Embed Size (px)
Citation preview

UNIVERSIDAD CENTRAL DE VENEZUELA
FACULTAD DE CIENCIAS
ESCUELA DE QUÍMICA
TRABAJO ESPECIAL DE GRADO
APROXIMACIÓN A LA SÍNTESIS DE LA ANDROST-5-EN-3,17-DIONA,
SÍNTESIS DE LA ANDROST-4-EN-3,6,17-TRIONA
Y SU POSTERIOR REDUCCIÓN
Caracas, octubre 2014
Trabajo Especial de Grado presentado
ante la Ilustre Universidad Central de
Venezuela por la Br. Daniela Antunez,
para optar por el título de Licenciada
en Química

UNIVERSIDAD CENTRAL DE VENEZUELA
FACULTAD DE CIENCIAS
ESCUELA DE QUÍMICA
TRABAJO ESPECIAL DE GRADO
APROXIMACIÓN A LA SÍNTESIS DELA ANDROST-5-EN-3,17-DIONA,
SÍNTESIS DELA ANDROST-4-EN-3,6,17-TRIONA
Y SU POSTERIOR REDUCCIÓN
Presentado por: Br. Daniela Antunez
Tutor: Dr. Gustavo Cabrera
Caracas, octubre de 2014

Yo, profesor Gustavo Cabrera, Investigador del Laboratorio de Productos
Naturales de la Escuela de Química de la Universidad Central de Venezuela
Certifico que el presente Trabajo Especial de Grado, titulado:
APROXIMACIÓN A LA SÍNTESIS DELA ANDROST-5-EN-3,17-DIONA,
SÍNTESIS DELA ANDROST-4-EN-3,6,17-TRIONA Y SU POSTERIOR
REDUCCIÓN
Que presenta la Br. Daniela C. Antunez Bracho, para aspirar al título de
Licenciada en Química, ha sido realizando en el Laboratorio de Productos
Naturales, de la Escuela de Química de la Universidad Central de Venezuela,
bajo mi dirección, durante los años 2013 y 2014, y con esta fecha autorizo su
presentación.
Caracas, octubre de 2014
____________________
Dr. Gustavo Cabrera
(Tutor)

Los abajo firmantes asignados por la Universidad Central de Venezuela, como
integrantes del jurado examinador del Trabajo Especial de Grado titulado:
APROXIMACIÓN A LA SÍNTESIS DE LA ANDROST-5-EN-3,17-DIONA,
SÍNTESIS DE LA ANDROST-4-EN-3,6,17-TRIONA Y SU POSTERIOR
REDUCCIÓN. Presentado por la Br. Daniela Coromoto Antunez Bracho,
certificamos que este trabajo cumple con los requisitos exigidos por nuestra
Magna Casa de Estudios para optar por el título de Licenciado en Química.
___________________________
Dr. Gustavo Cabrera
Director
___________________________ ___________________________
Dra. María Antonieta Ranaudo Dra. Alírica Suarez
(Jurado) (Jurado)

AGRADECIMIENTOS
A Dios y la virgen por protegerme en cada paso de mi vida, por estar presentes
a lo largo de mí camino, porque aunque no los vea creo plenamente en su
existencia.
A las personas más importantes en mi vida, mis padres, con ustedes aprendí
que las metas se logran con voluntad y disposición. Mamá, gracias por esa
palabra de aliento en los momentos difíciles, por tu amor y dedicación, una vida
entera no bastaría para darte las gracias y a ti papá gracias por siempre
depositar confianza y fe en mi. Gracias a mis hermanos Dayani y Daniel por
estar siempre a mi lado, por cuidarme y protegerme cada día.
Gracias a los profesores Gustavo Cabrera y Pablo Neacato, por todos los
conocimientos enseñados y la ayuda brindada a lo largo de este trabajo, sus
palabras fueron una gran motivación, ante cualquier dificultad.
Infinitas gracias a todo el grupo de Productos Naturales, prof. María, Prof. Jairo,
Reine, Luis e Ynés, gracias por el apoyo, por darme ánimo y ayudarme
siempre.
Gracias a mis amigas y amigos, Yeni, Flor, Roxy, Joel, Herli, Jeniree, Gustavo
Urdaneta, Maikel, Gilkar, Roger y Kizzy, por todas las alegrías compartidas, por
siempre ayudarme y estar pendientes, sin ustedes esta carrera se hubiese
hecho más cuesta arriba, gracias por todo su apoyo. Tenerlos como amigos ha
sido una bendición.

Un especial agradecimiento a Lourdes Gotopo quien más que ser una amiga es
mi hermana de corazón, gracias, porque entre caídas, chistes y risas, logramos
alcanzar nuestra meta, ha sido un privilegio trabajar a tu lado, cada vez que
algo salía mal siempre estabas a mi lado apoyándome, más que compartir un
mesón, compartimos un sueño, espero sigamos compartiendo nuevas metas,
eres un pilar fundamental en la realización de esta tesis, siempre ayudándome,
dándome ánimos, gracias por reírte conmigo de mis locuras, de corazón deseo
el mayor de los éxitos para ti. Mil gracias por formar parte de esta historia.

RESUMEN
En el presente Trabajo Especial de Grado se realizaron una serie de
modificaciones estructurales en los anillos A y B de la dehidroepiandrosterona
con la finalidad de obtener una serie de compuestos, los cuales serán utilizados
para la creación de una base de datos sobre la actividad antiviral de los
mismos a través de ensayos biológicos.
Las modificaciones estructurales realizadas se dividieron en dos rutas
sintéticas: La Ruta Sintética Nº1, donde sólo se realizó la bromación de la
dehidroepiandrosterona, esta ruta no fue terminada debido a los inconvenientes
obtenidos en el primer paso y la Ruta Sintética Nº2 la cual consistió en la
oxidación directa de la dehidroepiandrosterona con el Reactivo de Jones como
primer paso, para la posterior Reducción de Huang-Minlon del producto
obtenido.
Los productos y los intermediarios obtenidos fueron caracterizados a través de
Espectroscopia de UV, IR, RMN-1H y RMN-13C.

INDICE GENRAL
1. INTRODUCCION .......................................................................... 2
2. MARCO TEÓRICO ....................................................................... 4
2.1 Terpenos y esteroides .................................................................. 4
2.1.1 Terpenos ......................................................................... 4
2.1.2 Esteroides ....................................................................... 6
2.1.2.1 Reacciones de los esteroides .................................................... 8
2.1.2.2 Esteroides glucocorticoides ....................................................... 9
2.1.2.3 Origen de los glucocorticoides .................................................. 9
2.1.2.4 Acciones fisiológicas y farmacológicas de los
glucocorticoides………………………………………………………………10
2.1.2.5 Acciones antiinflamatorias e inmunodepresoras de los
glucocorticoides ..................................................................................... 11
2.2 Reacciones de adición electrofílica ............................................. 11
Dirección de la adición......................................................... 12
Estereoquímica de la reacción de adición ........................... 13
2.3 Adición de halógenos a dobles enlaces ...................................... 13
2.4 Deshalogenación de dihaluros vecinales .................................... 15
2.5 Reacciones de oxidación ............................................................ 18
2.5.1 Oxidación o deshidrogenación de alcoholes a aldehídos
y cetonas ……………………………………………………………..18
2.6 Reacciones de reducción ............................................................ 23
2.6.1 Reducciones de carbonilos a metilenos en aldehídos y
cetonas ....................................................................................... 23
2.6.1.1 Reducción de Clemmensen .................................................... 24
6.2.1.2 Reducción de Wolff Kisnher .................................................... 26
3. OBJETIVOS................................................................................ 28
3.1 Objetivo General: ........................................................................ 28
3.2 Objetivos Específicos: ................................................................. 28
4. ANTECEDENTES ....................................................................... 29

5. PARTE EXPERIMENTAL ........................................................... 42
5.1 Ruta Sintética Nº1 ....................................................................... 44
Síntesis del Δ5-androst-3,17-ona, a través de una secuencia de tres etapas
que implican, la protección del doble enlace por una bromación, oxidación a
la dibromo cetona y finalmente la desbromación con zinc en ácido acético. 44
5.1.1 Bromación del 3β -hidroxi-androst-5-en-17-ona. ........... 44
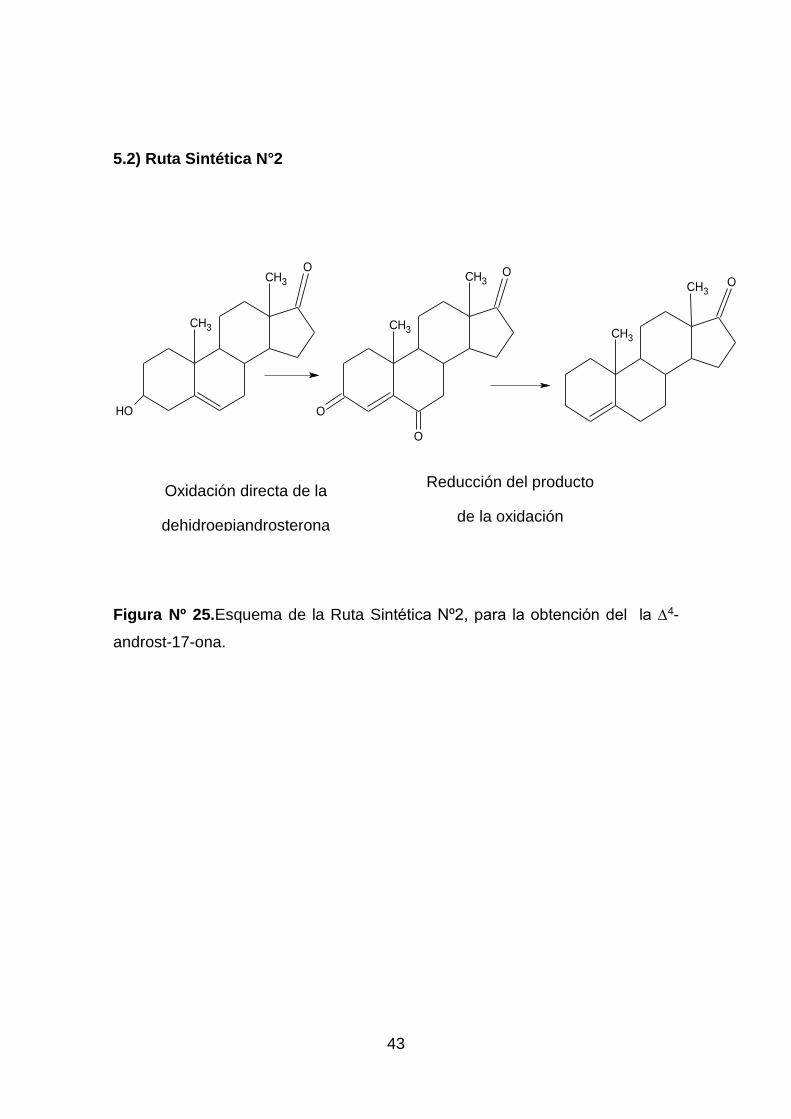
5.2 Ruta Sintética Nº2 ....................................................................... 45
Síntesis de laΔ5-androst-3,6,17-triona, a partir de la oxidación directa de la
dehidroepiandrosterona (3β -hidroxi-androst-5-en-17-ona), para la posterior
reducción de Huang-Minlon del producto obtenido. ...................................... 45
5.2.1 Oxidación del 3β-hidroxi-androst-5-en-17-ona, con el
Reactivo de Jones ....................................................................... 45
5.2.2 Reducción de laandrost-5-en-3,6,17-triona empleando
la Reducción de Huang-Minlon. .................................................. 46
5.2.2.1 Síntesis de la hidrazona de la Δ4-androst-3,6,17-triona .......... 46
5.2.2.2 Reducción de la hidrazonadel Δ4-androst-en-3,6,17-triona ..... 47
5.2.2.3 Reducción directa Δ4-androst-en-3,6,17-trionaempleando el
método de Huang –Minlon. .................................................................... 47
6. RESULTADOS Y DISCUSIÓN ................................................... 49
6.1 Síntesis del Δ5-androst-3,17-ona, a través de una secuencia de
tres etapas que implican, la protección del doble enlace por una bromación,
oxidación a la dibromo cetona y finalmente la desbromación con zinc en
ácido acético. ................................................................................................ 49
6.1.1 Bromación del 3β -hidroxi-androst-5-en-17-ona. ........... 49
6.2 Síntesis del Δ4-androst-3,6,17-diona, a partir de la oxidación
directa de la dehidroepiandrosterona (3β -hidroxi-androst-5-en-17-ona), para
su posterior reducción empleando el método de HuangMinlon .................... 57
6.2.1 Síntesis del Δ4-androst-3,6,17-diona, a partir de la
oxidación directa de la dehidroepiandrosterona con el reactivo de
Jones…………………………………………………………………..57
6.2.2 Reducción Huang-Minlon de la Δ4-androst-en-3,6,17-
triona…………………………………………………………………...67

