Embed Size (px)
Citation preview

REVIEW www.rsc.org/npr | Natural Product Reports
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
FView Article Online / Journal Homepage / Table of Contents for this issue
Total biosynthesis: in vitro reconstitution of polyketide and nonribosomalpeptide pathways
Elizabeth S. Sattely,a Michael A. Fischbachb and Christopher T. Walsh*a
Received 28th February 2008
First published as an Advance Article on the web 23rd May 2008
DOI: 10.1039/b801747f
Covering: 1990 to February 2008
This review surveys efforts to reconstitute key steps in polyketide and nonribosomal peptide
biosynthetic pathways with purified enzymes and substrates; 344 references are cited.
1 Introduction
2 Chemical logic and enzymatic machinery of NRPS and
PKS assembly lines
2.1 Chemical logic
2.2 Enzymatic machinery
2.3 Type I and type II enzymes
2.4 Four stages in NRP and PK biosynthesis
3 Phosphopantetheinyl transferases: the essential post-
translational priming catalysts
4 Just-in-time fashioning of dedicated monomers as
building blocks for assembly lines
4.1 Hydroxyphenylglycine and dihydroxyphenyglycine
building blocks for glycopeptide antibiotics
4.2 Halogenation of amino acid building blocks
4.3 b-Amino acid building blocks
4.4 Other nonproteinogenic building blocks
5 Assembly line action
5.1 Chain initiation: monomer selection, loading and
acylation
5.2 Chain elongation: Claisen, amide, and ester conden-
sations
5.2.1 C domains: condensation and heterocyclization
5.2.2 C domains that make ester linkages
5.3 NRP-PK hybrids: module switching and chain elon-
gation
6 Chain tailoring on the assembly line
6.1 Epimerization at Ca
6.2 N- and C-Methylations at Ca and Cb
6.3 Cb branches
6.4 Oxidative transformations on assembly lines
7 Chain release mechanisms and catalytic machinery
8 Post-assembly-line tailoring reactions
8.1 Post-assembly tailoring to produce glycopeptide and
lipoglycopeptide antibiotics
8.2 Tandem glycosylations of aromatic polyketide agly-
cones
8.3 C-Glycosylation: enterobactin to salmochelin
8.4 Post-assembly-line oxygenases
aDepartment of Biological Chemistry and Molecular Pharmacology,Harvard Medical School, 240 Longwood Ave., Boston, MA, 02115, USAbBroad Institute of MIT and Harvard, 7 Cambridge Center, Cambridge,MA, 02142, USA
This journal is ª The Royal Society of Chemistry 2008
9 In vitro reconstitution of complete pathways
9.1 Enterobactin to salmochelin to microcin E492: three
layers of secondary metabolism
9.2 Yersiniabactin reconstitution: an NRP-PK hybrid
9.3 Aromatic polyketide scaffolds from malonyl-CoA
monomers
9.4 Enterocin: a type II PKS pathway that builds a tricyclic
scaffold
9.5 Aminocoumarins: four-component assembly of the
novobiocin scaffold and seven-component assembly of
coumermycin
9.6 Rebeccamycin, staurosporine, violacein, and terrequi-
none: reconstruction of pathways involving tryptophan
oxidative dimerization
9.6.1 Rebeccamycin and staurosporine
9.6.2 Violacein
9.6.3 Terrequinone
10 Lessons learned
11 Acknowledgements
12 References
1 Introduction
Bacterial and fungal genome sequences have radically altered the
framework for the study of natural product biosynthesis. This is
particularly true for molecular scaffolds of nonribosomal peptide
(NRP) and polyketide (PK) origin,1 where biosynthetic genes are
clustered together and can be readily detected by bioinformatic
algorithms. It has long been known that some bacterial phyla are
more prolific producers of NRP and PK products than others,
but a recent review by Donadio et al. in this journal provides
a clear and comprehensive view of NRP, PK and hybrid
NRP-PK biosynthesis from the sequenced genomes of 223
bacteria.2 Donadio et al. noted that in the current genomic
database, g-proteobacteria, actinobacteria, b-proteobacteria,
and firmicutes, in decreasing order, have the highest number of
nonribosomal peptide synthetase (NRPS) and polyketide
synthase (PKS) genes. They also made the correlation that PK
and NRP natural products are rare in bacteria with genomes
#3 Mb. For bacteria with genomes >5 Mb, there is a linear
correlation between genome size and number of PK and NRP
gene clusters, as though these classes of molecules are luxury
items added after a basic self-sustaining metabolic capacity has
Nat. Prod. Rep., 2008, 25, 757–793 | 757

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
been achieved. NRP genes encoding siderophores3 are the most
common among all the NRPS and PKS encoding genes yet
detected, consistent with the observation that iron scarcity often
limits bacterial growth.4
The current bacterial genome database is biased towards the
genomes of pathogens, reflecting strong initial interest in using
genome information to identify new targets for antibiotics. As
bacteria with larger genomes are sequenced,5–9 it is likely that the
library of predicted novel PKS and NRPS biosynthetic gene
clusters will grow and serve as a beacon for selecting organisms
from which to isolate biologically active small molecules with
novel molecular scaffolds.
Microbial genomics and associated bioinformatic analyses
indicate the potential NRP and PK natural product biosynthetic
capacity of a producer organism, but they yield little information
about: (i) which pathways are active or cryptic under given
conditions, (ii) the structures of the encoded natural products,
and (iii) what chemical steps have been achieved by particular
protein catalysts. To close the gap between genome-guided
prediction and current chemical observation, two routes are
possible. One is to culture predicted secondary metabolite
producers, whose genomes have not yet been sequenced, under
a matrix of conditions to elicit the production of secondary
metabolites in the laboratory setting. This is the focus of much
current effort in PK and NRP discovery.10
A complementary approach is to overproduce some or even all
of the proteins in a predicted pathway, characterize their
catalytic activities, and look for intermediates and/or new
Elizabeth S: Sattely
Elizabeth S. Sattely received her
Ph.D. in 2006 from Boston
College with Amir Hoveyda,
where her research focused on
catalytic asymmetric olefin
metathesis and alkaloid total
synthesis through a collabora-
tion with Richard Schrock at
MIT. She is currently a Damon
Runyon Cancer Research Foun-
dation Fellow in the laboratory
of Christopher Walsh, where she
is studying siderophore biosyn-
thetic pathways.
Christopher T: Walsh
Christopher T. Walsh is the Hamilt
macology at HMS. He has served as
and the Department of BCMP at HM
Cancer Institute (1992–1995). His r
758 | Nat. Prod. Rep., 2008, 25, 757–793
products as a way to elucidate the metabolic logic of their
biosynthesis. This topic—the chemical transformations enacted
by NRPS and PKS catalysts—is the subject of this article and
should serve as a complement and companion piece to the recent
genomics review by Donadio et al. in this journal.2
In vitro studies on one or more enzymatic components of
a microbial natural product pathway allow the dissection of its
chemical logic. This approach also sheds light on the means by
which simple monomeric building blocks—typically acyl-CoAs
and amino acids—are forged into molecular scaffolds of
remarkable architectural diversity and functional group
complexity. Understanding the fundamental steps of acyl chain
initiation, elongation, termination, and tailoring is an essential
first step toward investigating how metabolic pathways evolve
and how they may be re-engineered to produce new molecules.
The catalytic rules for assembly line enzymes that have been
deciphered thus far allow the scaffolds of some natural products to
be predicted through bioinformatic analysis of gene sequences.11
However, the information in most gene clusters cannot yet be
read to predict end product structures, especially for gene clusters
with multiple tailoring enzymes, biosynthetic genes in
unusual orientations, or ‘missing’ genes, and for gene clusters that
encode novel small molecules or molecules produced by assembly
line enzymes that operate in an unconventional fashion.12
This review recounts some of the recent progress that has been
made to characterize enzymes in NRP, PK, and NRP-PK
pathways with emphasis on novel chemical transformations and
unusual functional groups that underlie the pharmacologic
Michael A: Fischbach
Michael A. Fischbach received
his Ph.D. in 2007 from Harvard
University, where he studied
siderophore-mediated iron
acquisition in the laboratories of
Christopher Walsh and David
Liu. He is currently an
Instructor of Medicine at
Harvard Medical School, where
he leads a collaborative effort
between HMS and the Broad
Institute focused on actinomy-
cete genome sequencing and
natural products research.
on Kuhn Professor of Biological Chemistry and Molecular Phar-
the Chair of the Department of Chemistry at MIT (1982–1987)
S (1987–1995), and the President and CEO of the Dana Farber
esearch has focused on enzymes, enzyme inhibitors, and antibiotics.
This journal is ª The Royal Society of Chemistry 2008

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
activity and therapeutic interest in many of these molecular
classes. While there have been many studies on individual
enzymes, our primary focus will be on sets of enzymes that act in
tandem to carry out a series of chemical transformations. In
a few cases, reconstitution of a complete pathway has been
achieved, providing insights into how the constituent enzymes
work together and enabling the in vitro biosynthesis of unnatural
analogs by mixing components from different pathways.
Hertweck and colleagues13 have recently reviewed type II PKSs
that construct aromatic natural products; in complementary
fashion, we will focus primarily on type I PKS and NRPS
assembly lines. Our intention is not to provide an exhaustive list
of examples; rather, we describe a few representative cases that
can serve as a gateway to the literature for interested readers.
Fig. 1 Monomers for FA, PK, and NRP biosynthesis. Malonyl-
coenzyme A (malonyl-CoA) is the monomer for fatty acid biosynthesis,
while malonyl-CoA and methylmalonyl-CoA are both monomers for PK
biosynthesis. Amino acids and aryl acids are the most common mono-
mers for NRP biosynthesis.
Fig. 2 Nascent PK and NRP chains grow as a series of elongating, protein-bo
the first four modules of the nystatin PKS are shown, featuring acyl or peptid
groups of thiolation (T) domains.
This journal is ª The Royal Society of Chemistry 2008
2 Chemical logic and enzymatic machinery of NRPSand PKS assembly lines
2.1 Chemical logic
The assembly of fatty acids (FAs), polyketides (PKs), non-
ribosomal peptides (NRPs), and NRP-PK hybrids involves
equivalent logic for converting simple acid and amino acid
building blocks into linear condensed polymers that may
undergo further elaboration.1,14 FA and PK chains grow by the
iterated addition of malonyl units (or methylmalonyl units for
PKs) in which the C–C bonds are forged during chain elongation
by decarboxylative thio-Claisen condensations. NRPs are
formed by the condensation of amino acids, by C–N (amide)
rather than C–C bond formation.
Both the C–C bond-forming enzymes in FASs and PKSs and
the C–N bond-forming catalysts in NRPs use acyl or aminoacyl
thioesters as activated monomer units (Fig. 1). Hydrolysis of one
of the reacting acyl thioesters drives the equilibrium in favor of
C–C or C–N bond formation with concomitant chain elonga-
tion. The process is repeated several times in succession; for
example, seven times by a FAS to build C16 fatty acids,15,16 or six
times by a PKS to create the fourteen-membered macrolactone
scaffold of erythromycin.17
The FA, PK and NRP chains thus grow as a series of
elongating acyl/peptidylthioesters in which the sulfur atom of
the thiol group is the terminus of a phosphopantetheine
arm covalently attached to carrier protein domains
embedded in the assembly line enzyme (Fig. 2; see also
Fig. 8 and Section 3). This process is sometimes referred to
as thiotemplating.18–21
und acyl or peptidylthioesters. Schematics of the echinomycin NRPS and
yl thioester intermediates tethered to the phosphopantetheine prosthetic
Nat. Prod. Rep., 2008, 25, 757–793 | 759

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
2.2 Enzymatic machinery
FAS and PKS enzymes have a core of catalytic domains along
with a carrier protein domain that constitutes the minimal
machinery for chain assembly, to which additional catalytic
domains can be added for tailoring purposes (Fig. 3).1 For PKSs,
the minimal domains are: (i) the C–C bond-forming ketosyn-
thase (KS); (ii) the acyltransferase (AT), which introduces
malonyl or methylmalonyl units during each cycle of chain
elongation; and (iii) the carrier protein domain, also known as
a thiolation (T) domain, on which the acyl chain is assembled and
elongated. Optional tailoring domains include ketoreductase
(KR), dehydratase (DH) and enoylreductase (ER).17,22
NRPS enzymes have a comparable minimal set of two cata-
lytic domains and one carrier protein domain.23 The peptide
bond-forming condensation (C) domain and the amino acid
Fig. 3 Core PKS and NRPS domains. Schematic abbreviations used
throughout the text are shown.
Fig. 4 Organization of assembly line enzyme domains. Type I PKSs and N
proteins. Type II PKSs and NRPSs have protein domains that are not covale
760 | Nat. Prod. Rep., 2008, 25, 757–793
adenylation (A) domain interact with a phosphopantetheine-
primed T domain to build the peptide chain as an elongating
thioester. Optional tailoring domains in NRPSs include epim-
erase (E) and methyltransferase (MT) domains.24 Since FASs,
PKSs, and NRPSs all build chains as elongating thioesters, they
must also have a catalytic domain to release full length acyl/
peptidyl chains from their covalent attachment to the assembly
line enzyme. This process is usually carried out by a thioesterase
(TE) domain.25,26
2.3 Type I and type II enzymes
Assembly line enzymes have been divided into two subclasses
known as type I and type II organizations.27 Type I enzymes have
their catalytic and carrier protein domains stitched together like
beads on a string, while the domains in type II enzymes are
separate proteins that interact only transiently during acyl/
peptidyl chain growth (Fig. 4). Yeast and humans use a type I
FAS,15,16 while bacteria use either a type I or a type II FAS.28,29
Assembly line enzymes are also divided into modular and
iterative systems (Fig. 5). In modular assembly lines, each
catalytic and carrier protein domain is used only once as the
growing chain passes from the N-terminal module to the
C-terminal module. The seven-module erythromycin PKS,
distributed over three enzymes, is a paradigm for modular
organization.30,31 Most NRPSs are also modular, type I assembly
lines.32 In contrast, many fungal type I enzymes33 and the
mammalian FAS34 comprise only a single module that is used
iteratively during cycles of chain elongation. For example, the
mammalian FAS catalyzes eight cycles of chain elongation—
with the growing chain anchored to a single carrier protein
domain—before releasing a C18 fatty acid. A more recently
characterized example is the bacterial protein family PKSE,
a group of type I modular PKSs that catalyze seven cycles of
elongation to synthesize a polyene fatty acid that is the precursor
to the highly reactive enediyne warhead of polyketides like
C-1027 and maduropeptin.35
Aromatic polyketides, such as the antibiotic tetracycline and
the anticancer drug daunomycin, are made by type II PKSs (see
Hertweck et al.13 for a recent review). The catalytic and carrier
protein domains are separate proteins that interact transiently.
The PK chain grows on the phosphopantetheine arm of the
carrier protein, which is visited iteratively by its catalytic
partners.36 One might conjecture that type II systems evolved
first, and subsequently became fused into type I enzymes
RPSs have covalently connected domains that form large, multi-domain
ntly connected; they are separate proteins that associate non-covalently.
This journal is ª The Royal Society of Chemistry 2008

Fig. 5 Mode of assembly line enzyme chain elongation. Modular PKSs and NRPSs use each catalytic and carrier protein domain once per molecule
synthesized, while iterative PKSs and NRPSs use catalytic and carrier protein domains for multiple cycles of chain elongation per molecule synthesized.
The lowercase ‘kr’ domain in DEBS 2 indicates an inactive ketoreductase domain.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
comprised of a single module that acts iteratively. Duplication of
these type I modules could then have led to the evolution of
modular type I assembly lines.37
A third category of PKSs, known as type III PKSs, differ from
type I and II PKSs primarily in that they use coenzyme A
thioesters directly as substrates—i.e., without the need for an
acyl carrier protein. While type III PKSs will not be covered in
detail in this review, readers are encouraged to read two recent
and interesting reviews on the topic.38,39
2.4 Four stages in NRP and PK biosynthesis
The logic of converting monomeric building blocks into
condensed natural product scaffolds by FAS, PKS, and NRPS
catalysts can be divided into four stages: (1) post-translational
priming of T domains to convert them from inactive apo to active
holo forms; (2) provision of unusual or dedicated monomeric
building block metabolites to feed into the assembly lines; (3) the
steps of chain initiation, chain elongation, and chain termination
during scaffold assembly; and (4) post-assembly-line tailoring of
nascent released products (Fig. 6). Enzymatic steps that have
been characterized from these four stages comprise the next four
sections of this review.
3 Phosphopantetheinyl transferases: the essentialpost-translational priming catalysts
The post-translational phosphopantetheinylation of all carrier
protein domains used in an FAS, PKS, or NRPS is the enabling
event that creates active protein machinery (Fig. 7).40–42 Coen-
zyme A (CoASH) is the phosphopantetheinyl donor and a serine
side chain in an elbow of the folded carrier protein domain is the
nucleophile, attacking CoASH at its pyrophosphate linkage.
This journal is ª The Royal Society of Chemistry 2008
This transfers the phosphopantetheinyl (Ppant) moiety of
CoASH onto the serine side chain, creating a stable phospho-
diester link to the 20 A long Ppant arm with a terminal thiol
group to which elongating FA, PK, and NRP chains are
covalently tethered (Fig. 8).
Given that phosphopantetheinyl transferase (PPTase)-
catalyzed priming is essential for assembly line function, it is not
surprising that PPTases are often encoded in natural product
gene clusters. Early on, it was discovered that the E. coli PPTase
that primed its FAS would not work on NRPS carrier protein
domains and only fitfully on PKS carrier protein domains.43 This
led to the discovery of different families of PPTases,40 with the B.
subtilis PPTase Sfp serving a particularly useful function since it
acts promiscuously on many types of carrier protein domains.44
Other characterized PPTases displaying relaxed specificity
include Svp from S. verticillus.45 Dozens of examples have now
been reported where either (i) coexpression of Sfp with PKS or
NRPS proteins in E. coli has led to primed, active assembly
lines;46,47 or (ii) incubation of purified carrier proteins with
purified Sfp has enabled in vitro assembly line priming.48–51
While Sfp will not accept dephospho-CoA as a Ppant donor
substrate, it will accept an almost limitless variety of acylated
CoA derivatives, therefore enabling the installation of a host of
acyl-S-pantetheinyl-phosphate moieties52–56 on almost any
carrier protein of interest. This has facilitated the investigation of
many alternate substrates in natural product assembly lines, both
for mechanistic study and for preparative uses.57,58 In a very real
sense, the discovery and characterization of Sfp and related
PPTases was a necessary precondition for in vitro studies of PKSs
and NRPSs. Prior to their availability, heterologous expression
of PKS and NRPS genes in E. coli was a poor substitute since it
yielded, at best, small fractions of an active assembly line enzyme
due to insufficient priming of the carrier proteins.
Nat. Prod. Rep., 2008, 25, 757–793 | 761
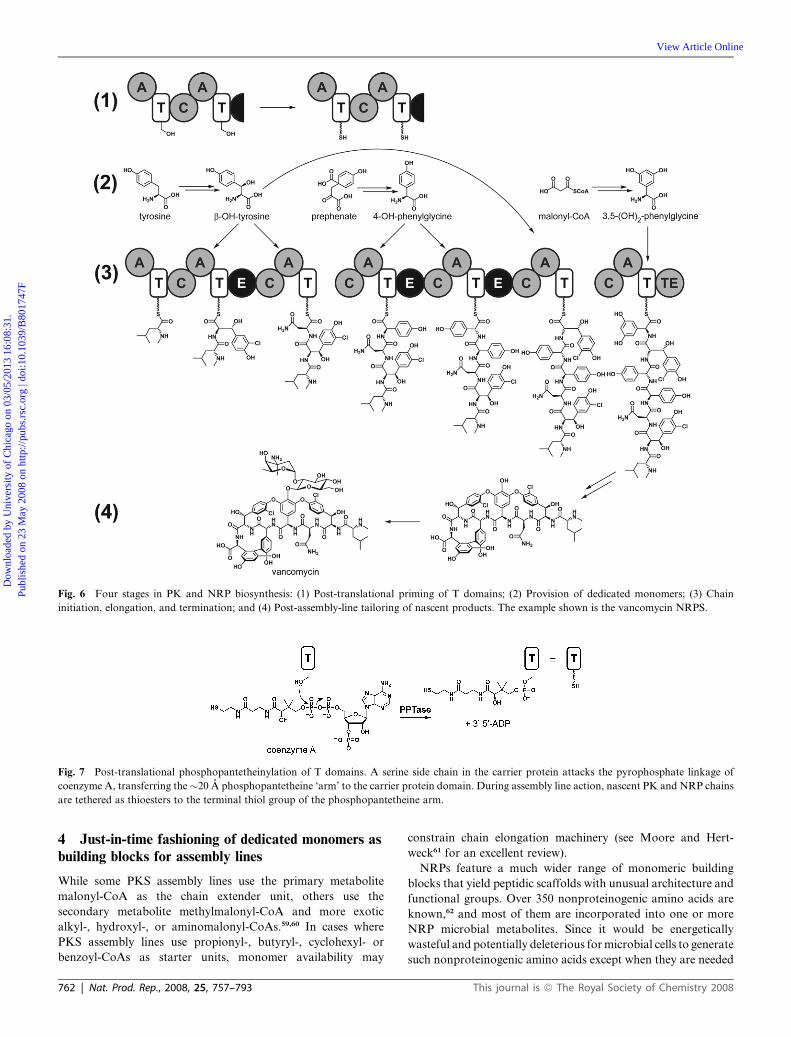
Fig. 6 Four stages in PK and NRP biosynthesis: (1) Post-translational priming of T domains; (2) Provision of dedicated monomers; (3) Chain
initiation, elongation, and termination; and (4) Post-assembly-line tailoring of nascent products. The example shown is the vancomycin NRPS.
Fig. 7 Post-translational phosphopantetheinylation of T domains. A serine side chain in the carrier protein attacks the pyrophosphate linkage of
coenzyme A, transferring the�20 A phosphopantetheine ‘arm’ to the carrier protein domain. During assembly line action, nascent PK and NRP chains
are tethered as thioesters to the terminal thiol group of the phosphopantetheine arm.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
4 Just-in-time fashioning of dedicated monomers asbuilding blocks for assembly lines
While some PKS assembly lines use the primary metabolite
malonyl-CoA as the chain extender unit, others use the
secondary metabolite methylmalonyl-CoA and more exotic
alkyl-, hydroxyl-, or aminomalonyl-CoAs.59,60 In cases where
PKS assembly lines use propionyl-, butyryl-, cyclohexyl- or
benzoyl-CoAs as starter units, monomer availability may
762 | Nat. Prod. Rep., 2008, 25, 757–793
constrain chain elongation machinery (see Moore and Hert-
weck61 for an excellent review).
NRPs feature a much wider range of monomeric building
blocks that yield peptidic scaffolds with unusual architecture and
functional groups. Over 350 nonproteinogenic amino acids are
known,62 and most of them are incorporated into one or more
NRP microbial metabolites. Since it would be energetically
wasteful and potentially deleterious formicrobial cells to generate
such nonproteinogenic amino acids except when they are needed
This journal is ª The Royal Society of Chemistry 2008

