T. Kan, V. Strezov, T.J. Evans, Lignocellulosic biomass pyrolysis: A review of product
properties and effects of pyrolysis parameters, Renew. Sustain. Energy Rev. 57 (2016)
126–1140. doi:10.1016/j.rser.2015.12.185.
[69] M. Balat, M. Balat, E. Kirtay, H. Balat, Main routes for the thermo-conversion of biomass
into fuels and chemicals. Part 1: Pyrolysis systems, Energy Convers. Manag. 50 (2009)
3147–3157. doi:10.1016/j.enconman.2009.08.014.
[70] P.S. Rezaei, H. Shafaghat, W.M.A.W. Daud, Production of green aromatics and olefins by
catalytic cracking of oxygenate compounds derived from biomass pyrolysis: A review,
Appl. Catal. A Gen. 469 (2014) 490–511. doi:10.1016/j.apcata.2013.09.036.
[71] J. Lehto, A. Oasmaa, Y. Solantausta, M. Kytö, D. Chiaramonti, Review of fuel oil quality
and combustion of fast pyrolysis bio-oils from lignocellulosic biomass, Appl. Energy. 116
(2014) 178–190. doi:10.1016/j.apenergy.2013.11.040.
[72] A. Krutof, K. Hawboldt, Blends of pyrolysis oil, petroleum, and other bio-based fuels: A
review, Renew. Sustain. Energy Rev. 59 (2016) 406–419. doi:10.1016/j.rser.2015.12.304.

39
[73] S.Y. No, Application of bio-oils from lignocellulosic biomass to transportation, heat and
power generation - A review, Renew. Sustain. Energy Rev. 40 (2014) 1108–1125.
doi:10.1016/j.rser.2014.07.127.
[74] S. Ayalur Chattanathan, S. Adhikari, N. Abdoulmoumine, A review on current status of
hydrogen production from bio-oil, Renew. Sustain. Energy Rev. 16 (2012) 2366–2372.
doi:10.1016/j.rser.2012.01.051.
[75] D. Shen, W. Jin, J. Hu, R. Xiao, K. Luo, An overview on fast pyrolysis of the main
constituents in lignocellulosic biomass to valued-added chemicals: Structures, pathways
and interactions, Renew. Sustain. Energy Rev. 51 (2015) 761–774.
doi:10.1016/j.rser.2015.06.054.
[76] K. Jacobson, K.C. Maheria, A. Kumar Dalai, Bio-oil valorization: A review, Renew.
Sustain. Energy Rev. 23 (2013) 91–106. doi:10.1016/j.rser.2013.02.036.
[77] S.K. Maity, Opportunities, recent trends and challenges of integrated biorefinery: Part I,
Renew. Sustain. Energy Rev. 43 (2015) 1427–1445. doi:10.1016/j.rser.2014.11.092.
[78] S.K. Maity, Opportunities, recent trends and challenges of integrated biorefinery: Part II,
Renew. Sustain. Energy Rev. 43 (2015) 1446–1466. doi:10.1016/j.rser.2014.08.075.
[79] L. Axelsson, M. Franzén, M. Ostwald, G. Berndes, G. Lakshmi, N.H. Ravindranath,
Perspective: Jatropha cultivation in southern India: Assessing farmers’ experiences,
Biofuels, Bioprod. Biorefining. 6 (2012) 246–256. doi:10.1002/bbb.
[80] E. Ten, W. Vermerris, Recent developments in polymers derived from industrial lignin, J.
Appl. Polym. Sci. 132 (2015) 1–13. doi:10.1002/app.42069.

40
[81] N. Wei, B.K. Via, Y. Wang, T. McDonald, M.L. Auad, Liquefaction and substitution of
switchgrass (Panicum virgatum) based bio-oil into epoxy resins, Ind. Crops Prod. 57
(2014) 116–123. doi:10.1016/j.indcrop.2014.03.028.
[82] Y. Celikbag, B.K. Via, S. Adhikari, Y. Wu, Effect of liquefaction temperature on
hydroxyl groups of bio-oil from loblolly pine (Pinus taeda), Bioresour. Technol. 169
(2014) 808–811. doi:10.1016/j.biortech.2014.07.075.
[83] M. Bertero, G. De La Puente, U. Sedran, Fuels from bio-oils: Bio-oil production from
different residual sources, characterization and thermal conditioning, Fuel. 95 (2012) 263–
271. doi:10.1016/j.fuel.2011.08.041.
[84] C. Zhang, M. Yan, E.W. Cochran, M.R. Kessler, Biorenewable polymers based on
acrylated epoxidized soybean oil and methacrylated vanillin, Mater. Today Commun. 5
(2015) 18–22. doi:10.1016/j.mtcomm.2015.09.003.
[85] A. Gandini, Polymers from Renewable Resources: A Challenge for the Future of
Macromolecular Materials, Macromolecules. 41 (2008) 9491–9504.
doi:10.1021/ma801735u.

41
CHAPTER II
Renewable Thermoset Copolymers from Tung Oil and Natural Terpenes
1. Introduction
The search for sustainable chemistry has led to the development of new synthetic routes to
produce polymers from renewable resources. Lately, particular attention has been paid to
biomass as a source of a wide range of biomaterials. Amongst many biomass derived materials,
it is important to highlight the potential use of vegetable oils. The employment of these materials
as platform chemical for polymer synthesis has several advantages, which include availability,
inherent biodegradability, high purity and low toxicity [1]. Vegetable oils or triglycerides are
fatty acid esters of the triol glycerol. Basically, a fatty acid consists of a hydrophobic
hydrocarbon chain with a hydrophilic group at one end. The degree of unsaturation varies
according to the composition of fatty acids presenting double bonds. This value, one of the most
important parameters in the chemistry of fatty acids and triglycerides, can be calculated through
the iodine value, a structural index which has been related to various physical and chemical
properties and has served as a quality control method in hydrogenation reactions [2].
Likewise, besides the carbon-carbon double bonds, some plant oils exhibit fatty acids containing
other functional groups such as hydroxyls, cyclic groups, epoxy groups, etc. which are amenable
for developing chemical reactions and structural modifications. According to the literature, there
are three main routes for the synthesis of polymers from plant oils. The first approach

42
corresponds to the direct polymerization through the C=C bonds [3,4]. Their presence allows the
polymerization of the oils into solid polymeric materials either through free radical or cationic
mechanisms [5–7]. The second route consists of the functionalization of the double bonds to
insert more reactive groups, usually epoxy, amine or hydroxyl, etc. [8,9]. They can be considered
as an intermediate product for subsequent reactions to yield bio-based polymers such as
polyurethane, polyester, etc. The third approach focuses on the chemical transformation of
vegetable oils to produce platform chemical which can be used to produce monomers for the
polymer synthesis [10].
Regarding the direct polymerization of the C=C, the free radical polymerization has received
little attention due to the presence of many side reactions. On the other hand, the cationic
polymerization has been studied by several researchers with successful results [11,12]. The
cationic copolymerization reaction usually requires an initiator. Several Lewis acids, such as
AlCl3, TiCl4, SnCl4 and BF3-OEt2 (BFE) amongst others, have been used as initiators to
copolymerize vinyl monomers cationically under relatively mild reaction conditions. As reported
by some researchers [13,14], BFE has proved to be a very efficient initiator in cationic
polymerization of vegetable oils. Usually, a trace of water or a diol, acting as a co-initiator is
needed to protonate the monomer. Triglycerides in general are cationically polymerizable
monomers. The accepted mechanism for such polymerizations is similar to that proposed for
cationic polymerization of vinyl monomers [15]. Each unsaturated fatty acid chain can
participate in the reaction, and the presence of several unsaturations leads to an extensive
crosslinking. Tung oil, containing a conjugated triene, is very reactive and highly susceptible to
polymerization via a cationic mechanism, because the intermediate carbocation can be stabilized

43
by resonance [1]. Likewise, vegetable oils can be polymerized with reactive monomers in order
to vary the properties of the oil-based matrix [16]. Due to the presence of multiple functional
sites per triglyceride molecule, which allow them to crosslink, oils can be considered as
analogous to vinyl esters and unsaturated polyesters [17].
Terpenes, natural monomers having carbon skeletons of isoprene units, are found in many
essential oils and represent a versatile chemical feedstock [18]. Even though the use of terpenes
in chemical synthesis is vast and well supported by a large number of publications, their
applications in polymer science are scarce and far between, usually limited to attempts to
polymerize bicylic monoterpenes, such as α and β pinene by Lewis acid and Ziegler-Natta
catalysis [19,20]. As stated by Norström [21], terpenes do not tend to polymerize, and their lack
of reactivity can be attributed to both steric hindrance and low stabilization energy between the
monomer and the chemical species in the transition state. However, since terpenes exhibit
carbon-carbon double bonds amenable to reactions, several works have been published
concerning the polymerization of terpenes with vinyl monomers in order to develop
biodegradable polymeric materials [22–24].
Envisioning the oil molecules as chains bearing multiple vinyl monomers, the main purpose of
this part of the project is to cover the major aspects related to the synthesis, characterization as
well as properties of new macromolecular bio-based thermoset polymers, produced through the
cationic copolymerization of tung (a natural oil) with three different terpenes; such as limonene,
myrcene and linalool as reactive comonomers. Also, in order to compare the properties of the
resulting materials, tung oil was copolymerized with styrene, a widely studied synthetic

44
monomer. The chemical, physical and mechanical properties of these new polymeric materials
were investigated as a function of the proportion and type of reactive monomer included in the
mixture. No other study has reported the copolymerization of plant oils and terpenes. The latter
can be considered as structural analogs to commercial vinyl monomers such as styrene, divinyl
benzene, etc.
2. Methods
2.1 Materials
Tung oil (TO), limonene (Lim), myrcene (Myr) and linalool (Lin) were purchased from VWR
International (US) and used with no further purification. Styrene (ST) was purchased from
Aldrich Chemical Company (US) and used as received. Tetrahydrofurane (THF) was used as an
initiator modifier and was supplied by VWR International (US). Boron Trifluoride Etherate
(BFE) was employed as an initiator and 1,4-butanediol was used as co-initiator; both were
acquired from VWR International (US).
2.2 Polymer Preparation Via Cationic Polymerization
The polymeric materials were prepared by cationic polymerization initiated by modified BFE.
The desired amount of the comonomer was measured out and added to the tung oil. The mixture
was vigorously stirred followed by the addition, drop by drop, of the appropriate amount of the
modified initiator. The modified catalyst was prepared by dissolving the boron trifluoride diethyl
etherate and 1,4-butanediol in the desired amount of THF. The reaction mixture was placed into
an aluminum mold and heated for a given time, usually 12 hrs. at room temperature, followed by
12 hrs. at 60 °C and finally for 12 hrs. at 110 °C.

45
2.3 Characterization
Soxhlet extraction was employed to characterize the structures of the bulk copolymers [25]. An
appropriate amount of the bulk polymer was extracted for 24 h with 200 mL of refluxing
dichloromethane with a Soxhlet extractor. Upon extraction, the resulting solution was
concentrated by rotary evaporation and subsequent vacuum drying. The soluble substances were
isolated for further characterization. The insoluble solid was dried in a vacuum oven for several
hours before it was weighed. FTIR spectra of both the soluble and insoluble fraction extracted by
Soxhlet were recorded. The densities of the copolymers were determined by pycnometry [26].
Mechanical properties of the resulting polymers were obtained by dynamic mechanical analysis
(DMA). To be able to explain the effects of the co-monomers on the mechanical properties, it is
useful to calculate the crosslinking density (n, mol/m³). The crosslinking density can be
estimated from the experimental data using the rubber elasticity theory. Thermosets behave as
rubbers above Tg. At small deformations, rubber elasticity predicts that the modulus storage (E’),
of an ideal elastomer with a network structure is proportional to the crosslinking density
according to Equation 2.1 [27]:
�� = 3��� = 3��/�� (2.1)
where E` is the rubbery storage modulus, R is the gas constant (8.314 J/mol K), T is the absolute
temperature (K), ρ is the density of the sample (g/m³), and Mc is the molecular weight between
crosslinks (g/mol). The temperature and rubbery modulus were determined for the calculation of
the equation at Tg + 30 °C. The temperature at which the peak of the tan delta presents a
maximum was considered the glass transition temperature of the material. In order to predict the
Tg based on the crosslinking density, the Fox and Loshaek model (which assumes specific

46
volume of the polymer at the glass transition as a linear function of Tg) was employed to study
the tung oil based copolymers. The Fox and Loshaek expression is shown in Equation 2.2 [17]:
� = � � + �/�� (2.2)
Where, K is a material constant, Tgu corresponds to the Tg predicted by the Fox equation, and Mc
corresponds to the molecular weight between crosslinking. The parameter K = 5.2 x 10³ K g/mol
was used to fit the model to the experimental results [17].
3. Results and Discussion
3.1. Cationic Polymerization of Vegetable Oils
Early in this research, several attempts to develop oil-based materials by cationic polymerization;
employing different types of vegetable oils, such as linseed oil, tung oil, soybean oil, corn oil,
etc. were carried out. Keeping in mind the preparation of completely bio-based polymers as the
major goal, the triglycerides were copolymerized with terpenes, natural monomers containing
alkenyl functional groups amenable to react.
Fruitful reactions leading to useful materials were achieved when tung oil, whose major
component is a fatty acid containing three conjugated unsaturations, was copolymerized with
limonene and myrcene as reactive comonomers (Figure 2.1).

47
Figure 2.1. Cationic polymerization of tung oil with limonene, myrcene and linalool
As pointed by Liu and Erham [11], the polymerization of 1,2 disubstituted ethylenes containing
small side groups easily occurs. Nevertheless, when bulky groups are attached to the double
bonds, the polymerization either does not occur or leads to low molecular weight products after
long reaction times. The steric hindrance plays a preponderant role during the propagation of the
growing chains. It introduces a high energy barrier between the monomer and the growing
species, allowing a very limited amount of polymer to be produced. Since fatty acids are 1,2
disubstituted ethylene monomers bearing bulky groups, their polymerization is considered
energetically unfavourable.
Unlike the other tested oils, tung oil was found to be highly reactive towards cationic and free
radical polymerizations. Its particular reactivity can be associated to its capacity to stabilize, by
resonance, the intermediate carbocation generated on the propagation step of the cationic
mechanism. In regards to the use of terpenes as comonomers, successful polymerizations were
R = α-Eleostearic Acid
=
Limonene Myrcene Linalool
Tung oil Crosslinked network

48
carried out when using limonene and myrcene. The polymerization of tung oil with linalool led
to highly porous materials. The gassing generated during curing could be attributed to side
reactions between the linalool and the boron trifluoride ethereate. All the copolymers appeared as
dark-brown rubbery materials at room temperature. For all the studied comonomers, suitable
materials for analysis were not obtained for terpene proportions above 60 wt%.
3.2. Characterization of the Polymer Networks
Once the reacting system has reached the gel point, the polymeric matrix is composed of two
fractions, the insoluble gel fraction and the soluble sol fraction. In order to characterize both
fractions, samples containing 80 wt% of tung oil (20 wt% comonomer) were Soxhlet extracted
using dichloromethane as a refluxing solvent. The study of the two phases, as well as the pure
tung oil (to track any possible change in its structure during curing) was developed through FTIR
spectroscopy.
The tung oil structure was characterized by the presence of a conjugated triene, which showed IR
absorption bands at 3010, 990 and 960 cm-1. The ester carbonyl exhibited two strong peaks at
1750 and 1150 cm-1. Methylene and methyl groups showed absorption bands at 2925 and 2854
cm-1, as well as at 1465 and 1370 cm-1. An inspection of the FTIR spectrum of the insoluble
fraction revealed that the major component in this phase was reacted oil (a cross-linked
triglyceride network). The spectrum presented similar absorption bands to those exhibited by the
tung oil. The most remarkable difference between them was the disappearance of the weak peak
at 3010 cm-1 (attributed to the presence of unsaturations in the tung oil structure). The lack of this
absorption peak in the FTIR spectrum of the insoluble fraction could be related to the fact that

49
the majority of double bonds had been consumed during the polymerization, either to react with
other fatty acid chains or with the comonomer, leading to the disruption of the conjugated triene,
responsible for the high reactivity of this oil. Upon extraction, for copolymers 80 wt% tung oil-
20 wt% comonomer, around 81-84 wt% of insoluble materials were retained. For all the
remaining copolymers, the retained insoluble materials ranged from 76 to 85 wt% after Soxhlet
extraction. FTIR analysis of the soluble fraction of the copolymer 80 wt% tung oil-20 wt%
comonomer suggested that the main component presented in this phase was unreacted oil.
3.2.1 Thermo-Mechanical Behavior
Dynamic mechanical analysis (DMA) measurements are intensively used to investigate the
amorphous phase transitions of polymers as they are stressed under periodic deformation. DMA
can be used to obtain the storage modulus and the loss modulus as a function of temperature at a
given frequency. Figure 2.2.a shows the temperature dependence of the storage modulus for the
copolymers tung oil-limonene. Results revealed that the materials behaved as thermosets. As
seen in the curves, the storage modulus initially remained approximately constant. As the
temperature increased, the storage modulus exhibited a drop over a wide temperature range
(glass transition), followed by a modulus plateau at high temperatures, where the material
behaved like a rubber. This region, called the rubbery plateau, evidences the presence of a stable
cross-linked network. The modulus drop is associated with the beginning of segmental mobility
in the cross-linked polymer network; whereas the constant modulus at temperatures above 30°C
is the result of the cross-linked structure of the copolymer [5]. The same behavior has been
reported by Li and Larock [7].
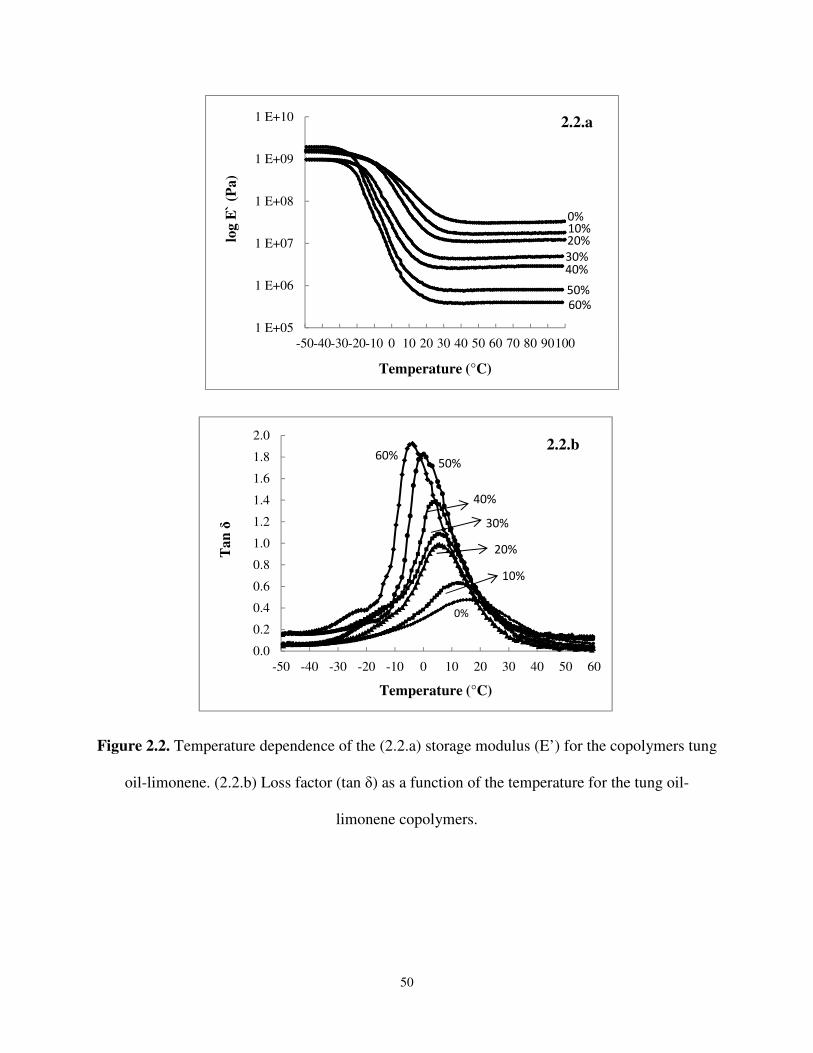
50
Figure 2.2. Temperature dependence of the (2.2.a) storage modulus (E’) for the copolymers tung
oil-limonene. (2.2.b) Loss factor (tan δ) as a function of the temperature for the tung oil-
limonene copolymers.
0% 10%20%
30% 40%
50%
60%
1 E+05
1 E+06
1 E+07
1 E+08
1 E+09
1 E+10
-50-40-30-20-10 0 10 20 30 40 50 60 70 80 90100
log
E`
(Pa
)
Temperature (°C)
0%
10%
20%
30%
40%
50% 60%
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
-50 -40 -30 -20 -10 0 10 20 30 40 50 60
Tan
δ
Temperature (°C)
2.2.b
2.2.a

51
The curves of the loss factor (tan δ) as a function of the temperature for the tung oil-limonene
copolymers are shown in Figure 2.2.b. The peak in tan δ corresponded to the main mechanical
relaxation of the matrix, which can be used as a criterion to determine the Tg of the copolymer.
As can be seen, the transition between the glassy to the rubbery state was wide and began below
room temperature for all the tested materials. Likewise, all the copolymers showed only one tan
δ peak, indicating that there was no phase separation. The results showed that the peaks shifted
to lower temperatures (indicating a decrease in the glass transition temperature), increased in
height and become narrower when increasing the co-monomer weight proportion. The peak
width at half height is a criterion used to indicate the homogeneity of the amorphous phase. A
higher value implies higher inhomogeneity of the amorphous phase. In this particular case, the
narrowing and increase in height of the loss factor peaks could be associated to less cross-linked
and heterogeneous networks with narrower molecular weight distribution of the chains between
crosslinking points [17].
Table 2.1 summarizes the properties of the tung oil-limonene copolymers. As shown, the storage
modulus, the crosslinking density and the Young`s modulus decreased when the tung oil
concentration increased. On the other hand, Tg decreased almost linearly with the increase in the
limonene content. Pycnometry showed variations in the density of the copolymers for
percentages of limonene higher than 30 wt%.
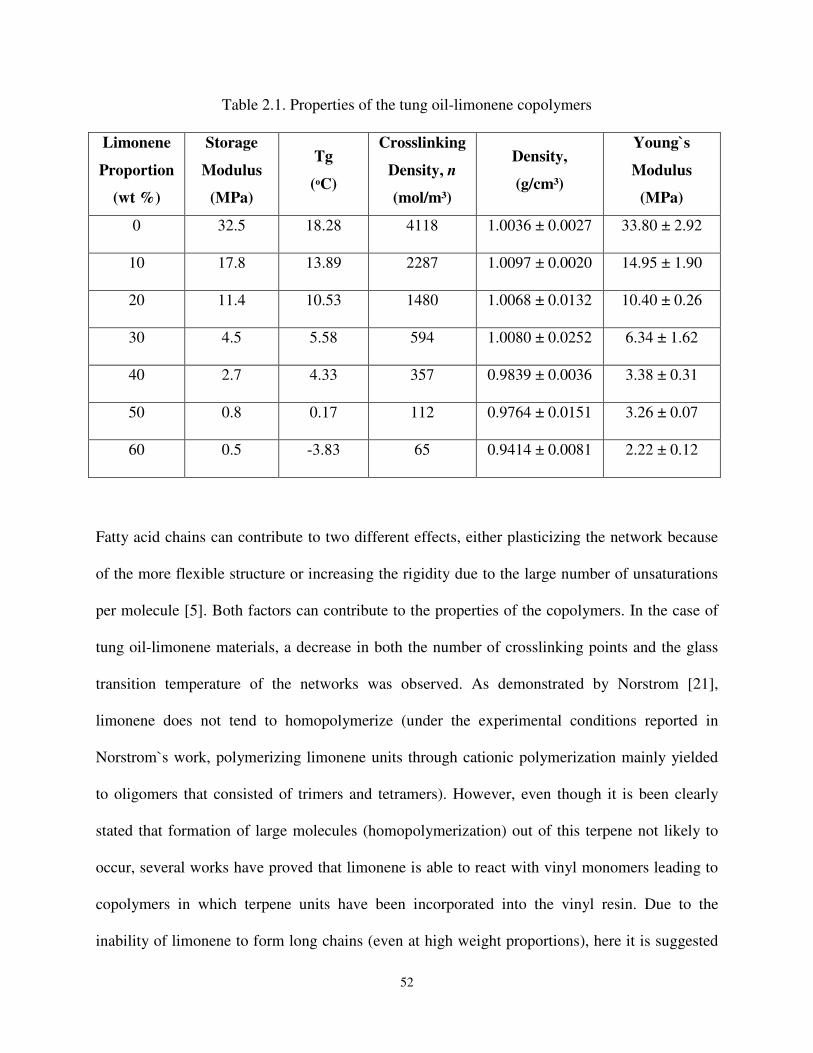
52
Table 2.1. Properties of the tung oil-limonene copolymers
Limonene
Proportion
(wt %)
Storage
Modulus
(MPa)
Tg
(ᵒC)
Crosslinking
Density, n
(mol/m³)
Density,
(g/cm³)
Young`s
Modulus
(MPa)
0 32.5 18.28 4118 1.0036 ± 0.0027 33.80 ± 2.92
10 17.8 13.89 2287 1.0097 ± 0.0020 14.95 ± 1.90
20 11.4 10.53 1480 1.0068 ± 0.0132 10.40 ± 0.26
30 4.5 5.58 594 1.0080 ± 0.0252 6.34 ± 1.62
40 2.7 4.33 357 0.9839 ± 0.0036 3.38 ± 0.31
50 0.8 0.17 112 0.9764 ± 0.0151 3.26 ± 0.07
60 0.5 -3.83 65 0.9414 ± 0.0081 2.22 ± 0.12
Fatty acid chains can contribute to two different effects, either plasticizing the network because
of the more flexible structure or increasing the rigidity due to the large number of unsaturations
per molecule [5]. Both factors can contribute to the properties of the copolymers. In the case of
tung oil-limonene materials, a decrease in both the number of crosslinking points and the glass
transition temperature of the networks was observed. As demonstrated by Norstrom [21],
limonene does not tend to homopolymerize (under the experimental conditions reported in
Norstrom`s work, polymerizing limonene units through cationic polymerization mainly yielded
to oligomers that consisted of trimers and tetramers). However, even though it is been clearly
stated that formation of large molecules (homopolymerization) out of this terpene not likely to
occur, several works have proved that limonene is able to react with vinyl monomers leading to
copolymers in which terpene units have been incorporated into the vinyl resin. Due to the
inability of limonene to form long chains (even at high weight proportions), here it is suggested

53
that the decrease in the number of crosslinking points arises as a result of limonene units that
have been incorporated into the fatty acid chains, lowering their ability to form an oil-based
cross-linked network. The decrease in the glass transition of the networks at high limonene
contents, in which crosslinking is very low, is driven by the glass transition of the limonene
oligomers (small linear segments) formed during heating of the polymeric matrices.
DMA of the tung oil-myrcene copolymers revealed a similar behavior to that shown by the tung
oil-limonene copolymers. When the temperature increased, the storage modulus exhibited a drop
over a wide temperature range, followed by a modulus plateau at high temperatures, evidencing
the presence of a stable cross-linked network. Also, the modulus decreased as the myrcene
proportion in the mixtures increased. The transition between the glassy to the rubbery state for
these copolymers was wide and began below room temperature. Likewise, for all the
copolymers, no phase separation was observed. The results showed that the tan δ peak increased
in height and become narrower as the myrcene content in the matrix increased. As in the case of
tung oil-limonene copolymers, this phenomenon could be attributed to the presence of less
heterogeneous networks. Unlike the tung oil-limonene materials, the maximum in the loss factor
shifted to higher temperatures when increasing the myrcene proportion above 10 wt%, indicating
that the glass transition temperature for these copolymers increased as the comonomer proportion
increased. Table 2.2 summarizes some of the properties of the tung oil-myrcene copolymers. As
shown, the glass transition temperature increased with the content of myrcene for percentages
higher than 10 wt%. Likewise, the increase in the comonomer proportion decreased the amount
of crosslinking points in the copolymers. The storage modulus, along with the Young`s modulus,
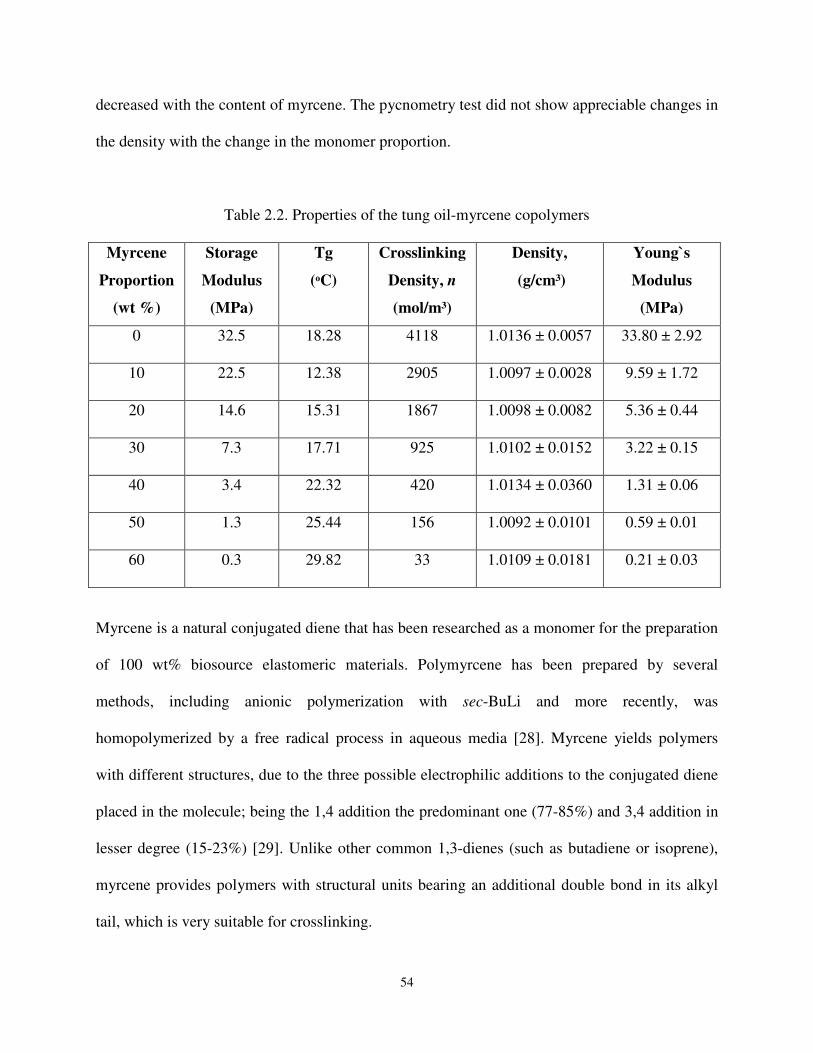
54
decreased with the content of myrcene. The pycnometry test did not show appreciable changes in
the density with the change in the monomer proportion.
Table 2.2. Properties of the tung oil-myrcene copolymers
Myrcene
Proportion
(wt %)
Storage
Modulus
(MPa)
Tg
(ᵒC)
Crosslinking
Density, n
(mol/m³)
Density,
(g/cm³)
Young`s
Modulus
(MPa)
0 32.5 18.28 4118 1.0136 ± 0.0057 33.80 ± 2.92
10 22.5 12.38 2905 1.0097 ± 0.0028 9.59 ± 1.72
20 14.6 15.31 1867 1.0098 ± 0.0082 5.36 ± 0.44
30 7.3 17.71 925 1.0102 ± 0.0152 3.22 ± 0.15
40 3.4 22.32 420 1.0134 ± 0.0360 1.31 ± 0.06
50 1.3 25.44 156 1.0092 ± 0.0101 0.59 ± 0.01
60 0.3 29.82 33 1.0109 ± 0.0181 0.21 ± 0.03
Myrcene is a natural conjugated diene that has been researched as a monomer for the preparation
of 100 wt% biosource elastomeric materials. Polymyrcene has been prepared by several
methods, including anionic polymerization with sec-BuLi and more recently, was
homopolymerized by a free radical process in aqueous media [28]. Myrcene yields polymers
with different structures, due to the three possible electrophilic additions to the conjugated diene
placed in the molecule; being the 1,4 addition the predominant one (77-85%) and 3,4 addition in
lesser degree (15-23%) [29]. Unlike other common 1,3-dienes (such as butadiene or isoprene),
myrcene provides polymers with structural units bearing an additional double bond in its alkyl
tail, which is very suitable for crosslinking.

55
When polymerizing myrcene and tung oil in the presence of BFE, the myrcene, contrarily to the
limonene, is not only able to react with the oil, but it is also capable of homopolymerizing and
creating linear myrcene-myrcene sequences. When increasing the amount of comonomer, the
diluting effect of the myrcene sequences immersed into the resin becomes predominant, and
hence, the crosslinking density decreases, consequently leading to a decrease in the storage
modulus. The increase in the glass transition of the networks when increasing the myrcene
content is attributed to the presence of these linear myrcene-myrcene sequences formed during
heating. As mentioned above, these linear polymyrcene sequences contain structural repeating
units bearing very short dangling chains having an extra double bond, which can be prompt to
react under the experimental conditions. At high myrcene concentrations, these additional
unsaturations can react and generate polymyrcene cross-linked networks. The bonding of these
polymyrcene linear segments (embedded in the matrix) through crosslinking, stiffs and strengths
the system and therefore; limits the segmental motion of the polymer chains, driving the glass
transition temperature to higher values whilst increasing the terpene content.
In order to compare the results obtained in this research with previous works dealing with
copolymerization of vegetable oils and synthetic vinyl monomers, tung oil was copolymerized
with styrene in the presence of boron trifluoride as an initiator, at room temperature. Table 2.3
summarizes the properties of the tung oil-styrene copolymers. The glass transition temperatures
(Tg) increased almost linearly with the content of styrene for percentages higher than 10 wt%.
The increase in tung oil concentration increased the amount of crosslinking points in the
copolymers. The storage modulus, along with the Young`s modulus, decreased with the content
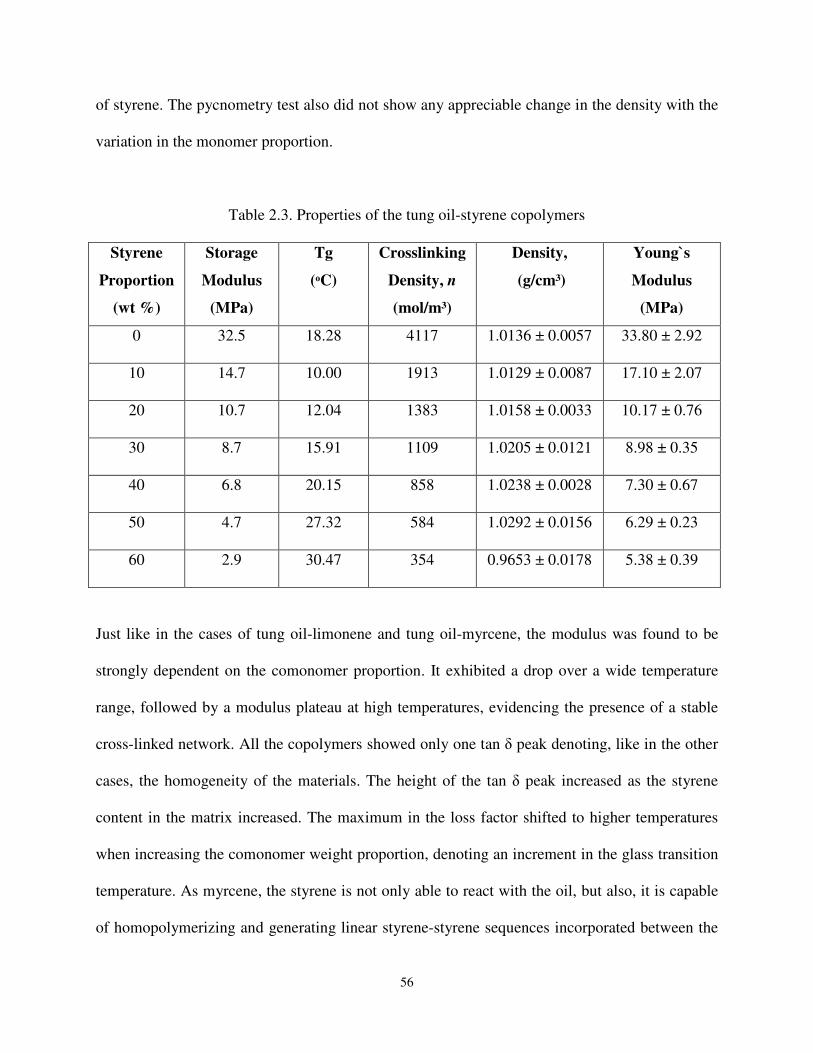
56
of styrene. The pycnometry test also did not show any appreciable change in the density with the
variation in the monomer proportion.
Table 2.3. Properties of the tung oil-styrene copolymers
Styrene
Proportion
(wt %)
Storage
Modulus
(MPa)
Tg
(ᵒC)
Crosslinking
Density, n
(mol/m³)
Density,
(g/cm³)
Young`s
Modulus
(MPa)
0 32.5 18.28 4117 1.0136 ± 0.0057 33.80 ± 2.92
10 14.7 10.00 1913 1.0129 ± 0.0087 17.10 ± 2.07
20 10.7 12.04 1383 1.0158 ± 0.0033 10.17 ± 0.76
30 8.7 15.91 1109 1.0205 ± 0.0121 8.98 ± 0.35
40 6.8 20.15 858 1.0238 ± 0.0028 7.30 ± 0.67
50 4.7 27.32 584 1.0292 ± 0.0156 6.29 ± 0.23
60 2.9 30.47 354 0.9653 ± 0.0178 5.38 ± 0.39
Just like in the cases of tung oil-limonene and tung oil-myrcene, the modulus was found to be
strongly dependent on the comonomer proportion. It exhibited a drop over a wide temperature
range, followed by a modulus plateau at high temperatures, evidencing the presence of a stable
cross-linked network. All the copolymers showed only one tan δ peak denoting, like in the other
cases, the homogeneity of the materials. The height of the tan δ peak increased as the styrene
content in the matrix increased. The maximum in the loss factor shifted to higher temperatures
when increasing the comonomer weight proportion, denoting an increment in the glass transition
temperature. As myrcene, the styrene is not only able to react with the oil, but also, it is capable
of homopolymerizing and generating linear styrene-styrene sequences incorporated between the

57
oil matrixes. As increasing the amount of comonomer, the diluting effect of these linear
segments immersed into the resin became predominant; leading to a decrease in the crosslinking
density (consequently conducing to a reduction in the storage modulus). The increase in the Tg is
also attributed to the presence of these stiff and strong linear Polystyrene segments embedded in
the resins.
3.3. Theoretical Prediction of Tg Based on the Fox-Loshaek Model.
In order to predict the Tg of the copolymers, the Fox-Loshaek model (which assumes specific
volume of the polymer at the glass transition as a linear function of Tg) was considered. The
model can be considered as the sum of two contributions; the linear contribution Tgu, predicted
by the Fox equation, where the system is assumed to be formed by hypothetical linear molecules;
and the crosslinking contribution K / Mc, calculated through the Nielsen equation, which takes
into account the crosslinking of those hypothetical linear molecules [30].
To apply the model, the relation between the Tg and the crosslinking density was studied.
Through a simple mathematical approach, the evolution of the glass transition temperature of the
copolymers was found to be linearly dependent of the crosslinking density. Thus, employing
both the Fox and Nielsen equations, the two contributions for different compositions were
calculated. Figure 2.3.a shows the experimental values of the Tg measured by DMA as a
function of the limonene content, the linear contribution (Fox equation) and the Tg predicted by
the model. As seen, the model was able to predict the general trend in which the Tg decreased as
a function of the comonomer content. For the tung oil polymer (0% limonene) the model
overestimated the crosslinking density. For comonomer contents above 40 wt%, the model
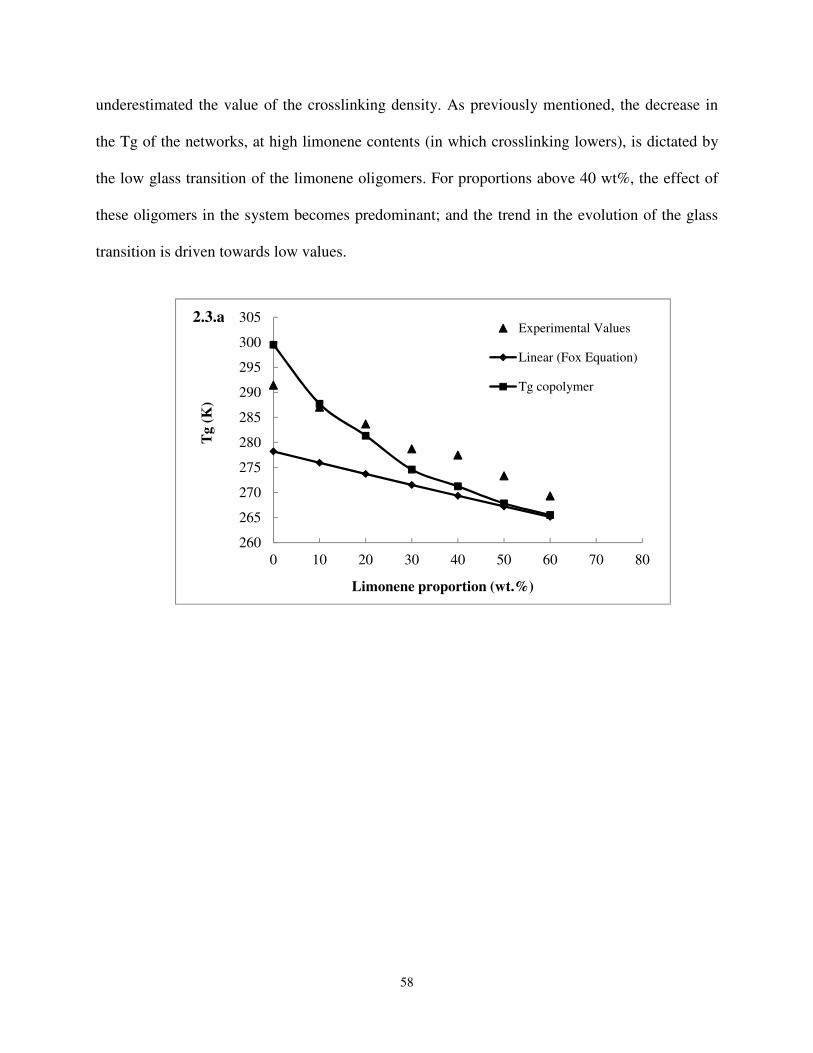
58
underestimated the value of the crosslinking density. As previously mentioned, the decrease in
the Tg of the networks, at high limonene contents (in which crosslinking lowers), is dictated by
the low glass transition of the limonene oligomers. For proportions above 40 wt%, the effect of
these oligomers in the system becomes predominant; and the trend in the evolution of the glass
transition is driven towards low values.
260
265
270
275
280
285
290
295
300
305
0 10 20 30 40 50 60 70 80
Tg (
K)
Limonene proportion (wt.%)
Experimental Values
Linear (Fox Equation)
Tg copolymer
2.3.a
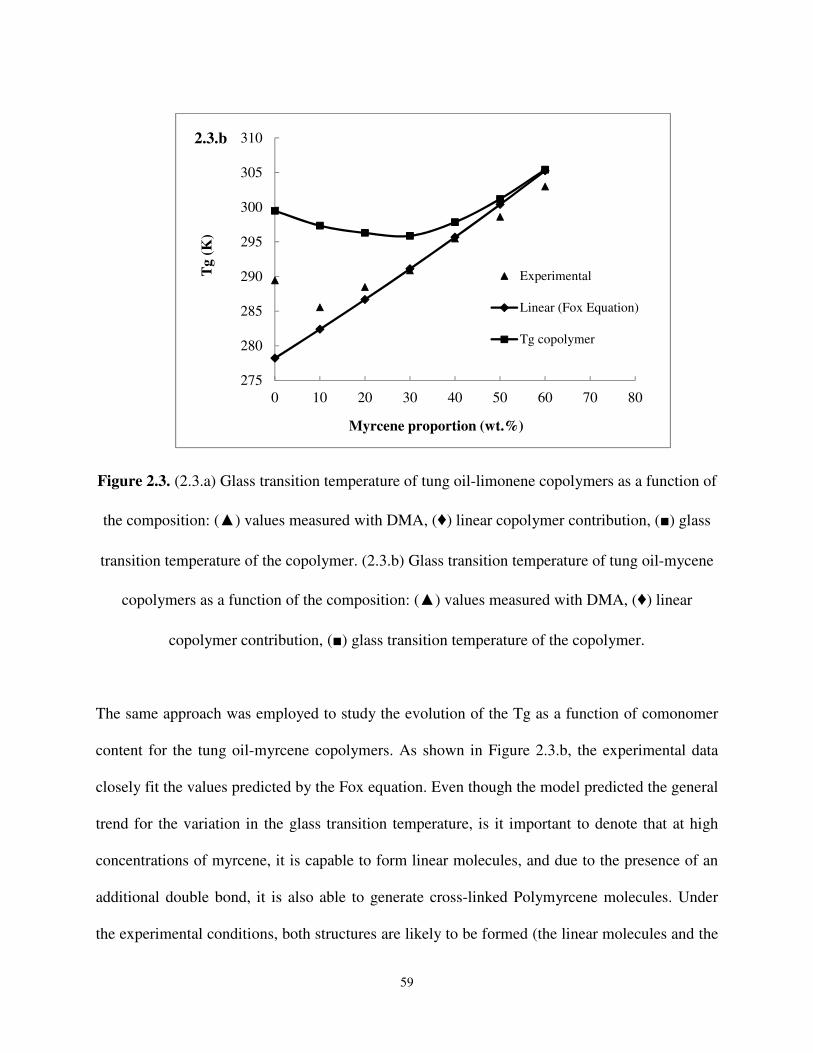
59
Figure 2.3. (2.3.a) Glass transition temperature of tung oil-limonene copolymers as a function of
the composition: (▲) values measured with DMA, (♦) linear copolymer contribution, (■) glass
transition temperature of the copolymer. (2.3.b) Glass transition temperature of tung oil-mycene
copolymers as a function of the composition: (▲) values measured with DMA, (♦) linear
copolymer contribution, (■) glass transition temperature of the copolymer.
The same approach was employed to study the evolution of the Tg as a function of comonomer
content for the tung oil-myrcene copolymers. As shown in Figure 2.3.b, the experimental data
closely fit the values predicted by the Fox equation. Even though the model predicted the general
trend for the variation in the glass transition temperature, is it important to denote that at high
concentrations of myrcene, it is capable to form linear molecules, and due to the presence of an
additional double bond, it is also able to generate cross-linked Polymyrcene molecules. Under
the experimental conditions, both structures are likely to be formed (the linear molecules and the
275
280
285
290
295
300
305
310
0 10 20 30 40 50 60 70 80
Tg
(K
)
Myrcene proportion (wt.%)
Experimental
Linear (Fox Equation)
Tg copolymer
2.3.b

60
cross-linked structure) and the generation of this reticulated Polymyrcene network is responsible
for the increase in the glass transition temperature observed in the experimental results. The
divergence between the experimental data and the values predicted by the model could be
associated to the fact that triglyceride-based polymers are very complex materials, containing
cross-linker molecules with a range of functionalities.

