Embed Size (px)
Citation preview

Universidad de Buenos Aires Facultad de Ciencias Exactas yNaturales Departamento de Computacion
Una comparacion mas eficiente y precisaentre Bonferroni y Benjamini-Hochberg
Tesis presentada para optar al tıtulo deLicenciado en Ciencias de la Computacion
TESISTA: Sebastian FerroLU 476/[email protected]
DIRECTOR: Dra. Diana [email protected] de Calculo, FCEyN, UBA, Buenos Aires
Buenos Aires, Julio 2011

Resumen
En este trabajo presentamos una revision de los procedimientos de test multiples, uncampo de investigacion de gran actividad en los ultimos anos, con aplicaciones importantescomo el analisis de los microarreglos, en Genomica.
Analizamos en detalle el algoritmo usado en Gordon, Glazko, Qiu y Yakovlev, “Control ofthe mean number of false discoveries, Bonferroni, and stability of multiple testing”, Annals ofApplied Statistics, 2007, para luego modificarlo y obtener una ganancia importante en tiempode proceso. Mostramos que con el nuevo algoritmo se pueden modelar situaciones mas realistasque las originales y mostramos como la precision alcanzada permite afirmar las conclusionesgenerales del artıculo original en estas situaciones.
Abstract
We present a review of multiple testing procedures, a research field of great activity inrecent years, with important applications such as microarray analysis, Genomics.
We analyze in detail the algorithm used in Gordon, Glazko, Qiu and Yakovlev, “ Controlof the mean number of false discoveries, Bonferroni, and Stability of multiple testing,”Annalsof Applied Statistics, 2007, and then modify it and obtain a significant gain in processingtime. We show that the new algorithm can model more realistic situations than the originaland show how the accuracy achieved allows us to affirm the general conclusions of the originalarticle in these situations.

Agradecimientos
Este trabajo representa el final de un gran esfuerzo hecho para retomar mi postergadacarrera y obtener el tıtulo de licenciado. Agradezco a mi novia, Flor, que tuvo una granpaciencia y comprension de lo importante que era para mı recibirme. A mi directora Diana,una excelente persona que logro llenarme del entusiasmo requerido para afrontar el esfuerzode desarrollar este trabajo y finalmente agradezco al Departamento de Computacion de laFacultad de Ciencias exactas y Naturales que me dio esta segunda oportunidad para completarmi carrera.

INDICE 4
Indice
1. Introduccion 71.1. Motivacion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.2. Bibliografıa de Test Multiples . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.3. Esquema de la tesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.4. El lenguaje R . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.5. El proyecto Bioconductor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.6. Investigacion reproducible . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
2. Procedimientos de Test Multiples 122.1. Introduccion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122.2. Distribucion generadora de los datos . . . . . . . . . . . . . . . . . . . . . . . . 122.3. Parametros . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132.4. Hipotesis nulas y alternativas . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.4.1. Hipotesis de submodelos . . . . . . . . . . . . . . . . . . . . . . . . . . . 142.4.2. Hipotesis Parametricas . . . . . . . . . . . . . . . . . . . . . . . . . . . . 142.4.3. Conjuntos de Hipotesis nulas verdaderas y falsas . . . . . . . . . . . . . 152.4.4. Hipotesis nula completa . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2.5. Estadısticos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152.6. Procedimientos de Test Multiples . . . . . . . . . . . . . . . . . . . . . . . . . . 162.7. P-Valores No Ajustados . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.7.1. Definicion de los p-valores no ajustados . . . . . . . . . . . . . . . . . . 162.7.2. Distribucion de los p-valores no ajustados . . . . . . . . . . . . . . . . . 17
2.8. Errores de Tipo I y Tipo II . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192.9. Errores de Tipo I en Test Multiples . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.9.1. Errores de Tipo I basados en la distribucion de los falsos positivos . . . 212.9.2. Errores de Tipo I basados en la distribucion de la proporcion de falsos
positivos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 222.9.3. Cantidad vs. proporcion de falsos positivos . . . . . . . . . . . . . . . . 222.9.4. Comparacion de errores de Tipo I . . . . . . . . . . . . . . . . . . . . . 23
2.10. P-Valores Ajustados . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 252.10.1. Definicion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 252.10.2. Representacion de un procedimiento de test multiples mediante sus p-
valores ajustados . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 252.10.3. Ventajas de la representacion por p-valores ajustados . . . . . . . . . . . 25
2.11. Potencia en Test Multiples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 262.12. Tipos de Procedimientos de Test Multiples . . . . . . . . . . . . . . . . . . . . 27
2.12.1. Procedimientos marginales descendentes y ascendentes . . . . . . . . . . 272.12.2. Comparacion de los p-valores ajustados ascendentes y descendentes . . . 29
2.13. Procedimiento de Bonferroni . . . . . . . . . . . . . . . . . . . . . . . . . . . . 302.13.1. Definicion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
2.14. Procedimiento de Benjamini-Hochberg . . . . . . . . . . . . . . . . . . . . . . . 312.15. Procedimiento de Benjamini-Hochberg . . . . . . . . . . . . . . . . . . . . . . . 31
2.15.1. Desigualdad de Simes . . . . . . . . . . . . . . . . . . . . . . . . . . . . 312.15.2. Definicion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.16. Aplicacion al analisis de los Microarreglos . . . . . . . . . . . . . . . . . . . . . 32

INDICE 5
3. Algoritmo GGQY 353.1. Introduccion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 353.2. Simulacion de P-Valores no ajustados . . . . . . . . . . . . . . . . . . . . . . . 36
3.2.1. Implementacion en R . . . . . . . . . . . . . . . . . . . . . . . . . . . . 373.2.2. Version vectorizada del test t . . . . . . . . . . . . . . . . . . . . . . . . 38
3.3. Ajuste de P-Valores . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 393.4. Ecualizacion de errores . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 393.5. Descripcion del algoritmo GGQY . . . . . . . . . . . . . . . . . . . . . . . . . . 42
3.5.1. Paso de Muestreo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 443.5.2. Pseudocodigo del algoritmo GGQY . . . . . . . . . . . . . . . . . . . . . 44
3.6. Analisis del algoritmo GGQY . . . . . . . . . . . . . . . . . . . . . . . . . . . . 453.6.1. Analisis de espacio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 453.6.2. Analisis de tiempo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 463.6.3. Sesgo por replicas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 473.6.4. Definicion de inversas . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
4. Nuevo algoritmo 524.1. Mejora en el Paso de Muestreo . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
4.1.1. Analisis de tiempo y espacio . . . . . . . . . . . . . . . . . . . . . . . . . 554.2. Mejora en la interpolacion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 554.3. Mejora en la eleccion de Grillas . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
5. Extension de los resultados originales 605.1. Nuevas condiciones mas realistas . . . . . . . . . . . . . . . . . . . . . . . . . . 605.2. Comparaciones bajo condiciones mas realistas . . . . . . . . . . . . . . . . . . . 60
5.2.1. Comparaciones de AvgPWR . . . . . . . . . . . . . . . . . . . . . . . . 625.2.2. Comparaciones de Desvıos de verdaderos positivos . . . . . . . . . . . . 635.2.3. Comparaciones de Desvıos de Positivos . . . . . . . . . . . . . . . . . . 64
6. Conclusiones 686.1. Trabajo futuro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
A. Librerıa de funciones 70
B. Reproduccion de resultados 74B.1. Comparacion de versiones del test t . . . . . . . . . . . . . . . . . . . . . . . . . 74B.2. Impacto en generacion de P-Valores no ajustados . . . . . . . . . . . . . . . . . 77B.3. Comparacion de versiones del Procedimiento de Benjamini-Hochberg . . . . . . 77B.4. Codigo para Medicion de tiempos del Algoritmo GGQY . . . . . . . . . . . . . 78B.5. Generacion de muestras . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80B.6. Reproduccion de figuras para comparacion de errores . . . . . . . . . . . . . . . 84
B.6.1. Codigo para la figura 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . 85B.6.2. Codigo para la figura 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . 85B.6.3. Codigo para la figura 3 . . . . . . . . . . . . . . . . . . . . . . . . . . . 86B.6.4. Codigo para la figura 4 . . . . . . . . . . . . . . . . . . . . . . . . . . . 86B.6.5. Codigo para la figura 5 . . . . . . . . . . . . . . . . . . . . . . . . . . . 87B.6.6. Codigo para la figura 6 . . . . . . . . . . . . . . . . . . . . . . . . . . . 88

INDICE 6
B.6.7. Codigo para la figura 7 . . . . . . . . . . . . . . . . . . . . . . . . . . . 88B.6.8. Codigo para la figura 8 . . . . . . . . . . . . . . . . . . . . . . . . . . . 89B.6.9. Codigo para la figura 9 . . . . . . . . . . . . . . . . . . . . . . . . . . . 89B.6.10. Codigo para la figura 12 . . . . . . . . . . . . . . . . . . . . . . . . . . . 89B.6.11. Codigo para la figura 11 . . . . . . . . . . . . . . . . . . . . . . . . . . . 90B.6.12. Codigo para la figura 10 . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
Referencias 91

1. Introduccion 7
1. Introduccion
1.1. Motivacion
Hoy en dıa, en areas como Astronomıa, Genomica y Marketing surgen problemas de in-ferencia estadıstica que involucran el testeo de miles, sino millones de hipotesis nulas. Estashipotesis pueden abarcar una amplia gama de parametros, para distribuciones con muy al-to grado de dimensiones y con estructuras desconocidas y complejas de dependencia entrevariables.
En Genomica, un ejemplo importante de estos problemas es la identificacion de genesdiferencialmente expresados en experimentos de microarreglos. Este nombre deriva del hechode que en estos experimentos se miden las expresiones de miles de genes dispuestos en unarreglo matricial de pocos cm. de lado (aprox. 0,5 x 0,5 cm2 o 1,3 x 1,3 cm2 o 2 x 6 cm2,dependiendo de la tecnologıa). Cada microarreglo corresponde a determinadas covariablesbiologicas o clınicas de interes y el problema consiste en determinar aquellas expresionesque dependen de las covariables investigadas. En general, en la sıntesis de una proteına y, enconsecuencia de un rasgo biologico, esta involucrada toda una red de genes. El efecto es que lasexpresiones exhiben un alto grado de correlacion y en general una estructura de dependenciacompleja como se ilustra en [KY07].
La teorıa clasica de Tests estadısticos se extendio para tratar estos problemas. No obstan-te, hay varias metodologıas propuestas y aun no hay consenso sobre cual es la mas indicada encada caso. La mas empleada es que la de Benjamini-Hochberg que mide el FDR, “False Dis-covery Rate”. En [GGQY07] se recupera el metodo de Bonferroni, que data de 1936 ([Bon36])y se muestra que compite con anterior.
Nuestro objetivo final sera mejorar los algoritmos usados para poder comprobar los resul-tados en situaciones mas realistas.
1.2. Bibliografıa de Test Multiples
Una introduccion clasica son los libros de [HT87] o [WY93]. Una revision es [Sha95]. Unamas actual y con aplicacion a la Genomica es: [DSB03]. En [PDvdL05] se describe la librerıamulttest, que es el software de codigo abierto mas popular para realizar test multiples.Seguiremos la notacion introducida en este artıculo.
Referencias sobre el “false discovery rate”(FDR), se puede encontrar en los websites de lossiguientes autores:
Yoav Benjamini www.math.tau.ac.il/ ybenja
Brad Efron www-stat.stanford.edu/ brad
Christopher Genovese www.stat.cmu.edu/ genovese/
John Storey faculty.washington.edu/ jstorey
Para los artıculos originales leer [BH95] y [BY01].Esta tesis se basa en la comparacion hecha en [GGQY07] entre los metodos de Bonferroni
y de Benjamini-Hochberg. La comparacion usa simulaciones anteriores, descritas en detalleen [QKY05].

1.3 Esquema de la tesis 8
1.3. Esquema de la tesis
Continuando con la introduccion vamos a describir el lenguaje utilizado y el modelo deinvestigacion reproducible adoptado. Luego, en la seccion 2 vamos a repasar rapidamente losconceptos de Test de Hipotesis estadısticas necesarios y presentaremos el marco teorico de laproblematica de los test multiples Presentaremos en detalle los procedimientos de Bonferroniy Benjamini-Hochberg y describiremos su aplicacion al analisis de microarreglos.
En la siguiente seccion 3 analizaremos en detalle el algoritmo presentado en [GGQY07] yampliaremos el marco teorico original. Lo cual nos permitira introducir mejoras y proponerun nuevo algoritmo en la siguiente seccion 4.
Luego, en 5 aplicaremos el algoritmo propuesto para construir simulaciones mas realistasque las originales y compararemos los resultados hallados con los originales de [GGQY07].
Despues de las conclusiones publicamos en el apendice no solo el codigo fuente de la librerıade funciones que implementan el nuevo algoritmo sino todo el codigo fuente necesario parareproducir cada cifra, tabla o figura de esta tesis.
1.4. El lenguaje R
El lenguaje R es a la vez un ambiente para la computacion estadıstica y grafica. Se haconvertido en un estandar de facto en el ambito academico. Es un proyecto GNU que essimilar al lenguaje S que fue desarrollado en los laboratorios Bell por John Chambers y colegas([VR00], [VR02], [IG96]). Se lo puede descargar libremente de http://www.r-project.org/
bajo los terminos de licencia publica GPL de la Free Software Foundation en forma de codigofuente. Compila y corre bajo una variedad de sistemas operativos, incluyendo Linux y muchosUnix, Windows y MacOS.
El lenguaje S es a menudo el elegido para realizar investigacion en metodologıas estadıs-ticas y el lenguaje R provee un acceso de codigo abierto para participar de esta actividad.
R provee una variedad de tecnicas estadısticas y graficas y es altamente extensible. Es unlenguaje interpretado con cierta orientacion a objetos y muy optimizado en la manipulacion devectores que son objetos naturales en las metodologıas estadısticas. Esta es una caracterısticadel lenguaje S y R que los programadores principiantes a menudo no toman en cuenta. Comodice [VR00] en p. 34:
Programmers coming to S from other languages are often slow to take advantageof the power of S to do vectorized calculations, that is calculations that operateon entire vectors rather than on individual components in sequence. This oftenleads to unnecessary loops. Explicit loops in S should be regarded as potentiallyexpensive in time and memory use and ways of avoiding them should be considered.
En esta tesis hicimos un fuerte uso de esta caracterıstica para optimizar tiempos y llegara tener resultados comparables a los de un codigo similar en lenguaje C.
1.5. El proyecto Bioconductor
Un proyecto GNU relacionado al proyecto R es el proyecto Bioconductor que provee he-rramientas para el analisis de los datos genomicos masivos. Bioconductor usa el lenguaje R ,tambien es de codigo abierto y de desarrollo abierto. Tiene dos actualizaciones por ano, 460librerıas y tiene una activa comunidad de usuarios. (ref: http://www.bioconductor.org/).

1.6 Investigacion reproducible 9
Dentro de este proyecto, la librerıa multtest ([PDvdL05]) es de referencia en procedi-mientos de test multiples. Fue desarrollada y es mantenida por un grupo de investigadoreslıderes en el campo de los test multiples.
Por eso, en nuestra tesis usamos funciones de esta librerıa y cuando desarrollamos imple-mentaciones propias, las comparamos con las de esta librerıa.
1.6. Investigacion reproducible
El concepto de investigacion reproducible se remonta al artıculo de [BD95] donde propu-sieron que en el ambito de la investigacion en wavelets todas las figuras publicadas deberıanser acompanadas de todo el ambiente de programas necesarios para generar dichas figuras.Mas recientemente [GL07] definen la investigacion reproducible como:
Artıculos de investigacion con las herramientas de software que permiten al lectorreproducir directamente los resultados y emplear los metodos que se presentan.
Esta combinacion entre codigo de software y texto tiene antecedentes en la ProgramacionLiteral introducida por [Knu83] donde la define como un modo de explicar a los programadoreshumanos que hace el codigo, mas que simplemente presentar las instrucciones en el ordencorrecto para el compilador. Ası, mas que presentar un codigo con comentarios el programadorescribe un texto pensado para lectores humanos integrado de segmentos de codigo y segmentosde documentacion. Los segmentos de texto proveen descripciones y detalles de que es lo quese supone que hace el codigo, mientras que los segmentos de codigo deben ser sintacticamentecorrectos pero no tienen que ser ordenados de acuerdo a lo requerido por el compilador.
Luego, mediante una operacion de “tangling” el autor puede extraer del texto integrado,los segmentos de codigo correctamente ensamblados para ser procesados por el compilador. Ymediante la operacion de“weaving”puede extraer los segmentos de texto y codigo ensambladosen un formato de lectura agradable para humanos. De esta manera, tanto el compilador comoel lector comparten una unica fuente.
Mientras la programacion literal nunca gano una gran aceptacion, muchos autores sena-laron que hay muy buenas razones para recomendarla a los estadısticos y bioinformaticos, yaque si bien la intencion original era de proveer un mecanismo para describir un programa oalgoritmo, tambien puede ser util para describir analisis de datos y metodologıas. Y mas aun,facilita la investigacion reproducible y mejora la comprension.
A la fecha existen varias implementaciones de este concepto usadas en el ambito de laestadıstica computacional que combinan un procesador de texto y un lenguaje, siendo elpaquete Sweave() es el mas popular.
Sweave: R y LATEX
odfWeave: R y Open Document Format (ODF)
R2HTML: R y HTML
SASweave: SAS y LATEX
StatWeave: R , SAS o Stata y LATEX o Open Document Format (ODF)
El paquete Sweave permite embeber codigo R en documentos LATEX. Y cuando se ejecutael paquete toda la salida del analisis de datos (tablas, figuras, etc.) se crea en el momento y es

1.6 Investigacion reproducible 10
insertada en el documento LATEX final. De esta manera el artıculo puede ser automaticamenteactualizado si los datos o el analisis cambian, lo que permite la investigacion reproducible.
Por ejemplo, un fragmento del codigo LATEX antes de procesar con Sweave de la seccionB.6 es:
Para ahorrar tiempo de proceso cargamos los objetos mr* y er* necesarios.
Ademas definimos las grillas originales del algoritmo GGQY y la funcion
plotGrids() que servira para simplificar en la seccion \ref{Ext:compar.algs}
la generacion de figuras que grafican las curvas superpuestas de las
4 semillas.
\begin{Scode}{keep.source=T}
load(file="D:/Uba/Test multiples/Tesis/v7/Objetos er y mr para comp.Robj")
gridGammas <-c(seq(from=1/100, to=1, by=1/100),
seq(from=1.1, to=10, by=1/10),
seq(from=11, to=100, by=1))
kDE <- 125
gridBetas <- gridGammas/(kDE + gridGammas)
...
\end{Scode}
Notemos con que naturalidad en R definimos los vectores gridGammas y gridBetas.Luego el paquete Sweave ejecuta el codigo R dentro del entorno “Scode” en una sesion
propia y los objetos R construidos, incluidas salidas como tablas y graficos quedan a disposiciondel autor para que se inserten en el documento LATEX final.
Siguiendo con el ejemplo, al tener los objetos mr* y er* cargados en la sesion los usamospara construir la primera figura (1) que aparece en esta tesis con el siguiente codigo embebido:
\subsubsection{Codigo para la figura \ref{Algo:ExplicCompErr1}}
\begin{Scode}{label=ExplicCompErr1, eval=T, keep.source=T, fig=T, include=F}
par(mfrow=c(2,2))
plot(x=mr.4.500.1255.280.1$bf$pvals, xlab="PFER Nominal",
y=mr.4.500.1255.280.1$bf$fdp.mean, type="l",
ylab="FDR estimado")
...
En esta tesis no adoptamos el enfoque mas radical que serıa proveer directamente el docu-mento LATEX con el codigo R embebido, sino que proveemos el documento LATEX final cuidandoque todo el codigo ejecutado sea publicado (esto se consigue con la opcion “keep.source=T”)para que el lector pueda reproducir los resultados. Tambien y de manera tradicional publica-mos el codigo en el apendice y proveemos una librerıa de funciones con codigo documentadopara ser usada independientemente del resto del codigo.
No adoptamos el enfoque mas radical por varias razones. Ademas de tener que obligar allector a que aprenda Sweave (aunque es sencillo), R y compile el documento generado en LATEX,el principal problema es que reproducir todo el codigo de esta tesis podrıa llevar mas de un dıade proceso y esto reducirıa la audiencia interesada. De todas formas, el lector interesado puede

1.6 Investigacion reproducible 11
tomarse el trabajo de ejecutar el codigo del apendice B.5 y reproducir todos los resultados deesta tesis. El lector avisado notara que solo es necesario ejecutar este codigo una sola vez yaque modificando previamente las rutas necesarias, este codigo graba en archivos los objetosgenerados. Luego, estos objetos se pueden recuperar con la operacion load() como se mostroen el ejemplo anterior.

2. Procedimientos de Test Multiples 12
2. Procedimientos de Test Multiples
2.1. Introduccion
Los Test de Hipotesis estadısticos se refieren a decisiones hechas en base a datos obser-vados sobre propiedades de la distribucion generadora de esos datos, que por lo general esdesconocida. Una hipotesis nula es una afirmacion de pertenencia de la distribucion de losdatos a un conjunto de submodelos, o sea, a un conjunto de posibles distribuciones. La hipo-tesis nula en un contexto frecuentista es verdadera o falsa. En este ultimo caso es cierta sunegacion, tambien llamada alternativa.
Frecuentemente las hipotesis nulas son parametricas. Esto es, son afirmaciones sobre unparametro de la distribucion, es decir sobre una funcion de la distribucion. Un ejemplo esafirmar que la media de la distribucion es cero. En este caso el conjunto de distribuciones queverifican la hipotesis nula es el conjunto de todas las distribuciones con media nula.
Un procedimiento de testeo es una regla para decidir en base a los datos observados si lahipotesis nula debe ser rechazada o no. Los datos observados se resumen por una funcion de losmismos, que por consecuencia es una variable aleatoria, llamada estadıstico. El procedimientode testeo divide los valores que toma el estadıstico en una region donde se rechaza la hipotesisnula (region de rechazo) y otra donde no se la rechaza (region de aceptacion). Las regionesde confianza y los p-valores que mas adelante detallaremos se asocian al concepto de regionde rechazo
Un procedimiento de test multiples, MTP (“Multiple Testing Procedure”), para M hipo-tesis nulas simultaneas provee regiones de rechazo para cada una de las M hipotesis. Luegoproduce un conjunto aleatorio (debido a que depende de los datos) de hipotesis rechazadasque estimaran el conjunto de hipotesis nulas falsas. Tambien extenderemos el concepto dep-valor para una unica hipotesis al concepto de p-valor ajustado para las M hipotesis.
Por supuesto no esperamos que el procedimiento no cometa errores. Si la hipotesis nula escierta pero el test la rechaza cometemos un error de Tipo I o falso positivo. En cambio, si lahipotesis nula es falsa pero no la rechazamos cometemos un error de Tipo II o falso negativo.En general no se pueden minimizar ambos tipos de errores a la vez y se elige minimizar elerror de Tipo II (de forma equivalente maximizar la potencia), sujeto a una restriccion sobreel error de Tipo I. Ası un MTP es una regla que a partir de los datos observados decide cualesde las M hipotesis rechazar controlando que el error de Tipo I cumpla la restriccion. En 2.8precisaremos esta restriccion y que medidas de errores usaremos.
Pero para empezar queda claro que lograremos un control probabilıstico del error de Tipo Isolo si conocemos la distribucion del estadıstico. En general no la conoceremos absolutamentesino que estara acotada por la distribucion bajo la hipotesis nula que si conoceremos conmas precision. Por propiedades de monotonıa, el error de Tipo I de la distribucion real perodesconocida sera menor que el de la distribucion nula.
Por ultimo en el Cuadro 1 adelantamos el Diagrama de Flujo de un procedimiento deTests Multiples y luego terminaremos esta seccion explicando cada paso.
2.2. Distribucion generadora de los datos
Sea Xn ≡ {Xi : i = 1, . . . , n} una muestra aleatoria de n variables aleatorias independien-tes e identicamente distribuidas (IID) de una distribucion generadora de los datos P , o sea,
XiIID∼ P , i = 1, . . . , n. Notemos con Pn la correspondiente distribucion empırica, que asigna

2.3 Parametros 13
probabilidad 1/n a cada realizacion de X.Cuando se necesite notaremos la distribucion acumulada (CDF), la funcion de supervi-
vencia y la densidad (PDF) correspondientes a P como F , F , y f , respectivamente.Asumiremos que la distribucion generadora de los datos P es un elemento de un cierto
modelo estadıstico M, o sea, un conjunto de distribuciones (parametricas o no).En muchos problemas de interes practico, la estructura de los datos consiste en vectores
aleatorios J-dimensionales: X = (X(j) : j = 1, . . . , J) ∼ P . Aquı un modelo no parametricopuede ser el conjunto de todas las distribuciones J-dimensionales continuas con media cero,mientras que un modelo parametrico podra ser el conjunto de todas las distribuciones normalesJ-variadas con media cero. En general, los modelos parametricos son mas faciles de usar.
En el caso particular de los microarreglos nos encontramos con un vector aleatorio dedimension J con dos componentes: Un vector de dimension G, X = (X(j) : j = 1, . . . , G),donde cada X(j) mide la expresion de un gen y un vector de dimension J −G, X((G + 1) :J) = (X(j) : j = G + 1, . . . , J) de covariables clınicas y resultados/fenotipos. Por ejemplo,estado del tumor, tiempo de supervivencia, nivel de insulina, etc.
Notemos que G es del orden del tamano del genoma, por lo que J tıpicamente supera los10000, mientras que el tamano de la muestra n es un numero muy inferior, posiblemente delorden de la centena. Esta diferencia entre el tamano de la muestra y el tamano del vector nosimpide usar las tecnicas clasicas del analisis multivariado.
2.3. Parametros
Definimos los parametros como cualquier funcion (integrable) de la distribucion generadorade los datos P : Ψ(P ) = ψ = (ψ(m) : m = 1, . . . ,M) ∈ RM , donde ψ(m) = Ψ(P )(m) ∈ R. Engeneral podemos clasificar los parametros usuales en microarreglos en clases de locacion, deescala y de regresion.
Ejemplo 2.1. Ejemplos de parametros de interes en analisis de microarreglos
Parametros de Locacion. Los parametros de locacion incluyen (funciones de:) medias ycuantiles.
ψ = Ψ(P ) = E[X(1 : G)]: G-vector de medias de expresiones, ψ(g) = E[X(g)],para cada gen g, g = 1, . . . , G
ψ = Ψ(P ) = E[X(1 : G)|Y = 1] − E[X(1 : G)|Y = 0]: G-vector de diferenciasde medias condicionales de expresiones, ψ(g) = E[X(g)|Y = 1] − E[X(g)|Y = 0],para cada gen g, g = 1, . . . , G, donde Y = X(G+ 1) ∈ {0, 1} es una indicadora dela pertenencia a una poblacion. Por ejemplo puede indicar dos tipos de leucemia(ALL B-cell vs. ALL T-cell) e interesa hallar los g tal que ψ(g) es significativamentedistinto de cero. Esto es hallar las expresiones que se expresan en una poblacionpero no en la otra. Este es el fenomeno llamado expresion diferencial y es uno delos objetivos del analisis de microarreglos.
Parametros de Escala. Incluyen (funciones de:) matrices de covarianzas y coeficientes decorrelacion para expresiones de genes.
σ = Σ(P ) = Cov[X(1 : G)]: G×G matriz de covarianza de X(1 : G), con elementosσ(g, g′) = Cov[X(g), X(g′)] que denotan la covarianza entre las expresiones de losgenes g y g′, g, g′ = 1, . . . , G. Podremos abreviar σ2(g) = σ(g, g) = Var[X(g)] paralos elementos diagonales de σ, o sea, las varianzas.

