Embed Size (px)
Citation preview

CHIMIE THÉORIQUE ET PHYSIQUE APPLIQUÉE À
L’ANALYSE STRUCTURALE DES BIOMOLÉCULES (CG 204)
Prof. B. Wathelet
UNITÉ DE CHIMIE BIOLOGIQUE INDUSTRIELLE
Prof. M. Paquot
NOTES DE TRAVAUX PRATIQUES
Ir. S. Gillet
Dr. A. Richel

*********************************************
Remarque préliminaire
Ces notes de cours sont destinées aux étudiants de l’Université de Liège-Gembloux Agro-Bio Tech.
Elles servent à l’apprentissage des méthodes spectroscopiques et constituent un support et une
illustration pour l’étude de la « Chimie Théorique et Physique appliquée à l’analyse structurale des
biomolécules » (B. Wathelet).
Par ailleurs, ces notes sont exploitées comme base de données pour l’interprétation des spectres.
Ces notes de cours sont donc volontairement inspirées de la littérature et notamment des ouvrages
suivants :
- Silverstein, Webster, Kiemle. Identification spectrométrique de composés organiques 2ème
édition. De boeck université 2007. 502P.
- Skoog, Holler, Nieman. Principes d’analyse instrumentale 1ère édition. De boeck université
2003. 956P.
- Rouessac, Rouessac. analyse chimique – méthodes et techniques instrumentales modernes
5ème édition. Dunod 2000. 430P.
- Cours de chimie Organique - G. Dupuis France
- Chim 1302. Prof. R. Giasson. Université de Monttréal.
Ces notes en format pdf ne sont pas destinées à être diffusées.
************************************************

TP de chimie théorique et physique
S. Gillet - 3 - IR
CHAPITRE 1. LA SPÉCTROMÉTRIE INFRAROUGE (IR)

TP de chimie théorique et physique
S. Gillet - 4 - IR
1. INTRODUCTION
La spectrométrie est l’étude des rayonnements électromagnétiques émis, absorbés ou
diffusés par la matière.
Les rayonnements électromagnétiques émis ou absorbés par les atomes et les molécules
s’étendent des rayons X jusqu’aux ondes hertziennes, en passant par l’ultraviolet, le visible et
l’infrarouge (figure 1).
Figure 1: le spectre électromagnétique
La grandeur caractéristique de chaque radiation est la fréquence υ, exprimée en hertz.
Les grandeurs suivantes, qui se déduisent de la fréquence, sont également utilisées : la
longueur d’onde dans le vide λ = c/υ, où c est la vitesse de propagation dans le vide ; le
nombre d’onde ν~ = 1/λ, exprimé en cm-1 ; l’énergie hυ du photon associé à l’onde, exprimé
en électronvolts (1 eV = 1,6 . 10-19J).
Il existe plusieurs principes de subdivision du spectre électromagnétique selon la
propriété étudiée (spectres d’émission, d’absorption, de réflexion de diffusion…), selon
l’origine de sa production et le mécanisme mis en jeu (spectre atomique, moléculaire,
électronique, de vibration, de rotation…), d’après son aspect (spectre de raies, de bandes,
continu).

TP de chimie théorique et physique
S. Gillet - 5 - IR
Selon la mécanique quantique, chaque atome ou chaque molécule peut être caractérisé
par une suite discontinue d’états d’énergie, qui vont de l’état fondamental E jusqu’aux
niveaux excités Ee. Le mécanisme fondamental de l’émission et de l’absorption d’un
rayonnement est celui qui a été décrit par Bohr. Lors d’une transition énergétique entre deux
états Em et En, il y a émission ou absorption d’une radiation de fréquence υ telle que la
variation d’énergie du système : ∆E = hυ = hc/λ.
Le spectre électromagnétique d’une espèce chimique correspond donc aux transitions
permises entre les différents niveaux énergétiques.
1.1. LES SPECTRES ATOMIQUES
En mécanique quantique, l’énergie de l’électron lié au noyau atomique est définie à
l’aide de quatre nombres quantiques : n, l, m, s. Pour chaque atome, les spectroscopistes ont
établi les diagrammes des différents niveaux d’énergie et les transitions possibles
correspondant aux fréquences des radiations émises ou absorbées. Ainsi, on distingue
habituellement les spectres optiques des spectres de rayon X. Les premiers correspondent à
des transitions entre les niveaux d’énergie possibles des électrons de valence (les plus
éloignés du noyau) qui interviennent dans les réactions chimiques. Les radiations émises vont
en général du domaine de l’ultraviolet à celui du proche infrarouge. Les rayons X sont
produits par des transitions qui intéressent les électrons proches du noyau.
1.2. LES SPECTRES MOLÉCULAIRES
A l’état de vapeur, les molécules, comme les atomes, ont des spectres constitués de
fréquences distinctes mais les raies spectrales sont beaucoup plus nombreuses et dans
certaines régions du spectre, elles forment des groupes ou bandes. Les niveaux énergétiques
quantifiés caractéristiques d’une molécule dépendent des états énergétiques des électrons, de
l’énergie de vibration de ses atomes les uns par rapport aux autres, et aussi de l’énergie de
rotation des groupes d’atomes par rapport aux axes de symétrie ou de rotation de cette
molécule. On peut considérer, sans tenir compte de l’énergie de translation, qu’un niveau
énergétique E d’une molécule résulte de la somme de trois termes, son énergie électronique
(Ee), son énergie de vibration (Ez), son énergie de rotation (Er) : E = Ee + Ez + Er.
Mais une transition ne correspond pas nécessairement à une variation des trois sortes
d’énergie. Trois types de spectres peuvent être distingués : les spectres de rotation pure, les

TP de chimie théorique et physique
S. Gillet - 6 - IR
spectres de rotation-vibration et les spectres électroniques, qui s’étendent du domaine des
radiofréquences à celui de l’ultraviolet.
La grande diversité des molécules ne permet pas de classer leurs spectres aussi
facilement que ceux, en petit nombre, des atomes. Cependant, l’expérience montre qu’ils ont
des bandes caractéristiques des groupes d’atomes qui les constituent, ce qui est très utile pour
déterminer leur structure.
2. LA SPECTROMÉTRIE INFRAROUGE
2.1. GÉNÉRALITÉS
La spectroscopie infrarouge est une spectroscopie d’absorption dont le principe repose
sur l’absorption d’un rayonnement infrarouge par la matière organique. La région comprise
entre 4000 et 400 cm-1 de nombre d’onde est particulièrement utile au chimiste. Les régions
du proche infrarouge (14290 – 4000 cm-1) et de l’infrarouge lointain (700 – 200 cm-1)
apportent parfois des informations intéressantes (figure 2).
Figure 2 : Le spectre électromagnétique et les différentes régions de l’infrarouge.
Bien que le spectre infrarouge soit caractéristique de la molécule entière, il est vrai que
certains groupes d’atomes donnent naissance à des bandes de fréquences identiques ou

TP de chimie théorique et physique
S. Gillet - 7 - IR
proches indépendamment de la structure du reste de la molécule. C’est la persistance de
bandes caractéristiques qui permet au chimiste d’obtenir des informations utiles sur la
structure par simple examen du spectre en se référant à des tables générales regroupant les
fréquences des groupes caractéristiques. Ces dernières nous seront des plus utiles.
Si le nombre d’onde (ν~ ) est inférieur à 100 cm-1, l’énergie absorbée est convertie en
énergie de rotation. Pour les nombres d’onde compris entre 10000 et 100 cm-1, cette énergie
est absorbée et convertie en énergie de vibration (figure 3). Le spectre de vibration apparaît
sous forme de bande (plutôt que sous forme de raies) car une unique variation d’énergie
vibratoire s’accompagne de nombreuses variations d’énergie rotationnelle. Nous travaillerons
donc à partir de ces bandes rotato-vibratoires, particulièrement celles comprises entre 4000 et
400 cm-1.
Il existe deux types de vibrations moléculaires : les élongations et les déformations
angulaires. Une vibration d’élongation est un mouvement rythmique le long de l’axe de la
liaison impliquant l’augmentation et la diminution de la distance inter-atomique. Une
déformation angulaire consiste en une variation de l’angle formé par deux liaisons
successives, c'est-à-dire ayant un atome en commun, ou bien le mouvement d’un groupe
d’atome par rapport au reste de la molécule, mais sans mouvement des atomes les uns par
rapport aux autres. Par exemple, une torsion un balancement et une rotation impliquent une
variation d’angle entre liaison par rapport à des coordonnées arbitraires fixées au sein de la
molécule (figure 3).
Figure 3 : Vibrations moléculaires d’élongation et de déformation angulaire.
Les absorptions dues aux élongations sont quantifiées : la fréquence d’oscillation
dépend des masses des atomes et de la force du lien quantifié par la loi de Hooke (figure 4).

TP de chimie théorique et physique
S. Gillet - 8 - IR
La loi de Hooke représente deux atomes et leur liaison comme un simple oscillateur
harmonique composé de deux masses reliées entre elles. Ainsi la fréquence de vibration des
atomes dépend de leurs masses et de la force de la liaison.
2
1
)/()(2
1~
+=
yxyx MMMM
f
cπν
Avec : ν~ le nombre d’onde, c la vitesse de la lumière, f la constante de force de la
liaison et Mx et My masses (en g) des atomes x et y.
Figure 4 : Effet de la force de la liaison et de la masse des atomes sur la fréquence de vibration
TP de chimie théorique et physique
S. Gillet - 9 - IR
Le modèle de l’oscillateur harmonique permet de déterminer, de manière théorique et
approximative, la « fréquence » de l’absorption correspondant à la vibration d’une liaison
entre deux atomes (exemple : C-H).
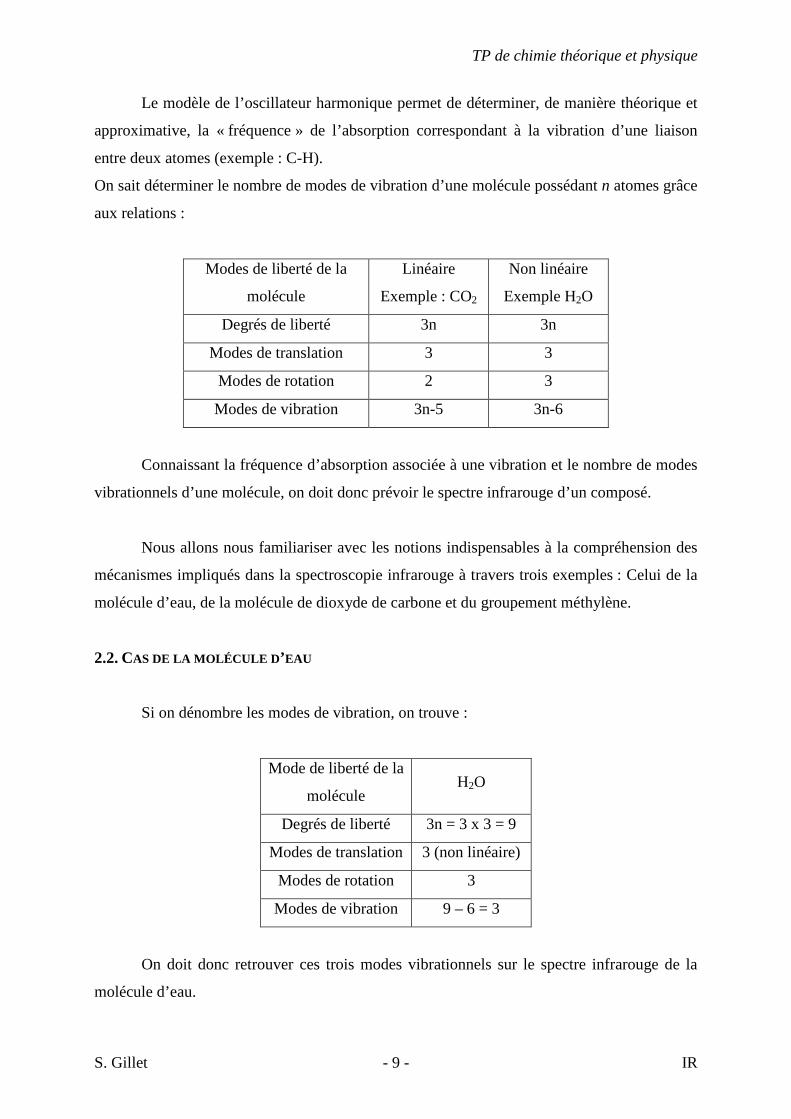
On sait déterminer le nombre de modes de vibration d’une molécule possédant n atomes grâce
aux relations :
Modes de liberté de la
molécule
Linéaire
Exemple : CO2
Non linéaire
Exemple H2O
Degrés de liberté 3n 3n
Modes de translation 3 3
Modes de rotation 2 3
Modes de vibration 3n-5 3n-6
Connaissant la fréquence d’absorption associée à une vibration et le nombre de modes
vibrationnels d’une molécule, on doit donc prévoir le spectre infrarouge d’un composé.
Nous allons nous familiariser avec les notions indispensables à la compréhension des
mécanismes impliqués dans la spectroscopie infrarouge à travers trois exemples : Celui de la
molécule d’eau, de la molécule de dioxyde de carbone et du groupement méthylène.
2.2. CAS DE LA MOLÉCULE D ’EAU
Si on dénombre les modes de vibration, on trouve :
Mode de liberté de la
molécule H2O
Degrés de liberté 3n = 3 x 3 = 9
Modes de translation 3 (non linéaire)
Modes de rotation 3
Modes de vibration 9 – 6 = 3
On doit donc retrouver ces trois modes vibrationnels sur le spectre infrarouge de la
molécule d’eau.
TP de chimie théorique et physique
S. Gillet - 10 - IR
Cependant, comme la molécule d’eau est symétrique, et qu’elle ne possède qu’un seul
type de vibrateur (O-H), on devrait s’attendre à n’observer qu’une seule absorption
d’élongation. L’utilisation du modèle de l’oscillateur harmonique permet de déterminer que
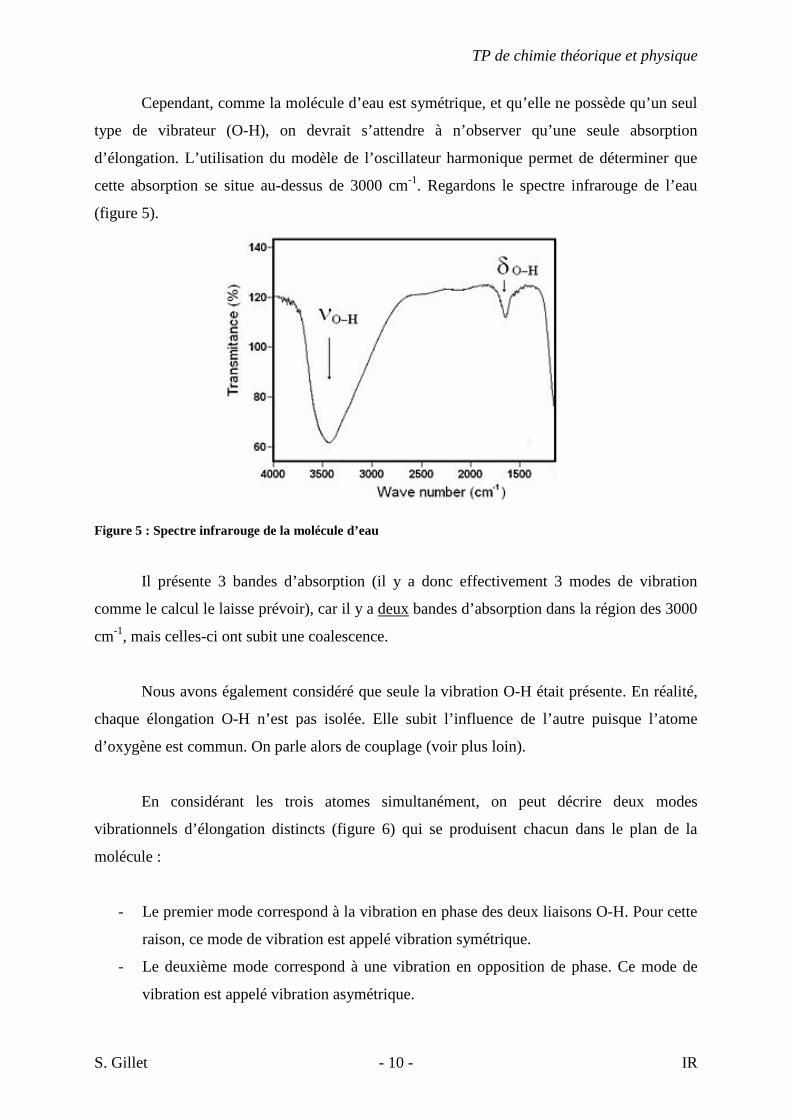
cette absorption se situe au-dessus de 3000 cm-1. Regardons le spectre infrarouge de l’eau
(figure 5).
Figure 5 : Spectre infrarouge de la molécule d’eau
Il présente 3 bandes d’absorption (il y a donc effectivement 3 modes de vibration
comme le calcul le laisse prévoir), car il y a deux bandes d’absorption dans la région des 3000
cm-1, mais celles-ci ont subit une coalescence.
Nous avons également considéré que seule la vibration O-H était présente. En réalité,
chaque élongation O-H n’est pas isolée. Elle subit l’influence de l’autre puisque l’atome
d’oxygène est commun. On parle alors de couplage (voir plus loin).
En considérant les trois atomes simultanément, on peut décrire deux modes
vibrationnels d’élongation distincts (figure 6) qui se produisent chacun dans le plan de la
molécule :
- Le premier mode correspond à la vibration en phase des deux liaisons O-H. Pour cette
raison, ce mode de vibration est appelé vibration symétrique.
- Le deuxième mode correspond à une vibration en opposition de phase. Ce mode de
vibration est appelé vibration asymétrique.

TP de chimie théorique et physique
S. Gillet - 11 - IR
Le couplage des deux vibrateurs a pour effet de donner deux bandes d’absorption,
l’une de fréquence supérieure, l’autre de fréquence inférieure à la fréquence calculée par le
modèle de l’oscillateur harmonique pour le vibrateur isolé.
L’intensité de ces deux bandes est différente. Pour comprendre cette différence
d’intensité d’absorption, il faut examiner les deux modes de vibration d’élongation du point
de vue de la variation du moment dipolaire de la molécule (figure 6).
Figure 6 : Les modes vibrationnels de la molécule d’eau.
Le mode de vibration d’élongation qui correspond à la plus forte variation du moment
dipolaire est la vibration d’élongation asymétrique. En effet, pour le mode de vibration en
phase (symétrique), seule l’intensité du moment dipolaire varie alors que pour le mode de
vibration en opposition de phase (asymétrique), on observe à la fois une variation d’intensité
mais aussi de direction.
Il reste une absorption dans la partie basse que nous n’avons pas encore associée à un
mode de vibration. Cette vibration correspond à une déformation de l’angle des liaisons.
L’intensité associée à cette vibration de déformation du lien O-H dépend de la
variation du moment dipolaire engendré par la vibration (figure 7). La vibration de liens
polarisés donnera lieu à des bandes intenses, alors que les bandes de lien non ou peu polarisé
(comme ceux retrouvés dans les molécules symétriques) seront peu ou pas visibles.
Cette variation du moment dipolaire due à la déformation angulaire est plus faible que dans le
cas de la vibration d’élongation asymétrique. Il n’est donc pas étonnant de trouver une
intensité faible à la vibration de déformation associée à la molécule d’eau.

TP de chimie théorique et physique
S. Gillet - 12 - IR
Figure 7 : Influence de la variation du moment dipolaire sur l’intensité des bandes d’absorption.
De manière générale,
- La partie haute (4000-1600 cm-1) correspond aux vibrations d’élongation et est
appelée la section des groupes fonctionnels car elle comporte la plupart des bandes qui
sont caractéristiques de ces groupes fonctionnels.
- La partie basse (1600-600 cm-1) correspond aux vibrations de déformation et est
appelée la section des empreinte digitales parce qu’elle comprend un très grand
nombre de bandes aux formes variées. Si toutes les bandes de cette région se
retrouvent dans deux spectres infrarouges, vous pouvez conclure qu’il s’agit de spectre
du même composé.
2.3 CAS DE LA MOLÉCULE DE DIOXYDE DE CARBONE
Le nombre de degrés de liberté de cette molécule est égal à 9 (puisqu’elle comporte 3
atomes : 3 x 3 = 9). Cette molécule étant linéaire, elle présente :
- 3 modes de translation
- 2 modes de rotation, et donc
- 9 – 3 – 2 = 4 modes de vibration
TP de chimie théorique et physique
S. Gillet - 13 - IR
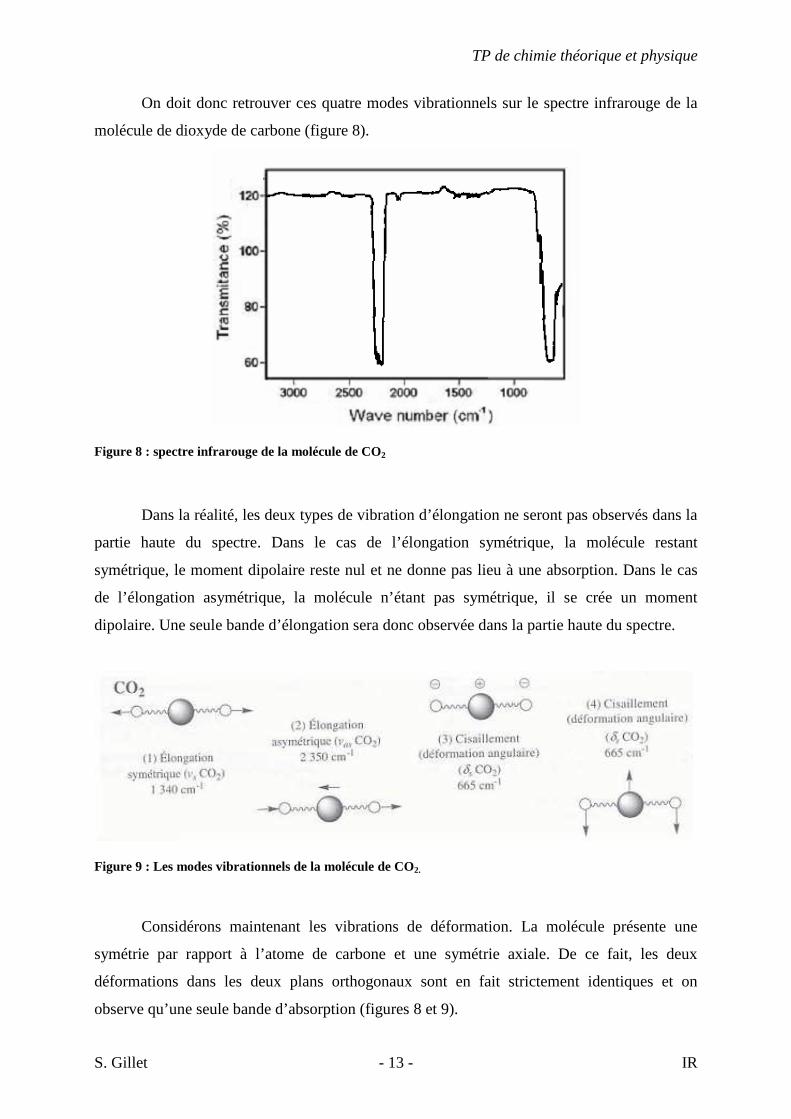
On doit donc retrouver ces quatre modes vibrationnels sur le spectre infrarouge de la
molécule de dioxyde de carbone (figure 8).
Figure 8 : spectre infrarouge de la molécule de CO2
Dans la réalité, les deux types de vibration d’élongation ne seront pas observés dans la
partie haute du spectre. Dans le cas de l’élongation symétrique, la molécule restant
symétrique, le moment dipolaire reste nul et ne donne pas lieu à une absorption. Dans le cas
de l’élongation asymétrique, la molécule n’étant pas symétrique, il se crée un moment
dipolaire. Une seule bande d’élongation sera donc observée dans la partie haute du spectre.
Figure 9 : Les modes vibrationnels de la molécule de CO2.
Considérons maintenant les vibrations de déformation. La molécule présente une
symétrie par rapport à l’atome de carbone et une symétrie axiale. De ce fait, les deux
déformations dans les deux plans orthogonaux sont en fait strictement identiques et on
observe qu’une seule bande d’absorption (figures 8 et 9).

TP de chimie théorique et physique
S. Gillet - 14 - IR
2.4. CAS D’UN GROUPEMENT CH2
Les divers modes d’élongation et de déformation d’un groupe AX2 pris comme une
fraction de la molécule, par exemple un groupe CH2 dans une molécule d’hydrocarbure, sont
présentés à la figure 10. La règle 3n-6 ne s’applique pas puisque le groupe CH2 ne représente
qu’une portion de la molécule.
Figure 10 : Les modes vibrationnels des groupements CH2.
***
En conclusion, il faut retenir que pour qu’une vibration moléculaire soit active en
infrarouge, une modification du moment dipolaire doit être observée pendant la vibration et la
fréquence d’absorption résultante doit être dans le domaine IR.

TP de chimie théorique et physique
S. Gillet - 15 - IR
2.4. L IMITES DU MODÈLE
Le nombre de vibrations fondamentales (fréquences d’absorption) est rarement
observé car les harmoniques (multiples d’une fréquence donnée) et les bandes de
combinaisons (somme de deux vibrations) accroissent le nombre de bandes, alors que le
nombre théorique de bande est réduit dans les cas suivants :
- Fréquences fondamentales tombant en dehors de la région comprise entre 4000 et 400
cm-1.
- Bandes fondamentales trop faibles pour être observées.
- Coalescences de vibrations fondamentales trop proches.
- Présence d’une bande dégénérée provenant de plusieurs absorptions de même
fréquence dans des molécules hautement symétriques.
- Vibrations fondamentales n’apparaissant pas en IR par absence de variation du
moment dipolaire de la molécule.
2.5. LES INTERACTIONS COUPLÉES
Quand deux oscillateurs couplés partagent un atome, ils se comportent rarement
comme des oscillateurs individuels à moins que les fréquences d’oscillations individuelles ne
soient vraiment différentes. Et ce parce qu’il existe une interaction de couplage mécanique
entre les oscillateurs.
La molécule de dioxyde de carbone, par exemple, a deux vibrations fondamentales
correspondant aux modes d’élongation symétrique et asymétrique. L’élongation symétrique
n’est pas active en infrarouge. Dans le mode d’élongation asymétrique, les deux liaisons C=O
s’étirent en opposition de phase ; pendant qu’une liaison C=O s’étire, l’autre se contracte.
Cette absorption se fait à une fréquence plus élevée que celle observée pour un groupe
carbonyle dans une cétone aliphatique. Cette différence dans les fréquences d’absorption du
carbonyle exemplifiée par la molécule de dioxyde de carbone résulte d’un fort couplage
mécanique (ou interaction). A l’opposé, deux groupes carbonyles cétoniques séparés par un
ou plusieurs atomes de carbone montrent une absorption carbonyle normale proche de 1715
cm-1 parce que le (ou les) atome(s) de carbone intermédiaire(s) empêche(nt) le couplage.

TP de chimie théorique et physique
S. Gillet - 16 - IR
Les conditions requises pour que le couplage soit effectif peuvent se résumer comme suit :
- Pour qu’une interaction ait lieu, les vibrations doivent être du même groupe de
symétrie.
- Un couplage fort entre des vibrations d’élongation nécessite le partage d’un atome
commun entre les oscillateurs.
- L’interaction est maximale quand les groupes couplés absorbent, individuellement, au
voisinage de la même fréquence.
- Le couplage entre les vibrations de déformation et d’élongation a lieu si la liaison en
élongation forme un coté de l’angle déformé.
- Le couplage entre les vibrations de déformation nécessite une liaison commune.
- Le couplage est négligeable quand les groupes sont séparés par un ou plusieurs atomes
de carbone et quand les vibrations sont perpendiculaires l’une à l’autre.
Le couplage de deux modes de vibrations fondamentales produira deux nouveaux
modes de vibrations avec des fréquences plus élevées et plus basses que celle observées
quand l’interaction est absente. Une interaction peut également avoir lieu entre des vibrations
fondamentales et des vibrations combinées ou harmoniques. De telles interactions sont
appelées « résonances de Fermi ».
2.6. LES LIAISONS HYDROGÈNES
La force de la liaison hydrogène est maximale quand le groupe donneur de proton et
l’axe de l’orbitale de la paire libre sont alignés. La force de la liaison décroît quand la distance
entre les deux groupes augmente.
Les liaisons hydrogènes modifient la constante de force des deux groupes, ainsi, les
fréquences des vibrations d’élongation et de déformation sont modifiées. Les bandes
d’élongation X-H (donneur de proton) se décalent vers les basses fréquences (longueur
d’ondes plus grandes), s’élargissent et leurs intensités augmentent. La fréquence d’élongation
du groupe accepteur, par exemple C=O est également réduite mais dans une moindre mesure
par rapport à celle du groupe donneur de proton. La vibration de déformation H-X, quant à
elle, se déplace habituellement vers les longueurs d’ondes plus courtes ; ce déplacement est
moins prononcé que celui des fréquences d’élongation.

TP de chimie théorique et physique
S. Gillet - 17 - IR
3. L’ APPAREILLAGE
3.1. SPECTROPHOTOMÈTRE TRADITIONNEL À DOUBLES FAISCEAUX
Figure 11 : Schéma d’un spectromètre traditionnel à doubles faisceaux.
La source est un filament incandescent qui émet sur l’ensemble du spectre infrarouge.
La radiation de la source est divisée en deux faisceaux par un miroir : les faisceaux de
référence et échantillon. Les faisceaux sont dirigés dans le compartiment à échantillon où ils
passent respectivement à travers les cellules de références et échantillon. A la sortie du
compartiment à échantillon, un obturateur permet de bloquer alternativement un et l’autre des
faisceaux. On obtient un seul faisceau composé de segments alternants des faisceaux de
référence et échantillon.
Le faisceau combiné passe à travers le monochromateur (double réseau) pour produire
un balayage de la bande de fréquences au détecteur. Ce détecteur (thermocouple) compare
l’intensité des segments de référence et échantillon pour chaque fréquence et produit un
spectre de la transmittance (%) ou de l’absorbance (UA) en fonction du nombre d’onde (cm-1)
ou de la longueur d’onde (µm).

TP de chimie théorique et physique
S. Gillet - 18 - IR
3.2. FTIR : SPECTROPHOTOMÈTRE MODERNE À TRANSFORMÉE DE FOURIER
Figure 12 : Schéma d’un spectromètre à transformée de Fourier.
La radiation de la source est divisée en deux faisceaux par un séparateur de faisceaux.
Un des faisceaux parcourt un chemin optique fixe, l’autre un chemin optique de longueur
variable à cause d’un miroir mobile, avant d’être recombinés, de traverser l’échantillon et de
frapper le détecteur.
Quand la différence de chemin optique entre les faisceaux correspond à un multiple
entier de la longueur d’onde d’une bande, on obtient une interférence constructive. Une
interférence négative est obtenue lorsque la différence correspond à un multiple entier impair
du quart de la longueur d’onde.
L’ensemble des interférences positives et négatives produit un interférogramme.
Celui-ci contient toutes les informations requises pour produire un spectre suite à une
opération mathématique appelée transformée de Fourier.
Les avantages de ce système sont une haute précision sur la fréquence, une acquisition
des données très rapide, un traitement informatique des données ainsi qu’un coût raisonnable.

TP de chimie théorique et physique
S. Gillet - 19 - IR
3.3. LA PRÉPARATION DES ÉCHANTILLONS
On peut obtenir un spectre infrarouge à partir de gaz, de liquide ou de solide. Pour les
gaz et les liquides à bas points d’ébullition, ils sont placés dans une cellule sous vide où il y
aura expansion de l’échantillon.
Quant aux liquides, ils peuvent être analysés purs ou en solution. Les échantillons purs
(figure 13A) sont placés entre deux fenêtres de sel d’épaisseur variable (1 à 10 mg). Pour les
échantillons en solutions (figures 13A et B), on place 0,1 à 1 ml de la solution de 0,05%
jusqu’à 10% dans les cellules mesurant 0,1 à 1 mm d’épaisseur. Le solvant utilisé doit être sec
et transparent dans la région observée (généralement CCl4 et CS2).
Les échantillons solides sont sous forme de pâtes ou de pastilles. La pâte est formée
par broyage de 2 à 5 mg d’échantillon avec 1 à 2 gouttes d’huile (nujol). On place alors le
film obtenu entre deux fenêtres de sel. Les pastilles sont réalisées à l’aide d’environ 1 mg de
produit à analyser avec 100 mg de bromure de potassium broyés puis mis sous pression
(figure 13C).
Figure 13 : (A) échantillon sous forme de film entre des pastilles de NaCl (liquide) ; (B) échantillon en
solution dans les cellules (liquides et solides) ; (C) Échantillon solide dans une pastille de KBr (solides).
A B C

TP de chimie théorique et physique
S. Gillet - 20 - IR
4. ABSORPTION CARACTÉRISTIQUE DES GROUPES DANS LES MOLÉCULES
ORGANIQUES
4.1. INTRODUCTION
Il n’y a pas de règles strictes pour l’interprétation d’un spectre infrarouge. Cependant,
avant toute interprétation, certaines conditions doivent être remplies :
- Le spectre doit présenter une résolution et une intensité adéquate ;
- Le spectre devrait être celui d’un composé raisonnablement pur ;
- Le spectrophotomètre doit être calibré afin d’observer les bandes à leur juste nombre
d’onde. Un bon calibrage doit être fait avec des standards fiables tels que par exemple,
un film de polystyrène ;
- Le conditionnement de l’échantillon doit être spécifique. Pour une solution, le solvant,
la concentration et l’épaisseur du film doivent être indiqués.
Un spectre infrarouge est un graphique représentant en abscisses la position des
bandes sous la forme de nombre d’onde ν~ (cm-1) et en ordonnée l’intensité de ces bandes.
Les intensités de bandes peuvent être exprimées soit en transmittance (T) soit en absorbance
(A). Les chimistes notent habituellement les intensités de manière semi-quantitative : s =
strong ; m = medium et w = weak.
Remarque : dans la suite du présent document, le sigle ∼ fait référence à un doublet d’électron
partagé entre deux liaisons. En particulier, C∼ C représentera une liaison entre deux carbones
appartenant à un cycle aromatique (Ar), ces carbones étant stabilisés par résonance.
Les sigles ν, δ, ρ se rapportent respectivement aux vibrations d’élongation, de cisaillement et
de rotation.
4.2. MARCHE À SUIVRE
La marche à suivre lors de l’analyse d’un spectre infrarouge consiste à identifier, dans
la section des « empreintes digitales » et dans la section des « groupes fonctionnels », les pics
d’absorption caractéristiques de certaines fonctions des molécules (voir point 5.). Les

TP de chimie théorique et physique
S. Gillet - 21 - IR
paragraphes suivants seront ainsi consacrés à la description des spectres IR des principales
familles de molécules organiques.
4.3. LES ALCANES LINÉAIRES
Les spectres des alcanes normaux sont interprétés en fonction de quatre vibrations qui
sont l’élongation et la déformation angulaire des liens C-H et C-C. Les plus caractéristiques
sont celles provenant de l’élongation et de la déformation angulaire des liens C-H. Les quatre
vibrations de déformation sont appelées cisaillement, rotation plane, balancement et torsion.
Les vibrations provenant des torsions et des balancements hors du plan des méthylènes ont
peu d’intérêt à cause de leur faible intensité et de leur variabilité.
La vibration angulaire de C-C apparaît à de très bas nombres d’onde (ν~ ) et donc ne se
situe pas dans nos spectres. L’élongation C-C est entre 1200 et 800 cm-1. Elle est faible et
présente donc peu d’intérêt.
Figure 14 : Dodécane. Elongation C-H : 2953 cm-1 ννννasCH3, 2870 cm-1 ννννsCH3, 2922 cm-1 ννννasCH2, 2853 cm-1
ννννsCH2. Deformation C-H : 1464 cm-1 δδδδsCH2, 1450 cm-1 δδδδasCH3, 1379 cm-1 δδδδsCH3. Rotation plane CH2: 724
cm-1 ρρρρCH2.
4.4. LES ALCANES RAMIFIÉS
Les changements proviennent de modification des vibrations d’élongation du squelette
et de déformation des méthyles. L’élongation d’un C-H tertiaire apparaît vers 2890 cm-1,
perdue dans celles des autres C-H et la déformation de C-H est observée sous forme d’un fort
doublet à 1385-1380 cm-1 et 1370-1365 cm-1 d’intensité égale pour l’isopropyle, de deux

TP de chimie théorique et physique
S. Gillet - 22 - IR
bandes à 1395-1385 cm-1 et 1370 cm-1 pour un ter-butyle et d’un doublet dans la même région
que l’isopropyle et t-butyle pour un gem-diméthyle qui n’est pas aux extrémités.
Figure 15 : 2,2,4-triméthylpentane. Elongation C-H. Déformation C-H : Des doublets se chevauchent pour
les groupes isopropyle et tertiobutyle vers 1400-1300 cm-1.
4.5. LES ALCANES CYCLIQUES
Les élongations des C-H sont les mêmes que celles des autres alcanes. Pour les
déformations des C-H, la cyclisation diminue la fréquence (nombre d’onde) de cisaillement
des CH2 : n-hexane 1468 cm-1, cyclohexane 1452 cm-1, cyclopentane 1455 cm-1, cyclopropane
1442 cm-1.
4.6. LES ALCÈNES
Pour les alcènes, de nouveaux modes de vibration apparaissent qui sont l’élongation
C=C, l’élongation C-H où C fait partie du lien alcène et la déformation dans et hors du plan
des C-H. L’élongation C=C, pour les alcènes linéaires non conjugués est moyenne à faible, de
1667 à 1640 cm-1. Les doubles liaisons internes absorbent moins fortement que les terminales.
L’élongation de C-H d’alcène apparaît à des nombres d’onde supérieurs à 3000 cm-1
pour les aromatiques, les hétéroatomes, les alcynes et les alcènes. La déformation de C-H
alcène est située dans le même plan ou perpendiculaire à C=C.
TP de chimie théorique et physique
S. Gillet - 23 - IR
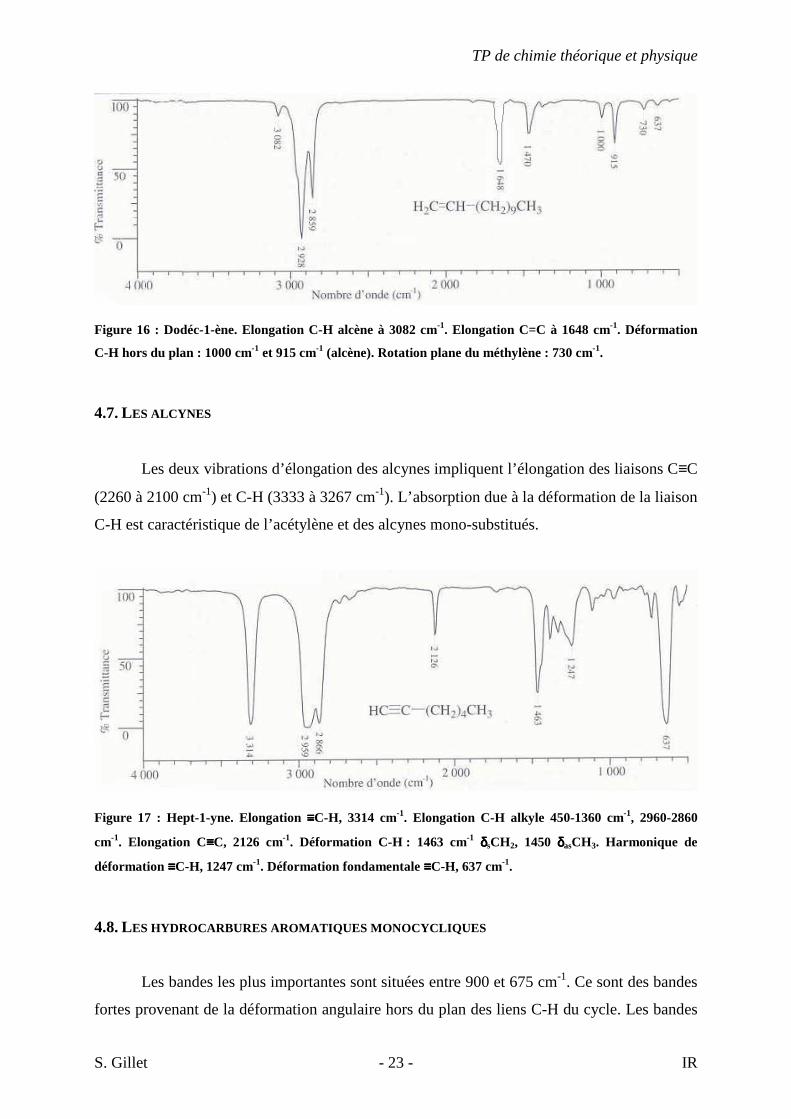
Figure 16 : Dodéc-1-ène. Elongation C-H alcène à 3082 cm-1. Elongation C=C à 1648 cm-1. Déformation
C-H hors du plan : 1000 cm-1 et 915 cm-1 (alcène). Rotation plane du méthylène : 730 cm-1.
4.7. LES ALCYNES
Les deux vibrations d’élongation des alcynes impliquent l’élongation des liaisons C≡C
(2260 à 2100 cm-1) et C-H (3333 à 3267 cm-1). L’absorption due à la déformation de la liaison
C-H est caractéristique de l’acétylène et des alcynes mono-substitués.
Figure 17 : Hept-1-yne. Elongation ≡≡≡≡C-H, 3314 cm-1. Elongation C-H alkyle 450-1360 cm-1, 2960-2860
cm-1. Elongation C≡≡≡≡C, 2126 cm-1. Déformation C-H : 1463 cm-1 δδδδsCH2, 1450 δδδδasCH3. Harmonique de
déformation ≡≡≡≡C-H, 1247 cm-1. Déformation fondamentale ≡≡≡≡C-H, 637 cm-1.
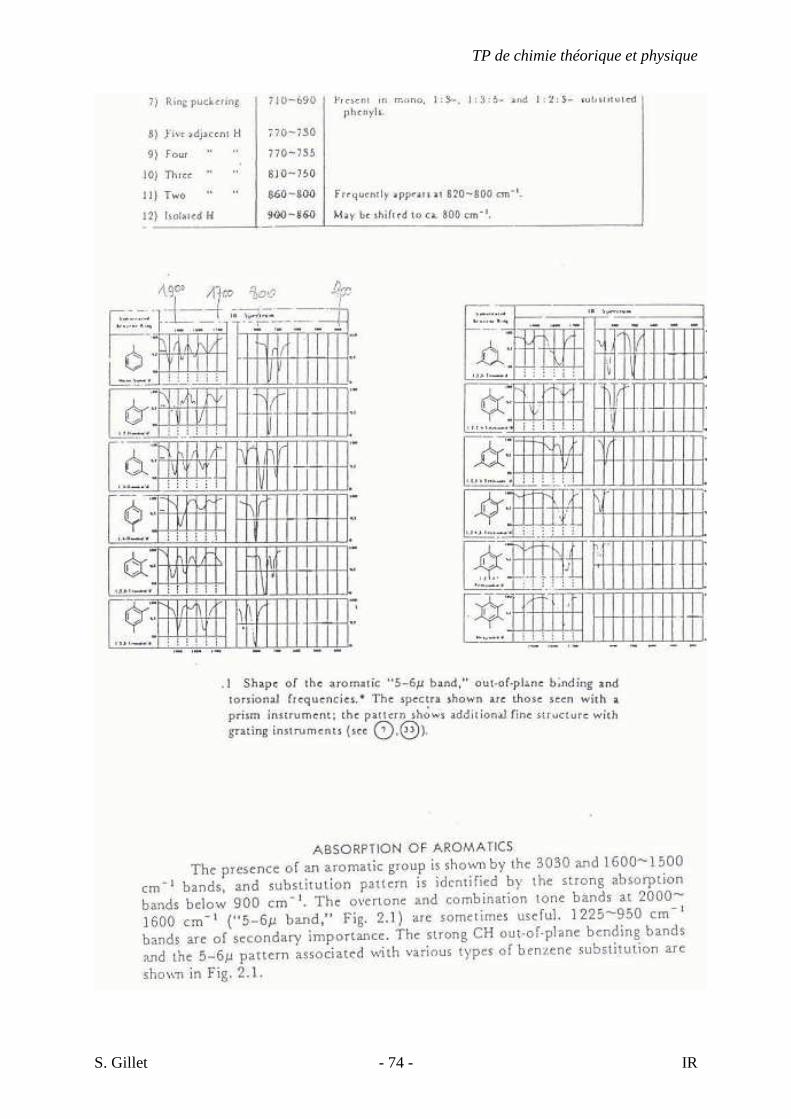
4.8. LES HYDROCARBURES AROMATIQUES MONOCYCLIQUES
Les bandes les plus importantes sont situées entre 900 et 675 cm-1. Ce sont des bandes
fortes provenant de la déformation angulaire hors du plan des liens C-H du cycle. Les bandes
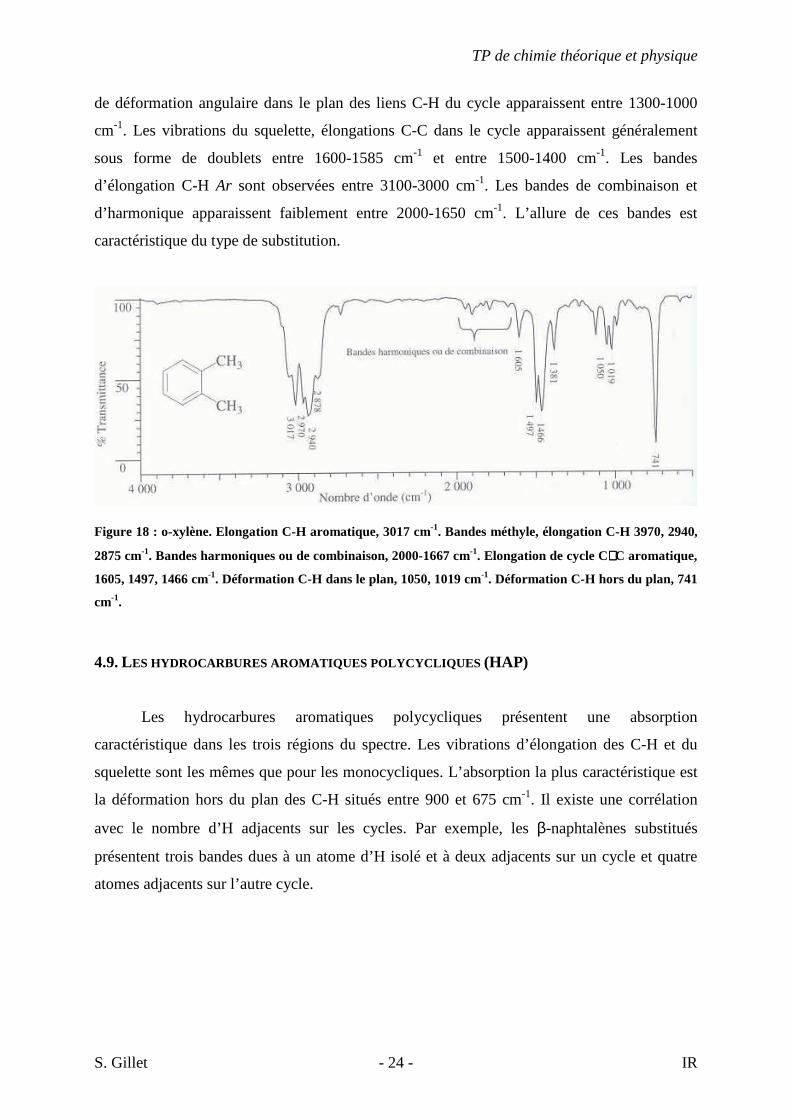
TP de chimie théorique et physique
S. Gillet - 24 - IR
de déformation angulaire dans le plan des liens C-H du cycle apparaissent entre 1300-1000
cm-1. Les vibrations du squelette, élongations C-C dans le cycle apparaissent généralement
sous forme de doublets entre 1600-1585 cm-1 et entre 1500-1400 cm-1. Les bandes
d’élongation C-H Ar sont observées entre 3100-3000 cm-1. Les bandes de combinaison et
d’harmonique apparaissent faiblement entre 2000-1650 cm-1. L’allure de ces bandes est
caractéristique du type de substitution.
Figure 18 : o-xylène. Elongation C-H aromatique, 3017 cm-1. Bandes méthyle, élongation C-H 3970, 2940,
2875 cm-1. Bandes harmoniques ou de combinaison, 2000-1667 cm-1. Elongation de cycle C∼∼∼∼C aromatique,
1605, 1497, 1466 cm-1. Déformation C-H dans le plan, 1050, 1019 cm-1. Déformation C-H hors du plan, 741
cm-1.
4.9. LES HYDROCARBURES AROMATIQUES POLYCYCLIQUES (HAP)
Les hydrocarbures aromatiques polycycliques présentent une absorption
caractéristique dans les trois régions du spectre. Les vibrations d’élongation des C-H et du
squelette sont les mêmes que pour les monocycliques. L’absorption la plus caractéristique est
la déformation hors du plan des C-H situés entre 900 et 675 cm-1. Il existe une corrélation
avec le nombre d’H adjacents sur les cycles. Par exemple, les β-naphtalènes substitués
présentent trois bandes dues à un atome d’H isolé et à deux adjacents sur un cycle et quatre
atomes adjacents sur l’autre cycle.
TP de chimie théorique et physique
S. Gillet - 25 - IR
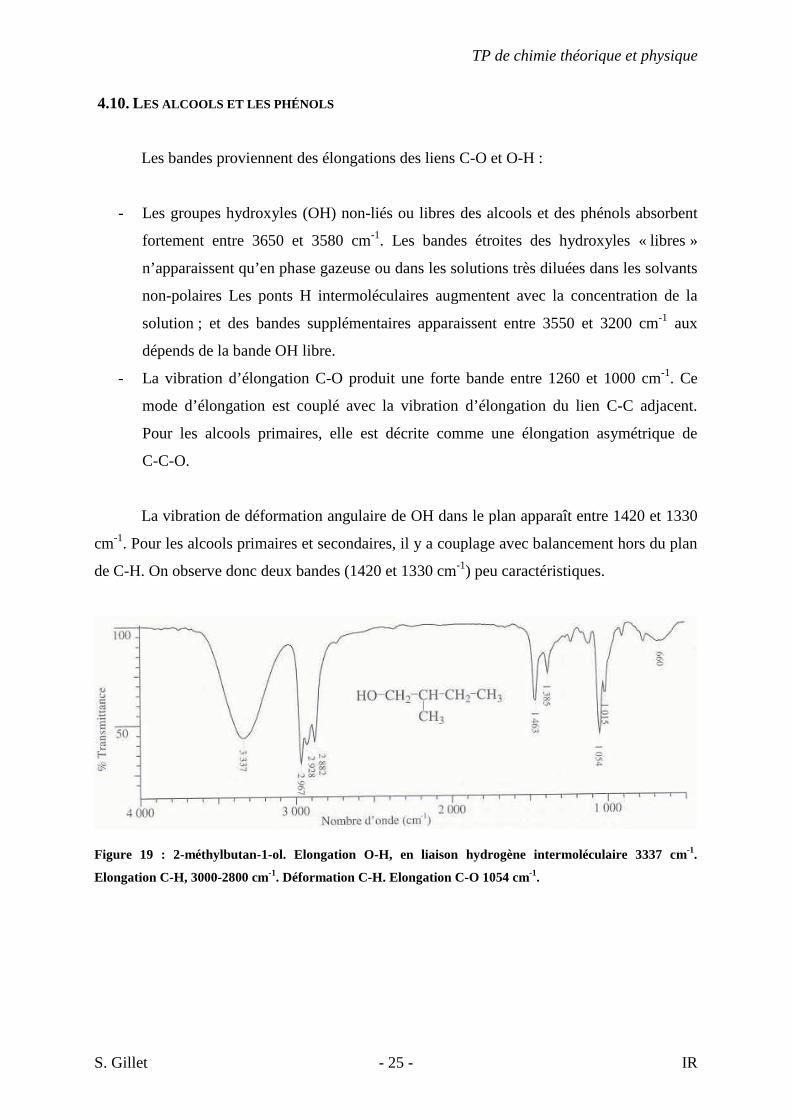
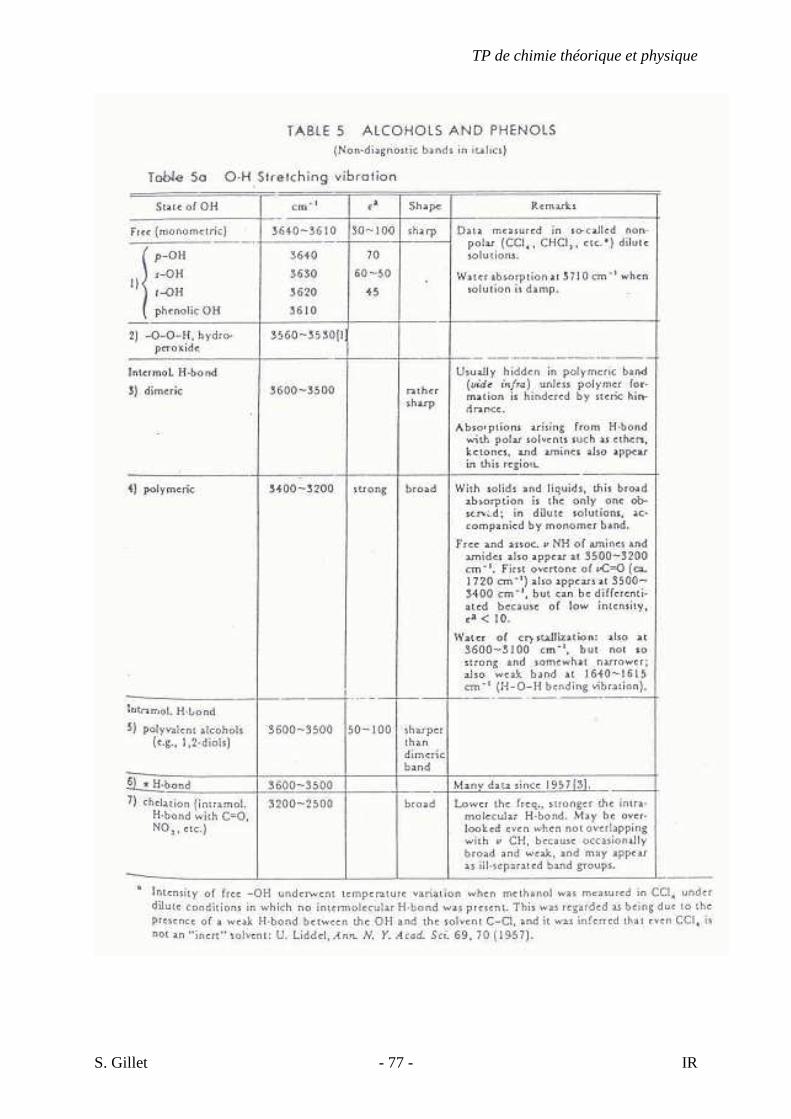
4.10. LES ALCOOLS ET LES PHÉNOLS
Les bandes proviennent des élongations des liens C-O et O-H :
- Les groupes hydroxyles (OH) non-liés ou libres des alcools et des phénols absorbent
fortement entre 3650 et 3580 cm-1. Les bandes étroites des hydroxyles « libres »
n’apparaissent qu’en phase gazeuse ou dans les solutions très diluées dans les solvants
non-polaires Les ponts H intermoléculaires augmentent avec la concentration de la
solution ; et des bandes supplémentaires apparaissent entre 3550 et 3200 cm-1 aux
dépends de la bande OH libre.
- La vibration d’élongation C-O produit une forte bande entre 1260 et 1000 cm-1. Ce
mode d’élongation est couplé avec la vibration d’élongation du lien C-C adjacent.
Pour les alcools primaires, elle est décrite comme une élongation asymétrique de
C-C-O.
La vibration de déformation angulaire de OH dans le plan apparaît entre 1420 et 1330
cm-1. Pour les alcools primaires et secondaires, il y a couplage avec balancement hors du plan
de C-H. On observe donc deux bandes (1420 et 1330 cm-1) peu caractéristiques.
Figure 19 : 2-méthylbutan-1-ol. Elongation O-H, en liaison hydrogène intermoléculaire 3337 cm-1.
Elongation C-H, 3000-2800 cm-1. Déformation C-H. Elongation C-O 1054 cm-1.

TP de chimie théorique et physique
S. Gillet - 26 - IR
Figure 20 : Phénol (fondu). Large pic de liaison hydrogène intermoléculaire, élongation O-H, 3244 cm-1.
Elongation aromatique, 3052 cm-1. Bandes harmoniques ou de combinaison, 2000-1667 cm-1. Elongation
de cycle C∼∼∼∼C aromatique, 1601, 1501, 1478 cm-1. Déformation OH dans le plan, 1378 cm-1. Elongation
C-O, 1231 cm-1. Déformation C-H hors du plan, 815, 753 cm-1. Déformation du cycle C∼∼∼∼C aromatique
hors du plan, 699 cm-1. Déformation O-H en liaison hydrogène hors du plan, environ 650 cm-1.
4.11. LES ÉTHERS
Pour les éthers, la vibration d’élongation de la liaison C-O-C se situe dans la même
région que celle de C-C-C mais elle est plus intense et peut se coupler avec d’autres
vibrations. Pour les éthers aliphatiques, on observe une bande à 1125 cm-1 et s’il y a une
ramification sur les atomes adjacents, il y aura éclatement de la bande C-O-C.
Figure 21 : Anisole. Elongation C-H aromatique, 3067, 3030, 3005 cm-1. Elongation C-H des méthyles,
2950, 2843 cm-1. Région d’harmonique et de combinaison, 2000-1650 cm-1. Elongation C∼∼∼∼C de cycle
aromatique, 1601, 1501 cm-1. Elongation C-O-C asymétrique 1254 cm-1. Elongation C-O-C symétrique,
1046 cm-1. Déformation C-H hors du plan, 784, 761 cm-1. Déformation C∼∼∼∼C de cycle hors du plan, 699
cm-1.
TP de chimie théorique et physique
S. Gillet - 27 - IR
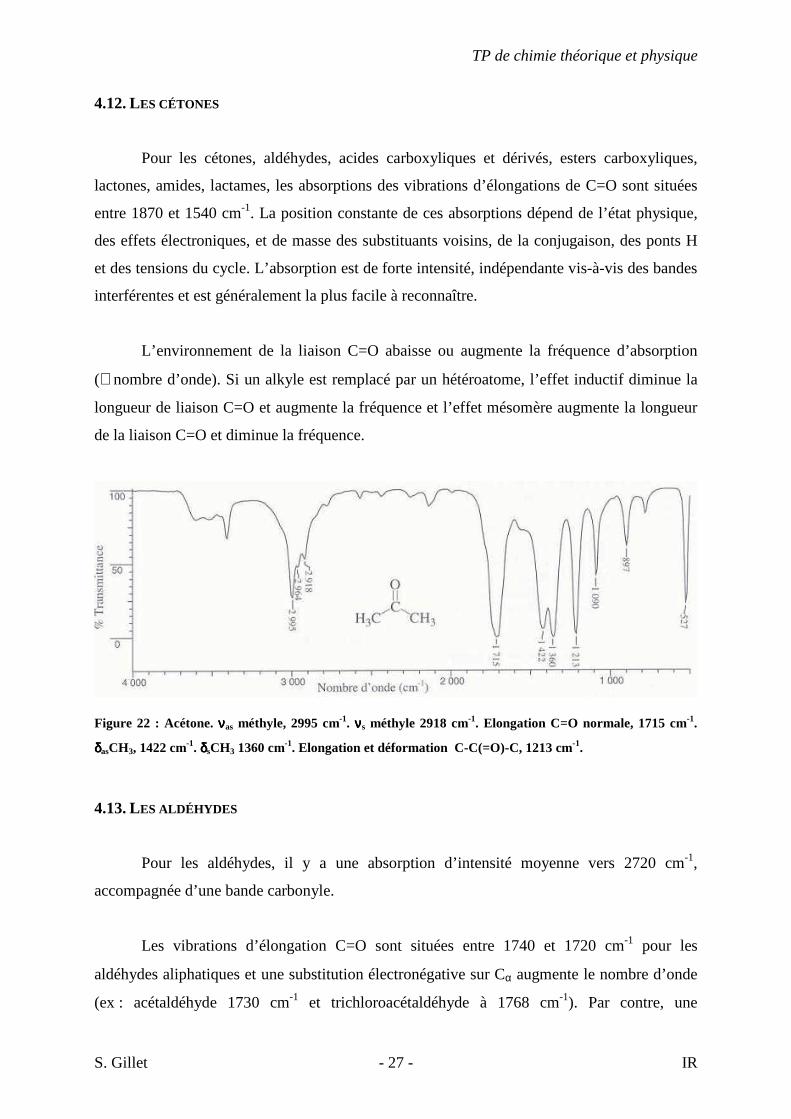
4.12. LES CÉTONES
Pour les cétones, aldéhydes, acides carboxyliques et dérivés, esters carboxyliques,
lactones, amides, lactames, les absorptions des vibrations d’élongations de C=O sont situées
entre 1870 et 1540 cm-1. La position constante de ces absorptions dépend de l’état physique,
des effets électroniques, et de masse des substituants voisins, de la conjugaison, des ponts H
et des tensions du cycle. L’absorption est de forte intensité, indépendante vis-à-vis des bandes
interférentes et est généralement la plus facile à reconnaître.
L’environnement de la liaison C=O abaisse ou augmente la fréquence d’absorption
(≅ nombre d’onde). Si un alkyle est remplacé par un hétéroatome, l’effet inductif diminue la
longueur de liaison C=O et augmente la fréquence et l’effet mésomère augmente la longueur
de la liaison C=O et diminue la fréquence.
Figure 22 : Acétone. ννννas méthyle, 2995 cm-1. ννννs méthyle 2918 cm-1. Elongation C=O normale, 1715 cm-1.
δδδδasCH3, 1422 cm-1. δδδδsCH3 1360 cm-1. Elongation et déformation C-C(=O)-C, 1213 cm-1.
4.13. LES ALDÉHYDES
Pour les aldéhydes, il y a une absorption d’intensité moyenne vers 2720 cm-1,
accompagnée d’une bande carbonyle.
Les vibrations d’élongation C=O sont situées entre 1740 et 1720 cm-1 pour les
aldéhydes aliphatiques et une substitution électronégative sur Cα augmente le nombre d’onde
(ex : acétaldéhyde 1730 cm-1 et trichloroacétaldéhyde à 1768 cm-1). Par contre, une

TP de chimie théorique et physique
S. Gillet - 28 - IR
insaturation conjuguée diminue le nombre d’onde tout comme les ponts H internes. Les
vibrations d’élongation de C-H apparaissent sous forme de deux bandes d’intensité moyenne
entre 2830 et 2695 cm-1.
Figure 23 : Octanal. Elongation C-H aldéhydique, 2715 cm-1. Elongation C=O aldéhydique, 1728 cm-1.
Déformation C-H aldéhydique, 1381 cm-1.
4.14. LES ACIDES CARBOXYLIQUES
Les vibrations d’élongation de O-H sont observables pour des solutions très diluées
dans les solvants polaires ou en phase gazeuse à 3520 cm-1 et pour les liquides, solides ou
solutions diluées dans du CCl4 > 0,01 M on observe une bande large et intense entre 3300 et
2500 cm-1, généralement centrée sur 3000 cm-1. L’élongation des liens C-H est donc
superposée à la bande du O-H.
Les vibrations d’élongation de C=O pour les acides carboxyliques sont plus intenses
que pour les cétones.
Pour les acides aliphatiques, l’absorption se situe vers 1760 cm-1 et si le dimère
possède un centre de symétrie, seul le mode d’élongation de C=O s’observe entre 1720 et
1706 cm-1. Les ponts H internes diminuent la fréquence et une insaturation provoque une
faible diminution de la fréquence.
Les vibrations d’élongation de C-O et de déformation angulaire O-H apparaissent sous
forme de deux bandes de 1320 à 1210 cm-1 pour C-O et de 1440 à 1395 cm-1 pour O-H. La
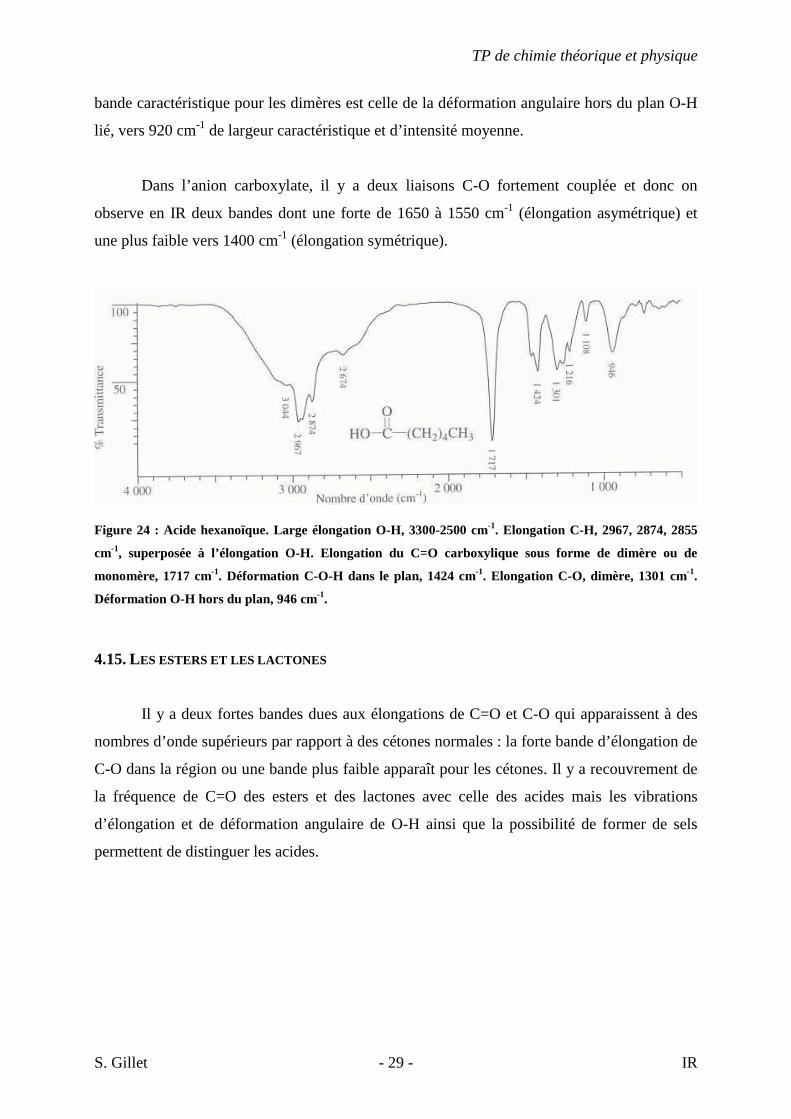
TP de chimie théorique et physique
S. Gillet - 29 - IR
bande caractéristique pour les dimères est celle de la déformation angulaire hors du plan O-H
lié, vers 920 cm-1 de largeur caractéristique et d’intensité moyenne.
Dans l’anion carboxylate, il y a deux liaisons C-O fortement couplée et donc on
observe en IR deux bandes dont une forte de 1650 à 1550 cm-1 (élongation asymétrique) et
une plus faible vers 1400 cm-1 (élongation symétrique).
Figure 24 : Acide hexanoïque. Large élongation O-H, 3300-2500 cm-1. Elongation C-H, 2967, 2874, 2855
cm-1, superposée à l’élongation O-H. Elongation du C=O carboxylique sous forme de dimère ou de
monomère, 1717 cm-1. Déformation C-O-H dans le plan, 1424 cm-1. Elongation C-O, dimère, 1301 cm-1.
Déformation O-H hors du plan, 946 cm-1.
4.15. LES ESTERS ET LES LACTONES
Il y a deux fortes bandes dues aux élongations de C=O et C-O qui apparaissent à des
nombres d’onde supérieurs par rapport à des cétones normales : la forte bande d’élongation de
C-O dans la région ou une bande plus faible apparaît pour les cétones. Il y a recouvrement de
la fréquence de C=O des esters et des lactones avec celle des acides mais les vibrations
d’élongation et de déformation angulaire de O-H ainsi que la possibilité de former de sels
permettent de distinguer les acides.

TP de chimie théorique et physique
S. Gillet - 30 - IR
Figure 25 : Acétate de phényle. Elongation C-H, 3075, 3052 cm-1. Elongation C=O, 1771 cm-1 : Cette
fréquence est supérieure à celle de l’élongation normale du C=O d’un ester du fait de la conjugaison du
phényle avec l’oxygène de l’alcool ; la conjugaison d’un groupe aryle ou d’une autre insaturation avec le
groupe carbonyle provoque le décalage de l’élongation C=O vers les basses fréquences. Elongation C∼∼∼∼C
du cycle aromatique, 1601 cm-1. δδδδasCH3, 1493 cm-1. δδδδsCH3, 1378 cm-1. Elongation de l’acétate C(=O)-O,
1223 cm-1. Elongation asymétrique O-C∼∼∼∼C aromatique, 1200 cm-1.
La vibration d’élongation C=O est observée entre 1750-1715 cm-1 pour les esters
aliphatiques saturés et entre 1730 et 1715 cm-1 pour les formates, les esters α, β-insaturés et
les benzoates. Une conjugaison en plus a peu ou pas d’effet.
S’il y a une insaturation adjacente à C-O pour des esters vinyliques et phényliques, il y
a une augmentation marquée du nombre d’onde du carbonyle et une diminution de C-O. Une
substitution en α par un halogène augmente la fréquence. Les δ-lactones saturées absorbent
dans la même région que les esters non conjugués à chaîne linéaire et une insaturation en α
diminue la fréquence (nombre d’onde). Quant à la vibration C-O, il y a deux vibrations
asymétriques couplées C-C(=O)-O et O-C-C entre 1300 et 1000 cm-1.
4.16. LES HALOGÉNURES D’ACIDE
Les vibrations d’élongation de C=O pour les halogénures d’acide conduisent à une
forte absorption qui apparaît entre 1815 et 1785 cm-1 pour les halogénures non-conjugués et à
un nombre d’onde inférieur pour les conjugués. Pour les aromatiques, on observe deux
absorptions dont une forte entre 1800-1770 cm-1 et une faible entre 1750-1735 cm-1.