Fig. 8 Crystal structure of E. coli ACP, a T domain, showing the
phosphopantetheine arm in black (PDB entry 1L0I). This figure was
created using PyMOL (http://pymol.sourceforge.net/).
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
as a monomer for natural product assembly, their synthesis is
coordinately regulated.63
While space limitations prevent a complete discussion of
dedicated building block synthesis, we consider below three
examples of the enzymatic construction of dedicated monomers
for (i) the glycopeptide antibiotics vancomcyin and teicoplanin,
(ii) halogenated natural products such as the antitumor agent
rebeccamycin and the phytotoxin syringomycin, and (iii)
b-amino acid-containing natural products such as the antibiotic
Fig. 9 Provision of nonproteinogenic amino acid monomers during chloroe
tyrosine (b-OH-Tyr), 3,5-dihydroxyphenylglycine (3,5-Dpg), and 4-hydroxyp
encoded by the chloroeremomycin gene cluster.
This journal is ª The Royal Society of Chemistry 2008
andrimid and the enediyne antitumor agent C-1027. Studies with
purified enzymes were required to decipher the mechanisms by
which these monomers are synthesized. These efforts have
revealed examples of novel biological catalysis.
4.1 Hydroxyphenylglycine and dihydroxyphenyglycine building
blocks for glycopeptide antibiotics
Most of the NRPS gene clusters that encode unusual NRP
scaffolds also encode genes for nonproteinogenic building
blocks. For example, five of the seven residues in the antibiotic
chloroeremomycin64,65 are nonproteinogenic: two b-hydroxy-3-
chlorotyrosines,66 two 4-hydroxyphenylglycines (Hpg),67 and one
3,5-dihydroxyphenylglycine (Dpg)68,69 (Fig. 9). When the
chloroeremomycin gene cluster is transcribed, these non-
proteinogenic amino acids are produced in a ‘just-in-time’
inventory control fashion because nine of its ORFs encode
enzymes to construct these monomers from primary metabolites.
To produce b-hydroxy-3-chloro-Tyr, tyrosine is first activated
and tethered as thioester on the Ppant arm of a carrier protein,
and then hydroxylated at the benzylic carbon by a dedicated
cytochrome P450 oxygenase.70 Chorismate is diverted from the
aromatic amino acid biosynthetic pathway toward Hpg by
oxygenation and rearrangement of its scaffold to yield
p-hydroxyphenylglyoxalate, with a final transamination to form
Hpg.67 The pathway to Dpg is particularly remarkable: DpgA,
a type III PKS,38 condenses four molecules of malonyl-CoA
using decarboxylative thio-Claisen condensations to yield the
eight-carbon product dihydroxyphenylacetyl-CoA.71 DpgC,
a rare metal-independent dioxygenase, then catalyzes a four-
electron oxidation of the a-methylene group to the ketone to
form dihydroxyphenylglyoxalate,72 which is transaminated to
Dpg. During DpgC catalysis, one atom from O2 ends up in the
ketone of dihydroxyphenylglyoxalate and the other in its
carboxylate, suggesting a novel mechanism of oxygenation that
may proceed through a dioxetane intermediate.73
remomycin biosynthesis. The nonproteinogenic amino acids b-hydroxy-
henylglycine (4-Hpg) are synthesized by dedicated enzymatic pathways
Nat. Prod. Rep., 2008, 25, 757–793 | 763

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
4.2 Halogenation of amino acid building blocks
Around 4000 halogenated natural products are known.74 For
almost all of them, the halogen is installed by enzymes
conducting oxidative chemistry to transfer the halide as Xc or
X+.75 When the cosubstrate oxidant is hydrogen peroxide, the
enzymes are haloperoxidases, which use either heme coenzymes
or vanadyl cofactors to convert hydrogen peroxide to the halo-
genating species HOX. When the cosubstrate oxidant is molec-
ular oxygen, O2 gets reductively activated and ultimately
fragmented. Two kinds of cofactors can be utilized, flavin or
mononuclear nonheme iron. FADH2 reacts with O2 to yield an
FAD-OOH on the way to nascent HOCl.76 HOCl may be trap-
ped by a lysine in the halogenase active site to yield a chloramine
as the proximal donor of a Cl+ equivalent.77 A separate
O2-consuming halogenase family uses mononuclear nonheme
iron as a cofactor and generates a high-valent oxoiron interme-
diate that can create substrate radicals and transfer Clc to form
the C–Cl bond in the product.78,79
While haloperoxidases are often involved in the chlorination of
isoprenoid natural products,80,81 O2-dependent halogenase genes
predominate in NRPS and PKS gene clusters, suggesting that
these chemistries are used to halogenate NRP and PK scaffolds.75
Studies with purified enzymes have validated that chlorination of
tryptophan at the 5, 6, or 7 positions of the indole ring is enacted
Fig. 10 Flavin-dependent halogenases. PltA catalyzes the 4,5-dichlori-
nation of pyrrolyl-S-PltL during pyoluteorin biosynthesis.
Fig. 11 Fe/a-KG dependent halogenases. BarB2 and BarB1 catalyze the
trichlorination of Leu-S-BarA during barbamide biosynthesis.
Fig. 12 Cryptic chlorination as a strategy for cyclopropane formation. Follo
chloride-displacing cyclopropanation as the penultimate step of coronamic a
764 | Nat. Prod. Rep., 2008, 25, 757–793
by three distinct FADH2-dependent halogenases.82–85 Bischlori-
nation of a pyrrole ring at positions 4 and 5 during pyoluteorin
biosynthesis is likewise mediated by an FADH2-utilizing
halogenase,86 working on a pyrrolyl-2-carboxyl-S-carrier protein
species (Fig. 10). Additionally, chlorination of a b-Tyr-S-carrier
protein substrate occurs during C-1027 assembly.87
These studies, garnered from purified enzyme incubations,
allow the generalization that electron-rich aromatic rings such as
phenols, pyrroles, and indoles will be halogenated by FADH2-
utilizing enzymes that deliver Cl+ equivalents. Bioinformatic
analysis predicts that the gene clusters for halogenated natural
products such as the NRP antibiotic chloroeremomycin64 and the
PK antitumor agent ansamitocin harbor genes encoding flavin-
dependent halogenases.88,89 However, bioinformatic analysis
does not reveal the timing of halogenation. During rebeccamycin
biosynthesis, chlorination of a free amino acid is an initiating
step,82 while in pyoluteorin86 and C-102787 construction, the
substrate for chlorination is an aminoacyl/peptidyl-S-carrier
protein. Each system must be investigated biochemically to
understand the specificity of its tailoring halogenases and the
timing of their action in a pathway.
The biosynthetic gene clusters for some halogenated natural
products harbor no FADH2-dependent halogenases. Instead,
there are homologs of a-ketoglutarate-dependent mononuclear
nonheme iron oxygenases.78,90 These enzymes have turned out to
be halogenases for unactivated carbon centers, which are not
electron-rich and are therefore unreactive with Cl+ equivalents.
To generate a more potent halogenating species, FeII centers in
the iron halogenases have chloride (or bromide) ions in the first
coordination shell; when the FeIV]O intermediate is generated,
Clc rather than OHc is transferred to unactivated aliphatic
carbon sites (methyls or methylenes) in aminoacyl substrates,
presented as aminoacyl-S-carrier protein species. The remark-
able trichlorination of a Val-S-carrier protein substrate during
barbamide biosynthesis is mediated by the tandem action of two
such halogenases (Fig. 11).91
Studies on a purified nonheme iron halogenase in the
biogenesis of the aminocarboxycyclopropane coronamic acid
(a building block for the NRP-PK phytotoxin coronatine) has
established that cryptic chlorination is one of Nature’s chemical
strategies for cyclopropane construction.57 An allo-Ile-S-carrier
protein is chlorinated on the unactivated methyl side chain by the
nonheme iron halogenase CmaB (Fig. 12). The resultant Cl-allo-
Ile-S-carrier protein is then a substrate for CmaC, which
catalyzes the remarkable intramolecular cyclization of C2 onto
C4 with the displacement of chloride ion as the leaving group,
forming a cyclopropane ring.92 The substrate C2 carbanion
required for this intramolecular C–C bond to form is enabled by
wing CmaB-catalyzed chlorination of allo-Ile-S-CmaD, CmaC catalyzes
cid biosynthesis.
This journal is ª The Royal Society of Chemistry 2008

Fig. 14 The kutznerides, a family of NRPs entirely comprised of non-
proteinogenic amino acids. Kutzneride 2 is shown with the building
blocks b-hydroxyglutamate, O-methylserine, 3-chloropiperazate, tert-
butylglycoylate, 2-(1-methylcyclopropyl)glycine, and dichlorohydrox-
yhexahydropyrroloindole-2-carboxylate highlighted.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
its thioester linkage, since the carbanion is a stabilized thioester
enolate, and therefore kinetically accessible in the active site of
CmaC.
4.3 b-Amino acid building blocks
SomeNRP scaffolds contain isopeptide bonds in place of peptide
bonds, arising from the incorporation of b-amino acid mono-
mers rather than the usual a-amino acid building blocks. Notable
examples include the anticancer drug taxol,93 the enediyne
antibiotic C-1027,94 the protease inhibitor bestatin,95 and the
antibiotic andrimid,96 all of which contain either b-Phe or b-Tyr.
The gene clusters for C-102794 and for andrimid96 harbor ORFs
with homology to phenylalanine ammonia lyase, an enzyme
prevalent in plants that converts Phe to cinnamic acid and
ammonia as the initial step in phenylpropanoid biosynthetic
pathways (Fig. 13).97 Phe and His ammonia lyases were the first
examples of enzymes found to contain an autocatalytic modifi-
cation of the enzyme’s primary structure in which an Ala-Ser-Gly
sequence in the enzyme’s backbone is converted into the cyclic
4-methylideneimidazol-5-one (MIO) cofactor.98 MIO has an
electrophilic enone functional group that enables the fragmen-
tation of Phe or His to cinnamate or urocanate via the loss of Hb
and ammonia. With this precedent, it was anticipated that SgcC4
from the C-1027 pathway,99–102 AdmH from the andrimid gene
cluster,103 and the Taxus PAM enzyme in taxol biosynthesis93
would be aminomutases, generating cinnamate transiently and
then catalyzing re-addition of ammonia with opposite
regiochemistry. These expectations have been validated with
purified enzymes, and an X-ray structure of the tyrosine
aminomutase SgcC4 clearly shows the auto-modified MIO
cofactor in the active site.99 The three-step branch of the C-1027
pathway that builds the Cl-b-tyrosine building block has now
been reconstituted: aminomutase-catalyzed conversion of Tyr to
b-Tyr, its installation as a b-Tyr-S-carrier protein substrate, and
finally its FADH2-dependent chlorination.87,100–102,104
4.4 Other nonproteinogenic building blocks
Much of the useful functional group diversity and the attendant
biological activity of nonribosomal peptides arise from their
unusual nonproteinogenic amino acid building blocks. The three
Fig. 13 Aminomutases. AdmH converts phenylalanine to b-phenylala-
nine during andrimid biosynthesis, and SgcC4 converts tyrosine to b-
tyrosine as a monomer for C-1027 biosynthesis.
This journal is ª The Royal Society of Chemistry 2008
examples noted above are only a partial list of what could be
enumerated. They illustrate the diverse chemistry practised by
enzymes encoded in the biosynthetic gene clusters to carry out
just-in-time inventory control as natural product biosynthetic
pathways are activated in microbial producers. Understanding
the chemical mechanisms for building block construction will
enable components from different pathways to be mixed to
create architectural diversity in natural product libraries.
Attention should continue to be focused on the chemical
strategies and types of enzymes needed for nonproteinogenic
amino acid elaboration. In this context, the kutznerides (Fig. 14),
antifungal agents from a Scandanavian soil actinomycete, are
cyclic hexapeptidolactones, notable for the diversity of their
building blocks.105 One residue is tert-butylglycolate, a hydroxy
acid that engages in the ester linkage in the scaffold. The other
five building blocks are all non-proteinogenic amino acids,
ranging from the relatively prosaic O-methylserine and
b-hydroxyglutamate to the downright unusual 3-chloropiper-
azate, 2-(1-methylcyclopropyl)glycine, and dichlorohydroxy-
hexahydropyrroloindole-2-carboxylate. Characterization of the
enzymes that fashion these monomers for assembly line use will
surely turn up new chemistry and strategies for combinatorial
biosynthesis.
In passing, we note that knowledge of the scope of reactions
catalyzed by the two types of O2-consuming halogenases
discussed in the previous section suggested that both types
should be encoded in the kutzneride biosynthetic gene cluster.
Indeed, gene probes for FADH2-halogenases and mononuclear
iron halogenases facilitated cloning of the �50 kb kutzneride
gene cluster105 and points the way for cloning of other gene
clusters encoding halogenated and cyclopropane-containing
natural product scaffolds,106 e.g. from environmental DNA.
5 Assembly line action
The enzyme components of type I PKS and NRPS assembly lines
have been a more difficult set to study than the enzymes, just
discussed, that fashion the monomeric building blocks to be
incorporated by assembly line action. The difficulties arise from
Nat. Prod. Rep., 2008, 25, 757–793 | 765

Fig. 15 The size of assembly line enzymes. The daptomycin NRPS comprises 13 modules distributed over three proteins to form a�1.7 MDa assembly
line, while the avermectin PKS is composed of 13 modules distributed over four proteins to form a �2.2 MDa assembly line.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
the multimodular nature and imposing size of many type I
assembly line proteins (Fig. 15). The thirteen-module assembly
line for the PK avermectin is distributed over four proteins,
AVES 1–4, each in the 400–700 kDa size range.107 These proteins
contain catalytic and carrier protein domains in each module.
Analogously, the thirteen modules of the daptomycin synthetase
are distributed in a five-, six-, and two-module array across three
large enzymes.108 At one limit, all catalytic and carrier protein
domains in an assembly line could be stapled together in a single
megaenzyme. Such is the case for the cyclosporine synthetase,
which has 47 domains arrayed in tandem in a single polypeptide
of�15,000 amino acids.109 It is a challenging prospect to express,
purify and characterize such megasynthases. At the other end of
the spectrum are type II FASs and PKSs in which each domain,
catalytic and carrier, are separate proteins.13 Historically, these
type II systems were the first ones characterized, enabling the
elucidation of fundamental chemical steps, partial reactions, and
iterative catalytic cycles.110–113
In this section, we focus on type I PKS, NRPS, and NRPS-
PKS assembly lines to illustrate approaches to (i) deconvolute
the role of individual catalytic and carrier protein components,
(ii) analyze the process of chain elongation from one carrier
protein to the next, and (iii) to understand how full-length acyl/
peptidyl chains are disconnected from the assembly line when
Fig. 16 Loading/initiation modules in PKSs and NRPSs. The two-domain
shown; the two domains are adjacent in the bacitracin NRPS, while they are
766 | Nat. Prod. Rep., 2008, 25, 757–793
they have reached the final carrier protein domain in an assembly
line.
Type I assembly line enzymes are multidomain proteins. While
modular type I enzymes require a module-by-module examina-
tion for reconstitution, their architecture makes possible the
evaluation of intermediates tethered transiently to each carrier
protein domain, e.g. by mass spectroscopic analysis.114 Fungal
iterative type I enzymes such as LovF (lovastatin)33 and PKS4
(bikaverin)115 require purification of only a single module, but
since they act iteratively, it has been much more difficult to stop
catalysis at intermediate cycles to examine the tailoring reactions
occurring during chain growth. In either type I variant (modular
or iterative), full reconstitution from purified components can be
daunting. First, we examine studies to decipher parts of assembly
line action before turning in a subsequent section to look at
examples of full assembly line reconstitution. To that end, we
take up chain initiation, chain elongation, and chain termination
processes in that order.116
5.1 Chain initiation: monomer selection, loading and acylation
The first modules of PKS and NRPS assembly lines are the way-
stations where acyl/peptidyl chain growth is initiated. Sometimes
these are referred to as loading modules to emphasize their role
loading modules of the bacitracin NRPS and the stigmatellin PKS are
separated in the stigmatellin PKS.
This journal is ª The Royal Society of Chemistry 2008

Fig. 18 Noncanonical starter units for NRPSs. A fatty acid such as
decanoic acid is the starter unit for the daptomycin NRPS, while 2,3-
dihydroxybenzoate (2,3-DHB) is the starter unit for the vibriobactin
NRPS.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
as the starting point for synthesis. NRPS initiation modules
comprise, at a minimum, an adenylation (A) domain for amino
acid selection and activation, and a carrier protein domain that
bears the Ppant thiol group (Fig. 16). We will use the term T
(thiolation) for carrier protein domains in the following discus-
sions. By analogy, PKS loading modules would have—at
a minimum—an acyltransferase (AT) domain and a T domain to
be loaded by the AT domain, e.g. with malonyl-CoA as the
substrate. Many PKS assembly lines have a KS domain in the
loading module (KS-AT-T) that decarboxylates a malonyl unit
to an acetyl group to start the PK chain.117
More interesting are variations from the canonical acetyl
and aminoacyl starter units for PKS and NRPS assembly lines
(Fig. 17). Acyl-CoAs such as benzoyl-CoA118 or dihydroxy-
cyclohexenyl-CoA119 initiate enterocin and rapamycin biosyn-
thesis, while ansa-bridged scaffolds such as rifamycin and
geldanamycin use 3-amino-5-hydroxybenzoate (AHBA) as
a starter unit.120–123 At the end of the assembly line, the 3-amino
moiety of AHBA engages in intramolecular amide bond
formation to construct the ansa bridge.
A large family of NRPs are N-acylated peptide scaffolds
(Fig. 18). The A-T didomain of the chain initiation module
selects and loads the first aminoacyl group, which undergoes
acylation before chain elongation commences. A condensation
(C) domain in the initiation module (C1-A1-T1) may catalyze
N-acylation.124 Simple saturated or unsaturated fatty acyl
groups, diverted from the producer organism’s fatty acid
biosynthetic pathway, may serve as acyl group donors.125 During
the biosynthesis of the Bacillus subtilisNRPmycosubtilin, a fatty
acid is activated by the assembly line’s first A domain to the acyl-
AMP intermediate for capture by aminoacyl-S-T1.126 During the
fermentation of daptomycin, decanoic acid is added exogenously
and is preferentially incorporated into the lipopeptidolactone.125
A variety of N-acylpeptides start with b-hydroxy fatty acids,
which are also intermediates in fatty acid biosynthesis re-routed
towards the NRPS assembly line. The b-hydroxy group may
subsequently participate in chain-terminating macrocyclizations,
as will be noted below.127,128 A variant of functionalization of the
Fig. 17 Noncanonical starter units for PKSs. Benzoyl-CoA is the starter
unit for the enterocin PKS, while 3-amino-5-hydroxybenzoate (3,5-
AHBA) is the starter unit for the geldanamycin PKS.
This journal is ª The Royal Society of Chemistry 2008
long-chain fatty acid occurs duringmycosubtilin biosynthesis: the
chain initiation module uses a b-ketoacyl-CoA as an N-acylating
reagent, and the ketone undergoes reductive amination to the
b-amino group.129 This reductive amination is achieved by
a pyridoxamine-phosphate-utilizing transaminase fused into the
initiation module. This example demonstrates a rare occurrence
of the intersection of pyridoxal chemistry and PKS chemistry, but
it suggests that transaminase domains may be portable and
capable of being fused into other PKS modules to introduce
nitrogen into PK backbones. In the mycosubtilin scaffold,
subsequent chain termination occurs by intramolecular partici-
pation of this amino group,130 to form a macrolactam rather than
a macrolactone bond, with increased hydrolytic stability.
A large family of NRPs are iron-chelating molecules that
bacteria produce in response to iron starvation. These natural
products, known as siderophores, are virulence-conferring
factors for their bacterial producers.3 Siderophores contain
different varieties of ferric iron-coordinating groups, including
catechols, phenols, hydroxamates, and heterocycles (thiazolines
and oxazolines). The phenols and catechols are selected in chain
initiation steps in which salicylate or 2,3-dihydroxybenzoate are
aryl acids used toN-acylate aminoacyl1-S-T1 intermediates at the
start of the siderophore assembly lines.131,132
One additional example to note is the occurrence of pyrrole-2-
carboxy moieties at the N-termini of some nonribosomal
peptides. These do not arise as a result of N-pyrrolylation of an
aminoacyl1-S-T1, but instead by the selection and loading of
proline by A1-T1 to yield the prolyl1-S-T1 intermediate.133 This is
subjected to two cycles of dehydrogenation on the assembly line
to yield pyrrolyl1-S-T1.134 During pyoluteorin biosynthesis this
pyrrolyl-thioester is the substrate for dichlorination by an
FADH2-utilizing halogenase, as noted in Section 4.2.86
As a transition to the chain elongation section, we note that
the myxobacterial antitumor agent epothilone is a hybrid NRP-
PK that has a thiazole group fashioned from acetyl and cysteinyl
Nat. Prod. Rep., 2008, 25, 757–793 | 767