2.4 Hipotesis nulas y alternativas 14
σ∗ = Σ∗(P ) = Cor[X(1 : G)]: G × G matriz de correlacion de X(1 : G), conelementos σ∗(g, g′) = Cor[X(g), X(g′)] = σ(g, g′)/σ(g)σ(g′) que denotan la corre-lacion entre las expresiones de los genes g y g′, g, g′ = 1, . . . , G.
Coeficientes de regresion. Ejemplos de parametros de regresion usuales en microarreglosson los que miden la asociacion entre la expresion de un gen y las covariables clınicasy/o de resultado, incluyendo:
ψ = Ψ(P ): G-vector de combinaciones lineales ψ(g) = a>λ(g), donde λ(g) denotaun (J −G)-vector de parametros de regresion en un modelo lineal que relaciona laexpresion X(g) del gen g a un (J −G)-vector de covariables Z = X((G+ 1) : J) =(X(j) : j = G+ 1, . . . , J), E[X(g)|Z] = Z>λ(g), g = 1, . . . , G.
2.4. Hipotesis nulas y alternativas
2.4.1. Hipotesis de submodelos
En muchos problemas de interes podremos definir el modeloM como una coleccion de Msubmodelos : M(m) ⊆ M, m = 1, . . . ,M , para la distribucion generadora de datos P Ası,podemos definir M hipotesis nulas y las correspondientes alternativas como:
H0(m) ≡ I (P ∈M(m)) y H1(m) ≡ I (P /∈M(m)) , (2.1)
respectivamente. Aquı, I (·) es la funcion indicadora, igual a uno si la condicion entre parentesises verdadera y cero si no.
Entonces, H0(m) es cierta (o sea, H0(m) = 1) si la distribucion generadora de datos Ppertenece al submodelo M(m) y H0(m) es falsa si no (o sea, H0(m) = 0).
Esta representacion general de submodelos cubre tests de medias, cuantiles, covarianzas,coeficientes de correlacion y coeficientes de regresion en modelos lineales y no lineales (logıs-ticos, de supervivencia y series de tiempo).
Por ejemplo, para el vector aleatorio J dimensional X ∼ P , los submodelos de interespueden ser de la forma M(m) = {P ∈ M : X(m) ∼ N(0, 1)}, m = 1, . . . ,M = J , dondeN(0, 1) denota la distribucion normal estandar con media cero y varianza uno
2.4.2. Hipotesis Parametricas
En muchos problemas de testeo, los submodelos son condiciones sobre parametros, es decir,funciones Ψ(P ) = ψ = (ψ(m) : m = 1, . . . ,M) ∈ RM de la distribucion generadora de datosP , donde cada hipotesis nula H0(m) se refiere a un solo parametro, ψ(m) = Ψ(P )(m) ∈ R.
Para estas hipotesis parametricas se pueden distinguir entre dos tipos de problemas detesteo: Tests unilaterales y Tests bilaterales.
Test Unilaterales H0(m) = I (ψ(m) ≤ ψ0(m)) (2.2)
vs. H1(m) = I (ψ(m) > ψ0(m)) , m = 1, . . . ,M.
Test Bilaterales H0(m) = I (ψ(m) = ψ0(m)) (2.3)
vs. H1(m) = I (ψ(m) 6= ψ0(m)) , m = 1, . . . ,M.
En general los valores nulos, ψ0(m), son cero. Por ejemplo, en analisis de microarreglos po-demos testear la hipotesis nula H0(m) de no diferencia en medias de expresion de genes entredos poblaciones de pacientes.

2.5 Estadısticos 15
2.4.3. Conjuntos de Hipotesis nulas verdaderas y falsas
Definimos el conjunto de las hipotesis nulas verdaderas como:
H0 = H0(P ) ≡ {m : H0(m) = 1} = {m : P ∈M(m)} (2.4)
y la cantidad de hipotesis nulas verdaderas como: h0 ≡ |H0| La notacion extendida H0(P )enfatiza la dependencia de este conjunto de la distribucion generadora de datos P .
Asimismo definimos el conjunto de las hipotesis nulas falsas como:
H1 = H1(P ) ≡ {m : H1(m) = 1} = {m : P /∈M(m)} = Hc0(P ) (2.5)
y definimos la cantidad de hipotesis nulas falsas como: h1 ≡ |H1| = M − h0El objetivo de un procedimiento de Test Multiples es estimar (o sea, rechazar) con precision
el conjunto H1, mientras controlamos con una alta probabilidad los falsos positivos.
2.4.4. Hipotesis nula completa
Definimos la hipotesis nula completa HC0 como
HC0 ≡
M∏m=1
H0(m) =M∏m=1
I (P ∈M(m)) = I(P ∈ ∩Mm=1M(m)
). (2.6)
Ası, la hipotesis nula completa es cierta si todas las M hipotesis nulas individuales H0(m)son ciertas. En otras palabras, es cierta si y solo si la distribucion de los datos P pertenece ala interseccion ∩Mm=1M(m) de los M submodelos.
2.5. Estadısticos
Un procedimiento de testeo es un algoritmo basado en datos observados o aleatorios paradecidir que hipotesis nulas deben ser rechazadas. Es decir, cual H0(m) debe ser declaradafalsa (cero), para concluir que P /∈M(m).
Las decisiones para rechazar o no las hipotesis nulas estan basadas en el M -vector deestadısticos, Tn = (Tn(m) : m = 1, . . . ,M), que son funciones Tn(m) = T (m;Xn) = T (m;Pn)de los datos Xn, es decir, de la distribucion empırica Pn.
Notaremos la usualmente desconocida distribucion M -multivariada del estadıstico T porQ = Q(P ). Para la muestra aleatoria de n observaciones, la distribucion conjunta de los es-tadısticos Tn usaremos la notacion Qn = Qn(P ). La distribucion del estadıstico T bajo lahipotesis nula completa, llamada tambien distribucion nula, la escribiremos Q0 y la distribu-cion del estadıstico Tn bajo la hipotesis nula completa sera la distribucion nula empırica Q0n.Por ultimo, la distribucion marginal de la distribucion Q0 para la hipotesis m, la escribiremosQ0,m. Por ultimo, la estimacion muestral de la misma la podemos escribir como Q0n,m.
Mas adelante (ver (2.14)) veremos que para controlar el Tipo I de error requeriremos quela distribucion marginal de T (m) este dominada por la distribucion marginal de Q0.
Como ejemplo de estadısticos para hipotesis nulas de la forma H0(m) = I (ψ(m) ≤ ψ0(m))o H0(m) = I (ψ(m) = ψ0(m)), m = 1, . . . ,M , podemos considerar los estadısticos de diferen-cias:
Tn(m) ≡ Estimador−Valor Nulo =√n(ψn(m)− ψ0(m)), (2.7)

2.6 Procedimientos de Test Multiples 16
y los estadısticos t (es decir, de diferencias estandarizadas),
Tn(m) ≡ Estimador−Valor Nulo
Error Estandar=√nψn(m)− ψ0(m)
σn(m). (2.8)
Aquı, Ψ(Pn) = ψn = (ψn(m) : m = 1, . . . ,M) denota un estimador para el parametroΨ(P ) = ψ = (ψ(m) : m = 1, . . . ,M) y (σn(m)/
√n : m = 1, . . . ,M) denota los errores
estandar estimados para los elementos ψn(m) de ψn.
2.6. Procedimientos de Test Multiples
Un procedimiento de test multiples (MTP) genera regiones de rechazo Cn(m), es decir,conjunto de valores para cada estadıstico Tn(m) que derivan en la decision de rechazar lahipotesis nula correspondiente H0(m) y declarar que P /∈ M(m), m = 1, . . . ,M . En otraspalabras, un MTP produce un conjunto (aleatorio porque depende de los datos observados)Rn de hipotesis rechazadas que estiman el conjunto H1 de hipotesis nulas falsas,
Rn = R(Tn, Q0n, α) ≡ {m : Tn(m) ∈ Cn(m)} = {m : H0(m) es rechazada} , (2.9)
donde Cn(m) = C(m;Tn, Q0n, α), m = 1, . . . ,M , denotan las posiblemente aleatorias regionesde rechazo. La notacion larga R(Tn, Q0n, α) y C(m;Tn, Q0n, α) enfatiza que un MTP dependelos tres siguientes ingredientes:
1. Los datos, Xn = {Xi : i = 1, . . . , n}, a traves del M -vector de los estadısticos, Tn =(Tn(m) : m = 1, . . . ,M);
2. una distribucion (estimada) nula M -variada, Q0n, de los estadısticos, para derivar lasregiones de rechazo, regiones de confianza y p-valores ajustados.
3. El nivel nominal de error de Tipo I, α, es decir, una cota superior provista por el usuariopara el Tipo de error I predefinido.
2.7. P-Valores No Ajustados
En general los MTPs se basan en regiones de rechazo anidadas:
C(m;Tn, Q0n, α1) ⊆ C(m;Tn, Q0n, α2), si α1 ≤ α2. (2.10)
Las regiones de rechazo estan tıpicamente definidas en terminos de intervalos: Cn(m) =(un(m),+∞), Cn(m) = (−∞, ln(m)), or Cn(m) = (−∞, ln(m))∪(un(m),+∞), donde ln(m) =l(m;Tn, Q0n, α) y un(m) = u(m;Tn, Q0n, α) son valores crıticos inferiores y superiores (cut-offs) calculados bajo la distribucion nula Q0n para los estadısticos Tn.
Asumiremos que grandes valores de los estadısticos Tn(m) proveen evidencia contra lacorrespondiente hipotesis nula H0(m).
2.7.1. Definicion de los p-valores no ajustados
Muchos procedimientos de test multiples son extensiones de procedimientos para un solotest y el primer paso consiste en realizar M procedimientos de test por separado. Por eso esutil el concepto de p-valor no ajustado que resume el resultado de un unico test.

2.7 P-Valores No Ajustados 17
En el contexto de un solo test para una sola hipotesis nula, digamos H0(m), la region derechazo Cn(m;α) = Cn(Q0,m;α) solo se basa en la distribucion marginal Q0,m. Es decir, seelige α que cumpla que
PrQ0,m
(Tn(m) ∈ Cn(m;α)) ≤ α (2.11)
Si la distribucion del estadıstico es continua se podra elegir una region de rechazo de nivelexacto α.
Los p-valores no ajustados, P0n(m) = P (Tn(m), Q0n,m), tambien llamados marginales ocrudos, para la sola hipotesis nula H0(m), se definen por:
P0n(m) ≡ ınf {α > 0 : Rechazar H0(m) al nivel nominal α}= ınf {α > 0 : Tn(m) ∈ Cn(m;α)} , m = 1, . . . ,M. (2.12)
Ası, el p-valor no ajustado P0n(m), para la sola hipotesis nula H0(m), es el menor α,o sea,el menor nivel nominal de error de Tipo I tal que uno rechazarıa H0(m), dado que observamosTn(m). A menor p-valor no ajustado P0n(m), mayor es la evidencia contra la correspondientehipotesis nula H0(m). Veremos luego como esta definicion se puede extender inmediatamenteal caso de test multiples.
Dos representaciones el conjunto de Rechazados Bajo la condicion de regiones anida-das, (2.10), se tienen dos representaciones equivalentes del test de la sola hipotesis nula H0(m):En terminos de las regiones de rechazo y en terminos de p-valores no ajustados.
Para verlo, notemos que la sola hipotesis nula H0(m) es rechazada a nivel nominal α siTn(m) ∈ Cn(m;α) o P0n(m) ≤ α. Entonces, el conjunto de hipotesis nulas rechazadas si lasconsideramos cada una por separado es:
Rn(α) = {m : Tn(m) ∈ Cn(m;α)} = {m : P0n(m) ≤ α} . (2.13)
2.7.2. Distribucion de los p-valores no ajustados
El test para la sola hipotesis nula H0(m) controla el error de Tipo I a nivel α si
PrQn,m
(Tn(m) ∈ Cn(m;α)) ≤ α.
La practica estandar consiste en elegir estadısticos cuya distribucion nula domina la distribu-cion verdadera bajo la hipotesis nula.
PrQn,m
(Tn(m) ∈ Cn(m;α)) ≤ PrQ0,m
(Tn(m) ∈ Cn(m;α)) ∀m ∈ H0 (2.14)
Esto es, la probabilidad de rechazar la hipotesis nula H0(m) es mayor bajo la distribucionnula que bajo la distribucion verdadera cuando la hipotesis nula es cierta.
Ciertamente, la distribuciones de los estadısticos t, definidos en (2.8) cumplen esta pro-piedad. Mas claramente, en este caso la distribucion marginal nula es simetrica:
Q0,m(z) = 1−Q0,m(z) = Q0,m(−z) ∀z ∈ R
yQn,m(z) ≤ Q0,m(z), ∀ z ∈ R.

2.7 P-Valores No Ajustados 18
Luego,
PrQn,m
(|Tn(m)| > z) = 2Qn,m(z) ≤ 2Q0,m(z) = PrQ0,m
(|Tn(m)| > z) .
El control del error de Tipo I se deriva de:
PrQn,m
(P0n(m) ≤ α) = PrQn,m
(|Tn(m)| > cn(m;α))
≤ PrQ0,m
(|Tn(m)| > cn(m;α)) ≤ α.
Proposicion 2.2. Distribucion de los p-valores no ajustados Consideremos un M -vector deestadısticos Tn = (Tn(m) : m = 1, . . . ,M), con distribucion conjunta verdadera Qn = Qn(P ) ydistribucion conjunta nula Q0. Tambien consideremos las M regiones de rechazo {Cn(m;α) :m = 1, . . . ,M} y el M -vector de los p-valores no ajustados (P0n(m) : m = 1, . . . ,M) quecumplen las ecuaciones (2.14) y (2.12) respectivamente. Esto es, sean Cn(m;α) y P0n(m)tales que:
PrQ0,m
(Tn(m) ∈ Cn(m;α)) ≤ α, (2.15)
Cn(m;α1) ⊆ Cn(m;α2), siempre que α1 ≤ α2,
P0n(m) = ınf {α > 0 : Tn(m) ∈ Cn(m;α)} , m = 1, . . . ,M.
Entonces, los p-valores no ajustados P0n(m) satisfacen la siguiente desigualdad con respectoa las distribuciones marginales Q0,m,
PrQ0,m
(P0n(m) ≤ z) ≤ z, ∀ z ∈ [0, 1]. (2.16)
Para distribuciones marginales continuas Q0,m, los p-valores no ajustados P0n(m) estan uni-formemente distribuidos en el intervalo [0, 1], esto es, P0n(m) ∼ U(0, 1) y
PrQ0,m
(P0n(m) ≤ z) = z, ∀ z ∈ [0, 1]. (2.17)
Mas aun, la ecuacion (2.16) se cumple bajo las distribuciones marginales verdaderas Qn,m
PrQn,m
(P0n(m) ≤ z) ≤ z, ∀ z ∈ [0, 1]. (2.18)
Demostracion. de Proposicion 2.2Asumiendo la condicion sobre regiones anidadas (2.10) se tiene que:⋃
α≤zCn(m;α) = Cn(m; z), ∀ z ∈ [0, 1].
Entonces,
PrQ0,m
(P0n(m) ≤ z) = PrQ0,m
(ınf {α > 0 : Tn(m) ∈ Cn(m;α)} ≤ z)
= PrQ0,m
Tn(m) ∈⋃α≤zCn(m;α)
= Pr
Q0,m
(Tn(m) ∈ Cn(m; z))
≤ z,

2.8 Errores de Tipo I y Tipo II 19
donde la ultima desigualdad se sigue de la definicion de las regiones de rechazo (2.15). Paradistribuciones marginales continuas Q0,m,
PrQ0,m
(Tn(m) ∈ Cn(m; z)) = z ⇒ PrQ0,m
(P0n(m) ≤ z) = z.
Por ultimo, para las distribuciones marginales verdaderas Qn,m, la condicion (2.14) implicaque:
PrQn,m
(P0n(m) ≤ z) = PrQn,m
(Tn(m) ∈ Cn(m; z))
≤ PrQ0,m
(Tn(m) ∈ Cn(m; z))
≤ z.
2.8. Errores de Tipo I y Tipo II
En cualquier test de hipotesis podemos cometer dos tipos de errores. Cuando rechazamosuna hipotesis nula que resulta cierta producimos un error de Tipo I o falso positivo (Rn∩H0).En cambio, cometemos un error de Tipo II o falso negativo si no rechazamos una hipotesisnula falsa (Rcn ∩H1). Vamos a introducir la notacion usual en test multiples. La cantidad dehipotesis nulas rechazadas es:
Rn ≡ |Rn| =M∑m=1
I (Tn(m) ∈ Cn(m)) , (2.19)
La cantidad de errores de Tipo I o falsos positivos es:
Vn ≡ |Rn ∩H0| =∑m∈H0
I (Tn(m) ∈ Cn(m)) , (2.20)
La cantidad de errores de Tipo II o falsos negativos es:
Un ≡ |Rcn ∩H1| =∑m∈H1
I (Tn(m) /∈ Cn(m)) , (2.21)
La cantidad de verdaderos negativos es:
Wn ≡ |Rcn ∩H0| =∑m∈H0
I (Tn(m) /∈ Cn(m)) (2.22)
= M −Rn − Un = h0 − Vn,
y la cantidad de verdaderos positivos es:
Sn ≡ |Rn ∩H1| =∑m∈H1
I (Tn(m) ∈ Cn(m)) (2.23)
= Rn − Vn = h1 − Un.

2.8 Errores de Tipo I y Tipo II 20
Cuadro 1: Diagrama de Flujo de Procedimiento de Test Multiples.
Especificar distribucion de los datos y parametros de interesP , ψ = (ψ(j) : j = 1, . . . , J)
⇓Definir hipotesis nulas y alternativas
H0(m) = I (P ∈M(m)) y H1(m) = I (P /∈M(m))⇓
Especificar EstadısticosTn = (Tn(m) : m = 1, . . . ,M)
⇓Estimar distribucion nula de los Estadısticos
Q0n
⇓Seleccionar medida de eror de Tipo I
Θ(FVn,Rn)⇓
Aplicar MTP⇓
Resumir resultadosp-valores ajustados, regiones de rechazo y regiones de confianza
Cuadro 2: Errores de Tipo I y Tipo II en test multiples. Esta tabla resume los dife-rentes tipos de error y decision en test multiples. La cantidad de hipotesis rechazadases Rn = |Rn|, la cantidad de errores de Tipo I o falsos positivos es Vn = |Rn ∩ H0|,la cantidad de errores de Tipo II o falsos negativos es Un = |Rcn ∩ H1|, la cantidadde verdaderos negativos es Wn = |Rcn ∩ H0|, y la cantidad de verdaderos positivos esSn = |Rn ∩H1|. Las celdas enmarcadas corresponden a errores.
Hipotesis NulasNo Rechazadas, Rcn Rechazadas, Rn
Verdaderas, H0 Wn = |Rcn ∩H0| Vn = |Rn ∩H0| h0
Hipotesis Nulas
Falsas, H1 Un = |Rcn ∩H1| Sn = |Rn ∩H1| h1
M −Rn Rn M

2.9 Errores de Tipo I en Test Multiples 21
Notemos que el numero de hipotesis rechazadas, Rn es una variable aleatoria observablepero como la distribucion generadora de los datos, P , es desconocida, las cantidades h0 = |H0|y h1 = |H1| = M − h0 de hipotesis nulas ciertas y falsas son parametros desconocidos y lasvariables aleatorias Sn, Un, Vn, y Wn son no observables.
Podemos agrupar las cantidades definidas en la tabla 2Idealmente queremos minimizar ambos tipos de error pero en la practica eso no es posible
y buscamos un compromiso entre ambos. La practica estandar es especificar un nivel α paracierto error de Tipo I predefinido y luego minimizar el error de Tipo II (es decir, maximizarla potencia) dentro de los procedimientos cuyo error de Tipo I es a lo sumo α.
2.9. Errores de Tipo I en Test Multiples
Ahora estamos en condiciones de definir los errores de Tipo I en test multiples. A partirde las cantidades definidas en la seccion anterior surgen varias posibilidades para extender loserrores de Tipo I y Tipo II para el caso de una hipotesis al caso de multiples hipotesis.
En general podemos definir el error de Tipo I de test multiples como un parametro θn =Θ(FVn,Rn) de la distribucion conjunta FVn,Rn de la cantidad de falsos positivos Vn = |Rn∩H0|e hipotesis rechazadas Rn = |Rn|.
2.9.1. Errores de Tipo I basados en la distribucion de los falsos positivos
La representacion general Θ(FVn,Rn) cubre los errores de Tipos I usuales, que son para-metros Θ(FVn) de la distribucion FVn de la cantidad de falsos positivos Vn.
El error (FWER), “family-wise error rate” es la probabilidad de al menos un falso posi-tivo:
FWER ≡ Pr(Vn > 0) = 1− FVn(0). (2.24)
Este tipo de error se generaliza naturalmente al error (gFWER), “generalized family-wise error rate”, que es la probabilidad de al menos (k + 1) falsos positivos, dondek ∈ {0, . . . ,M}, es un argumento entero provisto por el usuario. Esto es:
gFWER(k) ≡ Pr(Vn > k) = 1− FVn(k). (2.25)
Por supuesto, cuando k = 0, el error gFWER se reduce al error clasico FWER.
El error (PCER), “per-comparison error rate” es la esperanza de la proporcion de falsospositivos entre los M tests.
PCER ≡ 1
ME[Vn] =
1
M
∫vdFVn(v). (2.26)
El error (PFER),“per-family error rate”es la esperanza de la cantidad de falsos positivos:
PFER ≡ E[Vn] =
∫vdFVn(v). (2.27)
Una alternativa a la media es la mediana de la cantidad de falsos positivos, (mPFER):
mPFER ≡ Median[Vn] = F−1Vn(1/2). (2.28)
Hasta hace poco la mayorıa de los procedimientos se enfocaban en controlar el errorFWER, del cual se tomaba como referencia el metodo clasico de Bonferroni.