61
3.4. Conclusions
Thermosets were prepared through copolymerization of tung oil with limonene, myrcene and
styrene. The Tg was found to be a function of both the type and comonomer weight content. The
Young`s modulus and the experimental crosslinking densities decreased as comonomer weight
content increased. Based on the thermo-mechanical evaluation, these materials behave as
elastomers at room temperature. The Fox and Loshaek model showed a relatively good
prediction of the Tg values for tung oil-limonene copolymers. For myrcene, Tg values matched
those predicted by the Fox equation. Even though triglyceride-based polymers are complex
materials, some of the thermo-mechanical properties were found to be predictable by simple
mathematical approaches and models developed for polyvinyl resins.

62
References
[1] Y. Xia, R.C. Larock, Vegetable oil-based polymeric materials: synthesis, properties, and
applications, Green Chem. 12 (2010) 1893. doi:10.1039/c0gc00264j.
[2] G. Knothe, Structure indices in FA chemistry. How relevant is the iodine value?, J. Am.
Oil Chem. Soc. 79 (2002) 847–854. doi:10.1007/s11746-002-0569-4.
[3] M. Sacristán, J.C. Ronda, M. Galià, V. Cádiz, Rapid soybean oil copolymers synthesis by
microwave-assisted cationic polymerization, Macromol. Chem. Phys. 211 (2010) 801–
808. doi:10.1002/macp.200900571.
[4] M. Galià, L.M. de Espinosa, J.C. Ronda, G. Lligadas, V. Cádiz, Vegetable oil-based
thermosetting polymers, Eur. J. Lipid Sci. Technol. 112 (2010) 87–96.
doi:10.1002/ejlt.200900096.
[5] C. Meiorin, M.I. Aranguren, M.A. Mosiewicki, Vegetable oil/styrene thermoset
copolymers with shape memory behavior and damping capacity, Polym. Int. 61 (2012)
735–742. doi:10.1002/pi.3231.
[6] D.D. Andjelkovic, R.C. Larock, Novel rubbers from cationic copolymerization of soybean
oils and dicyclopentadiene. 1. Synthesis and characterization, Biomacromolecules. 7
(2006) 927–936. doi:10.1021/bm050787r.
[7] F. Li, R.C. Larock, Synthesis, structure and properties of new tung oil-styrene-
divinylbenzene copolymers prepared by thermal polymerization., Biomacromolecules. 4
(2003) 1018–25. doi:10.1021/bm034049j.
[8] G. Lligadas, J.C. Ronda, M. Galias, V. Cadiz, Plant oils as platform chemicals for
polyurethane synthesis: Current state-of-the-art, Biomacromolecules. 11 (2010) 2825–
2835. doi:10.1021/bm100839x.

63
[9] T. Saurabh, M. Patnaik, S.L. Bhagt, V.C. Renge, Epoxidation of Vegetable Oils : a
Review, Int. J. Adv. Eng. Technol. 2 (2011) 491–501.
[10] J.C. Ronda, G. Lligadas, M. Gali??, V. C??diz, Vegetable oils as platform chemicals for
polymer synthesis, Eur. J. Lipid Sci. Technol. 113 (2011) 46–58.
doi:10.1002/ejlt.201000103.
[11] Z. Liu, S.Z. Erhan, Preparation of soybean oil polymers with high molecular weight, J.
Polym. Environ. 18 (2010) 243–249. doi:10.1007/s10924-010-0179-y.
[12] F. Li, M.. Hanson, R.. Larock, Soybean oil–divinylbenzene thermosetting polymers:
synthesis, structure, properties and their relationships, Polymer (Guildf). 42 (2001) 1567–
1579. doi:10.1016/S0032-3861(00)00546-2.
[13] A. V Radchenko, S. V Kostjuk, L. V Gaponik, BF3OEt2-coinitiated cationic
polymerization of cyclopentadiene in the presence of water at room temperature, Polym.
Bull. 67 (2011) 1413–1424. doi:DOI 10.1007/s00289-011-0455-6.
[14] Z. Liu, S.Z. Erhan, Ring-opening polymerization of epoxidized soybean oil, JAOCS, J.
Am. Oil Chem. Soc. 87 (2010) 437–444. doi:10.1007/s11746-009-1514-0.
[15] N.G. McCrum, C.P. Buckley, C.B. Bucknall, Principles of Polymer Engineering, Oxford
University Press, 1997. https://books.google.com/books?id=UX-sAQAAQBAJ&pgis=1
(accessed March 23, 2016).
[16] S. Miao, P. Wang, Z. Su, S. Zhang, Vegetable-oil-based polymers as future polymeric
biomaterials, Acta Biomater. 10 (2014) 1692–1704. doi:10.1016/j.actbio.2013.08.040.
[17] J. La Scala, R.P. Wool, Property analysis of triglyceride-based thermosets, Polymer
(Guildf). 46 (2005) 61–69. doi:10.1016/j.polymer.2004.11.002.

64
[18] M. Firdaus, L. Montero De Espinosa, M.A.R. Meier, Terpene-based renewable monomers
and polymers via thiol-ene additions, Macromolecules. 44 (2011) 7253–7262.
doi:10.1021/ma201544e.
[19] C. Guo, L. Zhou, J. Lv, Effects of expandable graphite and modified ammonium
polyphosphate on the flame-retardant and mechanical properties of wood flour-
polypropylene composites, Polym. Polym. Compos. 21 (2013) 449–456. doi:10.1002/app.
[20] S. Sharma, A.K. Srivastava, Synthesis and characterization of copolymers of limonene
with styrene initiated by azobisisobutyronitrile, Eur. Polym. J. 40 (2004) 2235–2240.
doi:10.1016/j.eurpolymj.2004.02.028.
[21] E. Norström, Terpenes as renewable monomers for biobased materials, Master Sci.
(2011).
[22] A.K. Srivastava, P. Pandey, Synthesis and characterisation of copolymers containing
geraniol and styrene initiated by benzoyl peroxide, Eur. Polym. J. 38 (2002) 1709–1712.
doi:10.1016/S0014-3057(02)00028-9.
[23] S. Sharma, A.K. Srivastava, Free radical copolymerization of limonene with butyl
methacrylate: Synthesis and characterization, Indian J. Chem. Technol. 12 (2005) 62–67.
[24] R.P.F. Guiné, J.A.A.M. Castro, Polymerization of β-pinene with ethylaluminum
dichloride (C2H5AlCl2), J. Appl. Polym. Sci. 82 (2001) 2558–2565.
doi:10.1002/app.2107.
[25] W.S. Precision, E.S. Waste, T. Edition, Standard Practice for Extraction of Solid Waste
Samples for Chemical Analysis, 93 (2008) 1–7. doi:10.1520/D5369-93R08E01.2.

65
[26] ASTM D891-09, Standard Test Methods for Specific gravity, apparent, of liquid industrial
chemicals, ASTM Int. 99 (2015).
[27] M. Fache, R. Auvergne, B. Boutevin, S. Caillol, New vanillin-derived diepoxy monomers
for the synthesis of biobased thermosets, Eur. Polym. J. 67 (2015) 527–538.
doi:10.1016/j.eurpolymj.2014.10.011.
[28] S. Loughmari, A. Hafid, A. Bouazza, A. El Bouadili, P. Zinck, M. Visseaux, Highly
stereoselective coordination polymerization of β-myrcene from a lanthanide-based
catalyst: Access to bio-sourced elastomers, J. Polym. Sci. Part A Polym. Chem. 50 (2012)
2898–2905. doi:10.1002/pola.26069.
[29] A. Behr, L. Johnen, Myrcene as a natural base chemical in sustainable chemistry: A
critical review, ChemSusChem. 2 (2009) 1072–1095. doi:10.1002/cssc.200900186.
[30] M.L. Auad, M. Aranguren, J. Borrajo, Epoxy-Based Divinyl Ester Resin / Styrene
Copolymers : Composition Dependence of the Mechanical and Thermal Properties, J.
Appl. Polym. Sci. 66 (1997) 1059–1066.

66
CHAPTER III
Triglycerides and Phenolic Compounds as Precursors of Bio-based Thermosetting Epoxy
Resins
1. Introduction
In recent years, there has been an increasing interest for the development and application of
biomass derived products to address issues related to the depletion of non-renewable resources.
Currently, the chemical industry is pursuing to substitute a growing part of fossil feedstocks with
renewable carbon. This trend is not only driven by environmental concerns, but it is also fueled
by the increase in the general demand for green products from costumers; as well as efforts from
government agencies providing funding to increase the present-day knowledge and technology to
process and transforming biomass into high-value chemicals [1]. In order to fulfill the needs of
the current industry, the polymer industry also demands a renewable chemical platform; and the
main challenge lies in developing materials with properties matching or even improving those of
resins in current use. In the particular case of thermosetting materials, which have played an
important role in the modern civilization, these properties include high modulus, strength,
durability, as well as the thermal and chemical resistances provided by the high crosslinking
density assembling their macromolecular structure [2].
In non-food applications, the most widely applied renewable resources include plant oils (only
castor and linseed oil are exclusively used of industrial applications) [3], polysaccharides

67
(mainly starch and cellulose), and proteins [2]. Ronda et al. [4] have considered three main
different approaches to convert oils and their derivatives into useful polymeric materials; the
direct polymerization of the double bonds, the chemical modification and posterior
polymerization, and the polymerization of monomers synthesized using oil-derived platform
chemicals. It is clear however, that this scheme can be extended not only to oils, but also to any
other type of biomass-derived compounds, leading to three different areas to generate
macromolecular materials, including direct use, chemical functionalization, and biorefinery.
As seen in Chapter II, direct polymerization of oils leads to obtain materials with marked
elastomeric nature, showing low Tg and modulus. Thus, chemical modification stands as a
common strategy for obtaining high performance polymeric materials. Carbon-carbon double
bonds can be converted into epoxides through a catalyzed oxidation using peracids, process that
is commonly known as the Prisleschajew reaction. The epoxidation takes place in a one-step,
concerted reaction that maintains the stereochemistry of any substituent on the double bond [5].
The epoxidized products could be diversified, because of the possibility of mono-, di-, or tri-
epoxides according to the origin of the oilseed [6]. Likewise, chemo-enzymatic epoxidation has
been study, mainly because it avoids the ring-opening of epoxide and the formation of
bioproducts, which are the most common drawbacks of the oxidation method [7].
Several studies have investigated the development of thermoset plastics and coatings from plant-
based oils. Zhu et al. [8] studied the development of inexpensive epoxy resins using soybean oil
derivatives cured with a commercial cycloaliphatic amine. Likewise, Shah and Ahmad
developed waterborne vegetable epoxy coatings, also curing the oil-based epoxy monomers with

68
anhydrides [9]. Also, epoxidized soybean oil composites have been prepared by reinforcing the
oil matrix, cured with commercial amines, with flax fibers [10]. A complete comparison of
curing agents for epoxidized oils is given by Espinosa-Perez et al. [11]. It is important to
highlight that the above mentioned studies employed the step-growth process to formulate the
epoxy resin, in which a commercial crosslinking agents such as amines, anhydrides, etc. are
required to crosslink the epoxy monomers. This approach generates partially bio-based resins.
Scarce studies have dealt with chain-growth copolymerization, approach that requires a small
amount of an initiator to generate the polymer, leading to final materials with lesser contribution
of commercial compounds per weight basis. Kim and Sharma [12] reported the
homopolymerization of oil-based epoxy resins by using N-benzyl pyrazinium
hexafluorantimonate. It is also important to point out that the inherit long chain aliphatic
structure of oil-based thermoset monomers limits the polymer glass transition temperatures
compared to commercial, aromatic based monomers, such as DGEBA [13]. This outcome stands
up as a key finding, because opens the possibility for the study of the copolymerizing of oil-
based monomers with low molecular aromatic compounds; evaluating whether or not these
compounds can improve the mechanical properties of the oil-based resins. Indeed, the thermal
and mechanical performance properties of organic polymers are generally linked to the presence
of rigid aromatic structures [14].
Hence, biomass is also a potential source of aromatic compounds. Lignin is the second most
abundant naturally occurring macromolecule, and due to its phenolic nature, it has been
considered as a promising source of high value aromatic compounds. Some novel technologies
based on biomass refinery appear as a good alternative to produce high valued chemical from the

69
residues of pulping industries [15]. Likewise, cashew nut shell, an agricultural by-product
abundantly available in tropical countries, is one of the major resources of naturally occurring
phenols [2]. It contains four major aromatic components, namely cardanol, cardol, anacardic acid
and 2-methylcarbol. The viability of using these compounds as building blocks for epoxy resins
has been already a subject of scientific studies [16]. In a lesser degree, polyphenols, more
specifically from condensed tannins extracted from wastes produced by the wood and wine
industries can also be an alternative to produce epoxy resins [17].
In the previous chapter, the direct polymerization of the double bonds present in the fatty acid
chains of the triglycerides was studied, and the copolymerization with terpenes, cyclic
compounds structurally resembling commercial monomers such as styrene and divinyl benzene,
was also a subject of evaluation. The first part of this chapter focuses on the chemical
modification of the double bonds of plant oils. These chemical modifications were addressed to
introduce more reactive functional groups able to polymerize and to produce materials with a
better thermo-mechanical performance. Three chemical modifications (epoxy, hydroxyl and
acrylate) are preliminary evaluated. Efforts are centered on the epoxy functional groups, and the
feasibility of using step-growth or chain-growth polymerizations for their conversion into
macromolecular materials is also assessed.
The second part of this research covers the copolymerization of oil-based epoxy monomers with
an aromatic low molecular weight comonomer, chemically modified with oxirane groups, to
evaluate whether or not the increase in the density of aromatic structures in the crosslinked
network can improve the mechanical properties of the resulting materials. Thus, by employing

70
linseed oil and α-resorcylic acid as a model, functionalized with epoxy groups (via oxidation and
nucleophilic addition respectively); the main purpose of this chapter is to cover the major aspects
related to the chemical synthesis, physical-chemical characterization and study of thermo-
mechanical properties of a bio-based epoxy resins. α-resorcylic acid is a constituent of peanuts
(Arachis hypogea), chickpeas (Cicer arietinum), red sandalwood (Pterocarpus santalinus), and
hill raspberry (Rubus niveus) [18]. Likewise, α-resorcylic acid derivatives have been found as a
secondary metabolite in cultures of the fungus Sporotrichum laxum ATCC 15155 [19].
2. Experimental Section
2.1 Materials
Linseed oil (LO) was purchased from VWR International (USA) and used with no further
purification. Glacial acetic acid (99.5 %), hydrogen peroxide (30 %), sodium carbonate, toluene,
petroleum ether, ethyl acetate, iodine monochloride, Wijs’ solution, 33 % HBr in glacial acetic
acid, and Amberlite ion-exchange resin were all AR grade and also acquired from VWR
International (USA). α-Resorcylic acid (α-RA) ( (≥ 98%), epichlorohydrin (99.0 %), sodium
hydroxide (≥ 98%), and dichloromethane were purchased from VWR (US).
Benzyltriethylammonium chloride (≥ 98%), used as a phase transfer agent, was also acquired
from VWR (US). 4-dimethylaminopyridine (DMAP, 99.0 %), used as initiator for the chain
homopolymerization of epoxy monomers, and EPON 828 (with an epoxy equivalent weight =
185-192 eq/gr [20]) were purchased from VWR and used as received.

71
2.2 Synthesis procedures
2.2.1 Synthesis of epoxidized triglycerides.
Linseed oil (LO) was epoxidized using a peracetic acid generated in situ from reaction between
acetic acid and hydrogen peroxide, with ion-exchange resin catalyst, at 60 °C in toluene, during 8
h. [21,22] . A solution of linseed oil (20.1 g, 67.0 mmol) and acetic acid (1.0 mL, 26.2 mmol) in
toluene, using Amberite as an ion exchange catalyst, was stirred at 35 °C, and H2O2 30% w/v
was added dropwise. After H2O2 addition was complete, the temperature was raised to 60 °C and
the flask contents were stirred vigorously for 8 h. Once the reaction time was completed the
mixture was first dissolved in ethyl acetate. The Amberlite was filtered off, and the filtrate was
poured into a separation funnel, and washed with brine until the pH was neutral. The oil phase
was further dried above anhydrous sodium sulfate and then filtered. Finally, the solvent was
removed using a rotary evaporator.
2.2.2 Synthesis of polyol.
Polyol was synthesized from epoxidized linseed oil, with an oxirane oxygen content of 0.653
mol/100g, through a ring opening reaction with boiling methanol in the presence of
tetrafluoroboric acid as acid catalyst following the procedure described by Dai et al. [23]. The
molar ratio of epoxy groups to methanol was 1:11, and the concentration of the catalyst was 1
wt% of the total weight of oil and methanol. Methanol and catalyst were placed into the reactor
and heated to approx. 60 °C using an oil bath. Once the temperature was reached, the epoxidized
linseed oil was added dropwise. The reaction mixture was kept for 1 hour. After cooling to room
temperature, the acid was neutralized with sodium bicarbonate aqueous solution (approx. 30
wt%) and the remaining solvent was removed on a rotatory evaporator under low vacuum.

72
2.2.3 Functionalization of phenolic compounds with epoxy groups. Synthesis of
triglycidylated ether of α-resorcylic acid.
The glycidylation of bio-oil was conducted following the method and conditions established by
[24,25]. The described process leads to the synthesis of epoxyalkylaryl ethers by the reaction
between compounds containing acid hydroxyl groups and haloepoxyalkanes in the presence of a
strong alkali. A reactor was loaded with the desired amount of bio-oil (containing a certain
amount of moles of -OH / gram of oil) and epichlorohydrin (5 moles epichlorohydrin / mole -
OH). The mixture was heated at 100 °C and benzyltriethylammonium chloride (0.05 moles
BnEt3NCl / mole -OH) was added. After 1 h, the resulting suspension was cooled down to 30 °C,
and an aqueous solution of NaOH 20 wt. % (2 moles NaOH / mole –OH and also containing 0.05
moles of BnEt3NCl / mole –OH) was added dropwise. Once the addition of the alkaline aqueous
solution was completed, the mixture was vigorously stirred for 90 min at 30 °C, and then the
reaction was stopped. After that, the mixture was poured into an extraction funnel and the
organic layer was separated. Through numerous liquid-liquid extractions with deionized water,
the organic phase was abundantly washed until neutral pH was reached, and then vacuumed
concentrated. The crude resulting product was purified by silica gel chromatography using
petroleum ether/ethyl acetate solvent system.
2.2.4. Synthesis of bio-based epoxy resins.
In order to generate a polymeric material, epoxy monomers can undergo step-growth
polymerization, using a crosslinking agent (such as anhydrides, amines, etc.) and chain
homopolymerization in the presence of both, Lewis acids and bases, such as tertiary amines
(known as anionic homopolymerization). As a first approach, epoxidized linseed oil was cured

73
with a commercial hardener (SC-15 part B by Applied Poleramic Inc. composed of
cycloaliphatic amine and polyoxyl-alkylamine) in different weight ratios. The mixture was cured
for 6 hrs. at 60 °C, 6 hrs. at 80 °C, 2 hrs. at 120 °C and finally 1 hr. at 160 °C. In order to
minimize the use of commercial agents in the formulation, epoxidized linseed oil and epoxidized
α-resorcylic acid were mixed in different weight proportions and cured by means of a chain
Homopolymerization. 4-dimethylaminopyridine or DMPA (a Lewis base) was added to the
mixtures in a ratio of 0.08 mol/mol epoxy groups [26]. The mixtures were then poured in an
aluminum mold and cured at 120 °C for 12 hrs and post cured for 2 hrs at 160 °C.
2.2.5. Preparation of the linseed oil based polyurethane.
The PUR plastic sheets were prepared following the procedure described by Kong and Narine
[27] by reacting the resulting polyol with aromatic TDA. As a first approach, a 1.0/1.1 molar
ratios of the -OH group to the isocyanate (NCO) group (Mratio), was chosen for the formulation.
The desired OH/NCO molar ratio satisfies the Equation 3.1:
������ = � ������ �� �������
(� ������ !"#$ %� ������)�� '(�)�#$#! �
(3.1)
Here Wpolyol is the weight of the polyol, EWpolyol is the equivalent weight of polyol, WPU is the
total weight of polyurethane to produce, and EWisocyanate is the equivalent weight of the
isocyanate. The equivalent weights of polyol were determined using Equation 3.2:
�* +�,-�, = ./001%23 $�45 � (3.2)
The weight of the polyol and isocyanate were calculated using the above calculated equivalent
weight. Suitable amounts of polyol and MDI were weighed in a plastic container and stirred
slowly for 2 min. The mixture was cast directly into a metallic mold previously greased with

74
silicone release agent and placed in an oven at 80 °C for polymerization. The samples were
postcured at 120 °C for 12 h.
2.3. Characterization of Starting and Modified Materials
2.3.1 Fourier Transform Infrared Spectroscopy (FTIR)
With the aim to study the functional groups present in the product, FTIR was performed. Infrared
spectra of the starting materials and epoxidized products were measured by attenuated total
reflection (ATR) method using a Thermo Nicolet 6700 Fourier transform infrared spectrometer
connected to a PC with OMNIC software analysis. All spectra were recorded between 400 and
4000 cm-1 over 64 scans with a resolutions of 16 cm-1.
2.3.2 Epoxide equivalent weight determination.
Epoxide equivalent weight (EEW, g/eq.) of the epoxy products was determined by epoxy
titration according to Pan et al. [28]. Basically, a solution of 0.1 M HBr in glacial acetic acid was
used as a titrant, 1 wt. % crystal violet in glacial acetic acid was used as indicator, and toluene as
solvent for the sample. The EEW was calculated using Equation 3.3:
��* = 0111 6 �7 6 8 (3.3)
Where W is the sample weight (g), N is the molarity of HBr solution, and V is the volume of HBr
solution used for titration (mL).
For triglycerides, from the oxirane content values, the relative fractional conversion to oxirane
can be calculated by means of the following expression:
Relative conversion to oxirane = OOexp
OOth (3.4)

75
Where OOexp is the experimentally determined content of oxirane oxygen in 100 grs of oil, and
OOth is the theoretical maximum oxirane oxygen in 100 grs, which is given by:
99�ℎ = {(<= ̥)/2@)/[100 + DE8̥FGH @̥]} K @̥ K 100 (3.5)
Where A (126.9) and A0 (16.0) are the atomic weights of iodine and oxygen, respectively, and
IVo is the initial iodine value of the samples.
2.3.3 Iodine value determination
Iodine value (IV, g I2/g oil.) of linseed oil was determined by titration according to the Wijs
method [29]. Around 0.5 grs of the sample were placed into the flask. 10 mL of toluene was
added to the sample. After that, 15 mL of the Wijs iodine solution was added. Following a
similar procedure, a blank solution was prepared. The mixtures were stored in the dark for 30
minutes at room temperature and then 10 mL of 15 wt. % potassium iodide solution, as well as
50 mL of distilled water, were added to the mixtures. The iodine content was determined by
titration using 0.1 N of sodium thiosulfate solution, using starch aqueous solution as an indicator,
until the color of the solution disappeared. The iodine value was then calculated using Equation
3.6:
<= = (L%M)6 7N 0F./P� (3.6)
Where W is the sample weight (g), N is the normality of Na2S2O3 solution, B is the volume of
solution Na2S2O3 solution used for titration (mL) of the blank, and S is the volume of solution
Na2S2O3 solution used for titration (mL) of the samples.

76
2.3.4 Hydroxyl number determination.
The hydroxyl number was evaluated following the procedure described by the ASTM D4274-11
[30]. Around 0.5 grs of the sample were placed into a pressure bottle, and then 20.0 mL of
acetylation solution (acetic anhydride in pyridine) was added. The bottle was swirled until the
sample was completely dissolved. The containers were placed in a water bath at 98°C and kept
for two hours. The excess of reagent is hydrolyzed with water and the acetic acid is titrated with
standard sodium hydroxyl solution 0.5 N using phenolphthalein as indicator. The hydroxyl
content is calculated from the difference in titration of the blank and sample solutions. The
hydroxyl number, mg KOH/g, of sample was calculated as follows:
Q-R��K-, �STUV� = [(W%X)(Y)(./.0)]Z (3.7)
Where A = NaOH required for titration of the sample (mL), B = NaOH required for titration of
the blank (mL), N = normality of the NaOH, and W = sample used (g).
2.4 Characterization of the epoxy networks
2.4.1 Soxhlet extraction.
Soxhlet extraction was employed to characterize the structures of the bulk copolymers following
the ASTM D5369-93 [31]. An appropriate amount of the bulk polymer was extracted for 24 h
with 200 mL of refluxing dichloromethane with a Soxhlet extractor. Upon extraction, the
resulting solution was concentrated by rotary evaporation and subsequent vacuum drying. The
soluble substances were isolated for further characterization. The insoluble solid was dried in a
vacuum oven for several hours before it was weighed.

77
2.4.2 Scanning Electron Microscopy (SEM)
SEM study was performed with the purpose of obtaining a topological characterization of the
materials. The fractured surfaces of pre-chilled samples in liquid nitrogen were studied by
scanning electron microscope (SEM, Cambridge 360). Samples were sputtered with gold prior to
SEM observations.
2.4.3 Thermal Properties
The thermal properties of the IPNs, as well as the parent resins, were investigated by Differential
Scanning Calorimetry (DSC) on a TA DSC Q2000. Nitrogen was used as the purge gas and the
samples were placed in aluminum pans. For each material sample, the thermal properties were
recorded at 5 °C/min. in a heat/cool/heat cycle from 30 °C to 300 °C.
2.4.4 Thermo-mechanical properties of the networks.
Mechanical properties of the resulting polymers were obtained by dynamic mechanical analysis
(DMA), using a TA Instruments RSA3 DMA. To be able to explain the effects of the co-
monomers on the mechanical properties, it is useful to calculate the crosslinking density (n,
mol/m³). The crosslinking density can be estimated from the experimental data using the rubber
elasticity theory. Thermosets behave as rubbers above Tg. At small deformations, rubber
elasticity predicts that the modulus storage (E’), of an ideal elastomer with a network structure is
proportional to the crosslinking density according to Equation 3.8 [32]:
�� = 3��� = 3��/�� (3.8)
where E` is the rubbery storage modulus, R is the gas constant (8.314 J/mol K), T is the absolute
temperature (K), ρ is the density of the sample (g/m³), and Mc is the molecular weight between

78
crosslinks (g/mol). The temperature and rubbery modulus were determined for the calculation of
the equation at Tg + 30 °C. The temperature at which the peak of the tan delta presents a
maximum was considered as the glass transition temperature of the material. The Fox equation
was employed to fit the experimental Tg data reported in the results. It serves as a way to
describe Tg of the multicomponent networks as a function of relative epoxy monomer content.
The Fox expression is shown in Equation 3.9 [33]:
0
[\ = �0[\0 + �F
[\F (3.9)
In this expression, W1 and W2 represent weight fractions of components 1 and 2 (α-resorcylic and
linseed oil, respectively), and Tg1 and Tg2 are Tg values of polymer samples containing only
components 1 and 2.
3. Results and Discussion
3.1 Synthesis of epoxidized triglycerides
Previous investigations (Chapter II) revealed that at room temperature, all the polymers are in the
transition from the glassy state to the rubbery state, when using direct polymerization as an
approach to obtained triglycerides-based polymeric materials (without any chemical
modification of the starting materials). Since one of the main goals of this research is to develop
and synthetize polymeric materials from renewable resources with high performance and
outstanding properties (such as high modulus, high strength, high glass transition, chemical
resistance, etc.); the driving force behind this project is to explore and study the chemical
functionalization of plant oils. The modification of the triglyceride double bonds to introduce
readily polymerizable functional groups has been used as a common strategy for obtaining high
performance polymeric materials. The unsaturated fatty acid content of commercially available
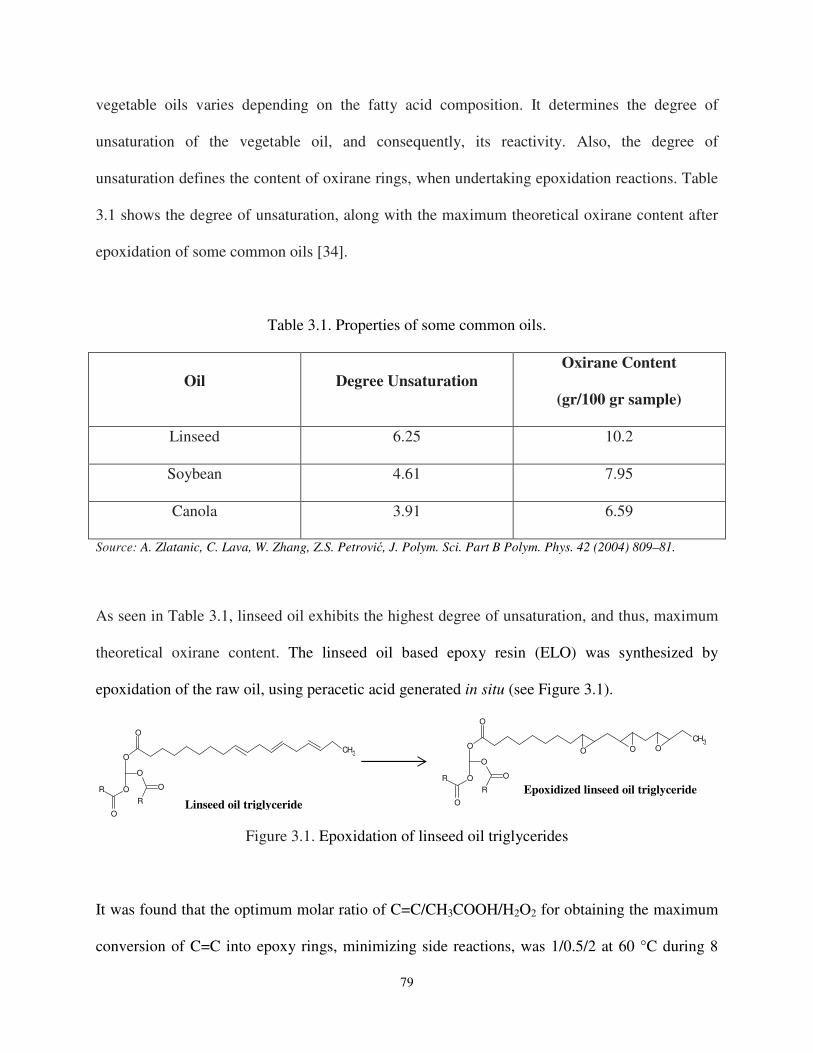
79
vegetable oils varies depending on the fatty acid composition. It determines the degree of
unsaturation of the vegetable oil, and consequently, its reactivity. Also, the degree of
unsaturation defines the content of oxirane rings, when undertaking epoxidation reactions. Table
3.1 shows the degree of unsaturation, along with the maximum theoretical oxirane content after
epoxidation of some common oils [34].
Table 3.1. Properties of some common oils.
Oil Degree Unsaturation
Oxirane Content
(gr/100 gr sample)
Linseed 6.25 10.2
Soybean 4.61 7.95
Canola 3.91 6.59
Source: A. Zlatanic, C. Lava, W. Zhang, Z.S. Petrović, J. Polym. Sci. Part B Polym. Phys. 42 (2004) 809–81.
As seen in Table 3.1, linseed oil exhibits the highest degree of unsaturation, and thus, maximum
theoretical oxirane content. The linseed oil based epoxy resin (ELO) was synthesized by
epoxidation of the raw oil, using peracetic acid generated in situ (see Figure 3.1).
Figure 3.1. Epoxidation of linseed oil triglycerides
It was found that the optimum molar ratio of C=C/CH3COOH/H2O2 for obtaining the maximum
conversion of C=C into epoxy rings, minimizing side reactions, was 1/0.5/2 at 60 °C during 8
O
O
O
O
O
R O
R
CH3
O O O
O
O
O
O
O
R O
R
CH3
Linseed oil triglyceride
Epoxidized linseed oil triglyceride

80
hrs. The iodine value and the oxirane value are important characteristics in the epoxidation of
oils. The iodine value specifies the unsaturation degree of the oils, whereas the oxirane value
determines epoxy content of the modified oil. Once the reaction was completed and the product
isolated and purified, both parameters were quantified through titrations. According to the
experimental results, the conversion of the double bonds to epoxy groups during the epoxidation
reaction was found to be around 90%. From iodine values before and after epoxidation, it was
found that around 98% of the total double bonds were removed during reaction. This indicates
that approximately 8% of epoxy groups were lost due to side reactions. Titration with HBr in
acetic acid revealed that this material has an average molar epoxy content of (5.8 ± 0.7) x 10-3
moles per gram of resin. FTIR analysis supported the grafting process of epoxy functions onto
the fatty acids chains of the triglyceride (Figure 3.2). The FTIR spectrum of triglyceride is
characterized by the absorption band of -C=C- at 3100 cm-1 and the carbonyl group at 1750 cm-1.
After epoxidation, the peak at 3100 cm-1 is removed due to the transformation of the double
bonds into oxirane groups; which is also evident by the presence of the absorption peaks at
approximately 820 cm-1.
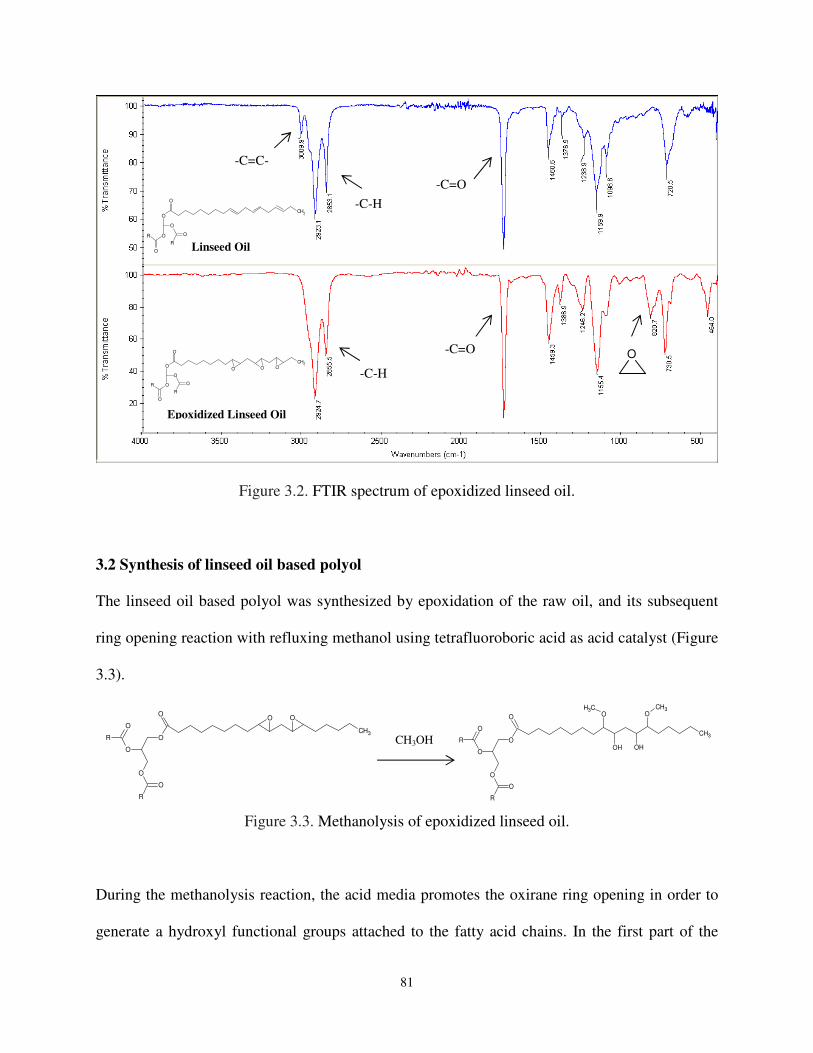
81
O
O
O
O
R
O
O
R
CH3
O O O
O
O
O
R
O
O
R
CH3
OH
CH3O
OH
O
CH3
CH3OH
Figure 3.2. FTIR spectrum of epoxidized linseed oil.
3.2 Synthesis of linseed oil based polyol
The linseed oil based polyol was synthesized by epoxidation of the raw oil, and its subsequent
ring opening reaction with refluxing methanol using tetrafluoroboric acid as acid catalyst (Figure
3.3).
Figure 3.3. Methanolysis of epoxidized linseed oil.
During the methanolysis reaction, the acid media promotes the oxirane ring opening in order to
generate a hydroxyl functional groups attached to the fatty acid chains. In the first part of the
O
O
O
O
O
R O
R
CH3
-C=C-
-C-H
-C=O
O
O
O
O
O
R O
R
CH3
O O O
-C-H
-C=O O
Linseed Oil
Epoxidized Linseed Oil

82
mechanism, the acid reacts with the epoxide to produce a protonated epoxide, a highly
electrophilic intermediate susceptible to the nucleophilic attack of the methanol molecules. Once
the reaction is completed and the product isolated and purified, the number of –OH groups
generated during reaction can be quantified through titrations. Table 3.2 summarizes the
properties of the obtained polyol.
Table 3.2. Properties of the resulting linseed oil based polyol.
Parameters
Hydroxyl Value HV
(mg KOH/g) (*)
Conversion
(%)
Theoretical
Molecular Weight
MW (g/mol)
Functionalitya
(*)
Values 230 (307) 75.0 % 1072 4.2 (5.8)
a. Functionality = [(MW x HV)/56110] * Parenthesis indicates theoretical values
Using the composition of the linseed oil, the theoretical molecular weight and the theoretical
hydroxyl number were calculated. Both parameters were based on the assumption that no dimers
or timers were formed, and that one hydroxyl group was formed per double bond. As seen, the
conversion from epoxy groups to hydroxyl groups reached approx. 75.0% of yield. Just like in
the case of the epoxidation reaction, there is side reactions involved in the ring opening reaction.
During the hydroxylation stage, the newly formed –OH groups, attached to the fatty acid chains,
might attack the protonated epoxy ring leading to the formation of dimers and trimmers, which
lowers the reaction yield. Another important parameter of a polyol, obtained from the hydroxyl
value, is the functionality; which allows calculating the correct stoichiometric ratios when
mixing polyols with multifunctional urethanes in order to produce polyurethanes. The
functionality of the resulting polyol is lower than the number of double bonds per molecule of

