Embed Size (px)
Citation preview

145
REACTIVOS NUCLEÓFILOS Y ELECTRÓFILOS
Un reactivo nucleófilo es un anión o una molécula con pares de electrones libres, que se une a un átomo de carbono con densidad electrónica baja (base de Lewis):
:Cl
::
_H O
::
_: : R O
::
_: N C :
_H2O
:: NH3:
Un reactivo electrófilo es un catión que se une a un par de electrones de un átomo de carbono con densidad electrónica alta (ácido de Lewis):
H3O:+ ++
NO2 Br RCH2+
Ejemplo de reacción nucleófila (el reactivo que provoca la reacción es un nucleófilo):
NCK + C6H5CH2_Br C6H5CH2
_CN + KBr
N C:_
CH2 BrC6H5
N C CH2
C6H5
+: Br: :_
Ejemplo de reacción electrófila (el reactivo que provoca la reacción es un electrófilo):
+ HO3NH2SO4
NO2H
HNO3 + H2SO4 NO2+
+ SO4H_
+ H2O
H
NO2+ NO2H
+SO4H
_ NO2
+ H2SO4
TIPOS DE REACCIONES ORGÁNICAS REACCIONES DE SUSTITUCIÓN REACCIONES DE ELIMINACIÓN REACCIONES DE ADICIÓN En las reacciones de sustitución y adición, el reactivo que interviene puede ser electrófilo o nuclófilo:
El reactivo es electrófilo: SUSTITUCIÓN ELECTRÓFILA REACCIONES DE SUSTITUCIÓN El reactivo es nucleófilo: SUSTITUCIÓN NUCLEÓFILA
El reactivo es electrófilo: ADICIÓN ELECTRÓFILA REACCIONES DE ADICIÓN El reactivo es nucleófilo: ADICIÓN NUCLEÓFILA
VI

146
Por otra parte, en las reacciones orgánicas intervienen átomos de carbono saturados o átomos de carbono no saturados. Existen seis tipos básicos de procesos:
Sustitución nucleófila en un átomo de carbono no saturado
Sustitución nucleófila en un átomo de carbono saturado
Sustitución electrófila en un átomo de carbono no saturado
Adición nucleófila a un átomo de carbono no saturado
Adición electrófila a un átomo de carbono no saturado
Reacciones de eliminación en un átomo de carbono saturado o no saturado
REACCIONES DE SUSTITUCIÓN NUCLEÓFILA EN UN ÁTOMO DE CARBONO NO SATURADO
C
O
Z
RNu
_C
O
Z
R
Nu + _
Los grupos salientes Z más comunes son: Cl−, R-CO-O−, HO−, RO−, NH2
−, RNH−. Es decir, son reacciones en las que están implicados los siguientes compuestos: cloruros de ácido, anhidridos de ácido, ácidos, ésteres y amidas.
eliminación C
O
CH3 Cl_
+
Cloruro de acetiloÁcido acético
C ClCH3
O
HO_
:adición C Cl
CH3
O:
HO
:_
HO
: :
eliminaciónC
O
CH3 Cl_
+
Cloruro de acetilo
C ClCH3
O:adición C Cl
CH3
O:
H3N
:_
H3N
: :H3N: + +
acetamida
+H NH2 C
O
CH3B: H2N C
O
CH3 + B H+
REACCIONES DE SUSTITUCIÓN NUCLEÓFILA EN UN ÁTOMO DE CARBONO SATURADO
Nu_
CH2____Z
R
Nu___CH2__R + Z:
_
NC RO HO Br C6H5O R-CO2 R-OH H2O: R3N:_ _ _ _ _ _
Los reactivos nucleófilos más empleados son:
::
:
VI

147
Los sustratos (RCH2 ⎯ Z) más comunes en estas reacciones son los derivados halogenados (Z= Cl, Br, I) La reacción entre un haluro de alquilo y amoníaco o anión cianuro son dos ejemplos:
H3N: CH2
CH3
Cl H3N CH2
CH3
Cl+_+
Cloruro de etilo
_ +NH2 CH2H
CH3CH3 CH2 NH2 + H2O
EtilaminaHO
CH2 CH CH2 I + NC_
CH2 CH CH2 CN + I_
Yoduro de alilo
3-Yodo-1-propeno3-Butenonitrilo
REACCIONES DE SUSTITUCIÓN ELECTRÓFILA EN UN ÁTOMO DE CARBONO NO SATURADO El benceno y la mayoría los hidrocarburos bencenoides reaccionan con electrófilos (E+) dando lugar a hidrocarburos aromáticos en los que el reactivo electrófilo sustituye a un átomo de hidrógeno:
+ E+
E
+
H
H+
Los reactivos (E) implicados normalmente en las reacciones de sustitución electrófila son los siguientes:
+++E = R R C
O
NO2 SO3 X +
RCOClAlCl3
COR
+ HCl
CO
R_ Cl : AlCl3 CO
R_ Cl AlCl3_+
CO
R_ AlCl4_
+
+ +CO_RH
H Cl3Al Cl_
CO_R
+ HCl + AlCl3R CO
VI

148
REACCIONES DE ADICIÓN NUCLEÓFILA A GRUPOS CARBONILO
Los sustratos más comunes en este tipo de reacciones son los aldehidos y las cetonas:
δ+
δ−
δ+
δ−
μ = 2,73 D μ = 2,84 D
C
O
Me HC
O
Me Me
Los nucleófilos que se utilizan son, entre otros, los siguientes:
H R_OH R_NH2 NC R_C CNu_ _ _ _:
:
:
:= Esquema general de la reacción:
Nu
R
R
C O_ Nu C_
R'
R
O HB Nu C
R'
R
OH
Los aldehidos y las cetonas reaccionan, por ejemplo, con anión cianuro (NaCN o KCN), dando lugar a un ión alcóxido que en medio ácido se transforma en una cianhidrina:
R
RO_ C__R
O_ H___OH2
+
C__R
OH
+ H2O
R Rión alcóxido cianhidrina
N C N C N C
CH2CHO + CN_
CH2__CH__CN
O_
H3O+CH2
__CH__CN
OH
Fenilacetaldehido 2-Hidroxi-3-fenilpropanonitrilo
(67 %)
REACCIONES DE ADICIÓN ELECTRÓFILA A UN ÁTOMO DE CARBONO NO SATURADO En este tipo de reacciones, el enlace π de una olefina se transforma en dos enlaces σ:
+
_R CH CH R
ER C
HHC R
E
+Nu
R CH
CH
R
E Nu
VI

149
R CH CH2
H Brδ+ δ−
lenta R CH CH3+
+ Br_
[carbocatión secundario]
R CH CH3+
+ Br_ rápida
R CH CH3
Br
2-Bromopropano
REACCIONES DE ELIMINACIÓN EN UN ÁTOMO DE CARBONO SATURADO O NO SATURADO Las reacciones de eliminación son, esencialmente, lo contrario de las reacciones de adición electrófila: formación de un enlace π a partir de dos enlaces σ. El desencadenante de la reacción es una base (nucleófilo):
H CHR
CH
Br
RHO_
CHR
CH R + H2O Br+_
H CHCH3
CH
Br
CH3HO_
CHCH3
CH CH3 + H2O Br+_
H C
CH3
C
Br
CH3HO_
CCH3
C CH3 + H2O Br+_
EJERCICIOS ADICIONALES 6.1 Los aniones alcóxido, empleados en la síntesis de éteres, se obtienen a partir del alcohol correspondiente por reacción con un metal (Na o K) o una base. Explica si las siguientes reacciones son adecuadas para la obtención de los alcóxidos que se indican, calculando en cada caso la variación de energía libre estandar y la constante de equilibrio (utiliza la tabla de pKa que aparece en los apéndices)
1. CH3CH2OH + NaOH CH3CH2O Na+ + H2O
2. 2CH3OH + 2Na 2CH3O Na+ + H2
3. Me3C-OH + NH3(liq.) Me3CO NH4+
4. Me2CH-OH + Na+H Me2CHO Na+ + H2
_
_
_
__
6.2 Los carbaniones son intermedios de reacción muy útiles en la síntesis de compuestos orgánicos. Explica si las siguientes reacciones son adecuadas para la obtención de los carbaniones que se indican, calculando en cada caso la constante de equilibrio.
VI

150
H CO
CH2 CO
H + HO_
H CO
CH CO
H
:
_+ H2O1.
CH4 + CH3CH2O_
CH3 + CH3CH2OH:_
2.
CH3 CN + CH3O_
CH2 CN + CH3OH:_
3.
CH3 +NO2 NH2:
:
_
CH2 +NO2 NH3::_
4.
CH3 C CH2 CO2C2H5 + HCO3
_CH3 C CH CO2C2H5 + H2CO3
:
_5.
O O
6.3 Considera el siguiente equilibrio conformacional:
H
H
CH3(a)
CH3(e) mol/cal1700G0298 −=Δ
Apartado 1: Calcula la constante de equilibrio a 250 C y una atmósfera de presión. Apartado 2: Averigua los porcentajes molares de las dos conformaciones, una vez que el sistema alcanza el
equilibrio. Apartado 3: Explica brevemente (sin realizar ningún cálculo) qué sucederá si, una vez alcanzado el equilibrio a
250C y una atmósfera de presión, la temperatura se eleva hasta 1500C manteniendo la presión constante
VI

7. REACCIONES DE OLEFINAS Y ALQUINOS
ESTABILIDADES DE LOS ALQUENOS
REACCIONES DE HIDROGENACIÓN
CH3CH2 CH CH2H2
[Pt]CH3CH2CH2CH3 ΔH0 = -30.3 kcal/mol
Butano
C CH
Me Me
H
H2
[Pt]CH3CH2CH2CH3
Butano
ΔH0 = -28.6 kcal/mol
(Z)-2-Buteno
C CH
Me H
Me
H2
[Pt]CH3CH2CH2CH3
Butano
ΔH0 = -27.6 kcal/mol
(E)-2-Buteno El alqueno con el doble enlace al final de la cadena (1-buteno, alqueno terminal) es menos estable que cual-quiera de los dos alquenos con el doble enlace en el interior de la cadena (2-buteno) El alqueno (E) con los grupos más voluminosos del doble enlace alejados entre sí, es más estable que el al-queno (Z) en el que dichos grupos están próximos.
CH3CH CH CH2H2
[Pt]CH3CHCH2CH3 ΔH0 = -30.3 kcal/mol
2-Metilbutano
CH3 CH3
3-Metil-1-buteno
alqueno terminal conun sustituyente (i-Pr) en el doble enlace
CH3CH2 C CH2H2
[Pt]CH3CH2CHCH3 ΔH0 = -28.5 kcal/mol
2-Metilbutano2-Metil-1-buteno
alqueno terminal condos sustituyentes (Me,Et) en el doble enlace
CH3 CH3
H2
[Pt]CH3CH2CHCH3 ΔH0 = -26.9 kcal/mol
2-Metilbutano2-Metil-2-buteno
alqueno con tres sustituyentes (Me) en el doble enlace
CH3CH3CH C CH3
CH3
VII

152
Los alquenos con el doble enlace en el interior de la cadena son más estables que los alquenos terminales. La estabilidad de un alqueno aumenta con el número de grupos alquilo que están unidos a los átomos de carbono del doble enlace. Los alquenos con los sustituyentes más voluminosos en posición trans, son más estables que los correspondientes diastereoisómeros cis.
En los alquenos, los términos cis y trans se utilizan para describir la posición relativa de dos grupos cualquiera unidos a átomos distintos del doble enlace. La terminología (Z) / (E) hace referencia a la configuración absoluta del alqueno.
REACCIONES DE ADICIÓN ELECTRÓFILA
REACCIONES CON HALUROS DE HIDRÓGENO (HCl, HBr, HI) Las adiciones electrófilas a enlaces múltiples implican la conversión de un enlace π en dos enlaces σ. El átomo de hidrógeno es el electrófilo más común en estas reacciones. En general, este tipo de reacciones es energéti-camente favorable, es decir, son exotérmicas. Las olefinas sustituidas simétricamente sólo pueden dar lugar a un producto de reacción:
CH3 CH CH CH3HX CH3 CH
HCHX
CH3
C6H5 CH CH C6H5HX C6H5 CH
HCHX
C6H5 En general:
R1 C C R1 HXR2 R2
R1 C C R1R2 R2
XH Sin embargo, cuando el doble enlace del alqueno no tiene los mismos sustituyentes, existe la posibilidad de que se formen dos productos diferentes:
CH3 CH CH2
(CH3CH2CH2Cl ; 1-Cloropropano)
(CH3_CH_CH3 ; 2-Cloropropano)
Cl
HCl
CH3 CH CH2
Cl H
CH3 CH CH2
H Cl
La adición electrófila de haluros de hidrógeno al doble enlace de una olefina transcurre a través de carbocatio-nes (Tema 6): Primer paso: reacción del alqueno con el protón procedente de H-X:
CH3 CH CH2
H Brδ+ δ−
lenta CH3 CH CH3+
+ Br_
[carbocatión secundario]
CH3 CH CH2
H Brδ+ δ−
lenta CH3 CH2 CH2+
+ Br_
[carbocatión primario]
VII

153
Segundo paso: reacción del catión carbonio con el anión bromuro (nucleófilo):
CH3 CH CH3+
+ Br_
[carbocatión secundario]
rápidaCH3 CH CH3
Br
2-Bromopropano
CH3 CH2 CH2+
+ Br_
[carbocatión primario]
rápidaCH3 CH2 CH2Br
1-Bromopropano
EJERCICIO 7.1La reacción del propeno con HBr da lugar únicamente a bromuro de isopropilo.
1. ¿Cuál de los dos equilibrios estará más desplazado hacia la derecha? :
2. Calcula las constantes de equilibrio correspondientes a las dos reacciones.
3. Calcula los porcentajes de 1-bromopropano y 2-bromopropano una vez alcanzado el equilibrio.
4. ¿Por qué no se obtiene nada de 1-bromopropano?
Datos: ΔG(1-propeno)= +14,99 kcal/mol; ΔG(1-bromopropano)= 5,37 kcal/mol
ΔG(2-bromopropano)= 6,51 kcal/mol; ΔG(1-bromopropano)= 12,73 kcal/mol
(g) + HBr (g) CH3 CH
CH3 (g)Br
CH3 CH CH2 (g) + HBr (g) (g)CH3 CH2 CH2Br
CH3 CH CH2
_
_ _
En la reacción entre propeno y HBr, el 2-bromopropano se forma más rápidamente que el 1-bromopropano (ver la respuesta del Ejercicio 1) ¿Por qué?
CH3 CH CH2
lenta CH3 CH CH3+
+ Br_
[carbocatión secundario]
lentaCH3 CH2 CH2
++ Br
_
[carbocatión primario]
HBr
rápidaCH3 CH CH3
Br
2-Bromopropano
rápidaCH3 CH2 CH2Br
1-Bromopropano La velocidad total de reacción es proporcional a la energía libre de activación del paso más lento que, en este caso, corresponde a la formación de los dos carbocationes (ecuación de Eyring, Tema 6) El carbocatión iso-propilo (secundario) es más estable que el catión propilo (primario) y la energía de activación necesaria para su formación (ΔG1
≠) es bastante menor que la correspondiente al primario (ΔG2≠) Por consiguiente, el carbocatión
secundario se forma más rápidamente que el primario y esto se refleja en el resultado final: no se obtiene nada de 1-bromopropano, que proviene del carbocatión primario. La reacción está sometida a control cinético.
VII

154
ΔG1
ΔG2
ΔG
CH3 CHBr
CH3
CH3 CH CH2
HBr
CH3CH2CH2
Br
CH3CHCH3
Br
CH3CH2CH2Br
curso
+
+
+ _
+
+ _
-7.63 kcal/mol
-8.77 kcal/mol
≅ 16 kcal/mol
≅ 1 kcal/mol
REGIOSELECTIVIDAD EN LA ADICIÓN DE ÁCIDOS PRÓTICOS A OLEFINAS. REGLA DE MARKOVNIKOV En este caso, el término regioselectividad hace referencia al hecho de que el reactivo electrófilo (H+) se une preferentemente al átomo de carbono del doble enlace que tiene más hidrógenos. O dicho de otro modo: el reactivo electrófilo se une al doble enlace dando lugar con preferencia al carbocatión más estable.
R1 C
R2
CH2
12
R1 C
R2
CH3+
[terciario]
R1 C
R2
CH3
Br
R1 CH
R2
CH2+
[primario]
Br_
(producto mayoritario)
Br_
R1 CH
R2
CH2Br
HBr
1
2(producto minoritario)
VII

155
R1 C
R2
CH
12
R1 C
R2
CH2+
[terciario]
R1 C
R2
CH2
Br
R1 CH
R2
CH+
Br_
(producto mayoritario)
Br_
R1 CH
R2
CH
HBr
1
2(producto minoritario)
R3
R3
R3
R3
BrR3
[secundario] Las olefinas que no tienen átomos de hidrógeno unidos a los carbonos del doble enlace, dan lugar siempre a dos carbocationes terciarios:
R1 C
R2
C
12
R1 C
R2
CH+
[terciario]
R1 C
R2
CH
X
R1 CH
R2
C+
X_
(1)
X_
R1 CH
R2
C
HX
1
2(2)
R4
R4
R4
R4
R3
R4
R3
R3 R3
[terciario]
R3
Br
En una situación así, resulta muy difícil (si no imposible) predecir cuál de los dos productos obtenidos (1) o (2) será el mayoritario. Lo habitual es que se obtenga una mezcla que contiene aproximadamente el 50 % de (1) y el 50 % de (2)
REACCIONES DE TRANSPOSICIÓN EN CARBOCATIONES Hay ocasiones en las que una reacción de adición electrófila da lugar a productos inesperados, en los que la estructura de la molécula inicial cambia de forma sustancial (Tema 6) Por ejemplo:
CH3 CHCH3
CH CH2HCl
250CCH3 C
CH3CH2 CH3
Cl+ CH3 CH
CH3CHCl
CH3
2-Cloro-2-metilbutano 2-Cloro-3-metilbutano
50 % 50 % En principio, no debería obtenerse 2-cloro-2-metilbutano:
_
[primario]
12
CH3 CHCH3
CH CH2HCl
250C
+
[secundario]
CH3 CHCH3
CH CH3
CH3 CHCH3
CH2 CH2+
1
2
Cl
_Cl
CH3 CHCH3
CH CH3
Cl
CH3 CHCH3
CH2 CH2Cl
2-Cloro-3-metilbutano
1- Cloro-3-metilbutano
(no se obtiene)
VII

156
¿De dónde procede el 2-cloro-2-metilbutano?
CH3 CHCH3
CH CH2
H Cl+
[secundario]
CH3 CHCH3
CH CH3
En el carbocatión secundario tiene lugar una transposición de anión hidruro (H:−):
+CH3 CCH3
CH CH3
H
transposición
+CH3 C
CH3
CHH
CH3
[secundario]
_Cl
[terciario; más estable]
CH3 CCH3
CH2 CH3
Cl
2-Cloro-2-metilbutano En estas reacciones de transposición pueden estar implicados, además del anión hidruro, grupos alquilo. El ejemplo siguiente es un “anión” metilo el que se desplaza:
CH3 CCH3
CH3
CH CH2HCl
-780CCH3 C
CH3
ClCHCH3
CH3 + CH3 CCH3
CH3
CHCl
CH3
3,3-Dimetil-1-buteno 2-Cloro-2,3-dimetilbutano 2-Cloro-3,3-dimetilbutano
61 % 37 %
EJERCICIO 7.2Escribe un mecanismo de la reacción anterior que permita explicar la formación de los dos haluros de alquilo.
ESTEREOQUÍMICA DE LAS REACCIONES QUE TRANSCURREN A TRAVÉS DE CARBOCATIONES Ya se ha dicho que los carbocationes tienen geometría plana (Tema 6) Este hecho da lugar a la aparición de mezclas racémicas en muchas reacciones de adición electrófila.
C6H5 CH CH2HBr C6H5 CH
BrCH3
* mezcla de dosestereoisómeros
C6H5 CH CH2
H BrCH CH3+
**
*
*
carbocatiónestabilizado
C6H5 CH3
H+
VII

157
C6H5CH3
H
C6H5
Br
CH3H
C6H5
Br
CH3
H
+
1
2
Br_
1
2
Br
C6H5 CH3
H(S)
H
C6H5 CH3
Br(R)
C6H5 CH CH2HBr
EJERCICIO 7.3 Escribe el mecanismo de la reacción que tiene lugar entre (Z)-3,4-dimetil-3-hexeno y HCl. Utiliza representa-ciones tridimensionales y comprueba que se obtiene una mezcla equimolecular de los cuatro estereoisómeros posibles del 3-cloro-3,4-dimetilhexano.
REACCIÓN CON AGUA EN MEDIO ÁCIDO FUERTE
CH3 CCH3
CCH3
CH3H2SO4
H2OCH3 CH
CH3
CCH3
CH3
OH
2,3-Dimetil-2-butanol2,3-Dimetil-2-buteno
H2O: H O S OHO
OH3O + O SO3H:
_+
CH3 CCH3
CCH3
CH3
H2O H+
CH3 CCH3
CCH3
CH3++ H2O:
H
CH3 CCH3
CCH3
CH3+:
HOH2
CH3 CCH3
CCH3
CH3H OH2:
+
:+CH3 CH
CH3CCH3
CH3OH
H :
CH3 CHCH3
CCH3
CH3OH
2,3-Dimetil-2-butanol
+ H3O+
OH2 El carbocatión que se forma, además de reaccionar con una molécula de agua, puede hacerlo con el anión hidrogenosulfato presente en el medio:
CH3 CCH3
CCH3
CH3+
:H
O
CH3 CCH3
CCH3
CH3
H OSO3H
_ SO3H
hidrogenosulfato de alquilo
éster de un alcohol y ácido sulfúrico
VII

158
Estos hidrogenosulfatos de alquilo reaccionan con agua dando lugar al mismo alcohol:
CH3 CHCH3
CCH3
CH3OH
2,3-Dimetil-2-butanol
CH3 CCH3
CCH3
OH CH3
hidrogenosulfato de alquilo
S OHO
O+ H OH + HO S OH
O
O
EJERCICIO 7.4Averigua que olefinas se deben utilizar para obtener los siguientes alcoholes: alcohol terc-butílico, 1-feniletanol,1-metilciclobutanol y 2-metil-2-butanol.
La reacción de olefinas cíclicas, con haluros de hidrógeno o con agua en medio ácido fuerte, transcurre tam-bién a través de carbocationes:
MeHCl
MeCl
Me
HBr
EtH EtH
Me
Br
MeH2O
Me
OH
HMe
H2SO4
HMe
EJERCICIO 7.5Escribe el mecanismo de las tres reacciones anteriores utilizando representaciones tridimensionales y averigua la configuración absoluta de los estereoisómeros que se obtienen.
REACCIONES DE LOS CARBOCATIONES CON ALQUENOS. POLIMERIZACIÓN CATIÓNICA DE OLEFINAS Los carbocationes son reactivos electrófilos, capaces de reaccionar con las olefinas:
CH3 CCH3
CH2 + H B+
CH3 CCH3
CH3++ :B
VII

159
CH3 CCH3
CH2 CCH3
CH3CH3
CH3 CCH3
CH2 C CH3
CH3
CH3+
catión dímero El proceso puede continuar indefinidamente, mientras existan moléculas de alqueno en el medio de reacción:
CH3 CCH3
CH2 C CH2
CH3
CH3+
catión trímero
CH3 CCH3
CH2CH3
CCH3
CH2 C CH3
CH3
CH3
catión dímero
CCH3
CH3CH3
CH3
CCH3
CH2 C CH2
CH3
CH3
+
catión trímero
CCH3
CH3CH3
CH3 CCH3
CH2 CH3 CCH3
CH2 C CH2
CH3
CH3+
catión tetrámero
CCH3
CH2CH3
CCH3
CH3CH3
La reacción se detiene cuando la base (:B) sustrae un protón del átomo contiguo al carbono con carga:
CH3 CCH3
CH C CH2
CH3
CH3+
CCH3
CH2CH3
CCH3
CH3CH3
HB:
CH3 CCH3
CH C CH2
CH3
CH3
CCH3
CH2CH3
CCH3
CH3CH3
+ H B+
Si se permite que la reacción avance, se obtiene un polímero (“plástico”) es decir, un compuesto de peso mole-cular elevado en el que se repite la estructura carbonada de la olefina de partida:
C
CH3
CH2 H B
polimerizaciónC
CH3
CH CCH3 CH3
CH3
CH3
CH2
n
polímero del metilpropeno
EJERCICIO 7.6Escribe la reacción de polimerización del etileno cuando se emplea un catalizador ácido, p.ej. H2SO4.
REACCIÓN DE LOS ALQUENOS CON HALÓGENOS Las olefinas reaccionan con cloro o bromo dando lugar a derivados dihalogenados:
CH3 CH CH CH3Cl2 CH3 CH
ClCHCl
CH3
2-Buteno 2,3-Diclorobutano
VII

160
CH2 CH2 + Br2NaCl / H2O
disolución saturada
BrCH2CH2Br + BrCH2CH2Cl
1,2-Dibromoetano 1-Bromo-2-cloroetano54 % 46 %
CH2 CH2 + Br2 BrCH2CH2Br + BrCH2CH2OH
1,2-Dibromoetano 2-Bromoetanol37 % 54 %
H2O
00C
Cuando se hace reaccionar el (E)-2-buteno con bromo se obtiene únicamente la forma meso del 1,2-dibromobutano:
H Me
Me H
(E)-2-Buteno
Br2
Br
BrH
Me
Me
H Br Br
H HMe Me
(2R,3S)
2 3
Si la reacción se lleva a cabo con el (Z)-2-buteno se obtiene una mezcla equimolecular de dos enantiómeros (racémico):
Me Me
H H
(Z)-2-Buteno
Br2
Br
BrMe
Me
H
H+
Br
Br Me
Me
H
H
(2S,3S) (2R,3R)
2 3 2 3
EJERCICIO 7.7Comprueba que si las dos reacciones anteriores transcurrieran a través de carbocationes, se obtendrían los mismos productos partiendo del estereoisómero (Z) o del (E)
Para poder explicar la estereoquímica de estas reacciones se planteó la siguiente hipótesis: la reacción con la olefina debe dar lugar a un catión en el que el halógeno está unido simultáneamente a los dos átomos de carbono del doble enlace (catión halogenonio):
R CH CH R'
X
X
:
::
:
1 2
3
R C C R'
X
H H
: :+
X ::_
(catión halogenonio) El catión halogenonio reacciona posteriormente con el anión del halógeno (nucleófilo) por el lado menos impe-dido estéricamente (adición anti):
R R'HH
X: :+
X::_
R
R'X
XH
H
(1)
VII

161
R R'HH
X: :+
X::_
R'
R X
X H
H
(2) Se obtiene una mezcla de dos enantiómeros: (1) y (2) Veamos si la hipótesis del catión halogenonio, permite dar cuenta de los resultados que se obtienen en la reac-ción del bromo con los dos estereoisómeros del 2-buteno: (Z)-2-buteno:
2 3
_
Me Me
HH Br2
Me Me
HHBr+
Me
Me Br
Br H
H
Me
MeBr
BrH
H
(1) (2S,3S)
Br
(2) (2R,3R)
HBr Me
H
Me Br
2
3
HBrMe
H
MeBr
2
3
32
1
2
1 2
Se obtiene una mezcla equimolecular (racémico) de la pareja de enantiómeros del 2,3-dibromobutano. (E)-2-buteno:
_
Me H
MeH Br2
Me H
MeHBr+
H
Me Br
Br Me
H
Me
HBr
BrH
Me
(1) (2S,3R)
Br
(2) (2R,3S)
HBr Me
Me
H Br
2
3
HBrMe
Me
HBr
2
3
2 3
2 3
1 2
1
2
Ahora se obtiene únicamente la forma meso: (1) = (2)
Si la reacción de una molécula da lugar a un solo estereoisómero, se dice que la reacción es estereoespecífica.Cuando el sustrato conduce a una mezcla de estereoisómeros en la que predomina uno de ellos, la reacciónrecibe el nombre de estereoselectiva.
La reacción del (E)-2-buteno con bromo es estereoespecífica, ya que se obtiene un solo estereoisómero: la forma meso.
VII