2.9 Errores de Tipo I en Test Multiples 22
2.9.2. Errores de Tipo I basados en la distribucion de la proporcion de falsospositivos
La representacion general Θ(FVn,Rn) tambien cubre los siguientes errores que se basan enparametros Θ(FVn/Rn) de la distribucion FVn/Rn . Es decir de la proporcion Vn/Rn de los falsospositivos entre las hipotesis rechazadas con la convencion de que Vn/Rn ≡ 0 si Rn = 0.
Dada una constante q ∈ (0, 1) provista por el usuario, el error (TPPFP), “tail probabilityfor the proportion of false positives“ se define como:
TPPFP (q) ≡ Pr
(VnRn
> q
)= 1− FVn/Rn(q). (2.29)
El error (FDR), “false discovery rate”, es la esperanza de la proporcion de los falsospositivos:
FDR ≡ E
[VnRn
]=
∫qdFVn/Rn(q). (2.30)
Podemos escribir el error FDR de varias maneras.
FDR = E
[Vn
max {Rn, 1}
](2.31)
= E
[VnRn
∣∣∣∣Vn > 0
]Pr(Vn > 0)
= E
[VnRn
∣∣∣∣Rn > 0
]Pr(Rn > 0).
Notemos que bajo la hipotesis nula completa HC0 = I
(P ∈ ∩Mm=1M(m)
), todas las Rn
hipotesis rechazadas son falsos positivos, luego Vn/Rn = 1 y FDR = FWER = Pr(Vn >0). De esta manera, bajo la hipotesis nula completa, los procedimientos que controlan elerror FDR tambien controlan el error FWER. No obstante, en la practica Vn/Rn ≤ 1,y en consecuencia el error FDR es menor o igual que el error FWER para cualquierprocedimiento de test multiples.
2.9.3. Cantidad vs. proporcion de falsos positivos
Los errores Θ(FVn/Rn) son mas difıciles de controlar porque estan basados en la distribu-cion conjunta de Vn y Rn, en vez de basarse solo en la distribucion marginal de Vn. Recordemosque Sn = |Rn ∩ H1| = Rn − Vn, por lo que conocer la distribucion conjunta de Vn y Rn im-plica conocer la potencia y esto en general es mucho mas difıcil. Sin embargo estos erroresson atractivos para los problemas de testeo en genomica donde el numero de hipotesis, M , esmuy grande debido a que no crecen con M , como si lo hacen en el caso de los errores basadosen controlar los parametros Θ(FVn) de la distribucion de falsos positivos, como son el errorFWER y el PFER.
Para ver esto consideremos el caso simplificado de estadısticos Tn(m) : m = 1, . . . ,M in-dependientes y donde cada hipotesis se testea individualmente a nivel α. Es decir, Pr(Tn(m) ∈Cn(m)) ≤ α. Supongamos ademas que c = h0/M se mantiene constante.
Entonces, de (2.20) el error PFER es:
E (Vn) =∑m∈H0
E (I (Tn(m) ∈ Cn(m))) ≤∑m∈H0
α = h0α = cMα (2.32)

2.9 Errores de Tipo I en Test Multiples 23
En el caso del error FWER, la relacion con M es exponencial:
Pr(Vn > 0) = 1− Pr(Vn = 0) (2.33)
= 1− Pr
⋂m∈H0
{Tn(m) /∈ Cn(m)}
= 1−
∏m∈H0
Pr (Tn(m) /∈ Cn(m))
< 1− (1− α)h0
= 1− (1− α)cM .
Luego, si fijamos un error de Tipo I, fijo, digamos 0.05, y no tenemos en cuenta M, estamosforzando a que el procedimiento sea extremadamente conservativo cuando M es muy grandecomo sucede en los estudios de experimentos de microarreglos.
En cambio, los errores basados en la proporcion de los falsos positivos son especialmenteatractivos en estas situaciones porque la proporcion permanece estable bajo un incrementode la cantidad de hipotesis M .
Estas consideraciones llevaron a Benjamini y Hochberg en su ya famoso artıculo [BH95] aproponer el error FDR y el procedimiento 2.4 para controlarlo que detallaremos en la seccion2.14
Hay muchas mas clases de errores propuestos pero dimos los ingredientes con los queconstruyen todos ellos. Para mas detalles ver [DSB03], [Sha95] y [HT87].
En la practica, primero se popularizo el uso del error FWER y luego el uso del FDR, quesigue siendo el mas popular. En esta tesis continuamos el trabajo de [GGQY07], que comparaeste error con el error PFER.
2.9.4. Comparacion de errores de Tipo I
Los procedimientos clasicos de test multiples se usaron para controlar el error FWER.Ası se aplico el procedimiento mas antiguo debido a Bonferroni ([Bon36]) presentado en laseccion: 2.13.
Como notamos en 2.31, FDR ≤ FWER. En el caso de la hipotesis nula completa (M =h0), Vn = Rn y por lo tanto FDR = FWER. Tambien podemos relacionar el error PFERcon el FWER, notando que:
VnM
=VnM
I(Vn > 0) ≤ I(Vn > 0) (2.34)
Luego,
PCER = E(VnM
) ≤ E(I(Vn > 0)) = Pr(Vn > 0) = FWER (2.35)
Por otro lado si aplicamos la desigualdad de Markov a la variable nonegativa Vn:
gFWER(k) = Pr(Vn > k) = Pr(Vn ≥ k + 1) ≤ E(Vn)
k + 1=PFER
k + 1(2.36)
En particular, cuando k = 0, obtenemos que FWER ≤ PFER.Luego tenemos que para cualquier procedimiento de test multiples, PCER ≤ FWER ≤
PFER y FDR ≤ FWER. Entonces, los procedimientos disenados para controlar por ejemplo

2.9 Errores de Tipo I en Test Multiples 24
el error FWER son en general mas conservativos que los disenados para controlar el errorPCER o el error FDR. Esto tambien muestra que no debemos usar la misma escala al compararun procedimiento que controla, digamos, el error PFER con un procedimiento que controla elerror FDR. Un procedimiento que controla el error PFER puede ser muy pobre en su controldel error FDR sin dejar de ser util. Dando un ejemplo un poco extremo, si un procedimientopara un nivel α determinado analizando 10000 hipotesis produce 3 falsos positivos y solo 10rechazos, entonces tiene un error aceptable PFER de 3 y un error FDR de 0.3, que es muyalto comparado con los valores usados en la practica de 0.05 y 0.01.
Para cerrar la seccion ilustraremos en detalle estas relaciones en el caso particular de losprocedimientos marginales, extendiendo un poco el ejemplo usado en 2.32.
PCER =E(Vn)
M(2.37)
=1
M
∑m∈H0
E (I (Tn(m) ∈ Cn(m)))
≤ 1
h0
∑m∈H0
E (I (Tn(m) ∈ Cn(m)))
≤ 1
h0
∑m∈H0
Pr (Tn(m) ∈ Cn(m))
≤ maxm∈H0
Pr (Tn(m) ∈ Cn(m))
≤ Pr
⋃m∈H0
{Tn(m) ∈ Cn(m)}
(2.38)
= Pr
⋃m∈H0
{I (Tn(m) ∈ Cn(m)) = 1}
= Pr
⋃m∈H0
{I (Tn(m) ∈ Cn(m)) > 0}
= Pr
∑m∈H0
I (Tn(m) ∈ Cn(m))
> 0
= Pr (Vn > 0)
= FWER
FWER ≤∑m∈H0
Pr (Tn(m) ∈ Cn(m)) por 2,38
=∑m∈H0
E (I (Tn(m) ∈ Cn(m)))
= PFER.
Como se explico en 2.9.3, al depender el error FDR de la distribucion conjunta de Vn y Sn,el error FDR no tiene una expresion simple en este caso.

2.10 P-Valores Ajustados 25
2.10. P-Valores Ajustados
2.10.1. Definicion
La nocion de p-valor se extiende directamente al caso de test multiple como sigue. Sea unprocedimiento de test multiples Rn(α) = R(Tn, Q0, α), con regiones de rechazo Cn(m;α) =C(m;Tn, Q0, α). Entonces, podemos definir un M -vector de p-valores ajustados:
P0n = (P0n(m) : m = 1, . . . ,M) = P (Tn, Q0) = P (R(Tn, Q0, α) : α > 0)
, como
P0n(m) ≡ ınf {α > 0 : Rechazar H0(m) a nivel nominal α del MTP } (2.39)
= ınf {α > 0 : m ∈ Rn(α)}= ınf {α > 0 : Tn(m) ∈ Cn(m;α)} , m = 1, . . . ,M.
Esto es, el p-valor ajustado P0n(m), para la hipotesis nula H0(m), es el menor nivel nominal deerror de Tipo I (por ej., FWER, PCER, FDR) que comete el procedimiento de test multiplesal cual sigue rechazando H0(m), dado Tn. Como sucede en el caso de una sola hipotesis,a menor valor del p-valor ajustado P0n(m), mayor es la evidencia en contra de la hipotesisnula H0(m). Ası, rechazamos H0(m) para p-valores ajustados pequenos P0n(m). Notar queel p-valor no ajustado P0n(m), para la sola hipotesis H0(m) corresponde al caso especialM = 1. Recalcamos que la principal diferencia entre p-valores no ajustados y ajustados esel procedimiento usado para rechazar H0(m), que se manifiesta en las diferentes regiones derechazo C(m;Tn, Q0, α).
Tambien notemos que ambos, P0n(m) y P0n(m) son variables aleatorias, o sea, funcionesde los datos, Xn a traves de los estadısticos Tn. Como es usual, denotaremos las realizacionesde los estadısticos, p-valores no ajustados y p-valores ajustados para la hipotesis nula H0(m)en letras minusculas por tn(m), p0n(m), y p0n(m), respectivamente.
2.10.2. Representacion de un procedimiento de test multiples mediante sus p-valores ajustados
Similar al caso de una sola hipotesis, bajo la condicion de regiones anidadas (2.10) setienen dos representaciones equivalentes de un procedimiento de test multiples: en terminosde regiones de rechazo o en terminos de p-valores ajustados. En detalle, el conjunto de hipotesisnulas rechazadas al nivel nominal α de error de Tipo I multiple es:
Rn(α) = {m : Tn(m) ∈ Cn(m;α)} ={m : P0n(m) ≤ α
}. (2.40)
Para los procedimientos multi-paso seran utiles los p-valores ajustados ordenados conındices On(m). Ası, P0n(On(1)) ≤ · · · ≤ P0n(On(M)). Luego, por el ejemplo, el conjunto dehipotesis rechazadas Rn(α) consiste de los ındices de las Rn(α) = |Rn(α)| hipotesis con losmenores p-valores ajustados. Esto es: Rn(α) = {On(m) : m = 1, . . . , Rn(α)}.
2.10.3. Ventajas de la representacion por p-valores ajustados
La representacion de los resultados de un MTP en terminos de p-valores ajustados ofrecevarias ventajas frente a expresar los resultados en terminos de regiones de rechazo.

2.11 Potencia en Test Multiples 26
Los p-valores se pueden definir para cualquier error de Tipo I multiple. (FWER, PCER,FDR).
Reflejan la fuerza de la evidencia en contra de la hipotesis nula en terminos del error deTipo I multiple.
Son resumenes flexibles de un MTP, en el sentido de que los resultados son provistospara cualquier nivel nominal α de error de Tipo I multiple. Es decir, el nivel α no esnecesario especificarlo de antemano.
Permiten comparar diferentes MTPs que miden el mismo error de Tipo I multiple, dondemenores p-valores ajustados indican un procedimiento menos conservativo.
2.11. Potencia en Test Multiples
Los conceptos de errores de Tipo II o Potencia pueden extenderse del caso de una hipotesisa multiples hipotesis de manera similar a lo hecho con los errores de Tipo I en 2.9. Ası definimospotencia como un parametro ϑn = Θ(FUn,Rn) de la distribucion conjunta FUn,Rn de los falsosnegativos Un = |Rcn ∩ H1| y las hipotesis rechazadas Rn = |Rn|. Recordemos que el numerode los verdaderos positivos Sn y los falsos negativos Un cumplen que Sn + Un = h1 (Cuadro2).
Las medidas mas usuales son:
El error AnyPwr, que se define como la probabilidad de rechazar al menos un verdaderopositivo:
AnyPwr ≡ Pr(Sn ≥ 1) = Pr(Un ≤ h1 − 1) = FUn(h1 − 1). (2.41)
El error AllPwr es la probabilidad de que no haya falsos negativos:
AllPwr ≡ Pr(Sn = h1) = Pr(Un = 0) = FUn(0). (2.42)
El error AvgPwr es la esperanza de la proporcion de hipotesis rechazadas entre lashipotesis nulas falsas:
AvgPwr ≡ 1
h1E[Sn] =
1
h1E[h1 − Un] = 1− 1
h1
∫udFUn(u). (2.43)
El error TDR, “true discovery rate”, es la esperanza de la proporcion de los verdaderospositivos:
TDR ≡ E
[SnRn
]= E
[Rn − VnRn
], (2.44)
con la convencion de que (Rn − Vn)/Rn ≡ 0 if Rn = 0. Podemos escribir el error TDRde varias maneras.
TDR = E
[Rn − VnRn
∣∣∣∣Rn > 0
]Pr(Rn > 0) = Pr(Rn > 0) (1− FDR) .
El error de tipo II TDR es analogo al error FDR de tipo I. Si todas las hipotesis nulasson falsas (h1 = M), entonces el error TDR es igual al error AnyPwr. Para mas detallesver [Sha95]

2.12 Tipos de Procedimientos de Test Multiples 27
2.12. Tipos de Procedimientos de Test Multiples
Podemos clasificar los procedimientos de test multiples segun dos criterios:
Procedimientos Marginales vs. Conjuntos. Los procedimientos de test multiples mar-ginales se basan unicamente en la distribucion marginal de los estadısticos. En cambio,los procedimientos de test multiples conjuntos toman en cuenta hasta cierto punto laestructura de dependencia entre los estadısticos (Tn(m)). Para esto recurren a metodosde remuestreo y requieren propiedades mas fuertes que (2.14) para mantener el controldel Tipo I de errores (ver [DSB03] y [Sha95]). En general son mas potentes que los me-todos marginales y son mucho mas costosos computacionalmente pero hay objecionessobre su efectividad [QKY05].
Procedimientos de Paso Unico y Multi-Paso. En los procedimientos de paso unico(“single-step”) cada hipotesis nula se testea usando una region de rechazo que es in-dependiente de los resultados del testeo de otras hipotesis. Notemos que este es unprocedimiento paralelizable. En cambio en los procedimientos multipaso (“stepwise”) setestea una hipotesis nula en cada paso. Y la region de rechazo depende del resultadode los tests de los pasos anteriores. En general, se consideran las hipotesis ordenadassegun los valores de los estadısticos Tn(m). Los metodos que empiezan rechazando lashipotesis cuyos estadısticos proveen la mayor evidencia contra la hipotesis nula se deno-minan descendentes (“step-down”). Mientras que los metodos que empiezan aceptandolas hipotesis con menor evidencia contra las hipotesis nulas se denominan ascenden-tes (“step-up”). A continuacion detallaremos esta clase de procedimientos, ya que esnecesario para comprender el procedimiento de Benjamini-Hochberg 2.4.
2.12.1. Procedimientos marginales descendentes y ascendentes
Los dos procedimientos siguientes muestran los pasos generales de los procedimientosdescendentes y ascendentes en terminos de secuencias crecientes de valores crıticos de p-valoresno ajustados.
Sean P0n(m) los p-valores no ajustados para las hipotesis nulas H0(m), m = 1, . . . ,M ,donde P0n(m) se calculan segun (2.12), bajo la distribucion nula Q0 del estadıstico Tn. SeanOn(m) los ındices para los p-valores no ajustados ordenados. Luego, P ◦0n(m) ≡ P0n(On(m)) yP0n(On(1)) ≤ · · · ≤ P0n(On(M)). Nos referiremos con la m-esima hipotesis nula mas signi-ficante a H0(On(m)) donde m es el rango (numero de orden) del menor p-valor no ajustadoP ◦0n(m).
Procedimiento 2.1 (Procedimiento generico marginal descendente). Dados los va-lores crıticos de los p-valores no ajustados a1(α) ≤ · · · ≤ aM (α), un procedimiento de testmultiples marginal descendente puede ser expresado como:
Rn(α) ≡ {On(m) : P0n(On(h)) ≤ ah(α), ∀ h ≤ m} . (2.45)
El numero de hipotesis nulas rechazadas Rn(α) esta dado por
Rn(α) ≡{M, Si P0n(On(m)) ≤ am(α) ∀ mmın {m : P0n(On(m)) > am(α)} − 1, Si no
. (2.46)

2.12 Tipos de Procedimientos de Test Multiples 28
Los p-valores ajustados correspondientes estan dados por:
P0n(On(m)) = maxh=1,...,m
{mın
{a−1h (P0n(On(h))), 1
}}, m = 1, . . . ,M, (2.47)
donde a−1m son las inversas de las funciones que asignan los valores crıticos am : α→ am(α),que asumimos por simplicidad continuas y estrictamente crecientes en α.
Procedimiento 2.2 (Procedimiento generico marginal ascendente). Dados los valorescrıticos de los p-valores no ajustados a1(α) ≤ · · · ≤ aM (α), un procedimiento de test multiplesmarginal ascendente puede ser expresado como:
Rn(α) ≡ {On(m) : ∃ h ≥ m tal que P0n(On(h)) ≤ ah(α)} . (2.48)
El numero de hipotesis nulas rechazadas Rn(α) esta dado por
Rn(α) ≡{
0, Si P0n(On(m)) > am(α) ∀ mmax {m : P0n(On(m)) ≤ am(α)} , Si no
. (2.49)
Los p-valores ajustados correspondientes estan dados por:
P0n(On(m)) = mınh=m,...,M
{mın
{a−1h (P0n(On(h))), 1
}}, m = 1, . . . ,M, (2.50)
donde a−1m son las inversas de las funciones que asignan los valores crıticos am : α→ am(α),que asumimos por simplicidad continuas y estrictamente crecientes en α.
Para derivar los p-valores ajustados usamos la definicion general dada en (2.39). En detalle,para el caso del procedimiento descendente 2.1,
P0n(On(m)) = ınf {α > 0 : On(m) ∈ Rn(α)}= ınf {α > 0 : Rn(α) ≥ m}= ınf {α > 0 : mın {h : P0n(On(h)) > ah(α)} > m}= ınf
{α > 0 : mın
{h : a−1h (P0n(On(h))) > α
}> m
}= ınf
{α > 0 : max
h=1,...,ma−1h (P0n(On(h))) ≤ α
}= max
h=1,...,m
{mın
{a−1h (P0n(On(h))), 1
}}.
Asimismo para el procedimiento ascendente 2.2,
P0n(On(m)) = ınf {α > 0 : On(m) ∈ Rn(α)}= ınf {α > 0 : Rn(α) ≥ m}= ınf {α > 0 : max {h : P0n(On(h)) ≤ ah(α)} ≥ m}= ınf
{α > 0 : max
{h : a−1h (P0n(On(h))) ≤ α
}≥ m
}= ınf
{α > 0 : mın
h=m,...,Ma−1h (P0n(On(h))) ≤ α
}= mın
h=m,...,M
{mın
{a−1h (P0n(On(h))), 1
}}.

2.12 Tipos de Procedimientos de Test Multiples 29
Notar que tomar el maximo de las cantidades mın{a−1h (P0n(On(h))), 1
}sobre los subconjun-
tos {1, . . . ,m} en (2.47) garantiza la propiedad descendente y la monotonıa de los p-valoresajustados. Luego, uno solo puede rechazar una hipotesis nula en particular solo cuando todasla hipotesis nulas con menores p-valores no ajustados han sido ya rechazadas y P0n(On(1)) ≤· · · ≤ P0n(On(M)). Asimismo, tomar mınimo de las cantidades mın
{a−1h (P0n(On(h))), 1
}so-
bre los subconjuntos {m, . . . ,M} en (2.50) garantiza la propiedad ascendente y la monotonıade los p-valores ajustados. Luego, apenas uno rechaza una hipotesis nula, todas las hipotesiscon menores p-valores no ajustados son rechazadas y P0n(On(1)) ≤ · · · ≤ P0n(On(M)).
Finalmente, notemos que podemos usar los mismos valores crıticos para los p-valores noajustados si probamos el control del error de Tipo I en los dos casos, siendo el caso ascendenteel mas difıcil. La mayor diferencia aquı serıa el orden en que las hipotesis nulas se testean:
Descendente: Desde la hipotesis mas significante a la menos significante
Ascendente: Desde la hipotesis menos significante a la mas significante
Esta distincion se ve reflejada en tomar maximo o mınimo en las ecuaciones (2.47) y (2.50),respectivamente.
2.12.2. Comparacion de los p-valores ajustados ascendentes y descendentes
Si elegimos los mismos valores crıticos de p-valores no ajustados para un procedimientoascendente y otro descendente, podemos escribir los p-valores ajustados como
P ↓0n(On(m)) = maxh=1,...,m
P−0n(On(h)) [descendente] (2.51)
P ↑0n(On(m)) = mınh=m,...,M
P−0n(On(h)) [ascendente],
para convenientemente definidos p-valores “pre-ajustados” P−0n(On(m)).Se sigue inmediatamente de la ecuacion (2.51) que para cada m = 1, . . . ,M ,
P ↓0n(On(m)) = maxh=1,...,m
P−0n(On(h)) (2.52)
≥ P−0n(On(m))
≥ mınh=m,...,M
P−0n(On(h)) = P ↑0n(On(m)).
Luego, un procedimiento ascendente rechaza tantas o mas hipotesis nulas que su contrapartedescendente basada en los mismos valores crıticos.
R↓n(α) ={On(m) : P ↓0n(On(m)) ≤ α
}⊆{On(m) : P ↑0n(On(m)) ≤ α
}= R↑n(α).
Podemos interpretar este resultado notando que al empezar por la hipotesis nula menos signi-ficativa y rechazar las restantes hipotesis mas significativas apenas uno rechaza una hipotesisnula, un procedimiento ascendente otorga a cada hipotesis nula varias oportunidades de re-chazo.
En esta tesis compararemos el procedimiento de Bonferroni que es marginal de paso unicocon el de Benjamini-Hochberg que es marginal ascendente.

2.13 Procedimiento de Bonferroni 30
2.13. Procedimiento de Bonferroni
2.13.1. Definicion
Procedimiento 2.3 (Procedimiento de Bonferroni, [Bon36]). Para controlar el error PFERa nivel α, el procedimiento marginal de paso unico de Bonferroni, [Bon36] rechaza cualquierhipotesis nula H0(m) con p-valor no ajustado P0n(m) menor o igual a (el valor crıtico comunde) am(α) ≡ α/M . Entonces, el conjunto de hipotesis nulas rechazadas es
Rn(α) ≡{m : P0n(m) ≤ 1
Mα
}. (2.53)
y los correspondientes p-valores ajustados estan dados entonces por
P0n(m) = MP0n(m), m = 1, . . . ,M. (2.54)
Los p-valores ajustados se derivan directamente de la definicion general dada en la ecuacion(2.39):
P0n(m) = ınf {α > 0 : m ∈ Rn(α)}
= ınf
{α > 0 : P0n(m) ≤ 1
Mα
}= MP0n(m).
Proposicion 2.3. Control de error PFER por el procedimiento de Bonferroni 2.3 Conside-remos un M -vector de estadısticos Tn = (Tn(m) : m = 1, . . . ,M) y el M -vector asociado dep-valores no ajustados P0n = (P0n(m) : m = 1, . . . ,M), definido segun la ecuacion (2.12) ydependiente de la distribucion nula de Tn, Q0. Entonces, el procedimiento de Bonferroni 2.3controla el error PCER, bajo cualquier distribucion verdadera del estadıstico Tn, Qn. Esto es,
E(Vn) ≤ α
Demostracion de Proposicion 2.3. Notemos que de acuerdo a 2.7.1
P0n(m) ≤ 1
Mα⇐⇒ Tn(m) ∈ Cn(m;
1
Mα)
Y por la definicion de Vn (2.20),
Vn =∑m∈H0
I
(Tn(m) ∈ Cn(m;
1
Mα)
)=∑m∈H0
I
(P0n(m) ≤ 1
Mα
)(2.55)
Luego,
E(Vn) =∑m∈H0
PrQn,m
(P0n(m) ≤ 1
Mα
)y aplicando la Proposicion 2.2, en particular la ecuacion (2.18) se llega a que:
E(Vn) ≤∑m∈H0
1
Mα =
h0M
α ≤ α

2.14 Procedimiento de Benjamini-Hochberg 31
Notar que cuanto menor a 1 sea la proporcion de hipotesis nulas verdaderas, h0/M , masconservativo sera el procedimiento.
Como se explico al principio de la seccion el primer error de Tipo I multiple usado fue elerror FWER. Y se comparaban los MTPs propuestos con el metodo de Bonferroni perdiendode vista que este metodo controla el error FWER a traves del control del error PFER. Comoresultado los procedimientos propuestos competıan con facilidad contra este.
2.14. Procedimiento de Benjamini-Hochberg
Benjamini y Hochberg en [BH95] presentaron el procedimiento 2.4 para controlar el errorFDR en el caso de estadısticos independientes.
En [BY01] probaron que el procedimiento 2.4 controla el error para el caso mas generalde estadısticos con estructura de dependencia de regresion positiva. Tambien en este artıculointrodujeron una variante a su procedimiento que controla el error FDR para distribucionesarbitrarias, con el costo de ser mas conservativo. En la practica se usa la version originalaun a riesgo de que puede ser anti-conservativa si no se cumple las condiciones como la dedependencia de regresion positiva.
Otros autores desarrollaron modelos Bayesianos empıricos para controlar el error FDR([EST01], [ETST01], [ST03]). La mayorıa de estas propuestas asumen que los estadısticosson independientes e identicamente distribuidos segun un modelo no parametrico mixto. En[QKY05] se elabora una crıtica de este enfoque senalando la debilidad de presuponer inde-pendencia o dependencia lo suficientemente debil.
2.15. Procedimiento de Benjamini-Hochberg
Como adelantamos en la seccion 2.9.3, [BH95] propusieron el control del error FDR, “falsediscovery rate”, o sea, la esperanza de la proporcion entre los falsos positivos y las hipotesisrechazadas. Ver ecuacion (2.30) y subsiguientes. El procedimiento se basa en la siguienteDesigualdad de Simes.
2.15.1. Desigualdad de Simes
Los procedimientos ascendentes como el de Benjamini-Hochberg tıpicamente se basan enla desigualdad de Simes [Sim86] para probar el control de errores de Tipo I.
Proposicion 2.4 (Desigualdad de Simes. [Sim86]). Consideremos un M -vector aleatorioZ = (Z(m) : m = 1, . . . ,M) con distribucion conjunta Q0, p-valores no ajustados P0 =(P0(m) : m = 1, . . . ,M), definidos de acuerdo a la ecuacion (2.12), basados en Q0 y finalmentelos p-valores no ajustados ordenados P ◦0 (m) tales que P ◦0 (1) ≤ · · · ≤ P ◦0 (M). Entonces, laDesigualdad de Simes afirma que:
PrQ0
(M⋂m=1
{P ◦0 (m) >
m
Mα})≥ 1− α (2.56)
o, de forma equivalente,
PrQ0
(M⋃m=1
{P ◦0 (m) ≤ m
Mα})≤ α.

2.16 Aplicacion al analisis de los Microarreglos 32
Simes [Sim86] probo esta desigualdad para estadısticos independientes con igualdad parael caso continuo, pero a traves de estudios de simulacion en el mismo artıculo sugirio quela desigualdad es conservativa para una variedad de distribuciones Gaussianas y Gammas.Luego, [SC97] demostro que la desigualdad se cumple para estadısticos con distribucionesmultivariadas con dependencia positiva intercambiable, incluyendo distribuciones multivaria-das Gaussianas equi-correlacionadas y ciertas distribuciones multivariadas F , t y Gamma quecomprenden a las estudiadas en las simulaciones de Simes.
Una de las primeros procedimientos que usaron la desigualdad de Simes fue el procedi-miento marginal ascendente de Hochberg para control del error FWER, [Hoc88].
Luego, [BH95] usan esta desigualdad para probar que el procedimiento 2.4 controla el errorFDR para estadısticos independientes. Luego, el siguiente artıculo [BY01] prueba el controlpara estadısticos con dependencia de regresion positiva
2.15.2. Definicion
Procedimiento 2.4 (Procedimiento ascendente de [BH95] para control del errorFDR). Para controlar el error FDR a nivel α, los valores crıticos de los p-valores no ajustadospara el procedimiento ascendente [BH95] son los siguientes:
am(α) ≡ m
Mα, m = 1, . . . ,M, (2.57)
Y el conjunto de hipotesis nulas rechazadas es
Rn(α) ≡{On(m) : ∃ h ≥ m tal que P0n(On(h)) ≤ h
Mα
}. (2.58)
Luego, los correspondientes p-valores ajustados estan dados por
P0n(On(m)) = mınh=m,...,M
{mın
{M
hP0n(On(h)), 1
}}, m = 1, . . . ,M. (2.59)
Los p-valores ajustados pueden derivarse directamente de la ecuacion (2.50) derivada delProcedimiento generico ascendente 2.2.
Notar que el Procedimiento 2.4 puede ser conservativo, incluso en el caso de estadısticosindependientes, ya que [BH95] probaron que se satisface E[Vn/Rn] ≤ h0α/M , y como en elcaso del Procedimiento de Bonferroni, esta cota es mas conservativa cuanto mas pequeno que1 sea h0/M . Ver [BH95] y [BY01] para las pruebas detalladas.
2.16. Aplicacion al analisis de los Microarreglos
Como se anticipo en la seccion 1.1, en los ultimos anos una cantidad de biotecnologıas no-vedosas han permitido a los investigadores examinar simultaneamente niveles de expresion degran parte del genoma. Por ejemplo, los experimentos en microarreglos proveen un alto volu-men de experimentos simultaneos para medir en diferentes muestras de celulas la abundanciade acidos deoxiribonucleicos (ADN) y acidos ribonucleicos (ARN).
La tecnologıa de microarreglos esta siendo aplicada de manera creciente en la investigacionmedica y biologica, por ejemplo para la clasificacion de tumores o para la respuesta genomicadel huesped ante infecciones bacterianas (ref. [Spe03] y [BAD+02]).