83
the starting oil, because a part of the functional groups was lost in the side reactions during the
preparation of the polyol [35].
Besides analytical quantification, the chemical transformation of carbon-carbon double bonds
into hydroxyl groups was also studied and traced through FTIR. As seen in Figure 3.4, the
absorption bands of epoxidized triglycerides appeared within 1720-1700 cm-1 for C=O, at 825
and 845 cm-1 for epoxy groups, and the absence of the C=C stretching band at approximately
3070 cm-1. The final polyol product showed a broad peak within 3500-3200 cm-1, indicating the
presence of –OH groups and the disappearance of the epoxy bands at 825 and 845 cm-1.
Figure 3.4. FTIR spectrum of hydroxylated linseed oil.
O
O
O
O
O
R O
R
CH3
O
O
O
O
O
R O
R
CH3
O O O
O
O
O
O
R
O
O
R
CH3
OH
CH3
O
OH
O
CH3
Linseed Oil
Epoxidized Linseed Oil
Hydroxylated Linseed Oil

84
3.3 Functionalization of phenolic compounds with epoxy groups.
3.3.1 Synthesis of triglycidylated ether of α-resorcylic acid
Regarding the coupling of epoxy groups to the α-resorcylic acid, it was found that the treatment
of this compound with epichlorohydrin in alkaline aqueous medium (see Figure 3.5), in the
presence of benzyltriethylammonium chloride as a phase transfer catalyst, allowed the total
glycidylation of this phenolic acid to generate the triglycidylated derivative of α-resorcylic acid
or TRA (also referred to as ERA, a short version of epoxidized α-resorcylic acid).
Figure 3.5. Glycidylation reaction of α-resorcylic acid with epichlorohydrin.
The reaction yield, after purification through silica gel chromatography, was around 80 %. The
remaining 20 % probably correspond to mono and di glycidilated products of α-resorcylic acid,
as well as, unreacted material. Because phenols are more acidic than alcohols, they can be
converted to phenolates through the use of a strong base. These intermediates are strong
nucleophiles and can react with primary alkyl halides to form ethers. According to the literature
[36], once the phenolate anions are formed, they can react with epichlorohydrin following two
competitive mechanisms: one-step nucleophilic substitution (mechanism SN2) with cleavage of
the C−Cl bond and a two-step mechanism based on ring opening of epichlorohydrin followed by
intramolecular cyclization (through SN1 mechanism), with the release of chloride, to reform the
epoxy ring. Titration with HBr showed that resulting mixture after modification has an average
OHOH
O OH
O O
O O
O
O
O
O
Cl
α- Resorcylic Acid Triglycidylated ether of α-Resorcylic Acid (TRA)
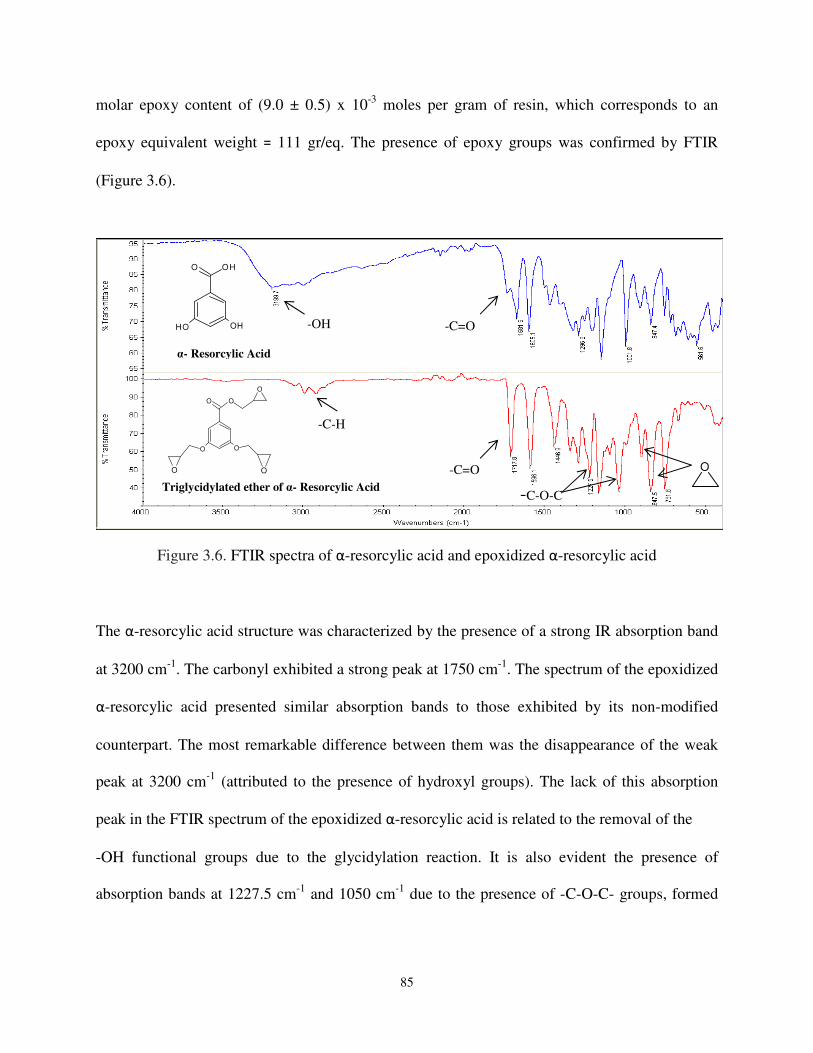
85
molar epoxy content of (9.0 ± 0.5) x 10-3 moles per gram of resin, which corresponds to an
epoxy equivalent weight = 111 gr/eq. The presence of epoxy groups was confirmed by FTIR
(Figure 3.6).
Figure 3.6. FTIR spectra of α-resorcylic acid and epoxidized α-resorcylic acid
The α-resorcylic acid structure was characterized by the presence of a strong IR absorption band
at 3200 cm-1. The carbonyl exhibited a strong peak at 1750 cm-1. The spectrum of the epoxidized
α-resorcylic acid presented similar absorption bands to those exhibited by its non-modified
counterpart. The most remarkable difference between them was the disappearance of the weak
peak at 3200 cm-1 (attributed to the presence of hydroxyl groups). The lack of this absorption
peak in the FTIR spectrum of the epoxidized α-resorcylic acid is related to the removal of the
-OH functional groups due to the glycidylation reaction. It is also evident the presence of
absorption bands at 1227.5 cm-1 and 1050 cm-1 due to the presence of -C-O-C- groups, formed
O OH
OHOH
O O
OO
O
OO
-OH
-C-H
-C=O
-C=O
O
-C-O-C
α- Resorcylic Acid
Triglycidylated ether of α- Resorcylic Acid

86
during the reaction, as well as the characteristic bands for oxirane rings at 847.5 cm-1 and 910
cm-1.
3.3.2. Synthesis of diglycidylated ether of α-resorcinol
With the aim of correlating the mechanical properties of the resulting resins with the number of
functionalities of the epoxy monomers, resorcinol (containing two phenolic –OH groups) was
also modified with epichlorohydrin (see Figure 3.7). The reaction yield, after purification
through silica gel chromatography, was around 85%. The remaining 15% probably corresponded
to monoglycidylated products and unreacted resorcinol. Titration with HBr showed that the
resulting mixture after chemical modification has an average molar epoxy content of (8.7 ± 0.5)
x 10-3 moles per gram of resin, resulting in an epoxy equivalent weight = 115 g/eq. The presence
of epoxy groups was also confirmed by FTIR (Figure 3.8). The lack of the absorption peak at
around 3200 cm-1 evidenced the transformation of phenolic hydroxyl groups into glycidyl
moieties. Bands for oxirane rings at 847.5 cm-1 and 910 cm-1 confirmed the presence of epoxy
groups.
Figure 3.7. Glycidylation reaction of resocinol with epichlorohydrin.
O
Cl
Resorcinol Diglycidylated ether of resorcinol
OHOH
OO
O O
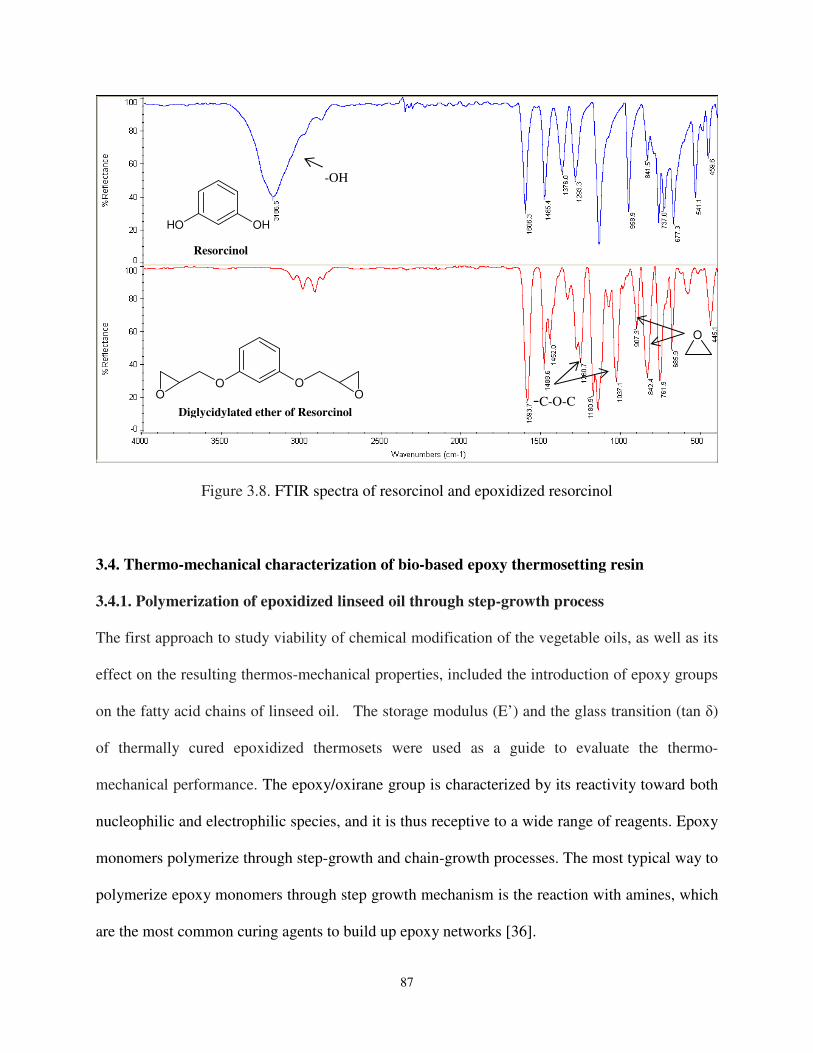
87
Figure 3.8. FTIR spectra of resorcinol and epoxidized resorcinol
3.4. Thermo-mechanical characterization of bio-based epoxy thermosetting resin
3.4.1. Polymerization of epoxidized linseed oil through step-growth process
The first approach to study viability of chemical modification of the vegetable oils, as well as its
effect on the resulting thermos-mechanical properties, included the introduction of epoxy groups
on the fatty acid chains of linseed oil. The storage modulus (E’) and the glass transition (tan δ)
of thermally cured epoxidized thermosets were used as a guide to evaluate the thermo-
mechanical performance. The epoxy/oxirane group is characterized by its reactivity toward both
nucleophilic and electrophilic species, and it is thus receptive to a wide range of reagents. Epoxy
monomers polymerize through step-growth and chain-growth processes. The most typical way to
polymerize epoxy monomers through step growth mechanism is the reaction with amines, which
are the most common curing agents to build up epoxy networks [36].
OHOH
OO
O O
Resorcinol
Diglycidylated ether of Resorcinol
O
-C-O-C
-OH
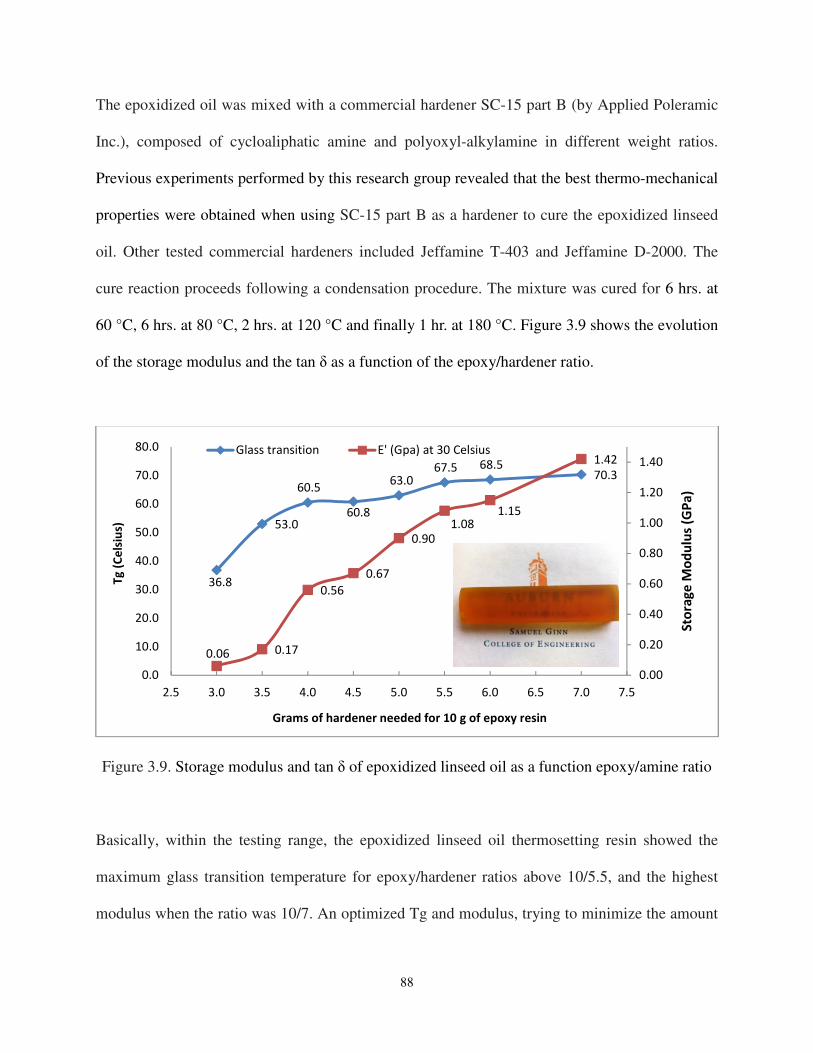
88
The epoxidized oil was mixed with a commercial hardener SC-15 part B (by Applied Poleramic
Inc.), composed of cycloaliphatic amine and polyoxyl-alkylamine in different weight ratios.
Previous experiments performed by this research group revealed that the best thermo-mechanical
properties were obtained when using SC-15 part B as a hardener to cure the epoxidized linseed
oil. Other tested commercial hardeners included Jeffamine T-403 and Jeffamine D-2000. The
cure reaction proceeds following a condensation procedure. The mixture was cured for 6 hrs. at
60 °C, 6 hrs. at 80 °C, 2 hrs. at 120 °C and finally 1 hr. at 180 °C. Figure 3.9 shows the evolution
of the storage modulus and the tan δ as a function of the epoxy/hardener ratio.
Figure 3.9. Storage modulus and tan δ of epoxidized linseed oil as a function epoxy/amine ratio
Basically, within the testing range, the epoxidized linseed oil thermosetting resin showed the
maximum glass transition temperature for epoxy/hardener ratios above 10/5.5, and the highest
modulus when the ratio was 10/7. An optimized Tg and modulus, trying to minimize the amount
36.8
53.0
60.5
60.8
63.067.5 68.5
70.3
0.06 0.17
0.56
0.67
0.901.08
1.15
1.42
0.00
0.20
0.40
0.60
0.80
1.00
1.20
1.40
0.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
80.0
2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5
Tg
(C
els
ius)
Grams of hardener needed for 10 g of epoxy resin
Glass transition E' (Gpa) at 30 Celsius
Sto
rag
e M
od
ulu
s (G
Pa
)

89
of hardener, is attained when using an epoxy/amine ratio of ~10/6. Materials with low amine
proportions contain unreacted groups or even free monomers. These unreacted species are
trapped inside of the polymer network and act as plasticizers lowering both the modulus and Tg.
3.5. Preparation of the linseed oil based polyurethane.
The second approach comprised the modification of the linseed oil structure by introducing
secondary –OH groups that can be polymerized by means of a reaction with isocyanates. The
linseed oil-based polyurethane was prepared by mixing the resulting polyol with aromatic TDA.
As a first approach to evaluate the thermo-mechanical behavior of this polyurethane, a 1.0/1.1
molar ratios of the -OH group to the isocyanate group was chosen for the formulation. Using
Equation a polyol equivalent weight = 244 g/eq. was obtained. Storage modulus (E`) and tan δ
were recorded as a function of the temperature. Results revealed the presence of a crosslinked
network, with a glass transition temperature of around – 6.5°C and a modulus with a magnitude
of approx. 3 MPa at ambient conditions. This is a clear indication of the elastomeric nature of the
resulting materials, and its poor capacity to withstand deformation when stress is applied.
3.6 Polymerization of acrylated soybean oil through free radical mechanism
The third approach covered the study of the acrylated oils. Acrylated soybean oil was employed
to explore the feasibility of using oils modified with acrylate groups to generate suitable
materials for different polymer applications. The supplier states that the product has a molecular
weight of approximately 1800-7200 g/mol; and a viscosity at 25°C of 18000-32000 cps. With the
aim of improving the solubility of the radical initiator (AIBN), the acrylate soybean oil was
diluted with methyl methacrylate, a low molecular weight comonomer. DMA was also used to
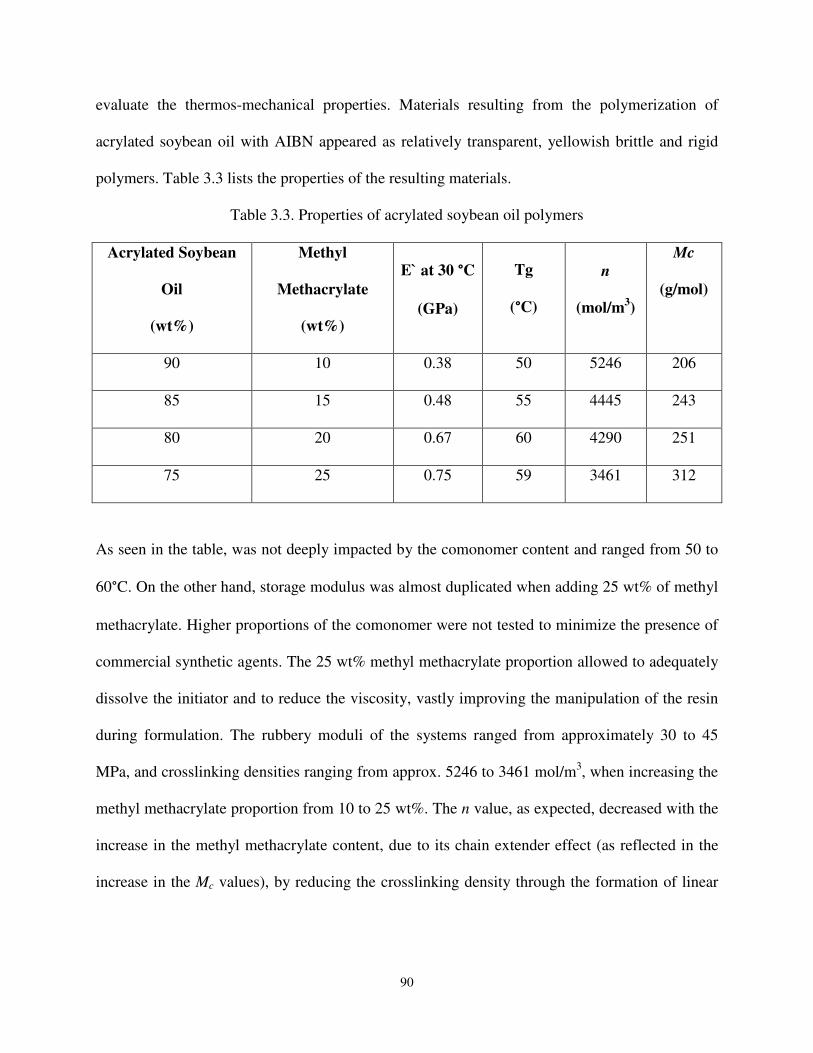
90
evaluate the thermos-mechanical properties. Materials resulting from the polymerization of
acrylated soybean oil with AIBN appeared as relatively transparent, yellowish brittle and rigid
polymers. Table 3.3 lists the properties of the resulting materials.
Table 3.3. Properties of acrylated soybean oil polymers
Acrylated Soybean
Oil
(wt%)
Methyl
Methacrylate
(wt%)
E` at 30 °C
(GPa)
Tg
(°C)
n
(mol/m3)
Mc
(g/mol)
90 10 0.38 50 5246 206
85 15 0.48 55 4445 243
80 20 0.67 60 4290 251
75 25 0.75 59 3461 312
As seen in the table, was not deeply impacted by the comonomer content and ranged from 50 to
60°C. On the other hand, storage modulus was almost duplicated when adding 25 wt% of methyl
methacrylate. Higher proportions of the comonomer were not tested to minimize the presence of
commercial synthetic agents. The 25 wt% methyl methacrylate proportion allowed to adequately
dissolve the initiator and to reduce the viscosity, vastly improving the manipulation of the resin
during formulation. The rubbery moduli of the systems ranged from approximately 30 to 45
MPa, and crosslinking densities ranging from approx. 5246 to 3461 mol/m3, when increasing the
methyl methacrylate proportion from 10 to 25 wt%. The n value, as expected, decreased with the
increase in the methyl methacrylate content, due to its chain extender effect (as reflected in the
increase in the Mc values), by reducing the crosslinking density through the formation of linear

91
chains embedded in the acrylated oil network. Physical densities were around 1.08 g/cm3, and
did not statically changed as a function of the methyl methacrylate proportion
As a summary of results, so far three chemical modifications (epoxy, hydroxyl and acrylate)
have been preliminary evaluated. As seen, the step-growth process employed to formulate the
epoxy resin implies the use of a commercial amine to crosslink the epoxy monomers. This
generates a partially bio-based (around 60 wt% of the total weight of the formulation) resin that
showed a considerably better thermo-mechanical performance than the materials produced in
Chapter II, where non-modified tung oil was cationically polymerized to yield elastomeric
materials. Clearly, this modification improves the mechanical properties of oil-based materials,
yielding thermosets with modulus in the order of GPa and glass transitions temperatures around
70°C.
Concerning the second approach, the inclusion of hydroxyl groups to produce polyurethanes; it is
evident that the elastomeric nature of these resins cannot make them suitable candidates to
replace materials intended to structural or composite applications. Due to their flexibility, these
polyurethanes could be used as matrices for thermal insulation, packing or cushioning. However,
the resulting materials were extremely flexible, and lacked of mechanical resistance; therefore,
no further investigations were carried out using polyurethanes. Regarding acrylated oils
polymerized through free radical mechanism, it can be concluded that acrylated oils can be used
to make rigid thermosetting polymers and composites. These materials exhibited high
crosslinking densities, resulting in higher Tg (approx. 60°C) and improved storage modulus (~0.7
GPa) than their non-functionalized counterparts. From these findings, the epoxy and acrylate

92
moieties are promising approaches to more reactive monomers able to generate materials with
good mechanical properties than their non-modified counterparts. Epoxy resins are the main
topic on the remaining sections of this chapter, meanwhile acrylated oils will be considered in
chapter IV as a source of monomers to generate high performance materials.
It is clear however, that all of the above mentioned modifications lead to not fully, partially bio-
based resins; which still lacking of the desire properties to simulate the performance of existing
counterparts from non-renewable resources. At this point of the research, the efforts were focus
on improving the thermo-mechanical properties of the epoxy resins by introducing aromatic, low
molecular weight comonomers able to increase the chain stiffness and thus, enhancing the
mechanical performance of the oil-based epoxy resins to simulate and even to improve the
properties of commercial systems. It was also extremely important to increase the contribution of
renewable materials into the formulations, by minimizing the use of commercial compounds.
Therefore, this chapter also studies the chain-growth copolymerization (which reduces the
amount of commercial agents in the formulation per weight basis) of epoxy monomers. The
details of the copolymerization of epoxidized linseed oil and a low molecular weight aromatic
epoxy comonomer are summarized in the following sections.
3.7 Copolymerization of epoxidized linseed oil and epoxidized α-resorcylic acid through
chain-growth process
Another possible ways to polymerize epoxy monomers is through the use of small amounts of
Lewis bases that act as initiators of the anionic polymerization. Commonly used initiators
include tertiary amines such as 4-dimethylaminopyridine (DMAP). The mechanism for the

93
anionic polymerization of epoxy monomers initiated by tertiary amines is highly complex and
has not been fully described yet. Figure 3.10 depicts a simplified scheme of the anionic
polymerization of epoxy monomers.
Figure 3.10. Anionic polymerization of epoxy monomers.
In order to prepare the resins, epoxidized linseed oil and triglycidylated ether of α-resorcylic acid
(TRA) or epoxidized α-resorcylic acid, were mixed in different weight proportions. The idea of
using this aromatic trifunctional monomer lies upon the principle that the flexibility of the
polymer backbone may be markedly decreased with the insertion of groups that stiffen the chain
by impeding rotation, since more thermal energy is then required to set the chain in motion. The
presence of rigid aromatic structures confers to the materials the thermo-mechanical properties
required in their industrial and high performance applications. Dimethylamino pyridine or
DMPA was added to the mixtures in a ratio of 0.08 mol DMAP/mol epoxy. After the curing
process, the polymeric materials appeared as dark-brown stiff materials at room temperature. For
all the studied systems, suitable materials for analysis were not obtained for epoxidized linseed
oil proportions above 30 wt%; due to the lack of miscibility of the two compounds.
+ n
DMAP Epoxy Monomer
O
R R1
N+
CH3
CH3
N
O
R1
O-
R1
R
R
R = H or Alkyl Group R1
= O-Ar
NN
CH3
CH3
n

94
3.8. Scanning Electron Microscopy
The fracture surfaces of the thermosets were studied by scanning electron microscopy (SEM).
The results of the morphological study are shown in Figure 3.11. The 100 wt% α-epoxy
resorcylic acid formulation possesses a fracture surface and an internal structure of a brittle
material, characterized by a highly glassy surface; where crack marks, ridges, and planes are
dispersed all over the surface [37]. Regarding the 100 wt% epoxy linseed oil resin, it shows the
morphology of a ductile material, with a smooth and featureless surface denoting a highly plastic
deformation [38]. The 80-20 and 70-30 wt% copolymers present a very irregular surface,
characterized by randomly distributed cavities, possibly denoting the presence of a non-
homogeneous structure, through the formation of rubbery domains as a result of micro-phase
separation of the components.
3.11.a
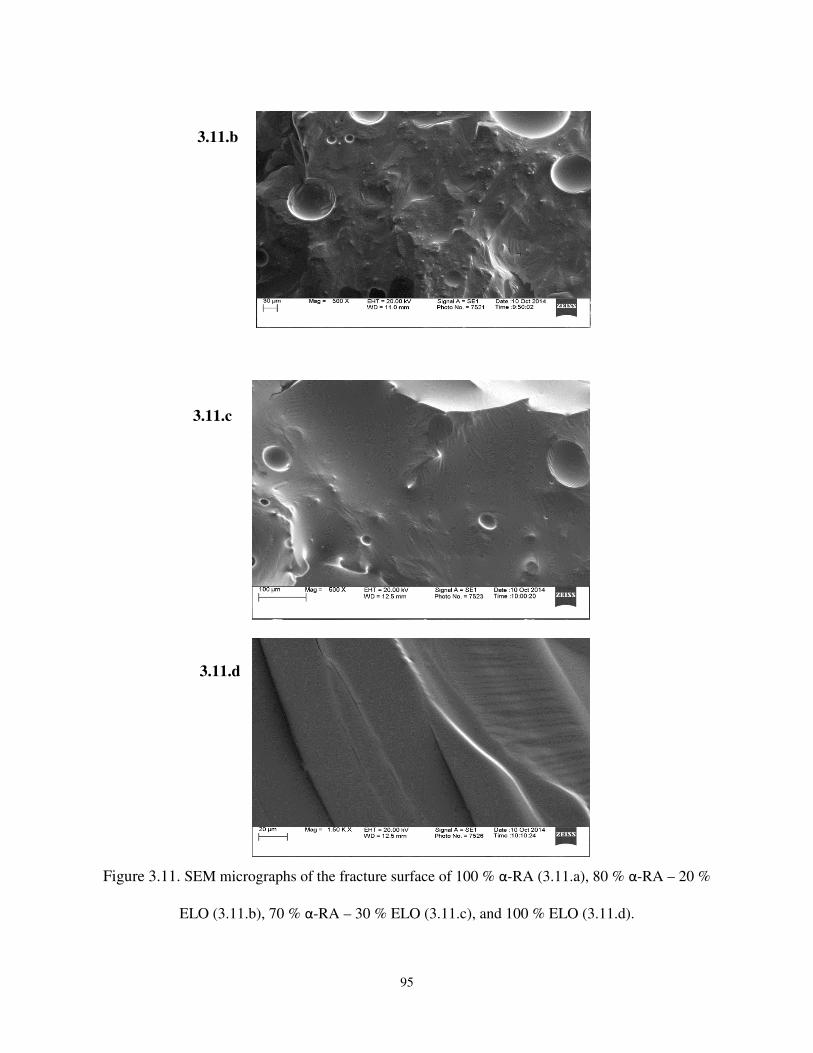
95
Figure 3.11. SEM micrographs of the fracture surface of 100 % α-RA (3.11.a), 80 % α-RA – 20 %
ELO (3.11.b), 70 % α-RA – 30 % ELO (3.11.c), and 100 % ELO (3.11.d).
3.11.b
3.11.c
3.11.d

96
3.9. Thermo-mechanical characterization of the epoxidized linseed oil (ELO) and
epoxidized α-resorcylic acid (ERA) copolymers.
Figure 3.12 shows the temperature dependence of the storage modulus (E`) and the tan δ. The
peak in tan δ corresponds to the main mechanical relaxation of the matrix, which was used as a
criterion to determine the Tg of the polymer.
Figure 3.12. Temperature dependence of the storage modulus (3.12.a) and loss factor or tan δ
(3.12.b) as a function of the composition of the linseed oil – α-RA epoxy resins
100% ERA
90% ERA- 10% ELO
80% ERA - 20% ELO
70% ERA - 30%
ELO
100% ELO
1E+05
1E+06
1E+07
1E+08
1E+09
1E+10
-50 -25 0 25 50 75 100 125 150 175 200
Sto
rag
e M
od
ulu
s (P
a)
Axis Title
100% ERA90% ERA - 10%
ELO
80% ERA - 20%
ELO
70% ERA - 30%
ELO
100% ELO
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2
-50 -25 0 25 50 75 100 125 150 175 200
Ta
n δ
Temperature (°C)3.12.b
3.12.a
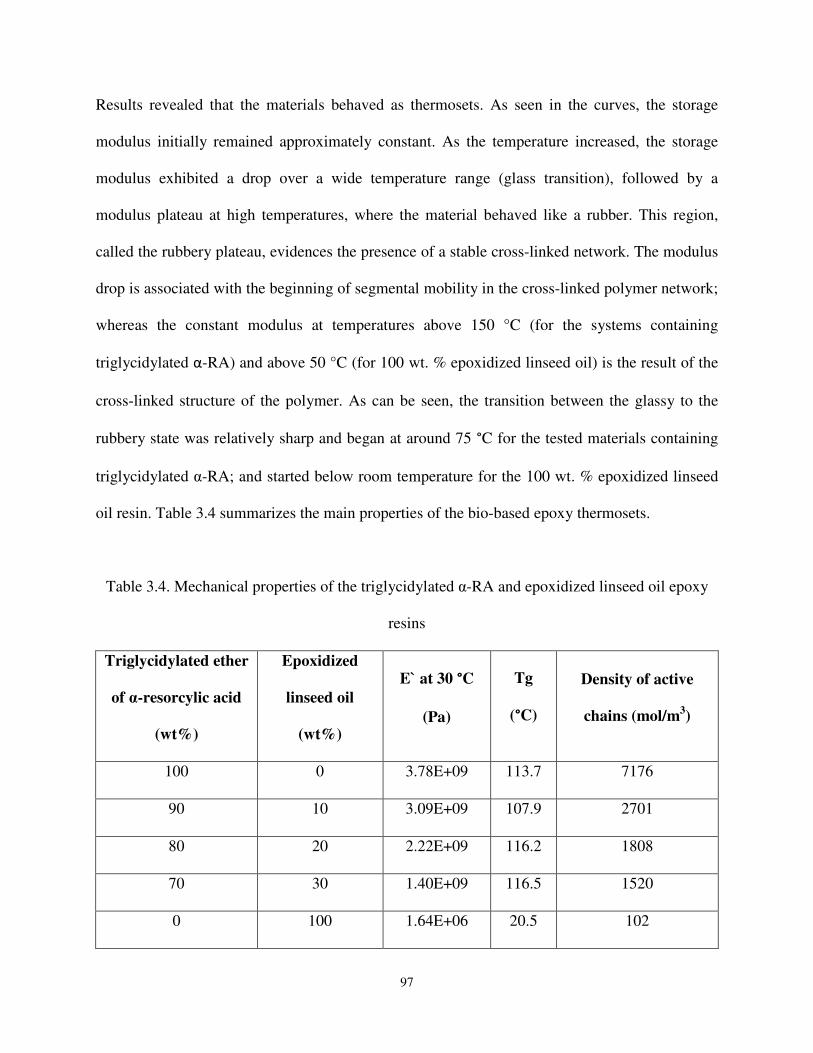
97
Results revealed that the materials behaved as thermosets. As seen in the curves, the storage
modulus initially remained approximately constant. As the temperature increased, the storage
modulus exhibited a drop over a wide temperature range (glass transition), followed by a
modulus plateau at high temperatures, where the material behaved like a rubber. This region,
called the rubbery plateau, evidences the presence of a stable cross-linked network. The modulus
drop is associated with the beginning of segmental mobility in the cross-linked polymer network;
whereas the constant modulus at temperatures above 150 °C (for the systems containing
triglycidylated α-RA) and above 50 °C (for 100 wt. % epoxidized linseed oil) is the result of the
cross-linked structure of the polymer. As can be seen, the transition between the glassy to the
rubbery state was relatively sharp and began at around 75 °C for the tested materials containing
triglycidylated α-RA; and started below room temperature for the 100 wt. % epoxidized linseed
oil resin. Table 3.4 summarizes the main properties of the bio-based epoxy thermosets.
Table 3.4. Mechanical properties of the triglycidylated α-RA and epoxidized linseed oil epoxy
resins
Triglycidylated ether
of α-resorcylic acid
(wt%)
Epoxidized
linseed oil
(wt%)
E` at 30 °C
(Pa)
Tg
(°C)
Density of active
chains (mol/m3)
100 0 3.78E+09 113.7 7176
90 10 3.09E+09 107.9 2701
80 20 2.22E+09 116.2 1808
70 30 1.40E+09 116.5 1520
0 100 1.64E+06 20.5 102

98
As shown, the storage modulus (E’) at room temperature considerably decreased when the
triglycidylated α-RA wt. % content was reduced. The decline in the modulus values is directly
correlated to the decrease in the density of active chains or n; parameter that is often associated
to the crosslinking density of the material. For the calculation of n, it was assumed that all of the
components in the systems entered into the network, which is in reality impossible. However,
after the Soxhlet extraction of samples with acetone for 6 hours, it was observed that the total
amount of monomers that did not polymerize ranged from 14 % to 19 % (from epoxidized
resorcylic acid 100 wt% to epoxidized linseed oil 100 wt%). The glass transition temperature
was not highly influenced by the reduction in the α-RA wt. % content and it showed its
maximum value for the 80 epoxidized α-RA wt. % - 20 wt. % epoxidized linseed oil systems. As
expected, the poorest mechanical properties where exhibited by the 100 % wt. epoxidized LO
system. It is of worth to highlight that there are two opposite mechanical behaviors present in the
two epoxy monomers. On the one hand, resins containing α-resorcylic acid present the typical
behavior of glassy, brittle polymers, with modulus in the order of GPa at room temperature and
glass transition temperatures above 100°C. On the other hand, the linseed oil proves to be
completely elastomeric and ductile material, having a crosslinking density even lower than a
common rubber band (typically n = 190 mol/m3 [39]).
As previously stated, suitable materials for analysis were not obtained for epoxidized linseed oil
proportions above 30 wt%. As a result of this observation, concerns about the miscibility of the
two phases arose during the characterization of the resulting materials. In order to study this
phenomenon, the correlation between the glass transition temperature and the composition,
through the Fox model was employed to evaluate the miscibility of the phases. This model
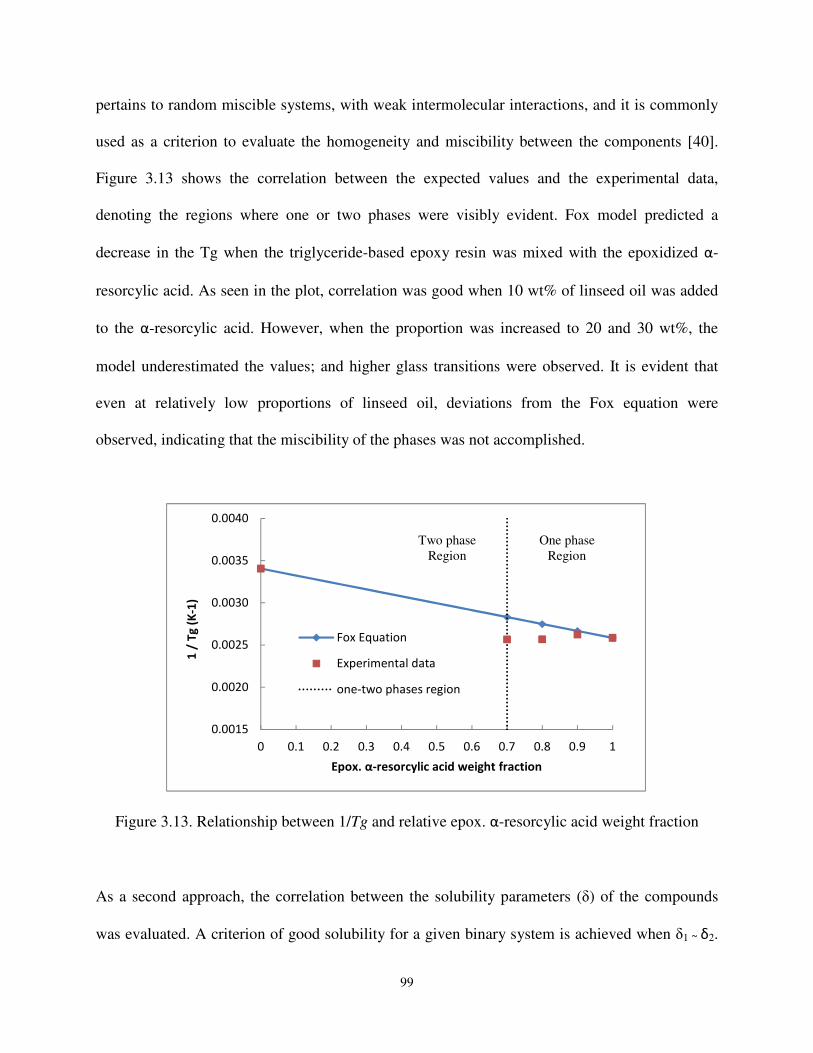
99
pertains to random miscible systems, with weak intermolecular interactions, and it is commonly
used as a criterion to evaluate the homogeneity and miscibility between the components [40].
Figure 3.13 shows the correlation between the expected values and the experimental data,
denoting the regions where one or two phases were visibly evident. Fox model predicted a
decrease in the Tg when the triglyceride-based epoxy resin was mixed with the epoxidized α-
resorcylic acid. As seen in the plot, correlation was good when 10 wt% of linseed oil was added
to the α-resorcylic acid. However, when the proportion was increased to 20 and 30 wt%, the
model underestimated the values; and higher glass transitions were observed. It is evident that
even at relatively low proportions of linseed oil, deviations from the Fox equation were
observed, indicating that the miscibility of the phases was not accomplished.
Figure 3.13. Relationship between 1/Tg and relative epox. α-resorcylic acid weight fraction
As a second approach, the correlation between the solubility parameters (δ) of the compounds
was evaluated. A criterion of good solubility for a given binary system is achieved when δ1 ~ δ2.
0.0015
0.0020
0.0025
0.0030
0.0035
0.0040
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
1 /
Tg
(K
-1)
Epox. α-resorcylic acid weight fraction
Fox Equation
Experimental data
one-two phases region
One phase Region
Two phase Region

100
It provides a useful practical guide for non-polar systems [41]. As an approximation, the reported
solubility parameters of a bisphenol A type epoxy resin and linseed oil have been considered
[42,43]. In the case of the epoxy resin, the influence of the dipolar interactions, as well as
hydrogen bonding, can been neglected; thus reducing the Hansen three component model to only
the dispersion contribution. Therefore, approximating δD ~ δt for non-polar compounds, the
reported value of the solubility parameter for a Bisphenol A type resin is 20.0 MPa1/2. On the
other hand, the solubility parameter for linseed oil is 16.4 MPa1/2. A difference of around 3.6
units in this parameter indicates that the linseed oil is sparingly soluble in the epoxy resin, which
structurally resembles the epoxidized derivative of α-resorcylic acid.
It is also important to discuss aspects related to the structural characteristics of the monomers,
which highly influence the reaction. On the one hand, epoxidized oils contain oxirane
functionalities that are located near the middle of the 18-carbon length fatty acid chains. As a
consequence of this feature, these groups are sterically hindered at both carbons; heavily
diminishing their reactivity. On the other hand, the triglycidylated ether of α-resorcylic acid
contains pending oxirane functionalities that are non-sterically hindered, facilitating the attack of
the anionic growing species generated during the propagation stage of the reaction. Mechanistic
studies indicate that initiation of the reaction involves SN2 displacement to form alkoxide ion,
and the propagation occurs via nucleophilic displacement involving the new alkoxide ion [44]. It
is well known that the SN2 transition state involves partial bond formation between the incoming
nucleophile and the substrate. It seems reasonable to consider that a hindered, bulky substrate
prevents the approach of the nucleophile, making bond formation difficult. As a result of this
steric effect, during the polymerization stage, the reaction might have had more affinity for the

101
epoxy monomers derived from the triglycidylated ether of α-resorcylic acid; than for the epoxy
monomers obtained from the modification for the linseed oil. After the curing and subsequent
formation of a three-dimensional network, the long chain rubbery molecules of the triglyceride
derived epoxy resin can either participate in the phase separation (forming small rubbery
domains), or taking part in the epoxy matrix by chemical bonding [45]. This process might
happen randomly, fact that is reflected in the lack of tendency observed in the evolution of the
glass transition temperature as a function of the linseed oil weight fraction.
Recalling from the discussion about the thermo-mechanical properties and the resulting effect of
adding a low molecular weight aromatic comonomer, it is very clear that the presence of rigid
aromatic structures do confer to the materials the typical behavior of glassy, brittle polymers; by
providing a highly crosslinked structure, able to withstand deformation when stresses are
applied. Compared to the triglyceride-based epoxy monomers, aromatic epoxy monomers
provide a considerably better option to formulate materials with good thermo-mechanical
performance.
In order to compare the results obtained in this research, the triglycidylated ether of α-resorcylic
acid was mixed in different weight proportions with the well-known commercial epoxy resin
EPON 828. This is an undiluted clear di-functional bisphenol A/epichlorohydrin derived liquid
epoxy resin. It is synthesized following a similar chemical approach to the one used in this
research to attach epoxy groups to the α-resorcylic acid. The triglycidylated ether of α-resorcylic
acid (TRA) and the EPON 828 were mixed in different weight proportions, and were also cured
using DMAP as an initiator.