TP de chimie théorique et physique
S. Gillet - 31 - IR
4.17. LES ANHYDRES D’ACIDES CARBOXYLIQUES
Les vibrations d’élongation de C=O sont observables sous forme de deux bandes
correspondant aux élongations asymétriques et symétriques de C=O. Pour les acycliques
saturés elles sont situées entre 1818-1750 cm-1, pour les acycliques conjugués entre 1775-
1720 cm-1 et pour les molécules cycliques à cinq chaînons, la fréquence est supérieure. Les
vibrations d’élongation de C-O présentent des bandes intenses résultant de l’élongation du
groupe C-C(=0)-O-C(=O)-C.
4.18. LES AMIDES
Pour les amides, il existe plusieurs bandes caractéristiques dont la « bande I » qui est
la bande d’absorption du carbonyle et dont la position dépend des ponts H ainsi que la
« bande II » qui correspond à la déformation angulaire de NH2 ou NH. Pour les amides
primaires, secondaires et quelques lactames, elle apparaît entre 1650 et 1515 cm-1. Cette
absorption implique le couplage entre la déformation angulaire de N-H et d’autres vibrations
fondamentales et nécessite une géométrie trans. Le balancement hors du plan de N-H produit
une large bande d’intensité moyenne entre 800 et 666 cm-1.
- Les vibrations d’élongation de N-H : Une solution diluée dans des solvants non-
polaires donne deux bandes d’intensité moyenne pour les amides primaires vers 3520
et 3400 cm-1 et entre 3500 et 3400 cm-1 pour les amides secondaires trans. Pour les
solides, il y a absorption vers 3350 et 3180 cm-1 pour les primaires et des bandes
multiples vers 3330 et 3060 cm-1 (dimères) pour les secondaires trans.
- Les vibrations d’élongation de C=0 (« bande I ») : pour les primaires, une bande forte
vers 1650 cm-1 (solide) ou vers 1690 cm-1 (solution diluée). Pour les secondaires
simples, une bande vers 1640 cm-1 (solide) ou vers 1680 cm-1 (solution diluée). Pour
les tertiaires, l’absorption est indépendante de l’état physique car il n’y a pas de
liaisons H possibles entre deux amides. Il y en a cependant avec le solvant, ce qui
donne une absorption entre 1680 et 1630 cm-1.
- Les vibrations de déformation angulaire de N-H (« bande II ») : Pour les amides
primaires, en solution diluée, il y a une bande étroite d’intensité égale à la moitié ou au
tiers de celle de C=O, vers 1620-1590 cm-1. Pour les pâtes ou pastilles, les vibrations
de déformation angulaire N-H sont situées entre 1655 et 1620 cm-1, sous la bande I.
TP de chimie théorique et physique
S. Gillet - 32 - IR
De plus, la nature du groupe R a peu d’effet. Pour les acycliques secondaires solides, il
y a absorption entre 1570 et 1515 cm-1 et pour les solutions diluées, entre 1550 et 1510
cm-1.
- On note également une élongation de C-N qui, pour les amides primaires, apparaît
vers 1400 cm-1. La large bande visible entre 800 et 666 cm-1 est due aux balancements
hors du plan de N-H.
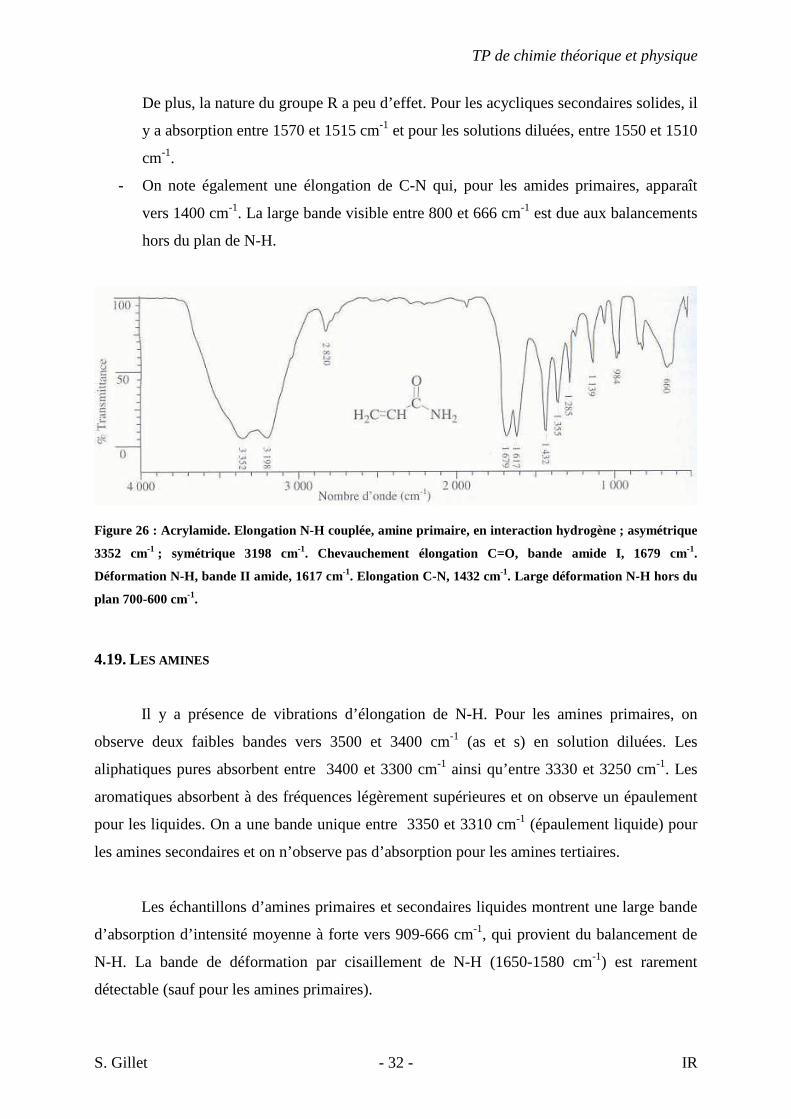
Figure 26 : Acrylamide. Elongation N-H couplée, amine primaire, en interaction hydrogène ; asymétrique
3352 cm-1 ; symétrique 3198 cm-1. Chevauchement élongation C=O, bande amide I, 1679 cm-1.
Déformation N-H, bande II amide, 1617 cm-1. Elongation C-N, 1432 cm-1. Large déformation N-H hors du
plan 700-600 cm-1.
4.19. LES AMINES
Il y a présence de vibrations d’élongation de N-H. Pour les amines primaires, on
observe deux faibles bandes vers 3500 et 3400 cm-1 (as et s) en solution diluées. Les
aliphatiques pures absorbent entre 3400 et 3300 cm-1 ainsi qu’entre 3330 et 3250 cm-1. Les
aromatiques absorbent à des fréquences légèrement supérieures et on observe un épaulement
pour les liquides. On a une bande unique entre 3350 et 3310 cm-1 (épaulement liquide) pour
les amines secondaires et on n’observe pas d’absorption pour les amines tertiaires.
Les échantillons d’amines primaires et secondaires liquides montrent une large bande
d’absorption d’intensité moyenne à forte vers 909-666 cm-1, qui provient du balancement de
N-H. La bande de déformation par cisaillement de N-H (1650-1580 cm-1) est rarement
détectable (sauf pour les amines primaires).
TP de chimie théorique et physique
S. Gillet - 33 - IR
Il y a également présence de vibrations de d’élongation de C-N. Pour les amines
primaires, secondaires et tertiaires aliphatiques, les vibrations de d’élongation de C-N
apparaissent entre 1250 et 1020 cm-1 et entre 1342 et 1266 cm-1 pour les aromatiques.
Enfin, l’élongation C-N donne des bandes d’absorption moyenne à faible entre 1250 et
1020 cm-1 chez les amines primaires, secondaires et tertiaires aliphatiques.
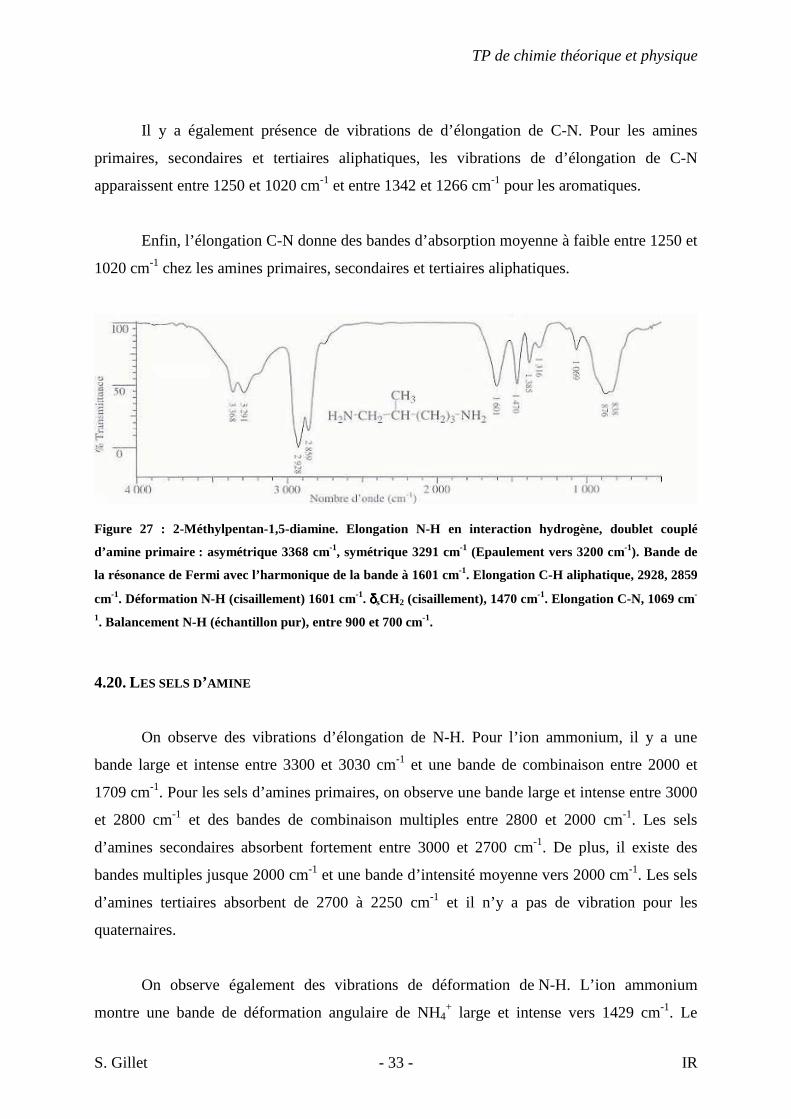
Figure 27 : 2-Méthylpentan-1,5-diamine. Elongation N-H en interaction hydrogène, doublet couplé
d’amine primaire : asymétrique 3368 cm-1, symétrique 3291 cm-1 (Epaulement vers 3200 cm-1). Bande de
la résonance de Fermi avec l’harmonique de la bande à 1601 cm-1. Elongation C-H aliphatique, 2928, 2859
cm-1. Déformation N-H (cisaillement) 1601 cm-1. δδδδsCH2 (cisaillement), 1470 cm-1. Elongation C-N, 1069 cm-
1. Balancement N-H (échantillon pur), entre 900 et 700 cm-1.
4.20. LES SELS D’AMINE
On observe des vibrations d’élongation de N-H. Pour l’ion ammonium, il y a une
bande large et intense entre 3300 et 3030 cm-1 et une bande de combinaison entre 2000 et
1709 cm-1. Pour les sels d’amines primaires, on observe une bande large et intense entre 3000
et 2800 cm-1 et des bandes de combinaison multiples entre 2800 et 2000 cm-1. Les sels
d’amines secondaires absorbent fortement entre 3000 et 2700 cm-1. De plus, il existe des
bandes multiples jusque 2000 cm-1 et une bande d’intensité moyenne vers 2000 cm-1. Les sels
d’amines tertiaires absorbent de 2700 à 2250 cm-1 et il n’y a pas de vibration pour les
quaternaires.
On observe également des vibrations de déformation de N-H. L’ion ammonium
montre une bande de déformation angulaire de NH4+ large et intense vers 1429 cm-1. Le
TP de chimie théorique et physique
S. Gillet - 34 - IR
groupe NH3+ de sel d’amine primaire absorbe de 1600 à 1575 et de 1550 à 1504 cm-1, les sels
d’amine secondaire entre 1620 et 1560 cm-1 tandis que les tertiaires ont des absorptions de
faibles intensités, sans aucune valeur pratique.
4.21. LES ACIDES AMINÉS ET LEURS SELS
Les acides aminés primaires libres sont caractérisés par les absorptions suivantes :
- Une bande d’élongation large et intense du groupe NH3+ entre 3100 et 2600 cm-1. Les
bandes de combinaisons multiples et les bandes harmoniques étendent l’absorption
jusqu’aux environs de 2000 cm-1. Cette région des harmoniques contient, en général,
une bande prédominante vers les 2222 – 2000 cm-1 attribuée à une combinaison de la
déformation asymétrique du groupe NH3+ avec l’oscillation de torsion de ce même
groupe. L’oscillation de torsion apparaît vers 500 cm-1. La bande à 2000 cm-1 est
absente si l’atome d’azote du groupe amino est substitué.
- Une faible bande de déformation asymétrique du groupe NH3+, vers 1660 – 1610 cm-1
et une bande de déformation symétrique plus intense vers 1550 et 1485 cm-1
- L’ion carboxylate –C(∼O)2- absorbe fortement entre 1600 et 1590 cm-1 et plus
faiblement vers 1400 cm-1. Ces bandes résultent, respectivement, des élongations
asymétriques et symétriques du groupe C(∼O)2.
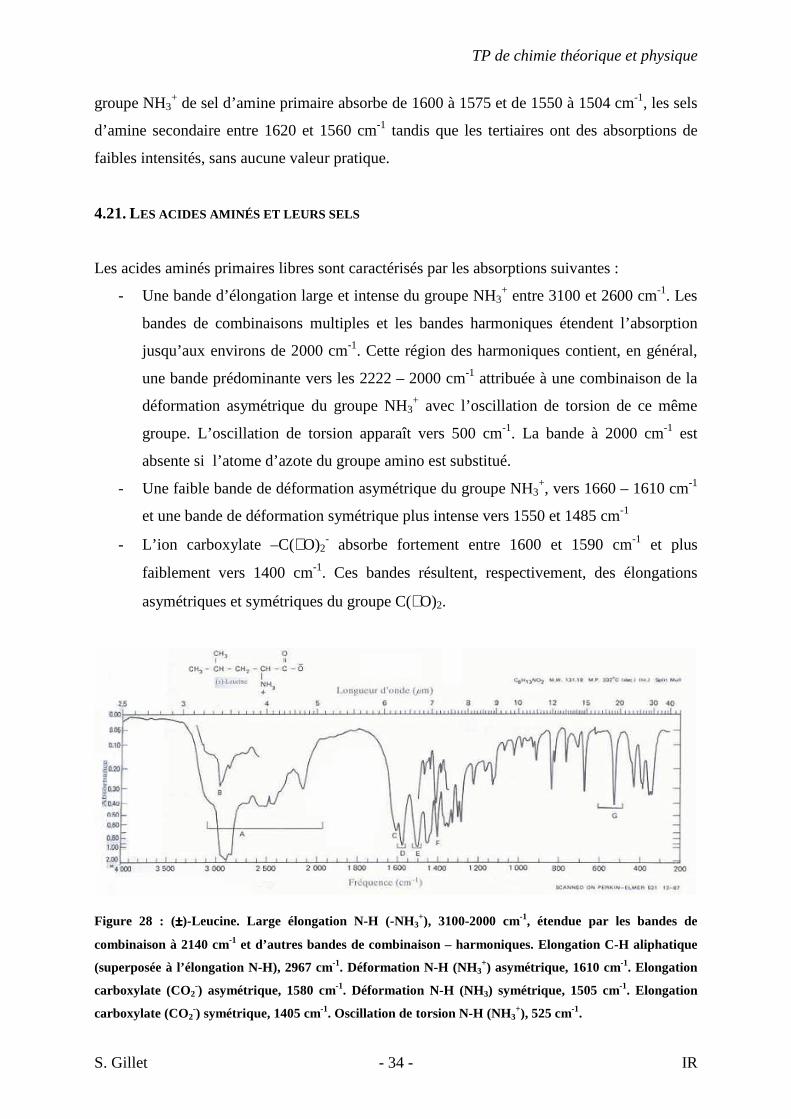
Figure 28 : (±±±±)-Leucine. Large élongation N-H (-NH3+), 3100-2000 cm-1, étendue par les bandes de
combinaison à 2140 cm-1 et d’autres bandes de combinaison – harmoniques. Elongation C-H aliphatique
(superposée à l’élongation N-H), 2967 cm-1. Déformation N-H (NH3+) asymétrique, 1610 cm-1. Elongation
carboxylate (CO2-) asymétrique, 1580 cm-1. Déformation N-H (NH3) symétrique, 1505 cm-1. Elongation
carboxylate (CO2-) symétrique, 1405 cm-1. Oscillation de torsion N-H (NH3
+), 525 cm-1.
TP de chimie théorique et physique
S. Gillet - 35 - IR
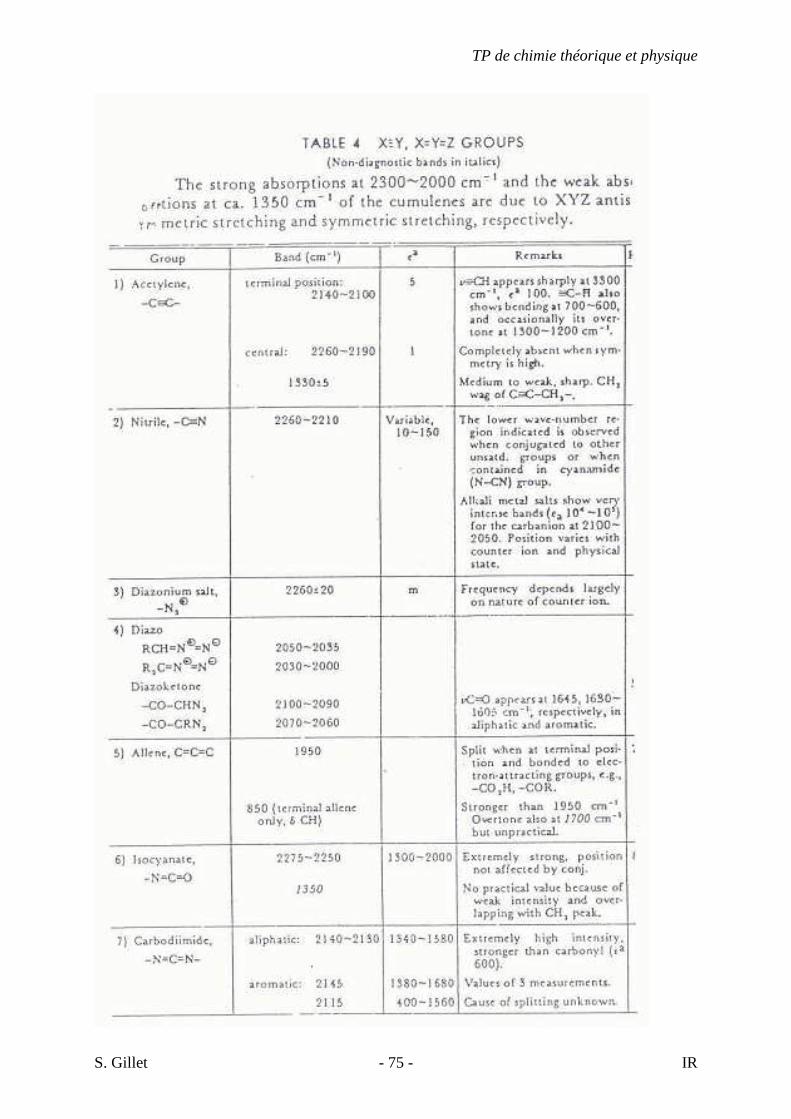
4.22. LES NITRILES
Les spectres de nitriles (R-C≡N) sont caractérisés par une absorption faible à moyenne
dans la région d’élongation des triples liaisons. Les nitriles aliphatiques absorbent vers 2260-
2240 cm-1. Les atomes électro-attracteurs tels que l’oxygène ou le chlore, liés à l’atome de
carbone en α du groupe C≡N réduisent l’intensité de l’absorption. La conjugaison, comme
dans les nitriles aromatiques, abaisse le nombre d’onde d’absorption vers 2240-2222 cm-1 et
accentue l’intensité de la bande.
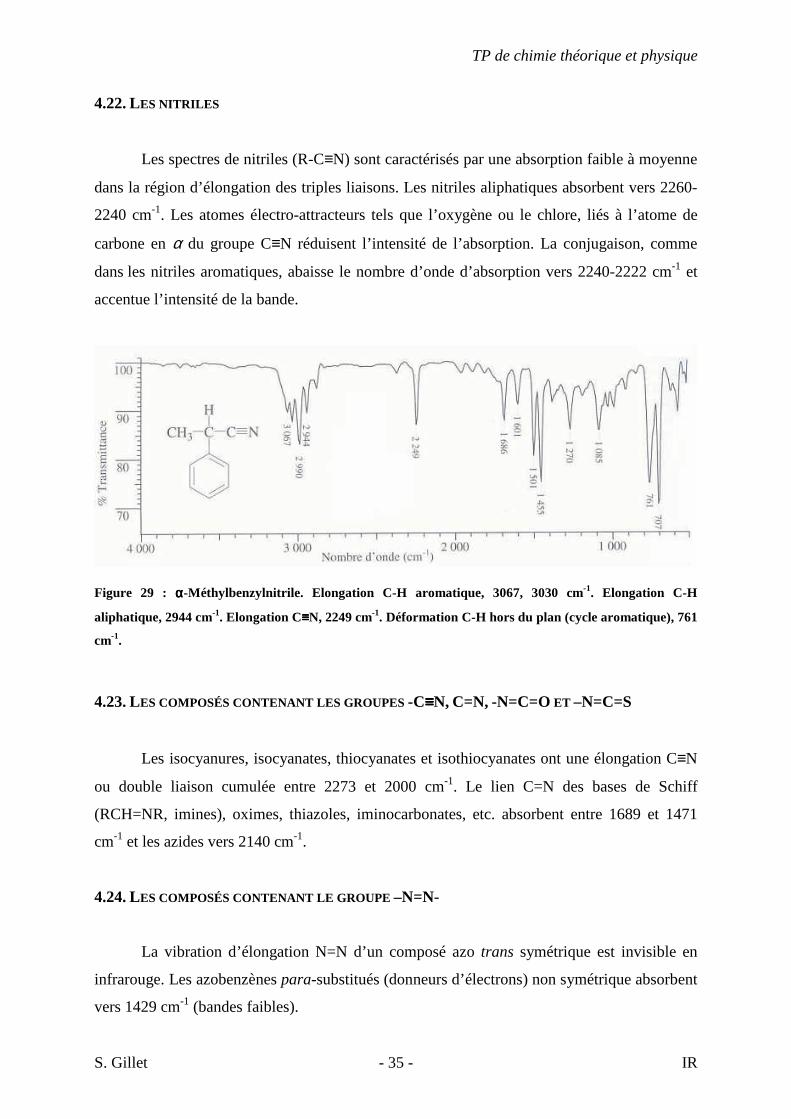
Figure 29 : αααα-Méthylbenzylnitrile. Elongation C-H aromatique, 3067, 3030 cm-1. Elongation C-H
aliphatique, 2944 cm-1. Elongation C≡≡≡≡N, 2249 cm-1. Déformation C-H hors du plan (cycle aromatique), 761
cm-1.
4.23. LES COMPOSÉS CONTENANT LES GROUPES -C≡≡≡≡N, C=N, -N=C=O ET –N=C=S
Les isocyanures, isocyanates, thiocyanates et isothiocyanates ont une élongation C≡N
ou double liaison cumulée entre 2273 et 2000 cm-1. Le lien C=N des bases de Schiff
(RCH=NR, imines), oximes, thiazoles, iminocarbonates, etc. absorbent entre 1689 et 1471
cm-1 et les azides vers 2140 cm-1.
4.24. LES COMPOSÉS CONTENANT LE GROUPE –N=N-
La vibration d’élongation N=N d’un composé azo trans symétrique est invisible en
infrarouge. Les azobenzènes para-substitués (donneurs d’électrons) non symétrique absorbent
vers 1429 cm-1 (bandes faibles).

TP de chimie théorique et physique
S. Gillet - 36 - IR
4.25. LES COMPOSÉS COVALENTS CONTENANTS DES LIENS AZOTE-OXYGÈNE
Les nitro, nitrates, nitramines contiennent un groupe NO2 dont l’absorption est due à
l’élongation asymétrique (bande intense entre 1661 et 1499 cm-1) et symétrique de NO2 (entre
1389 et 1259 cm-1). La position dépend de la substitution et de l’insaturation vicinale au
groupe NO2.
Figure 30 : Nitrobenzène. Elongation C-H aromatique, 3113, 3082 cm-1. Elongation asymétrique (ArNO2)
(N∼∼∼∼O)2, 1532 cm-1. Elongation symétrique (ArNO2) (N∼∼∼∼O)2, 1355 cm-1. Elongation C-N pour ArNO2, 853
cm-1. Les bandes à basses fréquences sont peu utiles à la détermination de la nature de la substitution du
cycle puisque ce motif d’absorption provient de l’interaction des fréquences de déformation hors du plan
de NO2 et de C-H. L’incapacité de la région des harmoniques à révéler des informations structurales est
typique des composés aromatiques portants des substituants fortement polaires.
Les vibrations les plus intéressantes sont celles d’élongation du lien N-O. Les
nitroalcanes absorbent vers 1550 et 1372 cm-1 et s’il y a conjugaison, on observera deux
bandes, de 1550 à 1500 et de 1360 à 1290 cm-1. Les groupes électronégatifs sur Cα
provoquent une augmentation de la fréquence de la bande asymétrique et une diminution de la
fréquence de la bande symétrique de NO2.
Les composés nitro aromatiques absorbent à la même fréquence que les conjugués
aliphatiques. Il y a interaction entre la déformation angulaire hors du plan de NO2 et des C-H
du cycle mais il n’y a pas d’interprétation fiable du mode de substitution. La vibration
d’élongation C-N est située vers 870 cm-1.
TP de chimie théorique et physique
S. Gillet - 37 - IR
Les nitrates ont une vibration d’élongation de N-O asymétrique forte entre 1660 et
1625 cm-1 et symétrique forte entre 1300 et 1255 cm-1. Les élongations des liaisons π du lien
N-O absorbent de 870 à 833 cm-1 et entre 763 et 690 cm-1 pour les vibrations de déformation
angulaire de NO2.
Les nitrites possèdent les absorptions les plus fortes en infrarouge. On observe deux
fortes bandes d’élongation N=O entre 1680 et 1650 cm-1 pour l’isomère trans et entre 1625 et
1610 cm-1 pour l’isomère cis. L’élongation N-O est entre 850 et 750 cm-1.
Les composés nitroso primaires et secondaires aliphatiques sont instables. Ils se
réarrangent en oximes (dimérisation). Par contre, les tertiaires et aromatiques sont stables. Ils
forment des monomères en phase gazeuse ou en solution diluée et des dimères dans les
échantillons purs. Les monomères aliphatiques absorbent entre 1585 et 1539 cm-1 et les
monomères aromatiques entre 1511 et 1495 cm-1.
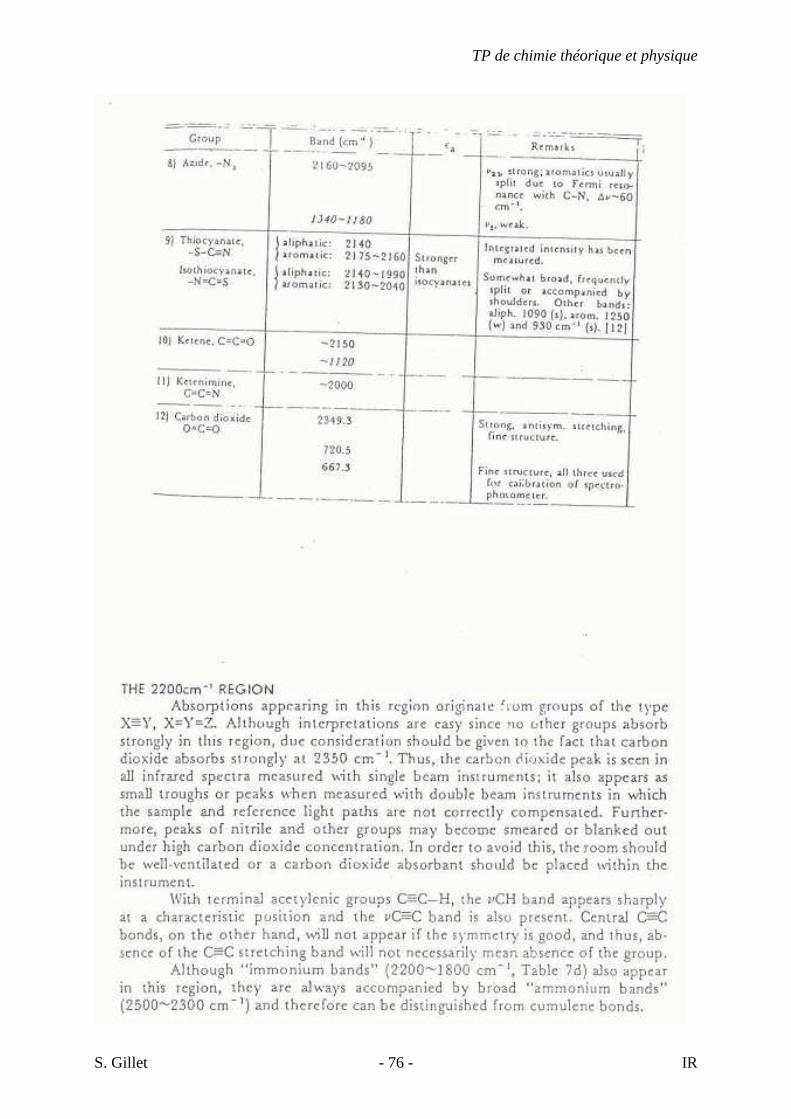
4.26. LES COMPOSÉS ORGANIQUES SOUFRÉS
- Les vibrations d’élongation S-H (mercaptans) : pour les mercaptans aliphatiques,
thiophénols, liquides ou en solution, ces vibrations sont entre 2600 et 2550 cm-1. Elles
sont faibles et ne sont pas détectées pour des solutions diluées ou en films minces.
C’est une des seules absorptions dans cette région, mais elle peut être cachée par la
forte absorption d’un carbonyle. Par ailleurs, les ponts H sont plus faibles que pour O-
H et N-H.
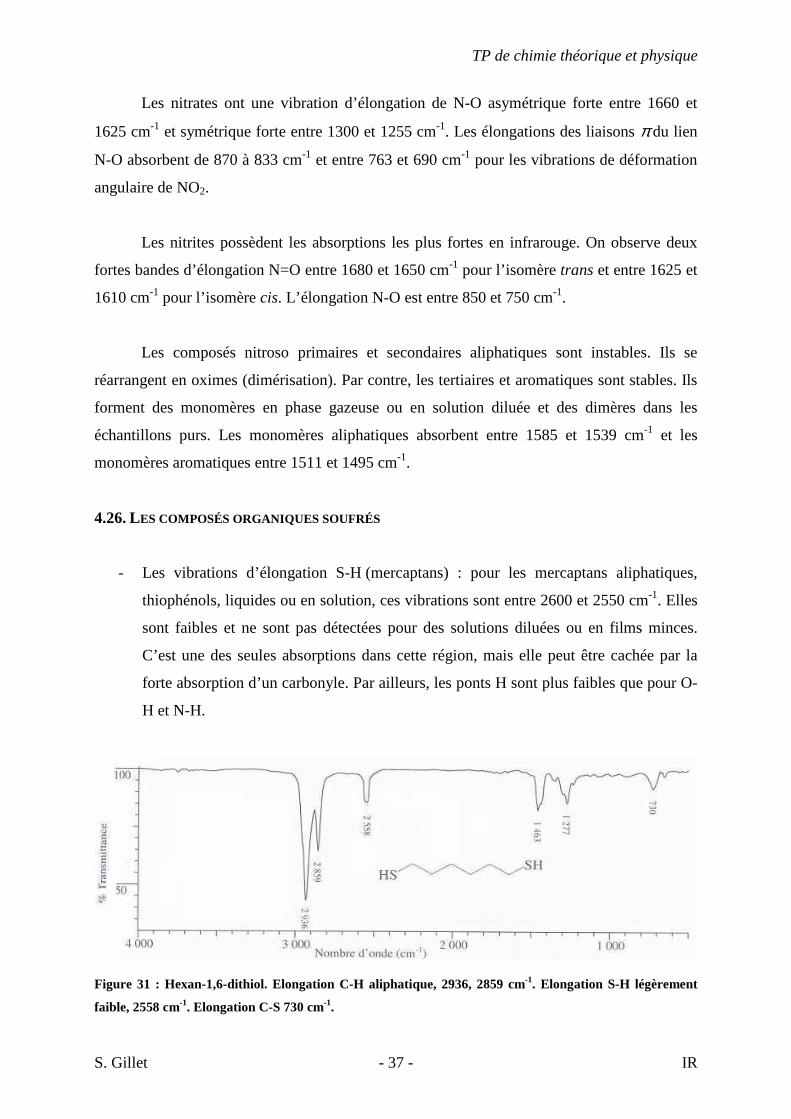
Figure 31 : Hexan-1,6-dithiol. Elongation C-H aliphatique, 2936, 2859 cm-1. Elongation S-H légèrement
faible, 2558 cm-1. Elongation C-S 730 cm-1.
TP de chimie théorique et physique
S. Gillet - 38 - IR
- Les vibrations d’élongation C-S et C=S : les vibrations d’élongation des liens C-S et
des sulfures sont situées entre 700 et 600 cm-1. Elles sont de faibles intensités et leurs
position est variable et donc présente peu d’intérêt. Pour les disulfures, ces vibrations
sont très faibles et situées entre 500 et 400 cm-1. La liaison C=S est plus faible que la
C=O et donc les vibrations apparaissent à des fréquences plus basses et sont plus
sensibles aux effets de couplages. L’identification est donc plus difficile et incertaine.
L’absorption des vibrations d’élongation des liens C-S pour les thiocarbonyles
apparaît entre 1250 et 1020 cm-1, dans la même région que les élongations de C-O et
C-N. Des interactions considérables peuvent apparaître entre ces vibrations au sein
d’une même molécule.
- Les vibrations d’élongation de S=O : pour les sulfoxydes, les aryles et alkyles liquides
ou en solution absorbent entre 1070 et 1030 cm-1 et une conjugaison provoque de
faibles variations de la fréquence. Les diallyles absorbent à 1047 cm-1. Pour les
sulfones, il y a l’élongation de SO2. On observe de fortes bandes entre 1350 et 1300
cm-1 et entre 1160 et 1120 cm-1. Les ponts H donnent une absorption entre 1300 et
1125 cm-1. Les chlorures de sulfonyles absorbent fortement de 1410 à 1380 cm-1 et de
1204 à 1177 cm-1. Pour les sulfonamides, il y a une forte absorption de 1370 à 1335 et
de 1170 à 1155 cm-1. Pour les sulfonates, les sulfates et les acides sulfoniques, il existe
des élongations asymétriques et symétriques et des bandes situées entre 1230 et 1120
cm-1 sont dues à l’hydratation.
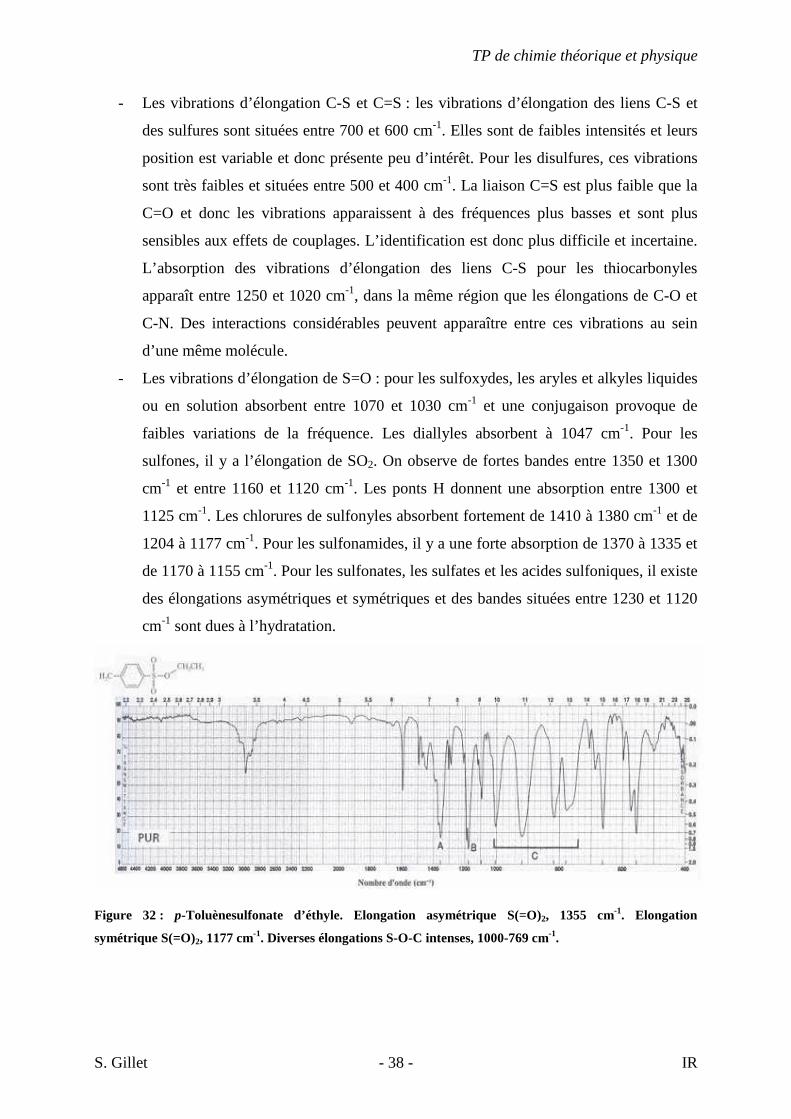
Figure 32 : p-Toluènesulfonate d’éthyle. Elongation asymétrique S(=O)2, 1355 cm-1. Elongation
symétrique S(=O)2, 1177 cm-1. Diverses élongations S-O-C intenses, 1000-769 cm-1.

TP de chimie théorique et physique
S. Gillet - 39 - IR
4.27. LES COMPOSÉS ORGANIQUES HALOGÉNÉS
L’absorption des vibrations d’élongation C-Hal est forte. Pour une molécule
aliphatique possédant une liaison C-Cl, l’absorption est située entre 850 et 550 cm-1 et si il y a
plusieurs Cl au même C, on a une bande plus intense (CCl4 : 797 cm-1). Pour les bromés, il y a
absorption entre 690 et 515 cm-1, entre 600 et 500 cm-1 pour les iodés et pour les liaisons C-F
entre 1400 et 730 cm-1. L’absorption des vibrations d’élongation C-Hal pour les
chlorobenzènes est entre 1096 et 1089 cm-1. Sa position dépend de la substitution. Pour les
fluorures d’aryles, entre 1250 et 110 cm-1 et pour un cycle benzénique monofluoré, on
observe une bande étroite et intense vers 1230 cm-1. Le balancement hors du plan CH2 est
applicable pour CH2X et situé entre 1300 et 1150 cm-1.
4.28. LES SILICONES
L’élongation de Si-H est située vers 2200 cm-1 et la déformation angulaire de Si-H de
950 à 800 cm-1. Si un groupe électronégatif est lié au Si, la fréquence augmente. Il existe
également des vibrations SiO-H et Si-O. Ces vibrations sont dans la même région que les
alcools, c'est-à-dire de 3700 à 3200 cm-1. Les bandes de Si-O sont intenses et situées entre
1110 et 830 cm-1 (importance des ponts H).
4.29. LES COMPOSÉS HÉTÉROATOMIQUES
On observe les mêmes modes de vibration que ceux observés pour les aromatiques :
- L’élongation de C-H : pyridines, pyrazines, pyrroles, furanes et thiophènes : entre
3077 et 3003 cm-1.
- L’élongation N-H est située entre 3500 et 3220 cm-1. Pour les pyroles et indoles, en
solution diluée dans les solvants non-polaires, on observe une bande étroite vers 3495
cm-1 et une bande élargie vers 3400 cm-1 pour les solutions concentrées.
- Les vibrations d’extension du cycle (élongation) absorbent entre 1600 et 1300 cm-1.
Elles correspondent à l’élongation et à la contraction de toutes les liaisons du cycle.
L’apparence et l’intensité relative des bandes dépendent du mode de substitution et de
la nature des substituants.

TP de chimie théorique et physique
S. Gillet - 40 - IR
- Les déformations angulaires hors du plan de C-H : L’apparence de ce type
d’absorption est déterminée par le nombre d’atomes d’H se déformant en phase.

TP de chimie théorique et physique
S. Gillet - 41 - IR
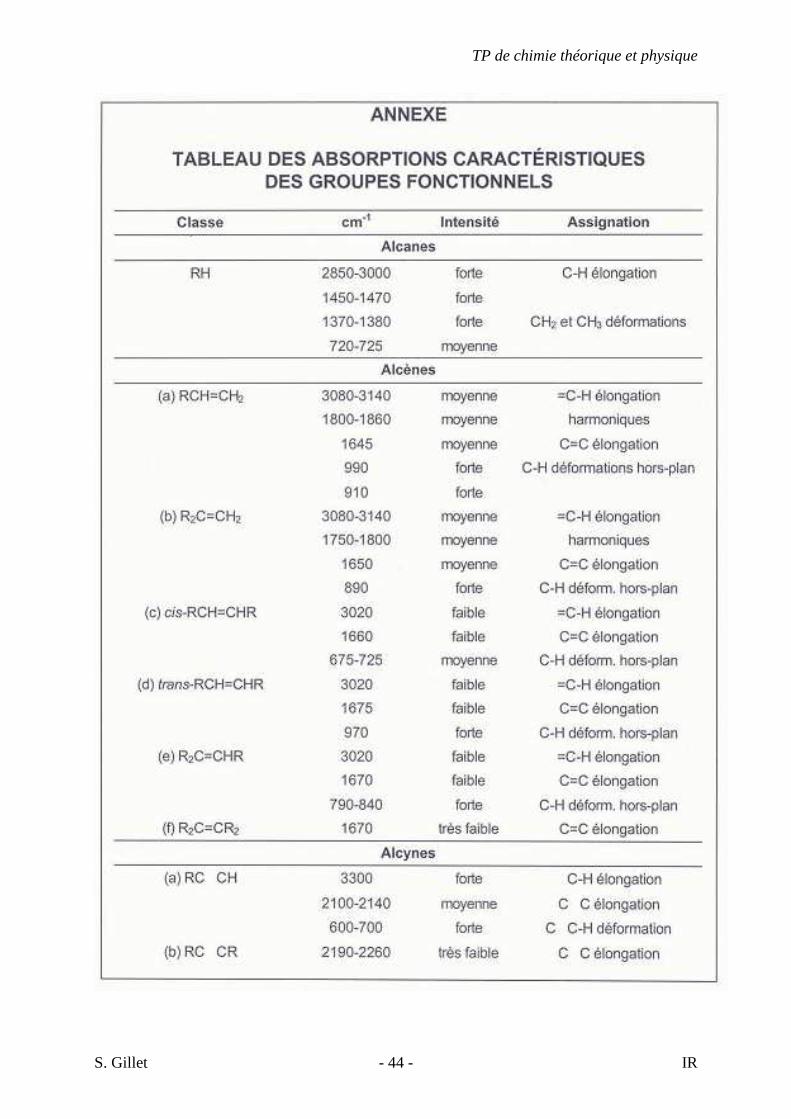
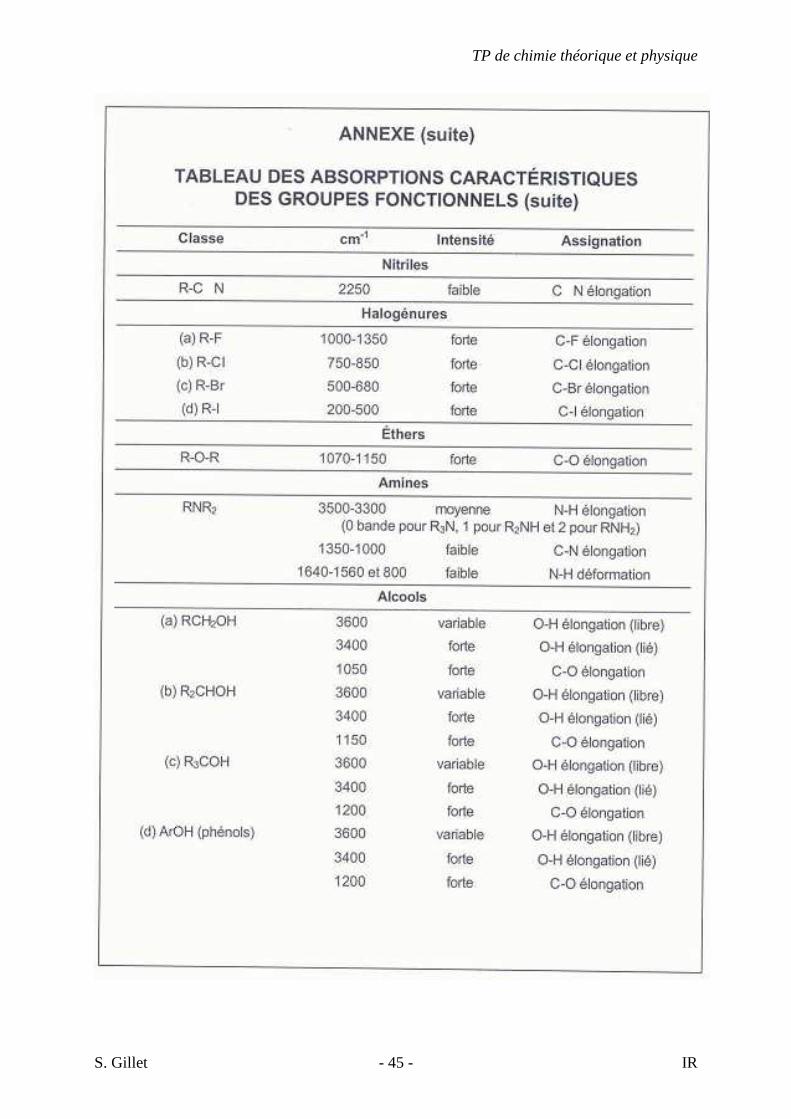
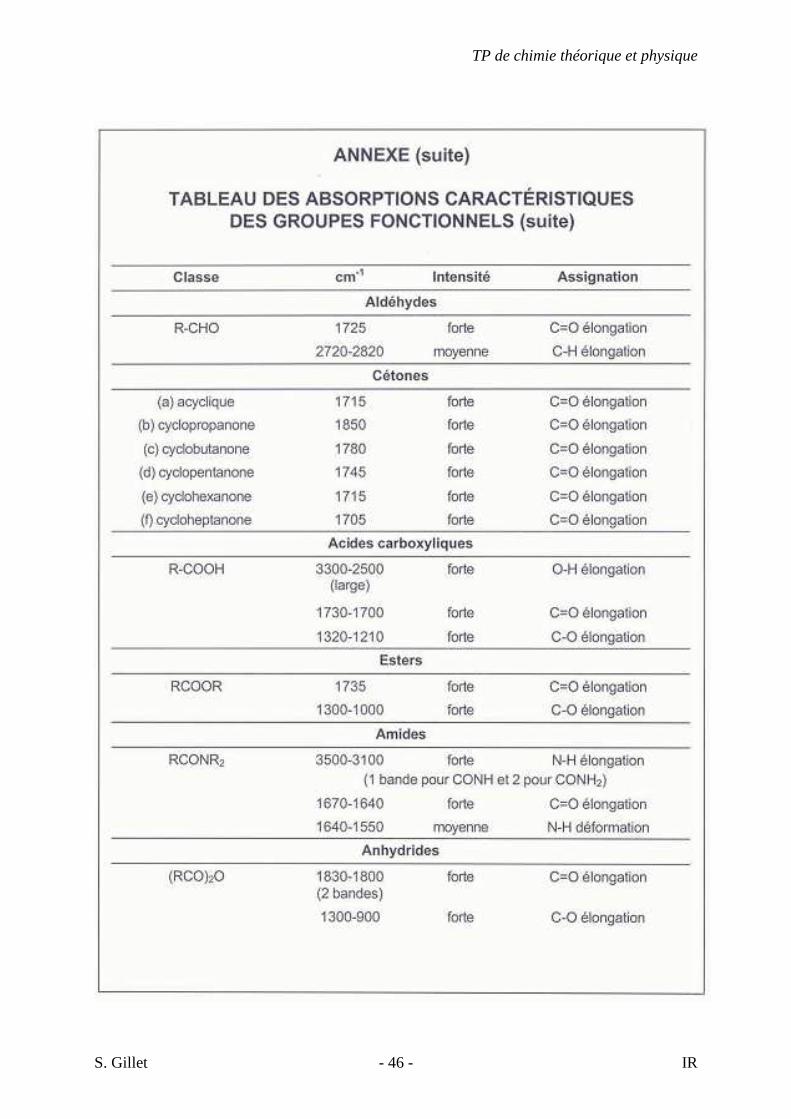
5. EXERCICES
5.1. DÉMARCHE

TP de chimie théorique et physique
S. Gillet - 42 - IR

TP de chimie théorique et physique
S. Gillet - 43 - IR
TP de chimie théorique et physique
S. Gillet - 44 - IR
TP de chimie théorique et physique
S. Gillet - 45 - IR
TP de chimie théorique et physique
S. Gillet - 46 - IR
TP de chimie théorique et physique
S. Gillet - 47 - IR
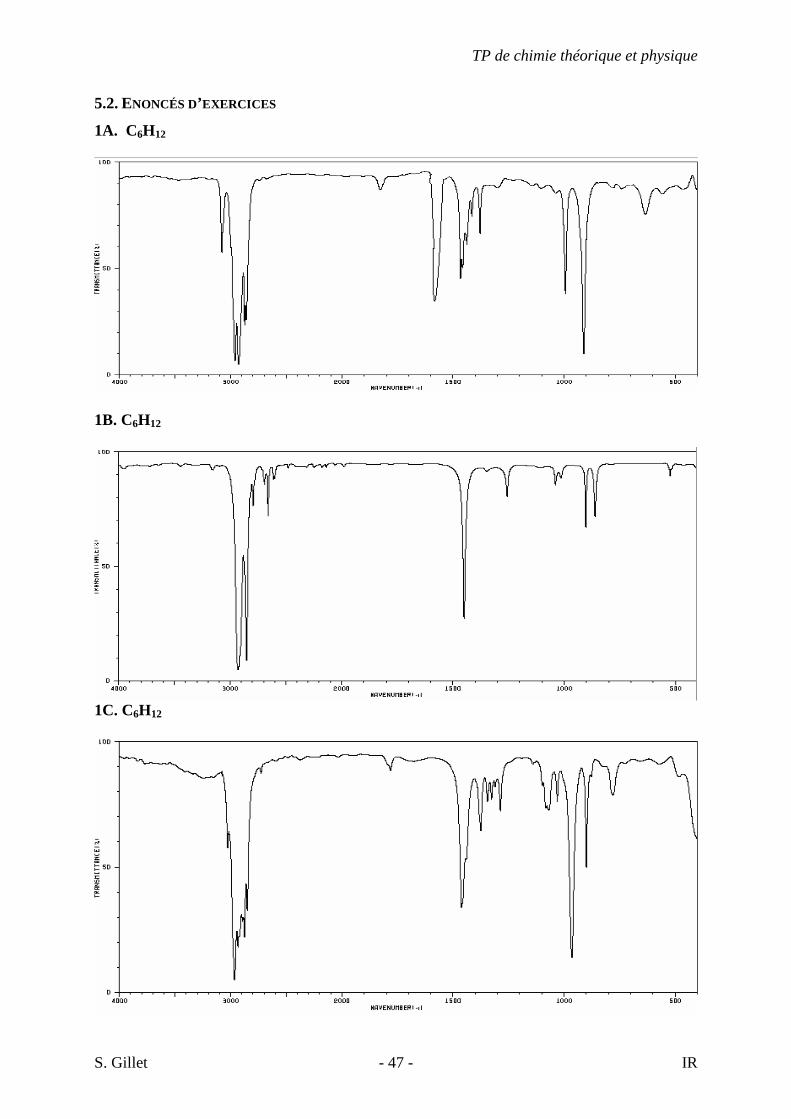
5.2. ENONCÉS D’EXERCICES
1A. C6H12
1B. C6H12
1C. C6H12
TP de chimie théorique et physique
S. Gillet - 48 - IR
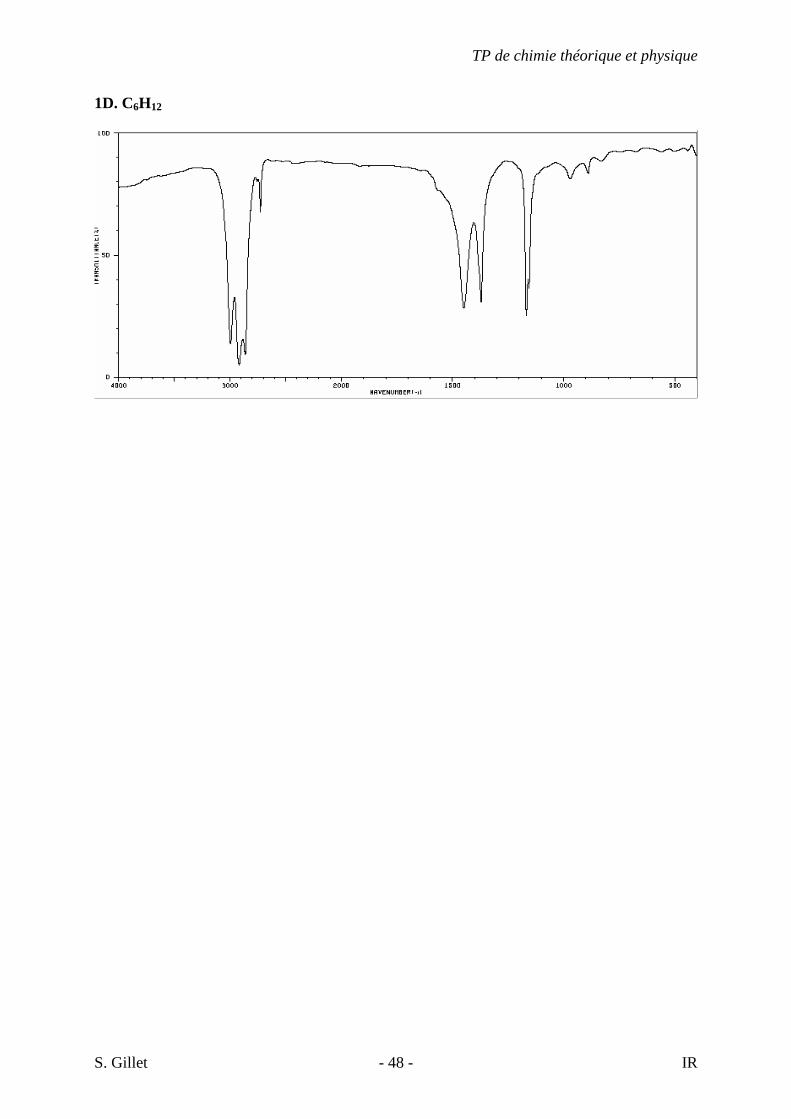
1D. C6H12
TP de chimie théorique et physique
S. Gillet - 49 - IR
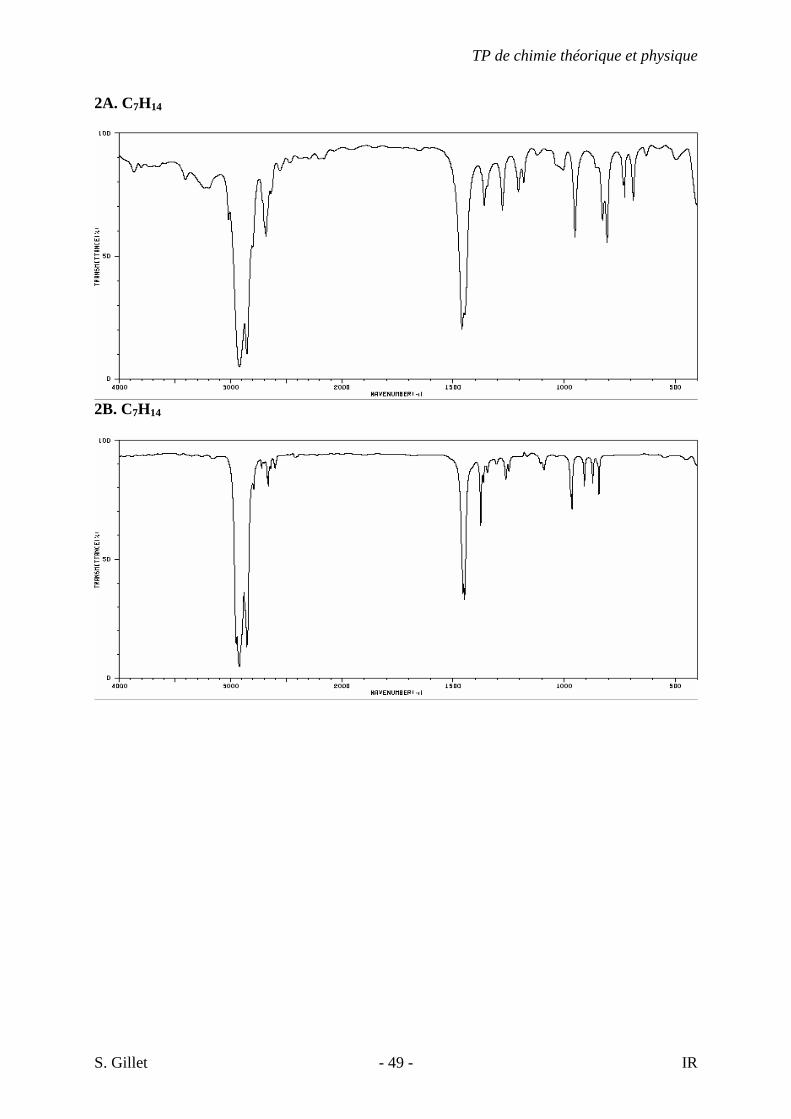
2A. C7H14
2B. C7H14
TP de chimie théorique et physique
S. Gillet - 50 - IR
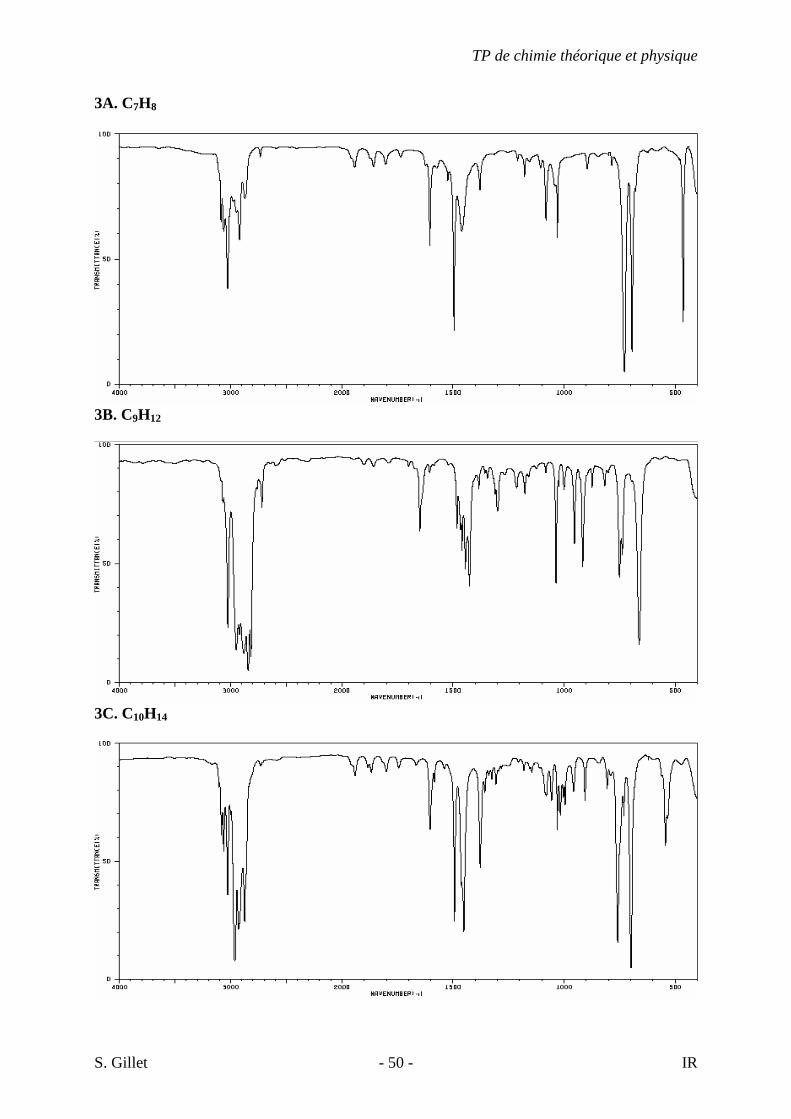
3A. C7H8
3B. C9H12
3C. C10H14
TP de chimie théorique et physique
S. Gillet - 51 - IR
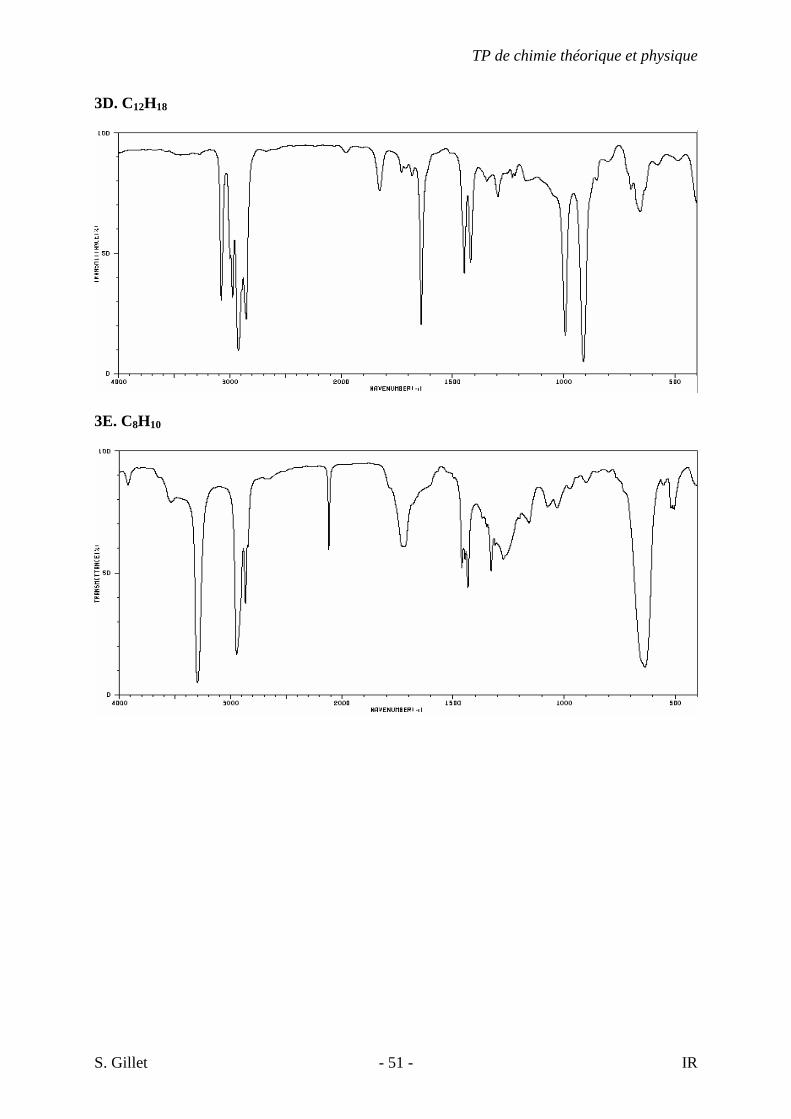
3D. C12H18
3E. C8H10
TP de chimie théorique et physique
S. Gillet - 52 - IR
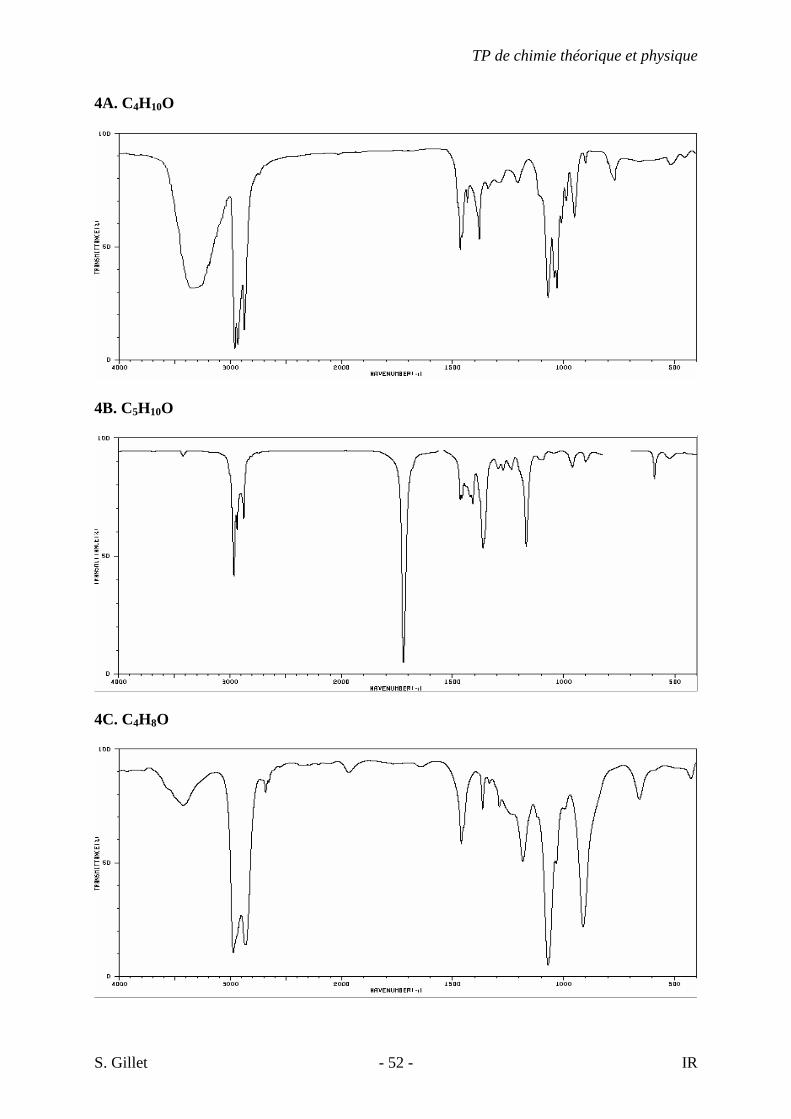
4A. C4H10O
4B. C5H10O
4C. C4H8O
TP de chimie théorique et physique
S. Gillet - 53 - IR
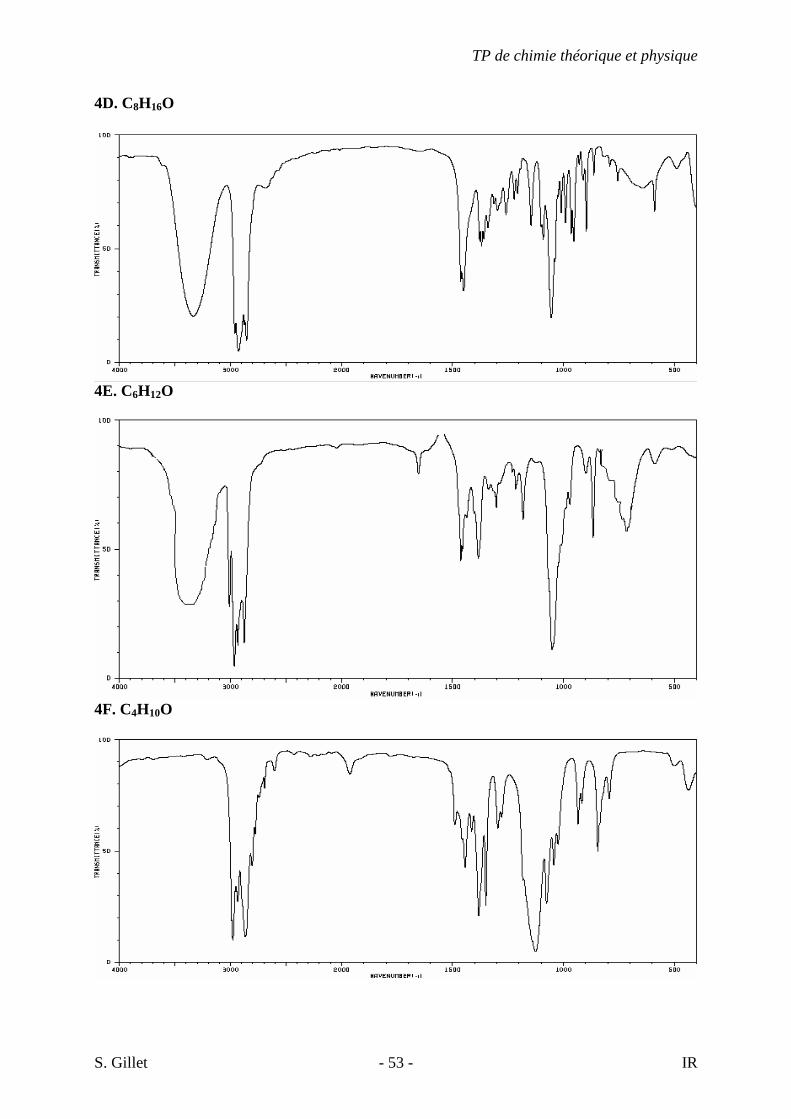
4D. C8H16O
4E. C6H12O
4F. C4H10O
TP de chimie théorique et physique
S. Gillet - 54 - IR
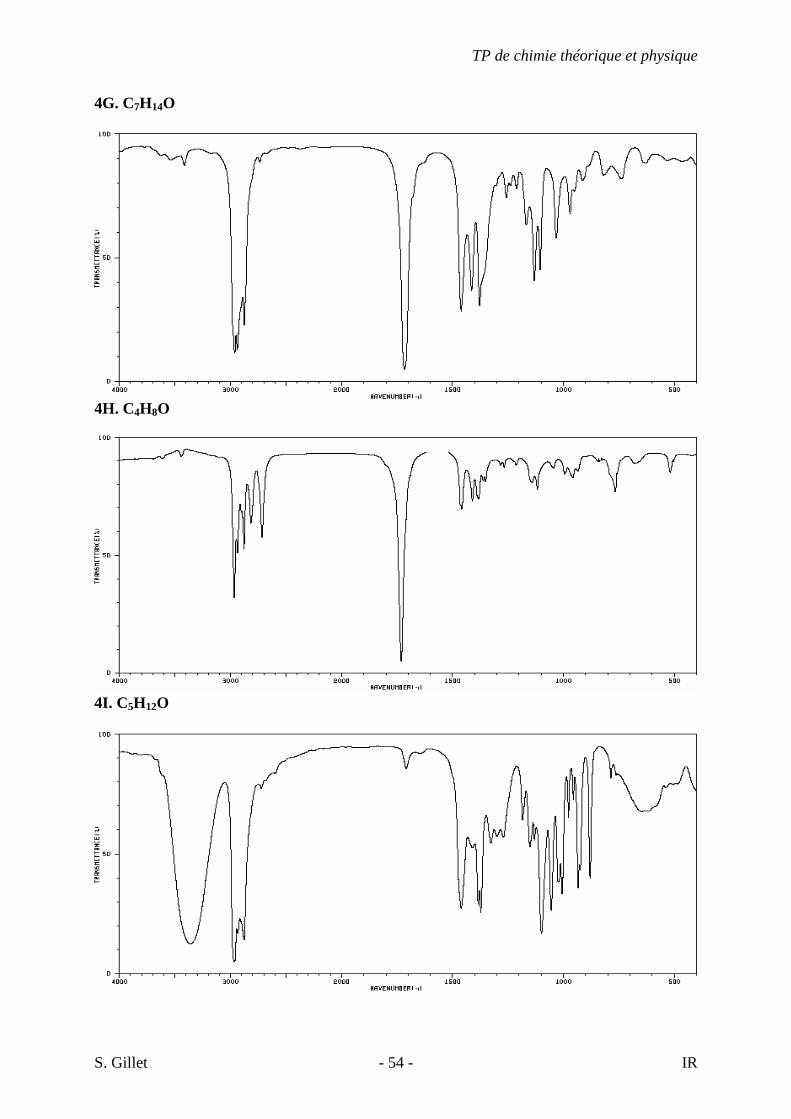
4G. C7H14O
4H. C4H8O
4I. C5H12O
TP de chimie théorique et physique
S. Gillet - 55 - IR
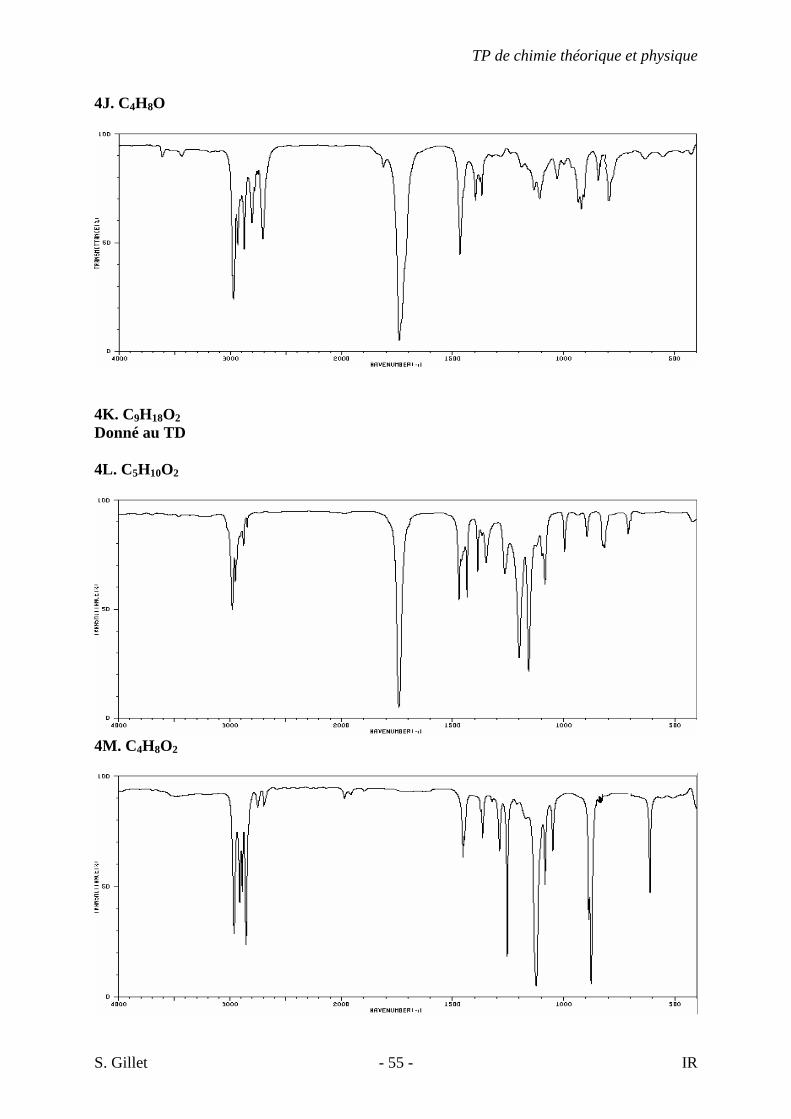
4J. C4H8O
4K. C9H18O2 Donné au TD 4L. C5H10O2
4M. C4H8O2
TP de chimie théorique et physique
S. Gillet - 56 - IR
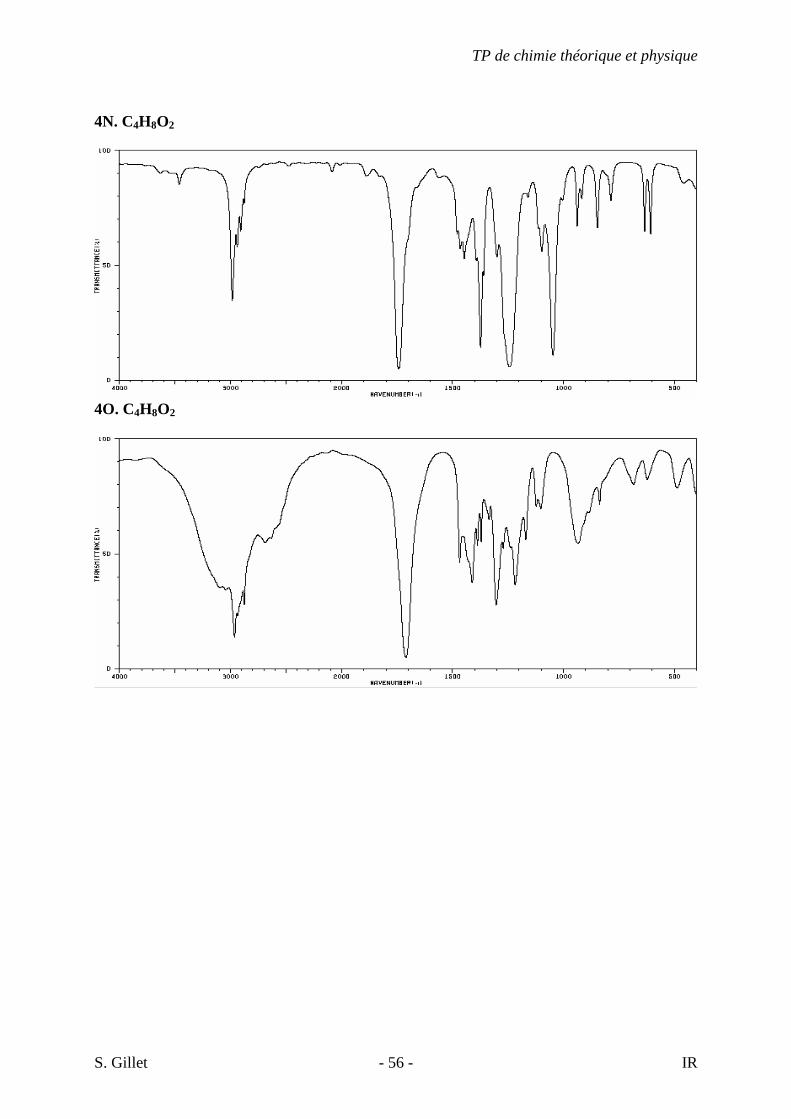
4N. C4H8O2
4O. C4H8O2
TP de chimie théorique et physique
S. Gillet - 57 - IR
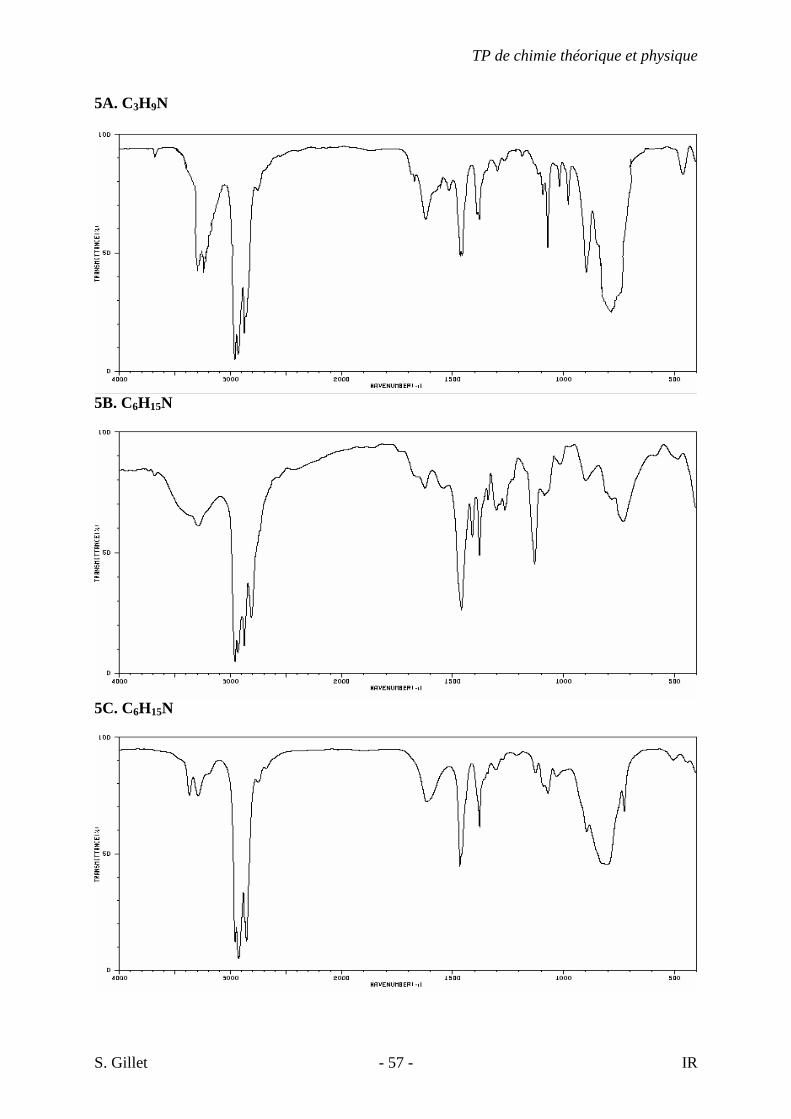
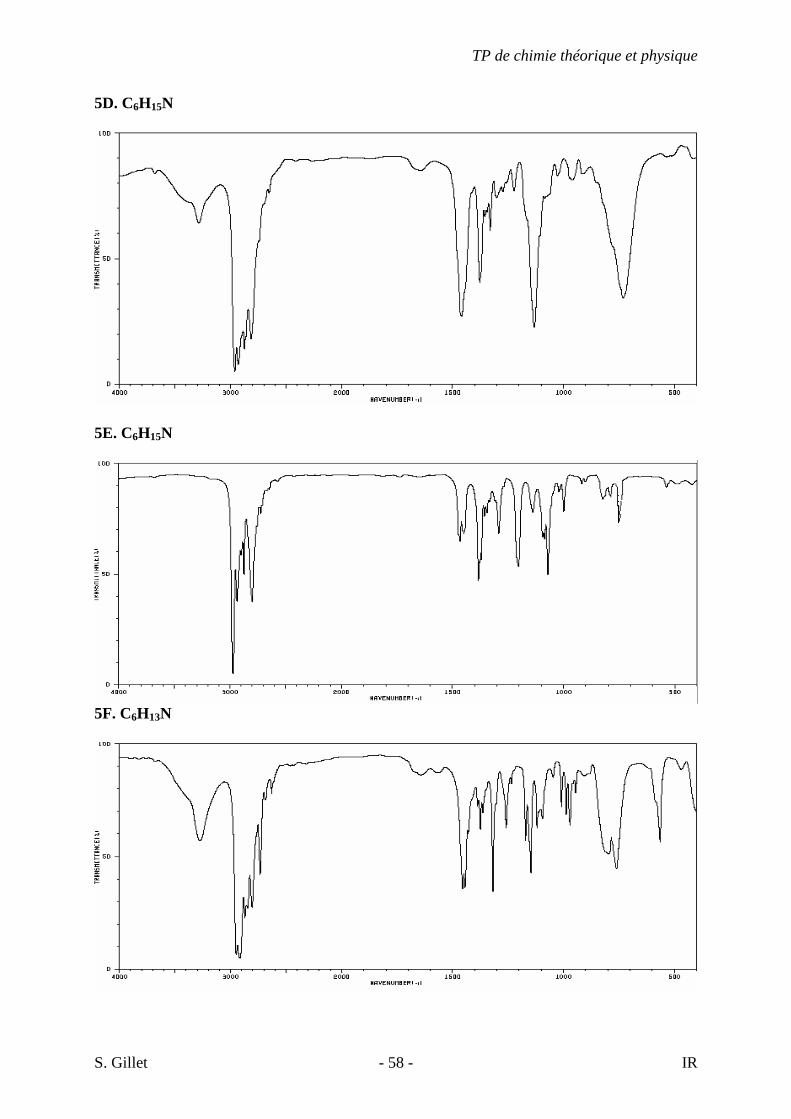
5A. C3H9N
5B. C6H15N
5C. C6H15N
TP de chimie théorique et physique
S. Gillet - 58 - IR
5D. C6H15N
5E. C6H15N
5F. C6H13N
TP de chimie théorique et physique
S. Gillet - 59 - IR
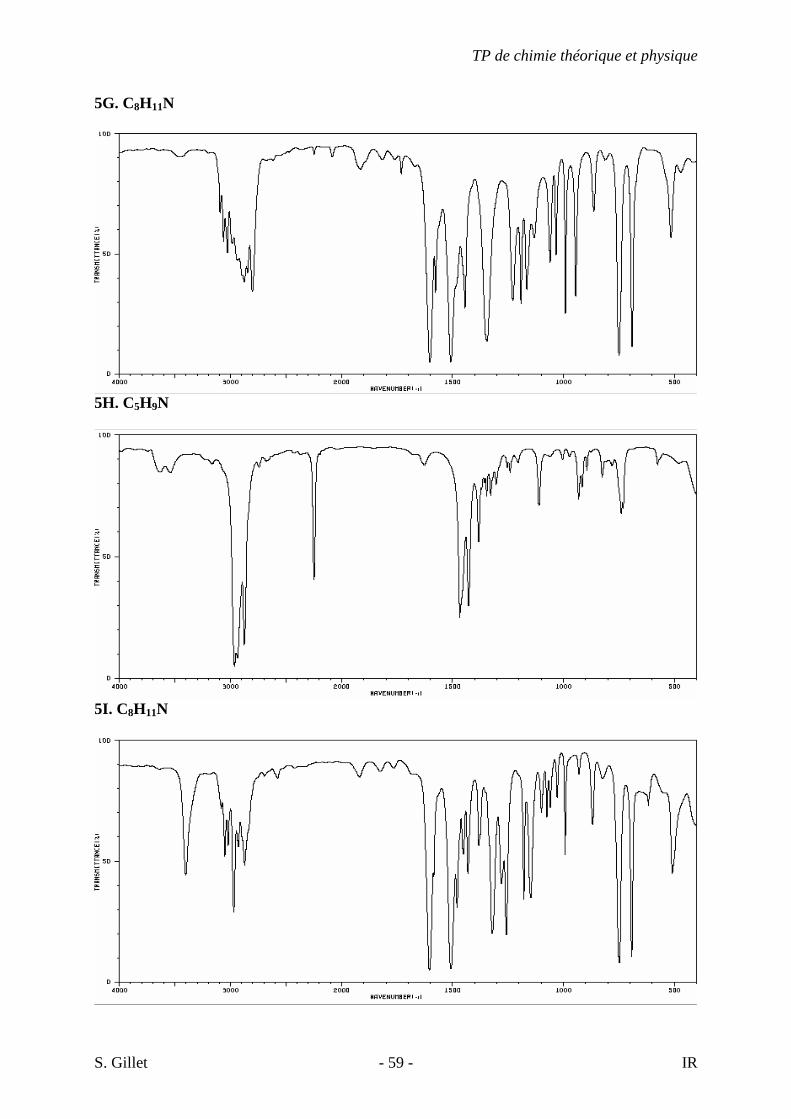
5G. C8H11N
5H. C5H9N
5I. C8H11N
TP de chimie théorique et physique
S. Gillet - 60 - IR
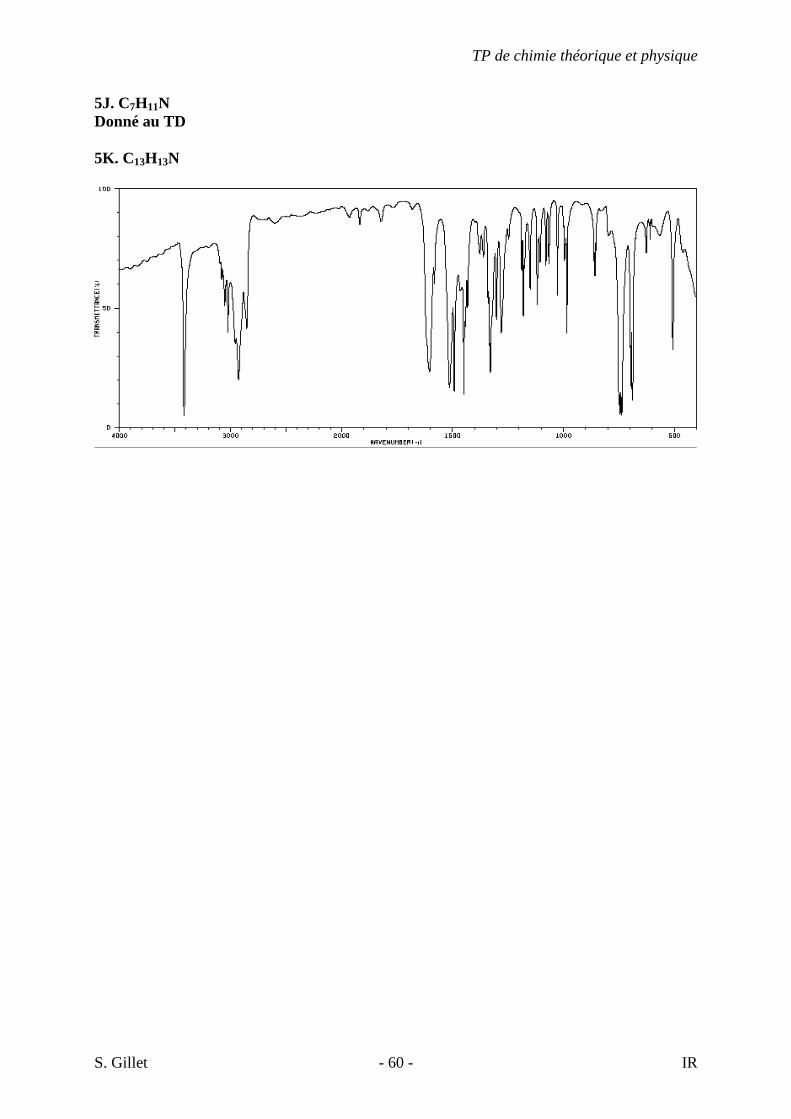
5J. C7H11N Donné au TD 5K. C13H13N
TP de chimie théorique et physique
S. Gillet - 61 - IR
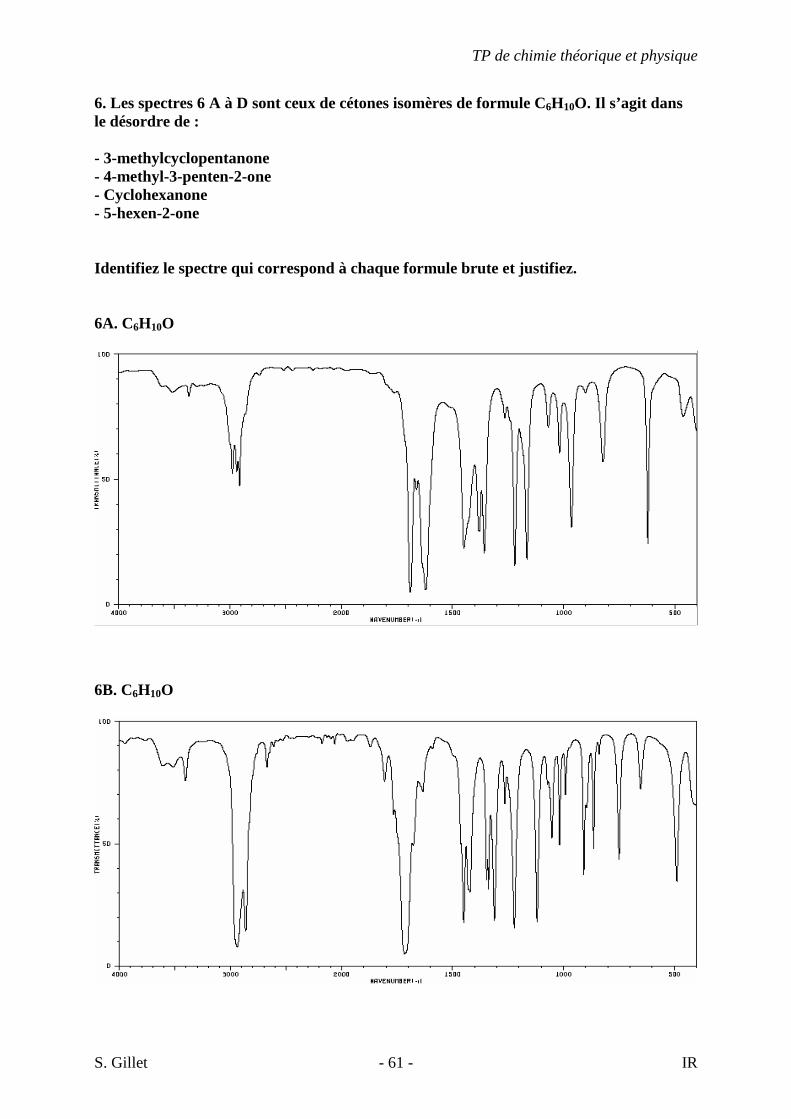
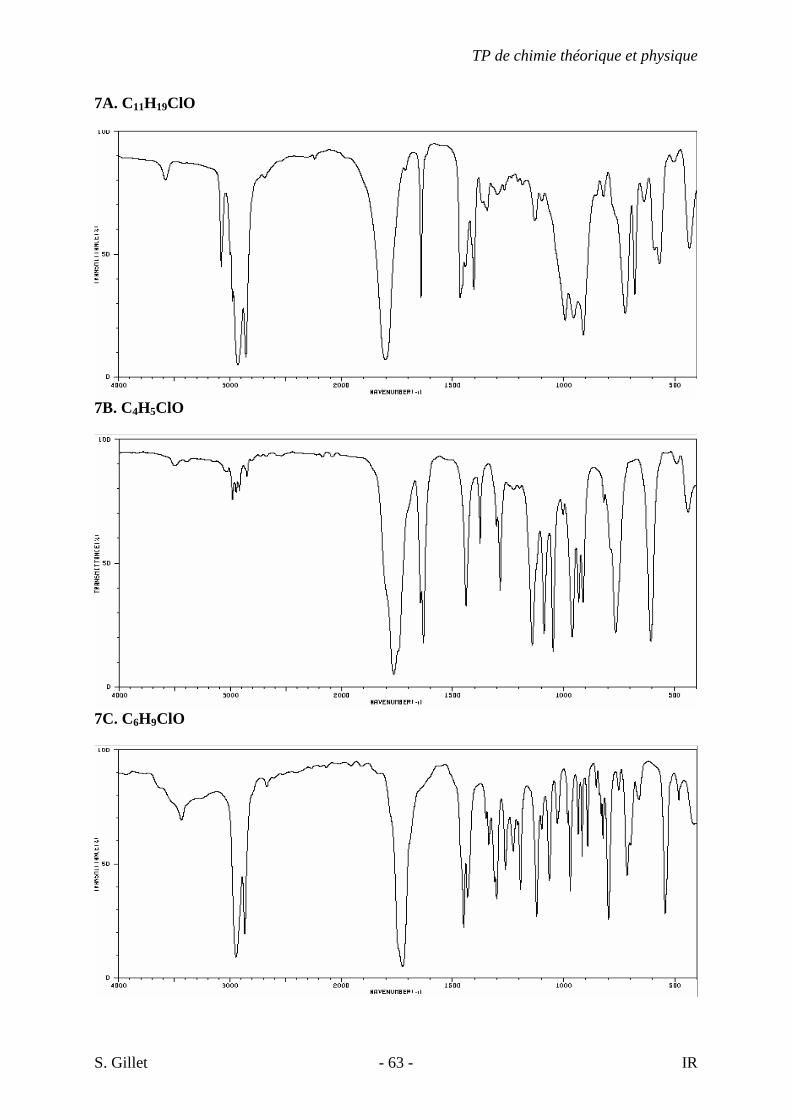
6. Les spectres 6 A à D sont ceux de cétones isomères de formule C6H10O. Il s’agit dans le désordre de : - 3-methylcyclopentanone - 4-methyl-3-penten-2-one - Cyclohexanone - 5-hexen-2-one Identifiez le spectre qui correspond à chaque formule brute et justifiez. 6A. C6H10O
6B. C6H10O
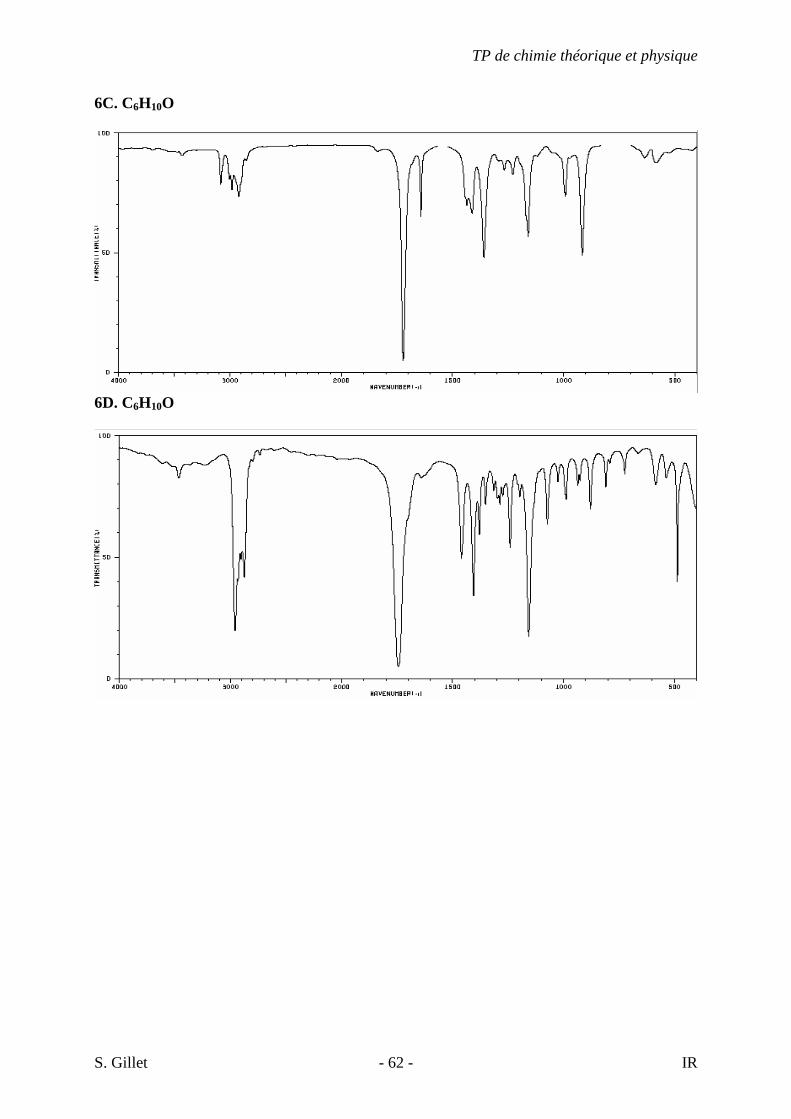
TP de chimie théorique et physique
S. Gillet - 62 - IR
6C. C6H10O
6D. C6H10O
TP de chimie théorique et physique
S. Gillet - 63 - IR
7A. C11H19ClO
7B. C4H5ClO
7C. C6H9ClO
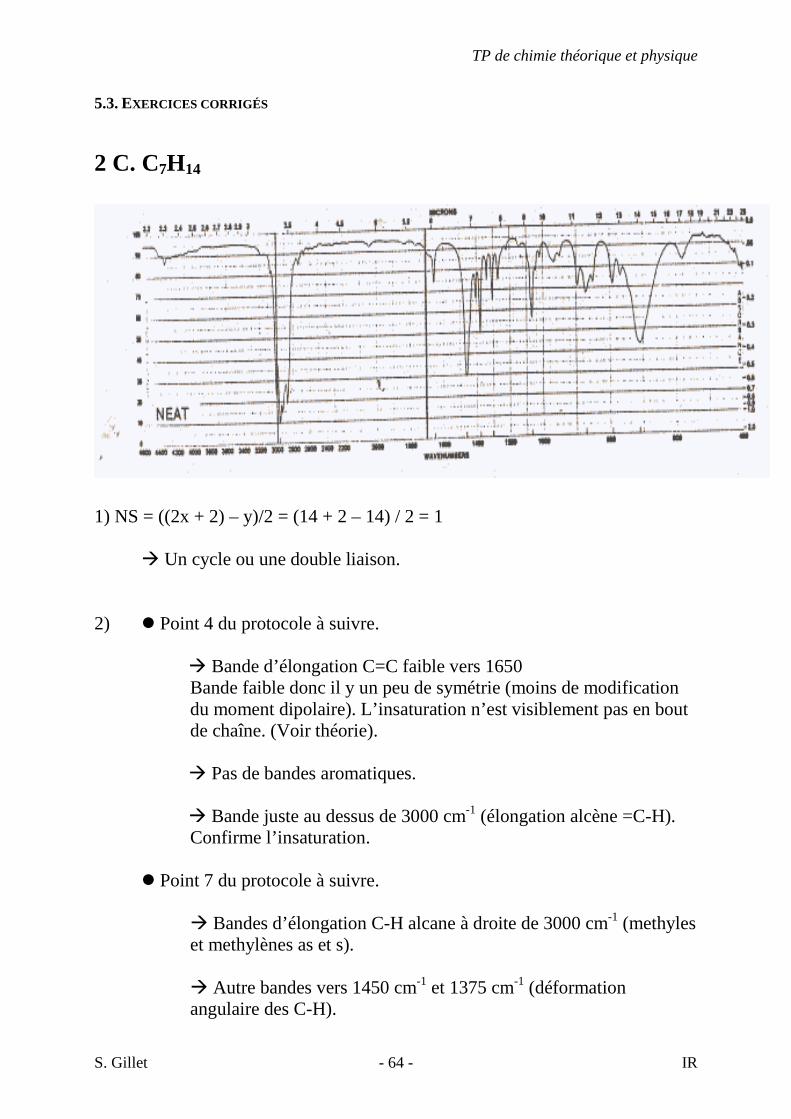
TP de chimie théorique et physique
S. Gillet - 64 - IR
5.3. EXERCICES CORRIGÉS
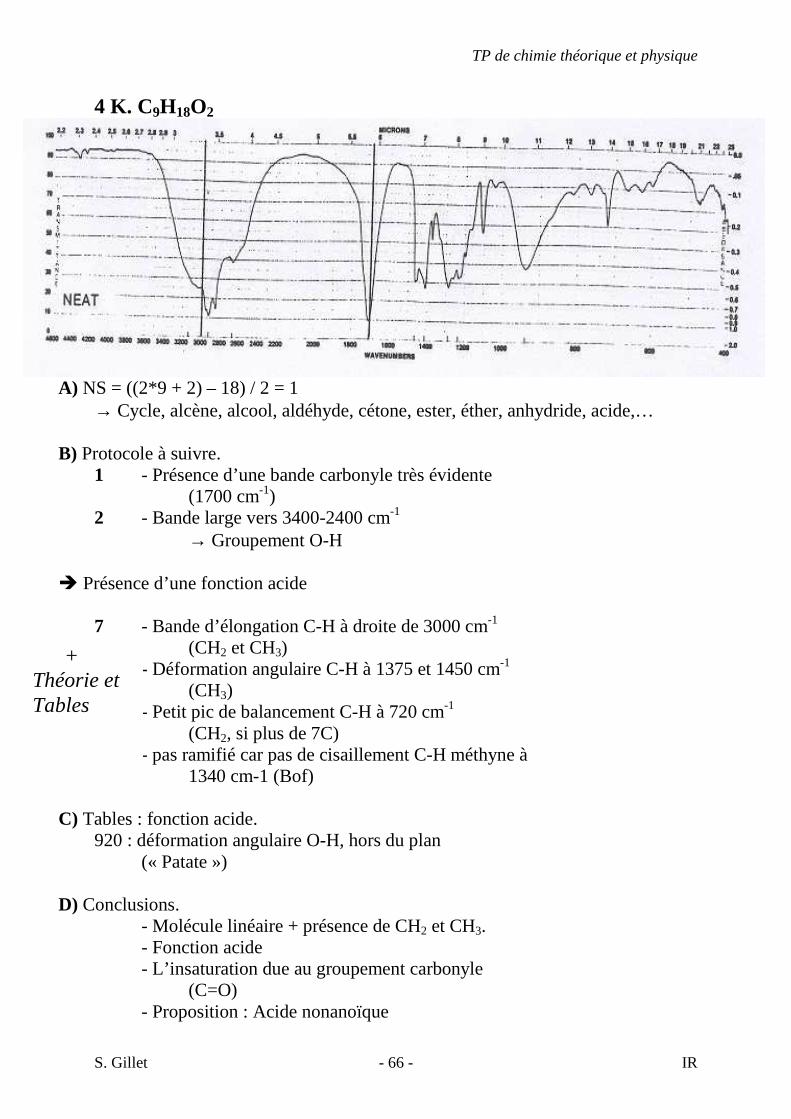
2 C. C7H14
1) NS = ((2x + 2) – y)/2 = (14 + 2 – 14) / 2 = 1 � Un cycle ou une double liaison. 2) � Point 4 du protocole à suivre. � Bande d’élongation C=C faible vers 1650
Bande faible donc il y un peu de symétrie (moins de modification du moment dipolaire). L’insaturation n’est visiblement pas en bout de chaîne. (Voir théorie). � Pas de bandes aromatiques. � Bande juste au dessus de 3000 cm-1 (élongation alcène =C-H). Confirme l’insaturation.
� Point 7 du protocole à suivre. � Bandes d’élongation C-H alcane à droite de 3000 cm-1 (methyles
et methylènes as et s). � Autre bandes vers 1450 cm-1 et 1375 cm-1 (déformation
angulaire des C-H).