2.16 Aplicacion al analisis de los Microarreglos 33
Una pregunta tıpica en el analisis de microarreglos es la identificacion de genes diferen-cialmente expresados, es decir, genes cuyos niveles de expresion estan asociados a covariablesclınicas o biologicas. Esta pregunta puede ser reformulada como un problema de test de mul-tiples hipotesis: Testear simultaneamente para cada gen la hipotesis nula de no asociacionentre los niveles de expresion y las covariables.
Entonces, para identificar los genes diferencialmente expresados entre dos grupos identifi-cados por digamos la covariable Z, (por ejemplo indica con 0 al grupo de celulas sanas y con 1al otro grupo de celulas con determinada condicion biologica), debemos testear para cada unode los M experimentos cuales tienen una diferencia significativa en los niveles de expresionmedios.
En el modelo mas simple de los experimentos, que es el llamado aditivo homocedasticoy que es el considerado por [GGQY07] podemos suponer que la expresion tiene distribucionlog-normal con varianza constante σ2 para todo m. Bajo la hipotesis nula de no diferencia,la diferencia entre las log-expresiones tendra distribucion normal con media cero y varianzaσ2. La practica usual para testear este tipo de hipotesis es considerar la diferencia entre lasmedias de las log-expresiones.
Entonces el parametro de interes para el experimento m, (o sea, la m-esima hipotesis) esla diferencia entre los niveles de expresion medios ψ(m) entre los dos grupos:
ψ(m) ≡ E[X(m)|Z = 1]− E[X(m)|Z = 0], m = 1, . . . ,M. (2.60)
Podemos considerar test bilaterales para cada experimento m de la hipotesis nula H0(m) =I (ψ(m) = 0) de no diferencias entre las medias de los niveles de expresion vs. la hipotesisalternativa H1(m) = I (ψ(m) 6= 0) de diferentes medias de niveles de expresion.
Al asumir la misma varianza y la distribucion normal de las log-expresiones el test maspotente sera el clasico test t a dos muestras:
Tn(m) ≡ ψn(m)− ψ0(m)
σψn(m)=
(X1(m)− X0(m)
)− 0√(
1n0(m) + 1
n1(m)
)S2p(m)
(2.61)
Notemos que ψ0(m) = 0 y que las cantidades de cada grupo son:
nk(m) ≡n∑i=1
I (Zi = k) ∀k ∈ {0, 1}
Por supuesto, n(m) = n0(m) + n1(m). Las medias muestrales de cada grupo estan definidaspor:
Xk(m) ≡ 1
nk(m)
n∑i=1
I (Zi = k)Xi(m) ∀k ∈ {0, 1}
y por ultimo S2p(m) es la estimacion de la varianza comun definida por:
(n0(m) + n1(m)− 2)S2p(m) = (n0(m)− 1)S0(m)2 + (n1(m)− 1)S1(m)2
=
n∑i=1
I (Zi = 0)(Xi(m)− X0(m)
)2+
n∑i=1
I (Zi = 1)(Xi(m)− X1(m)
)2

2.16 Aplicacion al analisis de los Microarreglos 34
En los cursos de Estadıstica se prueba que S2p(m)/σ2(m) ∼ χ2
n0(m)+n1(m)−2/(n0(m)+n1(m)−2), que S2
p(m) es independiente de X0(m) y de X1(m) y que en consecuencia:
Tn(m) ∼ tn0(m)+n1(m)−2
ψ(m)√( 1n0(m) + 1
n1(m))σ2
Bajo la hipotesis nula, ψ(m) = 0, por ende Tn(m) ∼ tn0(m)+n1(m)−2 ≡ tn(m)−2. Luego, elp-valor no ajustado de la m-esima hipotesis se calcula como:
P0n(m) = 2Q0,m(|Tn(m)|) = 2 Pr(tn(m)−2 > |Tn(m)|
)(2.62)
Entonces los estimadores de las diferencias entre las log expresiones, ψ(m), son simple-mente las correspondientes diferencias entre las medias muestrales de cada grupo:
ψn(m) ≡ X1(m)− X0(m)
Y la hipotesis nula H0(m) es rechazada, es decir, el gen m es declarado con expresion di-ferencial para valores absolutos significativos del estadıstico Tn(m) o cuando su p-valor noajustado es significativamente bajo.
Por ultimo en el ambito del analisis de los microarreglos se usa una terminologıa propiaque conviene adoptar. Un gen declarado diferencialmente expresado (abr. d.e.) se lo llamadescubrimiento. Luego rechazo, descubrimiento y positivo son sinonimos en este contexto. Asıse denominan falsos descubrimientos a los falsos positivos Vn y verdaderos descubrimientos alos verdaderos positivos Sn (ref. Cuadro 2).

3. Algoritmo GGQY 35
3. Algoritmo GGQY
3.1. Introduccion
Los autores de [GGQY07] bien senalan que gran parte de la investigacion en test mul-tiples se oriento a la busqueda de alternativas al error FWER debido a que en el contextodel analisis de microarreglos, el numero de hipotesis, M , es del orden de 104 y como se no-to en 2.9.3, la relacion con M es exponencial y esto fuerza a que el control del error porcualquier procedimiento, incluyendo el procedimiento de Bonferroni 2.3 sea extremadamenteconservativo.
Dentro de las alternativas propuestas, casi todas ellas explicadas en la seccion 2.9, el errorFDR se torno el mas popular y el procedimiento de Benjamini-Hochberg 2.4 con sus varianteslos mas usados. Los autores de [GGQY07] comentan que es difıcil hallar algun estudio demicroarreglos que no afirme que el procedimiento de Bonferroni para control del error FWERes extremadamente conservativo por lo que se favorece el control del error FDR. Argumentanque esto se debe a que el conservatismo se debe al control de error FWER a traves del controlde error PFER (ref. 2.9.4), que es el verdadero error controlado por el procedimiento deBonferroni y no debido al procedimiento en sı.
Para probar esta afirmacion realizan un estudio de simulacion que compara los dos proce-dimientos ecualizando los errores (debido a que son muy distintos ya en su rangos mismos: elerror FDR es un valor entre 0 y 1 mientras que el error PFER es un numero entre 0 y h0 ≤M)y estudiando la variabilidad (estabilidad) de los resultados asociados a los dos procedimientos.
Afirman que se trata del primer estudio de estabilidad de los resultados de los dos pro-cedimientos y concluyen que la ventaja del procedimiento de Benjamini-Hochberg no es taly proponen que los usuarios decidan que metodo usar en base al error que se desea contro-lar. Por ejemplo, si el investigador establece que tolera hasta 2 falsos positivos en promedio,entonces puede usar el procedimiento de Bonferroni con error nominal 2. En cambio, si elinvestigador no quiere que en promedio la proporcion de falsos positivos entre las hipotesisrechazadas exceda el 10 %, entonces puede usar el procedimiento de Benjamini-Hochberg conerror nominal de 0.1.
Por ultimo, los autores concluyen la “restauracion” del procedimiento de Bonferroni no-tando:
La generalidad del procedimiento de Bonferroni que es valido para cualquier estructu-ra de dependencia, a diferencia del de Benjamini-Hochberg que es valido bajo ciertascondiciones (ref. 2.15.1)
La simplicidad del procedimiento de Bonferroni que permite que su computo sea casitrivial una vez computados los p-valores no ajustados.
La mayor estabilidad en el numero total de rechazos. Una propiedad que es importanteen la presencia de estadısticos fuertemente correlacionados [QKY05].
Si bien los resultados son prometedores nos ha llamado la atencion algunas deficienciasdel estudio. Mientras los mismos autores observan que en la practica las correlaciones de apares observadas son altas variando entre 0.84 y 0.97 (ref: [KY07]), la simulacion se realizacon el supuesto de una correlacion de a pares de 0.4.
No menos importante, la comparacion usa un estudio anterior de los mismos autores (ref:[QKY05]), donde se estudian datos reales y publicos de pacientes pediatricos con leucemia

3.2 Simulacion de P-Valores no ajustados 36
(mas especıficamente con ALL, “acute lymphoblastic leukemia”). Los datos estan disponiblesen http://www.stjuderesearch.org y representan las expresiones de 12558 genes de 335pacientes. Para la simulacion en cambio se simulan 1255 genes cuando ser realista en lacantidad de hipotesis M es importante.
Examinando el algoritmo usado en las simulaciones que llamaremos de aquı en adelanteGGQY de acuerdo a las iniciales de los autores notamos algunas deficiencias en tiempo yespacio, lo que nos dio la oportunidad de mejorarlo significativamente. Gracias a ello pudimosreplicar el estudio en condiciones mas realistas aumentando la correlacion a 0.9 y la cantidadde genes simulados a 12558.
A continuacion vamos a presentar el algoritmo GGQY y senalaremos los lugares donde sehicieron las mejoras. Empezamos por la materia prima comun a los dos MTPs: los p-valoresno ajustados.
3.2. Simulacion de P-Valores no ajustados
Como se explica en [QKY05] el estudio asume el modelo aditivo homocedastico donde laslog-expresiones tienen distribucion normal y la varianza σ2 se asumira constante 1. Asumentambien un modelo simplificado de estructura de dependencia, el de correlacion intercam-biable. Para construirlo primero consideremos una matriz V de M filas x n columnas dondecada celda sigue una distribucion normal estandar y es independiente de las demas. Esto es,V = (vij , i = 1, . . . ,M, j = 1, . . . n), con vij ∼ N (0, 1).
Vamos a suponer como en 2.16 una covariable Z que indique dos grupos de microarreglos,de acuerdo a si tienen una determinada condicion biologica. Como el estadıstico Tn(m) definidoen (2.61) no varıa de acuerdo al orden de los microarreglos, vamos a suponer simplementeque las primeras n1 columnas corresponden a los microarreglos sin determinada condicionbiologica a analizar, tambien llamado“normal” o“wild”mientras que las segundas n2 columnascorresponderan al segundo grupo de microarreglos con la condicion biologica, tambien llamado“target”.
Naturalmente la cantidad de genes diferencialmente expresados sera h1. En el codigousaremos la constante mas nemotecnica kDE. Nuevamente dado que el orden no importapodemos suponer que los primeros h1 genes son los d.e.. Luego, siguiendo a [GGQY07] paramodelar este conjunto de genes d.e. sumamos 1 (que serıa la media de las log-expresiones delos genes d.e. en el caso de independencia) a las celdas de las primeras h1 filas y las columnasdel grupo “target”. En sımbolos:
xij = vij + I (1 ≤ i ≤ h1, n1 < j ≤ n)) ∀i = 1 . . .M, j = 1 . . . n
Para simular la fuerte correlacion entre genes observada en la practica primero generamosun vector aleatorio de dimension n, A = (aj), j = 1 . . . n , donde cada componente sigue unadistribucion normal estandar independiente de las otras componentes y de las variables xij .Luego se construye la matriz Y = (yij), i = 1 . . .M, j = 1 . . . n, definiendo para ρ ∈ [0, 1):
yij =√ρ aj +
√1− ρ xij (3.1)
Las yij ası definidas tienen medias:
E(yij) =
{0 i = h1 + 1 . . .M ∨ j = 1 . . . n1,√
1− ρ i = 1 . . . h1 ∧ j = n1 + 1 . . . n(3.2)

3.2 Simulacion de P-Valores no ajustados 37
En otras palabras, las medias de las log-expresiones de los genes d.e es√
1− ρ. Notemosque cuando las correlaciones tienden a uno, este numero tiende a cero y en consecuencia elproblema de distinguir los genes d.e se hace mas difıcil.
Con respecto a las covarianzas:
Cov(yij , yhk) = pCov(aj , ak) + (1− p) Cov(xij , xhk)
= p I(j = k) + (1− p) I(j = k) I(i = h)
=
1 i = h ∧ j = k
p i 6= h ∧ j = k
0 i 6= h ∨ j 6= k
(3.3)
Ası Var(yij) = 1 y por ende las correlaciones son:
Cor(yij , yhk) = Cov(yij , yhk)
=
1 i = h ∧ j = k
p i 6= h ∧ j = k
0 i 6= h ∨ j 6= k
(3.4)
En palabras, las log-expresiones de dos microarreglos diferentes no estan correlacionadas perodentro de un mismo microarreglo las log-expresiones correspondientes a dos genes diferentestienen correlacion ρ.
Finalmente los p-valores se calculan para cada fila m, aplicando las formulas dadas en(2.61) y en (2.62).
3.2.1. Implementacion en R
Vamos a mostrar como implementamos en R el experimento simulado, o sea la generacionde los M p-valores. Tambien mostraremos como optimizamos el test t frente a la versionestandar de R y frente a la version estandar de la librerıa multtest.
En [GGQY07] se eligieron los siguientes valores para el experimento simulado M = 1255,h1 = 125, n = 86 y n1 = n2 = 43. Por eso definimos las siguientes constantes que siguiendoel “Google’s R Style Guide” llevan de prefijo la letra “k”:
> # Constants
> kGenes <- 1255
> kDE <- 125
> kDEl <- 1:kDE
> kSamp <- 86
> # True null hypothesis. "Normal or Wild type"
> kSampW <- 1:43
> # False null hypothesis. "Target type"
> kSampT <- 44:86
> kDist <- "Normal"
> kMeanDE <- 1
La generacion de variables aleatorias depende de la generacion de numeros (pseudo-) alea-torios que estan determinados por una semilla inicial. Al fijarla en el valor 101 garantizamos

3.2 Simulacion de P-Valores no ajustados 38
la reproducibilidad de los resultados. En nuestro estudio usamos las semillas iniciales 1, 2,101 y 102. Las otras dos constantes se explicaran luego.
> kSeed <- 101
> kGridSize <- 2500
> kRep <- 250
La siguiente funcion crea un objeto que encapsula las constantes definidas.
> base.par <- function() list(Genes=kGenes, DE=kDE, DEl=kDEl,
Samp=kSamp, SampW=kSampW, SampT=kSampT,
Dist=kDist, MeanDE=kMeanDE,
Seed=kSeed, GridSize=kGridSize, Rep=kRep)
Tanto el ρ de la formula (3.1) como el MTP seran variables. La variable mtp.id se usarapara identificar el procedimiento de Bonferroni 2.3 con el valor “BF” y el procedimiento deBenjamini-Hochberg 2.4 con el valor “BH”
> # Variables
> rho <- 0
> mtp.id <- "BF"
> # rhof(0.9) -> ".9"
> rhof <- function(rho) substring(format(rho, nsmall=1),2)
Para generar variables normales estandar independientes usamos la funcion rnorm() comoes estandar en R . Como se explico en la seccion 1.4 es mas eficiente generar la muestra totalde variables en una sola invocacion si no hay restricciones de espacio. La matriz Y ocupa1255 × 86 × 8 bytes ≈ 843Kb por lo que no hay restricciones de espacio y usamos plenaventaja del manejo eficiente de vectores de R . Para calcular el ultimo paso (3.2) primeroconstruimos una version vectorizada del test t que se revelo mas eficiente que la versionestandar de R y que la version implementada en la librerıa multtest (ver Apendice, seccion:B.1).
3.2.2. Version vectorizada del test t
Una de las fortalezas del lenguaje R es su manejo de vectores. Lo comprobamos al construirla siguiente version vectorizada del test t.
> t.test.v <- function(X,cols1,cols2) {
n1 <- length(cols1)
n2 <- length(cols2)
X1 <- X[,cols1]
X2 <- X[,cols2]
X1.m <- rowMeans(X1)
X2.m <- rowMeans(X2)
S2p <- (rowSums((X1-X1.m)*(X1-X1.m))
+ rowSums((X2-X2.m)*(X2-X2.m)))/(n1+n2-2)
t <- (X1.m-X2.m)/sqrt(S2p*(1/n1+1/n2))
res <- 2*pt(abs(t), df=n1+n2-2, lower.tail=F)

3.3 Ajuste de P-Valores 39
}
> rawPValues <- function(rho=rho, par=base.par()) {
P <- vector(length=par$Genes)
A <- rnorm(par$Samp)
X <- rnorm(par$Genes*par$Samp)
dim(X) <- c(par$Genes,par$Samp)
X[par$DEl, par$SampT] <- X[par$DEl, par$SampT] +
par$MeanDE
Y <- sqrt(rho)*rep(1,par$Genes)%*%t(A) +
sqrt(1-rho)*X
P <- t.test.v(Y,par$SampW,par$SampT)
P
}
3.3. Ajuste de P-Valores
Una vez obtenidos los p-valores no ajustados los ajustamos segun los procedimientos deBonferroni 2.3 o Benjamini-Hochberg 2.4. El primero es muy facil de implementar, bastamultiplicar por M los p-valores no ajustados para ajustarlos. El procedimiento de Benjamini-Hochberg esta implementado en la funcion mt.rawp2adjp de la librerıa multtest. Como sehizo en la seccion anterior construimos una version propia competitiva (ver Apendice seccionB.3). Como veremos mas adelante la mejora obtenida tendra un impacto significativo en laoptimizacion lograda.
El vector praw implementa los p-valores no ajustados ordenados, P ◦0n = (P ◦0n(m),m =1, . . . ,M), donde P ◦0n(m) ≡ P0n(On(m)). El vector pbf implementa los p-valores ajustadospor Bonferroni y el vector pbh implementa el ajuste por Benjamini-Hochberg de la ecuacion(2.59). El vector de identifica cuales son los genes d.e..
> adjustedPValues <- function(rp, par=base.par()) {
de <- vector(mode="logical", length=par$Genes)
pbf <- pbh <- vector(mode="numeric", length=par$Genes)
o <- order(rp)
de <- (o %in% par$DEl)
praw <- rp[o]
pbf <- praw*par$Genes
pbh <- rev(cummin(rev(pmin(praw*(par$Genes/(1:par$Genes))
,1))))
ap <- list(de=de, pbf=pbf, pbh=pbh)
}
3.4. Ecualizacion de errores
El objetivo principal de [GGQY07] es comparar los procedimientos de Bonferroni 2.3 y deBenjamini-Hochberg 2.4. Como vamos a comparar estos procedimientos si controlan erroresdiferentes? El de Bonferroni controla el error multiple de Tipo I PFER mientras que el deBenjamini-Hochberg controla el error multiple de Tipo I FDR (bajo ciertas hipotesis).

3.4 Ecualizacion de errores 40
Ante todo podemos observar que dado un nivel nominal α de error de Tipo I PFER, porejemplo 2, podemos realizar una simulacion del experimento, ejecutar el procedimiento deBonferroni y observar Vn pero tambien podemos medir la proporcion Vn/Rn o cualquier otravariable que nos interese. Por supuesto, el procedimiento nos garantiza que el Vn observadosera en promedio menor a α, esto es que el error PFER sera menor que α pero, que podemosafirmar en cuanto al error FDR?
En realidad si sabemos de la ecuacion (2.59) que los p-valores ordenados y ajustados porBenjamini-Hochberg estan en orden creciente de acuerdo a los p-valores ordenados no ajusta-dos. Pero estos ultimos son iguales a los p-valores ordenados y ajustados por Bonferroni salvouna constante multiplicativa. De aquı vemos que la relacion entre ambos p-valores ordenadosy ajustados es monotona creciente.
Recordando la definicion de los p-valores ajustados (ref: (2.39)) y que asumimos la hipotesisde regiones anidadas (ref: 2.7) podemos ver que la relacion entre los p-valores ajustados y loserrores correspondientes tambien es monotona creciente. Esto es, a menor p-valor ajustado deBonferroni menor sera el error PFER. Lo mismo vale para el p-valor ajustado de Benjamini-Hochberg y el error FDR. Entonces, de acuerdo a la relacion entre los p-valores ajustadostambien vale que a menor p-valor ajustado de Bonferroni menor sera el error FDR.
En la practica no conocemos la relacion exacta entre el p-valor ajustado y el error de TipoI, esto es entre el valor nominal del error y el valor actual del error ya que no conocemosel valor exacto del error de Tipo I. En cambio en la simulacion planteada por [GGQY07]podemos medir con precision el numero de falsos positivos Vn y rechazos o descubrimientosRn para un experimento de n observaciones ya que conocemos de antemano cuales son losgenes d.e y cuales no. Replicando este experimento un numero grande de veces podemos esti-mar la distribucion conjunta F (Vn, Rn) o cualquier parametro de esta, simplemente tomandopromedios del parametro sobre todas las replicas. En particular podemos estimar los erroresFDR, PFER y tambien seran de interes medidas de potencia como el AvgPwr, es decir lamedia de los verdaderos descubrimientos, ası como su mediana y desvıo. Recordando la doblerepresentacion dada en (2.40) podemos relacionar los niveles nominales de error multiple deTipo I con las estimaciones de los valores actuales de los errores PFER y FDR, ası como conlas caracterısticas de la potencia.
En la figura 1 mostramos un ejemplo de este procedimiento. Simulando con los mismosparametros que en [GGQY07], o sea, con M = 1255, n = 86, K = 500 y ρ = 0,4 obtuvimoslas funciones que relacionan los p-valores ajustados con las estimaciones de FDR y AvgPWR.Mas adelante veremos los detalles del algoritmo. Se puede reproducir en base al codigo deB.6.
Vemos claramente que no podemos comparar directamente las curvas de FDR o AvgPWR.En este contexto, los autores de [GGQY07] se plantearon ecualizar los errores para poder asıinvestigar las caracterısticas de potencia en pie de igualdad. Vamos a introducir la abreviaturausada en [GGQY07] para los niveles nominales de los errores de Tipo I. Con β designaremosal nivel nominal de error FDR y con γ designaremos al nivel nominal de error PFER. Lasestimaciones del verdadero error FDR y del verdadero error PFER, la notaremos como en elartıculo con FDR y PFER respectivamente.
Consideremos las curvas de la figura 1. Esto es consideremos que hemos hallado funciones
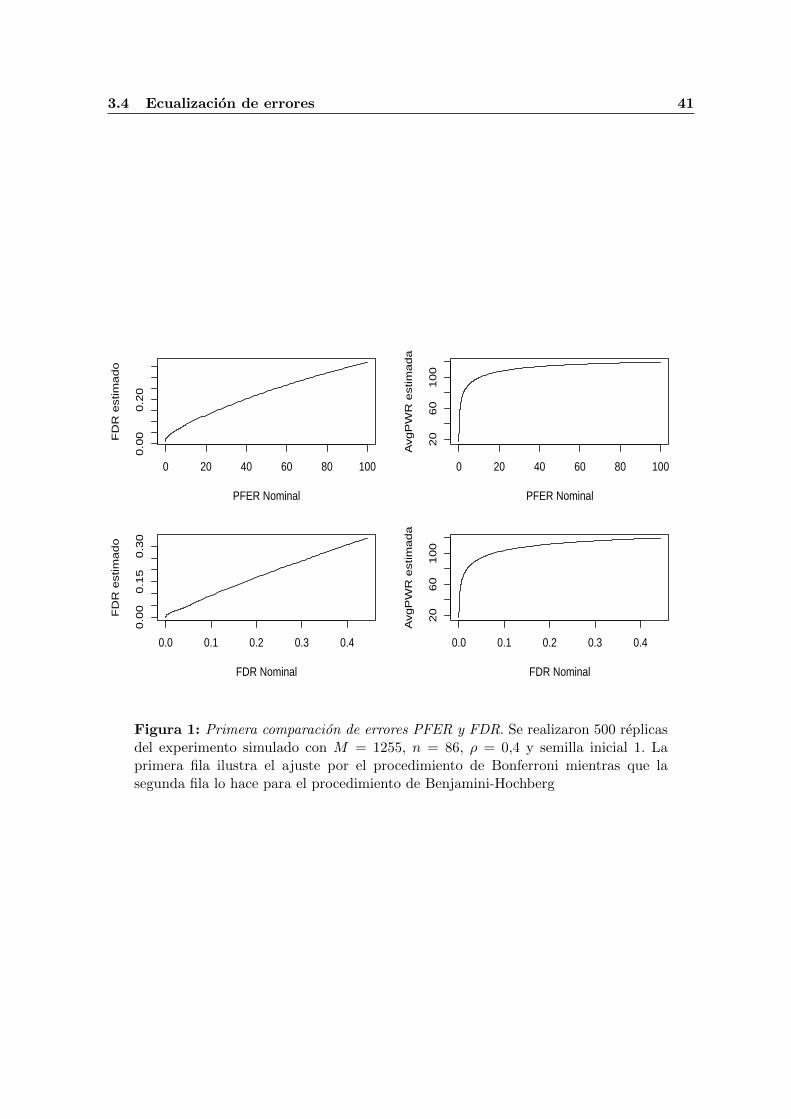
3.4 Ecualizacion de errores 41
0 20 40 60 80 100
0.0
00
.20
PFER Nominal
FD
R e
stim
ad
o
0 20 40 60 80 100
20
60
10
0
PFER Nominal
Avg
PW
R e
stim
ad
a
0.0 0.1 0.2 0.3 0.4
0.0
00
.15
0.3
0
FDR Nominal
FD
R e
stim
ad
o
0.0 0.1 0.2 0.3 0.4
20
60
10
0
FDR Nominal
Avg
PW
R e
stim
ad
a
Figura 1: Primera comparacion de errores PFER y FDR. Se realizaron 500 replicasdel experimento simulado con M = 1255, n = 86, ρ = 0,4 y semilla inicial 1. Laprimera fila ilustra el ajuste por el procedimiento de Bonferroni mientras que lasegunda fila lo hace para el procedimiento de Benjamini-Hochberg

