Embed Size (px)
Citation preview

of May 4, 2018.This information is current as
Fibrosis in MicePromotes Bleomycin-Induced PulmonaryChemokines by Respiratory Epithelial Cells TLR2-Mediated Production of IL-27 and
Hyun ChungHye Sung Kim, Heounjeong Go, Shizuo Akira and Doo
http://www.jimmunol.org/content/187/8/4007doi: 10.4049/jimmunol.1101654September 2011;
2011; 187:4007-4017; Prepublished online 19J Immunol
MaterialSupplementary
4.DC1http://www.jimmunol.org/content/suppl/2011/09/16/jimmunol.110165
Referenceshttp://www.jimmunol.org/content/187/8/4007.full#ref-list-1
, 16 of which you can access for free at: cites 45 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2011 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
TLR2-Mediated Production of IL-27 and Chemokines byRespiratory Epithelial Cells Promotes Bleomycin-InducedPulmonary Fibrosis in Mice
Hye Sung Kim,*,† Heounjeong Go,* Shizuo Akira,‡ and Doo Hyun Chung*,†
Idiopathic pulmonary fibrosis is a fatal disease characterized by progressive destruction of the lung. Although TLR2 bridges innate
and adaptive immunity by sensing tissue damage, its role in pulmonary fibrosis remains unclear. To address this issue, TLR22/2 and
WT mice were examined for bleomycin-induced pulmonary fibrosis (BIPF). Flow cytometric and immunohistochemical analysis
revealed that TLR2 expression in bronchial epithelial and immune cells of the lungs was upregulated in WT mice during BIPF.
Levels of IL-27, TGF-b, chemokines, and hydroxyproline were lower in lungs of TLR22/2 mice than in those of WT mice, but IL-
17 levels were higher in TLR22/2 mice. In in vivo experiments using bone marrow-chimeric mice, TLR2 expression on respiratory
epithelial cells, rather than immune cells, induced IL-27 and chemokine production in the lungs, further stimulating BIPF. This
effect of TLR2 depended on IRF complexes and MyD88. BIPF was more severe in IL-17A2/2 mice and in TLR22/2 mice treated
with anti–IL-17 mAb than in TLR22/2 and WT mice. Furthermore, IL-27 blockade in WT mice reduced hydroxyproline levels by
enhancing IL-17 production, whereas the treatment of TLR22/2 mice with a chemokine mixture increased hydroxyproline levels
by recruiting inflammatory cells into the lungs. TLR2 signaling promotes BIPF by inducing IL-27 and chemokine production by
respiratory epithelial cells, thereby inhibiting IL-17 production and recruiting inflammatory cells into the lungs. The Journal of
Immunology, 2011, 187: 4007–4017.
Idiopathic pulmonary fibrosis is a progressive and fatal diseasecharacterized by fibrosis-induced destruction of the lungparenchyma. The fibrosis, which is initiated by inflammation
consequent to lung tissue injury, induces the massive production ofcollagen in the alveolar septa and ultimately destroys pulmonaryfunction (1, 2). During pulmonary fibrosis, various immune cellsinfiltrate the lung tissues, which have a complicated cytokinenetwork that regulates inflammation and fibrosis (3). Th1-typecytokines, such as IFN-g, prevent fibroblast activation and pul-monary fibrosis (4, 5), whereas Th2-type cytokines, such as IL-4and IL-13, promote these processes (6, 7). Mice deficient in my-eloid differentiation factor 88 (MyD88), an adaptor protein in thesignal transduction pathway mediated by IL-1 and TLRs, exhibita profound defect in the activation of Ag-specific Th1 immuneresponses, but not in Th2 immune responses, suggesting that TLRsignaling has a critical role in balancing Th1/Th2 responses (8).
The TLRs are a family of conserved innate immune recep-tors that function in the first line of defense against pathogensby recognizing microorganism-associated molecular pattern (9).
These receptors also sense tissue injury by recognizing damage-associated molecular patterns of endogenous ligands generated
during inflammation and cell damage (10). Therefore, TLRs hasbeen proposed to regulate pulmonary fibrosis by sensing alveolar
epithelial cell damage. Consistent with this suggestion, severalstudies have shown that TLRs expressed in lung tissues regulatepulmonary fibrosis (11–14).TLR2, a TLR expressed on the surface of various types of
cells, recognizes conserved structure motif, including lipoteichoicacid (LTA), found on Gram-positive bacteria (15). Stimulationof respiratory epithelial (RE) cells with TNF-a, IFN-g, or corti-costeroid significantly increases the expression of TLR2 on themembranes of these cells (16–18), suggesting that TLR2 expres-sion on RE cells might be regulated by tissue damage–mediatedalteration to the cytokine milieu of the lungs. However, the func-tional role of TLR2 on RE cells has not been investigated inpulmonary fibrosis. Recently, Yang et al. (11) demonstrated thatbleomycin-induced inflammation and fibrosis are attenuated inTLR2-deficient mice and in wild type (WT) mice treated withTLR2-neutralizing mAb, suggesting that TLR2 promotesbleomycin-induced pulmonary fibrosis (BIPF). However, in theircomparison of the immune response phenotypes of WT andTLR2-targeted mice during BIPF, they did not examine the mech-anistic details of TLR2-mediated regulation of BIPF.In this study, we sought to identify the lung cell type with
a critical role in TLR2-mediated regulation of bleomycin-inducedtissue injury and to examine the mechanism by which TLR2promotes pulmonary fibrosis. Our results demonstrate that TLR2-mediated signaling induces the production of IL-27 and chemo-kines by RE cells, rather than immune cells, and that these effectsenhance BIPF by inhibiting IL-17 production and stimulating therecruitment of inflammatory cells into the lungs.
*Department of Pathology, Seoul National University College of Medicine, Seoul110-799, Korea; †Laboratory of Immune Regulation, Department of BiomedicalSciences, Seoul National University College of Medicine, Seoul 110-799, Korea; and‡Laboratory of Host Defense, World Premier International Immunology FrontierResearch Center, Osaka University, Osaka 565-0871, Japan
Received for publication June 7, 2011. Accepted for publication August 10, 2011.
This work was supported by a National Research Foundation of Korea grant fundedby the Korean government (Ministry of Education, Science and Technology; 2010-0017890) and Seoul National University Hospital (03-2009-0040).
Address correspondence and reprint requests to Dr. Doo Hyun Chung, Department ofPathology and Laboratory of Immune Regulation, Department of Biomedical Scien-ces, Seoul National University College of Medicine, 103 Daehak-ro, Jongno-gu,Seoul 110-799, Korea. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: BALF, bronchoalveolar lavage fluid; BIPF,bleomycin-induced pulmonary fibrosis; BM, bone marrow; IRF, IFN regulatory fac-tor; LTA, lipoteichoic acid; MyD88, differentiation factor 88; RE, respiratory epithe-lial; Treg, CD4
+CD25+Foxp3+ regulatory T; WT, wild type.
Copyright� 2011 by The American Association of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1101654
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Materials and MethodsMice
C57BL/6 (B6) mice were purchased from the Orient Company (Seoul,Korea) and Jackson Laboratory (Bar Harbor, ME). TLR22/2 mice werea gift from Shizuo Akira (Osaka University, Osaka, Japan). IL-17A2/2
mice (C57BL/6 background) were obtained from Y. Iwakura (University ofTokyo, Tokyo, Japan). Mice were bred and maintained in a specificpathogen-free environment at the Clinical Research Institute of SeoulNational University Hospital (accredited by the Association for Assess-ment and Accreditation of Laboratory Animal Care International) or theCenter of Animal Resource Development of Seoul National UniversityCollege of Medicine. All procedures involving animals were approved bythe Institutional Animal Care and Use Committee of the Clinical ResearchInstitute of Seoul National University Hospital.
Induction of pulmonary fibrosis using bleomycin
BIPF was induced by the instillation of a 50-ml bolus of bleomycin (NipponKayaku, Tokyo, Japan) into the trachea of young mice (8–10 wk old). Forexperiments using non-bone marrow (BM)-chimeric mice and for survivalstudies with BM-chimeric mice, bleomycin was dissolved in sterile PBS toproduce a total dose of 2 mg bleomycin per kilogram body weight. Forother experiments using BM-chimeric mice, the bleomycin dose was 1 mg/kg body weight. Mice were anesthetized with 2,2,2-tribromomethanol(Sigma-Aldrich, St. Louis, MO) before dosing with bleomycin. Seven, 14,or 21 d after the administration of bleomycin, the mice were sacrificed, andthe bronchoalveolar lavage fluid (BALF) and lung tissues were examined.
Bronchoalveolar lavage
After the mice were sacrificed, a 1-ml syringe containing 1 ml cold PBS wasinserted into the exposed trachea. The PBS was injected, aspirated back intothe syringe, and aside 4˚C. This processed was then repeated four times,and the five individual samples were pooled. After the pooled sampleswere centrifuged at 2000 rpm for 10 min at 4˚C, the supernatant fractionswere used for cytokine measurements. The pellets were reserved for thedetermination of total cell numbers and for flow cytometric analysis.