TP de chimie théorique et physique
S. Gillet - 65 - IR
3) Annexes p 26. � trans RCH=CHR pas de bande forte vers 970 (déformation C-H
hors plan). � cis RCH=CHR correspond assez bien. 4) Conclusions. � Alcène cis. � Double liaison centrale-symétrie. � Proposition cis hept-3-ène.
TP de chimie théorique et physique
S. Gillet - 66 - IR
4 K. C9H18O2
A) NS = ((2*9 + 2) – 18) / 2 = 1
→ Cycle, alcène, alcool, aldéhyde, cétone, ester, éther, anhydride, acide,… B) Protocole à suivre.
1 - Présence d’une bande carbonyle très évidente (1700 cm-1)
2 - Bande large vers 3400-2400 cm-1 → Groupement O-H
� Présence d’une fonction acide
7 - Bande d’élongation C-H à droite de 3000 cm-1 (CH2 et CH3)
- Déformation angulaire C-H à 1375 et 1450 cm-1 (CH3)
- Petit pic de balancement C-H à 720 cm-1 (CH2, si plus de 7C)
- pas ramifié car pas de cisaillement C-H méthyne à 1340 cm-1 (Bof)
C) Tables : fonction acide. 920 : déformation angulaire O-H, hors du plan
(« Patate »)
D) Conclusions. - Molécule linéaire + présence de CH2 et CH3. - Fonction acide - L’insaturation due au groupement carbonyle
(C=O) - Proposition : Acide nonanoïque
+ Théorie et Tables
TP de chimie théorique et physique
S. Gillet - 67 - IR
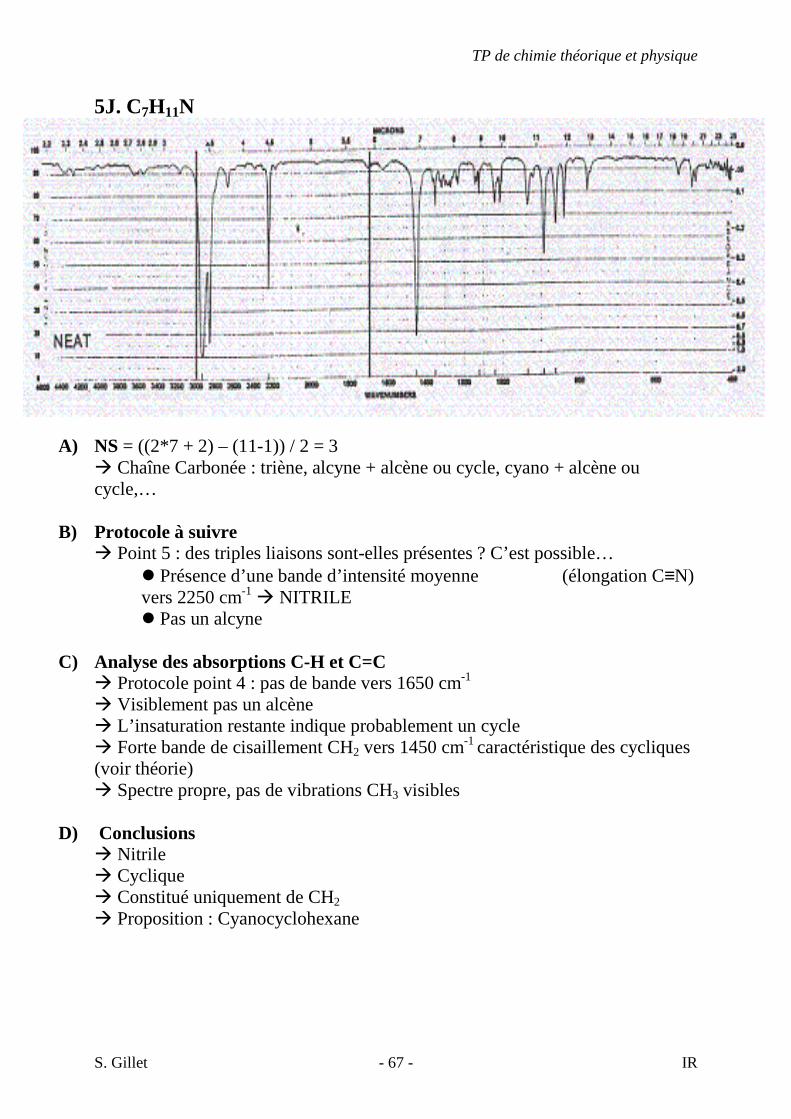
5J. C7H11N
A) NS = ((2*7 + 2) – (11-1)) / 2 = 3 � Chaîne Carbonée : triène, alcyne + alcène ou cycle, cyano + alcène ou cycle,…
B) Protocole à suivre
� Point 5 : des triples liaisons sont-elles présentes ? C’est possible… � Présence d’une bande d’intensité moyenne (élongation C≡N) vers 2250 cm-1 � NITRILE � Pas un alcyne
C) Analyse des absorptions C-H et C=C
� Protocole point 4 : pas de bande vers 1650 cm-1 � Visiblement pas un alcène � L’insaturation restante indique probablement un cycle � Forte bande de cisaillement CH2 vers 1450 cm-1 caractéristique des cycliques (voir théorie) � Spectre propre, pas de vibrations CH3 visibles
D) Conclusions � Nitrile � Cyclique � Constitué uniquement de CH2
� Proposition : Cyanocyclohexane
TP de chimie théorique et physique
S. Gillet - 68 - IR
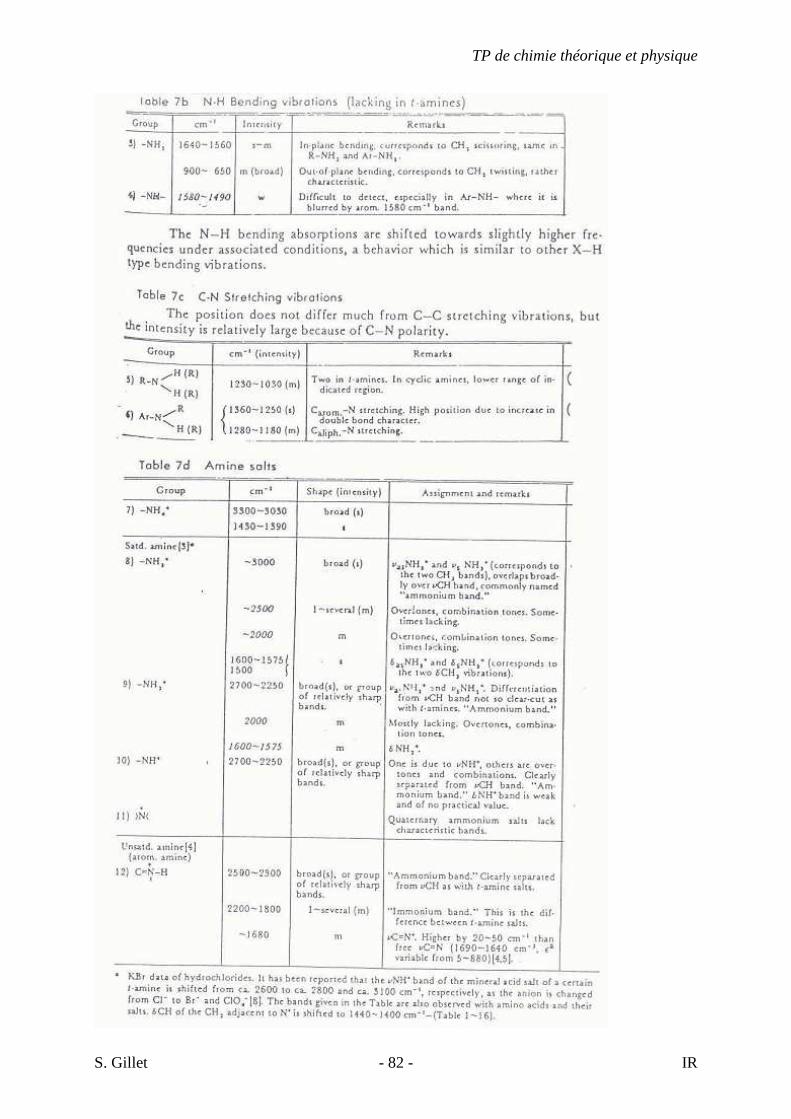
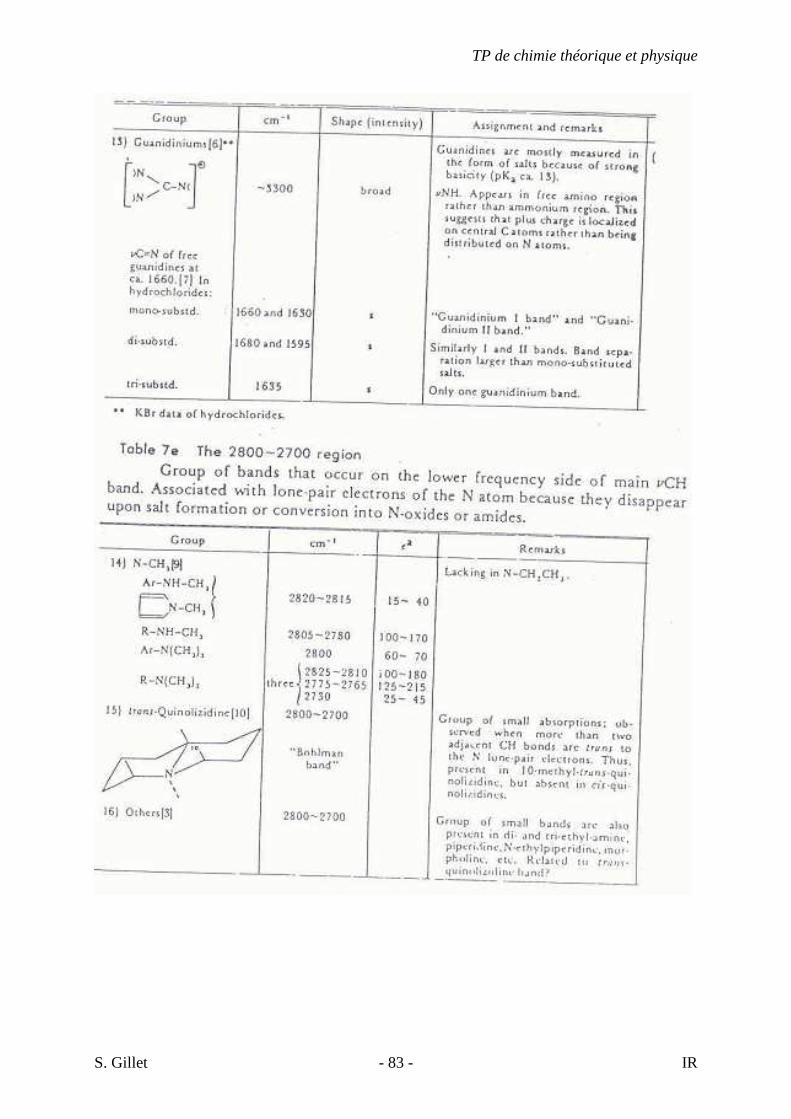
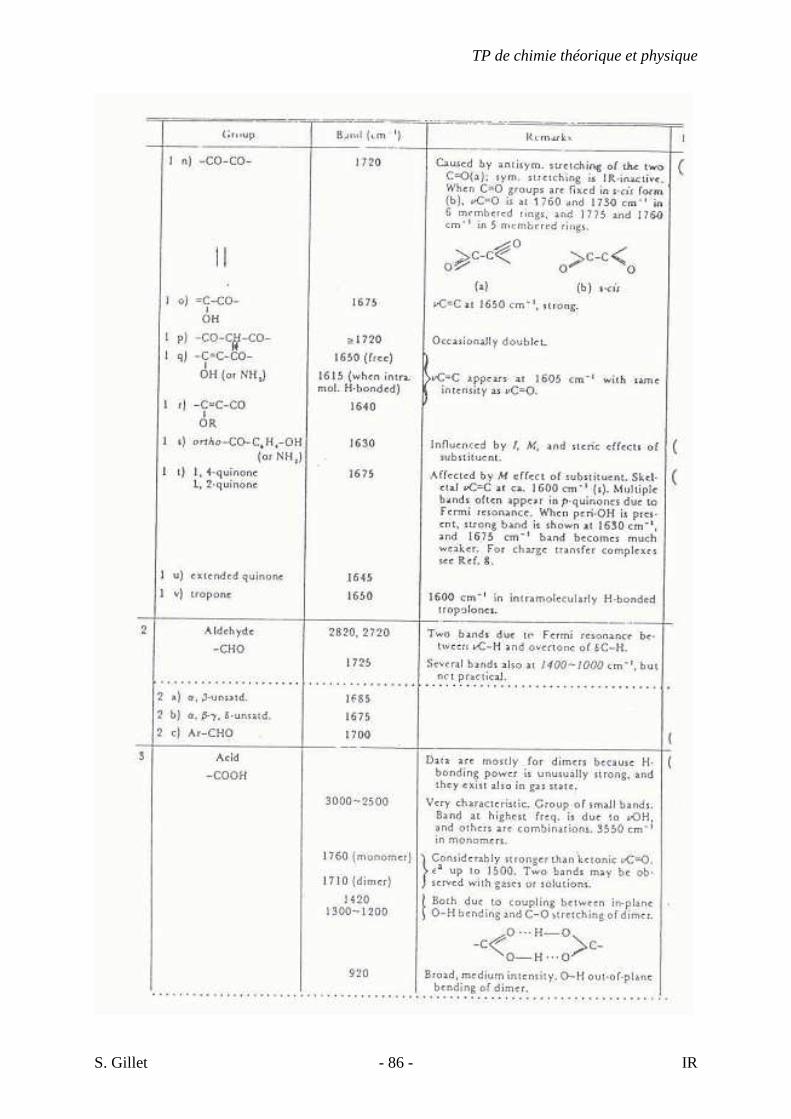
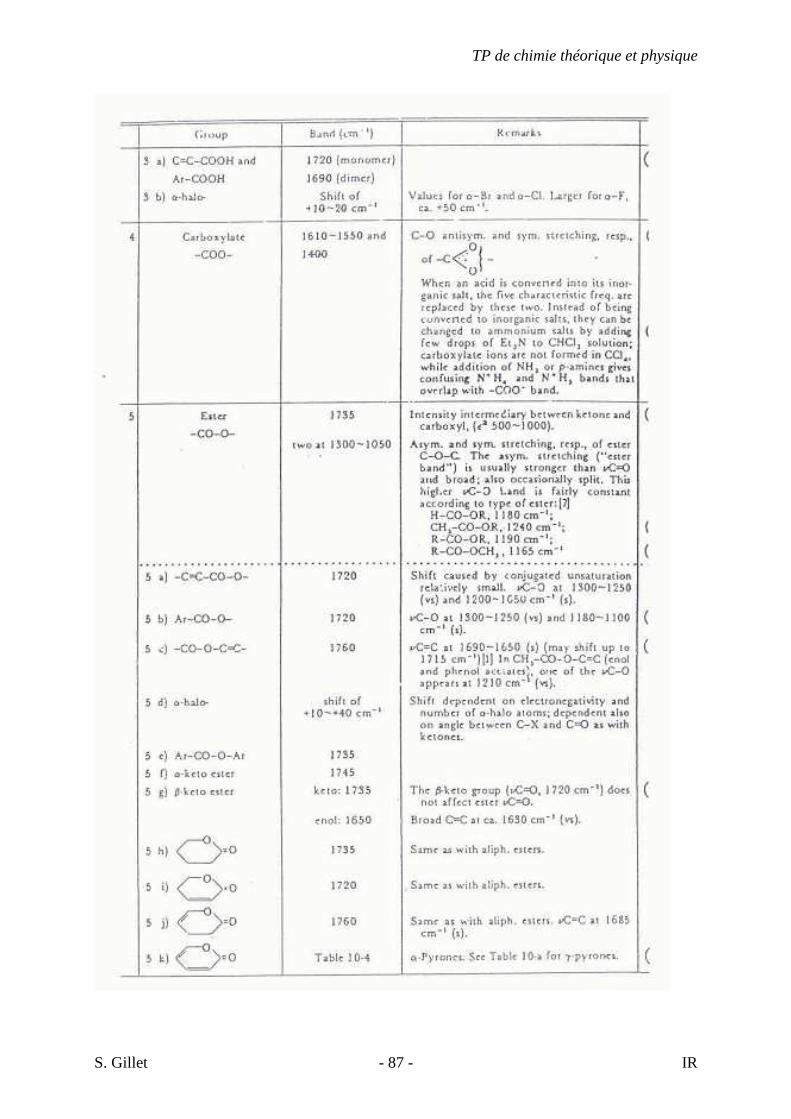
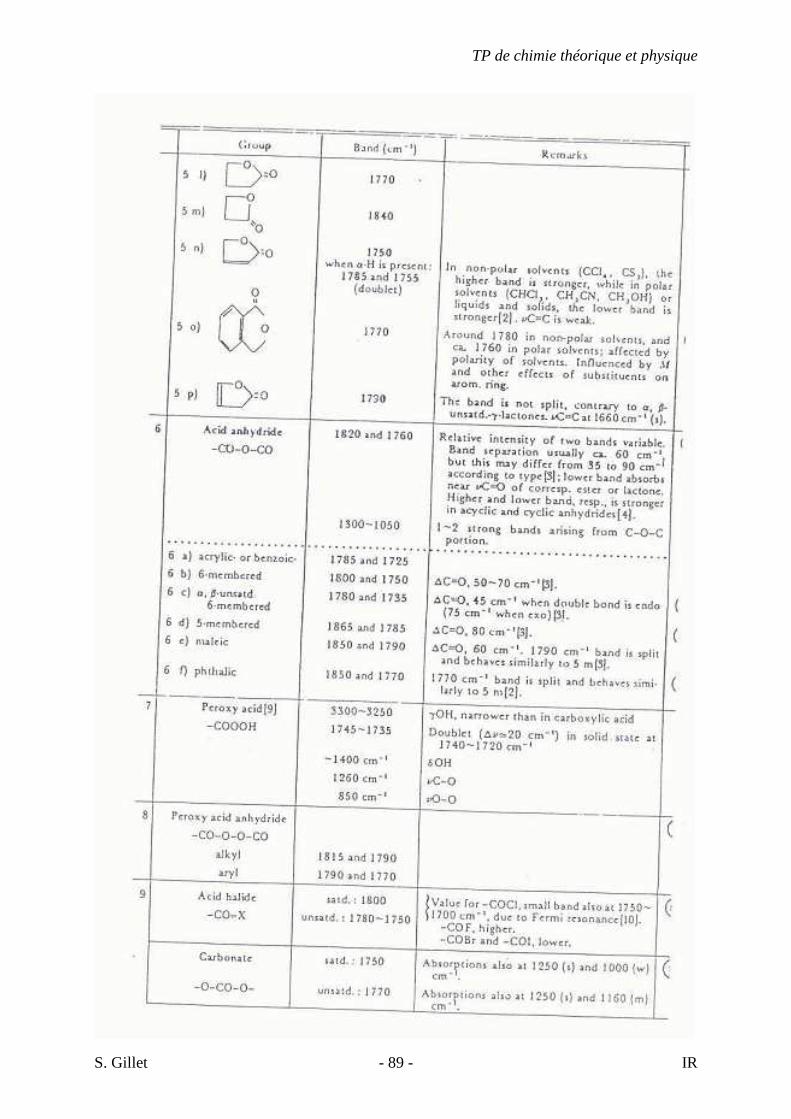
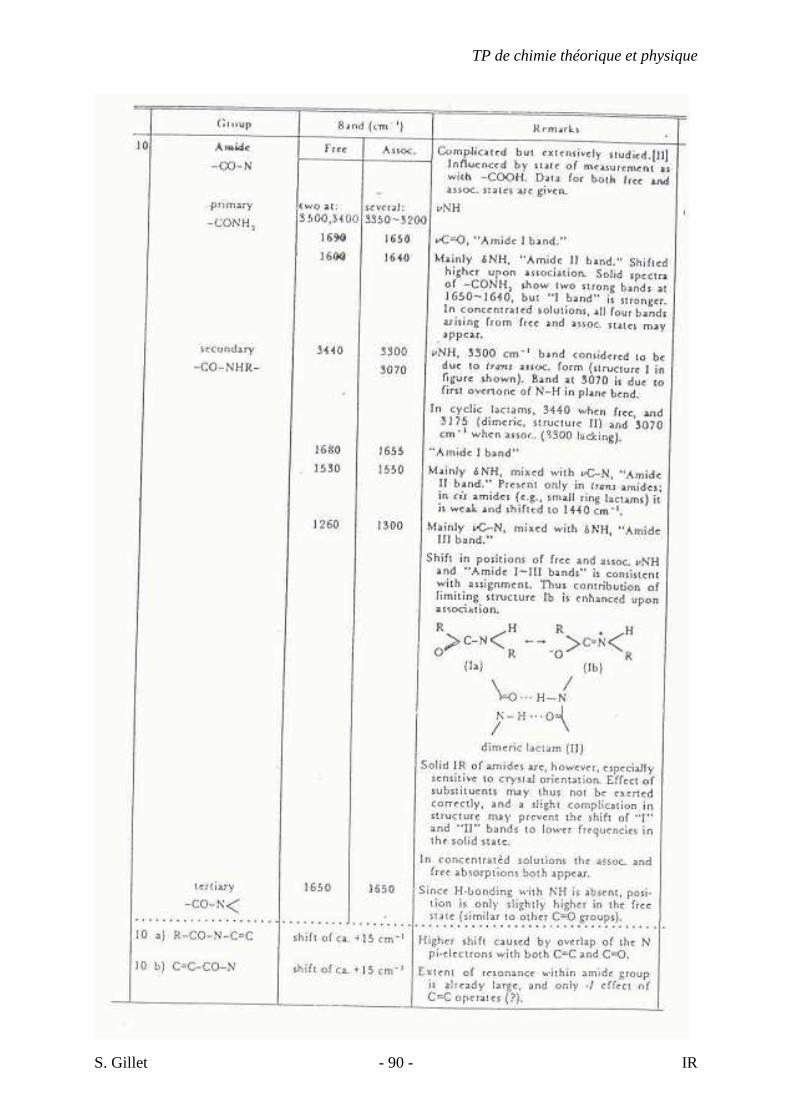
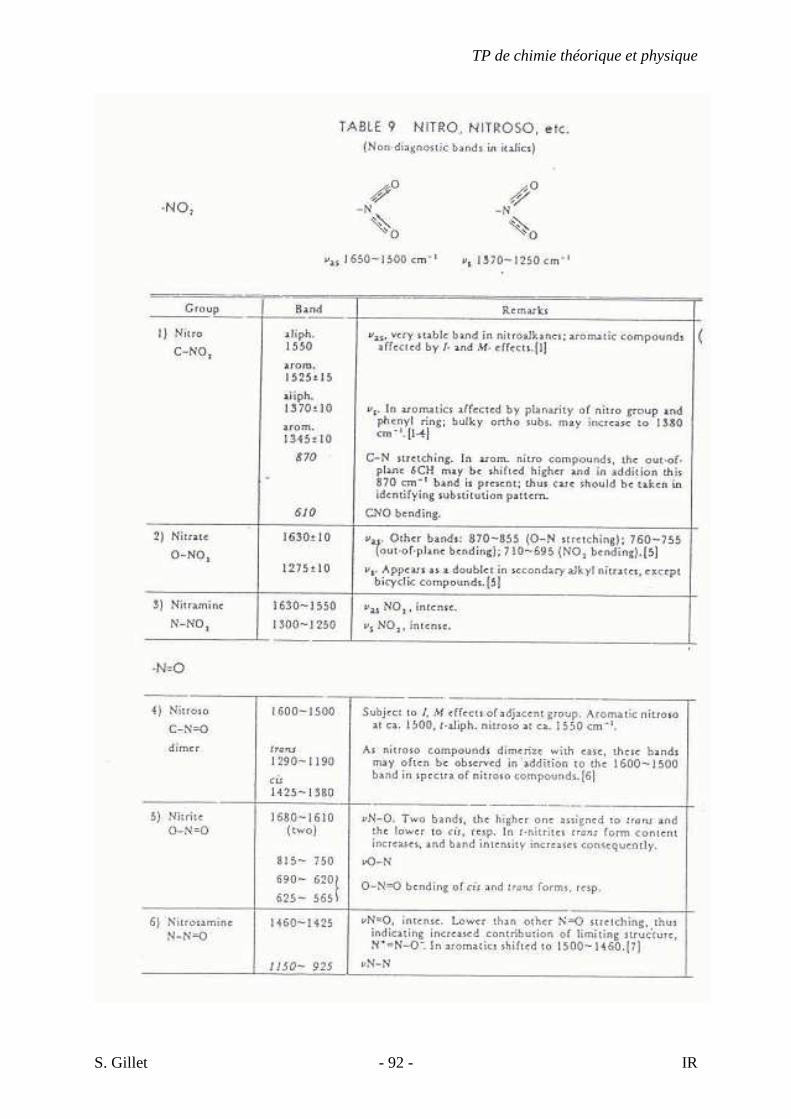
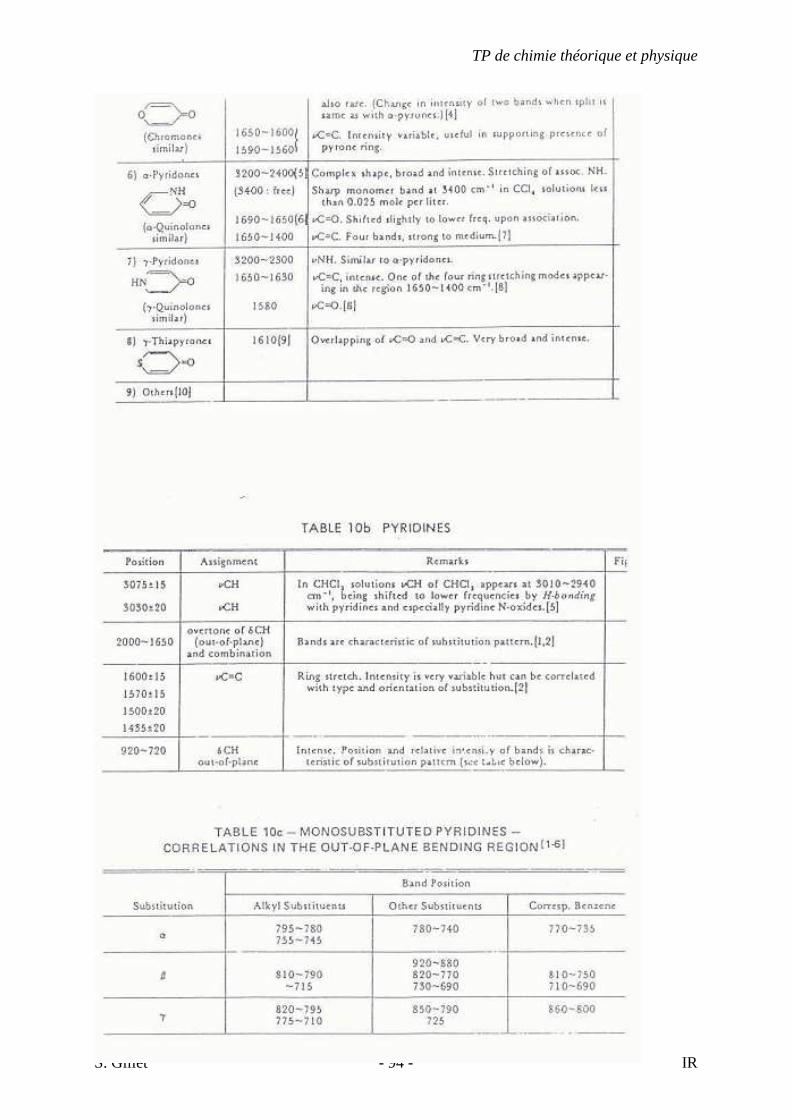
5.4. TABLES DÉTAILLÉES

TP de chimie théorique et physique
S. Gillet - 69 - IR
TP de chimie théorique et physique
S. Gillet - 70 - IR

TP de chimie théorique et physique
S. Gillet - 71 - IR

TP de chimie théorique et physique
S. Gillet - 72 - IR
TP de chimie théorique et physique
S. Gillet - 73 - IR
TP de chimie théorique et physique
S. Gillet - 74 - IR
TP de chimie théorique et physique
S. Gillet - 75 - IR
TP de chimie théorique et physique
S. Gillet - 76 - IR
TP de chimie théorique et physique
S. Gillet - 77 - IR

TP de chimie théorique et physique
S. Gillet - 78 - IR

TP de chimie théorique et physique
S. Gillet - 79 - IR

TP de chimie théorique et physique
S. Gillet - 80 - IR

TP de chimie théorique et physique
S. Gillet - 81 - IR
TP de chimie théorique et physique
S. Gillet - 82 - IR
TP de chimie théorique et physique
S. Gillet - 83 - IR

TP de chimie théorique et physique
S. Gillet - 84 - IR

TP de chimie théorique et physique
S. Gillet - 85 - IR
TP de chimie théorique et physique
S. Gillet - 86 - IR
TP de chimie théorique et physique
S. Gillet - 87 - IR

TP de chimie théorique et physique
S. Gillet - 88 - IR
TP de chimie théorique et physique
S. Gillet - 89 - IR
TP de chimie théorique et physique
S. Gillet - 90 - IR

TP de chimie théorique et physique
S. Gillet - 91 - IR
TP de chimie théorique et physique
S. Gillet - 92 - IR

TP de chimie théorique et physique
S. Gillet - 93 - IR
TP de chimie théorique et physique
S. Gillet - 94 - IR
TP de chimie théorique et physique
S. Gillet - 95 - IR
TP de chimie théorique et physique
S. Gillet - 96 - IR
TP de chimie théorique et physique
S. Gillet - 97 - IR

TP de chimie théorique et physique
S. Gillet - 98 - IR

TP de chimie théorique et physique
S. Gillet - 99 - IR
6. RÉFÉRENCES BIBLIOGRAPHIQUES
SILVERSTEIN, WEBSTER, KIEMLE. IDENTIFICATION SPECTROMÉTRIQUE DE COMPOSÉS
ORGANIQUES 2ÈME ÉDITION. DE BOECK UNIVERSITÉ 2007. 502P.
CHIMIE THÉORIQUE ET PHYSIQUE. PROF. M. MARLIER. COURS DE 1ÈRE INGÉNIEUR CHIMIE ET
BIOINDUSTRIES. FUSAGX.
CHIMIE THÉORIQUE ET PHYSIQUE APPLIQUÉE À L’ANALYSE STRUCTURALE DE BIOMOLÉCULES.
PROF. B. WATHELET. COURS DE 3ÈME BACHELIER CHIMIE ET BIOINDUSTRIES. FUSAGX.
CHIM 1302. PROF. R. GIASSON. UNIVERSITÉ DE MONTTRÉAL.
SKOOG, HOLLER, NIEMAN . PRINCIPES D’ANALYSE INSTRUMENTALE 1ÈRE ÉDITION. DE BOECK
UNIVERSITÉ 2003. 956P.
SPECTRAL DATABASE FOR ORGANIC COMPOUNDS, SDBS.
http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi
CHIMIE THÉORIQUE ET PHYSIQUE, NOTES DE TRAVAUX PRATIQUES 2003. FUSAGX.

TP de chimie théorique et physique
S. Gillet - 100 - MS
CHAPITRE 2 : LA SPÉCTROMÉTRIE DE MASSE (MS)

TP de chimie théorique et physique
S. Gillet - 101 - MS
1. INTRODUCTION
La spectrométrie de masse (MS) est une technique d’identification et de dosage des
molécules. Son concept est relativement simple, un composé est ionisé, les ions sont séparés
selon leur rapport masse/charge (m/z) et le nombre d’ions de chaque « unité » masse/charge
(m/z) est enregistré sous la forme d’un spectre.
Cette technique nous permet de déterminer la masse moléculaire d’un composé et nous
fournit également de précieuses informations sur sa structure. Cependant, contrairement aux
autres techniques présentées dans le cours de chimie théorique et physique appliquée à
l’analyse structurale de biomolécules, la spectrométrie de masse est une technique destructive.
L’échantillon est irrécupérable après analyse.
Un spectre de masse (figure 33) est un graphique représentant en abscisse le rapport
masse/charge (m/z) des différents ions ayant été séparés et en ordonnée leur abondance ou
abondance relative (intensité des pics verticaux).
On considère le pic le plus haut comme ayant 100% d’intensité. Il est appelé pic de
base et l’intensité des autres pics est déterminée par rapport à ce dernier. Le pic de plus haute
valeur masse/charge (m/z) est appelé ion moléculaire. Il représente la molécule intacte moins
un électron qui a été arraché. Une partie des ions moléculaires produisent par fragmentation
une série d’ions fragments (ou filles) dont l’analyse fournit des indications sur la structure du
composé. En effet, chaque composé, selon sa classe chimique et sa structure, va se fragmenter
de manière différente.
Figure 33 : Spectre de masse IE de la benzamide avec schéma de fragmentation expliquant les ions importants.

TP de chimie théorique et physique
S. Gillet - 102 - MS
2. INSTRUMENTATION
De manière générale, l’analyse en spectrométrie de masse (MS) repose sur les étapes
suivantes :
- Introduction de l’échantillon/mise en phase (soit directe ou infusion, soit couplée)
- Production d’ions (injection d’une charge – ionisation de la substance à analyser) –
Source
- Séparation des ions selon le rapport masse sur charge (m/z) – Analyseur
- Mesure de l’abondance de chaque ion – Détecteur
Dans ce chapitre, un instrument type sera divisé en méthodes d’ionisation, de
séparation d’ions et de détection (figure 34). Généralement, la méthode d’ionisation est
indépendante de la méthode de séparation et vice versa, bien qu’il y ait des exceptions.
Certaines méthodes reposent sur une introduction en sortie de ligne chromatographique alors
que d’autres excluent l’usage de la chromatographie dans l’introduction de l’échantillon.
Figure 34 : Schéma block des éléments d’un spectromètre de masse type.
Cependant, une caractéristique commune revient chez tous les appareils. En effet,
aussi bien l’analyseur que le détecteur sont sous vide ou à une pression fort faible (et parfois
la source également). On évite ainsi les interférences des composants de l’air. Si trop de
molécules sont présentes lors de l’analyse, il y a apparition de collisions, la trajectoire des
ions est modifiée et ils vont se décharger sur les parois de l’appareil. D’autre part, la collision
entre ions et molécules entraîne des réactions qui compliquent le spectre.

TP de chimie théorique et physique
S. Gillet - 103 - MS
2.1. LES TECHNIQUES D’ IONISATION
Il existe différents types de source. En fonction de la molécule à analyser ou de
l’objectif de l’analyse, il peut être intéressant d’utiliser l’une ou l’autre source.
2.1.1. L’ IONISATION PAR IMPACT ÉLECTRONIQUE
L’impact électronique (IE) est la technique de production d’ions la plus répandue en
spectrométrie de masse. Un filament de tungstène chauffé émet des électrons. Ils sont
accélérés et entrent en collision avec les molécules gazeuses de l’analyte situées dans la
source (figure 35).
Figure 35 : Source par impact électronique.
Des molécules en phase vapeur de l’échantillon, sont bombardées par des électrons
très énergétiques (généralement 70 eV), qui éjectent un électron d’une molécule de
l’échantillon en produisant un radical cation nommé ion moléculaire. Comme le potentiel
d’ionisation de composés organiques typiques est généralement inférieur à 15 eV, les
électrons d’impact transfèrent un excédent d’énergie de 50 eV (ou plus) à l’ion moléculaire
formé, qui le dissipe en partie par rupture de liaisons covalentes ayant une énergie comprise
entre 3 et 10 eV. Cela mène à la formation d’ions primaires qui pourront eux même se
fragmenter.
TP de chimie théorique et physique
S. Gillet - 104 - MS
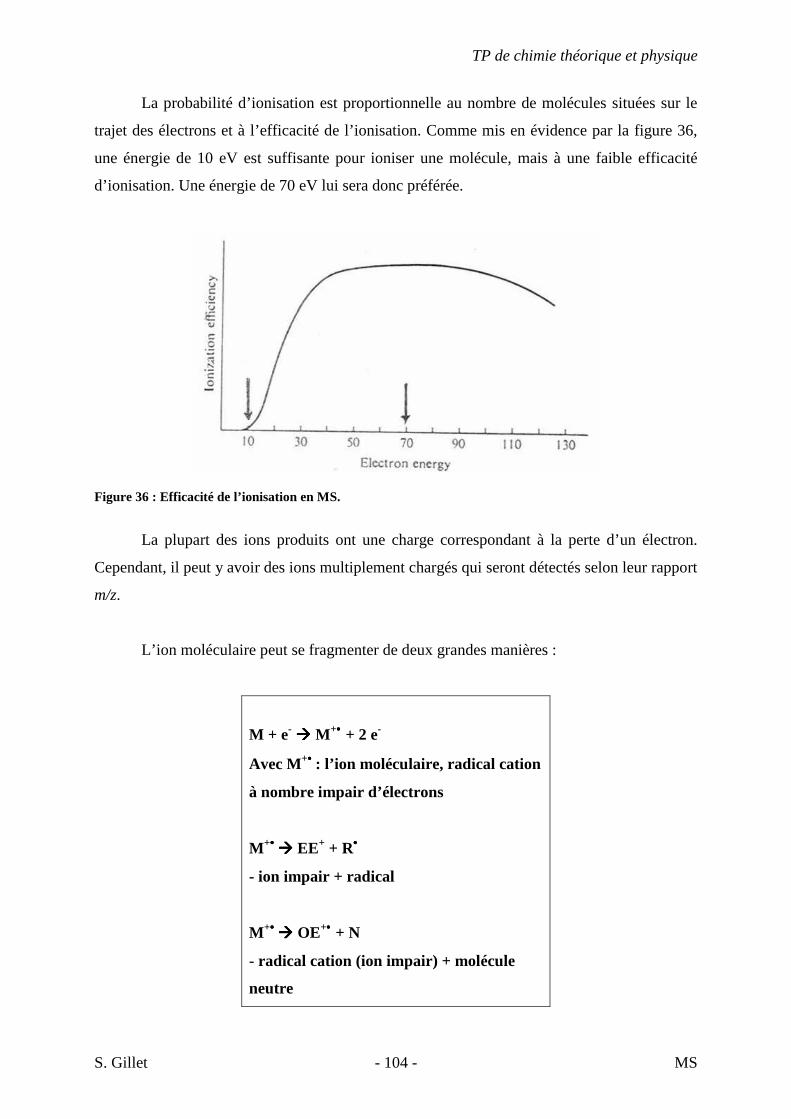
La probabilité d’ionisation est proportionnelle au nombre de molécules situées sur le
trajet des électrons et à l’efficacité de l’ionisation. Comme mis en évidence par la figure 36,
une énergie de 10 eV est suffisante pour ioniser une molécule, mais à une faible efficacité
d’ionisation. Une énergie de 70 eV lui sera donc préférée.
Figure 36 : Efficacité de l’ionisation en MS.
La plupart des ions produits ont une charge correspondant à la perte d’un électron.
Cependant, il peut y avoir des ions multiplement chargés qui seront détectés selon leur rapport
m/z.
L’ion moléculaire peut se fragmenter de deux grandes manières :
M + e- ���� M+•••• + 2 e-
Avec M+•••• : l’ion moléculaire, radical cation
à nombre impair d’électrons
M+•••• ���� EE+ + R••••
- ion impair + radical
M+•••• ���� OE+•••• + N
- radical cation (ion impair) + molécule
neutre

TP de chimie théorique et physique
S. Gillet - 105 - MS
En spectrométrie de masse par impact électronique, la rupture de la liaison est
habituellement totale et critique, hautement reproductible et caractéristique du composé. De
plus, ce procédé de fragmentation étant prédictible, il est à la base de la puissance de la
spectrométrie de masse dans l’élucidation de structure.
Les spectres obtenus par IE (uniquement) peuvent également constituer des bases de
données. Cela permet à un composé inconnu, d’être comparé à la base de données (par le
calcul d’un pourcentage de corrélation par rapport aux spectres de cette base), et d’être
identifié facilement, rapidement et efficacement (ce qui est fait couramment dans la pratique).
Cela étant dit, tous les spectres de tous les composés ne sont pas repris dans des bases de
données. Il est donc important d’étudier les mécanismes de fragmentation, qui donneront dans
tous les cas des informations structurales importantes.
Cette technique comporte de nombreux avantages :
- Une production facile d’électrons
- Une production de l’ion moléculaire et des ions fragments dans des domaines
d’énergie proches
- Le bombardement par des électrons est continu ce qui donne des spectres stables
- Les mécanismes de fragmentations sont bien connus
- Il existe des bases de données contenant plus de 390000 composés
Elle présente cependant certains inconvénients :
- Le pic de l’ion moléculaire est parfois non-visible car la fragmentation est totale
- Il y a des risques de décomposition thermique de l’échantillon, car les températures
utilisées pour vaporiser l’échantillon sont de l’ordre de 250 °C
- La technique ne convient donc pas aux composées non volatils
- Des réactions entre les ions et les molécules pourraient générer des pics à m/z
supérieur à celui de l’ion moléculaire.
- La technique n’est pas idéale pour les composés de haut poids moléculaire

TP de chimie théorique et physique
S. Gillet - 106 - MS
2.1.2. L’ IONISATION CHIMIQUE
L’ionisation par impact électronique provoque souvent une telle fragmentation qu’il
est impossible d’observer le pic moléculaire. Un moyen de contourner le problème est de
recourir à une technique d’ionisation plus douce, parmi lesquelles l’ionisation chimique (IC)
est la plus importante. En IC, les molécules de l’échantillon (en phase vapeur) ne sont pas
bombardées par un faisceau d’électrons énergiques. Un gaz réactif (habituellement le
méthane, l’isobutane, l’ammoniac ou autres) est introduit dans la source et ionisé par
bombardement électronique pour donner des ions primaires. Les molécules de l’échantillon
entrent en collision avec les molécules ionisée du gaz réactif (CH5+, C4H9
+, etc.) dans la
source IC où la pression est relativement élevée pour conduire à une ionisation secondaire par
transfert de proton en produisant un ion [M + 1]+, par addition électrophile en produisant des
ions [M + 15]+, [M + 24]+, [M + 43]+ ou [M + 18]+ (avec NH4+) ou (plus rarement) par
transfert de la charge en produisant un ion [M]+.
EI :
CH4 + e- ���� CH4+•••• + 2e-
Fragmentation :
CH4+•••• ���� CH3
+ + H••••
CH4+•••• ���� CH2
+•••• + H2
Collisions:
CH4+•••• + CH4 ���� CH5
+ + CH3••••
CH3+ + CH4 ���� C2H5
+ + H2
CH2+•••• + CH4 ���� C2H3
+ + H2 + H••••
C2H3+ + CH4 ���� C3H5
+ + H2
Ionisation de l’échantillon:
CH5+ + M ���� MH + + CH4
MH + = ion quasi ou pseudo moléculaire

TP de chimie théorique et physique
S. Gillet - 107 - MS
L’excès d’énergie transféré à l’échantillon durant la phase d’ionisation est
suffisamment faible, généralement inférieur à 5eV, pour réduire la fragmentation. Cela à
plusieurs conséquences importantes, les plus riches étant l’abondance d’ions moléculaires et
une sensibilité accrue due à un courant ionique total concentré dans un petit nombre d’ions.
En revanche, on en retire moins d’informations structurales. Les ions quasi-moléculaires sont
généralement assez stables et facilement détectés. Souvent, seul un ou deux fragments sont
produits et même parfois aucun.
La spectrométrie de masse par ionisation chimique n’est ni performante pour la
reconnaissance de pic (que ce soit manuellement ou automatiquement) ni particulièrement
utile en élucidation de structure ; son principal intérêt réside dans la détection de l’ion
moléculaire et donc de la masse moléculaire.
* * *
Dans les deux premières techniques qui viennent d’être présentées (impact
électronique et ionisation chimique), l’échantillon est sous forme gazeuse. Ces techniques ne
permettent donc pas, du moins telles quelles, d’analyser de composés thermosensibles ou non-
volatils. Pour y remédier, d’autres techniques d’ionisation ont été développées. Elles vont être
détaillées dans la suite de cette section.
2.1.3. L’ IONISATION PAR BOMBARDEMENT D’ATOMES RAPIDES (FAB)
Dans les techniques d’ionisation par désorption, les molécules à étudier passent
directement d’une phase condensée à la phase vapeur sous forme d’ions. Elles sont
principalement utilisées dans le cas de composés lourds, non-volatils ou ioniques. Elles
peuvent présenter des inconvénients importants Les techniques de désorption n’utilisent
généralement pas les échantillons de manière très efficace. Souvent, l’information retirée est
limitée. Pour des composés inconnus, ces techniques servent principalement à fournir la
masse moléculaire et parfois une masse exacte. Toutefois, même pour ces applications, il faut
rester prudent car l’ion moléculaire ou quasi-moléculaire n’est parfois pas très évident. Il en
résulte souvent des spectres compliqués par de nombreux ions provenant de la matrice.

TP de chimie théorique et physique
S. Gillet - 108 - MS
Le bombardement par atomes rapides (figure 37) met en jeu des atomes de xénon ou
d’argon très énergiques (6 à 10 keV) pour bombarder des échantillons dissous dans un liquide
de faible tension de surface (ex : glycérol). La matrice protège l’échantillon des dommages
excessifs dus aux radiations. On obtient des ions quasi-moléculaires (MH+, MNa+,…).
Figure 37 : Source par bombardement d’atomes rapides.
2.1.4. L’ IONISATION PAR DÉSORPTION LASER D’UNE MATRICE (MALDI)
L’ionisation par désorption laser assistée par matrice est une nouvelle méthode
d’ionisation qui permet d’obtenir des informations précises concernant les masses
moléculaires de biopolymères polaires. Ces masses peuvent varier de plusieurs centaines à
plusieurs milliers de Da.
On envoie le laser sur une goutte séchée de la solution, l’analyse se fait donc sur un
échantillon solide. La substance à analyser est mélangée à une solution de petites molécules
organiques appelée matrice. Ces petites molécules possèdent une forte absorption à la
longueur d’onde utilisée du laser. De ce fait, les molécules de la matrice vont subir une
excitation, et la matrice va donc ioniser l’échantillon (figure 38).

TP de chimie théorique et physique
S. Gillet - 109 - MS
Figure 38 : Source MALDI.
En utilisant une matrice, il n’est plus nécessaire d’adapter la longueur d’onde du laser
à la substance à analyser. C’est une méthode très sensible.
2.1.5. L’ IONISATION PAR ÉLECTROSPRAY
En électrospray (ES), les ions sont générés à pression atmosphérique (ou proche) ; la
technique s’appelle donc également ionisation à pression atmosphérique (API). L’échantillon
en solution (habituellement dans un solvant polaire) pénètre la source d’ions via un capillaire
en acier inoxydable entouré d’un flux coaxial d’azote, appelé gaz nébuliseur. L’extrémité du
capillaire est maintenue à une tension élevée par rapport à une contre électrode. La différence
de potentiel produit un gradient de champ pouvant aller jusqu’à 5 kV/cm. Un aérosol de
gouttelettes polychargées se forme lorsque la solution quitte le capillaire. Le flux de gaz
nébuliseur dirige les effluents vers le spectromètre de masse.
Dans l’aérosol, la taille des gouttelettes diminue avec l’évaporation du solvant et la
concentration en ions chargés augmentant en conséquence. Quand la répulsion électrostatique
entre les ions atteint un point critique, les gouttelettes subissent ce que l’on nomme une
« explosion coulombienne », qui libère les ions dans la phase vapeur (figure 39). On obtient
ainsi des ions en phase gazeuse qui ont une très faible énergie interne. Les ions désolvatés
sont focalisés par plusieurs échantillonneurs vers l’analyseur de masse.

TP de chimie théorique et physique
S. Gillet - 110 - MS
Figure 39 : Source électrospray.
2.2. LES TECHNIQUES DE SÉPARATION
Il existe différents types d’analyseurs. Les principaux vont être décrits dans la suite de
ce chapitre. L’analyseur va séparer le mélange d’ions générés à l’étape d’ionisation par ordre
de rapport masse/charge (m/z) afin d’obtenir un spectre. C’est le cœur du spectromètre.
La qualité d’un analyseur peut être évaluée selon différents critères :
- La limite de masse : est la valeur limite de m/z mesurable
- La transmission : est le rapport entre le nombre d’ions arrivant au détecteur et celui
dans la source
- La résolution : est la capacité de fournir des signaux distinguables entre les ions de
masses voisines (R = m/δm). On admet que deux pics sont résolus lorsque l’intensité
de la « vallée » entre ceux-ci vaut 10% de l’intensité du pic le plus faible (figure 40).
Figure 40 : La résolution en spectrométrie de masse.

TP de chimie théorique et physique
S. Gillet - 111 - MS
2.2.1. LES ANALYSEURS MAGNÉTIQUES ET MAGNÉTO-ÉLECTROSTATIQUES
Dans un spectromètre de masse à secteur magnétique, un champ magnétique dévie
circulairement la trajectoire des ions (figure 41). La séparation des ions est basée sur le
rapport m/z ; les ions les plus légers étant plus déviés que les ions les plus lourds. La
résolution dépend de chaque ion entrant dans le champ magnétique (depuis la source) avec la
même énergie cinétique, obtenue en accélérant les ions (de charge z) par une tension V.
chaque ion acquiert une énergie cinétique E = zV = mv2/2 (1). Quand un ion accéléré entre
dans le champ magnétique (B), il subit une force (Bzv) qui courbe sa trajectoire
perpendiculairement à sa direction initiale. L’ion évolue maintenant sur une trajectoire
circulaire de rayon r, donné par Bzv = mv2/r. De la, on peut tirer que mv = zBr (2). Comme le
montrent ces équations, un appareil à secteur magnétique sépare les ions sur la base de leur
moment, qui est le produit de la masse par la vitesse, plutôt que sur la seule masse. En
conséquence, des ions de même masse mais d’énergies différentes focaliseront en des points
différents. De (2), on tire v et on introduit sa valeur dans (1) : m/z = B2r2/2V. Puisque le rayon
de l’instrument est fixe, le champ magnétique est balayé pour focaliser successivement les
différents ions.
Figure 41 : Analyseur magnétique
Un analyseur électrostatique (ESA) peut considérablement réduire la distribution
d’énergie d’un faisceau d’ion en forçant les ions de même charge (z) et énergie cinétique
(indépendamment de la masse), à suivre la même trajectoire. Une fente à la sortie de l’ESA
focalise encore le faisceau d’ion avant qu’il n’entre dans le détecteur. La combinaison d’un
ESA et d’un secteur magnétique est appelée double focalisation (figure 42), car les deux
champs contrecarrent les effets dispersifs que chacun a sur la direction et la vitesse.

