Embed Size (px)
Citation preview

439 10.2217/FCA.12.12 © 2012 Future Medicine Ltd ISSN 1479-6678Future Cardiol. (2012) 8(3), 439–450
Futu
re C
ard
iolo
gy
part of
BackgroundCatecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare disorder, with an estimated prevalence of approximately one in 10,000 [1,2]. This number may under-represent the true prevalence of the disease, as CPVT still remains an under-recognized problem in the general population [3].
CPVT was first recognized in the 1970s [4]. After initial observations linked CPVT to chromosome 1 [5], two disease-causing genes were identified: the ryanodine receptor 2 gene (RYR2) [6,7] and the cardiac calsequestrin 2 gene (CASQ2) [7] , both involved in calcium homeos-tasis in cardiac cells. CPVT is characterized by life-threatening ventricular arrhythmias, usually bidirectional ventricular tachycardia, polymor-phic ventricular tachycardia or ventricular fibril-lation, especially under conditions of increased sympathetic activity, including physical exercise and emotional stress (Figure 1).
Sudden cardiac death (SCD) is the first pres-entation in up to 30% of cases [8]. The mean age at presentation is between 7 and 9 years of age, although an atypical form of CPVT (typically gene negative) may be observed in older patients and appears to be more sporadic [1,8]. Mortality is high when left untreated, reaching 30–50% by 30 years of age [1].
Diagnosis of CPVT remains challenging, especially as the cardiac evaluation is unremark-able at rest. The diagnosis is based on exercise or catecholamine-induced polymorphic or bidi-rectional ventricular tachycardia in the absence of structural heart disease, or a prolonged QT interval [1]. Taking b-adrenergic blocking agents protects most patients. However, b-blockers do not offer complete protection and many patients continue to have symptoms, documented exer-cise-induced ventricular tachycardia or both and remain at risk of sudden death.
Mechanism of CPVTMutations in genes encoding cardiac RYR2 [6,9] and CASQ2 [7,10] are recognized as caus-ing the autosomal dominant and autosomal recessive forms of CPVT, respectively. A total of 155 RYR2 mutations and 15 CASQ2 muta-tions [101] have been reported so far as causative for CPVT [11]. A gain-of-function mutation of the RYR2 receptor leads to a premature release of calcium. RYR2 in the sarcoplasmic reticu-lum is involved in the release of calcium ions at the time of excitation-contraction coupling [2]. Excitation-contraction coupling is mediated by calcium-induced calcium release at the time of cardiac depolarization. With cellular depolariza-tion, a small influx of calcium into the cytosol
Treatment of asymptomatic catecholaminergic polymorphic ventricular tachycardia
Manoj N Obeyesekere*, Raymond W Sy, Peter Leong-Sit, Lorne J Gula, Raymond Yee, Allan C Skanes, George J Klein & Andrew D KrahnThe University of Western Ontario, Division of Cardiology, London, Ontario, Canada *Author for correspondence: 339 Windermere Road, C6–110, London, Ontario, N6A 5A5, Canada n Tel.: +1 519 663 3746 n Fax: +1 519 663 3782 n [email protected]
Catecholaminergic polymorphic ventricular tachycardia is a rare genetic disorder caused by mutations in genes involved in the intracellular calcium homeostasis of cardiac cells. Affected patients typically present with l ife-threatening ventricular arrhythmias precipitated by emotional/physical stress. The diagnosis is based on the demonstration of polymorphic or bidirectional ventricular tachycardia associated with adrenergic stress. Genetic testing can be confirmatory in some patients. Treatment for catecholaminergic polymorphic ventricular tachycardia includes medical and surgical efforts to suppress the effects of epinephrine at the myocardial level and/or modulation of calcium homeostasis. Mortality is high when untreated and sudden cardiac death may be the first manifestation of the disease. First-degree relatives of a proband should be offered genetic testing if the causal mutation is known. If the family mutation is not known, relatives should be clinically evaluated with provocative testing. In the absence of rigorous trials, prophylactic treatment of the asymptomatic catecholaminergic polymorphic ventricular tachycardia patient appears to reduce morbidity and mortality.
Keywords
n asymptomatic n b-blocker n catecholaminergic polymorphic ventricular tachycardia n CASQ2 n CPVT n flecainide n left cardiac sympathectomy n polymorphic n ryanodine n RYR2 n sudden cardiac death
Revie
wFor reprint orders, please contact: [email protected]

Future Cardiol. (2012) 8(3)440 future science group
binds the RYR2 channel on the sarcoplasmic reticulum and opens the channel, leading to a larger release of calcium from the sarcoplasmic reticulum. This calcium binds to contractile proteins within the cardiac cell with resultant muscle contraction. Although the mechanism of calcium dysregulation in CPVT is not fully understood; spontaneous calcium release from the sarcoplasmic reticulum due to altered sen-sitivity/threshold of the RYR2 channel to cal-cium, calcium overload within the sarcoplasmic reticulum and second messenger regulation of the RYR2 channel play a role in the mecha-nism [12]. The RYR2 receptor is associated with CASQ2, a regulatory protein. CASQ2 mutations can also contribute to abnormal calcium regula-tion [13]. Thus, an increase in diastolic calcium is thought to cause delayed afterdepolarizations and the subsequent arrhythmia by activating a calcium-dependent inward current [14].
Mutations in two other genes (KCNJ2 and ANKB) may cause catecholamine-induced ven-tricular arrhythmia (including bidirectional ventricular tachycardia [VT]) and may resemble CPVT [15,16]. A large genomic deletion involving the third RYR2 exon, which leads to extended clinical phenotypes has also been described, with sinoatrial and atrioventricular node dys-function, atrial fibrillation, atrial standstill and dilated cardiomyopathy [17].
Diagnosing CPVT in the index case & asymptomatic relatives
The key element in the treatment of asympto-matic CPVT is to characterize the index case both clinically and genetically, and use this to direct the optimal diagnostic approach to iden-tifying typically asymptomatic or minimally symptomatic family members. This typically
involves clinical assessment, including exclusion of structural heart disease and targeted genetic testing when available. Arrhythmia in CPVT is induced by sympathetic stimulation, such as exercise or isoproterenol/epinephrine infusions. A systematic approach to clinical testing that includes provocative testing (exercise or phar-macological) results in unmasking of the cause of apparently unexplained aborted SCD in >50% of patients and is very helpful in direct-ing genetic testing of the index case and family to diagnose genetically mediated arrhythmia syndromes, such as CPVT [18,19]. In one study of patients with aborted SCD without evident cardiac disease, a systematic approach, includ-ing provocative testing and targeted genetic testing, unmasked the cause of aborted SCD in 35 out of 63 patients (eight of the 35 patients were diagnosed with CPVT).