102
Figure 3.14 shows the micrographs of the fracture surfaces. All the resins present homogeneous
morphology with no visible phase domains. The surfaces are characterized by smooth, glassy
microstructures, without any plastic deformation.
3.14.a
a
3.14.b

103
Figure 3.14. SEM micrographs of the fracture surface of 100 wt% α-RA (3.14.a), 75 wt% α-RA – 25
wt% EPON 828 (3.14.b), 50 wt% α-RA – 50 wt% EPON 828 (3.14.c), and 100 wt% EPON 828
(3.14.d).
3.14.c
3.14.d
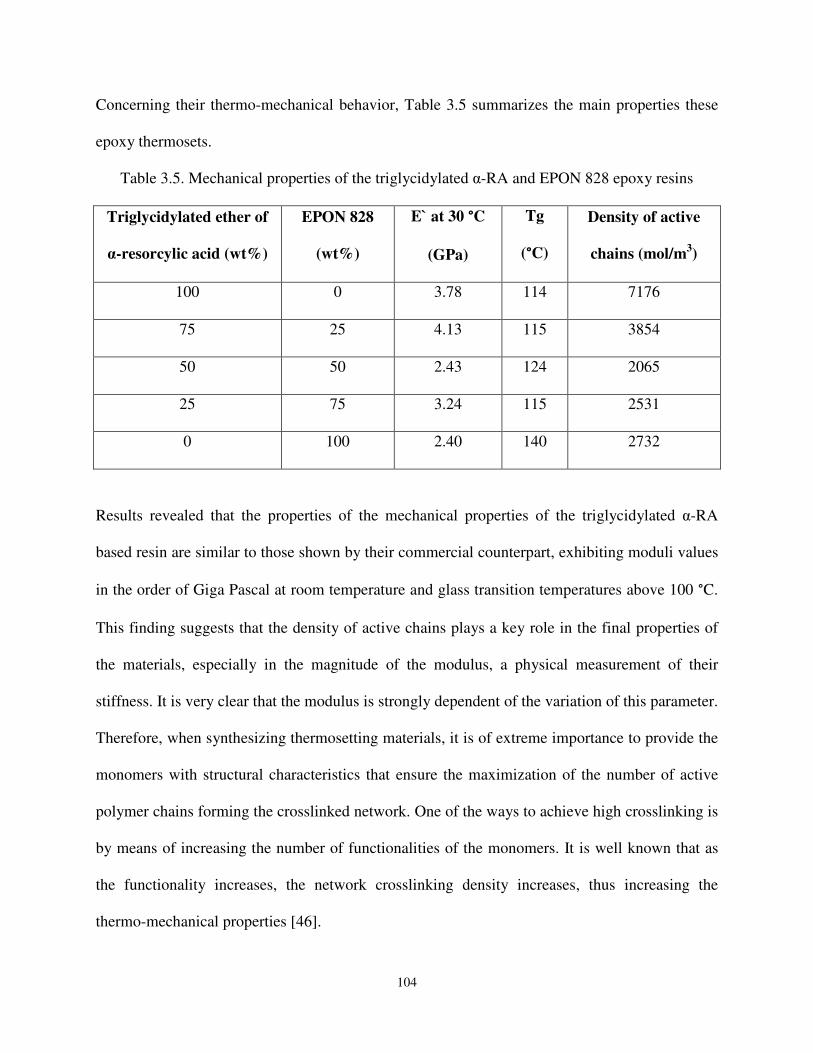
104
Concerning their thermo-mechanical behavior, Table 3.5 summarizes the main properties these
epoxy thermosets.
Table 3.5. Mechanical properties of the triglycidylated α-RA and EPON 828 epoxy resins
Triglycidylated ether of
α-resorcylic acid (wt%)
EPON 828
(wt%)
E` at 30 °C
(GPa)
Tg
(°C)
Density of active
chains (mol/m3)
100 0 3.78 114 7176
75 25 4.13 115 3854
50 50 2.43 124 2065
25 75 3.24 115 2531
0 100 2.40 140 2732
Results revealed that the properties of the mechanical properties of the triglycidylated α-RA
based resin are similar to those shown by their commercial counterpart, exhibiting moduli values
in the order of Giga Pascal at room temperature and glass transition temperatures above 100 °C.
This finding suggests that the density of active chains plays a key role in the final properties of
the materials, especially in the magnitude of the modulus, a physical measurement of their
stiffness. It is very clear that the modulus is strongly dependent of the variation of this parameter.
Therefore, when synthesizing thermosetting materials, it is of extreme importance to provide the
monomers with structural characteristics that ensure the maximization of the number of active
polymer chains forming the crosslinked network. One of the ways to achieve high crosslinking is
by means of increasing the number of functionalities of the monomers. It is well known that as
the functionality increases, the network crosslinking density increases, thus increasing the
thermo-mechanical properties [46].
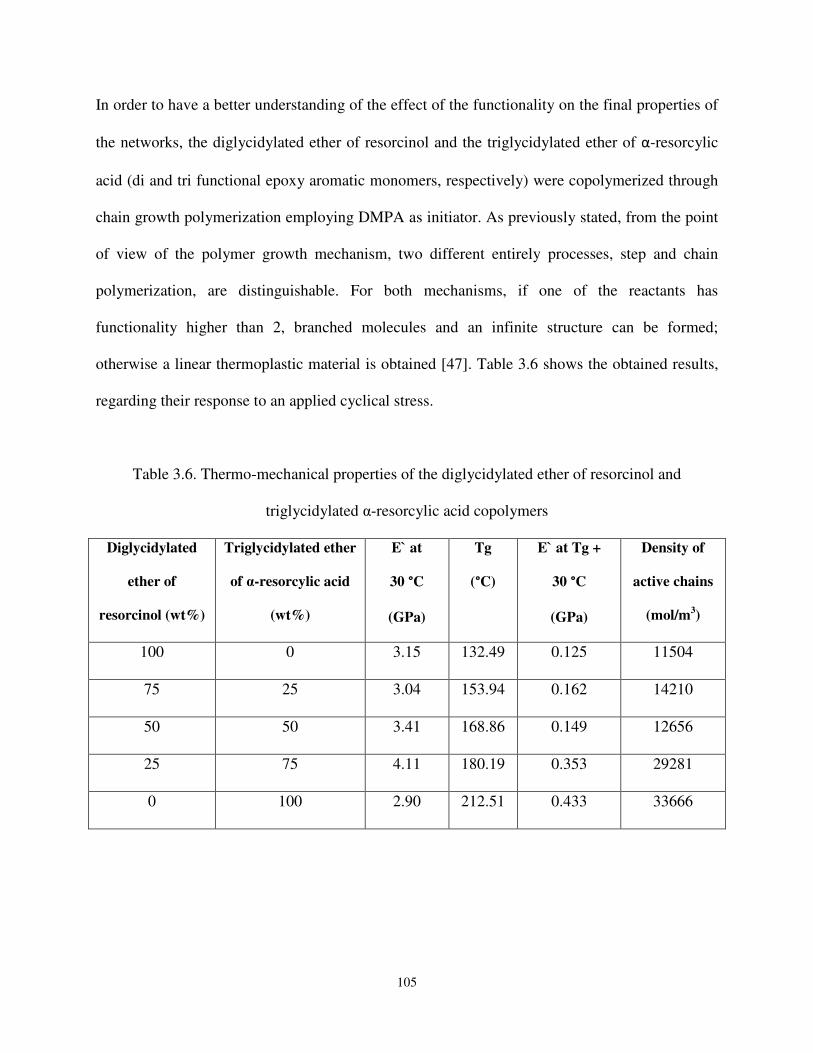
105
In order to have a better understanding of the effect of the functionality on the final properties of
the networks, the diglycidylated ether of resorcinol and the triglycidylated ether of α-resorcylic
acid (di and tri functional epoxy aromatic monomers, respectively) were copolymerized through
chain growth polymerization employing DMPA as initiator. As previously stated, from the point
of view of the polymer growth mechanism, two different entirely processes, step and chain
polymerization, are distinguishable. For both mechanisms, if one of the reactants has
functionality higher than 2, branched molecules and an infinite structure can be formed;
otherwise a linear thermoplastic material is obtained [47]. Table 3.6 shows the obtained results,
regarding their response to an applied cyclical stress.
Table 3.6. Thermo-mechanical properties of the diglycidylated ether of resorcinol and
triglycidylated α-resorcylic acid copolymers
Diglycidylated
ether of
resorcinol (wt%)
Triglycidylated ether
of α-resorcylic acid
(wt%)
E` at
30 °C
(GPa)
Tg
(°C)
E` at Tg +
30 °C
(GPa)
Density of
active chains
(mol/m3)
100 0 3.15 132.49 0.125 11504
75 25 3.04 153.94 0.162 14210
50 50 3.41 168.86 0.149 12656
25 75 4.11 180.19 0.353 29281
0 100 2.90 212.51 0.433 33666

106
Figure 3.15 shows the temperature dependence of the storage modulus (E`) and the tan δ. The
peak in tan δ corresponds to the main mechanical relaxation of the matrix, which was used as a
criterion to determine the Tg of the polymer.
Figure 3.15. Temperature dependence of the storage modulus (3.15.a) and loss factor or tan δ
(3.15.b) as a function of the composition of the resorcinol weight content for the resorcinol-α-
resorcylic acid epoxy resins.
1.E+07
1.E+08
1.E+09
1.E+10
30 55 80 105 130 155 180 205 230 255 280 305 330
Log
Sto
rag
e M
od
ulu
s (P
a)
Temperature (°C)
100% Resorcinol
75% Resorcinol
50% Resorcinol
25% resorcinol
0% Resorcinol
0.0
0.1
0.2
0.3
0.4
0.5
0 50 100 150 200 250 300 350
Ta
n δ
Temperature (°C)
100% Resorcinol
75% Resorcinol
50% Resorcinol
25% Resorcinol
0% Resorcinol
3.15.b
3.15.a

107
Considering the modulus at room temperature, the values are approximately constant (~ 3- 4
GPa), and are just a weak function of the weight ratio of the two monomers. This behavior is not
surprising, since the modulus for glassy polymers just below the glass transition temperature is
known for being approximately constant over a wide range of polymers; having a value of
around 3 x 109 Pa. In this state, molecular motions are restricted to vibrations and short-range
rotational motion. Classic explanations for this phenomenon imply the use of Lennard-Jones
potentials, as well as carbon-carbon bonding force fields [39]. Regarding the glass transition, it is
evident that is a strong function of the proportion of the monomers, as well as their chemical
functionality, producing a difference of approx. 80°C when switching from two to three epoxy
groups (di and tri glycidylated monomers). The rapid, coordinated molecular motion in this
region is governed by principles of reputation and diffusion. This phenomenon can be suppressed
or retarded (in terms of energy), if the crosslinking density is increased. As seen in the Table, the
higher the crosslinking density, the more constrained the chain motion; and thus the stiffer and
more thermally resistant the molecular structure is, fact that is reflected by the values of the
rubbery modulus (or E` at Tg + 30°C). Clearly, increasing crosslinking density approx. 3 times
led to an increase of around 80°C in the Tg. As the temperature is raised past the rubbery plateau
region for linear amorphous polymers, the rubbery flow region is reached. It has to be
emphasized that this region does not occur for crosslinked polymers. This behavior was expected
for the 100 wt% diglycidylated ether of resorcinol, however it was not clearly observed in the E`
vs temperature plots. After raising the temperature past the 250°C, the modulus dropped and
remained unstable, probably due to thermos-oxidative decomposition of the polymers.
Differential scanning calorimetry studies revealed signs of thermal decomposition for all of the
materials when approaching 300°C.

108
4. Conclusions
This chapter focuses on the chemical modification of two different functional groups of biomass
derived materials; the carbon-carbon double bonds of triglycerides, and the hydroxyl groups of
phenolic compounds. These chemical modifications were addressed to introduce more reactive
functional groups able to polymerize and to produce materials with a better thermo-mechanical
performance. Three chemical modifications (epoxy, hydroxyl and acrylate) were preliminary
evaluated. The step-growth process employed to formulate the epoxy resin implies the use of a
commercial amine to crosslink the epoxy monomers. It was observed that the epoxidized oil,
when mixed in an epoxy/amine ratio of ~10/6, produced resins with acceptable thermo-
mechanical properties, showing a Tg of around 70°C and a modulus of around 1.4 GPa at room
conditions. Regarding acrylated oils polymerized through free radical mechanism, it can be
concluded that acrylated oils can be used to make rigid thermosetting polymers and composites.
These materials exhibited high crosslinking densities, resulting in higher Tg (approx. 60°C) and
improved storage modulus (~0.7 GPa) than their non-functionalized counterparts. These two
approaches produced materials with considerably better thermo-mechanical performance than
the thermosets produced in chapter II.
This chapter also studied the chain-growth copolymerization (which reduces the amount of
commercial agents in the formulation per weight basis) of epoxy monomers. This approach
allowed increasing the contribution of renewable materials into the formulations, by minimizing
the use of commercial crosslinking compounds. Likewise, efforts were focus on improving the
thermo-mechanical properties of the epoxy resins by introducing triglycidylated ether of α-
resorcylic acid, a low molecular weight comonomer able to increase the chain stiffness and thus,

109
enhancing the mechanical performance of the oil-based epoxy resins. Testing showed that this
aromatic monomer is able to produce resins with high Tg and modulus (~ 114°C and 3.8 GPa).
When combined with epoxidized oil, a decrease in the storage modulus when higher amounts of
epoxidized linseed oil were added to the mixtures was observed. Tg appeared to be not strongly
dependent of the amounts of epoxidized linseed oil. SEM images confirmed the glassy brittle
nature of the materials and revealed possible thermodynamic incompatibility of these two
compounds. When compared to samples of commercial origin, testing showed the mechanical
properties of the triglycidylated α-RA based resins are similar to those shown by EPON 828
epoxy resin. Studies about effect of the functionality on the final properties of the networks,
employing the diglycidylated ether of resorcinol and the triglycidylated ether of α-resorcylic acid
as models, revealed that the crosslinking density rises as a functionality of the monomers
increases, thus increasing the thermo-mechanical properties.

110
References
[1] G. Lligadas, J.C. Ronda, M. Galià, V. Cádiz, Renewable polymeric materials from
vegetable oils: A perspective, Mater. Today. 16 (2013) 337–343.
doi:10.1016/j.mattod.2013.08.016.
[2] J.M. Raquez, M. Deléglise, M.F. Lacrampe, P. Krawczak, Thermosetting (bio)materials
derived from renewable resources: A critical review, Prog. Polym. Sci. 35 (2010) 487–
509. doi:10.1016/j.progpolymsci.2010.01.001.
[3] U. Biermann, U. Bornscheuer, M.A.R. Meier, J.O. Metzger, H.J. Schaefer, Oils and fats as
renewable raw materials in chemistry, Angew. Chemie - Int. Ed. 50 (2011) 3854–3871.
doi:10.1002/anie.201002767.
[4] J.C. Ronda, G. Lligadas, M. Galias, V. Cadiz, Vegetable oils as platform chemicals for
polymer synthesis, Eur. J. Lipid Sci. Technol. 113 (2011) 46–58.
doi:10.1002/ejlt.201000103.
[5] L.G. Wade, Organic Chemistry, 2006.
[6] A. Corma Canos, S. Iborra, A. Velty, Chemical routes for the transformation of biomass
into chemicals, Chem. Rev. 107 (2007) 2411–2502. doi:10.1021/cr050989d.
[7] C. Aouf, E. Durand, J. Lecomte, M.-C. Figueroa-Espinoza, E. Dubreucq, H. Fulcrand, et
al., The use of lipases as biocatalysts for the epoxidation of fatty acids and phenolic
compounds, Green Chem. 16 (2014) 17406–1754. doi:10.1039/c3gc42143k.
[8] J. Zhu, K. Chandrashekhara, V. Flanigan, S. Kapila, Curing and Mechanical
Characterization of a Soy-Based Epoxy Resin System, J. Appl. Polym. Sci. 91 (2004)
3513–3518. doi:10.1002/app.13571.

111
[9] M.Y. Shah, S. Ahmad, Waterborne vegetable oil epoxy coatings: Preparation and
characterization, Prog. Org. Coatings. 75 (2012) 248–252.
doi:10.1016/j.porgcoat.2012.05.001.
[10] Z. Liu, S.Z. Erhan, D.E. Akin, F.E. Barton, “Green” composites from renewable
resources: Preparation of epoxidized soybean oil and flax fiber composites, J. Agric. Food
Chem. 54 (2006) 2134–2137. doi:10.1021/jf0526745.
[11] J.D. Espinosa-Perez, B. Nerenz, D. HAageson, Z. Chen, Comparison of curing agents for
epoxidized vegetable oils applied to composites, Polym. Compos. 16 (2011) 1806–1816.
doi:10.1002/pc.
[12] J.R. Kim, S. Sharma, The development and comparison of bio-thermoset plastics from
epoxidized plant oils, Ind. Crops Prod. 36 (2012) 485–499.
doi:10.1016/j.indcrop.2011.10.036.
[13] R. Wang, T.P. Schuman, Vegetable oil-derived epoxy monomers and polymer blends: A
comparative study with review, Express Polym. Lett. 7 (2012) 272–292.
doi:10.3144/expresspolymlett.2013.25.
[14] C. Aouf, H. Nouailhas, M. Fache, S. Caillol, B. Boutevin, H. Fulcrand, Multi-
functionalization of gallic acid. Synthesis of a novel bio-based epoxy resin, Eur. Polym. J.
49 (2013) 1185–1195. doi:http://dx.doi.org/10.1016/j.eurpolymj.2012.11.025.
[15] A. Gandini, Polymers from Renewable Resources: A Challenge for the Future
of\rMacromolecular Materials, Macromolecules. 41 (2008) 9491–9504.
doi:10.1021/ma801735u.

112
[16] F. Jaillet, E. Darroman, A. Ratsimihety, R. Auvergne, B. Boutevin, S. Caillol, New
biobased epoxy materials from cardanol, Eur. J. Lipid Sci. Technol. 116 (2014) 63–73.
doi:10.1002/ejlt.201300193.
[17] H. Noualhas, C. Aouf, C. Le Guerneve, S. Caillol, B. Boutevin, H. Fulcrand. Synthesis
and properties of biobased epoxy resins. Part 1. Glycidylation of flavonoids by
epichlorohydrin, J. Polym. Sci. Part a-Polymer Chem. 49 (2011) 2261–2270.
doi:10.1002/pola.
[18] S. Khadem, R.J. Marles, Monocyclic Phenolic Acids; Hydroxy- and Polyhydroxybenzoic
Acids: Occurrence and Recent Bioactivity Studies, Molecules. 15 (2010) 7985–8005.
doi:10.3390/molecules15117985.
[19] S. Wang, S. Zhang, T. Zhou, J. Zhan, Three new resorcylic acid derivatives from
Sporotrichum laxum, Bioorganic Med. Chem. Lett. 23 (2013) 5806–5809.
doi:10.1016/j.bmcl.2013.08.109.
[20] Epon® Resin 828, (n.d.). http://www.polysciences.com/default/catalog-
products/monomers-polymers/polymers/epoxy-resins/eponsupsup-resin-828/ (accessed
March 30, 2016).
[21] Z.S. Petrović, A. Zlatanić, C.C. Lava, S. Sinadinović-Fišer, Epoxidation of soybean oil in
toluene with peroxoacetic and peroxoformic acids - Kinetics and side reactions, Eur. J.
Lipid Sci. Technol. 104 (2002) 293–299. doi:10.1002/1438-
9312(200205)104:5<293::AID-EJLT293>3.0.CO;2-W.
[22] G. Lligadas, J.C. Ronda, M. Galià, V. Cádiz, Bionanocomposites from renewable
resources: Epoxidized linseed oil-polyhedral oligomeric silsesquioxanes hybrid materials,
Biomacromolecules. 7 (2006) 3521–3526. doi:10.1021/bm060703u.

113
[23] H. Dai, L. Yang, B. Lin, C. Wang, G. Shi, Synthesis and Characterization of the Different
Soy-Based Polyols by Ring Opening of Epoxidized Soybean Oil with Methanol, 1,2-
Ethanediol and 1,2-Propanediol, J. Am. Oil Chem. Soc. 86 (2009) 261–267.
doi:10.1007/s11746-008-1342-7.
[24] C. Aouf, S. Benyahya, A. Esnouf, S. Caillol, B. Boutevin, H. Fulcrand, Tara tannins as
phenolic precursors of thermosetting epoxy resins, Eur. Polym. J. 55 (2014) 186–198.
doi:10.1016/j.eurpolymj.2014.03.034.
[25] S. Benyahya, C. Aouf, S. Caillol, B. Boutevin, J.P. Pascault, H. Fulcrand, Functionalized
green tea tannins as phenolic prepolymers for bio-based epoxy resins, Ind. Crops Prod. 53
(2014) 296–307. doi:10.1016/j.indcrop.2013.12.045.
[26] I.E. Dell`Erba, J.J. Williams, Homopolymerization of epoxy monomers initiated by 4-
(dimethylamino)pyridine, Poly. Eng. Sci. 46 (2006) 351–359. doi:10.1002/pen.
[27] X. Kong, S.S. Narine, Physical Properties of Polyurethane Plastic Sheets Produced from
Polyols from Canola Oil Physical Properties of Polyurethane Plastic Sheets Produced
from Polyols from Canola Oil, (2007) 2203–2209. doi:10.1021/bm070016i.
[28] V. V. Goud, A. V. Patwardhan, S. Dinda, N.C. Pradhan, Epoxidation of karanja
(Pongamia glabra) oil catalysed by acidic ion exchange resin, Eur. J. Lipid Sci. Technol.
109 (2007) 575–584. doi:10.1002/ejlt.200600298.
[29] E. Milchert, A. Smagowicz, G. Lewandowski, Optimization of the epoxidation of
rapeseed oil with peracetic acid, Org. Process Res. Dev. 14 (2010) 1094–1101.
doi:10.1021/op900240p.
[30] D. Esterification, Standard Test Methods for Testing Polyurethane Raw Materials :
Determination of Hydroxyl Numbers of Polyols 1, (2010) 1–9. doi:10.1520/D4274-11.

114
[31] W.S. Precision, E.S. Waste, T. Edition, Standard Practice for Extraction of Solid Waste
Samples for Chemical Analysis, 93 (2008) 1–7. doi:10.1520/D5369-93R08E01.2.
[32] M. Fache, R. Auvergne, B. Boutevin, S. Caillol, New vanillin-derived diepoxy monomers
for the synthesis of biobased thermosets, Eur. Polym. J. 67 (2015) 527–538.
doi:10.1016/j.eurpolymj.2014.10.011.
[33] F. Hu, J.J. La Scala, J.M. Sadler, G.R. Palmese, Synthesis and characterization of
thermosetting furan-based epoxy systems, Macromolecules. 47 (2014) 3332–3342.
doi:10.1021/ma500687t.
[34] A. Gandini, M.N. Belgacem, Lignins as components of macromolecular materials, in
M.N. Belgacem, A. Gandini (EDs.), Monomers, Polymers and Composites, Elsevier, UK,
2008, pp. 243-272.
[35] A. Zlatanic, C. Lava, W. Zhang, Z.S. Petrović, Effect of Structure on Properties of Polyols
and Polyurethanes Based on Different Vegetable Oils, J. Polym. Sci. Part B Polym. Phys.
42 (2004) 809–819. doi:10.1002/polb.10737.
[36] R. Auvergne, S. Caillol, G. David, B. Boutevin, J.P. Pascault, Biobased thermosetting
epoxy: Present and future, Chem. Rev. 114 (2014) 1082–1115. doi:10.1021/cr3001274.
[37] G. Yang, S.-Y. Fu, J.-P. Yang, Preparation and mechanical properties of modified epoxy
resins with flexible diamines, Polymer (Guildf). 48 (2007) 302–310.
doi:10.1016/j.polymer.2006.11.031.
[38] H. Yahyaie, M. Ebrahimi, H.V. Tahami, E.R. Mafi, Toughening mechanisms of rubber
modified thin film epoxy resins, Prog. Org. Coatings. 76 (2013) 286–292.
doi:10.1016/j.porgcoat.2012.09.016.

115
[39] L.H. Sperling, Introduction to Physical Polymer Science, 2006.
doi:10.1021/ed078p1469.1.
[40] C. Aouf, E. Durand, J. Lecomte, M. Figueroa-Espinosa, E. dubreucq, H. Fulcrand, P.
Villeneuve. The use of lipases as biocatalysts for the epoxidation of fatty acids and
phenolic compounds. Green Chem. 16 (2014) 1740-1754.
[41] J.M. Prausnitz, R.N. Lichtenthaler, E.G. de Azevedo, Molecular Thermodynamics of
Fluid-Phase Equilibria, Pearson Education, 1998.
https://books.google.com/books?id=VSwc1XUmYpcC&pgis=1 (accessed June 1, 2016).
[42] Hansen solubility parameters, (n.d.). http://www.stenutz.eu/chem/solv24.php (accessed
June 1, 2016).
[43] F. Cataldo, O. Ursini, G. Angelini, Biodiesel as a Plasticizer of a SBR-Based Tire Tread
Formulation, ISRN Polym. Sci. 2013 (2013) 9.
[44] M. P. Stevens, Polymer Chemistry an Introduction. Oxford University Press, 1999.
[45] S. Sprenger, Fiber-reinforced composites based on epoxy resins modified with elastomers
and surface-modified silica nanoparticles, J. Mater. Sci. 49 (2014) 2391–2402.
doi:10.1007/s10853-013-7963-8.
[46] D.L. Safranski, K. Gall, Effect of chemical structure and crosslinking density on the
thermo-mechanical properties and toughness of (meth)acrylate shape memory polymer
networks, Polymer (Guildf). 49 (2008) 4446–4455. doi:10.1016/j.polymer.2008.07.060.
[47] J.-P. Pascault, H. Sautereau, J. Verdu, R.J.J. Williams, Thermosetting Polymers, CRC
Press, 2002. https://books.google.com/books?id=OrnJdwIYLA4C&pgis=1 (accessed
April 1, 2016).

116
CHAPTER IV
Synthesis and Characterization of Interpenetrating Polymer Networks (IPNs) Using
Biomass Derived Materials.
1. Introduction
Thermosetting polymers are generally stronger than thermoplastic materials due to the presence
of a three-dimensional network of bonds (or cross-linking). This structural characteristic makes
them suitable for high-temperature applications, showing a number of advantages over
thermoplastics, such as elevated heat distortion temperatures, good solvent resistance and high
modulus. These outstanding properties have resulted in their use in a wide range of commodity,
engineering and specialty applications [1].
Nowadays, there is an increasing growing interest to develop novel bio-based products. The
synthesis of bioplastics, which are intended to simulate the performance of existing counterparts
from non-renewable sources, also goes hand in hand with the improvement of the current
available materials; most of them traditionally produced from petroleum based monomers. To
expand the physical and chemical performance of polymeric materials, multicomponent
macromolecular systems have been proposed and extensively used for several decades [2].
Amongst several type of systems, interpenetrating polymer networks or IPNs are a particular
class of hybrid polymer consisting of two (or more) crosslinked polymers, in which the
macromolecular networks are held together by permanent entanglements with only few covalent

117
bonds between the polymers [3]. IPNs are an intimate combination of two or more polymers in
network form, where at least one of the polymers is polymerized and/or crosslinked in the
immediate presence of the other [4].
Ideally, IPNs interlace or interpenetrate on a molecular scale; however, the actual
interpenetration process may be limited by phase separation. When two polymers are mixed, the
most frequent result is a system that exhibit almost total phase separation. From a
thermodynamical point of view, this phenomenon arises as a result of the reduction on the
combinatorial entropy of mixing, compared to the enthalpic contribution of the mixing process,
of the two types of polymer chains [4]. However, if mixing is accomplished simultaneously with
crosslinking, phase separation may be kinetically controlled by permanent interlocking of
entangled chains [3]. Also, in recent years, with the aim of minimizing phase separations, there
has been an increasing interest in developing graft-IPNs, in which the two networks are
chemically cross-linked [5]. These multicomponent polymer systems seek to combine the best
properties of two or more different polymer networks in order to achieve a material with better
properties compared to their not-interpenetrated counterparts. It has been found that the network
interlock has measurable influences on the curing behaviors and on the mechanical properties of
the resulting materials [6].
Over the years, IPNs have been categorized into different ways, mainly according to their
synthesis method and also by their structure. A detailed description is given by Sperling [7],
however, the description provided in this document will principally cover the synthesis method,
which is the main focus of this research. According to the chemistry of preparation, IPNs can be

118
classified into: (i) Simultaneous IPN, when the precursors of both networks are mixed and the
two networks are synthesized at the same time by independent, non-interfering reactions. (ii)
Sequential IPNs, in which polymer network I is synthesized first and then monomer II, plus
crosslinking agents and catalyzers, are swollen into network I and polymerized in situ. If a
crosslinker is present, fully IPN results, while in the absence of a crosslinker, a network having
linear polymer embedded within the first network is formed (semi-IPN) [8].
In the last decades, few research groups have focused their work on the synthesis and
characterization of IPNs from natural products, beginning with the pioneering work of Sperling
and his coworkers, who developed IPNs from castor oil-based elastomers and styrene monomers
[9]. This fact stands up as a key finding, giving the opportunity to perform research on a field
lacking of recent developments. Also, other naturally functionalized triglyceride oils and their
use in producing semi-IPNs is reviewed by the same group [10]. After this work, other groups
have used castor oil as a source of polyurethanes to prepare oil-based IPNs [11,12]. Other oil-
based IPNs include the use of oligomerized soybean oil [13], tung oil [14], and canola-oil based
polyol with primary terminal functional groups (ref semi and full IPN). It is of worth to highlight
that most of these oil-based IPNs structures are not completely, but partially, derived from
biological raw materials and fossil fuels. IPNs between commercial resins, such as epoxy, and
elastomeric materials of natural origin are known in literature. Examples include acrylate and
methacrylate monomers derived from vernonia oil combined with bisphenol A-type resin [15],
flexible methacrylate network based on camelina oil and a rigid aliphatic glycidylether modified
bisphenol A based liquid epoxy resin [16].

119
On the other hand, lignin, which annual production on earth has been estimated to be in the range
of 5 – 36 x 108 tons, is undoubtedly the most promising natural source of aromatic monomers for
the chemical industry. For years, synthesis strategies addressed to modify lignins have struggle
with low reactivity associated to steric hindrance [17]. However, some novel technologies based
on biomass refinery appear as a good alternative to overcome this problem. Modern pyrolysis
techniques stand as a sustainable way for aromatics biosourcing, by providing low molecular
weight fragments of natural lignin, which can be used as platform chemical for organic synthesis.
A vast amount of lignin-based compounds, including phenol and more complex guaiacol and
syringol derivatives, has been identified in pyrolysis oils [18–20]. Tannins have also been
explored as a source of aromatic macromonomers to produce thermosetting resins [21]. To the
best knowledge of the authors of this project, interpenetrated polymer systems employing
monomers, or their precursors, that can be potentially obtain from biorefinery have not been
reported in literature.
In this Chapter, the formulation of interpenetrating polymer networks, comprising epoxy and
acrylate phases, is extensively studied; mainly focusing on the thermo-mechanical properties of
the resulting materials (Figure 4.1). Two types of compounds were tested as the epoxy phase of
the system, epoxidized linseed oil (containing fatty acid chains bearing epoxy moieties) and
glycidyl ether of α-resorcylic acid (or epoxidized α-resorcylic acid), crosslinked with
commercial multifunctional amines. The acrylate phase was synthetized using acrylated soybean
oil, slightly diluted with methyl methacrylate. As seen in Chapter III, the copolymerization of
epoxidized vegetable oil with epoxidized α-resorcylic acid vastly improved the thermo-
mechanical properties of the materials. However, phase separation occurred for mixtures

120
containing epoxidized triglycerides in weight contents higher than 30 wt%. The idea of
developing an interpenetrated system containing an epoxy phase derived from α-resorcylic acid,
and an acrylate phase derived from vegetable oil arises as an approach to not only improve the
properties by including an aromatic monomer in the system, but also to minimize the phase
separation phenomena by kinetically controlling this thermodynamical process by permanent
interlocking of entangled chains, avoiding phase separation.
Figure 4.1. Generic scheme of the IPNs containing epoxy and acrylate networks.
The strategy to prepare the IPNs can be divided into two sections. The first one comprises the
chemical modification of the raw materials, as well as a quantitative analysis of the targeted
functional groups. In a second step, the modified monomers are polymerized and the resulting
materials characterized, mainly focusing on their thermo-mechanical behavior. α-resorcylic acid
(a phenolic acid with two hydroxyl and one carboxylic acid functional groups) was modified
CH2O
CH3
CH3
O
O
O
O
O
R
O
O
R
CH3
OH
O O
CH2
O
O
O
O
O
R O
R
CH3
O O O
O O
O O
O
O
O
Epoxy Phase
Acrylate Phase
Acrylated Soybean Oil Methyl
Methacrylate
Epoxidized Linseed Oil Epoxidized α-Resorcylic Acid
+
OR
IPN
* Crosslinked with commercial amine curing agents
* Polymerized with AIBN

121
with reactive epoxy moieties, and used as low molecular weight comonomer to enhance the
density of aromatic structures in the crosslinked network. From the literature review, it was
found that most of the current IPN are not fully, but partially, prepared from biological raw
materials and fossil fuels. The novelty of the present study is not only derived from the selection
of the macromolecular components, which try to provide a response for the need of IPNs
prepared from formulations including a higher contribution of biological materials; but also from
the final properties that can be generated when associating the phases in the IPN structure.
2. Experimental Section
2.1 Materials
Linseed oil (LO) was purchased from VWR International (USA) and used with no further
purification. Glacial acetic acid (99.5 %), hydrogen peroxide (30 %), sodium carbonate, toluene,
petroleum ether, ethyl acetate, iodine monochloride, Wijs’ solution, 33 % HBr in glacial acetic
acid, methyl methacrylate, 2,2′-Azobis(2-methylpropionitrile) (AIBN), and Amberlite ion-
exchange resin were all AR grade and also acquired from VWR International (USA). α-
Resorcylic acid (α-RA, ≥ 98%), epichlorohydrin (99.0 %), sodium hydroxide (≥ 98%), and
dichloromethane were purchased from VWR (US). Benzyltriethylammonium chloride (≥ 98%),
used as a phase transfer agent, was also acquired from VWR (US).
Acrylated soybean oil (Figure 2) was purchased from Sigma Aldrich (USA). In Figure 4.2, R
represents the fatty acid chain modified with the acrylate group. The supplier states that the
product has a molecular weight of approximately 1800-7200 g/mol. The average number of
acrylate groups per triglyceride has not been determined by the supplier or Sigma. However, it

122
O
O
O
O
R
O
O
R
CH3
OH
O O
CH2
has been established that it contains in average 4.5 double bonds, that can be first converted into
epoxy groups, and then into acrylate moieties [22].
Figure 4.2. Chemical structure of the acrylated soybean oil.
2.2 Synthesis procedures
2.2.1 Synthesis of epoxidized triglycerides
Refer to Chapter III for the synthesis of epoxidized linseed oil.
2.2.2 General procedure for the glycidylation of α-resorcylic acid.
Refer to Chapter III for the glycidylation of α-resorcylic acid.
2.2.3 Synthesis of IPNs
As previously detailed, IPNs can be prepared according to two different basic synthesis methods,
known as Simultaneous IPNs (SIN) and as Sequential IPNs. During the simultaneous approach,
the monomers or polymer constituents, plus crosslink agents and catalyzers are mixed together.
After mixing, the two polymerization reactions are carried out simultaneously as indicated by its
name. In terms of the chemical mechanism they follow to polymerize, there is no interference
whatsoever between reactions. In the case of sequential method, polymer network I is

123
synthesized first and then monomer II plus crosslinking agents and catalyzers are swollen into
the network I and polymerize in situ. For this work, the simultaneous method was chosen to
prepare the IPNs. Two types of IPNs were synthesized and studied, both of them composed of
two different phases, the epoxy phase and the acrylate phase. The epoxy phase is polymerized
through a step-reaction polymerization, by curing the epoxy monomers with a commercial
amine. The acrylate phase, on the other hand, is polymerized through a chain reaction that
requires an initiator to generate reacting species and then propagate the reaction. The types of
IPNs are detailed as follows.
System A: Epoxidized linseed oil/acrylated soybean oil IPNs
The first type of IPN consisted of combining triglyceride-derived materials, the epoxidized
linseed oil (polymerized through step-growing mechanism) and the acrylated soybean oil
(polymerized through chain-growth mechanism). First, the epoxy phase was prepared by mixing
epoxidized linseed oil with the commercial hardener SC-15 part B (Applied Poleramic Inc.),
composed of cycloaliphatic amine and polyoxyl-alkylamine, in a 10/6 weight ratio. Even though
technical information for this agent is very limited, previous experiments performed by this
research group revealed that the best thermo-mechanical properties were obtained when using
SC-15 part B as a hardener to cure the epoxidized linseed oil (refer to Chapter III).
The components of the acrylated phase, acrylated soybean oil, methyl methacrylate (added to
reduce the high viscosity of the acrylate oil in a 20 wt% of the total weight of acrylated oil) and
2,2-azobis(2-methylpropionitrile) or AIBN as initiator (1 wt% of the total weight of the acrylate
phase) were mixed separately and then added to the epoxy phase precursor solution in different

124
weight percentages. After mixing these solutions, all samples were placed in glass moulds and
then cured in the oven following the next heating sequence: 50°C for 1 hr., 60°C for 1 hr., 75°C
for 2 hours, 120°C for 12°C hours, and finally 180°C for 1 hour.
System B: Epoxidized α-resorcylic acid/acrylated oil IPNs
The second type of IPN consisted of combining a phenolic-based epoxy resin with the
triglyceride-based acrylate resin. The epoxy phase was prepared by mixing the triglycidylated
derivative of the α-resorcylic acid (TRA) or epoxidized α-resorcylic acid with the commercial
hardener Jeffamine T-403, a well-known trifunctional amine, in a 10/7 weight ratio. The
acrylated phase is exactly as the one described for above for the type A IPN. After mixing these
solutions, all samples were also placed in glass moulds and then cured in the oven following the
next heating sequence: 50°C for 1 hr., 60°C for 1 hr., 75°C for 2 hours, 120°C for 12°C hours,
and finally 160°C for 1 hour.
2.3. Characterization of Materials and Networks
2.3.1 Fourier Transform Infrared Spectroscopy (FTIR)
With the aim of studying the chemical functionalization of both, the linseed oil and the α-
resorcylic, FTIR was performed. Infrared spectra of the starting materials, as well as the
epoxidized products, were measured by attenuated total reflection (ATR) method using a
Thermo Nicolet 6700 Fourier transform infrared spectrometer connected to a PC with OMNIC
software analysis. All spectra were recorded between 400 and 4000 cm-1 over 64 scans with a
resolutions of 16 cm-1.

125
2.3.2 Kinetics of Gelation
The characteristic peaks in the infrared region for the acrylic unsaturation (1638 cm-1) and epoxy
ring (915 cm-1) respectively were used to calculate the conversion of the acrylic and epoxy
groups. The band at approx. 2960 cm-1, assigned to the C-H stretch absorption, was used as a
reference because it does not suffer any chemical modification during the process. The progress
of curing reaction was evaluated using Equation (4.1):
]��^V�_��� (%) =abbbc1 −
(G���G� e)!f
(G���G� e)1f
ghhhi
∗ 100 (4.1)
Where, Apol = 915 cm-1 or 1638 cm-1 peak areas, Aref = 2960 cm-1 peak area, t = reaction time, and
0 = initial moment of reaction (uncured mixture) [23].
2.3.3 Determination and Measurement of the Extent of Reaction at the Gel Time.
Gel time was determined following the method described by the ASTM D2471-99 [24]. This test
method covers the determination of the time from the initial mixing of the reactants of a
thermosetting plastic composition to the time when solidification commences. For each system,
at least three sets of experiments were performed. The extent of the reaction at the gel point can
be calculated by means of two theories, and is of especial interest. Two theories were compared.
According to Carothers [25], the critical extent of the reaction, Pc, at the gel point is given by
Pc = 2 / favg (4.2)
where, favg represents the average functionality of the system, and is given by
favg = ∑Nifi / ∑Ni (4.3)
and where, Ni is the number of molecules of monomer I with functionality fi.