6.2.2.1 Síntesis de la hidrazona de la Δ4-androst-3,6,17-
triona…………………………………………………………………..67
6.2.2.2 Reducción de la hidrazona de la Δ4-androst-3,6,17-triona ...... 70
6.2.2.3 Reducción directa del producto oxidado empleando en método
de Huang –Minlon. ................................................................................ 81
7. CONCLUSIONES ........................................................................................ 85
8. ESPECTROS ............................................................................................... 87
9. BIBLIOGRAFÍA ......................................................................................... 103

INDICE DE FIGURAS
Figura Nº 1. Molécula de isopreno .................................................................... 5
Figura Nº 2. Sistema de anillos ciclopentano-perhidrofenantreno ..................... 6
Figura Nº 3. Sistemas básicos de anillos de las series de esteroides 5α (unión
trans) y 5β (unión cis). ................................................................................. 7
Figura Nº 4. Estructuras de los diferentes grupos de esteroides. ...................... 8
Figura Nº 5. Mecanismo general para una reacción de adición electrofílica. .. 12
Figura Nº 6. Orientación Markownicoff y anti- Markownicoff de las adiciones 12
Figura Nº 7. Adiciones syn y anti en dobles enlaces ....................................... 13
Figura Nº 8. Secuencia de pasos para la bromación de una olefina ............... 15
Figura Nº 9.Deshalogenación del trans-1,2-dibromociclohexano con zinc ...... 15
Figura Nº 10. Deshalogenación de dihaluros vecinales................................... 16
Figura Nº 11. Mecanismo de bromación del colesterol.................................... 17
Figura Nº 12. Secuencia de pasos para la oxidación de un alcohol ................ 19
Figura Nº 13. Mecanismo de Oxidación de un alcohol con dicromato ............ 20
Figura Nº 14. Oxidación del hidrato de aldehído al ácido carboxílico .............. 21
Figura Nº 15. Reacciones generales para la formación del Reactivo de Jones
y del reactivo de Sarett.............................................................................. 22
Figura Nº 16. Reducción de la acetofenona empleando el método de
Reducción de Clemmensen. ..................................................................... 24
Figura Nº 17. Mecanismos de reducción de Clemmensen, planteado por
Nakabayashi para la obtención de un metileno a partir de un carbonilo. [16]
[21] .............................................................................................................. 25
Figura Nº18. Mecanismo de reducción de Wolff-Kishner de un grupo carbonilo
a un metileno ............................................................................................. 26
Figura Nº 19. Productos obtenidos durante la oxidación del Colesterol con el
complejo de óxido de cromo (VI)-piridina en diclorometano ...................... 35
Figura Nº 20. Complejos monopiridina (17) y dipiridina (18) .......................... 38

Figura Nº 21. Esquema de síntesis para la obtención Δ5-Colesten-3-ona,
mediante una bromación del doble enlace, oxidación del alcohol del C-3 y
desbromación del doble enlace. ................................................................ 39
Figura Nº 22. Esquema de síntesis para la conversión directa del grupo OH
de esteroides Δ5-3β-alcohol a Δ5- y Δ4-3-cetona, utilizando el reactivo de
Jones......................................................................................................... 40
Figura Nº 23. Esquema de síntesis para conversión del grupo carbonilo de C-
17 en la dehidroepiandrosterona a través de la reducción de Wolff-Kishner.
.................................................................................................................. 41
Figura Nº 24. Esquema de la Ruta Sintética Nº1, para la obtención de la ∆5-
androst-17-ona .......................................................................................... 42
Figura Nº 25. Esquema de la Ruta Sintética Nº2, para la obtención del la ∆4-
androst-17-ona. ......................................................................................... 43
Figura Nº 26. Bromación de la Dehidroepiandrosterona ................................. 49
Figura Nº 27. Isómeros del bromociclohexano. ............................................... 52
Figura Nº 28.Esquema de del mecanismo de reacción para la síntesis de α-
halocetona. ................................................................................................ 53
Figura Nº 29. Síntesis del 3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona a
través de la bromación de la dehidroepiandrosterona .............................. 54
Figura Nº 30. Esquema de síntesis de la Δ4-androst-3,6,17-triona.................. 57
Figura Nº 31. Segunda derivada del espectro de IR Nº4 en la región de
absorción de 1735,1 cm-1 hasta 1685,1 cm-1 ............................................ 59
Figura Nº 32. Estructuras de la testosterona y el colesterol ........................... 62
Figura Nº 33. Síntesis de esteroides 4-en-3,6-dionas a partir de esteroides 5-
en-3β-ol, utilizando el Reactivo de Jones .................................................. 63
Figura Nº 34. Esquema de síntesis de 4-en-3,6-dionas a partir de 5-en-3β-ol 65
Figura Nº 35. Síntesis de la Δ4-androst-3,6,17-triona ...................................... 66
Figura Nº 36. Deconvolución del espectro de IR Nº7 en la región de absorción
de 1736,1 cm-1 hasta 1666,9 cm-1. ........................................................... 69
Figura Nº 37. Posibles Estructura de la Hidrazona sintetizada. ...................... 70

Figura Nº 38. Reducción de la hidrazona de la colestanona a la mezcla de
alcoholes (α y β)- colestanol ..................................................................... 74
Figura Nº 39. Esquema de reducción del carbonilo al alcohol, empleando el
método de Wolff-Kishner. .......................................................................... 74
Figura Nº 40. Secuencia de mecanismos para la reducción del carbonilo al
alcohol, durante la reducción de Wolff-Kishner. ........................................ 75
Figura Nº 41. Posible estructura (21) para el producto de reducción de la
hidrazona de la Δ4-androst,3,6,17-triona. .................................................. 76
Figura Nº 42. Estructura del 3ξ,4ξ6ξ,17ξ-tetrahidroxi-androstano .................. 77
Figura Nº 43. Posible mecanismo de reacción para la sustitución del grupo
hidroxilo en la posición C-4 de la posible hidrazona (19) sintetizada. ..... 78
Figura Nº 44. Deconvolución del Espectro IR del producto de reducción directa
de la Δ4-androst-3,6,17-triona. ................................................................. 83
Figura Nº 45. Posibles producto de la reducción directa de la Δ4-androst-
3,6,17-triona. ............................................................................................. 84

INDICE DE TABLAS
Tabla N° 1. Solubilidad del complejo oxido de cromo (VI)- piridina a 25 ºC .... 31
Tabla N° 2. Oxidación de alcoholes empleando el complejo CrO3-piridina en
diclorometano a una temperatura de 25 ºC ............................................... 32
Tabla Nº 3. Oxidación de alcoholes con trióxido de cromo (VI) –piridina en
diclorometano (preparado in situ). ............................................................. 34
Tabla N° 4. Oxidación del colesterol (1) con el complejo óxido de cromo (VI)-
piridina (CrO3 2C5H5N) en diclorometano. ................................................. 36
Tabla N°5. Datos espectroscópicos de RMN-13C (Espectro N°3, CDCl3), del
producto de la bromación del 3β -hidroxi-androst-5-en-17-ona................. 55
Tabla N°6. Datos espectroscópicos de RMN-13C, CDCl3, para la testosterona y
el colesterol. .............................................................................................. 61
Tabla Nº 7. Datos espectroscópicos de RMN-13C (Espectro N° 7, CDCl3), del
producto de oxidación, empleando el reactivo de Jones ........................... 64
Tabla Nº 8 . Datos espectroscópicos del espectro de RMN-13C, CDCl3, para
algunos carbonos de la 4ξ,17β-dihidroxi5ξ-androstano y la 3β,6ξ-dihidroxi-
5ξ-androst-17-ona. .................................................................................... 72
Tabla Nº 9.Datos espectroscópicos de RMN-C13 (Espectro N°10 , CDCl3), del
producto de reducción. .............................................................................. 79
Tabla N°10 Datos espectroscópicos de RMN-C13 (Espectro N°10 , CDCl3), del
posible producto de reducción (22), el cual corresponde al 3ξ,4ξ,6ξ,17ξ-
tetrahidroxi-androstano………………………………………………………………81

ÍNDICE DE ESPECTROS
Espectro Nº 1. IR de la3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona .......... 88
Espectro Nº2. RMN-1H de la 3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona . 89
Espectro Nº 3. RMN- 13 C de la3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona
.................................................................................................................. 91
Espectro Nº 4. IR de la Δ4-androst-3,6,17-triona. ............................................ 91
Espectro Nº 5. Espectro UV, tomado en diclorometano de la Δ4-androst-
3,6,17-triona. ............................................................................................. 93
Espectro Nº 6. RMN-1H de la Δ4-androst-3,6,17-triona ................................... 94
Espectro Nº 7. RMN- 13C de la Δ4-androst-3,6,17-triona ................................. 96
Espectro Nº 8. IR de la Hidrazona de la Δ4-androst-3,6,17-triona ................... 97
Espectro Nº 9. RMN-1H para de la Reducción de la Hidrazona de la Δ4-
androst-3,6,17-triona ................................................................................. 98
Espectro Nº10.RMN-13C para de la Reducción de la Hidrazona de laΔ4-
androst-3,6,17-triona ................................................................................. 99
Espectro Nº 11. Espectro UV(CH2Cl2) del producto de la reducción directa del
Δ4-androst-3,6,14-triona empleando en método de Huang –Minlon ....... 101
Espectro Nº 12. Reducción directa del Δ4-androst-3,6,14-triona empleando en
método de Huang –Minlon ...................................................................... 102

2
1. INTRODUCCION
La síntesis orgánica ha sido utilizada como una herramienta fundamental para
el diseño de nuevos y mejores fármacos, mucho más efectivos y con menor
riego de toxicidad humana. Gracias a las distintas modificaciones estructurales
que pueden realizarse a moléculas mediante la utilización de diversas
reacciones, se puede lograr obtener moléculas biológicamente activas, para la
preparación de sustancias terapéuticas.
Fármaco es, en sentido amplio, toda sustancia química capaz de interactuar
con un organismo vivo. En sentido más restringido, es toda sustancia química
utilizada en el tratamiento, la curación, la prevención o el diagnóstico de una
enfermedad o para evitar la aparición de un proceso fisiológico no deseado
[1].Los efectos de casi todos los fármacos son consecuencia de su interacción
con componentes macromoleculares del organismo; dichas interacciones
modifican la función del componente pertinente y con ello inician los cambios
bioquímicos y fisiológicos que caracterizan la reacción al fármaco [2].
Los esteroides son productos naturales de gran importancia, ya que muchos
de ellos son reguladores de funciones biológicas en organismos vivos, además
de presentar efectos fisiológicos sorprendentes cuando se suministran a los
mismos [3].Dicha característica ha permitido su utilización como sustancias
terapéuticas, causando un poderoso interés en el ámbito de la medicina y la
farmacología, el cual ha generado una serie de investigaciones científicas para
determinar la actividad biológica de esteroides naturales así como propiciar el
estudio y desarrollo de una serie de esteroides sintéticos, con el propósito de
ser empleados como posibles agentes medicinales.

3
Los virus son parásitos intracelulares que tienen la capacidad de invadir a las
células, liberando los componentes de su ácido nucléico para redirigir las
actividades biosintéticas de la célula hacia la producción de partículas del virus
progenitor. Un virus en particular es adecuado para su replicación en algunos
tipos de células y especies, pero no en otros, incluso si una célula no es
permisiva para la replicación viral, el virus puede todavía ser capaz de invadir la
célula y a través de la de los genes virales, alterar las propiedades de la
misma, siendo tal característica una causa importante de las enfermedades en
humanos [4]. Por tal razón el tratamiento de enfermedades virales representa
todo un desafío científico en el campo de la medicina ya que se deben
desarrollar herramientas efectivas para combatir y prevenir dichas
enfermedades.
El padecimiento de enfermedades virales como el dengue, la fiebre amarilla, el
sarampión, la hepatitis B, entre otras, se ha hecho común dentro de la
población mundial. Por tal razón este trabajo de investigación forma parte de
un proyecto que se fundamenta en la realización de modificaciones
estructurales en androstanos, con el fin de generar una serie de compuestos, a
los que posteriormente se le realizarán ensayos biológicos y estudios de
modelaje molecular. Dicho proyecto se basa en los resultados previamente
obtenidos del trabajo en conjunto entre el Laboratorio de Productos Naturales
de la Facultad de Ciencias-UCV y el Instituto de Biomedicina de la Facultad de
Medicina-UCV; sobre el uso de androstanos como antivirales, contra el dengue
y la fiebre amarilla.