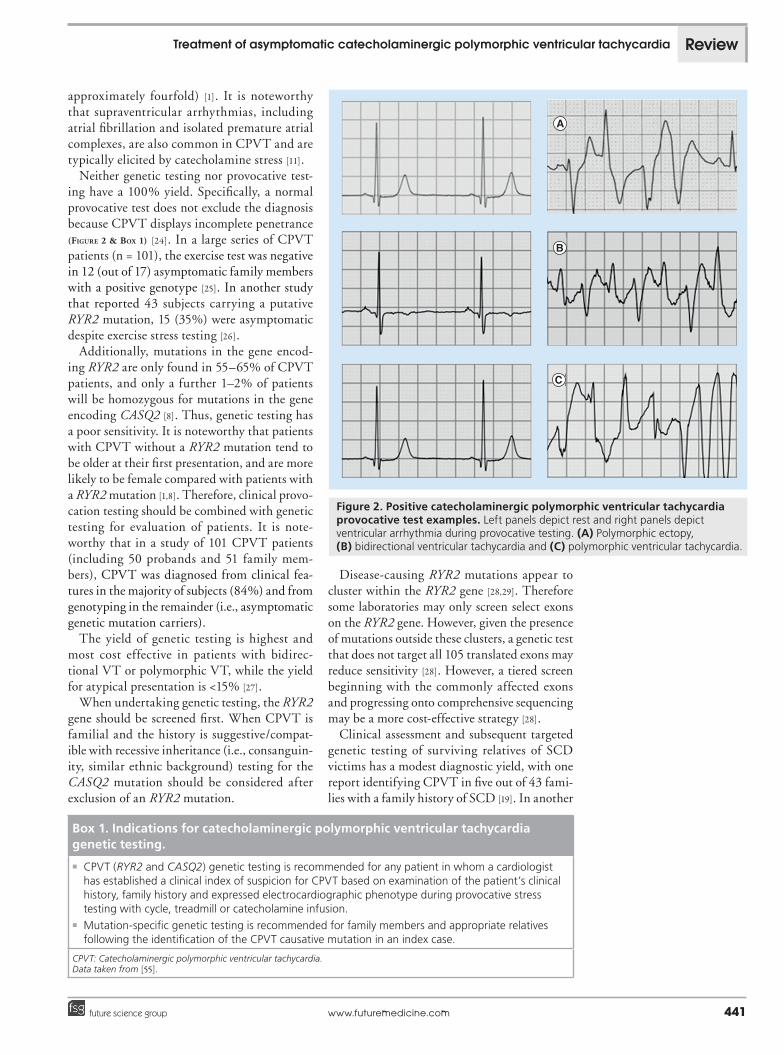
An exercise or epinephrine/isoproterenol provocation test is considered positive when ventricular ectopy, bidirectional ventricular tachycardia or polymorphic VT occurs, typi-cally in a progressive order with increasing heart rate (Figure 2). Continuation of testing may lead to sustained arrhythmia and syncope. The tachyarrhythmias resolve with discontinuation of exercise or ceasing the infusion. Intravenous b-blocker therapy can be utilized to suppress ventricular arrhythmias in CPVT testing. Polymorphic or bidirectional VT is provoked by exercise in 63% and epinephrine in 82% of patients with CPVT [1]. Interestingly, the heart rate threshold for induction of ventricu-lar arrhythmias has been reported to be lower for epinephrine infusion than exercise testing [1]. However, exercise/catecholamine-induced premature ventricular contractions (or bigem-iny) may be the only manifestation of CPVT [11,20]. These premature ventricular contractions are typically of right ventricular outflow tract origin [1,21]. In a majority of patients the initiat-ing beats in CPVT also have a right ventricular outflow origin [21]. However, a strategy of ablat-ing ‘premature ventricular contraction triggers’ may not be feasible in CPVT [22], as optical mapping of bidirectional ventricular tachy-cardia in a mouse model revealed CPVT that has an origin in the His–Purkinje system [23]. Testing for CPVT should be discontinued if systolic blood pressure rises above 200 mmHg or ventricular arrhythmias develop. Holter monitoring may be useful for evaluation of CPVT by bringing out the progressive arrhyth-mia with exercise. However, provocative testing significantly improves the diagnostic yield (by
I
II
III
II
aVF
aVL
V3 V6
V5V2
V1 V4aVR
Figure 1. Catecholaminergic polymorphic ventricular tachycardia.
Review Obeyesekere, Sy, Leong-Sit et al.
www.futuremedicine.com 441future science group
approximately fourfold) [1]. It is noteworthy that supraventricular arrhythmias, including atrial fibrillation and isolated premature atrial complexes, are also common in CPVT and are typically elicited by catecholamine stress [11].
Neither genetic testing nor provocative test-ing have a 100% yield. Specifically, a normal provocative test does not exclude the diagnosis because CPVT displays incomplete penetrance (Figure 2 & Box 1) [24]. In a large series of CPVT patients (n = 101), the exercise test was negative in 12 (out of 17) asymptomatic family members with a positive genotype [25]. In another study that reported 43 subjects carrying a putative RYR2 mutation, 15 (35%) were asymptomatic despite exercise stress testing [26].
Additionally, mutations in the gene encod-ing RYR2 are only found in 55–65% of CPVT patients, and only a further 1–2% of patients will be homozygous for mutations in the gene encoding CASQ2 [8]. Thus, genetic testing has a poor sensitivity. It is noteworthy that patients with CPVT without a RYR2 mutation tend to be older at their first presentation, and are more likely to be female compared with patients with a RYR2 mutation [1,8]. Therefore, clinical provo-cation testing should be combined with genetic testing for evaluation of patients. It is note-worthy that in a study of 101 CPVT patients (including 50 probands and 51 family mem-bers), CPVT was diagnosed from clinical fea-tures in the majority of subjects (84%) and from genotyping in the remainder (i.e., asymptomatic genetic mutation carriers).