Fig. 20 b-Carbon tailoring domains in PKSs. The ketoreductase (KR)
domain catalyzes the two-electron reduction of the b-keto group to
a b-hydroxyl, the dehydratase (DH) domain dehydrates the b-hydroxyl to
an a,b-enoyl species, and the enoylreductase (ER) domain reduces the
a,b-enoyl to a fully saturated acyl group.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
moieties.135 This thiazole is cyclized during chain elongation
rather than chain initiation.
5.2 Chain elongation: Claisen, amide, and ester condensations
The transfer of growing chains from module to module is cata-
lyzed by C (condensation) domains in NRPS assembly lines and
KS (ketosynthase) domains in FAS and PKS assembly lines.1
C and KS domains make amide and carbon–carbon bonds
between upstream and downstream substrates tethered as Ppant
thioesters on T domains (Fig. 19). Condensation is directional,
from upstream where the growing acyl/peptidyl chain acts as the
electrophilic donor, to downstream where the thioester-tethered
monomer acts as the nucleophilic acceptor. In NRPS assembly
lines, the deprotonated amino group of an aminoacyl-S-Tn
attacks the peptidyl thioester on Tn�1, and amide bond forma-
tion is concomitant with chain translocation from Tn�1 to Tn.32
Correspondingly, in FAS and PKS assembly lines, the
downstream malonyl-S-Tn is decarboxylated to yield a two-
carbon thioester enolate nucleophile that attacks the KS-bound
thioester previously on Tn�1. Carbon–carbon bond formation
moves the growing chain from Tn�1 to Tn.17
Tailoring of the growing chain depends on which optional
catalytic domains are embedded within a module. In PKS
assembly lines, the minimal KS-AT-T module can also contain
a ketoreductase (KR), a dehydratase (DH), and an enoylre-
ductase (ER), which can respectively process the initial
b-ketoacyl-S-Tn moiety to the b-hydroxyl, then to the a,b-enoyl,
and finally to the fully saturated acyl chain elongated by two
carbons (Fig. 20).1 If one or more of the KR, DH, or ER domains
is absent or catalytically defective, intermediate oxidation states
persist and are subsequently carried forward as chain elongation
proceeds to the next module.136
Studies on KR domains and on purified PKS modules con-
taining KR domains have revealed that reduction to either the
S-alcohol or the R-alcohol is possible, and stereoselectivity is
correlated with the presence or absence of a key aspartyl residue
in the KR domain active site.137–139 There has been much interest
Fig. 19 Action of ketosynthase (KS) and condensation (C) domains in PK
cysteine with the upstream chain, decarboxylate the downstream (methyl)ma
enolate on their acyl-S-enzyme intermediate, forming a C–C bond and trans
attack of the downstream aminoacyl group on the upstream peptidyl group, tr
species.
768 | Nat. Prod. Rep., 2008, 25, 757–793
in evaluating the role of the b-KR domains in setting the adjacent
a-methyl stereochemistry when methylmalonyl-CoA is the
building block, as in the erythromycin PKS.140–144 Proposals for
epimerase and reductase activities in KR domains have been
made to explain syn and anti dispositions of the b-hydroxy-a-
methyl during chain elongation.144 Subsequent dehydration
would remove the b-substituent but leave the a-CH3 groups: in
erythromycin the methyls are 2R, 4R, 6S, 8R, 10R, and 12S,
respectively.145
5.2.1 C domains: condensation and heterocyclization. In
analogy to PKSs, the minimal domain set for NRPS elongation
modules is C-A-T, and only rarely are additional catalytic
S and NRPS chain elongation. KS domains autoacylate an active site
lonyl-S-T monomer, and then catalyze attack of the resulting thioester
locating the chain to the downstream T domain. C domains catalyze the
anslocating the chain without the known intermediacy of an acyl–enzyme
This journal is ª The Royal Society of Chemistry 2008

Fig. 21 Action of cyclization (Cy) domains in NRPS chain elongation and heterocycle formation. Cy domains are variants of C domains; both domains
catalyze peptide bond formation with concomitant chain translocation. The Cy domain then catalyzes the attack of the b-nucleophile (either a hydroxyl
as shown, or a thiol) on the upstream amide carbonyl, forming a five-membered adduct that dehydrates to an oxazoline or a thiazoline.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
domains found in elongation modules, although we shall note
methyltransferase domains below. Perhaps the most chemically
intriguing and consequential aspect of NRPS elongation
modules is that sometimes the C domain is substituted by
a variant known as a cyclization (Cy) domain (Fig. 21).146 This C
domain subclass operates on Cys, Ser, and Thr-S-Tn as down-
stream partner nucleophiles to yield acyl-Cys/Ser/Thr-S-Tn
adducts from the amide bond-forming condensation step. Then,
before the chain is translocated to the next module, cyclo-
dehydration is catalyzed in a second Cy-mediated step to yield
the thiazolinyl-S-Tn, oxazolinyl-S-Tn, or methyloxazolinyl-S-Tn
as product.147 This cyclization to the five-membered dihydro-
heterocyclic rings radically alters peptide backbone connectivity
and also creates chelating groups for FeIII, so one sees such
oxazolines and thiazolines in siderophores such as mycobactin
fromMycobacterium tuberculosis,148 yersiniabactin fromYersinia
pestis,149 and vibriobactin from Vibrio cholerae.150
5.2.2 C domains that make ester linkages. A large number of
PK and NRP natural products have macrolactone or macro-
lactam linkages. As will be noted in a subsequent section, mac-
rocyclization is catalyzed by the chain-terminating thioesterase
domain at the end of NRPS and PKS assembly lines. A subset of
nonribosomal peptide scaffolds have multiple ester linkages
replacing the normal amide backbone linkages: these are made
by condensation domains that accept a-hydroxyacyl-S-Tn as
a downstream acceptor, thereby acting as ester synthases rather
than amide synthases (Fig. 22).151
The a-hydroxyacyl units can be installed by two different
routes. The cyclodepsipeptides of the enniatin152–154 and beau-
vericin155 family are cyclic molecules in which N-methyl-L-amino
acids alternate with D-hydroxyisovalerate moieties (D-HIV). One
module of the two-module enniatin NRPS activates the L-amino
Fig. 22 Formation of an ester bond by a C domain during cereulide biosynth
is an a-hydroxyacyl-S-T rather than an a-aminoacyl-S-T, forming an ester b
This journal is ª The Royal Society of Chemistry 2008
acid and has an MT domain for N-methylation. The other
module activates D-HIV rather than an amino acid. One C
domain in the two-module enniatin synthetase is an amide bond-
forming catalyst, while the other C domain is an ester synthase.
A second route for a-hydroxyacyl unit installation occurs
during the biosynthesis of the potassium ionophores valinomy-
cin156 and cereulide.151,157 In their assembly lines, the modules
incorporating the hydroxy acid monomers have the four-domain
architecture C-A*-KR-T. The A* domain activates a-keto acids
(rather than a-hydroxy or a-amino acids) and installs them on
the downstream T domain as thioesters. The embedded KR
domain is an a-ketoreductase, converting the achiral a-ketoacyl-
S-Tn in situ to a chiral D- or L-a-hydroxyacyl-S-Tn intermediate.
These are the donor species for the downstream Cn+1 domains,
which use a-aminoacyl-S-Tn+1 species as nucleophiles. The C
domains in these modules are chiral ester synthases rather than
amide synthases, and in accordance with the nomenclature from
Section 6.1 below, they act as DCL catalysts.
5.3 NRP-PK hybrids: module switching and chain elongation
Natural products that are hybrid NRP-PKs have been known for
decades; these include the clinically-used immunosuppressant
FK506 and antitumor agent bleomycin. Given that NRP and PK
metabolites can be made by modular type I assembly lines using
similar chemical logic, it appeared that the two types of assembly
lines must be able to mix and match components to form hybrid
assembly lines.14 Sequences of the gene clusters for these hybrid
molecules confirmed module mixing,158,159 and raised questions
of catalyst specificity and domain portability to create new
hybrid assembly lines and variant hybrid products. A common
blueprint appears to involve switching between NRPS and PKS
assembly lines and back by interchanges of whole modules
esis. The terminal C domain of CesA uses a nucleophilic co-substrate that
ond rather than an amide bond.
Nat. Prod. Rep., 2008, 25, 757–793 | 769

Fig. 23 PKS-NRPS interfaces in hybrid assembly line enzymes. Bottom: a PKS-to-NRPS interface from the myxalamid synthase, in which the
upstream co-substrate of the MxaA C domain is a PK rather than a NRP. Top: an NRPS-to-PKS interface from the bleomycin synthetase, in which the
upstream co-substrate of the BlmVIII KS is a NRP rather than a PK.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
(Fig. 23). The bleomycin assembly line is mostly an NRPS
assembly line with one PKS module functioning in the middle.159
At the other end of the continuum, the myxalamid assembly line
is entirely composed of PKS modules until the end where MxaA,
a four-domain NRPS module with the domain structure C-A-
T-Re, is found.160 The A domain of this module selects and
installs alanine, and the C domain uses the alanyl amine as
a nucleophile and inserts it through an amide linkage into the
growing PK chain. This NRPS module is like a plug-in device for
inserting an amino acid into the middle of a PK scaffold and
represents one mode for introducing nitrogen into the backbone
of PK molecules.
Another example of note occurs in the epothilone PKS-
NRPS.135,161,162 The first three enzymes, EpoA, EpoB, and EpoC,
are a PKS module, an NRPS module, and a PKS module,
respectively. EpoA generates acetyl-S-T1 as starter unit using
typical PKS logic. EpoB (Cy-A-Ox-T2) activates cysteine, and in
a step that constitutes the PKS-NRPS interface, the Cy domain
transfers the acetyl group to make N-acetyl-Cys-S-T2.49 Since
this is a Cy domain rather than a C domain, the N-acetyl-Cys
moiety is subsequently cyclodehydrated to form thiazolinyl-
S-T2.163 EpoC (KS-AT-DH-KR-T3) is a typical PKS module,
transferring methylmalonyl onto T3. The KS domain acts as the
NRPS to PKS interface, forming a carbon–carbon bond and
transitioning the growing chain back onto a PKS track.164 Study
of the EpoA-EpoB and the EpoB-EpoC interactions with
purified proteins164 has revealed mechanisms and specificity for
prototypic PKS-NRPS and NRPS-PKS interfaces and the
characteristics of the key C and KS domains that facilitate bond
formation at the hybrid junctions. The collective action of
purified EpoABC yields the methylthiazolyl-methylacrylyl-S-T3
chain that persists in the final epothilone scaffold.164 Variant
heterocycles can be assembled by the use of different substrates
770 | Nat. Prod. Rep., 2008, 25, 757–793
for EpoA and EpoB,165 providing an instructive lesson for
engineering NRPS and PKS interfaces in future hybrid systems.
Many other examples of NRP-PK hybrids and their gene
clusters have been reported in recent years, including two
molecules in which valine is extended by a malonyl unit
(barbamide166 and andrimid).96 We will note the complete
reconstitution of the yersiniabactin NRPS-PKS in Section 9.2.
Some recently characterized PKSs andNRPSs deviate from the
conventional rules of domain organization or action described
above, for example by using AT domains that act in trans rather
than in cis, or by harboring modules that act iteratively or are
skipped entirely. While these unconventional but increasingly
prevalent enzymes are not described in detail here, readers are
encouraged to read two interesting reviews on the topic.12,27
6 Chain tailoring on the assembly line
In Section 5.2, we noted that most PKS modules contain one or
more of the optional KR, DH, and ER domains that tailor the
redox state of the b-ketoacyl-S-Tn chain in each PKS module
before the chain passes to the next module. It is the rule rather
than the exception that PKS modules contain one, two, or three
of these chain-tailoring domains.
In NRPSs, chain-tailoring domains are an exception rather
than a rule. Up to this point, we have noted one kind of tailoring
step during NRP chain elongation: the Cy variant of C domains
that catalyze cyclodehydration to form thiazolinyl, oxazolinyl,
and methyloxazolinyl-S-T species (see Section 5.2.1). In several
cases, the modules containing Cy domains also have embedded
FAD-dependent oxidase (Ox) domains to convert the
dihydroaromatic thiazolines and oxazolines to thiazoles and
oxazoles (Fig. 24). This is the case for EpoB (mentioned above
in Section 5.3), which creates a methylthiazole species,167 and
This journal is ª The Royal Society of Chemistry 2008

Fig. 24 Conversion of a thiazoline to a thiazole by a flavin-dependent oxidase domain during epothilone biosynthesis. After the EpoB Cy domain
catalyzes condensation, cyclization, and dehydration to form methylthiazolinyl-S-EpoB, the EpoB Ox domain catalyzes its FAD-dependent oxidation
to the fully aromatic methylthiazolyl-S-EpoB.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
more famously in the bleomycin NRPS-PKS,159 in which
a tandem thiazolinyl-thiazolinyl-S-T intermediate is doubly
oxidized to the planar bithiazole. The bithiazole moiety can
intercalate between base pairs in DNA and is the DNA-targeting
pharmacophore of this drug.168 In principle, the knowledge
gained from the enzymatic studies on Cy and Ox catalysts could
allow the insertion of a pair of Cys-specific modules (Cyn-An-
Oxn-Tn-Cyn+1-An+1-Oxn+1-Tn+1) into an NRP, PK, or hybrid
assembly line to produce a bithiazole moiety that should target
the new natural product to DNA.
6.1 Epimerization at Ca
In addition to the use of nonproteinogenic amino acid
monomers, one of the hallmarks of nonribosomal peptides is the
common occurrence of D-residues in the backbone scaffolds.
While in principle it could be the A domains in those modules
that select and activate D-amino acids rather than the available
L-amino acids,169 most of the D-residues in NRP scaffolds arise
by activation of L-amino acid monomers followed by the
epimerization of assembly line-tethered intermediates.51,170,171 In
some assembly lines, e.g. from Bacillus strains,172 there are
50 kDa epimerization (E) domains embedded in modules that
elongate D-residues (Fig. 25). The E domains epimerize either the
free L-aminoacyl-S-Tn to D-aminoacyl-S-Tn species before
condensation or act after condensation by Cn and before action
of Cn+1 on the peptidyl-S-Tn intermediate.173 Epimerization of C2
chirality is enabled by the kinetic acidity of the C2-H in
aminoacyl and peptidyl thioesters, since the resultant carbanion
is a stabilized thioester enolate.
Coupled to such on-line epimerizations must be chiral recog-
nition by the downstream C domain (Cn+1) to accept the
D-stereocenter in the upstream donor. That is, the Cn+1 must be
a DCL amide bond-forming catalyst. These predictions have been
borne out by studies with purified modules elongating D-residue
peptide chains.174 There are many other NRPS assembly lines
This journal is ª The Royal Society of Chemistry 2008
that also elongate D-residues but do not contain epimerization
domains. Studies on the purified modules have shown that the
C domains have both epimerization and condensation activity
(see Fig. 25).175 Again, the pairwise cooperation of tandem
C domains in assembly lines sets the chirality of the elongating
residues in the NRP chain. Any efforts in combinatorial
biosynthesis of NRPs to mix and match D- and L-amino acid
residues in backbone scaffolds must take into account the
existence of LCL and DCL chirality in C domains.176 To date noDCD condensation domains have been detected, suggesting that
D-D residues, e.g. as found at residues 4 and 5 in the vancomycin
scaffold (see Fig. 27), are generated by tandem action of DCL
domains with epimerization of a D-L- to a D-D-peptidyl-S-T
intermediate occurring before the second DCL domain acts.
6.2 N- and C-Methylations at Ca and Cb
Two other ‘‘on-assembly-line’’ tailoring events are of note: (1) N-
andC-methylations and (2) b-branching in polyketide backbones
(Fig. 26). A variety of NRP natural products are produced by
assembly lines with N-methyltransferase (N-MT) domains
embedded between the A and T domains (C-A-MT-T) in
modules that carry out N-methylation during amino acid
incorporation and elongation.32 The cosubstrate for methylation
is S-adenosylmethionine (SAM), which provides an electrophilic
‘‘CH3+’’ equivalent that is captured by the NH2 of the aminoacyl-
S-Tn domain to yield N-Me-aminoacyl-S-Tn before the next
elongation step.177
Less common are MT domains that effect C-methylation of
PK and hybrid PK-NRP products during chain elongation.1,178
The required nucleophile tethered to the growing substrate
chain is provided just after a KS domain has acted to produce
a b-ketoacyl-S-Tn intermediate. The methylene hydrogens on Ca
are acidic since the resultant carbanion is delocalized as the
thioester enolate. Studies with purified yersiniabactin synthetase
components established that mono- and di-C-methylation occur
Nat. Prod. Rep., 2008, 25, 757–793 | 771

Fig. 25 Generation of D-amino acid residues on NRP assembly lines. (a) Timing of epimerization and condensation in tyrocidine biosynthesis. (b)
Epimerase activity of C domain embedded in arthrofactin synthetase.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
to yield the a,a-dimethyl-b-ketoacyl-S-Tn species by C-MT
domain action, prior to reduction of the ketone to the alcohol by
the KR domain in that module (Fig. 37). A comparable
C-methylation occurs in the PK scaffold of epothilones
(Fig. 26).135 Anexample ofC-methylation of anNRP fragment by
an embedded C-MT domain occurs in the last module of yersi-
niabactin synthetases where C2 of the thiazolinyl-S-Tn, derived
from Cy domain action on an an acyl-Cys-S-Tn, is methylated
prior to release of the full length yersiniabactin chain.179
The SAM-dependent mechanism just noted leads to C-methyl-
ations at Ca of polyketide chains. A more common route to Ca
methyl substitution is the use of methylmalonyl-CoA monomers
by KS domains in PKS elongation. That is the strategy by which
DEBS 1, 2, and 3 introduce a-branching at carbons 2, 4, 6, 8, 10,
12 in the erythromycin scaffold.31
Although not discussed in detail here, post-assembly-line
methylation has also been noted as a point of structural
diversification, for exampleN-methylation in vancomycin,180 and
O-methylation of sugars181,182 and carboxylic acids.183
6.3 Cb branches
There are a small number of polyketides, including mupirocin,184
jamaicamide,185 bacillaene,186 and myxovirescin,187 that contain
b-methyl branches (Fig. 26). In vitro studies with five purified
enzymes, encoded as a cassette in the bacillaene and myxovir-
escin gene clusters, have revealed a SAM-independent pathway
772 | Nat. Prod. Rep., 2008, 25, 757–793
that represents an intersection of isoprene and polyketide
biochemistry.188,189
The first enzyme of the cassette is a homolog of b-hydroxy-
methylglutaryl (HMG)-CoA synthase (HCS) which is found in
isoprenoid biosynthetic pathways. HCS condenses the C2 anion
of acetyl-CoA onto the C3 ketone of acetoacetyl-CoA, making
the C–C bond in the branched chain HMG-CoA product. In
analogy, HCS acts on a b-ketoacyl intermediate on PKS
assembly lines, adding the C2 acetyl anion onto the 3-ketoacyl-S-
Tn intermediate to yield a product with a CH2COOH branch.
This intermediate then undergoes elimination to the conjugated
enoyl-S-Tn species. The resulting vinylogous carboxylate is lost
through decarboxylation by the next enzyme. Regiospecific
protonation of the thioester dienolate yields a D2-isoprenyl-S-Tn
tethered chain. The methyl group, derived ultimately from C2 of
the initial acetyl moiety, is in the b-position of the growing PK
scaffold. Transfer of this branched chain to the next module of
the PKS assembly line allows for continued chain elongation.
This machinery is likely responsible for subsequent elaboration
of b-branched species to the cyclopropane group in curacin190
and the vinyl chloride branch in jamaicamide.185
6.4 Oxidative transformations on assembly lines
In Sections 5.1 and 6, we have noted in passing two oxidative
dehydrogenations that occur while monomers or growing chains
are tethered on assembly lines. The first example is the double
This journal is ª The Royal Society of Chemistry 2008

Fig. 26 Strategies for methylation during NRP and PK biosynthesis. (a) SAM-dependent N-methylation. (b) Two sources of a-methyl substituents in
PKs: SAM-dependent methyl transfer in epothilone biosynthesis and incorporation of methylmalonyl CoAmonomers by the erythromycin synthase. (c)
SAM-independent b-carbon methylation by HMG-CoA synthase homologs.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
dehydrogenation of prolyl-S-T intermediates to pyrrolyl-S-T191
which occurs not only during pyoluteorin biosynthesis (Fig. 10)
but also in the biogenesis of the first pyrrole ring of the tripyrrolic
prodiginine pigments192 and in the pyrrolyl moieties of the
aminocoumarin antibiotics clorobiocin and coumermycin193
(Section 9.5), as established by studies with purified enzymes.
The second is the desaturation of thiazoline and oxazoline rings
in NRP assembly lines for making epothilone or oxazole analogs
of epothilone (Fig. 24).165 All of the above desaturations to
heteroaromatic systems are effected by flavoproteins, either
embedded in the samemodules as Cy domains (epothilones) or as
partner proteins to the proline activation adenylation domains.
Oxidations of elongating chains involving molecular oxygen
are at present best characterized by a set of three to four
This journal is ª The Royal Society of Chemistry 2008
hemeproteins of the cytochrome P450 superfamily which
perform the aryl–ether and aryl–aryl coupling chemistry in the
vancomycin (3 crosslinks) and teicoplanin (4 crosslinks) family of
antibiotics65 (Fig. 27). The biosynthetic gene clusters for this
antibiotic family encode three or four hemeproteins respectively,
OxyABC192 or StaFGHJ.194–196 Each of these O2-dependent
enzymes successively makes one of the 2–4 (Tyr2–Hpg4), 4–6
(Hpg4–Tyr6) or 1–3 (Hpg1–Hpg3) aryl ether linkages before
formation of the 5–7 direct C–C linkage between Hpg5 and Dpg7.
In the vancomycin series, the 4–6 ether crosslink is made first,
then the 2–4, and finally the 5–7.197 In the teicoplanin series, the
4–6 and then the 1–3 aryl ether crosslinks are formed in that
order.198 The net result is the conversion of the acyclic
heptapeptide to the highly rigidified, cup-shaped architecture of
Nat. Prod. Rep., 2008, 25, 757–793 | 773