Histologic examination and immunohistochemistry
To examine lung tissues for histologic alterations, whole lungs were fixed in10% formalin, embedded in paraffin, and sectioned onto slides. The slideswere stained with H&E and scored for the extent of pathology on a scale of0 to 5, where 0 was defined as no lung abnormality, and 1, 2, 3, 4, and 5were defined as the presence of inflammation involving 10%, 10–30%, 30–50%, 50–80%, or .80% of the lungs, respectively.
For immunohistochemistry, the lung sections were deparaffinized withxylene for 20 min and thoroughly dehydrated with serially diluted ethanol.Immunohistochemical staining was performed using an Envision kit (Dako,Ely, U.K.) containing a peroxidase-conjugated anti-mouse Ig polymer. AmAbagainst TLR2 (1:100 dilution; Novusbio, Littleton, CO) was used as primaryAb. To visualize the reaction, diaminobenzidine was used for 5 min.
Treatment of mice with TLR2 agonists in vivo
Five micrograms of LTA (Sigma-Aldrich) or Pam3csk4 (Invivogen, SanDiego, CA) was injected i.p. into the mice immediately after bleomycininstillation.
Treatment of cultured cells with TLR2 agonists, recombinantIL-27, MyD88-inhibitory peptide, and concanavalin A
For in vitro experiments, cells in culture were treated with 1 mg LTA or 100ng Pam3csk4 for 24 h. MyD88 signaling was inhibited by the addition ofan MyD88-inhibitory peptide or control peptide (both from Invivogen) intothe culture medium at 20 mM. Cells in BALF obtained from mice sacri-ficed 7 d after bleomycin injection were incubated with concanavalin A (1mg/ml) or recombinant mouse IL-27 (100 ng/ml; Prospec-Tany, Rehovot,Israel), or both, for 24 h.
Quantitation of hydroxyproline in the lungs
Whole lungs were obtained 21 d after bleomycin injection, homogenized in2 ml PBS, and dried in a vacuum dryer for 6 h. The dried powder for eachlung was mixed with 1 ml 6 N HCl, incubated overnight at 110˚C, andfiltered through a 45-mm syringe filter (Millipore, Bedford, MA). A 50-mlaliquot of the filtered sample was mixed with 50 ml citrate-acetate bufferand 1 ml chloramine T solution for 15 min at room temperature. Next, 1 mlEhrlich’s solution was added, and the mixture was incubated for 15 min at65˚C to allow the conversion of pyrrole to a red chromogen. This chro-
mogen was detected spectrophotometrically at 550 nm used as a measureof the amount of hydroxyproline in the lung. All reagent used in thepreparation of citrate-acetate buffer prepared as 5 g citric acid, 1.2 mlglacial acetic acid, 7.24 g sodium acetate, 3.4 g NaOH, and 98 ml dis-tilled water and Ehrlich’s solution prepared as 4.5-g r-dimethylamino-benzaldehyde, 18.6 ml n-propanol, and 7.8 ml 70% perchloric acid werepurchased from Sigma-Aldrich, with the exception of n-propanol, whichwas purchased from Junsei Chemical (Tokyo, Japan).
Real-time PCR quantitation of cytokine expression
Total RNA was isolated from mouse lungs using an RNeasy Mini Kit(Qiagen, Hilden, Germany) according to the manufacturer’s instructions.RNA yield and purity were determined using a NanoDrop spectropho-tometer (NanoDrop Technologies, Wilmington, DE). The total RNA wasreverse-transcribed into cDNA and amplified with MMLV-RT Taq poly-merase (Promega, Madison, WI). Gene-specific PCR was performed usingprimers and probes from Applied Biosystems (Foster City, CA) and fol-lowed using an Applied Biosystems 7500 Sequence Detection System. Theresults for each cytokine were normalized to that for GAPDH expression inthe sample. The following TaqMan assay reagents from Applied Bio-systems were used: for GAPDH, 4352339E; for TLR2, Mm00442346_m1; for IL-6, Mm99999064_m1; for IL-17, Mm00439619_m1; forIL-23, Mm00518984_m1; for IL-27, Mm00461164_m1; for MIP-1a,Mm 99999057_m1; for MCP-1, Mm 99999056_m1; for IP-10, Mm00445235_m1; and for RANTES, Mm 01302428_m1.
ELISAs
To quantitate cytokines and chemokines, ELISAs were performed using kitsfrom BD Pharmingen (for MCP-1, IL-4, IFN-g, IL-10, and TGF-b; SanDiego, CA) and R&D Systems (for all others; Minneapolis, MN)according to the manufacturer’s instructions. Colorimetric reactions wereneutralized using 3 N HCl, and the sample absorbance at 450 and 570 nmwas measured on a Biotek Powerwave HT 96/384 Microplate reader(BioTek Instruments, Colmar, France).
Isolation of epithelial cells from mouse lung
For isolation of alveolar RE cells, lungs were perfused via the pulmonarycirculation with sterile PBS, lavaged three times with PBS, and then filled (2ml per lung) via the airway with RPMI 1640 with 2.5% FBS (HyCloneLaboratories, Logan, UT), 80 U elastase (Worthington Biochemical, LorneDiagnostics; Reading, U.K.), and 0.05 mg/ml trypsin (Sigma-Aldrich).After 20 min incubation at 37˚C, lungs were homogenized in a 7-ml ho-mogenizer (Wheaton Scientific, Millville, NJ). After the homogenate wascentrifuged at 2000 rpm, the supernatant fraction containing the cell sus-pension was layered on top of a sterile, discontinuous Percoll (GEHealthcare, Little Chalfont, U.K.) gradient and centrifuged for 20 min at4˚C and 1500 rpm. The cells at the interface between Percoll layers wereremoved and cultured in plates coated with anti-Fc receptor mAb (BDPharmingen) at 37˚C for 20 min. Next, the nonadherent cells were re-moved, centrifuged, and resuspended in DMEM containing with 10% FBSand 1% penicillin-streptomycin (Life Technologies BRL, Paisley, U.K.).
To determine the purity of the isolated RE cells, the cells were stainedwith PE-conjugated anti-mouse pan-cytokeratin mAb (Abcam, Cambridge,U.K.) and fluorescein isothiocyanate-conjugated anti-mouse FcgIII/II re-ceptor mAb (BD Pharmingen). Flow cytometric analysis of the stainedcells established the purity of the RE cell preparation as .90%.
Western blot analysis
Mice were sacrificed 7 d after bleomycin administration with or withoutLTA or Pam3csk4 injection. To estimate the level of IRF1, IRF3, and IRF7gene transcription, the proteins were eluted from the cells using extrac-tion reagent (GenDepot, Barker, TX) and analyzed by Western blotting,as described previously (19), with rabbit anti-mouse IFN regulatory fac-tors (IRFs) 1, 3, or 7 (all from Cell Signaling Technology, Beverly, MA)or an mAb against b-actin (control; Sigma-Aldrich). Appropriate HRP-conjugated secondary Abs were then applied, and the detected proteinswere visualized using an LAS-4000 Mini imaging system (Fujifilm, Tokyo,Japan).
Intracellular staining and flow cytometric analysis
For flow cytometric analysis, cells were incubated with an mAb for 15 minon ice to block FcgRII/III, washed, and stained in a 150-ml reactionmixture containing 0.2 mg of an mAb mixture containing mAbs specificfor TLR2, F4/80, Gr-1(Ly6C+Ly6G+), CD11c, CD4, CD8, and gd-TCR(BD Bioscience or eBioscience, San Diego, CA). CD1d tetramers wereobtained from the Tetramer Core Facility of the National Institutes of
4008 TLR2-MEDIATED IL-27 AND CHEMOKINES PROMOTE PULMONARY FIBROSIS
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Health (Bethesda, MD) and used for detection of NKT cells. After the cellswere incubated with the Abs for 15 min at 4˚C, they were washed twicewith PBS and analyzed by flow cytometry on a BD FACSCalibur in-strument (Becton Dickinson, Mountain View, CA).
For intracellular cytokine staining, cells were collected from BALFobtained 7 d after bleomycin administration, incubated with concanava-lin A (1 mg/ml; Sigma-Aldrich) for 24 h at 37˚C, surface-stained withmAbs specific for CD4, CD25, or gd-TCR, fixed, and permeabilizedwith Cytofix/Cytoperm (BD Biosciences or Miltenyi Biotec, Bergisch-Gladbach, Germany) according to the manufacturer’s instructions. Anti-mouse IL-17 and anti-mouse Foxp3 Abs were purchased from BD Bio-sciences and Miltenyi Biotec, respectively.