The yield of genetic testing is highest and most cost effective in patients with bidirec-tional VT or polymorphic VT, while the yield for atypical presentation is <15% [27].
When undertaking genetic testing, the RYR2 gene should be screened first. When CPVT is familial and the history is suggestive/compat-ible with recessive inheritance (i.e., consanguin-ity, similar ethnic background) testing for the CASQ2 mutation should be considered after exclusion of an RYR2 mutation.
Disease-causing RYR2 mutations appear to cluster within the RYR2 gene [28,29]. Therefore some laboratories may only screen select exons on the RYR2 gene. However, given the presence of mutations outside these clusters, a genetic test that does not target all 105 translated exons may reduce sensitivity [28]. However, a tiered screen beginning with the commonly affected exons and progressing onto comprehensive sequencing may be a more cost-effective strategy [28].
Clinical assessment and subsequent targeted genetic testing of surviving relatives of SCD victims has a modest diagnostic yield, with one report identifying CPVT in five out of 43 fami-lies with a family history of SCD [19]. In another
Box 1. Indications for catecholaminergic polymorphic ventricular tachycardia genetic testing.
n CPVT (RYR2 and CASQ2) genetic testing is recommended for any patient in whom a cardiologist has established a clinical index of suspicion for CPVT based on examination of the patient’s clinical history, family history and expressed electrocardiographic phenotype during provocative stress testing with cycle, treadmill or catecholamine infusion. n Mutation-specific genetic testing is recommended for family members and appropriate relatives
following the identification of the CPVT causative mutation in an index case.
CPVT: Catecholaminergic polymorphic ventricular tachycardia. Data taken from [55].
Figure 2. Positive catecholaminergic polymorphic ventricular tachycardia provocative test examples. Left panels depict rest and right panels depict ventricular arrhythmia during provocative testing. (A) Polymorphic ectopy, (B) bidirectional ventricular tachycardia and (C) polymorphic ventricular tachycardia.
Treatment of asymptomatic catecholaminergic polymorphic ventricular tachycardia Review

Future Cardiol. (2012) 8(3)442 future science group
study, relatives of six unrelated CPVT patients were tested for the RYR2 gene [30]. Thirty family members were subsequently identified as mutation carriers. Previously undiagnosed CPVT-related symptoms were reported by only eight subjects (thus 22 out of 30 patients were asymptomatic). Furthermore, cascade screening in families with CPVT, based on genetic testing and stress testing results in successful prophy-lactic treatment in a substantial proportion of mutation carriers [31]. Therefore, the high success rates in diagnosing and protecting affected sur-viving relatives of sudden death victims justifies a systematic evaluation of surviving relatives in the authors’ opinion.
In general, the first step is to characterize the parents of the proband (Figure 3), if available. If genetic screening in parents is negative, the mutation is usually considered to be de novo and the proband’s siblings are not tested. An excep-tion to this rule is a rare mode of inheritance known as germline mosaicism [32]. Sporadic cases of CPVT are usually thought to be caused by the occurrence of a de novo mutation. RYR2 mutations were recently observed to be de novo in 20% [28]. However, cases identified as de novo in the proband may arise through the existence of mosaicism in the germ cells of one of the asymptomatic parents [28,33]. When mosaicism is present in the germ cells of one of the asymptomatic parents of the proband, the mutation can either occur in a germ cell alone
that continues to divide (thus the mutation-negative parent is unaffected but can transmit the mutation), or the mutation may occur very early before the separation to germ cells and, therefore, be present in both somatic and germ cells (mutation-positive parent). Therefore, the phenomenon of germline mosaicism may jus-tify genetic screening of siblings of a sporadic proband, even if the causal mutation was not detected in the parents.
Germline mosaicism likely explains a very small proportion of de novo RYR2 mutations (<1%). There have only been two published reports of germline mosaicism involving the RYR2 gene causing CPVT [28,32]. When sib-lings of a proband are also phenotypically and genotypically affected, despite the absence of the mutation in the biological parents, germline mosaicism is highly likely. Routine genotyping of siblings of mutation-positive probands in the absence of the mutation in the biological parents is likely to be cost prohibitive. A suitable alter-native is to clinically assess all the siblings and screen the siblings with provocative testing even in the absence of the causative mutation in the biological parents of the gene-positive proband. This is a rapidly evolving field where the clini-cal approach to this dilemma may become sim-plified with rapidly developing technology and understanding.
Screening and treating asymptomatic gen-otype-negative CPVT families is challenging due to the limitation of stress testing to diag-nose CPVT with absolute accuracy (i.e., lack of sensitivity). It is noteworthy that the cardiac ryanodine receptor, encoded by 105 exons, is one of the largest ion channels, comprising 4967 amino acids. Most initial mutational analyses target three ‘hot spot’ domains that only encode <40% of the translated exons. Up to 17% of putative mutations may localize outside these domains [28]. Thus, it is critical that a com-prehensive mutational analysis is performed prior to diagnosing genotype-negative CPVT. Compound heterozygosity of polymorphisms as well as somatic mutations may be another mechanism of genotype-negative CPVT, although this is rare. Large genomic deletions/rearrangements also require additional genetic analysis. Testing of the CASQ2 gene may also be considered in select circumstances. It is also notable that long QT syndrome (LQTS) may mimic CPVT – in one study of 11 unrelated patients with suspected CPVT, four possessed LQTS-associated mutations (LQT5 and LQT7 in four unrelated females with exercise-induced
Index case or relative
Clinical assessment
Exercise testing
± Epinephrine infusion
Genetics clinicPedigree/family history
Discuss genotyping
Reassurance/initiatetherapy
Risk discussion± Genetic testing†
Initiate β-blockerInitiate Flecainide if no contraindication
Exclude structural heart disease
Figure 3. An algorithmic approach to investigating suspected cases of catecholaminergic polymorphic ventricular tachycardia.†See Box 1 for indications for genetic testing.
Review Obeyesekere, Sy, Leong-Sit et al.

www.futuremedicine.com 443future science group
bidirectional VT [34]. Thus, in certain circum-stances LQT genetic testing should be con-sidered prior to diagnosing genotype-negative CPVT. Thus, probands with genotype-negative CPVT where limited RYR2 exon sequencing was performed some years ago may require repeat and/or additional testing (and/or specific LQT genetic testing).