Fig. 27 On assembly-line oxidative crosslinking of aglycone vancomycin and teicoplanin scaffolds by trans-acting cytochrome P450 oxidases.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
the vancomycin/teicoplanin aglycone. Enzymatic studies by
Robinson and colleagues199–201 have indicated that the cross-
linking steps occur while the heptapeptide is still tethered in
thioester linkage to T7. Chain release by the adjacent TE domain
may be controlled by the architectural changes introduced by the
crosslinks. The mechanism of these P450 crosslinking enzymes is
not yet deconvoluted, although phenoxy radical intermediates
are reasonable candidates as intermediates for the action of each
of the regioselective hemeprotein tailoring enzymes.202
7 Chain release mechanisms and catalytic machinery
When the growing ketidyl/peptidyl chains reach the most
downstream T domain and all the ‘‘on-assembly-line’’ tailoring
steps have been completed, the mature chain must be discon-
nected from its covalent thioester linkage to the Ppant prosthetic
group. The vast majority of PKSs and NRPSs have a thioesterase
(TE) domain at the C-terminus of their final module. The TE is
the catalyst for acyl/peptidyl thioester disconnection.
Two major routes of PK- and NRP-S-T disconnection are
known for subclasses of TE domains (Fig. 28). The first is
hydrolysis, where the linear natural product acid is released by
TE-catalyzed intermolecular attack of water on the ester linkage.
For example, this is the outcome for vancomycin,65 yersinia-
bactin,203 and the ACV tripeptide precursor of b-lactam anti-
biotics.204,205 A more intriguing subclass of TE domains
encompasses those that carry out intramolecular regiospecific
macrocyclization by a nucleophilic hydroxyl or amine in the PK/
NRP chain to create a cyclic lactone or lactam.25,26This is the fate
of the chain tethered to DEBS 3 in erythromycin biosynthesis206
and of the daptomycin chain207 tethered to DptD.
Among the most spectacular of the TE-mediated cyclizations
are the actions of the TE domain at the end of the four-domain
(C-A-T-TE) protein EntF which comprises the second (of two)
modules in enterobactin synthetase.208 The E. coli siderophore3
enterobactin is a cyclic trilactone of N-(2,3-dihydroxy-
benzoyl)serine, sent out to scavenge ferric iron, which it binds
using three pairs of catechol oxygens. EntF loads a 2,3-dihy-
droxybenzoyl-Ser (DHB-Ser) moiety covalently on the T domain
and then passes DHB-Ser to the active site serine side chain of the
TE domain where it is protected from adventitious hydrolysis
long enough for another DHB-Ser to accumulate on the T
774 | Nat. Prod. Rep., 2008, 25, 757–793
domain. Next, the two tethered DHB-Ser chains are condensed
using the side chain of the Ser moiety of one of the DHB-Ser
residues to build up a DHB-Ser-O-DHB-Ser dimer on the TE
domain. The T domain reloads a third time, builds the linear
(DHB-Ser)3-O-TE, and cyclizes it to release the trilactone
enterobactin. Studies with purified forms of EntF with mutant
TE domains have detected accumulation of the (DHB-Ser)2-O-
TE intermediate, and such results were central in formulation of
this mechanism.208
Since regiospecific macrocyclizations build in conformational
constraints in the released product and contribute dramatically
to medicinal activity, this class of TE cyclization catalysts has
generated much interest.209 Excision of the 30–35 kDa TE
domains from megasynthase assembly lines indicates they retain
their regioselective cyclization capacities and they can be
used for chemoenzymatic syntheses of macrocycles with soluble
acyl/peptidyl-thioester substrates to make new molecules
(Fig. 28). Use of a hybrid PK-NRP acyl donor with the excised
TE from the tyrocidine synthetase led to enzymatic production
of novel hybrid PK-NRP heterocycles,210,211 while studies with
the daptomycin synthetase TE have led to novel antibiotic
analogs.125,212
An intriguing variant of TE domain catalysis has been
deciphered by recent reconstitution of the fungal metabolite
terrequinone from purified enzymes,213 discussed in Section 9.6.3.
The TE domain of the lone NRPS module in the pathway
performs a remarkable feat: the double Claisen condensation
of two tethered indolepyruvyl–enzyme intermediates to construct
a benzoquinone ring during release of the dimerized indolyl-
benzoquinone.
A third and fundamentally distinct mode of chain release is
seen in PK and NRP assembly lines where the C-terminal TE
domain is replaced with a domain homologous to short chain
dehydrogenases214 and so has been annotated as a reductase
(Red) domain (Fig. 29). Reductive release requires NAD(P)H
as a cosubstrate. Hydride transfer from NADPH to the acyl/
peptidyl thioester linkage yields a hemithioacetal linkage which
unravels spontaneously to the released PK/NRP aldehyde and
the free thiolate of the pantetheinylated T domain. This is the
route for the reductive release of aminoadipoyl-S-T during lysine
biosynthesis in yeast.215 Intramolecular capture of the newly-
formed aldehyde by an amino group is proposed for the safracin
This journal is ª The Royal Society of Chemistry 2008

Fig. 28 Two modes of thioesterase (TE)-mediated chain termination: (a) TE-catalyzed hydrolysis by the ACV synthetase to yield the linear ACV-
tripeptide-carboxylic acid. (b) TE-catalyzed attack by an internal nucleophile: macrocylization yielding the daptomycin depsipeptide, and selective
trimerization by EntF to deliver the lactone enterobactin. (c) Excised TE domains can be used to generate novel hybrid macrocylic products of the
tyrocidine and daptomycin family.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
and saframycin antitumor antibiotics,216 and has been charac-
terized in vitro in the case of nostocylopeptide imines217 (Fig. 29).
There are some examples where reductive chain release is a net
four-electron rather than a two-electron process, taking the acid
oxidation state down to that of an alcohol. In lyngbyatoxin
biosynthesis, the reductase domain terminus of LtxA has been
shown to catalyze two iterations of hydride transfer from
NADPH.218 Reductive cleavage of the thioester of N-methyl-
Val-Trp-S-T to the aldehyde is followed by a second addition of
hydride to give the alcohol. For myxochelin A biosynthesis, net
four-electron reduction occurs through an analogous route.219
Reconstitution of this system has been accomplished through
incubation of DHB and L-Lys with the myxochelin synthetase as
shown in Fig. 29.
Not all modes of PK and NRP chain release have yet been
characterized (Fig. 30). In gliotoxin biosynthesis220 and ergota-
mine formation221 release of chains as cyclic diketopiperazines
occur.222 In the myxobacterial aurafuranone pathway223 the last
PKS module is lacking a TE domain so an oxidative Baeyer–
Villiger process followed by chain release has been proposed but
not yet validated.
This journal is ª The Royal Society of Chemistry 2008
8 Post-assembly-line tailoring reactions
In Section 6 we have noted sets of PK and NRP chain modifi-
cations that can occur by domains embedded within specific
modules of assembly lines. But this does not exhaust the catalytic
repertoire of the proteins encoded within the PK and NRP
biosynthetic gene clusters.224 In many cases, genes in these
clusters encode tailoring enzymes that act after release of the PK,
NRP, NRP-PK hybrid nascent product from assembly lines.24
The product that rolls off the assembly line is properly consid-
ered a late-stage intermediate rather than the final product. Some
of the subsequent tailoring enzyme steps can dramatically
resculpt the scaffold connectivity and decorate the periphery.225
We describe below examples of post-assembly tandem
glycosylations of both NRP and PK aglycones.226 One of the
characteristics of glycosylated PK and NRP natural products is
that the hexoses are often not the normal D-glucose, mannose, or
galactose monomers of primary metabolism.227,228 Instead,
nucleoside diphospho-esters of deoxy and aminodeoxyhexose
variants with balanced hydrophobic and charge properties
contribute important pharmacophores to natural products.
Nat. Prod. Rep., 2008, 25, 757–793 | 775

Fig. 30 Twomodes of presently uncharacterized chain release. (a) Spontaneous diketopiperazine formation exemplified by the gliotoxin synthetase. (b)
Baeyer–Villiger-type oxidation, followed by hydrolysis of the resulting hemiacetal, sets the stage for heterocycle formation in aurafuron A biosynthesis.
Fig. 29 Reductive chain release is catalyzed by a C-terminal reductase domain (Re) with NAD(P)H as the cosubstrate. Intramolecular capture of the
resulting aldehyde by an amine nucleophile delivers the nostocyclopeptide class of macrocyclic imines, while further reduction results in the alcohol of
lyngbyatoxin and myxochelin.
Fig. 31 Conversion of UDP-D-glucose to TDP-L-epivancosamine by
a dedicated, multi-step pathway.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
Thus, biosynthetic gene clusters for glycosylated PKs and NRPs
typically encode genes for making these dedicated NDP-hexoses
from the abundant metabolites UDP-glucose or TDP-glucose
(Fig. 31). For example, the gene cluster for chloroeremomycin,
discussed in Section 8.1, encodes six enzymes that convert TDP-
D-glucose to TDP-L-epivancosamine.229 While efforts over the
past 20 years have revealed the mechanisms and strategies for
many such NDP-deoxysugar-forming enzymes to make 2-deoxy,
3-deoxy, 4-deoxy, and 6-deoxy sugars,227,230 the in vitro
reconstitution of the five-step pathway from TDP-glucose
to TDP-L-epivancosamine is one of few examples where purified
776 | Nat. Prod. Rep., 2008, 25, 757–793 This journal is ª The Royal Society of Chemistry 2008

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
enzymes have been shown to recapitulate the in vivo pathway to
the dedicated L-aminodeoxysugar building block for the post-
assembly-line tailoring of antibiotics. In vitro analysis of the
TDP-L-mycarose pathway has also been described.231
8.1 Post-assembly tailoring to produce glycopeptide and
lipoglycopeptide antibiotics
Among the best-studied late-stage tailoring enzymes are those
involved in the maturation of two clinically used glycopeptide
antibiotics, teicoplanin and vancomycin, and a close relative,
chloroeremomycin.232 We have noted the enzymatic construction
of the dedicated monomers incorporated at residues 4,5,7 of the
heptapeptide scaffold (Section 4.1) and their oxygenative cross
linking (Section 6.4) that account for 14 of the tailoring enzymes
encoded in these biosynthetic gene clusters.
The most notable remaining features of vancomycin and its
close relative chloroeremomycin are the disaccharide chain
attached to the phenolic hydroxyl of Hpg4 of each crosslinked
heptapeptide scaffold and the monosaccharide attached to the
b-hydroxyl of Tyr6 of chloroeremomycin. The three glycosyl-
transferases (Gtfs) in the chloroeremomycin cluster64 and the two
Gtfs in the vancomycin cluster233 have been purified and char-
acterized both kinetically234,235 and structurally236–238 to establish
order of action, mechanism, and selectivity (Fig. 32A). The
glucosyl moiety is added by the first Gtf (GtfB or GtfE) from
UDP-glucose to the Hpg4-hydroxyl of the crosslinked hepta-
peptide aglycone. The 2-hydroxyl of the glucosyl group is then
the nucleophile for GtfD-mediated transfer of L-vancosaminyl
moiety from TDP-L-vancosamine (vancomycin) or GtfC-
promoted addition of the epimeric L-epivancosaminyl sugar
in chloroeremomycin maturation. GtfA also uses TDP-L-epi-
vancosamine as a sugar donor, but is regiospecific for transfer
to the benzylic alcohol of b-hydroxy-Tyr6 of the crosslinked
Fig. 32 Action of post-assembly-line tailoring glycosyltransferases. (a) Site-s
and teicoplanin aglycones to the mature glycopeptide antibiotics. (b) Glycosy
This journal is ª The Royal Society of Chemistry 2008
scaffold of chloroeremomycin. X-Ray structures reveal the
location of substrates and the basis for altered regiochemistry
between GtfA and GtfC.237 The monoglucosylated vancomycin
intermediate has also been a testing ground for chemoenzymatic
glycorandomization and neoglycosylation strategies to build
semisynthetic glycopeptide antibiotic variants.239–241
Teicoplanin, the second glycopeptide family member used as
a human antibiotic, has three differences from vancomycin.195,196
First is the replacement of Val1 and Asn3 of vancomcyin with
Tyr1 and Hpg3 in teicoplanin, allowing a fourth crosslink and so
requiring a fourth tailoring oxygenase (see discussion in Section
6.4). Second, there is only a monosaccharide, D-2-aminoglucose,
in glycosidic linkage to HPG4. There is an N-acetylglucosamine
attached to residue six, and finally there is a long-chain acyl
group linked to the 2-amino group of the D-glucosamine residue.
Studies with the two purified teicoplanin glycosyltransferases
and the acyltransferase242 have established the order of those
three tailoring steps: glucosamine addition to HPG4 first, then
N-acylation and subsequent installation of the N-acetylglucos-
amine moiety at residue six of the crosslinked peptide scaffold.
Knowledge of the order of action of the tailoring Gtfs and
acyltransferase enable strategies for design and enzymatic
synthesis of lipoglycopeptide variants of teicoplanin.
There are several NRP and PK lipid-containing natural
products that have undergone prenylation reactions at some
stage in their biosynthesis rather than acylation. Two examples
(Fig. 33) reflect the capture of electrophilic prenyl units by
cosubstrate nucleophiles. The aminocoumarin antibiotics, noted
in Section 9.5, all contain a prenylated hydroxybenzoate unit
which arises by attack of the C3 carbanion, generated through
the adjacent C4-hydroxyl of 4-hydroxybenzoate, on C1 of D2-
isopentenyl diphosphate.243,244 Lyngbyatoxin, a cyanobacterial
metabolite, is assembled from an N-methyl-Val-Trp core by
a two-module NRPS with reductive cleavage of the Trp moiety
elective glycosylation converts the NRP chloroeremomycin, vancomycin
l-transfer to aromatic PKs.
Nat. Prod. Rep., 2008, 25, 757–793 | 777

Fig. 33 Action of prenyltransferases during the maturation of novobi-
ocin and lyngbyatoxin.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
(see Fig. 29) to the alcohol mentioned in Section 7. This is
followed by cytochrome P450-mediated addition of theN-methyl
group to C4 of the indole side chain of the tryptophanol
(presumably via an arene oxide intermediate) and finally
geranylation at C7 of the indole ring (Fig. 33).245
8.2 Tandem glycosylations of aromatic polyketide aglycones
The daunomycin and aclacinomycin family of aromatic poly-
ketides are clinically used antitumor agents246 whose tetracyclic
aromatic ring scaffold is assembled by type II PKSs.13 Subsequent
regiospecific glycosylation of the 7-hydroxy substituent is cata-
lyzed by tailoring PKGtfs (Fig. 32B).24 Daunomycin has a single
L-aminodeoxysugar, L-daunosamine, while aclacinomycin A has
tethered L-rhodosamine, 2-deoxy-L-fucose, and L-cinerulose. This
trisaccharide chain is involved in binding to the minor groove of
DNA, while the anthracycline intercalates between base
pairs.247,248 There is one Gtf in the daunomycin-tailoring
Fig. 34 C-Glycosylation of enterob
778 | Nat. Prod. Rep., 2008, 25, 757–793
pathway36 and two, AknS and AknK, in the aclacinomycin-
tailoring pathway. With purified enzymes it has been possible
to show that AknS acts first but only in the presence of a second
protein AknT,249,250 to add L-rhodosamine from TDP-L-rhodos-
amine to the polyketide aglycone. Then AknK251 can add the
second and thirddeoxysugar residues. There are other examples in
polyketide maturation where a single Gtf will add tandem
deoxyhexose residues to build an oligosaccharide chain.252 AknS
joins a small family of other polyketide glycosyltransferases,
among them theGtfs that glycosylate the erythromycin scaffold at
the C3 hydroxyl,253,254 where a second (regulatory?) subunit (here
AknT) is required for active catalysis, a feature that could only be
gleaned from in vitro reconstitutions. It will be possible to use
variant TDP-L-aminodeoxysugars to alter the sugar residues in
the tailoring steps as a tool to evaluate structure activity
relationships for antitumor efficacy.253
8.3 C-Glycosylation: enterobactin to salmochelin
Although the vast majority of glycosides in primary metabolites
and secondary natural products are O-glycosides, there are
a small number of C-glycoside natural products in which C1 of
the glycosyl unit is attached by a direct C–C bond to a carbon of
the aglycone that is presumed to have acted as a nucleophile
during C–C bond formation.255 C-Glycosides would be resistant
to hydrolysis by the suite of glycosidase enzymes and so represent
a metabolically stable linkage. AC-glycosyltransferase, IroB, has
been purified from a uropathogenic E. coli strain that converts
the catecholic NRP siderophore enterobactin to diglucosyl-
enterobactin (DGE), known as salmochelin S4 because of its
initial detection as a Salmonella-derived iron chelator (Fig. 34).256
Both glucosyl units are connected by C-glycosidic bonds, from
C1 of glucose to C5 of two of the three DHB moieties. The
regioselectivity of C-glucosylation, meta or para to the existing
actin to form the salmochelins.
This journal is ª The Royal Society of Chemistry 2008

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
catechol hydroxyls of the DHB units, make direct C-glucosyla-
tion more likely than O-glucosylation followed by a sterically
demanding O-to-C glucosyl transfer across the face of the DHB
ring. UDP-glucose is the glucose donor and enterobactin the
acceptor, suggesting that ionization of the C2-hydroxyl of a DHB
ring to the phenolate would initiate catalysis. The C5 anion,
a resonance contributor to the phenolate anion, is the proposed
nucleophile that attacks C1 of the glucosyl donor UDP-glucose.
The C-glucosyl units in DGE do not compromise the ferric
iron-scavenging function but the modified siderophore no longer
binds to siderocalin, an innate immune protein from the
vertebrate host. Siderocalin binds ferric enterobactin, preventing
its uptake by bacteria, and thus reducing virulence as part of the
innate immune response. The C-glucosyl units in DGE sterically
block sequestration by siderocalin and allow ferric-DGE uptake
by its Gram-negative enteric producers.1,257
8.4 Post-assembly-line oxygenases
Most enzymes that have evolved to catalyze the reductive
activation and fragmentation of O2 with insertion of one or both
oxygen atoms into a cosubstrate utilize either the conjugated
organic cofactor FAD258 or the redox active transition metal
iron, in both heme and nonheme active site environments.259–261
Both categories are found as tailoring enzymes for PK and NRP
natural products as illustrated by some type I examples
(Fig. 35).24
In passing we note that metal-independent oxygenases,
exemplified by DpgC72,73,262 (Fig. 9), are also found in quinone-
forming steps of aromatic polyketides where phenolic ring-
containing polycyclics are prone to one electron oxidation,
generating phenoxy radicals reactive with triplet O2.
FAD-dependent enzymes that carry out Baeyer–Villiger ring
expansion reactions of cyclic ketones in type II PKS products
Fig. 35 Post-assembly-line tailoring by oxygenases of the (a) flavin-dep
This journal is ª The Royal Society of Chemistry 2008
appear to be prevalent in maturation steps of aromatic polyke-
tide families.13 In the biosynthesis of the anticancer agent
mithramycin, a late-stage tetracyclic intermediate, premi-
thramycin B, is substrate for purified MtmOIV.263,264 This FAD
enzyme performs a Baeyer–Villiger ring expansion of a six-
membered ring enone to a seven-membered ring lactone.
Subsequent hydrolytic ring opening of the lactone and decar-
boxylation may be spontaneous or enzyme-assisted. Post-
assembly-line oxidation of a secondary alcohol catalyzed by
flavin-dependent dehydrogenase SpnJ has also been recently
described in the context of spinosyn biosynthesis.265
In vitro studies with purified oxygenases have allowed evalu-
ation of catalytic parameters and specificity determinants for
maturation of both polyketide and nonribosomal peptide
scaffolds. In the erythromycin pathway, 6-deoxyerythronolide B
released by the PKS assembly line undergoes stereo- and regio-
specific hydroxylation at C6 by the cytochrome P450 EryF.266
This product is then a substrate for the two glycosyltransferases
that attach deoxysugars to the 3-hydroxyl and 5-hydroxyl of the
macrolactone scaffold.254 At this juncture a second cytochrome
P450 monooxygenase, EryK,267 acts to hydroxylate C12,
completing a four-step tailoring sequence of two oxygenations
and two glycosylations of the macrolide. In the maturation of the
polyene macrolide pimaricin, a late-step regioselective epoxida-
tion of the C4–C5 double bond is catalyzed by the hemeprotein
epoxidase PimD.268 In analogy, the epoxidase EpoK acts in the
last step of epothilone tailoring to introduce the C12,C13 epoxide
in epothilones A and B.269
In post-assembly-line oxidative tailoring of nonribosomal
peptide scaffolds requiring dioxygen, the most celebrated trans-
formations are the generation of the 5,4- (penicillin) and 6,4-
(cephalosporin) fused ring systems of the b-lactam antibiotics
from the acyclic precursor aminoadipoyl-cysteinyl-valine (ACV).
ACV synthetase204 is a ten-domain, three-moduleNRPSassembly
endent, (b) cytochrome P450, and (c) Fe/a-KG-dependent classes.
Nat. Prod. Rep., 2008, 25, 757–793 | 779