162
EJERCICIO 7.8Para averiguar qué estereoisómeros se forman al reaccionar cualquier olefina con cloro o bromo, sólo es preciso trabajar con uno de los dos cationes halogenonio posibles: (1) o (2)
R H
R'H X2
R H
R'HX+
R H
R'H
X+
o
(1)
(2)
Comprueba que esto es cierto utilizando el catión bromonio (2) del (E)-2-buteno.
REACCIÓN DE LOS ALQUENOS CON ÁCIDOS HIPOHALOSOS (HOCl Y HOBr)
Los ácidos hipohalosos se obtienen haciendo reaccionar sus sales sódicas con un ácido fuerte disuelto en agua:
NaOCl + H3O+ HOCl + Na+ + H2O
hipoclorito sódico
ácidohipocloroso
δ+ δ−
En los ácidos hipohalosos el enlace que une el oxígeno y el halógeno está polarizado: HO X La baja densidad electrónica sobre el átomo de halógeno hace que se comporte como un electrófilo potencial. El resultado de la interacción entre el par de electrones del doble enlace y el halógeno electrófilo, conduce también a un catión halogenonio, igual que en la reacción de halógenos con una olefina:
C C
OH
X ::
:
1 2
3
(catión halogenonio)
R R
HH
(Z)
C C
X::
R R
HH
+
+ HO_
(nucleófilo)
Ahora, el catión halogenonio reacciona en anti con el anión hidroxilo. Si la olefina tiene los mismos sustituyen-tes en los dos carbonos del doble enlace, el catión halogenonio que se forma es simétrico. En una situación como ésta, la reacción posterior con el nucleófilo tiene la misma probabilidad de ocurrir por cualquiera de los dos carbonos.
_
R R
HHX+
R
R OH
X H
H
R
RHO
XH
H
(1)
HO
(2)
1
2
1 2
(Z)
(halogenohidrina)
(halogenohidrina)
En este caso se obtiene un racémico de las halogenohidrinas (1) y (2)
VII

163
R R
HH X-OHR R
HHX+
R R
HH
X+
(Z)
1 2
1 2
HO_
....
HO_
....
EJERCICIO 7.9Dibuja los estereoisómeros que se obtienen en las siguientes reacciones:
R H
RH X-OHR H
RHX+
R H
RH
X+
(E)
1 2
1 2
HO_
....
HO_
....
(dos estereoisómeros)
(dos estereoisómeros)
(dos estereoisómeros)
(dos estereoisómeros)
Cuando los sustituyentes son distintos, en particular si uno de los carbonos del doble enlace tiene unidos dos átomos de hidrógeno, el catión halogenonio deja de ser simétrico:
R H
HR X-OHR H
HRX+
1 2 Ahora, la reacción por 1 o 2 con el nucleófilo (HO−) no tiene lugar con la misma facilidad. Veamos dos ejemplos concretos:
CH3CH2 CCH3
CH2Cl-OHH2O CH3CH2 C
CH3CH2Cl
OH
2-Metil-1-buteno 1-Cloro-2-metil-2-butanol
C6H5 CH CH2Cl2, H2ONa2CO3
C6H5 CHOH
CH2Cl
Feniletileno (Estireno)
2-Cloro-1-feniletanol
72 % El átomo de halógeno se une al carbono con más hidrógenos (el menos sustituido) y el grupo HO− al carbono más sustituido. ¿Por qué la reacción tiene una regioselectividad de tipo Markovnikov, cuando no existe ningún carbocatión como intermedio?
VII

164
Supongamos que el catión halogenonio no simétrico reacciona con el nucleófilo por el átomo de carbono al que están unidos los dos hidrógenos:
R H
HRX+
1
HO_
R H
HRX
OH
δ+
δ−
carbocatión incipiente
H
R OH
X H
R
(minoritario)
La aproximación del HO− al átomo de carbono debilita el enlace C-X, que comienza a romperse. Este hecho da lugar a la aparición de un carbocatión primario incipiente, que no llega a formarse porque el nucleófilo (HO−) se une a él. Si por el contrario, la reacción transcurre por el otro átomo de carbono, el carbocatión incipiente será terciario y más estable:
R H
HRX+
2
HO_
R H
HRX
OH
δ+
δ−
carbocatión incipiente
H
R X
HOH
R
(mayoritario)
Una consideración importante: el hecho de que los grupos alquilo unidos a un carbono con carga positiva estabilicen el carbocatión, significa que son cesores de electrones. Cualquier carbocatión es inestable debidoa la carga positiva; si un grupo cede electrones al carbono con carga, disminuye su inestabilidad, ya que hace aumentar la densidad electrónica sobre él.
La reacción entre ácidos hipohalosos y olefinas no simétricas, como la que se muestra a continuación, da lugar a una mezcla de estereoisómeros de dos compuestos diferentes:
R CH CH R'XOH R CH
OHCHX
R' + R CHX
CHOH
R'
(Z) o (E) Para obtener los estereoisómeros de los dos compuestos es necesario trabajar con los dos cationes halogeno-nio posibles:
R R'
HHX+
R R'
HH
X+
yEstereoisómero (Z):
1 2
3 4
R H
R'HX+
R H
R'H
X+
yEstereoisómero (E):
1 2
3 4
VII

165
EJERCICIO 7.10Utiliza representaciones tridimensionales para escribir la reacción entre ácido hipobromoso y 1-etil-2-metil- ciclohexeno. Nombra todos los compuestos que se obtienen y averigua la configuración absoluta de los este- reoisómeros
REACCIONES DE LOS ALQUENOS CON ELECTRÓFILOS OXIGENADOS
OXIDACIÓN DE ALQUENOS CON PEROXIÁCIDOS
R CO
OH R CO
O OH
ácido carboxílico ácido peroxicarboxílico Ácidos peroxicarboxílicos empleados habitualmente:
H CO
O OH CH3 CO
O OH C6H5 CO
O OH
CO O OH
Ác. peroxifórmico Ác. peroxiacético Ác. peroxibenzoico Ác. m-cloroperoxibenzoicoÁc. m-cloroperbenzoico
Cl
La reacción de las olefinas con peroxiácidos da lugar a derivados de oxirano:
R R
HH
C OR
O
OH
R R
HH
C OR
HO
O
Siguen un par de ejemplos:
H C6H5
HC6H5
H C6H5
HC6H5
O1
2 3CH3CO3H
CH2Cl, 30-350C
1,2-Difeniletileno (2S,3S)-2,3-Difeniloxirano
H H
O
HCO3H
Ciclohexeno Epoxiciclohexeno
VII

166
El anillo de oxirano se abre fácilmente en medio ácido:
H C6H5
HC6H5
O ::
H2O H+
H C6H5
HC6H5
O:H
+
H C6H5
HC6H5
O :H
+
OH2: H2O
OH
C6H5
H
H
C6H5
+
OH
OH
C6H5
H
H
C6H5
+HH2O : HO
OH
C6H5
H
H
C6H5
1 2+ H3O+
(1S,2R)-1,2-Difenil-1,2-etanodiol
(forma meso)
: :
El proceso completo da lugar a una adición neta anti de los dos grupos OH al doble enlace de la olefina. Es decir, permite obtener 1,2-dioles a partir de olefinas:
R R
HHR R
HHO
HO
OH
R
R
H
H
(Z)
R R
HHO
RCO3H H3O+
H+
1 2 OH
HO
R
R
H
H
H2O
1
2
La reacción de la olefina con el peroxiácido transcurre con una estereoselectividad superior al 99 % (si lo hicie-se con un 100 % sería estereoespecífica) ya que la geometría de los grupos R y H unidos a C1 y C2 en el 1,2-diol es la misma que la del oxirano de partida.
Comprueba que cuando en la reacción anterior se parte del estereoisómero (E) se obtiene la forma meso.
Igual que sucede con los cationes halogenonio la apertura de los oxiranos no simétricos, catalizada por ácidos, tiene lugar con regioselectividad Markovnikov. En este sentido, la reacción es parecida a una SN1 (ver Tema 9), en la que el carbocatión intermedio más estable reacciona con el nucleófilo antes de que tenga lugar su rota-ción interna:
VII

167
Et Me
HMeEt Me
HMeO
1)RCO3H
2)H3O+
3-Metil-2-penteno
H
+
Et Me
HMeO
H2O
H+ OH
H2O
Et
Me
Me
HEt Me
HMeO
H
+
H2O:
: +
OH
HO
Et
Me
Me
H
23
(1) (2S,3S)
3-Metil-2,3-pentanodiol
H2O::H2O
OH
H2O
Et
Me
Me
H+
(1') (2R,3R)
3-Metil-2,3-pentanodiol
Et Me
HMe
OH
+ Et Me
HMe
OH
+
OH
HO
Et
Me
Me
H
En esta reacción se obtendrá una mezcla mayoritaria de (1) y (1’), ya que la reacción transcurre preferentemen-te a través del carbocatión incipiente más estable (en este caso el carbocatión terciario).
Emplea representaciones tridimensionales para escribir la reacción entre propilidenciclohexano y un peroxi-ácido, seguida de acidificación. Indica cuáles serán los productos mayoritarios y averigua la configuraciónabsoluta de los estereoisómeros.
EJERCICIO 7.11
OXIDACIÓN DE ALQUENOS CON TETRAÓXIDO DE OSMIO El tetraóxido de osmio (OsO ) reacciona con las olefinas, dando lugar a ésteres del ácido osmico: 4
R R'
HHR R'
HH OOsO4
Et2O, piridina
O
OsOO
(adición syn) La descomposición en medio básico del éster da lugar a un glicol en el que los dos grupos OH están en posi-ción syn (al contrario de lo que sucede en la reacción con peroxiácidos, que sitúa los dos grupos OH en posi-ción anti)
VII

VII
168
Para averiguar el número total de estereoisómeros que se obtienen en la reacción hay que trabajar con los dos ésteres posibles: (1) y (2):
eriguar el número total de estereoisómeros que se obtienen en la reacción hay que trabajar con los dos ésteres posibles: (1) y (2):
R R'
HH OKOH
O
OsOO
H2OR'R
OHHO
HH(1) (1')
R R'HH
O
KOH
O
OsOO
H2O
R'R
OHHO
HH
(2)
(2')
EJERCICIO 7.12Utiliza representaciones tridimensionales para escribir la rea sNombra todos los compuestos que se obtienen y averigua la configuración abso
En la reacción del OsO4 con cada estereoisómero de la olefina se obtiene una pareja de enantiómeros. En el ejemplo los enantiómeros son (1’) y (2’).
cción entre O O4 y 1-etil-2-metilciclohexeno. luta de sus estereoisómeros.
EJERCICIO 7.13Averigua la estereoquímica de los productos que se obtienen en las siguientes reacciones:
CH3 CH
CH CH2CH3
(Z)
1)RCO3H
2)H3O+CH3 CH CH CH2CH3
CH3 CH CH CH2CH3
(E) OCH3 CH
1)OsO4
2)KOH, H2
CH CH2CH3
HO
HO HO
HO
OLEFINAS CON DOBLES ENLACES CONJUGADOS
CH2 CH CH CH2 CH3 CH CH CH CH CH3
1,3-Butadieno 2,4-Hexadieno 1,3-Ciclohexadieno
CH2 CH
REACCIÓN CON HALUROS DE HIDRÓGENO Y HALÓGENOS A TRAVÉS DE CARBOCATIONES
CH CH2
1,3-Butadieno
1 2 4
HCl
AcOH, 00CCH3 CH CH CH2
ClCH3 CH CH CH2Cl+
3-Cloro-1-buteno 1-Cloro-2-buteno
(adición 1,2 ; 80 %) (adición 1,4 ; 20 %)

169
VII
RCO3H
RCO3H
OsO4
OsO4
12
1
1 1
1
22
22
[1]
[2]
[4]
[3]
(Z)(E)
anti syn
antisyn
HO
OH
R'
HR
H
OH
HO
R
HR'
H
HO
RH
OH
R'H
HO
RH
OH
R'H
R R'
HH
R H
R'H
R R'
HH
O
1
2
R R'
HH
2
1
R H
R'H
21
R H
R'H
O
enantiómeros: [1] y [2] Cuando R = R´ forma meso: [3] = [4]
Enantiómeros: [1] y [2] ; [3] y [4]
HIDROXILACIÓN DE OLEFINAS CON PEROXIÁCIDOS Y TATRAÓXIDO DE OSMIO

170
Primer paso: reacción del electrófilo (H+) con el par de electrones del doble enlace.
CH2 CH CH CH2
1,3-Butadieno
1 2 CH3 CH CH CH2
CH2 CH2 CH CH2
+CH3 CH CH CH2
+
(carbocatión estabilizado por deslocalización de la carga)
+(carbocatión no estabilizado)
HCl
1
2 La reacción transcurre a través del carbocatión estabilizado. El carbocatión primario no estabilizado carece de importancia, ya que se forma en cantidades tan pequeñas que no es preciso tenerlo en cuenta.
−Segundo paso: reacción del nucleófilo (Cl ) con el carbocatión estabilizado:
CH3 CH CH CH2
ClCH3 CH CH CH2Cl
3-Cloro-1-buteno 1-Cloro-2-buteno
(adición 1,2 ; 80 %) (adición 1,4 ; 20 %)
CH3 CH CH CH2+
CH3 CH CH CH2+
Cl_
Cl_
Cuando la reacción se realiza a baja temperatura el producto mayoritario es la olefina menos sustituida (olefina terminal) esto es, la menos estable:
CH2 CH CH CH2
1,3-Butadieno
HBrEt2O, -800C
CH3 CH CH CH2
BrCH3 CH CH CH2Br+
(80 %) (20 %)
CH2 C CH CH2
2-Metil-1,3-Butadieno
HCl -150C
CH3 C CH CH2
CH3CH3 C CH CH2Cl+
(73 %) (27 %)
CH3
Cl
CH3
(Isopreno) Estos resultados experimentales indican que, en el carbocatión estabilizado, la carga positiva no está distribui-da simétricamente entre los dos carbonos, es decir, cada carbono no tiene la misma carga (+1/2) Si el átomo de carbono con mayor carga está unido al metilo (cesor de electrones) el carbocatión es más estable que cuando la carga máxima está en el carbono terminal.
CH3 CH CH CH2+
CH3 CH CH CH2+
mayor carga; catiónestabilizado por CH3
mayor carga; catión no estabilizado
La energía de activación es menor en el primer caso que en el segundo:
VII

171
CH3 CH CH CH2+
+ X_
CH3 CH CH CH2
X
δ+
δ−
ΔG (1,2)
CH3 CH CH CH2+
+ X_
CH3 CH CH CH2
X δ−
δ+ΔG (1,4)
ΔG (1,2) ΔG (1,4)<
Es importante fijarse que el producto obtenido a través del estado de transición de menor energía no tiene por qué ser el más estable. De hecho no lo es, ya que se trata de la olefina menos sustituida en el doble enlace. Cuando la reacción se realiza a temperaturas bajas, el producto mayoritario es el que proviene del estado de transición de menor energía y el proceso está sometido a control cinético. Se obtiene la olefina que se forma más rápidamente, a través del estado de transición de menor energía, es decir, la olefina menos estable (olefina terminal: CH3-CHX-CH=CH2) En esta situación, el sistema no ha tenido tiempo de alcanzar el equilibrio, y por este motivo se dice que la reacción está controlada cinéticamente. Sin embargo, cuando la reacción se hace a temperatura elevada está sometida a control termodinámico, es decir, se alcanza el equilibrio. Los porcentajes de los productos de reacción se invierten: la olefina mayoritaria es la más estable (adición 1,4) y la minoritaria la menos estable (adición 1,2):
CH2 CH CH CH2
1,3-Butadieno
1 2 4
HCl1000 C
CH3 CH CH CH2
ClCH3 CH CH CH2Cl +
(adición 1,4)
75 %
(adición 1,2)
25 % Ahora, la mezcla de isómeros refleja sus estabilidades termodinámicas relativas. Al aumentar la temperatura, aumenta el número de moléculas capaces de alcanzar el estado de transición de mayor energía y, si se pro-longa el tiempo de reacción, el sistema alcanza el equilibrio (control termodinámico)
CH2 CH
H
CH CH2
δ+δ+
CH3 CH CH CH2
ΔG
CH3 CH CH CH2X
ΔG (1,4)
CH3 CH CH CH2
X δ−
δ+
CH3 CH CH CH2
X
δ+
δ−
+CH3 CH CH CH2
CH3 CH CH CH2+
XCH2 CH CH CH2+HX
ΔG (1,2)
ΔG(1,2)
ΔG(1,4)
curso
(menos estable)
(más estable)
VII

172
EJERCICIO 7.15En la reacción del 1-fenil-4-metoxi-1,3-ciclopentadieno con HBr pueden formarse cuatro carbocationes diferen-tes. Elige el menos inestable de los cuatro para averiguar los estereoisómeros que se obtienen, cuando el proceso está sometido a control termodinámico o a control cinético.
REACCIONES DE LOS ALQUINOS
ACIDEZ DE LOS ALQUINOS TERMINALES Los alquinos con un triple enlace terminal tienen valores de pKa ≈ 26 (Apéndice II):
R C C Hácido
R C C:_
+ H + pKa 26
base Son compuestos poco ácidos que sólo se desprotonan en presencia de bases muy fuertes, por ejemplo el anión amiduro (NH2
−) cuyo ácido conjugado (NH3) tiene un pKa ≈ 36. El anión amiduro se obtiene por reacción entre NH3 líquido (¡no NH3 disuelto en agua!) y sodio metálico; se trata de una reacción de oxidación-reducción:
2NH3 (liq.) + 2Na -330 C 2Na+ :NH2_
+ H2
Amiduro sódico
R C C H + :NH2_
R C C:_
+ :NH3 (liq.)
pKa 26 pKa 36Keq. 1010
anión acetiluro El equilibrio está desplazado a la derecha de forma prácticamente total. Los aniones acetiluro son nucleófilos muy efectivos, que dan lugar a reacciones de sustitución nucleófila cuan-do se utilizan haluros de alquilo como sustratos (Tema 6):
H C C H
Acetileno
_Na+:NH2
-330 CH C C:
_
H C C:_
CH2 XR
H C C CH2 R + X_
alquino con triple enlace terminal
Si se parte de acetileno, esta reacción permite obtener alquinos terminales con mayor número de carbonos:
H C C H + CH3 Br
_Na+:NH2
-330 CCH3 C CH
Propino
H C C H + CH3CH2 C CH
1-Butino
CH3CH2 Cl
_Na+:NH2
-330 C
VII

173
H C C H +
1-Pentino
CH3CH2CH2 Br CH3CH2CH2 C CH
_Na+:NH2
-330 C
Cuando se utiliza cualquier alquino terminal que no sea el acetileno, el triple enlace queda situado en el interior de la cadena:
CH3CH2 C CH CH3CH2 C C:_
_Na+:NH2
-330 C
CH3CH2 C C:_
CH2 BrCH3
CH3CH2 C C CH2CH3 + Br_
3-Hexino
REACCIONES DE ADICIÓN ELECTRÓFILA
ADICIÓN DE HALUROS DE HIDRÓGENO
CH3CH2 C CH HBr
150 CCH3CH2 C CH2
Br+ CH3CH2 C CH3
Br
Br
2-Bromobuteno 2,2-Dibromobutano La reacción transcurre a través de carbocationes con regioselectividad Markovnikov:
CH3CH2 C CH
H Br
CH3CH2 C CH2+
Br_
CH3CH2 C CH2
Br
2-Bromopenteno
CH3CH2 C CH2
Br
H Br
CH3CH2 C CH3
Br
+
Br_
CH3CH2 C CH3
Br
Br
2,2-Dibromopentano La interpretación de estos resultados no es evidente. Observa qué sucede cuando la reacción se hace con acetileno:
H C C HHCl, ZnCl2
1000 CCH2 CHCl
Cloruro de vinilo
HCl, HgCl2250 C
CH3 CH ClCl
1,1-Dicloroetano Reacción 1:
HC CH
H Cl
+
Cl_
CH2 CH CH2 CHClCloruro de vinilo
VII

174
Reacción 2:
CH2 CH Cl
1 2
HCl
CH3 CH Cl+
+ Cl_
CH2 CH2Cl+
+ Cl_
CH3 CHCl
Cl
Cl CH2 CH2 Cl
(1)
(2)
1,1-Dicloroetano
1,2-Dicloroetano
1
2
En el carbocatión (1) el cloro está unido al átomo de carbono con carga. El átomo de cloro es más electronega-tivo que el de carbono y cabría esperar que su efecto electroatrayente disminuyera la carga del carbono, des-estabilizando aún más el carbocatión. Sin embargo, esto no sucede, puesto que el carbocatión (2) que no tiene el cloro unido al carbono con carga, es menos estable que (1) La estabilización de (1) puede explicarse admitiendo que existe una interacción entre el OA pz lleno del cloro y el OA pz vacío del carbono:
ClH
CH3 +
(1)
HCH3
+
pz vacio pz lleno
(1)
HCH3 +
enlace π de dos electrones aportados por el cloro
Si esta interpretación se realiza utilizando enlaces de valencia, es necesario dibujar dos estructuras del carbo-catión (1):
ClH
CH3 +
(1)
: ClH
CH3:
+
(1) La deslocalización de la carga entre los átomos de carbono y cloro estabiliza el carbocatión (1)
EJERCICIO 7.16Considera las siguientes reacciones sucesivas:
Haz un esquema en el que aparezcan todos los carbocationes posibles y justifica la formación de los productos, empleando un razonamiento semejante al utilizado en el caso del acetileno.
R C C H HCl R C CH2
ClHBr
R C CH3
Cl
Br
ADICIÓN DE AGUA CON CATÁLISIS ÁCIDA El acetileno reacciona con agua, en presencia de un ácido fuerte y sales mercúricas (Hg2+) dando lugar a ace-taldehído:
HC CHH2O, H2SO4
HgSO4CH3 C
OH
Acetaldehido
VII

175
La reacción transcurre del modo siguiente (ver equilibrio ceto-enólico, Tema 9, p. 229):
HC CHH OH2
+ H2C CH+
OH2:
H2C CHHO H OH2:
+
CH2 CHOH
+ H3O+
enol del acetaldehido
CH2 CH
OH
CH3 CO
H Acetaldehido
(equilibrio ceto-enólico)
El resto de los alquinos se transforman en cetonas; los que están sustituidos en los dos carbonos del triple enlace son bastante reactivos y no se necesita utilizar catalizadores de mercurio para que tenga lugar la reac-ción:
CH3CH2CH2CH2 C CHH2O, H2SO4
HgSO4CH3CH2CH2CH2 C CH3
O
1-Hexino 2-Hexanona80 %
C C CH2CH3H2O, H2SO4
00 C, 10 min.CO
CH2CH3 + CO
CH3
3-Pentanona 2-Pentanona50 % 50 %
2-Pentino
CH3 CH2CH3 CH3CH2CH2
REDUCCIÓN DE ALQUINOS CON SODIO EN AMONÍACO LÍQUIDO Los alquinos se reducen a olefinas cuando reaccionan con sodio o litio metálicos en amoníaco líquido. La reac-ción es estereoselectiva, ya que conduce mayoritariamente al estereoisómero (E) del alqueno:
R C C R'Na, NH3(liq.)
-330 C
HR
H R'(E)
En la reacción están implicados radicales libres, debido a la ruptura homolítica del triple enlace: 1. Reacción de oxidación-reducción:
R C C R'
Na
R C C R'
_
anión radical
+ Na+
2. Reacción del anión radical con amoníaco:
R C C R'_
H NH2
R C CR'
H+ :NH2
_(anión amiduro)
radical
VII

176
3. Reacción de oxidación-reducción:
Na
R C C
R'
Hradical
R C C
R'
H
_
carbanión
+ Na+
4. La reacción del carbanión con amoníaco da lugar a la olefina (E):
R
C C
R'
H
_
carbanión
HH2N
R
C C
R'
H
H
(E)
+_
:NH2
:
:
:
Por ejemplo:
CH3CH2 C C CH2CH2CH2CH3Na, NH3 (liq.)
-330 CH
CH3CH2
CH2CH2CH2CH3
H
3-Octino
(E)-3-Octeno
SÍNTESIS DE COMPUESTOS ORGÁNICOS A PARTIR DE OLEFINAS Y ALQUINOS Los alquenos y los alquinos se utilizan como sustancias de partida para la obtención de otros compuestos or-gánicos. A continuación aparece una síntesis de la 2-butanona empleando acetileno como único producto orgánico. Retrosíntesis de la 2-butanona:
CH3CH2 CO
CH3 CH3CH2 COH
CH2 CH3CH2 C CH
CH3CH2 X
+H C C:
_
CH2 CH2
HC CH
Síntesis de la 2-butanona:
HC CHH2
[Pt]CH2 CH2
HBrCH3CH2Br
HC CH HC C:_
HC C:_
CH2
CH3
Br HC C CH2CH3
HC C CH2CH3H2O
H3O+CH3CH2 C
OHCH2
CH3CH2 CO
CH3
2-Butanona
_:
Na+:NH2
-330 C
VII

177
EJERCICIO 7.17Utiliza acetileno como único producto orgánico para obtener 3,4-hexanodiol y (E)-3-hexeno
R CH2CH2 R
R CH2CH RX
R CH2CH ROH
R CH__CH RX X
R CH__CH RX OH
R CH__CH ROH
OH
R CH__CH ROH OH
RCO3H
H2
[Pt]
HX
H2O
H3O+
X2
XOH
OsO4
R CH CH R
ESQUEMA GENERAL DE LAS REACCIONES DE OLEFINAS
HX
H2O
H3O+
R C CH
1)Na+NH2_
2)R'-XR C C R'
R CX
CH2HX R C
XCH3
X
R COH
CH2 R CO
CH3
R CH CH2 R CH2 CH3H2
[Pt]
H2
[Pt]
ESQUEMA GENERAL DE LAS REACCIONES DE ALQUINOS
VII

178
EJERCICIOS ADICIONALES
7.1 La reacción de un compuesto A(C4H8) con ácido hipocloroso, da lugar a una mezcla de (2S,3R) y (2R,3S)-3-cloro-2-butanol. Averigua la estructura de A y escribe el mecanismo a través del que discurre el proceso. 7.2 En el esquema siguiente se muestran los productos a los que da lugar una olefina A, cuando reacciona con ácido peroxifórmico o tetraóxido de osmio:
A
1)HCO3H
2)H2O
1)OsO4
2)H2O
[(2R,3R) + (2S,3S)] 2,3-pentanodiol
[(2R,3S) + (2S,3R)] 2,3-pentanodiol (racémico)
Averigua la estereoquímica de A, e indica qué productos se obtendrán cuando dicha olefina reacciona con ácido hipocloroso a través de iones onio. 7.3 Escribe las reacciones del 1,2-dimetilciclohexeno con tetraóxido de osmio, ácido peroxibenzoico y ácido clorhídrico (vía carbocationes). Averigua la configuración absoluta de todos los productos que se obtienen y nombra cada uno de ellos. 7.4 El (E)-1-fenil-2-penteno reacciona con HBr dando lugar a una mezcla de dos racémicos. Uno de ellos corresponde al 1-bromo-1-fenilpentano y el otro al 3-bromo-1-fenil pentano. Apartado 1: Averigua si la reacción transcurre a través de carbocationes o de iones onio y haz un esquema con todas las reacciones. Apartado 2: Escribe las reacciones del (E)-1-fenil-2-penteno con ácido peroxibenzoico y con tetraóxido de osmio, indicando en cada caso la configuración absoluta de los productos que se obtienen. 7.5 La reacción del (4S)-4-fenil-2-penteno con HBr da lugar a los siguientes resultados:
(4S)-4-fenil-2-penteno
2-bromo-4-fenilpentano (mezcla de dos estereoisómeros)
1-bromo-1-fenil-2-metilbutano (mezcla de cuatro estereoisómeros)
HBr
Averigua si la reacción transcurre a través de carbocationes o de iones onio y haz un esquema con todas las reacciones. 7.6 La reacción de una olefina A(C6H9OCl) con ácido hipocloroso, da lugar a la siguiente mezcla de cuatro este-reoisómeros:
[(1S,2R) + (1S,2S)] 3,3-dicloro-1,2-ciclohexanodiol + [(1R,2S,3S) + (1S,2R,3S)] 1,2-dicloro-1,3-ciclohexanodiol Averigua la estereoquímica de la olefina A y escribe el mecanismo de todas las reacciones.
VII

