Embed Size (px)
DESCRIPTION
Academic summary looking at the potential to develop a vaccine for the Ebola Virus (written before the 2014 outbreak).
Citation preview

To What Extent Is It Possible To Develop A Prophylactic for Ebola? James Baxter
ABSTRACT Ebola is a virus with the capability of inducing a lethal viral haemorrhagic fever, causing massive internal bleeding in humans and non-‐human primates. It was first encountered in 1976 in the Democratic Republic of the Congo (formerly Zaire) in which the fatality rate was 88%. Further outbreaks have occurred since with fatality rates ranging from 25%-‐100%, yet there is currently no approved course of treatment nor licensed vaccine. The report contains detail on the pathogenesis of Ebola (EBOV) before pursuing ways in which it could be prevented. Using research carried out by the London School of Hygiene and Tropical Medicine, the American military and Médecins Sans Frontières, it was found that whilst several prototype vaccines are being researched, it is not expected that any will be ready for large scale administration within the next 5 years.

The first recorded outbreaks of Ebola occurred simultaneously in Zaire, now the Democratic Republic of the Congo, and Sudan, now South Sudan, in 1976. The emergence in the towns of Yambuku, Zaire and Nzara, Sudan killed 280 of 318 and 151 of 284 cases respectively. Named after a river near Yambuku, Ebola returned again the following year, killing one, and again to the Democratic Republic of the Congo in 1995 in Kikwit – a town of population in excess of 500,000 with a mortality rate equally as shocking the first outbreaks (Fleck, 2011). More recently it has been reported that there is currently an on-‐going outbreak in Guinea, West Africa (BBC, 2014). It was the persistent 90% death rate of the major outbreaks of Ebola (WHO, 2012) that brought about the initial interest in Ebola, primarily to look at how the condition was treated and, as a keen traveller and a prospective medical student, whether vaccination was a viable method of protection against the virus. Upon discovering that there was at present no vaccination programme for any strain of Ebola, it was decided to research more about the pathogenesis of the virus and examine the current efforts made by international medical organisations to develop effective treatments and a vaccine. In order to establish a common ground of knowledge, the report opens with an introduction to fundamental virology.
An Introduction to Viruses Viruses are unlike any other organism on earth: their origins are unknown and there is still significant debate as to whether they are even ‘alive.’ Yet these less than microscopic organisms pose some of the biggest health threats to humans including: Human Immunodeficiency Virus (HIV), SARS coronavirus and Rabies. The latter stimulated the discovery of viruses when Louis Pasteur was unable to find a pathogen responsible for the onset of the disease and proposed that there may be an instigator too small to be viewed under a light microscope (Wikipedia, 2013). At a similar time, the Russian Botanist Dmitri Ivanovsky showed that after passing an infectious sample of Tobacco plants, suffering from Mosaic Disease, through a filter with pores able to prevent the flow of bacteria, the solution retained its potential to infect (Soper, 1997a). Dutch microbiologist, Martinus Beijerinck repeated this experiment, using a diluted sample that regained its original concentration after the infection of a second plant, demonstrating that the infectious agent could reproduce, defining it as a living organism. In 1903 the director of the Pasteur Institute, Pierre Roux, defined the characteristics of ‘filterable viruses,’ into 3 properties:
• Filterable: Organisms small enough to pass through a Chamerberland Filter • Invisible: Organisms not possible to be viewed under a light microscope • Non – cultivable: Organisms that cannot be grown on a bacterial culture plate.
Throughout the early 20th Century, more was discovered about the morphology of viruses, but very little was appreciated about their mechanisms. In an attempt to understand this, chicken eggs were infected with a pathogen, after which a plaque upon the embryo membrane developed. It was first observed on the inoculated membranes of chick embryos that viruses multiplied inside the cells, often killing the infected cell (Crawford, 2000a). The Invention of the electron microscope in 1938 revealed the identities of such pathogens:
Figure 1: Electron Micrograph Images of Some Viruses (Stannard, 1995)
Influenza Virus Rotavirus (common cause of diarrhoea)
Hepatitis B Virus Adenovirus (causes conjunctivitis)

Whilst viruses could now be seen, many questions still remained over their construction. Experiments whereby the Tobacco Mosaic Virus was crystalized suggested that it consisted of pure protein. However as understanding of the biological significance of chromosomes and DNA grew, it became apparent that viruses also contained genetic material. This discovery distinguished the differences between bacteria and viruses in a way that was finally unequivocal (Crawford, 2000b).
Figure 2: Comparing the size of a virus (Openstax, 2013)
Virus Anatomy Viruses are the simplest known organisms in existence, consisting of solely a protein shell and containing a genetic code. Virions are mature viruses and range from 20nm – 400nm in size, a minor fraction the size of any cell (Martin & Hine, 2000a). As there are no provisions for respiration, viruses cannot perform any metabolic reactions such as protein synthesis or reproduction, which leads to significant debate as to whether they are deemed to be truly living (Crawford, 2000b). The protein ‘capsid’ that encases the inner core of nucleic acid consists of many identical protein subunits arranged into an isometric 3-‐D shape. This is typically a 20-‐sided icosahedron or a thin spiral tube. In some cases a second capsid called the viral envelope, is also present on the exterior of the capsid. This is formed from the surface membrane of the host cell and can be used to evade the host organism’s immune response (Smith, 1995a). The genetic information in the interior of the virus may be stored either as deoxyribonucleic acid (DNA), where typically a double helix structure consisting of two polynucleotide strands is present, or ribonucleic acid (RNA), which is a single-‐stranded polynucleotide (Smith 1995a). In viruses, single stranded DNA (ssDNA) is more common than its double-‐stranded (dsDNA) counterpart, as found in more complex organisms. Double-‐stranded RNA (dsRNA) viruses are also an anomaly to the standard form of single stranded RNA (ssRNA). The nucleic acids can be further categorised by a characteristic known as ‘sense.’ This is defined as ‘the concept used to compare the polarity of nucleic molecules, such as DNA or RNA, to other nucleic molecules’ (Wikipedia, 2014). In DNA in general, there are two antiparallel strands of nucleotides that are bonded together by inter-‐molecular forces between the complementary nitrogenous bases. One of these strands, which has the same base code as the messenger RNA (mRNA) involved in protein synthesis, is labelled as ‘positive sense,’ whilst the complimentary strand of DNA is called ‘negative sense.’ The gene on the positive sense strand is the same permutation that is seen on mRNA during the translation phase of protein synthesis, but is not actually used during transcription. As the mRNA must be an exact copy of the coding DNA positive sense strand, the complimentary pairs of the DNA negative sense strand (also called the transcribed strand) are used in transcription so that an identical mRNA strand of the DNA positive sense strand is formed. The same principle exists for RNA, whereby the negative strand is complimentary to the mRNA whereas the positive strand is a copy. This can be used to classify viruses, because if a positive sense nucleic acid is present, once it enters the host cell it may undergo direct replication and then translation, utilising the host cell’s ribosomes. For nucleic acid negative sense strands however, RNA-‐dependent RNA polymerase or DNA-‐dependent DNA polymerase enzymes are required to replicate the respective nucleic acid and convert the produced genetic material into the positive sense strand. Ambisense viruses also exist in which the nucleic acid has a double strand intermediate preceding transcription. In this instance, transcription could occur on either strand (Wikipedia 2014). Retroviruses have a different way again of storing their genome and store all the genetic information for the virus as mRNA. Upon entering the host cell, the
mRNA is turned into either RNA or DNA using the virus’ reverse transcriptase enzyme (Martin & Hine, 2000b). A useful method of classifying viruses thus transpires: (Baltimore, 1971):
I Double strand DNA viruses II Single strand DNA viruses III Double strand RNA viruses IV Positive sense single strand RNA viruses V Negative sense single strand RNA viruses VI RNA retroviruses VII DNA retroviruses
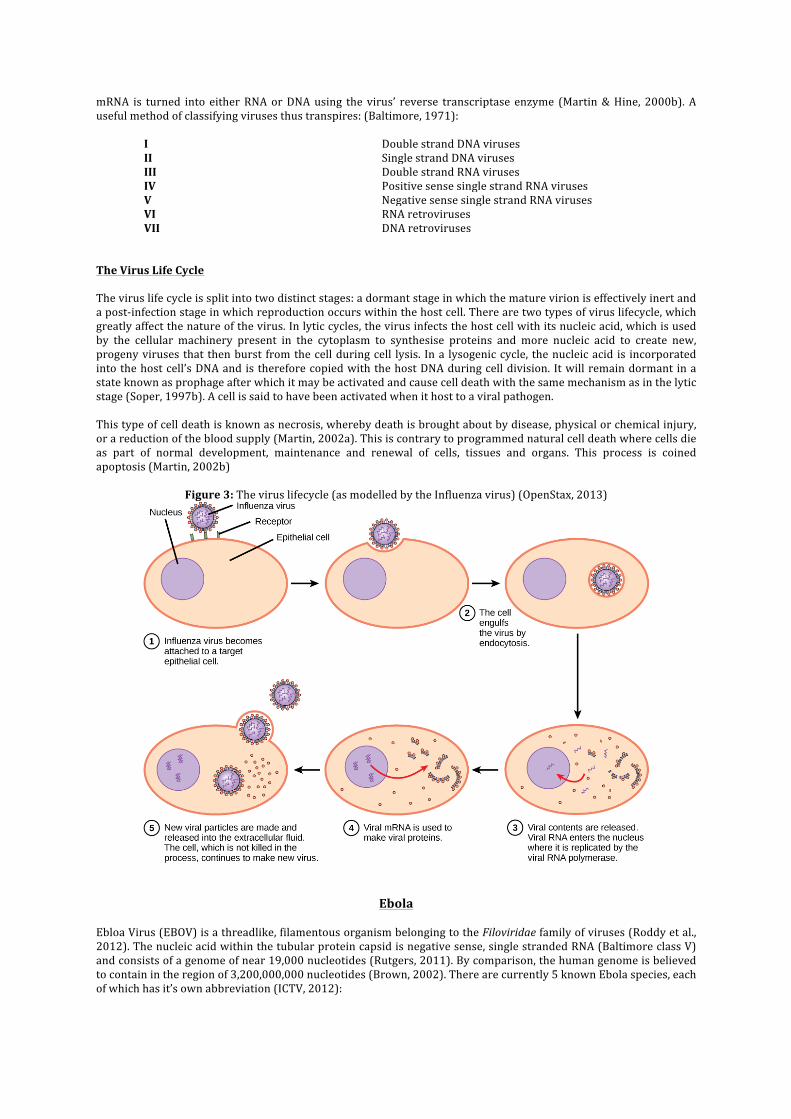
The Virus Life Cycle The virus life cycle is split into two distinct stages: a dormant stage in which the mature virion is effectively inert and a post-‐infection stage in which reproduction occurs within the host cell. There are two types of virus lifecycle, which greatly affect the nature of the virus. In lytic cycles, the virus infects the host cell with its nucleic acid, which is used by the cellular machinery present in the cytoplasm to synthesise proteins and more nucleic acid to create new, progeny viruses that then burst from the cell during cell lysis. In a lysogenic cycle, the nucleic acid is incorporated into the host cell’s DNA and is therefore copied with the host DNA during cell division. It will remain dormant in a state known as prophage after which it may be activated and cause cell death with the same mechanism as in the lytic stage (Soper, 1997b). A cell is said to have been activated when it host to a viral pathogen. This type of cell death is known as necrosis, whereby death is brought about by disease, physical or chemical injury, or a reduction of the blood supply (Martin, 2002a). This is contrary to programmed natural cell death where cells die as part of normal development, maintenance and renewal of cells, tissues and organs. This process is coined apoptosis (Martin, 2002b)
Figure 3: The virus lifecycle (as modelled by the Influenza virus) (OpenStax, 2013)
Ebola
Ebloa Virus (EBOV) is a threadlike, filamentous organism belonging to the Filoviridae family of viruses (Roddy et al., 2012). The nucleic acid within the tubular protein capsid is negative sense, single stranded RNA (Baltimore class V) and consists of a genome of near 19,000 nucleotides (Rutgers, 2011). By comparison, the human genome is believed to contain in the region of 3,200,000,000 nucleotides (Brown, 2002). There are currently 5 known Ebola species, each of which has it’s own abbreviation (ICTV, 2012):

• Zaire ebolavirus (ZEBOV) • Sudan ebolavirus (SEBOV) • Reston ebolavirus (REBOV) • Côte d’Ivoire ebolavirus (CIEBOV) • Bundibugyo ebolavirus (BEBOV)
The ZEBOV, SEBOV and BEBOV strains of Ebola are known to induce a viral haemorrhagic fever (VHF), which is similar to the other virus of the filoviridae family, Marburg virus (WHO, 2012). Both have very similar attributes whereby they cause their respective VHFs – Ebola haemorrhagic fever (EHF) and Marburg haemorrhagic fever (MHF), and both originate from sub-‐Saharan Africa. There have been 24 recorded outbreaks of EHF in humans, but only 11 in MHF (Roddy et al., 2012), although given the remote location and relatively primordial medical resources, it is likely that both these figures are underestimates of the actual number more recent outbreaks. Both EHF and MHF are described as ‘severe acute viral illness[es] often with the loss of blood,’ by World Health Organisation (Fleck, 2011). Transmission Ebola Virus is considered to be zoonotic because it must be transmitted from a non-‐human vertebrate to a human in order to start an epidemic. A zoonotic virus is said to be capable of infecting both non-‐human vertebrates as well as humans. Once a member of the human population has been infected, the virus is quickly spread through