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
line protein that releases theACVwhere the L-valine unit has been
epimerized to D-Val on the assembly line by an epimerization
domain (see Fig. 28). The next enzyme is isopenicillin N synthase
(IPNS), an a-ketoglutarate- and O2-requiring nonheme
mononuclear FeII enzyme that makes both the four-ring b-lactam
and the five-ring thiolane while O2 is reduced by four electrons to
two molecules of H2O.270 The subsequent conversion of the
penicillin scaffold to the ring-expanded cephem nucleus is then
mediated by another member of the FeII monooxygenase
superfamily, deacetoxycephalosporinC synthase (DAOCS).271,272
The structural and mechanistic scrutiny these two enzymes have
received is due, in large part, to the central importance of b-lactam
antibiotics in anti-infective therapy.
9 In vitro reconstitution of complete pathways
In this closing section we note six case studies in which full
reconstitution of NRP, PK, or hybrid pathways have been
achieved in vitro with purified enzymes. We conclude with
a discussion of lessons learned from the characterization of
catalysts that function in tandem.
9.1 Enterobactin to salmochelin to microcin E492: three layers
of secondary metabolism
Siderophores are secreted by many genera of bacteria as
a response to a variety of iron-limited microenvironments,
ranging from the ocean to mammalian serum.3 NRP genes
encoding siderophore biosynthesis are among the most
widespread of the thiotemplated biosynthetic pathways found in
the 223 sequenced bacterial genomes,2 emphasizing the vital role
that the iron-chelating molecules play in bacterial survival. The
Fig. 36 Reconstitution of the microcin E492 biosynthetic pa
780 | Nat. Prod. Rep., 2008, 25, 757–793
reconstituted biosynthesis of the DHB-derived siderophore
myxochelin219 has outlined one such pathway (Section 7).
In our discussion of assembly-line chain release mechanisms
(Section 7) we have also noted that the Gram-negative bacterial
siderophore enterobactin is made on a two-module NRPS
assembly line and that, in some virulent bacteria, it is C-gluco-
sylated at C5 of two of the three DHB residues to yield
salmochelin (DGE) as a second layer of maturation256 (Section
8.3). A third layer of secondary metabolism occurs in enteric
bacteria elaborating the 84-residue polypeptide microcin E492
(MccE492) (Fig. 36). The C-terminal Ser of this peptide is
enzymatically modified by a linearized monoglucosylenter-
obactin via an oxoester bond between the Ser84 carboxyl and the
C6-hydroxyl of glucose. The resultant MccE492 can now be
taken up by neighboring cells that express siderophore-specific
outer membrane permeases. Once internalized into the
periplasmic space, MccE492 molecules self-assemble into inner
membrane pores that depolarize target cells.273
Eight enzymes are required to reconstitute the three layers
of secondary metabolism. Four purified proteins, the two-
module NRPS assembly line composed of EntEBF and the
phosphopantetheinyltransferase EntD, are required to make
enterobactin.131 Two enzymes, MceC (an IroB homolog) and
MceD, are a C-glucosyltransferase and a regiospecific trilactone
hydrolase, respectively. MceI and MceJ form a heterodimer that
catalyzes the attachment of the linearized MGE to the MccE492
C-terminus.274 Among the unusual chemistry revealed in these
three enzymatic layers of natural product biosynthesis are: (1)
identification of the EntF TE domain as a cyclotrimerizing
macrolactone synthase; (2) demonstration of C–C bond
formation by C-glucosyltransfer (IroB/MceC); and (3)
construction of a glucose bridge between a ribosomal peptide
thway illustrating three layers of secondary metabolism.
This journal is ª The Royal Society of Chemistry 2008

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
(pre-MccE492) and a nonribosomal peptide scaffold (enter-
obactin). Close inspection of MceIJ biochemistry revealed that
the initial product is the MccE492 ester attached to the
4-hydroxyl of the glucosyl bridge; nonenzymatic rearrangement
at physiological pH moves the 84-residue peptide from the
4-hydroxyl to the 6-hydroxyl of the bridging glucose.
9.2 Yersiniabactin reconstitution: an NRP-PK hybrid
Full reconstitution of the NRP-PK hybrid siderophore yersi-
niabactin,149 a virulence-conferring metabolite made by the
plague bacterium Yersinia pestis, has been achieved from four
purified proteins, High Molecular Weight Proteins 1 and 2
(HMWP1 and HMWP2), YbtE (a salicylate-adenylating
domain) and YbtU, a thiazoline reductase203 (Fig. 37). YbtE
activates salicylate as salicyl-AMP then transfers it to T1 of the
next protein, the 220 kDa HMWP2. HMWP2 is a two-module
NRPS with three carrier protein (T) domains and two cyclization
domains, building a hydroxyphenyl-thiazolinyl-thiazolinyl-S-T3
intermediate.275,276 HMWP1 is a 330 kDa two module hybrid,
with a PKS module followed by an NRPS module.48 Transfer of
the bisthiazolinyl chain from T3 on HMWP2 to malonyl-S-T4 in
the PKS module constitutes an NRPS-PKS interface where the
KS is the catalytic domain. Double Ca-methylation by an MT
domain in the PKS module occurs on the b-ketoacyl-S-T4
condensation product before reduction to the b-hydroxy,
a,a-dimethyl intermediate. The chain is then transferred to the
downstream NRPS module of HMWP1 where the Cy domain
constitutes the PKS to NRPS interface. This third NRPS
module, like the first two on HMWP2, activates cysteine and
cyclizes the chain after amide bond formation to a thiazolinyl-S-
T5. Another C-methylation occurs by a second embedded MT
domain, this time at Ca of the third thiazolinyl ring before
hydrolytic chain release.179 The fourth protein, YbtU, acts in
trans as a tailoring catalyst to reduce the middle thiazoline ring to
the tetrahydrothiazolidine oxidation state. All told, 22 chemical
operations happen on the five T domains of HMWP2/1 with
a net throughput of mature yersiniabactin of 1.4 product
molecules per minute.203
Reconstitution of an NRP-PK-NRP hybrid assembly line
from purified proteins and building blocks revealed the chemical
logic and plasticity of KS and Cy domains for switching growing
chains from one type of building block (acyl-CoAs) to the other
Fig. 37 Reconstitution of the yersiniabactin biosynthetic pathw
This journal is ª The Royal Society of Chemistry 2008
(amino acids). Characterization of this assembly line also
illuminated the tandem construction of bisthiazolines and their
redox adjustment as well as two modes of C-methylation.
9.3 Aromatic polyketide scaffolds from malonyl-CoA
monomers
Polycyclic aromatic scaffolds are assembled both by type I
(iterative) fungal PKS17 and by bacterial type II PKS
enzymes.13,277 The separate subunits of minimal PKS catalysts
have been purified and reconstituted from several streptomycete
systems (Fig. 38). The type II minimal set consists of the KS
catalytic subunit, KSa and its regulatory partner KSb (also
termed CLF (chain length factor) because it controls polyketide
length), that form a KS-CLF (KSa-KSb) heterodimer. A malonyl
acyl transferase (AT) delivers malonyl units from malonyl-CoA
to a fourth protein, the holo form of the acyl carrier protein
(T domain).278 When these four proteins from the actinorhodin
system279 are combined with the aromatase/cyclase and a sixth
protein, the free-standing ketoreductase (KR),280 chain elonga-
tion to an octaketide is observed.281 The first ring is regiospe-
cifically cyclized with the anticipated C7–C12 connectivity, but
the remaining polyketones undergo off-pathway intramolecular
cyclization and dehydration.
Mechanistic282 and structural studies283 of the CLF subunits
have revealed that volume control in the active site pocket is
a major factor in determining how many cycles of chain elon-
gation occur before chain release (typically 8–16 for aromatic
polyketides from bacterial type II PKSs). For control of
subsequent cyclization chemistry, it has been suggested that the
regioselective reduction of the C9 keto group in growing
polyketone chains may be a crucial step in biasing subsequent
ring closure aldol chemistry.280,284 Conversion of the sp2 ketone
to sp3 alcohol is likely to introduce a kink in the polyketone
(polyenolate) chain and constrain the acyclic product in
a conformer poised for intramolecular ring formation. Cyclases
may also have a chaperone like function in addition to aldol
condensation catalysis by binding the polyketone-S-T
intermediates, before and after C9 keto reduction, to prevent off
pathway intramolecular cyclizations.13 The role of cyclases for
control of topologies of the second, third, and fourth ring
cyclizations in tetracyclic aromatic polyketides was originally
determined by genetic studies but has since been examined by
ay demonstrates catalysis across an NRPS/PKS interface.
Nat. Prod. Rep., 2008, 25, 757–793 | 781

Fig. 38 Reconstitution of the biosynthetic pathways of aromatic polyketides. (a) Action of minimal Type II PKS in the early steps of actinorhodin
biosynthesis. (b) Chain elongation, aromatization, and tailoring to produce tetracenomycin C. (c) Mechanism of chain initiation and elongation in the
biosynthesis of the R1128 polyketides. (d) Bikaverin synthase and the use of alternative starter units.
782 | Nat. Prod. Rep., 2008, 25, 757–793 This journal is ª The Royal Society of Chemistry 2008
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
purified cyclases from the tetracenomycin,285 nogalamycin,286
aclacinomycin,287 and doxorubicin288 type II PKS pathways.
Full reconstitution of the tetracyclic backbones of the scaf-
folds in doxorubicin36 and oxytetracycline289 purified systems
remain to be established, although studies with specific genes on
plasmids in engineered strains of streptomyces have gone a long
way to facilitate identification of the roles of particular protein
components, accumulation of intermediates in blocked strains,
and re-engineering to produce novel products.13 In the case of
tetracenomycin, biochemical characterization of the Tcm poly-
ketide synthase in vivo290 has been supplemented with detailed
mechanistic studies of isolated proteins.291 TcmN has been
shown to initiate aromatization of the decaketide interme-
diate,292 while TcmI functions to close the last ring of the tetra-
cycle.285,293 TcmH,294 N,292 OP and G295 then act to tailor the
tetracenomycin core. In the R1128 Type II PKS system, studies
with purified proteins that carry out chain initiation296,297 and
then chain elongation steps298 show orthogonal specificity of the
KS components for the initiation or elongation T domain,
indicating approaches to use an alternate starter acyl-CoA to
reprogram those pathways.299,300
The fungus Gibberella fujikuroi has an iterative type I PKS
known as PKS4 that elaborates the tetracyclic metabolite bika-
verin, which has anticancer activity.115 The single PKS4 module
is used nine times to build a tetracyclic nonaketide scaffold from
malonyl-CoA monomers. PKS4 can be expressed in and purified
from E. coli in the active, holo form. Addition of malonyl-CoA
monomers leads to the tetracyclic product SMA76a, a non-
aketide that has undergone two dehydrations. In addition to full
chain length control, purified PKS4 catalyzes the four regiospe-
cific cyclizations found in the bikaverin scaffold: an aldol
condensation to connect C2 and C7, a Claisen condensation to
connect C10 to C1, and attack of the C9 phenolate on the C13
carbonyl to yield the tricyclic naphthopyrone. This three-ring
connectivity is also seen in action of the Aspergillus nidulans wA
synthase.301 The last ring then presumably arises by C12–C17
aldol-type condensation. Use of different starter acyl-CoAs such
as octanoyl-CoA reroutes flux to a benzopyrone hexaketide,
reminiscent of volume control of chain length by CLFs in the
type II bacterial PKSs noted above. The PKS4 module can be
mutated in its terminal cyclase domain and then reconstituted in
trans with purified cyclases to establish alternate ring-closing
topologies to generate other aromatic scaffolds.302
Fig. 39 Complete reconstitution of a type II PKS biosynthetic pathway. The c
with the enterocin synthase results in formation of the complex tricyclic scaf
This journal is ª The Royal Society of Chemistry 2008
While we do not cover them in detail here, we note that much
progress has been made in reconstituting the biosynthesis of type
III PKs in vitro, including structural studies, the identification of
residues that control chain length, and successful efforts to
engineer these enzymes to produce novel products.38,39,303–307
9.4 Enterocin: a type II PKS pathway that builds a tricyclic
scaffold
The antibiotic enterocin, produced by Streptomyces maritimus, is
derived from an octaketide scaffold that is enzymatically
processed after chain elongation to a tricyclic core308 (Fig. 39). It
is assembled by a type II PKS system that uses benzoate as an
unusual starter unit309 followed by seven malonyl CoAs as
extender units. The minimal PKS requires the KS and a regula-
tory CLF type subunit (EncAB), the holo form of the T domain
(EncC), and an acyltransferase (FabD) borrowed from the
organism’s type II FAS. The ATP-dependent benzoate ligase
(EncN) generates benzoyl-AMP as the thermodynamically
activated starter unit. In the absence of tailoring enzymes, the
octaketone intermediate is rerouted to cyclic off-pathway struc-
tures,310 as is typical of type II PKS systems in the absence of
their cognate-tailoring enzymes. When EncM, EncK, and EncR
are provided, full reconstitution of enterocin is enabled with the
eight purified enzymes.310 EncM is the key rearrangement
catalyst, a proposed ‘‘Favorskiiase’’ that converts the C13 ketone
into an ester and yields the tricyclic scaffold.311 Further tailoring
involvesO-methylation (EncK) and installation of the 5-hydroxy
group (provided by the CytP450-type monoxygenase EncR).
EncM presumably uses a triketonic intermediate and makes the
C6–C11 and C7–C14 bonds by aldol chemistry. This test tube
recapitulation of the enterocin pathway installs the ten C–C
bonds, the five C–O bonds, and creates the seven chiral centers in
the natural product; most remarkable is the Favorskii-style
rearrangement. Only in this context can studies with purified
EncM shed light on the precise mechanism of tricyclic scaffold
generation.
9.5 Aminocoumarins: four-component assembly of the
novobiocin scaffold and seven-component assembly of
coumermycin
The aminocoumarin antibiotics (novobiocin, clorobiocin, and
coumermycin) act by targeting bacterial type II topoisomerases,
ombination of a benzoate starter unit and seven malonyl CoAmonomers
fold.
Nat. Prod. Rep., 2008, 25, 757–793 | 783

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
thereby blocking DNA replication.312–314 Novobiocin and
clorobiocin differ only in their aminocoumarin C8 substituents
(chlorine vs. methyl) and their L-noviosyl 30-substituents
(carbamoyl vs. 5-methylpyrrole-2-carboxyl) (Fig. 40 and Fig. 41).
Coumermycin is a pseudosymmetric dimer built around
a pyrrole scaffold with the CH3 substituent on the amino-
coumarins and the methylpyrrolyl-ester on the noviose rings.
Cloning of the gene clusters315–317 has enabled subsequent
reconstitution studies with purified Nov, Clo, and Cou enzymes
such that all but one step in the four-part assembly of the three
aminocoumarin antibiotics have been described.
The eponymous bicyclic aminocoumarin ring is formed on
a free-standing A-T didomain enzyme (NovH) which activates
L-Tyr and installs it as the Tyr-S-T thioester in preparation for
b-hydroxylation by a partner cytochrome P450 type mono-
oxygenase (NovI).318 The newly introduced benzylic hydroxyl is
then oxidized by a heterodimeric NAD enzyme (NovJK) to yield
the b-keto-Tyr-S-T intermediate.319 A presumed hydroxylation,
by an as yet uncharacterized enzyme, sets up intramolecular
lactonization as the bicyclic aminocoumarin gets released from
the pantetheinyl thiol tether. The second building block is the
Fig. 40 (a) Biosynthesis of the aminocoumarin ring. (b) Production of th
novobiocin and clorobiocin biosynthetic pathways is centered on elaboration
784 | Nat. Prod. Rep., 2008, 25, 757–793
3-prenyl-4-hydroxybenzoate, derived from tyrosine by oxidative
enzymology (CloR)320 and prenyltransferase (CloQ) catalysis.244
The aminocoumarin and the prenylated hydroxybenzoate frag-
ments are ligated (NovL)321 by an ATP-dependent ligase that
generates the acyl-AMP intermediate that is captured by the
amine of the aminocoumarin to yield novobiocic acid. This
product must be enzymatically tailored at C8 of the
aminocoumarin before the hexosyl unit (L-noviose) is added to
the phenolic hydroxyl. C-methylation requires the carbanion at
C8, which is generated on deprotonation of the phenol hydroxyl
in the active site of NovO.322 Subsequent attack of the activated
methyl group of SAM by the resonance-stabilized carbanion
results in the new C–C bond. Glycosylation is then effected by
NovM, using TDP-L-noviose as glycosyl donor.323 The enzymes
converting TDP-glucose to the dedicated building block TDP-
L-noviose are also encoded in the biosynthetic gene cluster, and
several of the steps have been characterized.324,325 The product
from NovM action requires two final tailoring steps to yield the
active antibiotic. NovP326 is a 40-O-methyltransferase, again
using SAM as cosubstrate methyl donor. The last step is cata-
lyzed by the carbamoyl transferase NovN, using carbamoyl
e 3-prenyl-4-hydroxybenzoate building block. (c) Reconstitution of the
of the eponymous aminocoumarin ring.
This journal is ª The Royal Society of Chemistry 2008

Fig. 41 Ligation of two aminocoumarin fragments to a methyl pyrrole is demonstrated in the reconstitution of the coumermycin biosynthetic pathway.
Characterization of the aminocoumarin enzyme set has enabled the synthesis of several structural analogs through combinatorial biosynthesis.
Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
phosphate as the carbamoyl donor.327 Carbamoylation results in
a 200-fold enhancement of antibiotic activity, indicating the
30-O-acyl substituent on the novose sugar moiety is a key
pharmacophore.328 Ten purified enzymes have been studied to
reconstruct the conversion of two molecules of tyrosine,
carbamoyl phosphate, D2-isopentenyl diphosphate, and TDP-L-
noviose to the antibiotic novobiocin, with one enzymatic step as
yet uncharacterized.
The clorobiocin pathway differs in that the C-methyl-
transferase NovO (and CouO) is replaced by the FADH2-
dependent halogenase, Clo halogenase, which is presumed to add
a Cl+ equivalent analogously at the clorobiocic acid stage.329 The
other main difference is that the 30-O-carbamoyl transferase
gene, novN, is replaced by seven contiguous ORFs in both the
clorobiocin and coumermycin clusters: cloN1–7 and couN1–7.193
Studies with purified CouN3, Clo/CouN4, and Clo/CouN5 have
confirmed that Clo/CouN4 is the proline-specific adenylyl-
transferase, Clo/CouN5 is the T domain, and Clo/CouN3 is the
flavin-dependent dehydrogenase that catalyzes the double
dehydrogenation of prolyl-S-T (where T is Clo/CouN5) to
pyrrolyl-S-T.134 The proteins Clo/CouN2 and Clo/CouN7 have
been predicted to be acyl transferases and Clo/CouN1 a variant
of a T domain. These three proteins presumably act in a sequence
where Clo/CouN2 transfers the pyrrolyl moiety fromClo/CouN5
to Clo/CouN1. Clo/CouN7 would then act as the final pyrrolyl
transferase to move the pyrrole-2-carboxyl moiety to the
clorobiocin/coumermycin penultimate intermediate where the
30-hydroxyl of the 40-O-Me-noviosyl ring is the nucleophile.
Indeed, the apo form of CouN1 can be primed with a Ppant
prosthetic group and the pyrrolyl moiety installed via Sfp action
with pyrrolyl-CoA as its substrate. Pyrrolyl-S-CouN1 is a good
This journal is ª The Royal Society of Chemistry 2008
substrate for coupling with the clorobiocin, coumermycin and
novobiocin scaffolds in a regiospecific acyl transfer catalyzed by
purified CouN7.58,330 With purified apo CouN1, CouN7 and
a variety of synthetic pyrrolyl-CoA analogs, the Sfp-mediated
loading of CouN1 has demonstrated that CouN7 will transfer
two dozen heterocyclic variants of the pyrrole-2-carbonyl moiety
to make variant antibiotics.58 Thus, in the Clo/CouN1–7 arm of
the pathway, five of the seven proteins have been purified and
characterized. To date, Clo/CouN2 has been insoluble on
heterologous expression but can be bypassed by using Sfp to load
CouN1. Clo/CouN6 is predicted to be a novel C-methylation
catalyst making the CH3–C5 bond in the 5-methylpyrrole-
2-carboxyl pharmacophore; Clo/CouN6 have yet to be
characterized but now can be approached.
Tracing the reconstitution of clorobiocin, 15 purified proteins
have so far been characterized and only the Clo halogenase,329
and the missing lactone-forming enzyme in aminocoumarin
construction are not yet evaluated. Finally, it is worth noting that
the more potent family member coumermycin is built from both
carboxyl groups of 2,4-dicarboxy-3-methylpyrrole as the central
scaffold. The ligase CouL adds 8-methylnovobiocic acid to both
arms of the dicarboxypyrrole to make two amide linkages.331
Both phenols in the two aminocoumarins are then noviosylated
by CouM and the noviosyl sugars are likewise eachO-methylated
by CouP.332 Use of the CouN1–CouN7 pair with Sfp and
pyrrolyl-CoAs allows enzymatic construction of coumermycin
variants.58,333 In one representative study, five different sites were
varied in the Cou scaffold by use of alternate substrate fragments
with the tandem action of the four enzymes CouLMP and
NovN.332 Clearly, one can tinker with very complex antibiotic
natural product scaffolds with purified biosynthetic enzymes.
Nat. Prod. Rep., 2008, 25, 757–793 | 785

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
9.6 Rebeccamycin, staurosporine, violacein, and terrequinone:
reconstruction of pathways involving tryptophan oxidative
dimerization
Microbial oxidation of tryptophan at the C2 amino group is the
first step in a number of pathways which divert part of the pool of
this proteinogenic amino acid to bisindole-containing structures
with diverse biological functions.334 For example, the six-ring
indolocarbazole scaffold is found in the antitumor agent
rebeccamycin and the protein kinase inhibitor staurosporine. A
different connectivity is found in the purple bisindole pigment
violacein that gives Chromobacterium violaceum its name
(Fig. 42). These three metabolites have a pyrrole ring connector
between the two indole moieties. In contrast, the cytostatic
fungal metabolite terrequinone consists of a benzoquinone
scaffold between the indole pair. Recent studies with the purified
biosynthetic enzymes for these four natural products have
allowed deconvolution of some remarkable enzyme-mediated
C–C bond-forming redox chemistry.
Fig. 42 Reconstitution of biosynthetic pathways based on oxidative coupling
staurosporine and rebeccamycin, while violacein displays a rearranged scaffol
Claisen condensation.
786 | Nat. Prod. Rep., 2008, 25, 757–793
9.6.1 Rebeccamycin and staurosporine. A combination of
cloning, gene cluster sequencing, and genetic knockout studies
yielded hypotheses about the roles of several enzymes encoded in
the reb and sta gene clusters,334,335with further illumination of the
remarkable chemistry provided by studies with purified
RebHODPC or their Sta counterparts.336,337 The first reaction in
the Reb pathway, but not the Sta pathway, is mediated by the
FADH2-dependent halogenase RebH76,77,82 (noted in Section
4.2), to yield 7-chloro-Trp as the first committed substrate.
Oxidation of 7-Cl-Trp by the flavoenzyme amino acid oxidase
RebO yields the imino acid form of the 7-chloroindolopyruvate.
While the imine could hydrolyze after release from the active site
of RebO, two such molecules can be intercepted by the next
enzyme, the hemeprotein RebD, which appears to mediate C–C
bond formation by benzylic coupling. Loss of one NH3 and
intramolecular cyclization of the initial adduct yields the pyrrole
ring that is the key connecting element in the observed RebD
product (dichloro)-chromopyrrolic acid. This oxidative
chemistry,337 initiated and controlled by RebD, has built
of tryptophan. Direct coupling results in the chromopyrrolic acid core of
d. Dimerization to produce the terrequinones proceeds through a double
This journal is ª The Royal Society of Chemistry 2008