179
7.7 El estereoisómero ópticamente activo de una olefina A(C H12 13Cl) reacciona con HOCl, dando lugar a cuatro productos distintos. Uno de ellos es el (1S,2S,3R) 1,3-Dicloro-1-fenil-2-hidroxiciclohexano. Apartado 1: Averigua la estructura de la olefina A y escribe únicamente la reacción que conduce al producto indicado. Apartado 2: La olefina A se somete a una reacción de eliminación en medio básico, obteniéndose un compues-to B(C H12 12) El tratamiento de B con HCl origina una mezcla de dos racémicos, uno de cuyos componentes es el compuesto de partida A. Escribe el mecanismo de la reacción entre B y HCl utilizando representaciones tridimensionales. 7.8 La reacción del 1,3-Ciclohexadieno con HCl, a través del carbocatión más estable, da lugar a un racémico. Utiliza representaciones tridimensionales para averiguar la estereoquímica de los productos resultantes y expli-ca por qué se obtienen los mismos productos cuando la reacción está sometida a control termodinámico o a control cinético. 7.9 Averigua la estructura de la olefina A:
A1)OsO4
2)H3O+[(1R,3S) + (1R,3R)]-3-hidroximetil-1,3-ciclohexanodiol
1. Escribe el mecanismo de la reacción empleando representaciones tridimensionales y asigna la configuración
a los estereoisómeros que se obtienen. 2. ¿Se llegaría al mismo resultado si la olefina A se trata con ácido peroxibenzoico, seguida de hidrólisis en
medio ácido? 7.10 Averigua la estructura de la olefina A, teniendo en cuenta que la reacción transcurre a través del carboca-tión incipiente más estable:
A [(1R,3R) + (1R,3S)]-3-clorometil-1,3-ciclohexanodiolClOH
7.11 Se hace reaccionar 3-metilenciclohexeno con HCl, dejando que la reacción alcance el equilibrio. Escribe el mecanismo de la reacción utilizando representaciones tridimensionales y averigua la estereoquímica de las olefinas monocloradas que se obtienen. 7.12 Averigua la estereoquímica del compuesto A(C5H10) y escribe el mecanismo de todas las reacciones que aparecen en el esquema.
3-cloro-2-pentanol (racémico) + 2-cloro-3-pentanol (racémico)
2,3-pentanodiol (racémico)
[(2R,3S) + (2S,3R)] 2,3-pentanodiol
HOCl
1)HCO3H
2)H3O+ / H2O
1)OsO4
2)H2O
A
7.13 El 1,4-difenil-1,3-butadieno reacciona con HBr a través del carbocatión más estable, dando lugar a una mezcla de dos racémicos. Uno de ellos corresponde al (E)-3-bromo-1,4-difenil-1-buteno y el otro al (E)-1-bromo-1,4-difenil-2-buteno.
VII

180
Apartado 1: Averigua si la configuración del 1,4-difenil-1,3-butadieno de partida es (1E,3E) o (1E,3Z) y escribe el mecanismo a través del que transcurren todas las reacciones. Apartado 2: El racémico del (E)-1-bromo-1,4-difenil-2-buteno se hace reaccionar con HCl acuoso, obteniéndose una mezcla de 1-bromo-1,4-difenil-4-hidroxibutano y 1-bromo-1,4-difenil-4-clorobutano. Explica mediante qué tipo de mecanismo tiene lugar la reacción, indicando la configuración absoluta de todos los estereoisómeros que se obtienen. 7.14 Las reacciones que aparecen a continuación transcurren a través de carbocationes y están sometidas a control termodinámico (equilibrio) Indica en cada caso la estereoquímica absoluta de los productos que se obtienen y ordénalos de acuerdo con su abundancia relativa (los estereoisómeros del mismo compuesto están colocados entre corchetes)
Reacción 1: 3-Fenilpropeno + Cl2
Reacción 2: 1-Fenilpropeno + HBr
Reacción 3: 1,3-Ciclopentadieno + Br2 [A + B + C + D] + [E + F + G]
[A + B ] + [ C + D]
[A + B ]
7.15 Apartado 1: Utiliza el esquema que aparece a continuación para identificar los estereoisómeros A y B del 2-buteno:
1)C6H5CO3H
2)H3O+
1)OsO4
2)H3O+
A(¿Z o E?)
B(¿Z o E?)
[(2R,3R) + (2S,3S)] 2,3-butanodiol
Escribe el mecanismo de todas las reacciones Apartado 2: Escribe el mecanismo de la reacción del estereoisómero A con HCl acuoso y asigna la configura-ción absoluta a los compuestos que se obtienen. 7.16 Averigua la configuración absoluta de los estereoisómeros que se forman en los siguientes procesos y escribe el mecanismo de todas las reacciones:
Reacción 1:(E)-2-hexeno1)OsO4
2)H3O+(1) + (2)
Reacción 2: (3) + (4)feniletileno HBr
Reacción 3: 1,2-dimetilciclohexeno HCl (5) + (6) + (7) + (8)
Reacción 4: 1-metilciclopenteno1) HCO3H
2) H3O+(9) + (10)
7.17 Uno de los estereoisómeros del 1-metil-3-hidroxiciclohexeno (A) se somete a una reacción de hidroxilación con ácido peroxibenzoico, obteniéndose una mezcla de (1R,2R,3S) y (1S,2S,3S)-1-metil-1,2,3-ciclohexanotriol.
VII

181
Apartado 1: Averigua la configuración absoluta del estereoisómero de partida (A) y escribe el mecanismo de la reacción utilizando representaciones tridimensionales. Apartado 2: Escribe la reacción del enantiómero de (A) con bromuro de hidrógeno acuoso utilizando de nuevo representaciones tridimensionales y asigna la configuración absoluta de todas las moléculas. 7.18 Averigua la configuración absoluta de los estereoisómeros que se forman en los siguientes procesos y escribe el mecanismo de todas las reacciones
Reacción 1:
Reacción 2:
Reacción 3:
Reacción 4:
(E) 2-hepteno1) OsO4
2) H3O+(1) + (2)
1,2-dietilciclohexenoHCl
(3) + (4)1-fenilpropenoHBr
1-etilciclopenteno1) HCO3H
2) H3O+(9) + (10)
(5) + (6) + (7) + (8)
7.19 Uno de los estereoisómeros del 3-fenil-1-buteno se hace reaccionar, primero con ácido m-cloroperoxibenzoico, y el compuesto que resulta se trata con HCl acuoso. El producto final es una mezcla de estereoisómeros del (3R)-3-fenil-1,2-butanodiol. Apartado 1: Averigua la configuración absoluta del 3-fenil-1-buteno de partida y escribe el mecanismo de las reacciones empleando representaciones tridimensionales. Apartado 2: La reacción del estereoisómero del Apartado 1 (3-fenil-1-buteno) con HBr, da lugar a una mezcla de estereoisómeros de dos compuestos distintos: el 2-bromo-3-fenilbutano y el 1-bromo-1-fenil-2-metilpropano. Escribe el mecanismo de las reacciones empleando representaciones tridimensionales y averigua la configura-ción absoluta de los estereoisómeros que se obtienen. 7.20 Uno de los estereoisómeros del 4-fenil-2-penteno se hace reaccionar con tetraóxido de osmio, y el com-puesto que resulta se trata con KOH acuoso. El producto final es una mezcla de los estereoisómeros (2S,3R,4S) y (2R,3S,4S) del 4-fenil-2,3-pentanodiol. Apartado 1: Averigua la configuración absoluta del 4-fenil-2-penteno de partida. Apartado 2: La reacción del estereoisómero del 4-fenil-2-penteno con bromo da lugar a una mezcla de diaste-reoisómeros del 2,3-dibromo-4-fenilpentano. Escribe el mecanismo de las reacciones empleando representa-ciones tridimensionales y averigua la configuración absoluta de los productos que se obtienen.
VII

8. REACCIONES DE SUSTITUCIÓN NUCLEÓFILA EN GRUPOS CARBONILO
INTRODUCCIÓN Las reacciones de sustitución nucleófila en moléculas con grupos carbonilo (C=O) transcurren de acuerdo con el siguiente esquema general:
_ _R C
OZ +
ONu R C + ZNu
Los grupos salientes Z más comunes son: Cl−, R–CO–O−, HO−, RO−, NH2
−, RNH−. Es decir, son reacciones en las que están implicados los siguientes compuestos: cloruros de ácido, anhidridos de ácido, ácidos, ésteres y amidas. Estas reacciones transcurren en dos pasos: en el primero, el nucleófilo (Nu) se adiciona al grupo carbonilo, y en el segundo se elimina el grupo saliente Z:
Nu_
CO
RZ
:
CO
RZ
:
Nu
:_
CO
R
:
Nu
:
+ Z:_adición eliminación
Por ejemplo:
Cloruro de acetilo
adición eliminaciónCO
CH3
Cl
:
CO
CH3
Cl
:
H3N
:_
CO
CH3
:
H3N
:
H3N: +Cl
_+
+ :B CO
CH3
:
H2N
:
+ HB
Acetamida
Cloruro de acetilo
adición eliminaciónCO
CH3
Cl
:
CO
CH3
Cl
:
HO
:_
CO
CH3
:
HO
:
HO Cl_
+
Ácido acético
_
CORRELACIONES ESTRUCTURA – REACTIVIDAD En el proceso adición-eliminación, la constante de equilibrio global está determinada por las constantes de equilibrio correspondientes a las reacciones individuales de adición y de eliminación. El valor de dichas constantes depende de las características estructurales del grupo carbonilo, del tipo de nucleófilo (Nu) y de la naturaleza del grupo saliente Z. Con respecto al grupo carbonilo, su interacción electrónica con el grupo saliente Z, es el factor determinante del valor de la constante de equilibrio en el paso de la adición. Un grupo Z que sea atractor de electrones, a través de un efecto inductivo -I, hace disminuir la densidad electrónica en el átomo de carbono, aumentando su reactividad. Debido a que todos los grupos Z tienen pares de electrones sin compartir, existe un efecto mesómero +M, opuesto al efecto inductivo -I, que provoca la deslocalización de dichos electrones, estabilizando la molécula, es decir, disminuyendo su reactividad.
VIII

183
Disminución de la densidad electrónica sobre el carbono del C=O:
efecto inductivo −I la reactividad aumentaR C
OZ
::δ
_
δ+
Deslocalización de los pares de electrones de Z:
efecto mesómero +M la reactividad disminuyeR C
O
Z
::
:: R C
O
Z
::
:
: _
+
Cuanto más electronegativo es el átomo del grupo Z que está unido al carbonilo, menor será la densidad electrónica sobre el carbono del C=O, y mayor su reactividad frente a los nucleófilos:
átomo máselectronegativo
átomo menoselectronegativo
R C
O
Cl : R C
O
O R C
O
NH:C
O
R R'(cloruro de ácido) (éster)(anhidrido de ácido) (amida)> > R C
O
O R' >
Por otra parte, la deslocalización de los pares de electrones presentes en Z, estabiliza la molécula, disminuyendo la reactividad del C=O. El efecto +M será tanto mayor cuanto menor sea la electronegatividad de Z:
R C
O
Cl
::
R C
O
Cl
::_
+::
R C
O
O
::
C
O
R R C
O
O
::
C
O
R
_
+R C
O
O
::
C
O
R
_
+::
R C
O
O
::
R C
O
O
::_
+R' R'
R C
O::
R C
O::_
NH R' N R'+
La combinación de los efectos -I y +M varia de un grupo a otro, como puede apreciarse en la tabla siguiente:
combinación de efectos reactividad del C=O
-I > +M
-I > +M
-I +M
-I < +M
muy grande
semejante a
muy grande
pequeña
sustrato
R C
O
Cl
R C
O
O C
O
R
R C
O
O R'
R C
O
NH2
R C
O
R'~_
VIII

184
Orden de reactividad en la etapa de adición
+ Nu_
R C
O
Cl
R C
O
Cl
_
+ Nu_
R C
O
O
R C
O
O
_
CO R'
CO R'Nu Nu
+ Nu_
R C
O
OR'
R C
O
OR'
_
Nu
+ Nu_
R C
O
NHR'
R C
O
NHR'
_
Nu Con respecto al grupo saliente Z, su facilidad de eliminación está directamente relacionada con la basicidad: cuanto menor sea su basicidad más fácilmente es eliminado:
grupo saliente (Z)
NH2
_
_
pKa
5
R CO
Z
R CO
Cl Cl_
-7
R CO
O CO
R R CO
O
R CO
O R' R'O_
15-19
R CO
NH2 36 Orden de reactividad en la etapa de eliminación:
>R C
O
Nu
Cl
_
R C
O
Nu
O
_
R C
O
Nu
OR'
_
R C
O
Nu
NHR'
_
> >
CO R'
R CO
Nu R CO
Nu R CO
Nu R CO
Nu
Cl_
R'CO_O_
R'O_
R'NH2_ _ _ _
Combinando las constantes de equilibrio de las etapas de adición y eliminación, es evidente que la constante del equilibrio global disminuye en el mismo orden:
>
[cloruro de ácido] [anhidrido de ácido]
[disminución de la reactividad]
[éster]
R C
O
Cl R C
O
O CO
R R C
O
OR'> R C
O
NHR'
[amida]
>
VIII

185
Este orden de reactividad implica que cada uno de los cuatro compuestos se transforma fácilmente en cualquiera de los que están situados a su derecha, pero no al revés. Por ejemplo, a partir de un cloruro de ácido se pueden obtener un anhidrido, un éster o una amida; sin embargo, no es posible sintetizar directamente un anhidrido partiendo de un éster o una amida. Los nucleófilos implicados en este tipo de reacciones son los siguientes: Nucleófilos de halógenos: Cl (generado a partir de SOCl2, PCl3 Y PCl5)
_
Nucleófilos de oxígeno: H2O: (agua)
HO (anion hidroxilo):_
R__OH (alcoholes)
R__O (aniones alcóxido):_
(ácidos carboxílicos)R CO
OH
R CO
O (aniones carboxilato)_:
Nucleófilos de nitrógeno: H3N: (amoniaco)
R__NH2 (aminas primarias) Nucleófilos de ésteres y cetonas: R__CH__CO2Et (aniones enolato de ésteres)
_
R__CH__CO__R (aniones enolato de cetonas)_
Nucleófilos de hidrocarburos (aniones alquilo y arilo): RCH2 (aniones de radicales alquilo; generados a partir de RCH2Li)
_
:_
(aniones de radicales arilo; generados a partir de C6H5Li)
Anión hidruro: H (generado a partir de LiAlH(OCMe3)3 y LiAlH(OEt)3):
_
VIII

186
Las reacciones de sustitución nucleófila en compuestos carbonílicos, tienen lugar entre los nucleófilos mencionados y cada uno de los siguientes sustratos: cloruros de ácido, anhidridos de ácido, ésteres y amidas. La adición del nucleófilo (Nu) es el primer paso y la eliminación del grupo saliente (Z) el segundo:
Nu_
CO
RZ
:
CO
RZ
:
Nu
:_
CO
RZ
:
Nu
:
+ Z:_adición eliminación
Conviene aclarar que los grupos liberados Z no siempre tienen carga negativa; el mecanismo general de adición-eliminación transcurre con frecuencia en presencia de ácidos, y este hecho da lugar a la liberación de moléculas neutras, que son mejores grupos salientes. Si la reacción se hace en medio básico lo más frecuente es que el grupo Z tenga carga negativa, pero el anión amiduro (NH2
−) es una excepción. Se trata de una base muy fuerte (pKa 36) y el mecanismo de la reacción transcurre de tal modo que el grupo realmente liberado es amoníaco o una amina primaria. En la tabla siguiente aparecen las reacciones más comunes de cloruros de ácido, anhidridos de ácido, esteres y amidas, con los nucleófilos que se utilizan habitualmente en este tipo de procesos.
Cl_
H2O
producto obtenidonucleófilo grupo liberado (Z)
:
:
R CO
R CO
Cl
Z
R CO
OH
:
R' OH R CO
OR'
R' O:
:: :
_R C
OOR'
Cl_
Cl_
N NR' C
OOH
_R' C
OO R C
OO C
OR'
R CO
O CO
R'
Cl_
R CO
NH2
R CO
NHR'
H3N:
:
R NH2
Cl_
R' CH CO2R''
:_R C
OCH CO2R''
R'
Cl_
Cl_
H_
R CO
H Cl_
VIII

187
producto obtenidonucleófilo grupo liberado (Z)R CO
Z
H2O (H3O+)
:
:
HO_ _
R CO
O CO
R R CO
OH R CO
OH
R CO
O_
R CO
O
:: (H3O+)
_
R' OH R CO
OR' R CO
OH
::R' O: R C
OOR'
_R C
OO
H3N: R CO
NH2
_R C
OO
:
R NH2 R CO
NHR'_
R CO
O
R CO
OR'' R CO
OHH2O (H3O+)
:
: R'' OH
HO_
R CO
O_
R'' OH
R CO
OR''
R CO
NHR'
:
R' NH2
H3N:
R'' O_
R CO
NH2R'' O
_
(H3O+)R' OH:
: R CO
OR'R'' OH
:_
R' CH CO2R'' R CO
CH CO2R''
R'R'' O
_
H2OR C
OOHLi R' (Ar) R C
OR' (Ar)
H2O (H3O+)
:
:
HO_
R CO
NHR' R CO
OH
:
R' NH2
R CO
O_
:
R' NH2
H_
R CO
H
:
R' NH2
VIII

188
APLICACIONES DE LA SUSTITUCIÓN NUCLEÓFILA EN SÍNTESIS
FORMACIÓN DE ENLACES C–X
SÍNTESIS DE CLORUROS DE ÁCIDO Los reactivos que se utilizan habitualmente son: cloruro de tionilo (SOCl2), tricloruro de fósforo (PCl3) y pentacloruro de fósforo (PCl5):
+ SOCl2 SO2 + HClR CO
OH R CO
Cl +
+R CO
OH3 R CO
Cl +PCl3 PO3H33
POCl3 + HCl+R CO
OH PCl5 R CO
Cl + Esquema de la reacción con cloruro de tionilo:
+ Cl_+
R CO
OH
: S Cl
Cl
OR C
OOH
SO
Cl
Cl_
adición del nucleófilo:+
RC
O
O S
O
Cl
H
RC
O
O S
O
ClCl
H
eliminación : + SO2 + (Cl_
+ H+) [HCl]RC
O
O S
O
ClCl
H
R CO
Cl
EJERCICIO 8.1Cuando se utiliza PCl3 para sintetizar un cloruro de ácido, se obtiene ácido fosforoso (PO3H3) como subproducto de la reacción. Su estructura no es (1) como podría pensarse, sino (2):
(1) (2)
PHO
OH
OH PHO OHH
O
Escribe el mecanismo completo de la reacción que tiene lugar entre un ácido carboxílico y el tricloruro de fósforo utilizando como ayuda la reacción con SOCl2. Idea un mecanismo que explique la transformación de (1) (éste es el compuesto que se obtiene inicialmente) en ácido fosforoso (2)
El empleo de cada uno de los tres reactivos, en la preparación de cloruros de ácido, depende de los puntos de ebullición de las sustancias que están presentes en el medio. Se elige aquél que permita separar más fácilmente el cloruro de ácido de la mezcla de reacción:
VIII
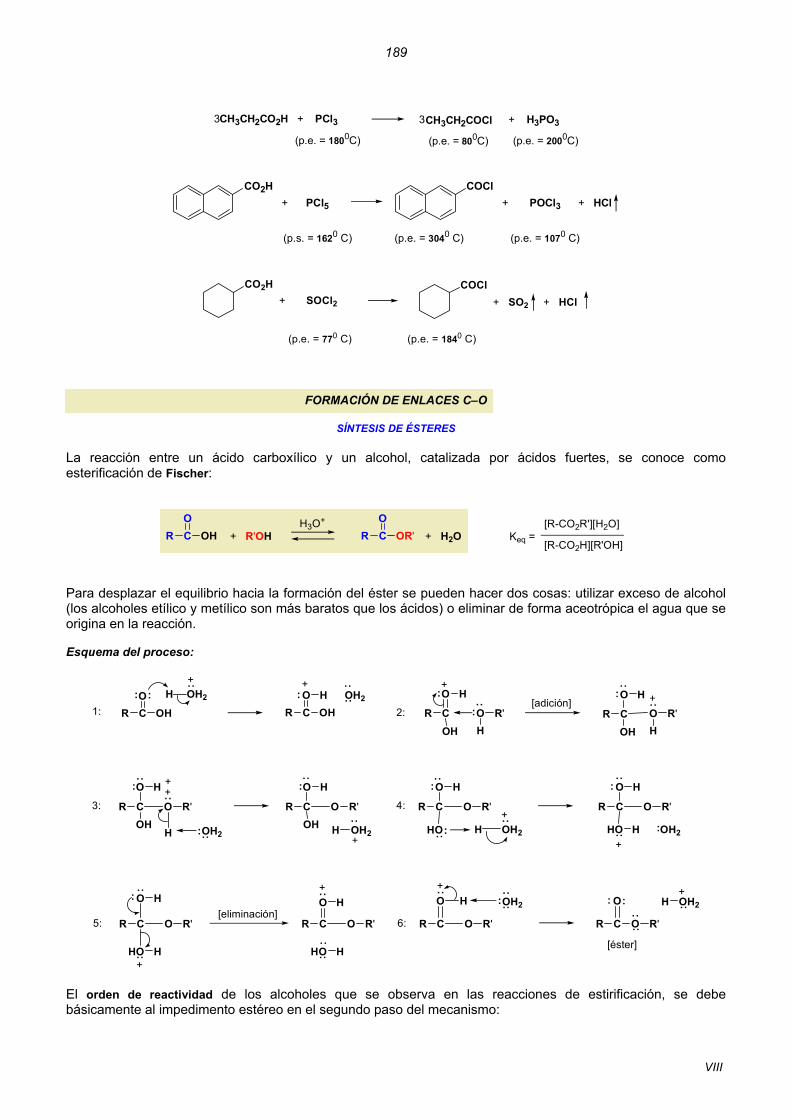
189
CH3CH2CO2H + PCl3
(p.e. = 1800C)
CH3CH2COCl
(p.e. = 800C)
+ H3PO3
(p.e. = 2000C)
3 3
CO2H COCl+ PCl5
(p.s. = 1620 C)
+ POCl3 + HCl
(p.e. = 3040 C) (p.e. = 1070 C)
(p.e. = 770 C)
CO2H+ SOCl2
COCl
(p.e. = 1840 C)
+ SO2 + HCl
FORMACIÓN DE ENLACES C–O
SÍNTESIS DE ÉSTERES La reacción entre un ácido carboxílico y un alcohol, catalizada por ácidos fuertes, se conoce como esterificación de Fischer:
+ R'OHH3O+ [R-CO2R'][H2O]
[R-CO2H][R'OH]Keq =R C
O OOH + H2OR C OR'
Para desplazar el equilibrio hacia la formación del éster se pueden hacer dos cosas: utilizar exceso de alcohol (los alcoholes etílico y metílico son más baratos que los ácidos) o eliminar de forma aceotrópica el agua que se origina en la reacción. Esquema del proceso:
H___OH2
:
: [adición]1: 2:R C
OOH
::R C
OOH
: H OH2
R C
O
OH
:
O R'H
H
R C
O
OH
:
O R'H
H
3: 4:
+
R C
O
OH
:
O R'
H
H
OH2:
R C
O
OH
:
O R'
H
OH2H
R C
O
HO
:
O R'
H
OH2H:
R C
O
HO
:
O R'
H
H OH2:
[eliminación]5: 6:
[éster]
R C
O
HO
:
O R'
H
H
R C
O
HO
O R'
H
H
R C
O
O R'
H OH2:
R C
O
O R'
OH2:: H
:+ + ::
+ :
:+
:
:
:
:
:
:
::
: :
::
::: ::
: :
+
+
+
+ +
+
+
+
: :
El orden de reactividad de los alcoholes que se observa en las reacciones de estirificación, se debe básicamente al impedimento estéreo en el segundo paso del mecanismo:
VIII

190
R CH2OH (primario) > R CHOH
R
(secundario) > R C
R
(terciario)
ROH
El impedimento estéreo en el carbono α, es el factor que determina la reactividad de los ácidos carboxílicos:
α
αα
CH3 CO2H > CH2 CO2HR > CH CO2HR
R
> C CO2HR
R
R
El método más utilizado en el laboratorio para sintetizar ésteres , es la acilación de alcoholes con cloruros o anhidridos de ácido: Con cloruros de ácido:
R CO
Cl +O
NaOH, H2OR' OH R C O R' + Cl
_+ H2O
10 %HO
_+
Esquema de la reacción con un cloruro de ácido:
+
_
[adición]R' OH C
O
Cl
R
R' O C
O
Cl
R
H
+
_
R' O C
O
Cl
R
H
R' O C
OH
Cl
R
HO_
[eliminación] _H O
C O
Cl
R R' C OR R'
O
+ Cl + H2O
Con anhidridos de ácido:
H3O+ (trazas)+R C
OO C
OR + R C
OOH
OR' OH R C O R'
Esquema de la reacción con un anhidrido de ácido
H2O HCO
RO C
OR
+ ::CO
R
O CO
R:H
+ H2O
+
VIII

191
[adición]C
O
R
O CO
R
:H+
R' OH C
OH
R
O C
O
R
:
R' O
H
+
H2O:
:: +H OH2
C
OH
R
O C
O
R
:
R' O
: H+
+ H2O
H2O:[eliminación]
:
+
ácido delanhidrido
éster
+ H3O+C
O
R
O C
O
R
:
R' O
: H+
H
R' O CO
R O C
O
R
H
Cuando la esterificación se realiza con un anhidrido de ácido, se suele utilizar piridina como catalizador; la piridina reac-ciona con el anhidrido dando lugar a una sal de acilamonio muy reactiva. Escribe el mecanismo completo de la siguiente reacción:
OH
N+
_
N
H+
+ CH3 CO O CO CH3
O CO
CH3
CH3 CO
O +
EJERCICIO 8.2
Si se trabaja con cantidades muy pequeñas de un ácido, su éster metílico se sintetiza habitualmente empleando Diazometano (CH2N2); esta reacción no es una sustitución nucleófila en el grupo carbonilo:
R CO
OH +Et2O
R CO
CH2N O CH3 + N22 Esquema de la reacción:
_:
+ _
[diazometano]
H2C N N :+
H2C N N
_+
CH3
R CO
O H_
:+
H2C N N R CO
O :+N NCH3
++ N N:: (N2)
[éster metílico]
_R C
OO + CH3
+R C
OO CH3
VIII

192
HIDRÓLISIS DE ÉSTERES Los ésteres pueden hidrolizarse en medio ácido o básico:
O
R CO
O R' R C R' OH+OH+ H OH
OOR C R C OO R' + HO− −
R' OH+ En medio ácido el equilibrio está parcialmente desplazado hacia la derecha, pero en medio básico la hidrólisis del éster es prácticamente total:
H3O+(50-700C)CH3CH2OHCH3CH2CH2 C
OOEt CH3CH2CH2 C
OOH +
(100 %)
NaOH (20%)+ CH3CH2OH
_CH3 C
OOEt CH3 C
OO Na+
triglicérido (éster de la glicerina con los ácidos Esteárico, Oleico y Linoleico)
NaOH / H2OQ
CH2 O CO
(CH2)16_CH3
CH O CO
(CH2)7_CH CH (CH2)7
_CH3
CH2 O CO
(CH2)7_CH CH CH2 CH CH (CH2)4
_CH3
OCO
CH3_(CH2)16
CHCH3_(CH2)4
[sal sódica del ácido Esteárico]
[sal sódica del ácido Oleico (Z)]
[sal sódica del ácido Linoleico (Z,Z)]
JABÓN
+
CH2 OH
CH OH
CH2 OH
_
CH CH CH (CH2)7 CO
O_
CHCH3_(CH2)7 CH (CH2)7 C
OO
_
Na+
Na+
Na+
ceras: ésteres derivados de ácidos grasos de 24 a 28 átomos de carbono y alcoholes primarios de 16 a 36 átomos de carbono.
aceites: ésteres de glicerina y ácidos carboxílicos (la mayoría insaturados) con 12 o más átomos de carbono.
grasas: ésteres de glicerina y ácidos carboxílicos (la mayoría saturados) con 12 o más átomos de carbono.
VIII