The extent to which empiric b-blocker ther-apy is utilized in asymptomatic relatives despite a negative stress test will, to some extent, depend on the degree of certainty of the diagnosis in the proband and the extent of screening under-taken in the relative (additional factors, such as expressivity in other relatives may also assist the physician in this difficult situation). A strong CPVT phenotype can be defined as exertional syncope with bidirectional and/or polymorphic VT. Whereas a possible CPVT phenotype may be established on the basis of exertional syncope and stress-induced ventricular ectopy but not bidirectional or polymorphic VT. In addition to screening with exercise stress testing, relatives of a genotype-negative CPVT proband should also be considered for pharmacological testing when exercise testing is negative. In the absence of data on outcomes of genotype and provoca-tive test negative first-degree relatives of the proband, relatives may be offered and choose b-blocker therapy due to the lack of sensitivity of the screening tests and tolerability of b-blockers. Having said this, we do not typically advocate b-blockers if all phenotype testing is negative in an asymptomatic carrier.
Natural history of CPVTThe aforementioned multicenter observational study of 101 subjects with CPVT (50 probands and 51 family members), also reported a high rate of cardiac symptoms in patients with CPVT before the diagnosis was made. CPVT-related symptoms occurred early in life and were reported to occur in approximately 35 and 72% by the age of 10 and 20 years, respectively. In 23 of the 40 family members without any prior cardiac symptoms, reproducible ventricular arrhythmias were recorded during exercise. The overall estimated 8-year cardiac event rate was 32%, with a 13% rate of fatal or near-fatal events [25]. No difference was observed in the cardiac event rates between the probands and family members. Interestingly, event rates dur-ing follow-up were not increased in patients with a prior history of syncope.
These results suggest the need for treatment in all CPVT patients identified during cascade
screening, independent of preceding symp-toms [25]. This includes genetically positive fam-ily members even with a negative provocative test. Similarly, genetically negative patients with a positive provocative test should also receive prophylactic therapy, given the potential uncer-tainty in genetic mechanism and potential for digenic inheritance. Furthermore, given the early age of symptoms, it is reasonable to con-sider genetic testing at birth, which allows early initiation of therapy.
Prognostic featuresMales may be at a higher risk for syncope, although this has not been validated across studies [8]. A younger age at diagnosis, a his-tory of aborted cardiac arrest, and the absence of b-blockers predict future events [25]. However, b-blocker therapy is not completely protective. Furthermore, suppression of exercise-induced ventricular arrhythmias with b-blocker therapy does not necessarily translate into long-term effectiveness of therapy [30]. Studies have also demonstrated the occurrence of SCD despite a negative exercise stress test in asymptomatic genotype-positive CPVT patients in the absence of b-blockers [25,26].
There is no established genotype-based risk stratification scheme in CPVT (based on the presence or the absence of a mutation or poly-morphism). Therefore, there is no differential treatment strategy for CPVT gene positive versus gene negative index case.
Treatment of CPVTArrhythmogenesis in CPVT is due to enhanced release of calcium in response to catechol-aminergic stimulation with resultant ventricu-lar arrhythmia. Therefore, treatments for CPVT include medical and surgical efforts to suppress the release and/or effects of epinephrine at the myocardial level or modulate calcium regulation by inhibiting RYR2 function. Implantable car-dioverter defibrillators (ICD) seek to treat the ventricular arrhythmia when the aforementioned measures fail (TaBle 1).
Lifestyle measuresIn general, all symptomatic CPVT patients should avoid intensive sports and exercise. Asymptomatic patients may undertake low intensity exercise. Exercise restriction can be guided by response observed during exer-cise stress testing. However, as stated earlier, suppression of exercise-induced ventricular arrhythmias with b-blocker therapy does not
Treatment of asymptomatic catecholaminergic polymorphic ventricular tachycardia Review

Future Cardiol. (2012) 8(3)444 future science group
necessarily translate into long-term effectiveness of therapy [30].
b-blockersb-blockers are considered first-line therapy for preventing ventricular arrhythmias in patients diagnosed with CPVT. b-blockers should be prescribed in every CPVT patient (diagnosed by a positive genetic test and/or a positive exercise/infusion test) regardless of the pres-ence or absence of symptoms. Although the 2006 ACC/AHA guidelines recommend that b-blockers may be considered for patients with CPVT who were genetically diagnosed in adult-hood and have never manifested clinical symp-toms of tachyarrhythmias as a class IIb recom-mendation (with level of evidence: C)[35], the authors advocate the use of b-blockers in this population, due to their tolerability and the reported potential for ventricular arrhythmias even in the asymptomatic adult patient (despite a negative exercise stress test) [25,26,30].
The eff icacy of b-blocker therapy for treating CPVT patients is well recognized. However, b-blockers are not completely effec-tive in preventing life-threatening arrhythmias [8,21,24–26,30]. The reported proportion of CPVT patients in whom b-blocker therapy fails to pre-vent life-threatening arrhythmic events ranges from 0 to 59% over a follow-up range between 1 and 8 years in these studies [36].
In the largest series (n = 101), with a mean follow-up of 8 years, the cardiac event rates were 27% in the patients with b-blockers and 58% in those without (including in two muta-tion carriers with normal exercise tests who were not treated with b-blockers; hazard ratio:
5.48; 95% CI: 1.80–16.68) [25]. The estimated 8-year fatal or near-fatal event rates were 11%, in patients with b-blockers and 25% in those without. Therefore, event rates were relatively high despite b-blocker therapy and also occurred despite a normal exercise stress test in the absence of b-blockers. In addition, there were 37 asymptomatic patients with a positive genotype. During follow-up, cardiac events were observed in nine of the 37 patients (24%) and in 18 of the other 64 patients (28%). No differences were observed in cardiac event rate and fatal or near-fatal event rate between these two groups. In this study, cardiac events were observed in 19% with nadolol and in 39% with b-blockers other than nadolol [25].