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
a pyrrole connector between the two indoles in a five-ring
pathway intermediate. The sixth ring is then formed by RebP,
a second hemeprotein, which acts to make the bond between the
two C2 positions in the indole moieties, probably via one-electron
chemistry.336,338 Remarkably, three products differing in the
oxidation state of the pyrrole ring are found in incubations of
chromopyrrolic acid with purified RebP/StaP. Addition of the
flavoenzyme RebC reroutes the flux to the aglycone of rebecca-
mycin. On the other hand, StaC sends the flux towards the
stauosporine amide, which lacks the second carbonyl of rebec-
camycin, installed through four-electron oxidation.
From the studies with the purified enzymes, we now know that
the four enzymes RebODPC catalyze a net fourteen-electron
oxidation as the two Trp substrate molecules are converted to the
six-ring indolocarbazole scaffold. Each enzyme has a redox
cofactor: FAD, heme, heme, and FAD, respectively, and O2 is
a cosubstrate for each of the four enzymes. This is a short but
impressive lesson in Nature’s redox catalyst capacity. The
aglycones in the rebeccamycin and staurosporine pathway then
get N-glycosylated (and O-methylated) by Gtfs that have not yet
been characterized biochemically. The staurosporine pathway
has one final twist in which C5 of the N-glycosyl deoxysugar
moiety is subjected to one more hemeprotein oxidative reaction,
setting up a second N–C glycosidic linkage. The resulting bridged
system of the staurosporine protein kinase inhibitor features
attachment of the deoxysugar to both indole nitrogens.
9.6.2 Violacein. Chromobacterium violaceum is found glob-
ally distributed in tropical waters and it is likely that the deep
violet color of violacein protects the producer organism’s
genome against UV damage.339 Violacein is most noteworthy
for a 1,2-indole shift that has occurred on the pyrrole ring
connecting scaffold340 as compared to the rebeccamycin and
staurosporine pathways.
The Vio pathway starts the same way as the Reb and Sta
pathways: with enzymatic oxidation of Trp to the iminopyruvate
by a flavoenzyme oxidase VioA. VioB is a hemeprotein analog of
RebD/StaD, and indeed, tandem action of purified VioA and
VioB yields chromopyrrolic acid (CPA). In violacein biosyn-
thesis, however, CPA is an off-pathway side product.
On-pathway, flux requires a third purified protein, VioE,341 that
has been found to mediate the 1,2-indole shift to yield
prodeoxyviolacein. Finally, action of purified VioD and VioC
lead to the final indole ring hydroxylations/oxidations to produce
violacein. Even more impressively, VioE will reroute the inter-
mediate made by RebOD away from chromopyrrolic acid to the
rearranged violacein scaffold. As yet, the mechanism of the
remarkable 1,2-indole shift is not understood, but the purified
VioBE combination offers the chance for mechanistic dissection
in both the Reb/Sta and Vio systems.
9.6.3 Terrequinone. The five-enzyme pathway to the Asper-
gillus metabolite terrequinone (a member of a family of asterri-
quinones) has recently been identified by a combination of fungal
genomics342,343 and purified enzyme analysis.213,344 Three
unanticipated features of enzyme chemistry have emerged. First,
the usual oxidation at the C2-amino group of Trp occurs;
however, in the terrequinone pathway, a PLP enzyme (TdiD),
rather than a flavoenzyme, mediates this transformation. The
This journal is ª The Royal Society of Chemistry 2008
PLP enzyme is a transaminase, and therefore the direct product is
the phenylpyruvate rather than the iminophenylpyruvate
generated by the flavoenzymes RebO and VioA. This detail
becomes relevant as the nitrogen of the rebeccamycin pyrrole is
lost before enzymatic dimerization occurs.
The second enzyme TdiA is a single module NRPS with an
A-T-TE tridomain organization. However, the A domain is
specific for selection and activation of the a-keto acid
phenylpyruvate, rather than the a-amino acid Trp. Such keto
acid activating domains are unusual but have now been detected
in the NRPS modules of valinomycin, cereluide, and barbamide
biosynthesis (vide supra). TdiA has some analogies to the enter-
obactin synthetase EntF in its TE domain: they both act as
cyclization catalysts, holding one acyl group as a thioester on the
Ppant arm of the T domain and the other as an oxoester at the
active site Ser residue of the TE domain. Because the two
tethered acyl chains here are phenylpyruvyl moieties, they can
react as enolates in C–C bond formation. Double Claisen-type
condensation can happen in head-to-tail fashion to release the
bisindolylbenzoquinone. This is novel cyclization chemistry for
an NRPS TE domain, set up by the A domain that activates
a keto acid rather than an amino acid. Note that the scaffold
connecting the two indole moieties is a six-membered benzo-
quinone rather than the five-membered pyrrole found in chro-
mopyrrolic acid in the Reb and Sta pathways. This harks back to
the use of a PLP transaminase rather than an FAD-oxidase in the
initial oxidation of the two Trp substrate molecules.
The remaining three proteins have the following roles. TdiC is
an NADH-dependent quinone reductase creating the hydroqui-
none oxidation state. The hydroquinone is required for C3 of the
ring to act as nucleophile in the active site of the next enzyme
TdiB, a prenyltransferase. TdiB catalyzes C–C bond formation
between C3 of the hydroquinone and C1 of the D2-isopentenyl
diphosphate partner subsbtrate. The C-monoprenyl product is
the substrate for a second prenyl transfer by TdiB, this time to C2
of one of the indole rings. The double prenylation by TdiB has
one additional novel feature: the second prenylation proceeds
with reverse regiochemistry (SN20), that results in capture of the
of the second D2-isopentenyl-PP moiety at C3, rather than at C1,
for C–C bond formation. Finally, TdiE appears to be a
chaperone which directs flux in the first prenylation step to
on-pathway C–C bond formation. In the absence of TdiE,
incubations of TdiC/TdiB result in off-pathwayO-prenylation of
the quinone. In loose analogy to VioE, noted in the violacein
pathway above, TdiE is a protein factor that keeps reactant flux
on-pathway by an as-yet unknown mechanism.
In summary, the studies of rebeccamycin, staurosporine,
violacein, and terrequinone biosynthesis with purified enzymes
have revealed some remarkable chemical strategies and mecha-
nisms for dimerizing tryptophan to a variety of scaffolds. They
also show the versatility of enzymes with FAD, PLP, and heme
iron redox cofactors to control and direct the flux of highly
reactive intermediates for regiospecific construction of the diverse
molecular frameworks of these biologically active metabolites.
10 Lessons learned
In vitro studies with purified enzymes bridge the gap between
bioinformatic predictions, resulting from sequence annotation
Nat. Prod. Rep., 2008, 25, 757–793 | 787

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
and gene knockouts, and the direct observation of gene product
function in NRPS, PKS, or hybrid NRPS-PKS biosynthetic
pathways. Experiments with encoded enzymes can uncover novel
chemistry and set the stage for rational manipulation of partic-
ular steps in natural product biosyntheses. Characterization of
enzymes in the four stages of type I PK and NRP synthesis have
revealed mechanisms and selectivity in: (1) the T domain priming
steps, (2) the pre-assembly line provision of dedicated monomer
units, (3) the multiple steps of assembly line operations, including
chain initiation, elongation, and termination, and (4) post-
assembly-line tailoring reactions to create mature natural
product scaffolds.
Investigation of the phosphopantetheinyl transferase
superfamily has greatly facilitated in vitro PKS and NRPS
reconstitutions by enabling the reliable priming of apo T domains
with the essential phosphopantetheinyl arm. PPTase studies have
revealed the molecular basis for discrimination of subclasses of T
domains—FAS and PKS acyl carrier proteins vs NRPS peptidyl
carrier proteins—and have shown that these enzymes can load
many forms of acyl-S-pantetheinyl chains onto a range of apo T
domains for mechanistic analysis, biosynthetic reconstitutions,
and the generation of novel natural product derivatives.
Work with purified enzymes that provide dedicated monomer
units has indicated which ORFs need to be transferred to cells for
effective reconstitution of functional assembly lines in new
producer organisms. In addition, the study of monomer-unit
provision has illuminated novel chemical strategies for the
construction of nonproteinogenic amino acid building blocks;
for example, the action of the metal-free dioyxgenase DpgC in
the vancomycin/teicoplanin pathways.
Efforts to reconstitute the action of assembly line modules
have also decifered mechanisms for chain initiation, including
the intersection of PLP-enzymes and PKS modules for the
introduction of nitrogen into polyketide scaffolds. Studies of
subsets of condensation domains have demonstrated variations
of C domain function both as chiral (DCL) amide and ester
synthases during chain elongations and epimerization catalysts.
The Cy subset of C domains are extraordinary catalysts,
converting acyl-Cys-S-Tn and acyl-Ser-S-Tn to tethered hetero-
cyclic thiazolinyl and oxazolinyl-S-Tn.
Insight into how isoprenoid and polyketide metabolism can
intersect on assembly lines has come from examination of a six-
protein cassette that includes an HMG CoA synthase homolog.
Experiments with the HMG synthase and a pair of tandem-
acting enoyl CoA hydratase family members that catalyze
dehydration, then decarboxylation, have revealed the strategy
and mechanism for in trans tailoring of elongating polyketides
that results in the introduction of b-branches in the chain
backbone.
Characterization of hemeproteins OxyA and OxyB has shown
how the peptide scaffold of the vancomycin family of glyco-
peptide antibiotics is rigidified through crosslinking. This mode
of peptide tailoring exemplifies enzymatic installation of archi-
tecturally constraining ether and C–C crosslinks. Examination of
the SyrB1,B2, and SyrC enzymes in the syringomycin pathway
has revealed Nature’s strategy for chlorination of threonine on
a side module of the assembly line followed by transfer to the
ninth module of the megasynthetase SyrE. Study of the RapP
NRPSmodule has shown how a pipecolyl unit is inserted into the
788 | Nat. Prod. Rep., 2008, 25, 757–793
middle of the polyketide backbone in rapamycin and FK506
immunosuppressants. Characterization of the chain-terminating
TE domains of NRPS and PKS assembly lines have illustrated
the regio- and stereospecific incorporation of macrocyclic
constraints into nascent scaffolds as they are released.
Investigation of the post-assembly-line tailoring reactions of
acylation, glycosylation, and oxidation has revealed the timing
and mechanism of natural product maturation and provided
information required for the manipulation of these late-stage
transformations for combinatorial engineering of new structures.
Finally, full reconstitution of natural product pathways in
both type I and type II contexts with pure enzymes in test tubes
has allowed the elaboration of remarkable complexity
from simple building blocks, as in enterocin, coumermycin,
rebeccamycin, and terrequinone scaffold construction. These
efforts enable future investigation of some remarkable, unsolved
catalytic transformations such as the Favorskii reaction in
enterocin assembly and the 1,2-indole shift reaction during
violacein biosynthesis. The studies on syringomycin and on
coronamic acid formation have revealed a striking class of
biological halogenases that utilize nonheme mononuclear high-
valent oxoiron intermediates for selective C–H activation and
halogen transfer to unactivated substrates. Undoubtedly, more
novel enzyme chemistry remains to be deciphered through the
deconvolution of NRP and PK biosynthetic pathways.
11 Acknowledgements
We acknowledge all of our colleagues whose names are listed in
the specific references for their efforts in natural product enzyme
characterization and pathway reconstitution.
12 References
1 M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496.
2 S. Donadio, P. Monciardini andM. Sosio,Nat. Prod. Rep., 2007, 24,1073–1109.
3 J. H. Crosa and C. T. Walsh, Microbiol. Mol. Biol. Rev., 2002, 66,223–249.
4 S. C. Andrews, A. K. Robinson and F. Rodriguez-Quinones, FEMSMicrobiol. Rev., 2003, 27, 215–237.
5 S. Schneiker, O. Perlova, O. Kaiser, K. Gerth, A. Alici,M. O. Altmeyer, D. Bartels, T. Bekel, S. Beyer, E. Bode,H. B. Bode, C. J. Bolten, J. V. Choudhuri, S. Doss,Y. A. Elnakady, B. Frank, L. Gaigalat, A. Goesmann,C. Groeger, F. Gross, L. Jelsbak, L. Jelsbak, J. Kalinowski,C. Kegler, T. Knauber, S. Konietzny, M. Kopp, L. Krause,D. Krug, B. Linke, T. Mahmud, R. Martinez-Arias,A. C. McHardy, M. Merai, F. Meyer, S. Mormann, J. Munoz-Dorado, J. Perez, S. Pradella, S. Rachid, G. Raddatz, F. Rosenau,C. Ruckert, F. Sasse, M. Scharfe, S. C. Schuster, G. Suen,A. Treuner-Lange, G. J. Velicer, F. J. Vorholter, K. J. Weissman,R. D. Welch, S. C. Wenzel, D. E. Whitworth, S. Wilhelm,C. Wittmann, H. Blocker, A. Puhler and R. Muller, Nat.Biotechnol., 2007, 25, 1281–1289.
6 M. Oliynyk, M. Samborskyy, J. B. Lester, T. Mironenko, N. Scott,S. Dickens, S. F. Haydock and P. F. Leadlay,Nat. Biotechnol., 2007,25, 447–453.
7 D. W. Udwary, L. Zeigler, R. N. Asolkar, V. Singan, A. Lapidus,W. Fenical, P. R. Jensen and B. S. Moore, Proc. Natl. Acad. Sci.U. S. A., 2007, 104, 10376–10381.
8 H. Ikeda, J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi,T. Shiba, Y. Sakaki, M. Hattori and S. Omura, Nat. Biotechnol.,2003, 21, 526–531.
This journal is ª The Royal Society of Chemistry 2008

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
9 S. Omura, H. Ikeda, J. Ishikawa, A. Hanamoto, C. Takahashi,M. Shinose, Y. Takahashi, H. Horikawa, H. Nakazawa,T. Osonoe, H. Kikuchi, T. Shiba, Y. Sakaki and M. Hattori, Proc.Natl. Acad. Sci. U. S. A., 2001, 98, 12215–12220.
10 H. B. Bode, B. Bethe, R. Hofs and A. Zeeck,ChemBioChem, 2002, 3,619–627.
11 I. de Bruijn, M. J. de Kock, M. Yang, P. de Waard, T. A. van Beekand J. M. Raaijmakers, Mol. Microbiol., 2007, 63, 417–428.
12 S. C. Wenzel and R. Muller, Curr. Opin. Chem. Biol., 2005, 9, 447–458.
13 C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat.Prod. Rep., 2007, 24, 162–190.
14 D. E. Cane and C. T. Walsh, Chem. Biol., 1999, 6, R319–325.15 I. B. Lomakin, Y. Xiong and T. A. Steitz, Cell, 2007, 129, 319–332.16 S. Jenni, M. Leibundgut, D. Boehringer, C. Frick, B. Mikolasek and
N. Ban, Science, 2007, 316, 254–261.17 J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416.18 O. Froyshov, Acta Microbiol. Acad. Sci. Hung., 1975, 22, 427–432.19 S. G. Laland and T. L. Zimmer, Essays Biochem., 1973, 9, 31–57.20 P. Zuber, Curr. Opin. Cell Biol., 1991, 3, 1046–1050.21 H. Kleinkauf and H. von Dohren, Eur. J. Biochem., 1990, 192, 1–15.22 D. A. Hopwood, Chem. Rev., 1997, 97, 2465–2498.23 D. Schwarzer, R. Finking and M. A. Marahiel, Nat. Prod. Rep.,
2003, 20, 275–287.24 C. T. Walsh, H. Chen, T. A. Keating, B. K. Hubbard, H. C. Losey,
L. Luo, C. G. Marshall, D. A. Miller and H. M. Patel, Curr. Opin.Chem. Biol., 2001, 5, 525–534.
25 R. M. Kohli and C. T. Walsh, Chem. Commun., 2003, 297–307.26 F. Kopp and M. A. Marahiel, Nat. Prod. Rep., 2007, 24, 735–749.27 B. Shen, Curr. Opin. Chem. Biol., 2003, 7, 285–295.28 Y. J. Lu, Y. M. Zhang and C. O. Rock, Biochem. Cell Biol., 2004, 82,
145–155.29 E. Schweizer and J. Hofmann, Microbiol. Mol. Biol. Rev., 2004, 68,
501–517, table of contents.30 C. Khosla, Chem. Rev., 1997, 97, 2577–2590.31 C. M. Kao, L. Katz and C. Khosla, Science, 1994, 265, 509–512.32 S. A. Sieber and M. A. Marahiel, Chem. Rev., 2005, 105, 715–738.33 C. R. Hutchinson, J. Kennedy, C. Park, S. Kendrew, K. Auclair and
J. Vederas, Antonie Van Leeuwenhoek, 2000, 78, 287–295.34 T. Maier, S. Jenni and N. Ban, Science, 2006, 311, 1258–1262.35 J. Zhang, S. G. Van Lanen, J. Ju, W. Liu, P. C. Dorrestein, W. Li,
N. L. Kelleher and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2008,105, 1460–1465.
36 C. R. Hutchinson, Chem. Rev., 1997, 97, 2525–2536.37 H. Jenke-Kodama, A. Sandmann, R. Muller and E. Dittmann,Mol.
Biol. Evol., 2005, 22, 2027–2039.38 K. Watanabe, A. P. Praseuth and C. C. Wang, Curr. Opin. Chem.
Biol., 2007, 11, 279–286.39 M. B. Austin and J. P. Noel, Nat. Prod. Rep., 2003, 20, 79–110.40 R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle,
M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, Chem. Biol.,1996, 3, 923–936.
41 C. T. Walsh, A. M. Gehring, P. H. Weinreb, L. E. Quadri andR. S. Flugel, Curr. Opin. Chem. Biol., 1997, 1, 309–315.
42 A. C.Mercer andM. D. Burkart,Nat. Prod. Rep., 2007, 24, 750–773.43 J. Crosby, D. H. Sherman, M. J. Bibb, W. P. Revill, D. A. Hopwood
and T. J. Simpson, Biochim. Biophys. Acta, 1995, 1251, 32–42.44 L. E. Quadri, P. H. Weinreb, M. Lei, M. M. Nakano, P. Zuber and
C. T. Walsh, Biochemistry, 1998, 37, 1585–1595.45 C. Sanchez, L. Du, D. J. Edwards, M. D. Toney and B. Shen, Chem.
Biol., 2001, 8, 725–738.46 K. Watanabe, M. A. Rude, C. T. Walsh and C. Khosla, Proc. Natl.
Acad. Sci. U. S. A., 2003, 100, 9774–9778.47 S. Gruenewald, H. D.Mootz, P. Stehmeier and T. Stachelhaus,Appl.
Environ. Microbiol., 2004, 70, 3282–3291.48 Z. Suo, C. C. Tseng and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A.,
2001, 98, 99–104.49 H. Chen, S. O’Connor, D. E. Cane and C. T. Walsh, Chem. Biol.,
2001, 8, 899–912.50 M. R. Mofid, R. Finking, L. O. Essen and M. A. Marahiel,
Biochemistry, 2004, 43, 4128–4136.51 D. B. Stein, U. Linne andM. A.Marahiel, FEBS J., 2005, 272, 4506–
4520.52 J. Yin, A. J. Lin, D. E. Golan and C. T. Walsh,Nat. Protocols, 2006,
1, 280–285.
This journal is ª The Royal Society of Chemistry 2008
53 J. Yin, F. Liu, X. Li and C. T. Walsh, J. Am. Chem. Soc., 2004, 126,7754–7755.
54 J. Yin, F. Liu, M. Schinke, C. Daly and C. T. Walsh, J. Am. Chem.Soc., 2004, 126, 13570–13571.
55 J. Yin, P. D. Straight, S. Hrvatin, P. C. Dorrestein, S. B. Bumpus,C. Jao, N. L. Kelleher, R. Kolter and C. T. Walsh, Chem. Biol.,2007, 14, 303–312.
56 J. Yin, P. D. Straight, S. M. McLoughlin, Z. Zhou, A. J. Lin,D. E. Golan, N. L. Kelleher, R. Kolter and C. T. Walsh, Proc.Natl. Acad. Sci. U. S. A., 2005, 102, 15815–15820.
57 F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. E. O’Connor andC. T. Walsh, Nature, 2005, 436, 1191–1194.
58 M. Fridman, C. J. Balibar, T. Lupoli, D. Kahne, C. T. Walsh andS. Garneau-Tsodikova, Biochemistry, 2007, 46, 8462–8471.
59 J. Ligon, S. Hill, J. Beck, R. Zirkle, I. Molnar, J. Zawodny, S.Moneyand T. Schupp, Gene, 2002, 285, 257–267.
60 Y. A. Chan, M. T. Boyne, 2nd, A. M. Podevels, A. K. Klimowicz,J. Handelsman, N. L. Kelleher and M. G. Thomas, Proc. Natl.Acad. Sci. U. S. A., 2006, 103, 14349–14354.
61 B. S. Moore and C. Hertweck, Nat. Prod. Rep., 2002, 19, 70–99.62 S. Caboche, M. Pupin, V. Leclere, A. Fontaine, P. Jacques and
G. Kucherov, Nucleic Acids Res., 2008, 36, D326–331.63 N. J. Ryding, T. B. Anderson and W. C. Champness, J. Bacteriol.,
2002, 184, 794–805.64 A. M. van Wageningen, P. N. Kirkpatrick, D. H. Williams,
B. R. Harris, J. K. Kershaw, N. J. Lennard, M. Jones, S. J. Jonesand P. J. Solenberg, Chem. Biol., 1998, 5, 155–162.
65 B. K. Hubbard and C. T. Walsh, Angew. Chem., Int. Ed., 2003, 42,730–765.
66 O. Puk, P. Huber, D. Bischoff, J. Recktenwald, G. Jung,R. D. Sussmuth, K. H. van Pee, W. Wohlleben and S. Pelzer,Chem. Biol., 2002, 9, 225–235.
67 B. K. Hubbard, M. G. Thomas and C. T. Walsh, Chem. Biol., 2000,7, 931–942.
68 V. Pfeifer, G. J. Nicholson, J. Ries, J. Recktenwald, A. B. Schefer,R. M. Shawky, J. Schroder, W. Wohlleben and S. Pelzer, J. Biol.Chem., 2001, 276, 38370–38377.
69 H. Chen, C. C. Tseng, B. K. Hubbard and C. T. Walsh, Proc. Natl.Acad. Sci. U. S. A., 2001, 98, 14901–14906.
70 O. Puk, D. Bischoff, C. Kittel, S. Pelzer, S. Weist, E. Stegmann,R. D. Sussmuth and W. Wohlleben, J. Bacteriol., 2004, 186, 6093–6100.
71 C. C. Tseng, S. M. McLoughlin, N. L. Kelleher and C. T. Walsh,Biochemistry, 2004, 43, 970–980.
72 C. C. Tseng, F. H. Vaillancourt, S. D. Bruner and C. T. Walsh,Chem. Biol., 2004, 11, 1195–1203.
73 P. F. Widboom, E. N. Fielding, Y. Liu and S. D. Bruner, Nature,2007, 447, 342–345.
74 G. W. Gribble, Chemosphere, 2003, 52, 289–297.75 F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova
and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378.76 E. Yeh, L. J. Cole, E. W. Barr, J. M. Bollinger, Jr., D. P. Ballou and
C. T. Walsh, Biochemistry, 2006, 45, 7904–7912.77 E. Yeh, L. C. Blasiak, A. Koglin, C. L. Drennan and C. T. Walsh,
Biochemistry, 2007, 46, 1284–1292.78 F. H. Vaillancourt, J. Yin and C. T. Walsh, Proc. Natl. Acad. Sci.
U. S. A., 2005, 102, 10111–10116.79 D. P. Galonic, E. W. Barr, C. T. Walsh, J. M. Bollinger, Jr. and
C. Krebs, Nat. Chem. Biol., 2007, 3, 113–116.80 A. Butler and J. N. Carter-Franklin, Nat. Prod. Rep., 2004, 21, 180–
188.81 J. N. Carter-Franklin, J. D. Parrish, R. A. Tschirret-Guth,
R. D. Little and A. Butler, J. Am. Chem. Soc., 2003, 125, 3688–3689.82 E. Yeh, S. Garneau and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A.,
2005, 102, 3960–3965.83 C. Dong, S. Flecks, S. Unversucht, C. Haupt, K. H. van Pee and
J. H. Naismith, Science, 2005, 309, 2216–2219.84 S. Zehner, A. Kotzsch, B. Bister, R. D. Sussmuth, C. Mendez,
J. A. Salas and K. H. van Pee, Chem. Biol., 2005, 12, 445–452.85 C. Seibold, H. Schnerr, J. Rumpf, A. H. Kunzendorf, C. T. Wage,
A. J. Ernyei, D. Changjiang, J. H. Naismith and K. H. Van Pee,Biocatal. Biotransform., 2006, 24, 401–408.
86 P. C. Dorrestein, E. Yeh, S. Garneau-Tsodikova, N. L. Kelleherand C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102,13843–13848.
Nat. Prod. Rep., 2008, 25, 757–793 | 789