TP de chimie théorique et physique
S. Gillet - 112 - MS
Figure 42 : Analyseur double focalisation
La résolution d’un appareil à secteur magnétique à double focalisation (figure) peut
être extrêmement élevée. Cela permet la mesure de «masses exactes», offrant un accès
univoque à la formule moléculaire.
2.2.2. LES ANALYSEURS QUADRIPOLAIRES
Un quadripôle se compose de quatre barreaux cylindriques parallèles entre eux, placés
aux sommets d’un carré (figure 43). Une analyse mathématique complète du quadripôle est
complexe mais son fonctionnement peut-être décrit en termes simples. Une tension continue,
constante, modulée en radiofréquence est appliquée aux barreaux. Les ions sont introduits à
une extrémité du « tunnel » formé par les quatre barreaux, au centre du carré et se déplacent le
long de son axe.
Pour chaque combinaison de tension continue et de modulation à la fréquence
appropriée, seuls les ions d’un m/z donné ont une trajectoire stable et donc traversent le
quadripôle pour atteindre le détecteur. Tous les ions de m/z différents ont des trajectoires
instables ou erratiques et percutent un des barreaux ou sortent du quadripôle. On peut se
représenter facilement le quadripôle comme un filtre de masse réglable. En d’autres termes,
tous les ions entrent d’un coté mais seuls les ions d’un m/z donné sortent de l’autre.

TP de chimie théorique et physique
S. Gillet - 113 - MS
Figure 43 : Analyseur quadripolaire.
Du point de vue de la résolution, le quadripôle est généralement inférieur au secteur
magnétique. Par contre, la sensibilité est généralement très élevée puisqu’il n’est pas
nécessaire d’employer une fente qui éliminerait une fraction des ions.
Il existe des appareils possédant plusieurs quadripôles en série. Le premier va servir à
piéger un ion donné. Cet ion sera ensuite refragmenté par collision avec un gaz inerte ou
réactionnel. Les ions ainsi formés seront analysés dans un second quadripôle.
2.2.3. LES ANALYSEURS À TRAPPE IONIQUE
La trappe ionique est parfois considérée comme une variante du quadripôle car leurs
aspects et leurs fonctionnements sont liés. Toutefois, la trappe ionique est potentiellement
bien plus polyvalente et est certainement promise à un développement important, car les
spectres IE obtenus avec une trappe ionique sont maintenant compatibles avec les bases de
données commerciales.
La trappe ionique a la capacité de piéger les ions pour une période relativement
longue. Elle se comporte généralement de trois électrodes, l’une annulaire de face interne
hyperbolique et les deux autres (électrodes d’entrée et de sortie), hyperboliques aux
extrémités (figure 44). L’électrode annulaire est soumise à un champ de radiofréquence
sinusoïdal alors que les électrodes d’entrée et de sortie sont placées à l’un des trois états
suivants : potentiel nul, tension continue ou alternative.

TP de chimie théorique et physique
S. Gillet - 114 - MS
Figure 44 : Analyseur trappe ionique.
La trappe ionique peut fonctionner selon trois modes de base :
- Premièrement, tension RF fixe et pas de polarisation entre les électrodes entrée/sortie
et l’électrode annulaire : tous les ions dans la limite d’un m/z donné seront piégés.
Quand la tension RF est augmentée, la limite de m/z diminue de façon contrôlée et les
ions sont successivement éjectés et détectés. Il en résulte qu’un spectre de masse
standard et cette procédure est nommée opération en mode « instabilité sélective de
masse ». Dans ce mode, le potentiel RF maximum applicable entre les électrodes fixe
limite la masse maximum. Les ions de masses supérieures à cette limite sont éliminés
quand le potentiel est ramené à zéro.
- Deuxièmement, tension continue entre les électrodes entrée/sortie ; globalement, cela
fait apparaître deux limites, basse et haute, de m/z. Le champ d’application de ce mode
est gigantesque et les trappes ioniques sont la plupart du temps utilisées dans cette
configuration. Il est possible de sélectionner jusqu’à une seule masse ionique. Un des
usages important de ce mode est le suivi sélectif d’ion. Il n’y a pas de limites pratiques
au nombre de masses ioniques qu’il est possible de sélectionner.
- Le troisième mode est similaire au deuxième avec en plus, un champ oscillant
auxiliaire entre les électrodes d’entrée/sortie, cela permet d’augmenter sélectivement
l’énergie cinétique d’un ion donné. Avec un champ auxiliaire faible, l’énergie
cinétique des ions choisis augmente lentement, durant ce temps ils peuvent
s’entrechoquer puis se fragmenter ; on fonctionne alors parfois à près de 100% en SM-
SM.

TP de chimie théorique et physique
S. Gillet - 115 - MS
2.2.4. LES ANALYSEURS À TEMPS DE VOL
Le concept de spectromètre de masse à temps de vol (TOF) est assez simple (figure
45). Les ions sont accélérés par un potentiel (V), on les laisse ensuite « dériver » le long d’un
tube jusqu’au détecteur. En supposant que tous les ions se présentant à l’entrée du tube ont la
même énergie, donnée par zeV = mv2/2, alors des ions de masse différentes auront des
vitesses différentes : v = (2zeV/m)1/2. Si le tube du spectromètre à une longueur L, le temps de
vol d’un ion est donné par : t = (L2m/2zeV)1/2, d’où on extrait facilement la masse d’un ion
donné.
L’aspect critique de cet instrument par ailleurs très simple, est la nécessité de produire
les ions à des positions et temps initiaux précisément connus. Ces contraintes limitent les
spectromètres TOF à des techniques d’ionisation pulsées, dont la désorption laser et plasma
(MALDI).
La résolution des instruments TOF est généralement inférieure à 20000 du fait de
l’inévitable distribution de l’énergie de l’ion. En outre, la gamme de masse de ces instruments
est illimitée et ils ont une excellente sensibilité. Cette technique est donc particulièrement
adaptée aux grosses biomolécules.
Figure 45 : Analyseur TOF.

TP de chimie théorique et physique
S. Gillet - 116 - MS
2.2.5. LES SPECTROMÈTRES DE MASSE À TRANSFORMÉE DE FOURIER
Dans un spectromètre de masse à transformée de Fourier, les ions sont confinés dans
une cellule par un potentiel électrique piège, sous un intense champ magnétique. A l’intérieur
de la cellule, chaque ion orbite sur une trajectoire perpendiculaire au champ magnétique, à
une fréquence proportionnelle à m/z. Une impulsion radiofréquence appliquée à la cellule,
place simultanément toutes les fréquences cyclotroniques en résonance pour donner un
interférogramme, conceptuellement similaire à la FID d’un signal RMN. L’interférogramme
qui est un spectre en temps est converti par transformée de Fourier en spectre en fréquence,
qui aboutit enfin au spectre m/z conventionnel.
2.3. LES DÉTECTEURS
Le rôle du détecteur est de transformer un courant ionique faible en un courant
mesurable. Il existe différents types de détecteurs les plus courant seront décrit dans la suite
de ce chapitre.
2.3.1. LES PLAQUES PHOTOGRAPHIQUES
Les plaques photographiques au bromure d’argent sont sensibles aux ions
énergétiques. Les ions accélérés par une différence de potentiel de quelques kilovolts
possèdent une énergie suffisante pour impressionner une émulsion photographique déposée en
couche mince sur une plaque de verre. Le point d’impact de l’ion est fonction du rapport m/z.
Le noircissement relatif de la plaque est fonction de la quantité d’ions la frappant. Un
étalonnage préalable est nécessaire.
2.3.2. LES CYLINDRES DE FARADAY
Le cylindre de Faraday est une boîte cylindrique dans laquelle les ions entrent et
heurtent le fond (figure 46). Ils communiquent de cette manière leur charge. Le fond est relié
à la terre par une résistance de grande valeur ce qui génère une différence de potentiel aux
bornes de cette résistance. Le courant généré est amplifié et mesuré. Il n’y a pas d’émission
d’électrons secondaires.

TP de chimie théorique et physique
S. Gillet - 117 - MS
Figure 46 : Détecteur cylindre de Faraday.
2.3.3. LES MULTIPLICATEURS D’ÉLECTRONS
Un multiplicateur d’électron se présente comme un dispositif en forme de trompe
façonné en un verre fortement dopé en plomb (figure 47). Contrairement au cylindre de
Faraday, on s’arrange ici pour avoir beaucoup d’émission secondaire d’électrons. Cela va
avoir pour effet d’amplifier le signal.
Une tension de courant est appliquée entre les deux extrémités. Le champ créé va accélérer les
électrons vers l’intérieur du tube. Chaque ion frappant la surface près de l’entrée éjecte des
électrons qui, rebondissant le long de la surface éjectent de plus en plus d’électrons à chaque
impact.
Le gain de conversion (nombre d’électrons en sortie pour un ion à l’entrée) de ce type
de transducteur est d’environ 105, mais dans certains cas, on peut atteindre des gains de 108.
Figure 47 : Détecteur multiplicateur d'électron

TP de chimie théorique et physique
S. Gillet - 118 - MS
2.3.4. LES MULTIPLICATEUR DE PHOTONS
Le système est constitué de deux dynodes, d’un écran phosphorescent et d’un
photomultiplicateur (figure 48).
Ce dispositif permet la détection d’ions positifs et négatifs. Des électrons secondaires
sont émis par les dynodes, lorsqu’elles sont frappées par des ions. Ces électrons sont accélérés
vers l’écran phosphorescent où ils sont convertis en photons. L’écran phosphorescent est
conducteur. De cette manière, il ne se charge pas de plus en plus négativement avec l’arrivée
d’électrons supplémentaires. Les photons ainsi produits sont ensuite détectés et quantifiés par
le photomultiplicateur.
Figure 48 : Détecteur multiplicateur de photons.
2.3.5. LES DÉTECTEURS À MICROCANAUX
Un détecteur à microcanaux se présente comme une plaque percée de canaux
cylindriques parallèles (figure 49). Chaque canal est recouvert d’un matériau semi-conducteur
émettant des électrons secondaires à chaque impact d’ion. La géométrie de la plaque est
analogue à celle d’une plaque photographique. Les ions de m/z différents arrivent en
différents endroits du détecteur et peuvent être compté simultanément pendant le balayage du
champ magnétique de l’analyseur. Cette technique offre un gain de conversion de 105 à 108.

TP de chimie théorique et physique
S. Gillet - 119 - MS
Figure 49 : Détecteur multicanaux
3. INTERPRÉTATION DES SPECTRES DE MASSE
3.1. GÉNÉRALITÉS
Notre discussion sur les spectres de masse se limitera aux spectres IE. Dans les
spectres de masse IE, la fragmentation est riche en informations structurales.
Les spectres de masse (par impact électronique) s’obtiennent en routine avec un
faisceau d’électrons de 70 eV. L’événement le plus simple est l’arrachement, par un des
électrons de ce faisceau, d’un électron d’une molécule en phase gazeuse pour former l’ion
moléculaire qui est un radical cation (M•+).
NB : Quand la charge peut être localisée sur un atome particulier, elle est indiquée sur cet
atome.
Plusieurs de ces ions moléculaires se désintègrent dans les 10-10 à 10-3 secondes pour
donner, dans le plus simple des cas, un fragment chargé positivement et un radical. On forme
ainsi de nombreux ions fragments et chacun d’eux peut se scinder à nouveau pour donner des
fragments plus petits.
Si certains des ions moléculaires parents restent intacts assez longtemps pour atteindre
le détecteur, on observe un pic correspondant à l’ion moléculaire. Il est important de
reconnaître le pic moléculaire car il donne le poids moléculaire du composé. Avec une
résolution unitaire, ce poids est le poids moléculaire arrondi au nombre entier le plus proche.
Un spectre de masse est une représentation des concentrations relatives des fragments chargés
positivement (incluant l’ion moléculaire) en fonction de leur masse. On attribue une valeur de
100% au pic le plus intense du spectre, le pic de base, et les intensités des autres pics

TP de chimie théorique et physique
S. Gillet - 120 - MS
(hauteurs x facteur de sensibilité), incluant celui de l’ion moléculaire, sont rapportées en
pourcentage du pic de base. Bien entendu quelque fois, le pic de base peut être celui de l’ion
moléculaire.
3.2. IDENTIFICATION DU PIC DE L ’ ION MOLÉCULAIRE
Bien souvent en impact électronique (EI), l’identification du pic de l’ion moléculaire
pose problème. Ce pic peut être très faible ou même ne pas apparaître du tout. Pour remédier à
ce problème, la meilleure solution est d’obtenir le spectre par une autre technique d’ionisation
telle que l’ionisation chimique. Dans ce cas, le résultat habituel est un pic intense à [M+1]
avec peu de fragmentation.
L’identification de l’ion moléculaire est la première étape d’une analyse spectrale. Elle
permettra la détermination de la masse moléculaire du composé. Une fois la masse connue, il
est possible d’utiliser des tables indiquant toutes les formules brutes possibles pour cette
masse moléculaire donnée.
L’identification du pic de l’ion moléculaire dépend de la stabilité de l’ion moléculaire.
Les ions moléculaires les plus stables sont ceux des systèmes purement aromatiques. Si des
groupements labiles sont présents, le pic de l’ion moléculaire sera moins intense et les pics
des fragments, relativement plus intenses.
De manière générale, on observe que :
1- Les groupes de composés suivants donneront en ordre décroissant des pics
moléculaires dominants : composés aromatiques > alcènes conjugués > composés
cycliques > sulfures organiques > alcanes normaux à chaîne courte > mercaptans.
2- Les ions moléculaires reconnaissables sont produits habituellement par les composés
suivant par ordre décroissant de stabilité : cétones > amines > esters > éthers > acides
carboxyliques ∼ aldéhydes ∼ amides ∼ halogénures.
3- L’ion moléculaire est souvent indétectable pour les alcools aliphatiques, les nitrites,
les nitrates, les composés nitros, les nitriles et les composés très ramifiés.

TP de chimie théorique et physique
S. Gillet - 121 - MS
3.3. RÈGLE DE L ’AZOTE
La « règle de l’azote » rend souvent service. Elle stipule qu’une molécule dont la
masse moléculaire est paire doit contenir un nombre nul ou pair d’atomes d’azote ; une masse
moléculaire impaire indique un nombre impair d’atomes d’azote.
Cette règle s’applique à tous les composés contenant du carbone, de l’hydrogène, de
l’oxygène, de l’azote, du soufre et des halogènes, ainsi que des atomes moins courants comme
le phosphore, le bore, le silicium, l’arsenic et les alcalino-terreux.
Partant de cette considération, il est possible d’éliminer certaines des possibilités de
formules moléculaires brutes fournies par les tables.
3.4. DÉTERMINATION DE LA FORMULE MOLÉCULAIRE À PARTIR DES INTENSITÉS DES P ICS
ISOTOPIQUES [M + 1] ET [M + 2]
La masse d’une molécule est obtenue en additionnant les masses des isotopes les plus
abondants pour chaque atome.
La présence d’isotopes pour certains éléments génère l’apparition de pics isotopiques.
En général, ces pics ont une intensité inférieure à celle de l’ion moléculaire sauf dans le cas
ou des atomes tels que le chlore ou le brome sont présent dans la molécule (cas particulier).
Les espèces moléculaires contenant des isotopes moins abondants donnent donc des « pics
isotopiques » à [M + 1], [M + 2], etc.
Si seuls C, H, N, O, F, P et I composent une molécule, les intensités relatives
approchées attendues pour [M + 1] et [M + 2] peuvent être calculées en utilisant les équations
suivantes :
- % [M + 1] = 100 [M + 1] / M ≅≅≅≅ 1,1 x nombre d’atomes de carbone + 0,36 x
nombre d’atomes d’azote
- % [M + 2] = 100 [M + 2] / M ≅≅≅≅ (1,1 x nombre d’atomes de carbone)2 / 200 + 0,2 x
nombre d’atomes d’oxygène

TP de chimie théorique et physique
S. Gillet - 122 - MS
Si ces pics isotopiques sont suffisamment intenses pour être mesurés avec précision, le
calcul ci-dessus peut permettre de déterminer la formule moléculaire.
Toutefois, cette méthode comporte certaines limites car les intensités observées sont
souvent imprécises. De plus, pour une masse moléculaire supérieure à 250, la méthode est
inadaptée.
Une formule moléculaire unique peut souvent être déduite d’une simple mesure de
masse suffisamment précise (4 décimales). Cela est possible parce que les masses atomiques
ne sont pas des nombres entiers. Cette approche est cependant fastidieuse et l’utilisation de
logiciels spécifiques peut faciliter les choses :
http://www.ch.cam.ac.uk/magnus/EadFormW.html
3.5. CALCUL DU NOMBRE D ’ INSATURATIONS
En plus du nombre et du type d’atomes, la formule moléculaire donne un indice de
déficience en hydrogène. Cet indice est le nombre de paires d’atomes d’hydrogène qui
doivent être retirées d’une formule « saturée » (c'est-à-dire CnH2n+2 pour les alcanes) pour
donner la formule moléculaire du composé que l’on considère. Cet indice de déficience en
hydrogène est également appelé le nombre de « sites d’insaturation ». Cette description est
incomplète puisque la déficience en hydrogène peut être due à des structures cycliques aussi
bien qu’à des liaisons multiples. L’indice est donc la somme du nombre de cycles, du nombre
de liaisons doubles et de deux fois le nombre de liaisons triples.
Cet indice peut être calculé pour des composés contenant carbone, hydrogène, azote,
halogène, oxygène et soufre de formule générale : CnHmXxNyOz à partir de l’équation :
Indice = (n) – (m / 2) – (x / 2) + (y / 2) + 1
Un cycle aromatique comportera par exemple quatre « sites insaturés » : trois pour les
doubles liaisons et un pour le cycle.

TP de chimie théorique et physique
S. Gillet - 123 - MS
La formule brute donne donc rapidement des informations sur la structure. De plus,
par simple observation du nombre d’oxygènes ou d’azotes, par exemple, il est également
possible de restreindre le choix des classes de molécules organiques possibles pour le cas
envisagé, et faciliter de cette manière l’analyse spectrale. Ainsi, une molécule ne comportant
qu’un seul oxygène et aucune insaturation sera forcément soit un alcool soit un éther.
3.6. LES RÈGLES DE FRAGMENTATION
La fragmentation est initiée par l’impact électronique. Seule une faible partie de la
force motrice de fragmentation est de l’énergie transférée lors de l’impact. La majeure partie
de cette force est le radical-cation qui est imposé à la structure.
La fragmentation d’un ion moléculaire à nombre impair d’électrons (radical-cation
M•+) peut se faire par clivage homo- ou hétérolytique d’une liaison simple. Dans le clivage
homolytique (I ), chaque électron se déplace indépendamment comme indiqué par les demi-
flèches : les fragments sont un cation à nombre pair d’électrons et un radical libre (un électron
non-apparié). Par souci de clarté, seule une flèche de chaque paire est représentée (II ).
Dans un clivage hétérolytique (III ), une paire d’électrons « migre » vers le site chargé comme
indiqué par la flèche courbe conventionnelle ; les fragments sont à nouveau un cation à
nombre pair d’électron et un radical, mais ici la charge finale est localisée sur le produit
alkyle.
En absence de cycle (dont la fragmentation requiert le clivage de deux liaisons au
plus), la plupart des fragments majeurs dans un spectre de masse sont des cations à nombre
pair d’électrons formé par un unique clivage. Une fragmentation supplémentaire de ce genre
de cation (IV ) donne en général un autre cation à nombre pair d’électrons et un fragment, ou
une molécule, à nombre pair d’électrons.

TP de chimie théorique et physique
S. Gillet - 124 - MS
Des clivages consécutifs ou simultanés de plusieurs liaisons peuvent se produire quand
un avantage énergétique dérive de la formation d’un cation fortement stabilisé et/ou d’un
radical stable, ou d’une molécule neutre, souvent par l’intermédiaire d’une voie de faible
énergie bien définie.
La probabilité de rupture d’une liaison particulière est reliée à la force de cette liaison,
à la possibilité de transitions de faibles énergies et à la stabilité des fragments chargés et non
chargés formés dans le processus de fragmentation. Notre connaissance des clivages
pyrolytiques peut être utilisée, jusqu’à un certain point, pour prédire les modes probables de
clivage de l’ion moléculaire.
Etant donné la pression extrêmement basse dans un spectromètre de masse, il y a très
peu de collisions entre fragments ; nous avons surtout affaire à des décompositions
unimoléculaires. Cette supposition qui s’appuie sur un grand nombre de spectres de
références, est à la base de l’énorme quantité d’informations disponibles à partir des motifs de
fragmentation d’une molécule.
Certaines règles générales permettant de prédire les pics majeurs dans les spectres EI,
peuvent être écrites et rationalisées en utilisant les concepts standards de la chimie organique
physique.
Ces règles sont énoncées ci-dessous :
1- La hauteur relative du pic de l’ion moléculaire est plus élevée pour un composé à
chaîne linéaire et décroît à mesure que le degré de ramification augmente (voir règle
3).
2- La hauteur relative du pic de l’ion moléculaire, en général, décroît quand le poids
moléculaire augmente dans une série homologue. Les esters gras semblent être une
exception.
3- Le clivage est favorisé au niveau des atomes de carbone substitués d’alkyles. Plus ils
sont substitués, plus le clivage est probable. Ceci est une conséquence de la stabilité
croissante d’un carbocation tertiaire par rapport à un secondaire, lui-même plus stable
qu’un primaire. Ordre de stabilité des cations : CH3+ < R2CH2
+ < R3CH+ < R3C+.
Généralement, le plus gros substituant d’un embranchement est éliminé très

TP de chimie théorique et physique
S. Gillet - 125 - MS
facilement comme radical, probablement parce qu’un radical à longue chaîne peut être
stabilisé par délocalisation de l’électron célibataire.
4- Les liaisons doubles, les structures cycliques et particulièrement les cycles
aromatiques (ou hétéroaromatiques) stabilisent l’ion moléculaire et augmentent ainsi
la probabilité de son apparition.
5- Les doubles liaisons favorisent le clivage allylique et donnent le carbocation allylique
stabilisé par résonance. Cette règle n’est pas valable pour les alcènes simples à cause
de la migration de la liaison double mais elle l’est pour les cyclo-alcènes.
6- Les cycles saturés tendent à perdre les chaînes alkyles latérales au niveau de la liaison
α. Il s’agit simplement d’un cas particulier de ramification (règle 3). La charge
positive tend à rester sur le fragment cyclique.
Les cycles insaturés peuvent subir une réaction de rétro-Diels-Alder.
7- Dans les composés aromatiques substitués par des alkyles, le clivage est le plus
probable au niveau de la liaison en β par rapport au cycle, donnant un ion benzyle
stabilisé par résonance ou plus probablement l’ion tropylium.
8- Les liaisons C-C proches d’un hétéroatome sont fréquemment clivées (scission α),
laissant la charge sur le fragment contenant l’hétéroatome dont les électrons non liants
permettent une stabilisation par résonance (schéma).

TP de chimie théorique et physique
S. Gillet - 126 - MS
9- Le clivage est souvent associé à l’élimination de petites molécules neutres stables,
telles que le monoxyde de carbone, les oléfines, l’eau, l’ammoniac, le sulfure
d’hydrogène, le cyanure d’hydrogène, des mercaptans, des cétènes ou des alcools,
souvent avec réarrangement.
3.7. LES RÉARRANGEMENTS
Les ions ayant subi un réarrangement sont des fragments dont l’origine ne peut être
décrite par simple clivage des liaisons dans l’ion moléculaire, mais sont le résultat d’un
réarrangement atomique intramoléculaire pendant la fragmentation. Les réarrangements
impliquant la migration d’atomes d’hydrogène dans les molécules qui contiennent un
hétéroatome sont très courants. Un exemple important est le réarrangement de McLafferty.
Pour subir un réarrangement de McLafferty, une molécule doit posséder un
hétéroatome (ex : O) placé de façon appropriée, un système π (en général une double liaison)
et un atome d’hydrogène labile en γ par rapport au motif C=O. De tels réarrangements rendent
souvent comptes de pics caractéristiques important et sont donc très utiles. Ils peuvent être
fréquemment rationalisés sur base des transitions de basse énergie et accroissent la stabilité
des produits. Les réarrangements résultant de l’élimination d’une molécule neutre stable sont
courants (par exemple l’alcène produit dans le réarrangement de McLafferty).
Les pics de réarrangement peuvent être reconnus en considérant le nombre de masse
m/z des fragments ioniques et des ions moléculaires correspondants. Un clivage simple (aucun
réarrangement) d’un ion moléculaire de masse paire donne un fragment ionique de masse
impaire et le clivage simple d’un ion moléculaire impair donne un fragment pair.
L’observation de la masse d’un fragment ionique différant d’une unité par rapport à la masse
attendue pour un fragment résultant d’un simple clivage (ex : un fragment de masse paire
provenant d’un ion moléculaire de masse paire) indique qu’un réarrangement d’hydrogène
accompagne la fragmentation.
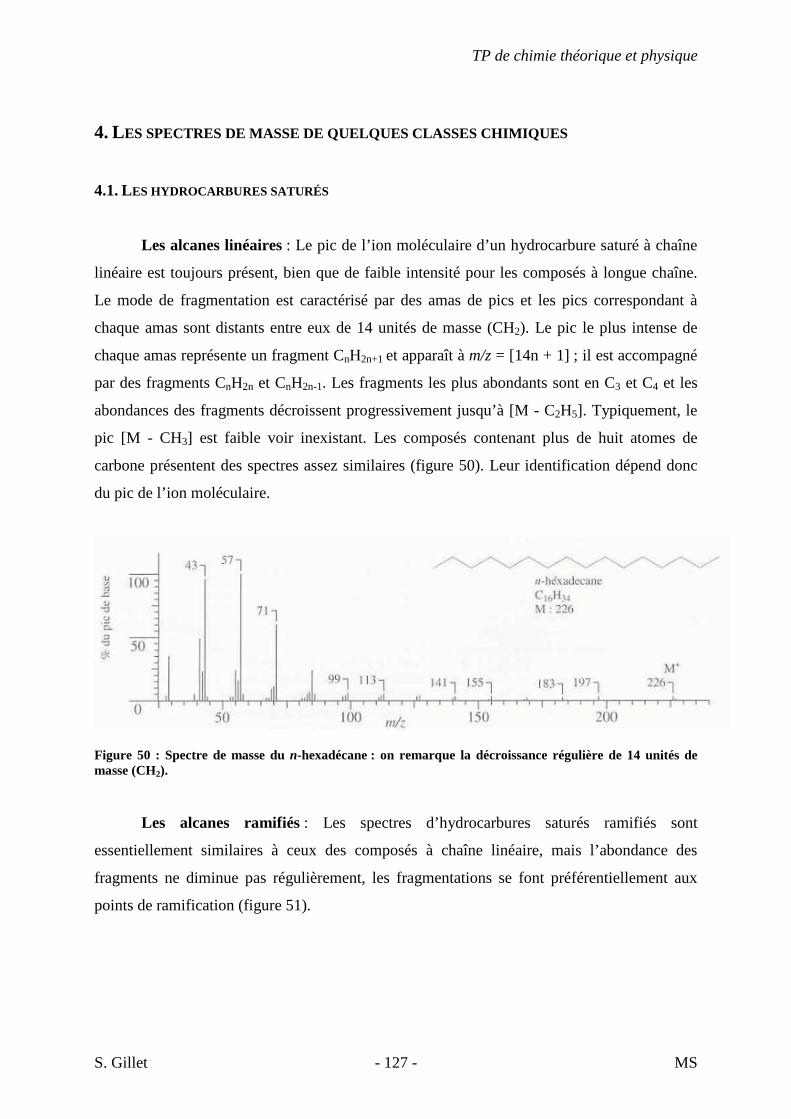
TP de chimie théorique et physique
S. Gillet - 127 - MS
4. LES SPECTRES DE MASSE DE QUELQUES CLASSES CHIMIQUES
4.1. LES HYDROCARBURES SATURÉS
Les alcanes linéaires : Le pic de l’ion moléculaire d’un hydrocarbure saturé à chaîne
linéaire est toujours présent, bien que de faible intensité pour les composés à longue chaîne.
Le mode de fragmentation est caractérisé par des amas de pics et les pics correspondant à
chaque amas sont distants entre eux de 14 unités de masse (CH2). Le pic le plus intense de
chaque amas représente un fragment CnH2n+1 et apparaît à m/z = [14n + 1] ; il est accompagné
par des fragments CnH2n et CnH2n-1. Les fragments les plus abondants sont en C3 et C4 et les
abondances des fragments décroissent progressivement jusqu’à [M - C2H5]. Typiquement, le
pic [M - CH3] est faible voir inexistant. Les composés contenant plus de huit atomes de
carbone présentent des spectres assez similaires (figure 50). Leur identification dépend donc
du pic de l’ion moléculaire.
Figure 50 : Spectre de masse du n-hexadécane : on remarque la décroissance régulière de 14 unités de masse (CH2).
Les alcanes ramifiés : Les spectres d’hydrocarbures saturés ramifiés sont
essentiellement similaires à ceux des composés à chaîne linéaire, mais l’abondance des
fragments ne diminue pas régulièrement, les fragmentations se font préférentiellement aux
points de ramification (figure 51).

TP de chimie théorique et physique
S. Gillet - 128 - MS
Figure 51 : Spectre de masse du 5-méthylpendadécane.
La discontinuité en C12 (168) indique que la branche la plus longue du 5-
méthylpentadécane possède 10 atomes de carbone.
Les pics à m/z 85 et 169 représentent le clivage de part et d’autre de la ramification
avec une rétention de charge sur le carbone substitué. En soustrayant du poids moléculaire la
somme de ces fragments, on rend compte du fragment –CH-CH3. De nouveau, nous
remarquons l’absence de fragment en C11 qui ne peut se former par un clivage unique.
Enfin, la présence d’un pic [M - 15] distinct indique également une ramification
méthyle. Le fragment résultant du clivage à un point de ramification tend à perdre un seul
atome d’hydrogène, et donc le pic résultant CnH2n est prédominant et quelque fois plus intense
que le pic CnH2n+1 correspondant.
Les alcanes cycliques : Un cycle saturé dans un hydrocarbure accroît l’intensité
relative du pic de l’ion moléculaire et favorise le clivage au niveau de la liaison entre le cycle
et le reste de la molécule (figure 52). La fragmentation du cycle est caractérisée en général par
la perte de deux atomes de carbone sous forme de C2H4 (28) et de C2H5 (29). Cette tendance à
perdre des fragments pairs tels que C2H4, donne un spectre qui contient des ions de masse
paire en plus grand nombre qu’un spectre d’hydrocarbure acyclique. Comme dans les
hydrocarbures ramifiés, le clivage C-C s’accompagne de la perte d’un atome d’hydrogène.
Les pics caractéristiques appartiennent donc aux séries CnH2n-1 et CnH2n-2.

TP de chimie théorique et physique
S. Gillet - 129 - MS
Figure 52 : Spectre de masse du cyclohexane.
Le spectre de masse du cyclohexane montre un ion moléculaire bien plus intense que
ceux des composés acycliques, puisque la fragmentation requiert le clivage de deux liaisons
carbone-carbone. Ce spectre a son pic de base à m/z 56 (dû à la perte de C2H4 : m/z 28) et un
grand pic à m/z 41, qui est un fragment de la série CnH2n-1 avec n = 3. On note également la
présence d’un pic à m/z 55 dû à la perte d’un C2H5 (29).
4.2. LES ALCÈNES
Le pic de l’ion moléculaire des alcènes, surtout des poly-alcènes, est en général visible
(figure 53). La localisation de la double liaison dans les alcènes acycliques est rendue difficile
par sa migration aisée dans les fragments. Dans les alcènes cycliques (surtout polycycliques),
la localisation de la double liaison est souvent évidente du fait de la forte tendance au clivage
allylique avec peu de migration de la double liaison. La position de la double liaison peut
également être fixée par conjugaison avec un carbonyle. Comme avec les hydrocarbures
saturés, les alcènes acycliques sont caractérisés par des amas de pics à intervalles de 14 unités.
Dans ces amas, les pics CnH2n-1 et CnH2n sont plus intenses que les pics CnH2n+1.
Le pic important à m/z 41 que l’on retrouve sur les spectres d’alcène est dû au
groupement +CH2-CH=CH2.

TP de chimie théorique et physique
S. Gillet - 130 - MS
Figure 53 : spectre de masse du ββββ-myrcène.
Dans le spectre du β-myrcène, les pics à m/z 41, 55, et 69 correspondent à la formule
CnH2n-1 avec n = 3, 4 et 5, respectivement.
La formation du pic à m/z 41 doit impliquer une isomérisation. Les pics à m/z 67 et 69
sont les fragments provenant de la rupture d’une liaison bi-allylique.
Le pic à m/z 93 est une structure C7H9+ formée par isomérisation (provoquant une
conjugaison accrue), suivi d’un clivage allylique. L’ion à m/z 93 possède au moins deux
formes de résonance importantes contribuant à sa stabilité.
Les alcènes cycliques montrent, en général, un pic d’ion moléculaire marqué. La
réaction de clivage rétro-Diels-Alder est caractéristique.

TP de chimie théorique et physique
S. Gillet - 131 - MS
Dans cet exemple, la réaction conduit à deux molécules d’isoprène. Puisque cette
réaction est un exemple de réarrangement, l’un des fragments est une molécule neutre.
4.3. LES HYDROCARBURES AROMATIQUES
Un cycle aromatique dans une molécule stabilise l’ion moléculaire, dont le pic est, en
général, suffisamment important pour que des mesures d’intensité précises puissent être faites
sur les pics [M + 1] et [M + 2].
Un pic prédominant (souvent le pic de base) à m/z 91 (C6H5CH2+) indique un benzène
substitué par un alkyle. La ramification sur le carbone en α conduit à des masses supérieures à
91 par incrément de 14, le plus gros substituant étant éliminé en premier. Cependant la simple
présence d’un pic de masse 91 ne veut pas dire qu’il y a ramification sur le carbone en α car
ce fragment hautement stabilisé peut provenir de réarrangements. Un pic [M - 1] marqué, et
parfois prédominant, résulte d’un clivage benzylique similaire de la liaison C-H.
On a proposé que, dans la plupart des cas, l’ion de masse 91 soit un cation tropylium
plutôt qu’un cation benzylique. Cela explique la facilité avec laquelle les xylènes perdent un
méthyle alors que cela est difficile dans le cas du toluène. L’ion moléculaire à peine créé se
réarrange en ion radical méthylcycloheptatriène parent, qui se clive à son tour pour donner
l’ion tropylium (C7H7+). Le pic souvent observé à m/z 65 résulte de l’élimination d’une
molécule d’acétylène neutre à partir de l’ion tropylium.

TP de chimie théorique et physique
S. Gillet - 132 - MS
La migration d’hydrogène avec élimination d’une molécule d’alcène neutre rend
compte du pic à m/z 92 que l’on observe quand le groupe alkyle contient plus de deux
carbones.
Un amas caractéristique d’ions dû à un clivage en α et à la migration d’hydrogène
dans les mono-alkylbenzènes apparaît à m/z 77 (C6H5+), 78 (C6H6
+) et 79 (C6H7+). Les
polyphényles alkylés et les hydrocarbures aromatiques polycycliques alkylés montrent le
même clivage en β que les composés alkyl benzène.
4.4. LES COMPOSÉS HALOGÉNÉS
Lorsque des composés halogénés sont présents dans la molécule, il n’est plus possible
de déterminer la formule moléculaire à partir des intensités des pics isotopiques [M + 1] et [M
+ 2] (section 3.4.).
Par contre, l’amas isotopique généré par la présence d’halogènes fournit de précieuses
informations quant à la nature de ces composés (figures 54 et 55). Ainsi, un composé qui
contient un atome de chlore aura un pic [M + 2] dont l’intensité vaudra approximativement un
tiers de celle du pic de l’ion moléculaire.
En général, le nombre d’atomes de chlore et/ou de brome peut-être déduit du nombre
de pics au-delà du pic moléculaire.
Figure 54 : Intensité relative des pics isotopiques pour des combinaisons de chlore et de brome.
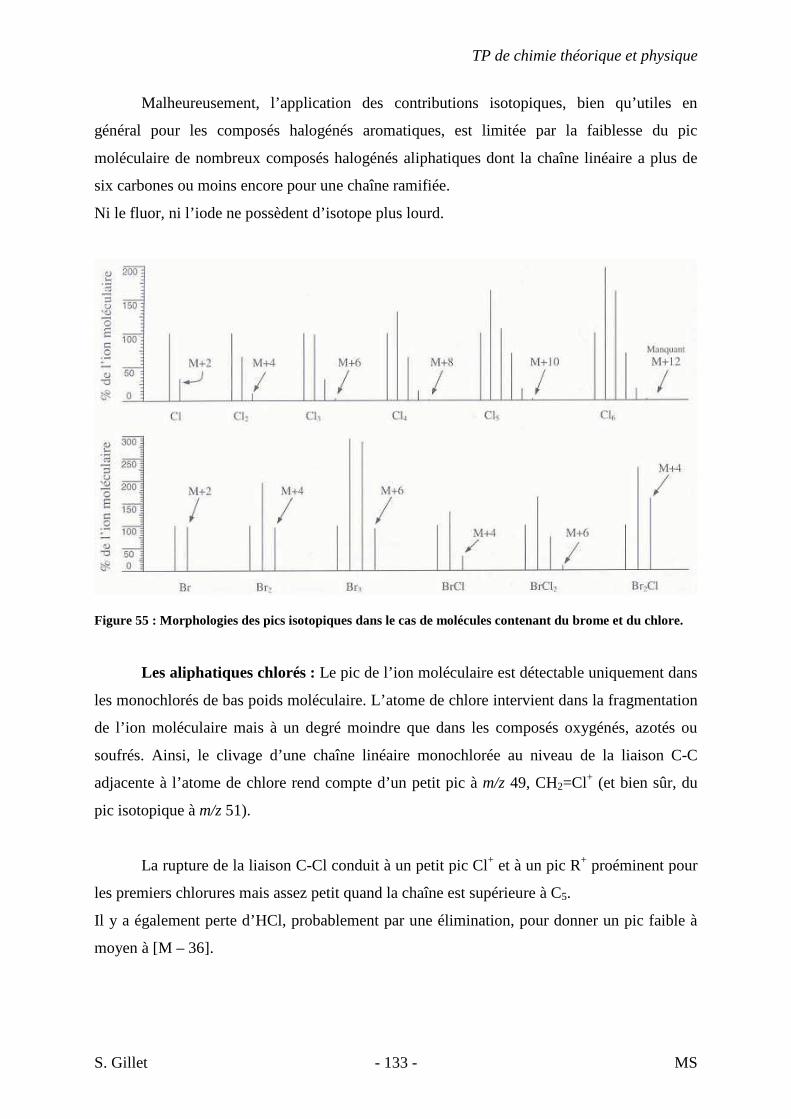
TP de chimie théorique et physique
S. Gillet - 133 - MS
Malheureusement, l’application des contributions isotopiques, bien qu’utiles en
général pour les composés halogénés aromatiques, est limitée par la faiblesse du pic
moléculaire de nombreux composés halogénés aliphatiques dont la chaîne linéaire a plus de
six carbones ou moins encore pour une chaîne ramifiée.
Ni le fluor, ni l’iode ne possèdent d’isotope plus lourd.
Figure 55 : Morphologies des pics isotopiques dans le cas de molécules contenant du brome et du chlore.
Les aliphatiques chlorés : Le pic de l’ion moléculaire est détectable uniquement dans
les monochlorés de bas poids moléculaire. L’atome de chlore intervient dans la fragmentation
de l’ion moléculaire mais à un degré moindre que dans les composés oxygénés, azotés ou
soufrés. Ainsi, le clivage d’une chaîne linéaire monochlorée au niveau de la liaison C-C
adjacente à l’atome de chlore rend compte d’un petit pic à m/z 49, CH2=Cl+ (et bien sûr, du
pic isotopique à m/z 51).
La rupture de la liaison C-Cl conduit à un petit pic Cl+ et à un pic R+ proéminent pour
les premiers chlorures mais assez petit quand la chaîne est supérieure à C5.
Il y a également perte d’HCl, probablement par une élimination, pour donner un pic faible à
moyen à [M – 36].

TP de chimie théorique et physique
S. Gillet - 134 - MS
Les aliphatiques bromés : Les remarques faites au sujet des aliphatiques chlorés
s’appliquent dans l’ensemble aux composés bromés correspondant.
Les aliphatiques iodés : Les aliphatiques iodés donnent le pic moléculaire le plus
intense parmi les aliphatiques halogénés. Puisque l’iode et mono-isotopique, il n’y a pas de
pic isotopique distinct. On peut remarquer sur les spectres un pic à 127 correspondant à I•
ainsi qu’un pic important à [M – 127].
Les aliphatiques fluorés : Les aliphatiques fluorés donnent le pic moléculaire le
moins intense parmi les aliphatiques halogénés. La rupture de la liaison C-C α,β est moins
importante que dans les autres mono-halogénés, mais la rupture de la liaison C-H sur le
carbone en α est plus importante. Sur le spectre, on note la présence d’un pic important à [M
– HF].
4.5 LES ALCOOLS
Les alcools aliphatiques : Le pic de l’ion moléculaire d’un alcool primaire ou
secondaire est très faible et souvent indétectable pour un alcool tertiaire. Le recours à d’autres
techniques d’ionisation est donc parfois nécessaire.
En général, il y a clivage du lien C-C voisin de l’atome d’oxygène. Ainsi, les alcools
primaires montrent un pic prédominant dû à +CH2-OH (m/z 31). Les alcools secondaires et
tertiaires se clivent de manière analogue pour donner un pic principal dû respectivement à +CHR-OH (m/z 45, 59, 73, etc.) et CRR’-OH (m/z 59, 73, 87, etc.). Le substituant le plus gros
est expulsé le plus rapidement. Occasionnellement, la liaison C-H en α de l’atome d’oxygène
est rompue ; ce chemin, moins favorable donne naissance à un pic à [M – 1].
Les alcools primaires (figure 56), en plus du clivage principal C-C en α de l’atome
d’oxygène, présentent une série de pics homologues d’intensité progressivement décroissante
résultant des clivages successifs de liaisons C-C à partir de l’atome d’oxygène. Dans les
alcools à longue chaîne (> C6), la fragmentation est dominée par le motif de fragmentation des
hydrocarbures ; de fait, le spectre ressemble à celui de l’alcène correspondant. Au voisinage

TP de chimie théorique et physique
S. Gillet - 135 - MS
du pic de l’ion moléculaire, très faible, voir inexistant, le spectre d’un alcool primaire est
parfois compliqué par de faibles pics [M – 2] et [M – 3].
Habituellement, un pic [M – 18] marqué, et parfois prédominant, résulte de la perte
d’une molécule d’eau. On le remarque plus dans les spectres d’alcools primaires. Cette
élimination par impact électronique est justifiée par la perte d’un hydrogène en δ.
Le pic [M – 18] est souvent amplifié par la décomposition thermique des alcools
supérieurs sur les surfaces chaudes de l’injecteur. L’élimination d’eau, en même temps que
celle d’un alcène à partir des alcools primaires, rend compte de la présence d’un pic à [M -
(alcène + H2O)], ce qui donne des pics à [M – 46], [M – 74], [M – 102], etc.
Les alcools cycliques : Les alcools cycliques subissent une fragmentation complexe,
On constate sur de telles molécules des pertes d’eau, d’éthène et d’un méthyle.
Figure 56 : spectre de masse du pentanol.
Les alcools benzyliques : Chez les alcools benzyliques, le pic parent est en général
intense. Un pic benzylique d’intensité modérée [M – OH] peut-être présent comme on peut
l’attendre à partir du clivage en β du cycle. Une séquence complexe conduit aux pics
principaux [M – 1], [M – 2] et [M – 3]. L’alcool benzylique lui-même se fragmente pour

TP de chimie théorique et physique
S. Gillet - 136 - MS
donner dans l’ordre, l’ion [M – 1], l’ion C6H7+ par élimination de CO et l’ion C6H5
+ par perte
de H2. Il est également courant d’avoir un pic marqué à [M - 18] dû à la perte de H2O.
Les phénols : Au niveau des phénols, un pic d’ion moléculaire très visible facilite leur
l’identification (figure 57). Dans le cas du phénol proprement dit, le pic de l’ion moléculaire
est le pic de base et le pic [M – 1] est petit. Les phénols présentent habituellement un pic de
réarrangement à m/z 77 et également des pics résultants de la perte de CO [M-28] et de CHO
[M-29].
Figure 57 : spectre de masse de l’o-éthylphénol.
4.6. LES ÉTHERS
Les éthers aliphatiques : Le pic de l’ion moléculaire (situé deux unités de masse plus
haut que l’analogue hydrocarboné) est petit, mais des échantillons plus important rendent, en
général, le pic moléculaire, ou le pic [M + 1], évident (transfert de H• au moment de la
collision ion-molécule). La présence d’un atome d’oxygène peut être déduite des pics intenses
à m/z 31, 45, 59, 73, etc. (figure 58). Ces pics représentent les fragments RO+ et ROCH2+. La
fragmentation se fait selon deux voies principales :
1. Rupture de la liaison C-C voisine de l’atome d’oxygène. L’un ou l’autre des ions
contenant l’atome d’oxygène peut être le pic de base.
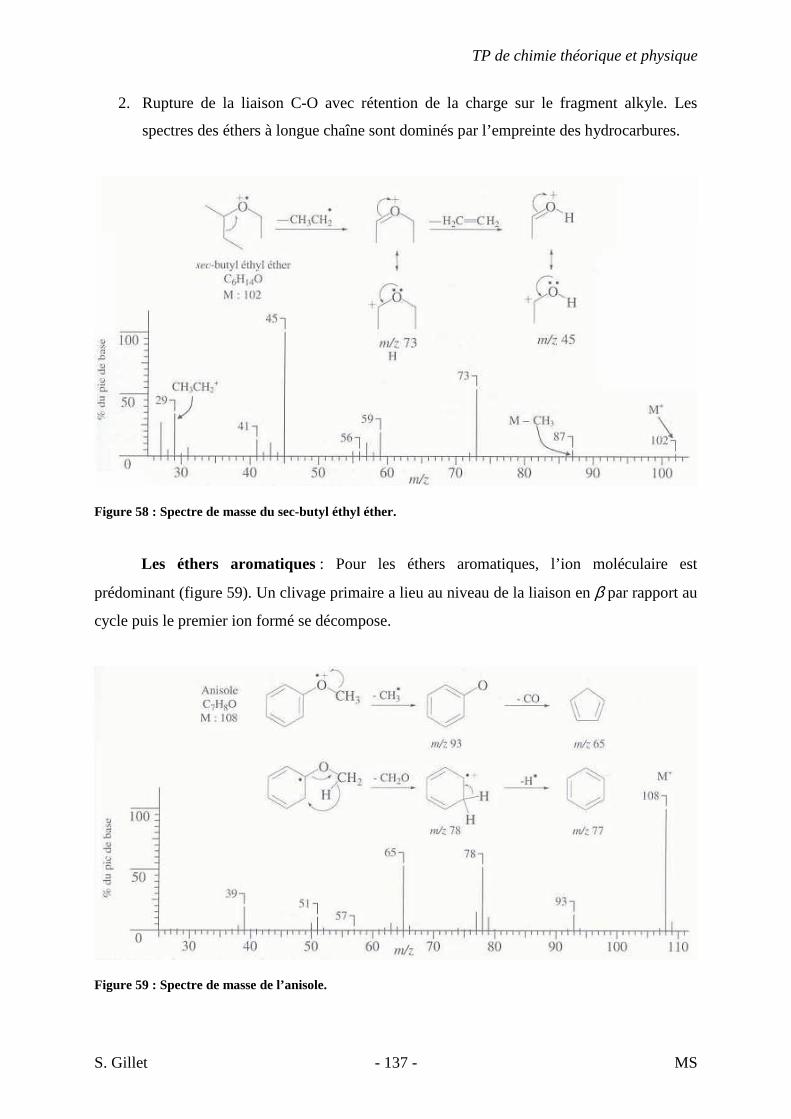
TP de chimie théorique et physique
S. Gillet - 137 - MS
2. Rupture de la liaison C-O avec rétention de la charge sur le fragment alkyle. Les
spectres des éthers à longue chaîne sont dominés par l’empreinte des hydrocarbures.
Figure 58 : Spectre de masse du sec-butyl éthyl éther.
Les éthers aromatiques : Pour les éthers aromatiques, l’ion moléculaire est
prédominant (figure 59). Un clivage primaire a lieu au niveau de la liaison en β par rapport au
cycle puis le premier ion formé se décompose.
Figure 59 : Spectre de masse de l’anisole.

TP de chimie théorique et physique
S. Gillet - 138 - MS
4.7. LES CÉTONES
Les cétones aliphatiques : Le pic de l’ion moléculaire des cétones, est généralement
assez prononcé. Les pics de fragmentation majeurs des cétones aliphatiques résultent du
clivage des liaisons C-C adjacentes à l’atome d’oxygène, la charge restant sur l’ion acylium
stabilisé par résonance.
Ainsi, comme avec les alcools et les éthers, le clivage est facilité par la présence de
l’atome d’oxygène. Ces clivages donnent naissance à des pics à m/z 43, 57 ou 71, etc. Le pic
de base résulte très souvent de la perte du groupe alkyle le plus important.
Quand une des chaîne alkyle liées au groupe C=O, est en C3 ou plus longue, la rupture
du lien C-C à une liaison du groupe C=O (liaison α,β), à lieu avec migration d’hydrogène
pour donner un pic principal. C’est le réarrangement de McLafferty. Le simple clivage du
lien α,β qui n’a jamais lieu, donnerait un ion de faible stabilité car il aurait deux centres
adjacents positifs.
Il faut également noter que les pics hydrocarbonés des cétones à longues chaînes ne
peuvent être distingués des pics acyles, puisque la masse d’une unité C=O (28) est la même
que celle de deux unités méthylène.
Les cétones cycliques : Pour les cétones cycliques, l’ion moléculaire est également
assez prononcé (figure 60). Comme pour les cétones aliphatiques, le premier clivage des
cétones cycliques est adjacent au groupe C=O, mais l’ion ainsi formé doit subir d’autres
clivages pour produire un fragment.

TP de chimie théorique et physique
S. Gillet - 139 - MS
Figure 60 : Spectre de masse de la cyclohexanone.
Les cétones aromatiques : Chez les cétones aromatiques, l’ion moléculaire est lui
aussi bien évidement très visible. Leur clivage se produit au niveau de la liaison en β par
rapport au cycle, laissant le fragment caractéristique ArC≡O+ (m/z 105) qui donne
généralement le pic de base. La perte d’un CO à partir de ce fragment donne l’ion aryle. La
rupture de la liaison adjacente au cycle pour former un fragment R≡CO+ est moins
importante.
4.8. LES ALDÉHYDES
Les aldéhydes aliphatiques : Le pic de l’ion moléculaire des aldéhydes aliphatiques
est généralement apparent. Les ruptures des liaisons C-H et C-C voisines de l’atome
d’oxygène résultent en un pic [M – 1] et [M – R] (m/z 29, CHO+). Le pic [M – 1] est
caractéristique même pour les aldéhydes à longues chaînes (figure 61), mais le pic à m/z 29
présent dans les aldéhydes en C4 et plus est dû à l’ion hydrocarboné C2H5+.
Dans ces aldéhydes, le clivage de McLafferty de la liaison C-C α,β donne un pic
majeur à m/z 44, 58 ou 72, etc. selon les substituants en α. Il s’agit de l’ion stabilisé par
résonance et formé durant l’état de transition cyclique.

TP de chimie théorique et physique
S. Gillet - 140 - MS
Dans les aldéhydes à chaîne linéaire, les autres pics caractéristiques sont à [M – 18]
(perte d’eau), [M – 28] (perte d’éthylène), [M – 43] (perte de CH2=CH-O•) et [M – 44] (perte
de CH2=CH-OH).
Figure 61 : Spectre de masse du nonanal.
Les aldéhydes aromatiques : Quant aux aldéhydes aromatiques, ils sont caractérisés
par un pic d’ion moléculaire grand et par un pic [M – 1] (Ar-C≡O+) toujours important
pouvant être plus grand que le pic de l’ion moléculaire. L’ion [M – 1], C6H5C≡O+ libère CO
pour donner l’ion phényle (m/z 77), qui à son tour élimine une molécule d’acétylène pour
donner l’ion C4H3+ (m/z 51).
4.9. LES ACIDES CARBOXYLIQUES
Les acides carboxyliques aliphatiques : Le pic de l’ion moléculaire d’un acide
carboxylique linéaire est faible bien que généralement présent. Le pic le plus caractéristique
(parfois le pic de base) à m/z 60 est dû à un réarrangement de McLafferty (figure 62). Une
ramification sur le carbone en α accentue ce clivage.

TP de chimie théorique et physique
S. Gillet - 141 - MS
Dans les acides à chaîne courte, les pics à [M – OH] et [M - CO2H] sont
prédominants : ils représentent le clivage des liaisons voisines du C=O. Dans les acides à
longue chaîne, le spectre consiste en deux séries de pics résultant de la rupture de chaque
liaison C-C avec rétention de la charge soit sur le fragment contenant l’atome d’oxygène (m/z
45, 59, 73, 87, etc.) soit sur le fragment alkyle (m/z 29, 43, 57, 71, 85, etc.). Le motif de
fragmentation des hydrocarbures montre aussi des pics à m/z 27, 28, 41, 42, 55, 56, 69, 70,
etc. En résumé, à coté du pic de réarrangement de McLafferty, le spectre des acides à
longue chaîne ressemble aux séries d’amas des hydrocarbures à intervalle de masse de 14
unités.
Figure 62 : Spectre de masse de l’acide décanoïque.
Les acides carboxyliques aromatiques : Chez les acides aromatiques, le pic de l’ion
moléculaire prédomine. Les autres pics majeurs sont formés par la perte de OH [M – 17] et de
COOH [M – 45].
Notons que si un groupe en ortho porte un hydrogène, il y a majoritairement perte de
H2O [M – 18].

TP de chimie théorique et physique
S. Gillet - 142 - MS
4.10. LES ESTERS
Les esters aliphatiques : Le pic de l’ion moléculaire d’un ester méthylique dérivant
d’un acide aliphatique linéaire est généralement marqué (même si un peu plus faible entre m/z
130 et 200). Le pic le plus caractéristique est du au réarrangement de McLafferty et au
clivage d’une liaison αβ par rapport au carbone du groupe C=O. Ainsi, un ester ramifié sur le
carbone en α donne un pic important à m/z 74 (figure 63). Le fragment portant la fonction
alcool et/ou le substituant en α peut souvent être déduit de la position du pic résultant de ce
clivage.
Pour un ester quelconque, quatre ions peuvent résulter d’une scission au voisinage de
C=O.
L’ion R+ domine dans le cas des esters courts mais décroît rapidement lorsque la
longueur de la chaîne augmente. L’ion R-C≡O+ donne un pic caractéristique des esters. Les
ions [OR’]+ et [C(=O)OR’]+ sont généralement peu importants.
Lorsque la partie acide est prédominante, on constate un clivage à chaque liaison C-C
qui donne un ion alkyle (m/z 29, 43, 57, etc.) et un ion contenant les atomes d’oxygène CnH2n-
1O2+ (m/z 59, 73, 87, etc.) Le pic (m/z 87) de l’ion [CH2CH2COOCH3]
+ est toujours plus
intense que ses homologues sans explication claire à cela (figure 31).
Figure 63 : Spectre de masse de l'octanoate de méthyle.

TP de chimie théorique et physique
S. Gillet - 143 - MS
Lorsque la partie alcool est prédominante, il y a élimination d’une molécule d’acide de
la même manière que les alcools éliminent l’eau. Les esters d’alcools à longue chaîne
montrent un pic caractéristique à m/z 61, 75 ou 89, etc. provenant de l’élimination de la partie
alkyle et du transfert de deux atomes d’hydrogène vers le fragment contenant les atomes
d’oxygène, qui est par essence l’acide carboxylique protoné.
Les esters aromatiques : Les esters d’acides aromatiques ont un pic d’ion moléculaire
prédominant. Plus la taille de la partie alcool augmente, plus l’intensité du pic de l’ion
moléculaire décroît rapidement. Le pic de base résulte de l’élimination de RO•, et
l’élimination de •COOR rend compte d’un autre pic prédominant.
Quand la partie alkyle s’allonge, trois modes de clivages deviennent importants :
- le réarrangement de McLafferty
- Le réarrangement de deux atomes d’hydrogènes avec élimination d’un radical alkyle
- La rétention de la charge positive par le groupe alkyle
4.11. LES AMINES
Les amines aliphatiques : Le pic de l’ion moléculaire d’une amine aliphatique est
impair, généralement plutôt faible, et indétectable dans les amines à longues chaînes ou
fortement ramifiée. Le pic de base résulte souvent d’un clivage C-C proche de l’atome
d’azote ; pour les amines primaires non ramifiées sur le carbone en α, il est à m/z 30
(CH2NH2+) Ce clivage rend compte du pic de base pour toutes les amines primaires ainsi que
pour les amines secondaires et tertiaires non ramifiées sur le carbone en α. La perte de la
ramification la plus importante du carbone en α est favorisée.
En l’absence de ramification sur le carbone en α, un pic [M – 1] est généralement
détecté. Il s’agit du même type de clivage que celui des alcools.

TP de chimie théorique et physique
S. Gillet - 144 - MS
Les amines primaires à chaîne linéaire présentent une série de pics homologues dont
l’intensité décroît progressivement à m/z 30, 44, 58, etc. résultant des clivages des liaisons C-
C de plus en plus éloignées de l’atome d’azote avec rétention de la charge sur le fragment
contenant l’azote. Ces pics sont accompagnés du motif de fragmentation des ions
hydrocarbures CnH2n+2, CnH2n, CnH2n-1.
Des fragmentations cycliques semblent également apparaître pendant la fragmentation
des amines à longue chaîne.
Bien que non concluant, un pic à m/z 30 permet de mettre en évidence une amine
primaire linéaire. La décomposition supplémentaire du premier ion formé à partir d’une amine
secondaire ou tertiaire, conduit à un pic à m/z 30, 44, 58, 72, etc. C’est un processus
semblable à celui décrit pour les alcools et les éthers aliphatiques.
Les amines cycliques : Les pics d’ions moléculaires des amines cycliques sont
habituellement intenses à moins qu’il n’y ait substitution en position α. Le clivage primaire
des liaisons voisines de l’atome d’azote conduit soit à la perte d’un atome d’hydrogène α pour
donner un pic [M – 1] important, soit à l’ouverture du cycle. Cette dernière possibilité est
suivie d’élimination d’éthylène pour donner •CH2N+H=CH2 (m/z 43, pic de base) puis par la
perte d’un atome d’hydrogène pour donner CH2=N+=CH2 (m/z 42).
Les amines aromatiques : Le pic d’ion moléculaire d’une amine aromatique est
intense. Dans le cas d’une amine primaire, on constate la perte d’un proton et d’une molécule
neutre HCN conduisant à des pics prédominants à m/z 66 et 65.
4.12. LES AMIDES
Les amides aliphatiques : Le pic de l’ion moléculaire des amides aliphatiques linéaire
est généralement présent. Les modes de clivage dominants dépendent de la longueur de la
partie acyle et de la longueur et du nombre des groupes alkyles liés à l’atome d’azote.

TP de chimie théorique et physique
S. Gillet - 145 - MS
Le pic de base (m/z 59, H2NC(=OH+)CH2•) présent pour toutes les amides primaires à
chaîne linéaires supérieures à la propionamide résulte du réarrangement de McLafferty. La
ramification sur le carbone en α (CH3, etc.) donne un pic homologue à m/z 73 ou 87, etc.
Les amides primaires donnent un pic important à m/z 44 dû à la rupture de la liaison
R-CONH2 (O=C=+NH2), c’est le pic de base dans les amides primaires en C1-C3. Un pic
modéré à m/z 86 résulte d’un clivage C-C γ,δ, pouvant s’accompagner d’une cyclisation.
Les amides secondaires et tertiaires portant un hydrogène sur le carbone en γ de la
partie acyle et des groupes méthyles sur l’atome d’azote présentent un pic dominant résultant
du réarrangement de McLafferty. Quand les groupes N-alkyles sont en C2 ou plus et que la
partie acyle est plus courte que C3, un autre mode de clivage prédomine. C’est le clivage du
groupe N-alkyle en β de l’atome d’azote, et le clivage de la liaison C-N (C du carbonyle) avec
migration d’un atome d’hydrogène en α de la partie acyle et libération de +NH2=CH2 (m/z
30).
Les amides aromatiques : Pour les amides aromatiques, dont la benzamide est un
exemple type, on note la perte de NH2 à partir de l’ion moléculaire conduisant à un cation
benzoyle stabilisé par résonance qui subit à son tour un clivage pour donner le cation phényle.
Une autre voie de fragmentation donne naissance à un pic d’intensité modeste à m/z 44.
4.13. LES NITRILES
Les nitriles aliphatiques : Les pics d’ion moléculaires des nitriles aliphatiques sont
faibles ou absents, mais le pic [M + 1] peut être repéré, en général, par son comportement en
augmentant la quantité d’échantillon. Un pic [M – 1] faible, mais utile pour le diagnostique,
est formé par perte d’un hydrogène en α avec formation de l’ion stable : RCH=C=N+.

TP de chimie théorique et physique
S. Gillet - 146 - MS
Le pic de base des nitriles à chaîne linéaire comprise entre C4 et C9 est à m/z 41. Ce pic
correspond à l’ion résultant d’un transfert d’hydrogène dans un état de transition formant un
cycle à six centres. La présence de ce pic ne permet pas de conclure quant à la présence d’un
nitrile ou non, car le pic C3H5, présent dans toutes les chaînes carbonées a le même rapport
m/z.
Un pic à m/z 97 est caractéristique et intense dans les nitriles à chaîne linéaire en C8 et
plus.
Un simple clivage de chaque liaison C-C tout au long de la chaîne (sauf celle voisine
de l’atome d’azote) donne une série caractéristique de pics homologues de masse paire (m/z
40, 54, 68, 82, etc.) due aux ions (CH2)nC≡N+.
4.14. LES COMPOSÉS NITRÉS
Les composés nitrés aliphatiques : Le pic de l’ion moléculaire (nombre pair) d’un
composé aliphatique mono-nitré est faible ou absent. Les pics principaux sont attribuables aux
fragments hydrocarbonés jusqu’à [M - NO2]. La présence du groupe nitro est indiquée par un
pic appréciable à m/z 30 (NO+) et un pic plus faible de masse 46 (NO2).
Les composés nitrés aromatiques : Les composés nitrés aromatiques ont un pic d’ion
moléculaire intense (nombre impair quand il n’y a qu’un atome d’azote). Les pics
prédominants résultent de l’élimination d’un radical NO2 ([M – 46], pic de base du
nitrobenzène), et d’une molécule neutre de NO avec réarrangement pour former le cation
phénoxy [M – 30].
La perte d’acétylène à partir de l’ion [M – 46] justifie le pic intense à [M – 72] et la
perte de CO à partir de l’ion [M – 30] donne un pic à [M – 58].

TP de chimie théorique et physique
S. Gillet - 147 - MS
4.15. LES NITRITES
Le pic de l’ion moléculaire (nombre impair) des nitrites aliphatiques (contenant un N)
est faible ou absent. Le pic à m/z 30 (NO+) est toujours intense et est souvent le pic de base. Il
y a un grand pic à m/z 60 (CH2=+ONO) pour tous les nitrites non ramifiés sur le carbone α.
Une ramification en α peut être identifiée par un pic à m/z 74, 88 ou 102, etc.
L’absence d’un grand pic à m/z permet de les différencier des composés nitrés. Les
pics hydrocarbonés sont prédominants et leurs distribution et intensités décrivent
l’arrangement de la chaîne carbonée.
4.16. LES NITRATES
Le pic de l’ion moléculaire (nombre impair) d’un nitrate aliphatique (contenant un seul
N) est faible ou absent. Un pic prédominant (souvent le pic de base) est formé par rupture de
la liaison C-C voisine du groupe ONO2 avec perte du groupe alkyle attaché au carbone α, le
plus lourd. Le pic NO2+ à m/z 46 est également dominant. Comme dans le cas des nitrites
aliphatiques, les fragments ioniques des hydrocarbures sont présents.
4.17. LES COMPOSÉS SOUFRÉS
La contribution de 4,4% de l’isotope 34S au pic [M + 2] et souvent au pic [fragment +
2], permet de reconnaître facilement les composés contenant du soufre. Une série homologue
de fragments soufrés est décalée de plus de quatre unités de masse par rapport à la série des
fragments hydrocarbonés. Le nombre d’atome de soufre peut être déterminé par l’importance
de la contribution de l’isotope 34S au pic [M + 2].
Les mercaptans aliphatiques (Thiols) : le pic de l’ion moléculaire des mercaptans
aliphatiques est intense sauf dans le cas des mercaptans tertiaires supérieurs. Le pic [M + 2]
est de ce fait mesurable. Les modes de clivages sont souvent proches de ceux des alcools. La
rupture de la liaison C-C (liaison α,β) voisine du groupe SH donne l’ion caractéristique
CH2=SH+ (m/z 47). Le soufre stabilise nettement moins ce fragment que l’azote mais mieux

TP de chimie théorique et physique
S. Gillet - 148 - MS
que l’oxygène. Le clivage de la liaison β,γ donne un pic à m/z 61 d’une intensité moitié
moindre que celle du pic à m/z 47. Le clivage au niveau de la liaison γ,δ donne un petit pic à
m/z 75 et le clivage au niveau de la liaison δ,ε donne un pic à m/z 89 qui est plus intense que
celui à m/z 75. Il est probable que l’ion à m/z 89 est stabilisé par cyclisation :
Les mercaptans primaires éliminent également H2S pour donner un pic intense à m/z
34, l’ion résultant perd ensuite une molécule d’éthylène : donnant naissance aux séries
homologues [M - H2S - (CH2=CH2)n].
Les mercaptans secondaires et tertiaires se clivent au niveau du carbone en α avec
perte du groupe le plus gros pour donner un pic dominant [M - CH3], [M - C2H5], [M - C3H7],
etc. Cependant, un pic à m/z 47 peut aussi apparaître comme un pic de réarrangement de
mercaptans secondaires et tertiaires. Un pic à [M – 33] (perte de HS) est habituellement
présent dans les mercaptans secondaires.
Dans les mercaptans à longue chaîne, le motif hydrocarboné se superpose au motif
mercaptan.
Les sulfures aliphatiques : Le pic de l’ion moléculaire des sulfures aliphatiques est
généralement suffisamment intense pour pouvoir mesurer le pic [M + 2] avec précision. Les
modes de clivages sont proches de ceux des éthers. La rupture de l’une ou l’autre des liaisons
C-C α,β avec perte du groupe le plus gros est favorisée. Ces ions formés en premier lieu
évoluent par transfert d’hydrogène et élimination d’un alcène. Les étapes rencontrées pour les
éthers aliphatiques se présentent également dans le cas des sulfures. Le résultat final est l’ion
RCH=SH+.
Pour un sulfure non ramifié sur un des carbone δ, cet ion est CH2=SH+ (m/z 47), et son
intensité peut prêter à confusion avec le même ion provenant d’un mercaptan ; cependant,
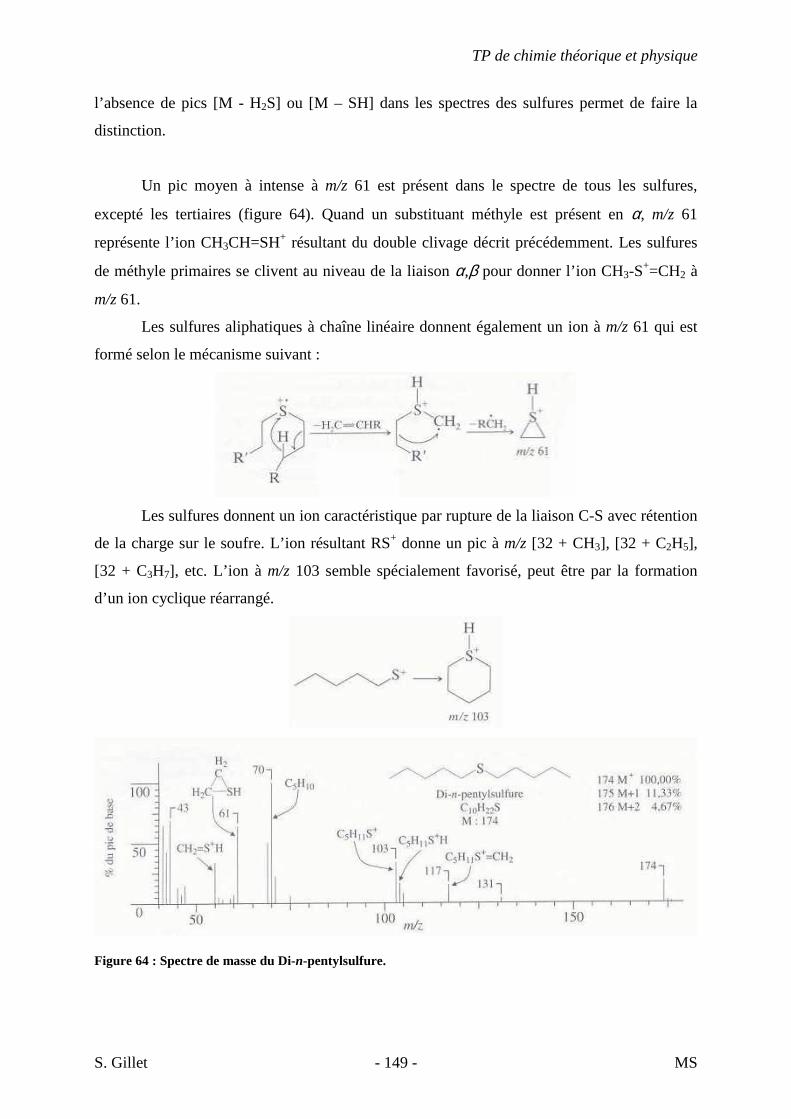
TP de chimie théorique et physique
S. Gillet - 149 - MS
l’absence de pics [M - H2S] ou [M – SH] dans les spectres des sulfures permet de faire la
distinction.
Un pic moyen à intense à m/z 61 est présent dans le spectre de tous les sulfures,
excepté les tertiaires (figure 64). Quand un substituant méthyle est présent en α, m/z 61
représente l’ion CH3CH=SH+ résultant du double clivage décrit précédemment. Les sulfures
de méthyle primaires se clivent au niveau de la liaison α,β pour donner l’ion CH3-S+=CH2 à
m/z 61.
Les sulfures aliphatiques à chaîne linéaire donnent également un ion à m/z 61 qui est
formé selon le mécanisme suivant :
Les sulfures donnent un ion caractéristique par rupture de la liaison C-S avec rétention
de la charge sur le soufre. L’ion résultant RS+ donne un pic à m/z [32 + CH3], [32 + C2H5],
[32 + C3H7], etc. L’ion à m/z 103 semble spécialement favorisé, peut être par la formation
d’un ion cyclique réarrangé.
Figure 64 : Spectre de masse du Di-n-pentylsulfure.