The efficacy of nadolol in the quoted study may be related to its strong b-blocking action, long half-life (although some other b-blockers can also be given with reduced frequency due to their long half-life), nonselectivity and excre-tion in an unmetabolized form (which may offer advantages over other b-blockers in spe-cific patients). Furthermore, nadolol has no intrinsic sympathomimetic activity (similar to most other b-blockers). Additional pharmaco-logical characteristics, such as solubility and membrane-stabilizing properties, may also con-tribute towards the varied efficacies. The lack of cardioselectivity of nadolol, however, may be disadvantageous in some patients. Despite the suggestion that nadolol is superior in this single study, in the absence of large systematic stud-ies comparing the efficacy of varied b-blockers, the choice of an appropriate b-blocker needs to be based on preferred pharmacodynamic and pharmacok inetic effects (namely, long half-life,
Table 1. Summary table of therapeutic options.
Therapeutic strategy Recommendations
Lifestyle measures All symptomatic patients should avoid intensive sports and exercise. Asymptomatic patients may undertake low intensity exercise
b-blockers Considered first-line therapy independent of symptomatic state
Flecainide Utilized in conjunction with b-blockers in symptomatic patients despite b-blockers. May also be used as first line therapy in asymptomatic cases with b-blockers
Calcium channel blockers Limited evidence highlighting the utility of combined b-blocker and calcium blocker therapy
Left cardiac sympathetic denervation
Currently only used as a secondary prevention strategy
Implantable defibrillator No indication for primary prevention implantation. Class IIa indication for patients with syncope and/or documented sustained ventricular tachycardia while receiving b-blockers. Class I indication for secondary prevention
Review Obeyesekere, Sy, Leong-Sit et al.

www.futuremedicine.com 445future science group
tolerability, strong b-receptor selectivity and clinical evidence of drug effect). The authors per-form exercise testing to assess adequate b-blocker effect following commencement of b-blocker, aiming for a heart rate reduction of 30 beats per min at peak workload compared with prior to initiation, and the absence of arrhythmia.
In a more recent study that evaluated the effi-cacy of flecainide in CPVT patients, ten of 33 genotype-positive patients had no symptoms at diagnosis, but all subsequently had ventricu-lar arrhythmias while taking b-blockers (one patient was also on verapamil, see below) [37]. It is noteworthy that b-blocker therapy remains safe despite a marked resting bradycardia that may be observed in patients with CPVT [21,24].
Calcium channel blockersCPVT patients in whom ventricular arrhyth-mias are not well controlled with b-blockers pose a significant challenge, as the availability of additional treatment options is limited and/or has been established only in small studies. Verapamil added to b-blockers suppresses ven-tricular arrhythmias in CPVT patients during exercise stress testing, suggesting its beneficial effect in combination with b-blockers [38,39]. However, calcium channel blockers are not effective in all patients [39]. Despite a more effec-tive suppression of exercise-induced ventricular arrhythmias with verapamil plus b-blockers, this combination therapy failed to prevent clinically significant ventricular arrhythmias during long-term follow-up. In a small observational study with a mean follow-up of 3 years, three out of six patients experienced recurrent arrhythmic symptoms including cardiac arrest (not related to stress) in one, appropriate ICD shock in the second and syncope without documented arrhythmias in the third [40]. The authors do not support the use of calcium channel blockers due to the lack of evidence supporting their efficacy.
FlecainideA recent study evaluated the efficacy and safety of flecainide in addition to conventional drug therapy in patients with genotype-positive CPVT [37]. Flecainide directly targets the molec-ular defect in CPVT by inhibiting RYR2 chan-nels and preventing arrhythmogenic calcium waves [41,42]. Sodium channel blockade further reduces the rate of triggered beats [41,42]. In the aforementioned recent multicenter international study, prior to flecainide therapy, all patients had persistent physical or emotional stress-induced ventricular arrhythmias documented by exercise
testing, Holter recordings, or ICD interrogation and/or persistent symptoms of palpitations, syn-cope, aborted cardiac arrest or appropriate ICD shocks while taking b-blockers, with or without calcium channel blockers. Thirty-three patients received flecainide because of exercise-induced ventricular arrhythmias despite conventional therapy (for different reasons, not always opti-mal). Twenty-two patients had either partial (n = 8, 28%) or complete (n = 14, 48%) suppres-sion of exercise-induced ventricular arrhythmias with flecainide (p < 0.001). Complete suppres-sion of exercise-induced ventricular arrhythmias (48%) was defined as the presence of none or iso-lated ventricular premature beats during exercise following a stable dose of flecainide. Patients also achieved significantly higher heart rates before ventricular arrhythmias occurred during exercise and a significant reduction in maximum sinus rate during exercise, even though a higher mean workload was achieved. In the subset of patients already on optimal b-blocker therapy (n = 15), flecainide further significantly improved the ventricular arrhythmia score compared with b-blocker therapy alone (p < 0.003).
Patients without suppression of exercise-induced ventricular arrhythmias (24%, n = 7) were receiving a significantly lower dose of fle-cainide compared with patients with ventricu-lar arrhythmia suppression. During follow-up (median: 20 months), VT recurred in a single patient who experienced several ICD shocks for polymorphic VT after 8 months of flecainide treatment. Her serum flecainide level was low, suggesting noncompliance. The optimal dose of flecainide was reported to be between 150 and 200 mg/day (range: 100–300 mg/day). Daily doses <100 mg were associated with a lack of therapeutic response. These findings suggested that flecainide should be considered for CPVT patients with refractory ventricular arrhythmias on b-blocker therapy, and the combination could even be considered as a first-line therapy in patients with a malignant presentation.
Flecainide did not cause arrhythmic events during a median follow-up of 20 months in these patients with a structurally normal heart. The CAST trial reported an excess of deaths after acute recurrent myocardial infarction in patients treated with flecainide (n = 323; mean follow-up of 10 months) compared with placebo (n = 318) [43]. Therefore, in general, flecainide should not be considered dangerous in CPVT unless left ventricular function is decreased.
CPVT patients treated with flecainide were limited to patients with predominantly RYR2
Treatment of asymptomatic catecholaminergic polymorphic ventricular tachycardia Review

Future Cardiol. (2012) 8(3)446 future science group
(RYR2 n = 32, CASQ2 n = 1) mutations. Testing the efficacy of flecainide in non-RYR2 mutation-carrying CPVT patients will further assist in understanding the pathophysiology of ventricular arrhythmias in RYR2 mutation-negative CPVT patients. It remains to be seen whether flecainide could serve as a first-line therapy, in combination with b-blocker therapy or for patients intolerant of b-blockers.b-blocker therapy should be continued in
combination with flecainide in CPVT patients, because b-blockers and flecainide act synergisti-cally on the mechanism underlying ventricular arrhythmia. The authors support combination therapy in asymptomatic CPVT patients in the absence of structural heart disease given the inadequate long-term efficacy with b-blocker therapy alone and the safety of flecainide in patients with no structural heart disease.