4
2. MARCO TEÓRICO
2.1 Terpenos y esteroides
2.1.1 Terpenos
Los terpenos se forman con moléculas de isopreno, cuando estas se unen
entre si y producen cadenas y anillos de forma y tamaño variable. El isopreno
es la unidad fundamental que define el esqueleto de estos compuestos. Las
cadenas que resultan de la condensación de estas unidades son estructuras
hidrofóbicas, hecho que las incluye dentro del grupo de los lípidos [5].
Los terpenos constituyen un grupo de compuestos que, en su mayoría, se
encuentran en el reino vegetal, su frecuencia y abundancia está íntimamente
ligada a factores genéticos y climáticos. Desde el punto de vista funcional la
gran variedad de terpenos dificulta el resumen de sus características comunes
[6].
De manera general, los terpenos obedecen a lo que se conoce como regla del
isopreno y fue esbozada por primera vez por WALACH (1887), es decir, la
secuencia de los átomos que conforman un terpeno es tal que pueden
localizarse varias unidades consecutivas de isopreno. Sin embargo, esto no es
siempre cierto, por cuanto el número de átomos de carbono puede no ser un
múltiplo de cinco, pues son frecuentes las reacciones de degradación, o bien
suceden re-arreglos esqueletales que conducen a secuencias anormales de
C5[7].

5
Normalmente en los isoprenoides, la cabeza de una unidad de isopreno se une
a la cola de la más próxima, aunque se ha encontrado cierto número de
ejemplos de uniones cabeza con cabeza y cola con cola
Cabeza CH2 C CH CH
2 Cola
CH
3
Figura Nº 1. Molécula de isopreno
La mayoría de los hidrocarburos terpénicos naturales tienen la formula
molecular (C5H8)n utilizándose el valor de n como la base de la clasificación. Se
tienen los siguientes grupos:
Monoterpenos (C10H16)
Sesquiterpenos (C15H24)
Diterpenos (C20H32)
Triterpenos (C30H48)
Tetraterpenos (C40H64)
Politerpenos(C5H8)n
Además de los hidrocarburos terpénicos, existen en cada grupo principalmente
derivados oxigenados (alcoholes, cetonas, esteres, éteres, ácidos carboxílicos
etc.), le siguen funciones con azufre, halógenos (principalmente encontrados
en compuestos de origen marino) y nitrógeno, en cuyo caso la molécula se
clasifica como alcaloides. El descubrimiento de un gran número de
compuestos que presentan diferentes grupos funcionales ha generado el uso

6
del término “terpenoides” para designarlos en lugar de “terpeno” cuya
terminación (-eno) denota más bien un hidrocarburo [7].
Los triterpenos(compuestos de C30) han sido estudiados ampliamente, debido
en parte, a su relación con los esteroides, los cuales han llamado
poderosamente la atención desde hace más de medio siglo, cuando se conoció
que las hormonas sexuales humanas pertenecen a este grupo de compuestos.
Los triterpenosse encuentran ampliamente distribuidos en la naturaleza tanto
en el reino vegetal como en el reino animal [6].
2.1.2 Esteroides
Los esteroides son una familia de compuestos que contiene un sistema de
anillos ciclopentano-perhidrofenantreno, tal y como se muestra en la figura
Nº2(con algunas excepciones) que puede o no presentar una cadena lateral
en C-17, así como los metilos angulares en C-10 y C-13[8].
CH3
CH3
A B
C D1 9
2
3 7
4 65
8 151410
12
1113 17
16
Figura Nº 2. Sistema de anillos ciclopentano-perhidrofenantreno
Los cuatro anillos se identifican con las letras A, B, C y D, tal y como se indica
en a figura Nº2.En la mayoría de los esteroides, las uniones entre los anillos B,
C y C, D son trans. Sin embargo, la unión de los anillos A, B puede ser cis o

7
trans y esto da lugar a dos grupos generales de esteroidesque tiene las
siguientes estructuras mostradas en la figura Nº3 [3].
H
CH3
H H
R
CH3
CH3
H AH
CH3 R
Todos los anillos estan unidos trans Unión cis de los anillos A y B
A BC D C D
B
5 5
Figura Nº 3.Sistemas básicos de anillos de las series de esteroides 5α (unión
trans) y 5β (unión cis).
Los compuestos de este grupo de encuentran ampliamente distribuidos en
plantas y animales y son de los productos naturales más importantes. A esta
familia pertenecen los esteroles los cuales poseen 27 carbonos, ácidos biliares,
hormonas sexuales, las hormonas de la corteza suprarrenal y los aglicones
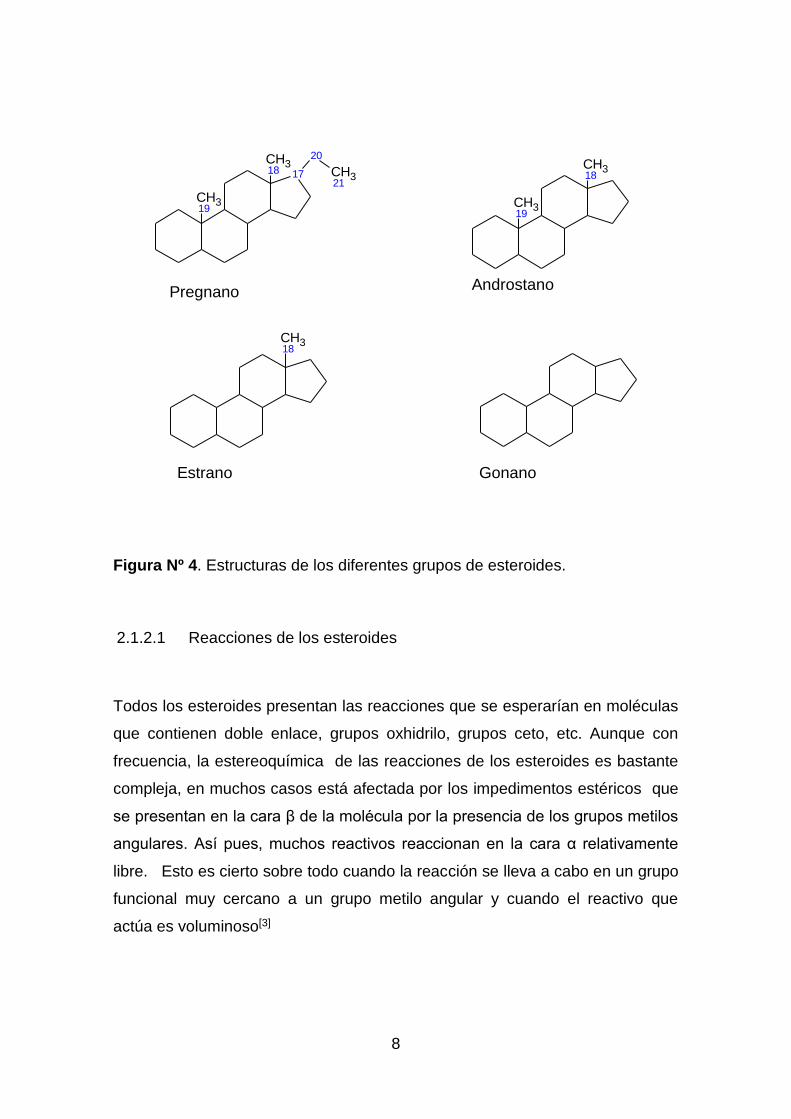
cardíacos (venenos), se prefiere designar como esteroide a los derivados del
preganano,que cuentan con 21 carbonosen su estructura, androstanocon 19
carbonos, estranocon 18 carbonos y gonano con 17 carbonos en su estructura,
los demás se designa como esteroles[8].
8
CH318
CH319
CH31817
CH319
CH318
20
CH321
GonanoEstrano
AndrostanoPregnano
Figura Nº 4. Estructuras de los diferentes grupos de esteroides.
2.1.2.1 Reacciones de los esteroides
Todos los esteroides presentan las reacciones que se esperarían en moléculas
que contienen doble enlace, grupos oxhidrilo, grupos ceto, etc. Aunque con
frecuencia, la estereoquímica de las reacciones de los esteroides es bastante
compleja, en muchos casos está afectada por los impedimentos estéricos que
se presentan en la cara β de la molécula por la presencia de los grupos metilos
angulares. Así pues, muchos reactivos reaccionan en la cara α relativamente
libre. Esto es cierto sobre todo cuando la reacción se lleva a cabo en un grupo
funcional muy cercano a un grupo metilo angular y cuando el reactivo que
actúa es voluminoso[3]

9
2.1.2.2 Esteroides glucocorticoides
Los esteroides Glucocorticoides aparentemente intervienen en la regulación de
un gran número de actividades biológicas incluyendo el metabolismo de
carbohidratos, proteínas y lípidos, el balance de agua y electrólitos y las
reacciones a los fenómenos alérgicos e inflamatorios. El efecto antiinflamatorio
de la cortisona y su utilidad en el tratamiento de la artritis reumatoide en 1949,
dio lugar a una intensa investigación en esta área[3][9].
2.1.2.3 Origen de los glucocorticoides
La corteza suprarrenal sintetiza toda clase de hormonas esteroideas: los
glucocorticoides cortisol y corticosterona, los mineralocorticoides aldosterona y
desoxicorticosterona y las hormonas gonadales dehidroepiandrosterona,
androstenodiona y testosterona.
A partir del esteroide natural cortisol se han obtenido numerosos derivados
sintéticos que mantienen algunas de sus propiedades y mejoran otras. El
número de derivados es muy amplio, así como las vías de administración por
las que se pueden utilizar. Con frecuencia se obtienen distintos ésteres de un
mismo producto para emplearlos por diferentes vías, pero con algunos que se
usan por vía tópica se consigue mantener su actividad antiinflamatoria y reducir
su capacidad de difusión con el fin de circunscribir su acción localmente y
restringir la acción sistémica [9].

10
2.1.2.4 Acciones fisiológicas y farmacológicas de los glucocorticoides
En ausencia completa de hormonas corticales se produce una depleción del
glucógeno hepático y muscular, disminuye la glucemia, se reduce la cantidad
de nitrógeno no proteico en la orina, aumenta la eliminación de sodio en orina,
desciende la presión arterial, disminuye la concentración de sodio en plasma y
aumenta la de potasio y se pierde la capacidad de concentrar o de diluir la
orina. La administración de corticosteroides restablece estas funciones y, si se
administran dosis excesivas, se aprecian expansión del volumen plasmático,
retención de sodio y pérdida de potasio, aumento de la presión arterial,
incremento del glucógeno en hígado y músculo, aumento de la glucemia,
reducción de la masa conjuntiva y muscular, y aumento de nitrógeno no
proteico en orina; en determinadas circunstancias, además, inhiben la
respuesta inflamatoria y ciertas manifestaciones de la respuesta inmunitaria.
Este conjunto de acciones suele clasificarse en dos tipos: las glucocorticoides
representadas por la capacidad de almacenar glucógeno hepático y por la
capacidad antiinflamatoria y las mineralocorticoides, representadas por la
capacidad de retener sodio y agua. Existe una clara disociación en la
capacidad de los corticoides naturales para activar unas u otras acciones: el
cortisol tiene mucha mayor actividad glucocorticoidea que mineralocorticoide,
mientras que con la aldosterona sucede lo contrario; entre estos dos extremos,
la cortisona y la corticosterona ocupan situaciones intermedias. Muchos
análogos sintéticos del cortisol muestran potencias crecientes de acción
glucocorticoidea y decrecientes de acción mineralcorticoide, lo que permite
una gran manejabilidad[9].