193
Observa que el mecanismo de la reacción de hidrólisis es exactamente el mismo que el de la esterificación, pero escrito al revés. Esto es cierto para cualquier reacción en equilibrio. Esquema de la hidrólisis en medio ácido:
R CO
O R'H3O+
R CO
OH + R' OH Primer paso:
[éster]
R C
O
O R'
H___OH2::
R C
O
O R'
OH2: H
Segundo paso:
R C
O
O R'
: H
O__HH
[adición]R C
O
O R'
: H
O HH
Tercer paso:
:
R C
O
O R'
: H
O HH
R C
O
O R'
: H
OH :OH2
Cuarto paso:
OH2H
R C
O
O R'
: H
OH : OH2
R C
O
O R'
: H
OH : H
Quinto paso:
[alcohol]R C
O
O R'
: H
OH : H
[eliminación]R C
O
O R'
H
OH : H
:
Sexto paso:
OH2:
[ácido]
R C
O
OH
: H :
R C
O
OH
: H OH2
VIII

194
Mecanismo de la reacción en medio básico:
R__C__OR' R__C__O Na+ + R'OHNaOHH2O
_O O
HO_
_
C
O
R
OR'[adición]
C
O
R
OR'HO
[eliminación]R'O+
_
_
C
O
R
OR'HO C
O
RHO
+ [anión del ácido]C
O
ROR'O_
H R' OH[alcohol]
R C
O
O_
H3O ++ (ácido fuerte: HCl)R CO
O_
R CO
OH
[ácido]
+ H2O
SÍNTESIS DE ANHIDRIDOS DE ÁCIDO El anhidrido acético es el más empleado de todos los anhidridos de ácido. Su síntesis industrial se realiza por deshidratación directa del ácido acético; la reacción se lleva a cabo haciendo pasar una corriente gaseosa de ácido acético por un tubo de cuarzo calentado a 8000 C que contiene porcelana pulverizada como catalizador:
8000CCH3 C
OOH H O C
OCH3
OO C
OCH3CH3 C H+ H O
Los anhidridos cíclicos que tienen un anillo de cinco o seis eslabones, se forman muy fácilmente por simple calefacción del ácido dicarboxilico correspondiente
Q+ H2O
Ácido maleico Ánhidrido maleico(89%)
C
C
H
H
O
O
OH
OHO
O
O
QCO2H
CO2H
O
O
O
+ H2O
Ácido ftálico Anhidrido ftálico
VIII

195
Los anhidridos se sintetizan también a partir de cloruros de ácido, por reacción con la sal sódica del ácido o con el mismo ácido libre:
_Na+
C+
C+ NaCl
Cloruro de benzoilo
Benzoato sódico Anhidrido benzoico
O O
O OClC O C
O
Esquema de la reacción con la sal sódica del ácido:
+ Cl_
R C
O
O_
CR
O
Cl[adición]
R C
O
O CR
O
Cl
_
[eliminación]R C
O
O C RO
Cuando se utilizan un cloruro de ácido y el ácido libre para obtener un anhidrido, la reacción se lleva a cabo empleando piridina como catalizador. La Piridina reacciona con el cloruro de ácido dando lugar a un catión acilamonio muy reactivo. Escribe el mecanismo completo de la siguiente reacción:
COCl
+
CO2H
N+ Cl
_
N +
C
O
O C
O
H
EJERCICIO 8.3
FORMACIÓN DE ENLACES C–N
SÍNTESIS DE AMIDAS Las amidas se pueden sintetizar a partir de cloruros de ácido, anhidridos o ésteres:
OR C Cl
ONH3
R'__NH2
R C NH2O O
R C O C RO
R C NH R'O
R C OR' Mecanismo de la reacción con cloruros de ácido:
+ Cl + NH4
_ +H3N: C
R
O
Cl
:[adición]
H2N C
H
R
Cl
O
+
: :_
H2N C
R
Cl
O: H :NH3[eliminación]
H2N C R
O: :
VIII

196
Mecanismo de la reacción con anhidridos de ácido:
C
O
R'
O C
O
R'
:
R2NH
:[adición]
C
O
R'
O C
O
R'
:
R2N
H
+
:_
C
O
R'
O C
O
R'R2N
:H
C
O
R'
O C
O
R'R2N
:HR2NH
:[eliminación]
C
O
R' OC
O
R'R2N
::
+_
+ R2N
H
H+
Escribe el mecanismo completo de la siguiente reacción:EJERCICIO 8.4
NH2:
+
HN
+ MeOHCH3CH2 CO
O Me
CO
CH2CH3
A continuación aparecen algunos ejemplos concretos de síntesis de amidas:
NH3 / H2O
(83 %)
CH3 CHCH3
COCl CH3 CHCH3
CONH2
+ NH3 +
Dietilacetamida (91 %)
bis-Dietilacetamida (9 %)
CH3CH2 CHCH2CH3
COClC6H6 CH3CH2 CH
CH2CH3CONH2 CH3CH2 CH
CH2CH3CO NH CO CH
CH2CH3CH2CH3
Cloroacetamida (84 %)
Cl CH2 CO2EtNH3 / H2O
Cl CH2 CONH2
COCl + 2Me2NHC6H6
CO
N,N-Dimetilciclohexano- carboxamida (89 %)
NMe2
Cloroformiato de etilo
CONH2
CO2Et
CO2H
CO2Et
Biciclo[2,2,2]octano-1,4-dicarboxilato de metilo
+ Cl CO
OEt -HCl
CO
CO2Et
O CO2Et
NH3 / H2O
VIII

197
HIDRÓLISIS DE AMIDAS Las amidas se hidrolizan en medio ácido o básico, transformándose en ácidos carboxílicos:
H3O+R CO
NH2 + R CO
OH + NH4+
_ _
R CO
NH2 + HO R CO
O + NH3 Mecanismo de la hidrólisis en medio ácido:
: H2O
[base]1 [ácido]2 [ácido]1 [base]2
R C O
NH2
H__OH2+
R C OH
NH2+
+
:+
H2O:
R
C
HO
NH2
+
[adición]:
R
C
HO
NH2H2O
[base]1 [ácido]2 [base]2[ácido]1
H2O: H O
H
C+
NH2
R
OH
H3O :+
+ HO C
OH
NH2
R
[base]1 [ácido]2 [ácido]1 [base]2
HO C
OH
NH2
R
H___OH2+
HO C
OH
NH3
R
++ H2O:
:[eliminación]
HO C
OH
NH3
R
+OHC
O
R
H+
:NH3+
:OH2 + H3O+
[ácido]
OH
C OR H+ OH
C OR
H___OH2+
H3N: NH4+ + H2O:
VIII

198
Escribe el mecanismo completo de la hidrólisis de una amida en medio básico:EJERCICIO 8.5
_ + NH3
_R C
ONH2 + HO R C
OO
Las lactonas y las lactamas son ésteres y amidas cíclicas que pueden obtenerse a partir de hidroxiácidos y aminoácidos:
CO2Hα
β
δγ
OH
-H2OO
O
-Valerolactona δÁc. 5-hidroxipentanoico (ác. Valérico)
α
β
δ
ε
γ
-H2O
NH2CO2H N O
H
-CaprolactamaεÁc. 6-aminohexanoico (ác. Caproico)
Escribe el mecanismo de las siguientes reacciones de hidrólisis:EJERCICIO 8.6
O
O
H2OHO_CH2CH2CH2
_CO2H
ác. γ-hidroxibutírico γ-butirolactona
H3O+
anión del ácido δ-aminopentanoico
H2N_CH2CH2CH2CH2_CO2
δvalerolactama
NO
H
_
H2OHO
_
FORMACIÓN DE ENLACES C–H
REDUCCIÓN DE CLORUROS DE ÁCIDO Y AMIDAS Los cloruros de ácido y las amidas se pueden reducir a aldehidos utilizando hidruro de litio y triterbutoxialuminio o de litio y trietoxialuminio:
C__Cl
O
+
Li
Al( OCMe3)3-780C
(MeOCH2CH2)2OC__H
O
+ LiCl + Al(OCMe3)3
(73 %)
H_
+
Hidruro de litio ytriterbutoxialuminio
Los hidruros metálicos actúan como dadores de anión hidruro (H−):
CLi
Al(
H
OCMe3)3Cl
O
C__H Li +Al( OCMe3)3
Cl
O[adición]_
+: ::_
++
VIII

199
[eliminación]C + LiCl + Al(OCMe3)3
OH
Hidruro de litio y trietoxialuminio
Butanal (85 %)
N,N-Dimetilbutiramida
CH3CH2CH2 CO
NMe2 + LiAlH(OCH2CH3)3 CH3CH2CH2 CO
H
FORMACIÓN DE ENLACES C–C
SÍNTESIS DE CETONAS A PARTIR DE ÁCIDOS CARBOXÍLICOS El litio reacciona con los derivados halogenados de alquilo o de arilo, dando lugar a compuestos organolíticos:
R__X: + Li [R__X:] + Li+_
[R__X:]_
R + :X:_
R + Li R__Li (organolítico)
CH3CH2CH2CH2_Br + 2Li
Tetrahidrofurano00C
CH3CH2CH2CH2_Li + LiBr
1-Butil-litio
Cl + 2Li Tetrahidrofurano
00CLi + LiCl
Fenil-litio Los compuestos organolíticos reaccionan con los ácidos carboxílicos dando lugar a cetonas:
CH3_(CH2)4_CO2H
Ác. hexanoico
1)CH3Li (2 eq.)
2)H3O+ CH3_(CH2)4_CO_CH3
2-Heptanona
(83 %)
Mecanismo de la reacción:
δ+
δ−CH3
LiR C
O
O
H
R C
O
O Li + CH4
δ+
δ−
2H3O+
+ H2O + 2LiC
O
R
O
H3C
Li
Li
[adición]CH3 C
O
O
R
Li
Li
CH3 C
O
O
R
H
H
+
VIII

200
:OH2[eliminación]
+ H3O+ + H2Ocetona
CH3 C
O
O
R
H
H H OH2+
:
CH3 C
O
O
R
H
H H+
CH3 C
O
R
EJERCICIO 8.7Escribe el mecanismo de las siguientes reacciones:
O
O
O
+ H2OH3O+ C__OH
C__OH
O
OAnhidrido ftálico
O
O
O
+ MeOHC__OMe
C__OH
O
O
H3O+
O
O
O
+ 2MeNH2
H3O+ C__NHMe
C__O
O
O
_ + MeNH3+
CONDENSACIÓN DE CLAISEN La condensación de Claisen es una reacción entre dos ésteres con hidrógenos en α, catalizada por bases, que conduce a β-cetoésteres:
β
-cetoésterβ
α
+ +R CH2 CO
OEtRCH
CO2EtO
H R CH2 C CH
RCO2Et EtOH
Los hidrógenos en α de los ésteres son ácidos y pueden ser extraídos por una base, originando aniones enolato:
α
+ H_B
[anión enolato]
+R CH2 C
O
OEt + :B R CH C
O
OEt_ :: *
*R CH C
O
OEt
::_
(1) (2)
De las dos formas resonantes dibujadas para representar el anión enolato, (2) es la que más se aproxima a la estructura electrónica de la molécula, ya que la carga está situada sobre el oxígeno, un átomo más electronegativo que el carbono.
VIII

201
Sin embargo, con el fin de simplificar los dibujos, utilizaremos normalmente la estructura (1) para escribir los mecanismos de reacción. Mecanismo simplificado:
[ácido]1 [base]2
R CH2 C
O
OEt + :B
[ácido]2[base]1
R CH C
O
OEt + H_B_ ::
+
+ EtO_
-cetoésterβ
R CH
CO2Et
_C
CH2R
O
OEt[adición]
R CH
CO2Et
C
CH2R
O
OEt
_
[eliminación]R CH
CO2Et
C CH2R
O
Él mecanismo completo de la reacción es el siguiente: Formación del anión enolato:
Keq = 10-6CH3_CO2Et + EtO CH2
_CO2Et + EtOH_ _
[ácido]1 [ácido]2[base]1[base]2
pKa 23 pKa 17 Adición del enolato (nucleófilo) al grupo carbonilo de otra molécula de éster, eliminación de un anión alcóxido y formación del β-cetoéster:
Keq = 1+ EtO__
C
CH3
O
OEt[adición]
CH2EtO2C C
CH3
O
OEt
_
[eliminación]CH2EtO2C C CH3
O
EtO2C CH2
Formación del enolato del β-cetoéster por reacción con la base. Este último paso es crucial, porque de todos los equilibrios es el único que está desplazado hacia la derecha:
[base]2
Keq = 106
pKa 11
_
[ácido]1 [base]1
pKa 17
+ EtO_
CH2EtO2C C CH3
O
CHEtO2C C CH3
O
+ EtOH
[ácido]2
Para recuperar el éster se añade un ácido fuerte a la mezcla de reacción:
+_CHEtO2C C CH3
O
+ H3O CH2EtO2C C CH3
O
+ H2O
[base]1 [ácido]2 [ácido]1 [base]2pKa -1,7 pKa 11
Keq = 5 x 1012
La formación del enolato del β-cetoéster determina el rendimiento global de la secuencia de reacciones. Si el éster empleado en la reacción tiene únicamente un átomo de hidrógeno en el carbonio α, no se puede formar el enolato final y la condensación de Claisen sólo tiene lugar si se utilizan bases muy fuertes:
VIII

202
[un sólo Hα]
x no hay reacciónCH3 CH
CH3
CO2Et
Para aumentar el rendimiento de la reacción se puede eliminar el alcohol según se va formando, ya que el equilibrio se desplaza entonces a la derecha.
CH3_CO2Et
1)EtO-Na+ ; EtOH
2)AcOH ; H2OCH3
_CO_CH2_CO2Et + EtOH
(75 %)
CH3_CO2CH2CHMe2
1)i-BuO-Na+ ; i-BuOH2)AcOH ; H2O
CH3_CO_CH2
_CO2CHMe2 + Me2CHCH2OH
Acetato de isobutilo (71 %)
CH3_(CH2)4_CO2Et
1)EtO-Na+ ; EtOH
2)AcOH ; H2O(80 %)
3-Oxooctanoato de etilo
CH3 (CH2)4 CO CHCO2Et
(CH2)3 CH3 + EtOH
Existe una forma alternativa de mejorar el rendimiento de la condensación de Claisen. Consiste en utilizar bases muy fuertes, de modo que el primer paso de la reacción (la formación del enolato del éster) sea prácticamente irreversible.
+Na NH2 NCHMe2
CHMe2
_
Li_ _
Hidruro sódico Amiduro sódico Diisopropilamiduro de litio (LDA)
Na H+ +
El amiduro sódico se obtiene por reacción entre sodio metálico y amoniaco líquido (no amoniaco disuelto en agua):
2 NH3(liq.) + Na 2 Na NH2 + H2
-330C + _
El diisopropilamiduro de litio se sintetiza a partir de n-butil-litio (ver p. 22):
LiCH3CH2CH2CH2_Li + H N
CHMe2
CHMe2
+N
CHMe2
CHMe2
_
+ CH3CH2CH2CH3
(LDA) Empleando cualquiera de estas bases la constante de equilibrio del primer paso es mucho mayor que uno:
CH3_CO2Et + Na H
Hidruro sódicopKa 23
CH2_CO2Et + H2
_
pKa 35
Keq = 1012+ _
VIII

203
CH3
_CO2Et + Na+ NH2
Amiduro sódicopKa 23
CH2-CO2Et + NH3
_
pKa 35
Keq = 1012_
CH3_CO2Et +
pKa 23
CH2_CO2Et +
_
pKa 36
Keq = 1013+Li NCHMe2
CHMe2
_
NCHMe2
CHMe2
H
Diisopropilamiduro de litio (LDA)
El diisopropilamiduro de litio (LDA) tiene la ventaja añadida de ser muy poco nucleófilo; debido a su gran volumen no reacciona con el átomo de carbono del C=O, más impedido estéricamente que los hidrógenos del Cα. Además, permite realizar la condensación de Claisen con ésteres que tienen un sólo átomo de hidrógeno en el carbono α:
[un sólo Hα]
LDA sí hay reacciónCH3 CHCH3
CO2Et
Las reacciones de condensación entre dos ésteres diferentes que tengan hidrógenos en α (condensaciones mixtas) dan lugar a mezclas de los cuatro β-cetoésteres posibles:
R_CH2_CO2Et + R'_CH2
_CO2Et
:B_
R CH2 CO
CHR
CO2Et + R' CH2 CO
CHR'
CO2Et + R CH2 CO
CHR'
CO2Et + R' CH2 CO
CHR
CO2Et
CH2 CO
OEt + :B R CH CO
OEt + H_B_ ::
+
_ R CHCO2Et
CO
CH2 + EtO_
-cetoésterβ
R CHCO2Et
CO
CH2R
OEt
Escribe el mecanismo simplificado de las reacciones que tienen lugar cuando se mezclan antidades equimole-culares de acetato de etilo y fenilacetato de etilo con etóxido sódico en etanol.
Mecanismo simplificado:
EJERCICIO 8.8
R
R
Las condensaciones mixtas son útiles en síntesis cuando uno de los ésteres carece de hidrógenos en α:
Formiato Carbonato Oxalato Benzoato
H CO
OR RO CO
OR RO CO
CO
OR CO
OR
VIII

204
Para minimizar la autocondensación del éster que sí tiene hidrógenos en α, se mezclan primero la base y el éster que no tiene hidrógenos en α y a continuación se añade lentamente el otro éster. Estos son algunos ejemplos de condensaciones mixtas con un éster sin hidrógenos en α:
Oxalato de dietilo Propanoato de etilo 3-Metil-2-oxobutanodioato de dietilo
(70 %)
α
EtO CO
CO
OEt + CH3CH2CO2Et1)EtONa ; EtOH
2)AcOH ; H2OEtO2C C
OCHCH3
CO2Et + EtOH
3-Fenil-2-metil-3-oxopropanoato de etilo
(56 %)
α
C6H5 CO
OMe +
Benzoato de metilo Propanoato de etilo
CH3CH2CO2Et1)NaH ; C6H6
2)AcOH ; H2OC6H5 C
OCHCH3
CO2Et + MeOH
H_CO2Et + C6H5_CH2
_CO2Et1)EtONa ; EtOH
2)H3O+
Formiatode etilo
Fenilacetato de etilo 2-Fenil-2-formilacetato de etilo
(90 %)
α
C6H5 CHCO2Et
CHO
En las condensaciones mixtas se puede utilizar LDA para preparar el enolato del éster (reacción totalmente desplazada a la derecha Keq >> 1) y a continuación añadir un cloruro de ácido:
3-Oxopentanoato de etilo(60 %)
CH3_CO2Et
2) CH3CH2COClCH3CH2 C
OCH2
1) LDACO2Et
CH3_CO2Et
LDATetrahidrofurano
; -780C
_
Keq = 1013H2C C
O
OEt
Li +
EtO C
O
CH2
_
CEt
O
Cl[adición]
EtO C
O
CH2 C
O
Et
Cl
_[eliminación]
CH3CH2 CO
CH2 CO2Et
EJERCICIO 8.9Escribe el mecanismo simplificado de las reacciones que aparecen a continuación, empleando la estructura resonante del enolato que tiene la carga en el átomo de oxígeno.
1. C6H5_CH2
_CO2Et + H_CO2Et1)EtO
_
2)H3O+A2.
Na+ NH2_
NH3 (liq.)EtO C
OOEtCH3 CH
CH3
CO2Et
3.LDA
BCH3COClC6H5 CH
CH3
CO2Et
VIII

205
ESTRATEGIA PARA LA SÍNTESIS DE β-CETOÉSTERES
esta parte de la molécula proviene del ataque nucleófilo al grupo C=Oen el éster RCH2CO2Et o en el cloruro de ácido RCH2COCl
esta parte de la molécula proviene del enolato del éster R'CH2CO2Et
RCH2 CO CH CO2EtR'
EJERCICIO RESUELTO¿A partir de qué compuestos se puede sintetizar el 4-Fenil-2-metil-3-oxobutanoato de etilo?
RESPUESTA
resto acilo en el fenilacetato de etiloo en el cloruro del ácido fenilacético
enolato del propanoato de etilo
C6H5CH2CO CHCH3
CO2Et
Alternativa 1: propanoato de etilo + fenilacetato de etilo
CH3CH2_CO2Et
NaHCH3CH_CO2Et
_
CH3 CH
CO2Et
_C
O
OEt
CH2C6H5
CH2 C
O
CH CO2Et
CH3
Alternativa 2: propanoato de etilo + cloruro del ácido fenilacético
C6H5
LDACH3 CH C
O
OEt
_Li+
CH3CH2_CO2Et
O C
CHCH3
_OEt
C
O
CH2C6H5
Cl CH3 CHCO2Et
CO CH2 + Cl_
C6H5
Los β-cetoésteres, como cualquier éster, se hidrolizan en medio ácido dando lugar a β-cetoácidos:
C6H5_CO_CH2
_CO2Et C6H5_CO_CH2
_CO2HH3O+
β
β−cetoácido
VIII

206
Los β-cetoácidos se descarboxilan (pierden CO2) fácilmente por calefacción:
CCH2
C
OH
O
C6H5
β-cetoácido
QC
C6H5
OH
CH2
+ O=C=O (CO2)
forma enólica de una cetona
O
Las cetonas están en equilibrio con sus correspondientes formas enólicas (ver detalles en Tema 9, p. 229):
enol
C6H5 C
O
CH2
H
C6H5 C
O
CH3
cetona Estas dos reacciones (hidrólisis del β-cetoéster y descarboxilación del β-cetoácido) permiten sintetizar cetonas a partir de β-cetoésteres:
C6H5_CH2
_CO2Et1)LDA
2)CH3CO2Etβ-cetoéster
H3O+
β-cetoácido
Q
-CO2C6H5
_CH2_CO_CH3
Bencil metil cetonaC6H5 CH
CO2EtCO CH3 C6H5 CH
CO2HCO CH3
REACCIÓN DE CLAISEN Los átomos de hidrógeno unidos al carbono α de las cetonas son algo más ácidos (pKa 19) que los de un éster (pKa 23) Este hecho permite realizar condensaciones mixtas empleando como nucleófilo el enolato de una cetona; el resultado de la reacción son 1,3-dicetonas:
1)NaH ; Et2O2)H3O+
CH3_CO_CH3 CH3
_CO_
2,4-Pentanodiona
(85 %)
CH2_CO_CH3CH3
_CO2Et +
CH3 CO CH3 + NaH CH3 C
O
CH2
_CH3 C
O
CH2
_
Na+ + 1/2 H2
CH3 C
O
CH2
_C
O
CH3
OEt[adición]
CH3 C
O
CH2 C
CH3
OEt
O_
[eliminación]CH3 CO CH2 CO CH3 + EtO
_
OH
α
+ H__CO2Et1)EtONa ; EtOH
2)H3O+
CHO
2-Formilciclohexanona
(59 %)
O
VIII

207
O
H1)NaH ; Et2O
2)H3O+
OCO2Et
(2-Oxocicloheptano)- carboxilato de etilo
(91-94 %)
Carbonatode dietilo
α
EtO CO
OEt+
Los γ- o δ-cetoésteres dan lugar a reacciones de Claisen intramoleculares en las que el enolato de la cetona reacciona con el grupo carbonilo del éster, originando 1,3-dicetonas cíclicas con cinco o seis eslabones:
CH3CH2_CO_CH2CH2
_CO2Et
α
γ
1)NaOMe ; MeOH
2)H3O+
Me
2-Metil-1,3-ciclopentanodiona
(70-71 %)
OO
O
Me
O O
MeOEt
OO
Me
OEtO
MeOEt
O
α
MeO_
_
_
+ EtO_[eliminación][adición]
O
α
δ
CH3CH2_CO_CH2CH2CH2
_CO2Et1)NaH ; Et2O
2)H3O+ O OMe
2-Metil-1,3-ciclohexanodiona
ENOLATOS TERMODINÁMICOS Y ENOLATOS CINÉTICOS Las cetonas que tienen dos tipos diferentes de hidrógenos en alfa pueden dar lugar a dos clases distintas de aniones enolato, dependiendo de cuál sea el hidrógeno extraído por la base. (ver detalles en Tema 9, p. 232) Cuando la base elimina el hidrógeno unido al carbono más sustituido, se obtiene el enolato más estable, ya que su doble enlace es también el más sustituido. Por el contrario, si la base extrae el hidrógeno del carbono menos sustituido se forma el enolato con el doble enlace también menos sustituido, es decir, el menos estable. Al enolato más estable se le llama enolato termodinámico y al menos estable enolato cinético.
enolato con el doble enlace menos sustituido / menos estable
enolato cinético
enolato con el doble enlace más sustituido / más estable
enolato termodinámico+ B_
α1α2
α2
α1
CH3 CO
CHCH3
CH3
CH3 CO
CCH3
CH3_ CH3 CO
CCH3
CH3
_
+ HB
CH2 CO
CHCH3
CH3
_CH2 C
OCHCH3
CH3
_
+ HB
VIII

208
El enolato termodinámico es el mayoritario cuando se deja que la reacción alcance el equilibrio (temperatura alta y bases moderadamente fuertes) El enolato cinético se forma más rápidamente que el termodinámico (menor impedimento estéreo), y es el mayoritario si los productos se aíslan antes que la reacción alcance el equilibrio, la temperatura es baja y la base muy fuerte e impedida estéricamente (LDA). El siguiente ejemplo es una reacción sometida a control cinético (se forma el enolato menos sustituido a partir del Hα del grupo metilo de la cetona, no del Hα del grupo isopropilo):
3-Metil-2-butanona
CH3 CO
CHCH3
CH3 + CH3 CHCH3
CO2Et
Isobutirato de etilo
LDA
2,6-Dimetil-3,5-heptanodiona
(70 %)
CH3 CHCH3
CO CH2 CO CHCH3
CH3
REACCIÓN DE DIECKMANN La reacción de Dieckmann es una condensación de Claisen intramolecular que tiene lugar cuando un diéster reacciona consigo mismo, originando β-cetoésteres cíclicos con anillos de cinco a siete eslabones.
O
CO2Et
2-Oxociclopentanocarboxilato de etilo
(86 %)
EtO CO
(CH2)4 CO
OEt
Hexanodioato de dietilo
1)KH ; Et2O
2)H3O+
Mecanismo de la reacción:
CO2Et
OEtO
CO2Et
_
α
OKH
CO2Et
OEt
O
_[adición] OEt
O
CO2EtOEt
_ + EtOHOEt
_
[eliminación]
CO2Et
O
H
O
CO2Et
_
O
CO2Et
_
00 C
CH3CO2H (10 %)O
CO2Et
H
enol
O
CO2Et
Puede suceder que el diéster sometido a la reacción de Dieckmann tenga dos tipos diferentes de Hα:
α α
EtO2C CHCH3
CH2CH2CH2 CH2 CO2Et
VIII

En un caso así, con independencia del tipo de base utilizada, siempre se forma el enolato correspondiente al carbono menos sustituido:
209
C__OEtMe
EtO_
Me O
CO2Et
2-Etoxicarbonil-6-metil-ciclohexanona
CO2Et_C
HC CO2Et
OOEt
MeO
EtO_
_
O
CO2Et
2-Etoxicarbonil-2-metil-
Me
XCHC CO2Et
Me
OOEt C
C CO2Et
Me
OOEt
EJERCICIO 8.10Escribe el meca e las e Dieckmann:
Reacción 1:
ciclohexanona (no se forma)
Este resultado puede deberse a las interacciones estéricas entre los sustituyentes situados en enlaces axiales en el estado de transición del anión enolato.
Reacción 3:
N
Reacción 2:
nismo simplificado d
MeO2C
MeO2C
siguientes reacciones d
EtO_
+ CO2
(68 %)
MeMe
O
H
H CO2Et
CH2_CO2Et
Me
Me
(60 %)
CH2CH2 CO2
MeOH
Me
CH CH2CH2
CH
CO2Me
CO2Et
(CH2)4__CO2Et
1)EtO_
2)H3O+ N
MeO
O
(
2C
O
O
CO2Me
27 %)
EtOHA
H3O+
MeONa
VIII