Left cardiac sympathetic denervationLeft cardiac sympathetic denervation is an effective and safe therapeutic option when symptoms persist despite pharmacological therapy. However, it requires at least minimally invasive endoscopic surgery, is not universally available and has only been tested in small cohorts [44–47]. Left cardiac sympathetic den-ervation interrupts the major source of epine-phrine released to the heart [48]. The surgery increases the threshold for ventricular fibrilla-tion [49] and increases ventricular refractoriness [50]. As preganglionic denervation precludes reinnervation, the effects of sympathetic den-ervation are permanent. This circumvents the limitations of medical therapy including poor compliance. As denervation can decrease the probability of arrhythmic events, left cardiac sympathetic denervation also compliments the use of an ICD. The ICD may also be more likely to interrupt ventricular fibrillation and restore sinus rhythm. Left cardiac sympathetic denervation does not preclude implantation of an ICD in patients experiencing VT despite optimal tolerated pharmacotherapy. When appropriate, the clinical strategy would be to use the ICD as a safety net, together with left cardiac sympathetic denervation, in con-junction with b-blockers and flecainide, to minimize the risk of further life-threatening arrhythmias.
Reports encompassing small numbers of patients have been described [44,47]. Left cardiac sympathetic denervation is predominantly used in both LQTS and CPVT patients experienc-ing arrhythmias despite pharmacotherapy. It has
been reported as a primary prevention strategy in patients with LQTS [47,51] and in a single patient with CPVT [52]. Although it appears difficult to identify a high-risk asymptomatic group of CPVT patients in whom primary left cardiac sympathetic denervation may be appropriate, it may be considered in select patients who are risk averse and/or have a contraindication to established pharmacotherapy.
Implantable cardioverter defibrillatorsArrhythmic events in CPVT patients treated with b-blocker may be attributed to noncompli-ance, underdosing due to the intolerability/side effects or incomplete efficacy. Thus, adjunctive therapies, such as ICDs, have been utilized to treat potential ventricular arrhythmias. The reported proportion of CPVT patients in whom b-blocker therapy fails to prevent life-threaten-ing arrhythmic events may be as high as 60% over a follow-up period of 8 years [8,21,24–26,30]. An ICD is indicated in such patients who con-tinue to have ventricular arrhythmias despite pharmacotherapy. However, ICDs are not fully protective and can be proarrhythmic in CPVT patients because both appropriate and inappro-priate ICD shocks can trigger catecholamine release, resulting in multiple shocks (arrhythmic storm), and death [53].
Current guidelines suggest that implantation of an ICD with use of b-blockers is indicated for patients with CPVT who are survivors of cardiac arrest [54]. Implantation of an ICD with use of pharmacotherapy can be effective for CPVT patients with syncope and/or docu-mented sustained VT while receiving b-block-ers (class IIa recommendation). Currently there is no guideline indication for an ICD in the asymptomatic patient [54]. Furthermore, there are no reliable genotype or phenotype prognosticators to identify a high-risk patient.
However, clinical circumstance may arise where patients and clinician may decide on a primary prevention ICD, based on patient and family expectations of managing the risks associated with CPVT compared with effi-cacy of pharmacological options alone. Prior to implanting ICDs in young asymptomatic patients with CPVT, clinicians should discuss the potential limitations, including inappropri-ate shocks (with the potential for catecholamine release), the need for ICD generator replace-ment and lead revision with possible compli-cation, and alternative therapeutic options, including the potential efficacy of combined b-blocker and flecainide therapy and primary
Review Obeyesekere, Sy, Leong-Sit et al.

www.futuremedicine.com 447future science group
left cardiac sympathetic denervation combined with b-blocker and flecainide therapy.
ConclusionCPVT is a potentially malignant inherited arrhythmia syndrome that can present with syncope, palpitations, cardiac arrest or sudden death. Asymptomatic CPVT is not uncom-mon when family screening of the proband is performed.
Identifying a disease-causing mutation is important to confirm the diagnosis, facilitate counseling and direct therapy in the proband. Additionally cascade genetic and/or provocative
testing enables identification of asymptomatic family members to initiate prophylactic therapy because such patients are at risk of cardiac events.
Although rigorous trials are lacking, prophy-lactic treatment of the asymptomatic CPVT patient appears to reduce morbidity and mor-tality. Lifestyle modification and drug therapy is indicated based on the diagnosis of CPVT, even in the absence of symptoms. Although evi-dence is lacking for the safety and efficacy of primary prevention ICDs and primary preven-tion left cardiac sympathectomy, well-informed patients and clinicians may decide on these therapeutic options based on patient and family
Executive summary
Mechanism of catecholaminergic polymorphic ventricular tachycardia n Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare genetic disorder caused by mutations in genes involved in the
calcium homeostasis of cardiac cells (autosomal dominant form caused by RYR2 and autosomal recessive form caused by CASQ2 mutations). Arrhythmia in CPVT is induced by sympathetic stimulation.
Diagnosing CPVT in the index case & asymptomatic relatives n Investigation of the index case should take into account the patient’s clinical and family history, ECG findings at rest and during
provocative testing. Genetic testing for CPVT in the index case is indicated when a clinical index of suspicion exists. Mutation-specific genetic testing is recommended for relatives following the identification of a causative mutation. Relatives of a mutation-negative CPVT index case should undergo clinical assessment that includes provocative testing.
Natural history of CPVT n Sudden cardiac death is the first presentation in up to 30% of cases. CPVT-related symptoms generally occur early in life and may be as
high as 35 and 70% by the age of 10 and 20 years, respectively. Mortality is high when untreated, reaching between 30 and 50% by 30 years of age.
Prognostic features n There are no reliable genotype or phenotype prognosticators to guide patient management.