TP de chimie théorique et physique
S. Gillet - 150 - MS
Les disulfures aliphatiques : Le pic moléculaire, au moins jusqu’aux disulfures en
C10, est important. Un pic majeur résulte de la rupture d’une des liaisons C-S avec rétention
de la charge sur le fragment alkyle. Un autre pic majeur est issu du même clivage avec
transfert d’un atome d’hydrogène pour former le fragment RSSH, qui conserve la charge. Les
autres pics résultent apparemment du clivage entre les atomes de soufre sans réarrangement et
avec migration d’un ou deux hydrogènes pour donner respectivement [RS+], [RS+ - 1] et
[RS+ - 2].
4.18. LES COMPOSÉS HÉTÉROAROMATIQUES
Le pic moléculaire des hétéroaromatiques alkylés et non alkylés est intense. La rupture
de la liaison en β du cycle est la règle générale.
La localisation de la charge de l’ion moléculaire sur l’hétéroatome plutôt que sur le
cycle aromatique, justifie de manière satisfaisante le mode de clivage observé.
Les cycles hétéroaromatiques à cinq chaînons présentent des schémas de clivage du
cycle très similaires. Dans tous les cas, la première étape est la rupture de la liaison carbone-
hétéroatome, suivie de la perte, soit d’une molécule neutre d’acétylène, soit d’un fragment
radicalaire.
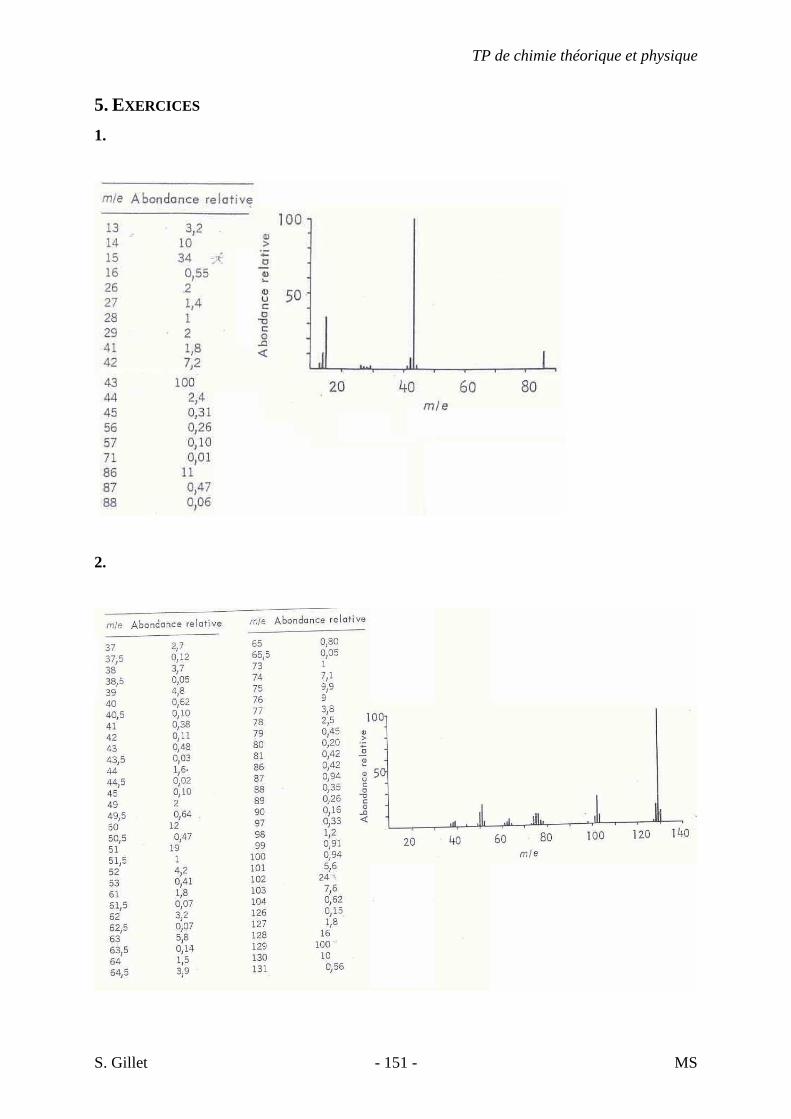
TP de chimie théorique et physique
S. Gillet - 151 - MS
5. EXERCICES
1.
2.
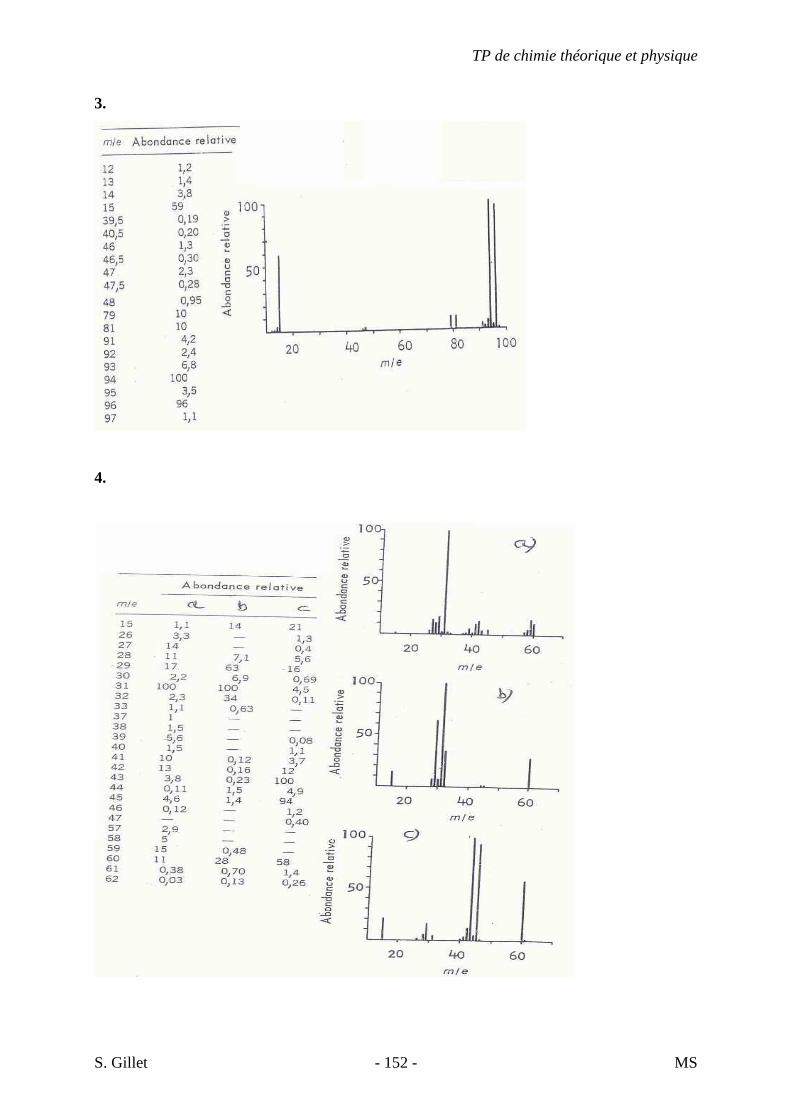
TP de chimie théorique et physique
S. Gillet - 152 - MS
3.
4.
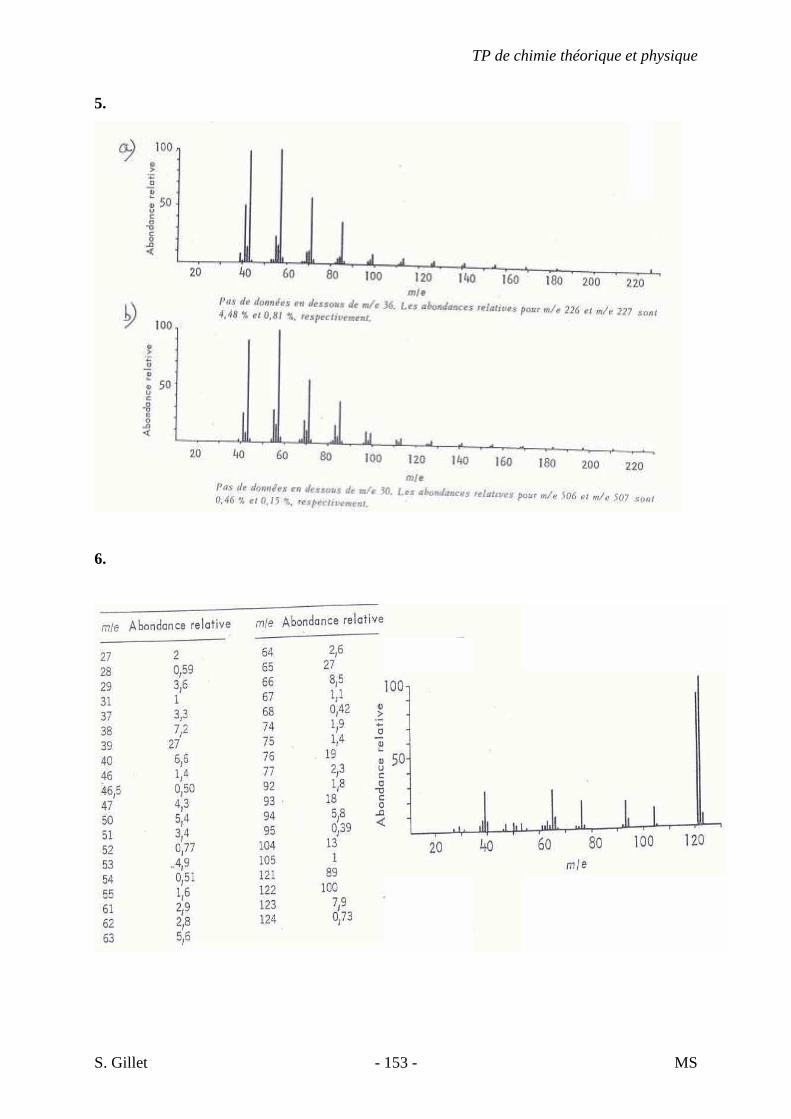
TP de chimie théorique et physique
S. Gillet - 153 - MS
5.
6.
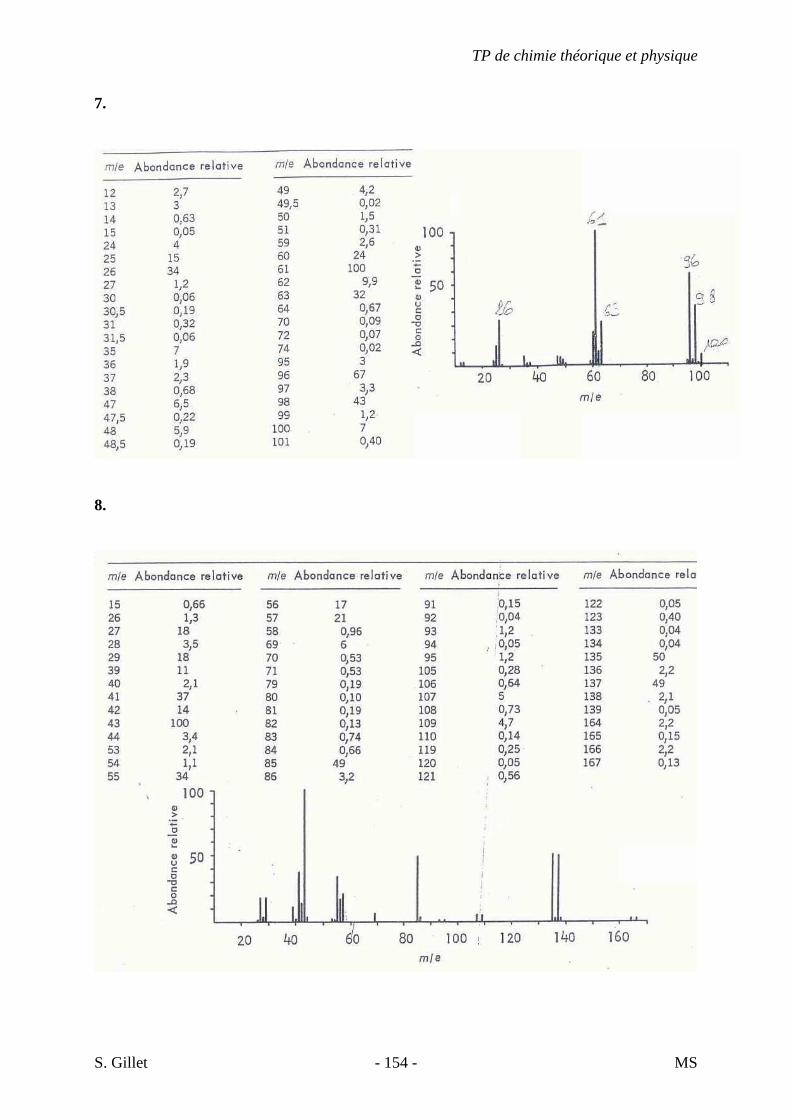
TP de chimie théorique et physique
S. Gillet - 154 - MS
7.
8.
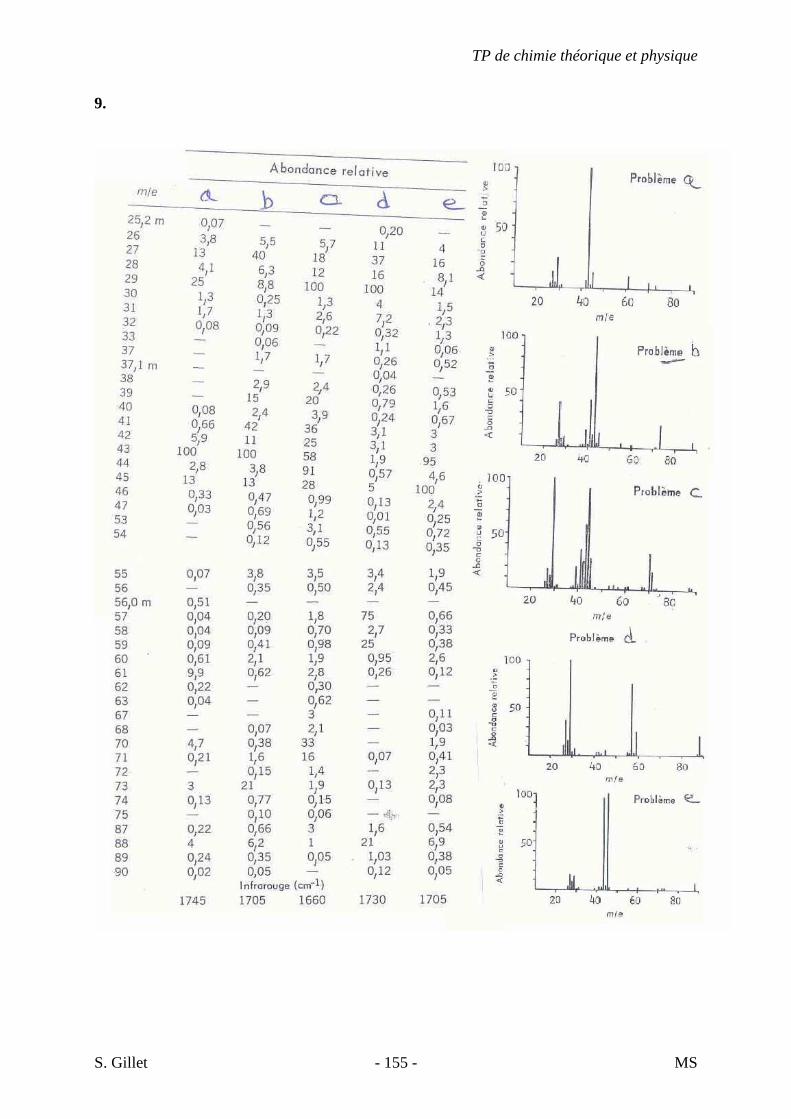
TP de chimie théorique et physique
S. Gillet - 155 - MS
9.
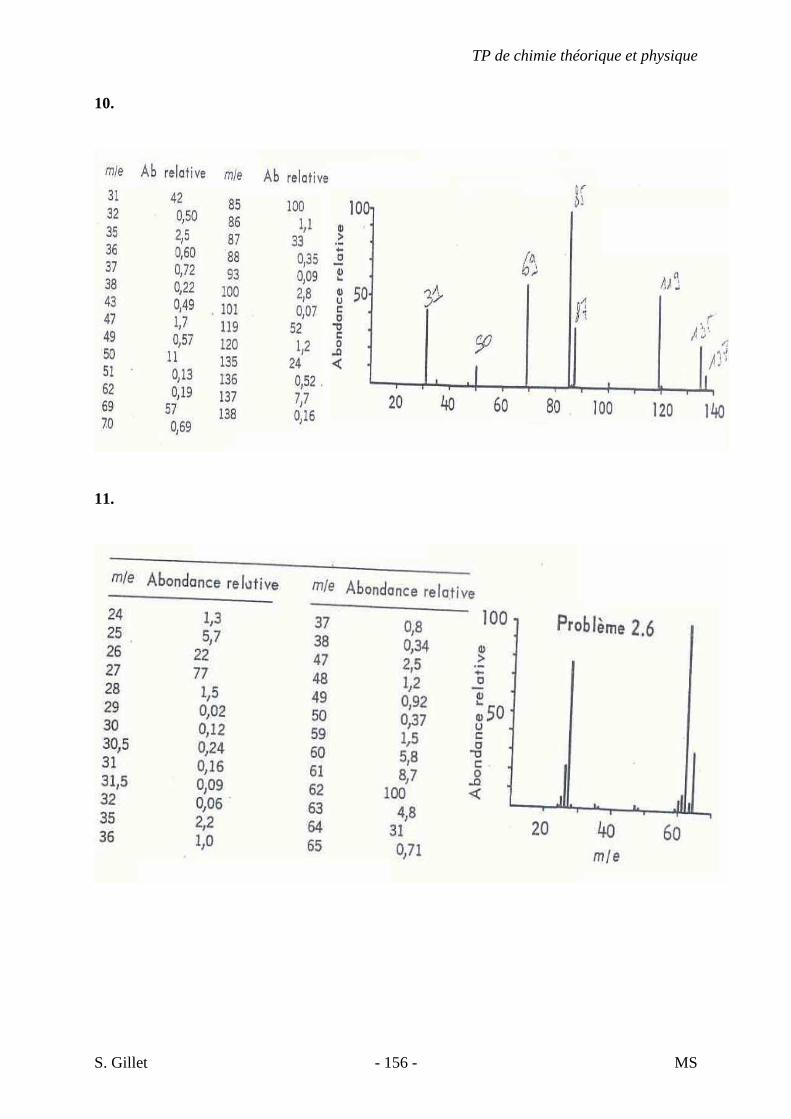
TP de chimie théorique et physique
S. Gillet - 156 - MS
10.
11.
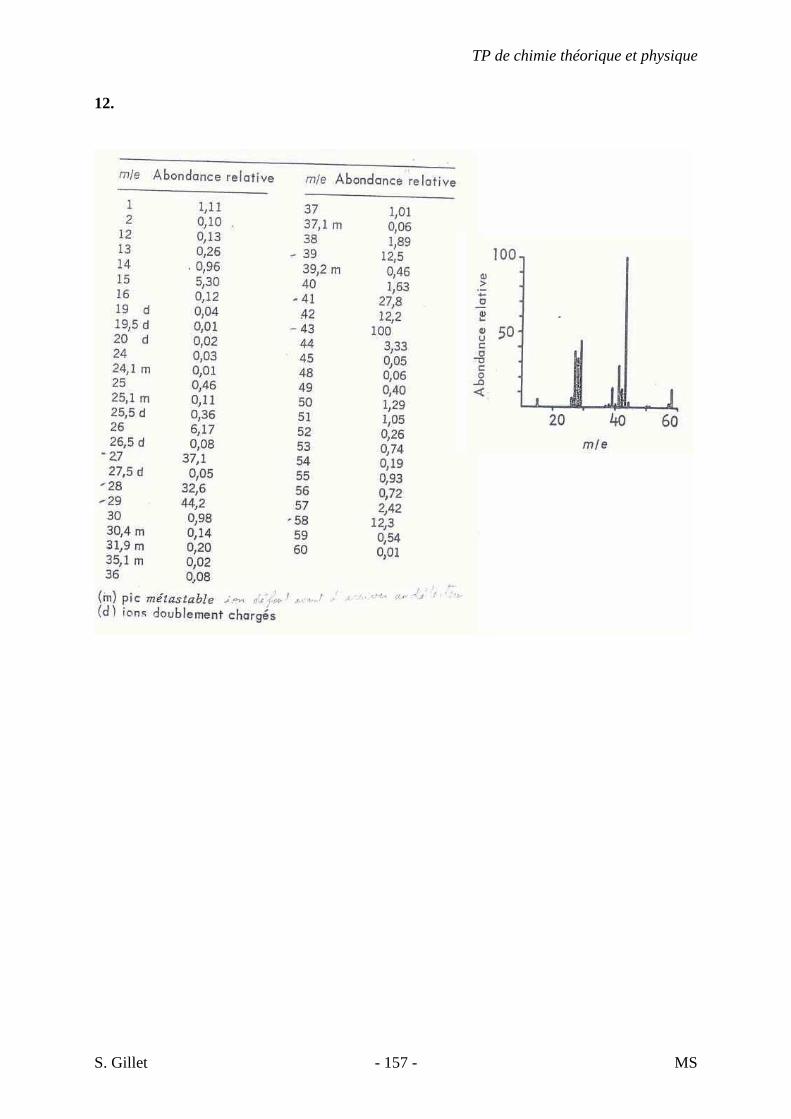
TP de chimie théorique et physique
S. Gillet - 157 - MS
12.
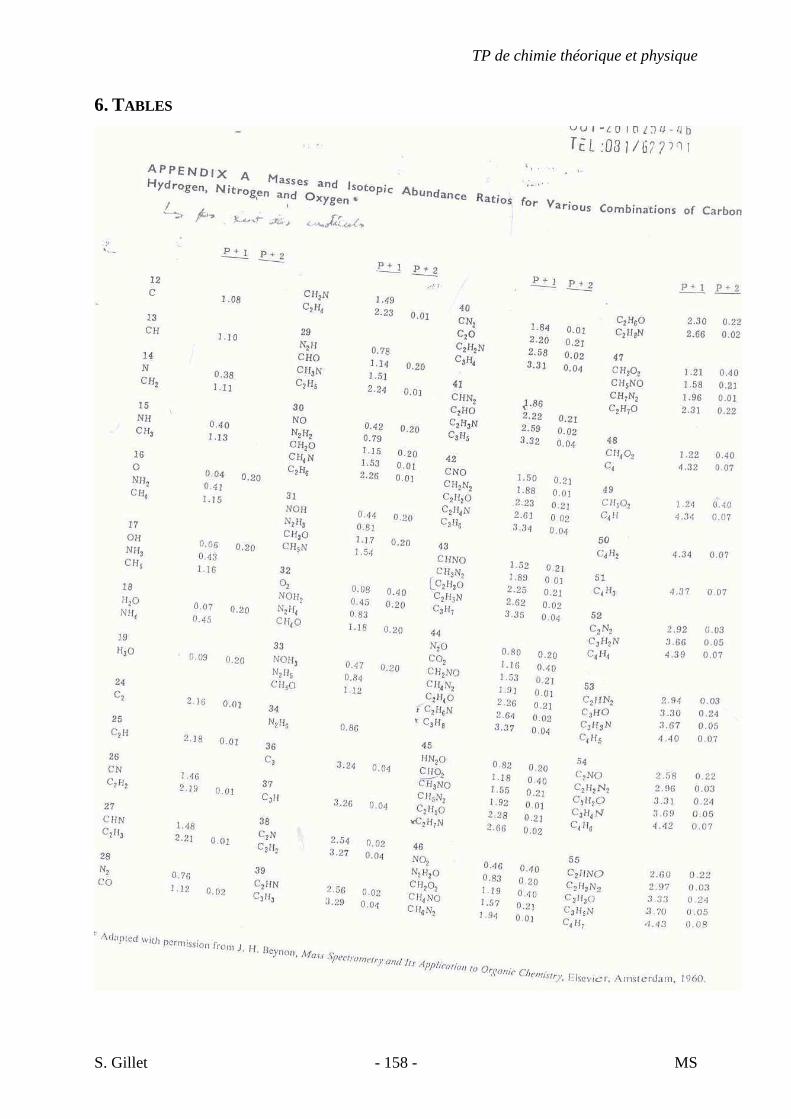
TP de chimie théorique et physique
S. Gillet - 158 - MS
6. TABLES

TP de chimie théorique et physique
S. Gillet - 159 - MS
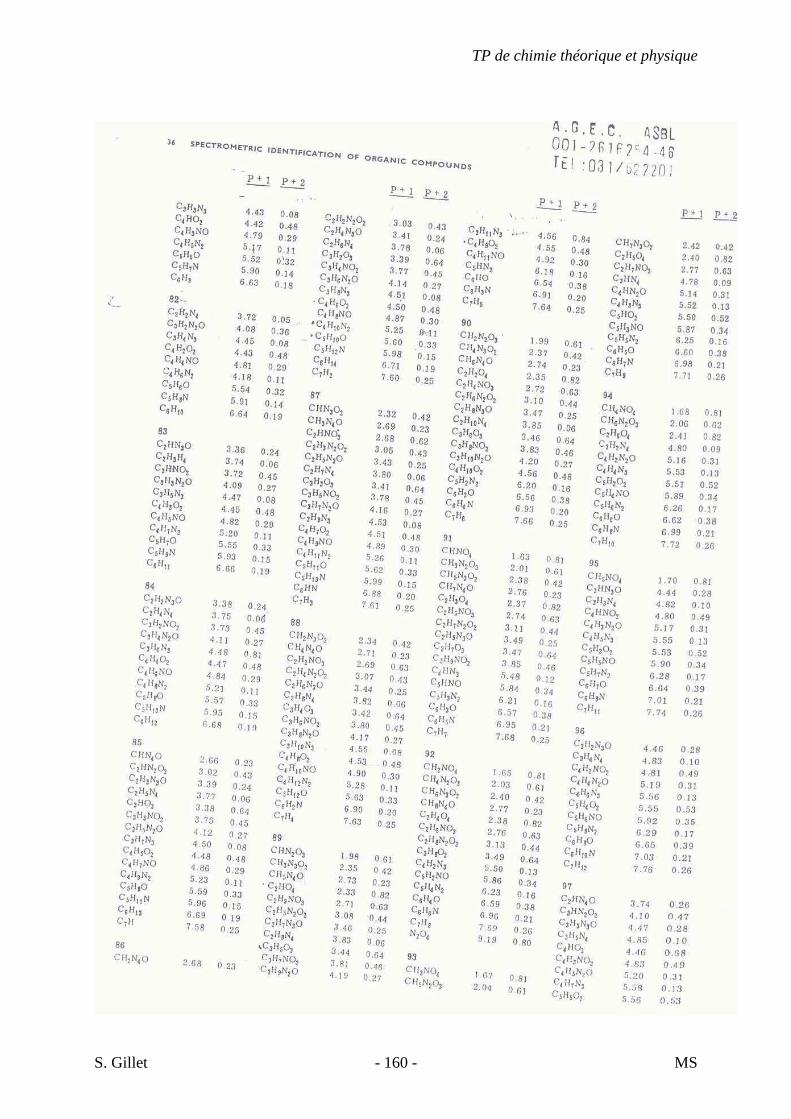
TP de chimie théorique et physique
S. Gillet - 160 - MS

TP de chimie théorique et physique
S. Gillet - 161 - MS
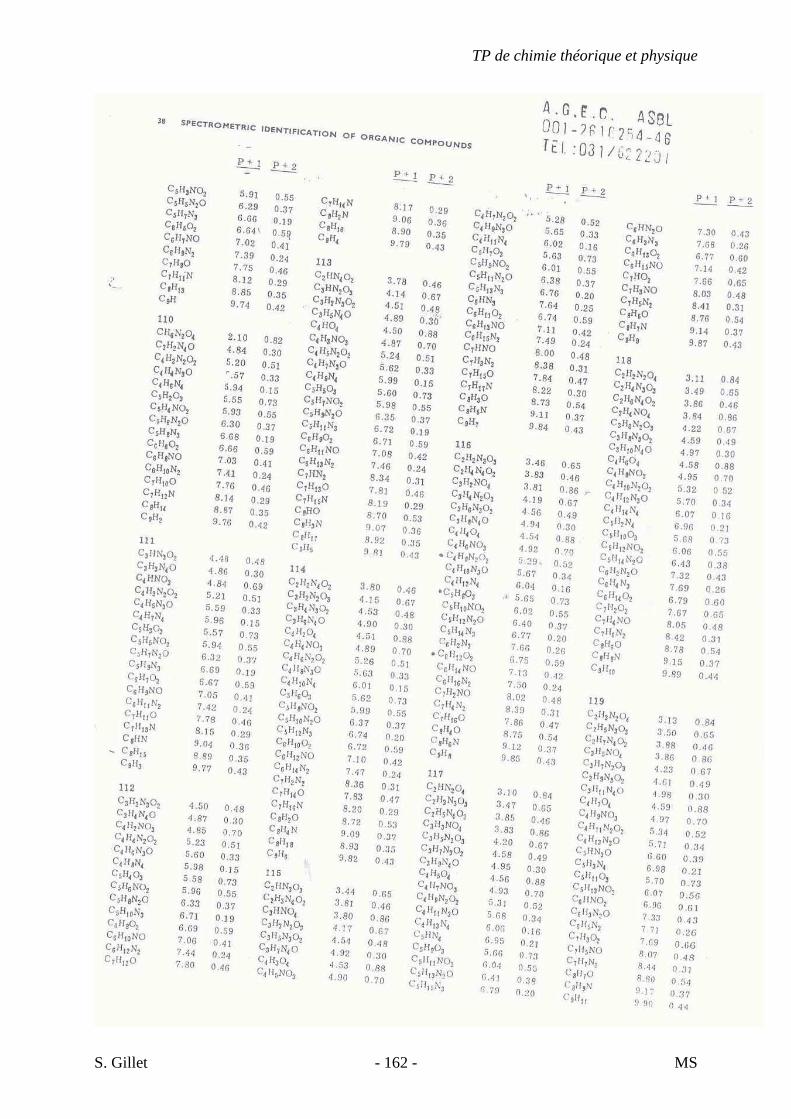
TP de chimie théorique et physique
S. Gillet - 162 - MS

TP de chimie théorique et physique
S. Gillet - 163 - MS
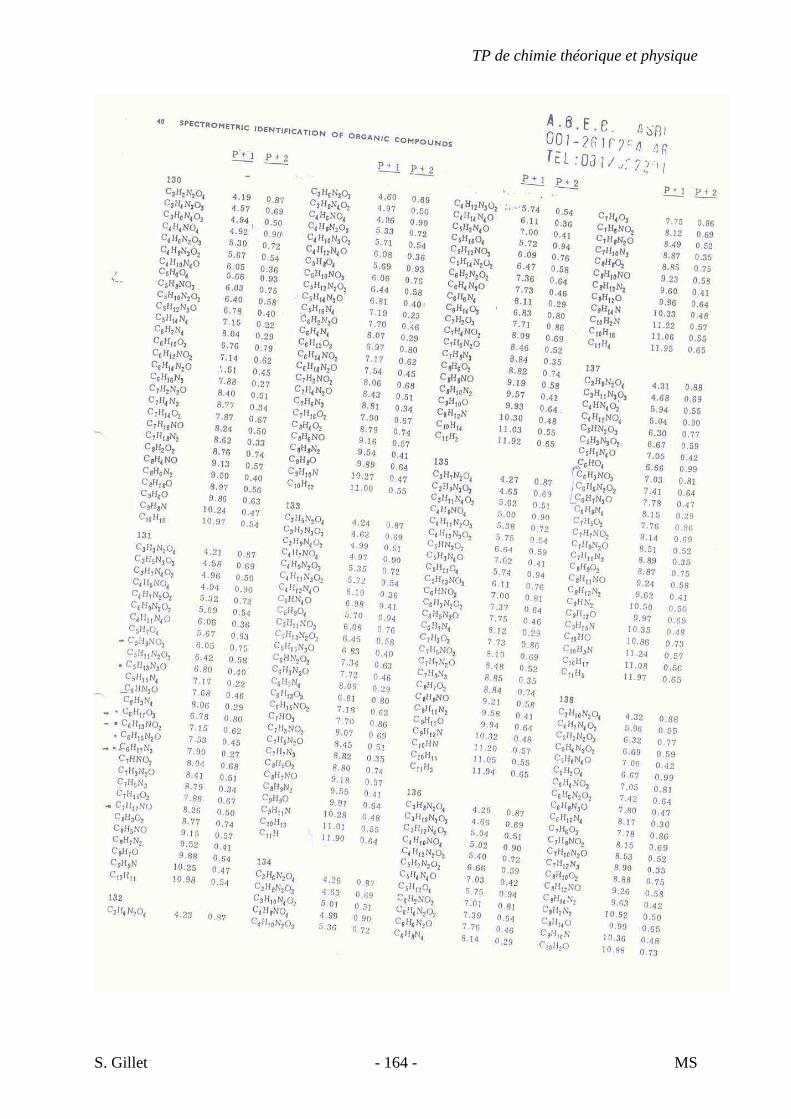
TP de chimie théorique et physique
S. Gillet - 164 - MS
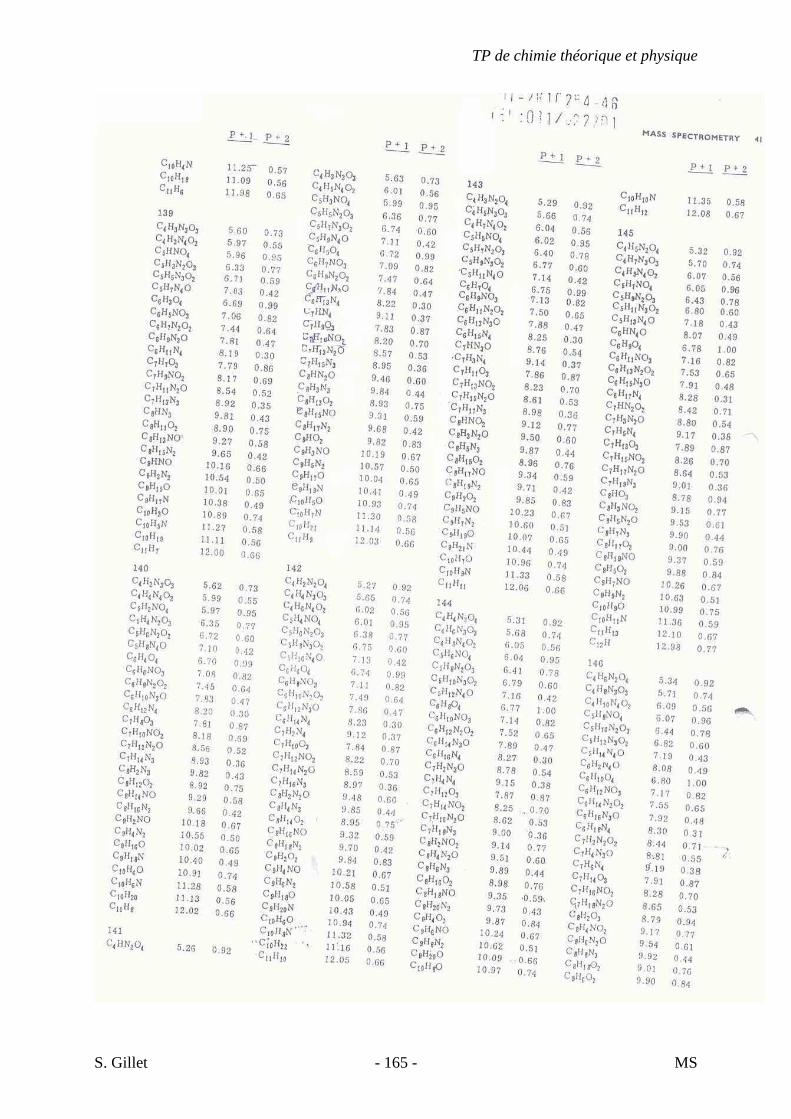
TP de chimie théorique et physique
S. Gillet - 165 - MS
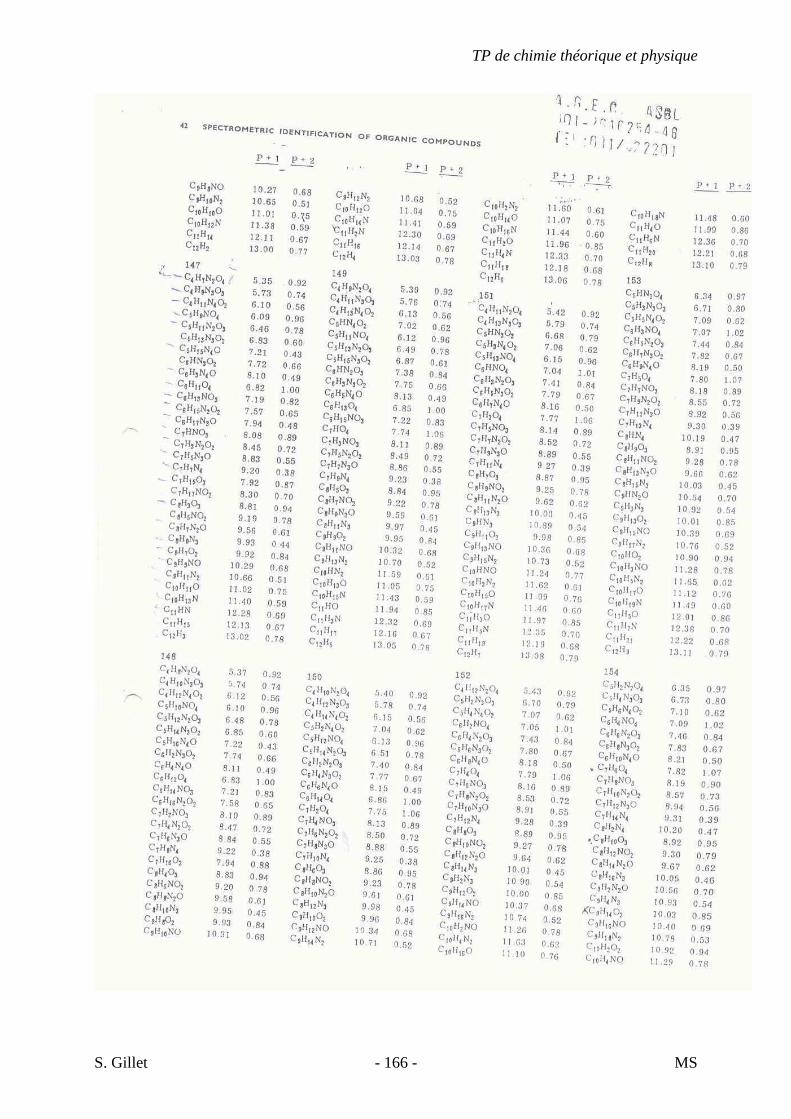
TP de chimie théorique et physique
S. Gillet - 166 - MS
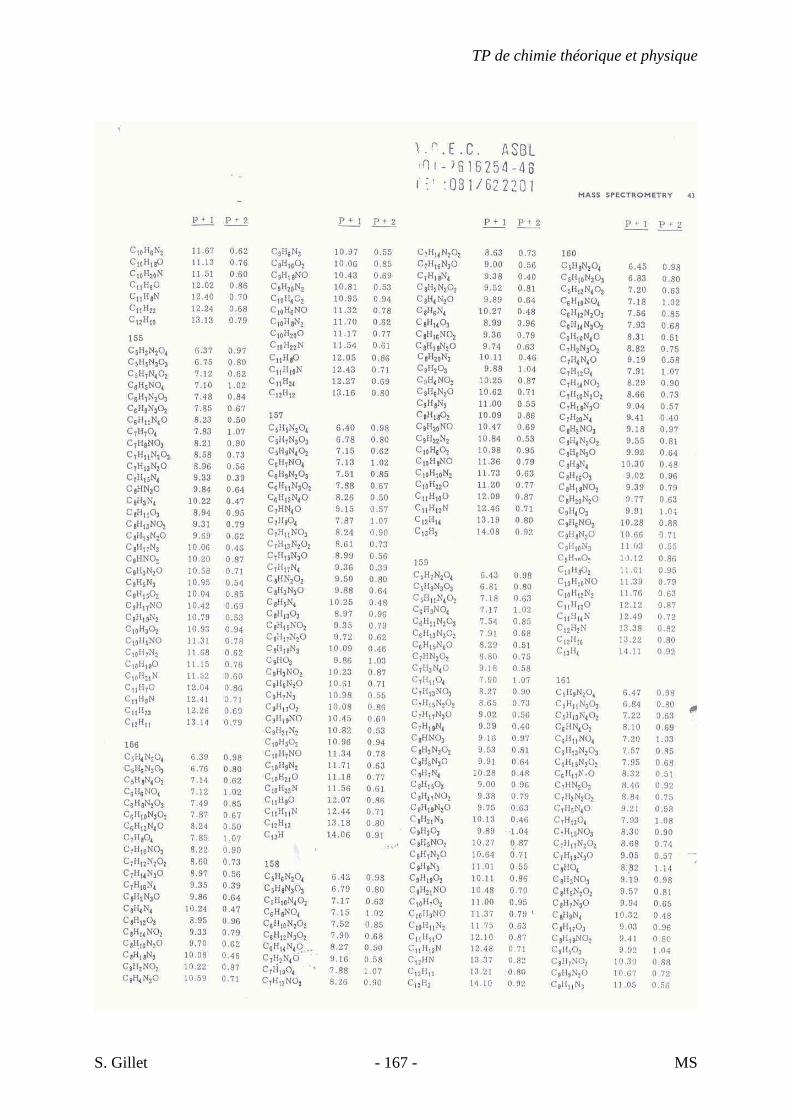
TP de chimie théorique et physique
S. Gillet - 167 - MS
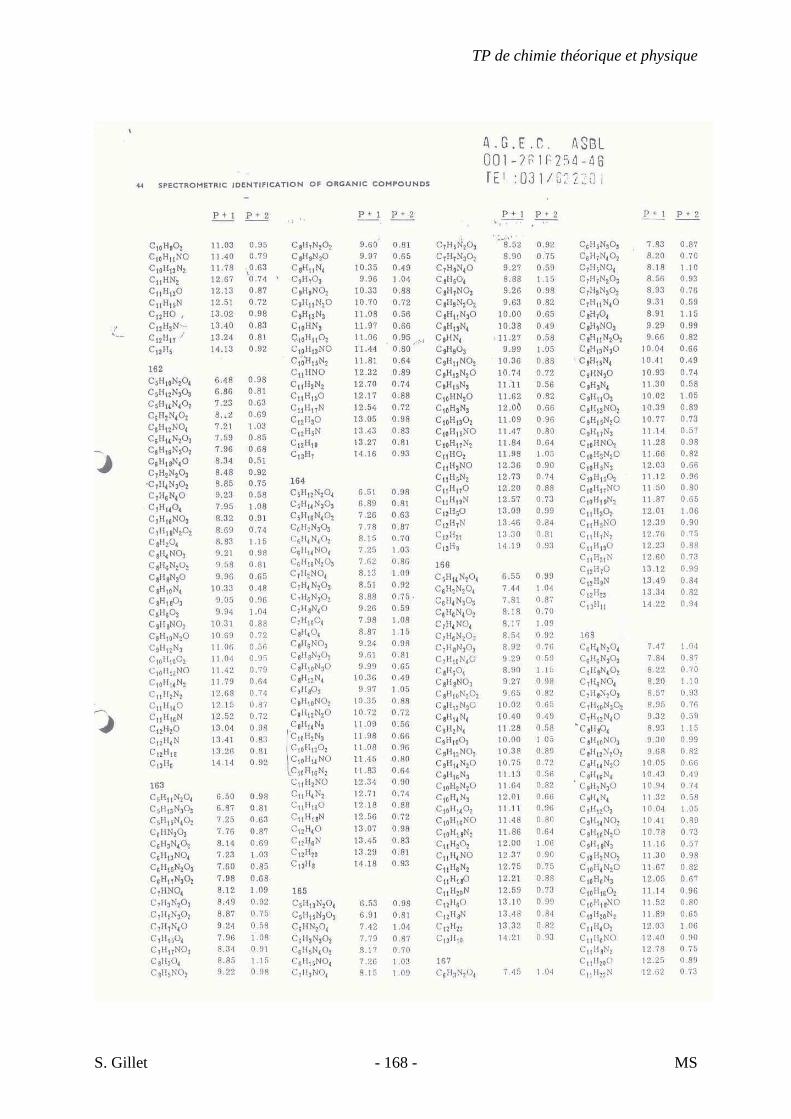
TP de chimie théorique et physique
S. Gillet - 168 - MS
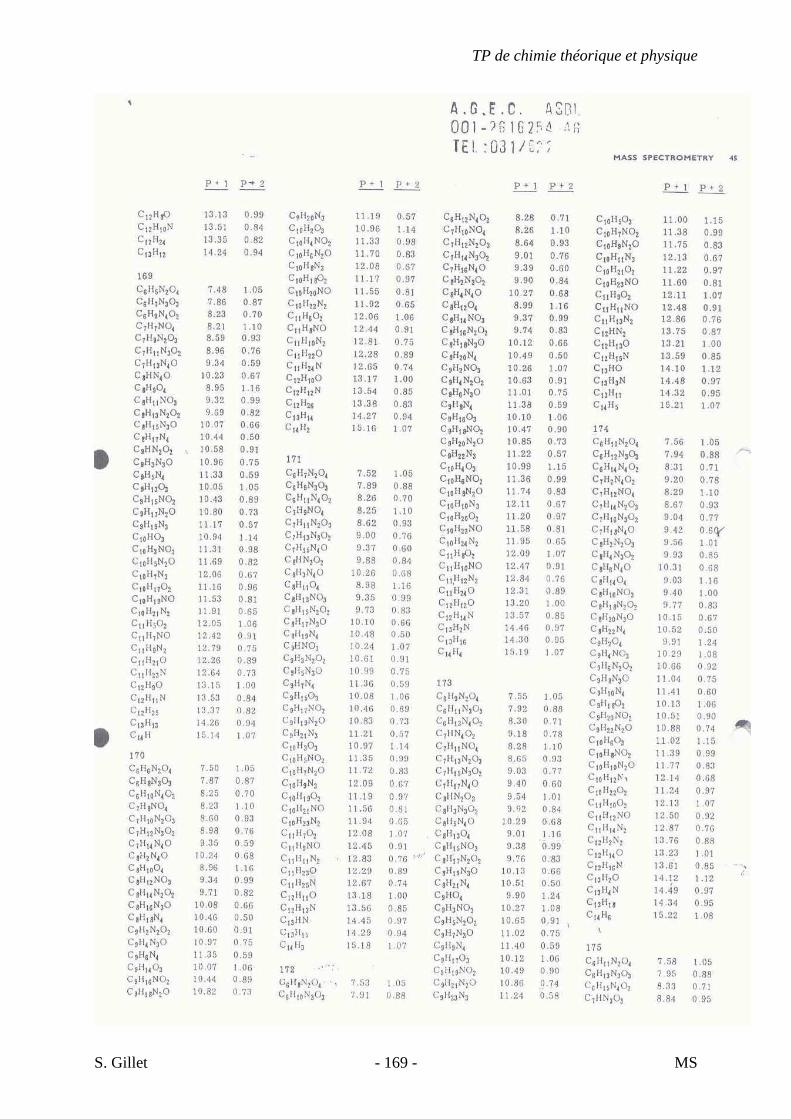
TP de chimie théorique et physique
S. Gillet - 169 - MS
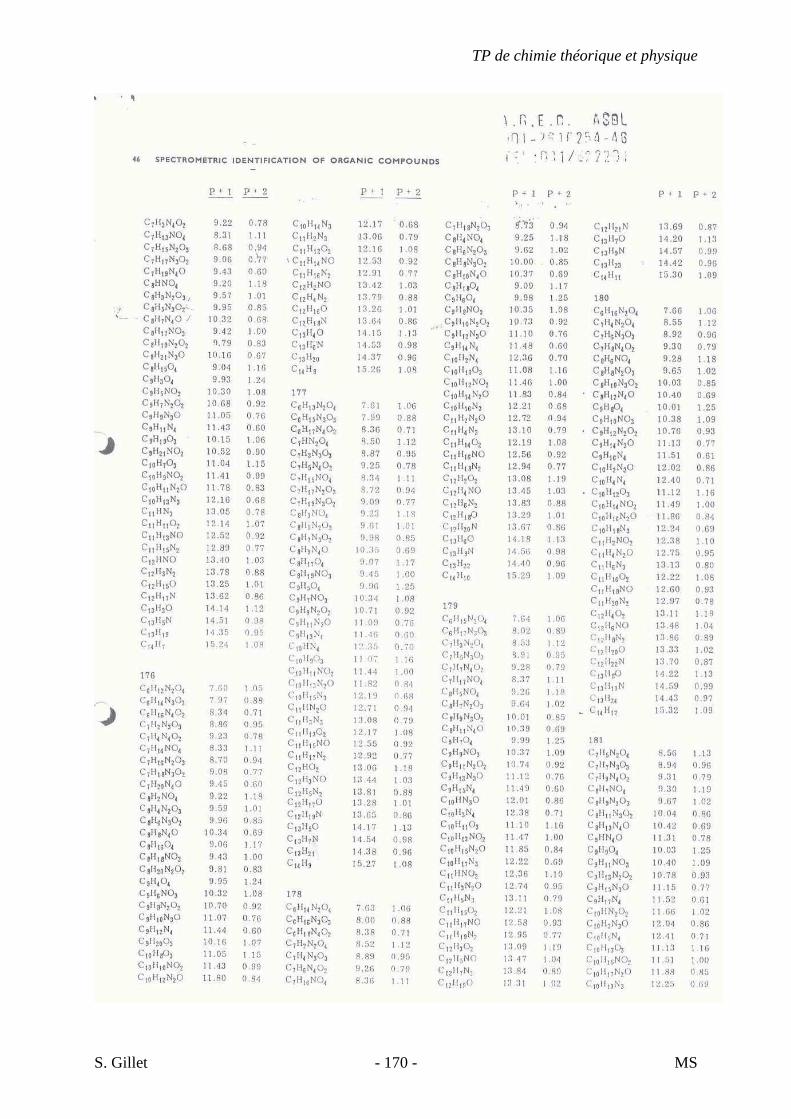
TP de chimie théorique et physique
S. Gillet - 170 - MS
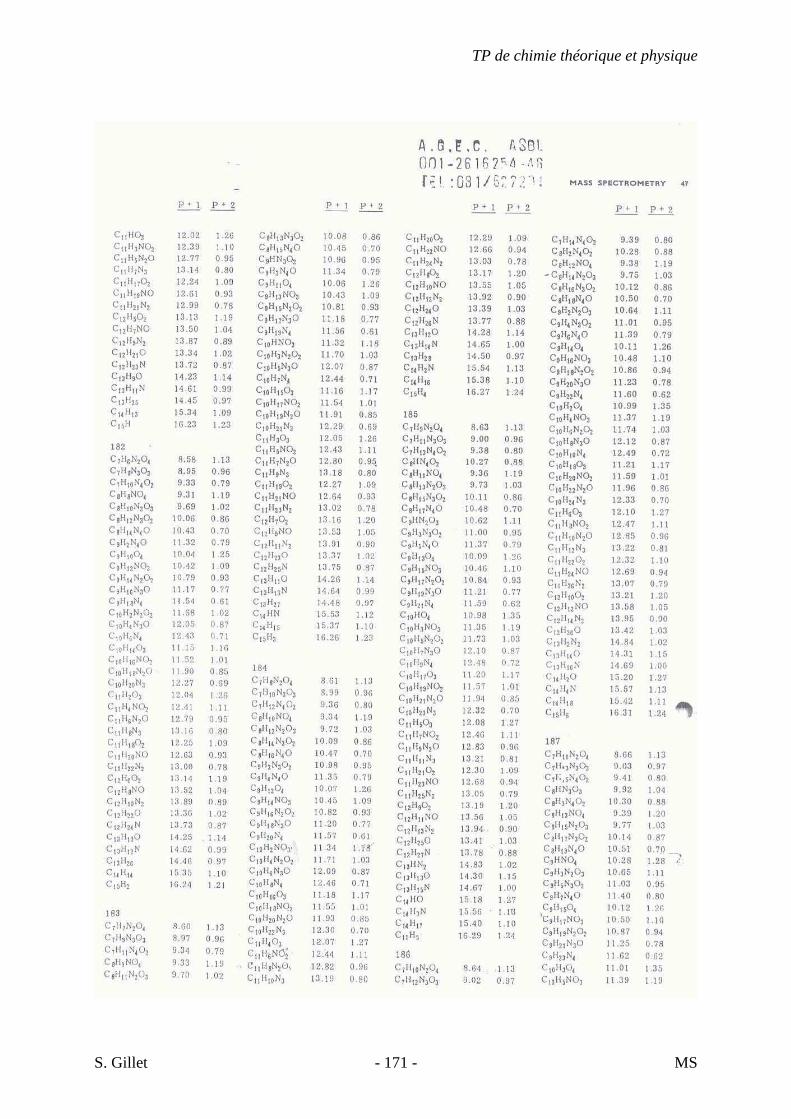
TP de chimie théorique et physique
S. Gillet - 171 - MS
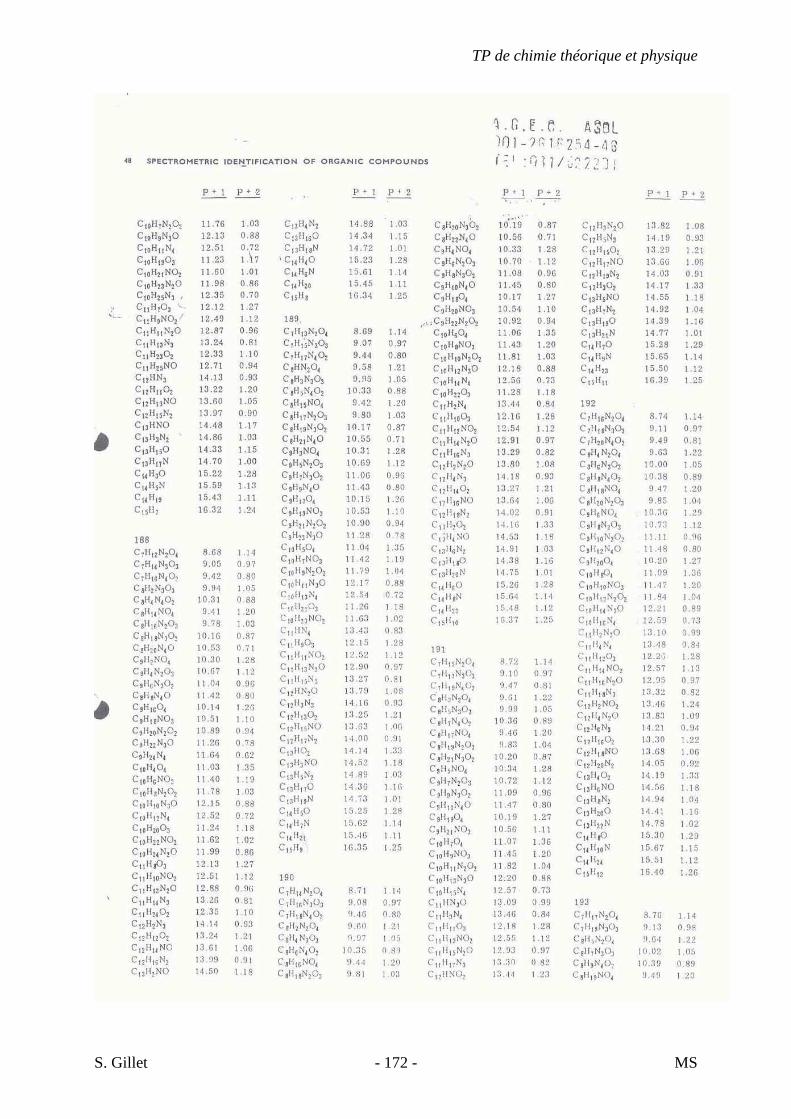
TP de chimie théorique et physique
S. Gillet - 172 - MS
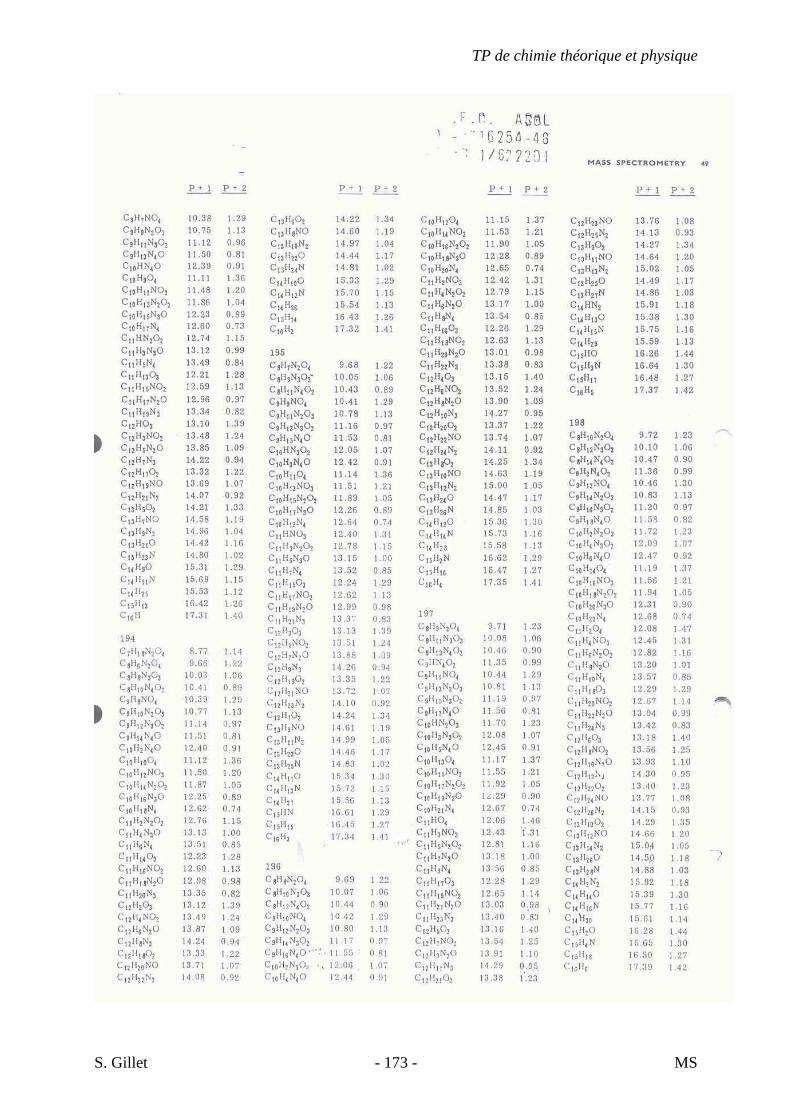
TP de chimie théorique et physique
S. Gillet - 173 - MS
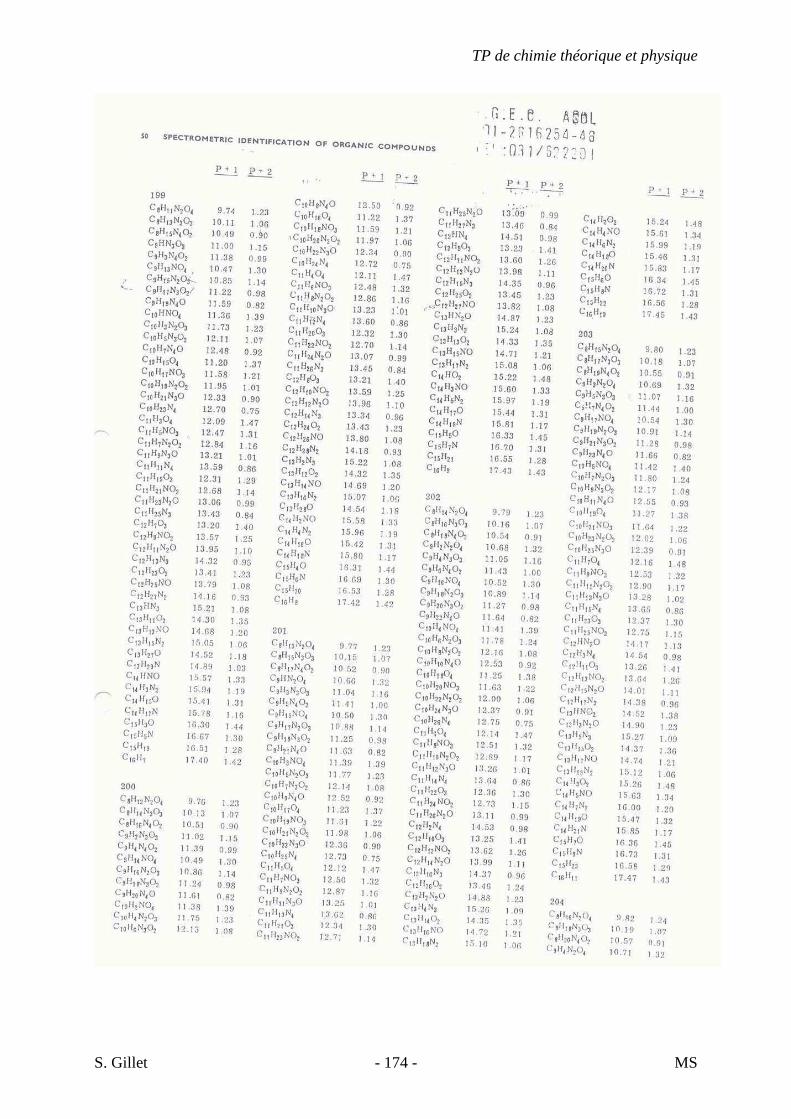
TP de chimie théorique et physique
S. Gillet - 174 - MS

TP de chimie théorique et physique
S. Gillet - 175 - MS

TP de chimie théorique et physique
S. Gillet - 176 - MS
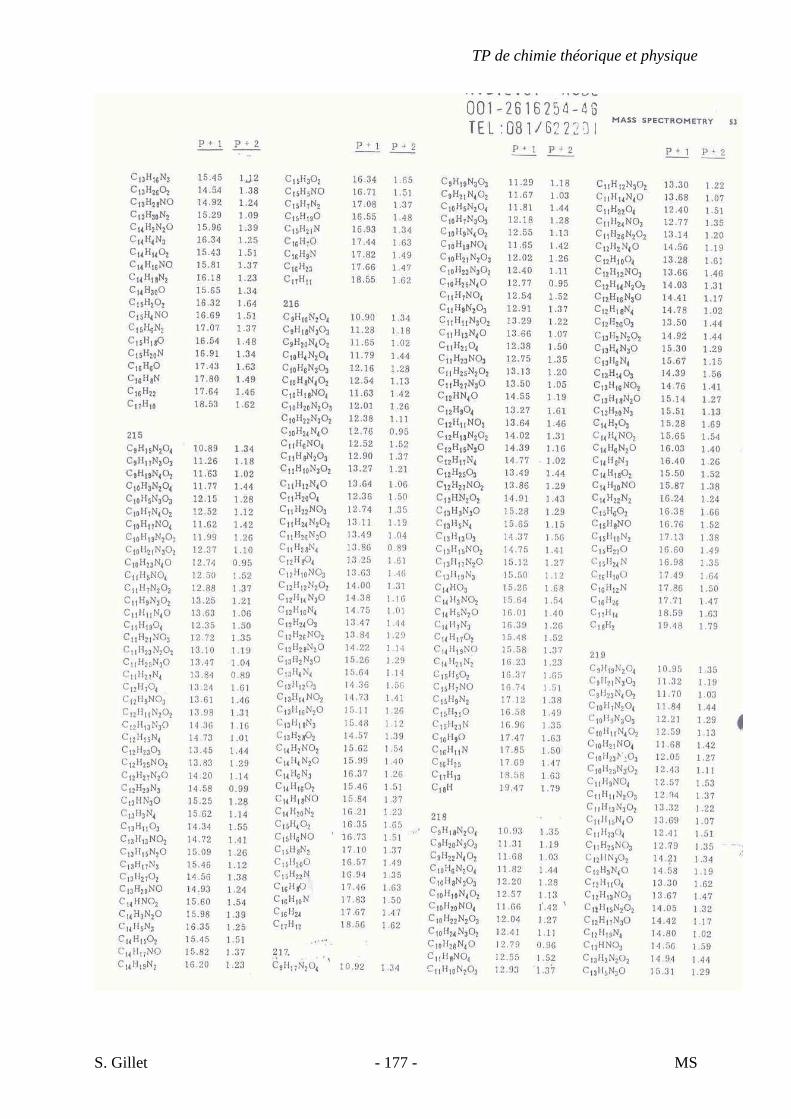
TP de chimie théorique et physique
S. Gillet - 177 - MS
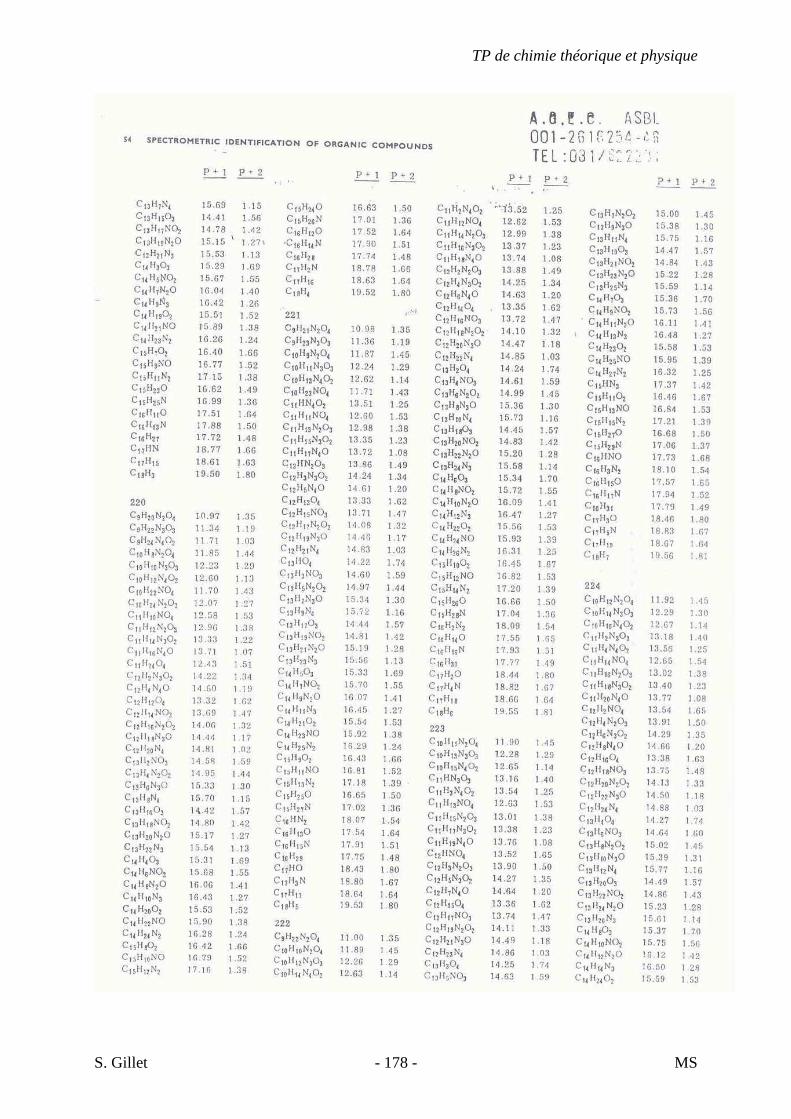
TP de chimie théorique et physique
S. Gillet - 178 - MS
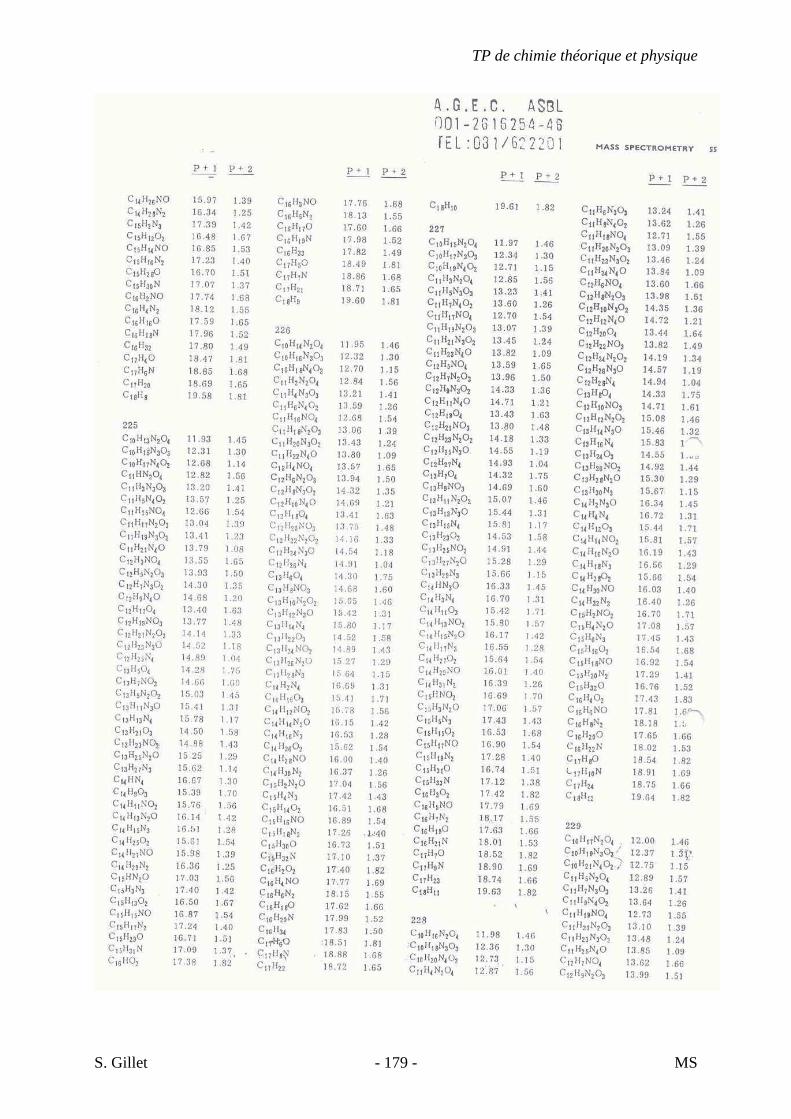
TP de chimie théorique et physique
S. Gillet - 179 - MS
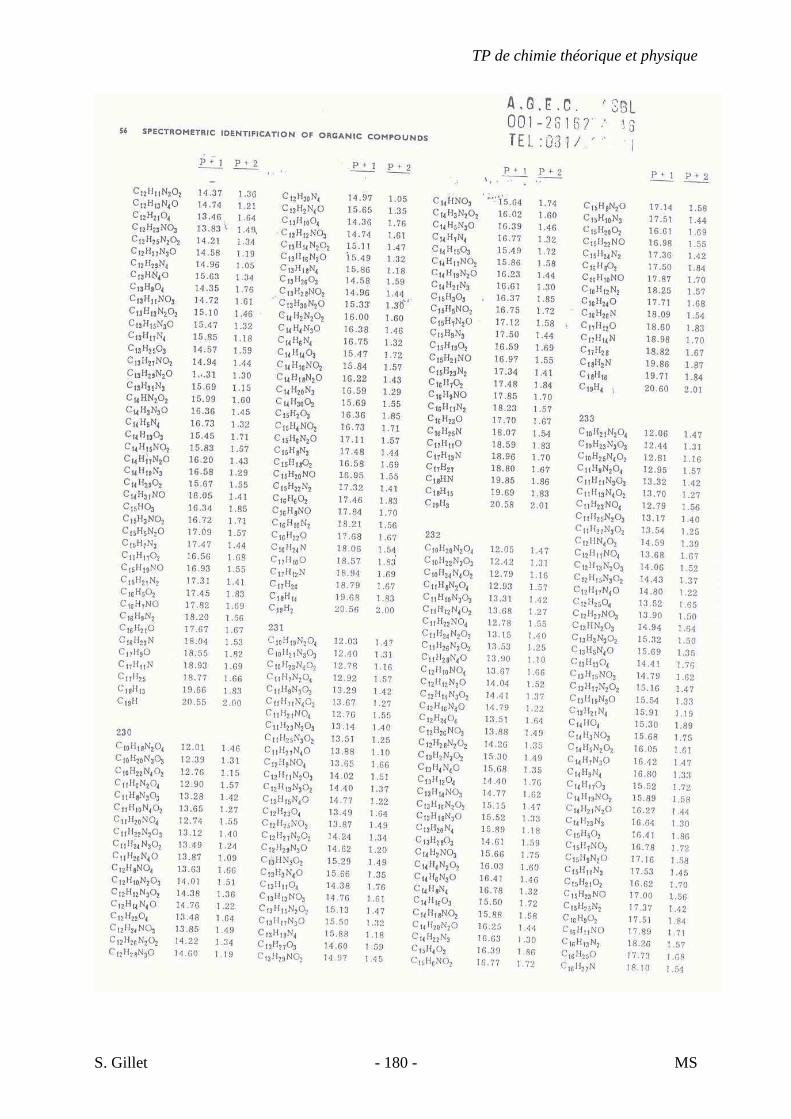
TP de chimie théorique et physique
S. Gillet - 180 - MS
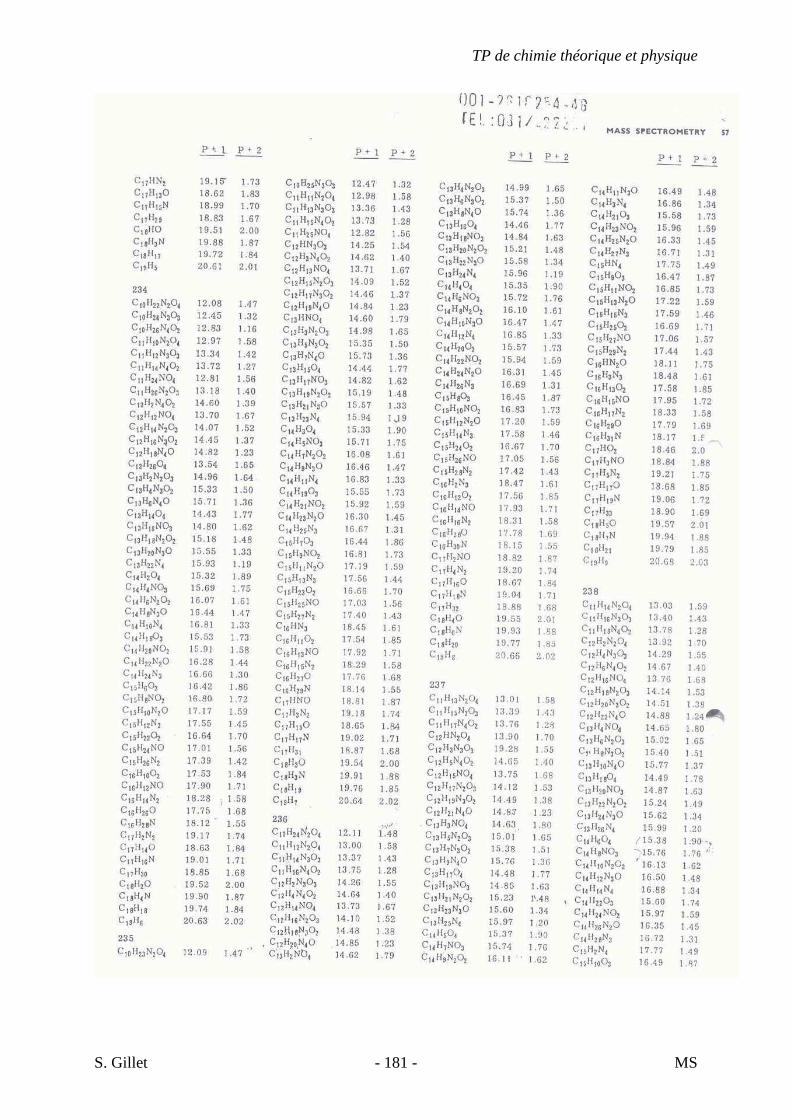
TP de chimie théorique et physique
S. Gillet - 181 - MS
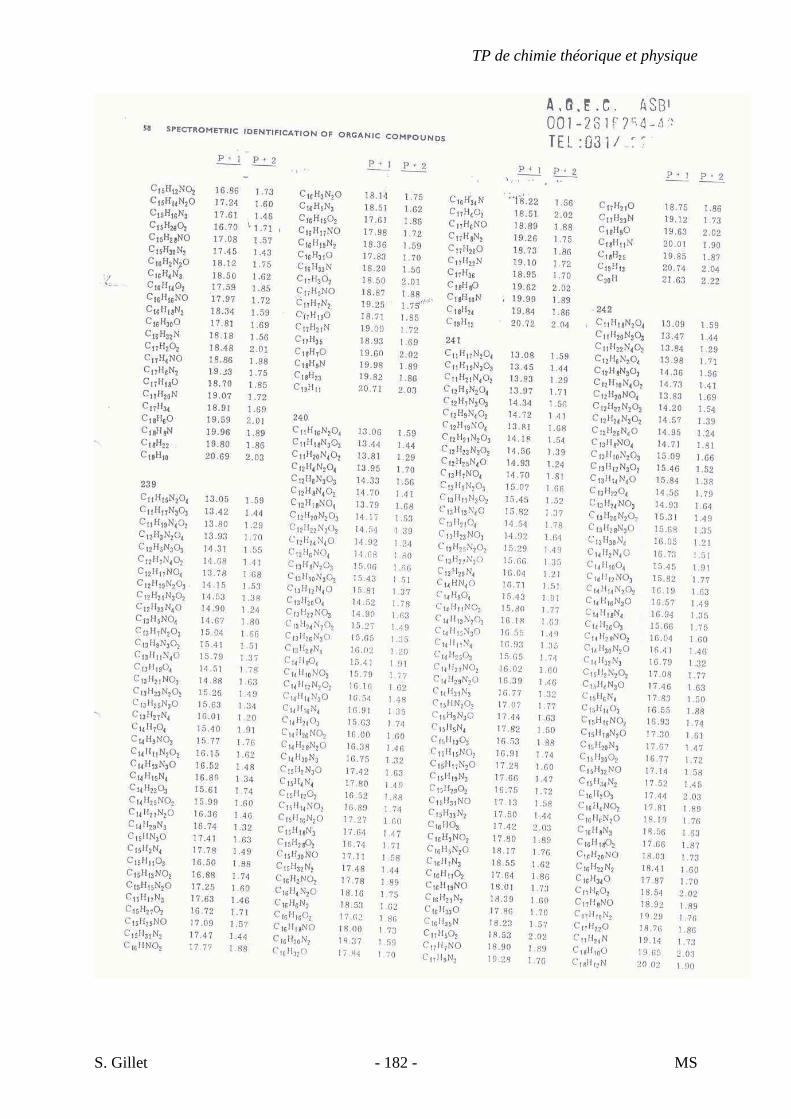
TP de chimie théorique et physique
S. Gillet - 182 - MS
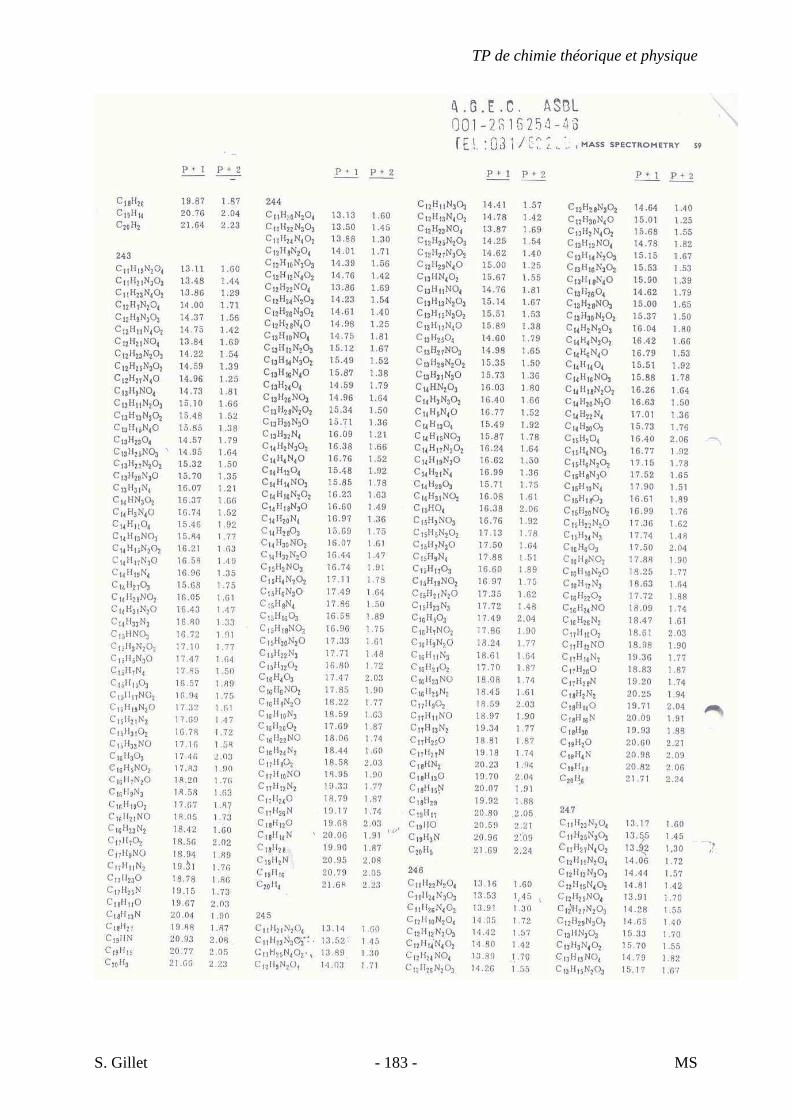
TP de chimie théorique et physique
S. Gillet - 183 - MS
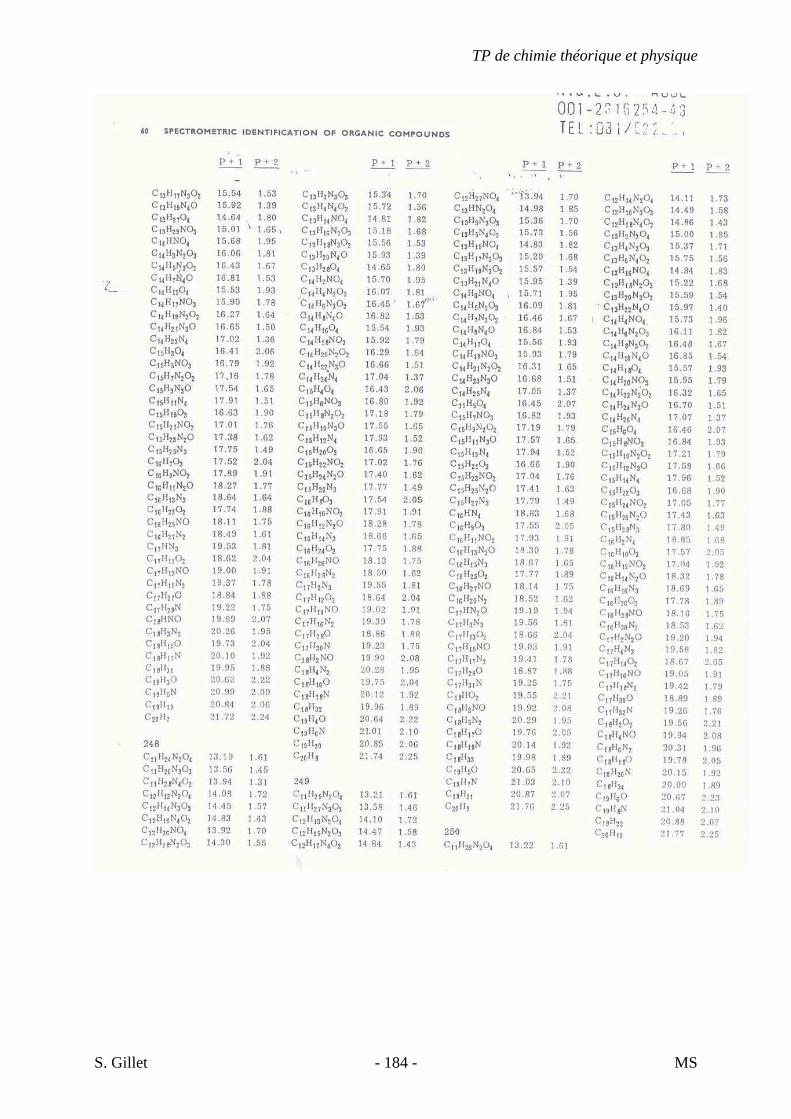
TP de chimie théorique et physique
S. Gillet - 184 - MS

TP de chimie théorique et physique
S. Gillet - 185 - MS
7. RÉFÉRENCES BIBLIOGRAPHIQUES
CHIM 1302. PROF. R. GIASSON. UNIVERSITÉ DE MONTTRÉAL.
CHIMIE THÉORIQUE ET PHYSIQUE APPLIQUÉE À L’ANALYSE STRUCTURALE DE BIOMOLÉCULES.
PROF. B. WATHELET. COURS DE 3ÈME BACHELIER CHIMIE ET BIOINDUSTRIES. FUSAGX.
CHIMIE THÉORIQUE ET PHYSIQUE, NOTES DE TRAVAUX PRATIQUES. FUSAGX.
CHIMIE THÉORIQUE ET PHYSIQUE. PROF. M. MARLIER. COURS DE 1ÈRE INGÉNIEUR CHIMIE ET
BIOINDUSTRIES. FUSAGX.
ROUESSAC, ROUESSAC. ANALYSE CHIMIQUE – MÉTHODES ET TECHNIQUES INSTRUMENTALES
MODERNES 5ÈME ÉDITION. DUNOD 2000. 430P.
SILVERSTEIN, WEBSTER, KIEMLE. IDENTIFICATION SPECTROMÉTRIQUE DE COMPOSÉS
ORGANIQUES 2ÈME ÉDITION. DE BOECK UNIVERSITÉ 2007. 502P.
SKOOG, HOLLER, NIEMAN . PRINCIPES D’ANALYSE INSTRUMENTALE 1ÈRE ÉDITION. DE BOECK
UNIVERSITÉ 2003. 956P.
SPECTRAL DATABASE FOR ORGANIC COMPOUNDS, SDBS.
http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi

TP de chimie théorique et physique
A. Richel - 186 - RMN
CHAPITRE 3. LA RÉSONANCE MAGNÉTIQUE NUCLÉAIRE (RMN)
PARTIE 1. SPECTROMÉTRIE RMN DU PROTON PARTIE 2. SPECTROMÉTRIE RMN DU CARBONE PARTIE 3. SPECTROMÉTRIE RMN DE CORRÉLATION : RMN 2D

TP de chimie théorique et physique
A. Richel - 187 - RMN
PARTIE 1. SPECTROMÉTRIE RMN DU PROTON
1. INTRODUCTION
La spectrométrie de résonance magnétique nucléaire (RMN) est basée sur les
propriétés magnétiques de certains noyaux atomiques.
Dans des conditions particulières, un échantillon placé dans un champ magnétique
peut absorber une onde électromagnétique dans la gamme des radiofréquences (rf) à des
fréquences propres et spécifiques de l’échantillon. L’absorption de l’onde électromagnétique
est fonction de certains noyaux présents dans la molécule.
Un spectre RMN à une dimension est donc un graphique représentant en ordonnée
l’intensité des pics et en abscisse la fréquence d’absorption carcatéristique de certains noyaux
et/ou groupements particuliers.