Generation of BM-chimeric mice
Chimeric mice were generated by adoptive transfer of donor BM cells intoirradiated recipient WT or TLR22/2 mice. BM cells were obtained byflushing the tibias and femurs of donor mice and then depleted of CD4+
and CD8+ T cells using a magnetic bead separation kit (Miltenyi Biotec)according to the manufacturer’s protocol. T cell-depleted BM cells (13106 cells per mouse) were injected into the tail veins of recipient micepretreated with a lethal dose (800 Gy) of radiation. Bleomycin was ad-ministered to the chimeric mice 8–12 wk after BM cell transplantation.
Neutralization of IL-17 and IL-27 in vivo and injection ofchemokine mixture
To neutralize IL-17 and IL-27 in vivo, mice were injected i.p. with anti-mouse IL-17 mAb (100 mg per mouse) or anti–IL-27 mAb (200 mg permouse), respectively, immediately after bleomycin administration. Mono-clonal rat IgG2A and goat IgG were used as negative controls for anti–IL-17 and anti–IL-27 mAb, respectively. The anti-mouse IL-17 and IL-27mAbs, rat IgG2A, and goat IgG were obtained from R&D Systems. Forexamination of the functional effects of chemokines in vivo, mice wereinjected i.p. with a mixture of recombinant mouse MIP-1a, MCP-1, andIP-10 (100 mg per mouse; all from R&D Systems) after bleomycin ad-ministration.
Depletion of CD4+CD25+ regulatory T cells in mice
To deplete CD4+CD25+Foxp3+ regulatory T (Treg) cells, WT and TLR22/2
mice were injected i.v. with 100 mg anti-CD25 mAb (3C7) or rat IgG2b,kisotype control (both from BD Pharmingen, San Diego, CA) 48 h beforebleomycin administration.
Statistical analysis
Statistical analysis was performed using the Prism 4.0 program (GraphPadSoftware, San Diego, CA). Unpaired t tests were used for two-group com-parisons. Paired one-way ANOVA tests were used for multigroup compar-isons. Differences were considered statistically significant at p , 0.05.
ResultsBIPF is attenuated in TLR2-deficient mice
To investigate whether TLR2 is involved in the development ofpulmonary fibrosis, we examined the effects of intratracheally in-stilled bleomycin on pulmonary inflammation and fibrosis inWTandTLR22/2 mice. Kinetic analysis of BALF revealed that the numberof cells in the lungs of WT mice reached a peak 1 wk after bleo-mycin administration and then gradually decreased, and it wasgreater than the number of cells in TLR22/2 mouse lungs (Fig. 1A).Subset analysis of BALF obtained 7 d after bleomycin admin-
istration revealed that the numbers of T, B, NK, and NKT cells, F4/80+ macrophages, CD11c+ dendritic cells, and Gr-1+ granulocyteswere all higher in BALF from WT mice than in BALF fromTLR22/2 mice (Fig. 1B). Histologic examination of WT mouselungs 7 d after bleomycin injection demonstrated multifocal fi-brotic pulmonary lesions with thickened alveolar septa, collapsedalveolar spaces, and massive infiltration of immune cells. Incontrast, inflammation and structural alteration in the lungs weredecreased in TLR22/2 mice (Fig. 1C, 1D). The amount of
FIGURE 1. TLR2-mediated signals promote BIPF. Lung and BALF were obtained from WT or TLR22/2 mice 7, 14, and 21 d after the intratracheal
instillation of bleomycin (BLM) or PBS (vehicle control) and analyzed for signs of BIPF. A, BALF was obtained from WT and TLR22/2 mice on days 7,
14, and 21, and the number of cells in the BALF was determined. B, Subset analysis of BALF cells obtained from WT and TLR22/2 mice on day 7. C,
Histologic examination of WT and TLR22/2 mouse lungs removed on day 7. H&E-stained paraffin sections are shown. Original magnification 3100. D,
The extent of inflammation and structural alteration in the pulmonary lesion was scored. E, Hydroxyproline content of lungs removed from WTand TLR22/2
mice on day 21. F and G, Effect of TLR2 agonists on BALF cell numbers (F) and hydroxyproline content (G) in WT and TLR22/2 mice. Mice were
injected i.p. with LTA or Pam3csk4 before BLM or PBS administration. F, BALF cell numbers were estimated on days 7, 14, and 21. G, The hydrox-
yproline content was determined on day 21. A–G, Data shown represent means 6 SEM for one experiment (n = 4 per group in A, B, F, and G; n = 3 per
group in C and D; n = 6 per group in E) of three independent experiments. *p , 0.05, **p , 0.01, ***p , 0.001. ns, not significant.
The Journal of Immunology 4009
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
hydroxyproline, a collagen component, was significantly lower inTLR22/2 mouse lungs than in WT mouse lungs 21 d after bleo-mycin injection (Fig. 1E). Furthermore, treatment of mice withTLR2 agonists, such as LTA or Pam3csk4, during BIPF increasedthe number of cells and the hydroxyproline level in the lungs ofWT mice (Fig. 1F, 1G) but not those of TLR22/2 mice. Thus,BIPF is attenuated in TLR2-deficient mice compared with WTmice, indicating that TLR2 promotes BIPF.
TLR2 expression on nonhematopoietic cells, rather thanimmune cells, of the lung contributes to BIPF
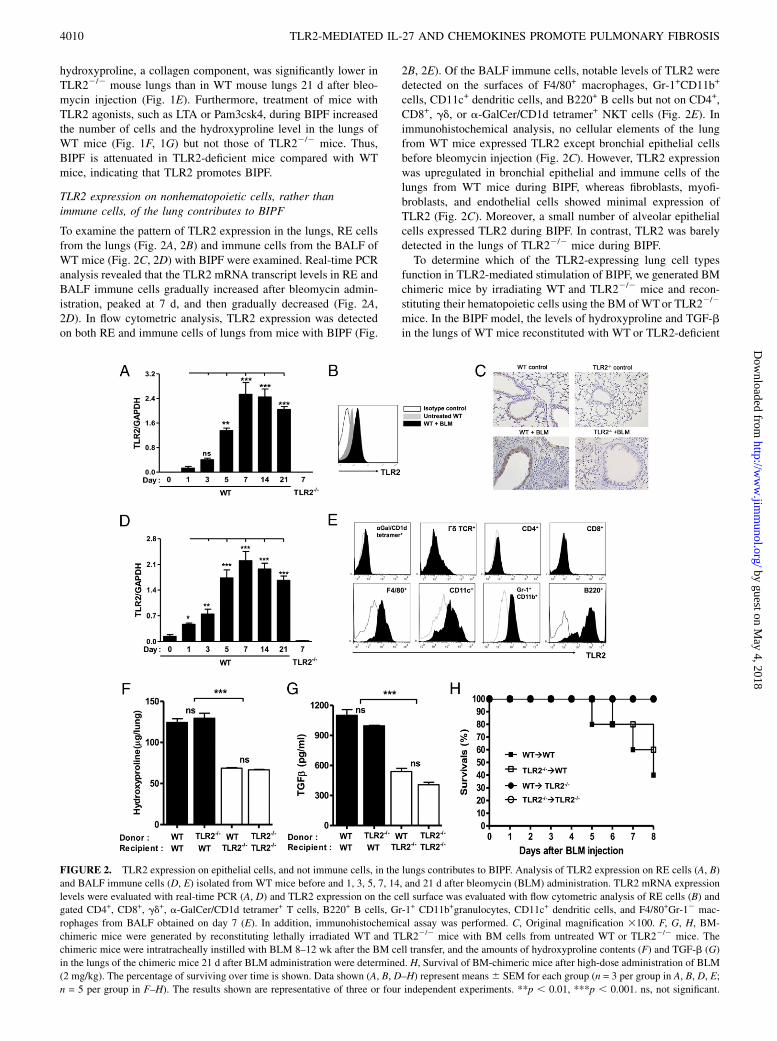
To examine the pattern of TLR2 expression in the lungs, RE cellsfrom the lungs (Fig. 2A, 2B) and immune cells from the BALF ofWT mice (Fig. 2C, 2D) with BIPF were examined. Real-time PCRanalysis revealed that the TLR2 mRNA transcript levels in RE andBALF immune cells gradually increased after bleomycin admin-istration, peaked at 7 d, and then gradually decreased (Fig. 2A,2D). In flow cytometric analysis, TLR2 expression was detectedon both RE and immune cells of lungs from mice with BIPF (Fig.