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
87 S. Lin, S. G. Van Lanen and B. Shen, J. Am. Chem. Soc., 2007, 129,12432–12438.
88 P. Spiteller, L. Bai, G. Shang, B. J. Carroll, T. W. Yu andH. G. Floss, J. Am. Chem. Soc., 2003, 125, 14236–14237.
89 T. W. Yu, L. Bai, D. Clade, D. Hoffmann, S. Toelzer, K. Q. Trinh,J. Xu, S. J. Moss, E. Leistner and H. G. Floss, Proc. Natl. Acad. Sci.U. S. A., 2002, 99, 7968–7973.
90 L. C. Blasiak, F. H. Vaillancourt, C. T. Walsh and C. L. Drennan,Nature, 2006, 440, 368–371.
91 D. P. Galonic, F. H. Vaillancourt and C. T. Walsh, J. Am. Chem.Soc., 2006, 128, 3900–3901.
92 W. L. Kelly, M. T. Boyne, 2nd, E. Yeh, D. A. Vosburg,D. P. Galonic, N. L. Kelleher and C. T. Walsh, Biochemistry,2007, 46, 359–368.
93 K. D. Walker, K. Klettke, T. Akiyama and R. Croteau, J. Biol.Chem., 2004, 279, 53947–53954.
94 W. Liu, S. D. Christenson, S. Standage and B. Shen, Science, 2002,297, 1170–1173.
95 H. Umezawa, T. Aoyagi, H. Suda, M. Hamada and T. Takeuchi,J. Antibiot. (Tokyo), 1976, 29, 97–99.
96 M. Jin, M. A. Fischbach and J. Clardy, J. Am. Chem. Soc., 2006,128, 10660–10661.
97 N. J. Bate, J. Orr, W. Ni, A. Meromi, T. Nadler-Hassar,P. W. Doerner, R. A. Dixon, C. J. Lamb and Y. Elkind, Proc.Natl. Acad. Sci. U. S. A., 1994, 91, 7608–7612.
98 L. Poppe, Curr. Opin. Chem. Biol., 2001, 5, 512–524.99 C. V. Christianson, T. J. Montavon, S. G. Van Lanen, B. Shen and
S. D. Bruner, Biochemistry, 2007, 46, 7205–7214.100 S. G. Van Lanen, P. C. Dorrestein, S. D. Christenson, W. Liu, J. Ju,
N. L. Kelleher and B. Shen, J. Am. Chem. Soc., 2005, 127, 11594–11595.
101 S. D. Christenson, W. Wu, M. A. Spies, B. Shen and M. D. Toney,Biochemistry, 2003, 42, 12708–12718.
102 S. D. Christenson, W. Liu, M. D. Toney and B. Shen, J. Am. Chem.Soc., 2003, 125, 6062–6063.
103 P. D. Fortin, C. T. Walsh and N. A. Magarvey, Nature, 2007, 448,824–827.
104 S. G. Van Lanen, S. Lin, P. C. Dorrestein, N. L. Kelleher andB. Shen, J. Biol. Chem., 2006, 281, 29633–29640.
105 D. G. Fujimori, S. Hrvatin, C. S. Neumann, M. Strieker,M. A. Marahiel and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A.,2007, 104, 16498–16503.
106 M. Piraee and L. C. Vining, J. Ind. Microbiol. Biotechnol., 2002, 29,1–5.
107 H. Ikeda, T. Nonomiya, M. Usami, T. Ohta and S. Omura, Proc.Natl. Acad. Sci. U. S. A., 1999, 96, 9509–9514.
108 V. Miao, M. F. Coeffet-Legal, P. Brian, R. Brost, J. Penn,A. Whiting, S. Martin, R. Ford, I. Parr, M. Bouchard, C. J. Silva,S. K. Wrigley and R. H. Baltz, Microbiology, 2005, 151, 1507–1523.
109 H. Kleinkauf, J. Dittmann and A. Lawen, Biomed. Biochim. Acta,1991, 50, S219–224.
110 F. Malpartida and D. A. Hopwood, Mol. Gen. Genet., 1986, 205,66–73.
111 G. D’Agnolo, I. S. Rosenfeld and P. R. Vagelos, J. Biol. Chem.,1975, 250, 5283–5288.
112 J. Elovson and P. R. Vagelos, J. Biol. Chem., 1968, 243, 3603–3611.113 C. R. Hutchinson, C. W. Borell, M. J. Donovan, F. Kato,
H. Motamedi, H. Nakayama, R. L. Rubin, S. L. Streicher,R. G. Summers, E. Wendt-Pienkowski and W. L. Wessel, PlantaMed., 1991, 57, S36–43.
114 P. C. Dorrestein and N. L. Kelleher, Nat. Prod. Rep., 2006, 23,893–918.
115 S.M.Ma, J. Zhan, K.Watanabe, X. Xie, W. Zhang, C. C.Wang andY. Tang, J. Am. Chem. Soc., 2007, 129, 10642–10643.
116 T. A. Keating and C. T. Walsh, Curr. Opin. Chem. Biol., 1999, 3,598–606.
117 C. Bisang, P. F. Long, J. Cortes, J. Westcott, J. Crosby,A. L. Matharu, R. J. Cox, T. J. Simpson, J. Staunton andP. F. Leadlay, Nature, 1999, 401, 502–505.
118 L. Xiang and B. S. Moore, J. Bacteriol., 2003, 185, 399–404.119 P. A. Lowden, B. Wilkinson, G. A. Bohm, S. Handa, H. G. Floss,
P. F. Leadlay and J. Staunton, Angew. Chem., Int. Ed., 2001, 40,777–779.
120 C. G. Kim, T. W. Yu, C. B. Fryhle, S. Handa and H. G. Floss,J. Biol. Chem., 1998, 273, 6030–6040.
790 | Nat. Prod. Rep., 2008, 25, 757–793
121 W. He, L. Wu, Q. Gao, Y. Du and Y. Wang, Curr. Microbiol., 2006,52, 197–203.
122 P. R. August, L. Tang, Y. J. Yoon, S. Ning, R. Muller, T. W. Yu,M. Taylor, D. Hoffmann, C. G. Kim, X. Zhang,C. R. Hutchinson and H. G. Floss, Chem. Biol., 1998, 5, 69–79.
123 A. Rascher, Z. Hu, N. Viswanathan, A. Schirmer, R. Reid,W. C. Nierman, M. Lewis and C. R. Hutchinson, FEMSMicrobiol. Lett., 2003, 218, 223–230.
124 V. Miao, M. F. Coeffet-Le Gal, K. Nguyen, P. Brian, J. Penn,A. Whiting, J. Steele, D. Kau, S. Martin, R. Ford, T. Gibson,M. Bouchard, S. K. Wrigley and R. H. Baltz, Chem. Biol., 2006,13, 269–276.
125 R. H. Baltz, V. Miao and S. K. Wrigley, Nat. Prod. Rep., 2005, 22,717–741.
126 D. B. Hansen, S. B. Bumpus, Z. D. Aron, N. L. Kelleher andC. T. Walsh, J. Am. Chem. Soc., 2007, 129, 6366–6367.
127 S. D. Bruner, T.Weber, R.M. Kohli, D. Schwarzer,M. A.Marahiel,C. T. Walsh and M. T. Stubbs, Structure, 2002, 10, 301–310.
128 C. C. Tseng, S. D. Bruner, R. M. Kohli, M. A. Marahiel,C. T. Walsh and S. A. Sieber, Biochemistry, 2002, 41, 13350–13359.
129 Z. D. Aron, P. C. Dorrestein, J. R. Blackhall, N. L. Kelleher andC. T. Walsh, J. Am. Chem. Soc., 2005, 127, 14986–14987.
130 E. H. Duitman, L. W. Hamoen, M. Rembold, G. Venema, H. Seitz,W. Saenger, F. Bernhard, R. Reinhardt, M. Schmidt, C. Ullrich,T. Stein, F. Leenders and J. Vater, Proc. Natl. Acad. Sci. U. S. A.,1999, 96, 13294–13299.
131 A. M. Gehring, I. Mori and C. T. Walsh, Biochemistry, 1998, 37,2648–2659.
132 T. A. Keating, C. G. Marshall and C. T. Walsh, Biochemistry, 2000,39, 15522–15530.
133 M. G. Thomas, M. D. Burkart and C. T. Walsh, Chem. Biol., 2002,9, 171–184.
134 S. Garneau, P. C. Dorrestein, N. L. Kelleher and C. T. Walsh,Biochemistry, 2005, 44, 2770–2780.
135 L. Tang, S. Shah, L. Chung, J. Carney, L. Katz, C. Khosla andB. Julien, Science, 2000, 287, 640–642.
136 C. Y. Kim, V. Y. Alekseyev, A. Y. Chen, Y. Tang, D. E. Cane andC. Khosla, Biochemistry, 2004, 43, 13892–13898.
137 R. Carvalho, R. Reid, N. Viswanathan, H. Gramajo and B. Julien,Gene, 2005, 359, 91–98.
138 R. Reid, M. Piagentini, E. Rodriguez, G. Ashley, N. Viswanathan,J. Carney, D. V. Santi, C. R. Hutchinson and R. McDaniel,Biochemistry, 2003, 42, 72–79.
139 P. Caffrey, ChemBioChem, 2003, 4, 654–657.140 R. Castonguay, W. He, A. Y. Chen, C. Khosla and D. E. Cane,
J. Am. Chem. Soc., 2007, 129, 13758–13769.141 A. Baerga-Ortiz, B. Popovic, A. P. Siskos, H. M. O’Hare,
D. Spiteller, M. G. Williams, N. Campillo, J. B. Spencer andP. F. Leadlay, Chem. Biol., 2006, 13, 277–285.
142 A. T. Keatinge-Clay and R.M. Stroud, Structure, 2006, 14, 737–748.143 I. E. Holzbaur, A. Ranganathan, I. P. Thomas, D. J. Kearney,
J. A. Reather, B. A. Rudd, J. Staunton and P. F. Leadlay, Chem.Biol., 2001, 8, 329–340.
144 I. E. Holzbaur, R. C. Harris, M. Bycroft, J. Cortes, C. Bisang,J. Staunton, B. A. Rudd and P. F. Leadlay, Chem. Biol., 1999, 6,189–195.
145 J. Staunton and B. Wilkinson, Chem. Rev., 1997, 97, 2611–2630.146 T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat.
Struct. Biol., 2002, 9, 522–526.147 C. G. Marshall, M. D. Burkart, T. A. Keating and C. T. Walsh,
Biochemistry, 2001, 40, 10655–10663.148 L. E. Quadri, J. Sello, T. A. Keating, P. H.Weinreb and C. T.Walsh,
Chem. Biol., 1998, 5, 631–645.149 C. Pelludat, A. Rakin, C. A. Jacobi, S. Schubert and J. Heesemann,
J. Bacteriol., 1998, 180, 538–546.150 E. E. Wyckoff, J. A. Stoebner, K. E. Reed and S. M. Payne,
J. Bacteriol., 1997, 179, 7055–7062.151 N. A. Magarvey, M. Ehling-Schulz and C. T. Walsh, J. Am. Chem.
Soc., 2006, 128, 10698–10699.152 R. Pieper, A. Haese, W. Schroder and R. Zocher, Eur. J. Biochem.,
1995, 230, 119–126.153 S. C. Feifel, T. Schmiederer, T. Hornbogen, H. Berg,
R. D. Sussmuth and R. Zocher, ChemBioChem, 2007, 8, 1767–1770.
This journal is ª The Royal Society of Chemistry 2008

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
154 M. Krause, A. Lindemann, M. Glinski, T. Hornbogen, G. Bonse,P. Jeschke, G. Thielking, W. Gau, H. Kleinkauf and R. Zocher,J. Antibiot. (Tokyo), 2001, 54, 797–804.
155 H. Peeters, R. Zocher and H. Kleinkauf, J. Antibiot. (Tokyo), 1988,41, 352–359.
156 Y. Q. Cheng, ChemBioChem, 2006, 7, 471–477.157 M. Ehling-Schulz, M. Fricker, H. Grallert, P. Rieck, M.Wagner and
S. Scherer, BMC Microbiol., 2006, 6, 20.158 K. Wu, L. Chung, W. P. Revill, L. Katz and C. D. Reeves, Gene,
2000, 251, 81–90.159 L. Du, C. Sanchez, M. Chen, D. J. Edwards and B. Shen, Chem.
Biol., 2000, 7, 623–642.160 B. Silakowski, G. Nordsiek, B. Kunze, H. Blocker and R. Muller,
Chem. Biol., 2001, 8, 59–69.161 B. Julien, S. Shah, R. Ziermann, R. Goldman, L. Katz and
C. Khosla, Gene, 2000, 249, 153–160.162 C. T. Walsh, S. E. O’Connor and T. L. Schneider, J. Ind. Microbiol.
Biotechnol., 2003, 30, 448–455.163 W. L. Kelly, N. J. Hillson and C. T. Walsh, Biochemistry, 2005, 44,
13385–13393.164 S. E. O’Connor, H. Chen and C. T. Walsh, Biochemistry, 2002, 41,
5685–5694.165 T. L. Schneider, C. T.Walsh and S. E. O’Connor, J. Am. Chem. Soc.,
2002, 124, 11272–11273.166 Z. Chang, P. Flatt, W. H. Gerwick, V. A. Nguyen, C. L. Willis and
D. H. Sherman, Gene, 2002, 296, 235–247.167 T. L. Schneider, B. Shen and C. T. Walsh, Biochemistry, 2003, 42,
9722–9730.168 T. T. Sakai, J. M. Riordan and J. D. Glickson, Biochemistry, 1982,
21, 805–816.169 K. Hoffmann, E. Schneider-Scherzer, H. Kleinkauf and R. Zocher,
J. Biol. Chem., 1994, 269, 12710–12714.170 T. Stachelhaus and C. T. Walsh, Biochemistry, 2000, 39, 5775–5787.171 H. M. Patel, J. Tao and C. T. Walsh, Biochemistry, 2003, 42, 10514–
10527.172 H. D. Mootz and M. A. Marahiel, J. Bacteriol., 1997, 179, 6843–
6850.173 L. Luo, R. M. Kohli, M. Onishi, U. Linne, M. A. Marahiel and
C. T. Walsh, Biochemistry, 2002, 41, 9184–9196.174 S. L. Clugston, S. A. Sieber, M. A. Marahiel and C. T. Walsh,
Biochemistry, 2003, 42, 12095–12104.175 C. J. Balibar, F. H. Vaillancourt and C. T. Walsh, Chem. Biol., 2005,
12, 1189–1200.176 U. Linne, S. Doekel and M. A. Marahiel, Biochemistry, 2001, 40,
15824–15834.177 T. Hornbogen, S. P. Riechers, B. Prinz, J. Schultchen, C. Lang,
S. Schmidt, C. Mugge, S. Turkanovic, R. D. Sussmuth,E. Tauberger and R. Zocher, ChemBioChem, 2007, 8, 1048–1054.
178 A. M. Hill, Nat. Prod. Rep., 2006, 23, 256–320.179 D. A. Miller, C. T. Walsh and L. Luo, J. Am. Chem. Soc., 2001, 123,
8434–8435.180 D. P. K. O’Brien, P.N., S. W. O’Brien, T. Staroske, J. B. Spencer,
D. H. Williams, T. I. Richardson, D. A. Evans and A. Hopkinson,Chem. Commun., 2000, 103–104.
181 J. A. Salas and C. Mendez, J. Mol. Microbiol. Biotechnol., 2005, 9,77–85.
182 E. P. Patallo, G. Blanco, C. Fischer, A. F. Brana, J. Rohr,C. Mendez and J. A. Salas, J. Biol. Chem., 2001, 276, 18765–18774.
183 I. Muller and R. Muller, FEBS J., 2006, 273, 3768–3778.184 A. K. El-Sayed, J. Hothersall, S. M. Cooper, E. Stephens,
T. J. Simpson and C. M. Thomas, Chem. Biol., 2003, 10, 419–430.
185 D. J. Edwards, B. L. Marquez, L. M. Nogle, K. McPhail,D. E. Goeger, M. A. Roberts and W. H. Gerwick, Chem. Biol.,2004, 11, 817–833.
186 J. Moldenhauer, X. H. Chen, R. Borriss and J. Piel, Angew. Chem.,Int. Ed., 2007, 46, 8195–8197.
187 Y. Paitan, E. Orr, E. Z. Ron and E. Rosenberg,Microbiology, 1999,145, 3059–3067.
188 C. T. Calderone, D. F. Iwig, P. C. Dorrestein, N. L. Kelleher andC. T. Walsh, Chem. Biol., 2007, 14, 835–846.
189 C. T. Calderone, W. E. Kowtoniuk, N. L. Kelleher, C. T. Walsh andP. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8977–8982.
This journal is ª The Royal Society of Chemistry 2008
190 Z. Chang, N. Sitachitta, J. V. Rossi, M. A. Roberts, P. M. Flatt,J. Jia, D. H. Sherman and W. H. Gerwick, J. Nat. Prod., 2004, 67,1356–1367.
191 C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat.Prod. Rep., 2006, 23, 517–531.
192 S. Pelzer, R. Sussmuth, D. Heckmann, J. Recktenwald, P. Huber,G. Jung and W. Wohlleben, Antimicrob. Agents Chemother., 1999,43, 1565–1573.
193 S. M. Li and L. Heide, Planta Med., 2006, 72, 1093–1099.194 J. Pootoolal, M. G. Thomas, C. G. Marshall, J. M. Neu,
B. K. Hubbard, C. T. Walsh and G. D. Wright, Proc. Natl. Acad.Sci. U. S. A., 2002, 99, 8962–8967.
195 M. Sosio, H. Kloosterman, A. Bianchi, P. de Vreugd, L. Dijkhuizenand S. Donadio, Microbiology, 2004, 150, 95–102.
196 T. L. Li, F. Huang, S. F. Haydock, T. Mironenko, P. F. Leadlay andJ. B. Spencer, Chem. Biol., 2004, 11, 107–119.
197 D. Bischoff, S. Pelzer, B. Bister, G. J. Nicholson, S. Stockert,M. Schirle, W. Wohlleben, G. Jung and R. D. Sussmuth, Angew.Chem., Int. Ed., 2001, 40, 4688–4691.
198 B. Hadatsch, D. Butz, T. Schmiederer, J. Steudle, W. Wohlleben,R. Sussmuth and E. Stegmann, Chem. Biol., 2007, 14, 1078–1089.
199 K. Zerbe, K. Woithe, D. B. Li, F. Vitali, L. Bigler andJ. A. Robinson, Angew. Chem., Int. Ed., 2004, 43, 6709–6713.
200 D. Bischoff, B. Bister, M. Bertazzo, V. Pfeifer, E. Stegmann,G. J. Nicholson, S. Keller, S. Pelzer, W. Wohlleben andR. D. Sussmuth, ChemBioChem, 2005, 6, 267–272.
201 K. Woithe, N. Geib, K. Zerbe, D. B. Li, M. Heck, S. Fournier-Rousset, O. Meyer, F. Vitali, N. Matoba, K. Abou-Hadeed andJ. A. Robinson, J. Am. Chem. Soc., 2007, 129, 6887–6895.
202 N. Geib, K. Woithe, K. Zerbe, D. B. Li and J. A. Robinson, Bioorg.Med. Chem. Lett., 2007.
203 D. A. Miller, L. Luo, N. Hillson, T. A. Keating and C. T. Walsh,Chem. Biol., 2002, 9, 333–344.
204 M. F. Byford, J. E. Baldwin, C. Y. Shiau and C. J. Schofield, Chem.Rev., 1997, 97, 2631–2650.
205 J. F. Martin, J. Antibiot. (Tokyo), 2000, 53, 1008–1021.206 C. Khosla, Y. Tang, A. Y. Chen, N. A. Schnarr and D. E. Cane,
Annu. Rev. Biochem., 2007, 76, 195–221.207 J. Grunewald, S. A. Sieber and M. A. Marahiel, Biochemistry, 2004,
43, 2915–2925.208 C. A. Shaw-Reid, N. L. Kelleher, H. C. Losey, A. M. Gehring,
C. Berg and C. T. Walsh, Chem. Biol., 1999, 6, 385–400.209 F. Kopp andM. A.Marahiel,Curr. Opin. Biotechnol., 2007, 18, 513–
520.210 R. M. Kohli, M. D. Burke, J. Tao and C. T. Walsh, J. Am. Chem.
Soc., 2003, 125, 7160–7161.211 R. M. Kohli, C. T. Walsh and M. D. Burkart, Nature, 2002, 418,
658–661.212 F. Kopp, J. Grunewald, C. Mahlert and M. A. Marahiel,
Biochemistry, 2006, 45, 10474–10481.213 C. J. Balibar, A. R. Howard-Jones and C. T. Walsh, Nat. Chem.
Biol., 2007, 3, 584–592.214 Y. Kallberg, U. Oppermann, H. Jornvall and B. Persson, Eur.
J. Biochem., 2002, 269, 4409–4417.215 D. E. Ehmann, A. M. Gehring and C. T. Walsh, Biochemistry, 1999,
38, 6171–6177.216 L. Li, W. Deng, J. Song, W. Ding, Q. F. Zhao, C. Peng, W. W. Song,
G. L. Tang and W. Liu, J. Bacteriol., 2008, 190, 251–263.217 F. Kopp, C. Mahlert, J. Grunewald and M. A. Marahiel, J. Am.
Chem. Soc., 2006, 128, 16478–16479.218 J. A. Read and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 15762–
15763.219 N. Gaitatzis, B. Kunze and R. Muller, Proc. Natl. Acad. Sci.
U. S. A., 2001, 98, 11136–11141.220 C. J. Balibar and C. T. Walsh, Biochemistry, 2006, 45, 15029–15038.221 T. Haarmann, I. Ortel, P. Tudzynski and U. Keller, ChemBioChem,
2006, 7, 645–652.222 D. M. Gardiner, P. Waring and B. J. Howlett, Microbiology, 2005,
151, 1021–1032.223 B. Frank, S. C. Wenzel, H. B. Bode, M. Scharfe, H. Blocker and
R. Muller, J. Mol. Biol., 2007, 374, 24–38.224 C. T. Walsh, Science, 2004, 303, 1805–1810.225 C. T. Walsh, Acc. Chem. Res., 2008, 41, 4–10.226 C. Walsh, C. L. Freel Meyers and H. C. Losey, J. Med. Chem., 2003,
46, 3425–3436.
Nat. Prod. Rep., 2008, 25, 757–793 | 791