person-‐to-‐person contact, which in the case of Ebola Virus involves the contact of bodily secretions and fluids. This is exacerbated in a healthcare environment where used needles and unsterilized equipment can harbour pathogens (NIAID, 2010). It has also been noted that cultural burials have been linked to the transmission of Ebola, where mourners have direct contact with the deceased (WHO, 2012). Pathogenesis Once EBOV has entered the body, it is swift to act and death is normally brought about in an incubation period of less than 21 days (Fleck, 2011). The early symptoms of the haemorrhagic fever include weakness, headache, vomiting and rash, all of which could lead to differential diagnosis (WHO, 2012). This is where several diseases share the same or very similar symptoms (Martin, 2002c) and as such several diagnoses could be made. This could mean that it is not beyond reasonable doubt that the small minority of survivors of earlier Ebola outbreaks were in fact misdiagnosed. The way in which EBOV brings about EHF is not entirely understood by the scientific community but the consensus is that death is a result of hypovolemic shock as a result of mass haemorrhaging or/and organ failure (Rutgers, 2011). Whereas most viruses target a specific type of cell for infection, there is evidence that in the case of EBOV, the initial site of viral replication is within dendritic cells and macrophages, before then proceeding to endothelial cells (Geisbert et al., 2003). The ability to infect a wide range of primate cells is considered to be a result of the way two identified glycoproteins on the virus capsid interact with target molecules on the cell surface, in particular the cholesterol transporter Niemann-‐Pick C1 (Carette et al., 2011)(Bray & Geisbert, 2004). The importance of the infection of dendritic cells should not be underestimated, as they are essential to the immune response of the mammalian body. Located within the skin, lungs and gastrointestinal tract, these cells react in the presence of foreign antigens, transferring them to lymph nodes where lymphocytes are located. These white blood cells produce antibodies, which are essential in the immune response to infection (Banchereau & Steinman, 1998). The death of dendritic cells due to necrosis prevents the passing of viral antigens to the lymphocytes, which results in the failure to produce antibodies against the virus. Likewise Natural Killer Cells, which engulf activated cells, do not receive the antigen signal and therefore do not act upon the viral infection and soon become depleted (Baize et al., 1999). Whilst the virus does not infect lymphocytes themselves, they are lost in high concentrations during the illness through apoptosis, inducing a condition known as lymphopenia, which is synonymous with symptoms of sepsis and septic shock (Bray & Mahanty, 2003). The second site of viral infection is in macrophages, which are white blood cells (leucocytes) that remove foreign pathogens through phagocytosis whereby the cell engulfs the target. As these cells are not fixed to lymph nodes, they become the vectors for the spread of the virus throughout the body. The infected macrophages release proinflammatory cytokines, chemokines and nitric oxide (NO) which all act as inter-‐cellular communicators (Bausch et al., 2008). It is thought that this response is brought about by the binding of viral products to pattern recognition molecules within the macrophage. The released proteins attract macrophages to the site of infection and at the same time mobilise immature neutrophils from the bone marrow and blood vessel walls (Bray & Geisbert, 2005). Neutrophils are a specific type of white blood cell called granulocytes and are capable of digesting foreign pathogens (Martin, 2002d), although it is assumed that these are summoned to ensure the maximum dispersal of the viral infection around the body is achieved. The chemokines and NO also remove inflammatory cells by increasing the permeability of the endothelium through a process known as vasodilatation. In effect, the virus hijacks a process that on a small scale is therapeutic, and implements it simultaneously throughout the patient as the activated leucocytes disperse, causing severe internal haemorrhaging (Bray & Geisbert, 2005). Another common observation during the
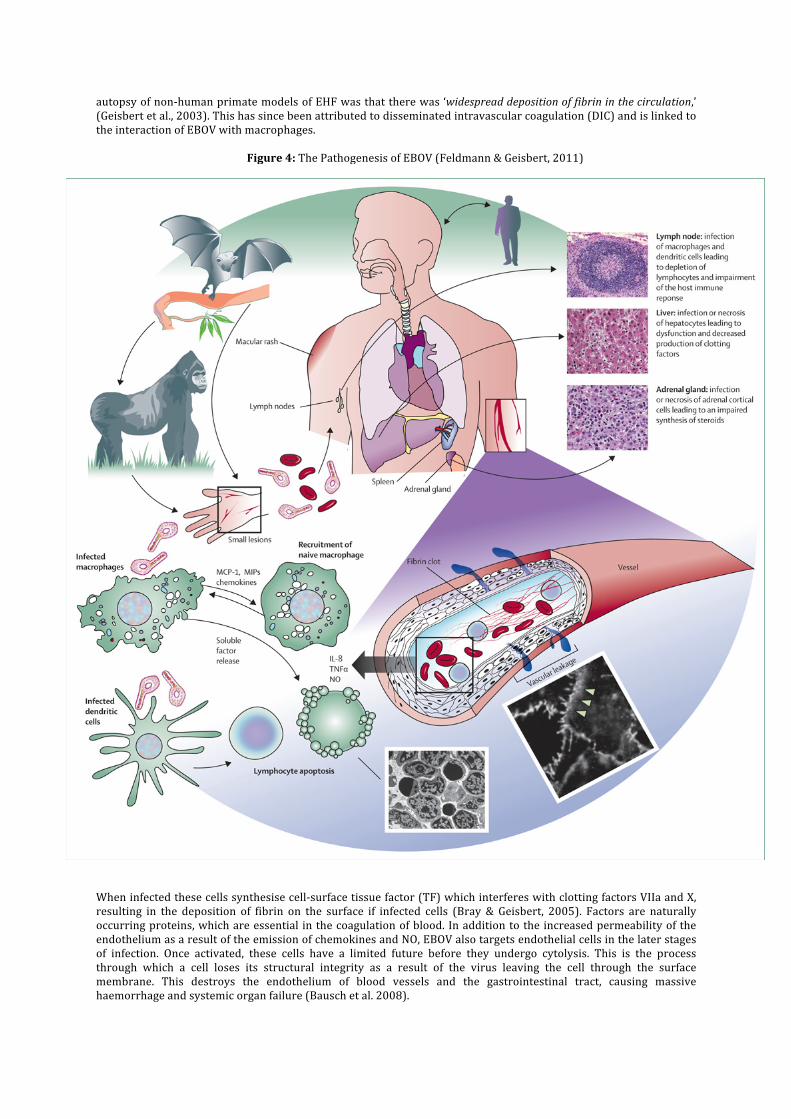
autopsy of non-‐human primate models of EHF was that there was ‘widespread deposition of fibrin in the circulation,’ (Geisbert et al., 2003). This has since been attributed to disseminated intravascular coagulation (DIC) and is linked to the interaction of EBOV with macrophages.
Figure 4: The Pathogenesis of EBOV (Feldmann & Geisbert, 2011)
When infected these cells synthesise cell-‐surface tissue factor (TF) which interferes with clotting factors VIIa and X, resulting in the deposition of fibrin on the surface if infected cells (Bray & Geisbert, 2005). Factors are naturally occurring proteins, which are essential in the coagulation of blood. In addition to the increased permeability of the endothelium as a result of the emission of chemokines and NO, EBOV also targets endothelial cells in the later stages of infection. Once activated, these cells have a limited future before they undergo cytolysis. This is the process through which a cell loses its structural integrity as a result of the virus leaving the cell through the surface membrane. This destroys the endothelium of blood vessels and the gastrointestinal tract, causing massive haemorrhage and systemic organ failure (Bausch et al. 2008).

Prognosis If a positive diagnosis of EBOV is made, then the outlook for a patient is relatively bleak as there is currently no specific therapeutic treatment. The standard practice is known as supportive treatment in which the patient is administered drugs, both intravenously and orally, that rehydrate and alleviate symptoms or pain caused as a result of the infection (Roddy et al., 2011). Whilst it is little comfort to neither patients nor their relatives, each outbreak of EBOV provides significant advances both in terms of pharmaceutical treatments and the quality of treatment delivered.