11
2.1.2.5 Acciones antiinflamatorias e inmunodepresoras de los
glucocorticoides
Los glucocorticoides ejercen una poderosa acción antiinflamatoria, sea cual
fuere la causa de la inflamación (infecciosa, química, física o inmunológica),
pudiendo inhibir tanto las manifestaciones inmediatas de la inflamación (rubor,
dolor, etc.) como tardías, entendiendo por tales ciertos procesos de
cicatrización y proliferación celular. Inhiben la dilatación vascular, reducen la
formación de edema y reducen el depósito de fibrina alrededor del área
inflamada[9].
2.2 Reacciones de adición electrofílica
La propiedad que presentan los compuestos con enlaces múltiples carbono-
carbono de dar lugar a reacciones de adición, se debe por una parte a la gran
polarizabilidad de la unión π y por otra parte a la poca energía que necesita la
disociación de esta unión.Para explicar estas reacciones se considera el hecho
de que debido a la superposición de los orbitales p, el doble enlace se hace
rico en electrones, permitiéndole esta propiedad a las olefinas actuar en
algunas condiciones como nucleófilos. [10] [11]
La densidad electrónica asociada con enlaces múltiples carbono-carbono de
las olefinas las convierte en sustratos ávidos de reactivos electrofílicos. La
especie ENu se aproxima a un doble enlace para formar un nuevo enlace σ con
la parte electrofílica E, de la especie ENu, a expensas del enlace π del
sustrato, lo cual desarrollará una carga positiva en el átomo vecino que será
saturada por el ataque del nucleófiloNu. [10]

12
E+ + C+
E
+ :Nu-
E Nu
Paso1 Paso 2
Figura Nº 5. Mecanismo general para una reacción de adición electrofílica.
Dirección de la adición
La repartición de la densidad electrónica en una olefina desigualmente
sustituida desplaza la nube electrónica hacia el carbono más receptor de
electrones, es decir, el menos sustituido, para generar una especie más
estable, donde la carga positiva pueda disiparse por efectos inductivos y de
resonancia de los sustituyentes, como consecuencia el electrófilo se unirá al
doble enlace de manera que forme el carbocatiónmás estable. Esto se conoce
como orientación Markownicoff, las adiciones también pueden ocurrir con
orientaciones anti-Markownicoff, en las cuales el nucleófilo se une al carbono
menos sustituido[10].
Figura Nº 6. Orientación Markownicoff y anti- Markownicoff de las adiciones

13
Estereoquímica de la reacción de adición
Las adición de las especies E y Nu de la molécula ENu pueden realizarse del
mismo lado de los sustituyentes de la olefina, siendo esta una adición cisó de
lado contrario al doble enlace y se denomina adición trans, también llamadas
adición syn y anti respectivamente[10].
+ ENu +E
Nu
Nu
E
adición
syn
+ ENuadición
anti
+
Nu
Nu
E
E
Figura Nº 7. Adiciones syn y anti en dobles enlaces
2.3 Adición de halógenos a dobles enlaces
Los halógenos se adicionan fácilmente a alquenos para formar dihalogenuros
vecinales. Bromo y Cloro son los halógenos más utilizados en síntesis,
generalmente se utiliza el halógeno propiamente dicho. La adición de una
molécula de Bromo eléctricamente neutra y apolar, a un doble enlace carbono-
carbono se genera debido a la interacción entre la molécula de bromo y el
enlace 𝜋 lo cual provoca la polarización de la misma y la salida del ion
bromuro, el cual es un buen grupo saliente [12].

14
La adición de X2(Cl2 o Br2) a una olefina se puede dividir en una serie de pasos
tal y como se ilustra a continuación donde se ejemplifica la adición de Br2a un
doble enlace:
Paso 1: Inicialmente la molécula del halógeno X2 se polariza al acercarse al
doble enlace, que es también una región de alta densidad electrónica. La
molécula del halógeno también puede ser polarizada por disolventes polares en
los cuales se lleve a cabo la reacción.
H2C=CH2+ Br-Br [Brδ+….Brδ-] [Br+ Br-]
Paso 2:El doble enlace reacciona atacando a la molécula polarizada del
halógeno, en este ejemplo seria al bromo molecular, obteniéndose como
resultado un anillo de tres miembros, que contiene una carga positiva sobre el
átomo de bromo, este anillo de tres miembros se conoce como ion bromonio.
IónBromonioIón Bromuro

15
Paso 3: El paso final es el ataque del ion halogenuro (ión bromuro Br-) por el
lado contrario al ataque inicial, formando el producto trans.El ataque
nucleofílico del ion bromuro da como resultado un vic-dibromuro
Br+
Br-
Br
Br
Vic-dibromuro
Figura Nº 8. Secuencia de pasos para la bromación de una olefina
2.4 Deshalogenación de dihaluros vecinales
Los dihaluros vecinales (o vic) son compuestos dihalogenados en los cuales los
halógenos están situados en átomos de carbonos adyacentes. Los vic
halogenuros sufren la pérdida de halógeno (Deshalogenación) cuando se les
trata con zinc. Los productos orgánicos de estas reacciones son alquenos [13].
Un ejemplo específico es la deshalogenación del 1,2-dibromociclohexano,
mostrada a continuación:
Figura Nº 9.Deshalogenación del trans-1,2-dibromociclohexano con zinc

16
Por lo general estas reacciones se realizan en un disolvente como la acetona,
que disuelve tanto el vic-dihaluro como el alqueno.El zinc casi siempre se
mantiene en suspensión (en forma de polvo) y la reacción se lleva a cabo sobre
su superficie. La deshalogenación de un vic-dihaluro es una reacción empleada
como un método útil en la purificación de alquenos y como un “grupo protector”
del doble enlace.
El zinc actúa como un agente reductor propiciando una adición oxidativa, que
genera una eliminación anti [14].En la siguiente figura se observa el posible
mecanismo de deshalogenación de un vic-dihaluro
Figura Nº 10.Deshalogenación de dihaluros vecinales
La halogenación–deshalogenación también puede utilizarse para proteger un
doble enlace mientras se lleva a cabo una reacciónen otra parte de la molécula.
Durante la síntesis de algunos esteroides que involucran reacciones de
oxidación con trióxido de cromo es necesaria la protección del doble enlace, ya
que desafortunadamente los dobles enlaces no son estables ante estas
oxidaciones y una oxidación directa provocaría la oxidación del doble enlace.
Zn
CH2 CH2
Br
Br
CH2 CH2 + ZnBr2
17
Sin embargo, al convertir el doble enlace en un vic-haluro se "protege" el
mismo transformándolo temporalmente en un grupo saturado. Posteriormente
el doble enlace puede regenerarse al colocar a reaccionar el vic-haluro con
zinc[13].
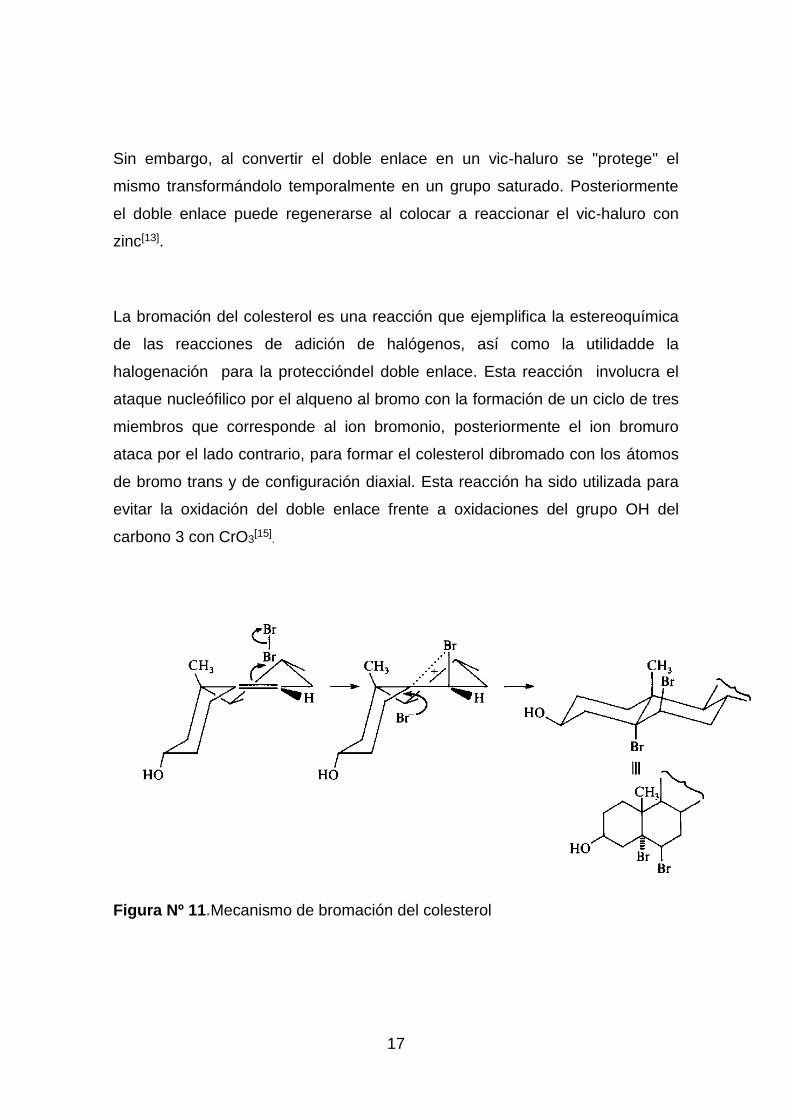
La bromación del colesterol es una reacción que ejemplifica la estereoquímica
de las reacciones de adición de halógenos, así como la utilidadde la
halogenación para la proteccióndel doble enlace. Esta reacción involucra el
ataque nucleófilico por el alqueno al bromo con la formación de un ciclo de tres
miembros que corresponde al ion bromonio, posteriormente el ion bromuro
ataca por el lado contrario, para formar el colesterol dibromado con los átomos
de bromo trans y de configuración diaxial. Esta reacción ha sido utilizada para
evitar la oxidación del doble enlace frente a oxidaciones del grupo OH del
carbono 3 con CrO3[15]
.
Figura Nº 11.Mecanismo de bromación del colesterol

18
2.5 Reacciones de oxidación
Se entiende por oxidación una reacción en la cual el sustrato gana oxígeno o
pierde hidrógeno (deshidrogenación).
2.5.1 Oxidación o deshidrogenación de alcoholes a aldehídos y cetonas
La oxidación de alcoholes a compuestos carbonílicos puede realizarse a través
de una gran variedad de reactivos. La elección del reactivo y el conjunto de
condiciones a utilizar depende de factores tales como la escala de reacción, la
velocidad de la reacción, el rendimiento de la misma y la facilidad de
aislamiento de los productos [16].
La utilización de agentes oxidantes fuertes como el ácido crómico ha permitido
la conversión de alcoholes a aldehídos y cetonas. Un ejemplo de esto es la
oxidación de alcoholes secundarios de forma sencilla a su correspondiente
cetona con ácido crómico a temperatura ambiente ó ligeramente más alta[17].
El mecanismo de oxidación de un alcohol con ácido crómico para dar un
compuesto carbonílico tiene como primera etapa la formación de un éster
crómico, mediante la esterificación del alcohol[16] [17].

19
Esterificación:
OH Cr
O
O
OH+ R O Cr
O
O
OH + OH2R OH
Ácido Crómico
Si el éster posee un átomo de hidrógeno en α puede ocurrir una eliminación,
siendo este el paso oxidativo.
Oxidación:
O Cr
O
O
OHHOH2 OH3
+++ O +
-CrO3H
Figura Nº 12. Secuencia de pasos para la oxidación de un alcohol
La oxidación de un alcohol con ácido crómico a menudo se realiza con
dicromato de potasio (o sodio) en medio ácido, el mecanismo de oxidación de
un alcohol a una cetona es el siguiente[16] [17]:

20
H2O Cr
2O
7
2- 2 HCrO4
- +
R-OH HCrO4
- H+ H2O+ + Cr
O
O
OHOR +
HOH
H
Cr
O
O
OHOR R O
Figura Nº 13. Mecanismo de Oxidación de un alcohol con dicromato
De los diferentes estados de oxidación de cromo (+2 a +6), elCr+6 es la especie
oxidante por excelencia y se reduce normalmente a Cr+3 en una reacción
Redox. Para facilitar el manejo, la oxidación con ácido crómico a menudo se
realiza con dicromato de potasio (o de sodio) en medio ácido. El equilibrio entre
ácido crómico-cromato-dicromato, depende de la concentración [16].
Existe una variedad de reactivos que emplean Cr+6 como un fuerte agente
oxidante, para la obtención de aldehídos y cetonas a partir de alcoholes,
entre estos reactivos destacan: El reactivo de Jones, el Reactivo de Collins y el
Reactivo de Sarett, los cuales se caracterizan por ser agentes oxidantes
selectivos[17].
El Reactivo Sarett se utiliza en la oxidación de alcoholes de manera efectiva
empleando el complejo de Oxido de cromo (VI)-piridina, en piridina. El complejo
fue sintetizado por primera vez en 1948 por Sislery col. cuya composición
empírica era CrO3·2C5H5N, esta oxidación permite la conversión del alcohol al
correspondiente grupo carbonilo.El complejo de piridina es útil para la oxidación

21
de sustancias que contienen grupos sensibles al medio ácido, este reactivo
permite la oxidación de alcoholes alílicos primarios y el alcohol bencílico a su
correspondiente aldehído, con un rendimiento entre el 50 y el 80 %[16] [17].
Para obtener un aldehído no volátil se ha utilizado la oxidación de alcoholes
primarios con CrO3 en piridina (Reactivo de Sarett) que conduce a rendimientos
muy aceptables. Se ha atribuido el éxito de este método al hecho de que bajo
estas condiciones la concentración de agua aumenta la formación del hidrato,
siendo este hidrato de aldehído, el que a continuación se oxida a su ácido
carboxílico a través de un éster crómico [18].
RCH2OH H
2CrO
4 RCHO+
RCH=O H2O +
OH
OH
H
R
OH
OHR
H
+ Cr
O
O
OH OH
OH2
Cr
O
O
OHO
OH
H
R + OH2
O
OHR+-CrO
4H+ OH3
+
Figura Nº 14.Oxidación del hidrato de aldehído al ácido carboxílico
El reactivo de Jones se caracteriza por la utilización de CrO3 como agente
oxidante disuelto en ácidosulfúrico, agua y acetona. Cuando alcoholes
secundarios se disuelven en acetona, la oxidación de estos a cetonas con el
reactivo de Jones ocurre rápidamente en un alto rendimiento, sin perturbar
algún doble o triple enlace que pudiese estar presente, permite la oxidación de
alcoholes primarios alílicos a su correspondiente aldehído aunque durante
este procedimiento se puede obtener el ácido carboxílico[16].