Treatment of CPVT n Management strategies for CPVT include lifestyle, medical and surgical interventions to suppress the release and/or effects of
catecholamines at the myocardial level, or correcting the abnormal calcium regulation. Implantable cardioverter defibrillators (ICDs) seek to treat the ventricular arrhythmia when the aforementioned measures fail.
Lifestyle measures n All symptomatic patients should avoid intensive sports/exercise. Asymptomatic patients may undertake low-intensity exercise.
b-blockers n b-blockers should be prescribed in every CPVT patient (diagnosed by a positive genetic test and/or a positive exercise/infusion test)
regardless of the presence or absence of symptoms. b-blockers are not completely effective in preventing life-threatening arrhythmias.
Calcium channel blockers n Evidence supporting the efficacy of calcium channel blockers in CPVT is lacking at present.
Flecainide n Flecainide, in addition to b-blocker therapy, should be considered for CPVT patients who otherwise have few alternative therapeutic
options. Flecainide may be utilized in conjunction with b-blockers as first-line therapy in asymptomatic cases.
Left cardiac sympathetic denervation n Left cardiac sympathetic denervation is an effective and safe therapeutic option when symptoms persist despite pharmacological
therapy. Left cardiac sympathetic denervation does not preclude implantation of an ICD in patients experiencing ventricular tachycardia despite optimal tolerated pharmacotherapy. When appropriate, the clinical strategy is to use the ICD as a safety net together with left cardiac sympathetic denervation, in conjunction with b-blockers and flecainide, to minimize the risk of life-threatening arrhythmias.
Implantable cardioverter defibrillator n An ICD is indicated in patients who continue to have ventricular arrhythmias despite pharmacotherapy. However, ICDs are not fully
protective and can be proarrhythmic because both appropriate and inappropriate ICD shocks can trigger catecholamine release, resulting in arrhythmic storm and death.
Treatment of asymptomatic catecholaminergic polymorphic ventricular tachycardia Review

Future Cardiol. (2012) 8(3)448 future science group
ReferencesPapers of special note have been highlighted as:n of interestnn of considerable interest
1. Sy RW, Gollob MH, Klein GJ et al. Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 8, 864–871 (2011).
n Characterizes arrythmia associated with provocative testing and presents data on presentation and long-term outcome.
2. Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca2+ regulation. Heart Rhythm 6, 1652–1659 (2009).
3. Tester DJ, Kopplin LJ, Will ML, Ackerman MJ. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm 2, 1099–1105 (2005).
4. Reid DS, Tynan M, Braidwood L, Fitzgerald GR. Bidirectional tachycardia in a child. A study using His bundle electrography. Br. Heart J. 37, 339–344 (1975).
5. Swan H, Piippo K, Viitasalo M et al. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J. Am. Coll. Cardiol. 34, 2035–2042 (1999).
6. Priori SG, Napolitano C, Tiso N et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200 (2001).
7. Lahat H, Pras E, Olender T et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 69, 1378–1384 (2001).
8. Priori SG, Napolitano C, Memmi M et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106, 69–74 (2002).
9. Laitinen PJ, Brown KM, Piippo K et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103, 485–490 (2001).
10. Postma AV, Denjoy I, Hoorntje TM et al. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 91, e21–e26 (2002).
11. Ackerman MJ, Priori SG, Willems S et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 8, 1308–1339 (2011).
12. Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 108, 871–883 (2011).
13. Wehrens XH. The molecular basis of catecholaminergic polymorphic ventricular tachycardia: what are the different hypotheses regarding mechanisms? Heart Rhythm 4, 794–797 (2007).
14. Liu N, Colombi B, Memmi M et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ. Res. 99, 292–298 (2006).
15. Tristani-Firouzi M, Jensen JL, Donaldson MR et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J. Clin. Invest. 110, 381–388 (2002).
16. Mohler PJ, Splawski I, Napolitano C et al. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl Acad. Sci. USA 101, 9137–9142 (2004).
17. Bhuiyan ZA, van den Berg MP, van Tintelen JP et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation 116, 1569–1576 (2007).
18. Krahn AD, Healey JS, Chauhan V et al. Systematic assessment of patients with unexplained cardiac arrest: Cardiac Arrest Survivors With Preserved Ejection Fraction Registry (CASPER). Circulation 120, 278–285 (2009).
n Utility of systematic testing for the diagnosis of sudden cardiac death.
19. Tan HL, Hofman N, van Langen IM, van der Wal AC, Wilde AA. Sudden unexplained death: heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 112, 207–213 (2005).
20. Allouis M, Probst V, Jaafar P, Schott JJ, Le Marec H. Unusual clinical presentation in a family with catecholaminergic polymorphic ventricular tachycardia due to a G14876A ryanodine receptor gene mutation. Am. J. Cardiol. 95, 700–702 (2005).
expectations of managing risk associated with CPVT compared with the risk associated with these therapeutic options.
Future perspectiveThe published literature is lacking in publica-tions addressing the role of primary preven-tion ICDs, primary prevention left cardiac sympathectomy and the primary prevention efficacy of combined b-blocker and flecainide therapy. Therefore, future studies should address whether flecainide may be utilized in conjunc-tion with b-blockers as first-line therapy in asymptomatic cases, and whether flecainide is efficacious in b-blocker-intolerant patients. The role of ICDs have not been systematically evalu-ated in patients as a primary prevention strategy and reports of the utility of ICDs as a secondary prevention strategy is also sparse. More studies
are needed to document the utility and potential limitations of ICDs in patients with CPVT. The effect of vagal stimulation might also have an effect on susceptibility to arrhythmia and should be evaluated. Novel and/or adjunctive agents to suppress the RYR2 receptor and its down-stream effects may be useful for arrhythmia suppression/prophylaxis in CPVT.
Financial & competing interests disclosureThe authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the sub-ject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Review Obeyesekere, Sy, Leong-Sit et al.

www.futuremedicine.com 449future science group
21. Sumitomo N, Harada K, Nagashima M et al. Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 89, 66–70 (2003).
22. Gopinathannair R, Olshansky B, Iannettoni M, Mazur A. Delayed maximal response to left cardiac sympathectomy for catecholaminergic polymorphic ventricular tachycardia. Europace 12, 1035–1039 (2010).