Existing Post-‐Exposure Treatments
Post exposure treatments are under intensive research and several lines of investigation are being made, looking at potential weaknesses in the pathogenesis of the virus and counteracting some of the effects. One of the main problems of developing both prophylactic and post exposure treatments is that clinical investigations are limited to in vitro and in vivo in non human primates. In some cases, the first in vivo testing is done on mice, for which EBOV must be altered to achieve a successful infection. The result is that while a treatment may be successful for this particular strain of EBOV, it may not be as effective, if at all, for human EBOV (Rutgers, 2011). As such there are currently no licensed antiviral drugs for EBOV (Bausch et al., 2008). One direction of current research is centred about the two glycoproteins on the exterior of the EBOV capsid. Together these proteins enable the infection of cells, binding to Niemann – Pick C1 (NPC1) Cholesterol Transporters, which are embedded into the phospholipid bilayer of the cell membrane. This is confirmed by means that cells which lack this molecule, such as those in NPC1 Disease patients, are resistant to the infection of EBOV (Carrette et al., 2011). Glycoprotein 1 (GP1) attaches the virion to the host cell membrane whereas Glycoprotein 2 (GP2) fuses the host membrane to the viral envelope, permitting the virus’ RNA to enter the cell through endocytosis. Essentially, each glycoprotein has two conformations – pre and post attachment (Rutgers, 2011). This fact is of particular importance when it is considered that EBOV antibodies, found in a survivor of the 1995 ZEBOV outbreak, in Kikwit, Democratic Republic of The Congo act upon the tertiary structure of these proteins. KZ52 prevents the change of conformation from pre –attachment to post –attachment, therefore removing the virion’s ability to infect a cell (Jeffrey et al., 2008). During the same outbreak, this was used as a form of treatment in which 8 patients, recently diagnosed with EBOV, were administered antibody-‐containing blood transfusions from ‘5 convalescent patients.’ This was a success because out of the sample, the fatality rate was only 12.5% (1 fatality), which is significantly lower than the 80% fatality rate of the ZEBOV Kikwit epidemic (Mupapa et al., 1999). Despite this initial success, there was significant failure in laboratory testing succeeding the outbreak on macaques, which lead to the dissolution of this course of treatment (Oswald et al., 2007). A successor to this is the administration of monoclonal KZ52 antibodies. These are clones of KZ52, which were isolated from recovering patients of the Kikwit outbreak. Clinical trials of this treatment in which 3 doses are administered 3 days apart, starting 24 hours post-‐infection have shown 100% success rates in Non-‐human primates, although this diminishes to 50% if the treatment starts at 48 hours post infection (Qiu et al., 2012). An alternative pathway for treating EBOV could be based on anti-‐apoptotic therapies designed to reduce the number of lymphocytes lost throughout the course of the infection. As B-‐lymphocytes produce antibodies specific to each pathogen, it would help to reinstate the otherwise defective immune system brought about by the Ebola virus (Parrino et al., 2007). This would appear to be another promising solution because the virus first infects the immune system before inducing any visible effect on any other tissue as shown in the table below. This means that given the therapeutic help, the immune system would retain sufficient strength to counteract the virus, before the activation of endothelial cells posed a life-‐threatening risk.
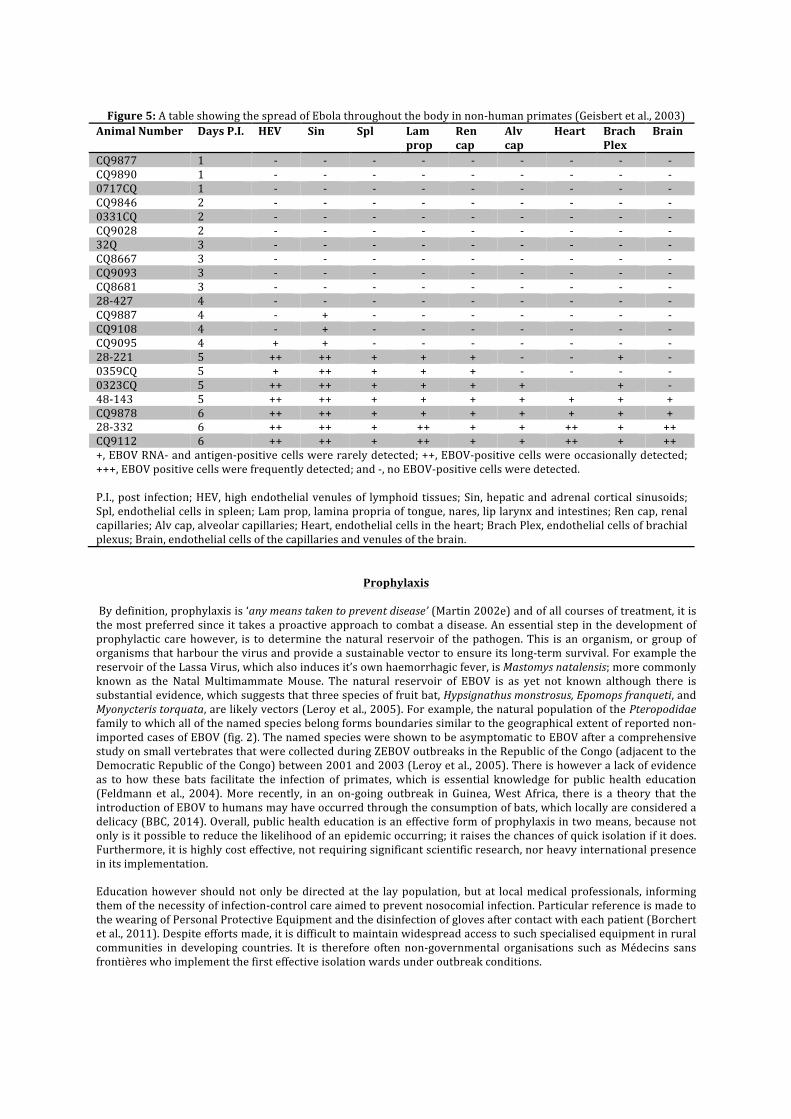
Figure 5: A table showing the spread of Ebola throughout the body in non-‐human primates (Geisbert et al., 2003)
Animal Number Days P.I. HEV Sin Spl Lam prop
Ren cap
Alv cap
Heart Brach Plex
Brain
CQ9877 1 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ9890 1 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ 0717CQ 1 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ9846 2 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ 0331CQ 2 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ9028 2 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ 32Q 3 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ8667 3 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ9093 3 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ8681 3 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ 28-‐427 4 -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ9887 4 -‐ + -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ9108 4 -‐ + -‐ -‐ -‐ -‐ -‐ -‐ -‐ CQ9095 4 + + -‐ -‐ -‐ -‐ -‐ -‐ -‐ 28-‐221 5 ++ ++ + + + -‐ -‐ + -‐ 0359CQ 5 + ++ + + + -‐ -‐ -‐ -‐ 0323CQ 5 ++ ++ + + + + + -‐ 48-‐143 5 ++ ++ + + + + + + + CQ9878 6 ++ ++ + + + + + + + 28-‐332 6 ++ ++ + ++ + + ++ + ++ CQ9112 6 ++ ++ + ++ + + ++ + ++ +, EBOV RNA-‐ and antigen-‐positive cells were rarely detected; ++, EBOV-‐positive cells were occasionally detected; +++, EBOV positive cells were frequently detected; and -‐, no EBOV-‐positive cells were detected. P.I., post infection; HEV, high endothelial venules of lymphoid tissues; Sin, hepatic and adrenal cortical sinusoids; Spl, endothelial cells in spleen; Lam prop, lamina propria of tongue, nares, lip larynx and intestines; Ren cap, renal capillaries; Alv cap, alveolar capillaries; Heart, endothelial cells in the heart; Brach Plex, endothelial cells of brachial plexus; Brain, endothelial cells of the capillaries and venules of the brain.
Prophylaxis
By definition, prophylaxis is ‘any means taken to prevent disease’ (Martin 2002e) and of all courses of treatment, it is the most preferred since it takes a proactive approach to combat a disease. An essential step in the development of prophylactic care however, is to determine the natural reservoir of the pathogen. This is an organism, or group of organisms that harbour the virus and provide a sustainable vector to ensure its long-‐term survival. For example the reservoir of the Lassa Virus, which also induces it’s own haemorrhagic fever, is Mastomys natalensis; more commonly known as the Natal Multimammate Mouse. The natural reservoir of EBOV is as yet not known although there is substantial evidence, which suggests that three species of fruit bat, Hypsignathus monstrosus, Epomops franqueti, and Myonycteris torquata, are likely vectors (Leroy et al., 2005). For example, the natural population of the Pteropodidae family to which all of the named species belong forms boundaries similar to the geographical extent of reported non-‐imported cases of EBOV (fig. 2). The named species were shown to be asymptomatic to EBOV after a comprehensive study on small vertebrates that were collected during ZEBOV outbreaks in the Republic of the Congo (adjacent to the Democratic Republic of the Congo) between 2001 and 2003 (Leroy et al., 2005). There is however a lack of evidence as to how these bats facilitate the infection of primates, which is essential knowledge for public health education (Feldmann et al., 2004). More recently, in an on-‐going outbreak in Guinea, West Africa, there is a theory that the introduction of EBOV to humans may have occurred through the consumption of bats, which locally are considered a delicacy (BBC, 2014). Overall, public health education is an effective form of prophylaxis in two means, because not only is it possible to reduce the likelihood of an epidemic occurring; it raises the chances of quick isolation if it does. Furthermore, it is highly cost effective, not requiring significant scientific research, nor heavy international presence in its implementation. Education however should not only be directed at the lay population, but at local medical professionals, informing them of the necessity of infection-‐control care aimed to prevent nosocomial infection. Particular reference is made to the wearing of Personal Protective Equipment and the disinfection of gloves after contact with each patient (Borchert et al., 2011). Despite efforts made, it is difficult to maintain widespread access to such specialised equipment in rural communities in developing countries. It is therefore often non-‐governmental organisations such as Médecins sans frontières who implement the first effective isolation wards under outbreak conditions.