22
Los reactivos de Jones (CrO3 en H2SO4y acetona) y Sarett (CrO3 en piridina)
son especialmente valiosos para la oxidación de alcoholes sin afectar a los
dobles enlaces que están presentes en la misma molécula. [19]
OH
R2
R
OH
R2
R
CrO3/ H
2SO
4/ H
2O R R2
O
R R2
O
N
CrO3
2+ CrO
N+
N+
O-
O-
+
Acetona
Reactivo de Jones
Reactivo de Sarett
Figura Nº 15. Reacciones generales para la formación del Reactivo de Jones
y del reactivo de Sarett.
El colesterol puede ser oxidado al 5-colesten-3-ona por el reactivo de Jones o
por el reactivo de Collins, el cual utiliza el complejo CrO3-piridina
(CrO3·2C5H5N) en diclorometano como medio de reacción, ambos reactivos
tienen la ventaja de que no promueven la isomerización del doble enlace ∆5 a
la posición ∆4[17].

23
2.6 Reacciones de reducción
La reducción es aquella reacción en la cual el sustrato gana hidrogeno o
pierde oxígeno (desoxigenación). El término reducción se asocia generalmente
con la saturación de un doble o triple enlace C-C o C-X donde X puede ser N,
O, S, etc. Sin embargo, también son procesos reductivos las sustituciones de
los enlaces C-X (X: halógenos, oxígeno, nitrógeno, azufre, etc.) por enlaces C-
C ó C-H [15].
2.6.1 Reducciones de carbonilos a metilenos en aldehídos y cetonas
Hay varias maneras de reducir el grupo C=O de aldehídos y cetonas a CH2.
Los dos más antiguosmétodos, pero todavía muy populares, son la reducción
de Clemmensen y la reducción de Wolff-Kishner. La reducción de
Clemmensen consiste en el calentamiento del aldehído o la cetona con una
amalgama de zinc en una solución de ácido clorhídrico. En la reducción de
Wolff-Kishner el aldehído o la cetona se calienta con hidrato de hidracina en
una base (usualmente hidróxido de sodio ó hidróxido de potasio) [16].

24
2.6.1.1 Reducción de Clemmensen
La Reducción de Clemmensen es un método utilizado para reducir carbonilos
a metilenos, permite la reducción de alquilaril cetonas a su correspondiente
hidrocarburo, como por ejemplo la acetofenona a etilbenceno[20].
COCH3 CH
2CH
3
Zn(Hg)-HCl
80%
Figura Nº 16. Reducción de la acetofenona empleando el método de
Reducción de Clemmensen.
Este método fue introducido por Clemmensen,como originalmente se aplicó, el
método consistía en reflujar la cetona con una amalgama de zinc y ácido
clorhídrico concentrado [20].
Los estudios de la reducción de Clemmnensen en monocetonashan dado lugar
a varias teorías concernientes al mecanismo de reacción. Los resultados
cinéticos obtenidos por Nakabayashi en 1960 mostraban que la velocidad de la
reacción depende tanto del ion cloruro como de la concentración de zinc, pero
era independiente del potencial del electrodo de la amalgama de zinc y de la
concentración de protones (H+).Este resultado llevo a la formulación del
siguiente esquema[20].

25
O + Zn C O-Zn
+
ClH
Cl
O-
Zn
H+
OH2
C+
Zn Cl
Zn
Zn2+
+C-
Zn Cl
H+
Zn Cl
HH
+
Cl-
H
H
+
gas
-
ZnCl2
Figura Nº 17. Mecanismos de reducción de Clemmensen, planteado por
Nakabayashi para la obtención de un metileno a partir de un carbonilo. [16] [21]
El complejo formado inicialmente determina la velocidad de adición del zinc al
carbonilo, el cual se protona y se pierde agua para dar un ion
carbonioorganometálico. El ataque por dos moléculas de zinc seguido de la
protonación daría el alcano correspondiente[21].
El sustrato posee dos sitios disponibles para el ataque inicial del metal uno de
ellos es el oxígeno del carbonilo y el otro es el átomo de carbono del carbonilo.
Sin embargo la velocidad de reacción de los compuestos disfuncionales, es
mucho más rápido que el observado en la reducción de las monocetonas, por
lo tanto para este caso se cree más probable el ataque inicial del zinc al
oxígeno. El mecanismo para las monocetonas de Nakabayashi postula el
ataque inicial del zinc al átomo de carbono del carbonilo.[21].

26
6.2.1.2 Reducción de Wolff Kisnher
La reducción de Wolff Kisnher, se utiliza con compuestos sensibles a un medio
acido, permite la transformación de un grupo carbonilo a su correspondiente
metileno y se basa en la descomposición de la hidrazona en medio fuertemente
básico y a elevadas temperaturas. El mecanismo de reducción consiste en una
primera etapa donde se forma la hidrazona correspondiente. Posteriormente
esta se descompone a través de la transposición de un protón catalizado por la
base y finalmente interviene un carbanión originado por la pérdida de la
molécula estable de nitrógeno N2, que es saturado con los protones del
disolvente[22].
Figura Nº18. Mecanismo de reducción de Wolff-Kishner de un grupo carbonilo
a un metileno

27
La Reducción Clemmensen suele ser más fácil de realizar, pero no para
sustratos sensibles a los ácidos y de alto peso molecular. Para estos casos la
reducción de Wolff-Kishner es bastante útil. Para sustratos de alto peso
molecular, una modificación de la reducción Clemmensen, utilizando zinc
activado y HCl gaseoso en un disolvente orgánico, tal como éter o
anhídridoacético, ha demostrado su eficacia.Las reacciones de Clemmsen y
Wolff-Kishner son complementarias, ya que la primera se realiza en
condiciones acidas, mientras que la segunda en un medio básico [17].

28
3. OBJETIVOS
3.1 Objetivo General:
• Sintetizar la androst-4-en-3,6,17-triona para su posterior reducción.
• Estudiar una posible ruta para la síntesis de la androst-5-en-3,17-diona a
partir de la dehidroepiandrosterona
3.2 Objetivos Específicos:
• Bromar la dehidroepiandrosterona (3β -hidroxi-androst-5-en-17-ona).
• Oxidar el compuesto bromado con el Reactivo de Jones.
• Sintetizar y caracterizar la Δ4-androst-3,6,17-triona a partir de la
oxidación de la dehidroepiandrosterona con el Reactivo de Jones
• Sintetizar y caracterizar la androst-4-en-17-ona a través de la reducción
de HuangMinlon de la Δ4-androst-3,6,17-triona.

29
4. ANTECEDENTES
La oxidación de grupos hidroxilos a aldehídos o cetonas había sido
conseguida durante mucho tiempo con el uso de ácido crómico y las sales del
mismo, las cuales eran utilizadas solo en medio ácido. Al tratar con moléculas
polifuncionales, las cuales presentaban en su estructura grupos sensibles al
medio ácido, estos se veían afectados por el reactivo, es por ello que surgió la
necesidad de trabajar con agentes oxidantes capaces de operar
selectivamente.
En 1952 Poos y col.[23]fueron los primeros en reportar el uso del complejo de
CrO3-piridina, para la oxidación selectiva de grupos hidroxilo a grupos
carbonilo, pertenecientes a estructuras tricíclicas, sin afectar los dobles
enlaces presentes. Estos creían que elcomplejo derivado deanhídridocrómicoy
una aminaterciariaestablepodría sercapaz de añadir aungrupo hidroxilo para
formarun éster decromato tal y como ocurría en la oxidación del alcohol con
ácido crómico y luegodebía descomponerse dela manera habitual paradarla
correspondiente cetonaó aldehído. De acuerdo con los resultados obtenidos el
complejo resultó ser moderadamente soluble en piridina y escasamente soluble
en otros solventes orgánicos como benceno, dioxano y acetona además de ser
inerte ante dobles enlaces y generar buenos rendimientos de reacción con los
distintos sustratos empleados.
En 1961 John Holum[24] estudió la efectividad del complejo de óxido de cromo
(VI)-piridina para la conversión de alcoholes alílicos y bencílicos a sus
correspondientes aldehídoso cetonas, así como la determinación de las
limitaciones del reactivo en un número sustancias. Los resultados
experimentales fueron obtenidos empleando las mismas condiciones y
proporciones relativas de reactivos reportadas por Poosy col. Estas

30
condiciones incluían el uso de una relación molar 3:1 de complejo a alcohol a
temperatura ambiente, la piridina como un medio dispersante para el complejo
y un tiempo de reacción que varió entre 15 y 22 horas. Los rendimientos se
determinaron en cada caso por alguna de estas tres formas: pesando
directamente el producto obtenido; usando el método de Smith y Mitchellel cual
involucraba la titulación del ácido clorhídrico liberado cuando reacciona el
clorhidrato de hidroxilamina con el aldehído o la cetona y por la determinación
del rendimiento víaobtención de la semicarbazona.En general se encontró que
el complejo óxido de cromo (VI)-piridina es un buen reactivo para la conversión
a temperatura ambiente de alcoholes bencílicos y alílicos a sus
correspondientes aldehídos o cetonas.
Experimentos realizados por Holumcon dicromato de amonio en una solución
acuosa de piridina indican que su poder oxidante es inferior al complejo CrO3-
piridina, obteniéndose el benzaldehído con rendimientos del 33% y 41 % en
comparación del 63% utilizando el complejo. La acetona fue utilizada como una
alternativa del medio dispersante para el complejo en sustitución de la piridina,
(El complejo trióxido de cromo-piridina es sólo ligeramente soluble en acetona
e insoluble en dimetilsulfóxido, nitrometano, acetato de etilo, bromuro de etilo,
nitrobenceno y cloroformo). Bajo condiciones donde la piridina es el medio
dispersante del complejo, se logró un rendimiento del 63% del benzaldehído a
partir del alcohol bencílico y un 71% de benzofenona a partir del benzidrol
mientras que durante la utilización de la acetona como medio dispersante se
generó un 41% de benzaldehído y un 45% de benzofenona (con un 49% de
recolección del benzidrol).
En 1968 con la finalidad de mejorar las propiedades oxidantes del complejo
Collins y col. [25]iniciaron la búsqueda de un solvente apropiado para llevar a

31
cabo la oxidación. Estosdeterminaron la solubilidad del complejo en diferentes
solventes, para posteriormente seleccionar uno en el cual se pudiese mejorar
las condiciones de la reacción de oxidación y de esta forma obtener mejores
rendimientos de reacción.En la siguiente tabla se muestran las solubilidades
obtenidas por Collins para el complejo dipiridina- anhídrido crómico en una
serie de solventes:
Tabla N° 1.Solubilidad del complejo oxido de cromo (VI)- piridina a 25 ºC
Solvente g/100mL Solvente g/100mL
Diclorometano 12,5 1,2-dicloroetano 3,2
Cis-1,2-dicloroetileno 7,1 1,1-dicloroetano 1,5
Piridina 6,1 tetraclorometano 1,4
Triclorometano 4,5 Trans-1,2 -dicloroetileno 0,5
Collins y colaboradores obtuvieronque el complejo, es apreciablemente soluble
en diclorometano siendola solubilidad del mismo en clorocarbonos
aproximadamente paralela a la polaridad delmedio. Por otra parte notó que el
complejo es hidrofílico, formando dicromato de dipiridino insoluble en
clorocarbonos, ya que se hidrata fácilmente, lo cual implica que se debelimitar
la exposicióndel mismo a la humedaddurante su uso y aislamiento del
producto.
Posteriormente Collins realizo una serie de oxidaciones en alcoholes primarios
y secundarios con el complejo de CrO3-piridinaen diclorometano, con dichas
reacciones obtuvo que los rendimientos más altos para la conversión del grupo
hidroxilo al grupo carbonilo se generan en una relación molar 6:1 de complejo
a alcohol.