2B, 2E). Of the BALF immune cells, notable levels of TLR2 weredetected on the surfaces of F4/80+ macrophages, Gr-1+CD11b+
cells, CD11c+ dendritic cells, and B220+ B cells but not on CD4+,CD8+, gd, or a-GalCer/CD1d tetramer+ NKT cells (Fig. 2E). Inimmunohistochemical analysis, no cellular elements of the lungfrom WT mice expressed TLR2 except bronchial epithelial cellsbefore bleomycin injection (Fig. 2C). However, TLR2 expressionwas upregulated in bronchial epithelial and immune cells of thelungs from WT mice during BIPF, whereas fibroblasts, myofi-broblasts, and endothelial cells showed minimal expression ofTLR2 (Fig. 2C). Moreover, a small number of alveolar epithelialcells expressed TLR2 during BIPF. In contrast, TLR2 was barelydetected in the lungs of TLR22/2 mice during BIPF.To determine which of the TLR2-expressing lung cell types
function in TLR2-mediated stimulation of BIPF, we generated BMchimeric mice by irradiating WT and TLR22/2 mice and recon-stituting their hematopoietic cells using the BM of WTor TLR22/2
mice. In the BIPF model, the levels of hydroxyproline and TGF-bin the lungs of WT mice reconstituted with WT or TLR2-deficient
FIGURE 2. TLR2 expression on epithelial cells, and not immune cells, in the lungs contributes to BIPF. Analysis of TLR2 expression on RE cells (A, B)
and BALF immune cells (D, E) isolated from WT mice before and 1, 3, 5, 7, 14, and 21 d after bleomycin (BLM) administration. TLR2 mRNA expression
levels were evaluated with real-time PCR (A, D) and TLR2 expression on the cell surface was evaluated with flow cytometric analysis of RE cells (B) and
gated CD4+, CD8+, gd+, a-GalCer/CD1d tetramer+ T cells, B220+ B cells, Gr-1+ CD11b+granulocytes, CD11c+ dendritic cells, and F4/80+Gr-12 mac-
rophages from BALF obtained on day 7 (E). In addition, immunohistochemical assay was performed. C, Original magnification 3100. F, G, H, BM-
chimeric mice were generated by reconstituting lethally irradiated WT and TLR22/2 mice with BM cells from untreated WT or TLR22/2 mice. The
chimeric mice were intratracheally instilled with BLM 8–12 wk after the BM cell transfer, and the amounts of hydroxyproline contents (F) and TGF-b (G)
in the lungs of the chimeric mice 21 d after BLM administration were determined. H, Survival of BM-chimeric mice after high-dose administration of BLM
(2 mg/kg). The percentage of surviving over time is shown. Data shown (A, B, D–H) represent means6 SEM for each group (n = 3 per group in A, B, D, E;
n = 5 per group in F–H). The results shown are representative of three or four independent experiments. **p , 0.01, ***p , 0.001. ns, not significant.
4010 TLR2-MEDIATED IL-27 AND CHEMOKINES PROMOTE PULMONARY FIBROSIS
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
BM (WT→WT and TLR22/2→WT chimeras) were higher thanthose of TLR22/2 mice treated with WT or TLR2-deficient BM(WT→TLR22/2 and TLR22/2→TLR22/2 chimeras; Fig. 2F,2G). Whereas high-dose administration of bleomycin killed halfof the WT→WT and TLR22/2→WT chimeric mice within 8 d,all WT→TLR22/2 and TLR22/2→TLR22/2 chimeric miceremained alive at 8 d (Fig. 2H). These findings indicate that TLR2expression on nonhematopoietic cells, rather than immune cells,of the lungs exacerbates BIPF.
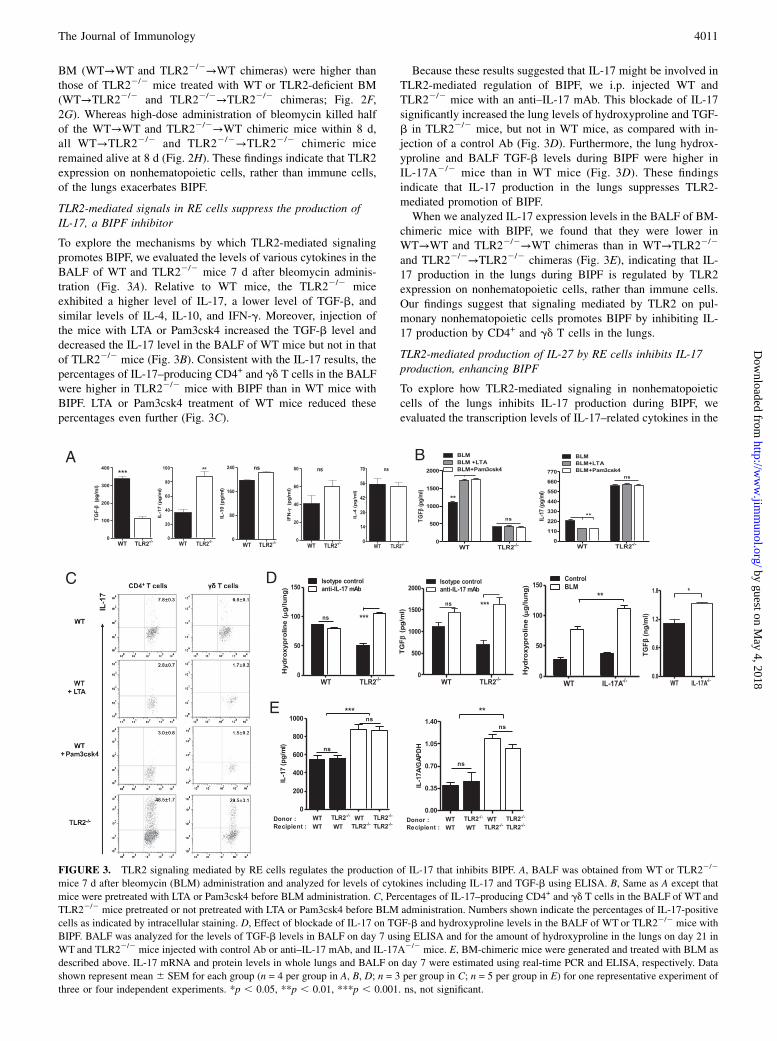
TLR2-mediated signals in RE cells suppress the production ofIL-17, a BIPF inhibitor
To explore the mechanisms by which TLR2-mediated signalingpromotes BIPF, we evaluated the levels of various cytokines in theBALF of WT and TLR22/2 mice 7 d after bleomycin adminis-tration (Fig. 3A). Relative to WT mice, the TLR22/2 miceexhibited a higher level of IL-17, a lower level of TGF-b, andsimilar levels of IL-4, IL-10, and IFN-g. Moreover, injection ofthe mice with LTA or Pam3csk4 increased the TGF-b level anddecreased the IL-17 level in the BALF of WT mice but not in thatof TLR22/2 mice (Fig. 3B). Consistent with the IL-17 results, thepercentages of IL-17–producing CD4+ and gd T cells in the BALFwere higher in TLR22/2 mice with BIPF than in WT mice withBIPF. LTA or Pam3csk4 treatment of WT mice reduced thesepercentages even further (Fig. 3C).
Because these results suggested that IL-17 might be involved inTLR2-mediated regulation of BIPF, we i.p. injected WT andTLR22/2 mice with an anti–IL-17 mAb. This blockade of IL-17significantly increased the lung levels of hydroxyproline and TGF-b in TLR22/2 mice, but not in WT mice, as compared with in-jection of a control Ab (Fig. 3D). Furthermore, the lung hydrox-yproline and BALF TGF-b levels during BIPF were higher inIL-17A2/2 mice than in WT mice (Fig. 3D). These findingsindicate that IL-17 production in the lungs suppresses TLR2-mediated promotion of BIPF.When we analyzed IL-17 expression levels in the BALF of BM-
chimeric mice with BIPF, we found that they were lower inWT→WT and TLR22/2→WT chimeras than in WT→TLR22/2
and TLR22/2→TLR22/2 chimeras (Fig. 3E), indicating that IL-17 production in the lungs during BIPF is regulated by TLR2expression on nonhematopoietic cells, rather than immune cells.Our findings suggest that signaling mediated by TLR2 on pul-monary nonhematopoietic cells promotes BIPF by inhibiting IL-17 production by CD4+ and gd T cells in the lungs.
TLR2-mediated production of IL-27 by RE cells inhibits IL-17production, enhancing BIPF
To explore how TLR2-mediated signaling in nonhematopoieticcells of the lungs inhibits IL-17 production during BIPF, weevaluated the transcription levels of IL-17–related cytokines in the
FIGURE 3. TLR2 signaling mediated by RE cells regulates the production of IL-17 that inhibits BIPF. A, BALF was obtained from WT or TLR22/2
mice 7 d after bleomycin (BLM) administration and analyzed for levels of cytokines including IL-17 and TGF-b using ELISA. B, Same as A except that
mice were pretreated with LTA or Pam3csk4 before BLM administration. C, Percentages of IL-17–producing CD4+ and gd T cells in the BALF of WT and
TLR22/2 mice pretreated or not pretreated with LTA or Pam3csk4 before BLM administration. Numbers shown indicate the percentages of IL-17-positive
cells as indicated by intracellular staining. D, Effect of blockade of IL-17 on TGF-b and hydroxyproline levels in the BALF of WT or TLR22/2 mice with
BIPF. BALF was analyzed for the levels of TGF-b levels in BALF on day 7 using ELISA and for the amount of hydroxyproline in the lungs on day 21 in
WT and TLR22/2 mice injected with control Ab or anti–IL-17 mAb, and IL-17A2/2 mice. E, BM-chimeric mice were generated and treated with BLM as
described above. IL-17 mRNA and protein levels in whole lungs and BALF on day 7 were estimated using real-time PCR and ELISA, respectively. Data
shown represent mean 6 SEM for each group (n = 4 per group in A, B, D; n = 3 per group in C; n = 5 per group in E) for one representative experiment of
three or four independent experiments. *p , 0.05, **p , 0.01, ***p , 0.001. ns, not significant.