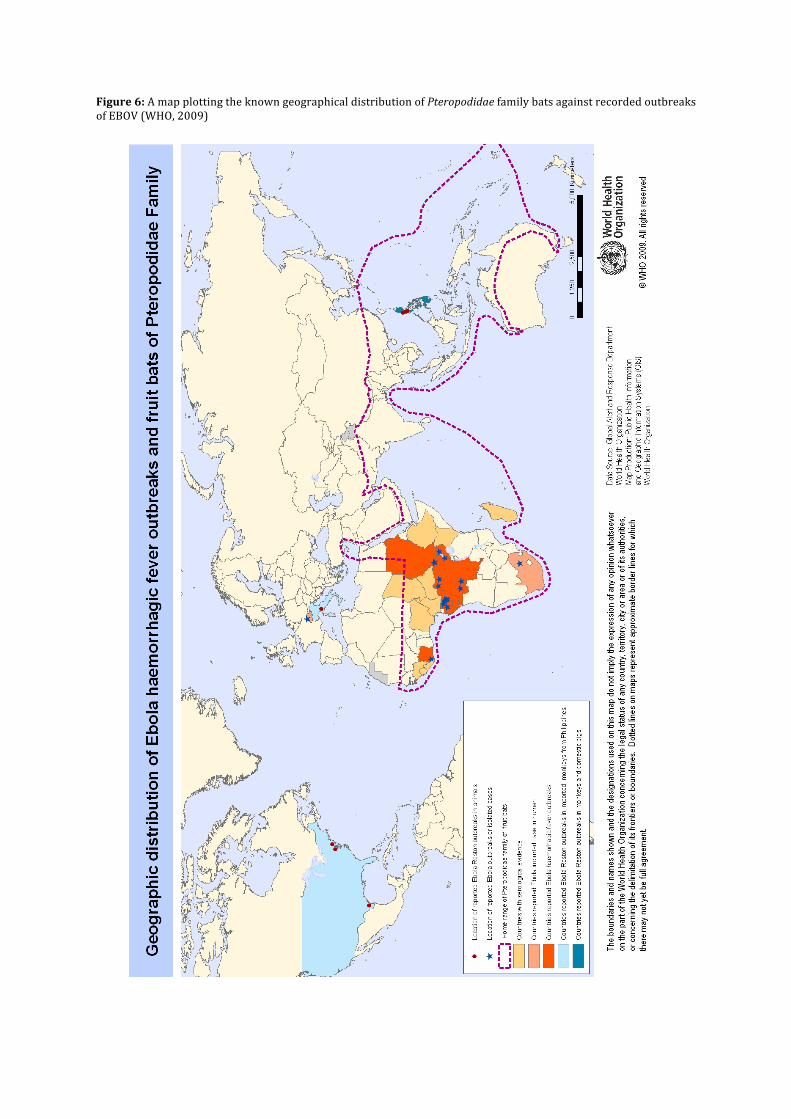
Figure 6: A map plotting the known geographical distribution of Pteropodidae family bats against recorded outbreaks of EBOV (WHO, 2009)

A more absolute prophylactic is vaccination, which is believed to predate the 16th Century with its origins in eastern Asia (Lombard et al., 2007), before Edward Jenner’s breakthrough with the development of a vaccination for Smallpox in 1796. The term vaccination was coined by Louis Pasteur in honour of Jenner, who used Cow Pox virus, Vaccina virus, to prevent the infection of Smallpox – Valoria major/minor. The latter was certified to be eradicated from the globe by WHO in 1979 (WHO, 2014). Vaccines function by providing antigens of a pathogen, triggering an immune response. In doing so, the body creates antibodies that can then be readily called upon to prevent an infection becoming serious. An antigen is a chemical found on the exterior surface of cells, which is used to recognise, whether a cell is foreign. If a foreign antigen is detected, an immune response, whereby a unique antibody to the antigen is synthesised in massive quantities, is triggered. These antibodies bind to the antigens and to other antibodies, isolating a number of pathogens, therefore enabling phagocytosis – the consumption of a pathogen by certain types of white blood cell. It is important to note that vaccination is significantly different to inoculation, which is the practice in which the real pathogen is administered in doses not considered to be life threatening. It is now however, thought to be unethical because there were a high percentage of cases where the pathogen could take a hold on the body and as a result, it is a discontinued form of prophylaxis.
Figure 7: A table displaying generic types of vaccine (NIAID, 2013) Vaccine Category Description Inactivated Vaccine: A pathogen is grown in a culture and then killed using
heat or formaldehyde. Whilst the dead pathogens cannot infect nor harm, the viral capsid or bacterial walls remain intact to enable recognition by the immune system.
Attenuated Vaccine: Live pathogens are administered, but only those with low or localised replication.
Virus-‐like Particle Vaccine: The viral proteins of the capsid will form to create ‘empty’ viruses that do not contain a nucleic acid.
Subunit Vaccine: Presents an antigen to the immune system without introducing the pathogen itself. This effectively tricks the immune system into believing there is a minor infection, creating antibodies in response.
DNA Vaccine: The genetic material of the vaccine is converted to dsDNA, which is then isolated and prepared in solution to create a vaccine that provokes an immune response upon administration.
Recombinant Vector Vaccine: Similar to DNA Vaccine, but a virus or bacteria acts as a carrier molecule or vector to the nucleic acid.
Although there are currently no licensed vaccines for any strain of EBOV, there is significant international interest in their development. There are two main categories of vaccine: replicating competent and non-‐replicating. The former has significant advantages over the latter in that the vaccine is more durable, typically requires fewer immunisations and provides longer-‐lasting immunity. On the other hand, replicating-‐competent vaccines are live and have the potential to become unsafe in the presence of another pathogen. This creates a dilemma over whether it is morally better to administer a more effective, yet potentially dangerous vaccine or one that is safer but also necessitates stronger doses, more immunisations and leads to a shorter period of immunisation (Falzarano et al., 2012). Replication-‐Competent Vaccines
Recombinant Vesicular Stomatitis Virus (rVSV) This virus has proven to be a useful attenuated and replication-‐competent vector for vaccines acting against a number of pathogens including influenza, hepatitis B and HPV amongst several others. It is genetically engineered so that the VSV glycoprotein is replaced by the GP of EBOV to create viral particles. These are structurally similar to VSV, but instead contain EBOV GP rather than VSV GP (Jones et al. 2005). Whilst initial clinical trials were successful, subsequent trials using a combination of SEBOV, ZEBOV and the other virus of the filoviridae family, Marburg (MARV) were less so. It was found that whilst rVSV-‐MARV GP adequately immunised against the different strains of MARV, rVSV-‐ZEBOV GP did not provide any immunity to SEBOV, which is a different species. The lack of cross-‐protection is a significant downfall, however it is feasible that an injection may contain several vaccines that offer protection for filoviridae as an entity. It is considered that the fact rVSV vaccines can be administered orally, nasally and intramuscularly would be an advantage in the extra-‐rural community (Falzarano et al., 2012). Recombinant Human Parinfluenza Virus 3 (rHPIV3) HPIV3 is a common paediatric respiratory virus, which can be genetically modified to express ZEBOV GP forming a rHPIV3-‐ZEBOV GP complex, in the same manner as rVSV-‐ZEBOV GP. It is known to only provide

immunity for ZEBOV, but studies using non-‐human primates have shown that 2 doses are required to obtain 100% survival (Bukreyev et al., 2007). The major disadvantage with rHPIV3 is that nearly all adults have gained immunity to it through childhood, which may restrict the function of rHPIV3 as a vaccine platform (Falzarano et al. 2012). The fact that rHPIV3 can be given in intranasal does however, is an advantage as it enables easier administration in rural areas (Falzarano et al., 2012).