32
Tabla N° 2. Oxidación de alcoholes empleando el complejo CrO3-piridina en
diclorometano a una temperatura de 25 ºC
Alcohol
% de rendimiento de la cetona
o del aldehído obtenido
2-Butanol 98
2-Octanol 97
Ciclohexanol 98
Bencidrol 96
Colesterola 64 (Δ5)
1-Heptanol 93
Alcohol bencílico 95
Alcohol 4-nitrobencilico 97
Alcohol 3-hidroxibencilico 87
aLa oxidación del colesterol fue conducida por 30 minutos a 10ºC, la solución del producto fue lavada con
ácido Clorhídrico al 3% para evitar la epimerización por trazas de piridina
Entre los resultados de Collins destacan la selectividad del reactivo en
diclorometano es ilustrada en la conversión del colesterol al colest-5-en-3-ona,
sin desplazar el doble enlace conjugado al grupo OH y en el rendimiento
razonable del 3-hidroxibenzaldehido, sin proteger el fenol. La oxidación del
colesterol generó otros dos productos: colest-4-en-3,6-diona en un 10% y la
colest-4-en-3-ol-6-ona en un 8% con pequeños rendimientos a diferencia de la
oxidación directa del colesterol con ácido crómicoen la que son los principales
productos.

33
En 1970 Ratcliffe y col. [26] en su trabajo con el complejo CrO3-piridina,
destacan que es el reactivo de elección para la oxidación selectiva de un
alcohol. Estos encontraron que las principales complicacionesy molestias en la
preparación del reactivo, su carácter higroscópico y su facilidad para
inflamarse durante su preparación pueden evitarse simplemente con la
preparación directadel complejo en soluciones de diclorometano utilizando una
relación molar 2:1 de piridina a CrO3. Por otra parte ratifican que la relación
molar 6:1 de complejo-alcohol obtenida por Collins proporciona la mayor
conversión del alcohol al aldehído o la cetona, en el menor tiempo posible.
En la tabla Nº3 se listan varios alcoholes que fueron oxidados por Ratcliffe y
col. utilizando soluciones del complejo trióxido de cromo-piridina preparadas in
situ, en diclorometano,la reacción fue hecha a temperatura ambiente, el
tiempo de duración fue 15 minutos y se empleóuna relación molar 6:1 de
complejo a alcohol.

34
Tabla Nº 3.Oxidación de alcoholes con trióxido de cromo (VI) –piridina en
diclorometano (preparado in situ).
Alcohol Mmol de alcohol
oxidado
% rendimiento del aldehído
o la cetona
(1)2-Octanol 5,0 97
(2)1-Octanol 5,0 90
(3)Alcohol bencílico 5,0 89
(4)Borneol 5,0 84
(5)3-fenil-2-propen-ol 5,0 96
(6) 137,0 94
(7) 26,4 99
(8) 1,4 95
(9) 42,6 85
(10) 11,5 90
O
O H
OH
OHH
OCH2ph OCH
2ph
O
O
OHH
O
O
OH
HH
OH
6 7 8 9 10
En 1976 Piers y col. [27]estudiaron el efecto de varios parámetros de reacción
durante la oxidación del colesterol (11) con el complejo de óxido de cromo
(VI)-piridina en diclorometano. La oxidación de (11) bajo una variedad de

35
condiciones proporcionó en cada caso el producto esperado, el colest-5-en-3-
ona (12), acompañado de una variedad de cantidades de colest-4-en-3,6-diona
(13) y cantidades trazas de 6β-hidroxi-colest-4-en-3-ona (14), además
cantidades variables del material de partida (11) fueron recuperadas,
encontrándose que la eficiencia en la oxidación del producto depende de la
atmósfera utilizada, temperatura, relación molar oxidante/alcohol, relación
molar CrO3/piridina y de la amina utilizada.
O
O
OH
O
O
OO
H
OOH
H+ H+
(16) (11) (15)
(13) (12) (14)
Figura Nº 19. Productos obtenidos durante la oxidación del Colesterol con el
complejo de óxido de cromo (VI)-piridina en diclorometano
En la tabla Nº 4 se muestran algunos de los resultados obtenidos durante la
oxidación del colesterol con el complejo trióxido de cromo-piridina en
diclorometano.
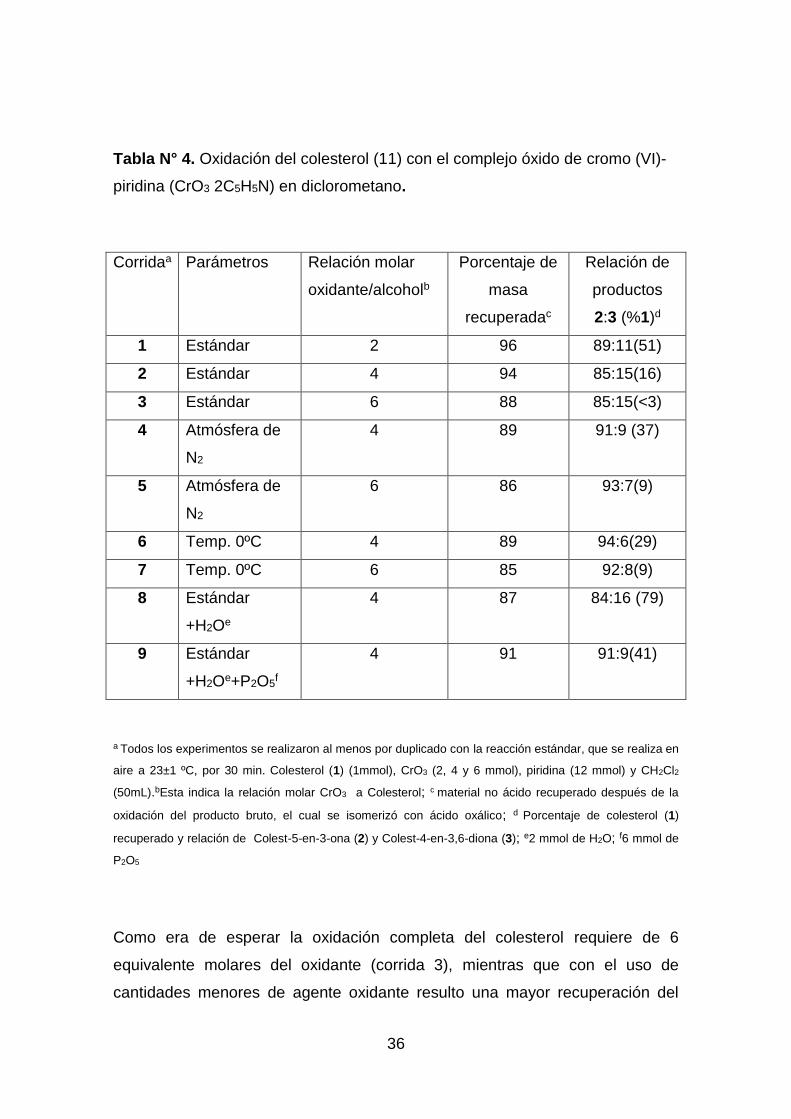
36
Tabla N° 4. Oxidación del colesterol (11) con el complejo óxido de cromo (VI)-
piridina (CrO3 2C5H5N) en diclorometano.
Corridaa Parámetros Relación molar
oxidante/alcoholb
Porcentaje de
masa
recuperadac
Relación de
productos
2:3 (%1)d
1 Estándar 2 96 89:11(51)
2 Estándar 4 94 85:15(16)
3 Estándar 6 88 85:15(<3)
4 Atmósfera de
N2
4 89 91:9 (37)
5 Atmósfera de
N2
6 86 93:7(9)
6 Temp. 0ºC 4 89 94:6(29)
7 Temp. 0ºC 6 85 92:8(9)
8 Estándar
+H2Oe
4 87 84:16 (79)
9 Estándar
+H2Oe+P2O5f
4 91 91:9(41)
a Todos los experimentos se realizaron al menos por duplicado con la reacción estándar, que se realiza en
aire a 23±1 ºC, por 30 min. Colesterol (1) (1mmol), CrO3 (2, 4 y 6 mmol), piridina (12 mmol) y CH2Cl2
(50mL).bEsta indica la relación molar CrO3 a Colesterol; c material no ácido recuperado después de la
oxidación del producto bruto, el cual se isomerizó con ácido oxálico; d Porcentaje de colesterol (1)
recuperado y relación de Colest-5-en-3-ona (2) y Colest-4-en-3,6-diona (3); e2 mmol de H2O; f6 mmol de
P2O5
Como era de esperar la oxidación completa del colesterol requiere de 6
equivalente molares del oxidante (corrida 3), mientras que con el uso de
cantidades menores de agente oxidante resulto una mayor recuperación del

37
material departida11(corridas 2).La caída en la eficiencia de la oxidación junto
con el aumento en la proporción de productos 2:3causada por el reemplazo del
aire atmosférico con nitrógeno (corridas 4 y 5) era desde un punto de vista
sintético una observación significativa y era presumible debido a la conocida
absorción de oxígeno en las oxidaciones de cromato y la susceptibilidad de 12
a la auto-oxidación. Además en contraste con las conclusiones de Collins una
menor temperatura de reacción pareció disminuir la eficiencia de la oxidación
(al menos dentro de una reacción de tiempo especificado) pero también
produce sintéticamente una más agradable relación 2:3 (corridas 6 y
7).Finalmente la adición de agua (corrida 8)tuvo el efecto esperado, ya que se
sabe que el complejo CrO3 2C5H5N se hidrata fácilmente para dar la especie
insoluble y no reactiva C10H12-Cr2N2O7.La adición de un agente deshidratante
como el P2O5(corrida 9), no puede restaurar eficientemente la oxidación
completa
Los experimentos realizados porPiers y col. variando las proporciones entre el
CrO3 y la piridina con el objetivo de determinar el efecto de la adición de
diferentes cantidades de piridina in situ, durante la oxidación de (1) con el
complejo de óxido de cromo (VI)-piridina en diclorometano, muestran
claramente que aunque la piridina es necesaria para la oxidación no hay
justificación de la práctica que emplea la relación molar 2:1 de piridina a CrO3
para la generación in situ del complejo de Sisler, así el uso de una proporción
1:1CrO3-piridina durante la oxidación da resultados similares a los obtenidos
durante el empleo de las dos sustancias en una relación1: 2 y 2:1.Estos
autores sugieren que la oxidación se lleva a cabo vía el complejo
monopiridina(17)en lugar del complejo dipiridina(18)generalmente postulado.
Las estructuras de estos complejos se muestran a continuación:

38
N+
Cr
O-
OO
O-
N+
CrN+
O
O-
(17) (18)
Figura Nº 20.Complejos monopiridina (17) y dipiridina (18)
En cuanto a los resultados obtenidos para la oxidación de 1(1mmol) con CrO3
(6mmol) en presencia de diferentes aminas heterocíclicas (6mmol) en
diclorometano por 30 minutos, la piridina pareció ser la amina más favorable
para la reducción del producto 13.Inesperadamente fue encontrado que la
oxidación no ocurre virtualmente cuando las aril aminas como la N,N-
dimetilanilina (pKb =5,15) son usadas, del mismo modo para la 2,2-bipiridina y
la 4,4- bipiridina donde ninguna cantidad del colesterol o casi nada era
oxidado.
En 1953 Fieser[28] realizó la bromación del colesterol, en éter con la adición de
una solución de bromo en ácido acético, obteniendo el producto dibromado con
un rendimiento del 72-74%, posteriormente logró la desbromacion del mismo
utilizando una suspensión del producto dibromado en éter la cual contenía
pequeñas cantidades de ácido acético, mezclando la misma con zinc a
temperatura ambiente, generándose un rendimiento del 93% de colesterol.
Con las mejoras al procedimiento de desbromación, Fieser logró su aplicación
durante la síntesis de la Δ5-Colesten-3-ona, la cual se llevó a cabo mediante
una secuencia de pasos que implicaban la bromación del colesterol hasta la
obtención del producto dibromado el cual fue oxidado con dicromato de sodio

39
(Na2Cr2O7) hasta la 5α,6β-dibromocolest-3-ona con un 96,5% de rendimiento y
finalmente dicho producto fue desbromado hasta generar la colest-5-en-3-ona
con un 88% de rendimiento
OBr
Br
OHBr
Br
C8H
17
C8H
17
C8H
17
C8H
17
Br2
HOAc/Et2O
Zn
Na2Cr
2O
7 / HOAc
O
OH
Figura Nº 21. Esquema de síntesis para la obtención Δ5-Colesten-3-ona,
mediante una bromación del doble enlace, oxidación del alcohol del C-3 y
desbromación del doble enlace.
En 1956Djerassi y col. [29]lograron obtener la conversión directa del grupo OH
de esteroides Δ5-3β-alcohol a Δ5-y Δ4-3-cetona, a la correspondiente cetona,
sin la necesidad de proteger el doble enlace. La conversión del Δ5-3β-alcohol
al Δ5-3-cetona, se lleva a cabo por una secuencia de tres etapas que implican,
la protección del doble enlace por una bromación, oxidación con trióxido de