23. Cerrone M, Noujaim SF, Tolkacheva EG et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 101, 1039–1048 (2007).
24. Postma AV, Denjoy I, Kamblock J et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 42, 863–870 (2005).
25. Hayashi M, Denjoy I, Extramiana F et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation 119, 2426–2434 (2009).
nn Largest clinical study of patients with catecholaminergic polymorphic ventricular tachycardia discussing b-blocker efficacy and arrythmic incidence.
26. Bauce B, Rampazzo A, Basso C et al. Screening for ryanodine receptor type 2 mutations in families with effort-induced polymorphic ventricular arrhythmias and sudden death: early diagnosis of asymptomatic carriers. J. Am. Coll. Cardiol. 40, 341–349 (2002).
27. Bai R, Napolitano C, Bloise R, Monteforte N, Priori SG. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ. Arrhythm Electrophysiol. 2, 6–15 (2009).
28. Medeiros-Domingo A, Bhuiyan ZA, Tester DJ et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J. Am. Coll. Cardiol. 54, 2065–2074 (2009).
29. George CH, Jundi H, Thomas NL, Fry DL, Lai FA. Ryanodine receptors and ventricular arrhythmias: emerging trends in mutations, mechanisms and therapies. J. Mol. Cell Cardiol. 42, 34–50 (2007).
30. Haugaa KH, Leren IS, Berge KE et al. High prevalence of exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia mutation-positive family
members diagnosed by cascade genetic screening. Europace 12, 417–423 (2010).
n Highlights the usefulness of cascade screening.
31. Hofman N, Tan HL, Alders M, van Langen IM, Wilde AA. Active cascade screening in primary inherited arrhythmia syndromes: does it lead to prophylactic treatment? J. Am. Coll. Cardiol. 55, 2570–2576 (2010).
32. Roux-Buisson N, Egea G, Denjoy I, Guicheney P, Lunardi J. Germline and somatic mosaicism for a mutation of the ryanodine receptor type 2 gene: implication for genetic counselling and patient caring. Europace 13, 130–132 (2011).
33. Zlotogora J. Germ line mosaicism. Hum. Genet. 102, 381–386 (1998).
34. Tester DJ, Arya P, Will M et al. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm 3, 800–805 (2006).
35. Zipes DP, Camm AJ, Borggrefe M et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 114, e385–e484 (2006).
36. Werf C, Zwinderman AH, Wilde AA. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: state of the art and future developments. Europace 14, 175–183 (2012).
37. van der Werf C, Kannankeril PJ, Sacher F et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol. 57, 2244–2254 (2011).
nn First clinical multicenter study evaluating the efficacy of flecainide.
38. Rosso R, Kalman JM, Rogowski O et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 4, 1149–1154 (2007).
39. Swan H, Laitinen P, Kontula K, Toivonen L. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in
catecholaminergic polymorphic ventricular tachycardia patients with RYR2 mutations. J. Cardiovasc. Electrophysiol. 16, 162–166 (2005).
40. Rosso R, Kalman J, Rogowsky O et al. Long-term effectiveness of beta blocker and calcium channel blocker combination therapy in patinets with CPVT. Heart Rhythm 7, S423 (2010).
41. Watanabe H, Chopra N, Laver D et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat. Med. 15, 380–383 (2009).
42. Hilliard FA, Steele DS, Laver D et al. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J. Mol. Cell Cardiol. 48, 293–301 (2010).
43. Echt DS, Liebson PR, Mitchell LB et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression trial. N. Engl. J. Med. 324, 781–788 (1991).
44. Wilde AA, Bhuiyan ZA, Crotti L et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N. Engl. J. Med. 358, 2024–2029 (2008).
n Case series and discussion of left cardiac sympathetic denervation.
45. Odero A, Bozzani A, De Ferrari GM, Schwartz PJ. Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias: the surgical supraclavicular approach to cervicothoracic sympathectomy. Heart Rhythm 7, 1161–1165 (2010).
46. Makanjee B, Gollob MH, Klein GJ, Krahn AD. Ten-year follow-up of cardiac sympathectomy in a young woman with catecholaminergic polymorphic ventricular tachycardia and an implantable cardioverter defibrillator. J. Cardiovasc. Electrophysiol. 20, 1167–1169 (2009).
47. Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm 6, 752–759 (2009).
48. Schwartz PJ. The rationale and the role of left stellectomy for the prevention of malignant arrhythmias. Ann. NY Acad. Sci. 427, 199–221 (1984).
49. Schwartz PJ, Snebold NG, Brown AM. Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am. J. Cardiol. 37, 1034–1040 (1976).
Treatment of asymptomatic catecholaminergic polymorphic ventricular tachycardia Review

Future Cardiol. (2012) 8(3)450 future science group
50. Schwartz PJ, Verrier RL, Lown B. Effect of stellectomy and vagotomy on ventricular refractoriness in dogs. Circ. Res. 40, 536–540 (1977).
51. Harris KM BJ, Johnson JJ, Ackerma MJ. Abstract: analysis of failure: phenotype of patients with long QT syndrome and breakthrough events following left cardiac sympathetic denervation. Heart Rhythm 8, S38 (2011).
52. Moray A, Kirk EP, Grant P, Camphausen C. Prophylactic left thoracic sympathectomy to prevent electrical storms in CPVT patients needing ICD placement. Heart Lung Circ. 20, 731–733 (2011).
53. Mohamed U, Gollob MH, Gow RM, Krahn AD. Sudden cardiac death despite an implantable cardioverter-defibrillator in a young female with catecholaminergic ventricular tachycardia. Heart Rhythm 3, 1486–1489 (2006).
54. Zipes DP, Camm AJ, Borggrefe M et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With
Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J. Am. Coll. Cardiol. 48, e247–e346 (2006).
55. Ackerman MJ, Priori SG, Willems S et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 13, 1077–1109 (2011).
Website101. Genetic mutations and inherited arrhythmias.
www.fsm.it/cardmoc
Review Obeyesekere, Sy, Leong-Sit et al.
















![Catecholaminergic polymorphic ventricular tachycardia ... · ECG findings during exercise or emotional stress.[1,5] In this article, we report a case of CPVT detected by an ILR in](https://img.pdfslide.net/doc/110x75/5f02db0d7e708231d40658dd/catecholaminergic-polymorphic-ventricular-tachycardia-ecg-findings-during-exercise.jpg)