VIII
210
REACCIONES DE SUSTITUCIÓN NUCLEÓFILA EN UN CARBONO NO SATURADO
RCH2_CO_Cl
LiAlH(t-BuO)3R'OH
H_OHSOCl2
RCH2_COOH
H_OHRCH2COCl
RCH2_CO_O_COCH2R
H_OH/H+
R'NH2
R'NH2
RCH2_CO_OR'
R'OH
HO /H2O
R'OH
MeCOCH2_CO_CH2R
1):B
2)MeCOMe
RCH2_CO_NHR'
RCH2_CHO
1)LiAlH(OEt)32)H+/ H2O
RCH2_CO_R'
R'Li
RCH2 COCO2R'
CHR :B
_

VIII
211
EJERCICIOS ADICIONALES
8.1 Averigua la estructura de los compuestos que aparecen en las siguientes secuencias de reacciones. Secuencia 1:
Me OH
OCH2N2 A(C3H6O2)
C2H5NH2 B(C4H9ON)HLiAl(OEt)3 C(C2H4O)
Me
Secuencia 2:
OH
O
PCl3 AMe CO2H C6H5 NH2B(C6H10O3)
H3O+
H2O
D(C3H6O2) + E(C7H9N)C(C10H13ON)
H5
Secuencia 3:
C6 Cl
OH
H3O+
2OA
C6H5LiB
O
Secuencia 4:
ONH3
H2OA(C5H11O2N)
H3O+
H2OB(C5H10O3)
H3O+
H2OC(
C6H5
C5H8O2)
Secuencia 5:
O C6H5
O OC6H5 OH
A(C14H12O2)MeNH2 B(C8H9ON)
H3O+
H2OC(C7H6O2)
Me LiD(C11H14O)

VIII
212 8.2 Completa las reacciones siguientes, escribiendo la fórmula de los compuestos que intervienen y, en los casos no indicados, diciendo los reactivos necesarios para efectuar cada transformación.
CH3 CH COCH3
CO2Et 1)
2)CH3 CH2 CO2Et
1)LDA
2)ICH3
+H3O
NH3 CH3 CH CONH2
CH3
+H3O
CH3Li
1)
2)CH3 CH CO
CH3
CH2 CO CH3
1)
Q
1)HO_
+2)H3O
CH3CH2CO2H
CH3 CH2 CO O CO CH2 CH3CH3NH2
CH3 CH2 CHO
CH3 CH CHOCH3
HO (dil.)_
+2)H3O

213
8.3 Escribe un mecanismo razonable para las siguientes reacciones:
CO2Et
+ Cl CO
OEt
Cloroformiato de etilo
H2O
CO2Et
OOO OH OEt
ONH3
H2O
CO2Et
O NH2
8.4 Averigua la estructura de los compuestos que aparecen en las siguientes secuencias de reacciones. Secuencia 1:
C6H5 Me
O
H CO2Et
1)HNa
2)A(C9H8O2)
Secuencia 2:
C6H5 Me
O1)HNa
2)OEt
OEtO
A(C11H12O3)
Secuencia 3:
C6H5 Me
O
A(C12H12O4)1)HNa
2)EtO2C_CO2Et Secuencia 4:
C6H5 Me
O1)HNa
2)C6H5CO2EtA(C15H12O2)
8.5 Averigua la estructura de los compuestos que aparecen en las siguientes secuencias de reacciones. Secuencia 1:
CH3CO2EtMe
COCl2)
1)LDAA(C8H14O3)
H3O+
QB(C5H10O)
Secuencia 2:
CH3CH2CH2CO2EtCH3COCl
1)LDA
2)A(C8H14O3)
H3O+
QB(C5H10O)
Secuencia 3:
CH3CO2Et1)LDA
2)Me COClA(C9H16O3)
H3O+
QB(C6H12O)
VIII

214
Secuencia 4:
CH3CH2CH2CO2EtCH3CH2COCl
1)LDA
2)A(C9H16O3)
H3O+
Q B(C6H12O)
8.6 Averigua la estructura de los compuestos que aparecen en las siguientes secuencias de reacciones. Secuencia 1:
Me Me
O1)HNa
2)CH3CO2EtA(C5H8O2)
Secuencia 2:
1)HNa
2)Me Me
O
CH3CH2CO2EtA(C6H10O2)
Secuencia 3:
C6H5 Me
O
C6H5CO2Et
1)HNa
2)A(C15H12O2)
Secuencia 4:
C6H5
OMe
1)HNa
2)C6H5CO2EtA(C16H14O2)
Secuencia 5:
C6H5 Me
O1)HNa
2)C6H5CH2CO2EtA(C16H14O2)
Secuencia 6:
C6H5
OMe
1)HNa
2)C6H5CH2CO2EtA(C17H16O2)
8.7 Averigua la estructura de los compuestos que aparecen en las siguientes secuencias de reacciones. Secuencia 1:
A(C8H8O) + B(C9H10O2)EtO
_
C6H5 C6H5
O O
Secuencia 2:
2 A(C10H12O2)EtO
_
C6H5 CO2EtO
C6H5
VIII

215
Secuencia 3:
C6H5 CO2Et+EtO
_
A(C5H10O3) C6H5
CO2Et
CO2Et Secuencia 4:
MeO
+ A(C3H6O2)EtO
_ MeO
CHO
VIII

9. REACCIONES DE SUSTITUCIÓN NUCLEÓFILA EN UN CARBONO SATURADO
GENERALIDADES Los métodos más usados en el estudio de los mecanismos de este tipo de reacciones son los cinéticos (medida de las velocidades de reacción) y los estereoquímicos. Los estudios cinéticos muestran el tiempo que tarda la reacción en transcurrir y los estereoquímicos la relación que existe entre las configuraciones de las sustancias reaccionantes y los productos. La combinación de ambos tipos de información puede dar una idea detallada de cómo el reactivo y el sustrato interaccionan durante la reacción:
__CH2
____Z
R
Nu___CH2__R + Z:Nu
Los reactivos nucleófilos (Nu) son especies que tienen un par de electrones no compartido (bases de Lewis):
NC , RO , HO , Br , C6H5O , R-CO2 , R_OH , H2O: , R3N:_ _ _ _ _ _
Los sustratos (R-Z) más comunes en las reacciones de sustitución nucleófila son los derivados halogenados (R-X; X=Cl, Br, I)
R_X + Nu: R_Nu+ + X_
R_X + Nu R_Nu + X__
Por ejemplo:
+ ICH2 CH CH2I + CN_
CH2 CH CH2 CN_
CH3CH2Cl + :NH3 CH3CH2NH3 + Cl
+CH3CH2NH2
+ Cl + H2OHO__ _
MECANISMO DE LAS REACCIONES DE SUSTITUCIÓN NUCLEÓFILA En general, estas reacciones transcurren mediante dos tipos de mecanismos distintos. 1. El proceso transcurre en un solo paso, de una manera concertada, de forma que la ruptura del enlace R-Z es simultánea a la formación del enlace R-Nu; la cinética de estas reacciones es de primer orden respecto del sustrato y del nucleófilo, es decir, de segundo orden total. Durante la reacción, dos moléculas experimentan cambio de valencia, por consiguiente, se trata de una reacción bimolecular y se representa por SN2. La reacción pasa por un estado de transición intermedio en el que ambos grupos, entrante y saliente, están unidos al mismo átomo de carbono:
v = k2 [CH3Br][HO ]HO + CH3_Br
_ SN2[HO...CH3...Br]
δ− δ−
HO_CH3 + Br_ _
IX

217
2. El proceso transcurre en dos pasos: en el primero se produce la ionización de R-Z y se libera el catión carbonio R+
que, en la segunda fase, se une al nucleófilo:
v = k1 [R_X]R_Z lenta [R....Z]δ+ δ−
R+ + Z_
R+ + Nu_ rápida [R....Nu]
δ+ δ−
R_Nu La velocidad y la naturaleza de los productos quedan determinadas en fases diferentes: la fase determinante de la velocidad es la primera, que es relativamente lenta, pues supone la ionización de un compuesto covalente; su estado de transición será el punto de máximo contenido energético en el progresivo debilitamiento del enlace entre R y Z, y por ello, se considera el estado de transición del proceso total. La segunda fase fija la naturaleza de los productos y es rápida, ya que supone la neutralización de un carbocatión. Como en la fase limitante de la velocidad sólo una molécula experimenta cambio de valencia, la reacción se llama unimolecular y se representa por SN1. Por ejemplo:
v = k1[t-BuBr]CH3 C
CH3
CH3
Br + HO_ SN1
CH3 C
CH3
CH3
OH + Br_
ESTEREOQUÍMICA DE LAS REACCIONES DE SUSTITUCIÓN Las reacciones de sustitución nucleófila bimoleculares SN2 tienen lugar con una inversión completa de la geometría del sustrato:
Cl
Me
EtH
HO
Me
EtH
+ Cl
Me
Et Cl
H
Me
HO Et
H
2 2HO
_ _
(2R) (2S)
2 2
Si la reacción se lleva a cabo en un compuesto con dos átomos de carbono asimétricos, el proceso de inversión conduce a un diastereoisómero del sustrato:
1
(1R,3S) (1S,3S)NC
Br_ _Me
(cis)
13
2
4 5
_
Me
(trans)
3
Br
H
C3(CCH)(CHH)C4
H
CN
C3(CCH)(CHH)C4
Br
HH H
H
CN
La inversión de la configuración en las reacciones de sustitución nucleófila se denomina frecuentemente Inversión de Walden, debido a su descubridor.
IX

218
El que una reacción SN2 transcurra con inversión de la configuración, no significa necesariamente que cuando tiene lugar una inversión de la configuración el proceso transcurra mediante un mecanismo SN2. Esto lleva a suponer que el estado de transición de una reacción SN2 tiene la forma de bipirámide trigonal, con los grupos entrante (Nu) y saliente (Z) ocupando los extremos del eje principal de la bipirámide:
Z
R1
R3
R2
+ Z..... .....Nu ZC
R3
Nu_ _
R2
R1
Nu
R1
R3
R2
El hecho de que la inversión de la configuración acompañe necesariamente a las reacciones SN2, es debido a que este tipo de reacciones son concertadas. Las reacciones de sustitución nucleófila monomoleculares SN1 transcurren en dos etapas:
C6H5 CH
Cl
CH3 C6H5 CH CH3+
+ Cl_
C6H5 CH CH3+
+ H2O C6H5 CH CH3
OH2+
H2OC6H5 CH CH3
OH+ H3O+
Si el producto de partida es ópticamente activo, el producto final es un racémico:
+
Cl
MeC6H5H
MeC6H5
H
1
2
Me
C6H5
H
1
2
OH
C6H5 Me
H
(1S)
HO
Cl_ H_OH
Me
C6H5H
HO OH
C6H5 Me
H
(1R)(1R)
Cuando el sustrato tiene varios átomos de carbono asimétricos, el producto formado será una mezcla de diastereoisómeros.
El tratamiento del (2R)2-Yodooctano con anión yoduro radioactivo produce [(2R) + (2S)] 2-Yodooctano radio-activo. La velocidad de racemización es el doble que la de incorporación de iodo radioactivo. Explica mediante qué tipo de mecanismo transcurre la reacción.
EJERCICIO 9.1
IX

219
REACCIONES DE TRANSPOSICIÓN CARBOCATIONES
En las reacciones SN1 pueden tener lugar transposiciones de Wagner-Meerwein en el catión carbonio intermedio:
CH3 CCH3
CH3
CH2 Brlenta
CH3 CCH3
CH3
CH2+
+ Br_
rápida
2-Metil-2-butanol
CH3 C
Me
MeCH2 CH3 C
MeCH2
Me
+
+ H_OH CH3 COH
MeCH2 Me
El carbocatión formado inicialmente es primario, menos estable que uno terciario; la emigración (transposición) de un grupo metilo permite que se origine el carbocatión más estable. Este carbocatión reacciona posteriormente con agua para dar un alcohol con un esqueleto carbonado isómero del que poseía el sustrato inicial. Puesto que un carbocatión saturado es tanto más estable cuanto mayor número de grupos alquilo están unidos a él, el orden de reactividad será: terciarios > secundarios > primarios.
R_Z R+ + Z (en H-CO2H a 1000 C)
CH3CH2 + Br v = 1,00
v = 26,1
+
v = 108
_
_
CH3CHCH3
CH3CHCH3
++ Br
_Br
CH3CH2 Br
CH3C
CH3
CH3
Br CH3C
CH3
CH3
+ + Br_
Velocidades relativas de reacciones SN1
IMPEDIMENTO ESTÉREO DE LOS SUSTITUYENTES EN EL ESTADO DE TRANSICIÓN En las reacciones SN1 el carbocatión intermedio plano contiene un carbono unido a tres radicales, mientras que el sustrato posee un carbono tetraédrico. En el estado de transición entre ambos, los sustituyentes unidos a dicho carbono están menos comprimidos unos contra otros; por consiguiente, el paso del sustrato al estado de transición está energéticamente favorecido, y lo estará tanto más cuanto más voluminosos sean los sustituyentes del carbocatión. Como el hidrógeno es muy poco voluminoso, el proceso será tanto más fácil cuanto mayor sea el volumen de los sustituyentes unidos al carbono que da origen al carbocatión. Es decir: terciario > secundario > primario.
IX

220
estado de transición favorecido energéticamente(grupos menos comprimidos)
(SN1)δ+ δ−
Z
RH
R
C Z
RH
R
RH
R
+ + Z_
Por el contrario, en las reacciones SN2, el carbono en el que tiene lugar la sustitución está unido a cinco átomos, mientras que el sustrato lo está a cuatro. En el estado de transición la aglomeración crece y la velocidad de reacción debe disminuir cuando aumenta el tamaño de los sustituyentes unidos al carbono en el que ocurre la sustitución:
Z
R
RR
+ Z..... .....Nu ZC
R
Nu_ _
R
R
Nu
R
RR
estado de transición desfavorecido energéticamente (grupos más comprimidos)
(SN2)δ− δ−
R_Br + IK R_I + BrK (en acetona a 250C)
CH3_Br CH3CH2
_Br
12.500.000 85.000 650 50
CH3CH2_Br
85.000
CH3CH2CH2_Br
70.000 3.000 1
Velocidades relativas de reacciones SN2
CH3 CHCH3
Br CH3 CCH3
BrCH3
CH3 CHCH3
CH2 Br CH3 CCH3
CH2 BrCH3
APLICACIONES DE LAS REACCIONES DE SUSTITUCIÓN NUCLEÓFILA EN SÍNTESIS
FORMACIÓN DE ENLACES C-O
SÍNTESIS DE ALCOHOLES, ÉTERES Y ÉSTERES Las reacciones de solvolisis (el nucleófilo es el disolvente) transcurren a través de un mecanismo SN1:
(1)
2-Propanol
CH3 CHCH3
Br + H_OH QSN1
CH3 CHCH3
OH + HBr
IX

221
(CH3)3C_O_CH2CH3 + (2)
N+H
Cl_
terc-Butil etil éter
CH3CH2_OH + (CH3)3C_Cl N
SN1
(3)
Acetato de butilo
CH3 CO
OH CH3(CH2)3_Br+Q
SN1CH3 C
OO +(CH2)3 CH3 HBr
Si se emplea agua como disolvente (1) el producto obtenido es un alcohol; si el disolvente es un alcohol (etanol en el ejemplo) (2) se llega a un éter y cuando se trata de un ácido (acético en el ejemplo) (3) el producto final es un éster. En la etanolisis del cloruro de t-butilo se añade al medio de reacción piridina (una amina, básica) con el fin de eliminar el HCl que se desprende y desplazar el equilibrio en el sentido de la formación del éter. Otro método consiste en utilizar oxianiones como reactivos nucleófilos; este procedimiento se utiliza con preferencia al anterior y transcurre mediante un mecanismo SN2:
2-Propanol
Na+HO + + NaBr_
CH3 CHCH3
BrH2O
C2H5OHCH3 CH
CH3
OH
(CH3)3CO K+ + CH3CH2_Br
C6H6 (CH3)3C_O_CH2CH3 + BrK
terc-Butil etil éter
_
Na+ +C2H5OH
+ BrNa_
Butanoato de butilo
CH3CH2CH2 CO
O CH3(CH2)3_Br CH3CH2CH2 CO
O (CH2)3 CH3
La reacción de un estereoisómero ópticamente activo del 1,3-Dibromociclohexano con NaOH transcurre a través de un mecanismo SN2 y da lugar a (1S,3S) 1,3-Ciclohexanodiol. Dibuja una representación tridimensional del 1,3-Dibromociclohexano de partida.
EJERCICIO 9.2
La reacción SN2 se puede utilizar para provocar la inversión de la configuración en un determinado centro asimétrico:
HCONMe2
CH3
BrH
H
SN2CH3CO2
_CH3
HO
HOCH3C
H2OHO
_CH3
HHO
H
+ CH3CO2_
La ventaja fundamental de las reacciones SN2 frente a las SN1 (cuando ambas se emplean con fines sintéticos) es que en los procesos SN1 tienen lugar con cierta frecuencia transposiciones moleculares que dan lugar a mezclas de productos:
IX

222
1-Cloro-3-metil-2-buteno
CH3 CCH3
CH CH2Cl + H OHSN1
acetonaCH3 C
CH3CH CH2OH
3-Metil-2-buten-1-ol
+ CH3 CCH3
CH CH2OH
2-Metil-3-buten-2-ol En este caso se trata de una transposición alílica: el catión alilo formado inicialmente puede reaccionar con el nucleófilo (H2O) en dos posiciones distintas, obteniéndose dos productos de sustitución:
CH3 CCH3
CH CH2ClSN1
CH3 CCH3
CH CH2 CH3 CCH3
CH CH2+
+
La reacción del 3Fenil-1-cloro-2-buteno en acetona acuosa origina una mezcla de 3-Fenil-2-buten-1-ol y 2-Fenil-3-buten-2-ol ópticamente activo. Escribe el mecanismo de las reacciones que tienen lugar.
EJERCICIO 9.3
La competición entre las reacciones de sustitución y eliminación es un factor a tener en cuenta a la hora de idear la síntesis concreta de un compuesto. Por ejemplo, en la síntesis de éteres de Williamson, el empleo de halogenuros terciarios, junto con nucleófilos fuertes, suele dar lugar casi exclusivamente a reacciones de eliminación:
Br + CH3CH2OH
(100 %)
CH3CH2O H__CH2___C____Br
CH3
CH3Bromuro de terc-butilo
CH2 CCH3
CH3 +__
Si se recurre a una reacción de solvolisis (el alcohol como nucleófilo) entonces el haluro de etilo es muy poco reactivo y el proceso es demasiado lento para tener aplicación en síntesis:
CH3CH2Cl + (CH3)3C_OH (CH3)3C_O_CH2CH3 + HCl (muy lenta)X La única solución sería emplear un alcóxido terciario y un halogenuro primario:
CH3CH2Cl + (CH3)3C_O K+ (CH3)3C_O_CH2CH3 + KCl_
Los aniones alcóxido empleados en la síntesis de éteres se obtienen a partir del alcohol correspondiente, por reacción con un metal (Na o K) o una base muy fuerte. Explica si las siguientes reacciones son adecuadas para la síntesis de los alcóxidos que se indican.
EJERCICIO 9.4
1. CH3CH2OH + NaOH CH3CH2O Na+ + H2O
2. 2CH3OH + 2Na 2CH3O Na+ + H2
3. Me3C-OH + NH3(liq.) Me3CO NH4+
4. Me2CH-OH + Na+H Me2CHO Na+ + H2
_
_
__
IX

223
La síntesis de Williamson también permite obtener éteres cíclicos a partir de halogenohidrinas:
Oxido de etileno(Oxirano)
HOClH2C CH2 HO CH2 CH2 Cl
K2CO3H2C CH2
O
+ Cl_
SN2
HO_CH2_CH2
_CH2_CH2
_ClO
+ H2O + NaCl
Tetrahidrofurano(95 %)
H2ONaOH,SN2
Se trata de una reacción de sustitución nucleófila intramolecular:
HO_CH2_CH2
_ClK2CO3 _
O_CH2_CH2
_Cl
_CH2 CH2
O
Cl
H2C CH2
O
+ Cl_SN2
HO_CH2_CH2
_CH2_CH2
_ClNaOHH2O
O_CH2_CH2
_CH2_CH2
_Cl_
O+ Cl
OCH2
___Cl_
_SN2
Normalmente los éteres no son suficientemente reactivos como sustratos para hidrolizarse con facilidad. Sin embargo, el óxido de etileno y sus derivados son muy reactivos debido a la tensión del anillo. Estos compuestos se hidrolizan en presencia de cualquier ácido o base. También reaccionan con nucleófilos, en ausencia de ácidos:
H R
HH
O
reacción con nucleófilos con catálisis ácida
reacción con nucleófilos sin catálisis ácida
La reacción con nucleófilos sin catálisis ácida tiene lugar en el átomo de carbono menos sustituido del oxirano. Por el contrario, la reacción con catálisis ácida ocurre en el átomo de carbono más sustituido del oxirano. Reacción catalizada por ácidos:
H2C CH2
O
H2SO4
H2OHOCH2
__CH2OH
O: : H___OH2 O:H
+ + H2O+
IX

224
EtilenglicolO:H
+H2C_____CH2OH
+
OH2
H2C_____CH2OH
O___HH__
+OH2:
HO_CH2___CH2
_OH + H3O+
La apertura de los oxiranos no simétricos, catalizada por ácidos, tiene lugar con regioselectividad Markovnikov. En este sentido, la reacción es parecida a una S 1, en la que el carbocatión incipiente más estable reacciona con el nucleófilo antes de que tenga lugar su rotación interna:
N
Et Me
HMeO
H2O
H+ OH
H2O
Et
Me
Me
HEt Me
HMeO
H
+
H2O:
: +
OH
HO
Et
Me
Me
H
23
(1) (2S,3S)
3-Metil-2,3-pentanodiol
H2O::H2O
OH
H2O
Et
Me
Me
H+
(1') (2R,3R)
3-Metil-2,3-pentanodiol
Et Me
HMe
OH
+ Et Me
HMe
OH
+
OH
HO
Et
Me
Me
H
En esta reacción se obtendrá una mezcla mayoritaria de (1) y (1’), ya que la reacción transcurre preferentemente a través del carbocatión incipiente más estable (en este caso el carbocatión terciario).
Escribe el mecanismo de la siguiente reacción (el Metanol es el nucleófilo):EJERCICIO 9.5
2,2,3-Trimetiloxirano
Me CMe
CH MeR_C_O_OH
O
Me CMe
CH Me
OH2SO4, H2O
Me CMe
CH Me
3-Metil-3-metoxi-2-butanol
OH
OMe
CH3OH
El anillo de los oxiranos también se abre fácilmente con reactivos nucleófilos en ausencia de ácidos; la reacción SN2 tiene lugar sobre el carbono menos sustituido del anillo oxiranico (menor impedimento estéreo)
O
Me H
HH
MeO_
:: O
OMe
H
MeH
H::
_MeO___H HO
OMe
H
MeH
H
+ MeO_
IX

225
Escribe el mecanismo de la siguiente reacción:
Me
Me Me
2,2,3-Trimetiloxirano
CH3O Na+_
Me COH
CH MeOMe
Me
2-Metil-3-metoxi-2-butanol
C CH
OMeOH
EJERCICIO 9.6
FORMACIÓN DE ENLACES C-X
SÍNTESIS DE HALUROS DE ALQUILO A PARTIR DE ALCOHOLES Los alcoholes reaccionan con haluros de hidrógeno dando lugar a haluros de alquilo:
R_CH2__CH2OH
HBrSN2
alcohol primarioR_CH2
__CH2Br + H2O
HBrSN1 / SN2
alcohol secundario
+ H2OR CH2 CHMe
OH R CH2 CHMe
Br
HBrSN1
alcohol terciario
+ H2OR CH2 CMe
OH R CH2 CMe
BrMe Me
Mecanismo de la reacción SN2:
R__CH2OH H____Br R__CH2OH2 + Br_
Keq = 107
pKa -9 pKa -2
+
+
Br_ R__CH2Br + H2OCH2
OH2
R
Si la reacción es SN1 pueden tener lugar transposiciones de Wagner-Meerwein:
+
3-Pentanol 2-Bromopentano
CH3CH2 CHOH
CH2CH3HBrSN1
CH3CH2 CHBr
CH2CH3
3-Bromopentano
CH3CH2 CH2 CHCH3
Br
Mecanismo de la reacción SN1:
+CH3CH2 CH
EtO
H
: H____Br CH3CH2 CHEt
OH2 +_
Br
IX

226
+
CH3CH2 CH CH2CH3
OH2
H2O_CH3CH2 CH CHCH3
+H
transposición
_Br
_Br
CH3CH2 CH CH2CH3
Br
CH3CH2 CH2 CH CH3+
CH3CH2 CH2 CH CH3
Br
SÍNTESIS DE HALUROS DE ALQUILO A PARTIR DE TOSILATOS Otra forma de convertir un alcohol en un haluro de alquilo es a través del tosilato del alcohol (éster del ácido p-toluensulfónico)
+ HCl
(tosilato) Cloruro del ácido p-toluensulfónico
Me SO
OCl + R CH
OHR' Me S
O
OO CH
R'R
Me S_
p-toluensulfonilo
(tosilo Ts)
O
O
El tosilato se transforma en el haluro de alquilo por reacción con el anión del halógeno:
Br_
Na+MeSO
OOCH
R
R'
BrNa R CHR'
Br + Me SO
OO
_Br
_Na+MeS
O
OOCH
R
R'
BrNa R CHR'
Br + Me SO
OO
_
La reacción se escribe normalmente de forma abreviada:
Na+BrR CHOH
R' + TsCl HCl_R CH
OTsR'
_R CH
BrR' + TsO
_+ Na+
Los haluros de alquilo pueden obtenerse por tratamiento del alcohol con SOCl2, PCl3 o PCl5. La reacción se parece mucho a la síntesis de cloruros de ácido. Utiliza como ayuda aquella síntesis para escribir el mecanismo de la reacción entre el 2-Butanol y Cloruro de tionilo (en este caso, la Piridina es simplemente una base)
EJERCICIO 9.7
IX

227
FORMACIÓN DE ENLACES C–N
SÍNTESIS DE AMINAS El amoníaco y las aminas reaccionan con los haluros a través de un mecanismo SN2:
Cl_CH2_CO2H + 2NH3 H2N_CH2
_CO2H + Cl NH4+
_
La reacción de alquilación del amoniaco y las aminas da lugar a mezclas de productos, dependiendo de la proporción relativa entre el derivado halogenado y el sustrato.
Averigua la estructura de los compuestos que aparecen en la siguiente secuencia de reacciones:EJERCICIO 9.8
HOIMe AQ
_D(C4H8) + H2O + NMe3CH3 CH
CH3
CH2IMe B IMe CNH2
La síntesis de Gabriel es un proceso especial que, en la preparación de aminas primarias, evita la aparición de productos más alquilados (aminas secundarias, terciarias y sales de amonio):
O
O
O
Anhidrido ftálico
+ NH3calor
N_H
O
O
Ftalimida
KOH
Ftalimida potásica
N K+
O
O
_
El reactivo nucleófilo es el anión ftalimido:
N
O
O
_CH2C2H5
Br N
O
O
CH2CH2CH3KOHcalor
CH3CH2CH2_NH2 +
CO2
CO2
K_
_
+
K+
N_CH2CH2CH3
O
O
KOHcalor
CO2
CO2
CH3CH2CH2_NH2 +
_
_
+K
+K
EJERCICIO 9.9Utiliza el Ejercicio 5 del Tema 1 para escribir el mecanismo de la hidrólisis en medio básico de la imida N-sus-tituida que aparece en el último pasop de la síntesis de Gabriel:
IX