Non-‐replicating Vaccines
DNA Whereas the first recombinant vaccines have an EBOV GP within the fabric of their own protein capsid, the DNA vaccine contains 3 plasmids of DNA that code for the SEBOV GP and nucleoprotein (NP) (Martin et al., 2006). Whilst DNA vaccines bring many benefits including the fact that they can be adapted quickly as pathogens evolve, the level of immunity provided has been less than satisfactory. Therefore it is highly unlikely that a stand-‐a-‐lone DNA vaccine will be pursued in future research, but rather in combination with another vector (Falzarano et al., 2012) Recombinant Adenovirus 5 (rAd5) In many ways, this vaccine platform is better than the combined DNA/rAd5 approach due to the far more compact vaccination course. This means that in the event of an outbreak or bioterrorism event, the vaccine could be administered swiftly, providing 100% survival 4 weeks post vaccination in non-‐human primates. In an identical manner to other recombinant vaccine platforms, rAd5 is genetically modified so that it contains the ZEBOV GP. Similarly, the immunity offered by rAd5-‐ZEBOV GP is thought to be species specific although a pan-‐filovirus adenovirus platform has been developed containing SEBOV GP, EBOV GO and the GPs of different strains of MARV. A further disadvantage of rAd5-‐ZEBOV GP is that it requires a single large dose of 1010 particles in order to ensure survival (Falzarano et al., 2012). Combined DNA and rAd5 The combination platform of DNA/rAd5 has received significant scientific interest, as it was the first to demonstrate 100% immunogenicity in non-‐human primates. The significant benefit of combining the two vectors is that the immune response is significantly increased in comparison when they are applied separately. Within the combination, there was a ratio of 3:1 between the DNA vaccine, coding for ZEBOV GP, SEBOV GP, CIEBOV GP and ZEBOV NP; and the rAd5-‐ZEBOV GP vaccine. When presented with ZEBOV, SEBOV or CIEBOV, there was no development of disease. In addition to the species mentioned, despite a significant difference in the primary structure of its GP, BEBOV is also protected against, contrary to logical assumptions (Hensley et al., 2010). The principle disadvantage with this vaccine is that the course of vaccinations can take up to 18 months, which in filovirus endemic regions is verging on logistical impossibility (Falzarano et al., 2012).
Virus Like Particle There are several key advantages that VLP has over a traditional inactivated vaccine. These include a high level of safety because VLPs are non-‐replicating and an ability to engage the wider immune response. A VLP containing GP, NP and VP40 derived from EBOV will assume the approximate morphology of the original pathogen. In clinical trials, eVLP protected 100% of the nonhuman primate cases, which failed to display any of the characteristic symptoms of an EBOV infection (Warfield et al., 2007). In all the laboratory testing, eVLP was produced using mammalian cells. This is not however cost effective and alternative production methods such as using insect cells are currently being studied (Falzarano et al., 2012). ∆VP30 ZEBOV In order to obtain the vaccine platform, the gene coding for VP30 within the EBOV genome is replaced with a reporter gene called neomycin. A reporter gene acts a marker and can help determine the dispersal of a gene throughout an organism. The result of this replacement is the generation of Ebola ∆VP30-‐neo, which is then encouraged to replicate. When a vaccine consisting of Ebola ∆VP30-‐neo was administered to mice at a quantity of 106 particles, all the specimens were resistant to the modified ZEBOV strain. Although this is a success in the development of this vaccine platform, it is unlikely to be progressed due to the fact that 85% of the ZEBOV genome remains intact. This could potentially give rise to infection, which is considered to be a risk that outweighs the potential cross-‐protection benefits (Falzarano et al., 2012).
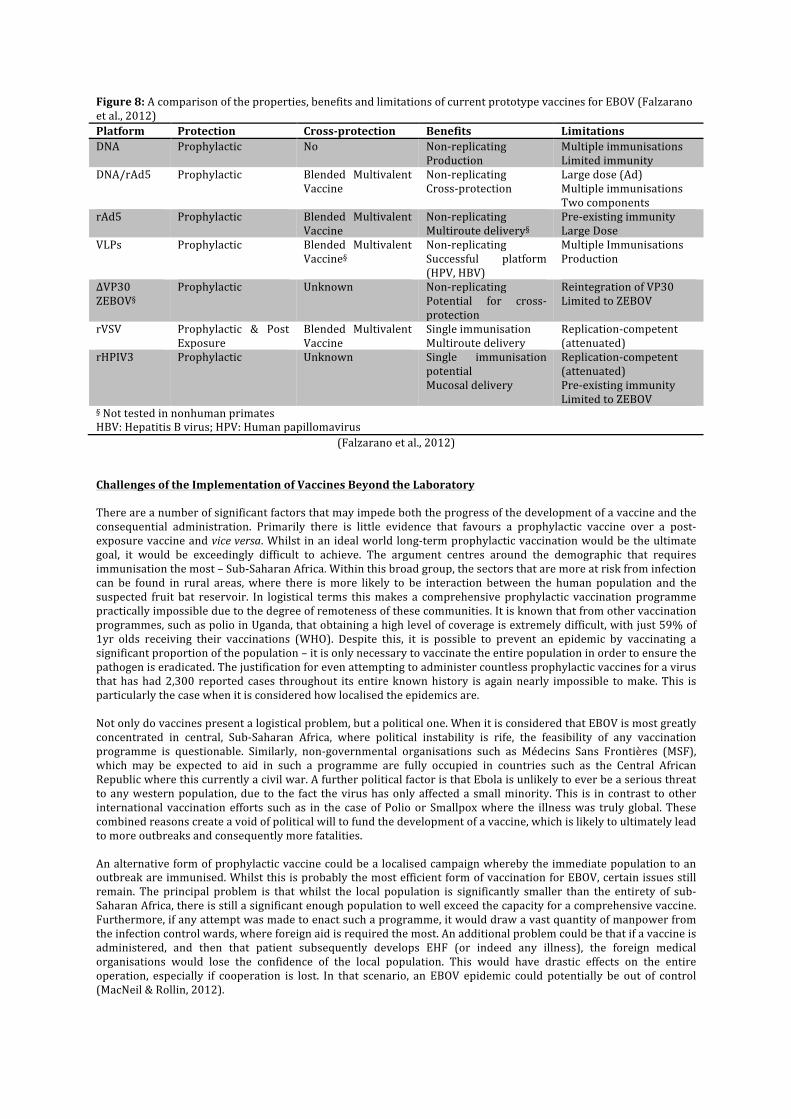
Figure 8: A comparison of the properties, benefits and limitations of current prototype vaccines for EBOV (Falzarano et al., 2012) Platform Protection Cross-‐protection Benefits Limitations DNA Prophylactic No Non-‐replicating
Production Multiple immunisations Limited immunity
DNA/rAd5 Prophylactic Blended Multivalent Vaccine
Non-‐replicating Cross-‐protection
Large dose (Ad) Multiple immunisations Two components
rAd5 Prophylactic Blended Multivalent Vaccine
Non-‐replicating Multiroute delivery§
Pre-‐existing immunity Large Dose
VLPs Prophylactic Blended Multivalent Vaccine§
Non-‐replicating Successful platform (HPV, HBV)
Multiple Immunisations Production
∆VP30 ZEBOV§
Prophylactic Unknown Non-‐replicating Potential for cross-‐protection
Reintegration of VP30 Limited to ZEBOV
rVSV Prophylactic & Post Exposure
Blended Multivalent Vaccine
Single immunisation Multiroute delivery
Replication-‐competent (attenuated)
rHPIV3 Prophylactic Unknown Single immunisation potential Mucosal delivery
Replication-‐competent (attenuated) Pre-‐existing immunity Limited to ZEBOV
§ Not tested in nonhuman primates HBV: Hepatitis B virus; HPV: Human papillomavirus
(Falzarano et al., 2012)
Challenges of the Implementation of Vaccines Beyond the Laboratory There are a number of significant factors that may impede both the progress of the development of a vaccine and the consequential administration. Primarily there is little evidence that favours a prophylactic vaccine over a post-‐exposure vaccine and vice versa. Whilst in an ideal world long-‐term prophylactic vaccination would be the ultimate goal, it would be exceedingly difficult to achieve. The argument centres around the demographic that requires immunisation the most – Sub-‐Saharan Africa. Within this broad group, the sectors that are more at risk from infection can be found in rural areas, where there is more likely to be interaction between the human population and the suspected fruit bat reservoir. In logistical terms this makes a comprehensive prophylactic vaccination programme practically impossible due to the degree of remoteness of these communities. It is known that from other vaccination programmes, such as polio in Uganda, that obtaining a high level of coverage is extremely difficult, with just 59% of 1yr olds receiving their vaccinations (WHO). Despite this, it is possible to prevent an epidemic by vaccinating a significant proportion of the population – it is only necessary to vaccinate the entire population in order to ensure the pathogen is eradicated. The justification for even attempting to administer countless prophylactic vaccines for a virus that has had 2,300 reported cases throughout its entire known history is again nearly impossible to make. This is particularly the case when it is considered how localised the epidemics are. Not only do vaccines present a logistical problem, but a political one. When it is considered that EBOV is most greatly concentrated in central, Sub-‐Saharan Africa, where political instability is rife, the feasibility of any vaccination programme is questionable. Similarly, non-‐governmental organisations such as Médecins Sans Frontières (MSF), which may be expected to aid in such a programme are fully occupied in countries such as the Central African Republic where this currently a civil war. A further political factor is that Ebola is unlikely to ever be a serious threat to any western population, due to the fact the virus has only affected a small minority. This is in contrast to other international vaccination efforts such as in the case of Polio or Smallpox where the illness was truly global. These combined reasons create a void of political will to fund the development of a vaccine, which is likely to ultimately lead to more outbreaks and consequently more fatalities. An alternative form of prophylactic vaccine could be a localised campaign whereby the immediate population to an outbreak are immunised. Whilst this is probably the most efficient form of vaccination for EBOV, certain issues still remain. The principal problem is that whilst the local population is significantly smaller than the entirety of sub-‐Saharan Africa, there is still a significant enough population to well exceed the capacity for a comprehensive vaccine. Furthermore, if any attempt was made to enact such a programme, it would draw a vast quantity of manpower from the infection control wards, where foreign aid is required the most. An additional problem could be that if a vaccine is administered, and then that patient subsequently develops EHF (or indeed any illness), the foreign medical organisations would lose the confidence of the local population. This would have drastic effects on the entire operation, especially if cooperation is lost. In that scenario, an EBOV epidemic could potentially be out of control (MacNeil & Rollin, 2012).

Although vaccines for any species of EBOV are unlikely to be approved in the nest 5 years (Falzarano et al., 2012), scientific advances will likely create an effective vaccine. Some of the prototype vaccines already show significant promise and could be highly effective when administered, but in the case of Ebola, the necessary clinical trials to licence a vaccine are considered to be unethical due to the high probability of death if the vaccine fails. The main issue however is that research is costly and there is not sufficient political desire amongst developed countries to fund the research of the vaccine. That is however of little relevance given the practical difficulties of the implementation of any vaccine for Ebola. It is highly likely therefore that any approved vaccine will be administered only to medical professionals who work in close quarters with EBOV. As is the way with the human race, it is probable that efforts will concentrate more on the solution, or in this case post-‐exposure treatment, rather than preventing it. Despite this, it is imperative that the natural reservoir for all the species of EBOV is known so that at least a certain degree of public health education can occur. For the human population of sub-‐Saharan Africa, that is likely the best method of prophylaxis they will receive in the foreseeable future.

References: Baize S, Leroy E, Georges-‐Courbot M, Capron M, Lansoud-‐Soukate J, et al. (1999) ‘Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-‐infected patients.’ Natural Medicine 5 (4) pp423-‐6 Baltimore D (1971) ‘Expression of Animal Virus Genomes.’ Bacteriological Reviews, 35 (3) pp235-‐238 Banchereau J, Steinman R (1998) ‘Dendritic cells and the control of immunity.’ Nature, 392 pp245-‐252 Bausch D, Sprecher A, Jeffs B, Boumandouki P (2008) ‘Treatment of Marburg and Ebola haemorrhagic fevers: A strategy fir testing new drugs and vaccines under outbreak conditions.’ Antiviral Research, 78 pp150-‐161 BBC (2014) ‘West Africa on Ebola virus high alert’ Available at http://www.bbc.co.uk/news/health-‐26740686 [Access date 25/3/14] Borchert M, Mutyaba I, Van Kerkhove M, Lutwama J, Luwaga H et al. (2011) ‘Ebola haemorrhagic fever outbreakin Msindi District, Uganda; outbreak description and lessons learned.’ BMC Infectious Diseases 11 Available at http://www.biomedcentral.com/content/pdf/1471-‐2334-‐11-‐357.pdf [Access date: 30/9/13] Bray M, Geisbert T (2005) ‘Ebola virus: The role of macrophages and dendritic cells in the pathogenesis of Ebola h[a]emorrhagic fever.’ The International Journal of Biochemistry and Cell Biology, 37 pp1560-‐1566 Bray M, Mahanty S (2003) ‘Ebola H[a]emorrhagic Fever and Septic Shock.’ The Journal of Infectious Diseases 188 pp1613-‐1617 Brown TA (2002) Genomes. 2nd edition. Oxford: Wiley-‐Liss. Chapter 1, The Human Genome. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21134/ [Access date 03/02/14] Bukreyev A, Rollin P, Tate M, Yang L, Zaki S et al. (2007) ‘Successful topical respiratory tact immunisation of primates against Ebola Virus.’ Journal of Virology 81 (12) Carette J, Raaben M, Wong A, Herbert S, Obernosterer G et al. (2011) ‘Ebola virus entry requires the cholesterol transporter Neimann-‐Pick C1.’ Nature 477 (7364) pp340-‐343 Crawford DH (2000a) The Invisible Enemy: A Natural History of Viruses Oxford University Press, Oxford pp13-‐14