126
Flory and Stockmayer [26], developed another relationship, correlating the extent of conversion
at the gel point for stoichiometric mixtures,
Pc = 1 / [(fw,epoxy -1 ) (fw,amine – 1) (4.4)
Where, fw,epoxy represents the weight average functionality of the epoxy monomers, and fw,amine
represents the weight average functionality of the amine monomers.
2.3.4 Soxhlet extraction.
Soxhlet extraction was employed to characterize the structures of the bulk copolymers following
the ASTM D5369-93 [27]. An appropriate amount of the bulk polymer was extracted for 24 h
with 200 mL of refluxing dichloromethane with a Soxhlet extractor. Upon extraction, the
resulting solution was concentrated by rotary evaporation and subsequent vacuum drying. The
soluble substances were isolated for further characterization. The insoluble solid was dried in a
vacuum oven for several hours before it was weighed.
2.3.5 Scanning Electron Microscopy (SEM)
SEM study was performed with the purpose of obtaining a topological characterization of the
materials. The fractured surfaces of pre-chilled samples in liquid nitrogen were studied by
scanning electron microscope (Zeiss EVO 50 variable pressure scanning electron microscope
with digital imaging and EDS). Samples were sputtered with gold prior to SEM observations
with an EMS 550X auto sputter coating device with carbon coating attachment.

127
2.3.6 Thermal Properties
The thermal properties of the IPNs, as well as the parent resins, were investigated by Differential
Scanning Calorimetry (DSC) on a TA DSC Q2000. Nitrogen was used as the purge gas and the
samples were placed in aluminum pans. For each material sample, the thermal properties were
recorded at 5°C/min. in a heat/cool/heat cycle from 30°C to 300°C.
2.3.7 Thermo-mechanical properties of the networks.
Dynamic mechanical analysis (DMA) on a TA Instruments RSA III was carried out to assess the
thermo-mechanical properties by three-point bending. The tests were performed at temperatures
ranging from 25 to 250°C with a heating rate of 10°C/min. The frequency was fixed a 1Hz and a
sinusoidal strain-amplitude of 0.1 % was used for the analysis. The dynamic storage modulus
(E′) and tan δ curves were plotted as a function of temperature. To be able to explain the effects
of the co-monomers on the mechanical properties, it is useful to calculate the crosslinking
density (n, mol/m³). The crosslinking density can be estimated from the experimental data using
the rubber elasticity theory. Thermosets behave as rubbers above Tg. At small deformations,
rubber elasticity predicts that the modulus storage (E’), of an ideal elastomer with a network
structure is proportional to the crosslinking density according to Equation (4.5) [28]:
�� = 3��� = 3��/�� Equation (4.5)
where E` is the rubbery storage modulus, R is the gas constant (8.314 J/mol K), T is the absolute
temperature (K), ρ is the density of the sample (g/m³), and Mc is the molecular weight between
crosslinks (g/mol). The temperature and rubbery modulus were determined for the calculation of
the equation at Tg + 30°C. The temperature at which the peak of the tan delta presents a
maximum was considered as the glass transition temperature of the material.

128
2.3.8 Linear Elastic Fracture Mechanics
In order to characterize the fracture toughness of the graft-IPNs synthesized, in terms of the
critical stress intensity factor, KIC, quasi-static fracture tests were performed following the
method described by the ASTM D5045-14 [29]. The cured IPN sheets were machined into
rectangular coupons of dimensions 70 mm x 20 mm and 2.8 mm thickness. An edge notch of 3
mm in length was cut into the samples, and the notch tip was sharpened using a razor blade. An
Instron 4465 universal testing machine was used for loading the specimen in tension and in
displacement control mode (crosshead speed = 1mm/min). The load-deflection data was recorded
up to crack initiation and during stable crack growth, if any. The crack initiation toughness or
critical stress intensity factor, KIC, was calculated using the load (F) recorded at crack initiation.
For each system, at least three sets of experiments were performed at laboratory conditions. The
mode-I stress intensity factor for a single edge notched (SEN) tensile strip using linear elastic
fracture mechanics is given by Equation (4.6) [5],
Equation (4.6)
where,
Equation (4.7)
and a is the edge crack length, w is the specimen width, B is the specimen thickness and F is the
peak load. All data are expressed as means ± standard deviations. Statistical analysis was
performed by one-way analysis of variance (ANOVA) in conjunction with Tukey`s post hoc test
for multiple comparisons using Minitab 17 software.
Ic
F a aK f
Bw w
π =
2 3 4
1.12 0.23 10.6 21.7 30.4a a a a a
fw w w w w
= − + − +

129
The critical energy release rate, GIc, can be related to the stress-intensity factor, KIc, in plane
strain conditions by Equation (4.8) [30]:
kᵢ� = (mᵢ))�
F (1 − ^F ) Equation (4.8)
where, v is the Poisson`s coefficient and E the Young`s modulus of the material. For all
formulation, v, was taken equal to 0.33 [31].
3. Results and Discussion
3.1 Chemical Functionalization of Raw Materials
3.1.1 Synthesis of Epoxidized Triglycerides
Refer to Chapter III for details about the synthesis of epoxidized linseed oil.
3.1.2 Functionalization of phenolic compounds with epoxy groups. Synthesis of
triglycidylated ether of α-resorcylic acid
Refer to Chapter III for details about the glycidylation of α-resorcylic acid.
3.2. Characterization of the IPNs.
3.2.1 System A: Epoxidized linseed oil/acrylated soybean oil IPNs
3.2.1.1 Scanning Electron Microscopy
Scanning electron microscopy was used to investigate the morphology of the systems. Figure 4.3
provides the obtained images of the fracture surfaces of the IPNs as well as the parent resins, pre-
chilled in liquid nitrogen.

130
4.3.a
4.3.b
4.3.c

131
Figure 4.3. SEM micrographs of the fracture surface of 100 % epoxidized linseed oil (4.3.a), IPN
80% epoxy - 20% acrylate (4.3.b), IPN 60% epoxy - 40% acrylate (4.3.c), IPN 40% epoxy - 60%
acrylate (4.3.d), IPN 20% epoxy-80% acrylate (4.3.e), 100% acrylated soybean oil (4.3.f).
4.3.d
4. 3.e
4.3.f
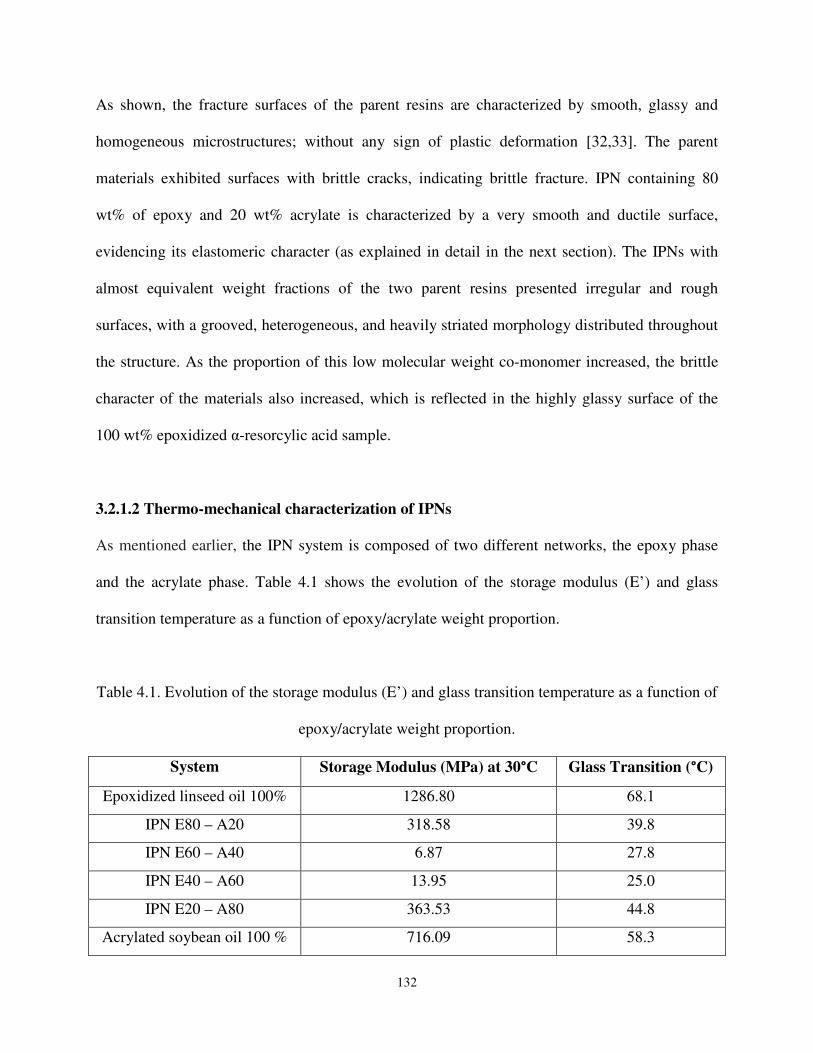
132
As shown, the fracture surfaces of the parent resins are characterized by smooth, glassy and
homogeneous microstructures; without any sign of plastic deformation [32,33]. The parent
materials exhibited surfaces with brittle cracks, indicating brittle fracture. IPN containing 80
wt% of epoxy and 20 wt% acrylate is characterized by a very smooth and ductile surface,
evidencing its elastomeric character (as explained in detail in the next section). The IPNs with
almost equivalent weight fractions of the two parent resins presented irregular and rough
surfaces, with a grooved, heterogeneous, and heavily striated morphology distributed throughout
the structure. As the proportion of this low molecular weight co-monomer increased, the brittle
character of the materials also increased, which is reflected in the highly glassy surface of the
100 wt% epoxidized α-resorcylic acid sample.
3.2.1.2 Thermo-mechanical characterization of IPNs
As mentioned earlier, the IPN system is composed of two different networks, the epoxy phase
and the acrylate phase. Table 4.1 shows the evolution of the storage modulus (E’) and glass
transition temperature as a function of epoxy/acrylate weight proportion.
Table 4.1. Evolution of the storage modulus (E’) and glass transition temperature as a function of
epoxy/acrylate weight proportion.
System Storage Modulus (MPa) at 30°C Glass Transition (°C)
Epoxidized linseed oil 100% 1286.80 68.1
IPN E80 – A20 318.58 39.8
IPN E60 – A40 6.87 27.8
IPN E40 – A60 13.95 25.0
IPN E20 – A80 363.53 44.8
Acrylated soybean oil 100 % 716.09 58.3

133
As seen in Table 4.1, compositional variations of epoxy/acrylate ratio deeply affected the
thermo-mechanical properties of the IPNs; and it is evident that the performance of the parent
systems (epoxy and acrylate networks) is considerably diminished when they are combined
forming IPNs. The storage modulus (E’) exhibited values of about 1.3 GPa and 0.7 GPa, and the
Tg was measured to be approx. 68°C and 58°C for the epoxy and acrylate systems, respectively,
indicating that materials behave as glassy polymers at ambient conditions. Since the parent resins
showed the highest thermo-mechanical performance, they can be easily visualized as the two
maximum reference points to correlated the thermo-mechanical properties of the IPNs. The
modulus of the IPNs varied as a function of their distance (in terms of composition) from the two
parent resins (the reference points) with values abruptly falling in between, reaching a minimum
for the IPN E60 – A40 and IPN E40 – A60. In the same manner, the Tg also considerably
decreased, reaching an average value of about 26°C for the IPNs with almost equivalent weight
fractions of the two parent resins. From the tan δ vs temperature plots, it was observed that there
was no phase separation, fact that was expected keeping in mind that the parent resins are both
triglyceride-derived materials.
The decline in the modulus values is directly correlated to the decrease in the density of active
chains or n; parameter that is often associated to the crosslinking density of the material (Figure
4.4). Conceptually, the higher the crosslinking density, the more constrained the chain motion;
and thus the stiffer and more thermally resistant the molecular structure is. Therefore, as the
crosslinking density increases, the hardness, modulus, mechanical strength, and chemical
resistance also increase [34].
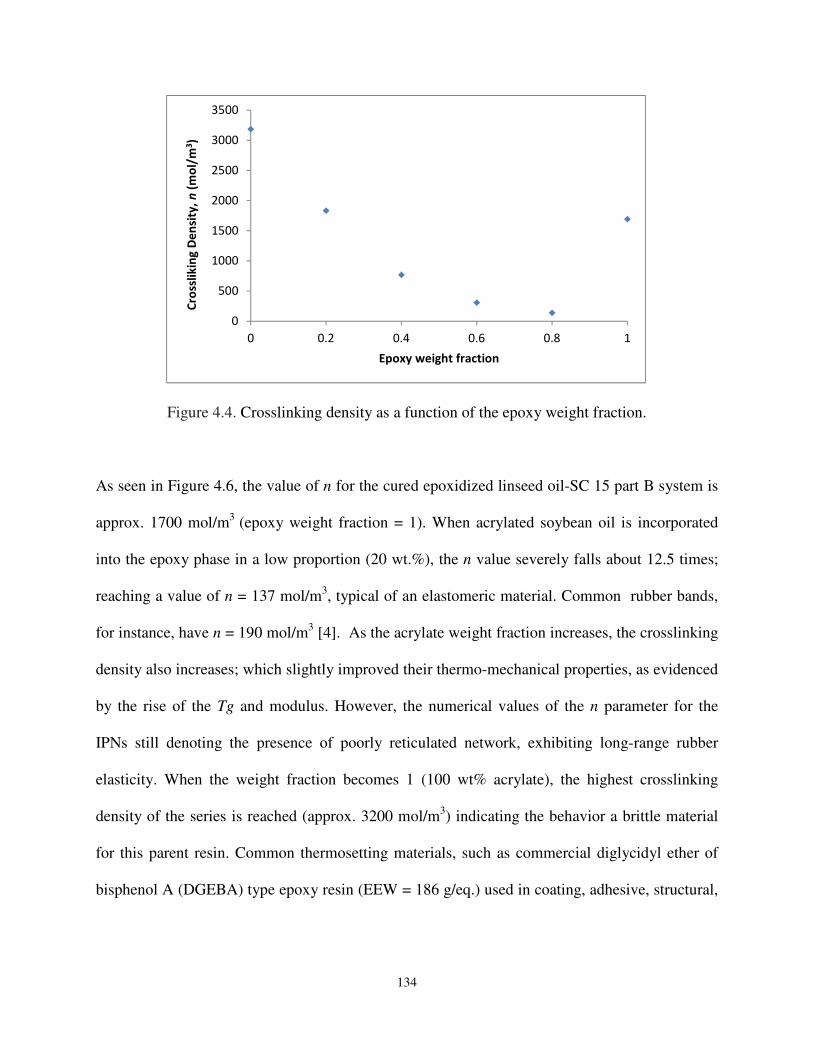
134
Figure 4.4. Crosslinking density as a function of the epoxy weight fraction.
As seen in Figure 4.6, the value of n for the cured epoxidized linseed oil-SC 15 part B system is
approx. 1700 mol/m3 (epoxy weight fraction = 1). When acrylated soybean oil is incorporated
into the epoxy phase in a low proportion (20 wt.%), the n value severely falls about 12.5 times;
reaching a value of n = 137 mol/m3, typical of an elastomeric material. Common rubber bands,
for instance, have n = 190 mol/m3 [4]. As the acrylate weight fraction increases, the crosslinking
density also increases; which slightly improved their thermo-mechanical properties, as evidenced
by the rise of the Tg and modulus. However, the numerical values of the n parameter for the
IPNs still denoting the presence of poorly reticulated network, exhibiting long-range rubber
elasticity. When the weight fraction becomes 1 (100 wt% acrylate), the highest crosslinking
density of the series is reached (approx. 3200 mol/m3) indicating the behavior a brittle material
for this parent resin. Common thermosetting materials, such as commercial diglycidyl ether of
bisphenol A (DGEBA) type epoxy resin (EEW = 186 g/eq.) used in coating, adhesive, structural,
0
500
1000
1500
2000
2500
3000
3500
0 0.2 0.4 0.6 0.8 1
Cro
ssli
kin
g D
en
sity
, n
(m
ol/
m3)
Epoxy weight fraction

135
and composite applications shows an approximate value of n = 5000 mol/m3 when crosslinked
with a tetrafunctional amine [35].
Based on the thermo-mechanical characterization, it is evident that there is a plasticizing effect
of one system into the other as soon as they are combined to form an interpenetrated network;
the chains are being separated from each other and hence making crosslinking harder. Therefore,
with the aim of studying how the presence of one system impacts the polymerization of the
other, differential scanning calorimetry (DSC) was employed to evaluate the curing behavior of
the resins. When analyzing the behavior of the parent resins (epoxy and acrylate) it can be
clearly seen that there is a big gap in temperature, and therefore energy, between the onsets of
the polymerizations. The calorimetry indicated that the acrylated soybean oil-methyl
methacrylate system, containing highly reactive acrylate functionalities that chemically behave
as vinyl groups, begins to react at around 70°C.
This value coincides with the expected initial decomposition temperature of the AIBN, which at
this stage, is able to generate a small number of radicals that subsequently initiate the
polymerization by reacting with the acrylate functional groups of the triglyceride monomer. The
reaction proceeds up to a temperature of about 120°C, point at which the reaction seems to stop
due to the complete consumption of the monomers. On the other hand, the ring opening reaction
between the epoxy groups of linseed oil and the commercial amine SC-15 part B begins at
around 150°C, almost 90°C higher than its acrylate counterpart. It was also observed that the
offset of the curing peak, and therefore the complexion of the reaction, was at a temperature of
approximately 275°C.

136
It is well known that the radical chain polymerization consists of a sequence of three steps,
commonly referred to as initiation, propagation, and termination [25]. Once the acrylated
soybean oil-methyl methacrylate system reaches a temperature of about 70°C, the homolytic
dissociation of the AIBN (an azo compound) takes place; generating resonance-stabilized
cyanopropyl radicals that initiate the polymerization. Subsequent additions of a large number of
monomers fuel the propagation reaction, and finally at around 120°C; the propagating polymer
network stops growing and terminates. The situation seems to be quite different for the epoxy
system. During epoxy-amine curing process, the general reaction occurs via a nucleophilic attack
of the amine nitrogen on the carbon of the epoxy function. The mechanism proceeds through a
SN2 type reaction, and thus the reaction rate obeys second-order kinetics [36].
The direction in which an unsymmetrical epoxide ring is opened (or which epoxide carbon is
attacked by the nucleophile) depends on its structure and on the reaction conditions. If a basic
nucleophile is used in a typical SN2 reaction, such as an amine, the attack takes place at the less
hindered epoxide carbon. Although an ether oxygen is normally a poor leaving group in a SN2
reaction, the reactivity of the three-membered ring is sufficient to allow epoxides to react with
nucleophiles at elevated temperatures (approx. 100°C) [37]. However, the structural
characteristics of the monomers highly influence the reaction, and more aggressive conditions
might be needed to start the polymerization depending on the chemical environment of the
oxirane ring. Epoxidized oils (such as linseed oil) contain oxirane functionalities that are located
near the middle of the 18-carbon length fatty acid chains. As a consequence of this feature, these
groups are sterically hindered at both carbons, and also experience the effect of two electron-
injecting alkyl groups; decreasing their affinity for nucleophiles. Due to these two different

137
chemical effects, these epoxides react sluggishly with nucleophilic curing agents, such as amines
[38]; often requiring higher temperatures to overcome the energy barrier imposed by structural
characteristics in order to obtain the desired product. It is important to highlight that the
temperature require to achieve the complexion of the reaction needs to be counterbalanced with
the degradation temperature of the reactants and products.
Kowalski [39] studied the thermal-oxidative decomposition of oil and fats by performing DSC
scans of edible oils by heating the samples up to 360°C in an atmosphere of oxygen. Results
revealed that at around 160°C, the onset of the oxidative decomposition was observed. The final
stage of curing for the epoxidized linseed oil-acrylated soybean oil consisted of heating the resins
up to 180°C. In spite that the results of the DSC study in inert atmosphere suggested that
temperatures up to 275°C were needed to fully cure the resins, higher temperatures were not
tested, because after 1 hour at 180°C; signs of thermo-oxidative degradation were already evident
in the materials.
Based on these findings, it is suggested that one reaction begins first than the other, and that
there is a big gap between the onsets of the reactions. Initially, for rich acrylate soybean oil
systems, the acrylate monomers begun to react at around 70°C; leading to the formation of a
crosslinked network; whilst the epoxy monomers remained swollen in it. As the temperature
increased, it was expected that the formation of covalent bonds between the epoxy and amine
hardener took place. However, it seems to be that, due to limited molecular diffusion required for
polymerization (which fuels condensation reactions) [25], as well as to the very high
temperatures required to polymerize the epoxy system; the epoxy monomers remain unreacted or

138
partially reacted in the acrylic network, plasticizing it and therefore; leading to a decrease in the
thermo-mechanical properties of the IPNs. On the other hand, for rich epoxy systems, it is
evident that even in small proportions; the acrylated system severely reduces the ability of the
epoxy system to react, and hence making the crosslinking process very hard to accomplish.
From the obtained results, the acrylate soybean oil system seems to offer a good candidate to
formulate the interpenetrated networks, due to its ability to react at rather low temperatures and
the relatively good mechanical properties of the final product. However, the epoxidized linseed
oil system, as a result of its low reactivity caused by structural factors, does not stand as a
feasible option to develop IPNs systems. Since the goal of this project is to produce polymeric
materials of high thermo-mechanical performance, it was also decided to explore using highly
reactive phenolic-based epoxy resins to formulate the IPNs, keeping also in mind that the
thermal and mechanical performance properties of organic polymers has been generally linked to
the presence of rigid aromatic structures.
3.2.2 System B: epoxidized α-resorcylic acid/acrylated oil IPNs
3.2.2.1 Scanning Electron Microscopy
The fracture surfaces of the thermosets were studied by scanning electron microscopy (SEM).
The results of the morphological study are shown in Figure 4.5.

139
4.5.a
4.5.b
4.5.c
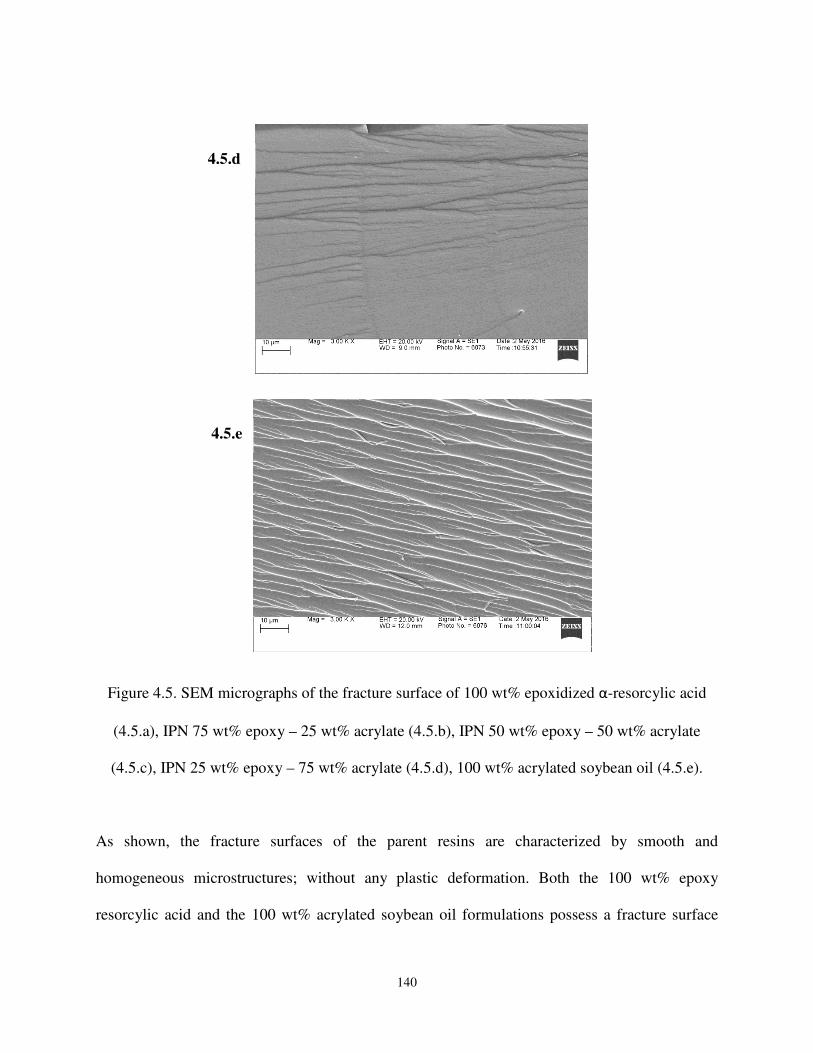
140
Figure 4.5. SEM micrographs of the fracture surface of 100 wt% epoxidized α-resorcylic acid
(4.5.a), IPN 75 wt% epoxy – 25 wt% acrylate (4.5.b), IPN 50 wt% epoxy – 50 wt% acrylate
(4.5.c), IPN 25 wt% epoxy – 75 wt% acrylate (4.5.d), 100 wt% acrylated soybean oil (4.5.e).
As shown, the fracture surfaces of the parent resins are characterized by smooth and
homogeneous microstructures; without any plastic deformation. Both the 100 wt% epoxy
resorcylic acid and the 100 wt% acrylated soybean oil formulations possess a fracture surface
4.5.d
4.5.e

141
and an internal structure of a brittle material, revealing a highly glassy surface; where crack
marks, ridges, and planes are dispersed all over the surface. The IPNs 75-25 wt% and 50-50 wt%
epoxy-acrylate are also characterized by a highly glassy surface. In the 25-75 wt%
epoxy/acrylate, the brittle character of the material seemed to decrease, and the surface exhibits
some degree of ductility and plastic deformation. As it will be discussed in the mechanical and
fracture studies, this sample showed the highest value for the critical energy release rate,
denoting its ability to dissipate energy due to its more ductile character. It was also evident from
the thermo-mechanical evaluation that this IPN presented the least brittle character of the series,
which was evident by its relatively high molecular weight between crosslinks. It is important to
highlight the morphology of the samples is consistent with the thermo-mechanical
characterization, which revealed a glass behavior at room conditions. Even though the starting
materials seemed to be heterogeneous in their chemical nature, there is no indication of phase
separation and no clear boundary between the phases. This indicated that the interpenetration
occurred at micro- or nano- size level [40], denoting that phase separation was kinetically
controlled by permanent interlocking of entangled chains.
3.2.2.2 Kinetics of gelation
The reaction between epoxy and amine groups is a step-growth reaction. The presence of three or
more epoxy groups leads to the formation of a tridimensional network when reacted with a
multifunctional amine. As a first approach to study the reactions, the gel times at 65°C were
measured (all measurement were performed three times). In order to study how the presence of
one system influences the crosslinking behavior of the other, a hypothetical epoxy system (in
which the acrylate phase has no initiator), and a hypothetical acrylate system (in which the epoxy
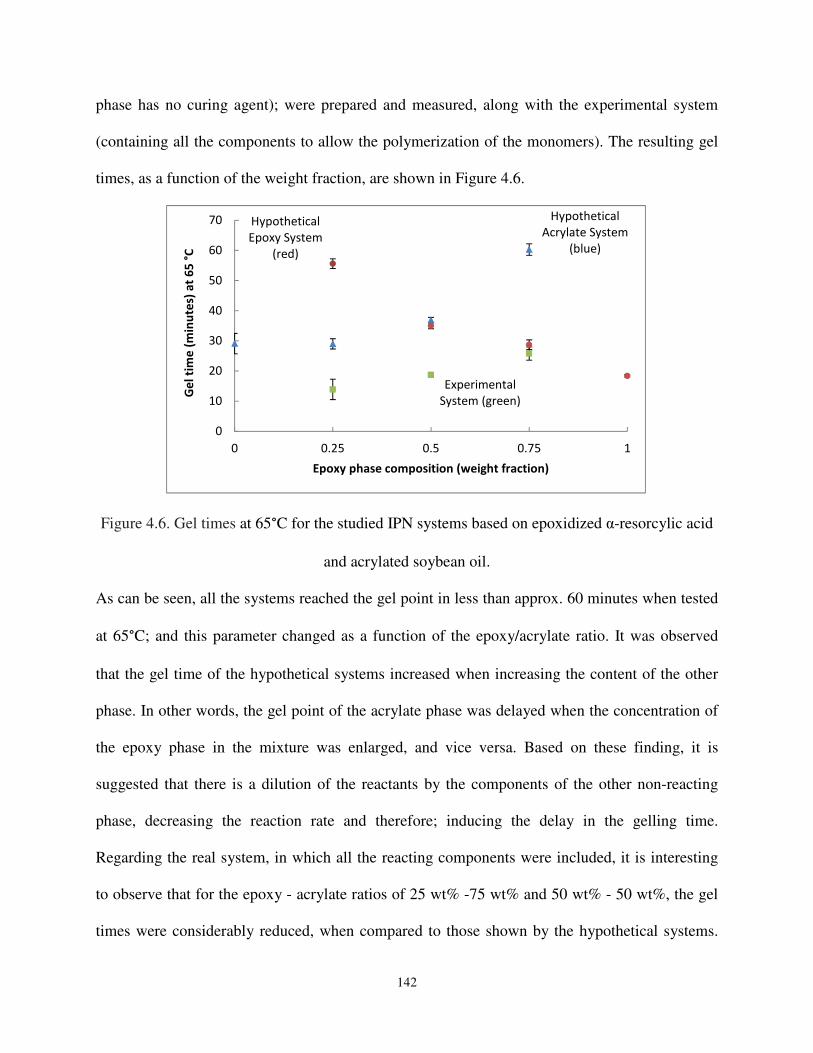
142
phase has no curing agent); were prepared and measured, along with the experimental system
(containing all the components to allow the polymerization of the monomers). The resulting gel
times, as a function of the weight fraction, are shown in Figure 4.6.
Figure 4.6. Gel times at 65°C for the studied IPN systems based on epoxidized α-resorcylic acid
and acrylated soybean oil.
As can be seen, all the systems reached the gel point in less than approx. 60 minutes when tested
at 65°C; and this parameter changed as a function of the epoxy/acrylate ratio. It was observed
that the gel time of the hypothetical systems increased when increasing the content of the other
phase. In other words, the gel point of the acrylate phase was delayed when the concentration of
the epoxy phase in the mixture was enlarged, and vice versa. Based on these finding, it is
suggested that there is a dilution of the reactants by the components of the other non-reacting
phase, decreasing the reaction rate and therefore; inducing the delay in the gelling time.
Regarding the real system, in which all the reacting components were included, it is interesting
to observe that for the epoxy - acrylate ratios of 25 wt% -75 wt% and 50 wt% - 50 wt%, the gel
times were considerably reduced, when compared to those shown by the hypothetical systems.
Hypothetical
Epoxy System
(red)
Hypothetical
Acrylate System
(blue)
Experimental
System (green)
0
10
20
30
40
50
60
70
0 0.25 0.5 0.75 1
Ge
l ti
me
(m
inu
tes)
at
65
°C
Epoxy phase composition (weight fraction)

143
The other ratios showed gelling times similar to the hypothetical counterparts. The system Epoxy
75 wt% - Acrylate 25 wt% reached the gel point at a similar time (~ 27.5 minutes), to that shown
by the hypothetical epoxy system (no initiator in the acrylate phase); denoting that the gelling of
this system is dominated by the epoxy phase.
As demonstrated by the DSC studies (detailed in the following section), the condensation
reaction of the epoxy monomers with the crosslinking agent (amine in this case) is highly
exothermic. It is important to mention that this is an expected behavior, because the ring opening
reaction of the highly strained three-member ring occurs easily, and is thermodynamically
favorable. On the other hand, it has been clearly stated by theory that the radical chain
polymerization rate is dependent on the initiator concentration [25]. Usually, thermal homolytic
dissociation of initiators is the most widely used method of generating radicals to start the
polymerization. Taking these facts into account, it is likely that the decrease in the gelling time
for the epoxy - acrylate ratios of 25 wt% - 75 wt% and 50 wt% - 50 wt% is due to a phenomenon
of auto-catalysis. The ring opening reaction of the highly strained three-member epoxy ring
releases extra energy in the system, favoring the dissociation of the initiator and therefore,
increasing the polymerization rate that leads to gelation at some point of the reaction. This
behavior, combined with the gelling of the epoxy phase, might have maximized the rate at which
the systems reached the gel point.
The theoretical extent of the reaction at the gel point was calculated by means of the Carothers
and Flory-Stockmayer approaches. According to Carothers, Equation (4.3) yields an average
functionality of 4.01 for an equimolar α-resorcylic acid/Jeffamine T-403 mix. Equation (4.2)
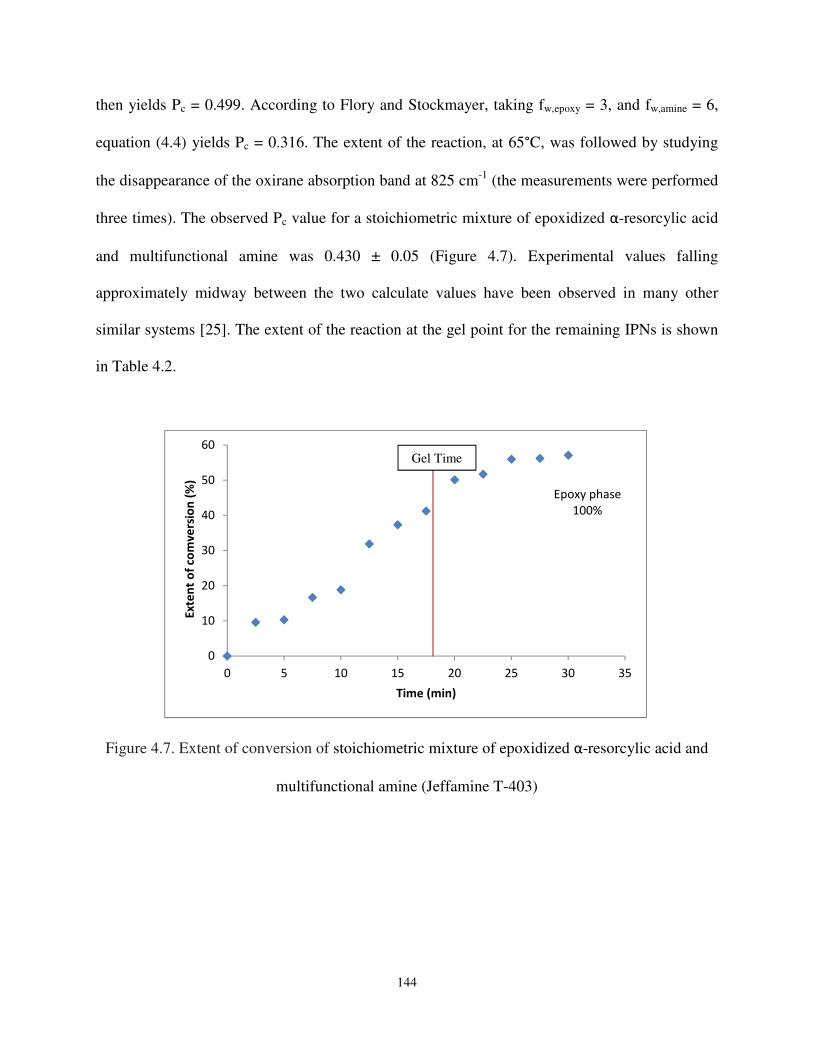
144
then yields Pc = 0.499. According to Flory and Stockmayer, taking fw,epoxy = 3, and fw,amine = 6,
equation (4.4) yields Pc = 0.316. The extent of the reaction, at 65°C, was followed by studying
the disappearance of the oxirane absorption band at 825 cm-1 (the measurements were performed
three times). The observed Pc value for a stoichiometric mixture of epoxidized α-resorcylic acid
and multifunctional amine was 0.430 ± 0.05 (Figure 4.7). Experimental values falling
approximately midway between the two calculate values have been observed in many other
similar systems [25]. The extent of the reaction at the gel point for the remaining IPNs is shown
in Table 4.2.
Figure 4.7. Extent of conversion of stoichiometric mixture of epoxidized α-resorcylic acid and
multifunctional amine (Jeffamine T-403)
Epoxy phase
100%
0
10
20
30
40
50
60
0 5 10 15 20 25 30 35
Ex
ten
t o
f co
mv
ers
ion
(%
)
Time (min)
Gel Time

145
Table 4.2. Extent of the reaction at the gel point for IPNs based on epoxidized α-resorcylic acid
and acrylated soybean oil.
IPN Epoxy phase conversion Acrylate phase conversion
Epoxy 100% 0.43 -------------------
Epoxy 75% - Acrylate 25% 0.40 0.47
Epoxy 50% - Acrylate 50% 0.55 0.43
Epoxy 25% - Acrylate 75% 0.50 0.20
Acrylate 100% ----------------------- 0.21
As can be seen, the conversion of the epoxy phase at the gel point was not deeply affected by the
presence of the acrylate phase, and it showed a medium conversion, when tested at 65°C. This
parameter ranged from 40 % to 55 %, and there was no clear trend in its behavior as a function
of the ratio epoxy/acrylate. On the other hand, the acrylate phase conversion was impacted by the
presence of the epoxy phase. Samples containing 100 wt% and 75 wt% of acrylate reached the
gel point at similar conversions (approx. 20 %), but at different times, with the Acrylate 75 wt%
system reaching the gelation almost two times faster than the Acrylate 100 wt%. When
increasing the weight content of the epoxy phase in the IPNs, the conversion of the acrylate at
the gel point also increased, reaching its maximum (approx. 96 %) for the system Epoxy 75 wt%
- Acrylate 25 wt%. The rise in the conversion values at the gel point for the IPNs Epoxy 50 wt%
- Acrylate 50 wt% and Epoxy 75 wt% - Acrylate 25 wt%, could be attributed to an autocatalysis
phenomenon in the system; due to the energy release of the ring opening reaction of the epoxy
ring when combined with the amine curing agent, as previously explained. As a general trend,
for large epoxy contents, the heat released during reaction might have triggered the conversion of

146
the acrylate phase; however, since its content is low, the gelling is dominated by the epoxy
counterpart.
3.2.2.3 Curing behavior of IPNs
In order to assess the curing of the epoxy – acrylate IPNs, about 5 mg of each mix was studied
by DSC (all measurements were performed three times). Likewise, the samples were cured at
65°C for 1 hour and then also subjected to calorimetric analysis to evaluate the extension of
curing after treatment at the above mentioned conditions. Table 4.3 shows the maximum values
of the curing peak temperatures for the IPNs, as well as, the onset of the peaks
Table 4.3. Onset and maximum of curing peak temperatures for the IPNs based on epoxidized α-
resorcylic acid and acrylated soybean oil.
System Onset of
Curing Peak
(°C)
Maximum of
Curing Peak
(°C)
Maximum of Curing Peak (°C)
After curing 1 hr. at 65 °C
Epoxy 100 wt% 65.82 102.66 89.89
Epoxy 75 wt% - Acrylate 25 wt% 68.80 103.65 103.53
Epoxy 50 wt% - Acrylate 50 wt% 73.98 100.43 97.79
Epoxy 25 wt% - Acrylate 75 wt% 74.99 93.40 115.14
Acrylate 100 wt% 76.62 86.42 116.61
The DSC of the IPNs, as well as the pure systems, showed only one exothermic peak,
corresponding to the curing of the resins. From Table 4.3, it can be observed that the onset of the
curing is shifted to higher temperatures when increasing the amount of acrylate resin in the
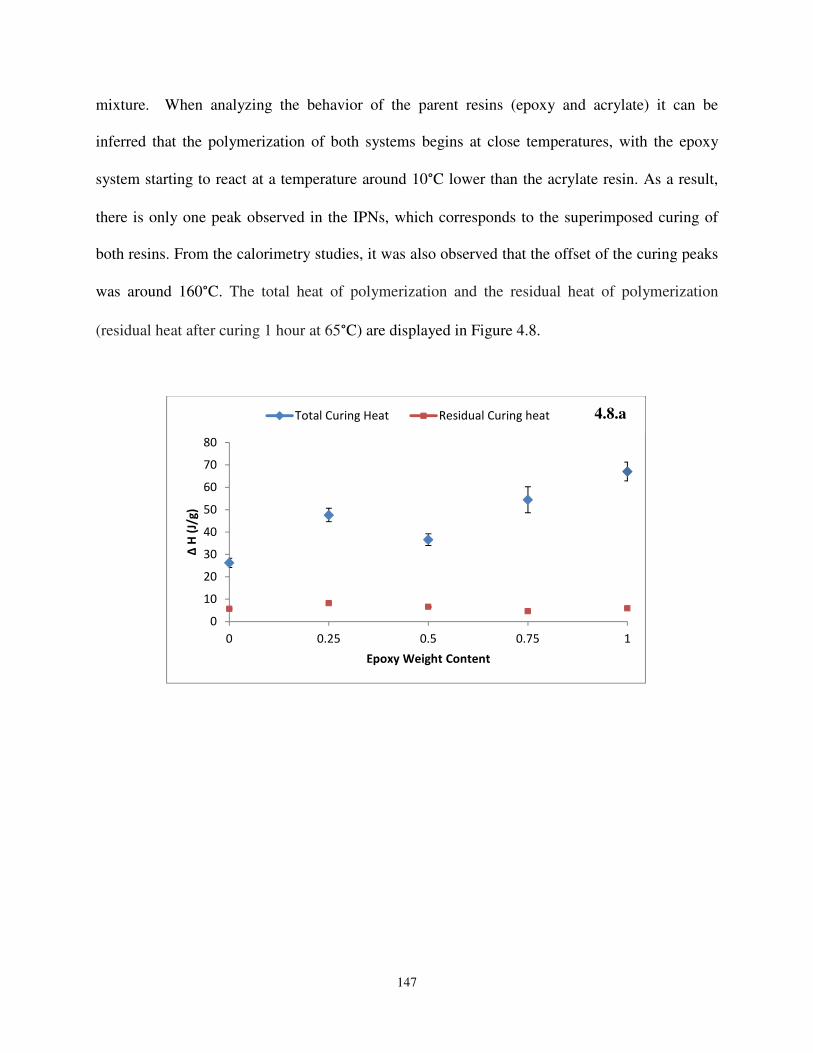
147
mixture. When analyzing the behavior of the parent resins (epoxy and acrylate) it can be
inferred that the polymerization of both systems begins at close temperatures, with the epoxy
system starting to react at a temperature around 10°C lower than the acrylate resin. As a result,
there is only one peak observed in the IPNs, which corresponds to the superimposed curing of
both resins. From the calorimetry studies, it was also observed that the offset of the curing peaks
was around 160°C. The total heat of polymerization and the residual heat of polymerization
(residual heat after curing 1 hour at 65°C) are displayed in Figure 4.8.
0
10
20
30
40
50
60
70
80
0 0.25 0.5 0.75 1
ΔH
(J/
g)
Epoxy Weight Content
Total Curing Heat Residual Curing heat 4.8.a