TP de chimie théorique et physique
A. Richel - 188 - RMN
2. ASPECTS THÉORIQUES
2.1. PROPRIÉTÉS MAGNÉTIQUES DES NOYAUX – NOTION DE SPIN
Tous les noyaux atomiques possèdent une charge. Pour certains, cette charge tourne
autour de l’axe nucléaire (Figure 1a). La rotation de cette charge induit un dipôle magnétique
le long de cet axe. Le moment angulaire de cette charge en rotation peut être décrit en termes
de nombre de spin I ayant pour valeur 0, 1/2, 1, 3/2, etc. (I = 0 signifie que le noyau ne
possède pas de spin). La valeur intrinsèque de dipôle généré est exprimée en termes de
moment magnétique nucléaire µµµµ.
(a) (b)
Figure 65. (a) Génération d’un dipôle magnétique par rotation de la charge du proton. (b) Mouvement de précession quand le noyau est placé dans un champ magnétique externe B0.
Le nombre de spin I peut être déterminé sur base des masses atomiques et des
nombres atomiques comme indiqué dans le Tableau 1.
Des noyaux possèdant un nombre de spin I de 1/2 donnent facilement des spectres
RMN (ils possèdent une distribution de charge sphérique uniforme comme illustrée dans la
Figure 1). C’est le cas principalement du 1H et du 13C, dont la spectrométrie RMN va être
décrite plus en détails.

TP de chimie théorique et physique
A. Richel - 189 - RMN
Tableau 1. Nombre de spin I pour différentes combinaisons de masses atomiques et de numéros atomiques.
I Nombre de masse Numéro atomique Exemple de noyaux
Impaire Impaire H1
1H
1
1H
3
1H
3
1N
15
7N
15
7F
19
9F
19
9P
31
15P
31
15 I =1/2 Impaire Paire C
13
6O
17
8O
17
8
I =1 Paire Impaire H2
1H
2
1N
14
7N
14
7
I =0 Paire Paire C12
6O
16
8O
16
8
2.2. EXCITATION DE NOYAUX DE NOMBRE DE SPIN I = 1/2
Pour un noyau avec I = 1/2 placé dans un champ magnétique externe, 2 niveaux
d’énergie existent : E0 et E1. Le niveau d’énergie le plus bas est le plus peuplé en accord avec
la distribution de Boltzmann.
La différence d’énergie (∆E) entre les niveaux E0 et E1 est donnée par la relation :
∆E = (hγ/2π)B0
Où : h = constante de Planck
γ = rapport gyromagnétique caractéristique de chaque noyau et définie par γ = 2πµ/hI
B0 = force du champ magnétique
Si une énergie est apportée sous la forme de radiation de type radiofréquence (avec
une fréquence ν1 suffisante), il est possible de provoquer une transition entre le niveau E0 et
E1 dans un champ magnétique stationnaire de force donnée B0.
Dans ces conditions, on dit que le système est en résonance ; l’énergie est absorbée
par le noyau (1H), l’amenant dans un état plus énergétique, et un spectre peut être enregistré.

TP de chimie théorique et physique
A. Richel - 190 - RMN
E0
E1
I = 1/2
Figure 66. Visualisation des 2 niveaux d’énergie E0 et E1 du proton dans un champ magnétique d’intensité B0. Le champ magnétique (B0) est dirigé vers le haut et la force du champ augmente vers la droite. Plus B0
est intense et plus ∆∆∆∆E est grand.
2.3. ENREGISTREMENT D ’UN SPECTRE PROTON
Quand il est soumis à un champ magnétique externe de force B0, le noyau s’aligne sur
le champ mais entre aussi en précession avec une fréquence ν appelée fréquence de Larmor
(Figure 1b).
Pour un ensemble de protons équivalents en précession avec une phase aléatoire
autour de l’axe z (dans la direction du champ magnétique), il en résulte une aimantation nette
M0 dans l’axe z (et nulle dans le plan xy) (Figure 3).

TP de chimie théorique et physique
A. Richel - 191 - RMN
B0
x
z
y
M0
Figure 67. Enemble de noyaux en précession et aimantation M 0 résultante alignée avec le champ magnétique stationnaire B0.
Le but lors de l’acquisition d’un spectre RMN est de faire basculer l’aimantation nette
M0 dans le plan xy et d’en mesurer la composante dans ce plan transversal. Pour ce faire, on
applique un second champ magnétique B1 tournant de direction convenable (càd ⊥ à B0)
pendant une durée limitée très courte (« technique pulsée »). On étudie ensuite la phase de
relaxation au cours de laquelle l'aimantation retrouve progressivement sa valeur d'équilibre
(Figure 4).
x
z
y
M0
x
z
y
M
B0
Projection de M sur y x
z
y
M0
M
(a) (b) (c)
Composante tounante de B1
Oscillateur de champ B1
Figure 68. (a) Application du second champ magnétique tournant de force B1. (b) Basculement de l’aimantation nette M0 en M dans le plan xy. (c) Relaxation de M vers M0 selon une spirale amortie.

TP de chimie théorique et physique
A. Richel - 192 - RMN
Le moment magnétique M généré dans le plan xy est mesuré par une bobine détectrice
montée dans le plan xy. Afin d’obtenir un spectre la fréquence de l’oscillateur ν1 est balayée
sur une gamme de fréquences. L’intensité de M est maximale quand la fréquence de
l’oscillateur est égale à la fréquence de précession de Larmor ν.
2.4. SEQUENCE PROTON CLASSIQUE
L’enregistrement d’un spectre 1H classique s’opère par technique pulsée (Figure 5).
Une impulsion radiofréquence (rf) de quelques µs et centrée sur la fréquence ν1 (ν de
résonance typique des protons) est appliquée le long de l’axe x. Son action est de faire
basculer l’aimantation nette M0 de 90° pour la positionner dans le plan xy. Les signaux
d’aimantation sont immédiatement détectés dans le plan xy après l’impulsion et collectés par
un ordinateur sur un laps de temps applée « période d’acquisition » t2. Durant cette période t2,
les projections sur l’axe y des signaux M en précession autour de l’axe z sont détectés (cfr
Figure 4c, spirale amortie de M) . Après l’impulsion, M est localisé sur l’axe y et l’intensité
du signal est maximale. Après un intervalle de temps, les signaux s’orientent sur l’axe x :
leurs projections sur y sont nulles. En poursuivant leur rotation, les signaux atteignent leur
minimum sur l’axe –y. Cette rotation se poursuit jusqu’à ce que M reviennent à l’équilibre
(Figure 5c). Le signal enregistré suit donc une sinusoidale amortie dénommée FID (Free
Induction Decay). L’application de la transformée de Fourier (transformation d’un signal en
fct du temps en un signal en fct de la fréquence) permet de convertir ce signal en un spectre
RMN conventionnel.
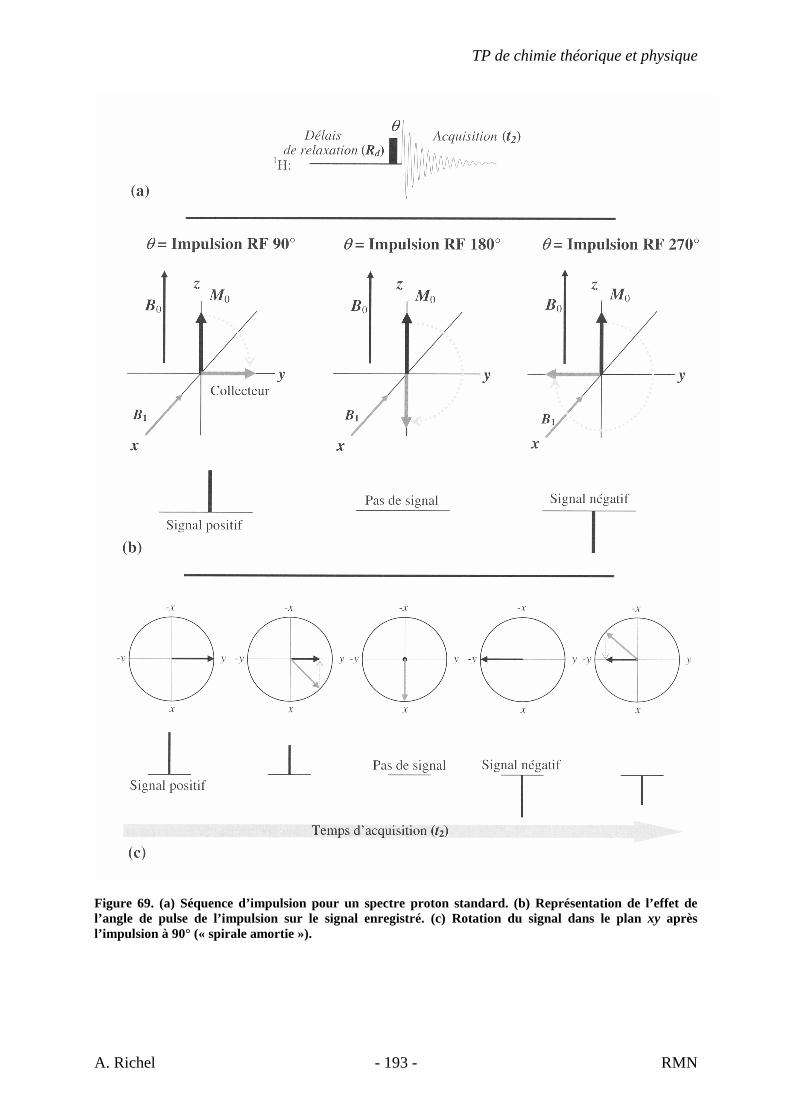
TP de chimie théorique et physique
A. Richel - 193 - RMN
Figure 69. (a) Séquence d’impulsion pour un spectre proton standard. (b) Représentation de l’effet de l’angle de pulse de l’impulsion sur le signal enregistré. (c) Rotation du signal dans le plan xy après l’impulsion à 90° (« spirale amortie »).
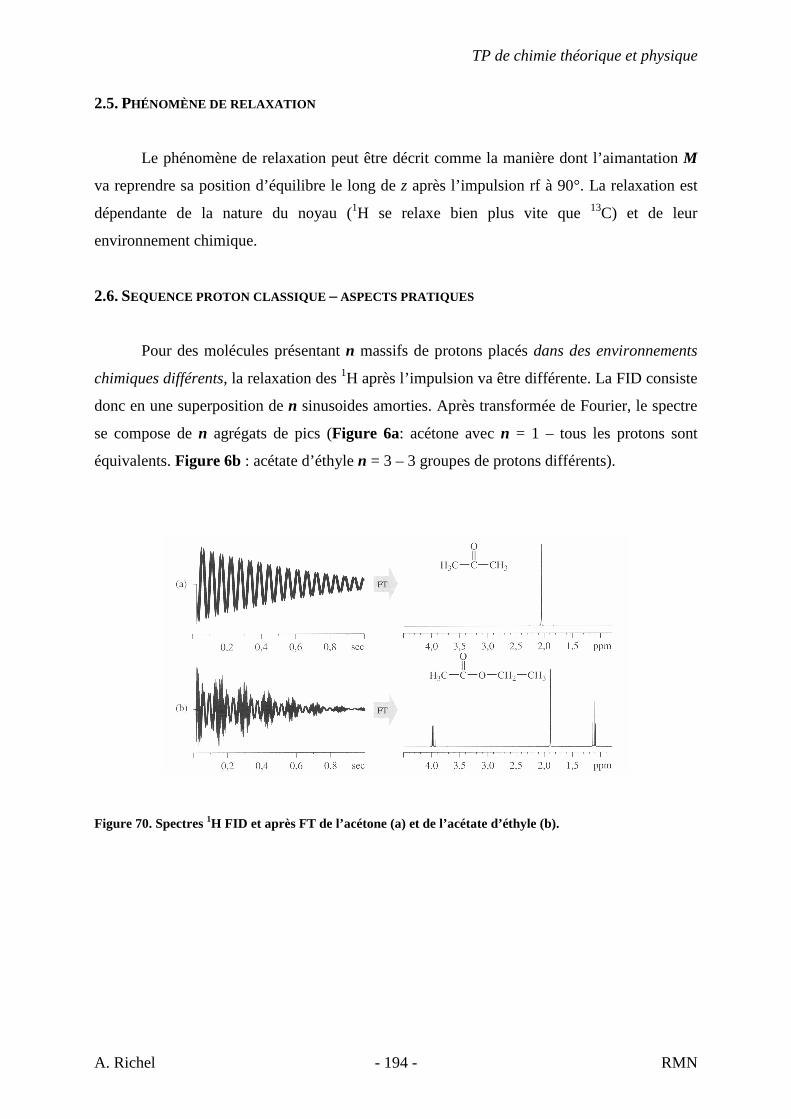
TP de chimie théorique et physique
A. Richel - 194 - RMN
2.5. PHÉNOMÈNE DE RELAXATION
Le phénomène de relaxation peut être décrit comme la manière dont l’aimantation M
va reprendre sa position d’équilibre le long de z après l’impulsion rf à 90°. La relaxation est
dépendante de la nature du noyau (1H se relaxe bien plus vite que 13C) et de leur
environnement chimique.
2.6. SEQUENCE PROTON CLASSIQUE – ASPECTS PRATIQUES
Pour des molécules présentant n massifs de protons placés dans des environnements
chimiques différents, la relaxation des 1H après l’impulsion va être différente. La FID consiste
donc en une superposition de n sinusoides amorties. Après transformée de Fourier, le spectre
se compose de n agrégats de pics (Figure 6a: acétone avec n = 1 – tous les protons sont
équivalents. Figure 6b : acétate d’éthyle n = 3 – 3 groupes de protons différents).
Figure 70. Spectres 1H FID et après FT de l’acétone (a) et de l’acétate d’éthyle (b).

TP de chimie théorique et physique
A. Richel - 195 - RMN
3. INSTRUMENTATION
Actuellement, les spectromètres RMN (250MHz-1.0GHz) employés sont à
transformée de Fourier avec aimant supraconducteur refroidi à l’hélium liquide et travaillent
en mode pulsé (Figure 7). Pour des analyses en phase liquide, l’échantillon (mis en solution
dans un solvant deutéré dans un tube en verre de 5 mm de diamètre) est positionné dans la
sonde (parallèle à l’axe z de l’aimant) contenant les bobines émettrices et réceptrices.
Figure 71. Spectromètre avec aimant supraconducteur de type Varian Unity opérant à 600 MHz.
Typiquement, des solvants comme le chloroforme deutéré (CDCl3), le méthanol-d4
(CD3OD), le DMSO-d6 ou l’eau deutérée (D2O) sont communs pour des analyses en routine.
Le deutérium a effectivement une fréquence de résonance bien différente de celle de
l'hydrogène, son rapport gyromagnétique γ valant 6,54 au lieu de 42,58 MHz/T pour
l'hydrogène.

TP de chimie théorique et physique
A. Richel - 196 - RMN
4. DÉPLACEMENTS CHIMIQUES - CONSTANTE D’ÉCRAN – BLINDAGE
4.1. SOURCE DU DÉPLACEMENT CHIMIQUE
Tous les noyaux 1H d’une même molécule n’ont pas exactement la même fréquence de
résonance. La gamme de fréquences est plus ou moins étendue selon le noyau examiné (de
l’ordre de 10 000 Hz pour le 1H et 45 000 Hz pour le 13C dans un champ magnétique de 11.6
T). En effet, dans cette gamme, les signaux apparaissent à des fréquences différentes si les
noyaux sont situés dans des environnements chimiques distincts. Le noyau n’étant pas isolé
mais situé dans un environnement électronique determiné par sa structure, l’équation
fondamentale de la RMN
ν1 = (γ/2π)B0
se réécrit : νeffectif = (γ/2π)B0 (1-σ)
La valeur σ est définie comme la constante d’écran dont la valeur est proportionelle
au degré de blindage du noyau par son nuage électronique. Des électrons sous l’influence
d’un champ magnétique circulent et, en circulant, génèrent leur propre champ magnétique qui
s’oppose au champ externe appliqué ; d’où le « blindage » (Figure 8).
Figure 72. Blindage des protons au sein d’une fonction aldéhyde. Circulation des électrons le long de la liaison C=O et formation d’un champ magnétique propre s’opposant à B0 externe.
La différence dans la position de l’absorption d’un proton donné par rapport à la
position d’absorption d’un proton de référence est appelée « déplacement chimique - δδδδ » de

TP de chimie théorique et physique
A. Richel - 197 - RMN
ce proton. Le composé de référence est le TMS (tétraméthylsilane: (CH3)4Si). Par convention,
le pic du TMS défini le zéro et est placé à droite du spectre.
Les déplacements chimiques s’expriment en unité sans dimension (représentées par δ
ou ppm) en partant du δ 0.0 du TMS et s’expriment selon la formule :
δ = ν - νref
νref. 106
Le déplacement chimique étant un rapport entre un numérateur et un dénominateur de
mêmes dimensions, ce terme n’a donc pas de dimension. Par contre, on voit souvent δ
exprimé en ppm, ceci étant dû à l’utilisation du facteur 106. Pour le calcul de δ, il est possible
de remplacer νref par ν0 (ν0 = fréquence du spectromètre) au dénominateur. Compte tenu des
écarts de fréquence observés en RMN, cela constitue une très bonne approximation.
On a alors :
δ = ν - νref
ν0. 106
TMS
ν (Hz) δ (ppm)
300010
27009
2400 8
21007
18006
15005
12004
900 3
600 2
300 1
0 0
TMS
ν (Hz) δ (ppm)
300010
27009
2400 8
21007
18006
15005
12004
900 3
600 2
300 1
0 0
300 MHz
600 MHz
600010
54009
4800 8
42007
36006
30005
24004
1800 3
1200 2
600 1
TMS
ν (Hz) δ (ppm)
0 0
600010
54009
4800 8
42007
36006
30005
24004
1800 3
1200 2
600 1
TMS
ν (Hz) δ (ppm)
0 0
Si
CH3
CH3
CH3CH3
Haute fréquence
Déblindé
(Champ faible)
Basse fréquence
Blindé
(Champ fort)
Figure 73. Echelle RMN à 300 et 600 MHz.
Ainsi, sur l’échelle de 600 MHz, un pic qui apparaitrait à 3000 Hz serait positionné à
3000 Hz/600MHz = 4 ppm (ppm = part par millon).

TP de chimie théorique et physique
A. Richel - 198 - RMN
4.2. RELATION ENTRE LE DÉPLACEMENT CHIMIQUE ET L ’ENVIRONNEMENT CHIMIQUE :
ÉLECTRONÉGATIVITÉ , ANISOTROPIE DIAMAGNÉTIQUE
La constante d’écran, et par conséquent le déplacement chimique, est fonction de
divers paramètres :
(a) l’électronégativité des substituants voisins du noyau étudié
Plus la couverture électronique est grande, plus le blindage est élevé et inversement.
Lorsque la couverture électronique augmente, le champ nécessaire pour produire la résonance
croît ainsi que l'illustre le dessin ci-dessous.
Plus la densité électronique autour du noyau est faible, plus la constante d’écran est
faible (déblindage) et plus le déplacement chimique est grand. L’exemple le plus significatif
est celui des halogénures d’alkyle (Tableau 2). Comme on pouvait le prévoir, le déplacement
chimique, c’est-à-dire le niveau de déblindage du proton suit l’électronégativité de
l’halogénure présent.
Tableau 2. Relation entre le déplacement chimique et l’électronégativité des halogènes (selon Pauling) pour une série d’halogénures d’alkyle.
CH3F CH3Cl CH3Br CH3I CH3H
Electronégativité 4.0 3.0 2.8 2.5 2.1
δ (CH3) 4.13 2.84 2.45 1.98 0.13
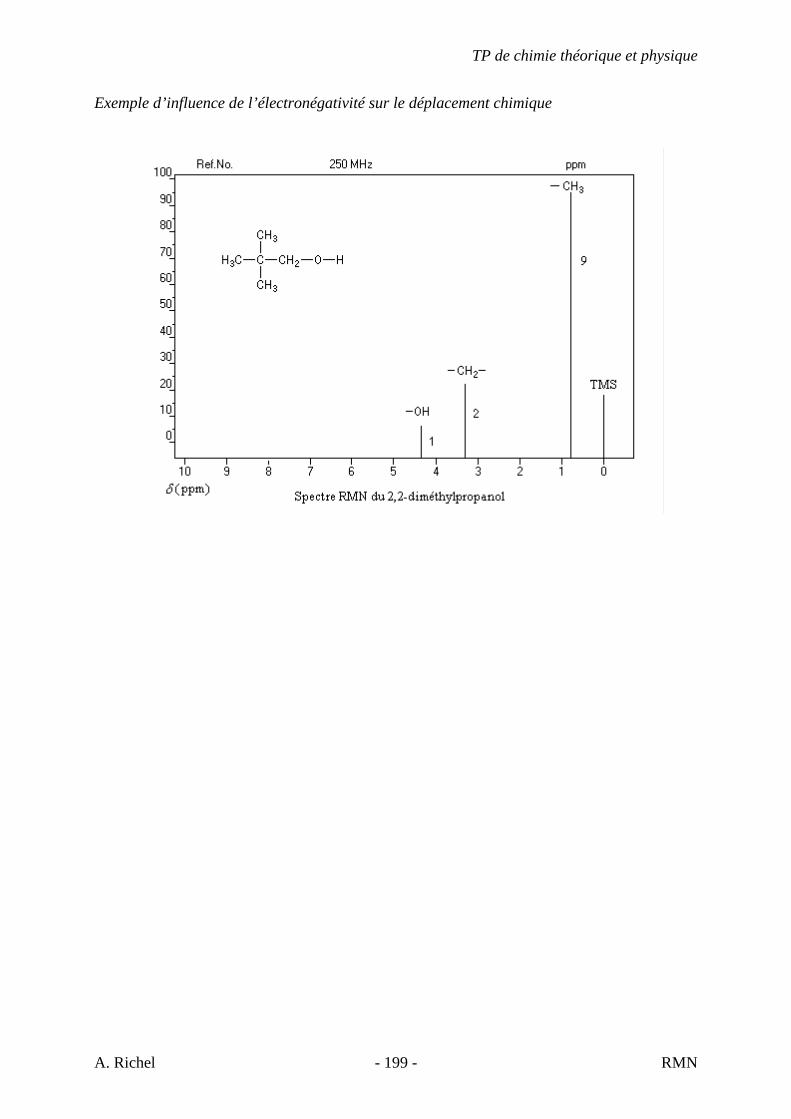
TP de chimie théorique et physique
A. Richel - 199 - RMN
Exemple d’influence de l’électronégativité sur le déplacement chimique

TP de chimie théorique et physique
A. Richel - 200 - RMN
(b) les effets d’anisotropie diamagnétique dus aux liaisons chimiques
magnétiquement anisotropes (liaisons doubles et triples, cycles...)
Il arrive couramment qu’il ne soit pas possible d’expliquer le déplacement chimique
uniquement à partir de l’électronégativité des groupements voisins. Ceci peut s’expliquer par
ce que l’on appelle l’anisotropie diamagnétique.
Les effets d’anisotropie diamagnétique signifient que le blindage (ou déblindage)
dépend de l’orientation de la molécule dans le champ magnétique externe.
Exemple 1: cas de la molécule d’acétylène (Figure 10)
Cette molécule est linéaire et présente une liaison triple le long de l’axe de la
molécule. Si l’axe de la molécule se trouve aligné avec le champ magnétique externe B0, les
électrons π des orbitales formant la liaison triple peuvent circuler perpendiculairement à B0.
Cette circulation d’électrons induit un champ magnétique propre qui s’oppose à B0. Les lignes
de champ magnétique induit par la circulation des électrons provoquent un blindage des
protons et on retrouve leur signal RMN à plus basses fréquences que l’electronégativité ne le
prédit.
(a) (b)
Figure 74. (a) Blindage des protons alcynes avec visualisation de la circulation des ékectrons ππππ. (b) Visualisation de l’apparition de zones indiquées par un (+) et propices au blindage alors que les zones symbolysées (-) favorisent un déblindage.

TP de chimie théorique et physique
A. Richel - 201 - RMN
Exemple 2: création de courant de cycle dans le benzène (Figure 11)
Cet exemple explique le déblindage important des protons situés dans le plan du cycle
benzénique. Typiquement, les protons relatifs à un cycle aromatique se présentent vers les δ
6-8 ppm.
(a) (b)
Figure 75. Déblindage des protons dans un cycle benzénique : effet de courant de cycle. (a) La circulation des électrons ππππ (trait continu) induit un champ de réaction (trait pointillé) ; (b) Anisotropie du bénzène avec visualisation des zones (+) propices au blindage ; tandis que les zones (-) favorisent un déblindage.
De manière générale, tous les groupements qui possèdent des électrons π au voisinage
du proton étudié vont générer de l’anisotropie diamagnétique. L’amplitude des effets de
l’anisotropie s’atténue lorsqu’un proton est plus éloigné des liaisons multiples. Ainsi, les
protons qui se trouvent sur des ramifications liées à un cycle aromatique seront peu influencés
par l’anisotropie, à mesure que la chaîne carbonée s’allonge.
Une bonne compréhension des concepts d’electronégativité et d’anisotropie
diamagnétique permet ainsi une rationalisation et une prédiction des déplacements chimiques
dans les spectres protons.
Exemple 3 : 3-penten-2-one
La mise en évidence de formes de résonance dans une cétone α,β-insaturée permet
d’illustrer le déblindage du proton en β (« appauvrissement » de sa densité électronique). Cet
effet explique pourquoi ce proton en β apparait à des déplacements chimiques plus
importants.

TP de chimie théorique et physique
A. Richel - 202 - RMN
CH3
H
H O
CH3 CH3
H
H O
CH3
_
+
α
β
H β β β β δ δ δ δ ~ 6.8 ppmH β β β β δ δ δ δ ~ 6.8 ppm H α α α α
δ δ δ δ ~ 6.2 ppmH α α α α δ δ δ δ ~ 6.2 ppm
4.3. TABLES DES DÉPLACEMENTS CHIMIQUES
Le déplacement chimique exprimé en ppm est indépendant du champ B0 appliqué, il
ne dépend que de la fonction chimique étudiée et de son environnement. La Figure 12 est une
table de déplacements chimiques proton pour les fonctions chimiques les plus courantes. Par
exemple, on observe que le déplacement chimique d'un hydrogène sur un cycle benzénique
est compris entre 6,8 et 8,4 ppm. La valeur exacte de son déplacement chimique dépendra des
autres substituants du cycle, du solvant, de la température, etc.
Figure 76. Tables des déplacements chimiques en RMN 1H pour des groupements chimiques classiques.

TP de chimie théorique et physique
A. Richel - 203 - RMN
5. NOTION D ’ INTÉGRATION
Les protons chimiquement identiques ont la même fréquence de précession (ou
fréquence de résonance) et ont de ce fait le même déplacement chimique. L’intensité du signal
considéré est directement proportionnelle au nombre de protons impliqués dans ce signal.
L’ intégration du signal (càd la mesure de l’aire sous le pic) par rapport à un signal de
référence permet alors de déterminer le nombre de protons par massif. Cette information sera
importante pour retrouver la structure de la molécule.
Un dispositif appelé intégrateur permet de mesurer la surface des différents pics ou
d'ensembles de pics proches (appelés massifs). Cette surface est proportionnelle au nombre de
protons correspondants. L'appareil trace alors sur le spectre une courbe possédant des marches
appelée courbe d'intégration. Le rapport des hauteurs entre deux paliers est égal au rapport des
nombre de protons impliqués dans les massifs correspondants. La courbe ne donne donc pas
le nombre absolu de protons d'un massif mais leur proportion relative dans ce massif.
La connaissance de la courbe d'intégration permet de préciser la structure de la
molécule comme le montrent les exemples ci-dessous.
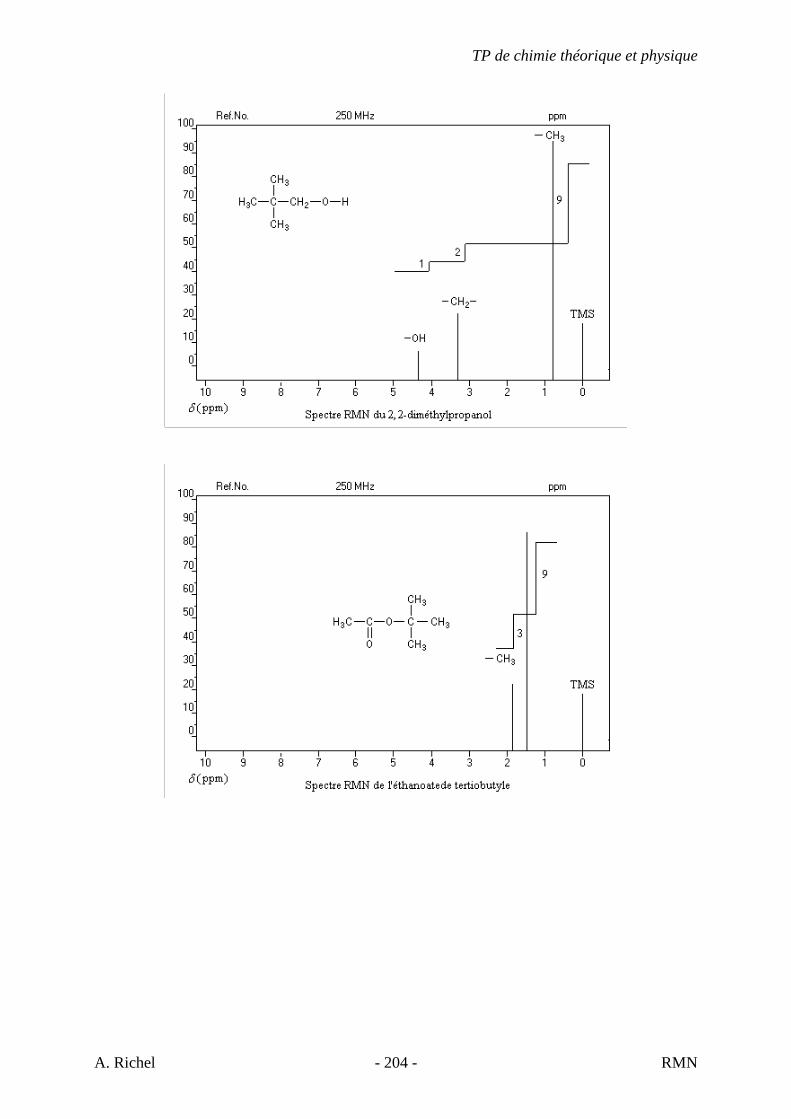
TP de chimie théorique et physique
A. Richel - 204 - RMN

TP de chimie théorique et physique
A. Richel - 205 - RMN
6. EQUIVALENCE ET NON -EQUIVALENCE DES DÉPLACEMENTS CHIMIQUES
Deux protons sont dits équivalents du point de vue du déplacement chimique s’ils
sont échangeables par une quelconque opération de symétrie ou par un mécanisme rapide (ex.
interconversion).
6.1. EQUIVALENCE DU DÉPLACEMENT CHIMIQUE PAR ÉCHANGE VIA UNE OPÉRATION DE
SYMÉTRIE
(a) Homotopie
Des noyaux (ou groupes de noyaux) sont dits homotopiques s’ils sont
interchangeables par une opération de rotation. Dans ce cas, les déplacements chimiques sont
identiques. C’est le cas, par exemple, des 3 protons du trichlorométhane qui sont
échangeables par rotation autour d’un axe de symétrie C3.
1
2
3
(b) Enantiotopie
Des noyaux (ou groupes de noyaux) sont dits énantiotopiques s’ils sont
interchangeables par une opération de rotation suivie d’une réflexion. Dans ce cas, les
déplacements chimiques sont identiques.

TP de chimie théorique et physique
A. Richel - 206 - RMN
Une combinaison entre homotopie et énantiotopie peut être pratiquement rencontrée.
C’est le cas du chlorométhoxyméthane. Les trois noyaux d’hydrogène homotopiques du
groupe méthyle donnent un signal unique, puisque l’échange conformationnel rapide, par
rotation autour de l’axe C—O, les rend équivalents en moyenne. Les deux noyaux
énantiotopiques du groupe méthylène donnent eux-aussi un signal unique
puisqu’échangeables par réflexion sur le plan contenant les atomes de carbone, d’oxygène et
de chlore.
6.2. NON-EQUIVALENCE DES DÉPLACEMENTS CHIMIQUES
Des noyaux (ou groupes de noyaux) sont dits diastéréotopiques s’ils ne sont pas
interchangeables par une opération de symétrie. Dans ce cas, les déplacements chimiques sont
différents. Le couplage entre les 2 protons Ha et Hb est visible sur le spectre.
Hc
Dans cette molécule, le groupement CH2 consiste en une paire de protons
diastéréotopiques Ha et Hb. Les protons Ha et Hb sont couplés entre eux par une constante de
couplage Jab. Un couplage avec un proton Hc adjacent est également observé avec des
constantes de couplage Jac = Jbc

TP de chimie théorique et physique
A. Richel - 207 - RMN
7. NOTIONS DE SYSTÈMES DE SPIN, DE COUPLAGE DE SPIN ET DE MULTIPLICITÉ
7.1. DÉFINITION GÉNÉRALE
Lorsque les spectres RMN sont observés avec une résolution suffisante, on observe
que certains signaux sont constitués de plusieurs pics. Ce phénomène est dû à une interaction
entre les noyaux qui porte le nom de couplage scalaire. On appelle système de spins un
ensemble de spins couplés de façons scalaire.
On distingue les couplages :
• hétéronucléaires : le système de spins est constitué de noyaux de nature différente (par
exemple 1H et 13C). Il s'agit de couplage 1J ;
• homonucléaires : le système de spins est constitué de noyaux de même nature (par
exemple des protons) il s'agit de couplages nJ avec n > 1.
Couplage Nombre de liaisons Nom 2J 2 géminal 3J 3 vicinal 4J 4 longue distance
L'interaction de contact de Fermi polarise le moment magnétique des électrons dans le
sens opposé au moment magnétique du noyau. Cette polarisation se transmet de proche en
proche aux autres noyaux grâce aux électrons de liaison.

TP de chimie théorique et physique
A. Richel - 208 - RMN
7.2. INTERACTION ENTRE DEUX PROTONS
Si l’on considère une molécule du type A2CH–HCX2 dans lequel A et X ne sont pas
identiques, une différenciation du déplacement chimique des protons HA et HX sera observée.
De plus, il est intéressant de constater que le spectre RMN ne montre pas uniquement deux
pics mais quatre, formés de deux paires de pics ayant la même intensité et séparés en
déplacement chimique (Figure 13). Les deux pics (attendus sur la base du déplacement
chimique) se présentent donc sous la forme de doublets. La même distance J mesurée en Hz
sépare les composantes de ces deux doublets et elle est indépendante du champ magnétique
appliqué. La multiplicité des signaux obtenus en RMN peut être beaucoup plus complexe et il
est courant d’observer la présence de doublets, triplets, quadruplets, etc.
Figure 77. Spectre résultant de deux spins A et X ayant des déplacements chimiques différents et couplés entre eux avec la constante J.
Cette démultiplication des pics provient du couplage spin-spin. Il s’agit d’interactions
magnétiques entre les protons qui sont transmises par les électrons à travers les liaisons
chimiques.
7.3. ORIGINES DU PHÉNOMÈNE DE COUPLAGE
On peut donner de ce phénomène l’interprétation suivante : le proton HX peut être
considéré comme un petit aimant qui induit un champ local ∆B au niveau du proton HA. Si ce
champ local s’ajoute au champ B0 lorsque HX est parallèle à B0, en revanche il se retranche de
B0 lorsque HX est antiparallèle à B0. Le proton HA est donc soumis, suivant l’orientation de

TP de chimie théorique et physique
A. Richel - 209 - RMN
HX, aux champs B0–∆B ou B0+∆B et il va résonner à deux fréquences différentes séparées par
la constante J (Figure 13). Le champ ∆B étant indépendant de B0, la constante J est
indépendante de B0 et on la mesure en Hz.
7.4 VALEUR DE LA CONSTANTE DE COUPLAGE J
La valeur de la constante de couplage dépend du nombre, de la nature et de la
géométrie des liaisons entre les noyaux couplés. De façon générale, plus le nombre de liaisons
est grand, plus le couplage est faible (Figure 14).
Figure 78. Gammes (Jab) des couplages 1H-1H pour différentes situations rencontrées dans les composés organiques.
Ainsi, les couplages entre les protons portés par deux carbones voisins, se font par le
biais de 3 liaisons chimiques, ils sont notés 3J. Sur une molécule sans insaturation, ils ont
généralement des valeurs comprises entre 6 et 8 Hz. Par contre, dans ce cas, les couplages 4J
sont trop petits pour être observés.

TP de chimie théorique et physique
A. Richel - 210 - RMN
7.5. CORRELATION ENTRE LA CONSTANTE DE COUPLAGE J ET LA GÉOMÉTRIE DE LA
MOLÉCULE
La valeur de la constante de couplage dépend également de la géométrie de la
molécule. Ainsi, il est possible de dire si deux protons sur une double liaison sont en position
cis, trans ou géminée (sur le même carbone) ; les constantes de couplage pour ces trois
géométries se situent respectivement dans les zones 6 à 10 Hz, 12 à 18 Hz, et 0 à 3 Hz.
7.6. NOMENCLATURE DES SYSTÈMES DE SPIN
Les lettres A, B, X, etc sont employées pour nommer des systèmes différents de noyaux en
interaction
L’indice sous chaque lettre désigne le nombre de protons isochrones (ayant le même
déplacement chimique).
7.7. EQUIVALENCE MAGNÉTIQUE
Heureusement, tous les noyaux ne sont pas couplés entre-eux car cela entraînerait une
très grande complication des spectres. Pour que les couplages entre noyaux n'apparaissent pas,
ceux-ci doivent être magnétiquement équivalents.
Considérons un ensemble de noyaux Ai. On dit que ces noyaux sont magnétiquement
équivalents si, et seulement si :
• ils sont chimiquement équivalents, c'est à dire qu'ils possèdent le même déplacement
chimique ; on dit que ce sont des noyaux isochrones ;
• ils sont couplés de façon identique (même constante de couplage) avec tous les
noyaux extérieurs à cet ensemble ; on dit que ces noyaux sont isogames.
Lorsque des noyaux sont chimiquement équivalents sans être magnétiquement
équivalents, les lettres correspondantes sont "primées".

TP de chimie théorique et physique
A. Richel - 211 - RMN
Dans l'exemple suivant, les noyaux A et A' ne sont pas isogames car ils n'ont pas les
mêmes constantes de couplages avec les noyaux X et X'. Il s'agit d'un système AA'XX' et non
d'un système A2X2.
De même dans l'exemple ci-dessous A et A' ne sont pas isogames.
En revanche, les noyaux A et A' sont isogames dans la molécule suivante. Leur
couplage est invisible dans le spectre. Il s'agit d'un système A2X.
Tout comme l'équivalence chimique, l'équivalence magnétique peut être le fruit d'une
moyenne qui résulte d'un mouvement interne de la molécule qui s'effectue avec une durée
caractéristique beaucoup plus courte que le temps de relaxation T2. Le spectre du système
suivant dépend de la température.

TP de chimie théorique et physique
A. Richel - 212 - RMN
• basse température : pas de rotation libre (structure "figée" I)
Protons H1H2 H3H4 H5
Système AA' BB' C
• température plus élevée : rotation libre (conformations I, II, III, etc.)
Protons H1H2 H3H4 H5
Système A2 B3
Remarque : dans les exemples précédents les protons H1 et H2 sont énantiotopiques et ils sont
donc isochrones (dans un solvant achiral). Il n'en serait pas de même si la présence voisine
d'un centre chiral les rendait diastéréotopiques.
7.8. ORDRE DES COUPLAGES
Le couplage est faible, lorsque :
On dit que le spectre est analysable au premier ordre. En pratique, on utilise le critère :
Lorsqu'un système de protons est analysable au premier ordre les groupes de protons
chimiquement équivalents sont désignés par des lettres éloignées dans l'alphabet. Par exemple
deux protons sont désignés par AX. Trois protons couplés par AMX.

TP de chimie théorique et physique
A. Richel - 213 - RMN
Si la condition précédente n'est pas réalisée, le couplage est fort. On dit que le spectre est du
second ordre. Le nombre et l'intensité des raies sont modifiées. Les déplacements chimiques
ne peuvent pas être lus directement sur le spectre. L'analyse spectrale ne peut être effectuée
que par le calcul. De même, la mesure directe de J n'est plus possible.
On peut revenir à un spectre du premier ordre en augmentant le champ magnétique. En
effet, la constante de couplage est indépendante du champ appliqué mais la différence de
fréquence entre deux noyaux croit avec lui.
• Système AMX
Le signal initial de A est d'abord dédoublé par son interaction avec M. Ces signaux
sont à leur tour dédoublés par l'interaction avec X. On obtient un doublet de doublet.
• Système AX2
Il s'agit d'un cas particulier de système AMX avec 3JAM = 3JAX. La raie centrale a donc
une hauteur double des deux autres.

TP de chimie théorique et physique
A. Richel - 214 - RMN
• On a de même des systèmes AX3 et plus généralement AXn.
Les hauteurs relatives des pics des multiplets sont données par le triangle de Pascal.
7.9. COUPLAGE VICINAL ET GÉMINAL
7.9.1. COUPLAGE GÉMINAL
On appelle couplage géminal, le couplage entre deux protons liés à un même atome de
carbone. Ce type de couplage n'est pas fréquemment observé car les protons géminés sont
souvent rendus magnétiquement équivalents en moyenne par les mouvements
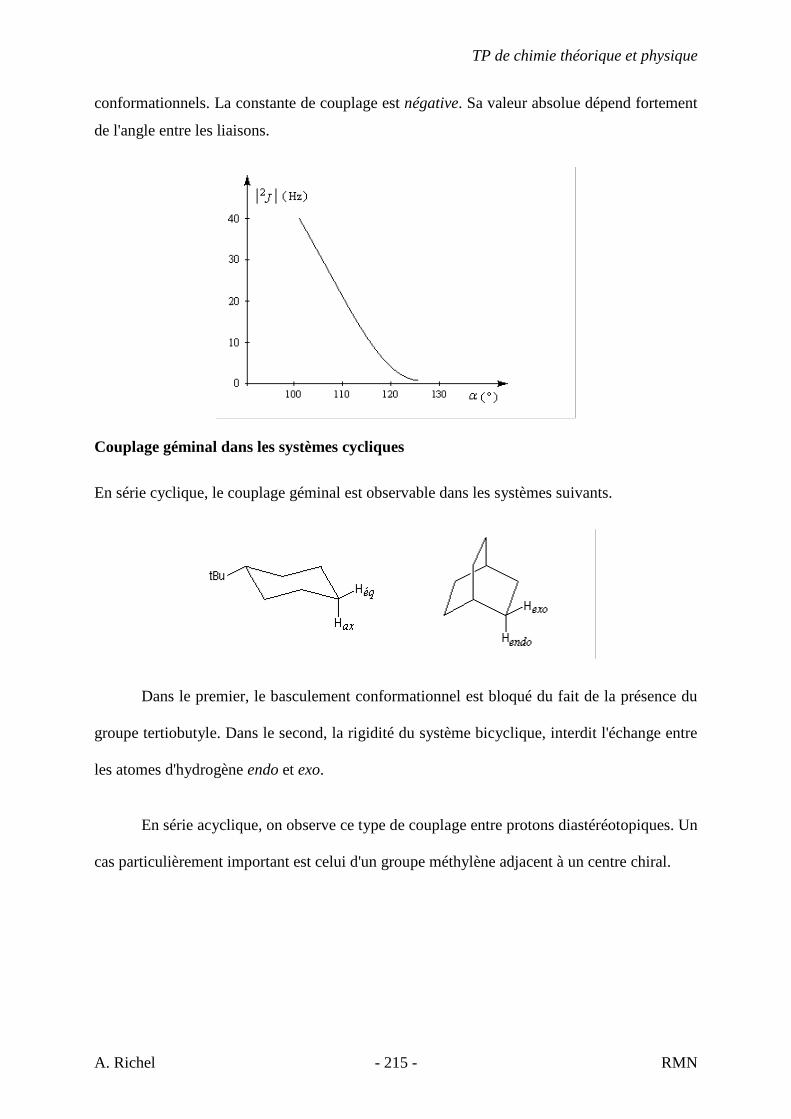
TP de chimie théorique et physique
A. Richel - 215 - RMN
conformationnels. La constante de couplage est négative. Sa valeur absolue dépend fortement
de l'angle entre les liaisons.
Couplage géminal dans les systèmes cycliques
En série cyclique, le couplage géminal est observable dans les systèmes suivants.
Dans le premier, le basculement conformationnel est bloqué du fait de la présence du
groupe tertiobutyle. Dans le second, la rigidité du système bicyclique, interdit l'échange entre
les atomes d'hydrogène endo et exo.
En série acyclique, on observe ce type de couplage entre protons diastéréotopiques. Un
cas particulièrement important est celui d'un groupe méthylène adjacent à un centre chiral.
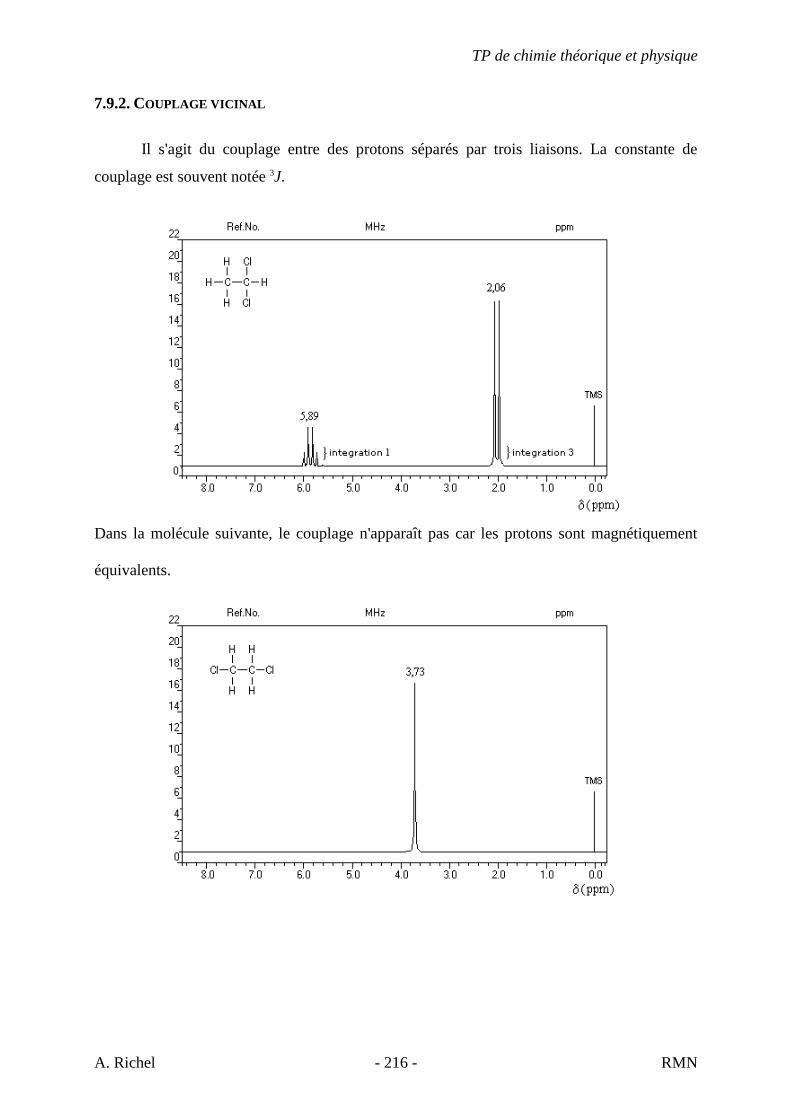
TP de chimie théorique et physique
A. Richel - 216 - RMN
7.9.2. COUPLAGE VICINAL
Il s'agit du couplage entre des protons séparés par trois liaisons. La constante de
couplage est souvent notée 3J.
Dans la molécule suivante, le couplage n'apparaît pas car les protons sont magnétiquement
équivalents.
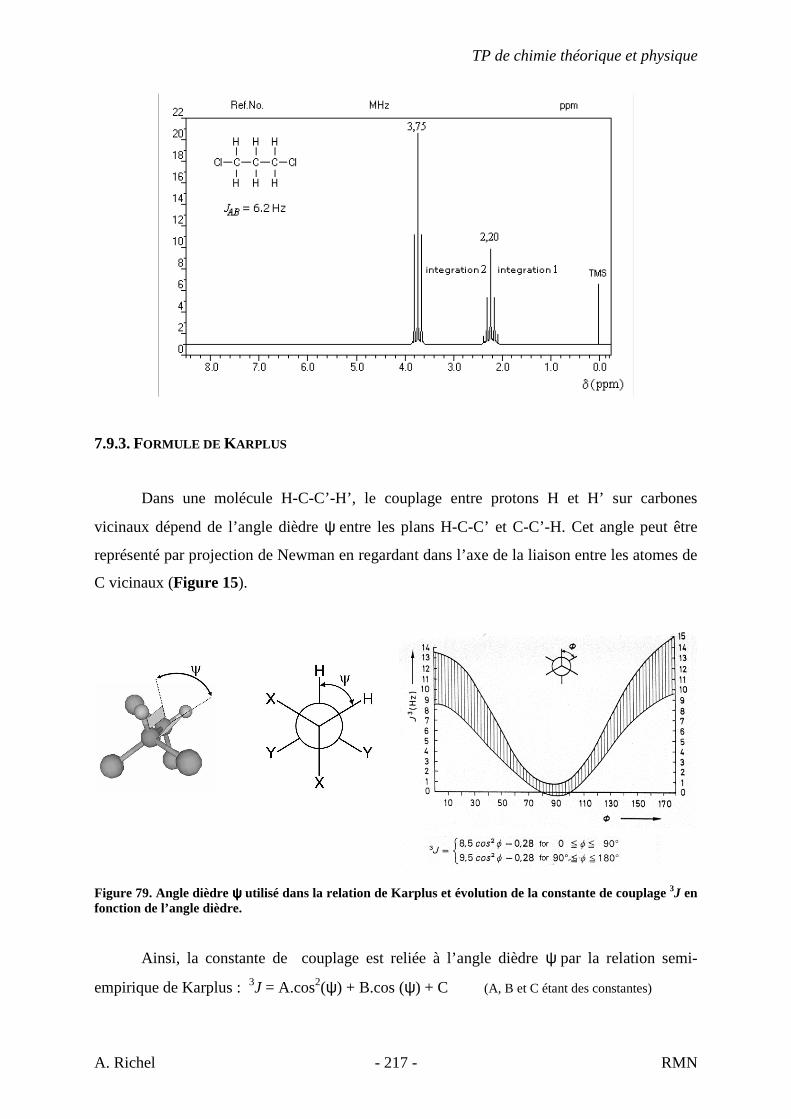
TP de chimie théorique et physique
A. Richel - 217 - RMN
7.9.3. FORMULE DE KARPLUS
Dans une molécule H-C-C’-H’, le couplage entre protons H et H’ sur carbones
vicinaux dépend de l’angle dièdre ψ entre les plans H-C-C’ et C-C’-H. Cet angle peut être
représenté par projection de Newman en regardant dans l’axe de la liaison entre les atomes de
C vicinaux (Figure 15).
Figure 79. Angle dièdre ψ ψ ψ ψ utilisé dans la relation de Karplus et évolution de la constante de couplage 3J en fonction de l’angle dièdre.
Ainsi, la constante de couplage est reliée à l’angle dièdre ψ par la relation semi-
empirique de Karplus : 3J = A.cos2(ψ) + B.cos (ψ) + C (A, B et C étant des constantes)

TP de chimie théorique et physique
A. Richel - 218 - RMN
Exemple : cas du cyclohexane (basculement conformationnel) (Tableau 3 et Figure 16)
Pour le cyclohexane : équilibre entre plusieurs conformations. Le passage d'une conformation
chaise à une autre, inverse les positions axiales et équatoriales. A 300 K le passage d'une
forme à l'autre s'effectue environ 100 000 fois par seconde. L'existence de deux catégories
d'atomes d'hydrogène peut être mise en évidence par spectroscopie RMN si la température est
suffisamment basse.
Tableau 3. Constantes de couplage J dans des cyclohexanes en fonction des angles de liaison
Angle dièdre ψ 3J (Hz)
Axial-axial 180° 8-14
Axial-équatorial 60° 1-7
Equatorial-équatorial 60° 1-7
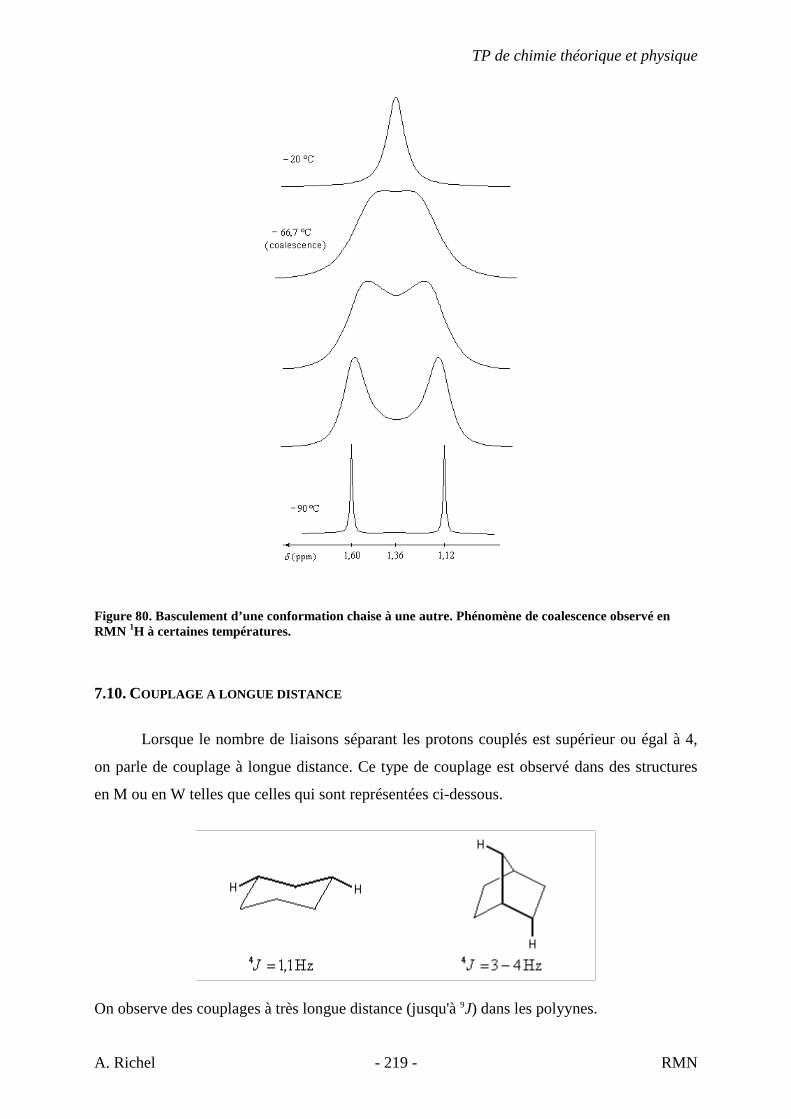
TP de chimie théorique et physique
A. Richel - 219 - RMN
Figure 80. Basculement d’une conformation chaise à une autre. Phénomène de coalescence observé en RMN 1H à certaines températures.
7.10. COUPLAGE A LONGUE DISTANCE
Lorsque le nombre de liaisons séparant les protons couplés est supérieur ou égal à 4,
on parle de couplage à longue distance. Ce type de couplage est observé dans des structures
en M ou en W telles que celles qui sont représentées ci-dessous.
On observe des couplages à très longue distance (jusqu'à 9J) dans les polyynes.

TP de chimie théorique et physique
A. Richel - 220 - RMN
7.11. COUPLAGE FORT
Des pics parfaitement symétriques n'apparaissent que dans le cas où la différence des
fréquences de résonance νA − νX des groupes de noyaux A et X est beaucoup plus élevée que la
constante de couplage J. On peut montrer par un calcul de mécanique quantique, que les
niveaux énergétiques des deux noyaux ne peuvent jamais s'intercaler. Afin d'éviter un tel
croisement des niveaux, les pics intérieurs ont une intensité qui croit aux dépens des pics
extérieurs dont l'intensité diminue. Les barycentres des doublets restent fixes à la valeur
(νA + νX)/2.
On donne le nom de quadruplet AB à ce type de spectre. Le rapprochement des lettres
A et B dans l’alphabet indique que la différence νA− νB n’est pas grande devant la constante de
couplage.
La figure ci-dessous montre l'évolution d'un système AX au fur et à mesure que le
rapport ∆ν/J, diminue. Lorsque la différence des déplacements chimiques augmente, le
système AB se rapproche graduellement d'un système AX. Au contraire quand elle tend vers
zéro, les pics externes disparaissent quelle que soit la valeur de J. On obtient un pic unique.
C'est pour cette raison que des protons magnétiquement équivalents apparaissent sous la
forme d’un signal unique.

TP de chimie théorique et physique
A. Richel - 221 - RMN
On rencontre ce type de spectres dans de nombreuses structures :
• chez les composés éthyléniques ;
• chez les composés aromatiques
8. PHENOMENES D'ECHANGE DE PROTONS
Le phénomène d'échange de protons est un cas particulier d'échange chimique qui
s'observe chaque fois qu'un proton est suffisamment mobile pour pouvoir être échangé entre
deux molécules. On va rencontrer ce phénomène dans les familles de composés où le proton
est lié à un hétéroatome : les alcools, les amines, les acides carboxyliques.
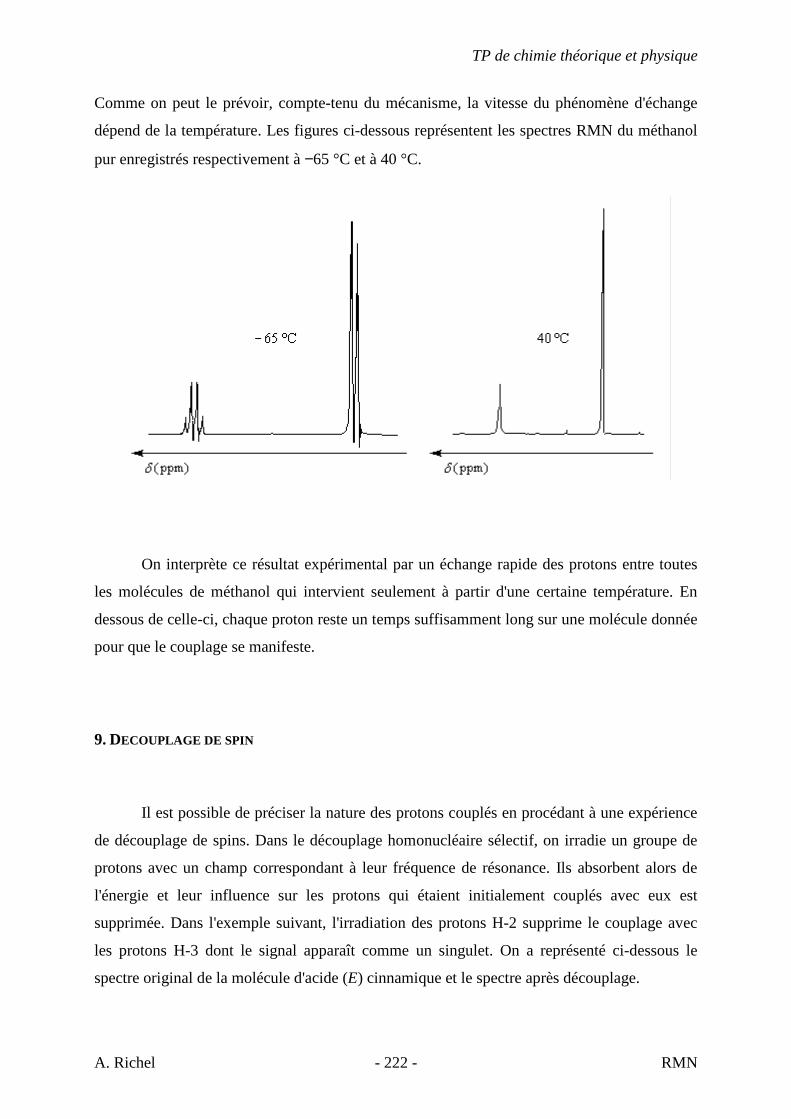
TP de chimie théorique et physique
A. Richel - 222 - RMN
Comme on peut le prévoir, compte-tenu du mécanisme, la vitesse du phénomène d'échange
dépend de la température. Les figures ci-dessous représentent les spectres RMN du méthanol
pur enregistrés respectivement à −65 °C et à 40 °C.
On interprète ce résultat expérimental par un échange rapide des protons entre toutes
les molécules de méthanol qui intervient seulement à partir d'une certaine température. En
dessous de celle-ci, chaque proton reste un temps suffisamment long sur une molécule donnée
pour que le couplage se manifeste.
9. DECOUPLAGE DE SPIN
Il est possible de préciser la nature des protons couplés en procédant à une expérience
de découplage de spins. Dans le découplage homonucléaire sélectif, on irradie un groupe de
protons avec un champ correspondant à leur fréquence de résonance. Ils absorbent alors de
l'énergie et leur influence sur les protons qui étaient initialement couplés avec eux est
supprimée. Dans l'exemple suivant, l'irradiation des protons H-2 supprime le couplage avec
les protons H-3 dont le signal apparaît comme un singulet. On a représenté ci-dessous le
spectre original de la molécule d'acide (E) cinnamique et le spectre après découplage.
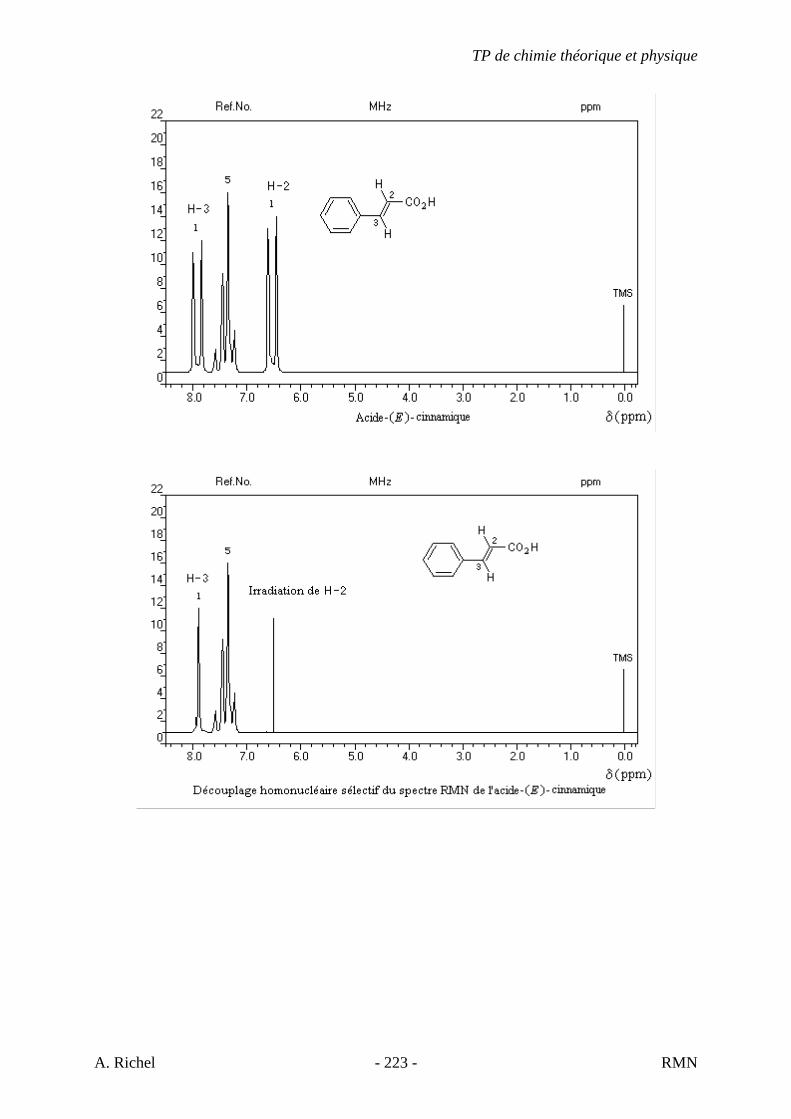
TP de chimie théorique et physique
A. Richel - 223 - RMN

TP de chimie théorique et physique
A. Richel - 224 - RMN
10. EFFET OVERHAUSER NUCLÉAIRE (EFFET NOE)
Considérons deux spins A et S suffisamment proches dans l'espace pour qu'une
interaction dipolaire entre-eux puisse se manifester. Cette distance doit être typiquement
inférieure à 500 pm car l'interaction, qui varie en 1/r6, s'amortit rapidement avec
l'éloignement.
Si on soumet le spin A à une irradiation à sa fréquence de résonance jusqu'à ce qu'il y
ait saturation. Cela a pour effet de faire disparaître le signal relatif à A. Mais, si les spins A et
S sont couplés, un phénomène de relaxation croisée va pouvoir transférer de l'aimantation, du
spin saturé A au spin S couplé avec lui. C'est l'effet Overhauser nucléaire (en anglais : nuclear
Overhauser effect ou nOe).
L'effet Overhauser nucléaire est très sensible à la distance entre les protons couplés.
C'est pourquoi il est à la base de méthodes très utiles pour la détermination de structures en
RMN. En particulier, il est utilisé pour différencier des diastéréoisomères. Dans l'exemple
suivant, les protons A et S manifestent un effet nOe beaucoup plus important dans la molécule
II que dans la molécule I du fait de leur plus grande proximité dans l'espace dans le second
cas.