3.5 Descripcion del algoritmo GGQY 42
f1, f2, g1 y g2 tales que:
γ 7−→f1
FDR γ 7−→f2
AvgPWR
β 7−→g1
FDR β 7−→g2
AvgPWR (3.5)
donde f1, f2, g1 y g2 son estimaciones de las versiones poblacionales f1, f2, g1 y g2 quecumplen:
γ 7−→f1
FDR γ 7−→f2
AvgPWR
β 7−→g1
FDR β 7−→g2
AvgPWR (3.6)
Notemos que si las distribuciones subyacentes son continuas y regulares, como es el caso delas simulaciones que consideramos, podemos esperar que las curvas poblacionales no solo seancrecientes sino estrictamente crecientes y en consecuencia tengan inversas bien definidas.
Los autores de [GGQY07] investigaron el comportamiento de la medida AvgPWR deforma “ecualizada” para los dos procedimientos investigando estimaciones de las curvas:{(
FDR, f2 ◦ f−11 (FDR))}
vs.{(FDR, g2 ◦ g−11 (FDR)
)}(3.7)
Expondremos el algoritmo en detalle y luego haremos una crıtica que sera origen de nuestronuevo algoritmo.
De todas formas queda claro que podemos emplear el mismo metodo para cualquier otravariable que dependa de β y γ.
3.5. Descripcion del algoritmo GGQY
Ahora estamos en condiciones de presentar el algoritmo usado en [GGQY07] en tododetalle. Como ya se adelanto se realizaron 500 replicas del experimento simulado con M =1255, n = 86 y ρ = 0,4.
Para muestrear las funciones f1 y f2 ejecutan el procedimiento de Bonferroni en las 500replicas para cada nivel nominal γi, donde γi pertenece a una grilla de 280 valores elegidos quese concentran en los valores nominales bajos, que son los de interes en la practica (si un inves-tigador testea 1255 genes no va a tolerar 250 falsos positivos por ejemplo). Especıficamente,definen:
γ1 = 0,01, γ2 = 0,02, . . . , γ100 = 1
γ101 = 1,1, γ102 = 1,2, . . . , γ190 = 10
γ191 = 11, γ192 = 12, . . . , γ280 = 100 (3.8)
De manera analoga, para muestrear las funciones g1 y g2 ejecutan el procedimiento deBenjamini-Hochberg en las 500 replicas para cada nivel nominal βj , donde βj pertenece a unagrilla de 280 que se define a partir de la grilla de los γ’s a traves de:
βj =γj
125 + γj∀j = 1, . . . 280 (3.9)

3.5 Descripcion del algoritmo GGQY 43
El justificativo es notar que bajo aproximaciones muy groseras:
β ≈ E(V
R) = E(
V
S + V) ≈ E(V )
E(S + V )≈ E(V )
125 + E(V )≈ γ
125 + γ
Estas aproximaciones por supuesto no se cumplen y estas grillas las podremos mejorar. Tam-bien veremos mas adelante que no es necesario ajustar para cada γi o para cada βj , basta conejecutarlo una sola vez para todas las replicas.
Al finalizar los muestreos se obtienen los siguientes valores que ademas los escribimosusando la notacion del artıculo:
FDRBonfγi = f1(γi) AvgPWRBonfγi = f2(γi) ∀i = 1, . . . 280
FDRBHβj = g1(βj) AvgPWR
BHβj = g2(βj) ∀j = 1, . . . 280 (3.10)
Para ecualizar por el error FDR, estiman
γ∗j ≈ f1−1 (
FDRBHβj
)∀j = 1, . . . 280
resolviendo:γ∗j = arg mın
i=1,...280
∣∣∣FDRBonfγi − FDRBHβj
∣∣∣ ∀j = 1, . . . 280 (3.11)
Luego, podemos investigar la potencia AvgPWR por ejemplo, comparandola con el errorecualizado FDR, como se definio en (3.7) graficando los pares de puntos:{(
FDRBHβj , f2 ◦ f1
−1(FDR
BHβj )), j = 1, . . . , 280
}y{(
FDRBHβj , g2 ◦ g1−1(FDRBHβj )
), j = 1, . . . , 280
}(3.12)
o lo que es lo mismo: {(FDR
BHβj , f2(γ∗j )), j = 1, . . . , 280
}y{(
FDRBHβj , g2(βj)
), j = 1, . . . , 280
}o tambien {(
FDRBHβj , AvgPWR
Bonfγ∗j
), j = 1, . . . , 280
}y{(
FDRBHβj , AvgPWR
BHβj
), j = 1, . . . , 280
}(3.13)
La primera figura de [GGQY07] se construyo graficando el interpolado lineal de este precisoconjuntos de puntos. Otras figuras emplean el mismo metodo para otras variables como lamediana y el desvıo de los verdaderos positivos. Esto es lo que se hizo para ecualizar por FDR,la ecualizacion por PFER es totalmente analoga.

3.5 Descripcion del algoritmo GGQY 44
3.5.1. Paso de Muestreo
Podemos dividir el algoritmo GGQY en un primer paso de muestreo, donde se obtienenlas medidas de las ecuaciones (3.10) y un segundo paso de ecualizacion donde se calculan losγ∗j definidos segun (3.11).
El primer paso lo podemos implementar con el siguiente algoritmo que es el que describenlos autores de [GGQY07].
Paso 1. (Muestreo).
Paso 1.1 (Generacion de Vn y Rn).Para cada replica
Generar p-valores no ajustados
Para cada γi de la grilla de Gammas
Ajustarlos por Bonferroni y medir Vn y RnPara cada βj de la grilla de Betas
Ajustarlos por Benjamini-Hochberg y medir Vn y RnPaso 1.2 (Estimacion errores).
Para cada βj de la grilla de Betas
Calcular estadısticos FDRBHβj y AvgPWR
BHβj
Para cada γi de la grilla de Gammas
Calcular estadısticos FDRBonfγi y AvgPWRBonfγi
En el Paso 1.1, es claro que basta medir Vn y Rn para calcular Sn y la proporcion Vn/Rn(ref. 2.9.2). Si solo vamos a considerar las medias en el Paso 1.2, bastara conservar 4 arreglosde numeros, enteros (entre 0 y M), cada uno del tamano de la grilla correspondiente. Porsimplicidad podemos asumir un unico tamano de grilla.
En cambio, si queremos otras medidas de error como la mediana de los falsos positivos ocualquiera que no sea fijado apriori, debemos conservar toda informacion que representa ladistribucion empırica FVn,Rn (ref: 2.9.1). Esto es, debemos conservar las 4 matrices de tipoentero, de dimensiones tamano de grilla x cantidad de replicas. Veremos que aun en este casoel tamano no es un problema sino el tiempo.
A modo de ejemplo, si usamos grillas de 2500 puntos y 5000 replicas para 12558 genes,los 4 arreglos ocuparan 191 Mb temporalmente. En cambio en la seccion 3.6.2 veremos que eltiempo puede ser de dıas.
En el Paso 1.2, simplemente medimos los estadısticos predefinidos (por ej. medias, media-nas, desvıos) sobre todas las replicas. El resultado sera un arreglo del tipo “double” o “float”del tamano de la grilla por cada estadıstico requerido. Siguiendo con el ejemplo, si queremos5 estadısticos, el resultado ocupara 5× 2500× 8b, que es menos que 100Kb. Al final de estepaso podemos liberar la memoria borrando los arreglos creados en el Paso 1.1.
En cuanto al tiempo, este paso es trivial en comparacion al Paso 1.1. El lenguaje R esoptimo para obtener los estadısticos usuales.
3.5.2. Pseudocodigo del algoritmo GGQY
Sumando el paso de ecualizacion al paso de muestreo presentamos nuestra version enpseudocodigo del algoritmo GGQY. Los parametros principales son la cantidad de genes M ,

3.6 Analisis del algoritmo GGQY 45
la cantidad de replicas K, las grillas de γ’s y β’s que podemos por simplicidad suponer delmismo tamano L.
Paso 1. (Muestreo).
Paso 1.1 (Generacion de Vn y Rn).Para cada k desde 1 a K
Generar replica k de p-valores no ajustados
Para cada γi de la grilla de Gammas
Ajustarlos por Bonferroni y medir Vn [γi, k] y Rn [γi, k]Para cada βj de la grilla de Betas
Ajustarlos por Benjamini-Hochberg y medir Vn [βj , k] y Rn [βj , k]Paso 1.2 (Estimacion errores).
Para cada βj de la grilla de Betas
Calcular estadısticos FDRBHβj y AvgPWR
BHβj
Para cada γi de la grilla de Gammas
Calcular estadısticos FDRBonfγi y AvgPWRBonfγiPaso 2. (Ecualizacion).
Para cada βj de la grilla de Betas
γ∗j <- ∞Para cada γi de la grilla de Gammas
dif <-∣∣∣FDRBonfγi − FDRBHβj
∣∣∣Si dif <γ∗j entonces γ∗j <- dif
fin Si
3.6. Analisis del algoritmo GGQY
En primer lugar podemos senalar que el Paso 2, de ecualizacion, a pesar de que es un dobleciclo, gracias al manejo de vectores de R se puede implementar eficientemente y su tiempoes trivial con respecto al Paso 1 de muestreo. El espacio requerido tambien es trivial. Poreso podremos obviar este paso en el analisis de tiempo y espacio. En segundo lugar vamos atratar de estimar el espacio y el tiempo requeridos si aumentamos en un orden de magnitudlos parametros M , K y L con respecto a los valores utilizados en [GGQY07], llevandolos aM = 12558,K = 5000, L = 2500, que es un objetivo mas realista. Para abreviar, vamos allamar a este conjunto de valores las condiciones objetivo mientras que al conjunto de valoresM = 1255,K = 500, L = 280 lo llamaremos las condiciones del artıculo. Esto nos permitiraestablecer el costo de una simulacion mas realista.
3.6.1. Analisis de espacio
No podemos olvidar senalar que los autores parecen emplear las simulaciones hechas en unartıculo anterior ([QKY05]), donde la estructura de datos dominante es una matriz Y = (yijk)de log-expresiones que siguen la definicion (3.1) para cada replica k. Si ası fuera, esta estructuradarıa cuenta por mucho por el espacio utilizado por el algoritmo ya que serıa una matriz de“floats” de dimensiones M × n × K. En las condiciones del artıculo esta matriz ocuparıa1255× 86× 500× 8b, que equivalen a 411Mb.

3.6 Analisis del algoritmo GGQY 46
En cambio, en el Paso 1.1 de 3.5.2 podemos ser mucho mas economicos construyendo unvector de “floats” de dimension M para almacenar los p-valores no ajustados de la replicaen proceso y 4 matrices de enteros (positivos entre 0 y M) de dimensiones L × K paraalmacenar las variables aleatorias Vn y Rn tanto para el metodo de Bonferroni como para elde Benjamini-Hochberg.
En el Paso 1.2 construimos un vector de “floats” de dimension L por cada medida/errordeseado.
Tanto en las condiciones del artıculo como las condiciones objetivo L ×K >> M por loque las estructuras de datos dominantes son las 4 matrices que insumen 4× L×K × 8b. Enlas condiciones del artıculo, esto es igual a 4,3Mb, mientras que en las condiciones objetivonecesitarıamos 381,5Mb, lo que no es demasiado. De aquı notamos que el espacio no es unalimitacion para realizar simulaciones mas realistas.
3.6.2. Analisis de tiempo
Si bien vamos a analizar los tiempos para el caso M = 12558 de todas maneras los algorit-mos involucrados son proporcionales a M . En el apendice B.4 se puede ver el codigo utilizadopara realizar las estimaciones. Aquı vamos a comentar los resultados. Los tiempos absolutospueden variar de maquina en maquina pero aquı nos interesa las performance relativa delalgoritmo GGQY vs. el nuevo algoritmo. Y las diferencias entre los algoritmos maquina amaquina fueron consistentes.
En el Paso 1.1, es claro que podemos separar el tiempo en tres terminos. El primero serael tiempo que lleva generar K muestras de p-valores no ajustados. El segundo sera el tiempoque lleva medir Vn y Rn para el caso de Bonferroni multiplicado por K × L veces. Y eltercer termino es sera el tiempo que lleva medir Vn y Rn para el caso de Benjamini-Hochbergmultiplicado por K × L veces.
Con respecto al primer termino, en la seccion B.1 se midieron los tiempos de diferentesimplementaciones para el caso de M = 12558.
La implementacion mas rapida -funcion rawPValues()- resulto ser la que usa una versionpropia del test t. En este caso tardo 2.34seg. por replica. Para mas detalles ver 3.2.2, B.1 ytambien B.4. En estas condiciones para K = 5000 este paso insumira 3,25 horas, lo que no esmucho.
Con respecto al segundo y tercer termino, esta claro que el tiempo es proporcional a K yL. Ajustar por Bonferroni y medir Vn y Rn para un determinado γi se estimo en 0,002 seg. Enlas condiciones objetivo estimamos el tiempo en 6,6 horas, lo cual todavıa no es muy costoso.
Ahora, en el caso del ajuste por Benjamini-Hochberg, la medicion de Vn y Rn para undeterminado βj insumio un tiempo de 0,022 seg. Que lleva a estimar que en las condicionesobjetivo el tiempo sea de 3,17 dıas, lo cual ya implica un costo importante. Y llegarıa a loimpracticable si se hubiera usado la versio del ajuste de Benjamini-Hochberg provista por lalibrerıa multtest.
El siguiente Paso 1.2. de medicion de estadısticos es despreciable en cuanto a los tiemposya que se tarda menos de 1s por estadıstico aun en las condiciones objetivo gracias al eficientemanejo de vectores de R (ver B.4).
En total para futura comparacion podemos estimar que en las condiciones objetivo elalgoritmo insumira 3,58 dıas.

3.6 Analisis del algoritmo GGQY 47
3.6.3. Sesgo por replicas
Para analizar el sesgo introducido por las replicas, los autores generaron otro conjuntode 500 replicas independientes, que llaman de control, mientras que a la primera la llaman
de entrenamiento. Indicamos con2 las estimaciones obtenidas con este conjunto. Los autores
observan que los resultados obtenidos en las comparaciones de{(FDR
BHβj ,2
f2(γ∗j )
), j = 1, . . . , 280
}y{(
FDRBHβj ,
2
g2(βj)
), j = 1, . . . , 280
}fueron similares a los obtenidos en (3.12) por lo que concluyen que el sesgo por replicas no essignificativo. Nosotros en cambio observamos una variacion significativa en el sesgo cuando elnumero de replicas es 500 y los valores nominales son bajos. Recordemos que estos son valoresinteresantes para el investigador.
Para mostrarlo, en la figura 2 graficamos las aproximaciones a f1 hechas con 5000 replicas.Ademas graficamos intervalos asintoticos alrededor de la semilla 2, por propositos didacticos.Los intervalos asintoticos alrededor de otras semillas son similares.
En la figura 3 graficamos los mismos intervalos asintoticos de la figura 2 y las aproxima-ciones hechas con solo 500 replicas. Vemos que estas son mucho menos monotonas y escapanfuera de los intervalos asintoticos excepto en el caso de la semilla inicial 2, que fue el motivopor el cual la elegimos en las figuras ya que eso permite mejores graficos.
Si naturalmente siguiendo a los autores hubieramos elegido la muestra de 500 replicasgenerada con la semilla inicial 1 como muestra de“entrenamiento”y la muestra de 500 replicasgenerada con la semilla inicial 2 como muestra de “control” verıamos que en la figura 3cometemos mucha mas imprecision que directamente generar una unica muestra de 5000replicas, que gracias a la mejora en eficiencia del nuevo algoritmo no es tan costosa.
3.6.4. Definicion de inversas
Investigamos las consecuencias de la definicion de γ∗j en (3.11). Para ello seleccionamosnueve integrantes de la grilla βj , j = 119, . . . , 127 definida segun (3.9) que se encuentran enuna region interesante de valores actuales de FDR estimados.
Graficamos en el primer panel de la figura 4 la porcion de la curva g1 obtenida en lascondiciones GGQY con semilla inicial 2. En el segundo panel graficamos la curva f1 obtenidaen estas condiciones con un trazo de lınea de guiones cortos y largos en color cyan. Ademasagregamos con trazo de lınea solido y en color negro la aproximacion a f1 hecha con K = 5000con sus intervalos asintoticos en trazo de linea de guiones en color rojo descritas en las figuras2 y 3.
Para investigar la precision en la construccion de los γ∗j agregamos al segundo panel de lafigura 4 los puntos de la grilla de γ’s original (ref: (3.8)) en la figura 5. Hay dos fuentes deimprecision, la imprecision en la aproximacion a f1 y por otro lado la imprecision debida alas grillas. Para ilustrarlo vemos que la eleccion de γ∗119 podrıa ser γ40 o γ47. Esta imprecisionno sucederıa si aumentasemos K a 5000. El otro tipo de imprecision lo podemos ver en laeleccion de γ∗122 que resulta ser igual a γ∗123 = γ52. Esta imprecision es mucho menor que laanterior. Se puede solucionar aumentando la densidad de las grillas y tambien notando que

3.6 Analisis del algoritmo GGQY 48
0.0 0.5 1.0 1.5 2.0 2.5 3.0
0.0
00
.01
0.0
20
.03
0.0
4
PFER Nominal
FD
R e
stim
ad
o
S 2S 2 + 2 SdS 2 − 2 SdS 1S 101S 102
Figura 2: Aproximaciones a f1 con 5000 replicas. Con el nuevo algoritmo se realiza-ron 5000 replicas del experimento simulado con M = 1255, n = 86, ρ = 0,4 y semillasiniciales 1,2,101 y 102 que en la figura se indican con S. Alrededor de la aproximacionhecha con semilla inicial 2 se graficaron intervalos de confianza asintoticos con lineasde guiones en color rojo. Notamos que las otras semillas originan aproximacionesdentro de estos intervalos.

3.6 Analisis del algoritmo GGQY 49
0.0 0.5 1.0 1.5 2.0 2.5 3.0
0.0
00
.01
0.0
20
.03
0.0
4
PFER Nominal
FD
R e
stim
ad
o
S 2S 2 + 2 SdS 2 − 2 SdS 1 K 500S 101 K 500S 102 K 500S 2 K 500
Figura 3: Aproximaciones a f1 con 500 replicas. Con el nuevo algoritmo se rea-lizaron 500 replicas del experimento simulado con M = 1255, n = 86, ρ = 0,4 ysemillas iniciales 1,2,101 y 102 que en la figura se indican con S y K = 500. Estasaproximaciones se superpusieron a la aproximacion hecha con semilla inicial 2 consus intervalos de confianza asintoticos mostradas en la figura 2. Las aproximacionesa f1 hechas con K = 500 son evidentemente mucho menos precisas que las hechascon K = 5000. Todas excepto la generada con semilla inicial 2 se escapan fuera delos intervalos asintoticos.

3.6 Analisis del algoritmo GGQY 50
0.022 0.024 0.026 0.028
0.0
17
0.0
18
0.0
19
0.0
20
0.0
21
FDR Nominal
FD
R e
stim
ad
o
●
●
●
●
●
●
●
●
●
●
●
β119
β120
β121
β122
β123
β124
β125
β126
β127
0.40 0.45 0.50 0.55 0.60 0.65
PFER Nominal
FD
R e
stim
ad
o
β119
β120
β121
β122
β123
β124
β125
β126
β127
Figura 4: Ilustracion de construccion de γ∗j . Primera parte. En el primer panelgraficamos la curva g1 obtenida con los parametros M = 1255, n = 86, ρ = 0,4,K = 500 y semilla inicial 2. En el segundo panel graficamos la curva f1 obtenidaen estas condiciones con un trazo de lınea de guiones cortos y largos en color cyan.Ademas agregamos con trazo de lınea solido y en color negro la aproximacion a f1hecha con K = 5000 con sus intervalos asintoticos en trazo de linea de guiones encolor rojo descritas en las figuras 2 y 3. Las lıneas nombradas con β119, . . . , β127indican los niveles de FDR estimado alcanzados para los niveles nominales iguales aβ119, . . . , β127, donde los βj se definen segun (3.9)
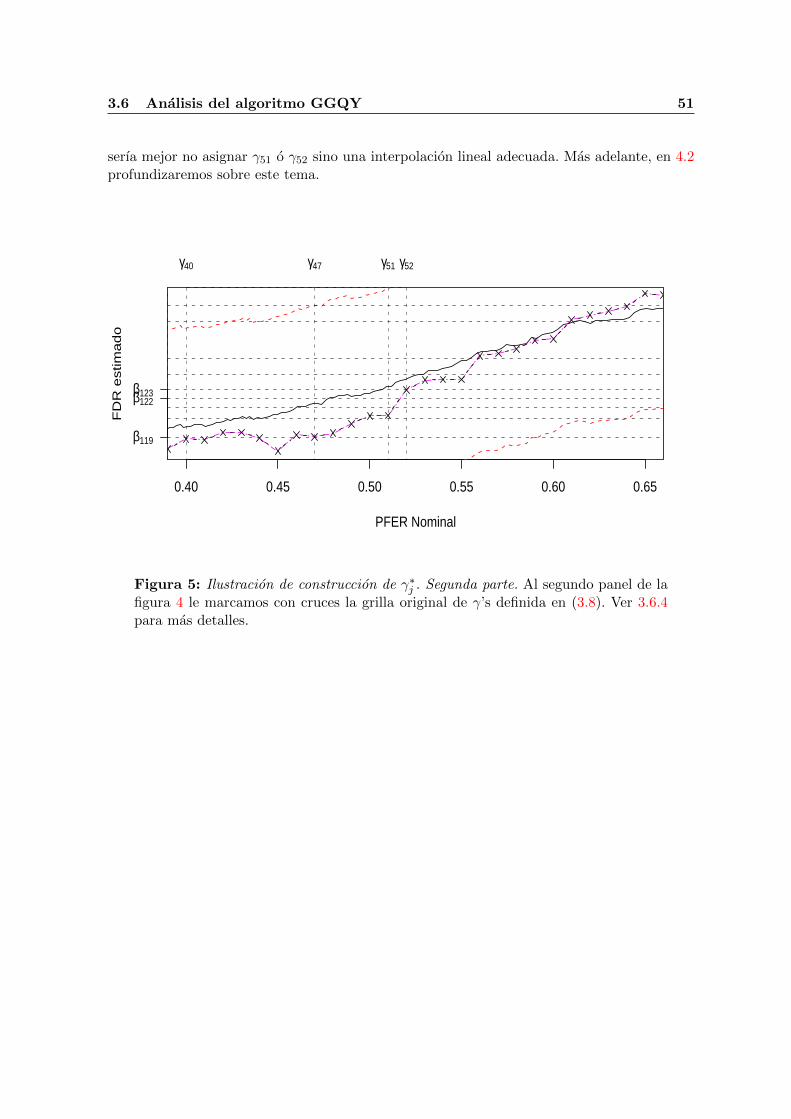
3.6 Analisis del algoritmo GGQY 51
serıa mejor no asignar γ51 o γ52 sino una interpolacion lineal adecuada. Mas adelante, en 4.2profundizaremos sobre este tema.
0.40 0.45 0.50 0.55 0.60 0.65
PFER Nominal
FD
R e
stim
ad
o
β119
β122β123
γ40 γ47 γ51 γ52
Figura 5: Ilustracion de construccion de γ∗j . Segunda parte. Al segundo panel de lafigura 4 le marcamos con cruces la grilla original de γ’s definida en (3.8). Ver 3.6.4para mas detalles.

4. Nuevo algoritmo 52
4. Nuevo algoritmo
Cuando empezamos a tratar de reproducir los resultados del artıculo [GGQY07] paracorrelaciones altas, vimos que el algoritmo era muy lento y los resultados eran muy imprecisos.De allı surgio la necesidad de mejorar el algoritmo GGQY. Obtuvimos exito al mejorar lospasos de Muestreo, la interpolacion y la eleccion de grillas.
4.1. Mejora en el Paso de Muestreo
Una mejora importante se puede conseguir notando que se pueden medir las variablesaleatorias Vn [αl, k] y Rn [αl, k] correspondientes a una replica k para toda una grilla de valoresordenados (αl, l = 1, . . . L) sin necesidad de ajustar los p-valores por cada valor nominal deerror multiple αl y sin tener que contar de forma independiente cada valor Vn [αl, k] yRn [αl, k].
Dado que estas variables son no decrecientes en αl (ver discusion en 3.4) sera suficientemedir la cantidad de positivos y falsos positivos que ocurrieron entre un valor de la grilla αly el siguiente αl+1 para calcular Vn [αl+1, k] y Rn [αl+1, k] en base a Vn [αl, k] y Rn [αl, k].
Para verlo en detalle y probarlo recordemos la ecuacion 2.40 dada en la seccion 2.10.2,que expresa el conjunto de rechazados o descubrimientos mediante sus p-valores ajustados.
Rn(α) = {m : Tn(m) ∈ Cn(m;α)} ={m : P0n(m) ≤ α
}. (4.1)
donde tambien definimosRn(α) = |Rn(α)|
Dado que nos basaremos en los p-valores ordenados P0n(On(m)) vamos a escribir lasvariables aleatorias Vn y Rn en funcion de estos y de los niveles de error multiple nominalesαl de una grilla de valores (αl, l = 1, . . . L).
Rn(αl) =∣∣∣{m : P0n(On(m)) ≤ αl
}∣∣∣ ∀j = 1, . . . L
Vn(αl) =∣∣∣{m : On(m) ∈ H0 ∧ P0n(On(m)) ≤ αl
}∣∣∣ (4.2)
Tambien definimos las variables auxiliares rx y vx que son facilmente calculables.
rx(i) = |{m : m ≤ i}| ∀i = 1, . . .M
vx(i) = |{m : On(m) ∈ H0 ∧m ≤ i}| (4.3)
Es inmediato ver que rx(i) = i para i entre 1 y M . Podemos expresar la variable vx, dado elvector de ındices (On(m),m = 1, . . .M) como:
vx(i) =
i∑m=1
I (On(m) ∈ H0)) (4.4)
Como adelanto, en la implementacion en R construimos el vector (vx(i), i = 1, . . . ,M)con:
> vx <- as.integer(cumsum(!ap$de))

4.1 Mejora en el Paso de Muestreo 53
Como los p-valores ordenados son crecientes:
m ≤ i⇔ P0n(On(m)) ≤ P0n(On(i)) (4.5)
Vn
(P0n(On(i))
)=∣∣∣{m : On(m) ∈ H0 ∧ P0n(On(m)) ≤ P0n(On(i))
}∣∣∣ ∀i = 1, . . .M
= |{m : On(m) ∈ H0 ∧m ≤ i}|= vx(i) (4.6)
Podemos afirmar un poco mas:
Proposicion 4.1. Calculo de Vn(αl) y Rn(αl) Si αl cumple que
P0n(On(i)) ≤ αl < P0n(On(i+ 1))
entonces
Vn (αl) = Vn
(P0n(On(i))
)= vx(i) y
Rn (αl) = Rn
(P0n(On(i))
)= rx(i)
Demostracion de Proposicion 4.1. Para probarlo notemos que:
Vn(αl) =∣∣∣{m : On(m) ∈ H0 ∧ P0n(On(m)) ≤ αl
}∣∣∣=∣∣∣{m : On(m) ∈ H0 ∧ P0n(On(m)) ≤ P0n(On(i))
}∣∣∣= Vn
(P0n(On(i))
)= vx(i) (4.7)
De manera similar se prueba para Rn(αl).
En base a esta proposicion podemos derivar el nucleo de nuestro algoritmo. Dado un ındicei ≤M − 1 tal que P0n(On(i)) ≤ αl podemos construir Vn(αl) y Rn(αl) con:
Mientras i+ 1 ≤M ∧ l ≤ L.Mientras αl < P0n(On(i+ 1)) ∧ l ≤ L.
Rn(αl)← rx(i)Vn(αl)← vx(i)l← l + 1
Fin Mientras
i← i+ 1
Observemos que la condicion P0n(On(i)) ≤ αl es un invariante del ciclo principal.Para ingresar a este ciclo debemos conseguir el primer l tal que P0n(On(1)) ≤ αl. Por otro
lado, cuando salimos del ciclo porque i = M entonces trivialmente se cumple que Rn(αl) =M ∧ Vn(αl) = vx(M) = h0 para todos los αl > P0n(On(M)).
Ası podemos escribir de forma completa el nuevo algoritmo para medir las variablesVn [αl, k] y Rn [αl, k] correspondientes a una replica k en toda una grilla de L valores no-minales (αl, l = 1, . . . L). Para alivianar la notacion omitimos el subındice k que es constanteaquı.