148
Figure 4.8. Heats of polymerization (4.8.a) and conversions (4.8.b) of the IPNs based on
epoxidized α-resorcylic acid and acrylated soybean oil.
When analyzing the behavior of the parent resins, it is clear that the ΔH of curing of the epoxy-
amine resin is highly exothermic (approx. 67 J/g), being about 2.5 times higher than that shown
by the acrylate phase (26 J/g). This means that mixtures richer in the epoxy phase are more
exothermic, behavior that is reflected in the experimental results, where the overall or total heat
of polymerization associated with the curing peaks is shifted to higher values when increasing
the content of epoxy phase in the systems. The total heat of polymerization approximately
increases as a linear function of the epoxy content; however this tendency is truncated by the
Epoxy 25 wt% - Acrylate 75 wt%, suggesting that the curing mechanism of each constituent is
affected by the presence of the other, probably due to autocatalysis or even chemical interactions
between the components [1].
75
77
79
81
83
85
87
89
91
93
95
0 0.25 0.5 0.75 1
Co
nv
ers
ion
(%
)
Epoxy Weight Fraction
4.8.b

149
Regarding the residual heat after curing 1 hour at 65°C, it seems to be not greatly affected by the
ratio epoxy-acrylate, and it ranged from around 4.000 J/g to 8.500 J/g. This finding suggests that
most of the components are reacted after 1 hour at 65°C, fact that is reflected in the conversions
(Figure 8); which ranged from around 78 % to 91 %. As happened with the curing heats, the
conversions shifted to higher values when increasing the proportion of the epoxy system;
probably due to the high chemical reactivity of non-sterically hindered glycidyl ethers, such as
epoxidized α-resorcylic acid, towards amines [41].
Concerning this reaction, the two amino hydrogens in primary amines, such as Jeffamine T-403,
have initially the same reactivity but once the first one has reacted, the secondary amine formed
may be less reactive [34]. As previously described, during epoxy-amine curing process, the
general reaction occurs via a nucleophilic attack of the amine nitrogen on the carbon of the
epoxy function. The mechanism proceeds through a SN2 type reaction, which is in turn highly
dependent on the structure and on the reaction conditions. Epoxidized α-resorcylic acid contains
oxirane functionalities that are non-sterically hindered, facilitating the nucleophilic attack of the
amino group. As a consequence of this feature, the reaction occurs rapidly at relatively low
temperatures (~ 65°C). It is important to mention that etherification products, arising from the
polymerization of epoxy groups and hydroxyl groups of acrylated soybean oil at elevated
temperatures, might be also present in the final polymer matrix. However, since stoichiometric
amounts of epoxy groups and amine comonomers are used, and also due to the low reactivity of
the secondary hydroxyl groups [42]; the effect of this side reaction on the final product can be
neglected.
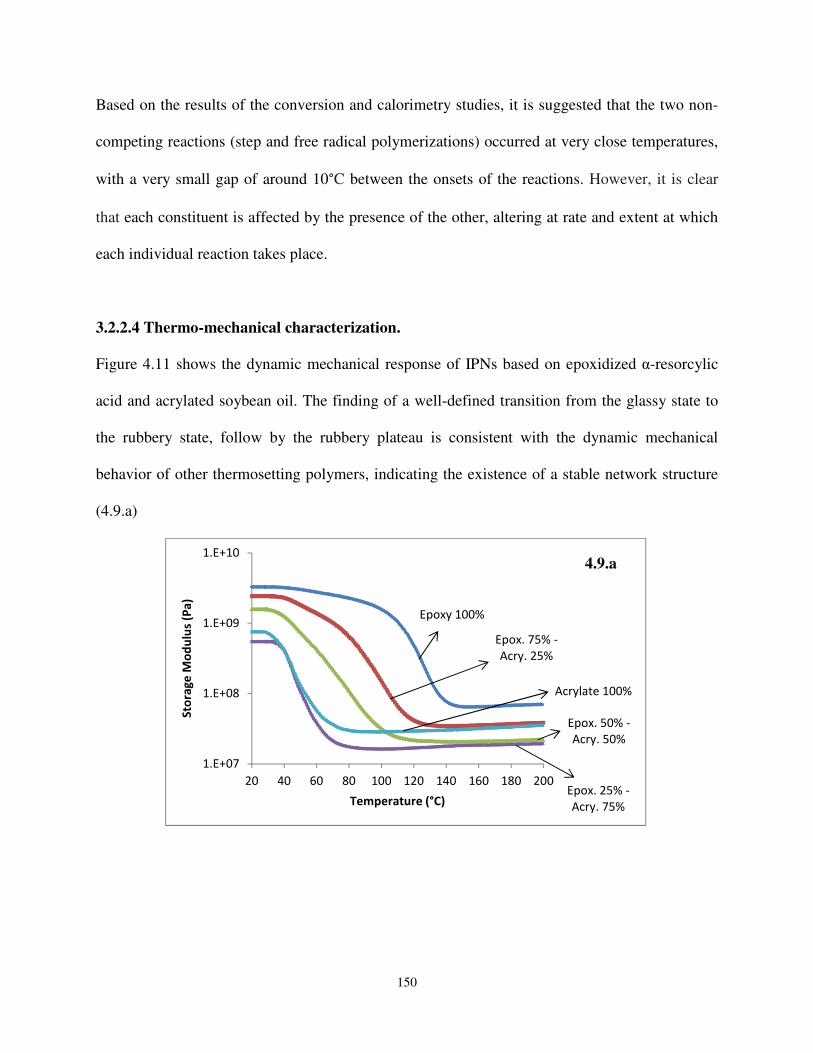
150
Based on the results of the conversion and calorimetry studies, it is suggested that the two non-
competing reactions (step and free radical polymerizations) occurred at very close temperatures,
with a very small gap of around 10°C between the onsets of the reactions. However, it is clear
that each constituent is affected by the presence of the other, altering at rate and extent at which
each individual reaction takes place.
3.2.2.4 Thermo-mechanical characterization.
Figure 4.11 shows the dynamic mechanical response of IPNs based on epoxidized α-resorcylic
acid and acrylated soybean oil. The finding of a well-defined transition from the glassy state to
the rubbery state, follow by the rubbery plateau is consistent with the dynamic mechanical
behavior of other thermosetting polymers, indicating the existence of a stable network structure
(4.9.a)
Epoxy 100%
Epox. 75% -
Acry. 25%
Epox. 50% -
Acry. 50%
Epox. 25% -
Acry. 75%
Acrylate 100%
1.E+07
1.E+08
1.E+09
1.E+10
20 40 60 80 100 120 140 160 180 200
Sto
rag
e M
od
ulu
s (P
a)
Temperature (°C)
4.9.a

151
Figure 4.9. Dynamic mechanical response of IPNs based on epoxidized α-resorcylic acid and
acrylated soybean oil. Storage modulus (4.9.a) and loss factor or tan δ (4.9.b) as a function of the
composition of the IPNs
In the glassy region, the systems showed a high modulus (~ 0.7 – 3 GPa). It is clearly seen that
with increasing the acrylated soybean oil content, the stiffness (E`) in the glassy state is
considerably decreased. Once the glass transition was surpassed, all of the resins exhibited a
rubbery plateau modulus that was insensitive to the temperature (~ 16 – 65 MPa). The plateau
moduli of the IPNs also decreased with increasing the acrylated oil content, which reduces the
crosslinking density or n values, as established by the proportionality between E` and this
parameter (Table 4.4). From the plots, it is evident that the epoxy parent system is more
crosslinked that the acrylate parent system, and that the crosslinking is also severely reduced
when the acrylate soybean oil content is increased in the systems. The crosslinking effect on Tg,
is an increasing function of the chain stiffness, which is under the dependence of molecular scale
factors, essentially aromaticity. The chain stiffness expressed by the flex parameter F which, for
Epoxy 100
Epox. 75% -
Acry. 25%
Epox. 50% -
Acry. 50%
Epox. 25% -
Acry. 75%Acrylate
100%
0.00
0.10
0.20
0.30
0.40
0.50
0.60
0.70
25 50 75 100 125 150 175 200
tan
δ
Temperature (°C)
4.9.b

152
a given chain of molar mass Me, is given by F = Me/Ne, where Ne is the number of elementary
(undeformable) segments. F is essentially an increase function of the content of aromatic nuclei
[43]. The α-resorcylic acid based epoxy matrix is structurally formed by aromatic units,
polyfunctional hard clusters that constitute a rigid and densely packed phase that largely reduces
the coordinated large scale motions in the network. It is therefore evident that mixtures richer in
the epoxy phase are stiffer, conferring support to the network when stress is applied.
The reduction in the stiffness of the systems is also accompanied by a similar change in the Tg,
as evidenced by the shift in the α-transition temperature peak from high to low temperatures.
The peaks of the parent resins were relatively narrow, whilst the IPNs (due to the multitude of
components and the great amount of possible relaxations modes acting in triglyceride-based
polymers) showed a wider transition [44]. Likewise, all the systems showed only one tan δ peak,
indicating that there was no phase separation and therefore, good compatibility between the
epoxy and acrylate phase. It is important to mention that there seems to be a decrease in the
intensity of the Tg peaks in the tan delta plots of the IPNs. This intensity reduction of the Tg
peak can be ascribed to some constraint effects on supramolecular level which supports the
presence of interpenetrating of the two networks [45]. Note that a reduction in Tg is usually due
to less tight crosslinking (co-network) which covers also the formation of IPN. Moreover, the
peak width at half height is a criterion used to indicate the homogeneity of the amorphous phase.
A higher value implies higher inhomogeneity of the amorphous phase. In this particular case, the
widening of the loss factor peak was observed for the IPNs, especially for the 50 wt% - 50 wt%
mixture. This behavior could be associated to less cross-linked and heterogeneous networks [46].

153
The resulting thermo-mechanical properties of the systems are listed in Table 4.4 The
crosslinking density (n) and molecular weight between crosslinks (Mc) were estimated from
experimental data using the rubber elasticity theory (Equation 4.5) to be able to explain the
effects of the phases on the mechanical properties of the resulting IPNs.
Table 4.4 Mechanical Properties of the IPNs based on epoxidized α-resorcylic acid and acrylated
soybean oil.
System
Tg
(°C)
E` at 30°C
(GPa)
n
(g/cm3)
n
(mol/m3)
Mc
(g/mol)
Epoxy 100% 129.10 3.28 1.1301 ± 0.0315 6026 188
E75% - A25% 104.18 2.43 1.1293 ± 0.0488 3431 329
E50% - A50% 89.11 1.56 1.1169 ± 0.0272 2235 500
E25% - A75% 56.41 0.55 1.0984 ± 0.0019 1872 587
Acrylate 100% 58.27 0.72 1.0815 ± 0.0084 3185 340
For the calculation of n, it was assumed that all of the components in the systems entered into the
network, which is in reality impossible. However, after the Soxhlet extraction of samples with
acetone for 6 hours, it was observed that the total amount of monomers that did not polymerize
ranged from 3 % to 7% (from Epoxy 100% to Acrylate 100%). These results allowed assuming
that the influence of low incorporation of monomers into the networks can be neglected.
As seen in the table, all of the systems behaved as brittle glassy polymers at room conditions.
The value of n for the epoxidized α-resorcylic acid cured with Jeffamine T-403 system is approx.
6026 mol/m3, denoting a highly crosslinked network (similar to a diglycidyl ether of bisphenol A
(DGEBA) type epoxy resin, having n = 5000 mol/m3). When acrylated soybean oil is

154
incorporated into the epoxy phase in a low proportion (25 wt.%), the n value fell about 2 times;
reaching a value of n = 3431 mol/m3, As the acrylate weight fraction increases, the crosslinking
density still decreasing; lowering the thermo-mechanical properties, as evidenced by the
numerical decrease of the Tg and modulus. For the 100 wt% acrylate soybean oil system, the
crosslinking density reached approx. 3185 mol/m3. It is clear from these results that the acrylated
soybean oil phase is plasticizing the epoxy network. The dynamic mechanic behavior previously
described comprises a balance between two factors, crosslinking density and plasticization. As
the crosslinking density increases the more constrained the chain motion; and thus the stiffer and
more thermally resistant the molecular structure is. Plasticization is due to the molecular nature
of the triglyceride. The positional distribution of the acrylate groups imply that dangling chain
ends remains after crosslinking of the acrylate moieties. Also, the soybean oil contains fatty acids
that are completely saturated and cannot be functionalized with acrylates. These inelastically
active fatty acid dangling chains do not support stress when a load is applied; therefore they act
as a plasticizing agent, introducing free volume and enabling the network to deform easily when
stress is applied to the material. This plasticizer effect is inherent to all natural triglyceride-based
materials; and it represents an issue for their potential use in high performance applications [47].
Likewise, based on property modeling of triglyceride-based thermosetting resins, La Scala and
coworkers [48] have also suggested that side reactions during crosslinking of fatty acids might
take place. Intramolecular cyclization of functional groups on the same triglyceride reduces the
number of effective attachment points of this triglyceride to the rest of the network. When a
triglyceride reacts with itself before reacting with another triglyceride, it forms a loop that is not
attached to the rest of the polymer network. Consequently, this reduces the crosslink density of

155
the resulting polymer. Every time two functional groups react in this way, the effective number
of functional groups per triglyceride is reduced by 2. As revealed by the results, the polymer
densities increased slightly with the increase in the epoxy phase content, and ranged from about
1.08 to 1.13 g/cm3. Similar values for triglyceride based materials have been reported by La
Scala and Wool [49]. The rubbery moduli of the systems ranged from approx. 16 to 65 MPa,
yielding molecular weight between crosslinks ranging from approx. 188 to 588 g/mol. The Mc,
as expected, increased with the increase in the acrylated soybean oil content, because the fatty
acid chains increase the free volume of the polymer matrix. Similar values have been obtained by
[44,50].
As a summary of the thermos-mechanical results, α-resorcylic acid based epoxy system provided
a considerable better option to formulate the interpenetrated networks, overcoming the
drawbacks given by its triglyceride-based epoxy counterpart, due to its ability to react at rather
low temperatures and the very good mechanical properties. However, no synergistic effect on the
thermo-mechanical properties was observed, and contrary to this a reduction in the mechanical
performance was observed in the IPN, when compared to their parent resins; mainly attributable
to the plasticizer effect conferred by the triglyceride-based acrylate matrix.
3.2.2.5 Linear Elastic Fracture Mechanics
In order to investigate the influence on the introduction of the acrylate phase on the epoxy phase,
and vice versa, the fracture toughness of the IPNs and parent resins was evaluated through the
critical stress intensity factor, KIc. Table 4.5 depicts the obtained results.
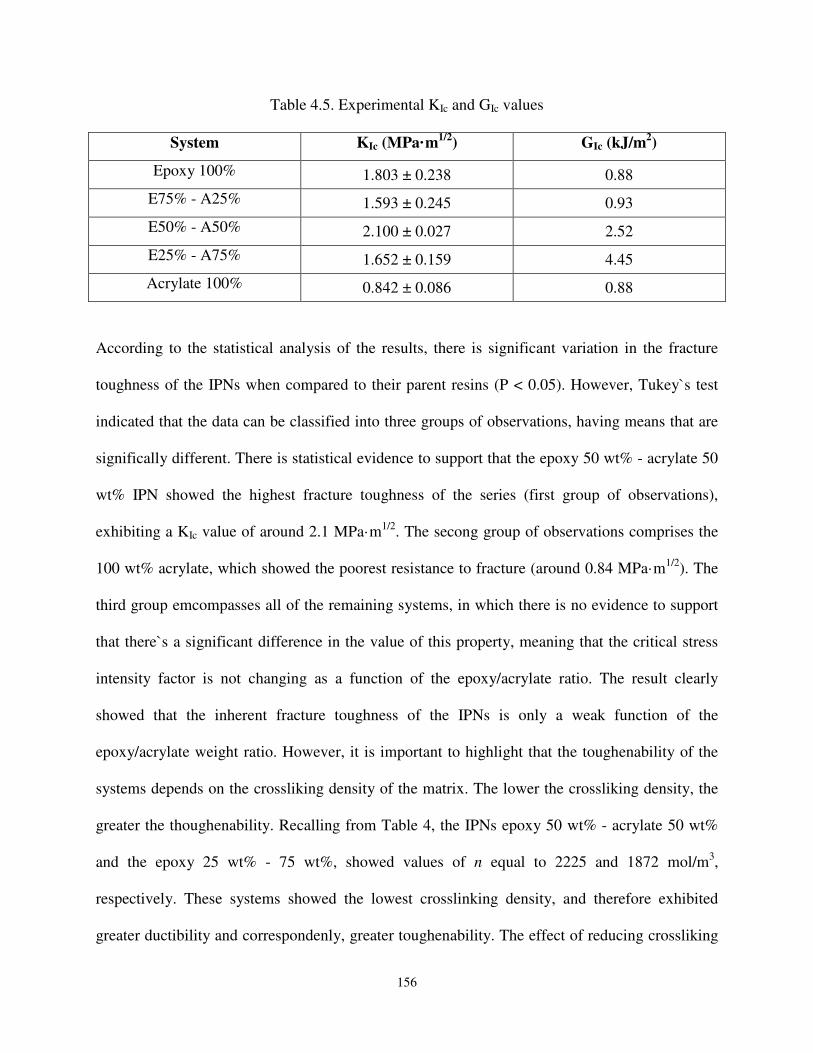
156
Table 4.5. Experimental KIc and GIc values
System KIc (MPa·m1/2
) GIc (kJ/m2)
Epoxy 100% 1.803 ± 0.238 0.88
E75% - A25% 1.593 ± 0.245 0.93
E50% - A50% 2.100 ± 0.027 2.52
E25% - A75% 1.652 ± 0.159 4.45
Acrylate 100% 0.842 ± 0.086 0.88
According to the statistical analysis of the results, there is significant variation in the fracture
toughness of the IPNs when compared to their parent resins (P < 0.05). However, Tukey`s test
indicated that the data can be classified into three groups of observations, having means that are
significally different. There is statistical evidence to support that the epoxy 50 wt% - acrylate 50
wt% IPN showed the highest fracture toughness of the series (first group of observations),
exhibiting a KIc value of around 2.1 MPa·m1/2. The secong group of observations comprises the
100 wt% acrylate, which showed the poorest resistance to fracture (around 0.84 MPa·m1/2). The
third group emcompasses all of the remaining systems, in which there is no evidence to support
that there`s a significant difference in the value of this property, meaning that the critical stress
intensity factor is not changing as a function of the epoxy/acrylate ratio. The result clearly
showed that the inherent fracture toughness of the IPNs is only a weak function of the
epoxy/acrylate weight ratio. However, it is important to highlight that the toughenability of the
systems depends on the crossliking density of the matrix. The lower the crossliking density, the
greater the thoughenability. Recalling from Table 4, the IPNs epoxy 50 wt% - acrylate 50 wt%
and the epoxy 25 wt% - 75 wt%, showed values of n equal to 2225 and 1872 mol/m3,
respectively. These systems showed the lowest crosslinking density, and therefore exhibited
greater ductibility and correspondenly, greater toughenability. The effect of reducing crossliking

157
density has been proposed to be an increase in the mainchain mobility which results in increased
ductility [51]. It is well known that plastic deformation is localized at the crack tip during crack
propagation in highly crosslinked thermosetting resins, and large plastic deformation occurs if
crossliking is light. Therefore, the increase in the fracture toughness with the decrease in the
croslinking density is mainly attributed to plastic deformation arising from easy chain flow [52].
For commercial diglycidyl ether of bisphenol A (DGEBA) type epoxy resin, crosslinked with
Jeffamine T-403, KIc was found to be approx. 0.80 MPa·m1/2 [53]. Regarding commercial
polymethylacrylate (PMMA), previous investigations performed by this group indicated a value
of around 1.1 MPa·m1/2 [5].
The critical energy release rate, GIc, can be related to the critical stress-intensity favor, KIc, in
plane strain conditions by the Equation (4.8). The storage modulus is a measure of the energy
stored elastically during deformation, and ideally is equivalent to Young`s modulus. However,
this is not true in real conditions, and their values might not be equal, mainly because the tests
are very different. The Young`s modulus is calculated from a stress-strain test, in which the
material is constantly stretched; whereas for the storage modulus, the material is oscillated in
dynamic test [4]. However, in spite this difference, storage modulus was used as an
approximation to Young`s modulus (E` ~ E) to establish some numerical values for the fracture
energy. As previously stated, in glassy polymers most of the energy necessary to propagate a
crack is due to the plastic deformation near the crack tip. This energy in macroscopically
measurable through the critical energy release rate, and the actual failure of the material require
molecular events such as chain pullout or chain scission within, or adjacent to, the plastic zone.
As seen in the table, the parent resins showed equal fracture energies, having values of GIc = 0.88

158
kJ/m2. Unlike the fracture toughness; the fracture energy showed a dependence on the
epoxy/acrylate ratio, increasing as the acrylate proportion is enlarged. The highest values were
exhibited by the IPNs epoxy 50 wt% - acrylate 50 wt% and the epoxy 25 wt% - 75 wt%, with GIc
= 2.5 kJ/m2 and GIc = 4.5 kJ/m2, respectively. The variation of GIc as a function of this relation
reflects both the increase in the toughness, due to its direct proportionality with KIc, and the
decrease in the storage modulus. The introduction of the acrylate phase into the epoxy phase
decreased the crosslinking density, favoring the chain flexibility as a consequence of having
higher Mc values. It is accepted that thermosetting resins undergo plastic deformation via shear
yielding rather than crazing, and this is attributed to high crosslinking density and small chain
contour length, le. However, when crosslinking decreases the chain contour length grows; the
materials become more ductile and tend to yield, dissipating energy through chain deformations
and molecular motion [52]. According to Karger-Kocsis and Gremmels (2000) [55], the fracture
toughness and energy toughness can be described by the rubber elasticity theory. It is established
that under plane strain conditions, KIc and GIc change as a function of the molecular weight
between crosslinks by linear and square root relations, respectively. Both relations were verified.
It was found that there is no a linear correlation between KIc and Mc for the systems under study.
The results showed that the KIc values did not increase as Mc increased, which seems to suggest
that a different mechanism is responsible for the increase in the fracture toughness displayed by
IPNs. It is clear however, that the high toughenability was correlated to high molecular weight
between crosslinks.
On the other hand, a similar analog was studied for the relation between the fracture energy and
Mc. Figure 4.10 depicts the results.
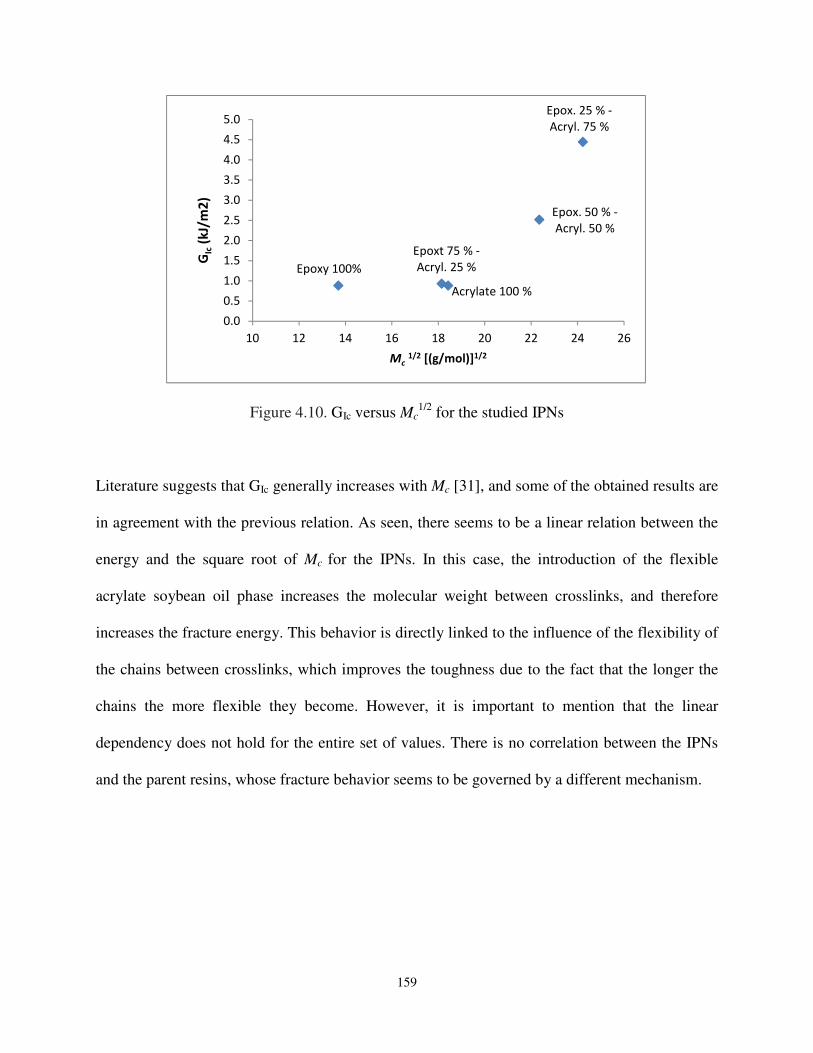
159
Figure 4.10. GIc versus Mc1/2 for the studied IPNs
Literature suggests that GIc generally increases with Mc [31], and some of the obtained results are
in agreement with the previous relation. As seen, there seems to be a linear relation between the
energy and the square root of Mc for the IPNs. In this case, the introduction of the flexible
acrylate soybean oil phase increases the molecular weight between crosslinks, and therefore
increases the fracture energy. This behavior is directly linked to the influence of the flexibility of
the chains between crosslinks, which improves the toughness due to the fact that the longer the
chains the more flexible they become. However, it is important to mention that the linear
dependency does not hold for the entire set of values. There is no correlation between the IPNs
and the parent resins, whose fracture behavior seems to be governed by a different mechanism.
Epoxy 100%
Epoxt 75 % -
Acryl. 25 %
Epox. 50 % -
Acryl. 50 %
Epox. 25 % -
Acryl. 75 %
Acrylate 100 %
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
10 12 14 16 18 20 22 24 26
GIc
(kJ/
m2
)
Mc1/2 [(g/mol)]1/2

160
4. Conclusions
IPNs have been prepared using an epoxy phase synthesized from linseed oil and/or α-resorcylic
acid and acrylate phase using acrylated soybean oil. The E.E.W. of the epoxy monomers after
chemical functionalization was found to be approx. 171 and 112 g/eq. for the epoxidized
derivatives of α-resorcylic acid and linseed oil, respectively. Grafting of epoxy functions onto
the monomer`s structure was confirmed by FTIR. DSC analysis revealed the presence of
exothermic peaks related to curing of the systems, and that the curing mechanism of each
constituent was affected by the presence of the other. The experimental Pc value for a
stoichiometric mixture of epoxidized α-resorcylic acid and multifunctional amine was 0.430 ±
0.100; falling in between the predicted by the Carothers and Flory theories. The mechanical
properties of the IPNs were not superior to those of the constituent polymers as a result of the
plasticizing effect of the triglyceride flexible chains in the macromolecular assembly of the IPNs.
However, IPNs based on epoxidized α-resorcylic acid and acrylated soy bean oil provided
considerably better mechanical properties than the system epoxidized linseed oil and acrylated
soy bean oil. For the α-resorcylic acid and soy bean oil system, modulus values ranged from 0.7
to 3.3 GPa at 30°C, and glass transition temperatures ranging from around 58 °C to approx.
130°C. Both parameters were affected by the epoxy/acrylate ratio, showing an increase as the
epoxy proportion was enlarged. Interpenetrating networks containing equivalent weight
proportions of the parent resins showed the highest fracture toughness of the series, exhibiting a
KIc value of around 2.1 MPa·m1/2. Likewise, FTIR and Soxhlet extractions indicated high degree
of conversion and incorporation of the monomers into the polymer networks. Based on the
results of this study, it is clear that the triglyceride-derived IPNs behave as elastomers at room
conditions; meanwhile the IPN in which modified α-resorcylic acid was introduced as the epoxy

161
phase correspond to glassy polymers at ambient conditions. The latter could be used as
thermosetting matrices for the development of composite materials. Likewise, they have
potential as partial replacements in the formulation of commercial thermosetting resins where
higher toughenability than their parent epoxy and acrylate resins is requiered.

162
References
[1] K. Dean, W.D. Cook, P. Burchill, M. Zipper, Curing behaviour of IPNs formed from
model VERs and epoxy systems: Part II. Imidazole-cured epoxy, Polymer (Guildf). 42
(2001) 3589–3601. doi:10.1016/S0032-3861(00)00745-X.
[2] M.R. Jean, I. Henry, M. Taha, Polyurethane Acrylate / Epoxy – Amine Acrylate Hybrid
Polymer Networks, (1999) 2711–2717.
[3] K.C. Frisch, D. Klempner, H.L. Frisch, Recent advances in interpenetrating polymer
networks, Polym. Eng. and Sci, 22 (1982) 1143-1152.
[4] L.H. Sperling, Introduction to Physical Polymer Science, 2006.
doi:10.1021/ed078p1469.1.
[5] R. Ballestero, Sequential graft-interpenetrating polymer networks based on polyurethane
and acrylic/ester copolymers, Express Polym. Lett. 10 (2015) 204–215.
doi:10.3144/expresspolymlett.2016.19.
[6] M.-S. Lin, S.-T. Lee, Mechanical behaviours of fully and semi-interpenetrating polymer
networks based on epoxy and acrylics, Polymer (Guildf). 38 (1997) 53–58.
doi:10.1016/S0032-3861(96)00484-3.
[7] L.H. Sperling, V. Mishra, The Current Status of Interpenetrating Polymer Networks,
Polym. Adv. Technol. 7 (1996) 197–208. doi:10.1002/(SICI)1099-
1581(199604)7:4<197::AID-PAT514>3.0.CO;2-4.
[8] E.S. Dragan, Design and applications of interpenetrating polymer network hydrogels. A
review, Chem. Eng. J. 243 (2014) 572–590. doi:10.1016/j.cej.2014.01.065.

163
[9] N. Devia-Manjarres, J. a. Manson, L.H. Sperling, A. Conde, Simultaneous
Interpenetrating Networks Based on Castor Oil Polyesters and Polystyrene.,
Macromolecules. 12 (1979) 360. http://www.scopus.com/inward/record.url?eid=2-s2.0-
0017937296&partnerID=tZOtx3y1.
[10] L.W. Barrett, L.H. Sperling, C.J. Murphy, Naturally Functionalized Triglyceride Oils in
Interpenetrating Polymer Networks, J. Am. Oil Chem. Soc. 70 (1993) 523–534.
doi:10.1007/bf02542588.
[11] Q.M. Jia, M. Zheng, H.X. Chen, R.J. Shen, Synthesis and characterization of
polyurethane/epoxy interpenetrating network nanocomposites with organoclays, Polym.
Bull. 54 (2005) 65–73. doi:10.1007/s00289-005-0372-7.
[12] P.L. Nayak, S. Lenka, S.K. Panda, T. Pattnaik, Polymers from renewable resources. I.
Castor oil-based interpenetrating polymer networks: thermal and mechanical properties, J.
Appl. Polym. Sci. 47 (1993) 1089–1096. doi:10.1002/app.1993.070470616.
[13] S. Dewasthale, D. Graiver, R. Narayan, Biobased interpenetrating polymers networks
derived from oligomerized soybean oil and polydimethylsiloxane, J. Appl. Polym. Sci.
132 (2015) 1–8. doi:10.1002/app.41709.
[14] Y. Yin, S. Yao, X. Zhou, Synthesis and dynamic mechanical behavior of crosslinked
copolymers and IPNs from vegetable oils, J. Appl. Polym. Sci. 88 (2003) 1840–1842.
doi:10.1002/app.11839.
[15] V. Kolot, S. Grinberg, Vernonia Oil – Based Acrylate and Methacrylate Polymers and
Interpenetrating Polymer Networks with Epoxy Resins, (2003).

164
[16] B. Balanuca, A. Lungu, I. Conicov, R. Stan, E. Vasile, D.M. Vuluga, et al., Novel bio-
based IPNs obtained by simultaneous thermal polymerization of flexible methacrylate
network based on a vegetable oil and a rigid epoxy, Polym. Adv. Technol. 26 (2015) 19–
25. doi:10.1002/pat.3413.
[17] A. Gandini, M.N. Belgacem, Lignins as components of macromolecular materials, in
M.N. Belgacem, A. Gandini (EDs.), Monomers, Polymers and Composites, Elsevier, UK,
2008, pp. 243-272.
[18] R. Lou, S. Wu, G. Lyu, Quantified monophenols in the bio-oil derived from lignin fast
pyrolysis, J. Anal. Appl. Pyrolysis. 111 (2015) 27–32. doi:10.1016/j.jaap.2014.12.022.
[19] J.E. Omoriyekomwan, A. Tahmasebi, J. Yu, Production of phenol-rich bio-oil during
catalytic fixed-bed and microwave pyrolysis of palm kernel shell, Bioresour. Technol. 207
(2016) 188–196. doi:10.1016/j.biortech.2016.02.002.
[20] G. Haarlemmer, C. Guizani, S. Anouti, M. Déniel, A. Roubaud, S. Valin, Analysis and
comparison of bio-oils obtained by hydrothermal liquefaction and fast pyrolysis of beech
wood, Fuel. 174 (2016) 180–188. doi:10.1016/j.fuel.2016.01.082.
[21] E.C. Ramires, E. Frollini, Tannin-phenolic resins: Synthesis, characterization, and
application as matrix in biobased composites reinforced with sisal fibers, Compos. Part B
Eng. 43 (2012) 2851–2860. doi:10.1016/j.compositesb.2012.04.049.
[22] S.H.L. and E.V. Santiago, Soybean - Bio-Active Compounds, InTech, 2013.
doi:10.5772/45866.
[23] A. Lungu, N.M. Florea, H. Iovu, Dimethacrylic/epoxy interpenetrating polymer networks
including octafunctional POSS, Polymer (Guildf). 53 (2012) 300–307.
doi:10.1016/j.polymer.2011.11.046.

165
[24] T. Resins, Standard Test Method for Gel Time and Peak Exothermic Temperature of
Reacting, 08 (2000) 1–3.
[25] G. Odian, Principles of Polymerization, John Wiley & Sons, 2004.
https://books.google.com/books?id=GbLrBgAAQBAJ&pgis=1 (accessed June 2, 2016).
[26] A.K. Higham, L.A. Garber, D.C. Latshaw, C.K. Hall, J.A. Pojman, S.A. Khan, Gelation
and cross-linking in multifunctional thiol and multifunctional acrylate systems involving
an in situ comonomer catalyst, Macromolecules. 47 (2014) 821–829.
doi:10.1021/ma402157f.
[27] W.S. Precision, E.S. Waste, T. Edition, Standard Practice for Extraction of Solid Waste
Samples for Chemical Analysis, 93 (2008) 1–7. doi:10.1520/D5369-93R08E01.2.
[28] M. Fache, R. Auvergne, B. Boutevin, S. Caillol, New vanillin-derived diepoxy monomers
for the synthesis of biobased thermosets, Eur. Polym. J. 67 (2015) 527–538.
doi:10.1016/j.eurpolymj.2014.10.011.
[29] ASTM D5045-14, Standard Test Methods for Plane-Strain Fracture Toughness and Strain
Energy Release Rate of Plastic Materials 1, ASTM Int. 99 (2013) 1–9.
doi:10.1520/D5045-14.priate.
[30] N.G. McCrum, C.P. Buckley, C.B. Bucknall, Principles of Polymer Engineering, Oxford
University Press, 1997. https://books.google.com/books?id=UX-sAQAAQBAJ&pgis=1
(accessed March 23, 2016).
[31] E. Urbaczewski-Espuche, J. Galy, J.F. Gerard, J.P. Pascault, H. Sautereau, Influence of
chain flexibility and crosslink density on mechanical properties of epoxy/amine networks,
Polym. Eng. Sci. 31 (1991) 1572–1580. doi:10.1002/pen.760312204.

166
[32] S. Ananda kumar, Z. Denchev, M. Alagar, Synthesis and thermal characterization of
phosphorus containing siliconized epoxy resins, Eur. Polym. J. 42 (2006) 2419–2429.
doi:10.1016/j.eurpolymj.2006.06.010.
[33] S. Gurusideswar, R. Velmurugan, N.K. Gupta, High strain rate sensitivity of epoxy/clay
nanocomposites using non-contact strain measurement, Polymer (Guildf). 86 (2016) 197–
207. doi:10.1016/j.polymer.2015.12.054.
[34] D. Ratna, Handbook of thermoset resins, 2009. doi:10.1002/0471743984.vse3011.pub2.
[35] Z.S. Pour, M. Ghaemy, Thermo-mechanical behaviors of epoxy resins reinforced with
silane-epoxide functionalized ??-Fe2O3 nanoparticles, Prog. Org. Coatings. 77 (2014)
1316–1324. doi:10.1016/j.porgcoat.2014.04.001.
[36] J. Ehlers, N.G. Rondan, L.K. Huynh, H. Pham, M. Marks, T.N. Truong, Theoretical Study
on Mechanisms of the Epoxy - Amine Curing Reaction, Macromolecules. 40 (2007)
4370–4377. doi:10.1021/ma070423m.
[37] J.E. McMurry, Organic Chemistry, Cengage Learning, 2011.
https://books.google.com/books?id=lM2VVe56iEAC&pgis=1 (accessed June 2, 2016).
[38] J.D. Earls, J.E. White, L.C. Lopez, Z. Lysenko, M.L. Dettloff, M.J. Null, Amine-cured-
epoxy fatty acid triglycerides: Fundamental structure-property relationships, Polymer
(Guildf). 48 (2007) 712–719. doi:10.1016/j.polymer.2006.11.060.
[39] B. Kowalski, Thermal-oxidative decomposition of edible oils and fats. DSC studies,
Thermochim. Acta. 184 (1991) 49–57. doi:10.1016/0040-6031(91)80134-5.
[40] L.Y. Jia, C. Zhang, Z.J. Du, C.J. Li, H.Q. Li, Preparation of interpenetrating polymer
networks of epoxy/polydimethylsiloxane in a common solvent of the precursors, Polym. J.
39 (2007) 593–597. doi:10.1295/polymj.PJ2006237.

167
[41] L. Shechter, J. Wynstra, R. Kurkjy, Glycidyl ether reactions with amines, Ind. Eng. …. 48
(1956) 94–97. doi:10.1021/ie50553a029.
[42] H. Dai, L. Yang, B. Lin, C. Wang, G. Shi, Synthesis and Characterization of the Different
Soy-Based Polyols by Ring Opening of Epoxidized Soybean Oil with Methanol, 1,2-
Ethanediol and 1,2-Propanediol, J. Am. Oil Chem. Soc. 86 (2009) 261–267.
doi:10.1007/s11746-008-1342-7.
[43] J.-P. Pascault, H. Sautereau, J. Verdu, R.J.J. Williams, Thermosetting Polymers, CRC
Press, 2002. https://books.google.com/books?id=OrnJdwIYLA4C&pgis=1 (accessed
April 1, 2016).
[44] J.J. La Scala, J.M. Sands, J.A. Orlicki, E.J. Robinette, G.R. Palmese, Fatty acid-based
monomers as styrene replacements for liquid molding resins, Polymer (Guildf). 45 (2004)
7729–7737. doi:10.1016/j.polymer.2004.08.056.
[45] S. Grishchuk, Hybrid thermosets from vinyl ester resin and acrylated epoxidized soybean
oil (AESO), eXPRESS Polym. Lett. 5 (2010) 2–11. doi:10.3144/expresspolymlett.2011.2.
[46] K.C. Manikandan Nair, S. Thomas, G. Groeninckx, Thermal and dynamic mechanical
analysis of polystyrene composites reinforced with short sisal fibres, Compos. Sci.
Technol. 61 (2001) 2519–2529. doi:10.1016/S0266-3538(01)00170-1.
[47] S.N. Khot, J.J. Lascala, E. Can, S.S. Morye, G.I. Williams, G.R. Palmese, et al.,
Development and application of triglyceride-based polymers and composites, J. Appl.
Polym. Sci. 82 (2001) 703–723. doi:10.1002/app.1897.
[48] J. La Scala, R.P. Wool, Fundamental thermo-mechanical property modeling of
triglyceride-based thermosetting resins, J. Appl. Polym. Sci. 127 (2013) 1812–1826.
doi:10.1002/app.37927.

168
[49] J. La Scala, R.P. Wool, Property analysis of triglyceride-based thermosets, Polymer
(Guildf). 46 (2005) 61–69. doi:10.1016/j.polymer.2004.11.002.
[50] J. Lu, S. Khot, R.P. Wool, New sheet molding compound resins from soybean oil. I.
Synthesis and characterization, Polymer (Guildf). 46 (2005) 71–80.
doi:10.1016/j.polymer.2004.10.060.
[51] R.A. Pearson, A.F. Yee, D. Building, A. Arbor, Toughening mechanisms in elastomer-
modified epoxies, 24 (1989) 2571–2580.
[52] K. Cho, D. Lee, C.E. Park, W. Huh, Effect of molecular weight between crosslinks on
fracture behaviour of diallylterephthalate resins, Polymer (Guildf). 37 (1996) 813–817.
doi:10.1016/0032-3861(96)87258-2.
[53] 828/T403: effect of 815C vs. 828, (n.d.). http://www.sandia.gov/polymer-properties/T16-
effect_of_815C.html (accessed April 4, 2016).
[54] A.. Fallis, No Title No Title, J. Chem. Inf. Model. 53 (2013) 1689–1699.
doi:10.1017/CBO9781107415324.004.
[55] J. Gremmels, Use of Hygrothermal Decomposed Polyester – Urethane, (2000) 1139–1151.

169
CHAPTER V
Fast Pyrolysis Bio-oil as Precursor of Thermosetting Epoxy Resins
1. Introduction
The use of biomass-based resources is currently a hot topic for both academic and industrial
research as an alternative to mitigate the strong dependence on fossil carbon in the chemical and
petrochemical industries. Lignocellulosic materials, being abundant and renewable (with annual
production arising to more than 1 billion dry tons in the U.S.A) [1], are the most favored
substrate for bioconversion. It is clear that the development of green polymers and materials is
growing, carrying along a vast and promising area for scientific research; that is supported not
just by the industrial or academic sectors, but also by governmental entities and agencies.
According to Effendi et al. [2], lignocellulosic biomass is made up of three main components:
hemicellulose, cellulose and lignin, of which the lignin fraction can account for up to 40% of its
dry weight. Lignin is an amorphous cross-linked polymer with very complicated structure. It can
be described as a three-dimensional, highly branched, polyphenolic substance consisting of an
irregular array of variously bonded “hydroxyl-“ and “methoxy”-substituted phenyl propane units.
These monomeric phenylpropane units exhibit p-coumaryl, coniferyl, and sinapsyl structures,
which are shown in Figure 5.1 [3].