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
227 X. M. He and H. W. Liu, Annu. Rev. Biochem., 2002, 71, 701–754.228 S. Blanchard and J. S. Thorson, Curr. Opin. Chem. Biol., 2006, 10,
263–271.229 H. Chen, M. G. Thomas, B. K. Hubbard, H. C. Losey, C. T. Walsh
and M. D. Burkart, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11942–11947.
230 C. Mendez and J. A. Salas, Ernst Schering Research Foundationworkshop, 2005, pp. 127–146.
231 H. Takahashi, Y. N. Liu, H. Chen and H. W. Liu, J. Am. Chem.Soc., 2005, 127, 9340–9341.
232 D. Kahne, C. Leimkuhler, W. Lu and C. Walsh, Chem. Rev., 2005,105, 425–448.
233 P. J. Solenberg, P. Matsushima, D. R. Stack, S. C. Wilkie,R. C. Thompson and R. H. Baltz, Chem. Biol., 1997, 4, 195–202.
234 H. C. Losey, M. W. Peczuh, Z. Chen, U. S. Eggert, S. D. Dong,I. Pelczer, D. Kahne and C. T. Walsh, Biochemistry, 2001, 40,4745–4755.
235 W. Lu, M. Oberthur, C. Leimkuhler, J. Tao, D. Kahne andC. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4390–4395.
236 A. M. Mulichak, W. Lu, H. C. Losey, C. T. Walsh andR. M. Garavito, Biochemistry, 2004, 43, 5170–5180.
237 A. M. Mulichak, H. C. Losey, W. Lu, Z. Wawrzak, C. T. Walsh andR. M. Garavito, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9238–9243.
238 A. M. Mulichak, H. C. Losey, C. T. Walsh and R. M. Garavito,Structure, 2001, 9, 547–557.
239 M. Oberthur, C. Leimkuhler, R. G. Kruger, W. Lu, C. T. Walsh andD. Kahne, J. Am. Chem. Soc., 2005, 127, 10747–10752.
240 H. C. Losey, J. Jiang, J. B. Biggins, M. Oberthur, X. Y. Ye,S. D. Dong, D. Kahne, J. S. Thorson and C. T. Walsh, Chem.Biol., 2002, 9, 1305–1314.
241 R. H. Baltz, Nat. Biotechnol., 2006, 24, 1533–1540.242 A. R. Howard-Jones, R. G. Kruger, W. Lu, J. Tao, C. Leimkuhler,
D. Kahne and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 10082–10083.
243 S. Keller, F. Pojer, L. Heide and D. M. Lawson, Acta Crystallogr.,Sect. F: Struct. Biol. Cryst. Commun., 2006, 62, 1153–1155.
244 F. Pojer, E.Wemakor, B. Kammerer, H. Chen, C. T.Walsh, S.M. Liand L. Heide, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2316–2321.
245 D. J. Edwards and W. H. Gerwick, J. Am. Chem. Soc., 2004, 126,11432–11433.
246 G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni,Pharmacol. Rev., 2004, 56, 185–229.
247 D. Yang and A. H. Wang, Biochemistry, 1994, 33, 6595–6604.248 G. A. Leonard, T. W. Hambley, K. McAuley-Hecht, T. Brown and
W. N. Hunter, Acta Crystallogr., Sect. D, 1993, 49, 458–467.249 C. Leimkuhler, M. Fridman, T. Lupoli, S. Walker, C. T. Walsh and
D. Kahne, J. Am. Chem. Soc., 2007, 129, 10546–10550.250 W. Lu, C. Leimkuhler, G. J. Gatto, Jr., R. G. Kruger, M. Oberthur,
D. Kahne and C. T. Walsh, Chem. Biol., 2005, 12, 527–534.251 W. Lu, C. Leimkuhler, M. Oberthur, D. Kahne and C. T. Walsh,
Biochemistry, 2004, 43, 4548–4558.252 A. Luzhetskyy, M. Fedoryshyn, C. Durr, T. Taguchi, V. Novikov
and A. Bechthold, Chem. Biol., 2005, 12, 725–729.253 Y. Yuan, H. S. Chung, C. Leimkuhler, C. T. Walsh, D. Kahne and
S. Walker, J. Am. Chem. Soc., 2005, 127, 14128–14129.254 H. Y. Lee, H. S. Chung, C. Hang, C. Khosla, C. T. Walsh, D. Kahne
and S. Walker, J. Am. Chem. Soc., 2004, 126, 9924–9925.255 M. Mittler, A. Bechthold and G. E. Schulz, J. Mol. Biol., 2007, 372,
67–76.256 M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Proc. Natl.
Acad. Sci. U. S. A., 2005, 102, 571–576.257 M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Nat. Chem.
Biol., 2006, 2, 132–138.258 W. J. van Berkel, N. M. Kamerbeek and M. W. Fraaije,
J. Biotechnol., 2006, 124, 670–689.259 T. D. Bugg and S. Ramaswamy, Curr. Opin. Chem. Biol., 2008.260 V. Joosten and W. J. van Berkel, Curr. Opin. Chem. Biol., 2007, 11,
195–202.261 M. J. Cryle, J. E. Stok and J. J. De Voss, Aust. J. Chem., 2003, 56,
749–762.262 E. N. Fielding, P. F. Widboom and S. D. Bruner, Biochemistry,
2007, 46, 13994–14000.263 M. Gibson, M. Nur-e-alam, F. Lipata, M. A. Oliveira and J. Rohr,
J. Am. Chem. Soc., 2005, 127, 17594–17595.
792 | Nat. Prod. Rep., 2008, 25, 757–793
264 D. Rodriguez, L. M. Quiros, A. F. Brana and J. A. Salas,J. Bacteriol., 2003, 185, 3962–3965.
265 H. J. Kim, R. Pongdee, Q. Wu, L. Hong and H. W. Liu, J. Am.Chem. Soc., 2007, 129, 14582–14584.
266 J. F. Andersen and C. R. Hutchinson, J. Bacteriol., 1992, 174, 725–735.
267 R. H. Lambalot, D. E. Cane, J. J. Aparicio and L. Katz,Biochemistry, 1995, 34, 1858–1866.
268 M. V. Mendes, N. Anton, J. F. Martin and J. F. Aparicio, Biochem.J., 2005, 386, 57–62.
269 H. Ogura, C. R. Nishida, U. R. Hoch, R. Perera, J. H. Dawson andP. R. Ortiz de Montellano, Biochemistry, 2004, 43, 14712–14721.
270 P. L. Roach, I. J. Clifton, C. M. Hensgens, N. Shibata,C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997, 387,827–830.
271 K. Valegard, A. C. van Scheltinga, M. D. Lloyd, T. Hara,S. Ramaswamy, A. Perrakis, A. Thompson, H. J. Lee,J. E. Baldwin, C. J. Schofield, J. Hajdu and I. Andersson, Nature,1998, 394, 805–809.
272 N. I. Burzlaff, P. J. Rutledge, I. J. Clifton, C. M. Hensgens,M. Pickford, R. M. Adlington, P. L. Roach and J. E. Baldwin,Nature, 1999, 401, 721–724.
273 D. Destoumieux-Garzon, X. Thomas, M. Santamaria, C. Goulard,M. Barthelemy, B. Boscher, Y. Bessin, G. Molle, A. M. Pons,L. Letellier, J. Peduzzi and S. Rebuffat, Mol. Microbiol., 2003, 49,1031–1041.
274 E. M. Nolan, M. A. Fischbach, A. Koglin and C. T. Walsh, J. Am.Chem. Soc., 2007, 129, 14336–14347.
275 T. A. Keating, Z. Suo, D. E. Ehmann and C. T.Walsh, Biochemistry,2000, 39, 2297–2306.
276 A. M. Gehring, I. Mori, R. D. Perry and C. T. Walsh, Biochemistry,1998, 37, 11637–11650.
277 B. Shen, Top. Curr. Chem., 2000, 209, 1–51.278 C. W. Carreras and C. Khosla, Biochemistry, 1998, 37, 2084–2088.279 C. W. P. Carreras, R. and C. Khosla, J. Am. Chem. Soc., 1996, 118,
5158–5159.280 A. T. Hadfield, C. Limpkin, W. Teartasin, T. J. Simpson, J. Crosby
and M. P. Crump, Structure, 2004, 12, 1865–1875.281 R. J. Zawada and C. Khosla, Chem. Biol., 1999, 6, 607–615.282 Y. Tang, S. C. Tsai and C. Khosla, J. Am. Chem. Soc., 2003, 125,
12708–12709.283 A. T. Keatinge-Clay, D. A. Maltby, K. F. Medzihradszky,
C. Khosla and R. M. Stroud, Nat. Struct. Mol. Biol., 2004, 11,888–893.
284 B. J. Rawlings, Nat. Prod. Rep., 2001, 18, 190–227.285 B. Shen and C. R. Hutchinson, Biochemistry, 1993, 32, 11149–11154.286 A. Sultana, P. Kallio, A. Jansson, J. S. Wang, J. Niemi, P. Mantsala
and G. Schneider, EMBO J., 2004, 23, 1911–1921.287 P. Kallio, A. Sultana, J. Niemi, P. Mantsala and G. Schneider,
J. Mol. Biol., 2006, 357, 210–220.288 S. G. Kendrew, K. Katayama, E. Deutsch, K. Madduri and
C. R. Hutchinson, Biochemistry, 1999, 38, 4794–4799.289 W. Zhang, K. Watanabe, C. C. Wang and Y. Tang, J. Biol. Chem.,
2007, 282, 25717–25725.290 B. Shen and C. R. Hutchinson, Science, 1993, 262, 1535–1540.291 W. Bao, E. Wendt-Pienkowski and C. R. Hutchinson, Biochemistry,
1998, 37, 8132–8138.292 B. Shen and C. R. Hutchinson, Proc. Natl. Acad. Sci. U. S. A., 1996,
93, 6600–6604.293 T. B. Thompson, K. Katayama, K. Watanabe, C. R. Hutchinson
and I. Rayment, J. Biol. Chem., 2004, 279, 37956–37963.294 B. Shen and C. R. Hutchinson, Biochemistry, 1993, 32, 6656–6663.295 B. Shen and C. R. Hutchinson, J. Biol. Chem., 1994, 269, 30726–
30733.296 Y. Tang, T. S. Lee, S. Kobayashi and C. Khosla, Biochemistry, 2003,
42, 6588–6595.297 Y. Tang, A. T. Koppisch and C. Khosla, Biochemistry, 2004, 43,
9546–9555.298 Y. Tang, H. Y. Lee, Y. Tang, C. Y. Kim, I. Mathews and C. Khosla,
Biochemistry, 2006, 45, 14085–14093.299 T. S. Lee, C. Khosla and Y. Tang, J. Am. Chem. Soc., 2005, 127,
12254–12262.300 Y. Tang, T. S. Lee and C. Khosla, PLoS Biol., 2004, 2, E31.301 I. Fujii, A. Watanabe, U. Sankawa and Y. Ebizuka, Chem. Biol.,
2001, 8, 189–197.
This journal is ª The Royal Society of Chemistry 2008

Dow
nloa
ded
by U
nive
rsity
of
Chi
cago
on
03/0
5/20
13 1
6:08
:31.
Pu
blis
hed
on 2
3 M
ay 2
008
on h
ttp://
pubs
.rsc
.org
| do
i:10.
1039
/B80
1747
F
View Article Online
302 S. M. Ma, J. Zhan, X. Xie, K. Watanabe, Y. Tang and W. Zhang,J. Am. Chem. Soc., 2008, 130, 38–39.
303 Y. Katsuyama, M. Matsuzawa, N. Funa and S. Horinouchi, J. Biol.Chem., 2007, 282, 37702–37709.
304 I. Abe, S. Oguro, Y. Utsumi, Y. Sano and H. Noguchi, J. Am. Chem.Soc., 2005, 127, 12709–12716.
305 I. Abe, Y. Utsumi, S. Oguro, H. Morita, Y. Sano and H. Noguchi,J. Am. Chem. Soc., 2005, 127, 1362–1363.
306 J. L. Ferrer, J. M. Jez, M. E. Bowman, R. A. Dixon and J. P. Noel,Nat. Struct. Biol., 1999, 6, 775–784.
307 M. B. Austin, M. Izumikawa, M. E. Bowman, D. W. Udwary,J. L. Ferrer, B. S. Moore and J. P. Noel, J. Biol. Chem., 2004, 279,45162–45174.
308 J. Piel, C. Hertweck, P. R. Shipley, D. M. Hunt, M. S. Newman andB. S. Moore, Chem. Biol., 2000, 7, 943–955.
309 M. Izumikawa, Q. Cheng and B. S. Moore, J. Am. Chem. Soc., 2006,128, 1428–1429.
310 Q. Cheng, L. Xiang, M. Izumikawa, D. Meluzzi and B. S. Moore,Nat. Chem. Biol., 2007, 3, 557–558.
311 L. Xiang, J. A. Kalaitzis and B. S. Moore, Proc. Natl. Acad. Sci.U. S. A., 2004, 101, 15609–15614.
312 A. Maxwell and D. M. Lawson, Curr. Top. Med. Chem., 2003, 3,283–303.
313 S. C. Kampranis, N. A. Gormley, R. Tranter, G. Orphanides andA. Maxwell, Biochemistry, 1999, 38, 1967–1976.
314 K. Drlica and X. Zhao, Microbiol. Mol. Biol. Rev., 1997, 61, 377–392.
315 F. Pojer, S. M. Li and L. Heide,Microbiology, 2002, 148, 3901–3911.316 M. Steffensky, A. Muhlenweg, Z. X. Wang, S. M. Li and L. Heide,
Antimicrob. Agents Chemother., 2000, 44, 1214–1222.317 Z. X. Wang, S. M. Li and L. Heide, Antimicrob. Agents Chemother.,
2000, 44, 3040–3048.318 H. Chen and C. T. Walsh, Chem. Biol., 2001, 8, 301–312.319 M. Pacholec, N. J. Hillson and C. T. Walsh, Biochemistry, 2005, 44,
12819–12826.320 F. Pojer, R. Kahlich, B. Kammerer, S. M. Li and L. Heide, J. Biol.
Chem., 2003, 278, 30661–30668.321 M. Steffensky, S. M. Li and L. Heide, J. Biol. Chem., 2000, 275,
21754–21760.322 M. Pacholec, J. Tao and C. T.Walsh, Biochemistry, 2005, 44, 14969–
14976.323 C. L. Freel Meyers, M. Oberthur, J. W. Anderson, D. Kahne and
C. T. Walsh, Biochemistry, 2003, 42, 4179–4189.
This journal is ª The Royal Society of Chemistry 2008
324 A. Freitag, S. M. Li and L. Heide, Microbiology (Reading, U. K.),2006, 152, 2433–2442.
325 T. T. Thuy, H. C. Lee, C. G. Kim, L. Heide and J. K. Sohng,Arch. Biochem. Biophys., 2005, 436, 161–167.
326 C. E. Stevenson, C. L. Freel Meyers, C. T.Walsh andD.M. Lawson,Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2007, 63,236–238.
327 C. L. Freel Meyers, M. Oberthur, H. Xu, L. Heide, D. Kahne andC. T. Walsh, Angew. Chem., Int. Ed., 2004, 43, 67–70.
328 M. Gellert, M. H. O’Dea, T. Itoh and J. Tomizawa, Proc. Natl.Acad. Sci. U. S. A., 1976, 73, 4474–4478.
329 A. S. Eustaquio, B. Gust, T. Luft, S. M. Li, K. F. Chater andL. Heide, Chem. Biol., 2003, 10, 279–288.
330 C. J. Balibar, S. Garneau-Tsodikova and C. T. Walsh, Chem. Biol.,2007, 14, 679–690.
331 E. Schmutz, M. Steffensky, J. Schmidt, A. Porzel, S. M. Li andL. Heide, Eur. J. Biochem., 2003, 270, 4413–4419.
332 C. L. Freel Meyers, M. Oberthur, L. Heide, D. Kahne andC. T. Walsh, Biochemistry, 2004, 43, 15022–15036.
333 C. Anderle, S. Hennig, B. Kammerer, S. M. Li, L. Wessjohann,B. Gust and L. Heide, Chem. Biol., 2007, 14, 955–967.
334 C. Sanchez, C. Mendez and J. A. Salas, Nat. Prod. Rep., 2006, 23,1007–1045.
335 C. Sanchez, I. A. Butovich, A. F. Brana, J. Rohr, C. Mendez andJ. A. Salas, Chem. Biol., 2002, 9, 519–531.
336 A. R. Howard-Jones and C. T. Walsh, J. Am. Chem. Soc., 2006, 128,12289–12298.
337 A. R. Howard-Jones and C. T. Walsh, Biochemistry, 2005, 44,15652–15663.
338 A. R. Howard-Jones and C. T. Walsh, J. Am. Chem. Soc., 2007, 129,11016–11017.
339 R. V. Antonio and T. B. Creczynski-Pasa, Genet. Mol. Res., 2004, 3,85–91.
340 C. J. Balibar and C. T. Walsh, Biochemistry, 2006, 45, 15444–15457.
341 K. S. Ryan, C. J. Balibar, K. E. Turo, C. T. Walsh andC. L. Drennan, J. Biol. Chem., 2008, 283, 6467–6475.
342 S. Bouhired, M. Weber, A. Kempf-Sontag, N. P. Keller andD. Hoffmeister, Fungal Genet. Biol., 2007, 44, 1134–1145.
343 J. W. Bok, D. Hoffmeister, L. A. Maggio-Hall, R. Murillo,J. D. Glasner and N. P. Keller, Chem. Biol., 2006, 13, 31–37.
344 P. Schneider, M. Weber and D. Hoffmeister, Fungal Genet. Biol.,2008, 45, 302–309.
Nat. Prod. Rep., 2008, 25, 757–793 | 793





