40
cromo a la dibromo cetona y finalmente la desbromación con zinc en ácido
acético
.
La utilización del procedimiento experimental originado por Jones y col.[30] el
cual involucró la adición de una solución de trióxido de cromo-ácido sulfúrico a
una solución del alcohol en acetona, permitió la obtención directa de la Δ5-3-
cetona en los esteroides utilizados. Entre los resultados reportados por
Djerassiy col.destacan la oxidación de la dehidroepiendrosterona (Δ5-
androsten-3β-ol-17-ona) con un 76% de rendimiento a la resultante Δ5-
androsten-3,17-diona.
OH
Ia R=COCH3
Ib R=COCH2OAc
Ic R=O
Id R=OCOC6H
5
O OCrO
3/H
2SO
4/H
2O
R
H+Acetona
Figura Nº 22. Esquema de síntesis para la conversión directa del grupo OH
de esteroides Δ5-3β-alcohol a Δ5- y Δ4-3-cetona, utilizando el reactivo de Jones

41
En 1949 HuangMinlon[31]realizó una modificación al procedimiento de
reducción de Wolff-Kishner, la cual consistía en eliminar el agua del medio,
evaporándola durante el período de calefacción para de esta forma obtener
mayores rendimientos en la conversión del grupo carbonílico al grupo
metilénico, ya que según Dutcher y col.[31]a través del procedimiento habitual
de Wolff-Kishner se obtenía el correspondientealcohol y en muy pequeñas
cantidades el producto metilénico. De acuerdo con los resultados obtenidos la
reacción procede en grupos ceto-esteroidales donde los grupos ceto se ubican
en las posiciones C3, C7, C12, C17 y C20.
OH
O
OH
Etilenglicol /KOH
NH2 NH2
Figura Nº 23. Esquema de síntesis para conversión del grupo carbonilo de C-
17 en la dehidroepiandrosterona a través de la reducción de Wolff-Kishner.
42
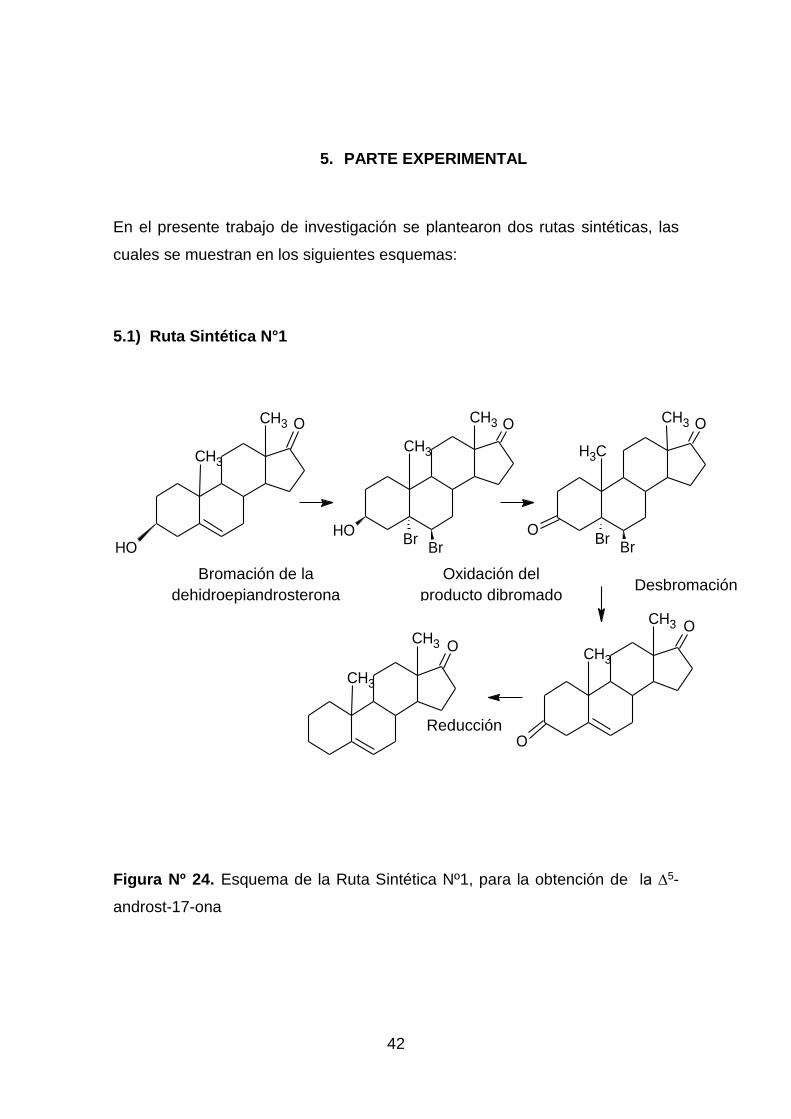
5. PARTE EXPERIMENTAL
En el presente trabajo de investigación se plantearon dos rutas sintéticas, las
cuales se muestran en los siguientes esquemas:
5.1) Ruta Sintética N°1
Figura Nº 24. Esquema de la Ruta Sintética Nº1, para la obtención de la ∆5-
androst-17-ona
O
OHBr
Br
CH3
CH3 O
OBr
Br
CH3
CH3
O
OH
CH3
CH3
O
O
CH3
CH3O
CH3
CH3
Bromación de la
dehidroepiandrosterona
Oxidación del
producto dibromado Desbromación
Reducción
43
5.2) Ruta Sintética N°2
Figura Nº 25.Esquema de la Ruta Sintética Nº2, para la obtención del la ∆4-
androst-17-ona.
O
O
CH3
OCH3
CH3
OCH3
CH3
CH3
O
OH
Oxidación directa de la
dehidroepiandrosterona
Reducción del producto
de la oxidación

44
5.1 Ruta Sintética Nº1
Síntesis del Δ5-androst-3,17-ona, a través de una secuencia de tres etapas que
implican, la protección del doble enlace por una bromación, oxidación a la
dibromo cetona y finalmente la desbromación con zinc en ácido acético, para la
posterior reducción del producto obtenido.
5.1.1 Bromación del 3β -hidroxi-androst-5-en-17-ona.
Se disolvió (1,04021 ± 0,00001g; 3,61mmol) de la 3β-hidroxi-androst-5-en-17-
ona en 20,0 ± 0,1mL de éter, en esta solución se le adicionó lentamente 5,4 ±
0,1mL de la solución de Br2 en ácido acético. La solución para bromar se
preparó de la siguiente manera: A 15,0 ± 0,1 mL de ácido acético colocados en
una fiola se le agregó 0,54253 ± 0,00001g de acetato de sodio con agitación y
2,7 ± 0,1mL de Bromo. La reacción se mantuvo con agitación constante y a
temperatura ambiente durante 10 horas, el curso de la reacción fue seguido
por cromatografía de capa fina. Finalizada la reacción se observó la
precipitación de un sólido de color amarillo correspondiente al producto, el cual
fue filtrado. Las aguas madres fueron diluidas para asegurar la precipitación
completa del producto dibromado, obteniéndose nuevamente la precipitación
del producto, el mismo fue lavado con metanol frio. El rendimiento de reacción
obtenido fue de un 80 %.

45
5.2 Ruta Sintética Nº2
Síntesis de laΔ5-androst-3,6,17-triona, a partir de la oxidación directa de la
dehidroepiandrosterona (3β -hidroxi-androst-5-en-17-ona), para la posterior
reducción de HuangMinlon del producto obtenido.
5.2.1 Oxidación del 3β-hidroxi-androst-5-en-17-ona, con el Reactivo de Jones
Se disolvió (1,04418 ± 0,00001g; 3,62mmol) de 3β-hidroxi-androst-5-en-17-ona
en 13,0 ± 0,1mL de acetona, posteriormente la solución fue llevada a un baño
de hielo (Temperatura = 0 ºC), seguidamente se le adicionó durante 15 minutos
9,0 ± 0,1mL del reactivo de Jones con agitación. El reactivo se preparó de la
siguiente forma; a 9,48324± 0,00001 g de Na2CrO3·2H2O, se le añadió 10,0 ±
0,1 mL de H2SO4 97-98% poco a poco y con enfriamiento, se observó la
formación de un sólido de color rojo correspondiente CrO3 el cual fue disuelto
con 15,0 ± 0,1mL de agua, luego la solución a temperatura ambiente, se llevó a
un volumen de 50,0 ± 0,1mL. La reacción se mantuvo por 8 horas en el baño
de hielo, siendo seguida por CCF. Finalizada la reacción, se procedió a
evaporar la acetona. La mezcla resultante cuya coloración era verde se
adicionó a una mezcla agua- hielo y se diluyó con 300 mL de agua,
obteniéndose la precipitación de un sólido de color amarillo.
El sólido obtenido fue recristalizado con una mezcla metanol-agua,
obteniéndose del mismo 0,56328 ± 0,00001g. El rendimiento de la reacción
obtenido fue de un 54%.

46
5.2.2 Reducción de laandrost-5-en-3,6,17-triona empleando la Reducción de
Huang-Minlon.
Para la reducción de la Δ4-androst-3,6,17-triona, se emplearon dos
procedimientos, el primero consistió en una pequeña modificación al método
de Huang Minlon la cual implicaba la formación inicial de la hidrazona de la Δ5-
androst-en-3,6,17-trionade manera aislada, sin emplear las condiciones
básicas (KOH) y la elevada temperatura, para posteriormente continuar con el
procedimiento original de Huang-Minlon.
En segundo método consistió en realizar la reducción directa del producto
oxidado siguiendo el procedimiento original de Huang-Minlon[30].
5.2.2.1 Síntesis de la hidrazona de la Δ4-androst-3,6,17-triona
Se disolvió (0,32481±0,0001g; 1,08mmol) del producto de la oxidación directa
en 20±0,01mL etanol en caliente con agitación constante, posteriormente se
adicionó gota a gota 0,05±0,01mL de Hidrato de hidracina al 67%,
correspondiente a una relación molar 1:1 para de esta forma evitar adicionar un
exceso del Hidrato de hidracina que pudiese generar la doble hidrazona. La
reacción se mantuvo a una temperatura de 65º C, un pH= 6 y con agitación
constante por 48 horas. Una vez finalizada la reacción la mezcla fue evaporada
a presión reducida y se obtuvo un sólido de color amarillo.
La baja solubilidad del compuesto dificultó la purificación del mismo, la cual se
realizó mediante cromatografía de columna, utilizando una mezcla de solventes
benceno-metanol (90-10v/v)

47
5.2.2.2 Reducción de la hidrazonadel Δ4-androst-en-3,6,17-triona
Se adicionó a un balón 15,0 ± 0,1 mL de etilenglicol y 1,0400 ± 0,00001 g de
hidróxido de potasio la solución se mantuvo en un reflujo hasta que todo el
hidróxido de potasio se disolvió, posteriormente se adicionó 0,23299 ± 0,00001
g de la hidrazona sintetizada disuelta en 20,0 ± 0,1mL de etanol, el reflujo se
mantuvo durante cinco horas. Finalizada la reacción, la mezcla fue neutralizada
con HCl(c) hasta obtener un pH = 7, posteriormente se realizó una extracción
con CH2Cl2, la capa orgánica fue secada con carbonato de calcio y el solvente
fue evaporado, obteniéndose como producto un aceite de color marrón el cual
fue purificado por cromatografía en columna empleando un mezcla de solvente
diclorometano-metanol (9:1; v/v)
5.2.2.3 Reducción directa Δ4-androst-en-3,6,17-trionaempleando el método
de Huang –Minlon.
Se adicionó a un balón 15,0 ± 0,1 mL de etilenglicol y 2,19035± 0,00001 g de
hidróxido de potasio, la solución se mantuvo en un reflujo conectado a una
trampa de agua por una hora hasta que todo el hidróxido de potasio se disolvió,
posteriormente se adicionó (0,28061 ± 0,00001g; 0,934mmol)la Δ4-androst-
3,6,17-triona, disuelta en 20±0,1mL de etanol, Luego se agregó gota a gota
0,1±0,01mL de Hidrato de hidracina al 67%.El reflujo se mantuvo durante tres
horas con la trampa de agua. Una vez finalizada la reacción, la mezcla fue
neutralizada con HCl(c) hasta obtener un pH = 7, posteriormente se realizó una
extracción con CHCl3,la capa orgánica fue secada con carbonato de calcio y el
solvente fue evaporado, obteniéndose como producto un aceite de color
marrón.

48
La purificación del producto obtenido se realizó empleando una placa
preparativa, la cual fue desarrollada en cloroformo, finalmente se obtuvo como
producto un aceite de color amarillo, el cual presentó un Rf=0,71 usando como
solvente cloroformo.