The Journal of Immunology 4011
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

lungs of WT and TLR22/2 mice with BIPF. Levels of IL-27 andIL-6 mRNA were lower in the lungs of TLR22/2 mice than inthose of WT mice, whereas the levels of IL-23 mRNA weresimilar in the two groups (Fig. 4A). Although IL-6 is knownto induce Th17 differentiation (20), it appears to exert a proin-flammatory effect on TLR2-mediated immune regulation, ratherthan an inductive effect on IL-17 production, based on lowerlevels of IL-6 in the lungs of TLR22/2 mice. Because IL-27 isknown to inhibit Th17 differentiation during various immuneresponses, we hypothesize that TLR2-mediated IL-27 productionby nonhematopoietic lung cells inhibits IL-17 production duringBIPF. Consistent with this hypothesis, IL-27 levels in BALFduring BIPF were higher in WT→WT and TLR22/2→WT chi-meras than in WT→TLR22/2 and TLR22/2→TLR22/2 chimeras(Fig. 4B).The difference in BALF IL-27 levels between WTand TLR22/2
mice during BIPF was enhanced by pretreatment of mice withLTA or Pam3csk4; this treatment further increased the amount ofIL-27 in the BALF of WT mice but not TLR22/2 mice (Fig. 4C).Furthermore, pretreatment with LTA or Pam3csk4 enhanced theexpression of IFN regulatory factors (IRFs) 1, 3, and 7 in the lungsof WT mice, but not in those of TLR22/2 mice (Fig. 4D). The
pattern of TLR2 expression we observed in the lungs suggestedthat RE cells might be a major subset of the nonhematopoieticcells responsible for the production of IL-27 during BIPF. Bleo-mycin stimulation of purified RE cells from WT mice enhancedtheir production of IL-27, which was further increased by theaddition of LTA or Pam3csk4 to the culture medium (Fig. 4E).This effect was decreased by the addition of a MyD88-inhibitorpeptide to the culture medium, as compared with a control peptide(Fig. 4E). However, bleomycin and TLR2-agonist treatment of REcells purified from TLR22/2 mice did not alter TLR2-mediatedIL-27 production in these cells. These findings suggest that REcells produce IL-27 during BIPF in a manner dependent onMyD88 and the IRF complexes.Next, to investigate whether TLR2-mediated induction of IL-27
production by RE cells inhibits IL-17 production by T cells, wemeasured IL-17 levels in the supernatant fractions of coculturesof RE and lung immune cells. Upon stimulation with LTA andbleomycin, the amount of IL-17 in the supernatant fractions fromcocultures ofWTRE cells andWTor TLR2-deficient immune cellswas lower than that from cocultures of TLR22/2 RE cells and WTor TLR2-deficient immune cells (Fig. 4F). This difference wasenhanced by the addition of a neutralizing anti–IL-27 mAb to the
FIGURE 4. TLR2-mediated induction of IL-27 production by RE cells inhibits IL-17 expression during BIPF. A, Levels of IL-27, IL-6, and IL-23 mRNA
in lungs obtained from WT or TLR22/2 mice on day 7 after bleomycin (BLM) administration were evaluated using real-time PCR. B, Levels of IL-27 in
BALF obtained from BM-chimeric mice on day 7 after BLM administration were estimated using ELISA. C, and D, WT and TLR22/2 mice were pre-
treated with LTA or Pam3csk4, treated with BLM, and sacrificed 7 d later. Next, the amount of IL-27 in the BALF was determined using an ELISA (C), and
the expression of IRF-1, IRF-3, and IRF-7 in the lungs was estimated using Western blotting (D). E, RE cells purified from lungs of WT or TLR22/2 mice
were cultured with BLM in the absence or presence of a TLR2 agonist (LTA or Pam3csk4) for 24 h. To estimate the effect of signaling through MyD88 on
IL-27 production by RE cells, MyD88-inhibitor or control peptide was added in culture medium. IL-27 levels in the culture supernatant fractions were then
determined using an ELISA. F, RE and immune cells from WT and TLR22/2 mice were cocultured for 24 h with or without an anti–IL-27 mAb or isotype
control Ab. The concentration of IL-17 in culture supernatant fractions was determined using an ELISA. G and H, WT or TLR22/2 mice were injected with
neutralizing anti–IL-27 mAb or an isotype-matched control Ab immediately after BLM administration and analyzed for the amount of hydroxyproline in
the lungs on day 21 (G) and for the levels of TGF-b (G) and IL-17 and the percentages of IL-17-producing cells in BALF on day 7 (H). I, Immune cells in
BALF from WTor TLR22/2 mice with BIPF were incubated with or without recombinant mouse IL-27 for 24 h and evaluated for IL-17 production and for
the percentage of IL-17–producing T cells. Data represent mean6 SEM for each group (n = 4 per group in A and G; n = 5 per group in B; n = 3 per group in
C–F, H, I) for one representative experiment of three or four independent experiments. *p , 0.05, **p , 0.01, ***p , 0.001. ns, not significant.
4012 TLR2-MEDIATED IL-27 AND CHEMOKINES PROMOTE PULMONARY FIBROSIS
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

cultures; blockade of IL-27 decreased the IL-17 level in cocultureswith WT but not TLR2-deficient RE cells (Fig. 4F). Furthermore,the injection of anti–IL-27 mAb into WT mice increased theamount of IL-17 in BALF and the percentages of IL-17–producingCD4+ and gd T cells in the lungs, whereas it reduced the lunglevels of hydroxyproline and TGF-b (Fig. 4G, 4H). In contrast,injection of anti–IL-27 mAb into TLR22/2 mice only minimallyaffected these parameters. Alternatively, recombinant mouse IL-27 reduced both the BALF IL-17 level and the percentages of IL-17–producing CD4+ and gd T cells in cultured BALF immunecells from TLR22/2 mice, whereas it did not significantly alterthese parameters in WT mice (Fig. 4I). These findings suggest thatTLR2-mediated IL-27 production by RE cells inhibits IL-17production in the lungs, thereby enhancing BIPF.
TLR2 signaling-induced production of chemokines by RE cellsalso promotes BIPF by recruiting immune cells into the lungs
Fewer immune cells were found in the lungs of TLR22/2mice withBIPF than in WT mice with BIPF (Fig. 1A, 1B), suggesting thatfewer immune cells were recruited into the lungs of TLR22/2
mice during BIPF. Therefore, we evaluated the mRNA expression
levels or various chemokines in the lungs of WT and TLR22/2
mice during BIPF. During BIPF, the levels of MCP-1, IP-10, andMIP-1a mRNA were higher in lungs of WT mice than in thoseof TLR22/2 mice, whereas the level of RANTES mRNA in thelungs was similar in the two groups (Fig. 5A). Similarly, MCP-1,IP-10, and MIP-1a levels were higher in the lungs of WT→WTand TLR22/2→WT chimeric mice than those of WT→TLR22/2
and TLR22/2→TLR22/2 chimeric mice during BIPF (Fig. 5B).These findings suggest that TLR2 signaling mediated by non-hematopoietic cells, rather than immune cells, of the lungs inducesMCP-1, IP-10, and MIP-1a production during BIPF. Stimulationof WT RE cells, but not TLR22/2 RE cells, with bleomycin andLTA or Pam3csk4 increased levels of MCP-1, IP-10, and MIP-1a,and this effect was suppressed by MyD88 inhibition (Fig. 5C).Injection of a mixture of recombinant mouse MCP-1, IP-10, andMIP-1a into TLR22/2 mice, but not WT mice, increased thehydroxyproline content in the lungs during BIPF (Fig. 6A, 6B),whereas it only minimally affected IL-27 and IL-17 levels in thelungs of WT or TLR22/2 mice (Fig. 6B, 6C). However, thischemokine mixture increased immune cell numbers in both WTand into TLR22/2 mice (Fig. 6A). These findings indicate that
FIGURE 5. RE cells produce chemokines in
response to TLR2 signaling during BIPF. A,
Seven days after the intratracheal administra-
tion of bleomycin (BLM) to WT and TLR22/2
mice, chemokine mRNA levels in the lungs
were evaluated using real-time PCR. B, Same
as A, except that BM-chimeric mice were
used. C, Purified RE cells from WT and
TLR22/2 mice were incubated with BLM in
the presence or absence of LTA or Pam3csk4
for 24 h. MyD88-mediated signaling was
suppressed by the addition of MyD88-in-
hibitory peptide to the culture medium, and
levels of MIP-1a, IP-10, and MCP in the su-
pernatant fraction were evaluated. Data rep-
resent mean 6 SEM for each group (n = 4 per
group in A; n = 5 per group in B; n = 3 per
group in C) for one representative experiment
of three or four independent experiments. *p,0.05, **p , 0.01. ns, not significant.