228
FORMACIÓN DE ENLACES C–C
SÍNTESIS DE NITRILOS
Un tipo de reacción muy útil es la que tiene lugar entre un halogenuro y un carbanión; los carbaniones alquilo sencillos no son satisfactorios, pero pueden emplearse muchas clases de carbaniones relativamente estables; el anión cianuro es un ejemplo:
CH3CH2CH2CH2_Br + Na+ NC CH3CH2CH2CH2
_CN + Na+Br_ _
Esta reacción (SN2) brinda un método fácil, para añadir un átomo de carbono más a una cadena carbonada:
C6H5_CH2
_Cl + Na+NC EtOH C6H5_CH2
_CN + Na+ClSN2
_ _
SN2Cl_CH2CH2CH2CH2
_Cl + 2Na+ NCacetona
NC_CH2CH2CH2_CN + 2Na+Cl
_ _
La hidrólisis de nitrilos conduce a ácidos carboxílicos; su reducción con hidruro de litio y aluminio (H4AlLi) da lugar a aminas:
C6H5_CH2
_CN
H4AlLi
C6H5_CH2
_CO2H
C6H5_CH2
_CH2_NH2
H3O+
La reacción del (R)(1-Bromoetil) metil éter con cianuro sódico conduce a (1-Cianoetil) metil éter ópticamente activo. Su configuración absoluta es la misma que la del producto de partida. Explica esta anomalía aparente.
EJERCICIO 9.10
SÍNTESIS DE ALQUINOS Los átomos de hidrógeno unidos a carbonos con triple enlace son lo suficientemente ácidos (pKa 25) para reaccionar con bases fuertes. El anión amiduro (pKa 35) es capaz de extraer este tipo de hidrógenos originando carbaniones:
+ NH3R C C H :NH2
_
Na
NH3 (liq.)R C C:
_
Estos carbaniones pueden reaccionar con derivados halogenados, dando lugar a alquinos:
R C C:_
CH2R'
X R C C CH2 R' + X_
IX

229
SÍNTESIS DE ALCOHOLES PRIMARIOS A PARTIR DE OXIRANOS Y COMPUESTOS ORGANOMAGNÉSICOS
El magnesio reacciona con los haluros de alquilo, dando lugar a compuestos organometálicos de magnesio (magnesianos, reactivos de Grignard)
RCH2__I + Mg RCH2
__Mg__IEt2O
magnesianoreactivo de Grignard
Se trata de una reacción de oxidación-reducción en la que el magnesio cede un par de electrones al carbono unido al halógeno:
RCH2 I + Mg: RCH2 + Mg2+ + Iδ+ δ− _
(carbanión)
_
El resultado es un carbanión que puede actuar como nucleófilo en una gran variedad de reacciones. Una de ellas es la reacción con oxiranos, que conduce a alcoholes primarios con dos átomos de carbono más que el haluro de partida:
RCH2__X + Mg RCH2
__Mg__X RCH2__CH2CH2OHOEt2O
dos átomos de carbono más que RCH2X
Mecanismo de la reacción:
+ H2OH3O+
RCH2 CH2 CH2OH + Mg2+ + X_Et2O
RCH2
Mg OX O
RCH2
XMg
Diseña una síntesis de la Pentilamina, empleando Etileno como único producto orgánico.EJERCICIO 9.11
ANIONES ENOLATO COMO INTERMEDIOS DE REACCIÓN La tautomería se refiere a una interconversión entre dos estructuras que se diferencian en la posición de un átomo o grupo (isómeros constitucionales) Los compuestos con grupos carbonilo están en equilibrio con sus formas enólicas correspondientes (tautomería ceto-enólica):
(99,999 %)
CH3 CO
CH3H2O
CH2 COH
CH3
(forma enólica)
(0,001 %)
H2OCH3 C
OCH2 C
OCH3
(83 %)
CH3 C CH CO
CH3
OH
(17 %)
IX

230
O
H
O O
HH2O
(50 %) (50 %)
OH
O OH
Me Me
H2O O O
Me Me
(5 %) (95 %)
H
La formación del enol está catalizada por bases:
Keq = 5,0 x 106H CH
CO
CO
Me
Me
HO_
pKa 9,0
H2O +
pKa 15,7
Me CO
CH CO
Me_
El anión enolato está estabilizado por deslocalización de la carga negativa:
enolato
Me C
O
CH C
O
Me_ :::
Me C
O
CH C
O
Me
::::_
Me C
O
CH C
O
Me
:::_
:
HO___H
enolenolato
Me C
O
CH C
O
Me
::::_
Me C
OH
CH C
O
Me
::
+ HO_
Reacción global:
cetona
Me CO
CH2 + HO_
enolatoMe C
O
CH C
O
Me
:::
:
:_
+ H2O
enol
Me C
OH
CH C
O
Me
::
+ HO_
CO
Me
Cuando la (2S)2-Metilciclohexanona se trata con cantidades catalíticas de NaOH se transforma en una mezcla de (2S) y (2R)2-Metilciclohexanona. Cómo puedes explicar la formación del estereoisómero R.
EJERCICIO 9.12
IX

231
Al aumentar la fuerza de la base en relación con el pKa del compuesto carbonílico, la concentración en el equilibrio del anión enolato también aumenta:
O
H
H
+ CH3O_
OH
+ MeOH
pKa 16pKa 15,5
Keq = 0,3_
O
H
H
+ HO_
OH
+ H2O
pKa 16pKa 15,7
Keq = 0,5_
O
H
H
+ EtO_
OH
+ EtOH
pKa 16pKa 17
Keq = 10_
O
H
H
+ Me3CO_
OH
+ Me3COH
pKa 16pKa 19
Keq = 103_
O
H
H
+ Na+ H_
OH
+ H2
pKa 16pKa 35
Keq = 1019_
O
H
H
+ Na+ NH2_
OH
+ NH3
pKa 16pKa 36
Keq = 1020_
O
H
H
+
OH
pKa 16 pKa 36
Keq = 1020_
[Me2C_N_CMe2] Li
_+
(LDA)
Me2C NH
CMe2+
IX

232
ALQUILACIÓN DE CETONAS
Los compuestos carbonílicos que no son simétricos, como es el caso de la 2-metilciclohexanona, pueden dar lugar a dos aniones enolato diferentes, dependiendo del átomo de hidrógeno extraído por la base:
OH
H
H
Me
1 2
:B :B
OHMe _
OHMe _
OMe
_O
Me
_
HH
(1) (2) La reacción de cada uno de los aniones enolato (nucleófilo) con un átomo de carbono electrófilo origina dos productos distintos:
(1)
OH
Me
_
CH2 BrR
SN2H
Me
CH2
O_
R + Br
_CH2R
Br
(2)
OMe
H
_
SN2Me
H
CH2
O
R Br+
Los aniones enolato son nucleófilos ambidentes que pueden reaccionar con un carbono electrófilo (de baja densidad electrónica) de dos formas distintas:
O
H
H
O
CH3O_
CH3OH
O
O
_
O
O
_
[nucleófilo ambidente]
IX

233
Reacción con el átomo de carbono (C-alquilación):
O
O
_
(CH2)2CH3
CH2
O
O
CH2CH2CH2CH3
(15 %)
δ+
SN2Br
Reacción con el átomo de oxígeno (O-alquilación):
O
O
_(CH2)2CH3
CH2 O
O
CH2CH2CH2CH3
(37 %)δ+
SN2Br
En general, los electrófilos de carbono reaccionan con preferencia a través del átomo de carbono del enolato y los electrófilos de silicio lo hacen exclusivamente por el átomo de oxígeno. Este hecho permitió estudiar las condiciones experimentales más adecuadas para controlar la regioselectividad de las reacciones de alquilación en moléculas no simétricas con grupos carbonilo:
OMe Et3N, Q
Dimetilformamida
OMe
SiMe3 OMe
SiMe3
+
(22 %) (78 %)
Me3SiCl
OMe
_O
Me
_
+
OMe (MeCH)2N Li+
00C
O Li+
Me
_O Li+
Me
_
+Me3SiCl
Et3N
O_SiMe3Me
O_SiMe3Me
+
(99 %) (1 %)
_
CONTROL TERMODINÁMICO Y CONTROL CINÉTICO DE LA REACCIÓN Cuando la reacción se hace a una temperatura elevada, la 2-metilciclohexanona se transforma más rápidamente en el enolato con el doble enlace menos sustituido (menor energía de activación) El enolato más sustituido se forma más lentamente porque la energía de activación es más alta. Sin embargo, conforme pasa el tiempo, la cantidad de enolato más sustituido va aumentando mientras que el enolato menos sustituido comienza a transformarse cada vez más rápido en la cetona, ya que la energía de activación ΔG (inversa) es menor que ΔG‡ (termodinámico) Si se deja que la reacción alcance el equilibrio, la concentración del enolato más sustituido será mayor que la del enolato menos sustituido. Por el contrario, si la reacción se lleva a cabo a temperatura baja, el número de moléculas de cetona con energía suficiente para pasar la barrera que conduce al enolato más sustituido [ΔG‡ (termodinámico)] es muy pequeño, comparado con el de moléculas capaces de traspasar la barrera que da lugar al enolato menos sustituido. Ahora, el tiempo requerido para que el sistema alcance el equilibrio es comparativamente mucho mayor que a temperatura elevada. En consecuencia, el enolato mayoritario será el menos sustituido.
IX

234
O
Me
_+ HB
enolato menos sustituido
O
Me
_+ HB
enolato más sustituido
O
Me
+ :B_
diferencia de energíaentre los dos enolatos
ΔG (cinético)
ΔG (termod.)
enolato termodinámicoenolato cinético
ΔG(inversa)
Control termodinámico: la reacción alcanza el equilibrio y el producto mayoritario es el que se obtiene a partir del enolato más estable, es decir, el que tiene el doble enlace más sustituido (1):
O
H
MeEt3N:
Et3NH+
OMe
_ CH2____X
R
OMe
CH2__R + X
_
OMe
(1)
_
SN2
Condiciones experimentales: temperatura elevada y base no muy fuerte (p.e. trietilamina)
IX

235
Control cinético: los productos se aíslan antes que la reacción alcance el equilibrio. El producto mayoritario es el que se forma más rápidamente y proviene del anión enolato menos estable, esto es, el que tiene el doble enlace menos sustituido (2):
O
H
HO
H_ CH2
____X
R
OH
CH2__R + X
_
OH
(2)
_
Me (Me2CH)2N Li+ Me
Me
MeSN2_
Condiciones experimentales: temperatura baja y base muy fuerte e impedida estéricamente (diisopropilamiduro de litio: LDA)
Escribe el mecanismo de las reacciones que aparecen a continuación:EJERCICIO 9.13
Reacción 1:
Reacción 2:
Reacción 3:
CH2
O
00CA + B
ICH3
250C
CH2 MeO
(76 %)
+
(6 %)
OMe
CH2_C6H5
(Me2CH)2N Li+_ C6H5
C__Me
CH2CH2CH2Br
Me3CO K+_
Q
CO_Me
(90 %)
O
C_Me
CH2CH2CH2Br-600C
CQ
O
(80 %)
(Me2CH)2N Li+_
O
ALQUILACIÓN DE ÉSTERES En la condensación de Claisen (Tema 8, p. 200) se dijo que el diisopropilamiduro de litio (LDA) permite obtener el enolato de un éster de forma cuantitativa, es decir, la reacción entre el éster y la base está totalmente desplazada hacia la formación del enolato:
IX

236
pKa 22
CH3CH2 CH2 CO
OMe + (MeCH)2N Li+_
CH3CH2 CH CO
OMe_
CH3CH2 CH CO
OMe
_Li+ + (MeCH)2NH
pKa 36
Keq = 1014
Utilizando derivados halogenados y LDA se pueden realizar reacciones de sustitución nucleófila con rendimientos muy elevados:
+ I_
2-Etilbutanoato de metilo
(92 %): :_
SN2CH3CH2 CH C
O
OMeCH2
CH3I
CH3CH2 CHCH2CH3
CO2Me
1)LDA, THF
2)CH3I
2,3-Dimetilbutanoato de etilo
(95 %)
CH3 CHCH3
CHCH3
CO2EtCH3 CHCH3
CH2 CO2Et
CH21)LDA, THF
2)CH3ICH
2-Fenilpropanoato de metilo
CO2MeCH3
CO2Me
Escribe el mecanismo de las siguientes reacciones:EJERCICIO 9.14
Reacción 1:
Reacción 2:
O
O1)LDA, THF
-780CA
H2C=CH-CH2Br
-780CB
O
O
1)LDA, THF
-780CC
HC C_CH2Br
-780CD
ALQUILACIÓN DE COMPUESTOS CON HIDRÓGENOS METILÉNICOS ÁCIDOS La presencia de ciertas funciones insaturadas como sustituyentes en un átomo de carbono saturado, tales como NO2, -CO- o -CN, provocan que los hidrógenos unidos a dicho átomo sean relativamente ácidos. En la tabla siguiente aparecen algunos ejemplos.
IX

237
COMPUESTOS CON HIDRÓGENOS ÁCIDOS Y OTROS REACTIVOS COMUNES
4,8
9
9
10
11
11
13
19
23
25
25
5,8
9
17NC-CH2−CO2Et
CH3CO2H
H2C(CO2Me)2
CH3CH2OH
CH3−CO−CH3
H2C(CN)2
CH3−CO2Et
CH3−CN
CH3−CONH2
CH3−NO2
Compuesto pKa pKaCompuesto
CH3CO−CH2−CO2Et
H2C(CO2Et)2O2N-CH2−CO2Et
CH3−CO−CH2−CO-CH3
Todos los compuestos con metilenos activos forman aniones enolato en medio básico:
Malonato de dietilo
pKa 13 pKa 10
_ +Keq = 103EtO C
OCH2 C
OOEt + Et3N: EtO C
OCH C
OOEt + Et3NH
Acetilacetato de etilo
pKa 11.0
CH3 CO
CH2 CO
OEt + HO_
CH3 CO
CH CO
OEt_
+ H2O
pKa 15,7
Keq = 5,0 x 104
Cianoacetato de etilo
pKa 9 pKa 15,5
N C CH2 CO
OEt + MeO_
N C CH CO
OEt + MeOH_
Keq = 3 x 106
Keq = 1,6 x 1011
Nitroacetato de etilo
pKa 5,8 pKa 17
O NO
CH2 CO
OEt + EtO_
O2N CH CO
OEt + EtOH:_
+
_
ALQUILACIÓN DE β-CETOÉSTERES (SÍNTESIS ACETILACÉTICA) Los sustratos más utilizados son los siguientes: 1. Derivados halogenados alifáticos (síntesis de metil alquil cetonas y ácidos monocarboxílicos) 2. α-Halocetonas (síntesis de 1,4-dicetonas) 3. α-Haloésteres (síntesis de cetoácidos y ácidos dicarboxílicos)
IX

238
1. SÍNTESIS DE METIL ALQUIL CETONAS Y ÁCIDOS MONOCARBOXÍLICOS
R−CO_C H R−CO_CH_CO2Et EtOH__
CO2Et
OEt
H
+
β−cetoéster
R−CO CH
CO2Et
_CH2
R'
SN2R−CO CH
CO2Et
CH2R'X
El β-cetoéster formado puede someterse a dos tipos de hidrólisis.
β-cetoéster
(CETONA)
R CO CH
CO2Et
CH2R'
R CO CH2 CH2R'base diluida o ácido diluido
base concentradaR CO2H R'−CH2CH2−CO2H+ (ÁCIDO)
1
2
1 HIDRÓLISIS CETÓNICA
HIDRÓLISIS ÁCIDA2 La hidrólisis cetónica da lugar a cetonas y se realiza con bases o ácidos fuertes diluidos en agua. La hidrólisis ácida origina ácidos carboxílicos y se lleva acabo empleando disoluciones acuosas concentradas de una base fuerte. HIDRÓLISIS CETÓNICA a) Con disoluciones diluidas de una base:
β-cetoéster
OH_
_
EtO__
β-cetoácido
R CO CHCH2R'
C
O
OEt
[adición]OHR CO CH
CH2R'C
O
OEt
[eliminación]OHR CO CH
CH2R'
CO
Como el medio de reacción es básico, el β-cetoácido se transforma en un anión carboxilato que pierde fácilmente CO2, transformándose en una cetona:
OH_ H2O_
H___OH_
enolato de la cetonaβ-cetoácido
OCH3 CO CH
CH2R
C
O
H OCH3 C CH
CH2R
C
O_
OCO2
_CH3 C CH
CH2R
O
enol de la cetona cetona
HO_ _CH3 C CH
CH2R
OH
CH3 CO
CH2CH2R
IX

239
b) Con disoluciones diluidas de un ácido fuerte:
β-cetoácido
_H3O+
EtOHR CO CH
CH2R'CO
OEt R CO CH
CH2R'
CO2H
HO
CR
CH2R'
O
O
CO2_
enol de la cetona
cetona
R C
OH
CH
CH2R'
R CO
CH2 CH2R'
HIDRÓLISIS ÁCIDA La reacción del β-cetoéster con la disolución concentrada de una base es una retrocondensación de Claisen, es decir, el proceso contrario a la condensación de Claisen (Tema 8, p. 200)) La hidrólisis ácida da lugar a ácidos carboxílicos:
β-cetoésterHO_
[adición]
_
[eliminación]
:_
R C
O
CH
CO2Et
CH2R'
OH
R C
O
CH
CO2Et
CH2R'
R CO
OH
CH
CO2Et
CH2R' El medio de reacción es básico:
R CO
OHHOH2O
_
__
(ac. fuerte: HCl)
H3O+
R CO
O R CO
OH
: _CH
CO2Et
CH2R'HO___H HO_
R'CH2CH2 C OEt
O+ R'CH2CH2 C O
O_
+ EtOH
_
(ac. fuerte: HCl)
H3O+
CO
O R'CH2CH2 CO
OHR'CH2CH2
12 HO dil. o H3O+
HO (conc.)
2
(cetona)
(ácido monocarboxílico)
Ruptura de un β−cetoéster (hidrólisis cetónica y ácida)
CH3CO
1CH3CO_
R
CH CO2EtR
_
_Ácetilacetato de etilo alquilado (R)
CH2
CH2
R
CO2H
IX

240
Ácetilacetato de etilo
CH3COCH2CO2Et1)EtO /EtOH
2)C4H9Br/EtOH
(69-72%)
1)NaOH
2)H3O+
(52-61%)
_
CHCO2Et
COCH3 CH2 COCH3C4H9C4H9
Estas reacciones de alquilación pueden aplicarse con éxito a la síntesis de compuestos cíclicos; en general, las velocidades relativas de cierre de anillos de diferente tamaño sigue el orden:
ciclos de 3, 5 y 6 eslabones > ciclos de 7 > ciclos de 4 = ciclos mayores de 7 = compuestos no cíclicos
CH3CO-CH-CO2Et
R
Producto principal Producto secundario
CH3COCO2Et
CH3COCO2Et
CH3CO
CO2Et
CH3CO
CO2Et
BrCH2_(CH2)_CH2Br
BrCH2_CH2Br
n = 0
R_X
n = 1
BrCH2_(CH2)2
_CH2Br
n = 2
BrCH2_(CH2)3
_CH2Br
n = 3
BrCH2_(CH2)4
_CH2Br
n = 4
BrCH2_(CH2)n
_CH2Br
CH3COCH2CO2Et
CO2EtCH3CO
EtO_
CH3CO CHCO2Et
(CH2)n+2 CHCO2Et
COCH3
CH3CO CHCO2Et
(CH2)2
CH3CO CHCO2Et
(CH3)3
CH3CO CHCO2Et
(CH2)4
CH3CO CHCO2Et
(CH2)5
CH3CO CHCO2Et
(CH2)6 CHCO2Et
COCH3
CHCO2Et
COCH3
CHCO2Et
COCH3
CHCO2Et
COCH3
CHCO2Et
COCH3
_ EtO /EtOH
1000CCH3COCH
CO2EtCH2CH2CH2CH2Br
Br _ CH2 CH2
CH2
CH2Br
HCCH3CO
CO2Et
EtO_
H2C
CCH3CO
EtO2C
_
Br
CH3CO
CO2Et
IX

241
Sintetiza la Ciclopentil fenil cetona empleando Benzoilacetato de etilo y el derivado halogenado que creas conveniente.
EJERCICIO 9.15
2. SÍNTESIS DE 1,4–DICETONAS A PARTIR DE α–BROMOCETONAS Las α-bromocetonas se obtienen por reacción de la cetona con bromo en medio básico:
_
cetona
R CH2 CO
R' + Br2HO
R CH CO
R'Br
α−bromocetona
enolcetona
CO
R':B
R CH C
OH
R'
:
:
+ Br_
+
enolR
HCC
OH
R' Br____Br
:
R
CHC
OH
R' Br
B:_
+ H_B
::+
:
R
CHC
O
R' Br
H
Br
CHC
O
R' R
R−CH2
_
1)HO (dil)
2)H3O+
1)EtO2)R'-X
SN2CH3CO CH
CO2EtCH2
CORBr CH3CO CH
CO2EtCH2 COR
_
_
CH3CO CHCO2H
CH2 COR
CO2Et
CCH3CO
R'
COR
QCH3CO CH
CO2H
CH2 CORCO2
_CH3CO CH2 CH2 COR
CO2Et
CCH3CO
R'
CORHO (dil.)
Q
_ CO2H
CCH3CO
R'
CORCO2
_CHCH3CO
R'
COR
Los β-cetoésteres pueden utilizarse también como precursores de aniones enolato en reacciones de sustitución nucleófila de moléculas con grupos carbonilo. El ejercicio 16 es un ejemplo de ello.
IX

242
1. Idea una síntesis de la 4-Metil-3,5-heptanodiona partiendo de Ácido propiónico como producto base.2. Diseña una síntesis de la Dibencil cetona empleando Fenilacetato de etilo como único producto orgánico. 3. Realiza la síntesis de la Etil fenil cetona utilizando Acetato de etilo y Benzoato de etilo como sustancias de partida.
EJERCICIO 9.16
3. SÍNTESIS DE CETOÁCIDOS Y ÁCIDOS DICARBOXÍLICOS A PARTIR DE α–BROMOÉSTERES La bromación en Cα de los ácidos carboxílicos (reacción de Hell-Volhard-Zelinsky) se realiza empleando tricloruro de fósforo como catalizador:
Br2R CH2 CO2H
PCl3R CH CO2H
Br
Los α-bromoésteres se obtienen por esterificación del correspondiente α-bromoácido:
EtOHH3O+R CH
BrCO2H R CH
BrCO2Et
_
1)HO-(conc.)
2)H3O+1)HO-(dil.)
2)H3O+ HO2C_CH2_CH2
_CO2H
CH3COCH2_CH2
_CO2H
1)EtO /EtOH
2)R_X
1)HO (conc.)
2)H3O+1)HO (dil.)
2)H3O+ HO2C_CH_CH2_CO2H
R
SN2CH3CO CH
CO2EtCH2
CO2Et
Br CH3CO CHCO2Et
CH2 CO2Et
_
CH3CO CCO2Et
CH2 CO2EtR
_
_
CH3COCH CH CO2HR
Averigua la estructura del β -cetoéster F y escribe el mecanismo simplificado de las reacciones que aparecen en los esquemas.
EJERCICIO 9.17
1)HO (dil.)
2)H3O+E(C7H10O2)
1)HO (conc.)
2)H3O+G(C8H12O5) Ácido 5-ceto-3-metilhexanoico
_
_
F(C10H14O4)
Q
Ác. 3-metilpentanodioico A(C10H18O4) ; Ác. propanodioicoSOCl2C2H5OH
H3O+B(C3H2O2Cl2)
1)HO (dil.)
2)H3O+E(C7H10O2)
1)EtOA(C10H18O4)
2)B(C3H2O2Cl2)
_ _QC(C13H18O6) D
IX

243
ALQUILACIÓN DE DIÉSTERES MALÓNICOS (SÍNTESIS MALÓNICA)
Los sustratos más empleados son los siguientes: 1. Derivados halogenados alifáticos (síntesis de ácidos monocarboxílicos) 2. Derivados dihalogenados alifáticos y α-haloésteres (síntesis de ácidos dicarboxílicos) 1. SÍNTESIS DE ÁCIDOS MONOCARBOXÍLICOS La síntesis malónica abre un camino, teóricamente inagotable, para la obtención de ácidos alifáticos. No obstante, presenta una limitación: no es posible obtener ácidos trisustituidos en el átomo de carbono α.
(80-90%)
CH2
CO2Et
CO2EtEtOHEtO
_
CHCO2Et
CO2Et
_ n-C4H9Br
EtOHCHCO2Et
CO2EtC4H9
KOH/H2OCHCO2 K+
CO2 K+C4H9
_
_
(80-90%)
CH2
CO2Et
CO2EtEtOHEtO
_
CHCO2Et
CO2Et
_ n-C4H9Br
EtOHCHCO2Et
CO2EtC4H9
KOH/H2OCHCO2 K+
CO2 K+C4H9
_
_
H2O
H2SO4CHCO2H
CO2HC4H9
CO2_
C4H9 CH2 CO2H
1)EtO /EtOH
(83-84%)2)C2H5CHBr
Me
_
CHMe
CHCO2Et
CO2EtC2H5KOHH2O
CHMe
CHCO2 K+
CO2 K+C2H5
__
H2C(CO2Et)2
H2SO4/H2O
(62-65%)CO2
_ CHMe
CH2 CO2HC2H5
Se pueden utilizar con éxito tanto los haluros primarios como los secundarios, y también haluros de alilo y bencilo primarios y secundarios. Sin embargo, los haluros de alquilo terciarios que tienen al menos un Hα son de poco valor, ya que se produce mayoritariamente una reacción de eliminación E2:
IX

244
EtOH(CH3)3C_Br + H2C(CO2Et)2 (CH3)3C_CH(CO2Et)2 +
(6%)
EtO_
CH3 CCH3
CH2
_
EtOH + H2C(CO2Et)2 +
(60%)
Br CH(CO2Et)2
EtO
Si la reacción de alquilación se realiza con un derivado dihalogenado, se llega a compuestos cíclicos; en tal caso, valen las mismas generalizaciones hechas en el caso de la síntesis acetilacética.
1)EtO /EtOH
2)Br(CH2)3BrCO2Et
CO2Et
+
(55-56%) (10%)
2H2C(CO2Et)2
_
CHEtO2C
CO2Et
(CH2)3 CH
CO2Et
CO2Et
EtOH2H2C(CO2Et)2 + BrCH2
_CH2Br
CO2Et
CO2Et
EtO_
Síntesis de un ácido espiroheptanocarboxílico:
EtOH H2C(CO2Et)2+
H4AlLi HBr H2C(CO2Et)2CH2
CH2Br
CH2Br
EtO_ CO2Et
CO2Et
CH2OH
CH2OH
CH2Br
CH2Br EtO_
1)HO /H2O
2)H3O+Q
CO2H
_
CO2_
CO2Et
CO2Et
CO2H
CO2H 2. SÍNTESIS DE ÁCIDOS DICARBOXÍLICOS Reacción del anión enolato con derivados halogenados que tengan los átomos de halógeno suficientemente alejados:
IX

245
CH
CO2Et
CO2Et
_CH2
Br
(CH2)n CH2
Br
CH
CO2Et
CO2Et
_CH
CO2Et
CO2Et
CH2 (CH2)n CH2 CH
CO2Et
CO2Et
H3O+CH
CO2H
CO2H
CH2 (CH2)n CH2 CH
CO2H
CO2H
QCO2
_ CH2 CH2 (CH2)n CH2 CH2HO2C C 2HO
Dependiendo del valor de n se pueden obtener ácidos dicarboxílicos cíclicos, como sucede en la síntesis acetilacética:
CH2
CO2Et
CO2Et
21)EtO
2)BrCH2CH2Br
_
CH
CO2Et
CO2Et
CH2 CH2 CH
CO2Et
CO2Et
1)EtO
2)BrCH2CH2Br
_ CO2EtCO2Et
CO2EtEtO2C
CO2EtCO2Et
CO2EtEtO2C
1)HO
2)H3O+
CO2H
CO2H
_ CO2HCO2H
CO2HHO2C
QCO2
_
Los α-haloésteres también pueden dar lugar a ácidos dicarboxílicos:
CO2Et
CH
CO2Et
_CH2
Br
CO2Et
CO2Et
CH
CO2Et
CH2 CO2EtH3O+
CO2H
CH
CO2H
CH2 CO2HQ
CO2_ CH2CH2 CO2HHO2C
El anión enolato del malonato de dietilo se utiliza también para sintetizar metil cetonas mediante reacciones de sustitución nucleófila en moléculas con grupos carbonilo:
CO2Et
CH
CO2Et
_C O
Cl
R
CO2Et
CH
CO2Et
C R
OH3O+
CO2H
CH
CO2H
C R
OQ
CO2_ R CO CH3
IX