171
Figure 5.1. Monomeric phenylpropane unit structures.
Source: D. Mohan, C.U. Pittman, P.H. Steele, Energy & Fuels. 20 (2006)
Lignin based materials can be obtained in very large quantities as by-products of the pulping
industry. These products are, as a matter of fact, fragments of the natural polymer; resulting from
different chemical splicing mechanisms during processing [4]. The exploitation of lignin as a
source of profitable chemicals or macromonomers for the polymer industry is not just backed up
by the huge amounts of this material available after the traditional pulping processes; but also by
the high content of aromatic moieties, containing aliphatic and phenolic hydroxyl groups
(present in different frequencies and proportions), which are of particular interest because they
serve as key monomers to synthesize high performance polymers [5].
The idea of using unmodified lignin for the preparation of epoxy resins was firstly reported in the
late 1990s; in which Kraft lignins were combined with different epoxy monomers and then
crosslinked with commercial aromatic diamines [6]. On the other hand, with the aim of obtaining
a modified lignin with enhanced reactivity (due to poor results with unmodified lignins), Glasser
et al. [6,7] reported the synthesis of lignin based epoxy resins by reacting partially oxypropylated
lignins (previously obtained by treating lignin with diethyl sulfate and then with propylene oxide
OH
OH
OH
OH
O
CH3
OH
OH
O O
CH3
CH3
p-Coumaryl alcohol Coniferyl alcohol Sinapyl alcohol

172
to “bring out” the -OH groups, and thus make them more available to react) with
epichlorohydrin. After modification, the lignin based material bearing epoxy moieties was also
reacted with an aromatic diamine. Even though studies like this concerning the use of lignin as a
component for macromolecular materials have been reported and published [8,9], often synthesis
strategies addressed to modify this material struggle with low reactivity associated to steric
hindrance; making difficult to insure good access to the targeted functional groups [4].
Some novel technologies based on biomass refinery appear as a good alternative to overcome
this problem, and stand as a sustainable way for aromatics biosourcing, by providing low
molecular weight fragments of natural lignin, whose functional groups are more exposed to the
reaction medium (due to their lesser steric crowding), and thus being readily reachable during
chemical synthesis. The thermochemical conversion of lignocellulosic biomass (i.e., combustion,
pyrolysis, or gasification) is now receiving an increasing interest as a method for production of
renewable energy, fuels, and high added value chemicals. When heated, the lignin component
depolymerizes to form monomeric and oligomeric phenolic compounds. It terminally
decomposes over a wide temperature range (approx. 160-900°C), and a possible free radical
mechanism for its decomposition has been proposed for several authors. Amongst them, Evans
et al. [10] suggested that alkyl hydroxyl groups in the α-position are first eliminated from the
propane side chain, followed by β-ether cleavage. Further details about the mechanism have been
recently described by Shen and others [11,12].
Amongst several thermal degradation processes, fast pyrolysis (also known as thermolysis) has
been developed relatively recent. Fast pyrolysis is a high-temperature process in which biomass
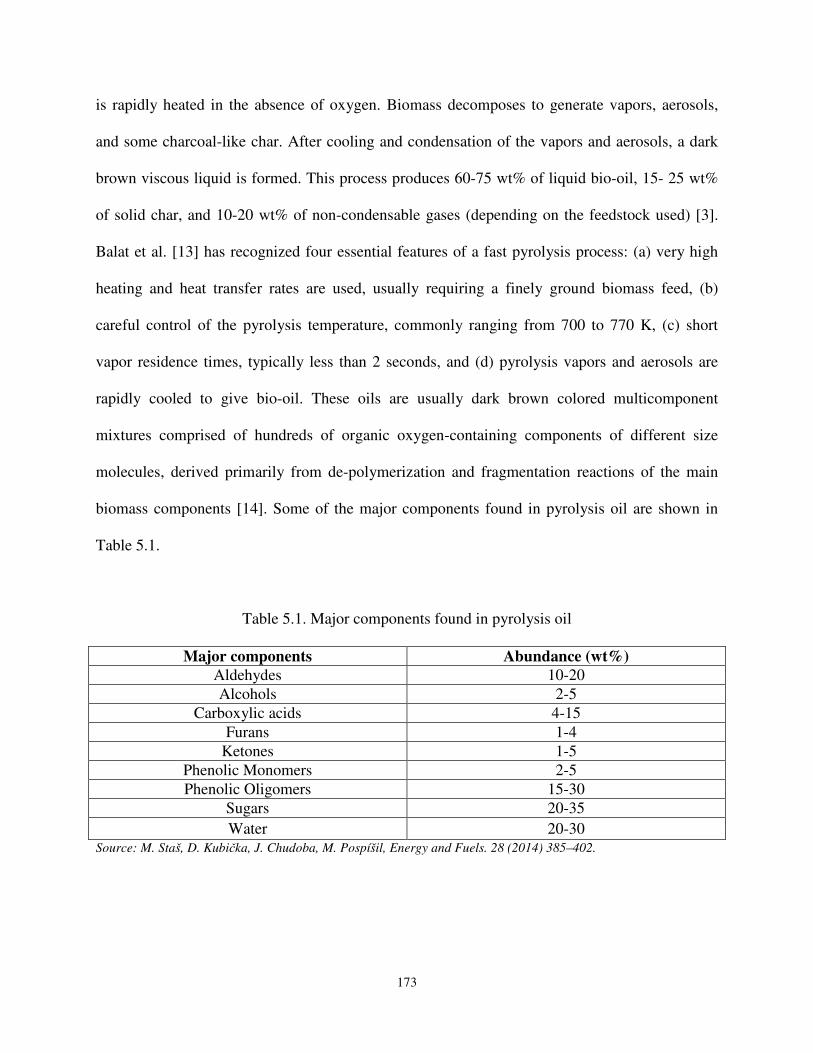
173
is rapidly heated in the absence of oxygen. Biomass decomposes to generate vapors, aerosols,
and some charcoal-like char. After cooling and condensation of the vapors and aerosols, a dark
brown viscous liquid is formed. This process produces 60-75 wt% of liquid bio-oil, 15- 25 wt%
of solid char, and 10-20 wt% of non-condensable gases (depending on the feedstock used) [3].
Balat et al. [13] has recognized four essential features of a fast pyrolysis process: (a) very high
heating and heat transfer rates are used, usually requiring a finely ground biomass feed, (b)
careful control of the pyrolysis temperature, commonly ranging from 700 to 770 K, (c) short
vapor residence times, typically less than 2 seconds, and (d) pyrolysis vapors and aerosols are
rapidly cooled to give bio-oil. These oils are usually dark brown colored multicomponent
mixtures comprised of hundreds of organic oxygen-containing components of different size
molecules, derived primarily from de-polymerization and fragmentation reactions of the main
biomass components [14]. Some of the major components found in pyrolysis oil are shown in
Table 5.1.
Table 5.1. Major components found in pyrolysis oil
Major components Abundance (wt%)
Aldehydes 10-20
Alcohols 2-5
Carboxylic acids 4-15
Furans 1-4
Ketones 1-5
Phenolic Monomers 2-5
Phenolic Oligomers 15-30
Sugars 20-35
Water 20-30 Source: M. Staš, D. Kubička, J. Chudoba, M. Pospíšil, Energy and Fuels. 28 (2014) 385–402.

174
Due to the presence of this vast amount of different and multifunctional compounds, the crude
bio-oil could serve as raw materials for the obtention and production (after further processing
and separation) of specialty chemicals and resins. As a renewable liquid fuel, bio-oil can be
easily stored and transported; however, compared to heavy petroleum fuel oil, bio-oils have
certain properties limiting their application as fuels. Some of these include high water content,
high viscosity, high ash content, high corrosiveness (acidity) and high oxygen content (low
heating value) [15]. Regarding this last feature, bio-oils contain up to 45% oxygen, and this
element is a component of most of the more than 400 compounds that have been identified in
pyrolysis oils. Phenolic compounds found in bio-oils are composed of both simple phenols and
oligomeric polyphenols; having multiple phenol structural units and containing varying number
of acidic, phenolic and carboxylic acid hydroxyl groups, as well as aldehyde, alcohol, and ether
functions [12]. The molecular weight of these oligomers (product of lignin decomposition)
ranges from several hundreds to 5000 g/mol, depending on the pyrolysis process [2].
Many commercial epoxy monomers usually have an aromatic structure, conferring thermo-
mechanical stability to the network. As seen in Chapters III and IV, the inclusion of α-resorcylic
acid considerable improved the mechanical properties of the resulting resins. It is clear that there
is a great potential to develop high-performance resins, and the problem of finding a renewable
source of aromatic materials needs to be addressed. Thus, in this context, bio-oil phenolic
derivatives are excellent candidates for the bio-sourcing of aromatic monomers that can be used
to synthesize epoxy resins and other resins. These compound present potential as fully or partial
replacements in the formulation of commercial epoxy resins used as coatings, adhesives or even
for the preparation of composite materials. Therefore, the aim of this work was to synthetize a

175
high performance bio-based epoxy polymer using fast pyrolysis bio-oil (containing lignin
fragments) as a source of phenolic compounds. The strategy to prepare bio-oil based polymers
can be divided into two sections. The first one comprises the chemical characterization of bio-
oils using chromatographic and spectroscopic methods, focusing on performing a qualitative
identification of the interest compounds, as well as a quantitative analysis of the targeted
functional groups. In a second step, the modified monomers are polymerized and the resulting
materials characterized, mainly focusing on their thermos-mechanical behavior. Likewise, α-
resorcylic acid (a phenolic acid with two hydroxyl and one carboxylic acid functional groups)
was also modified with reactive epoxy moieties, and used as low molecular weight comonomer
to enhance the density of aromatic structures in the crosslinked epoxy network.
2.Experimental section
2.1 Materials
Fast pyrolysis bio-oil, from hardwood (Oak), was supplied by the Department of Biosystems
Engineering, Auburn University, Alabama, United States. α-resorcylic acid or 3,5-
dihydroxybenzoic acid (α-RA, ≥ 98%), epichlorohydrin (99.0 %), and sodium hydroxide (NaOH,
≥ 98%), were purchased from VWR (US). Reagent grade chloroform, pyridine, and
dichloromethane were also purchased from VWR (US). Benzyltriethylammonium chloride
(BnEt3NCl, ≥ 98%), used as a phase transfer catalyst, was also acquired from VWR (US). 2-
chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP, 95.0 %) was used as
phosphorylating agent. 4-dimethylaminopyridine (DMAP, 99.0 %) was used as initiator for the
chain homopolymerization of epoxy monomers. Super Sap 100/1000 epoxy resin/hardener, by
Entropy Resins, with a bio-based carbon content of 37 %; and a mix of 2/1 wt% ratio [16].

176
2.2 Synthesis procedures
2.2.1 General method for the glycidylation (functionalization with epoxy groups) of bio-oil
The glycidylation of bio-oil was conducted following the method and conditions established by
[17,18]. The described process leads to the synthesis of epoxyalkylaryl ethers by the reaction
between compounds containing acid hydroxyl groups, and haloepoxyalkanes in the presence of a
strong alkali. A reactor was loaded with the desired amount of bio-oil (containing a certain
amount of moles of -OH / gram of oil) and epichlorohydrin (5 moles epichlorohydrin / mole -
OH). The mixture was heated at 100°C and benzyltriethylammonium chloride (0.05 moles
BnEt3NCl / mole -OH) was added. After 1 h, the resulting suspension was cooled down to 30°C,
and an aqueous solution of NaOH 20 wt% (2 moles NaOH / mole –OH and also containing 0.05
moles of BnEt3NCl / mole –OH) was added dropwise. Once the addition of the alkaline aqueous
solution was completed, the mixture was vigorously stirred for 90 min at 30°C, and then the
reaction was stopped. As soon as the reaction was completed, the crude product was filtered to
remove the insoluble matter. After that, the mixture was poured into an extraction funnel, the
organic layer was separated; and through numerous liquid-liquid extractions with deionized
water, the organic phase was abundantly washed until neutral pH was reached. Finally, the
organic phase was vacuum concentrated and the reaction yield was gravimetrically determined.
A similar procedure was carried out to functionalize the α-resorcylic acid (or α-RA) with epoxy
groups. Refer to Chapter III for more details about this synthesis.

177
2.3. Characterization of Starting and Modified Materials
2.3.1 Gas Chromatography - Mass Spectrometry (GC-MS)
Chemical composition of bio-oil was analyzed with an Agilent 7890 GC/5975MS using a DB-
1701 column (30 m; 0.25 mm i.d.; 0.25 mm film thickness). A representative bio-oil sample
(~150 mg) was weighed and mixed with 3 mL of methanol and diluted to 10 mL with
dichloromethane. The dilute sample was injected into the column and each sample was injected
twice. The initial temperature of the column, 40°C, was maintained for 2 min, and the
temperature was subsequently increased to 250°C at 5°C/ min, and the final temperature was held
for 8 min. Helium of ultra-high purity (99.99%) supplied from Airgas Inc. (Charlotte, NC) was
used as a carrier gas and flowed at 1.25 mL/min. The injector and the GC/MS interface were
kept at constant temperature of 280 and 250°C, respectively. Compounds were ionized at 70 eV
electron impact conditions and analyzed over a mass per change (m/z) range of 50–550. Bio-oil
compounds were identified by comparing the mass spectra with the NIST (National Institute of
Standards and Technology) mass spectral library [19].
2.3.2 Hydroxyl group analysis through 31
P-NMR spectroscopy.
Since the aim of this work is to functionalize the aromatic structures of the lignin fragments and
other phenolic compounds present in the bio-oil with reactive epoxy groups (by reaction with
epichlorohydrin), it is of great importance to quantify the content of phenolic hydroxyl groups
(mmol of phenolic -OH / gram of bio-oil). This determination allows establishing the amount of
epichlorohydrin needed for the reaction. The type of -OH groups present in the bio-oil was
determined and quantitatively calculated using 31P-NMR. For lignin and lignin derived products,
the most common used phosphytylation reagent is 2-chloro-4,4,5,5-tetramethyl-1,3,2-

178
dioxaphospholane (TMDP), because it provides better signal resolutions when collecting the
spectra; as well as higher stability of the phophitylated products. This compound reacts with
hydroxyl groups arising from aliphatic, phenolic, and carboxylic groups in the presence of an
organic base (such as pyridine) to give phosphylated products, whose presence can be quantified
through 31P-NMR spectroscopy [20]. Prior to 31P-NMR analysis, bio-oils were phosphitylated
with 150 mL of TMDP and then the spectra were acquired with a Bruker Avance II 250 MHz
according to the method reported by David et al. [21]. In order to assess the extent of the
functionalization of the aromatic structures of the lignin fragments and other phenolic
compounds present in the bio-oil with reactive epoxy groups, 31P-NMR was performed again to
the reaction product to verify the removal of phenolic hydroxyl groups after chemical reaction
with epichlorohydrin in alkaline conditions.
2.3.3 Fourier Transform Infrared Spectroscopy (FTIR)
With the aim to study the functional groups present in the product, FTIR was performed. Infrared
spectra of the starting materials and glycidylated products were measured by attenuated total
reflection (ATR) method using a Thermo Nicolet 6700 Fourier transform infrared spectrometer
connected to a PC with OMNIC software analysis. All spectra were recorded between 400 and
4000 cm-1 over 64 scans with a resolutions of 16 cm-1.
2.3.4 Epoxide equivalent weight determination.
Epoxide equivalent weight (EEW, g/eq.) of the epoxy products was determined by epoxy
titration according to Pan et al. [22]. Briefly, a solution of 0.1 M HBr in glacial acetic acid was

179
used as a titrant, 1 wt% crystal violet in glacial acetic acid was used as indicator, and toluene as
solvent for the sample. The EEW was calculated using Equation:
EEW = 0111 6 �7 6 8 (5.1)
where W is the sample weight (g), N is the molarity of HBr solution, and V is the volume of HBr
solution used for titration (mL).
2.4 General procedure for the formulation of crosslinked epoxy networks.
In order to generate a cross-linked polymeric network, epoxy monomers can undergo a chain
homopolymerization in the presence of both Lewis acids and bases such as tertiary amines
(process known as anionic homopolymerization). Glycidylated bio-oil (or epox. bio-oil) and
triglycidylated ether of α-resorcylic acid (or epox. α-RA), used as a comonomer, were mixed in
different weight proportions. After that, 4-dimethylaminopyridine or DMPA (a Lewis base) was
added to the mixtures in a ratio of 0.08 mole DMAP / mole epoxy [23]. The mixtures were then
poured in an aluminum mold and cured at 120°C for 12 hrs and post cured for 2 hrs at 160°C.
2.5 Characterization of the epoxy networks
2.5.1 Soxhlet extraction.
Soxhlet extraction was employed to characterize the structures of the bulk copolymers following
the ASTM D5369-93 [24]. An appropriate amount of the bulk polymer was extracted for 24 h
with 200 mL of refluxing dichloromethane with a Soxhlet extractor. Upon extraction, the
resulting solution was concentrated by rotary evaporation and subsequent vacuum drying. The
soluble substances were isolated for further characterization. The insoluble solid was dried in a
vacuum oven for several hours before it was weighed.

180
2.5.2 Scanning Electron Microscopy (SEM)
SEM study was performed with the purpose of obtaining a topological characterization of the
materials. The fractured surfaces of pre-chilled samples in liquid nitrogen were studied by
scanning electron microscope (Zeiss EVO 50 variable pressure scanning electron microscope
with digital imaging and EDS). Samples were sputtered with gold prior to SEM observations
with an EMS 550X auto sputter coating device with carbon coating attachment.
2.5.3 Thermo-mechanical properties of the networks.
Dynamic mechanical analysis (DMA) on a TA Instruments RSA III was carried out to assess the
thermo-mechanical properties by three-point bending. The tests were performed at temperatures
ranging from 25 to 250°C with a heating rate of 10°C/min. The frequency was fixed a 1Hz and a
sinusoidal strain-amplitude of 0.1 % was used for the analysis. The dynamic storage modulus
(E′) and tan δ curves were plotted as a function of temperature. To be able to explain the effects
of the co-monomers on the mechanical properties, it is useful to calculate the crosslinking
density (n, mol/m³). The crosslinking density can be estimated from the experimental data using
the rubber elasticity theory. Thermosets behave as rubbers above Tg. At small deformations,
rubber elasticity predicts that the modulus storage (E’), of an ideal elastomer with a network
structure is proportional to the crosslinking density according to Equation 5.2 [25]:
�� = 3��� = 3��/�� (5.2)
where E` is the rubbery storage modulus, R is the gas constant (8.314 J/mol K), T is the absolute
temperature (K), ρ is the density of the sample (g/m³), and Mc is the molecular weight between
crosslinks (g/mol). The temperature and rubbery modulus were determined for the calculation of

181
the equation at Tg + 30°C. The temperature at which the peak of the tan delta presents a
maximum was considered as the glass transition temperature of the material. The Fox equation
was employed to fit the experimental Tg data reported in the results. It serves as a way to
describe Tg of the multicomponent networks as a function of relative epoxy monomer content.
The Fox expression is shown in Equation 5.3 [26]:
0
[\ = �0[\0 + �F
[\F (5.3)
In this expression, W1 and W2 represent weight fractions of components 1 and 2 (bio-oil and α-
resorcylic acid, respectively), and Tg1 and Tg2 are Tg values of polymer samples containing only
components 1 and 2.
3. Results and Discussion
3.1 Gas Chromatography - Mass Spectrometry (GC-MS)
According to Laurichesse et al. [8], during the lignification process, the monolignol units are
linked together via radical coupling reactions to form a complex three-dimensional molecular
architecture, containing a great variety of bonds, with typically 50% of β-O-4 ether linkages.
During pyrolysis, biomass is heated at around 500°C without oxygen. Lignin starts to decompose
between 280-500°C by the cleavage of ether and carbon-carbon linkages. The components have
been classified into the main categories of hydroxyaldehydes, hydroxyketones, sugars,
carboxylic acids and phenolics. The concentration of phenol is typically very low (~ 0.1%),
while GC analyzable monomeric phenols commonly range around 1- 4%. Most of the phenolics
are too high in molecular weight to be quantified by GS analysis, and they are present as
oligomers containing varying numbers of acidic, phenolic and carboxylic acid hydroxyl groups,
as well as aldehyde, alcohol, and ether functions. The molecular weight of these oligomers
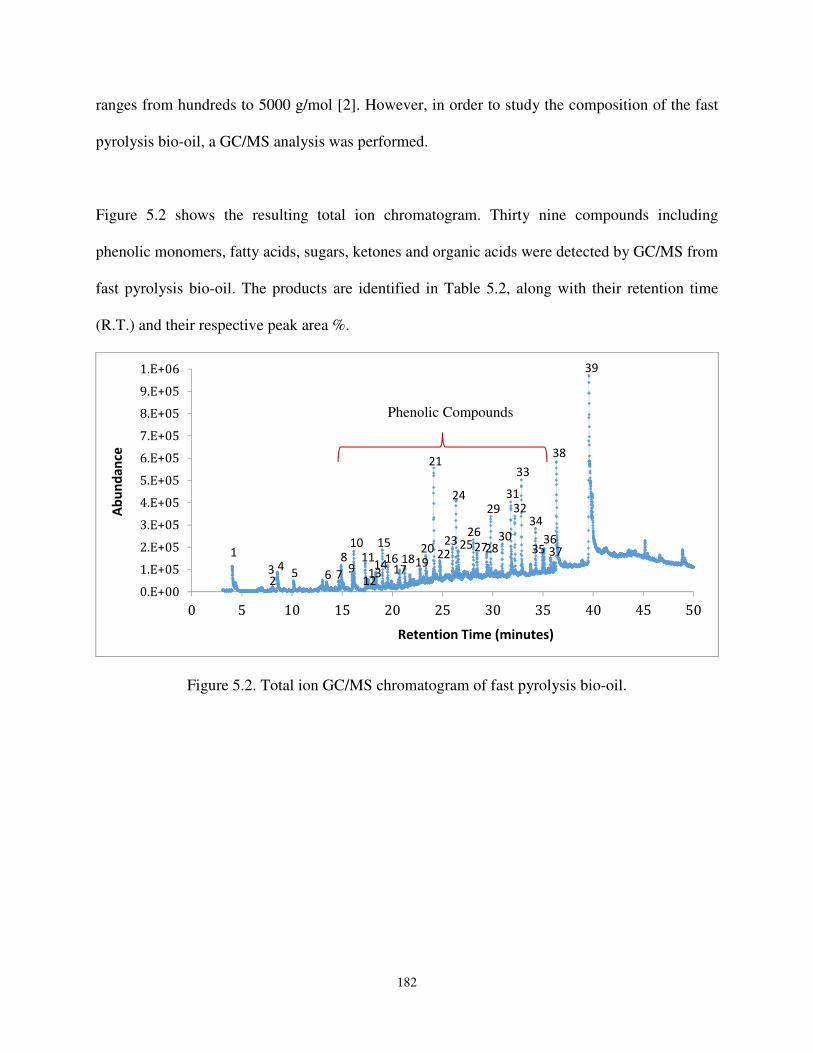
182
ranges from hundreds to 5000 g/mol [2]. However, in order to study the composition of the fast
pyrolysis bio-oil, a GC/MS analysis was performed.
Figure 5.2 shows the resulting total ion chromatogram. Thirty nine compounds including
phenolic monomers, fatty acids, sugars, ketones and organic acids were detected by GC/MS from
fast pyrolysis bio-oil. The products are identified in Table 5.2, along with their retention time
(R.T.) and their respective peak area %.
Figure 5.2. Total ion GC/MS chromatogram of fast pyrolysis bio-oil.
1
23 4
5 6 7
89
1011
12
1413
15
1617
181920
21
2223
24
2526
2728
29
30
3132
33
34
353637
38
39
0.E+00
1.E+05
2.E+05
3.E+05
4.E+05
5.E+05
6.E+05
7.E+05
8.E+05
9.E+05
1.E+06
0 5 10 15 20 25 30 35 40 45 50
Ab
un
da
nce
Retention Time (minutes)
Phenolic Compounds

183
Table 5.2. Major pyrolysis decomposition products identified by GS/MS
Peak Number Compound R.T. (min) Peak Area (%)
1 Acetic acid 4.402 3.36
2 Methoxycyclobutane 8.036 0.59
3 2-cyclopenten-1-one 8.494 0.58
4 Furfural 8.540 2.24
5 2-methyl-2-cyclopenten-1-one 10.159 0.87
6 4,5-dihydro-2-methyl-1H-imidazole 13.432 0.79
7 2,3- dimethyl-2-cyclopenten-1-one 14.662 0.68
8 3-methyl-1,2-cyclopentanedione 14.891 2.09
9 Phenol 15.996 1.56
10 2-methoxyphenol 16.156 2.65
11 2-methylphenol 17.289 1.65
12 2-methoxy-3-methylphenol 18.124 0.60
13 3-methylphenol 18.307 0.84
14 4-methylphenol 18.365 1.01
15 2-methoxy-4-methylphenol 19.000 2.32
16 2,4-dimethylphenol 19.520 1.09
17 5-methoxy-2,3-dimethylphenol 20.716 0.99
18 4-ethyl-2-methoxyphenol 21.231 1.15
19 m-methoxybenzamide 22.759 1.07
20 4-allyl-2-methoxyphenol 23.325 1.33
21 2,6-dimethoxyphenol 24.115 6.58
22 2-methoxy-4-[(1 Z)-1-propenyl]-phenol 24.727 1.56
23 2-methoxy-4-[(1 E)-1-propenyl)]-phenol 25.980 1.94
24 Dehydroacetic acid 26.335 3.96
25 4-Hydroxy-3-methoxybenzaldehyde 26.535 1.94
26 1,2,3-trimethoxy-5-methylbenzene 28.063 2.02
27 1-(3-hydroxy-4-methoxyphenyl)-ethanone 28.429 1.45
28 2,5-dimethoxy-4-ethylamphetamine 29.396 1.17
29 3-(4-hydroxy-3-methoxyphenyl)-2-propenoic acid 29.797 3.21
30 2-hydroxyimino-N-(4-methoxyphenyl)-acetamide 30.936 1.66
31 D-allose 31.800 5.20
32 2,6-dimethoxy-4-(2-propenyl-1)-phenol 32.206 2.88
33 4-hydroxy-3,5-dimethoxybenzaldehyde 32.847 5.71
34 1-(4-hydroxy-3,5-dimethoxyphenyl)-ethanone 34.254 2.49
35 4-Hydroxy-3-methoxycinnamaldehyde 34.975 1.24
36 1-(2,4,6-trihydroxy-3-methylphenyl)-1-butanone 35.113 1.39
37 2-methyl-9H-carbazole 35.759 0.70
38 Hexadecanoic acid 36.343 10.67
39 6-octadecenoic acid 39.582 16.78
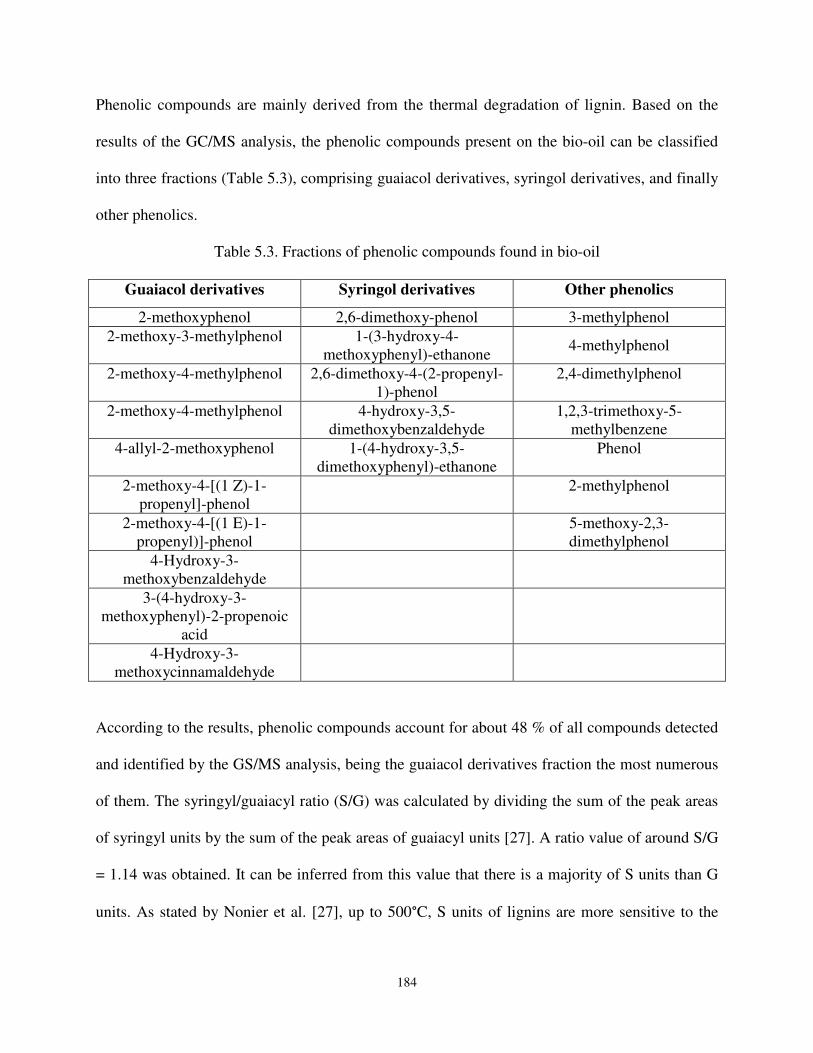
184
Phenolic compounds are mainly derived from the thermal degradation of lignin. Based on the
results of the GC/MS analysis, the phenolic compounds present on the bio-oil can be classified
into three fractions (Table 5.3), comprising guaiacol derivatives, syringol derivatives, and finally
other phenolics.
Table 5.3. Fractions of phenolic compounds found in bio-oil
Guaiacol derivatives Syringol derivatives Other phenolics
2-methoxyphenol 2,6-dimethoxy-phenol 3-methylphenol
2-methoxy-3-methylphenol 1-(3-hydroxy-4-methoxyphenyl)-ethanone
4-methylphenol
2-methoxy-4-methylphenol 2,6-dimethoxy-4-(2-propenyl-1)-phenol
2,4-dimethylphenol
2-methoxy-4-methylphenol 4-hydroxy-3,5-dimethoxybenzaldehyde
1,2,3-trimethoxy-5-methylbenzene
4-allyl-2-methoxyphenol 1-(4-hydroxy-3,5-dimethoxyphenyl)-ethanone
Phenol
2-methoxy-4-[(1 Z)-1-propenyl]-phenol
2-methylphenol
2-methoxy-4-[(1 E)-1-propenyl)]-phenol
5-methoxy-2,3-dimethylphenol
4-Hydroxy-3-methoxybenzaldehyde
3-(4-hydroxy-3-methoxyphenyl)-2-propenoic
acid
4-Hydroxy-3-methoxycinnamaldehyde
According to the results, phenolic compounds account for about 48 % of all compounds detected
and identified by the GS/MS analysis, being the guaiacol derivatives fraction the most numerous
of them. The syringyl/guaiacyl ratio (S/G) was calculated by dividing the sum of the peak areas
of syringyl units by the sum of the peak areas of guaiacyl units [27]. A ratio value of around S/G
= 1.14 was obtained. It can be inferred from this value that there is a majority of S units than G
units. As stated by Nonier et al. [27], up to 500°C, S units of lignins are more sensitive to the

185
pyrolysis reaction compared to G units, which require higher temperatures for thermal
decomposition. This behavior leads to mixtures of phenolic derivatives richer in syringyl
compounds. As previously stated, most of the phenolics present in the crude bio-oil are too high
in molecular weight to be quantified by GS analysis. They are present as oligomers containing
varying numbers of different organic functional groups.
3.2 Hydroxyl group analysis through 31
P-NMR spectroscopy.
31P-NMR method involves the phosphitylation of hydroxyl groups in a substrate using a 31P
reagent followed by quantitative 31P-NMR analysis. The type of -OH groups present in the bio-
oil was determined and quantitatively calculated using 31P-NMR. The results are shown in Table
5.4. According to the 31P-NMR analysis, the total hydroxyl number for freshly produced fast
pyrolysis bio-oil was (10.83 ± 1.08) mmol/g. It was found that aliphatic, phenolic and acidic
hydroxyl groups accounted for 49 %, 28 %, and 23 % of the total –OH number, respectively.
Aliphatic –OH are likely to arise as a result of the degradation of cellulose, hemicellulose and
lignin; whist the presence of phenolic –OH can be attributed to the cleavage of ether bonds in
linin during thermal decomposition.
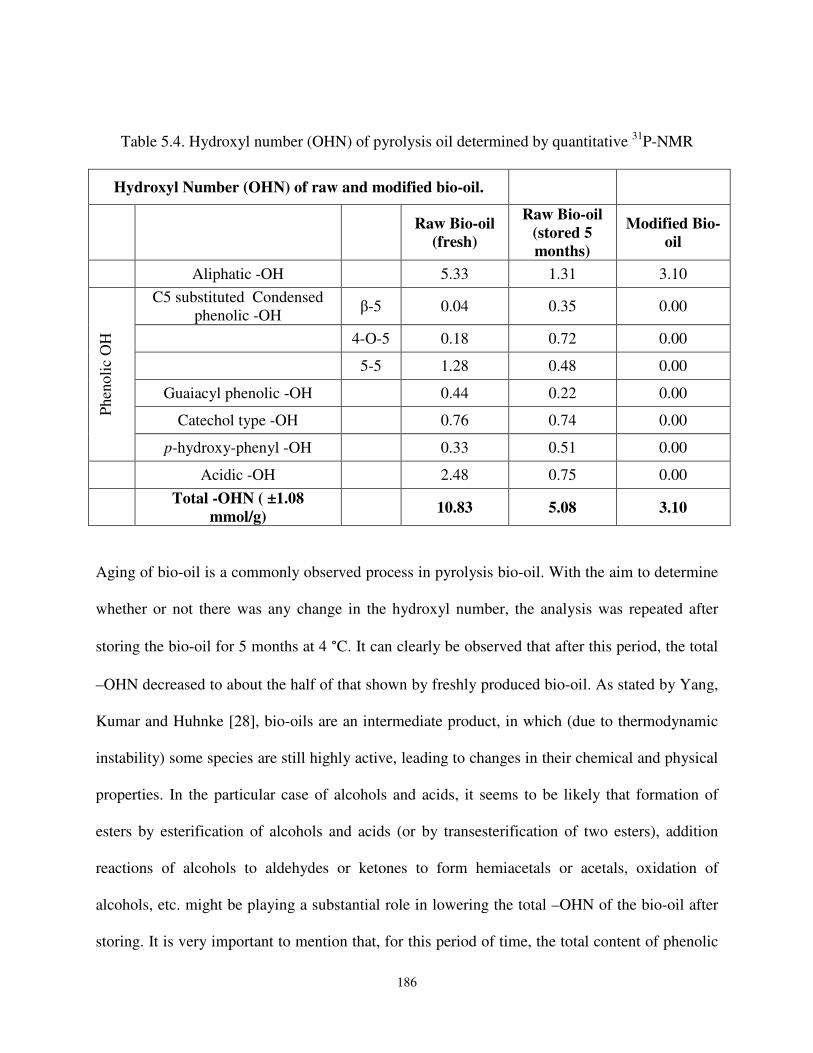
186
Table 5.4. Hydroxyl number (OHN) of pyrolysis oil determined by quantitative 31P-NMR
Hydroxyl Number (OHN) of raw and modified bio-oil.
Raw Bio-oil
(fresh)
Raw Bio-oil
(stored 5
months)
Modified Bio-
oil
Aliphatic -OH
5.33 1.31 3.10
Ph
eno
lic
OH
C5 substituted Condensed phenolic -OH
β-5 0.04 0.35 0.00
4-O-5 0.18 0.72 0.00
5-5 1.28 0.48 0.00
Guaiacyl phenolic -OH
0.44 0.22 0.00
Catechol type -OH
0.76 0.74 0.00
p-hydroxy-phenyl -OH
0.33 0.51 0.00
Acidic -OH
2.48 0.75 0.00
Total -OHN ( ±1.08
mmol/g) 10.83 5.08 3.10
Aging of bio-oil is a commonly observed process in pyrolysis bio-oil. With the aim to determine
whether or not there was any change in the hydroxyl number, the analysis was repeated after
storing the bio-oil for 5 months at 4 °C. It can clearly be observed that after this period, the total
–OHN decreased to about the half of that shown by freshly produced bio-oil. As stated by Yang,
Kumar and Huhnke [28], bio-oils are an intermediate product, in which (due to thermodynamic
instability) some species are still highly active, leading to changes in their chemical and physical
properties. In the particular case of alcohols and acids, it seems to be likely that formation of
esters by esterification of alcohols and acids (or by transesterification of two esters), addition
reactions of alcohols to aldehydes or ketones to form hemiacetals or acetals, oxidation of
alcohols, etc. might be playing a substantial role in lowering the total –OHN of the bio-oil after
storing. It is very important to mention that, for this period of time, the total content of phenolic

187
–OH remained invariant (around 3.03 mmol/g); suggesting that these compounds seemed to play
a secondary role in bio-oil aging.
3.3 General method for the glycidylation (functionalization with epoxy groups) of bio-oil.
3.3.1 Glycidylation of the α-resorcylic acid
In order to study the reaction between polyphenols and the epichlorohydrin, α-resorcylic acid
(containing two phenolic hydroxyl groups and one carboxylic acid moiety), was employed as a
representative model. For details about this reaction refer to Chapter III.
3.3.2 Glycidylation of the bio-oil.
Phenols can be converted into glycidyl ethers by reaction with 2,3-epoxyalkyl halides in alkaline
solution. The condensation of phenolic compounds with epichlorohydrin, catalyzed by bases, has
been the subject of study since long time ago [29]. In alkaline solutions, phenols exist as the
phenoxide ion. As stated by Auvergne et al. [30], the reaction between the strongly nucleophilic
phenoxide ions and epichlorohydrin can follow two competitive mechanisms: a one-step
nucleophilic substitution, in which the phenoxide ion attacks the halide and displaces a halide ion
(Clˉ in this case) through a SN2 mechanism (widely known as the Williamson synthesis of
ethers); and a two-step mechanism involving a nucleophilic attack of the epoxide (due to the ease
of opening of the strained three-membered ring) to form 1-aryloxy-3-chloropropan-2-ols. The
second step comprises the subsequent regeneration of the epoxide ring through intramolecular
cyclization of the previously formed 1-aryloxy-3-chloropropan-2-ols. From Tomita`s patent [31],
which covers and details the procedure to obtain epoxy resins derived from gallic acid and
tannins (by introducing glycidyl groups to carboxyl and phenols groups through reaction with

188
epichlorohydrin in alkaline media), it can be inferred that, under the disclosed experimental
conditions, the two-step reaction mechanism seems to be favored. In the first step, the addition of
epichlorohydrin to carboxyl and hydroxyl groups, at high temperature, takes place; and during
the second step of the process, the dehydrogenation of the halohydrin ether and ester moieties
obtained in the first step is achieved by adding an alkaline aqueous solution. It is also important
to highlight that the presence of a phase transfer catalyst is essential for the addition reaction in
the first step.
Since the main goal of this research lies in developing bio-based thermosetting materials, a
similar approach was used to functionalize the phenolic compounds found in the bio-oil. Once
the total –OHN was determined, the glycidylation reaction was performed. The following
scheme (Figure 5.3) depicts the reaction of a hypothetical lignin fragment with epichlorohydrin
in alkaline medium.
Figure 5.3. Glycidylation of bio-oil with epichlorohydrin in alkaline medium.
As previously detailed, most of the chemistry of phenols is similar to that of alcohols. Thus,
phenols can be converted into ethers by reaction with alkyl halides in the presence of a base.
Since the formation of the reactive phenoxide ion occurs more readily than the alkoxide ion, the
OH
O H
O H
O
CH3
O CH3
O
L
OH
OH
O
O
CH3
O CH3
O
L
OO
C l
1) 100 °C, 1h, BnEt
3NCl
2) NaOH (aq),
BnEt NCL = Lignin Fragments