The Journal of Immunology 4013
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

TLR2-induced chemokine production by RE cells recruits im-mune cells into the lungs, thereby promoting BIPF through asecond, additional mechanism.
TLR2-dependent Treg cells have minimal inhibitory effect onTh17 cells in the lungs, although Treg cells attenuate BIPF
In several in vivo studies, TLR2 has been demonstrated to controlthe expansion and function of Treg cells (21–23), which inhibitTh17 cells via TLR2 signals in a pulmonary fungal infectionmodel system (24). These findings led us to hypothesize that theTLR2-dependent expansion of Treg cells might inhibit Th17 cellsin the BIPF model. To address this hypothesis, we measurednumbers of Treg cells and levels of cytokines and hydroxyprolinein the lungs of WT and TLR22/2 mice. Numbers of Treg cells inthe lungs were greater in the WT mice than in the TLR22/2 mice,although the percentage of Treg cells in the lungs was higher inthe TLR22/2 mice (Fig. 7A). When we depleted Treg cells in WTand TLR22/2 mice using an anti-CD25 mAb before bleomycinadministration, TGF-b levels, cell numbers, and hydroxyprolinelevels in the lungs increased in both WT and TLR22/2 mice (Fig.7B, 7C). In contrast, the IL-17 and IL-27 levels in BALF andthe percentages of IL-17–producing T cells in the lungs of WTand TLR22/2 mice were similar to those of Treg cell-depleted WTand TLR22/2 mice during BIPF (Fig. 7D). Therefore, TLR2-dependent Treg cells probably have little inhibitory effect on Th17cells in the lungs, although Treg cells attenuate BIPF.
DiscussionTo explore TLR2-mediated regulatory effects in BIPF, we exam-ined the pattern of TLR2 expression in the lungs and identified thelung cell type critical to the development of BIPF. In ourexperiments, both RE and immune cells exhibited increased TLR2
expression during BIPF in WT mice, and pulmonary fibrosisduring BIPF was attenuated in TLR22/2 mice relative to WTmice. Among RE cells, bronchial epithelial cells highly expressedTLR2 compared with alveolar epithelial cells, but other non-hematopoietic cells such as fibroblasts, myofibroblasts, and en-dothelial cells showed minimal expression of TLR2 during BIPF,as analyzed using immunohistochemistry. Furthermore, BIPF wasenhanced by TLR2-signaling mediated by RE cell, rather thanimmune cells, of the lungs. Therefore, these findings suggest thatbronchial epithelial cells might be a major subset of the lung inTLR2-mediated regulation of BIPF. Consistent with these results,RE cells are known to have critical roles in the development andprogression of pulmonary fibrosis (25). However, two independentresearch groups have suggested that RE cells contribute onlyminimally to the TLR2-mediated promotion of pulmonary fibro-sis. Yang et al. (11) have proposed that bleomycin-induced tissueinjury causes release of HMGB1 protein, which is recognized byTLR2 on immune cells and induces Th2-dominant immune re-sponses that enhance pulmonary fibrosis. However, we observedthat BALF levels of Th1 and Th2 cytokines were similar inTLR22/2 and WT mice after bleomycin administration, suggest-ing that TLR2 does not contribute to BIPF by regulating the Th1/Th2 balance in the lungs. Jiang et al. (10, 12) also suggested thatRE cells exert protect against, rather than promote, bleomycin-induced lung injury by enhancing interaction between TLR andhigh-molecular-weight hyaluronan. However, their experimentalstrategy might not have been appropriate for evaluating the exactfunctions of TLR2 in RE cells during BIPF, given that they usedTLR22/2TLR42/2 and MyD882/2 mice instead of TLR22/2
mice. In contrast, BM-chimeric mice and an in vitro culture sys-tem allowed us to investigate the critical functions of TLR2 on REcells in BIPF. Our results clearly show that TLR2 expression on
FIGURE 6. TLR2-induced production of chemokines by RE cells enhances recruitment of immune cells into the lungs, thereby promoting BIPF. WT and
TLR22/2 mice were injected with a chemokine mixture of MIP-1a, IP-10, and MCP-1 immediately before bleomycin administration. BALF fluid cell
numbers (A) and hydroxyproline levels in the lungs (B) were estimated 7 and 21 d after bleomycin injection, respectively. The IL-17, IL-27, and TGF-b
levels in the BALF (B) and the percentages of IL-17–producing T cells (C) were also evaluated. Data represent mean 6 SEM for each group (n = 3 per
group) for one representative experiment of three or four independent experiments. *p , 0.05, **p , 0.01, ***p , 0.001. ns, not significant.
4014 TLR2-MEDIATED IL-27 AND CHEMOKINES PROMOTE PULMONARY FIBROSIS
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

RE cells does not protect mice from BIPF; rather, it promotesBIPF.IL-27 is a heterodimeric cytokine of the IL-12 family consisting
of p28 and EBI3 (26). TLR3, TLR4, and TLR7/8 signaling inducethe expression of EBI3 and p28 in macrophages or dendritic cells
in a MyD88-, NF-kB–, IRF-1–, and IRF-3–dependent manner(27–30). In humans, IL-27 production by macrophages is alsoregulated by IFN-a in an autocrine manner (31). However, theadministration of TLR2 agonists to human macrophages failsto stimulate IFN-a production, showing that unlike these TLRs,
FIGURE 7. Treg cells only minimally
affect TLR2-mediated IL-27 and IL-17
production in the lungs during BIPF. A,
Percentages and numbers of Treg cells
in BALF taken from WT and TLR22/2
mice 7 d after bleomycin administration.
B–D, To deplete Treg cells in vivo, anti-
CD25 mAb was injected i.v. into WT
and TLR22/2 mice 48 h before bleo-
mycin administration. BALF was taken
from these mice and from non–anti-CD25
mAb-treated WT and TLR22/2 mice 7
d after bleomycin administration and an-
alyzed for the number of cells (B), the
level of TGF-b and hydroxyproline (C),
and the levels of IL-17 and IL-27 and the
percentages of IL-17–producing CD4+
T cells (D). Data shown in A–C represent
mean 6 SEM for each group (n = 4 per
group in A; n = 3 per group in B–D) for
one representative experiment of three
or four independent experiments. *p ,0.05, **p , 0.01, ***p , 0.001. ns, not
significant.
FIGURE 8. TLR2 regulates pulmonary fibrosis via two axes; RE cells/IL-27/IL-17 and RE cells/chemokines/inflammatory cells. Injury to bronchial
epithelial cells triggers secretion of endogenous ligands for TLR2, which induces production of IL-27 and chemokines by RE cells via TLR2 engagement.
During repair processes of pulmonary injury, IL-27 inhibits IL-17 production by immune cells and chemokines recruit inflammatory cells into the lungs,
resulting in promotion of pulmonary fibrosis.