246
Efectividad de algunos nucleófilos habituales (reacciones SN2)
reactividad nucleófilo reactividad relativa nucleófilos muy activos I¯ HS¯ RS¯ >105
nucleófilos activos Br¯ HO¯ RO¯ NC¯ 104
nucleófilos aceptables :NH3 Cl¯ F ¯ RCO2¯ 103
nucleófilos débiles H2O ROH 1 nucleófilos mediocres RCO2H 10-2
(*) reactividades relativas respecto al metanol
Efecto del grupo saliente
Grupos salientes en orden creciente de facilidad para ser eliminados (reacciones SN2)
Clase de compuesto Grupo saliente R-OR' Dialquiéteres R'O− Alcóxido*
R-OH Alcoholes HO− Hidróxido
R-O-Ar Alquilariléteres ArO− Fenóxido R-OCOR' Ésteres R'CO-O− Anión carboxilato R-F Fluoruros de alquilo F− Fluoruro (v.r.= 0,001)**
R-Cl Cloruros de alquilo Cl− Cloruro (v.r.= 0,02)
Éteres O-protonados R'OH Alcohol Alcoholes protonados H2O Agua (v.r.= 1)
R
R-Br Bromuros de alquilo Br− Bromuro (v.r.= 1) R-I Ioduros de alquilo I− Ioduro (v.r.= 3)
_OH(R')+
R-OH2+
*Los grupos fuertemente básicos (RO-, HO-, H2N-, F-) no pueden ser desplazados en condiciones ordinarias de reacción. Esta es la razón por la que se usan catalizadores ácidos en reacciones de sustitución de alcoholes, éteres y aminas.**(v.r.) = velocidad relativa de eliminación
IX

247
REACCIONES DE SUSTITUCIÓN NUCLEÓFILA EN UN ÁTOMO DE CARBONO SATURADO
ENLACEFORMADO R_Z + Nu: R__Nu + Z:
R_Z Nu: R_Nu Z:
__
_ _
R_X
HO ; H2O
R'O ; R'OH
R'CO2
R__OH
R__OR'
R__O_COR'
[RCH=CH2 + H2SO4]
(tosilato)
H2O R_CH2__OH
R'_CO_O R_CH2__O-COR'
O O O
H2O ; H3O+
O; H3O+
HOCH2CH2__OH
ArO Na+
Cl_CH2CH2_OH
[Z_CH2CH2_Nu] O
Cl_CH2CH2CH2CH2_OH
[Z_CH2CH2CH2CH2_Nu] O
C__O
*
sulfato de dimetilo
+
N2
N2
(diazometano)
X
_
_ _
_
R CH
Me
O SO
OOH HO
_R CH
MeOH O S
O
OOH
_
R CH2 O SO
OMe _ O S
O
OMe
_
MeO SO
OOMe
_ArO__Me MeO S
O
OO Na+_
Cl
Cl
_
_
Me N N
R CO
OH R CO
O__Me
R CO
CH CO
R_
R CO
CH CO
R
_
R CO
CH CO
RMe
*éster del ácido p-toluensulfónico
IX

248
ENLACEFORMADO R_Z + Nu: R__Nu + Z:
R_Z Nu: R_Nu Z:
__
_ _
NH3
R'NH2
R2'NH
R3'N
R__NH2
R__NH_R'
R__NR2'
R__NR3'+
R_X
X = (Cl, Br, I)
X_
C−−N
O
HO_CH2CH2__NH2
HO_CH2CH2__NHR
OO
HO2C_CH2CH2__NHR
HO2C_CH2CH2__NH2
NH3
RNH2
NH3
RNH2
R_OH
(X = Cl , Br , I )
R__X
R_OH SOCl2, PCl3, PCl5 R__Cl
R_OMe R__I
X HO
MeO
__ _ _
_
_HI
R_X LiAlH4 (H ) R__H
OLiAlH4 (H )
R_OTs(tosilato)
LiAlH4 (H ) R__H TsO
X_
_
_
_
_
R CH CH
H OHR
R R
C−−H
R_X LiAlH4 (H ) R__H
OLiAlH4 (H )
R_OTs(tosilato)
LiAlH4 (H ) R__H TsO
X_
_
_
_
_
R CH CH
H OHR
R R
C−−X
IX

249
ENLACEFORMADO R_Z + Nu: R__Nu + Z:
R_Z Nu: R_Nu Z:
__
_ _
R_X
R__CN
C__C
HC(CO2Et)2
R'CO_CH_COR'
CH3CO_CH_CO2Et
_
_
_
O
(X = Cl, Br, I)
X_Mg_R (R )
NC_
X_
R' C C Na+_R' C C R
R CH(CO2Et)2
R' CO CH
R
COR'
CH3 CO CH
R
CO2Et
_R__CH2CH2OH
EJERCICIOS ADICIONALES 9.1 Considera la siguiente secuencia de reacciones:
2-Bromo-3-metil-3-pentanolNaOH, H2O
SN2A(C6H12O)
MeOH, H3O+B(C7H16O2)
Utiliza representaciones tridimensionales para escribir la secuencia de reacciones con cada uno de los estereoisómeros del 2-Bromo-3-metil-3-pentanol. Asigna la configuración absoluta a todos los compuestos que intervienen. 9.2 Averigua la configuración absoluta de los compuestos A y B.
(2S,3R)-3-Cloro-2-butanolNaOH, H2O
SN2A(C4H8O)
H2SO4
H2OB (C4H10O2)
9.3 Averigua la configuración absoluta de los compuestos A y B.
(2S,3R)-3-Cloro-2-butanolNaOH, H2O
SN2A(C4H8O)
HBr (48 %)
00CB(C4H9OBr)
9.4 Averigua la configuración absoluta de los compuestos A y B:
A(C4H9OBr) B(C4H8O) (2R,3S)-3-Amino-2-butanolHO_
SN2
NH3, H2O
SN2
IX

250
9.5 Dos compuestos isómeros A(C6H11OBr) y B(C6H11OBr) reaccionan con NaOH acuoso, a través de un mecanismo SN2, dando lugar a la misma sustancia ópticamente inactiva C(C6H10O). La reacción SN2 de C con metóxido sódico en metanol, conduce a una mezcla de (1R,2R) y (1S,2S)-2-Metoxiciclohexanol. Averigua la configuración absoluta de los compuestos A, B y C. 9.6 Utiliza representaciones tridimensionales para averiguar la configuración absoluta de todos los compuestos que aparecen en las siguientes secuencias de reacciones:
(1R,2R)-2-Bromo-1-metilciclohexanolNaOH, H2O
A(C7H12O)MeO Na+, MeOH
_
SN2SN2B(C8H16O2)
C(C8H16O2)
MeOHH3O+
9.7 Empleando Éster acetilacético o Malonato de dietilo, y cualquier otro compuesto orgánico o inorgánico que precises, diseña una síntesis de las siguientes sustancias:
CO2H
Me
MeCO2H
O O
(1) (2) (3) (4)
CO2H
CO2Et
CO2Et
EtO2C CO2Et
(5) (6) (7) (8)
CO2H
CO2H
9.8 Averigua la configuración absoluta del compuesto A:
SN2C(C13H15O2N)[(2S,3R) + (2R,3R)]
Ácido (3R)-3-Fenilbutanoico
A(C8H9Br) + B(C5H7O2N) EtO_
H3O+
D(C11H12O4) Q
E(C3H4O4) G(C9H14O4) Ácido ciclopropanocarboxílicoEtO + BrCH2CH2Br
_EtOHH3O+
H3O+F
Q
H3O+B
9.9 Apartado 1: Teniendo en cuenta las reacciones que aparecen a continuación, averigua la estructura de los β-
cetoésteres A, B, C, D, y E.
A(C8H14O3)
2)H3O+ CH3CO2H + CH3CH2CH2CO2H1)HO-(conc.)
C6H5COCH2COCH3 + CO22)H3O+
1)HO-(dil.)B(C13H14O4)
IX

251
1)HO-(conc.)2)H3O+ CH3CH2CO2H + HO2C-CH2CH2-CO2HC(C11H18O5)
1)HO-(dil.)2)H3O+ CH3COCH2CH2COCH3 + CO2D(C9H14O4)
E(C5H8O3) 1)HO-(dil.)
2)H3O+ CH3CHO + CO2
Apartado 2: Una vez conocida la estructura de los cinco β-cetoésteres, realiza su síntesis utilizando reacciones de sustitución nucleófila. Puedes emplear cualquier producto inorgánico, pero únicamente los siguientes productos orgánicos: Etanol Cloroacetona Formiato de etilo Cloruro de benzoilo Acetato de etilo Bromoacetato de etilo Acetilacetato de etilo Propionilacetato de etilo 9.10 Averigua la estructura del β-cetoéster E y escribe el mecanismo de las reacciones que aparecen en los esquemas.
E(C9H12O4)
1)HO (dil.)
2)H3O+
1)HO (conc.)
2)H3O+[F(C7H10O5) + G(C7H10O5)]
-2CO2
Ac. 4-ceto-3-metilpentanoico +Ac. 4-ceto-2-metilpentanoico
_
_
D(C6H8O2)
Q
1)EtO
2)BrCH2CO2Et2)MeCOCl
1)EtO_ _
MeCH2CO2EtNaOHH2O
H3O+
QA B C(C9H12O4) D(C6H8O2)
9.11 Apartado1: Empleando como sustancias de partida Heptanodioato de dietilo y Formiato de etilo, diseña
una síntesis de la 2-Formilciclohexanona, Apartado2: Utiliza Acetilacetato de etilo y Acetona como reactivos para sintetizar 3-Metil-2,5-hexadiona. Apartado3: Emplea Malonato de dietilo y Etilenglicol como reactivos para sintetizar ácido 1,4-Cicloexano-
dicarboxílico. NOTA: Además de las sustancias de partida indicadas en cada apartado, puedes utilizar cualquier reactivo
orgánico o inorgánico que necesites. 9.12 Indica cómo llevarías a cabo las siguientes reacciones estereoespecíficas:
OH Me
HH(2S)-1,2-Propanodiol
OH Me
HH(2R)-1,2-Propanodiol
9.13 Averigua la configuración absoluta del compuesto A:
A(C4H8O) NaCNEtOH, H2O
[(2R,3R) + (2S,3S)] 3-Ciano-2-butanol
IX

252
9.14 Averigua la configuración absoluta de los compuestos A, B y C:
A(C3H6O)1)C6H5MgBr
2)H3O+B(C9H12O) HBr
SN2C(C9H11Br)
Me C CH , NH2Na
SN2(4R)-5-Fenil-4-metil-2-pentino
9.15 Averigua la estructura de los compuestos que aparecen en las siguientes secuencias de reacciones. Secuencia 1:
A(C13H16O4) + C6H5 BrEtO
_
B(C20H22O4)H3O+
C6H5
CO2H
CO2H
C6H5 Q
C(C15H14O2) EtOHH3O+
D(C17H18O2)
Secuencia 2:
Me CHOO
+ A(C3H5Br)HO
_ MeO
CH2
Secuencia 3:
A(C7H12O4) + B(C7H7Br)EtO
_
C6H5CO2Et
CO2Et
1)EtO_
2)C6H5COClC
9.16 Idea un mecanismo razonable que justifique la siguiente transformación:
Me
Me
OH
H3O+
Me OH
Me
9.17 Averigua la estructura de los compuestos que aparecen en las siguientes secuencias de reacciones. Secuencia 1:
C6H5 Me
O
Br2
HO_ A
Me O
O_
Na+B(C10H10O3)
Secuencia 2:
Me2NH + A Me2NOH 1)EtO
_
2) ?
C6H5CH2ClC6H5 N
OOH
Me Me
+Cl
_B(C6H15NO2)
Secuencia 3:
MeMe
O
Br2
HBrA Me
MeO
OCO2Et
Me
EtO_
? +
IX

253
Secuencia 4:
O
MeCO2Et
O1)EtO
_
2)A(C6H8O3)(lactona)
H3O+
QB(C5H10O2) ?
MeBr
O
Secuencia 5:
CO2Et
(CH2)4
CO2Et
EtO_
A(C8H12O3)1)EtO
_
2)
O
C6H5
HClH2O
O
C6H5
ClB(C16H20O4)
Secuencia 6:
1)EtO_
2)H2C(CO2Et)2 A(C10H14O4)
H3O+
Q
H3O+, H2O
Hg2+Me CO2H
O
B(C5H6O2)BrCH2 C CH
9.18 Averigua la estructura de los compuestos que aparecen en la siguiente secuencia de reacciones.
MeCO2Et
OEtO
_
Me IA
OC6H5
EtO_
[B(C16H21O4)](anión) C(C14H16O3) HBrH3O+, Q
[D(C14H17O3Br)] E(C13H17OBr)HO
_Me
C6H5
MeO
IX

10. REACCIONES DE ELIMINACIÓN
GENERALIDADES En un sustrato saturado, el esquema general de cualquier reacción de eliminación es el siguiente:
π
:B_
H C C Z C C BH Z_
+ +
[:B- es una base (nucleófilo) que puede no llevar carga] Si la reacción ocurre en un sustrato insaturado:
π
:B_
H C C Z BH Z_
+ +C C
Los sustratos más comunes en las reacciones de eliminación son los haluros de alquilo y los alcoholes:
CH3 CH CH
H
Br
CH3
B_
CH3 CH CH CH3 + HB + Br_
H2O:
CH3 C OHCH3
CH3
H3O+H CH2 C
CH3OH2
CH3
+CH2 C CH3
CH3+ H3O+ H2O+
MECANISMO DE LAS REACCIONES DE ELIMINACIÓN De forma semejante a las reacciones SN, las reacciones de eliminación transcurren normalmente a través de dos tipos diferentes de mecanismos.
REACCIONES DE ELIMINACIÓN E1 El proceso transcurre en dos pasos: en el primero se produce la ionización del sustrato y se libera un catión carbonio que, en la segunda fase, reacciona con la base originando el producto no saturado. La reacción responde a una cinética de primer orden:
v = k1[R-Z]H C C Z lenta H C C Zδ+ δ−
H C C+
+ Z_
+ HBrápidaB
_H C C + C C
X

255
Resulta evidente que la etapa lenta es la misma que en las reacciones SN1; todas las consideraciones con respecto a la influencia de Z, R y el disolvente sobre el mecanismo SN1 son aplicables al mecanismo E1. En cambio, la naturaleza de la base influye sobre el producto obtenido. Por ejemplo, el bromuro de t-butilo reacciona en etanol originando el 81% de t-butil etil éter (SN1) y el 19% de isobutileno (E1):
CH3 C BrCH3
CH3 lentaCH3 C
CH3
CH3+ + Br
_
(81%)
t-Butil etil éter
SN1: C2H5 OH C CH3
CH3
CH3
+rápida
C2H5 O C CH3
CH3
CH3
H
+C2H5 O C CH3
CH3
CH3
H+_
E1: (19%)C2H5 OH CCH2
CH3
CH3
H +rápida
C2H5 OH2+
+ CH2 C CH3
CH3
Isobutileno La formación del carbocatión, por ionización del bromuro de t-butilo, es la etapa determinante de la velocidad en ambas reacciones. El etanol puede actuar como nucleófilo o como base; en el primer caso reacciona con el carbono cargado positivamente, dando lugar al producto de sustitución, y en el segundo lo hace con el hidrógeno en β, originando el alqueno. Puesto que las reacciones E1 y SN1 tienen lugar a partir del mismo catión carbonio, el tipo de estructura carbonada influye de igual forma en ambos tipos de procesos. El grupo saliente (Z) no suele jugar ningún papel en las cantidades relativas de productos E1 o SN1, ya que todos ellos se forman después de que Z se ha desprendido del sustrato. La temperatura es el factor que suele determinar la proporción relativa de productos de eliminación o sustitución. En general, una temperatura elevada favorece la reacción de eliminación. Los halogenuros terciarios reaccionan más fácilmente que los secundarios a través de reacciones E1, ya que dan lugar a olefinas más sustituidas y, por consiguiente, más estables (control termodinámico) Sin embargo, un disolvente polar y nucleófilo, y la ausencia de bases fuertes, favorecen la formación de productos de sustitución en vez de los de eliminación. La ruta E1 está favorecida por los reactivos fuertemente básicos y débilmente nucleófilos, y también por los sustituyentes atractores de electrones que aumentan la acidez del hidrógeno en β.
REACCIONES DE ELIMINACIÓN E2 El proceso transcurre en un solo paso de una manera concertada, es decir, la ruptura de los dos enlaces σ y la formación del enlace π son simultáneas. La reacción pasa por un estado de transición del tipo:
δ+δ− δ−
+ Z_
H C C ZB:_
B H C C Z HB + C C
v = k2[R-Z][:B- ]La cinética de la reacción es de segundo orden:
X

256
Lo mismo que la reacción E1 es competitiva con la SN1, las reacciones de sustitución SN2 pueden competir con las de eliminación E2. En este sentido, es importante la naturaleza primaria, secundaria o terciaria del carbono al que está unido Z. La formación de un alqueno mediante un mecanismo E2 va acompañada de una disminución de la aglomeración alrededor del átomo de carbono que contiene el grupo saliente Z, mientras que en el estado de transición que conduce a la sustitución SN2, la aglomeración en torno a este átomo aumenta.
........
δ−
Z
H
R
R'
R
R'Nu
R R'R'R
Z
H.....Nu
δ−
[la aglomeración disminuye]
E2:R R'
R'R + H_Nu + Z_
_
δ−
Z
H
R
R'
R
R'Nu
R'R'
Z
Nu
δ−
[la aglomeración aumenta]
SN2:H
RR
H
RR
Nu
R'R'
+ ZSN2
_
_
E2
La reacción E2 está favorecida si existe considerable impedimento estéreo en el carbono y si la base(nucleófilo) es grande y fuertemente básica. Inversamente, la reacción SN2 está favorecida si el impedimento estéreo es mínimo en el carbono y el nucleófilo (base) es pequeño y muy polarizable. Finalmente, lareacción E2 prevalece sobre la SN2 en disolventes que no sean muy polares (p.e. un alcohol)
α
α
Se comprende fácilmente que los dos factores actúan en el mismo sentido, para favorecer considerablemente la eliminación en los derivados terciarios y para entorpecerla en los primarios. En resumen, la competencia entre los caminos SN2 y E2 está gobernada por: 1 La magnitud del impedimento estereo en el carbono al que está unido el grupo saliente Z. 2 La basicidad, el tamaño y la polarizabilidad del nucleófilo. 3 La temperatura a la que se realiza la reacción. Por ejemplo, el bromuro de etilo (carbono en posición α no impedido estéricamente) reacciona con el metiltiolato sódico (nucleófilo pequeño y muy polarizable), dando lugar a sulfuro de etilo y metilo (SN2):
CH3CH2Br + CH3_S_Na CH3CH2
_S_CH3 + NaBrSN2
Sin embargo, el bromuro de t-butilo (carbono en α muy impedido) reacciona con el t-butóxido potásico (base nucleófila grande y poco polarizable) para dar como producto más abundante el isobuteno (E2):
(CH3)3C_Br + (CH3)3CO K+ E2 (CH3)3C_OH_
CH3 CCH3
CH2 + + KBr
X

257
REGIOSELECTIVIDAD EN LAS REACCIONES E1 Y E2
REACCIONES E1 Si la base puede reaccionar con dos hidrógenos en β que no son equivalentes, se obtiene una mezcla de olefinas. El alqueno más sustituido (producto Saytzev) suele predominar sobre el menos sustituido (producto Hofmann):
[Saytzev] (32%) [Hofmann](8%)
CH3CH2 CCH3
CH3
Br E1:B CH3 CH C CH3
CH3+ CH3 CH2 C
CH3CH2
[Saytzev] (32%)
[Hofmann] (8%)
=
:BB: 1 2+
CH3CH2 CCH3
BrCH3
E1:B CH3CH2 C
CH3
CH3
+ CH3 CH
H
C CH2
CH3
H
1
2
E1
CH3 CH CCH3
CH3
CH3 CH2 CCH3
CH2
[Saytzev] (81%) [Hofmann] (19%)E1
CH3CH2 CHBr
CH3EtO
_
CH3 CH CH CH3 + CH3 CH2 CH CH2
REACCIONES E2 La extensión en la que se cumple la regioselectividad Saytzev, en las reacciones E2, depende de la estructura de la cadena carbonada, de la naturaleza del grupo saliente y de la base que desencadena la eliminación. A medida que crece el impedimento estéreo del hidrógeno en β y el tamaño de la base, el producto Saytzev va siendo menos abundante:
[Saytzev] (81%) [Hofmann] (19%)
CH3CH2 CHBr
CH3E2
EtO
Me3CO
_
_
CH3 CH CH CH3 + CH3 CH2 CH CH2
[Saytzev] (47%) [Hofmann] (53%)
CH3 CH CH CH3 + CH3 CH2 CH CH2
+
[Saytzev] (14%) [Hofmann] (86%)
Me3CE2
EtO
Me3CO
_
_
Me3C CH
CH2 C CH3
Br
CH3
C CH3
CH3Me3C CH2 C CH2
CH3
+
[Saytzev] (2%) [Hofmann] (98%)
Me3C CH C CH3
CH3Me3C CH2 C CH2
CH3
X

258
ESTEREOQUÍMICA DE LAS REACCIONES DE ELIMINACIÓN
REACCIONES DE ELIMINACIÓN MONOMOLECULARES (E1)
Con independencia del tipo de molécula de partida, ninguna reacción E1 es estereoespecífica. El giro libre del enlace en el carbocatión intermedio da lugar a dos conformaciones distintas: una de ellas origina el estereoisómero Z de la olefina, y la otra el estereoisómero E. No obstante, la interacción entre los grupos de carbonos contiguos hace que uno de los estereoisómeros predomine sobre el otro. El mayoritario será aquél que surja del carbocatión con menor impedimento estéreo:
H H
H
Me
Me I
E1H
HMe
MeH+
:
MeHH
Me(Z)
H
HMe
HMe+
:
HMeH
Me(E)
H Me
H
H Me
H Me
H
Me H=
=
(minoritario)
(mayoritario)
B
B
Los ejemplos que siguen son todos reales, pero es probable que los tipos de moléculas que aparecen en ellos se transformen en olefinas a través de mecanismos E1 o E2, dependiendo de las siguientes variables: la naturaleza del grupo saliente (Z), la base concreta que desencadena la reacción, el impedimento estéreo en el hidrógeno β, el disolvente y la temperatura a la que tiene lugar el proceso. Por los mismos motivos, además de la reacción de eliminación (E1 o E2), pueden producirse reacciones de sustitución nucleófila competitivas (SN1 o SN2)
EJEMPLO 1
E1R
RCH2
HH
RRCH2
H+
(E)(Z)H
RCH2 CHZ
CH2R
H H
H
CH2R
R Z
E1CH2RHH
R(Z)
(E)
H R
H
H CH2R
=
=
HH
H CH2RR
+:
HCH2R
H HR
+:
H R
H
RCH2 H HCH2RH
R
B
B
Se obtiene una mezcla de los estereoisómeros (Z) y (E) de la olefina. El impedimento estéreo en el carbocatión que da lugar al isómero (Z) (interacción entre los grupos R / RCH2 y H / H) es mayor que el existente en el otro carbocatión (interacción entre los grupos R / H y RCH2 / H) Cabe esperar que el producto mayoritario sea el estereoisómero (E)
X

259
EJEMPLO 2
E1H
RR
Z
ZR
H+
(E)(Z)
R
R CHZ
CHZ
R
Prioridades de los grupos: Z > C(R, R’) > H
R Z
H
H R
=
=H R
ZR
(Z)
H
Z RZ
HR
(R,R)
E1
H
RZR
H +:
H
RZH
R +:
R Z
H
R HR R
ZH
(E)
B
B
Z R
H
R H
=
=R Z
RH
(Z)
H
Z ZR
RH
(S,S)
E1
H
ZRH
R +:
H
ZRR
H +:
Z R
H
H RH Z
RR
(E)
B
B
Z R
H
H R
=
=H Z
RR
(E)
H
Z ZR
HR
(R,S)
E1
H
ZRR
H +:
H
ZRH
R +:
Z R
H
R HR Z
RH
(Z)
B
B
Si el radical R es de mayor tamaño que el grupo saliente Z, el estereoisómero (Z) será el mayoritario cuando la pareja de enantiómeros (R,R) / (S,S) o la forma meso (R,S) se someten a una reacción de eliminación E1.
X

260
EJEMPLO 3
E1R'
HRCH2
HH
R'RCH2
H+
(E)(Z)
R
HR'CH2
HH
RR'CH2
H+
(E)(Z)
+RCH2 CHZ
CH2R'
Z R'
H
H CH2R
=
=R' CH2R
HH
(Z)
H
ZR'H
CH2RH
E1
H
R'H H
CH2R+:
H
R'H CH2R
H+:
H R'
H
RCH2 HR' H
CH2RH
(E)
(R) o (S)
B
B
Z R
H
H CH2R'
=
=R CH2R'
HH
(Z)
H
ZRH
CH2R'H
E1
H
RH H
CH2R'+:
H
RH CH2R'
H+:
H R
H
R'CH2 HR H
CH2R'H
(E)
(R) o (S)
B
B
El estereoisómero (E) será el mayoritario en ambos casos, con independencia de los tamaños relativos de los grupos R, R', RCH2 y R'CH2 .
EJEMPLO 4
E1H
R'R
ZR'
ZR
H+
(E)(Z)
Z
R'R
H
(Z)
+
R'
HR
Z
(E)
+R CHZ
CHZ
R'
Prioridades de los grupos: Z > C(R, R’) > H Si los radicales R y R' son de mayor tamaño que el grupo saliente Z, el estereoisómero (Z) será el mayoritario en los dos casos.
X

261
R' Z
H
H R
=
=H R'
ZR
(Z)
H1
Z R'Z
H2R
E1
H
R'ZR
H +:
H
R'ZH
R +:
R' Z
H
R HR R'
ZH
(E)
(R,R) (S,S)(R,S) (S,R)
R Z
H
H R'
=
=Z R'
HR
(Z)
H2
ZZR
R'H1
E1
H
ZR H
R'+:
H
ZR R'
H+:
R Z
H
R' HZ H
R'R
(E)
B
B
B
B
La deshidratación del 4-Metil-3-hexanol en medio ácido transcurre a través de un proceso E1. Escribe el mecanismo de la reacción que aparece seguidamente para cada uno de los cuatro estereoisómeros del alcohol.
EJERCICIO 10.1
E1CH3CH2 CHMe
CHOH
CH2CH3 CH3CH2 CMe
CH CH2CH3H3O+
REACCIONES DE ELIMINACIÓN BIMOLECULARES (E2) En las reacciones E2 asume enorme importancia la disposición relativa que adoptan los grupos eliminados. Normalmente, las reacciones E2 transcurren con mayor facilidad cuando dichos grupos están en relación antiperiplanar; por este motivo, el proceso se denomina anti-eliminación. Si son posibles dos o más conformaciones antiperiplanares, la más favorecida será la de menor contenido energético (interacción mínima entre grupos de carbonos contiguos):
1H
I2H
Me
Me
H
Me H2
H1
I
H Me E22H Me
HMe
[antiperiplanar](menos impedida)
(E)
[mayoritario]
2H
IMe
Me
1H
H
1H Me
H2
I
H Me E2
Me MeH1H
(Z)
[mayoritario] [antiperiplanar] (más impedida)
B:
B:
X