TP de chimie théorique et physique
A. Richel - 225 - RMN
11. TABLES UTILES POUR L ’ INTERPRÉTATION DES SPECTRES RMN 1H
Tables issues de « Identification spectrométrique de composés organiques » (2de édition)
Silverstein, Webster, Kiemle Ed. De Boeck Université 2007

TP de chimie théorique et physique
A. Richel - 226 - RMN
12. MARCHE À SUIVRE POUR L ’ INTERPRÉTATION D ’UN SPECTRE PROTON
La suite chronologique des informations à exploiter est :
(1) le nombre de signaux et la valeur du déplacement chimique correspondant, ce qui
permet d'identifier le nombre et la nature des groupes de protons équivalents ; (en utilisant
une table de données)
(2) la courbe d'intégration qui donne le nombre de protons de chaque type ; (l'aire de chaque
pic est proportionnelle au nombre de protons responsables du pic. Sur la courbe d'intégration
la distance entre deux paliers est proportionnelle à la surface du pic correspondant et donc au
nombre de protons.)
(3) la forme de chaque signal qui renseigne sur le nombre de protons voisins du proton
étudié. (un signal peut être constitué de plusieurs pics. Ce phénomène est lié à la présence des
protons voisins et est appelé couplage spin-spin. En pratique un proton ou un groupe de
protons équivalents ayant n protons voisins donnera un signal constitué de (n+1) pics, appelé
multiplet (singulet : 1 pic;doublet : 2 pics;triplet : 3 pics;quadruplet : 4 pics;quintuplet : 5
pics....)).
Les intensités relatives des pics constituant des multiplets sont entre elles comme les
coefficients du polynôme (a+b)n avec n représentant le nombre de protons voisins:
n = 1 doublet 1:1 n = 2 triplet 1:2:1 n = 3 quadruplet 1:3:3:1 n = 4 quintuplé 1:4:6:4:1
Attention! la multiplicité des pics ne dépend pas du nombre de protons qui résonnent, mais de
ceux de leurs voisins.
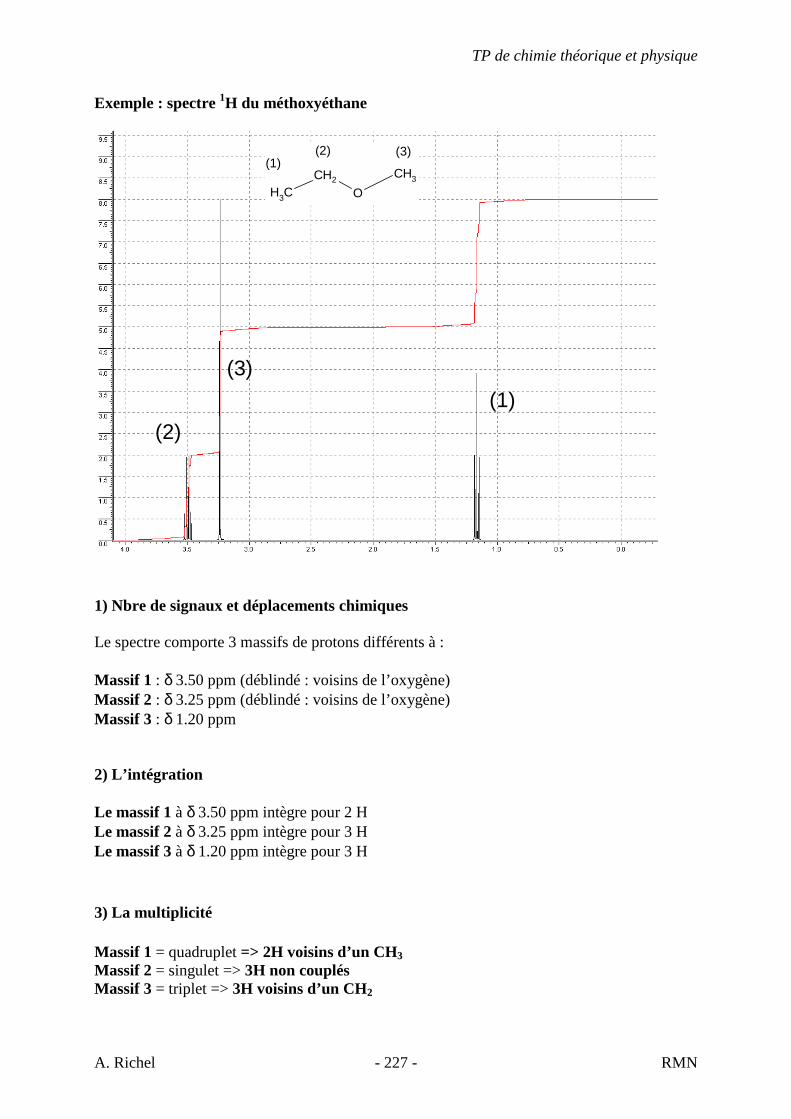
TP de chimie théorique et physique
A. Richel - 227 - RMN
Exemple : spectre 1H du méthoxyéthane
CH3 O
CH3CH2
(1)(2) (3)
(3)
(1)
(2)
1) Nbre de signaux et déplacements chimiques Le spectre comporte 3 massifs de protons différents à : Massif 1 : δ 3.50 ppm (déblindé : voisins de l’oxygène) Massif 2 : δ 3.25 ppm (déblindé : voisins de l’oxygène) Massif 3 : δ 1.20 ppm 2) L’intégration Le massif 1 à δ 3.50 ppm intègre pour 2 H Le massif 2 à δ 3.25 ppm intègre pour 3 H Le massif 3 à δ 1.20 ppm intègre pour 3 H 3) La multiplicité Massif 1 = quadruplet => 2H voisins d’un CH3 Massif 2 = singulet => 3H non couplés Massif 3 = triplet => 3H voisins d’un CH2
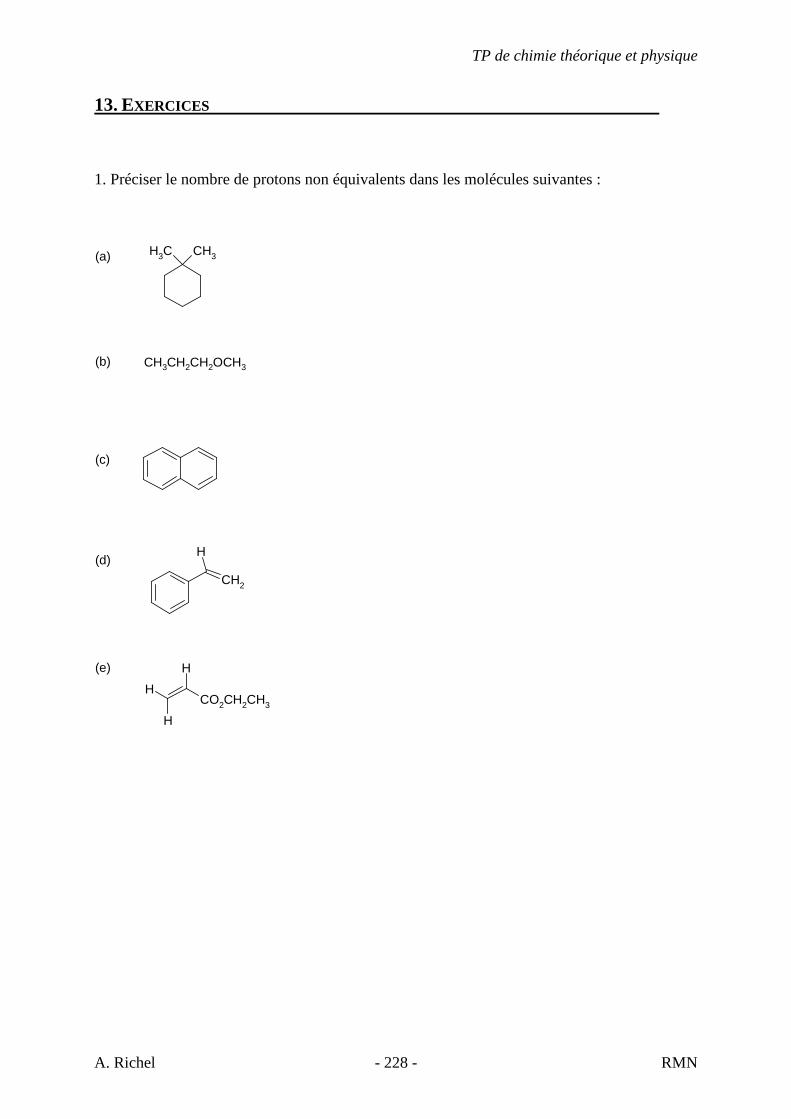
TP de chimie théorique et physique
A. Richel - 228 - RMN
13. EXERCICES
1. Préciser le nombre de protons non équivalents dans les molécules suivantes :
CH3 CH3
H
CH2
H
H
CO2CH2CH3
H
(a)
(b) CH3CH2CH2OCH3
(c)
(d)
(e)

TP de chimie théorique et physique
A. Richel - 229 - RMN
2. Préciser la multiplicité (nombre de protons par massif) dans les molécules suivantes : (a)
(b)
(c) trans-2-butène
CH3CH2COOCH3
(CH3)3CH
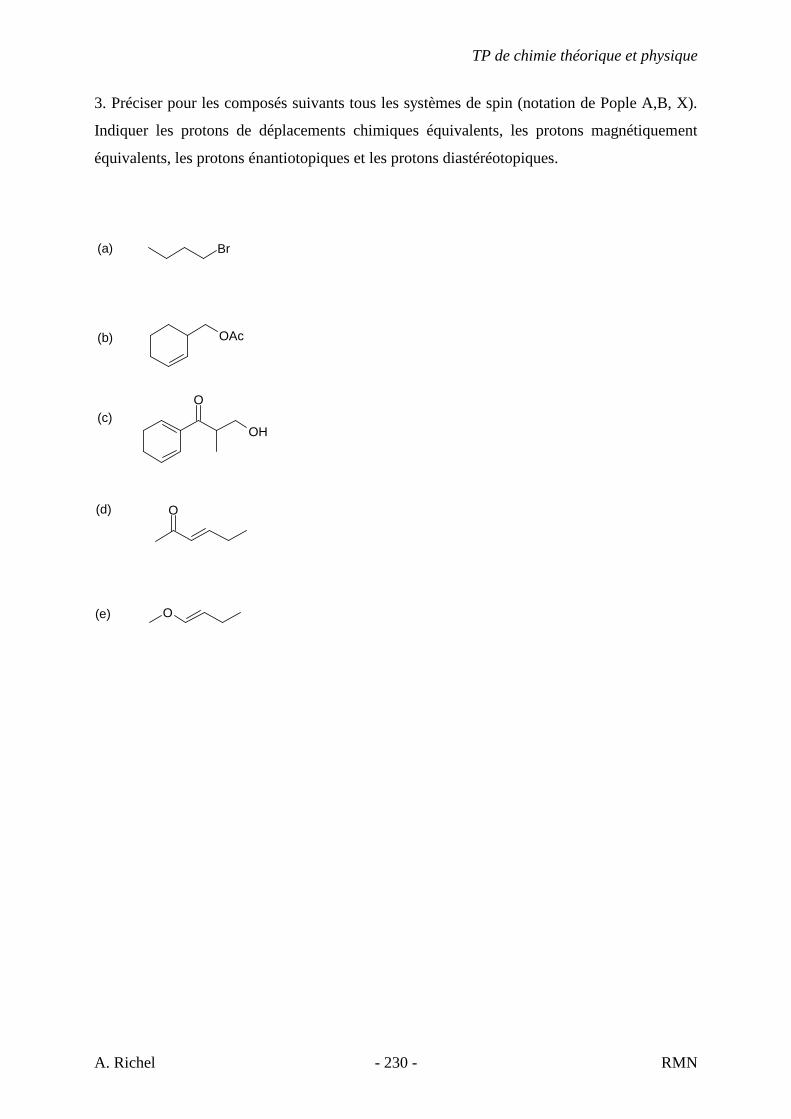
TP de chimie théorique et physique
A. Richel - 230 - RMN
3. Préciser pour les composés suivants tous les systèmes de spin (notation de Pople A,B, X).
Indiquer les protons de déplacements chimiques équivalents, les protons magnétiquement
équivalents, les protons énantiotopiques et les protons diastéréotopiques.
Br
OAc
O
OH
O
O
(a)
(b)
(c)
(d)
(e)

TP de chimie théorique et physique
A. Richel - 231 - RMN
O O
OH
O
OAc
FH
H H
Cl
O
(f)
(g)
(h)
(i)
(j)

TP de chimie théorique et physique
A. Richel - 232 - RMN
4. Comment expliquer que le 2-chloropropène possède des signaux relatifs à 3 types de
protons dans son spectre RMN 1H ?

TP de chimie théorique et physique
A. Richel - 233 - RMN
5. Chacun de ces composés présente un seul pic en RMN 1H. En se basant sur les tables de
corrélation, vers quel déplacement chimique approximatif se situe ce pic ?
(a) Cyclohexane (b) CH3COCH3 (c) Benzène (d) Glyoxal
O O
H H (e) CH2Cl2 (f) (CH3)3N
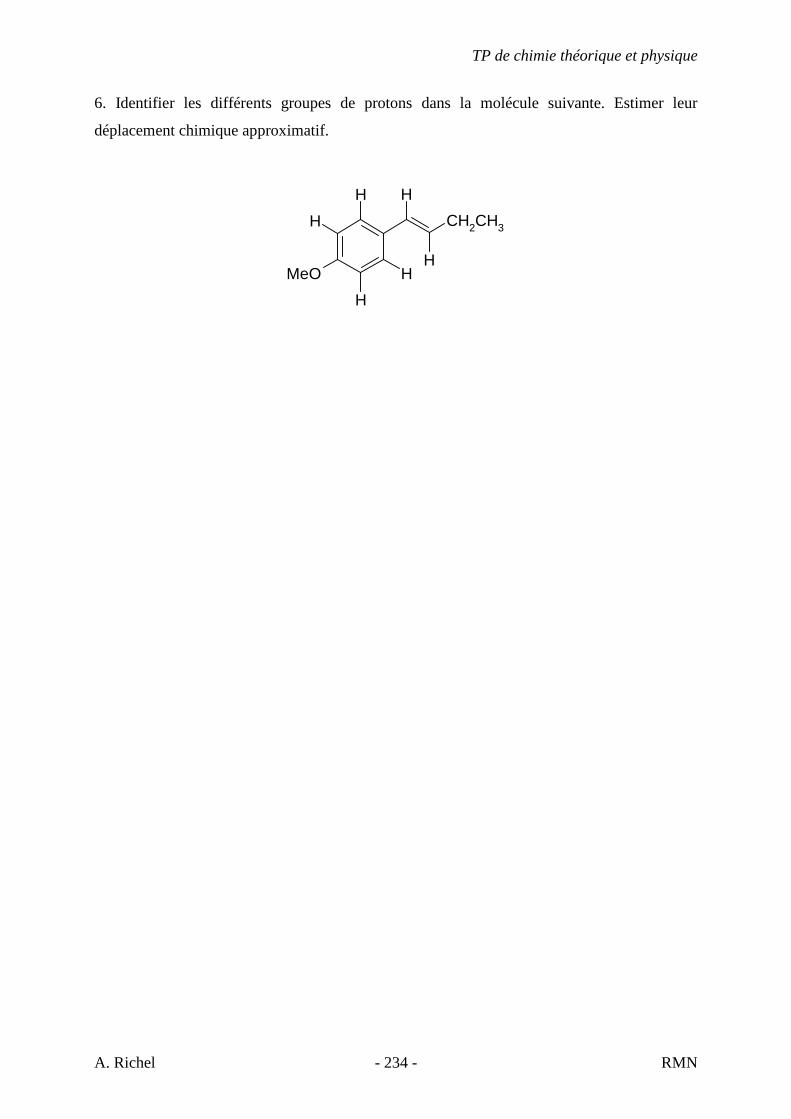
TP de chimie théorique et physique
A. Richel - 234 - RMN
6. Identifier les différents groupes de protons dans la molécule suivante. Estimer leur
déplacement chimique approximatif.
MeO
H
H
H
H H
H
CH2CH3

TP de chimie théorique et physique
A. Richel - 235 - RMN
7. Proposer une structure pour les composés ayant les données spectrales suivantes : Composé 1 : C5H10O 6H : doublet à δ 0.95 ppm, J = 7.0 Hz 3H : singulet à 2.10 ppm 1H : multiplet à 2.43 ppm Composé 2 : C3H5Br 3H : singulet à δ 2.32 ppm 1H : singulet (large) à 5.35 ppm 1H : singulet (large) à 5.54 ppm

TP de chimie théorique et physique
A. Richel - 236 - RMN
8. Attribuer les signaux dans les spectres RMN 1H suivants (a) 2-méthyl-2-butanol (CDCl3)
(b) cyclohexanone (CDCl3)
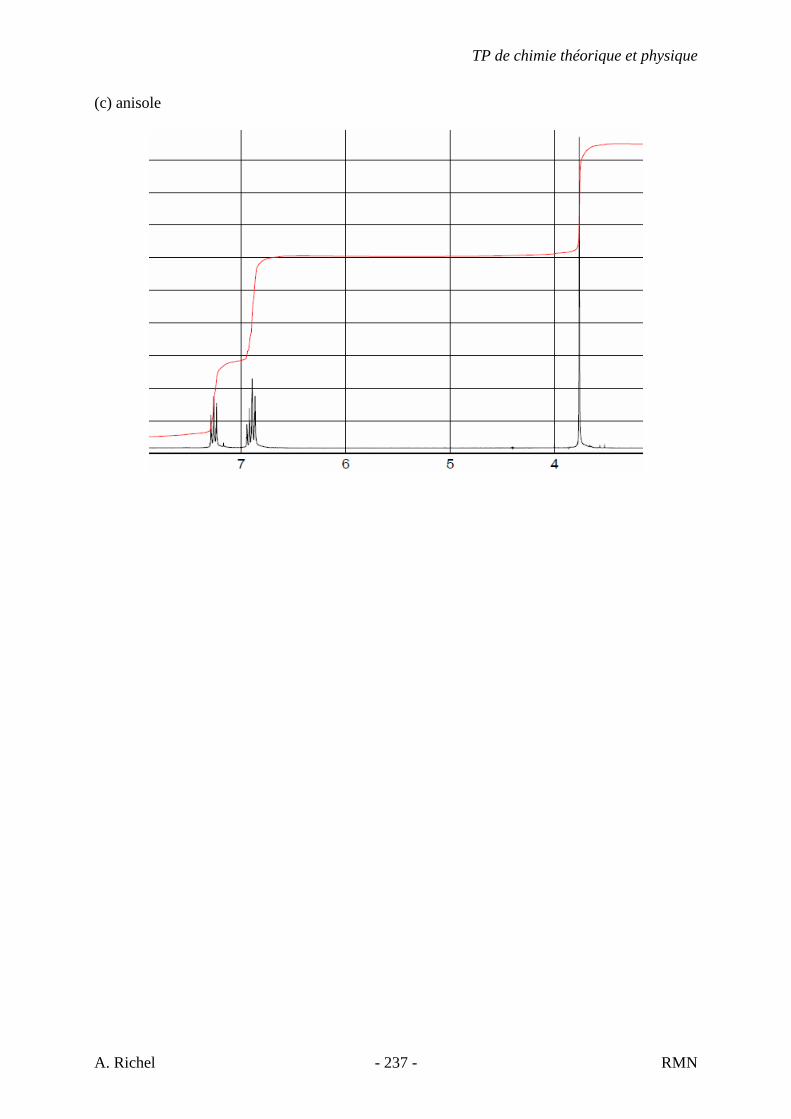
TP de chimie théorique et physique
A. Richel - 237 - RMN
(c) anisole
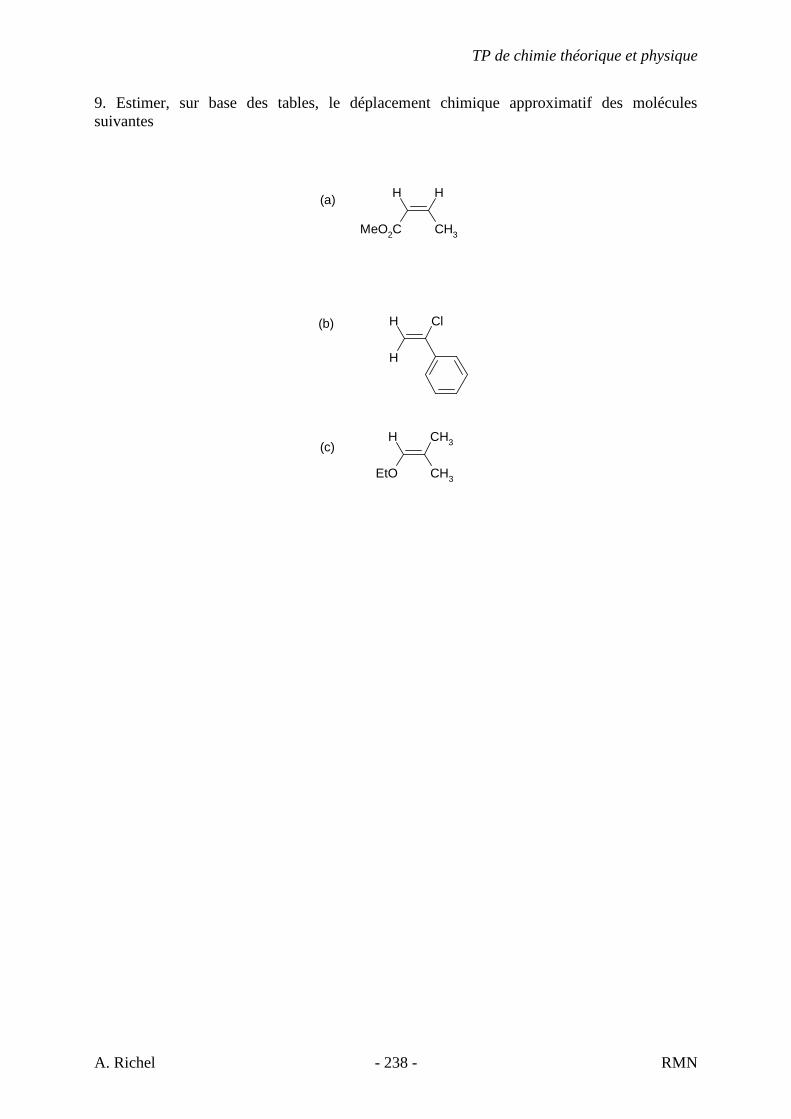
TP de chimie théorique et physique
A. Richel - 238 - RMN
9. Estimer, sur base des tables, le déplacement chimique approximatif des molécules suivantes
H
CH3
H
MeO2C
H
H
Cl
H
CH3
CH3
EtO
(a)
(b)
(c)
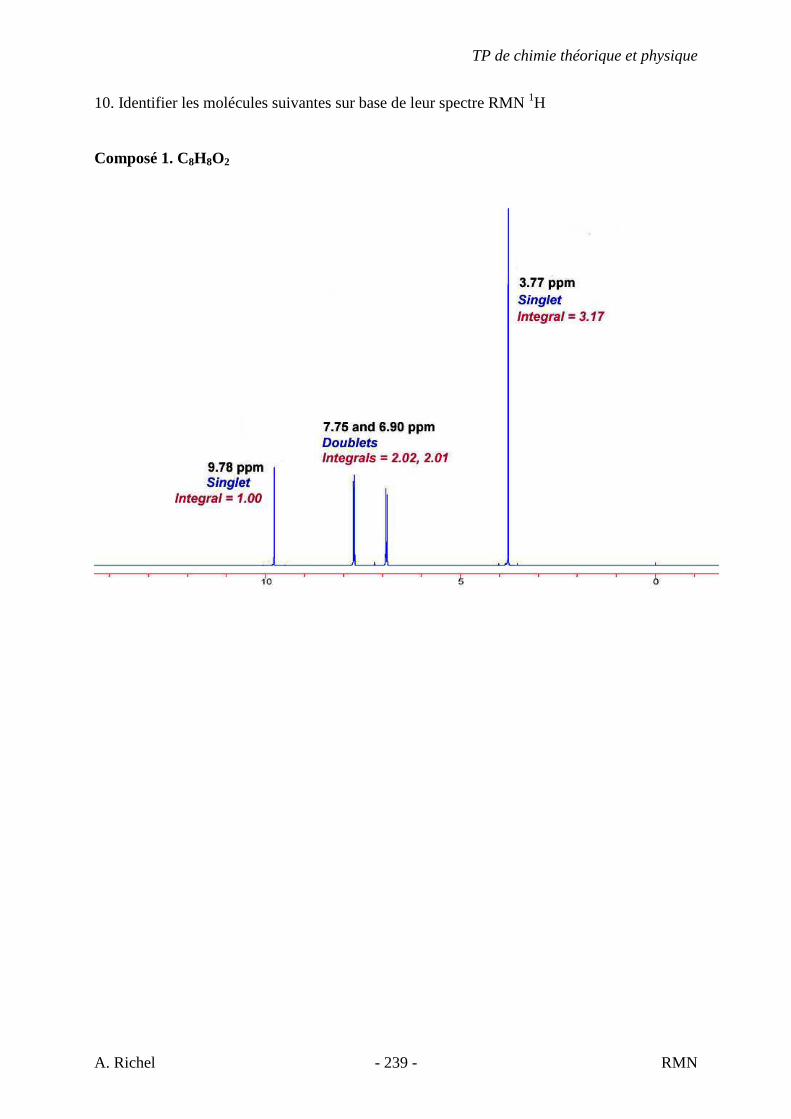
TP de chimie théorique et physique
A. Richel - 239 - RMN
10. Identifier les molécules suivantes sur base de leur spectre RMN 1H
Composé 1. C8H8O2
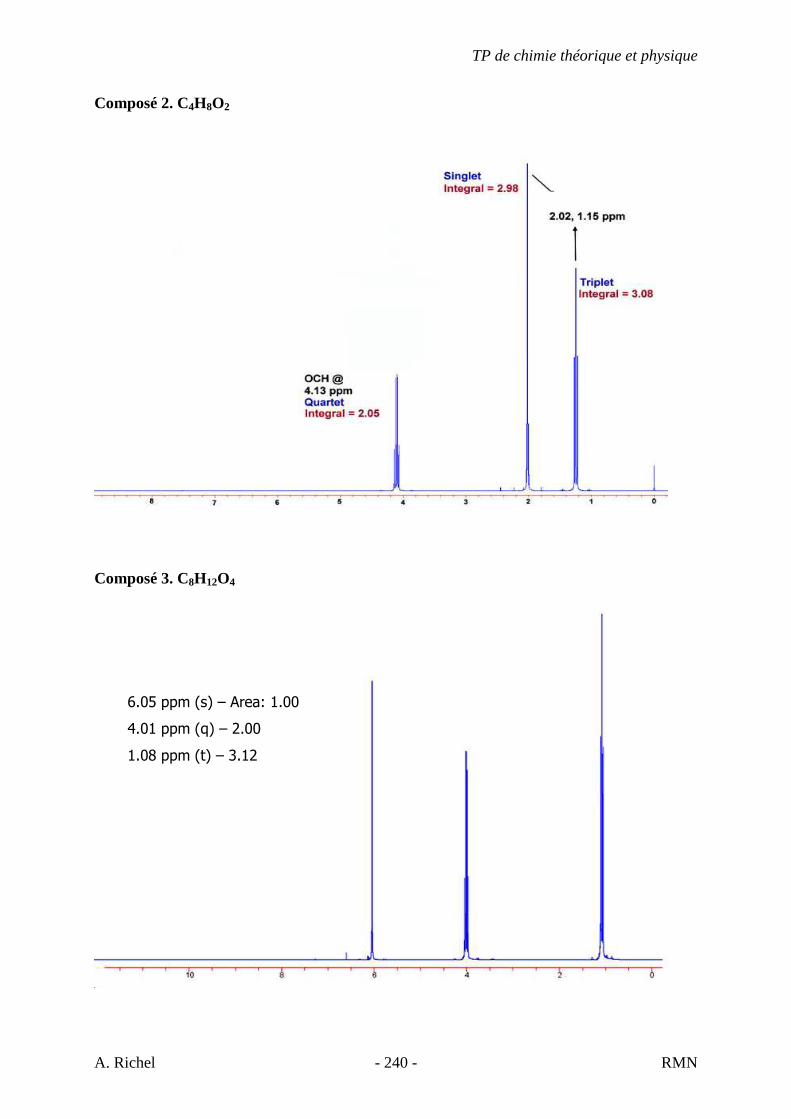
TP de chimie théorique et physique
A. Richel - 240 - RMN
Composé 2. C4H8O2
Composé 3. C8H12O4
6.05 ppm (s) – Area: 1.00
4.01 ppm (q) – 2.00
1.08 ppm (t) – 3.12
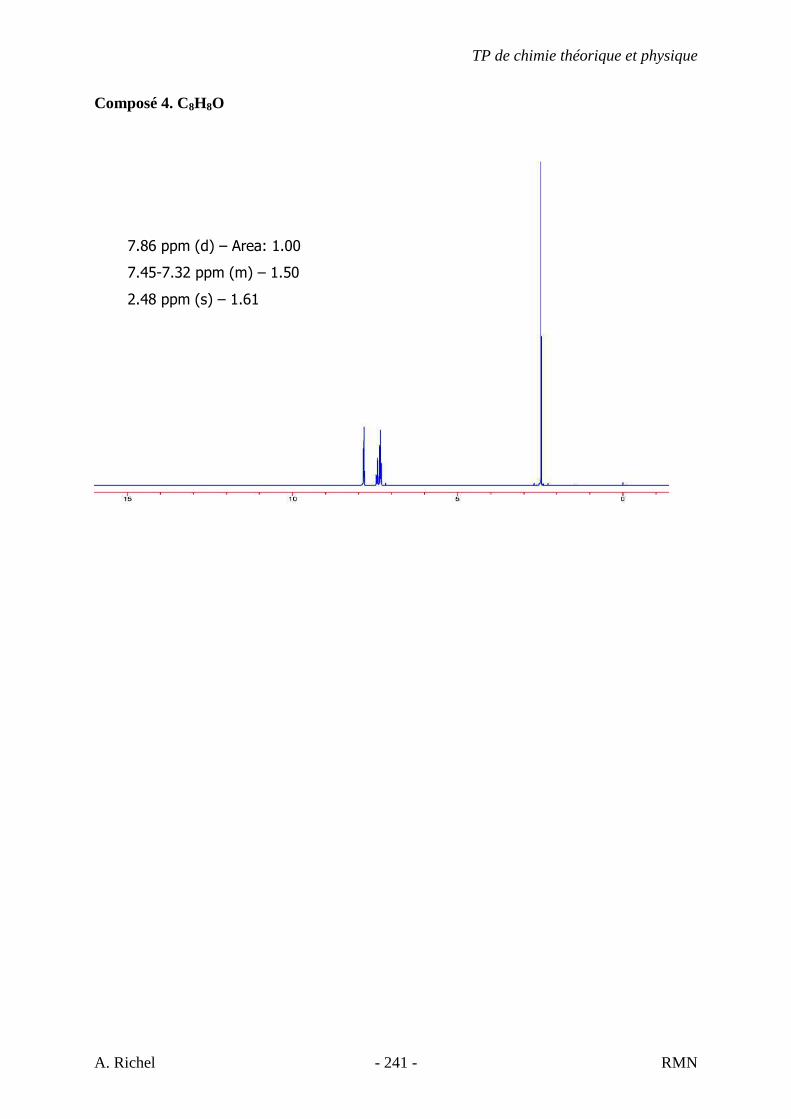
TP de chimie théorique et physique
A. Richel - 241 - RMN
Composé 4. C8H8O
7.86 ppm (d) – Area: 1.00
7.45-7.32 ppm (m) – 1.50
2.48 ppm (s) – 1.61

PARTIE 2. SPECTROMÉTRIE RMN DU 13C
1. INTRODUCTION
Les bases théoriques de la RMN 13C sont identiques à celles de la RMN 1H.
Cependant, certains aspects sont différents :
1) Dans les spectres 13C avec découplage proton, les pics sont systématiquement des
singulets (sauf si couplage avec 31P, 19F, etc). L’utilisation de techniques spéciales
d’enregistrement (Découplage par Impulsion Composite) permet de supprimer les
couplages 13C-1H.
2) La gamme de déplacements chimiques est plus importante (δ 0-220 ppm).
3) L’intensité des pics en 13C n’est pas corrélée au nombre d’atomes de carbone dans la
molécule (temps de relaxation plus longs).
4) Les noyaux 13C sont moins abondants (1.1% du 12C) et moins sensibles (1.6% du 1H)
que les 1H. Les quantités d’échantillon et les temps d’acquisition doivent donc être
augmentés.
5) Pour un solvant deutéré donné, les pics de solvant 13C et 1H diffèrent par leur
multiplicité.
En règle générale, les pics en RMN 13C sont plus fins et des effets de superposition sont
rarement observés.
2. DÉPLACEMENTS CHIMIQUES DE DIFFÉRENTES FAMILLES DE CO MPOSÉS
ORGANIQUES
Tableaux issus de « Identification spectrométrique de composés organiques » (2de édition)
Silverstein, Webster, Kiemle Ed. De Boeck Université 2007

TP de chimie théorique et physique
A. Richel - 243 - RMN
2.1. ALCANES LINÉAIRES ET RAMIFIÉS
En règle générale, les molécules hydrocarbonées saturées se manifestent dans un spectre
RMN 13C à des valeurs de déplacements chimiques comprises entre 0 et 45 ppm en fonction
du nombre d’atomes de C qu’ils comportent et du degré de ramification de la molécule.
Table RMN13C-1. Déplacements chimiques 13C de quelques alcanes linéaires et branchés
(en ppm par rapport au TMS)
Il est également possible d’estimer par calcul approximatif la modification des déplacements
chimiques lors de la substitution d’un H en position terminale ou interne par un autre atome
ou groupement fonctionnel Y. Ce calcul s’effectue en se reportant à la table ci-dessous et en
additionnant les différentes valeurs données à la valeur du déplacement chimique de l’atome
de carbone approprié donnée dans le tableau précédent.
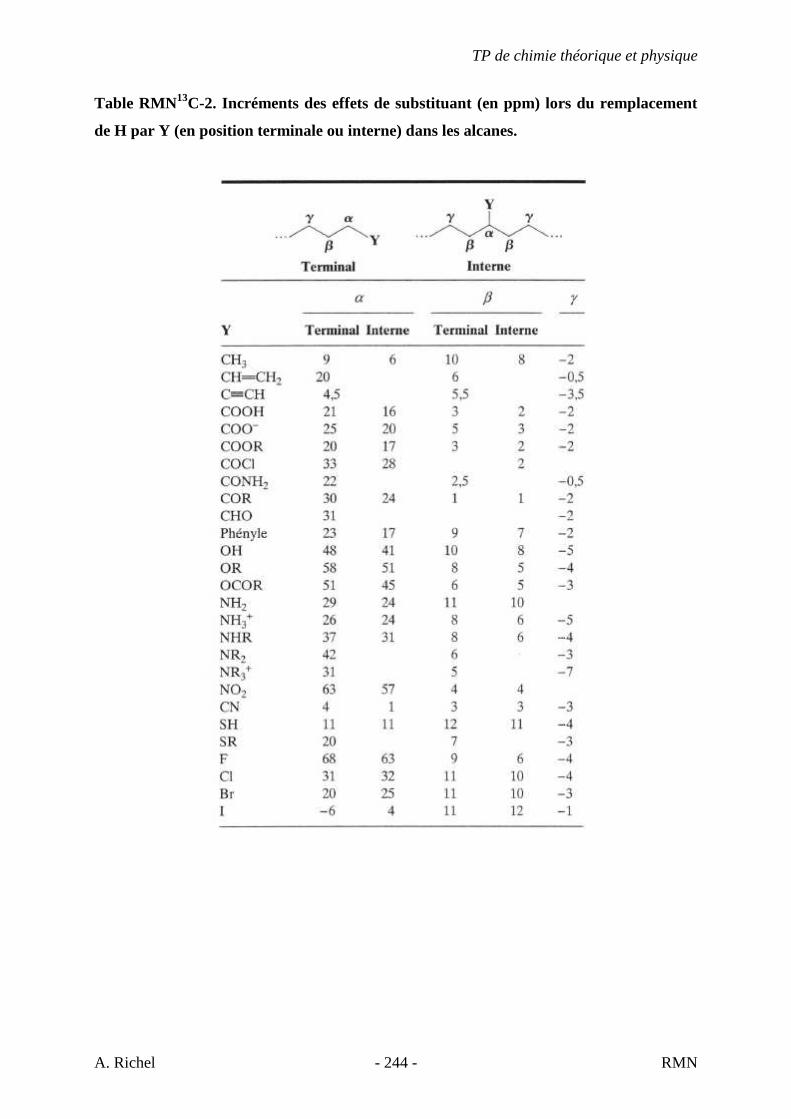
TP de chimie théorique et physique
A. Richel - 244 - RMN
Table RMN13C-2. Incréments des effets de substituant (en ppm) lors du remplacement
de H par Y (en position terminale ou interne) dans les alcanes.

TP de chimie théorique et physique
A. Richel - 245 - RMN
2.2. ALCÈNES ET ALCÈNES CYCLIQUES
De manière très générale, les alcènes s’identifient en RMN 13C entre 100 et 140 ppm.
Table RMN13C-3. Déplacements chimiques caractéristiques de certains alcènes linéaires
ou cycliques

TP de chimie théorique et physique
A. Richel - 246 - RMN
Table RMN13C-4. Effet de la substitution de la C=C interne ou terminale par un
groupement donneur ou attracteur sur les déplacements chimiques
2.3. ALCYNES
Les alcynes se manifestent approximativement entre 65 et 90 ppm. La triple liaison a pour
effet de décaler l’absorption ders atomes de carbone sp3 directement liés d’environ 5 à 15
ppm vers les champs forts par rapport à l’alcane correspondant.
Une liaison alcyne terminale se manifeste à champ plus élevé qu’une liaison triple interne.
Les atomes de carbone alcyniques portant un groupement polaire s’observent entre 20 et 95
ppm (par rapport au TMS).

TP de chimie théorique et physique
A. Richel - 247 - RMN
2.4. LES COMPOSÉS AROMATIQUES
Les 6 atomes de C d’un benzène (pur ou en solution dans le CDCl3) absorbent à δ 128.5 ppm.
Dans le cas d’une mono-substitution, le C portant le substituant peut avoir un déplacement
chimique décalé de ±35 ppm. Ce déplacement des valeurs de δ est corrélé à l’électronégativité
du substituant après correction de l’effet d’anisotropie magnétique.
Il est possible de calculer en première approximation, les valeurs de déplacements chimiques
de C (ipso, ortho, méta et para) en se référant à la méthodologie reprise ci-dessous.
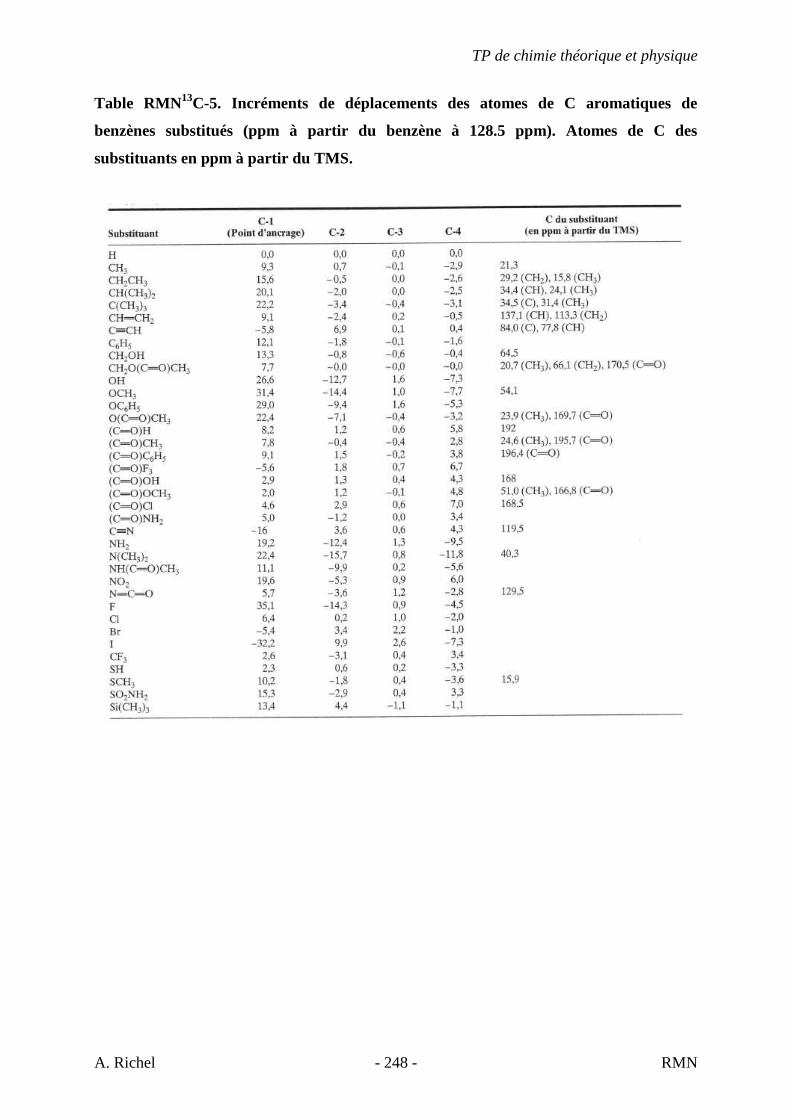
TP de chimie théorique et physique
A. Richel - 248 - RMN
Table RMN13C-5. Incréments de déplacements des atomes de C aromatiques de
benzènes substitués (ppm à partir du benzène à 128.5 ppm). Atomes de C des
substituants en ppm à partir du TMS.
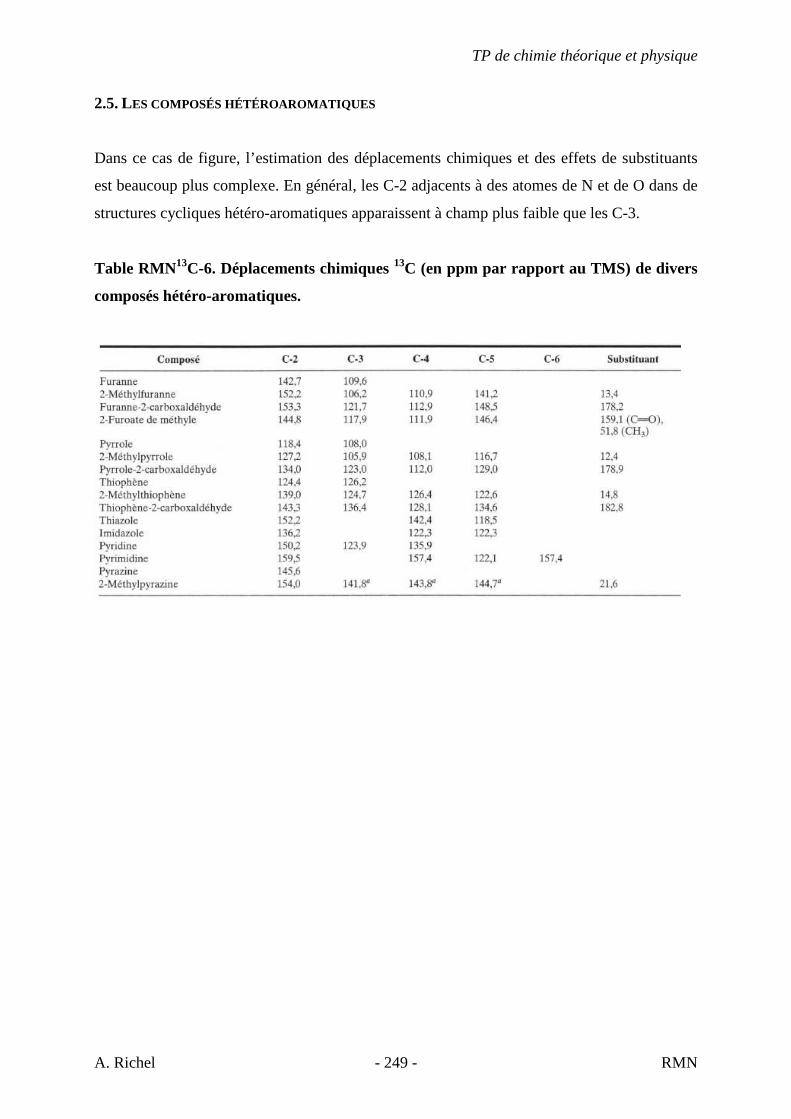
TP de chimie théorique et physique
A. Richel - 249 - RMN
2.5. LES COMPOSÉS HÉTÉROAROMATIQUES
Dans ce cas de figure, l’estimation des déplacements chimiques et des effets de substituants
est beaucoup plus complexe. En général, les C-2 adjacents à des atomes de N et de O dans de
structures cycliques hétéro-aromatiques apparaissent à champ plus faible que les C-3.
Table RMN13C-6. Déplacements chimiques 13C (en ppm par rapport au TMS) de divers
composés hétéro-aromatiques.

TP de chimie théorique et physique
A. Richel - 250 - RMN
2.6. LES ALCOOLS ALIPHATIQUES
La substitution d’un atome de H par un groupement OH dans un alcane décale à champ faible
les signaux de C-1 de 35 à 50 ppm et de C-2 de 5 à 15 ppm environ. Les signaux
correspondants au C-3 sont décalés de 0 à 6 ppm à champ fort.
Table RMN13C-7. Déplacements chimiques de certains alcools aliphatiques.

TP de chimie théorique et physique
A. Richel - 251 - RMN
2.7. LES ÉTHERS ALIPHATIQUES ET AROMATIQUES
La substitution d’un atome de C par un groupement alkoxy déplace le signal de C-1 à champ
faible (~10-15 ppm de plus) que lors de la substitution par un groupement OH.
Table RMN13C-8. Déplacements 13C de certains éthers aliphatiques (cycliques).
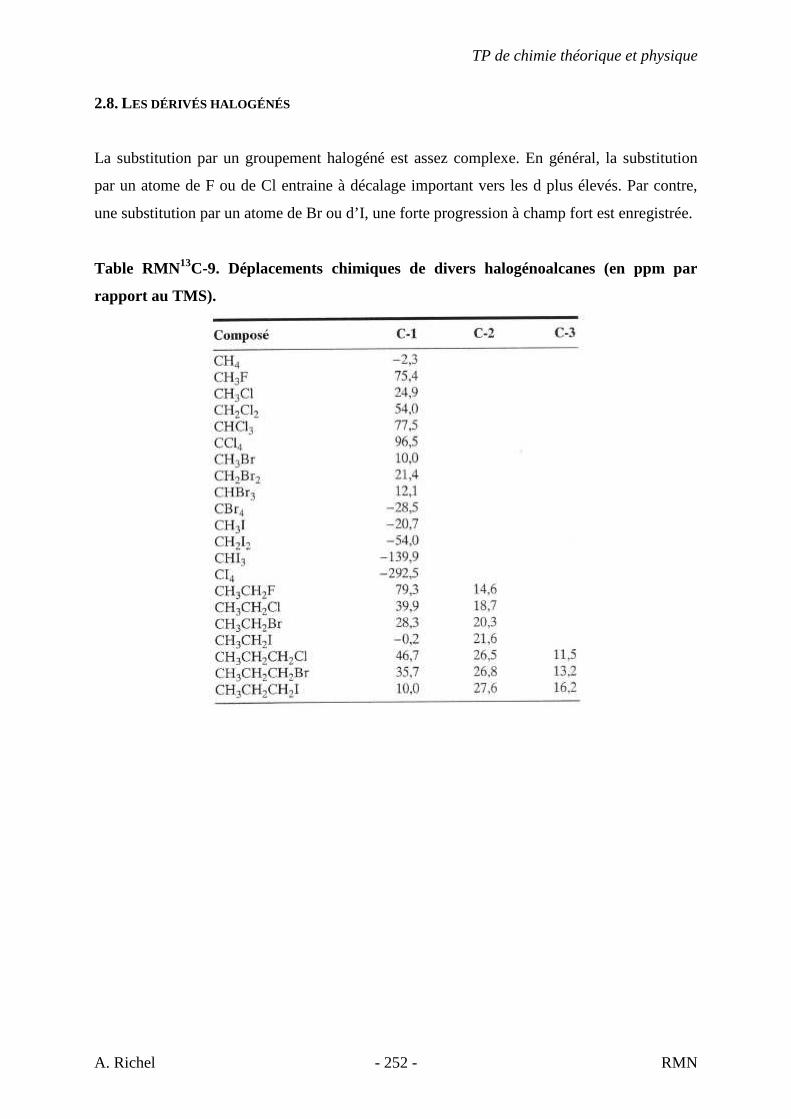
TP de chimie théorique et physique
A. Richel - 252 - RMN
2.8. LES DÉRIVÉS HALOGÉNÉS
La substitution par un groupement halogéné est assez complexe. En général, la substitution
par un atome de F ou de Cl entraine à décalage important vers les d plus élevés. Par contre,
une substitution par un atome de Br ou d’I, une forte progression à champ fort est enregistrée.
Table RMN13C-9. Déplacements chimiques de divers halogénoalcanes (en ppm par
rapport au TMS).

TP de chimie théorique et physique
A. Richel - 253 - RMN
2.9. LES CÉTONES, LES ALDÉHYDES ET LES ACIDES CARBOXYLIQUES
Les atomes de carbone des liaisons carbonyles C=O des cétones et aldéhydes absorbent dans
une région spectrale très caractéristique (entre 210 et 190 ppm).
Table RMN13C-10. Déplacements chimiques de la fonction C=O et autres atomes de C
adjacents dans le cas des cétones et des aldéhydes.
La C=O de l’acétone (CH3)2C=O se manifeste à d 203.3 ppm et la fonction C=O de
l’acétaldéhyde CH3C(O)H à 199.3 ppm.
• La substitution alkyle sur le Cα entraine décalage de 2 – 3 ppm de la C=O vers les
champs faibles.
• Par contre, la substitution par un groupement encombré stériquement (ex. phényle)
induit un déplacement du signal vers les champs forts (δ plus faible).
• L’introduction d’une insaturation C=C en a, b

TP de chimie théorique et physique
A. Richel - 254 - RMN
Résonance magnétique nucléaire (RMN) : RMN 13C Bases théoriques pour la RMN 13C identiques à celles de la RMN 1H 12C : I = 0
� Non « actif » en RMN 13C : I = ½
� Ok pour RMN � Mais abondance naturelle 13C = 1.1 % de 12C � Mais sensibilité 13C = 1.6 % sensibilité du 1H
RMN 13C Temps d’accumulation + importants et/ou concentration en échantillon + importante

TP de chimie théorique et physique
A. Richel - 255 - RMN
Résonance magnétique nucléaire (RMN) : RMN 13C
� Déplacements chimiques entre δδδδ 0 et 220 ppm
� Intensité des pics non corrélée aux nombre d’atomes de C présents
� Enregistrement des spectres en « découplage
proton » 1 Carbone = 1 pic Pas de notion de multiplicité Règle (2n I +1) non appliquée comme en RMN 1H
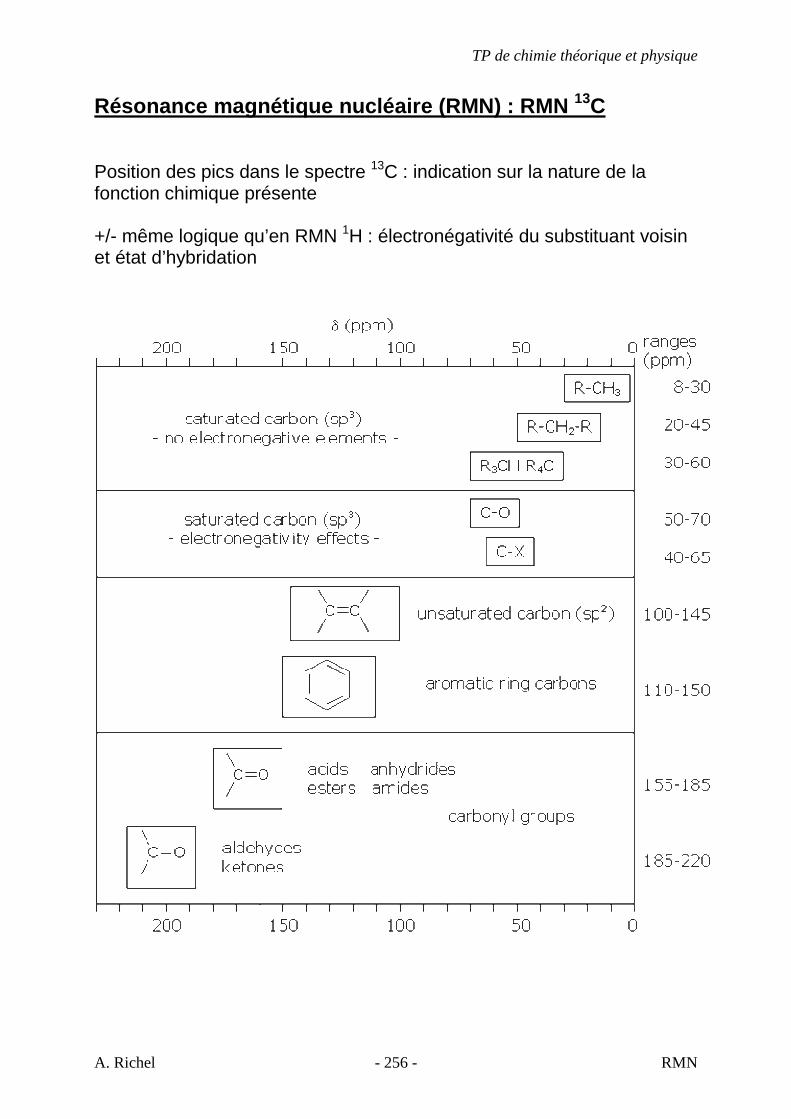
TP de chimie théorique et physique
A. Richel - 256 - RMN
Résonance magnétique nucléaire (RMN) : RMN 13C Position des pics dans le spectre 13C : indication sur la nature de la fonction chimique présente +/- même logique qu’en RMN 1H : électronégativité du substituant voisin et état d’hybridation
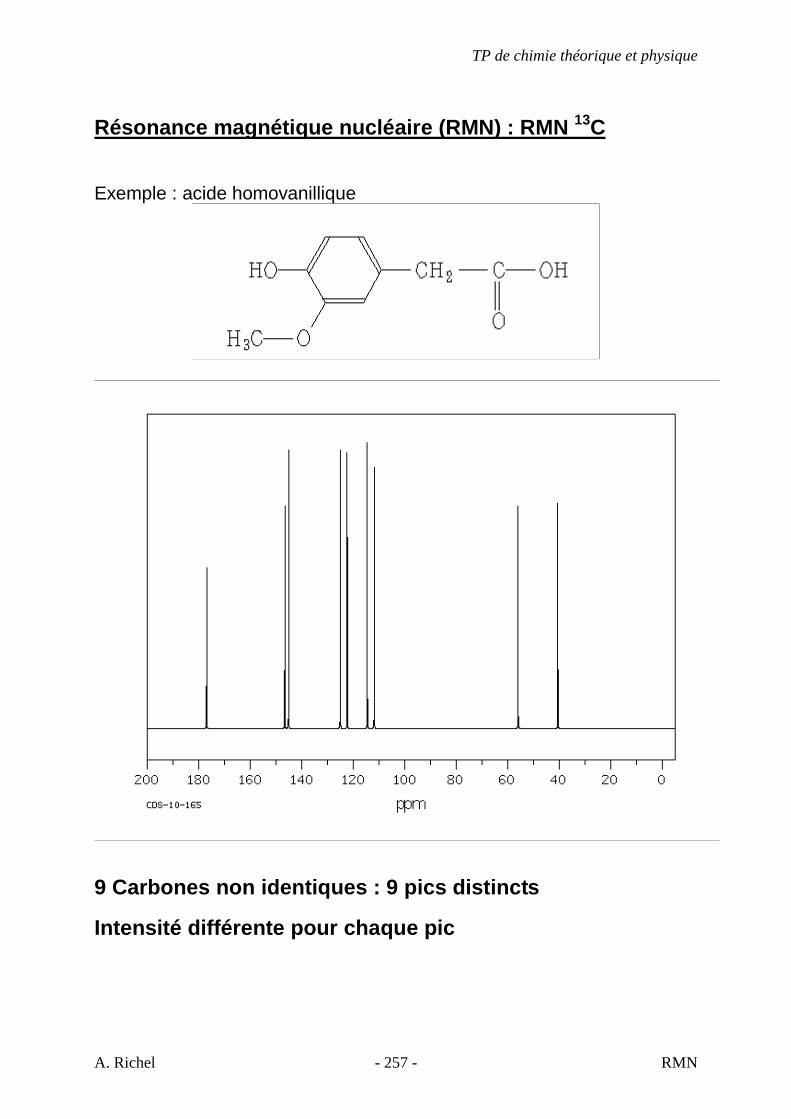
TP de chimie théorique et physique
A. Richel - 257 - RMN
Résonance magnétique nucléaire (RMN) : RMN 13C Exemple : acide homovanillique
9 Carbones non identiques : 9 pics distincts
Intensité différente pour chaque pic
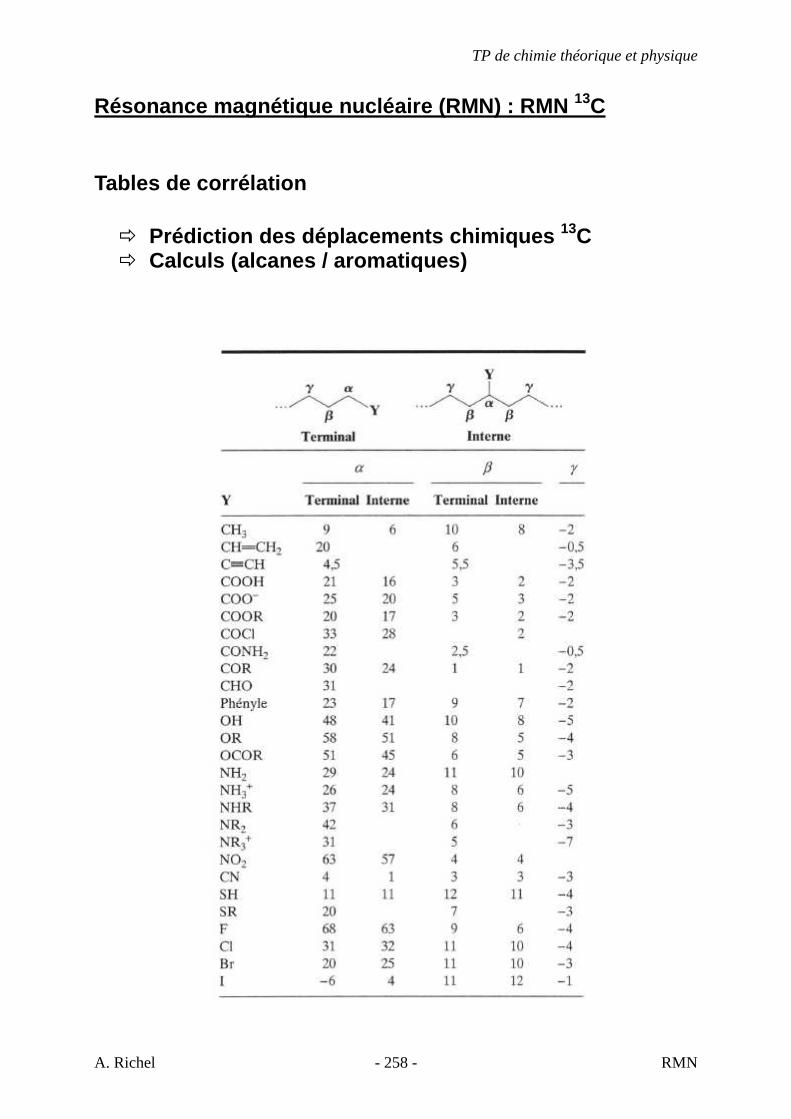
TP de chimie théorique et physique
A. Richel - 258 - RMN
Résonance magnétique nucléaire (RMN) : RMN 13C
Tables de corrélation
� Prédiction des déplacements chimiques 13C � Calculs (alcanes / aromatiques)
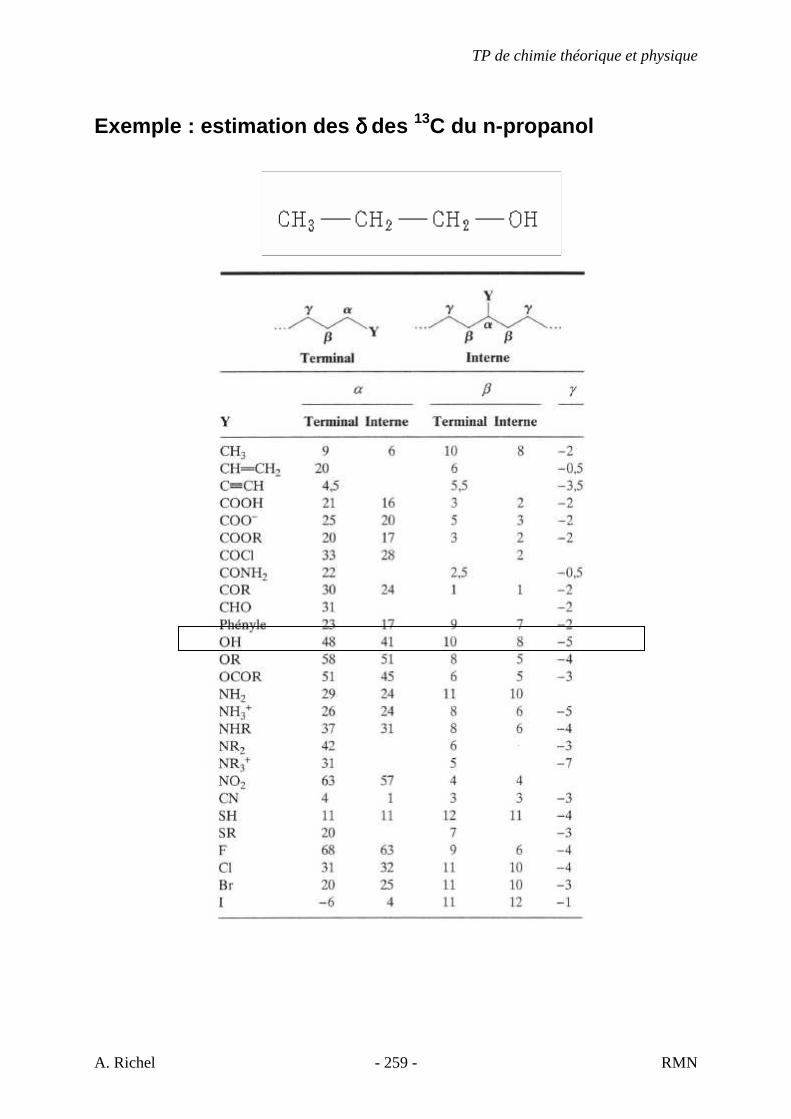
TP de chimie théorique et physique
A. Richel - 259 - RMN
Exemple : estimation des δ δ δ δ des 13C du n-propanol
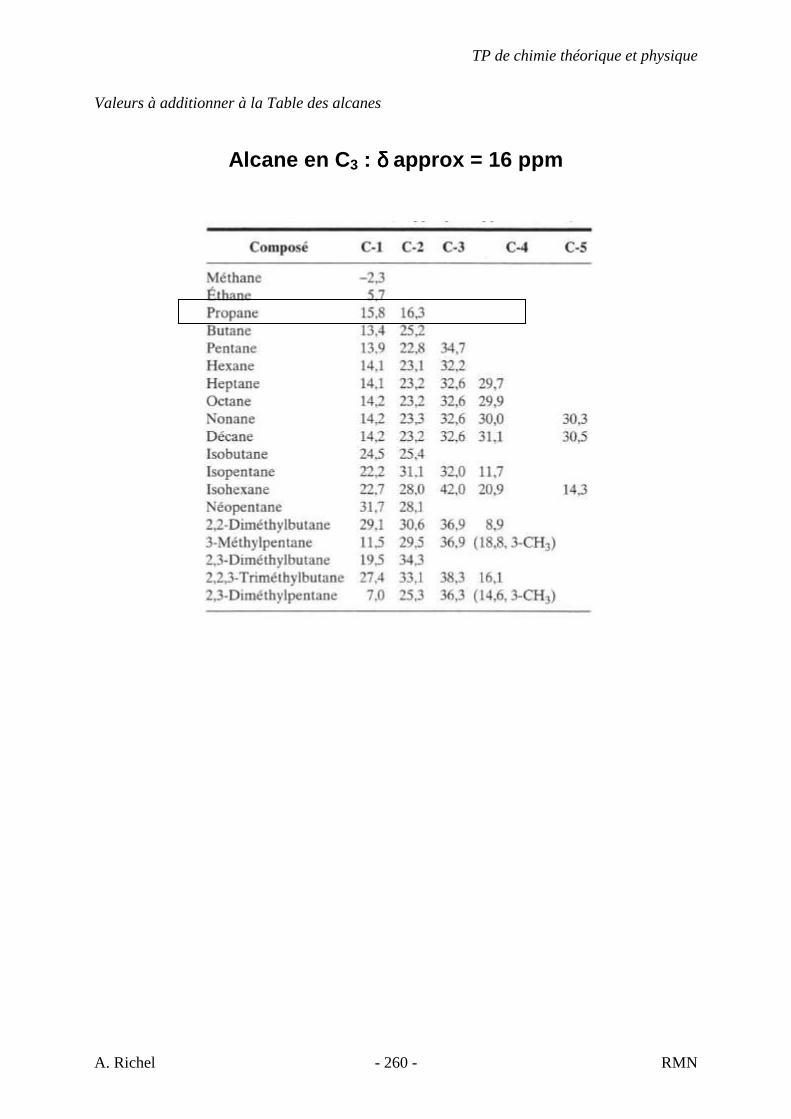
TP de chimie théorique et physique
A. Richel - 260 - RMN
Valeurs à additionner à la Table des alcanes
Alcane en C 3 : δδδδ approx = 16 ppm
TP de chimie théorique et physique
A. Richel - 261 - RMN
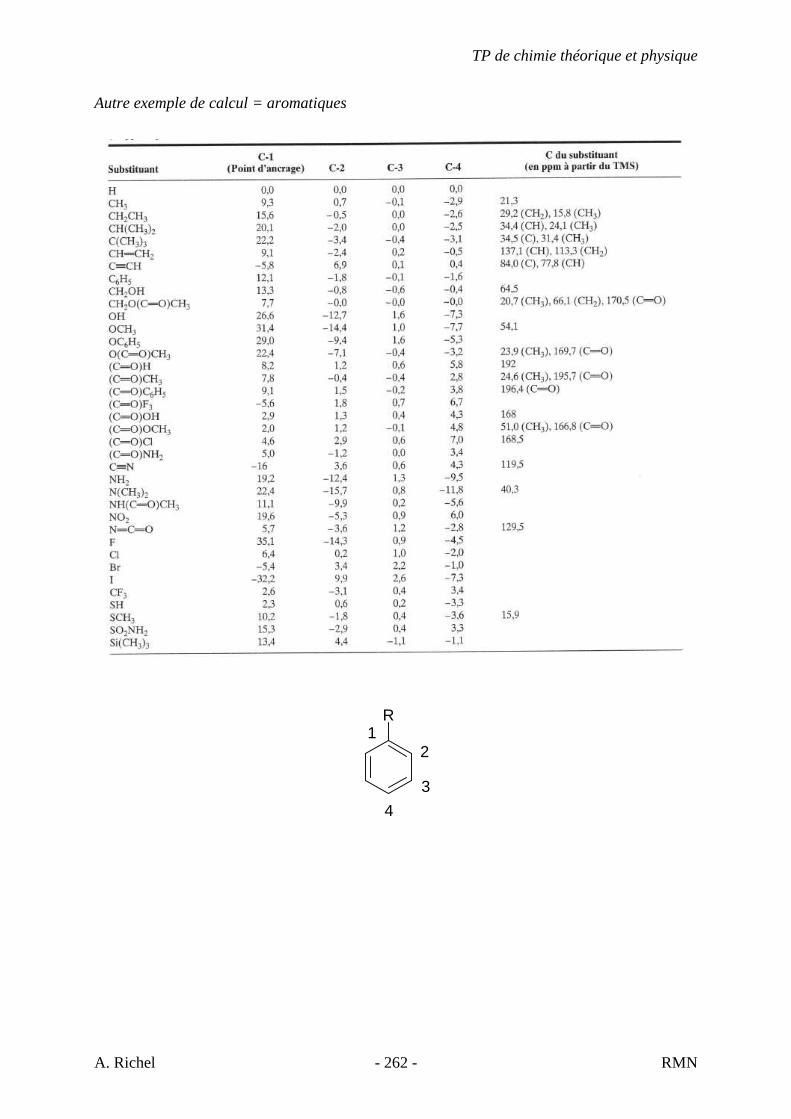
TP de chimie théorique et physique
A. Richel - 262 - RMN
Autre exemple de calcul = aromatiques
R1
2
3
4
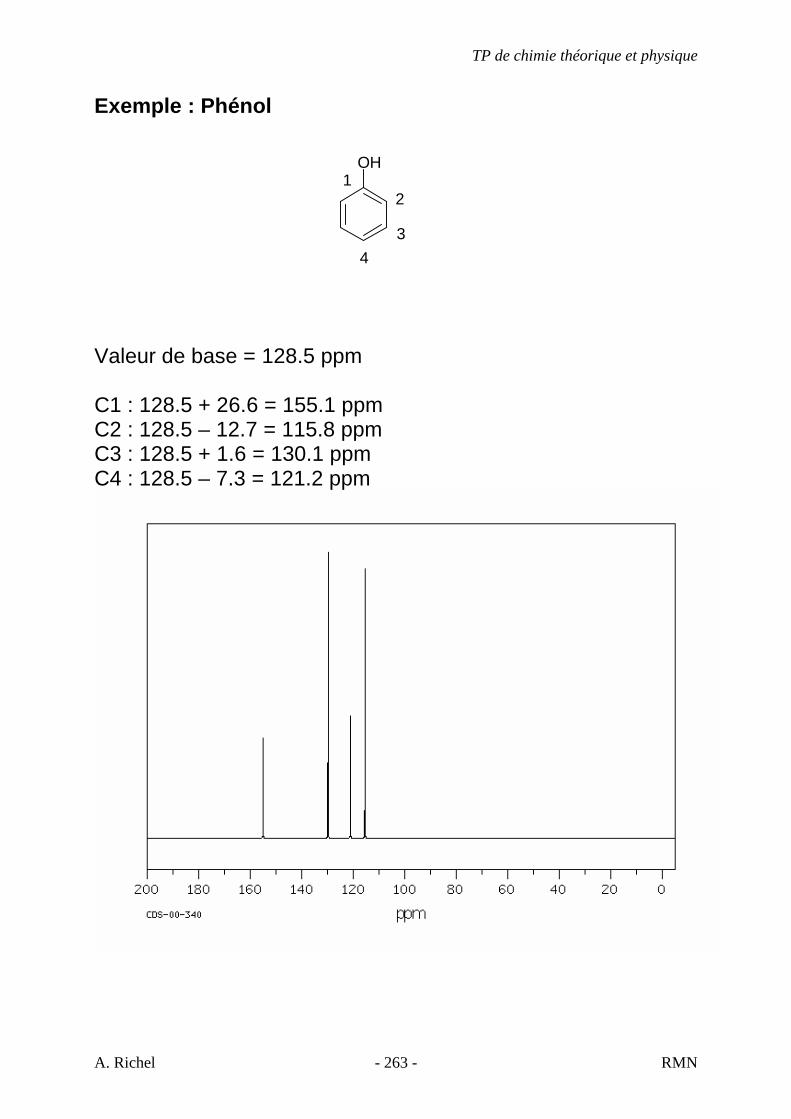
TP de chimie théorique et physique
A. Richel - 263 - RMN
Exemple : Phénol
Valeur de base = 128.5 ppm C1 : 128.5 + 26.6 = 155.1 ppm C2 : 128.5 – 12.7 = 115.8 ppm C3 : 128.5 + 1.6 = 130.1 ppm C4 : 128.5 – 7.3 = 121.2 ppm
OH1
2
3
4
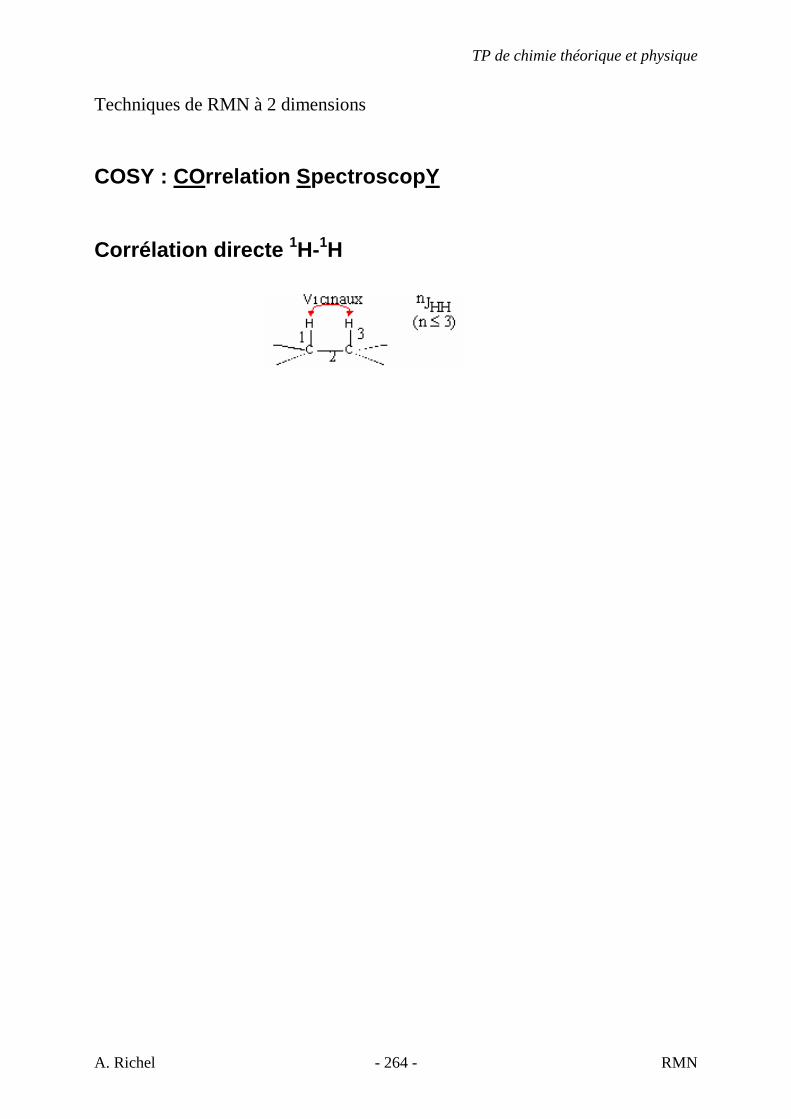
TP de chimie théorique et physique
A. Richel - 264 - RMN
Techniques de RMN à 2 dimensions
COSY : COrrelation S pectroscopY Corrélation directe 1H-1H
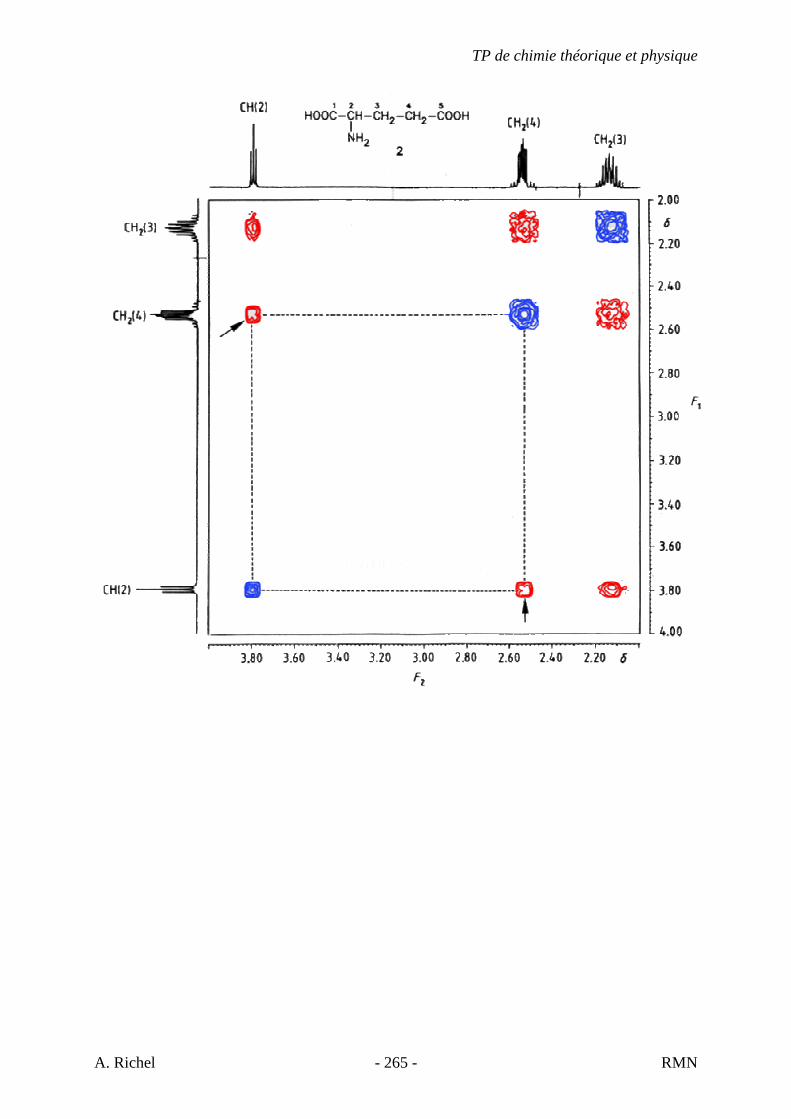
TP de chimie théorique et physique
A. Richel - 265 - RMN
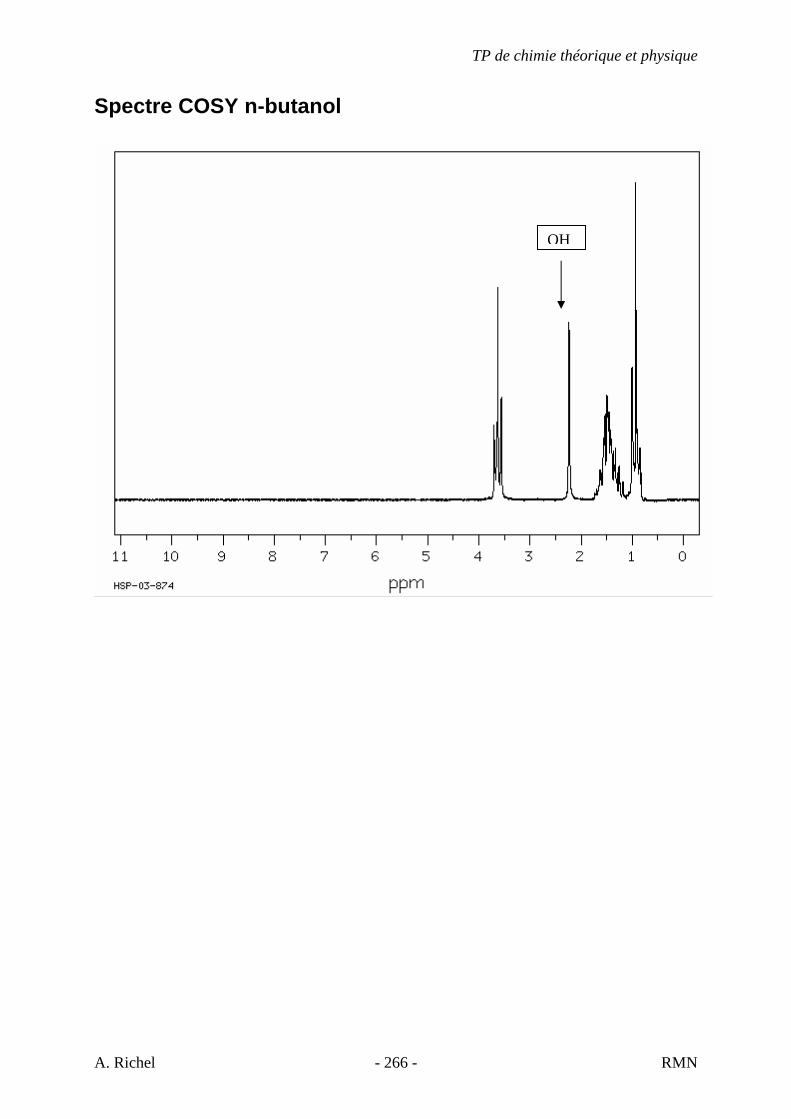
TP de chimie théorique et physique
A. Richel - 266 - RMN
Spectre COSY n-butanol
OH
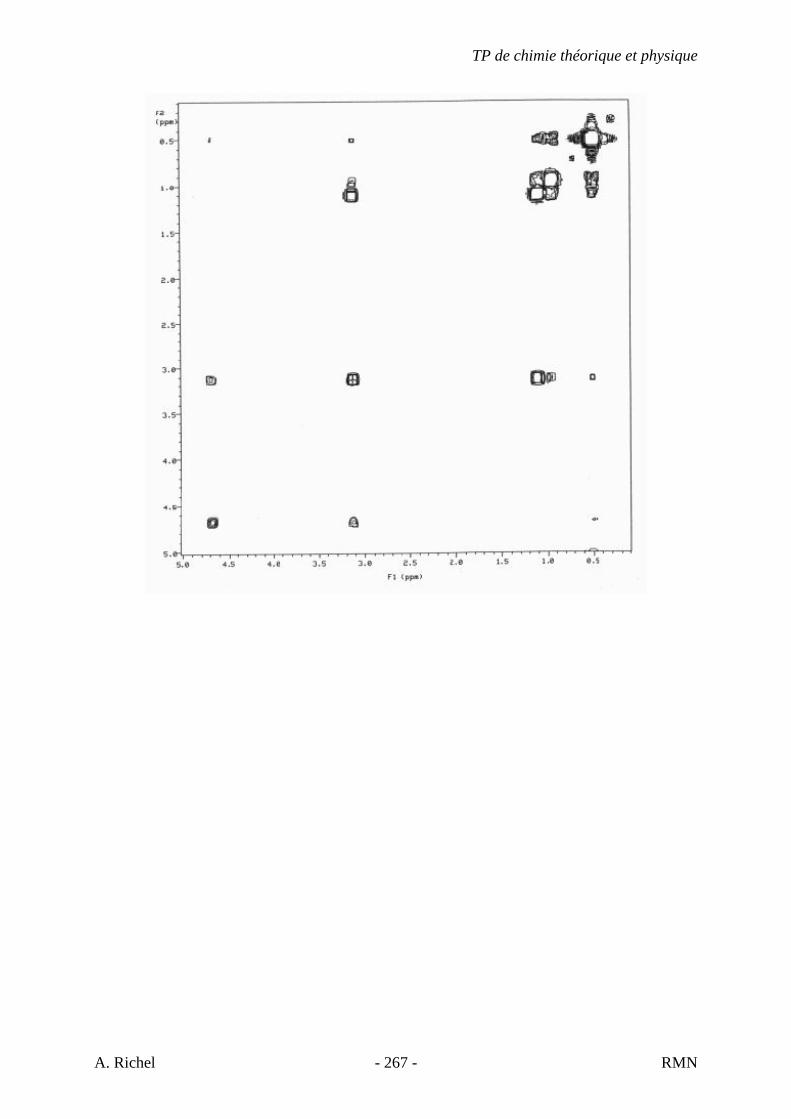
TP de chimie théorique et physique
A. Richel - 267 - RMN