The Journal of Immunology 4015
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

TLR2 does not activate p28 gene transcription in these cells (31).Thus, TLR2-mediated signaling in immune cells appears to min-imally induce IL-27 production.In contrast, our experiments produced several pieces of evidence
to suggest that TLR2 induces IL-27 production in RE cells duringBIPF. First, IL-27 production in the lungs was higher in WT→WTand TLR22/2→WT chimeric mice than in WT→TLR22/2 andTLR22/2→TLR22/2 mice, indicating that TLR2 expression onnonhematopoietic cells of the lungs regulates IL-27 productionduring BIPF. Second, TLR2 ligands enhanced IL-27 production byRE cells of WT mice, but not TLR22/2 mice, in the presence andabsence of bleomycin (Figs. 4E, 5C, Supplemental Fig. 1), whichwas in an MyD88-dependent manner. These findings suggestthat TLR2 agonists modulate IL-27–mediated immune responsesin the lungs even in the absence of bleomycin-induced injury.However, single intratracheal injection of TLR2 agonists into WTmice minimally modulated inflammation and cytokine environ-ment in the lungs (data not shown), suggesting that sustainedstimulation of TLR2 in RE cells for long term or additional en-vironmental modulation such as tissue damage in the lungs mightbe necessary to TLR2-mediated regulation of pulmonary fibro-sis. Third, blockade of IL-27 in vivo decreased the amount ofhydroxyproline in the lungs of WT mice, but not of TLR22/2
mice. We have thus demonstrated for the first time that TLR2-mediated IL-27 production by RE cells promotes BIPF.IL-27 has been demonstrated to inhibit Th17 cells in a manner
dependent on the orphan nuclear receptor RORgt, to promote Th1differentiation, and to enhance IL-10 production (32–34). IL-27thus regulates various immune diseases through its dual proin-flammatory and anti-inflammatory effects on immune responses(35). A role for IL-27 in pulmonary fibrosis has been suggested bythe demonstration that the serum level of IL-27 is elevated inpatients with systemic sclerosis and positively correlates with theextent of pulmonary and dermal fibrosis (36). We observed thatlevels of IL-17 and IL-27 in the lungs during BIPF were higherand lower, respectively, in TLR22/2 mice than in WT mice,suggesting that IL-27 and IL-17 production in the lungs are in-versely regulated by TLR2 on RE cells during BIPF. Consistentwith this suggestion, blockade of IL-27 enhanced IL-17 produc-tion in a TLR2-dependent manner in vivo and in vitro, indicat-ing that IL-27 produced by RE cells inhibits IL-17 productionduring BIPF. However, the regulatory effects of TLR2 on Th17cells have appeared to be contradictory in various studies. In pul-monary fungal infection and brain abscess, TLR2 was reported toact as a negative regulator for Th17 cells (24, 37), whereas inexperimental autoimmune encephalomyelitis, skin inflammation,and tuberculosis infection, TLR2 was reported to have the oppo-site effect (38–40). These findings suggest that the specific effectof TLR2 on Th17 cells varies with the type of immune responseand depends on the microenvironment of the target tissues.Therefore, our experiments suggest that bleomycin-induced tissuedamage in the lungs might produce a microenvironment that in-duces TLR2 to inhibit Th17 cells by inducing IL-27 production.In our experiments, IL-17 produced by CD4+ and gd T cells
suppressed BIPF, and IL-17 production was inhibited by the TLR2signaling-induced production of IL-27 by RE cells. Consistentwith our results, gd T cells in the lungs of WT mice were found topredominantly produce IL-17, and the amount of hydroxyprolinein the lungs increased in gd T cell-deficient mice during BIPF(41). In contrast, in a recent study, IL-17A produced by CD4+ andgd+ T cells promoted bleomycin and IL-1b–mediated pulmonaryfibrosis (42). Although the functions of IL-17 in BIPF in our studyappear inconsistent with this result, CD4+ and gd T cells were themajor cell subsets producing IL-17 during BIPF in both studies.
Moreover, the cytokine milieus of pro- and anti-fibrogenic cyto-kines make critical contributions to the process of fibrosis in targettissues (43). Differences in these milieus might explain the con-tradictory IL-17–mediated outcomes in BIPF. Further study willbe needed to define the exact function of IL-17 in pulmonary fi-brosis.Agonist stimulation of TLR2 causes RE cells to produce IL-6
and IL-8 (16, 44), suggesting that TLR2-mediated signals in REcells regulate the immune response by regulating the productionof various chemokines and cytokines. In our experiments, treat-ment of RE cells with TLR2 agonists induced the production ofMIP-1a, MCP-1, and IP-10, stimulating the recruitment of in-flammatory cells into the lungs during BIPF. Although IP-10 hasbeen reported to exert antifibrotic effect on the lungs (45), a che-mokine mixture increased the amount of hydroxyproline in thelungs of TLR22/2 mice, but not WT mice, and only minimallyaffected the production of IL-27 and IL-17 in the lungs of bothgroups of mice. These results suggest that TLR2 signaling-induced chemokines produced by RE cells promote BIPF by re-cruiting inflammatory cells into the lungs rather than by regulatingthe production of IL-27 and IL-17.In conclusion, the results of our study demonstrate that RE cells
produce IL-27 and chemokines in a TLR2-dependent manner thatpromotes BIPF by inhibiting IL-17 production and recruiting in-flammatory cells into the lungs. We suggest that TLR2 regulatespulmonary fibrosis through two axes; RE cells/IL-27/IL-17 and REcells/chemokines/inflammatory cells (Fig. 8). Our findings providevaluable insight needed for the design of therapeutic approachesfor idiopathic pulmonary fibrosis.
AcknowledgmentsWe thank Dr. Seung Yong Seong for support in obtaining TLR22/2 mice
and the members of the Department of Experimental Animals at Clinical
Research Institute of Seoul National University Hospital for animal man-
agement.
DisclosuresThe authors have no financial conflicts of interest.
References1. Hattori, N., J. L. Degen, T. H. Sisson, H. Liu, B. B. Moore, R. G. Pandrangi,
R. H. Simon, and A. F. Drew. 2000. Bleomycin-induced pulmonary fibrosis infibrinogen-null mice. J. Clin. Invest. 106: 1341–1350.
2. Swaisgood, C. M., E. L. French, C. Noga, R. H. Simon, and V. A. Ploplis. 2000.The development of bleomycin-induced pulmonary fibrosis in mice deficient forcomponents of the fibrinolytic system. Am. J. Pathol. 157: 177–187.
3. Lukacs, N. W., C. Hogaboam, S. W. Chensue, K. Blease, and S. L. Kunkel. 2001.Type 1/type 2 cytokine paradigm and the progression of pulmonary fibrosis.Chest 120(1 Suppl)5S–8S.
4. Segel, M. J., G. Izbicki, P. Y. Cohen, R. Or, T. G. Christensen, S. B. Wallach-Dayan, and R. Breuer. 2003. Role of interferon-gamma in the evolution ofmurine bleomycin lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 285:L1255–L1262.
5. Keane, M. P., J. A. Belperio, M. D. Burdick, and R. M. Strieter. 2001. IL-12attenuates bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell.Mol. Physiol. 281: L92–L97.
6. Gharaee-Kermani, M., Y. Nozaki, K. Hatano, and S. H. Phan. 2001. Lunginterleukin-4 gene expression in a murine model of bleomycin-induced pulmo-nary fibrosis. Cytokine 15: 138–147.
7. Huaux, F., T. Liu, B. McGarry, M. Ullenbruch, and S. H. Phan. 2003. Dual rolesof IL-4 in lung injury and fibrosis. J. Immunol. 170: 2083–2092.
8. Schnare, M., G. M. Barton, A. C. Holt, K. Takeda, S. Akira, and R. Medzhitov.2001. Toll-like receptors control activation of adaptive immune responses. Nat.Immunol. 2: 947–950.
9. Aderem, A., and R. J. Ulevitch. 2000. Toll-like receptors in the induction of theinnate immune response. Nature 406: 782–787.
10. Jiang, D., J. Liang, Y. Li, and P. W. Noble. 2006. The role of Toll-like receptorsin non-infectious lung injury. Cell Res. 16: 693–701.
11. Yang, H. Z., B. Cui, H. Z. Liu, Z. R. Chen, H. M. Yan, F. Hua, and Z. W. Hu.2009. Targeting TLR2 attenuates pulmonary inflammation and fibrosis by re-version of suppressive immune microenvironment. J. Immunol. 182: 692–702.
4016 TLR2-MEDIATED IL-27 AND CHEMOKINES PROMOTE PULMONARY FIBROSIS
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

12. Jiang, D., J. Liang, J. Fan, S. Yu, S. Chen, Y. Luo, G. D. Prestwich,M. M. Mascarenhas, H. G. Garg, D. A. Quinn, et al. 2005. Regulation of lunginjury and repair by Toll-like receptors and hyaluronan. Nat. Med. 11: 1173–1179.
13. Yoshizaki, A., Y. Iwata, K. Komura, F. Ogawa, T. Hara, E. Muroi, M. Takenaka,K. Shimizu, M. Hasegawa, M. Fujimoto, et al. 2008. CD19 regulates skin andlung fibrosis via Toll-like receptor signaling in a model of bleomycin-inducedscleroderma. Am. J. Pathol. 172: 1650–1663.
14. Trujillo, G., A. Meneghin, K. R. Flaherty, L. M. Sholl, J. L. Myers,E. A. Kazerooni, B. H. Gross, S. R. Oak, A. L. Coelho, H. Evanoff, E. Day,G. B. Toews, A. D. Joshi, M. A. Schaller, B. Waters, G. Jarai, J. Westwick,S. L. Kunkel, F. J. Martinez, and C. M. Hogaboam. 2010. TLR9 differentiatesrapidly from slowly progressing forms of idiopathic pulmonary fibrosis. Sci.Transl. Med. 2: 57ra82.
15. Schwandner, R., R. Dziarski, H. Wesche, M. Rothe, and C. J. Kirschning. 1999.Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274: 17406–17409.
16. Armstrong, L., A. R. Medford, K. M. Uppington, J. Robertson, I. R. Witherden,T. D. Tetley, and A. B. Millar. 2004. Expression of functional toll-like receptor-2and -4 on alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 31: 241–245.