262
En los ejemplos que siguen se utilizan los mismos tipos de moléculas que en las reacciones E1 (ver p.6-9) Así se pueden apreciar mejor las diferencias y semejanzas entre ambas clases de procesos (E1 y E2).
EJEMPLO 1
R
RCH2
HH
RRCH2
H+
(E)(Z)H
RCH2 CHZ
CH2R E2
E2CH3CH2 CH
BrCH2CH3
[(Z) + (E)]
CH3CH CH CH2CH3
1H
BrMe
Et
2H
H
2H Me
H1
Br
H Et E2
Me EtH2H
(Z)
[minoritario]
2H
Br1H
Et
Me
H
Me H1
H2
Br
H Et E21H Et
HMe
(E)
[mayoritario]
B:
B:
EJEMPLO 2
E2H
RR
ZR
ZR
H
(E)(Z)(R,R) o (S,S)
E2
(R,S)
R CHZ
CHZ
R R CHZ
CHZ
R
CH3 CHBr
CHBr
CH3E2
CH3 CH C CH3
Br
La reacción es estereoespecífica:
H
BrBr
Me
Me
H
Me Br
H
Br
H Me E2
Br MeHMe
(Z)
H
Br Me
H
Br
Me
Me Br
H
Br
H Me E2
H MeBrMe
2
3
(2R,3R)
(2R,3R) (Z)
2
3
B:
B:
X

263
H
BrMe
H
Br
Me
Br Me
H
Br
Me H E2
Me HMeBr
(Z)
H
Br Br
Me
Me
H
Br Me
H
Br
Me H E2
Me BrMeH
23
(2S,3S)
(2S,3S) (Z)
23
B:
B:
H
BrBr
H
Me
Me
Me Br
H
Br
Me H E2
Br HMeMe
(E)
H
Br Br
H
Me
Me
Br Me
H
Br
H Me E2
H BrMeMe
2
3
(2R,3S)
(2R,3S) (E)
2
3
B:
B:
EJEMPLO 3
R'
HRCH2
HH
R'RCH2
H+
(E)(Z)
R
HR'CH2
HH
RR'CH2
H+
(E)(Z)
+(R) o (S)
E2RCH2 CH
ZCH2R'
E2
[1] [2]
CH3CH2CH2 CHCl
CH2CH3 CH3CH2 CH CH CH2CH3 + CH3CH2CH2 CH CH CH3
3-Hexeno 2-Hexeno
3-Hexeno [1]:
1H
Cl H2
H
Et
Et
2H Et
H1
Cl
H Et E2
H H2EtEt
(Z)[minoritario]
2H
Cl Et
H
H1
Et
Et H1
H2
Cl
H Et E2
H EtH1Et
(E)[mayoritario]
32
32
B:
B:
(3R)
1H
Cl H2
H
Et
Et
2H Et
H1
Cl
H Et E2
H H2EtEt
2H
Cl Et
H
H1
Et
Et H1
H2
H Et
H EtH1Et
B:
B:
(3R)
X

264
1H
Cl H2
Et
Et
H
2H Et
H1
Cl
Et H E2
Et H2EtH
2H
Cl Et
Et
H1
H
Et H1
H2
Cl
Et Et E2
Et EtH1H
3
(3S) (E)
[mayoritario]
(Z)
[minoritario]
2
3
2
B:
B:
2-Hexeno [2]:
1H
Cl H2
Pr
Me
H
2H Me
H1
Cl
Pr H E2
Pr H2MeH
2H
Cl Me
Pr
H1
H
Me H1
H2
Cl
Pr H E2
Pr MeH1H
3
(E)
[mayoritario]
(Z)
[minoritario]
2
2
3
B:
B:
(3R)
1H
Cl H2
H
Me
Pr
2H Me
H1
Cl
H Pr E2
H H2
MePr
2H
Cl Me
H
H1
Pr
Me H1
H2
Cl
H Pr E2
H MeH1Pr
3
(3S) (Z)
(E)
[minoritario]
[mayoritario]
2
32
B:
B:
X

265
EJEMPLO 4
R'
HR
ZR'
ZR
H+
(Z)(Z)
H
R'R
ZZ
R'R
H+
(E)(E)
(R,R) o (S,S)
E2
(R,S) o (S,R)
E2R CHZ
CHZ
R' R CHZ
CHZ
R'
CH3 CH CHBr Br
CH2CH3E2
CH3 C CHBr
CH2CH3 + CH3 CH CBr
CH2CH3 + CH3 CHBr
CH CH CH3
[1] [2] [3] 2-Bromo-2-penteno [1] y 3-Bromo-2-penteno [2]:
Br
1H H2
Me
Et
Br
2H Et
Br
1H
Me Br E2
Me H2EtBr
2H
Br Et
1H
Br
Me
Et Br
H2
Br
1H Me E21H Et
BrMe
2
(Z)
(Z)
(2R,3R)[1]
3
23
[2]
B:
B:
Br
1H Et
Br
H2
Me
Et H2
Br
1H
Br Me E2
Br EtH2Me
2H
Br Br
Me
Et
1H
Br Et
H2
Br
Me H1E2
Me BrEt1H
2
(Z)
(Z)
(2S,3S)[1]
3
2
3 [2]
B:
B:
Br
1H Et
Me
H2
Br
Et H2
Br
1H
Me Br E2
Me EtH2Br
2H
Br Br
1H
Et
Me
Br Et
H2
Br
1H Me E21H Br
EtMe
2
(E)
(E)
(2R,3S)[1]
3
23
[2]
B:
B:
X

266
Br
1H H2
Br
Et
Me
2H Et
Br
1H
Br Me E2
Br H2EtMe
2H
Br Et
Me
Br
1H
Et Br
H2
Br
Me H1E2
Me EtBr1H
2
(E)
(E)
(2S,3R)[1]
3
23
[2]
B:
B:
Escribe el mecanismo de formación de los estereoisómeros del compuesto [3] (Ejemplo 4) utilizando los cuatro estereoisómeros del 2,3-Dibromopentano.
E2
[3]
CH3 CH
Br
CH
Br
CH2CH3 CH3 CHBr
CH CH CH3
EJERCICIO 10.2
TIPOS DE ESTEREOISÓMEROS OBTENIDOS EN LAS REACCIONES E1 Y E2
EJEMPLO 1
E1R
HRCH2
HH
RRCH2
H+
(E)(Z)
E2
R
HRCH2
HH
RRCH2
H+
(E)(Z)
[estereoselectiva]
RCH2 CH CH2RZ
EJEMPLO 2
E1
E2
H
RR
ZR
ZR
H+
(E)(Z)
(R,R) o (S,S)* *
R
ZR
H
(Z)
[estereoespecífica]
R CHZ
CHZ
R
X
267
E1
E2
H
RR
ZR
ZR
H+
(E)(Z)
(R,S)
(meso)
* *H
RR
Z
(E)
[estereoespecífica]
R CHZ
CHZ
R
EJEMPLO 3
E1
E2
R'
HRCH2
HH
R'RCH2
H+
(E)(Z)
R
HR'CH2
HH
RR'CH2
H+
(E)(Z)
+
(R) o (S)*
R'
HRCH2
HH
R'RCH2
H+
(E)(Z)
R
HR'CH2
HH
RR'CH2
H+
(E)(Z)
+
RCH2 CHZ
CH2R'
EJEMPLO 4
E1
E2
H
R'R
ZR'
ZR
H+
(E)(Z)
Z
R'R
H
(Z)
+
R'
HR
Z
(E)
+
R'
H2R
ZR'
ZR
H1
+
(Z)(Z)
* *(R,R) o (S,S)
R CH1 CH2 R'Z Z
E1
E2
H
R'R
ZR'
Z
H+
(E)(Z)
Z
R'
H
(Z)
+
R'
HR
Z
(E)
+
H2
R'R
ZZ
R'R
H1
+
(E)(E)
* *(R,S) o (S,R)
R CH1Z
CH2Z
R'
R R
X

268
ESTEREOQUÍMICA Y REGIOSELECTIVIDAD EN LAS REACCIONES E1 Y E2
REACCIONES E1 Si la base puede extraer dos hidrógenos en β que no son equivalentes, se pierde preferentemente el que da lugar al alqueno más sustituido:
CH3CH2 CHBr
CH3E1 CH3 CH CH CH2
1 2+
1
2
EtO_
CH3 CH CH CH3
(Z) + (E)
CH3CH2 CH CH2
(Saytzev)
(Hofmann)
H H
Productos Saytzev:
H
HMe
HMe Me
H
HMe
H
MeH
HMe
H
Me
H
HH Me
HMeMeH
MeMeHH
+
+
(E)
(Z)
(may.)
(min.)
B:
B:
Producto Hofmann:
H
HEt
HH H
H
HEt H
HHEtH+
B:
REACCIONES E2 La extensión en la que se cumple la regioselectividad Saytzev en las reacciones E2, depende de la estructura de la cadena carbonada, de la naturaleza del grupo saliente y de la base que desencadena la eliminación. A medida que crecen el impedimento estéreo del hidrógeno en β y el tamaño de la base, el producto Saytzev va siendo menos abundante:
+
[Saytzev] (81%) [Hofmann] (19%)
CH3CH2 CHBr
CH3E2
EtO
Me3CO
_
_
CH3 CH CH CH3 CH3 CH2 CH CH2
+
[Saytzev] (47%) [Hofmann] (53%)
CH3 CH CH CH3 CH3 CH2 CH CH2
X

269
Productos Saytzev:
H1
2HMe Me
H1
2HHMe
Me2H
(E)Br
HMe
H
H2
Me1H
H1
H2
MeH1H
MeMe
Br
HMe
HMe
Br
Br
(may.)
(min.)(Z)
MeB:
B:
Producto Hofmann:
H
HH H
H
H EtH
HHB:
Br
EtH EtH
Br
H
HH
Br
H
H
H
HH
H
1
2Saytzev
Hofmann
[Saytzev] (14%) [Hofmann] (86%)
Me3C CH2 CBr
CH3CH3
E2
EtO
Me3CO
_
_
Me3C CH C CH3
CH3+ Me3C CH2 C CH2
CH3
[Saytzev] (2%) [Hofmann] (98%)
Me3C CH C CH3
CH3+ Me3C CH2 C CH2
CH3
Producto Hofmann:
H
HH H
H
HCH2CMe3H
HHB:
Br
CH2CMe3Me
CH2CMe3Me
Br (Hofmann)
X

270
Productos Saytzev:
H1
2HMe3C CMe3
H1
2H MeMe3C
Me2H
B:
Br
MeMe
MeMe
H2
Me3C1H
H1
H2
Me3C Me1H
MeMe3C
B:
Br
MeMe
MeMe
Br
Br
(Saytzev)
(Saytzev)
H
HH
Br
H
H
Me
HH
H
1
2Saytzev
Hofmann
APLICACIONES DE LAS REACCIONES DE ELIMINACIÓN EN SÍNTESIS
REACCIONES E1 Los cationes carbonio que se forman en las reacciones de solvolisis dan lugar con frecuencia a mezclas de productos E1 y SN1. La proporción relativa de unos y otros está determinada en parte por la basicidad del disolvente y también por la presencia de otros nucleófilos en la disolución.
(27 %) (7 %)
CH3CH2 CBr
CH3
CH3
EtOH250 CH3CH2 C CH3
CH3
+ C2H5OH
SN1
E1
CH3CH2 CCH3
CH3
O CH2CH3 (66 %)
CH3 CH CCH3
CH3 + CH3CH2 CCH3
CH2
Por ejemplo, la solvolisis en piridina (básica) conduce en general a reacciones en las que predomina la eliminación E1, mientras que la solvolisis en presencia de tiosulfato (S2O3
2-) da lugar normalmente a reacciones SN1. Cuando aumenta la sustitución en el carbono β o se eleva la temperatura se incrementa la proporción del producto de eliminación. Las reacciones E1 tienden a originar productos en los que predomina la olefina más sustituida (producto Saytzev). Ya que el grupo saliente no interviene en el paso final que da lugar a los productos, sus porcentajes relativos son independientes de los sustratos de partida:
X

271
(36 %) (64 %)
CH3 CCH3
BrCH3
C2H5OH / H2O (80:20)
650H2C C
CH3CH3 + CH3 C
CH3
CH3
OH + CH3 CCH3
CH3
O CH2CH3
(catión sulfonio) (36 %) (64 %)
C2H5OH / H2O (80:20)
650H2C C
CH3CH3 + CH3 C
CH3
CH3
OH + CH3 CCH3
CH3
O CH2CH3CH3 CCH3
SCH3
CH3CH3
+
La deshidratación de alcoholes, catalizada por ácidos, sólo tiene aplicación en síntesis cuando no hay transposiciones de Wagner-Meerwein que den lugar a productos indeseables:
CH3 CH2OHH2SO4
1800CH3 CH2 OH2
+ H2O_CH3 CH2
+ SO4H_
CH2 CH2
H3PO4 (85 %)
OH
E1I2 (trazas) +
90-1000
(85 %) (15 %)
CH3 COH
CH3
CH2CH3 CH3 C CH2CH3CH3
CH3 CCH3
CH2CH3 + CH2 CCH3
CH2CH3
E1
H2SO4
(80 %)
C
OH
CH3
C
CH3
+C
CH2
REACCIONES E2 Las reacciones E2 se favorecen cuando la temperatura y la basicidad del reactivo nucleófilo son altas y el impedimento estéreo en el carbono β del sustrato es elevado. Si existen sustituyentes electronegativos unidos a dicho carbono, aumenta la acidez del hidrógeno susceptible de eliminación y la reacción E2 transcurre con mayor facilidad. Las bases (nucleófilos) empleados en las reacciones de eliminación son, entre otras, las siguientes:
H2O ; R3N ; CH3CO2 ; HO ; RO ; H2N ; CO3
2_ _ _ _ _
Los grupos eliminados son los mismos que en las reacciones SN2:
Cl ; Br ; I ; RSO3 ; RCO2 ; R3N: ; R2S ; HO ; RO_ _ _ _ _ _ _
La reactividad de los nucleófilos en las reacciones E2 es paralela a la basicidad de dichos nucleófilos en las reacciones de trasferencia protónica. En consecuencia, nucleófilos tales como RS¯ , que deben la mayor parte de su reactividad a una polarizabilidad elevada, tienden a producir reacciones de sustitución en lugar de eliminación. Las bases muy fuertes, como el anión amiduro H2N¯ provocan predominantemente reacciones de eliminación en sustratos primarios.
X

272
Algunas de estas tendencias aparecen en los ejemplos siguientes:
(90 %) (10 %)
CH3CH2Br + EtO_ EtOH
550CH3CH2 O CH2CH3 + CH2 CH2
EtOH
550
(21 %) (79 %)
CH3 CHCH3
Br +_
EtO CH3 CHCH3
O CH2CH3 + CH2 CH CH3
550CH3 CH
CH3Br + EtOH CH3 CH
CH3O CH2CH3 + CH2 CH CH3
(97 %) (3 %)
EtOH
550
(100 %)
CH3 CCH3
BrCH3
+_
EtO CH2 CCH3
CH3
Las reacciones de eliminación en haluros de alquilo tiene escaso valor en síntesis, ya que dichos sustratos se obtienen normalmente a partir de alcoholes, y éstos son fácilmente deshidratables a olefinas:
HBr
H3O+
R CH2 CHOH
R
R CH2 CHBr
R
R CH CH R
Sin embargo, hay situaciones en las que la deshidrohalogenación es útil:
EtOHCl2
NaOH+CH2 CH2 ClCH2 CH2Cl CH2 CHCl
Br2 NCH3CH2CH2 CO2HPBr3
CH3CH2 CHBr
CO2H CH3CH CH CO2H
La mayor parte de las reacciones E2 son estereoespecíficas:
Br
H
C6H5
C6H5H
Me
C6H5 C6H5
HMe
(Z)
12
(1R,2R)
HO_
EtOHKOH
Br
H
C6H5
HC6H5
Me
C6H5 HC6H5Me12
(1S,2R)(E)HO
_
EtOHKOH
X

273
Las reacciones de eliminación son muy rápidas en los ésteres β-sustituidos, ácidos, cetonas, aldehidos y nitrocompuestos. Los grupos electronegativos unidos al mismo carbono que el hidrógeno eliminado aumentan la acidez de dicho hidrógeno, facilitando la reacción.
NaOH _N
N-H
H2O
C6H5 CO
CH
H
CH2
HO_
NH2O
C6H5 CO
CH CH2 +
EtONa / H2O _Na+ CH(CO2Et)2C6H5 CH CH
CH(CO2Et)2
H
CO2Et_
EtO
C6H5 CH CH CO2Et +
La reacción de dihaluros vecinales con Zn da lugar a olefinas:
Br
Br
Me
MeH
H
Me MeHH
(Z)
2
(2S,3S)
3 + ZnBr2Me2CO
Zn
Br
Br
Me
HMe
H
Me H
MeH2
(2S,3R)
(E)
3 + ZnBr2
Zn
Me2CO
Apartado 1: ¿Qué estereoisómeros de A y B son coherentes con las reacciones del esquema?EJERCICIO 10.3
A(C5H10I2) (Z) 2-Penteno
Zn
SN2
Br_
B(C5H10Br2)
Apartado 2: ¿Cuáles son los estereoisómeros del 3-Metil-2-pentanol que conducen a los compuestos siguientes?
[A(C6H13Br) + B(C6H13Br)] 3-Metil-2-pentanolSN1 SN2
(2R,3S)
(2S,3S)
2-Amino-3-metilpentano+HBr NH3
Apartado 3: ¿Qué estereoisómeros del 2,3-Dibromobutano darán lugar a reacciones de eliminación estereo- específicas?
E2
X

274
COMPETITIVIDAD ENTRE LAS REACCIONES DE SUSTITUCIÓN Y DE ELIMINACIÓN
DERIVADOS ALQUÍLICOS PRIMARIOS SIN IMPEDIMENTO ESTÉREO Nucleófilo básico con impedimento estéreo: normalmente E2:
Me3CO_
H CHR
CH2 O SO
OMe R CH CH2 + Me3C OH + O S
O
OMe
_
+
Me3CO_
H CHR
CH2 NMe3 R CH CH2 + Me3COH + NMe3
Nucleófilo sin impedimento estéreo: normalmente SN2, con independencia de Nu, Z y disolvente. Estas tres variables influyen en la velocidad de reacción pero no en el tipo de mecanismo.
IK + RCH2ClMe2CO
RCH2I + ClK
OH + RCH2Br RCH2OH + Br_ _
H2S + RCH2NMe3 H2S_CH2R + NMe3++
Br + RCH2-OH2 RCH2Br + H2O+_
Me3N + RCH2I RCH2NMe3 + I+ _
Reactividad: MeCl >> MeCH2Cl > MeCH2CH2Cl ≅ Me(CH2)nCH2Cl [modelo]: RCl RIIK
Me2CO;
DERIVADOS ALQUÍLICOS PRIMARIOS CON IMPEDIMENTO ESTÉREO Dos sustituyentes β (tipo neopentilo): Normalmente SN1 + E1 (transposiciones de Wagner-Meerwein)
SN1
E1
transp. de W-MCH3 C
CH3
CH3
CH2BrH2O
CH3 CCH3
CH3
CH2+
CH3 CCH3
CH2 CH3+
CH3 COH
CH3
CH2CH3
CH3 CCH3
CH CH3
Un sustituyente β o dos γ: Normalmente SN2 (mayoritario) + E2 (minoritario)
_EtO+ CH3 CH
CH3CH2 O CH2CH3 + CH3 C
CH3CH2CH3 CH
CH3CH2Br
X

X
275
Normalmente S
CH3 CCH3
CH3
CH2CH2Br +_
EtO CH3 CCH3
CH3
CH2CH2 O CH2CH3 + CH3 CCH3
CH
CH3
CH2
Reactividad: R CH2CH2CH2 Z > R CHR'
CH2 Z R CR'
R''CH2CH2 Z >>> R C
R'
R''CH2 Z
DERIVADOS ALQUÍLICOS PRIMARIOS QUE ORIGINAN CARBOCATIONES ESTABILIZADOS
N1 muy rápida:
+SN1 NuCH2 CH CH2Cl CH2 CH CH2
+H2CHCH2C
_
CH2 CH CH2 Nu
+
+SN1 Nu...CH2 CH2*
*
*
*
Qmax
C6H5 CH2Cl
_
C6H5 CH2 Nu
DERIVADOS ALQUÍLICOS SECUNDARIOS
Nu poco y disolvente muy polar: SN1
Nu muy r y disolvente poco polar: SN2
reactivo
eactivoNormalmente:
_
_

276
SUSTITUCIÓN – ELIMINACIÓN [ I ]
se forma un alqueno por E2
¿es Nu- una base fuerte impedida? (p.e. t-BuO-)
dos sustituyentes β (R' y R'') (tipo neopentilo)
R-C-CH2-Z
R'
R''
un sustituyente β (R')
R-C-CH2-Z
R'
H
R-C-CH2CH2-Z
R'
dos sustituyentes γ (R' y R'')
carbocationesestabilizados del tipo:
SI
SN2 lenta
NO
(SN1 + E1)
transposiciones de Wagner-Meerwein endisolventes muy polares
SN2 (mayoritario)
E2 (minoritario)
SN1 muy rápida
(lo mismo para secundarios)
PRIMARIOcon impedimento estéreo
PRIMARIOsin impedimento estéreo R-C-Z
H
H
¿qué tipo de sustituciónpresenta el C unido a Z?
¿es Z un buen grupo saliente?
SI
NONO HAY REACCIÓN
R''
R-CH-CH=CH2+
+R-CH-C6H5
X

X
277
¿es Nu- una base fuerte? (p.e. RO-)
SI
SN2moderadamente rápida
¿qué tipo de sustituciónpresenta el C unido a Z?
¿es Z un buen grupo saliente?
SI
NONO HAY REACCIÓN
SECUNDARIOR Z
R HTERCIARIO
R Z
R Rse forma un alqueno por E1 o E2
¿el disolvente es polar? ¿es Nu- una base fuerte? (p.e. RO-)
¿es Nu- un buen nucleófilo, débil o moderadamente básico? (p.e. : I-, Br-, RS-)
SN1 rápida
reacción muy lenta(no útil en síntesis)
SI
NO
¿el disolvente es polar?SI
se forma un alqueno por E2
NO
SI
¿el disolvente es polar?
SN1 lenta
SI
SI
NO
NO NO
NO
SUSTITUCIÓN – ELIMINACIÓN [ II ]

278
EJERCICIOS ADICIONALES
10.1 Cuando el estereoisómero ópticamente inactivo del 2,3-Dibromobutano se somete a una reacción de eliminación E2, con metóxido sódico en metanol, se obtienen varios compuestos de fórmula molecular C4H7Br. Haz un esquema de las reacciones que tienen lugar y nombra de forma inequívoca todos los compuestos que se obtienen. 10.2 La reacción del 3-Fenil-2-yodo-butanoato de etilo con nitrato de plata en etanol, da lugar a una mezcla de cuatro estereoisómeros de fórmula molecular C12H O14 2. Sabiendo que la cinética de la reacción es de primer orden, averigua la estructura de dichos compuestos y explica cuáles de ellos serán mayoritarios. Teniendo en cuenta que la sustancia de partida tiene dos átomos de carbono asimétricos, indica si su configuración absoluta influye en la estereoquímica de los productos que se obtienen. 10.3 Averigua la configuración absoluta del compuesto A(C10H Cl): 13
A + IK
(2S,3S) + (2R,3S)-3-Fenil-2-yodobutano
2-Fenil-2-yodobutano (racémico)
A + EtONa (Z) 2-Fenil-2-buteno + (3R) 3-Fenil-1-buteno
H2O
EtOH
Una vez conocida la estructura de A, escribe todas las reacciones que conducen a los compuestos indicados en los esquemas. 10.4 Averigua la configuración absoluta de las moléculas que constituyen el racémico A(C9H10ClBr):
A (racémico)E2
(Z) B(C9H9Br) 1-Fenilpropino
(Z) C(C9H9Cl) 1-Fenilpropino
_EtO
HO
HO
_
_
H2O
H2O
MeCOCH2CO2Et + EtO_
Econc.HO
_C6H5CH2 CH CH2CO2H
MeB
H2
[Pd]D(C9H11Br)
MeCOCH2CO2Et + EtOconc.
_HO
_CH3CH2 CH CH2CO2H
C6H5
CH2
[Pd]F(C9H11Cl) H
X

279
10.5 Averigua la estereoquímica de los compuestos que aparecen en las siguientes secuencias de reacciones. Secuencia 1:
B(C4H9OBr) (racémico)SN2
C(C4H8IBr) (racémico)E2
(Z) 2-Bromo-2-buteno + (Z) 2-Yodo-2-butenoA EtO_
HOBr IH
Secuencia 2:
D(C6H10O3) + E(C4H9Br)SN2
HO (conc.)
HO (dil.)
Ácido (3S) 3-Metilpentanoico
(4S) 4-Metil-2-hexanona
EtO_
_
_
F(C10H18O3)
10.6 Averigua la estereoquímica de los compuestos que aparecen en las siguientes secuencias de reacciones (los estereoisómeros de cada compuesto están colocados entre corchetes) Secuencia 1:
(Z) 1-Bromo-3-fenil-2-butenoNaOH, H2O(20%) [A + B] + [C] (%A = %B = %C)
250C Secuencia 2:
(3R,4R) 3-Cloro-4-metilhexanoEtO , EtOH
_[A + B] + [C] (%A = %B %C)~_
650C Secuencia 3:
2,2-Dimetil-1-yodopentano [A + B] + [C] (%A > B >> %C) + [D + E] (%D = %E)H2O
500C 10.7 Averigua la estereoquímica de los compuestos que aparecen en el esquema y explica cuál será la abundancia relativa de los compuestos B, D y F.
A(C4H8BrI)
E2B [(Z) + (E)]
D [(Z) + (E)] + F(C4H7I)
H2
[Pd]1-Bromobutano
(2R)-1,2-ButanodiolOH-(dil.)
SN2 10.8 Apartado 1: Realiza la síntesis de los compuestos H y G:
1-Yodo-2-metilbutano H2C(CO2Et)2 + EtO 1)HO-
2)H3O+, QG
_
H
X

280
Apartado 2: Una vez conocidas las estructuras de H y G, averigua las de D, E y F:
MeCOCH2CO2Et + EtO
E(C5H11Cl)
1)HO- (conc.)
2)H3O+, QG
MeCOCH2CO2Et + EtO
_
_
D(C5H11I)
F
Apartado 3: ¿Qué estructura tienen los compuestos B y C?
(E) C(C5H9Cl)
(Z) B(C5H9I)
E(C5H11Cl)
D(C5H11I)H2
[Pt]
H2
[Pt] Apartado 4: Finalmente, averigua la configuración absoluta de los enantiómeros que constituyen el racémico A.
(Z) B(C5H9I)
(E) C(C5H9Cl)
A(C5H10ICl) EtONaEtOH
(único producto de reacción)
(único producto de reacción) 10.9 El tratamiento del (1S,2S)1-Fenil-1,2-propanodiol con HBr acuoso da lugar a un único producto de reacción (1) (C H Br9 10 2) Si (1) se somete a una reacción de eliminación bimolecular se obtienen dos compuestos ópticamente inactivos [(2) y (3)] y otro que presenta actividad óptica (4) Con estas tres sustancias se llevan a cabo las siguientes trasformaciones:
(1) (C9H10Br2) E2
(2)
(3)
(4)
[(5) + (6)]
[(9) + (10)]
E1
SN2
[(7) + (8)]
[(11) + (12)]
SN1[(13) + (14)] + [(15) + (16)]
H2
HO_
HO_
H2[Pd]
[Pd]NH3
Apartado 1: Ordena los compuestos (2), (3) y (4) de acuerdo con su abundancia relativa. Apartado 2: Escribe el mecanismo de todas las reacciones utilizando representaciones tridimensionales, y asigna la configuración absoluta a cada uno de los estereoisómeros que aparecen.
X



















![Hoja de calculo - COnnecting REpositories · de aminas primarias,[6] electrófilos,[7] ... artículo resume los aspectos claves de esta metodología, así ... The development of new](https://img.pdfslide.net/doc/110x75/604bbdd6188e1044e069f30f/hoja-de-calculo-connecting-repositories-de-aminas-primarias6-electrfilos7.jpg)