4.1 Mejora en el Paso de Muestreo 54
(Medicion de Vn y Rn para una replica k).i← 1j ← 1
Mientras αl < P0n(On(1)) ∧ l ≤ L.Rn(αl)← 0Vn(αl)← 0j ← j + 1
Fin Mientras
Mientras i+ 1 ≤M ∧ l ≤ L.Mientras αl < P0n(On(i+ 1)) ∧ l ≤ L.
Rn(αl)← rx(i)Vn(αl)← vx(i)j ← j + 1
Fin Mientras
i← i+ 1Fin Mientras
Mientras l ≤ L.Rn(αl)← rx(i)Vn(αl)← vx(i)j ← j + 1
Fin Mientras
El Paso de Muestreo general queda entonces:
Paso 1. (Muestreo).
Paso 1.1 (Generacion de Vn y Rn).Para cada k desde 1 a K
Generar replica k de p-valores no ajustados
Ajustarlos por Bonferroni.
Medir Vn [·, k] y Rn [·, k] en la grilla de Gammas
Ajustar la replica k de p-valores no ajustados por Benjamini-Hochberg.
Medir Vn [·, k] y Rn [·, k] en la grilla de Betas
Paso 1.2 (Estimacion errores).
Para cada βj de la grilla de Betas
Calcular estadısticos FDRBHβj y AvgPWR
BHβj
Para cada γi de la grilla de Gammas
Calcular estadısticos FDRBonfγi y AvgPWRBonfγi
Como introduccion a la implementacion en R mostramos el fragmento correspondiente:
> for (k in 1:par$Rep) {
ap <- adjustedPValues(rawPValues(rho, par), par)
res <- rv(ap, "BF", grid.bf)
R.bf[,k] <- res$r
V.bf[,k] <- res$v

4.2 Mejora en la interpolacion 55
res <- rv(ap, "BH", grid.bh)
R.bh[,k] <- res$r
V.bh[,k] <- res$v
}
4.1.1. Analisis de tiempo y espacio
En cuanto al espacio, los dos pares de matrices Vn y Rn son las mismas matrices que lasdel algoritmo original GGQY. Por lo visto en la seccion 3.6.1 estas son las estructuras dedatos dominantes e insumen L×K × 8b cada una.
En cuanto al tiempo, la generacion, ordenamiento y ajuste de una replica es del orden deM . La medicion de Vn y Rn para una replica de una grilla de valores de longitud L es unproceso lineal que recorre la grilla de valores nominales y el vector de p-valores ordenados yajustados. Entonces es del orden del maximo entre L y M , que es M tanto en las condicionesdel artıculo como en las condiciones objetivo. Vemos aquı una diferencia notable con respectoal algoritmo original donde el tiempo se multiplicaba por L.
Como detalle, notemos que se pueden ordenar a los p-valores no ajustados de la replica kuna sola vez y luego ajustarlos por Bonferroni o por Benjamini-Hochberg. Luego, no agrega-mos un costo adicional de ordenamiento con respecto al algoritmo original porque esta tareaes parte del ajuste de Benjamini-Hochberg.
Con las mejoras introducidas no fue necesario estimar el tiempo de procesamiento en lascondiciones objetivo sino que se lo pudo observar directamente (ver codigo utilizado en B.5).La media fue de 8720 segundos mientras que la mediana de los tiempos fue de 8721s. Es decirtardo 2 horas 25 min. Comparado con el tiempo estimado para el algoritmo GGQY, estetiempo representa una mejora de 35 veces!
4.2. Mejora en la interpolacion
En esta seccion vamos a profundizar el comentario que hicimos al final de la seccion 3.6.4donde observamos que en la figura 5 cometemos cierta imprecision en la eleccion de γ∗122.
En la seccion 3.5 vimos que podemos expresar las comparaciones “ecualizadas” propuestasen [GGQY07] como las interpolaciones lineales de los conjuntos:
{(FDR
BHβj , f2(γ∗j )), j = 1, . . . , L
}y{(
FDRBHβj , g2(βj)
), j = 1, . . . , L
}En nuestra notacion lo podemos escribir como:{(
g1(βj), f2(γ∗j )), j = 1, . . . , L
}y (4.8)
{(g1(βj), g2(βj)) , j = 1, . . . , L}(4.9)
Recordemos la definicion de los γ∗j dada en (3.11).
γ∗j = arg mıni=1,...L
∣∣∣FDRBonfγi − FDRBHβj
∣∣∣ ∀j = 1, . . . L (4.10)

4.2 Mejora en la interpolacion 56
Si no hubiera imprecision en la estimacion de γ∗j de acuerdo a (3.11), podrıamos definir
γ−1j de manera tal que:
f1(γ−1j ) = g1(βj) ∀j = 1, . . . L (4.11)
Como las funciones son regulares podemos suponer la existencia de los γ−1j . Vemos quecuanto mas precisa sea la estimacion en (4.10), el primer conjunto de (4.8) se aproximara a:
{(f1(γ
−1j ), f2(γ
−1j )), j = 1, . . . , L
}(4.12)
Ahora, una estimacion mas precisa de este conjunto teorico se logra con el interpoladolineal de: {(
f1(γ∗j ), f2(γ
∗j )), j = 1, . . . , L
}(4.13)
Para probar esta afirmacion, supongamos sin perdida de generalidad que:
f1(γ∗j ) ≤ f1(γ−1j ) < f1(γ
∗j+1)
entonces por la interpolacion lineal entre los puntos(f1(γ
∗j ), f2(γ
∗j ))
y(f1(γ
∗j+1), f2(γ
∗j+1)
)contiene al siguiente punto:(
f1(γ−1j ), f2(γ
∗j ) +
f2(γ∗j+1)− f2(γ∗j )
f1(γ∗j+1)− f1(γ∗j )
(f1(γ
−1j )− f1(γ∗j )
))(4.14)
Y por la regularidad de f1 y f2, este punto es una mejor aproximacion al punto(f1(γ
−1j ), f2(γ
−1j ))
que el punto de la interpolacion original ((4.8))(g1(βj), f2(γ
∗j ))
.
Por supuesto, cuanto menor sea la diferencia entre f1(γ−1j ) y f1(γ
∗j ) menor sera la diferencia
entre las dos aproximaciones. Cuanto mas densa sea la grilla mas precisas seran las dosaproximaciones. Por eso elegimos elevar en un orden de magnitud el tamano de la grilla enlas condiciones objetivo.
Podemos mejorar todavıa el interpolado de (4.13) notando que al ser los (γ∗j , j = 1, . . . , L)un subconjunto de (γi, i = 1, . . . , L), esto es, una grilla mas gruesa, el siguiente interpoladosera mas preciso:
{(f1(γi), f2(γi)
), i = 1, . . . , L
}(4.15)
Notemos que entonces hemos reducido el problema a graficar una simple curva de para-metro γ y evitamos la estimacion de las inversas γ∗j . Ademas de la precision, hemos facilitadoel calculo. El Paso 2 del algoritmo GGQY (ref: 3.5.2) no es necesario en consecuencia.

4.3 Mejora en la eleccion de Grillas 57
En la notacion del artıculo, hemos probado que un grafico mas preciso se obtiene alinterpolar los conjuntos:
{(FDRBonfγi , AvgPWRBonfγi
), i = 1, . . . , L
}y{(
FDRBHβj , AvgPWR
BHβj
), j = 1, . . . , L
}4.3. Mejora en la eleccion de Grillas
No solo la precision aumenta al crecer L sino que tambien es afectada por la distribucionde los valores dentro de la grilla. No es deseable una distribucion uniforme en todo el rangode valores por ejemplo porque no todas las regiones presentan el mismo interes. Como yadijimos es de interes conocer con precision el comportamiento de los errores y medidas depotencia para valores de error multiple estimados bajos. Por ejemplo deseamos conocer conprecision el comportamiento de f1, f2, g1 y g2 alrededor de un error FDR estimado igual a0.01 y alrededor de un PFER estimado de 3.
Por esta razon, los autores de [GGQY07] elaboraron las grillas dando preponderancia a losvalores bajos (ver (3.8) y (3.9)) y graficaron las comparaciones usando un rango logarıtmicopara el FDR estimado o el PFER estimado. En el caso del FDR usan el rango entre -6 y-1. De esta forma, para investigar lo que sucede alrededor de un error FDR estimado igual a0.01 inspeccionamos lo que sucede alrededor de −4,6 ≈ log(0,01). El extremo izquierdo delrango corresponde a un error estimado de exp(−6) ≈ 0,00249 mientras que el extremo derechocorresponde a un error estimado de exp(−1) ≈ 0,368.
En los paneles izquierdos de las figuras 6 y 7 podemos ver que valores se muestrearonusando la grilla del artıculo de las funciones g1 y f1 respectivamente. No usamos interpolacionentre puntos para no ocultar la distribucion de los puntos. La poca continuidad y defectosen la monotonıa observadas se deben al insuficiente numero de replicas. En los dos casosobservamos que es mejor directamente elegir los valores uniformemente distribuidos en escalalogarıtmica. De esta forma en escala logarıtmica le damos la misma importancia a todo elrango. Y esto tiene el efecto de lograr que los valores bajos esten bien representados. En elcaso de la figura 7 esto es especialmente notorio.
Esta propuesta de grillas la implementamos en la funcion pvals.gridStd() en R , cuyocodigo se explica por si mismo.
> pvals.gridStd <- function(mtp.id, gridSize=base.par()$GridSize)
switch(mtp.id,
BH = grid <- exp(seq(from=-8, to=0, length.out=gridSize)),
BF = grid <- exp(seq(from=-6, to=6, length.out=gridSize)),
stop("BF (Bonferroni) and BH (Benjamini-Hochberg) are the only MTPs implemented"))

4.3 Mejora en la eleccion de Grillas 58
−8 −6 −4 −2 0
0.0
00
.01
0.0
20
.03
0.0
40
.05
Grilla Original (L=280)Log FDR Nominal
FD
R E
stim
ad
o
−8 −6 −4 −2 0
0.0
00
.01
0.0
20
.03
0.0
40
.05
Grilla Nueva (L=2500)Log FDR Nominal
FD
R E
stim
ad
o
Figura 6: Ilustracion de mejora en el muestreo de g1 por la eleccion de grillas. Launica diferencia entre los dos paneles es la grilla usada. Los otros parametros sonM = 1255, rho = 0,4, K = 500 y semilla inicial 1. En el primer panel usamos lagrilla original del artıculo de 280 valores, mientras que en el segundo usamos la grillapropuesta de 2500 valores.

4.3 Mejora en la eleccion de Grillas 59
−6 −4 −2 0 2 4 6
0.0
00
.01
0.0
20
.03
0.0
40
.05
Grilla Original (L=280)Log PFER Nominal
FD
R E
stim
ad
o
−6 −4 −2 0 2 4 6
0.0
00
.01
0.0
20
.03
0.0
40
.05
Grilla Nueva (L=2500)Log PFER Nominal
FD
R E
stim
ad
o
Figura 7: Ilustracion de mejora en el muestreo de f1 por la eleccion de grillas. Launica diferencia entre los dos paneles es la grilla usada. Los otros parametros sonM = 1255, rho = 0,4, K = 500 y semilla inicial 1. En el primer panel usamos lagrilla original del artıculo de 280 valores, mientras que en el segundo usamos la grillapropuesta de 2500 valores.

5. Extension de los resultados originales 60
5. Extension de los resultados originales
En esta seccion aplicaremos el nuevo algoritmo desarrollado a condiciones mas realistasque serıan costosas para el algoritmo GGQY y extenderemos las comparaciones hechas en[GGQY07] al caso ρ = 0,9. Tambien usaremos los resultados del algoritmo GGQY bajo lascondiciones del artıculo como referencia para evaluar las mejoras de nuestro algoritmo.
5.1. Nuevas condiciones mas realistas
Como adelantamos en la seccion 3.1 replicamos las comparaciones hechas en [GGQY07]en condiciones mas realistas aumentando la correlacion a 0.9 y la cantidad de genes simula-dos a 12558. Allı mencionamos que este es el numero de genes original de la base de datosHYPERDIP del St. Jude Children’s Research Hospital (SJCRH) sobre pacientes pediatricoscon leucemia, que fue usada en un artıculo anterior de los autores ([QKY05]) como fuentede datos biologicos. En el mismo artıculo los autores trabajan con datos simulados de 1255genes. Y estas fueron usadas con pequenas modificaciones en [GGQY07].
Respetar la cantidad de genes es importante porque en general las relaciones estudiadasson muy complejas y no podemos asegurar a priori que sean lineales.
En cuanto a la correlacion, podemos esperar que las relaciones sean menos lineales todavıay que el comportamiento para ρ = 0,4 sea diferente al comportamiento para ρ = 0,9.
Cabe preguntarse porque simular para un ρ tan alto. El hecho resaltado por los autoresde [KY07] es que:
It is a well-known fact that pairwise correlations between gene expression levelsare overwhelmingly positive and strong [Qiu et al. (2005a, 2005b), Almudevar etal. (2006)]. When studying various data sets, we observed the average (over allgene pairs) of correlation coefficients ranging from 0.84 to 0.97. Another featureof the correlation structure of microarray data is that the correlations are long-ranged, that is, a particular gene may have very high correlation coefficients witha vast majority of other genes.
y mas adelante informando sobre los hallazgos del analisis que hicieron sobre los datos dela base HYPERDIP:
The mean (over the selected gene pairs) value of these correlation coefficients is0.942 with the corresponding standard deviation being equal to 0.036.
Por estas razones usaremos el nuevo algoritmo con los parametros mas realistas ρ = 0,9 yM = 12558 para actualizar las comparaciones entre los dos MTP’s hechas en [GGQY07].
5.2. Comparaciones bajo condiciones mas realistas
Para contrastar los resultados obtenidos simulamos tambien que hubieran obtenido losautores si hubieran usado ρ = 0,9 en vez de ρ = 0,4. Esta claro que no habrıa inconvenientesde performance en hacer esto con el algoritmo GGQY bajo las condiciones del artıculo. Esdecir, usando los parametros M = 1255, K = 500 y L = 280 (respetando las grillas originales).Si los resultados ası obtenidos no fueran mejorados por el algoritmo propuesto usando lascondiciones objetivo, M = 12558,K = 5000, L = 2500, entonces nuestro esfuerzo no tendraun resultado apreciable.

5.2 Comparaciones bajo condiciones mas realistas 61
Por lo tanto en todas las figuras de esta seccion mostraremos en panel izquierdo la compa-racion hecha con el algoritmo GGQY bajo las condiciones del artıculo y en el panel derechola comparacion hecha con el algoritmo propuesto bajo las condiciones objetivo. Usaremos elestilo de lınea solida y color negro para el MTP de Bonferroni y el estilo de lınea punteada ycolor azul para el MTP de Benjamini-Hochberg.
Para notar la variacion debida al cambio de semillas iniciales graficamos las mismas curvasusando cuatro semillas iniciales: 1,2,101 y 102. De esta manera apreciaremos la mejora en laprecision al aumentar el numero de replicas.
Naturalmente vamos a establecer la misma proporcion del 10 % para los genes d.e. en lascondiciones objetivo. Es decir, establecemos que los primeros 1256 genes de los 12558 son d.e..En la implementacion, usamos el parametro kDE igual a 1256 en este caso.
Por ultimo, nuestras simulaciones de los resultados bajo el algoritmo GGQY son algomejores que las originales ya que usan la mejora en la interpolacion descrita en la seccion 4.2.

5.2 Comparaciones bajo condiciones mas realistas 62
5.2.1. Comparaciones de AvgPWR
Como adelantamos en la seccion 3.5 la primera aplicacion del algoritmo GGQY que hi-cieron los autores fue analizar la medida AvgPWR ecualizando por FDR (ver (3.13)). Con-cluyeron que no habıa diferencias entre estas medidas de potencia una vez ecualizados losmetodos.
En nuestro caso obtuvimos la figura 8 donde vemos el mismo comportamiento de igualdadentre los dos metodos. El metodo de Bonferroni parece ser un poco mas potente a partir deun FDR estimado de exp(−2,5) ≈ 0,082. En general este es un valor muy alto comparado conlos usuales de la practica del orden de 0,01. Tambien vemos claramente en el panel derechoun aumento de precision con nuestro algoritmo.
−6 −5 −4 −3 −2 −1
01
02
03
04
05
0
rho 0.9 M 1255 K 500 L 280
Log FDR estimado
Me
dia
S
BFBH
−6 −5 −4 −3 −2 −1
01
00
20
03
00
40
05
00
rho 0.9 M 12558 K 5000 L 2500
Log FDR estimado
Me
dia
S
BFBH
Figura 8: Comparacion de AvgPWR ecualizando por FDR. Para ρ = 0,9 compa-ramos la medida de potencia AvgPWR,i.e., la media de los verdaderos positivosecualizando por el error FDR. El primer panel simula los resultados obtenidos para4 semillas iniciales (1,2,101 y 102) usando el algoritmo GGQY en las condiciones delartıculo. El segundo panel muestra los resultados obtenidos para las mismas semillascon el nuevo algoritmo en las condiciones objetivo. La lınea solida y negra denotael metodo de Bonferroni mientras que la punteada y azul, el metodo de Benjamini-Hochberg.

5.2 Comparaciones bajo condiciones mas realistas 63
5.2.2. Comparaciones de Desvıos de verdaderos positivos
Si las potencias de los dos metodos son parecidas en las regiones de interes, la siguientecaracterıstica a observar para decidir cual es preferible desde el punto de vista de la potencia esel desvıo de los verdaderos positivos Sn. Un metodo con alto desvıo tendera con alta frecuenciaa subestimar o sobreestimar la potencia AvgPWR, es decir la media de Sn.
En el artıculo original los autores observan que el metodo de Benjamini-Hochberg tieneun desvıo apenas mas alto que el de Bonferroni. En nuestro caso podemos tener esa sospechaal observar el panel izquierdo de la figura 9 pero lo vemos mucho mas claramente en el panelderecho correspondiente a nuestro algoritmo.
Entonces podemos afirmar que el metodo de Benjamini-Hochberg es mas inestable o menospreciso en la cantidad de verdaderos positivos o descubrimientos que el metodo de Bonferroni apesar de que en promedio la cantidad de verdaderos positivos es la misma cuando se ecualizanambos metodos.
−6 −5 −4 −3 −2 −1
01
02
03
04
05
0
rho 0.9 M 1255 K 500 L 280
Log FDR estimado
De
svío
S
BFBH
−6 −5 −4 −3 −2 −1
01
00
20
03
00
40
05
00
rho 0.9 M 12558 K 5000 L 2500
Log FDR estimado
De
svío
S
BFBH
Figura 9: Comparacion de los desvıos de los verdaderos positivos ecualizando porFDR. Para ρ = 0,9 comparamos el desvıo de los verdaderos positivos Sn ecualizandopor el error FDR. El primer panel simula los resultados obtenidos para 4 semillasiniciales (1,2,101 y 102) usando el algoritmo GGQY en las condiciones del artıculo.El segundo panel muestra los resultados obtenidos para las mismas semillas con elnuevo algoritmo en las condiciones objetivo. La lınea solida y negra denota el metodode Bonferroni mientras que la punteada y azul, el metodo de Benjamini-Hochberg.

5.2 Comparaciones bajo condiciones mas realistas 64
5.2.3. Comparaciones de Desvıos de Positivos
Una medida relacionada al desvıo de Sn es el desvıo de Rn, es decir el desvıo de todos losdescubrimientos o rechazados. Esta es una medida de importancia practica porque puede serobservable a diferencia de Sn que no lo es (ver 2.8). A mayor desvıo de Rn mas inestable serael MTP.
Hasta ahora vimos que el caso de ρ = 0,9 podemos mantener las conclusiones del artıculohechas sobre el caso de ρ = 0,4. Pero en el caso del desvıo de Rn veremos algunas diferenciaspor lo que vamos a incluir nuestras reproducciones de las figuras originales de [GGQY07].Como en las anteriores, en los paneles derechos graficamos las figuras obtenidas con el mismoρ bajo las condiciones objetivo.
En el panel izquierdo de la figura 10 reprodujimos la figura 2.C de [GGQY07] y en el de lafigura 11 reprodujimos la figura 2.D. Podemos estar de acuerdo con los autores en que a partirde Log FDR estimado de -3 los metodos divergen pero los autores van mas alla afirmandoque:
A very interesting observation is that the standard deviation of the total numberof rejections for both procedures has a minimum when considered as a functionof the corresponding threshold parameter (Figures 2C and 2D). The minimum isattained when the mean number of rejections becomes close to the total numberof true alternative hypotheses. This is not an entirely unexpected phenomenonif one thinks loosely of testing outcomes as a Bernoulli trial. What is important,however, is that the position of this minimum does not change much in the presenceof correlations.
El subrayado es nuestro. Podemos contrastar la afirmacion de que el mınimo no cambiaexaminando los paneles derechos de la figuras 10 y 11. En esta ultima notamos que las cur-vas debidas a Bonferroni no tienen un solo mınimo. Tambien observamos que el metodo deBenjamini-Hochberg tiene un desvıo mas alto en todo el rango del error FDR examinado.
Volviendo a nuestro objetivo principal de comparar bajo ρ = 0,9, veamos la figura 12. Eneste caso el metodo de Benjamini-Hochberg es mucho mas inestable que el de Bonferroni entodo el rango de error FDR y el mınimo si existe, es menor a exp(−6).

5.2 Comparaciones bajo condiciones mas realistas 65
−6 −5 −4 −3 −2 −1
05
10
15
rho 0 M 1255 K 500 L 280
Log FDR estimado
De
svío
R
BFBH
−6 −5 −4 −3 −2 −1
05
01
00
15
0
rho 0 M 12558 K 5000 L 2500
Log FDR estimado
De
svío
R
BFBH
Figura 10: Comparacion de los desvıos de todos los positivos ecualizando por FDRpara ρ = 0. Comparamos el desvıo de los positivos o rechazados Rn ecualizandopor el error FDR. El primer panel simula los resultados obtenidos para 4 semillasiniciales (1,2,101 y 102) usando el algoritmo GGQY en las condiciones del artıculo.El segundo panel muestra los resultados obtenidos para las mismas semillas con elnuevo algoritmo en las condiciones objetivo. La lınea solida y negra denota el metodode Bonferroni mientras que la punteada y azul, el metodo de Benjamini-Hochberg.

5.2 Comparaciones bajo condiciones mas realistas 66
−6 −5 −4 −3 −2 −1
05
01
00
15
02
00
rho 0.4 M 1255 K 500 L 280
Log FDR estimado
De
svío
R
BFBH
−6 −5 −4 −3 −2 −1
05
00
10
00
15
00
20
00
rho 0.4 M 12558 K 5000 L 2500
Log FDR estimado
De
svío
R
BFBH
Figura 11: Comparacion de los desvıos de todos los positivos ecualizando por FDRpara ρ = 0,4. Comparamos el desvıo de los positivos o rechazados Rn ecualizandopor el error FDR. El primer panel simula los resultados obtenidos para 4 semillasiniciales (1,2,101 y 102) usando el algoritmo GGQY en las condiciones del artıculo.El segundo panel muestra los resultados obtenidos para las mismas semillas con elnuevo algoritmo en las condiciones objetivo. La lınea solida y negra denota el metodode Bonferroni mientras que la punteada y azul, el metodo de Benjamini-Hochberg.

5.2 Comparaciones bajo condiciones mas realistas 67
−6 −5 −4 −3 −2 −1
05
01
00
15
02
00
rho 0.9 M 1255 K 500 L 280
Log FDR estimado
De
svío
R
BFBH
−6 −5 −4 −3 −2 −1
05
00
10
00
15
00
20
00
rho 0.9 M 12558 K 5000 L 2500
Log FDR estimado
De
svío
R
BFBH
Figura 12: Comparacion de los desvıos de todos los positivos ecualizando por FDRpara ρ = 0,9. Comparamos el desvıo de los positivos o rechazados Rn ecualizandopor el error FDR. El primer panel simula los resultados obtenidos para 4 semillasiniciales (1,2,101 y 102) usando el algoritmo GGQY en las condiciones del artıculo.El segundo panel muestra los resultados obtenidos para las mismas semillas con elnuevo algoritmo en las condiciones objetivo. La lınea solida y negra denota el metodode Bonferroni mientras que la punteada y azul, el metodo de Benjamini-Hochberg.