Crawford DH (2000b) The Invisible Enemy: A Natural History of Viruses Oxford University Press, Oxford pp14-‐15 Falzarano D, Geisbert T, Feldman H (2012) ‘Progress in filovirus vaccine development: evaluating the potential for clinical use.’ Expert Revue Vaccines 10 (1) pp63-‐77 Feldmann H, Geisbert T (2011) ‘Ebola Haemorrhagic Fever.’ Lancet 377 (9768) pp849-‐862 Feldmann H, Wahl-‐Jensen V, Jones S, Ströher U (2004) ‘Ebola virus ecology: a continuing mystery.’ Trends in Microbiology 12 (10) pp433-‐437 Fleck F (ed.) (2011) Bugs, drugs and smoke World Health Organisation pp121-‐122 Geisbert T, Young H, Jharling P, Davis K, Larsen T, et al. (2003) ‘Pathogenesis of Ebola H[a]emorrhagic Fever in Primate Models.’ American Journal of Pathology 163 (6) pp2371-‐2382 Hensley L, Mulangu S, Asiedu C, Johnson J, Honko A et al. (2010) ‘Deomstration of cross-‐protectivie vaccine immunity against an emerging pathogenic Ebolavirus Species.’ PLoS Pathogens 6 (5) Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2873919/pdf/ppat.1000904.pdf [Access date 24/02/14] International Committee on Taxonomy of Viruses (ICTV) (2012), Virus Taxonomy, Available at http://www.ictvonline.org/virusTaxonomy.asp [Access date: 04/02/2014] Jeffrey E, Fusco M, Hessell A, Oswald W, Burton D, Ollmann Saphire E (2008) ‘Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor.’ Nature 454 pp177-‐182 Jones S, Feldmann H, Ströher U, Geisbert J, Fernando L et al. (2005) ‘Live attenuated ecombinant vaccine protects non human primates against Ebola and Marburg viruses.’ Natural Medicine 11(7) pp786-‐790 Leroy E, Kumulungui B, Pourrut X, Rouquet P, Hassanin A et al. (2005) ‘Fruit Bats as reservoirs of Ebola Virus.’ Nature 438 pp575-‐576 Lombard M, Pastoret P, Moulin A (2007) ‘A Brief History of Vaccines and Vaccination.’ Revue Scientifique et technique 26 (1) pp29-‐48 MacNeil A, Rollin P (2012) ‘Ebola and Marburg H[a]emorrhagic Fevers: Neglected Tropical Diseases?’ PLoS Neglected Tropical Diseases 6 (6) Available at

http://www.plosntds.org/article/fetchObject.action?uri=info%3Adoi%2F10.1371%2Fjournal.pntd.0001546&representation=PDF [Access date 24/02/14] Martin E (ed.) (2002a) Concise Colour Medical Dictionary Oxford University Press, Oxford “necrosis” p459 Martin E (ed.) (2002b) Concise Colour Medical Dictionary Oxford University Press, Oxford “apoptosis” pp44-‐45 Martin E (ed.) (2002c) Concise Colour Medical Dictionary Oxford University Press, Oxford “differential diagnosis” p195 Martin E (ed.) (2002d) Concise Colour Medical Dictionary Oxford University Press, Oxford “neutrophil” p469 Martin E (ed.) (2002e) Concise Colour Medical Dictionary Oxford University Press, Oxford “prophylaxis” p469 Martin E, Hine RS (2000a) Dictionary of Biology Oxford University Press, Oxford “Virus” pp620-‐621 Martin E, Hine RS (2000b) Dictionary of Biology Oxford University Press, Oxford “Retrovirus” p519 Martin J, Sullivan N, Enama M, Gordon I, Roederer M et al. (2006) ‘A DNA Vaccine for Ebola Virus Is Safe and Immunogenic in a Phase 1 Clinical Trial.’ Clinical and Vaccine Immunology 13 (11) Mupapa K, Massamba M, Kibadi K, Kuvula K, Bwaka A et al. (1999) ‘Treatment of Ebola H[a]emorrhagic Fever with Blood Transfusions from Convalescent Patients.’ The Journal of Infectious Diseases 179 (suppl 1) ppS18-‐S23 National Institute of Allegry and Infectious Diseases (NIAID) (2010) Transmission of Ebola/Marburg Viruses, Available at http://www.niaid.nih.gov/topics/ebolaMarburg/understanding/Pages/transmission.aspx [Access date: 16/02/14] National Institute of Allegry and Infectious Diseases (NIAID) (2013) Types of Vaccine http://www.niaid.nih.gov/topics/vaccines/understanding/pages/typesvaccines.aspx [Access date: 22/02/14] OpenStax College (2013) ‘Viruses.’ Available at http://cnx.org/content/m45541/latest/#fig-‐ch17_01_02 [Access date: 22/03/14] Oswald W, Geisbert T, Davus K, Geisbert J, Sullivan N et al. (2007) ‘Neutralising Antibody Fails to Impact the Course of Ebola Virus Infection in Monkeys.’ PLoS Pathogens 3(1)
Available at http://www.plospathogens.org/article/fetchObject.action?uri=info%3Adoi%2F10.1371%2Fjournal.ppat.0030009&representation=PDF [Access date: 23/02/14] Parrino J, Hotchkiss R, Bray M (2007) ‘Immune Cell Apoptosis Prevention as Potentential Therapy for Severe Infections.’ Emerging Infectious Diseases 13 (Internet Serial) Available at: http://wwwnc.cdc.gov/eid/article/13/2/06-‐0963.htm [Access date: 22/02/14] Qui X, Audet J, Wong G, Pillet S, Bello A et al. (2012) ‘Successful treatment of ebola virus-‐infected cynomolgus macaques with monoclonal antibodies.’ Science Translational Medicine 4 (138) pp138-‐181 Roddy P, Colebunders R, Jeffs B, Palma P, Van Herp M, Borchert M (2011) ‘Filovirus H[a]emorrhagic Fever Outbreak Case Management: A Review of Current and Future Treatment Options.’ The Journal of Infectious Diseases 204 (suppl 3) pp791-‐795 Roddy P, Howard N, Van Kerkhove MD, Lutwama J, Wamala J, et al. (2012) ‘Clinical manifestations and case management of Ebola Haemorrhagic Fever caused by a newly identified virus strain, Bundibugyo, Uganda 2007-‐2008.’ PLoS ONE, 7(12) Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3532309/pdf/pone.0052986.pdf [Access date 20/08/13] Rutgers (2011) Molecular Anatomy Project, Ebola Virus, Available at http://maptest.rutgers.edu/drupal/?q=node/453 [Access date 03/02/14] Smith T (ed.) (1995a) Complete Family Health Encyclopaedia The British Medical Association, Dorling Kindersley, London “Viruses” p1055 Soper R (ed.) (1997a) Biological Science 1 & 2 Cambridge University Press, Cambridge 2.41 Soper R (ed.) (1997b) Biological Science 1 & 2 Cambridge University Press, Cambridge 2.43 Stannard LM (1995) Division of Medicial Virology, University of Cape Town. Available at http://web.uct.ac.za/depts/mmi/stannard/emimages.html [Access date 03/11/13] Wikipedia (2013) Virus. Available at http://en.wikipedia.org/wiki/Virus [Access date 03/11/13] Wikipedia (2014) Sense (Molecular Biology). Available at https://en.wikipedia.org/wiki/Sense_(molecular_biology) [Access date 05/01/14]

World Health Organisation (2009) ‘Geographic distribution of Ebola haemorrhagic fever outbreaks and fruit bats of Pteropodidae Family.’ Available at http://www.who.int/csr/disease/ebola/Global_EbolaOutbreakRisk_20090510.png?ua=1 [Access date: 29/9/13] World Health Organisation (2012). Ebola Haemorrhagic Fever. Fact sheet 103 Available at http://www.who.int/mediacentre/factsheets/fs103/en/ [Access date 21/08/13]






![BioDrugs Volume Issue 2013 [Doi 10.1007%2Fs40259-013-0046-1] Choi, Jin Huk; Croyle, Maria a. -- Emerging Targets and Novel Approaches to Ebola Virus Prophylaxis and Treatment](https://img.pdfslide.net/doc/110x75/55cf94ab550346f57ba3a0b5/biodrugs-volume-issue-2013-doi-1010072fs40259-013-0046-1-choi-jin-huk.jpg)






