189
acid phenolic –OH groups are the preferred target of the reaction. As seen in Table III, 31P-NMR
analysis of the modified pyrolysis oil revealed that the phenolic –OH groups were totally
removed. Titration with HBr showed that this compound has an average molar epoxy content of
(3.0 ± 0.1) x 10-3 mol per gram of resin, corresponding to an epoxy equivalent weight = 333 g/eq.
This value is approx. the half of the content shown by commercial epoxy resins, such as the well-
known EPON 828, having an average molar epoxy content of approx. 5.3 x 10-3 mol per gram of
resin, resulting in an epoxy equivalent weight = 185-192 g/eq. During purification and
recovering of the modified resin, around 40 wt% is recovered as epoxidized bio-oil. The content
of insoluble matter, mainly char, is around 30 wt% and the remaining 30 wt% is lost during
purification (water soluble compounds such as acids, low molecular weight alcohols and
carbonyl compounds, dehydrated sugars and other products of cellulose pyrolysis). This is also
reflected in the results of the 31P-NMR determination, where the highly hydrophilic acidic –OH
compounds (mainly acetic acid) are completely lost after modification and subsequent washing
and purification. Aliphatic –OH content increase is likely due to mass concentration, after
elimination of acid compounds and insoluble matter.
3.4 Fourier Transform Infrared Spectroscopy (FTIR)
With the aim to study the functional groups present in the product, FTIR was performed. The
results are shown in Figure 5.4. The FTIR spectrum of bio-oil was characterized by the presence
of a strong IR absorption band at 3370 cm-1 (due to the presence of hydroxyl groups from
alcohols, acids, and phenols); as well as an intense carbonyl strong peak at 1750 cm-1 (mainly
attributed to ketones, aldehydes and acids). In the case of the modified bio-oil, there is a decrease
in the intensity of the absorption band at 3370 cm-1, related to the removal of phenolic and acid -
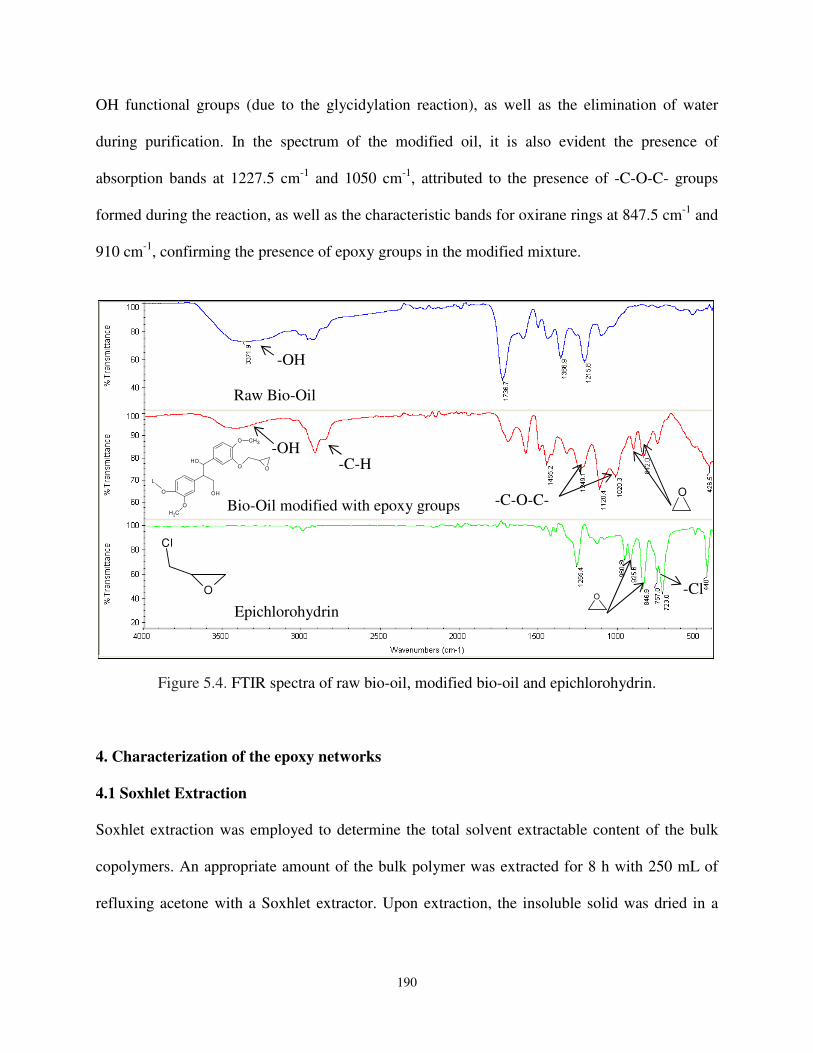
190
OH functional groups (due to the glycidylation reaction), as well as the elimination of water
during purification. In the spectrum of the modified oil, it is also evident the presence of
absorption bands at 1227.5 cm-1 and 1050 cm-1, attributed to the presence of -C-O-C- groups
formed during the reaction, as well as the characteristic bands for oxirane rings at 847.5 cm-1 and
910 cm-1, confirming the presence of epoxy groups in the modified mixture.
Figure 4. FTIR spectra of raw bio-oil, modified bio-oil and epichlorohydrin.
Figure 5.4. FTIR spectra of raw bio-oil, modified bio-oil and epichlorohydrin.
4. Characterization of the epoxy networks
4.1 Soxhlet Extraction
Soxhlet extraction was employed to determine the total solvent extractable content of the bulk
copolymers. An appropriate amount of the bulk polymer was extracted for 8 h with 250 mL of
refluxing acetone with a Soxhlet extractor. Upon extraction, the insoluble solid was dried in a
O
Cl
Epichlorohydrin
-Cl O
Bio-Oil modified with epoxy groups
Raw Bio-Oil
O
OH
OH
O
O
CH3
O CH3
O
L
O
-C-O-C-
-OH
-C-H -OH
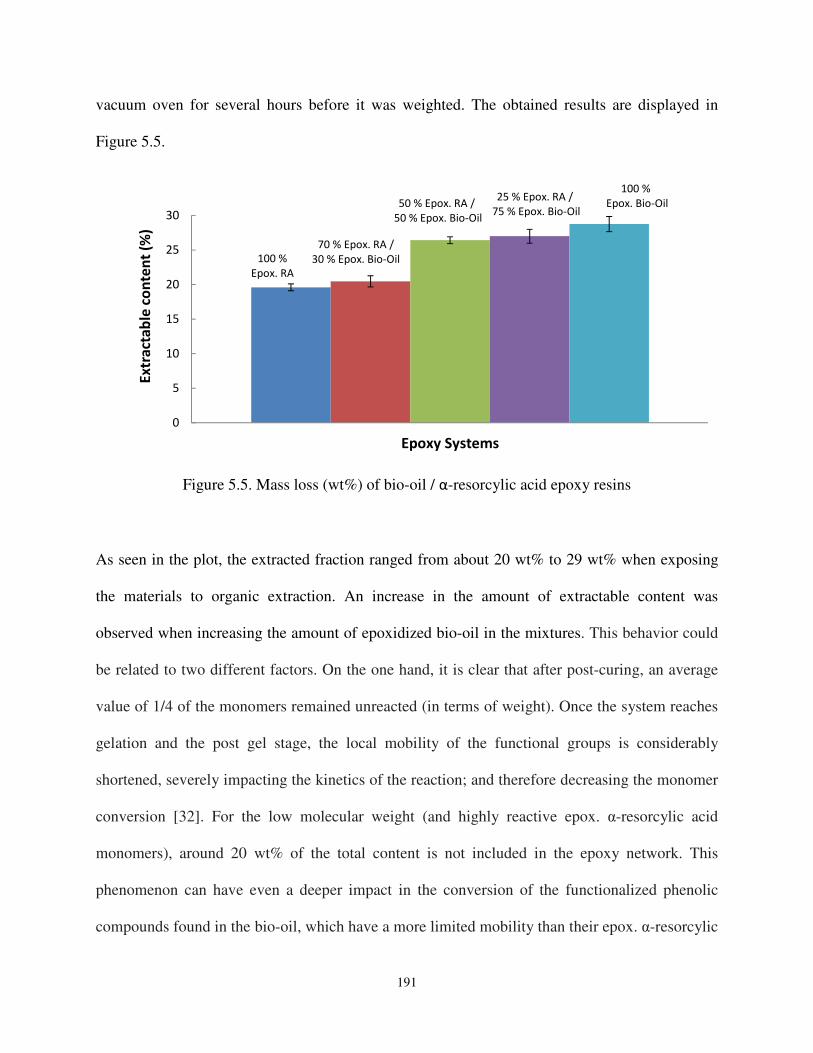
191
vacuum oven for several hours before it was weighted. The obtained results are displayed in
Figure 5.5.
Figure 5.5. Mass loss (wt%) of bio-oil / α-resorcylic acid epoxy resins
As seen in the plot, the extracted fraction ranged from about 20 wt% to 29 wt% when exposing
the materials to organic extraction. An increase in the amount of extractable content was
observed when increasing the amount of epoxidized bio-oil in the mixtures. This behavior could
be related to two different factors. On the one hand, it is clear that after post-curing, an average
value of 1/4 of the monomers remained unreacted (in terms of weight). Once the system reaches
gelation and the post gel stage, the local mobility of the functional groups is considerably
shortened, severely impacting the kinetics of the reaction; and therefore decreasing the monomer
conversion [32]. For the low molecular weight (and highly reactive epox. α-resorcylic acid
monomers), around 20 wt% of the total content is not included in the epoxy network. This
phenomenon can have even a deeper impact in the conversion of the functionalized phenolic
compounds found in the bio-oil, which have a more limited mobility than their epox. α-resorcylic
100 %
Epox. RA
70 % Epox. RA /
30 % Epox. Bio-Oil
50 % Epox. RA /
50 % Epox. Bio-Oil
25 % Epox. RA /
75 % Epox. Bio-Oil
100 %
Epox. Bio-Oil
0
5
10
15
20
25
30
Epoxy Systems
Ex
tra
cta
ble
co
nte
nt
(%)

192
acid counterpart (due to their larger size and higher molecular weight), leading to a lesser
incorporation of monomers into the growing network. On the other hand, as previously stated,
bio-oil consists is a complex mixture of a large number of pyrolysis compounds. After chemical
modification and subsequent purification, many non-functionalized compounds remain in the
polymerizable mixture. After curing, there is a population of compounds, not able to react,
showing chemical affinity for the acetone; and therefore, extractable, resulting in a higher
amount of soluble products when the proportion of bio-oil was increased in the mixtures.
4.2 Scanning Electron Microscopy (SEM)
Scanning electron microscopy was used to investigate the morphology of the epoxy systems.
Figure 5.6 provides the obtained images of the fracture surfaces of the bio-oil / α-RA materials,
pre-chilled in liquid nitrogen. As shown, the fracture surfaces of the materials containing
epoxidized α-resorcylic acid are characterized by smooth, glassy and homogeneous
microstructures; without any plastic deformation [33,34]. They exhibited surfaces with cracks
and different planes, indicating brittle fracture. As the proportion of this low molecular weight
co-monomer decreases, the brittle character of the materials also decreased, which is reflected in
the more ductile surface of the 100 wt% epoxidized bio-oil sample, which exhibits some degree
of plastic deformation. Likewise, globular cavities dispersed in the continuous epoxy matrix are
observed, probably due to evaporation of residual solvent used during the polymerization stage.

193
Figure 5.6. SEM micrographs of the 100 wt% epoxidized bio-oil (5.6.a), 50 wt% epoxidized bio-
oil – 50 wt% epoxidized α-RA (5.6.b), and 100 wt% epoxidized α-RA (5.6.c) resins.
5.6.a
5.6.b
5.6.c

194
4.3 Thermo-mechanical characterization of bio-oil-based epoxy thermosetting resin
With the aim of minimizing the use of commercial agents as a part of the epoxy resin
formulation, the polymeric materials were prepared by anionic polymerization initiated by
DMPA, in a ratio of 0.08 mol DMAP/mol epoxy. In order to prepare the resins, epoxidized bio-
oil and triglycidylated ether of α-resorcylic acid (as a low molecular weight reactive comonomer)
were mixed in different weight proportions. After curing, the polymeric materials appeared as
dark-brown stiff materials at room temperature. Dynamic mechanical analysis (DMA)
measurements are intensively used to investigate the amorphous phase transitions of polymers as
they are stressed under periodic deformation. Results are shown in Figure 5.7.
100 % Epox. RA
90% Epox. RA - 10%
Epox. Bio-Oil
70% Epox RA - 30%
Epox. Bio-oil
50% Epox. RA - 50%
Epox. Bio-oil
25% Epox. RA- 75%
Epox.Bio-Oil100% Epox. Bio-Oil
1.E+06
1.E+07
1.E+08
1.E+09
1.E+10
25 50 75 100 125 150 175 200 225 250
Sto
rag
e M
od
ulu
s (
Pa
)
Temperature (Celsius)
7-a
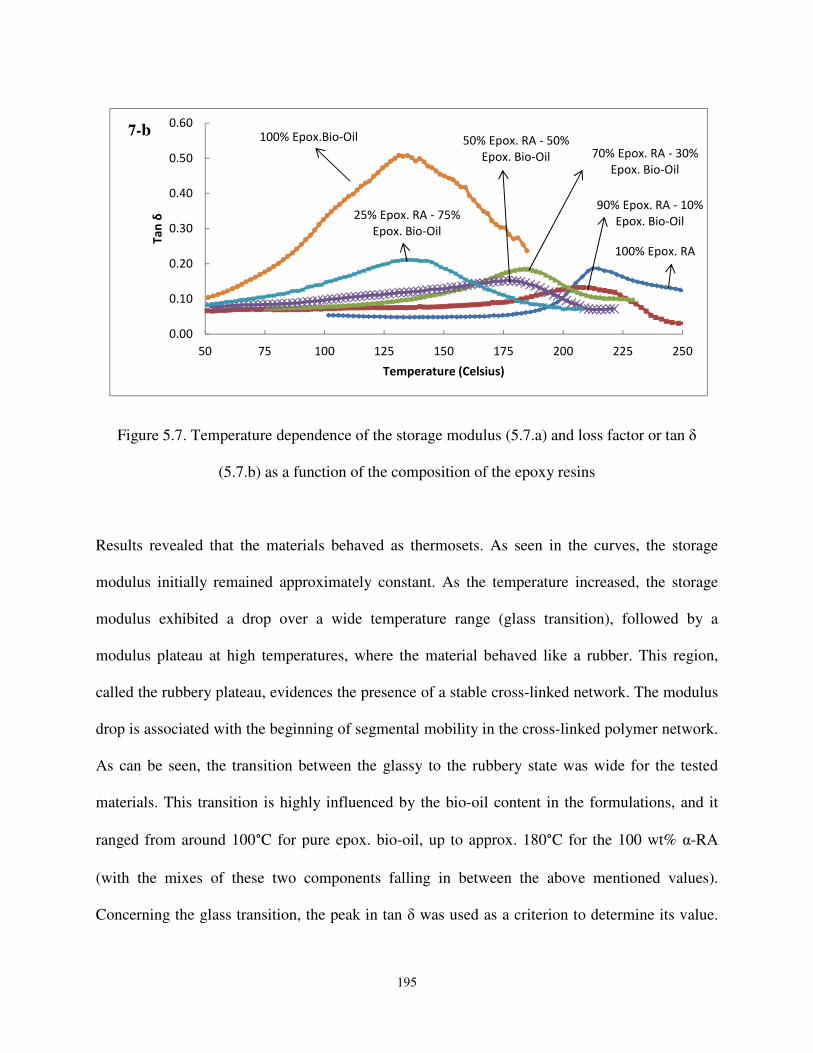
195
Figure 5.7. Temperature dependence of the storage modulus (5.7.a) and loss factor or tan δ
(5.7.b) as a function of the composition of the epoxy resins
Results revealed that the materials behaved as thermosets. As seen in the curves, the storage
modulus initially remained approximately constant. As the temperature increased, the storage
modulus exhibited a drop over a wide temperature range (glass transition), followed by a
modulus plateau at high temperatures, where the material behaved like a rubber. This region,
called the rubbery plateau, evidences the presence of a stable cross-linked network. The modulus
drop is associated with the beginning of segmental mobility in the cross-linked polymer network.
As can be seen, the transition between the glassy to the rubbery state was wide for the tested
materials. This transition is highly influenced by the bio-oil content in the formulations, and it
ranged from around 100°C for pure epox. bio-oil, up to approx. 180°C for the 100 wt% α-RA
(with the mixes of these two components falling in between the above mentioned values).
Concerning the glass transition, the peak in tan δ was used as a criterion to determine its value.
100% Epox. RA
90% Epox. RA - 10%
Epox. Bio-Oil
70% Epox. RA - 30%
Epox. Bio-Oil
50% Epox. RA - 50%
Epox. Bio-Oil
25% Epox. RA - 75%
Epox. Bio-Oil
100% Epox.Bio-Oil
0.00
0.10
0.20
0.30
0.40
0.50
0.60
50 75 100 125 150 175 200 225 250
Ta
n δ
Temperature (Celsius)
7-b

196
The results showed that the peaks shifted to lower temperatures (indicating a decrease in the
glass transition temperature), when the proportion of epoxidized bio-oil was increased. The peak
width at half height is a criterion used to indicate the interaction between the phases and
homogeneity of the amorphous phase. A higher value implies higher inhomogeneity of the
amorphous phase. In this particular case, the results showed that the tan δ peak became wider as
the bio-oil content in the matrix increased. This phenomenon could be associated to the presence
of less cross-linked and heterogeneous networks with wider molecular weight distribution of the
chains between crosslinking points [35]. Likewise, all the materials showed only one tan δ peak,
indicating that there was no phase separation. Table 5.5 summarizes the main properties of the
bio-oil-based epoxy thermosets.
Table 5.5. Thermo-mechanical properties of epoxidized bio-oil and the triglycidylated ether of α-
resorcylic acid (epoxidized α-resorcylic) resins
Epoxidized
bio-oil (wt%)
Triglycidylated ether
of α-resorcylic acid
(wt%)
E` at 30°C
(Pa)
Tg
(°C)
Density of active chains
(mol/m3)
100 0 1.48E+09 131.2 1006
75 25 1.58E+09 138.5 9169
50 50 2.18E+09 176.7 13702
30 70 2.32E+09 183.6 15238
10 90 2.74E+09 206.3 27150
0 100 3.78E+09 212.2 35709

197
As shown in Table 5.5, the storage modulus (E’) exhibited values in the order of GPa, indicating
that materials behave as glassy polymers at ambient conditions; and it considerably decreased
when the epoxidized bio-oil wt% content was increased. This parameter for glassy polymers
below the glass transition temperature is approx. constant, having a value of around 3 x 109 Pa
[36]. The decline in the modulus values is directly correlated to the decrease in the density of
active chains or n; parameter that is often associated to the crosslinking density of the material.
As the crosslinking density increases, the hardness, modulus, mechanical strength, and chemical
resistance increase [37]. For instance, slightly crosslinked materials, such as typical rubber
bands, have n = 190 mol/m3 [36]. When the same rubber is highly crosslinked, it generates a
brittle material. A common commercial diglycidyl ether of bisphenol A (DGEBA) type epoxy
resin (epoxy equivalent weight = 185-192 g/eq. [38]) used in coating, adhesive, structural, and
composite applications shows an approximate value of n = 5000 mol/m3 when crosslinked with a
tetrafunctional amine [39]. Conceptually, the higher the crosslinking density, the more
constrained the chain motion; and thus the stiffer and more thermally resistant the molecular
structure is. As seen in Table 5.5, the value of n increased approximately 37 times, when
comparing the pure epoxidized materials (100 wt% bio-oil and 100 wt% α-resorcylic acid).
When more α-RA is incorporated into the bio-oil, the n value increased, leading to more
crosslinked materials; which improved their thermo-mechanical properties, as evidenced by the
rise of the Tg and modulus. Such behavior could be related to the fact that the average epoxy
content of the epoxidized derivatives of the α-resorcylic acid is 3 times larger than the one shown
by the epox. bio-oil, leading to a higher capability to crosslink. Likewise, structural differences
between the epoxidized bio-oil monomers, as well as differences in the molecular weights, might
play a key role in the way they are incorporated into the network during polymerization.

198
The glass transition temperature was also highly influenced by the epox. bio-oil wt% content,
ranging from approx. 130 to 212 °C, showing its maximum value for the 100 α-resorcylic acid
wt%. The Fox equation was used to fit the experimental data. Figure 5.8 is a plot of 1/Tg versus
relative epoxy bio-oil content. This equation assumes uniform morphology; therefore, the good
agreement (R2 = 0.9654) with the bio-oil / α-resorcylic acid epoxy systems implies that the
networks are relatively homogeneous due to good miscibility between the components [40].
Figure 5.8. Relationship between 1/Tg and relative epoxy bio-oil weight content (Wbio-oil)
In order to compare the results obtained in this research, the obtained thermo-mechanical
properties of the 100 wt% epoxidized bio-oil and 100 wt% epoxidized α-RA were contrasted to
those shown by a commercial epoxy resin (Super Sap 100 Epoxy by Entropy Resins)
polymerized by both condensation with a commercial amine and by anionic polymerization with
DMPA. Table 5.6 summarizes the main properties these epoxy thermosets.
0.0000
0.0005
0.0010
0.0015
0.0020
0.0025
0.0030
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
1 /
Tg
(K-1
)
Epox. Bio-Oil Weight Fraction (Wbio-oil)
Fox Equation
Experimental Data
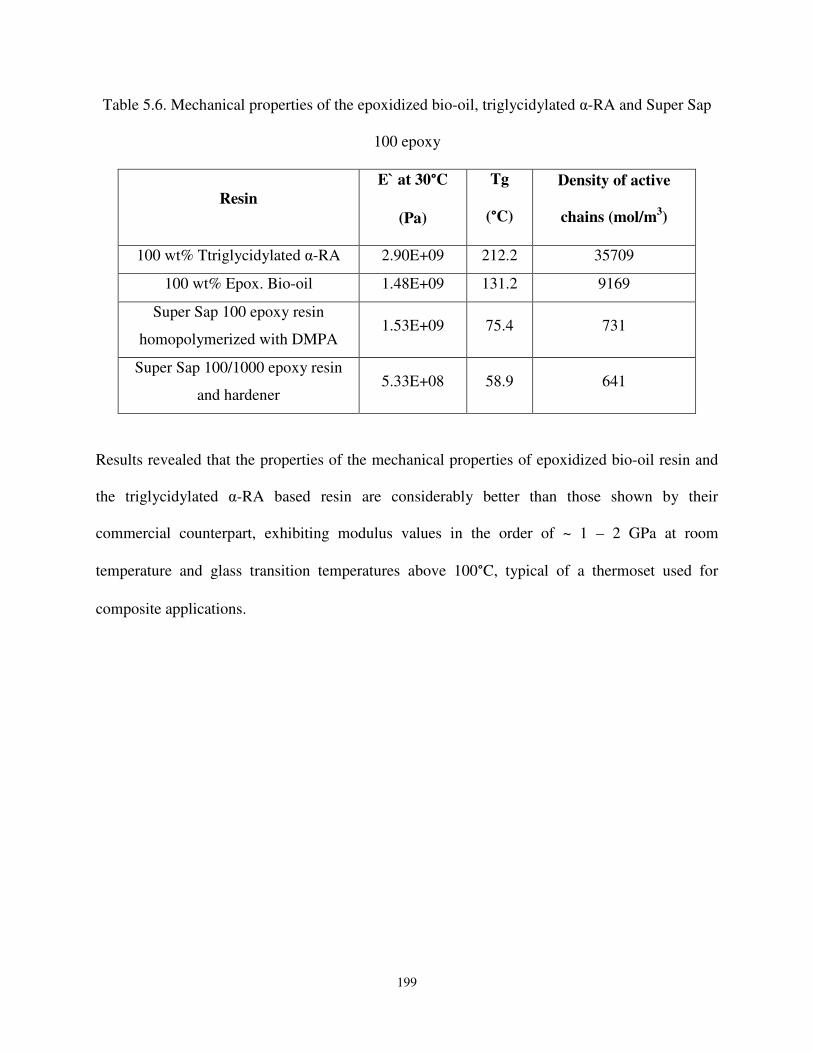
199
Table 5.6. Mechanical properties of the epoxidized bio-oil, triglycidylated α-RA and Super Sap
100 epoxy
Resin
E` at 30°C
(Pa)
Tg
(°C)
Density of active
chains (mol/m3)
100 wt% Ttriglycidylated α-RA 2.90E+09 212.2 35709
100 wt% Epox. Bio-oil 1.48E+09 131.2 9169
Super Sap 100 epoxy resin
homopolymerized with DMPA 1.53E+09 75.4 731
Super Sap 100/1000 epoxy resin
and hardener 5.33E+08 58.9 641
Results revealed that the properties of the mechanical properties of epoxidized bio-oil resin and
the triglycidylated α-RA based resin are considerably better than those shown by their
commercial counterpart, exhibiting modulus values in the order of ~ 1 – 2 GPa at room
temperature and glass transition temperatures above 100°C, typical of a thermoset used for
composite applications.

200
4. Conclusions
Bio-oil pyrolysis phenolic derivatives, as well as α-resorcylic acid, were chemically
functionalized with epoxy monomers through glycidylation with epichlorohydrin in alkaline
conditions. The phenolic compounds were identified by GS/MS, and the total hydroxyl number
(after and before chemical modification) was successfully calculated through 31P-NMR. The
epoxy content of the bio-oil upon chemical functionalization was measured by means of a
titration and found to be (3.0 ± 0.1) x 10-3 mol per gram of resin. Grafting of epoxy functions
onto the monomer`s structure was confirmed by FTIR. Testing showed an increase in the storage
modulus, from approx. 1.5 to 3.8 GPa at room temperature, when higher amounts of
triglycidylated ether of α-resorcylic acid were added to the mixtures. Glass transition
temperature appeared to be strongly dependent of the amounts of triglycidylated ether of α-
resorcylic acid when it was combined with the epoxidized bio-oil. When compared to samples of
commercial origin, testing showed the thermo-mechanical properties of the epoxidized bio-oil /
triglycidylated α-RA based resins are considerably higher to those shown by their commercial
counterpart. Based on the results of this study, it is clear that these materials behave as glassy
polymers at room conditions; and they could be used as thermosetting matrices for the
development of composite materials. Likewise, they have potential as partial replacements in the
formulation of commercial epoxy resins used as coatings, adhesives, etc. This study opens the
possibility of using these kinds of pyrolysis bio-oils as renewable precursors for polymer
synthesis.

201
References
[1] R. D. Perlack, L. L Wright, A. F. Turhollow, R. L. Graham, Biomass as a feedstock for a
bionergy and bioproducts industry: the technical feasibility of a billion-ton annual supply,
U.S. Department of Agriculture. (2005) 1–5.
[2] A. Effendi, H. Gerhauser, A. V. Bridgwater, Production of renewable phenolic resins by
thermochemical conversion of biomass: A review, Renew. Sustain. Energy Rev. 12 (2008)
2092–2116. doi:10.1016/j.rser.2007.04.008.
[3] D. Mohan, C.U. Pittman, P.H. Steele, Pyrolysis of Wood / Biomass for Bio-oil : A Critical
Review, Energy & Fuesl. 20 (2006) 848–889. doi:10.1021/ef0502397.
[4] A. Gandini, T.M. Lacerda, From monomers to polymers from renewable resources:
Recent advances, Prog. Polym. Sci. (2015). doi:10.1016/j.progpolymsci.2014.11.002.
[5] M. Fache, A. Viola, R. Auvergne, B. Boutevin, S. Caillol, Biobased epoxy thermosets
from vanillin-derived oligomers, Eur. Polym. J. 68 (2015) 526–535.
doi:10.1016/j.eurpolymj.2015.03.048.
[6] A. Gandini, M.N. Belgacem, Lignins as components of macromolecular materials, in
M.N. Belgacem, A. Gandini (EDs.), Monomers, Polymers and Composites, Elsevier, UK,
2008, pp. 243-272.
[7] W.G. Lora, J.H. Glasser, Recent application of lignin: A sustainable alternative to
nonrenewable materials., J. Polym. Environ. 10 (2002) 39–48.
[8] S. Laurichesse, L. Avérous, Chemical modification of lignins: Towards biobased
polymers, Prog. Polym. Sci. 39 (2014) 1266–1290.
doi:10.1016/j.progpolymsci.2013.11.004.

202
[9] H. Hatakeyama, T. Hatakeyama, Lignin structure, propeeties and applications, in A. Abe,
K. Dušek, S. Kobayashi (EDs.), Biopolymers Lignin, Proteins, Bioactive
Nanocomposites, Springer, Heidelberg, 2012, pp. 29–55.
[10] J.E. Thomas, A. Milne, N. Soltys, Direct mass-spectrometric of carbonaceous fuels studies
of the pyrolysis of lignin, J. Anal. Appl. Pyrolysis. 9 (1986) 207–236.
[11] D.K. Shen, S. Gu, K.H. Luo, S.R. Wang, M.X. Fang, The pyrolytic degradation of wood-
derived lignin from pulping process, Bioresour. Technol. 101 (2010) 6136–6146.
doi:10.1016/j.biortech.2010.02.078.
[12] J.-S. Kim, Production, separation and applications of phenolic-rich bio-oil – A review,
Bioresour. Technol. 178 (2015) 90–98. doi:10.1016/j.biortech.2014.08.121.
[13] M. Balat, M. Balat, E. Kirtay, H. Balat, Main routes for the thermo-conversion of biomass
into fuels and chemicals. Part 1: Pyrolysis systems, Energy Convers. Manag. 50 (2009)
3147–3157. doi:10.1016/j.enconman.2009.08.014.
[14] M. Staš, D. Kubička, J. Chudoba, M. Pospíšil, Overview of analytical methods used for
chemical characterization of pyrolysis bio-oil, Energy and Fuels. 28 (2014) 385–402.
doi:10.1021/ef402047y.
[15] S. Xiu, A. Shahbazi, Bio-oil production and upgrading research: A review, Renew.
Sustain. Energy Rev. 16 (2012) 4406–4414. doi:10.1016/j.rser.2012.04.028.
[16] Entropy Super Sap 100/1000 Epoxy Resin, (n.d.).
http://www.jamestowndistributors.com/userportal/show_product.do?pid=63933 (accessed
March 30, 2016).

203
[17] C. Aouf, S. Benyahya, A. Esnouf, S. Caillol, B. Boutevin, H. Fulcrand, Tara tannins as
phenolic precursors of thermosetting epoxy resins, Eur. Polym. J. 55 (2014) 186–198.
doi:10.1016/j.eurpolymj.2014.03.034.
[18] S. Benyahya, C. Aouf, S. Caillol, B. Boutevin, J.P. Pascault, H. Fulcrand, Functionalized
green tea tannins as phenolic prepolymers for bio-based epoxy resins, Ind. Crops Prod. 53
(2014) 296–307. doi:10.1016/j.indcrop.2013.12.045.
[19] S. Thangalazhy-Gopakumar, S. Adhikari, H. Ravindran, R.B. Gupta, O. Fasina, M. Tu, et
al., Physiochemical properties of bio-oil produced at various temperatures from pine wood
using an auger reactor, Bioresour. Technol. 101 (2010) 8389–8395.
doi:10.1016/j.biortech.2010.05.040.
[20] Y. Pu, S. Cao, A.J. Ragauskas, Application of quantitative 31P NMR in biomass lignin
and biofuel precursors characterization, Energy Environ. Sci. 4 (2011) 3154.
doi:10.1039/c1ee01201k.
[21] K. David, M. Kosa, A. Williams, R. Mayor, M. Realff, J. Muzzy, et al., P-NMR analysis
of bio-oils obtained from the pyrolysis of biomass, Biofuels. 1 (2010) 839–845.
doi:10.4155/bfs.10.57.
[22] X. Pan, P. Sengupta, D.C. Webster, Novel biobased epoxy compounds: epoxidized
sucrose esters of fatty acids, Green Chem. 13 (2011) 965. doi:10.1039/c0gc00882f.
[23] R. Sengupta, S. Chakraborty, S. Bandyopadhyay, S. Dasgupta, R. Mukhopadhyay, K.
Auddy, et al., A Short Review on Rubber / Clay Nanocomposites With Emphasis on
Mechanical Properties, Engineering. 47 (2007) 21–25. doi:10.1002/pen.
[24] W.S. Precision, E.S. Waste, T. Edition, Standard Practice for Extraction of Solid Waste
Samples for Chemical Analysis, 93 (2008) 1–7. doi:10.1520/D5369-93R08E01.2.

204
[25] M. Fache, R. Auvergne, B. Boutevin, S. Caillol, New vanillin-derived diepoxy monomers
for the synthesis of biobased thermosets, Eur. Polym. J. 67 (2015) 527–538.
doi:10.1016/j.eurpolymj.2014.10.011.
[26] F. Hu, J.J. La Scala, J.M. Sadler, G.R. Palmese, Synthesis and characterization of
thermosetting furan-based epoxy systems, Macromolecules. 47 (2014) 3332–3342.
doi:10.1021/ma500687t.
[27] M.F. Nonier, N. Vivas, N. Vivas de Gaulejac, C. Absalon, P. Soulié, E. Fouquet,
Pyrolysis–gas chromatography/mass spectrometry of Quercus sp. wood, J. Anal. Appl.
Pyrolysis. 75 (2006) 181–193. doi:10.1016/j.jaap.2005.05.006.
[28] Z. Yang, A. Kumar, R.L. Huhnke, Review of recent developments to improve storage and
transportation stability of bio-oil, Renew. Sustain. Energy Rev. 50 (2015) 859–870.
doi:10.1016/j.rser.2015.05.025.
[29] W. Bradley, J. Forrest, O. Stephenson, The catalysed transfer of hydrogen chloride from
chlorohydrins to epoxides. A new method of preparing glycidol and some of its
derivatives, J. Chem. Soc. 1589 (1971) 1589–1598. doi:10.1039/JR9510001589.
[30] R. Auvergne, S. Caillol, G. David, B. Boutevin, J.P. Pascault, Biobased thermosetting
epoxy: Present and future, Chem. Rev. 114 (2014) 1082–1115. doi:10.1021/cr3001274.
[31] K. Yonezawa, K. Kaisha, F. Application, P. Data, P.E.S. Milestone, 4,540,802, (1985) 2–
7.
[32] J.P. Pascault, R.J.J. Williams, General Concepts about Epoxy Polymers, Epoxy Polym.
New Mater. Innov. (2010) 1–12. doi:10.1002/9783527628704.ch1.

205
[33] S. Ananda kumar, Z. Denchev, M. Alagar, Synthesis and thermal characterization of
phosphorus containing siliconized epoxy resins, Eur. Polym. J. 42 (2006) 2419–2429.
doi:10.1016/j.eurpolymj.2006.06.010.
[34] S. Gurusideswar, R. Velmurugan, N.K. Gupta, High strain rate sensitivity of epoxy/clay
nanocomposites using non-contact strain measurement, Polymer (Guildf). 86 (2016) 197–
207. doi:10.1016/j.polymer.2015.12.054.
[35] K.C. Manikandan Nair, S. Thomas, G. Groeninckx, Thermal and dynamic mechanical
analysis of polystyrene composites reinforced with short sisal fibres, Compos. Sci.
Technol. 61 (2001) 2519–2529. doi:10.1016/S0266-3538(01)00170-1.
[36] L.H. Sperling, Introduction to Physical Polymer Science, fourth ed., Wiley Interscience,
New Jersey, 2006.
[37] D. Ratna, Handbook of thermoset resins, 2009. doi:10.1002/0471743984.vse3011.pub2.
[38] Epon® Resin 828, (n.d.). http://www.polysciences.com/default/catalog-
products/monomers-polymers/polymers/epoxy-resins/eponsupsup-resin-828/ (accessed
March 30, 2016).
[39] Z.S. Pour, M. Ghaemy, Thermo-mechanical behaviors of epoxy resins reinforced with
silane-epoxide functionalized ??-Fe2O3 nanoparticles, Prog. Org. Coatings. 77 (2014)
1316–1324. doi:10.1016/j.porgcoat.2014.04.001.
[40] A.M. Peterson, R.E. Jensen, G.R. Palmese, Room-temperature healing of a thermosetting
polymer using the diels-alder reaction, ACS Appl. Mater. Interfaces. 2 (2010) 1141–1149.
doi:10.1021/am9009378.

206
General conclusions
• Thermosetting polymers where obtained from biomass derived materials such as
vegetable oils, and terpenes, as well as from biorefinery products. The obtained resins
could be employed as partial replacements of thermosetting commercial resins, and/or in
the formulation of composite materials.
• From the experimental results, it was clear that the chemical modification of different
functional groups of biomass derived materials considerably improved the thermo-
mechanical performance of the obtained polymers, when compared to their non-modified
counterparts.
• Studies of the polymerization reactions revealed that the homopolymerization of epoxy
resins using anionic chemical initiators stands as a viable option to generate materials
with higher contributions of biomass-derived compounds per weight basis
• The vast thermo-mechanical analysis of the resulting thermosets revealed the deep
existing link between the thermo-mechanical response of the materials and their chemical
structure. Stiffer materials were obtained when the density of aromatic units were
increased in the crosslinked networks. Likewise, properties were also dependent on the
chemical nature and position of the polymerizable functional groups.
• A significant chemical analysis, both quantitative and qualitative, has been devoted to the
study of pyrolysis bio-oil of lignocellulosic biomass as a source of chemicals for the
synthesis of thermosetting resins.

207
• Overall results indicated that the materials presented thermo-mechanical behaviors
similar to those shown by commercial formulations.
Conclusions highlighting the major achievements of Chapter II:
• Thermo-mechanical analysis revealed that the cationic copolymerization of tung oil and
terpenes in presence of boron trifluoride yielded elastomeric materials at room
temperature. For all the studied comonomers, suitable materials for analysis were not
obtained for terpene proportions above 60 wt%.
• The obtained copolymers presented a maximum in their thermo-mechanical properties,
showing a modulus of approx. 38 MPa at room temperature and a glass transition
temperature of around 30 °C. The Tg was found to be a function of both the type and
comonomer weight content.
• Studies revealed that the properties of non-chemically modified vegetable oils can change
as a function of two different effects of the fatty acid chains, either by plasticizing the
network because of their flexible structure, or by increasing the rigidity due to the large
number of unsaturations per molecule.
• Some of the thermo-mechanical properties were found to be predictable by simple
mathematical approaches and models developed for polyvinyl resins.

208
Conclusions highlighting the major achievements of Chapter III:
• Thermo-mechanical tests indicated that the chemical incorporation of polymerizable
groups onto the structure of the vegetable oils considerably improved their mechanical
performance. The maximum glass transition obtained in Chapter II was doubled, and the
modulus increased by a fold of approximately 37 times.
• The inclusion of α-resorcylic acid as a low molecular weight comonomer increased the
chain stiffness and therefore improved the thermo-mechanical behavior, rendering resins
with properties matching those shown by glassy thermosetting commercial resins.
• When polymerizing the epoxidized derivatives of linseed oil and α-resorcylic acid,
thermodynamical incompatibility was found for oil proportions higher than 30 wt%. A
new method to improve the mixture of these two materials is exposed in Chapter IV.
• Studies also revealed that the chain-growth copolymerization allowed increasing the
contribution of renewable materials into the formulations, by minimizing the use of
commercial crosslinking compounds.

209
Conclusions highlighting the major achievements of Chapter IV:
• Results revealed that interpenetrating of the system containing an epoxy phase derived
from α-resorcylic acid, and an acrylate phase derived from vegetable oil arises as an
approach to not only improve the properties by including an aromatic monomer in the
system; but also to minimize the phase separation phenomena by kinetically controlling
this thermodynamical process by permanent interlocking of entangled chains, avoiding
phase separation.
• Thermo-mechanical characterization showed that the obtained materials behave as
thermoset amorphous polymers, and that both the modulus and glass transition are
extremely dependent on the epoxy/acrylate weight ratio, as well as on the chemical
structure of the epoxy monomers.
• Modulus values ranged from 0.7 to 3.3 GPa at 30 °C, and glass transition temperatures
ranged from around 58 °C to approx. 130 °C. No synergistic effect on these two
properties was observed.
• Interpenetrating networks containing equivalent weight proportions of the parent resins
showed the highest fracture toughness of the series, exhibiting a KIc value of around 2.1
MPa·m1/2. The results showed that the KIc values did not increase as Mc increased, which
seems to suggest that a different mechanism is responsible for the increase in the fracture
toughness displayed by IPNs. Also, there seems to be an exponential type increase in the
fracture energy with the Mc1/2
for the materials containing the epoxy phase

210
Conclusions highlighting the major achievements of Chapter V:
• Biomass refinery stands as a sustainable way for aromatics biosourcing, by providing low
molecular weight fragments of natural lignin, whose functional groups are more exposed
to the reaction medium (due to their lesser steric crowding), and thus being readily
reachable during chemical synthesis.
• According to the results, phenolic compounds account for about 48 % of all compounds
detected and identified by the GS/MS analysis, being the guaiacol derivatives fraction the
most numerous of them.
• According to the 31P-NMR analysis, the total hydroxyl number for freshly produced fast
pyrolysis bio-oil was (10.83 ± 1.08) mmol/g. It was found that aliphatic, phenolic and
acidic hydroxyl groups accounted for 49 %, 28 %, and 23 % of the total –OH number,
respectively.
• Testing showed an increase in the storage modulus, from approx. 1.5 to 3.8 GPa at room
temperature, when higher amounts of triglycidylated ether of α-resorcylic acid were
added to the mixtures. Glass transition temperature appeared to be strongly dependent of
the amounts of triglycidylated ether of α-resorcylic acid when it was combined with the
epoxidized bio-oil. 100 wt% epoxidized bio-oil was characterized by a glass transition of
approx. 130 °C.

211
Future Work
A considerable part of this document deals with the formulation of thermosetting materials,
especially with epoxy resins. Nature provides many bio-based feedstocks such as vegetable oils
and polyphenols to obtain bio-based epoxy resins; and research and studies have focused on the
formulation of epoxidized products, but no crosslinking or curing agents. It is essential that all
components of the formulation are both renewable and sustainable. Therefore, it is of great
importance to develop curing agents from renewable resources rather than petroleum-derived
materials for producing the next generation of bio-based epoxy formulations. There is a
considerable potential to perform research on this area, which can catapult bio-base epoxy resin
to the commercial markets, especially on amine hardeners, which production remains as a
challenge.
As explained in the general introduction, composite materials will be a sector highly influenced
by the evolution in the polymer and materials industry. Therefore, more research needs to be
performed concerning the application of new bio-based derived materials in the design and
formulation of composite materials.
Bio-based chemicals are already industrially available but most of them are aliphatic or
cycloaliphatic regarding their chemical structure. However, as stated in this document, many key
chemical compounds are aromatic, which in our current times are ultimately derived from
petroleum. Thus, there is a challenge for finding aromatic building blocks derived from biomass.

212
In nature, aromatics are mostly found in lignins; however some crucial aspects need to be
improved if these products want to be taken to the commercialization stage, such as:
• Due to the inherited variability of the chemical composition of lignocellulosic materials,
as well as pretreatment techniques, pyrolysis products and their composition are highly
variable, making hard the comparison of results and approaches to convert them into
added-value products. The field needs standard protocols for the extraction procedures,
pretreatments, catalysts, analysis techniques and procedures, so as to make results and
approaches possibly comparable.
• Further fundamental understanding of both the structure and the chemistry of the
products of fast pyrolysis of biomass is required for assessment of their reactivity under
different reaction conditions. This will allow extracting meaningful and valuable
information for the structure-property-reactivity relations of the obtained materials.
• In the case of pyrolysis techniques, more experimental work is required to determine
optimal operation conditions, such as catalysts, catalysts deactivations, temperature,
pressure to control product yield. Moreover, purification and separation of the chemical
compounds present in pyrolysis bio-oils remains as a major challenge. Adequate isolation
of certain products or fractions can vastly improve the analysis and study of their
structure and reactivity.

213
• Catalysis stands up as a key technology for biomass conversion, especially for the
valorization of lignin and its derivatives. Development of more efficient catalytic
technologies, increasing the selectivity of the conversion toward the obtention of
particular products or family of products remains as a challenge.
• Particularly for aromatic bio-sourcing, pyrolysis techniques should be applied directly to
lignins. In this way, more selective and richer fractions in aromatic can be obtained,
eliminating the influence and presence of decomposition products of the hemicellulose
and cellulose.





