6. Conclusiones 68
6. Conclusiones
La principal conclusion de nuestro trabajo es que verificamos las conclusiones generales del[GGQY07] bajo condiciones mas realistas. En particular, mostramos que el procedimiento deBonferroni es mas estable en el numero total de descubrimientos y en el numero de verdaderosdescubrimientos que el de Benjamini-Hochberg cuando se comparan los metodos ecualizandosus errores.
Tambien mostramos que una conjetura acerca del mınimo en las curvas que representanel desvıo en el numero total de descubrimientos no se cumple al inspeccionarlas con mayorprecision.
Realizar estas nuevas comparaciones hubiera requerido muchos dıas de proceso de un PCsi no se hubiera hecho ninguna mejora al algoritmo planteado en [GGQY07] y al metodode Benjamini-Hochberg implementado en el la librerıa multtest. Por lo que las mejorasintroducidas al algoritmo original representan un aporte significativo. Las podemos resumiren:
Modificamos el algoritmo para hacerlo mas eficiente en tiempo, lo que nos permitio unmuestreo mucho mas preciso.
Definimos con precision el concepto de“ecualizacion”de errores y dimos un marco teoricoadecuado, lo que nos permitio mejorar la interpolacion.
La mejora en grillas fue otro ingrediente que permitio incrementar la precision de manerauniforme en toda la region explorada.
A medida que avanza la tecnologıa aumenta el poder de computo y a la par tambien aumentala cantidad de datos a analizar. Las nuevas tecnologıas hacen prever que el numero de hipotesissea del orden de 105 y 106 por lo que la eficiencia en los algoritmos de muestreo no es un temamenor.
6.1. Trabajo futuro
Es de nuestro interes lograr una difusion de nuestro trabajo en la comunidad de investiga-dores por lo que cuidamos en darle a los lectores el codigo fuente en R y una introduccion almismo para que puedan investigar rapidamente en este tema. Los objetos mr* y er* construi-dos tienen muchas mas medidas que las usadas en esta tesis y en el trabajo original [GGQY07].En particular, se almacenan las medianas y otros cuantiles de la distribuciones de Vn, Sn, Rny de la proporcion Vn/Rn. Esto posibilita por ejemplo el trabajo interesante de medir losresultados de ecualizar bajo otras medidas de error como el error mPFER (ref. (2.28)) y elerror TPPFP (ref. (2.29)).
Estas medidas alternativas tienen mayor interes en modelos que no asumen normalidaden la distribucion de la funcion generadora de datos y en modelos donde las estructuras dedependencia son mas complejas que el relativamente simple modelo de dependencia asumidoen esta tesis (ref. (3.1)). Por lo que tambien una vıa interesante de investigacion serıa adoptarun modelo de dependencia mas complejo que imite las complejas interacciones que sucedenen las redes de genes.
En el analisis de microarreglos muchas veces se practican transformaciones para eliminarsesgos debido a errores asociados a distintas fuentes (puede haber sesgo asociado al lugar del

6.1 Trabajo futuro 69
spot m, correspondiente a la hipotesis m por ejemplo). Estas transformaciones hacen mascomplejo el modelo y habrıa que tenerlas en cuanta para hacer mas realistas las simulaciones.
Por ultimo, el codigo es facilmente adaptable para incorporar otros MTP’s y ası poderrealizar nuevas comparaciones ecualizadas.

A. Librerıa de funciones 70
A. Librerıa de funciones
A continuacion publicaremos el codigo fuente de la librerıa de funciones que implementanel nuevo algoritmo y permiten reproducir los resultados de esta tesis. Para que el lector ejecuteel codigo solo debe cambiar las rutas de acceso en las variables kPath y kFile. De aquı enadelante se asumira que la librerıa se ha ejecutado mediante por ejemplo el comando:
> load(file=paste(kPath, kFile, sep=""))
A continuacion publicamos el codigo fuente de la librerıa con el codigo comentado eningles.
> ##### Library of functions #####
>
> ### The pathnames and filenames should be changed
> kPath <- "D:/Uba/Test multiples/Tesis/v6/"
> kFile <- "Functions and constants.v6.Robj"
> ### All functions of this script can be loaded by
> # load(file=paste(kPath, kFile, sep=""))
>
> # Constants
> kGenes <- 1255
> kDE <- 125
> kDEl <- 1:kDE
> kSamp <- 86
> # True null hypothesis. "Wild type"
> kSampW <- 1:43
> # False null hypothesis. "Target type"
> kSampT <- 44:86
> kDist <- "Normal"
> kMeanDE <- 1
> # Not often changed variables
> kSeed <- 101
> kGridSize <- 2500 #Itt’s assumed that itt’s less than kGenes*kRep
> kRep <- 250
> base.par <- function() list(Genes=kGenes, DE=kDE, DEl=kDEl,
Samp=kSamp, SampW=kSampW, SampT=kSampT,
Dist=kDist, MeanDE=kMeanDE,
Seed=kSeed, GridSize=kGridSize, Rep=kRep)
> # Variables
> rho <- 0
> mtp.id <- "BF"
> # rhof(0.9) -> ".9"
> rhof <- function(rho) substring(format(rho, nsmall=1),2)
> # Derived variables
> RV.file <- function(rho)
paste(kPath, "RV",".RH",rhof(rho),".RP",kRep,
".GR",kGridSize,".Robj",sep="")

A. Librerıa de funciones 71
> ER.file <- function(rho)
paste(kPath, "ErrorRates",".RH",rhof(rho),
".RP",kRep,".GR",kGridSize,".Robj",sep="")
> # Statistics functions
> # t.test that assumes equal variance
> # Its more efficient for vectorized calculations than the usual t.test()
> t.test.v <- function(X,cols1,cols2) {
n1 <- length(cols1)
n2 <- length(cols2)
X1 <- X[,cols1]
X2 <- X[,cols2]
X1.m <- rowMeans(X1)
X2.m <- rowMeans(X2)
S2p <- (rowSums((X1-X1.m)*(X1-X1.m))
+ rowSums((X2-X2.m)*(X2-X2.m)))/(n1+n2-2)
t <- (X1.m-X2.m)/sqrt(S2p*(1/n1+1/n2))
res <- 2*pt(abs(t), df=n1+n2-2, lower.tail=F)
}
> means <- function(x) {
res <- sapply(c(0,.05,.1,.25), function(y) mean(x, trim=y))
names(res) <- list("0%","5%", "10%", "25%")
res
}
> sumariz <- function(x) {
res <- c(Mean=means(x),Std=sd(x), Mad=mad(x),
Q=quantile(x, probs=c(.25,.5,.75,.95,.99)))
names(res)[1] <- "Mean" # I change "Mean.0%" by "Mean"
res
}
> ##### Main functions #####
> # X is big a matrix of log-expressions of all genes across samples and replicates
> # P is a vector of raw p-values with the correlation structure determined by kRho
> # and KDEl
> rawPValues <- function(rho=rho, par=base.par()) {
P <- vector(length=par$Genes)
A <- rnorm(par$Samp)
X <- rnorm(par$Genes*par$Samp)
dim(X) <- c(par$Genes,par$Samp)
X[par$DEl, par$SampT] <- X[par$DEl, par$SampT] +
par$MeanDE
Y <- sqrt(rho)*rep(1,par$Genes)%*%t(A) +
sqrt(1-rho)*X
P <- t.test.v(Y,par$SampW,par$SampT)
P
}
> # pbf contains the list of p-values adjusted by Bonferroni
> # proceduce.

A. Librerıa de funciones 72
> # pbh contains the list of p-values adjusted by Benjamini-
> # Hochberg procedure.
> adjustedPValues <- function(rp, par=base.par()) {
de <- vector(mode="logical", length=par$Genes)
pbf <- pbh <- vector(mode="numeric", length=par$Genes)
o <- order(rp)
de <- (o %in% par$DEl)
praw <- rp[o]
pbf <- praw*par$Genes
pbh <- rev(cummin(rev(pmin(praw*(par$Genes/(1:par$Genes))
,1))))
ap <- list(de=de, pbf=pbf, pbh=pbh)
}
> pvals.gridStd <- function(mtp.id, gridSize=base.par()$GridSize)
switch(mtp.id,
BH = grid <- exp(seq(from=-8, to=0, length.out=gridSize)),
BF = grid <- exp(seq(from=-6, to=6, length.out=gridSize)),
stop("BF (Bonferroni) and BH (Benjamini-Hochberg) are the only MTPs implemented"))
> rv <- function(ap, mtp.id, grid=pvals.gridStd(mtp.id)) {
switch(mtp.id,
BH = pval <- ap$pbh,
BF = pval <- ap$pbf,
stop("BF (Bonferroni) and BH (Benjamini-Hochberg) are the only MTPs implemented"))
r <- v <- vector(mode="integer", length=length(grid))
n <- length(pval)
rx <- as.integer(cumsum(rep(1,length=n)))
vx <- as.integer(cumsum(!ap$de))
gridSize <- length(grid)
i <- 1
j <- 1
while (pval[1] > grid[j] && j <= gridSize) {
r[j] <- v[j] <- as.integer(0)
j <- j + 1
}
while (i+1 <= n && j <= gridSize) {
# Invariant: pval[i] <= grid[j]
while (pval[i+1] > grid[j] && j <= gridSize) {
r[j] <- rx[i]
v[j] <- vx[i]
j <- j + 1
}
i <- i + 1
}
# Only applies in the case of i==n
# in that case we ran out of pvalues and the
# the statistics equals the last calculated

A. Librerıa de funciones 73
while (j <=gridSize) {
r[j] <- rx[i]
v[j] <- vx[i]
j <- j + 1
}
list(r=r, v=v)
}
> # The big object is
> RV <- function(rho, par=base.par(),
grid.bf=pvals.gridStd("BF", gridSize=par$GridSize),
grid.bh=pvals.gridStd("BH", gridSize=par$GridSize)) {
R.bf <- vector(mode="integer",
length=length(grid.bf)*par$Rep)
dim(R.bf) <- c(length(grid.bf),par$Rep)
V.bf <- R.bf
R.bh <- vector(mode="integer",
length=length(grid.bh)*par$Rep)
dim(R.bh) <- c(length(grid.bh),par$Rep)
V.bh <- R.bh
set.seed(par$Seed)
for (k in 1:par$Rep) {
ap <- adjustedPValues(rawPValues(rho, par), par)
res <- rv(ap, "BF", grid.bf)
R.bf[,k] <- res$r
V.bf[,k] <- res$v
res <- rv(ap, "BH", grid.bh)
R.bh[,k] <- res$r
V.bh[,k] <- res$v
}
list(rho=rho, base.par=par,
bf=list(pvals=grid.bf, R=R.bf, V=V.bf),
bh=list(pvals=grid.bh, R=R.bh, V=V.bh))
}
> meanErrors <- function(RV) {
meanErrors.mtp <- function(l) {
R <- l$R
V <- l$V
S <- R - V
FDP <- V/pmax(R,1)
list(r.mean=rowMeans(R),
v.mean=rowMeans(V),
s.mean=rowMeans(S),
fdp.mean=rowMeans(FDP),
pvals=l$pvals)
}
list(base.par=RV$base.par, rho=RV$rho,

B. Reproduccion de resultados 74
bf=meanErrors.mtp(RV$bf),
bh=meanErrors.mtp(RV$bh))
}
> errorRates <- function(RV) {
errorRates.mtp <- function(l) {
R <- l$R
V <- l$V
S <- R - V
FDP <- V/pmax(R,1)
R.smz <- apply(R, 1, sumariz)
V.smz <- apply(V, 1, sumariz)
S.smz <- apply(S, 1, sumariz)
FDP.smz <- apply(FDP, 1, sumariz)
list(r=R.smz, v=V.smz, s=S.smz, fdp=FDP.smz,
pvals=l$pvals)
}
list(base.par=RV$base.par, rho=RV$rho,
bf=errorRates.mtp(RV$bf),
bh=errorRates.mtp(RV$bh))
}
> save(kGenes, kDE, kDEl, kSamp, kSampW, kSampT, kDist, kMeanDE, kSeed, kGridSize, kRep, base.par, rho, mtp.id, rhof, RV.file, ER.file , t.test.v, means, sumariz, rawPValues, adjustedPValues, pvals.gridStd, rv, RV, meanErrors, errorRates,
file=paste(kPath, kFile, sep=""), compress=T)
B. Reproduccion de resultados
En esta seccion cumplimos el compromiso dado en la seccion 1.6 de dar los instrumentosadecuados al lector para que reproduzca cualquier cifra, tabla o figura de esta tesis.
B.1. Comparacion de versiones del test t
En la seccion 3.2.2 se afirmo que nuestra funcion t.test.v optimiza la funcion estandar deR y la implementada en la librerıa multtest. Aquı presentamos las pruebas que son totalmentereproducibles para sostener dicha afirmacion.
> # Corro primero mi librerıa
> #source(file="D:/Uba/Test multiples/Tesis/v6/Functions library v6.R")
> load(file="D:/Uba/Test multiples/Tesis/v6/Functions and constants.v6.Robj")
Primero construımos la funcion rawPValues.st que genera los p-valores no ajustadosusando el test t estandar de R .
> rawPValues.st <- function(rho=rho, par=base.par()) {
P <- vector(length=par$Genes)
A <- rnorm(par$Samp)
X <- rnorm(par$Genes*par$Samp)
dim(X) <- c(par$Genes,par$Samp)

B.1 Comparacion de versiones del test t 75
X[par$DEl, par$SampT] <- X[par$DEl, par$SampT] +
par$MeanDE
Y <- sqrt(rho)*rep(1,par$Genes)%*%t(A) +
sqrt(1-rho)*X
for (i in 1:kGenes) { P[i] <-
t.test(Y[i,par$SampW],Y[i,par$SampT],var.equal=TRUE)$p.value }
P
}
Una mejora significativa se consiguio usando la version del test t implementada en lalibrerıa multtest mediante la funcion mt.teststat, que es el estandar en el ambito de testmultiples (ref. 1.4).
> library(multtest)
> rawPValues.mt <- function(rho=rho, par=base.par()) {
P <- vector(length=par$Genes)
A <- rnorm(par$Samp)
X <- rnorm(par$Genes*par$Samp)
dim(X) <- c(par$Genes,par$Samp)
X[par$DEl, par$SampT] <- X[par$DEl, par$SampT] +
par$MeanDE
Y <- sqrt(rho)*rep(1,par$Genes)%*%t(A) +
sqrt(1-rho)*X
cl <- rbind(cbind(par$SampW,0),cbind(par$SampT,1))[,2]
P <- 2*pt(abs(mt.teststat(Y, cl,test="t.equalvar")),
df=par$Samp-2, lower.tail=F)
P
}
Para decidir cuales de las tres versiones fue mas eficiente primero se compararon lostiempos en que tardo cada una en una situacion realista. Como la medicion de tiempos noes muy confiable generamos varias replicas y elegimos las medianas de las distribucionesobtenidas.
> ### Comparacion de resultados de test t
> set.seed(kSeed)
> reps <- 10
> M <- 12558
> n <- 86
> cl <- c(rep(0,n/2), rep(1,n/2))
> t2 <- t1 <- t0 <- rep(0.0, reps)
> rp.st <- rp.mt <- rp <- matrix(0,M,reps)
> for (k in 1:reps) {
X <- runif(M*n)
dim(X) <- c(M,n)
t0[k] <- system.time({
rp[,k] <- t.test.v(X,1:(n/2),(n/2+1):n)
})[1]

B.1 Comparacion de versiones del test t 76
t1[k] <- system.time({
rp.mt[,k] <- 2*pt(abs(mt.teststat(X,
cl,test="t.equalvar")), df=n-2,
lower.tail=F)
})[1]
t2[k] <- system.time({
for (i in 1:M) { rp.st[i,k] <-
t.test(X[i,1:(n/2)],X[i,(n/2+1):n],var.equal=TRUE)$p.value }
})[1]
}
> max(abs(rp-rp.mt))
[1] 1.554312e-15
> max(abs(rp-rp.st))
[1] 3.330669e-16
Verificamos que las implementaciones son correctas midiendo que el error absoluto en losM ∗ reps = 125580 p-valores no supera 1e-15.
Para compararar las performances ejecutamos:
> summary(t0)
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.4600 0.4700 0.4800 0.5460 0.4875 1.1400
> summary(t1)
Min. 1st Qu. Median Mean 3rd Qu. Max.
1.230 1.330 1.340 1.345 1.380 1.420
> summary(t2)
Min. 1st Qu. Median Mean 3rd Qu. Max.
54.12 54.62 54.89 54.91 55.13 56.04
> mean(t2)/mean(t0)
[1] 100.5604
> mean(t1)/mean(t0)
[1] 2.46337
> median(t2)/median(t0)
[1] 114.3438
> median(t1)/median(t0)
[1] 2.791667
Entonces, la version vectorizada nuestra es 114,344 veces mas rapida que la version conel test t estandar y 2,792 veces mas rapida que la obtenida con el test t implementado en elpaquete multtest, lo cual es notable ya que la funcion mt.multstat esta optimizada en C.Vemos aquı la potencia del lenguaje R si explotamos sus cualidades.

B.2 Impacto en generacion de P-Valores no ajustados 77
B.2. Impacto en generacion de P-Valores no ajustados
Para medir la verdadera ganancia en tiempos de la version propia frente a la de multtest
escribimos una version de la funcion rawPValues que usa la funcion de esta librerıa, aumen-tamos las replicas, verificamos una vez mas que el error absoluto entre las dos versiones esdespreciable y comparamos los tiempos.
> kGenes <- M <- 12558
> rho <- .9
> reps <- 50
> n <- 86
> t1 <- t0 <- rep(0.0, reps)
> rp.mt <- rp <- matrix(0,M,reps)
> for (k in 1:reps) {
set.seed(kSeed)
t0[k] <- system.time(rp[,k] <- rawPValues(rho, base.par()))[1]
set.seed(kSeed)
t1[k] <- system.time(rp.mt[,k] <- rawPValues.mt(rho, base.par()))[1]
}
> max(abs(rp-rp.mt))
[1] 6.661338e-16
Revisandos los tiempos
> summary(t0)
Min. 1st Qu. Median Mean 3rd Qu. Max.
2.220 2.285 2.340 2.356 2.377 2.860
> summary(t1)
Min. 1st Qu. Median Mean 3rd Qu. Max.
2.970 3.130 3.170 3.205 3.250 3.670
> mean(t1)/mean(t0)
[1] 1.360411
> median(t1)/median(t0)
[1] 1.354701
Entonces, usando nuestra version vectorizada tardamos aproximadamente 2,34 segundos,mientras que si usamos la version que usa multtest tardamos 3,17 segundos. Esto representauna mejora de 1,355 veces.
B.3. Comparacion de versiones del Procedimiento de Benjamini-Hochberg
En la seccion 3.3 se afirmo que nuestra version del ajuste es competitiva frente a la provistapor la librerıa multtest. Para probarlo procederemos como en la seccion B.1 donde medimosel tiempo en el caso que nos interesa de M = 12558.
> source(file="D:/Uba/Test multiples/Tesis/v6/Functions library v6.R")
> library(multtest)

B.4 Codigo para Medicion de tiempos del Algoritmo GGQY 78
Aquı probamos mediante la funcion identical que las dos versiones coinciden exacta-mente.
> set.seed(kSeed)
> reps <- 50
> M <- 12558
> t1 <- t0 <- rep(0.0, reps)
> ap.mt <- ap <- matrix(0,M,reps)
> for (i in 1:reps) {
rp <- runif(M)
t0[i] <- system.time({
o <- order(rp)
praw <- rp[o]
ap[,i] <- rev(cummin(rev(pmin(praw*
(M/(1:M)),1))))})[1]
t1[i] <- system.time({ap.mt[,i] <-
mt.rawp2adjp(rp, proc="BH")$adjp[,"BH"]})[1]
}
> identical(ap,ap.mt)
[1] TRUE
Medimos los tiempos
> summary(t0)
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.0300 0.0300 0.0300 0.0378 0.0400 0.1600
> summary(t1)
Min. 1st Qu. Median Mean 3rd Qu. Max.
1.170 1.200 1.210 1.215 1.220 1.370
> mean(t1)/mean(t0)
[1] 32.14286
> median(t1)/median(t0)
[1] 40.33333
y observamos que nuestra version tarda aproximadamente 0,03 segundos por replica, mien-tras que si usamos la version que usa multtest tardamos 1,21 segundos. Esto representa unamejora de 40,333 veces, que es importante y tiene un impacto significativo.
B.4. Codigo para Medicion de tiempos del Algoritmo GGQY
Aquı damos el codigo con el cual se midieron los tiempos del algoritmo GGQY. Notarque se estiman los tiempos del Paso 1.1 para el caso de interes de M = 12558, K = 5000 yL = 2500. Se tiene cuidado en evitar los errores por truncamiento debido a la imprecision dela funcion system.time(). El tiempo de ejecucion total fue menor a dos horas.

B.4 Codigo para Medicion de tiempos del Algoritmo GGQY 79
> source(file="D:/Uba/Test multiples/Tesis/v6/Functions library v6.R")
> kPath="D:/Uba/Test multiples/Tesis/v7/"
> kSeed <- 1
> set.seed(kSeed) # Se usa en la funcion RV
> kRho <- 0.4
> kGenes <- 12558
> kDE <- 1256
> kDEl <- 1:kDE
> kRep <- 500
> kGridSize <- 100
> t.rp <- rep(0.0,kRep)
> rp <- matrix(0.0, kGenes, kRep)
> for (k in 1:kRep)
t.rp[k] <- system.time(rp[,k] <- rawPValues(rho=kRho))[1]
> summary(t.rp)
Min. 1st Qu. Median Mean 3rd Qu. Max.
1.070 1.140 1.150 1.155 1.170 1.500
> # Varıa un poco con respecto a \ref{Ap:CompTestT}. Uso dicho numero por consistencia.
> (t.rp.est <- difftime(Sys.time() + 2.34*5000, Sys.time()))
Time difference of 3.25 hours
> t.bf <- rep(0.0, kRep)
> R <- S <- V <- matrix(0.0, kGridSize, kRep)
> # Medicion de Vn y Rn para cada gamma de la grilla gammas
> gammas <- exp(seq(from=-6, to=6, length.out=kGridSize))
> for (k in 1:kRep)
t.bf[k] <- system.time(
for (l in 1:kGridSize) {
R[l,k] <- sum(rp[,k]*kGenes<gammas[l])
S[l,k] <- sum(rp[1:kDE,k]*kGenes<gammas[l])
V[l,k] <- R[l,k] - S[l,k]
})[1]
> # Tiempos x cada replica
> summary(t.bf/kGridSize)
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.001800 0.001900 0.001900 0.001934 0.002000 0.002500
> # Resumo
> t.bf <- median(t.bf)/kGridSize
> # Luego para K=5000 y L=2500 tardarıa
> (t.bf.est <- difftime(Sys.time() + t.bf*5000*2500, Sys.time()))
Time difference of 6.597222 hours
> #### Medicion BH ####
> # Medicion de Vn y Rn para cada beta de la grilla betas
>
> betas <- exp(seq(from=-8, to=0, length.out=kGridSize))
> t.bh <- rep(0.0, kRep)

B.5 Generacion de muestras 80
> for (k in 1:kRep)
t.bh[k] <- system.time(
for (l in 1:kGridSize) {
op <- order(rp[,k])
praw <- rp[op,k]
deo <- (op %in% kDEl)[op]
pbh <- rev(cummin(rev(pmin(praw*(kGenes/(1:kGenes)),1))))
R[l,k] <- sum(pbh<betas[l])
S[l,k] <- sum(pbh[deo]<betas[l])
V[l,k] <- R[l,k] - S[l,k]
})[1]
> # Tiempos x cada replica
> summary(t.bh)/kGridSize
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.02140 0.02180 0.02190 0.02198 0.02210 0.02310
> # Resumo
> t.bh <- median(t.bh)/kGridSize
> # Luego para K=5000 y L=2500 tardarıa
> (t.bh.est <- difftime(Sys.time() + t.bh*5000*2500, Sys.time()))
Time difference of 3.168403 days
> ### Estimacion de tiempos de estadısticos ####
> X <- matrix(1/3, kGridSize, kRep)
> (t.estad <- system.time(rowMeans(X))+
system.time(apply(X,1,summary))+
system.time(apply(X,1,sd)))
user system elapsed
0.52 0.00 0.53
> # < 1s por estadıstico. Es despreciable.
>
> save(t.rp.est, t.bf.est, t.bh.est, t.rp, t.bh, t.bf,
file=paste(kPath, "Tiempos estimados de GGQY.Robj", sep=""),
compress=T)
B.5. Generacion de muestras
Aquı damos el codigo con el cual se generaron las muestras variando los parametros ρ,M , L y K, que en el codigo estan representados por las variables kRho, kGenes, kRep ykGridSize. Ademas variamos las semillas iniciales. Para no forzar ninguna eleccion usamoscuatro semillas naturales: 1,2,101 y 102. Ademas registramos los tiempos de proceso y acu-mulamos los resultados en el data frame laps.all para conveniencia del analisis posterior.
Cada llamada a la funcion gen.error.rates() puede tardar varios minutos para las casosde pocas replicas o algunas horas en los dos primeros casos que corresponden a 5000 replicasy 12558 genes. El proceso completo puede tardar varias horas en una PC con procesador IntelCore i5 y 2Gb de RAM por ejemplo.