17. Homma, T., A. Kato, N. Hashimoto, J. Batchelor, M. Yoshikawa, S. Imai,H. Wakiguchi, H. Saito, and K. Matsumoto. 2004. Corticosteroid and cytokinessynergistically enhance toll-like receptor 2 expression in respiratory epithelialcells. Am. J. Respir. Cell Mol. Biol. 31: 463–469.
18. Gribar, S. C., W. M. Richardson, C. P. Sodhi, and D. J. Hackam. 2008. No longeran innocent bystander: epithelial toll-like receptor signaling in the developmentof mucosal inflammation. Mol. Med. 14: 645–659.
19. Kim, H. Y., S. Kim, and D. H. Chung. 2006. FcgammaRIII engagement providesactivating signals to NKT cells in antibody-induced joint inflammation. J. Clin.Invest. 116: 2484–2492.
20. Veldhoen, M., R. J. Hocking, C. J. Atkins, R. M. Locksley, and B. Stockinger.2006. TGFbeta in the context of an inflammatory cytokine milieu supports denovo differentiation of IL-17-producing T cells. Immunity 24: 179–189.
21. Sutmuller, R. P., M. H. den Brok, M. Kramer, E. J. Bennink, L. W. Toonen,B. J. Kullberg, L. A. Joosten, S. Akira, M. G. Netea, and G. J. Adema. 2006.Toll-like receptor 2 controls expansion and function of regulatory T cells. J. Clin.Invest. 116: 485–494.
22. Liu, H., M. Komai-Koma, D. Xu, and F. Y. Liew. 2006. Toll-like receptor 2signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc. Natl.Acad. Sci. USA 103: 7048–7053.
23. Zanin-Zhorov, A., L. Cahalon, G. Tal, R. Margalit, O. Lider, and I. R. Cohen.2006. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell functionvia innate TLR2 signaling. J. Clin. Invest. 116: 2022–2032.
24. Loures, F. V., A. Pina, M. Felonato, and V. L. Calich. 2009. TLR2 is a negativeregulator of Th17 cells and tissue pathology in a pulmonary model of fungalinfection. J. Immunol. 183: 1279–1290.
25. Chapman, H. A. 2011. Epithelial-mesenchymal interactions in pulmonary fi-brosis. Annu. Rev. Physiol. 73: 413–435.
26. Pflanz, S., J. C. Timans, J. Cheung, R. Rosales, H. Kanzler, J. Gilbert, L. Hibbert,T. Churakova, M. Travis, E. Vaisberg, et al. 2002. IL-27, a heterodimeric cy-tokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+)T cells. Immunity 16: 779–790.
27. Liu, J., X. Guan, and X. Ma. 2007. Regulation of IL-27 p28 gene expression inmacrophages through MyD88- and interferon-gamma-mediated pathways.J. Exp. Med. 204: 141–152.
28. Molle, C., M. Nguyen, V. Flamand, J. Renneson, F. Trottein, D. De Wit,F. Willems, M. Goldman, and S. Goriely. 2007. IL-27 synthesis induced by TLRligation critically depends on IFN regulatory factor 3. J. Immunol. 178: 7607–7615.
29. Molle, C., M. Goldman, and S. Goriely. 2010. Critical role of the IFN-stimulatedgene factor 3 complex in TLR-mediated IL-27p28 gene expression revealinga two-step activation process. J. Immunol. 184: 1784–1792.
30. Wirtz, S., C. Becker, M. C. Fantini, E. E. Nieuwenhuis, I. Tubbe, P. R. Galle,H. J. Schild, M. Birkenbach, R. S. Blumberg, and M. F. Neurath. 2005. EBV-induced gene 3 transcription is induced by TLR signaling in primary dendriticcells via NF-kappa B activation. J. Immunol. 174: 2814–2824.
31. Pirhonen, J., J. Siren, I. Julkunen, and S. Matikainen. 2007. IFN-alpha regulatesToll-like receptor-mediated IL-27 gene expression in human macrophages.J. Leukoc. Biol. 82: 1185–1192.
32. Yoshida, H., S. Hamano, G. Senaldi, T. Covey, R. Faggioni, S. Mu, M. Xia,A. C. Wakeham, H. Nishina, J. Potter, et al. 2001. WSX-1 is required for theinitiation of Th1 responses and resistance to L. major infection. Immunity 15:569–578.
33. Stumhofer, J. S., A. Laurence, E. H. Wilson, E. Huang, C. M. Tato,L. M. Johnson, A. V. Villarino, Q. Huang, A. Yoshimura, D. Sehy, et al. 2006.Interleukin 27 negatively regulates the development of interleukin 17-producingT helper cells during chronic inflammation of the central nervous system. Nat.Immunol. 7: 937–945.
34. Fitzgerald, D. C., G. X. Zhang, M. El-Behi, Z. Fonseca-Kelly, H. Li, S. Yu,C. J. Saris, B. Gran, B. Ciric, and A. Rostami. 2007. Suppression of autoimmuneinflammation of the central nervous system by interleukin 10 secreted by in-terleukin 27-stimulated T cells. Nat. Immunol. 8: 1372–1379.
35. Yoshida, H., M. Nakaya, and Y. Miyazaki. 2009. Interleukin 27: a double-edgedsword for offense and defense. J. Leukoc. Biol. 86: 1295–1303.
36. Yoshizaki, A., K. Yanaba, Y. Iwata, K. Komura, A. Ogawa, E. Muroi, F. Ogawa,M. Takenaka, K. Shimizu, M. Hasegawa, et al. 2011. Elevated serum interleukin-27 levels in patients with systemic sclerosis: association with T cell, B cell andfibroblast activation. Ann. Rheum. Dis. 70: 194–200.
37. Nichols, J. R., A. L. Aldrich, M. M. Mariani, D. Vidlak, N. Esen, and T. Kielian.2009. TLR2 deficiency leads to increased Th17 infiltrates in experimental brainabscesses. J. Immunol. 182: 7119–7130.
38. Aliahmadi, E., R. Gramlich, A. Grutzkau, M. Hitzler, M. Kruger, R. Baumgrass,M. Schreiner, B. Wittig, R. Wanner, and M. Peiser. 2009. TLR2-activated humanlangerhans cells promote Th17 polarization via IL-1beta, TGF-beta and IL-23.Eur. J. Immunol. 39: 1221–1230.
39. Reynolds, J. M., B. P. Pappu, J. Peng, G. J. Martinez, Y. Zhang, Y. Chung, L. Ma,X. O. Yang, R. I. Nurieva, Q. Tian, and C. Dong. 2010. Toll-like receptor 2signaling in CD4(+) T lymphocytes promotes T helper 17 responses and reg-ulates the pathogenesis of autoimmune disease. Immunity 32: 692–702.
40. Teixeira-Coelho, M., A. Cruz, J. Carmona, C. Sousa, D. Ramos-Pereira,A. L. Saraiva, M. Veldhoen, J. Pedrosa, A. G. Castro, and M. Saraiva. 2011.TLR2 deficiency by compromising p19 (IL-23) expression limits Th 17 cellresponses to Mycobacterium tuberculosis. Int. Immunol. 23: 89–96.
41. Braun, R. K., C. Ferrick, P. Neubauer, M. Sjoding, A. Sterner-Kock, M. Kock,L. Putney, D. A. Ferrick, D. M. Hyde, and R. B. Love. 2008. IL-17 producinggammadelta T cells are required for a controlled inflammatory response afterbleomycin-induced lung injury. Inflammation 31: 167–179.
42. Wilson, M. S., S. K. Madala, T. R. Ramalingam, B. R. Gochuico, I. O. Rosas,A. W. Cheever, and T. A. Wynn. 2010. Bleomycin and IL-1b-mediated pul-monary fibrosis is IL-17A dependent. J. Exp. Med. 207: 535–552.
43. Yoshizaki, A., K. Yanaba, Y. Iwata, K. Komura, A. Ogawa, Y. Akiyama,E. Muroi, T. Hara, F. Ogawa, M. Takenaka, et al. 2010. Cell adhesion moleculesregulate fibrotic process via Th1/Th2/Th17 cell balance in a bleomycin-inducedscleroderma model. J. Immunol. 185: 2502–2515.
44. Becker, M. N., M. S. Sauer, M. S. Muhlebach, A. J. Hirsh, Q. Wu,M. W. Verghese, and S. H. Randell. 2004. Cytokine secretion by cystic fibrosisairway epithelial cells. Am. J. Respir. Crit. Care Med. 169: 645–653.
45. Tager, A. M., R. L. Kradin, P. LaCamera, S. D. Bercury, G. S. Campanella,C. P. Leary, V. Polosukhin, L. H. Zhao, H. Sakamoto, T. S. Blackwell, andA. D. Luster. 2004. Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am. J. Respir. Cell Mol. Biol. 31: 395–404.
The Journal of Immunology 4017
by guest on May 4, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from





