TP de chimie théorique et physique
A. Richel - 268 - RMN
HSQC : HETERONUCLEAR SINGLE -QUANTUM COHERENCE SPECTROSCOPY
Correlation directe 1H-13C (quel proton est porté par quel carbone...) « Mulitplicity edited » Par convention sur notre spectromètre 600 MHz (Vari an) CH et CH3 : traces rouges CH2 : traces bleues
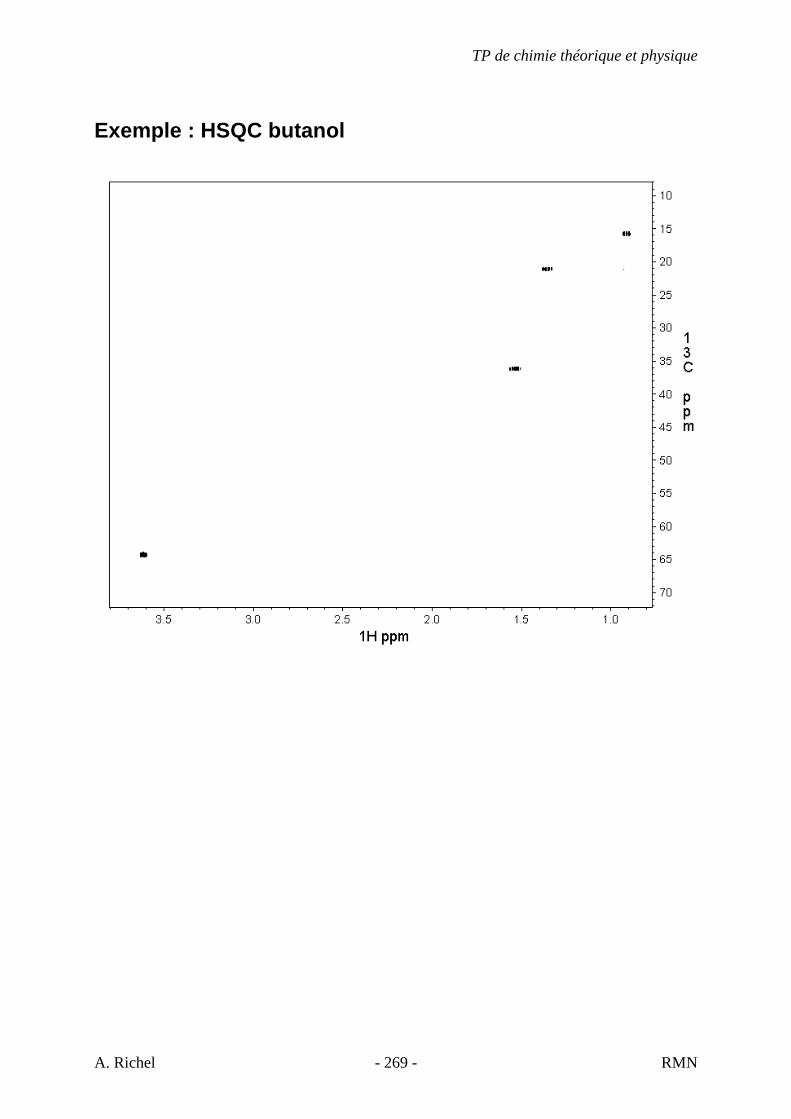
TP de chimie théorique et physique
A. Richel - 269 - RMN
Exemple : HSQC butanol
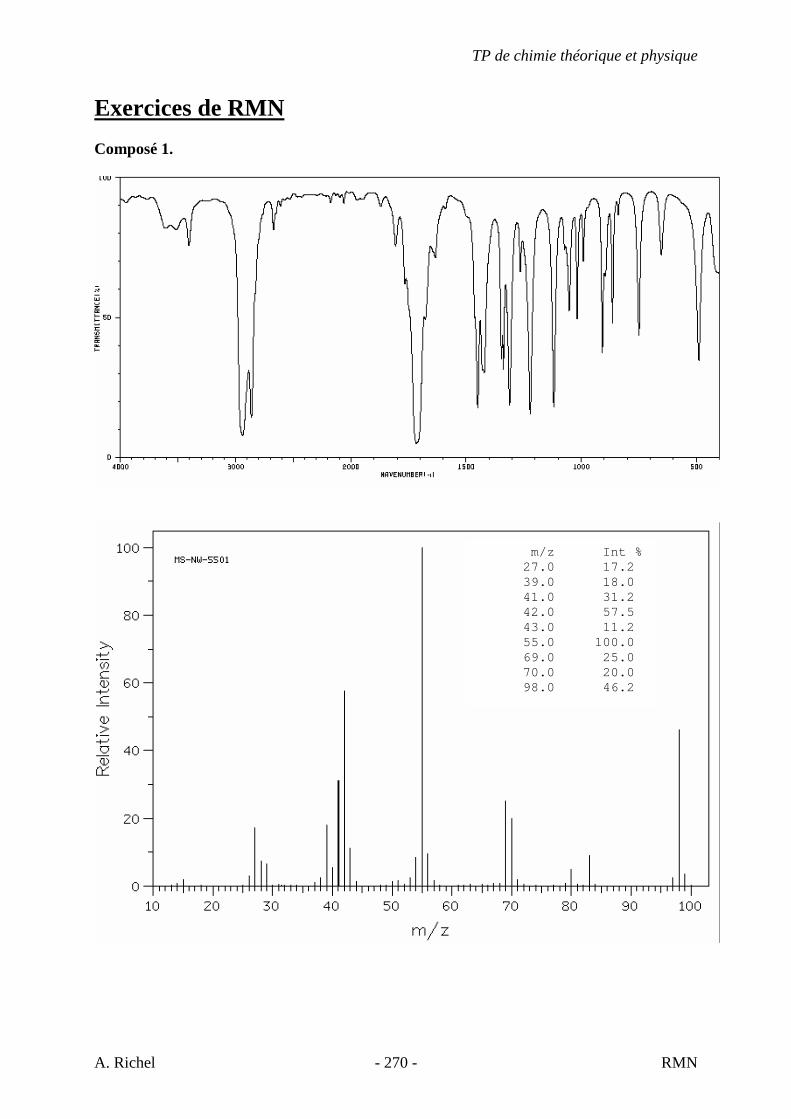
TP de chimie théorique et physique
A. Richel - 270 - RMN
Exercices de RMN Composé 1.
m/z Int % 27.0 17.2 39.0 18.0 41.0 31.2 42.0 57.5 43.0 11.2 55.0 100.0 69.0 25.0 70.0 20.0 98.0 46.2
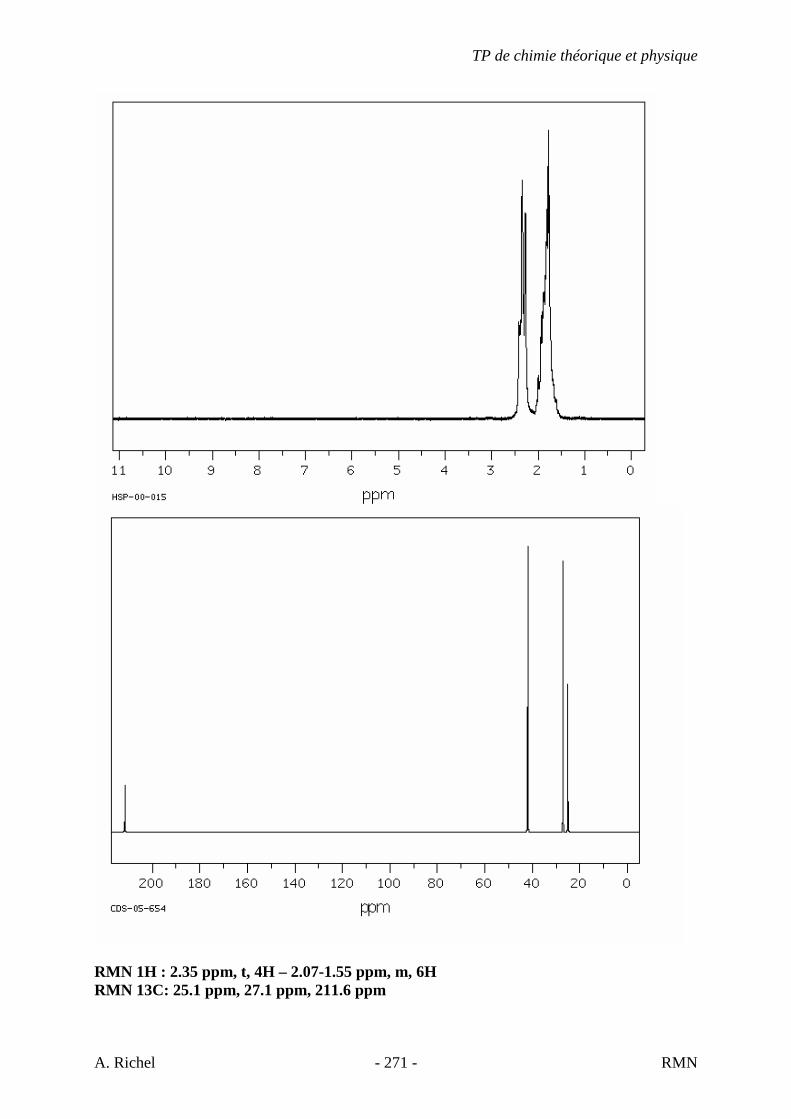
TP de chimie théorique et physique
A. Richel - 271 - RMN
RMN 1H : 2.35 ppm, t, 4H – 2.07-1.55 ppm, m, 6H RMN 13C: 25.1 ppm, 27.1 ppm, 211.6 ppm
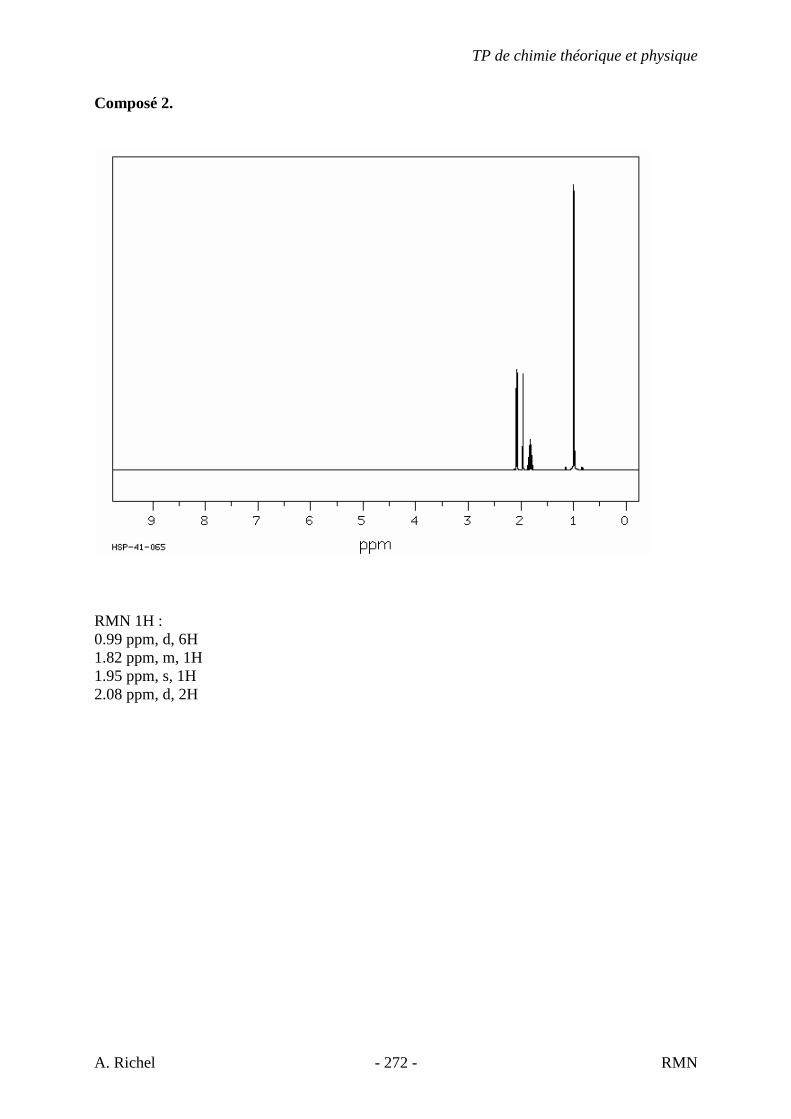
TP de chimie théorique et physique
A. Richel - 272 - RMN
Composé 2.
RMN 1H : 0.99 ppm, d, 6H 1.82 ppm, m, 1H 1.95 ppm, s, 1H 2.08 ppm, d, 2H
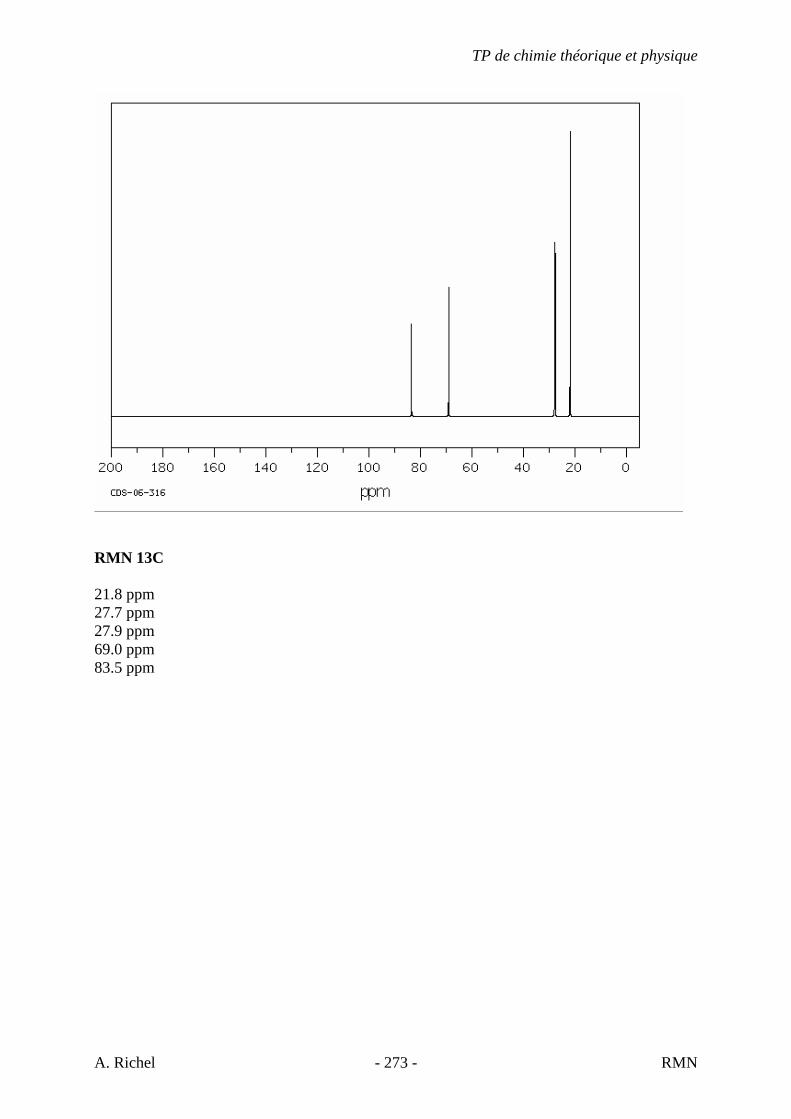
TP de chimie théorique et physique
A. Richel - 273 - RMN
RMN 13C 21.8 ppm 27.7 ppm 27.9 ppm 69.0 ppm 83.5 ppm
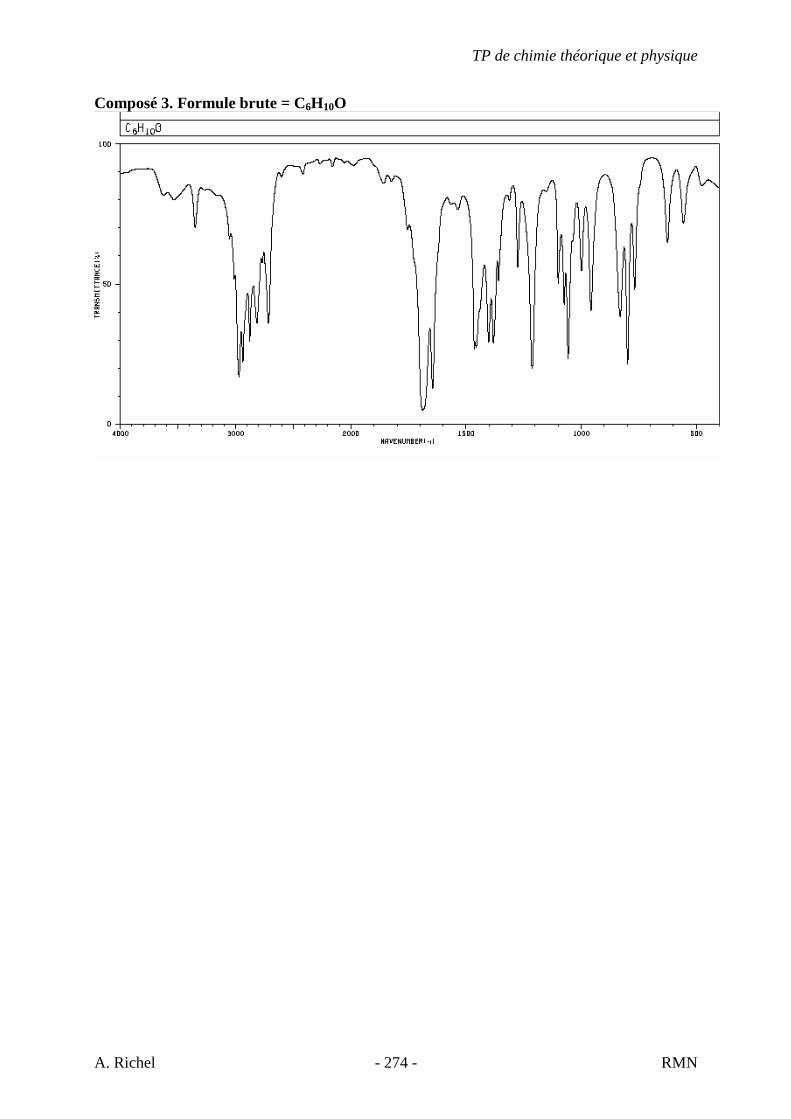
TP de chimie théorique et physique
A. Richel - 274 - RMN
Composé 3. Formule brute = C6H10O
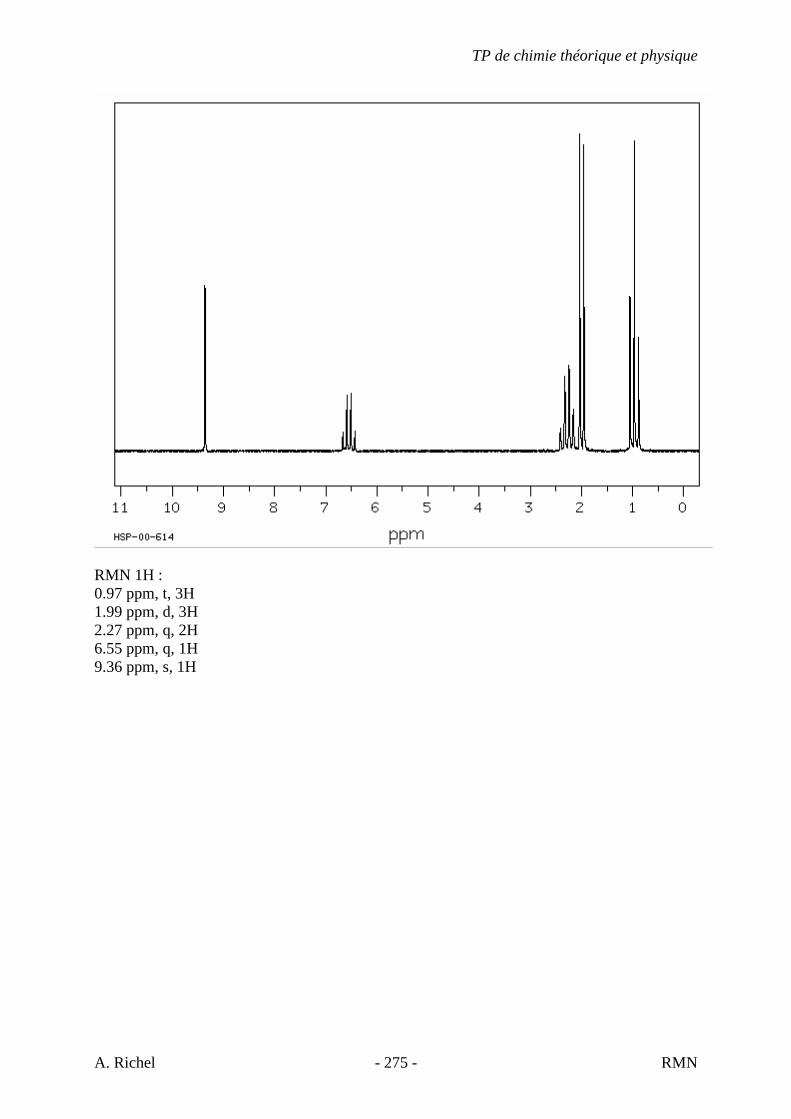
TP de chimie théorique et physique
A. Richel - 275 - RMN
RMN 1H : 0.97 ppm, t, 3H 1.99 ppm, d, 3H 2.27 ppm, q, 2H 6.55 ppm, q, 1H 9.36 ppm, s, 1H
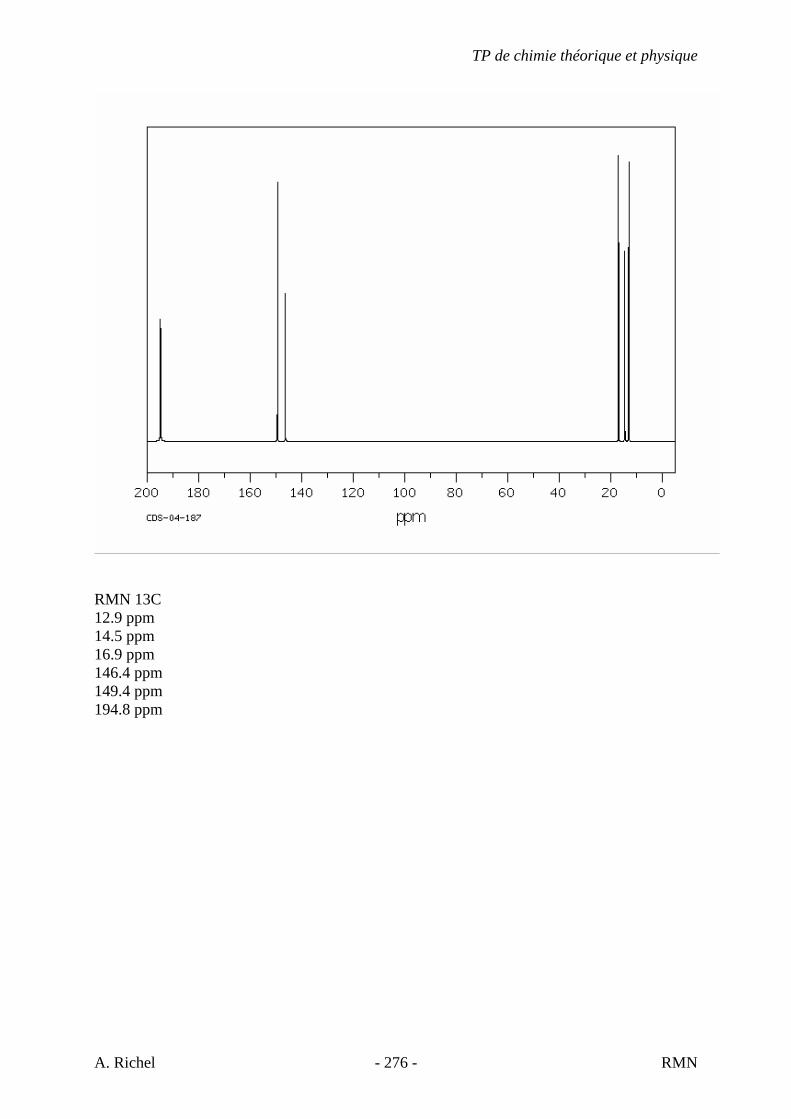
TP de chimie théorique et physique
A. Richel - 276 - RMN
RMN 13C 12.9 ppm 14.5 ppm 16.9 ppm 146.4 ppm 149.4 ppm 194.8 ppm
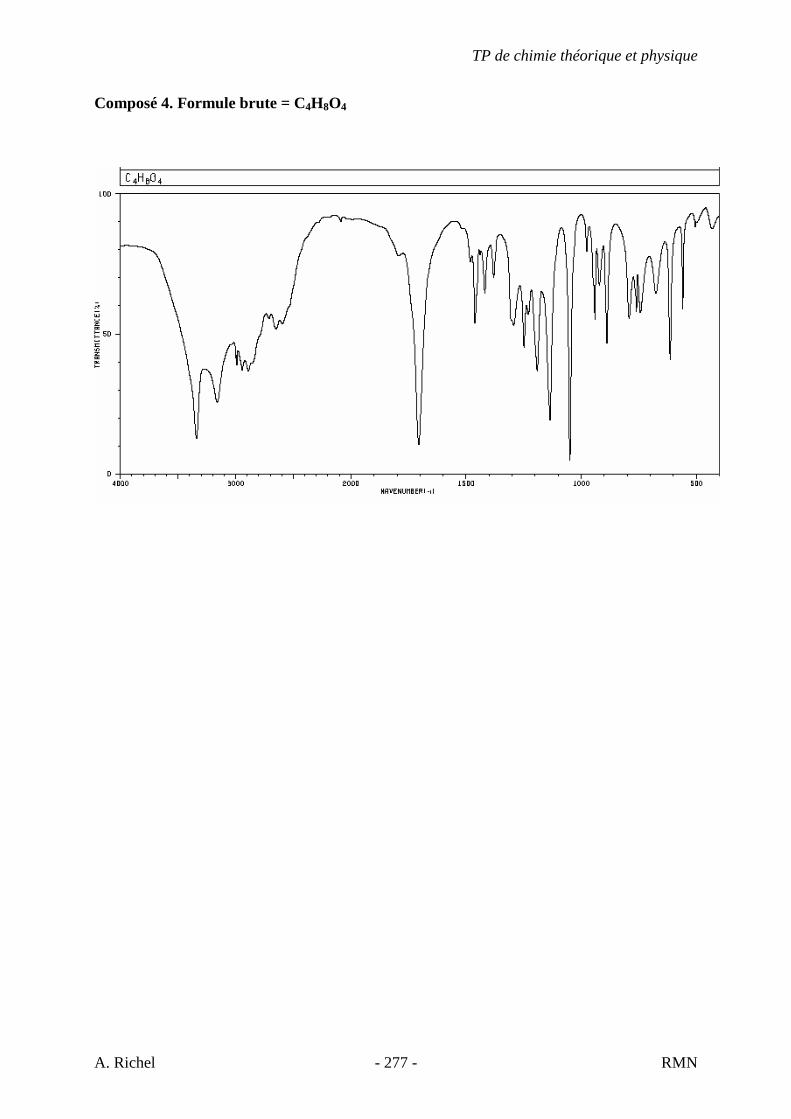
TP de chimie théorique et physique
A. Richel - 277 - RMN
Composé 4. Formule brute = C4H8O4
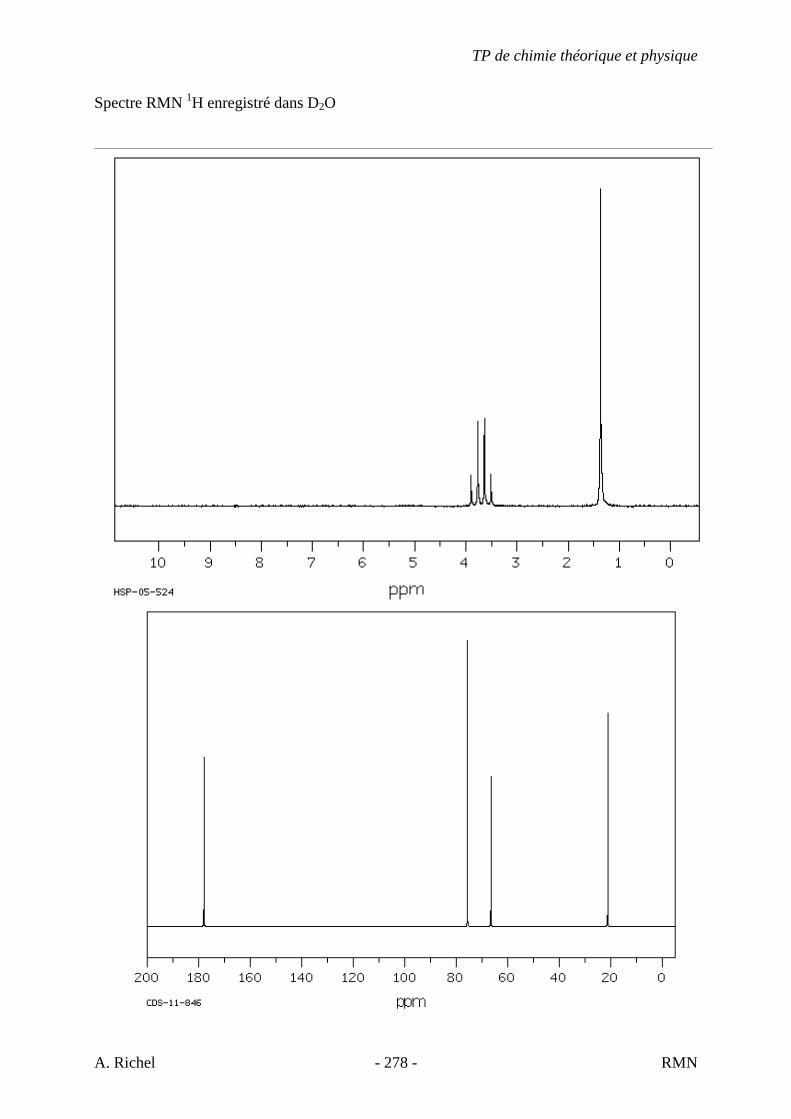
TP de chimie théorique et physique
A. Richel - 278 - RMN
Spectre RMN 1H enregistré dans D2O
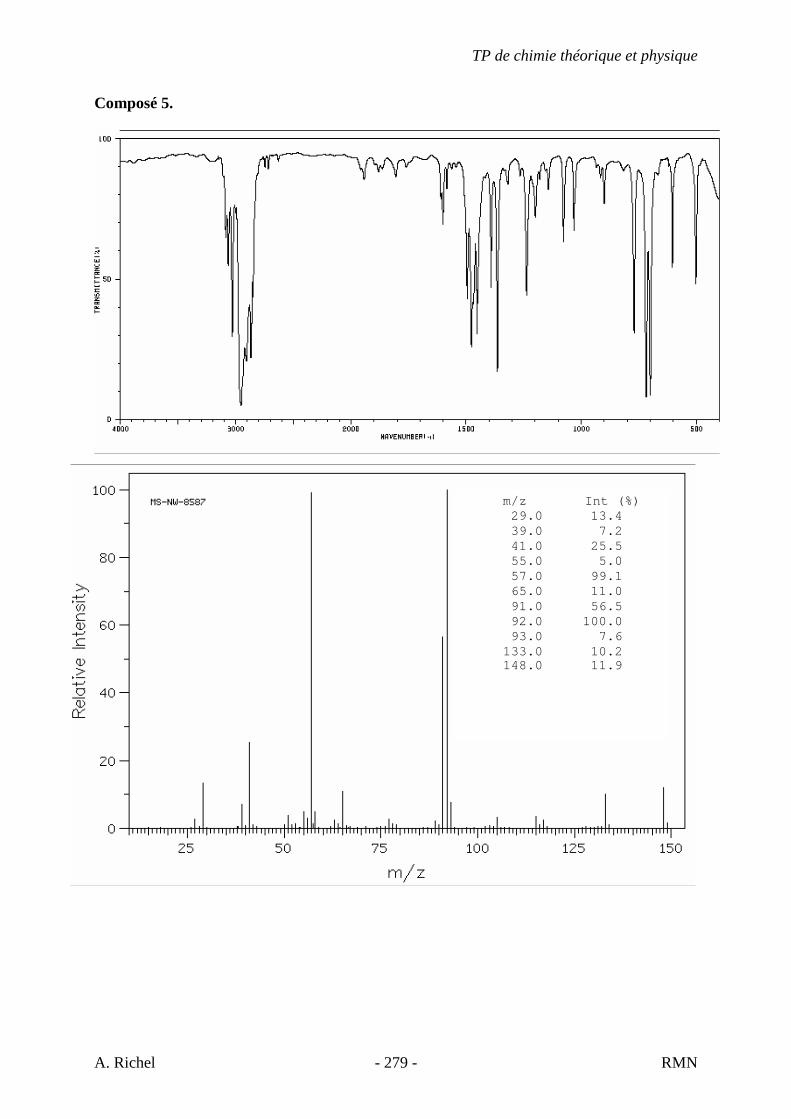
TP de chimie théorique et physique
A. Richel - 279 - RMN
Composé 5.
m/z Int (%) 29.0 13.4 39.0 7.2 41.0 25.5 55.0 5.0 57.0 99.1 65.0 11.0 91.0 56.5 92.0 100.0 93.0 7.6 133.0 10.2 148.0 11.9
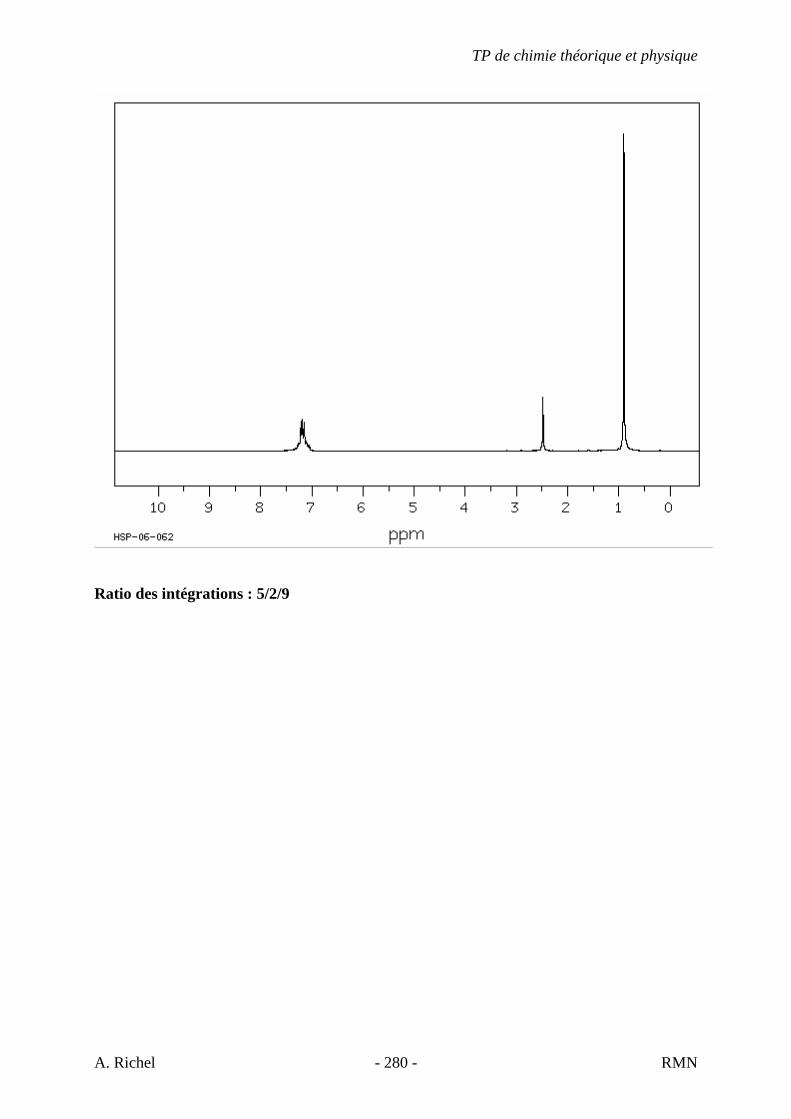
TP de chimie théorique et physique
A. Richel - 280 - RMN
Ratio des intégrations : 5/2/9
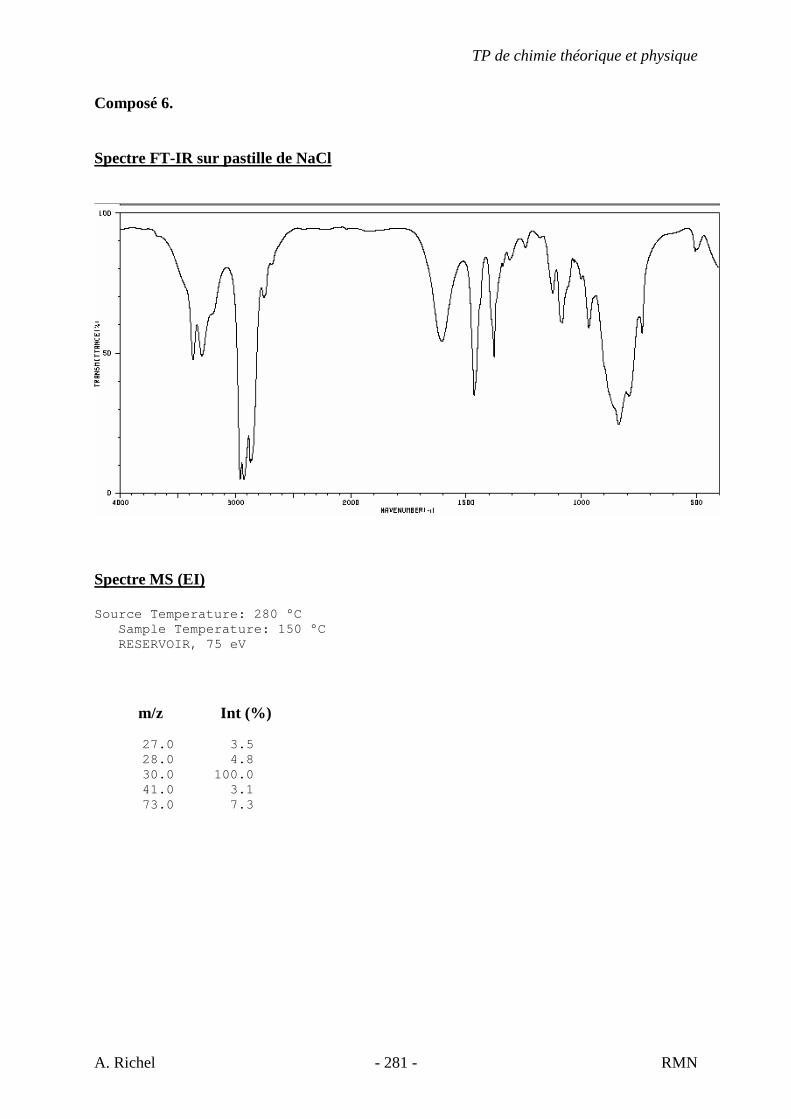
TP de chimie théorique et physique
A. Richel - 281 - RMN
Composé 6. Spectre FT-IR sur pastille de NaCl
Spectre MS (EI) Source Temperature: 280 °C Sample Temperature: 150 °C RESERVOIR, 75 eV
m/z Int (%) 27.0 3.5 28.0 4.8 30.0 100.0 41.0 3.1 73.0 7.3
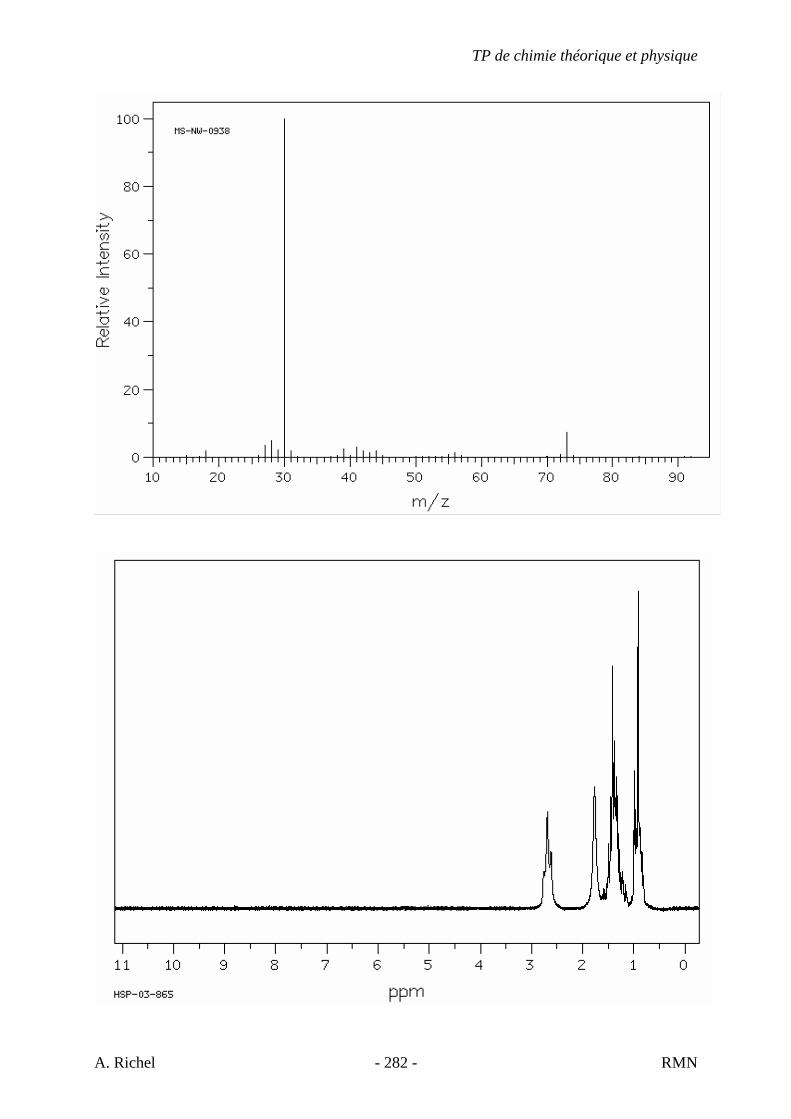
TP de chimie théorique et physique
A. Richel - 282 - RMN
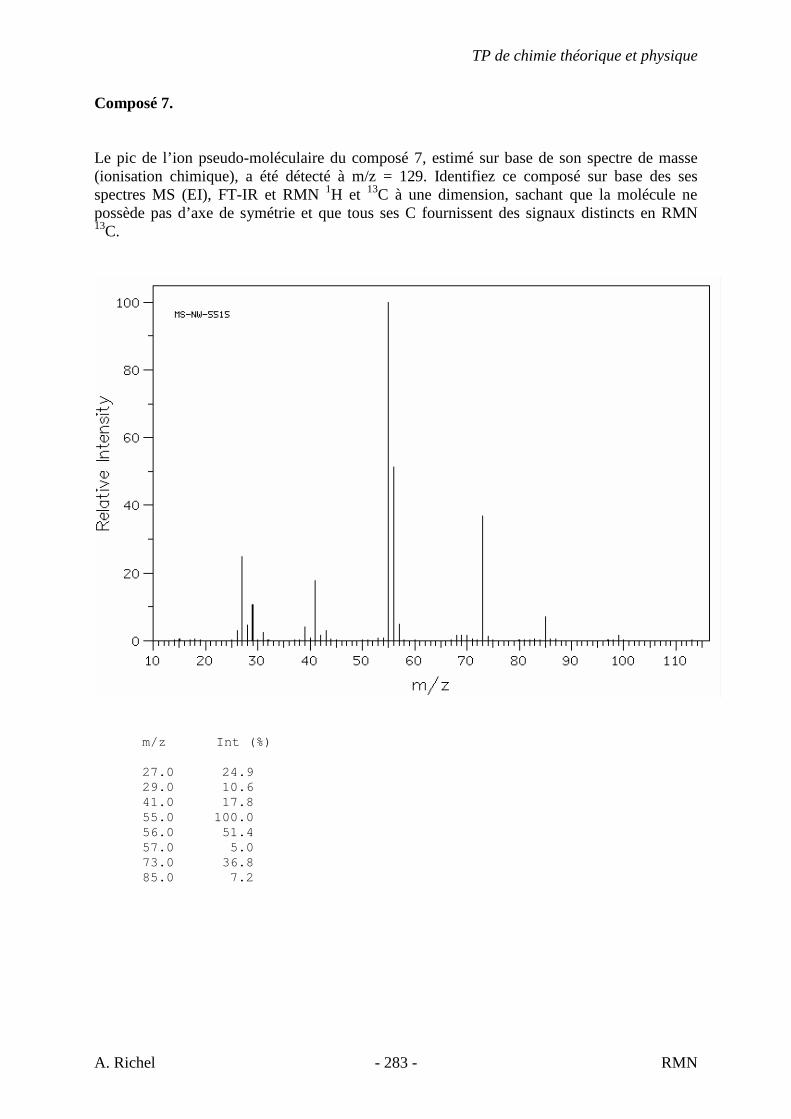
TP de chimie théorique et physique
A. Richel - 283 - RMN
Composé 7. Le pic de l’ion pseudo-moléculaire du composé 7, estimé sur base de son spectre de masse (ionisation chimique), a été détecté à m/z = 129. Identifiez ce composé sur base des ses spectres MS (EI), FT-IR et RMN 1H et 13C à une dimension, sachant que la molécule ne possède pas d’axe de symétrie et que tous ses C fournissent des signaux distincts en RMN 13C.
m/z Int (%) 27.0 24.9 29.0 10.6 41.0 17.8 55.0 100.0 56.0 51.4 57.0 5.0 73.0 36.8 85.0 7.2
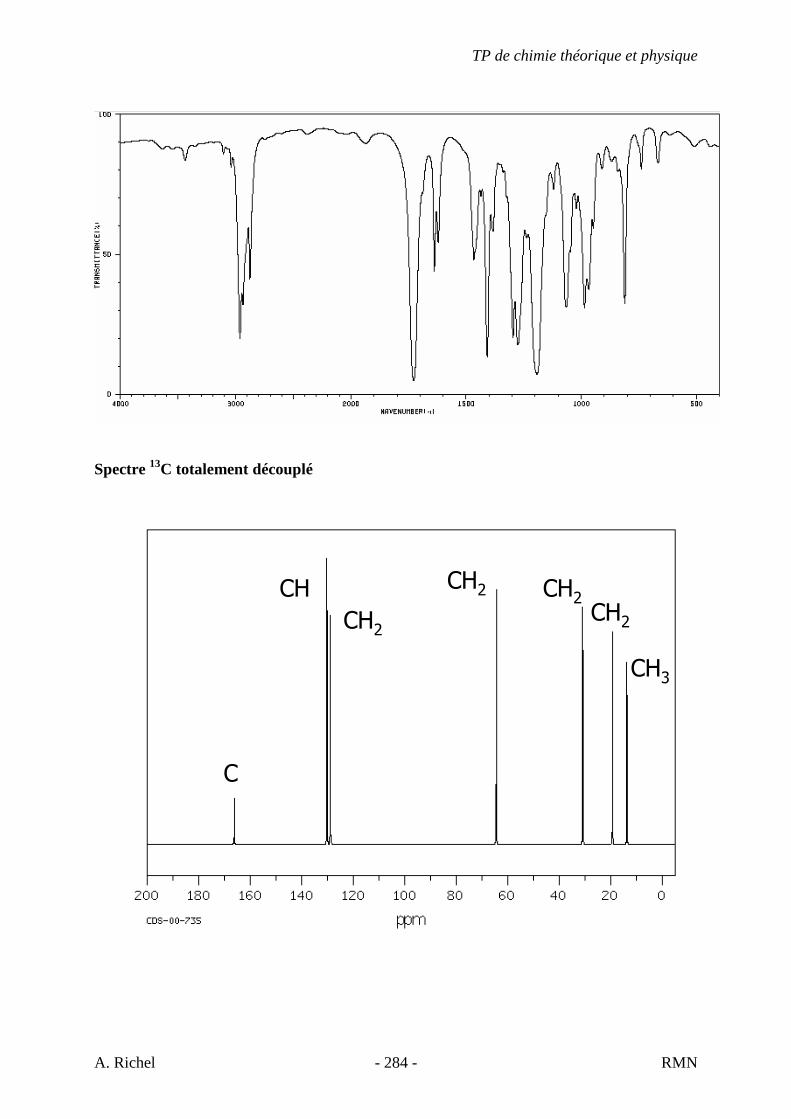
TP de chimie théorique et physique
A. Richel - 284 - RMN
Spectre 13C totalement découplé
C
CH
CH2
CH2 CH2CH2
CH3
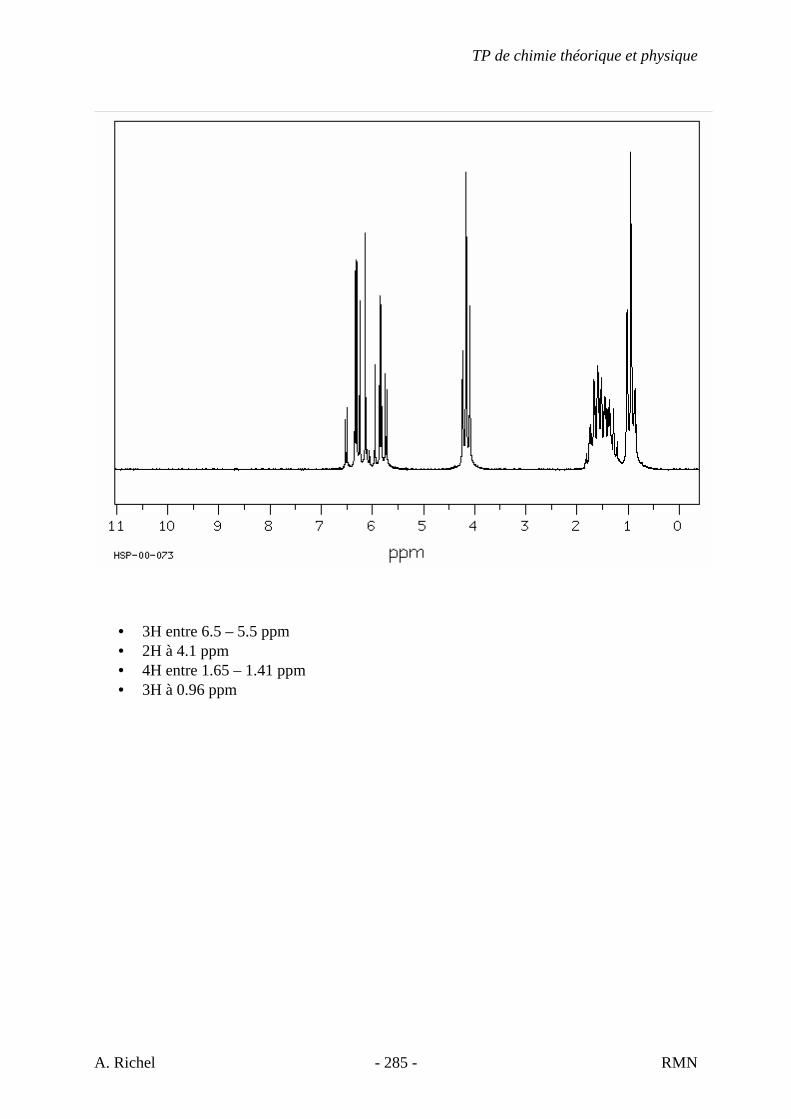
TP de chimie théorique et physique
A. Richel - 285 - RMN
• 3H entre 6.5 – 5.5 ppm • 2H à 4.1 ppm • 4H entre 1.65 – 1.41 ppm • 3H à 0.96 ppm
TP de chimie théorique et physique
A. Richel - 286 - RMN
Composé 8.
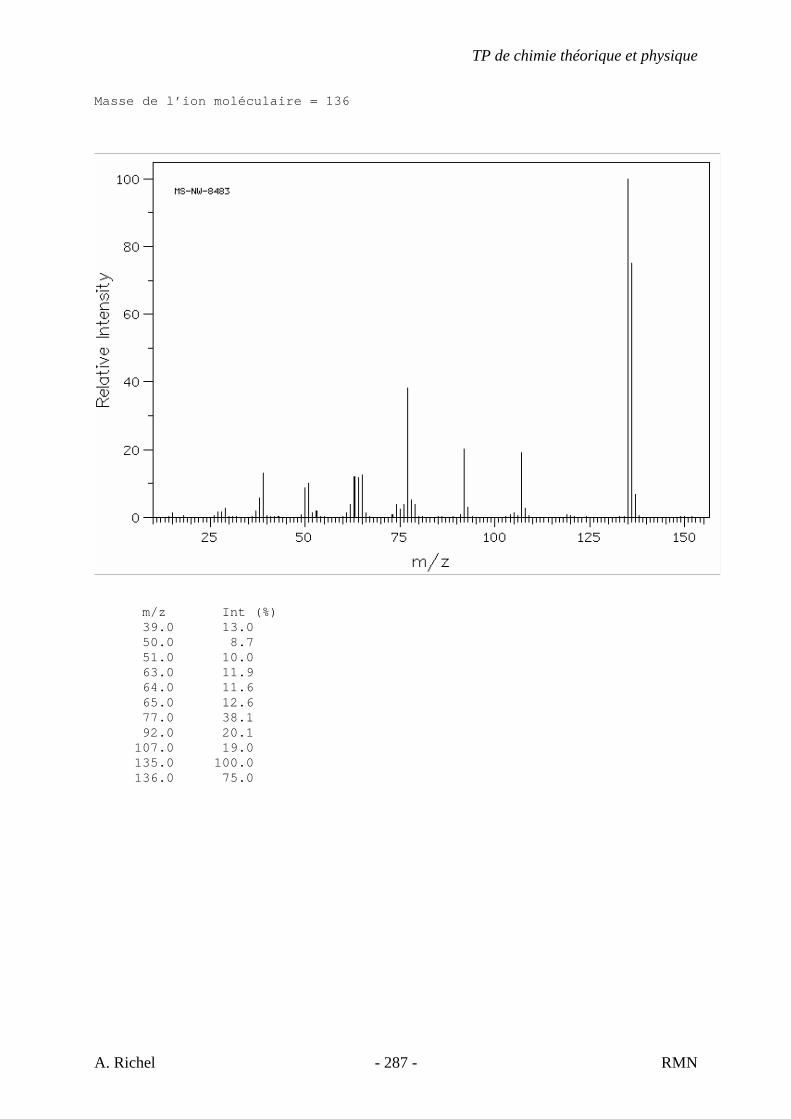
TP de chimie théorique et physique
A. Richel - 287 - RMN
Masse de l’ion moléculaire = 136
m/z Int (%) 39.0 13.0 50.0 8.7 51.0 10.0 63.0 11.9 64.0 11.6 65.0 12.6 77.0 38.1 92.0 20.1 107.0 19.0 135.0 100.0 136.0 75.0
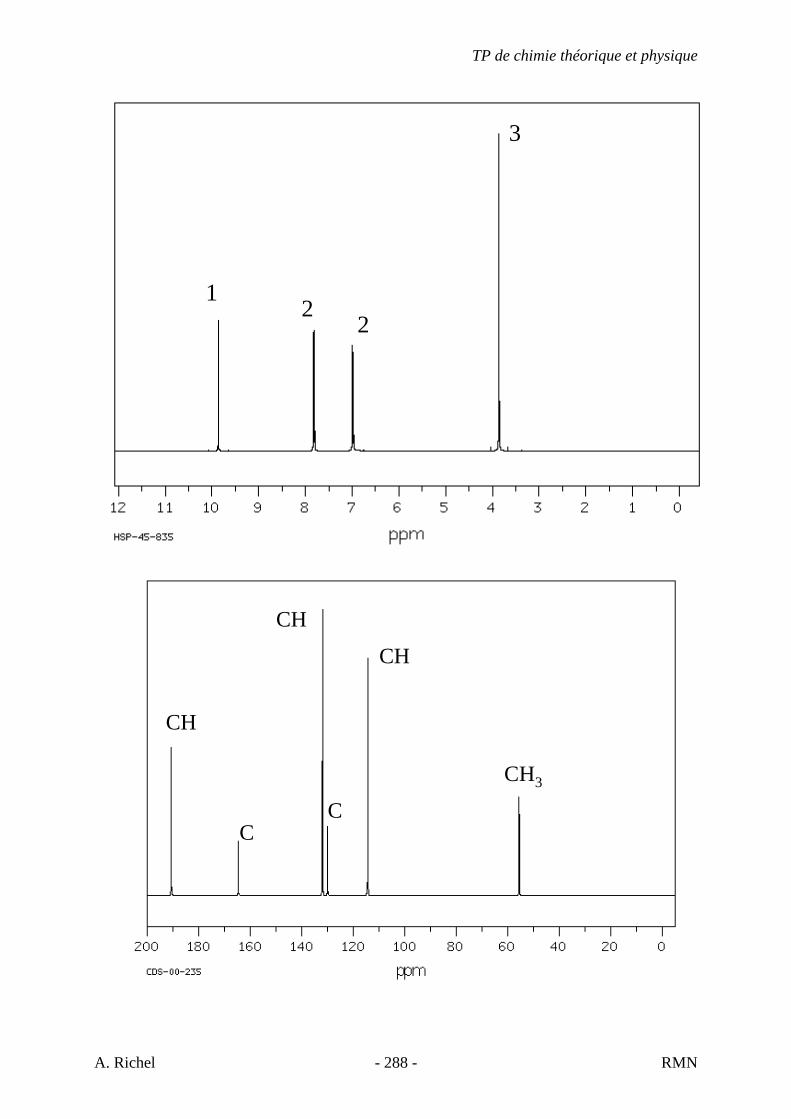
TP de chimie théorique et physique
A. Richel - 288 - RMN
12
2
3
CH
CH
C
CH
C
CH3
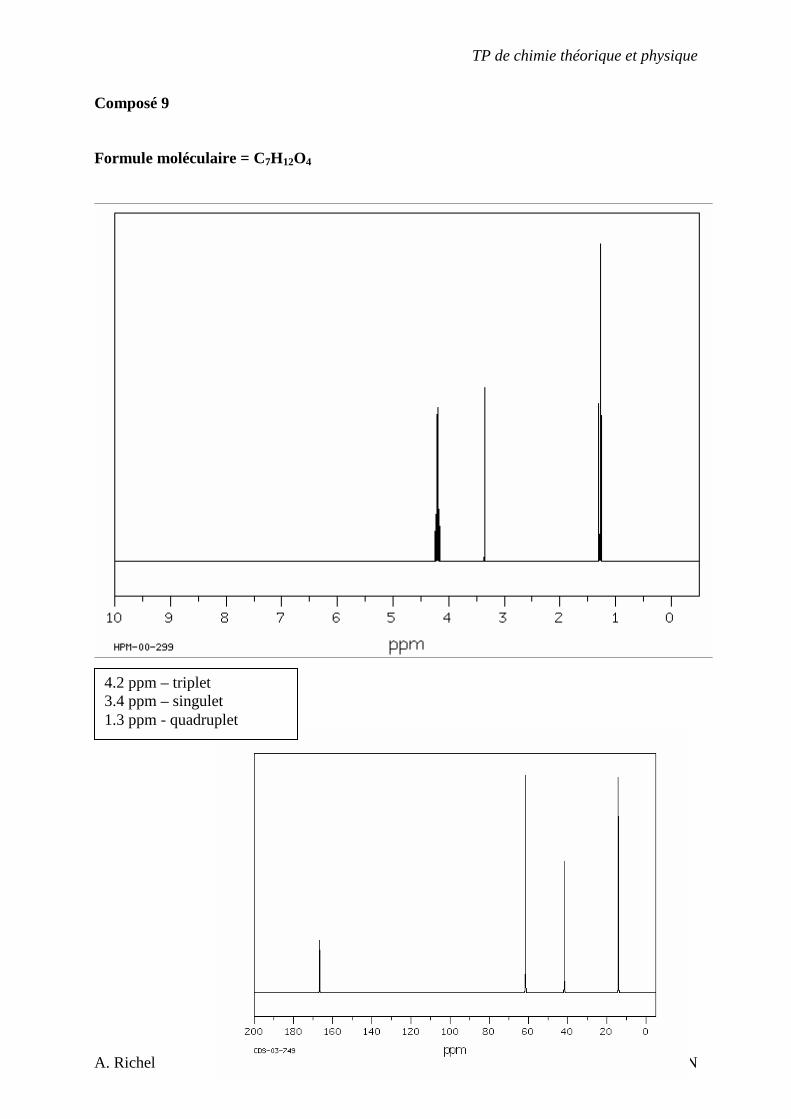
TP de chimie théorique et physique
A. Richel - 289 - RMN
Composé 9 Formule moléculaire = C7H12O4
4.2 ppm – triplet 3.4 ppm – singulet 1.3 ppm - quadruplet
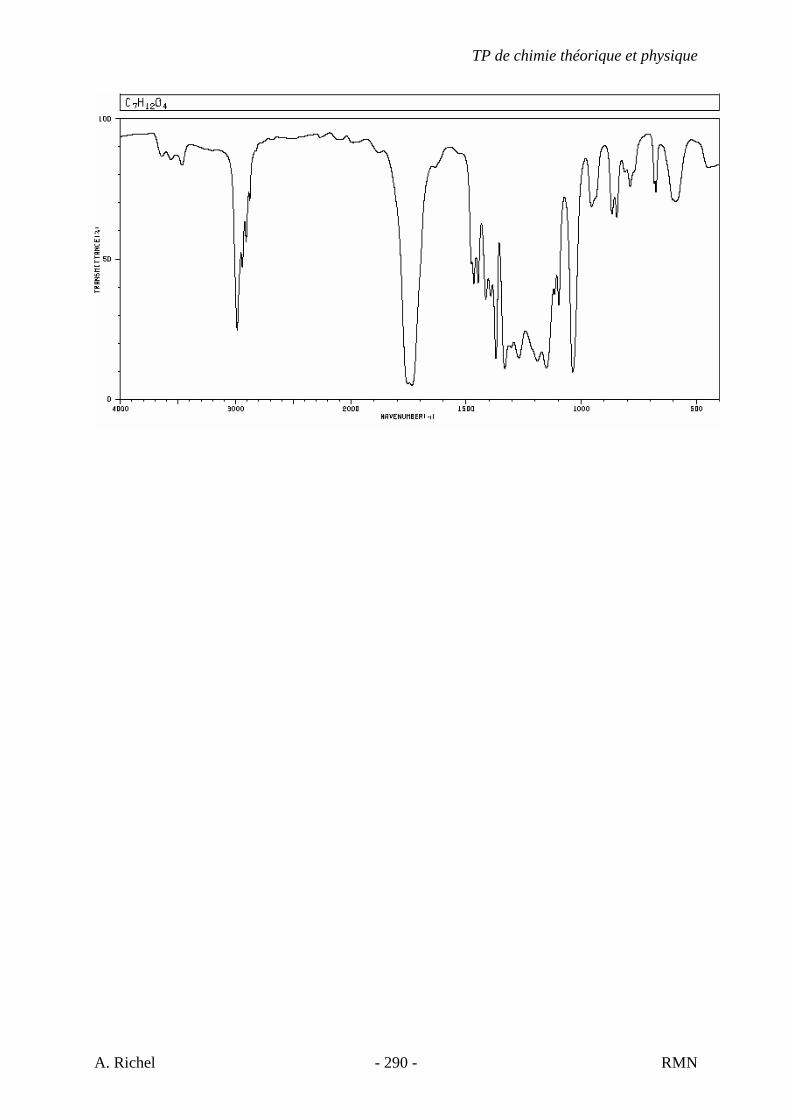
TP de chimie théorique et physique
A. Richel - 290 - RMN
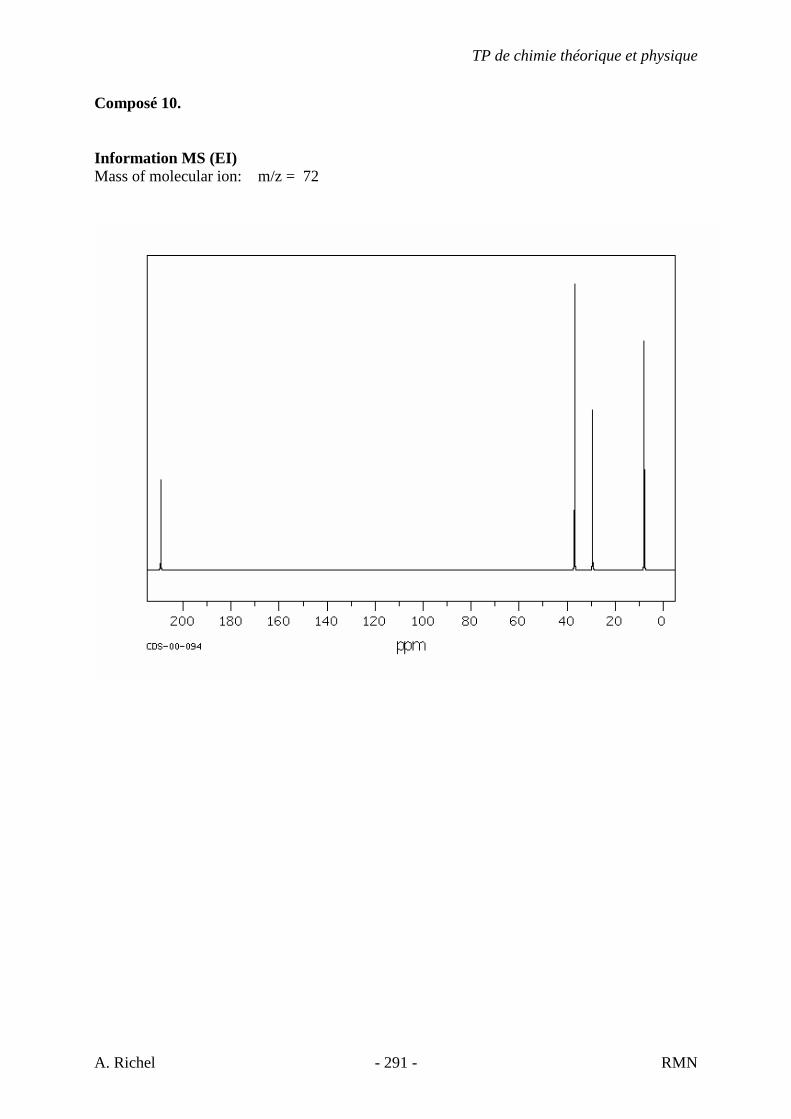
TP de chimie théorique et physique
A. Richel - 291 - RMN
Composé 10. Information MS (EI) Mass of molecular ion: m/z = 72

TP de chimie théorique et physique
A. Richel - 292 - RMN
Spectre COSY
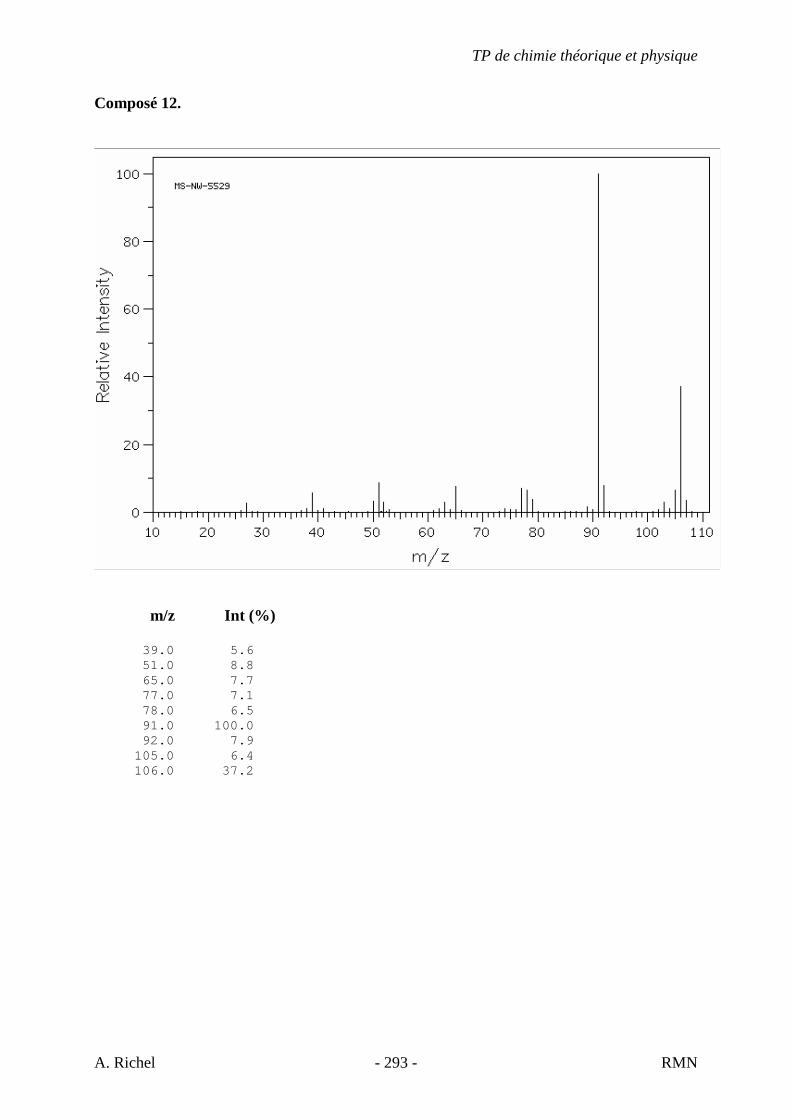
TP de chimie théorique et physique
A. Richel - 293 - RMN
Composé 12.
m/z Int (%) 39.0 5.6 51.0 8.8 65.0 7.7 77.0 7.1 78.0 6.5 91.0 100.0 92.0 7.9 105.0 6.4 106.0 37.2
TP de chimie théorique et physique
A. Richel - 294 - RMN
Spectre HSQC





