B.5 Generacion de muestras 81
> source(file = "D:/Uba/Test multiples/Tesis/v6/Functions library v6.R")
> kPath = "D:/Uba/Test multiples/Tesis/v7/"
> gen.error.rates <- function() {
objs <- c()
laps <- data.frame(Rho = NULL, Rep = NULL,
Genes = NULL, GridSize = NULL, Seed = NULL,
time.RV = NULL, time.mr = NULL, time.er = NULL)
for (rho in kRhos) {
for (kSeed in kSeeds) {
par <- base.par()
par["Seed"] <- kSeed
param.name <- paste(rhof(rho), kRep,
kGenes, kGridSize, kSeed, sep = ".")
rv.name <- paste("rv", param.name,
sep = "")
mr.name <- paste("mr", param.name,
sep = "")
er.name <- paste("er", param.name,
sep = "")
if (kGridSize == 280) {
gridGammas <- c(seq(from = 1/100,
to = 1, by = 1/100), seq(from = 1.1,
to = 10, by = 1/10), seq(from = 11,
to = 100, by = 1))
gridBetas <- gridGammas/(125 +
gridGammas)
time.RV <- system.time(assign(rv.name,
RV(rho = rho, par = par, grid.bf = gridGammas,
grid.bh = gridBetas)))[1]
}
else time.RV <- system.time(assign(rv.name,
RV(rho = rho, par = par)))[1]
time.mr <- system.time(assign(mr.name,
meanErrors(get(rv.name))))[1]
time.er <- system.time(assign(er.name,
errorRates(get(rv.name))))[1]
objs <- c(objs, c(mr.name, er.name))
cat("Se terminaron de generar: ",
c(mr.name, er.name), "\n")
lap <- data.frame(Rho = rho, Rep = kRep,
Genes = kGenes, GridSize = kGridSize,
Seed = kSeed, lap.RV = time.RV,
lap.mr = time.mr, lap.er = time.er)
laps <- rbind(laps, lap)
cat("El valor actual de lap es: \n")

B.5 Generacion de muestras 82
cat(str(lap), "\n")
}
}
save(laps, list = objs, file = kFile, compress = T)
cat("Se grabo con exito el archivo: ", kFile,
"\n")
objs
}
> objs.saved <- c()
> kGenes <- 12558
> kDE <- 1256
> kDEl <- 1:kDE
> kRep <- 5000
> kGridSize <- 2500
> kSeeds <- c(1, 2, 101, 102)
> kRhos <- list(0)
> kFile <- paste(kPath, "Objs.M12558.K5000.L2500.rho.0.Robj",
sep = "")
> objs.saved <- c(objs.saved, gen.error.rates())
> kRhos <- list(0.4)
> kFile <- paste(kPath, "Objs.M12558.K5000.L2500.rho.4.Robj",
sep = "")
> objs.saved <- c(objs.saved, gen.error.rates())
> kRhos <- list(0.9)
> kFile <- paste(kPath, "Objs.M12558.K5000.L2500.rho.9.Robj",
sep = "")
> objs.saved <- c(objs.saved, gen.error.rates())
> kGenes <- 1255
> kDE <- 125
> kDEl <- 1:kDE
> kRep <- 500
> kGridSize <- 280
> kSeeds <- c(1, 2, 101, 102)
> kRhos <- c(0, 0.4, 0.9)
> kFile <- paste(kPath, "Objs.M1255.K500.L280.Robj",
sep = "")
> objs.saved <- c(objs.saved, gen.error.rates())
> kGridSize <- 2500
> kFile <- paste(kPath, "Objs.M1255.K500.L2500.Robj",
sep = "")
> objs.saved <- c(objs.saved, gen.error.rates())
> kRep <- 5000
> kRhos <- list(0)
> kFile <- paste(kPath, "Objs.M1255.K5000.L2500.rho.0.Robj",
sep = "")
> objs.saved <- c(objs.saved, gen.error.rates())
> kRhos <- list(0.4)

B.5 Generacion de muestras 83
> kFile <- paste(kPath, "Objs.M1255.K5000.L2500.rho.4.Robj",
sep = "")
> objs.saved <- c(objs.saved, gen.error.rates())
> kRhos <- list(0.9)
> kFile <- paste(kPath, "Objs.M1255.K5000.L2500.rho.9.Robj",
sep = "")
> objs.saved <- c(objs.saved, gen.error.rates())
> objs.saved
> files.saved <- c("Objs.M12558.K5000.L2500.rho.0.Robj",
"Objs.M12558.K5000.L2500.rho.4.Robj", "Objs.M12558.K5000.L2500.rho.9.Robj",
"Objs.M1255.K500.L280.Robj", "Objs.M1255.K500.L2500.Robj",
"Objs.M1255.K5000.L2500.rho.0.Robj", "Objs.M1255.K5000.L2500.rho.4.Robj",
"Objs.M1255.K5000.L2500.rho.9.Robj")
> laps.all <- NULL
> for (f in files.saved) {
load(file = paste(kPath, f, sep = ""))
laps.all <- rbind(laps.all, laps)
}
> real.name <- function(obj.name) {
par <- get(obj.name)$base.par
r <- rhof(get(obj.name)$rho)
paste("xx", paste(r, par$Rep, par$Genes, par$GridSize,
par$Seed, sep = "."), sep = "")
}
> objs.to.save <- ls(pattern = glob2rx("mr.*"))
> objs.to.save <- c(objs.to.save, ls(pattern = glob2rx("er.*")))
> objs.to.save
> setdiff(objs.to.save, objs.saved)
> setdiff(objs.saved, objs.to.save)
> for (l in objs.to.save) {
cat(rbind(l, real.name(l)), "\n")
stopifnot(substring(l, 3) == substring(real.name(l),
3))
}
> for (l in objs.to.save) {
if (!(substring(l, 3) == substring(real.name(l),
3)))
cat(rbind(l, real.name(l)), "\n")
}
> save(laps.all, list = objs.to.save, file = paste(kPath,
"Objetos er y mr para comp.Robj", sep = ""),
compress = T)
> attach(laps.all)
> sel <- laps.all[Rep == 5000 & Genes == 12558 &
GridSize == 2500, ]
> sel
> summary(cbind(RV = sel$lap.RV, mr = sel$lap.mr,

B.6 Reproduccion de figuras para comparacion de errores 84
er = sel$lap.er, total = sel$lap.RV + sel$lap.mr +
sel$lap.er, RV.y.mr = sel$lap.RV + sel$lap.mr))
> t.nalg.est <- difftime(Sys.time() + median(sel$lap.RV +
sel$lap.mr), Sys.time())
> t.nalg.est
> round(as.numeric(t.nalg.est - 2, units = "mins"),
digits = 2)
B.6. Reproduccion de figuras para comparacion de errores
Aquı damos el codigo para reproducir las figuras que sirvieron para realizar la comparacionde errores.
Para ahorrar tiempo de proceso cargamos los objetos mr* y er* necesarios. Ademas de-finimos las grillas originales del algoritmo GGQY y la funcion plotGrids() que servira parasimplificar en la seccion 5.2 la generacion de figuras que grafican las curvas superpuestas delas 4 semillas.
> load(file="D:/Uba/Test multiples/Tesis/v7/Objetos er y mr para comp.Robj")
> gridGammas <-c(seq(from=1/100, to=1, by=1/100),
seq(from=1.1, to=10, by=1/10),
seq(from=11, to=100, by=1))
> kDE <- 125
> gridBetas <- gridGammas/(kDE + gridGammas)
> plotGrids <- function(grid, subtitle,
varx = "fdp[\"Mean\",]",
vary, ylab, ylim
) {
plot(x=seq(-6,-1), sub = subtitle, type="n",
xlim=c(-6,-1), xlab="Log FDR estimado", cex.sub=.6,
ylim=ylim, ylab=ylab)
for (sem in c(1,2,101,102)) {
xaxis.name <- paste(grid, ".", sem, "$bh$", varx, sep="")
xaxis <- eval(parse(text=xaxis.name))
yaxis.name <- paste(grid, ".",sem, "$bh$", vary, sep="")
yaxis <- eval(parse(text=yaxis.name))
lines(x=log(xaxis), y=yaxis, lty=2, col="blue")
}
for (sem in c(1,2,101,102)) {
xaxis.name <- paste(grid, ".", sem, "$bf$", varx, sep="")
xaxis <- eval(parse(text=xaxis.name))
yaxis.name <- paste(grid, ".",sem, "$bf$", vary, sep="")
yaxis <- eval(parse(text=yaxis.name))
lines(x=log(xaxis), y=yaxis)
}
legend("topleft", c("BF","BH"), lty=c(1,2), cex=.6,
col=c("black","blue"), bty="n")

B.6 Reproduccion de figuras para comparacion de errores 85
}
B.6.1. Codigo para la figura 1
> par(mfrow=c(2,2))
> plot(x=mr.4.500.1255.280.1$bf$pvals, xlab="PFER Nominal",
y=mr.4.500.1255.280.1$bf$fdp.mean, type="l",
ylab="FDR estimado")
> plot(x=mr.4.500.1255.280.1$bf$pvals, xlab="PFER Nominal",
y=mr.4.500.1255.280.1$bf$s.mean, type="l",
ylab="AvgPWR estimada")
> plot(x=mr.4.500.1255.280.1$bh$pvals, xlab="FDR Nominal",
y=mr.4.500.1255.280.1$bh$fdp.mean, type="l",
ylab="FDR estimado")
> plot(x=mr.4.500.1255.280.1$bh$pvals, xlab="FDR Nominal",
y=mr.4.500.1255.280.1$bf$s.mean, type="l",
ylab="AvgPWR estimada")
> par(mfrow=c(1,1))
B.6.2. Codigo para la figura 2
> # PRIMERA FIGURA PARA EXPLICAR MEJORA POR REPLICAS
> # Primero muestro que las de 5000 se encuentran dentro de
> # las bandas de confianza
> plot(x=mr.4.5000.1255.2500.2$bf$pvals,
y=mr.4.5000.1255.2500.2$bf$fdp.mean,
xlim=c(0,3),
ylim=c(0, 0.04),
type="l", xlab="PFER Nominal", ylab="FDR estimado")
> lines(x=mr.4.5000.1255.2500.2$bf$pvals, lty=2, col=2,
y=mr.4.5000.1255.2500.2$bf$fdp.mean +
2/sqrt(5000)*er.4.5000.1255.2500.2$bf$fdp["Std",])
> lines(x=mr.4.5000.1255.2500.2$bf$pvals, lty=2, col=2,
y=mr.4.5000.1255.2500.2$bf$fdp.mean -
2/sqrt(5000)*er.4.5000.1255.2500.2$bf$fdp["Std",])
> lines(x=mr.4.5000.1255.2500.1$bf$pvals, col=3, lty=3,
y=mr.4.5000.1255.2500.1$bf$fdp.mean)
> lines(x=mr.4.5000.1255.2500.101$bf$pvals, col=4, lty=4,
y=mr.4.5000.1255.2500.101$bf$fdp.mean)
> lines(x=mr.4.5000.1255.2500.102$bf$pvals, col=5, lty=5,
y=mr.4.5000.1255.2500.102$bf$fdp.mean)
> legend("bottomright", cex=.8, bty="n",
c("S 2", "S 2 + 2 Sd", "S 2 - 2 Sd",
"S 1", "S 101", "S 102" ),
col=c(1,2,2,3,4,5), lty=c(1,2,2,3,4,5))

B.6 Reproduccion de figuras para comparacion de errores 86
B.6.3. Codigo para la figura 3
> # En la segunda figura comparo con la de 500 replicas
> plot(x=mr.4.5000.1255.2500.2$bf$pvals,
y=mr.4.5000.1255.2500.2$bf$fdp.mean,
xlim=c(0,3),
ylim=c(0, 0.04),
type="l", xlab="PFER Nominal", ylab="FDR estimado")
> lines(x=mr.4.5000.1255.2500.2$bf$pvals, lty=2, col=2,
y=mr.4.5000.1255.2500.2$bf$fdp.mean +
2/sqrt(5000)*er.4.5000.1255.2500.2$bf$fdp["Std",])
> lines(x=mr.4.5000.1255.2500.2$bf$pvals, lty=2, col=2,
y=mr.4.5000.1255.2500.2$bf$fdp.mean -
2/sqrt(5000)*er.4.5000.1255.2500.2$bf$fdp["Std",])
> lines(x=mr.4.500.1255.2500.1$bf$pvals, col=3, lty=3,
y=mr.4.500.1255.2500.1$bf$fdp.mean)
> lines(x=mr.4.500.1255.2500.101$bf$pvals, col=4, lty=4,
y=mr.4.500.1255.2500.101$bf$fdp.mean)
> lines(x=mr.4.500.1255.2500.102$bf$pvals, col=5, lty=5,
y=mr.4.500.1255.2500.102$bf$fdp.mean)
> lines(x=mr.4.500.1255.2500.2$bf$pvals, col=6, lty=6,
y=mr.4.500.1255.2500.2$bf$fdp.mean)
> legend("bottomright", cex=.8, bty="n",
c("S 2", "S 2 + 2 Sd", "S 2 - 2 Sd",
"S 1 K 500", "S 101 K 500",
"S 102 K 500","S 2 K 500" ),
col=c(1,2,2,3,4,5,6), lty=c(1,2,2,3,4,5,6))
B.6.4. Codigo para la figura 4
> # En la tercera figura hacemos una lupa en el rango
> # 0.4 y 0.65
> par(mfrow=c(1,2))
> plot(x=mr.4.500.1255.280.2$bh$pvals,
y=mr.4.500.1255.280.2$bh$fdp.mean,
xlim=c(0.022,0.029),
ylim=c(0.017, 0.0212),
type="l", xlab="FDR Nominal",
ylab="FDR estimado")
> lines(x=mr.4.500.1255.280.2$bh$pvals,
y=mr.4.500.1255.280.2$bh$fdp.mean,
type="b", pch=1, lty=2)
> text(x=mr.4.500.1255.280.2$bh$pvals[119:127],
y=mr.4.500.1255.280.2$bh$fdp.mean[119:127],
adj=c(1.5,0), cex=.75,
labels=expression(beta[119], beta[120], beta[121]

B.6 Reproduccion de figuras para comparacion de errores 87
, beta[122], beta[123], beta[124], beta[125]
, beta[126], beta[127]))
> for (i in 119:127)
abline(h=mr.4.500.1255.280.2$bh$fdp.mean[i],lty=2)
> plot(x=mr.4.5000.1255.2500.2$bf$pvals,
y=mr.4.5000.1255.2500.2$bf$fdp.mean,
xlim=c(0.4,0.65),
ylim=c(0.017, 0.0212),
type="l", yaxt="n",
xlab="PFER Nominal", ylab="FDR estimado")
> lines(x=mr.4.5000.1255.2500.2$bf$pvals, lty=2, col=2,
y=mr.4.5000.1255.2500.2$bf$fdp.mean +
2/sqrt(5000)*er.4.5000.1255.2500.2$bf$fdp["Std",])
> lines(x=mr.4.5000.1255.2500.2$bf$pvals, lty=2, col=2,
y=mr.4.5000.1255.2500.2$bf$fdp.mean -
2/sqrt(5000)*er.4.5000.1255.2500.2$bf$fdp["Std",])
> lines(x=mr.4.500.1255.280.2$bf$pvals, col=6, lty=6,
y=mr.4.500.1255.280.2$bf$fdp.mean)
> axis(side=2, at=mr.4.500.1255.280.2$bh$fdp.mean[119:127],
tck=1, lty=2, lwd=.5, las = 1, cex=.5,
labels=expression(beta[119], beta[120], beta[121]
, beta[122], beta[123], beta[124], beta[125]
, beta[126], beta[127]))
> par(mfrow=c(1,1))
B.6.5. Codigo para la figura 5
> oldpar <- par(no.readonly = TRUE)
> par(mar = c(5.1, 4.1, 5.1, 2.1))
> plot(x=mr.4.5000.1255.2500.2$bf$pvals,
y=mr.4.5000.1255.2500.2$bf$fdp.mean,
xlim=c(0.4,0.65),
ylim=c(0.017, 0.0212),
type="l", yaxt="n",
xlab="PFER Nominal", ylab="FDR estimado")
> lines(x=mr.4.5000.1255.2500.2$bf$pvals, lty=2, col=2,
y=mr.4.5000.1255.2500.2$bf$fdp.mean +
2/sqrt(5000)*er.4.5000.1255.2500.2$bf$fdp["Std",])
> lines(x=mr.4.5000.1255.2500.2$bf$pvals, lty=2, col=2,
y=mr.4.5000.1255.2500.2$bf$fdp.mean -
2/sqrt(5000)*er.4.5000.1255.2500.2$bf$fdp["Std",])
> lines(x=mr.4.500.1255.280.2$bf$pvals, col=6, lty=6,
y=mr.4.500.1255.280.2$bf$fdp.mean)
> axis(side=2, at=mr.4.500.1255.280.2$bh$fdp.mean,
labels=F, tck=1, lty=2, lwd=.5)
> lines(x=mr.4.500.1255.280.2$bf$pvals,

B.6 Reproduccion de figuras para comparacion de errores 88
y=mr.4.500.1255.280.2$bf$fdp.mean,
type="b", pch=4, lty=2)
> axis(side=2, at=mr.4.500.1255.280.2$bh$fdp.mean[
c(119,122,123)],
labels=expression(beta[119],beta[122],beta[123]),
las=1, cex=.5 )
> axis(side=3, at=mr.4.500.1255.280.2$bf$pvals[
c(40,47,51,52)],
labels=expression(gamma[40], gamma[47],
gamma[51],gamma[52]),
las=1, cex=.5, tck=1, lty=2, lwd=.5 )
> par(oldpar)
B.6.6. Codigo para la figura 6
> par(mfrow=c(1,2))
> plot(x=log(mr.4.500.1255.280.1$bh$pvals),
y=mr.4.500.1255.280.1$bh$fdp.mean,
col="black", type="p", pch=".",
ylim=c(0,0.05), xlim=c(-8,0),
xlab = "Log FDR Nominal",
ylab = "FDR Estimado",
sub = "Grilla Original (L=280)"
)
> plot(x=log(mr.4.500.1255.2500.1$bh$pvals),
y=mr.4.500.1255.2500.1$bh$fdp.mean,
col="black", type="p", pch=".",
ylim=c(0,0.05), xlim=c(-8,0),
xlab = "Log FDR Nominal",
ylab = "FDR Estimado",
sub = "Grilla Nueva (L=2500)"
)
> par(mfrow=c(1,1))
B.6.7. Codigo para la figura 7
> par(mfrow=c(1,2))
> plot(x=log(mr.4.500.1255.280.1$bf$pvals),
y=mr.4.500.1255.280.1$bf$fdp.mean,
col="black", type="p", pch=".",
ylim=c(0,0.05), xlim=c(-6,6),
xlab = "Log PFER Nominal",
ylab = "FDR Estimado",
sub = "Grilla Original (L=280)"
)

B.6 Reproduccion de figuras para comparacion de errores 89
> plot(x=log(mr.4.500.1255.2500.1$bf$pvals),
y=mr.4.500.1255.2500.1$bf$fdp.mean,
col="black", type="p", pch=".",
ylim=c(0,0.05), xlim=c(-6,6),
xlab = "Log PFER Nominal",
ylab = "FDR Estimado",
sub = "Grilla Nueva (L=2500)"
)
> par(mfrow=c(1,1))
B.6.8. Codigo para la figura 8
> par(mfrow=c(1,2))
> plotGrids(grid = "er.9.500.1255.280",
subtitle = "rho 0.9 M 1255 K 500 L 280",
varx = "fdp[\"Mean\",]", vary = "s[\"Mean\",]",
ylab = "Media S", ylim = c(0,50))
> plotGrids(grid = "er.9.5000.12558.2500",
subtitle = "rho 0.9 M 12558 K 5000 L 2500",
varx = "fdp[\"Mean\",]",
vary = "s[\"Mean\",]",
ylab = "Media S", ylim = c(0,500))
> par(mfrow=c(1,1))
B.6.9. Codigo para la figura 9
> par(mfrow=c(1,2))
> plotGrids(grid = "er.9.500.1255.280",
subtitle = "rho 0.9 M 1255 K 500 L 280",
varx = "fdp[\"Mean\",]", vary = "s[\"Std\",]",
ylab = "Desvıo S", ylim = c(0,50))
> plotGrids(grid = "er.9.5000.12558.2500",
subtitle = "rho 0.9 M 12558 K 5000 L 2500",
varx = "fdp[\"Mean\",]",
vary = "s[\"Std\",]",
ylab = "Desvıo S", ylim = c(0,500))
> par(mfrow=c(1,1))
B.6.10. Codigo para la figura 12
> par(mfrow=c(1,2))
> plotGrids(grid = "er.9.500.1255.280",
subtitle = "rho 0.9 M 1255 K 500 L 280",

B.6 Reproduccion de figuras para comparacion de errores 90
varx = "fdp[\"Mean\",]", vary = "r[\"Std\",]",
ylab = "Desvıo R", ylim = c(0,200))
> plotGrids(grid = "er.9.5000.12558.2500",
subtitle = "rho 0.9 M 12558 K 5000 L 2500",
varx = "fdp[\"Mean\",]", vary = "r[\"Std\",]",
ylab = "Desvıo R", ylim = c(0,2000))
> par(mfrow=c(1,1))
B.6.11. Codigo para la figura 11
> par(mfrow=c(1,2))
> plotGrids(grid = "er.4.500.1255.280",
subtitle = "rho 0.4 M 1255 K 500 L 280",
varx = "fdp[\"Mean\",]", vary = "r[\"Std\",]",
ylab = "Desvıo R", ylim = c(0,200))
> plotGrids(grid = "er.4.5000.12558.2500",
subtitle = "rho 0.4 M 12558 K 5000 L 2500",
varx = "fdp[\"Mean\",]", vary = "r[\"Std\",]",
ylab = "Desvıo R", ylim = c(0,2000))
> par(mfrow=c(1,1))
B.6.12. Codigo para la figura 10
> par(mfrow=c(1,2))
> plotGrids(grid = "er.0.500.1255.280",
subtitle = "rho 0 M 1255 K 500 L 280",
varx = "fdp[\"Mean\",]", vary = "r[\"Std\",]",
ylab = "Desvıo R", ylim = c(0,15))
> plotGrids(grid = "er.0.5000.12558.2500",
subtitle = "rho 0 M 12558 K 5000 L 2500",
varx = "fdp[\"Mean\",]", vary = "r[\"Std\",]",
ylab = "Desvıo R", ylim = c(0,150))
> par(mfrow=c(1,1))

REFERENCIAS 91
Referencias
[BAD+02] J. C. Boldrick, A. A. Alizadeh, M. Diehn, S. Dudoit, C. L. Liu, C. E. Belcher,D. Botstein, L. M. Staudt, P. O. Brown, and D. A. Relman. Stereotyped andspecific gene expression programs in human innate immune responses to bacteria.Proc. Natl. Acad. Sci., 99(2):972–977, 2002. 2.16
[BD95] Jonathan B. Buckheit and David L. Donoho. Wavelab and reproducible research.Technical report, October 20 1995. 1.6
[BH95] Y. Benjamini and Y. Hochberg. Controlling the false discovery rate: A practicaland powerful approach to multiple testing. Journal of the Royal Statistical Society,Series B, 57:289–300, 1995. 1.2, 2.9.3, 2.14, 2.15, 2.15.1, 2.4, 2.15.2
[Bon36] C. E. Bonferroni. Teoria statistica delle classi e calcolo delle probabilita. Pubbli-cazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze,pages 3–62, 1936. 1.1, 2.9.4, 2.3
[BY01] Y. Benjamini and D. Yekutieli. The control of the false discovery rate in multipletesting under dependency. Annals of Statistics, 29(4):1165–1188, 2001. 1.2, 2.14,2.15.1, 2.15.2
[DSB03] S. Dudoit, J.P. Shaffer, and J.C. Boldrick. Multiple hypothesis testing in micro-array experiments. Statistical Science, 18:71–103, 2003. 1.2, 2.9.3, 2.12
[EST01] B. Efron, J. D. Storey, and R. Tibshirani. Microarrays, empirical Bayes methods,and false discovery rates. Technical Report 2001–218, Department of Statistics,Stanford University, Stanford, CA 94305, 2001. 2.14
[ETST01] B. Efron, R. Tibshirani, J. D. Storey, and V. Tusher. Empirical Bayes analysisof a microarray experiment. Journal of the American Statistical Association,96:1151–1160, 2001. 2.14
[GGQY07] A. Gordon, G. Glazko, X. Qiu, and A.Y. Yakovlev. Control of the mean number offalse discoveries, Bonferroni, and stability of multiple testing. Annals of AppliedStatistics, 1:179–190, 2007. 1.1, 1.2, 1.3, 2.9.3, 2.16, 3.1, 3.2, 3.2.1, 3.4, 3.4, 3.4,3.5, 3.5, 3.5.1, 3.6, 4, 4.2, 4.3, 5, 5.1, 5.2.3, 6, 6.1
[GL07] Robert Gentleman and Duncan Temple Lang. Statistical analyses and reprodu-cible research. Journal of Computational and Graphical Statistics, 16(1):1–23,March 2007. 1.6
[Hoc88] Y. Hochberg. A sharper Bonferroni procedure for multiple tests of significance.Biometrika, 75:800–802, 1988. 2.15.1
[HT87] Y. Hochberg and A. C. Tamhane. Multiple Comparison Procedures. Probabilityand Mathematical Statistics. Wiley, New York, 1987. 1.2, 2.9.3
[IG96] Ross Ihaka and Robert Gentleman. R: A language for data analysis and graphics.Journal of Computational and Graphical Statistics, 5(3):299–314, 1996. 1.4

REFERENCIAS 92
[Knu83] Donald E. Knuth. Literate programming. Technical report STAN-CS-83-981,Stanford University, Department of Computer Science, 1983. 1.6
[KY07] L. Klebanov and A.Y. Yakovlev. Diverse correlation structures in microarraygene expression data and their utility in improving statistical inference. Annalsof Applied Statistics, 1:538–559, 2007. 1.1, 3.1, 5.1
[PDvdL05] K. S. Pollard, S. Dudoit, and M. J. van der Laan. Multiple testing procedu-res: The multtest package and applications to genomics. In R. C. Gentleman,V. J. Carey, W. Huber, R. A. Irizarry, and S. Dudoit, editors, Bioinformatics andComputational Biology Solutions Using R and Bioconductor, chapter 15, pages249–271. Springer, New York, 2005. 1.2, 1.5
[QKY05] X. Qiu, L. Klebanov, and A.Y. Yakovlev. Correlation between gene expressionlevels and limitations of the empirical bayes methodology for finding differentiallyexpressed genes. Statistical Applications in Genetics and Molecular Biology, 4,2005. 1.2, 2.12, 2.14, 3.1, 3.2, 3.6.1, 5.1
[SC97] S. K. Sarkar and C-K. Chang. The Simes method for multiple hypothesis testingwith positively dependent test statistics. Journal of the American StatisticalAssociation, 92:1601–1608, 1997. 2.15.1
[Sha95] J.P. Shaffer. Multiple hypothesis testing: A review. Annual Review of Psychology,46:561–584, 1995. 1.2, 2.9.3, 2.11, 2.12
[Sim86] R. J. Simes. An improved Bonferroni procedure for multiple tests of significance.Biometrika, 73:751–754, 1986. 2.15.1, 2.4, 2.15.1
[Spe03] T. P. Speed, editor. Statistical Analysis of Gene Expression Microarray Data.Chapman & Hall/CRC, Boca Raton, FL, 2003. 2.16
[ST03] J. D. Storey and R. Tibshirani. SAM thresholding and false discovery rates underdependence, with applications to DNA microarrays. Technical report, Departmentof Statistics, Stanford University, Stanford, CA 94305, 2003. 2.14
[VR00] W. N. Venables and B. D. Ripley. S Programming. Springer, New York, 2000. 1.4
[VR02] W. N. Venables and B. D. Ripley. Modern Applied Statistics with S. Springer,New York, 4th edition, 2002. 1.4
[WY93] P. H. Westfall and S. S. Young. Resampling-Based Multiple Testing: Examplesand Methods for P -Value Adjustment. Wiley, New York, 1993. 1.2


![Improved Inclusion-Exclusion Identities and Bonferroni ...as Bonferroni-type inequalities [GS96a, GS96b] or as inequalities of Bonferroni-Galambos type [MS85, M ar89, TX89]. A new](https://img.pdfslide.net/doc/110x75/60f8023676d34610215f4403/improved-inclusion-exclusion-identities-and-bonferroni-as-bonferroni-type-inequalities.jpg)




















![Comparaci..[1] proyecto de investigacion 2](https://img.pdfslide.net/doc/110x75/559cc2481a28ab83788b4630/comparaci1-proyecto-de-investigacion-2.jpg)