49
6. RESULTADOS Y DISCUSIÓN
6.1 Síntesis del Δ5-androst-3,17-ona, a través de una secuencia de tres
etapas que implican, la protección del doble enlace por una bromación,
oxidación a la dibromo cetona y finalmente la desbromación con zinc en ácido
acético.
6.1.1 Bromación del 3β -hidroxi-androst-5-en-17-ona.
La bromación de la dehidroepiandrosterona se realizó con el objetivo de
proteger el doble enlace ubicado en el anillo B.Las condiciones de reacción
involucraron la utilización de éter como solvente y para bromar, una solución de
bromo en ácido acético y acetato de sodio. El ácido acético se utilizó con la
finalidad de polarizar la molécula de bromo y facilitar su adición al doble
enlace.
10
5
1
4
2
3
8
7
9
6
13
14
1211
17
16
15
OH
O
19
18
Br Br 10
5
1
4
2
3
8
7
9
6
13
14
1211
17
16
15
O
19
18
OHBr
Br
Et2O/CH
3COOH
Figura Nº 26. Bromación de la Dehidroepiandrosterona

50
Una vez finalizada la reacción y purificado el producto se determinaron algunas
de sus propiedades físicas, presentando éste un punto de fusión de (182-
185±1) ºC y una alta solubilidad en diclorometano y cloroformo, siendo
totalmente insoluble en agua y parcialmente soluble en metanol y etanol. El
compuesto presentó un Rf=0,66 utilizando como solvente diclorometano
(CH2Cl2). Su caracterización se hizo a través de las técnicas de Espectroscopia
de Infrarrojo (IR), Resonancia Magnética Nuclear de Protones (RMN-1H) y
Resonancia Magnética Nuclear de Carbono (RMN-13C), con el propósito de
determinar si era el compuesto esperado.
El espectro de IR (Espectro N°1, tomado en pastilla de KBr) mostró dos
señales agudas e intensas en 2948 cm-1 y 2871 cm-1 correspondiente a
vibraciones de tensión de enlaces C-H sp3. Centrada en 1759,9 cm-1 una banda
estrecha e intensa, corresponde a una vibración de tensión de un enlace
carbonílico C=O, por otra parte se observa en una vibración de tensión de un
enlace carbonílo de un éster en1732,9 cm-1, en 1240,1 cm-1 una banda intensa
correspondiente a las vibraciones de tensión del grupo acetato, hecho que
indica que ocurrió una esterificación del grupo OH, ya que no se aprecia la
banda ancha correspondientes a vibraciones de tensión de enlacesO–H. En
756 cm-1 se encuentra una señal intensa y aguda asignable a vibraciones de
tensión de enlaces halogenados del tipo C-Br.
En el espectro de RMN-1H (Espectro N°2, CDCl3) se puede observar un
singlete centrado en 1,46 ppm y otro en 1,12 ppm; ambas señales atribuibles a
los protones metílicos localizados en los carbonos 19 y 18 respectivamente.
Centrado en 2,02 ppm se logra observar un singlete deformado el cual
corresponde al metilo del acetato. En 5,47 ppm se aprecia un multiplete, dicho
desplazamiento químico corresponde al protón unido al carbono 3, el cual se
esterificó. En 4,81 y 4,80 se observa una banda, de la cual se presume que son

51
dos singletes solapados los cuales pueden atribuirse a protones unidos a un
carbono unido a un átomo de bromo (CHBr), estos desplazamientos se asignan
a los protones ubicados en los carbonos 6 y 16 respectivamente.
El espectro de RMN-13C, (Espectro N°3, CDCl3) muestra una serie de señales
entre las cuales destacan 170,35ppm valor que se atribuye al carbono
carbonílico del éster formado, por otra parte se observan los desplazamientos
de carbonos halogenados a 86,71ppm y 59,14 ppm lo cual indica que ocurrió
la bromación del doble enlace, estos desplazamientos químicos se asignan a
los carbonos 5 y 6 respectivamente. De acuerdo a lo reportado por la literatura,
el carbonilo de C-17 para la dehidroepiandrosterona posee un desplazamiento
químico de 219 ppm. En los datos espectroscópicos obtenidos se observó que
el carbonilo ubicado en el C-17 del compuesto sintetizado presentó un
desplazamiento a campo alto ubicándose esta señal a 205,75 ppm, este
desplazamiento a campo alto se originó como consecuencia de la bromación
del carbono 16, Cα a la cetona, a éste se le asignó un desplazamiento químico
de 47,31ppm.
De acuerdo a lo reportado por la literatura [13][14][27] y bajo las mismas
condiciones de reacción, la adición del bromo para el doble enlace del
colesterol genera el producto diaxial, el cual corresponde al 5α,6β-
dibromocolesterol. Debido a la similitud del anillo A y B de la
dehidroepiandrosterona y el colesterol, se espera que la estereoquímica de la
reacción haya seguido el mismo curso, obteniéndose en nuestro caso también
el 5α,6β-dibromo. Por otra parte de acuerdo a los datos espectroscópicos
reportados para el bromociclohexano, cuando el bromo se ubica en la posición
axial, el protón que está unido al mismo átomo de carbono donde éste se
encuentra, posee un desplazamiento químico de 4,81ppm y cuando el bromo
se ubica en la posición ecuatorial, el protón aparece a un desplazamiento
químico de 4,09ppm[33].

52
Figura Nº 27. Isómeros del bromociclohexano.
Dentro de los datos espectroscópicos obtenidos en el espectro de RMN-1H,
CDCl3, se obtuvo una señal en 4,81 ppm, la cual coincide con el
desplazamiento químico del protón cuando el bromo se ubica en la posición
axial del bromociclohexano, estos resultados indican que se habría obtenido
5α,6β-dibromo como producto, por lo que el ataque nucleofílico del ion
bromuro se realizó por la cara α de la molécula, la cual estaría menos impedida
estéricamente, el mecanismo de reacción de esta adición puede ser observado
en la Figura Nº11
Con respecto a la estereoquímica de la reacción de adición de bromo en el C-
16, durante la formación de α-halocetonas, las interferencias estéricas
existentes en la molécula causan ataques estéreoespecíficos. Por ejemplo la
bromación de la 3-colestanona en ácido acético genera exclusivamente la 2α-
bromocolestanona, proveniente del ataque de la molécula de bromo por la cara
α del enol, posición en la cual se evita las molestias estéricas con el metilo
angular (C-10) orientado hacia la cara β de la molécula, tal y como se observa
en la siguiente figura [34].
Br
HBr
H

53
Figura Nº 28.Esquema de del mecanismo de reacción para la síntesis de α-
halocetona.
Basados en estas evidencias, la halogenación de la cetona ubicada en el anillo
D (C-17) debería obedecer al mismo mecanismo de adición, ya que la
presencia del metilo angular C-18 genera interferencias estéricas que propician
el ataque a la molécula de bromo por la cara α, por lo que debería obtenerse la
16α-bromocetona. Por otra parte los protones ubicados en C-15, son
diasterotópicos y al determinar teóricamente los acoplamientos con el protón
ubicado en C-16, cuando el bromo se encuentra en la cara α de la molécula,
se obtiene una señal que se caracteriza por presentar dos dobletes para un
protón y un triplete para el otro, con un desplazamiento químico de 2,54 ppm.
Experimentalmente se obtuvo esta señal en el espectro de RMN-1H, CDCl3,
con esas mismas características centrada en 2,79 ppm debido al efecto del
bromo, el cual induce un desplazamiento a campo bajo, esta señal confirma la
estereoquímica α del bromo tal y como lo expresa la literatura.
La utilización de ácido acético durante la síntesis favoreció la bromación del
Cα a la cetona(C-16), ya que propicio la formación del equilibrio ceto-enolico,
para la cetona ubicada en el C-17; de manera que el enol reaccionó con el
bromo generando la α-bromocetona. Por otro lado la reactividad del grupo OH
CH3
OH
Br Br
O
Br
CH3
H
O
CH3

54
ubicado en la posición 3 de la estructura permitió la esterificación del mismo,
mediante la reacción de éste con el ácido acético, presente en el medio de
rección. La siguiente figura esquematiza el producto obtenido durante esta
reacción de bromación.
10
4
1
4
2
3
7
7
9
5
13
14
12
1117
16
15
19
18
OH
O
10
5
1
4
2
3
7
7
9
6
13
14
12
1117
16
15
19
18
O
O
20CH321
O
Br
BrBr
Br Br
Et2O/CH3COOH
Figura Nº 29. Síntesis del 3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona a
través de la bromación de la dehidroepiandrosterona
En base al análisis antes mencionado se procedió a realizar la asignación de
los carbonos según los desplazamientos químicos obtenidos, dichas
asignaciones se muestran en la tabla Nº5

55
Tabla N°5.Datos espectroscópicos de RMN-13C (Espectro N°3, CDCl3), del
producto de la bromación del 3β -hidroxi-androst-5-en-17-ona.
*Datos obtenidos a través de la simulación de la molécula con el programa ACD ChemSketch
versión 3.5
Señal Experimental
δ(ppm)
Señal teórica
δ(ppm)*
Asignación
205,75 213,20 C-17
170,35 170,20 C-20
86,71 82,94 C-5
71,65 71,61 C-3
59,14 55,79 C-6
54,64 48,52 C-14
47,80 47,69 C-13
47,31 47,55 C-16
46,63 47,06 C-9
46,48 51,70 C4
42,03 39,96 C-10
41,3 36,03 C-7
36,41 32,67 C-15
36,20 32,05 C-8
32,44 32,43 C-12
29,53 29,14 C-1
26,06 26,19 C-2
21,30 21,30 C-21
20,13 21,13 C-11
20,04 18,57 C-19
16,36 14,80 C-18

56
La acetilación del grupo hidroxilo, ubicado en la posición 3 del anillo A no
permitió el siguiente paso en la ruta sintética Nº1, el cual corresponde a la
oxidación del grupo hidroxilo, ubicado en C-3, empleando el reactivo de Jones
como agente oxidante. Por tal razón fue descartada la ruta sintética Nº1.

57
6.2 Síntesis del Δ4-androst-3,6,17-diona, a partir de la oxidación directa de
la dehidroepiandrosterona (3β -hidroxi-androst-5-en-17-ona), para su posterior
reducción empleando el método de HuangMinlon
6.2.1 Síntesis del Δ4-androst-3,6,17-diona, a partir de la oxidación directa de
la dehidroepiandrosterona con el reactivo de Jones.
El producto obtenido posee un punto de fusión de (226-228 ±1) ºC y una alta
solubilidad en éter, acetona, diclorometano y cloroformo, siendo totalmente
insoluble en agua. Por otra parte, el rendimiento de la reacción fue de un 64%
y el compuesto presentó un Rf= 0,48 utilizando como solvente éter.
10
4
1
4
2
3
7
7
9
5
13
14
12
1117
16
15
19
18
OH
O
10
5
1
4
2
3
7
7
9
6
13
14
12
1117
16
15
19
18
O
O
O
CrO3/H2SO4/H2O
Acetona
Figura Nº 30. Esquema de síntesis de la Δ4-androst-3,6,17-triona
Una vez sintetizado dicho compuesto fue caracterizado mediante un análisis
espectroscópico, a través de las técnicas de Espectroscopia de Infrarrojo (IR),
Resonancia Magnética Nuclear de Protones (RMN-1H) y Resonancia
Magnética Nuclear de Carbono (RMN-13C), con el propósito de determinar si
era el compuesto esperado.

58
El espectro de IR (Espectro N°4, tomado en pastilla de KBr) mostró una señal
poco intensa en 3012,53cm-1 correspondiente a vibraciones de tensión de
enlaces C(sp2)-H tres señales agudas e intensas en 2954 cm-1, 2916 cm-1 y
2856 cm -1 correspondiente a vibraciones de tensión de enlaces C(sp3)-H.
Centrada en 1735,9 cm-1 una banda estrecha e intensa, corresponde a una
vibración de tensión de un enlace carbonílico (C=O), por otra parte se observa
en 1685,1 cm-1 una señal muy intensa la cual corresponde a la vibración de
otro enlace carbonílico (C=O), perteneciente a la nueva cetona formada en la
posición 3 del anillo A. La disminución de la frecuencia de vibración del nuevo
enlace carbonílico puede atribuirse a la existencia del doble enlace en la
molécula, alfa al grupo C=O el cual por ser un grupo atractor de electrones
baja la frecuencia de absorción. La banda para las vibraciones de tensión de
enlaces C=C se centra en 1600 cm-1 y presenta una intensidad media.
Es importante destacar que en el espectro de IR sólo se logra apreciar dos
señales correspondientes a la frecuencia de absorbancia de los grupos
carbonilos. Para determinar la presencia de otro grupo carbonilo en la
estructura se procedió a realizar la deconvolución del espectro IR en la región
de absorbancia de 1735,1 cm-1 hasta 1685,1cm-1 con la finalidad de obtener la
segunda derivada de la frecuencia de absorbancia para dichos grupos
carbonílicos, ya que si existen señales solapadas estas se desdoblan en la
cantidad de máximos correspondientes a la vibración de tensión de cada grupo
carbonilos. De acuerdo con los resultados obtenidos, la deconvolución en la
región de 1735,1cm-1 genero un solo máximo, el cual corresponde al carbonilo
de C-17, mientras que en la región de 1685,1cm-1 se generaron dos máximos
el primero en 1694,52cm-1 el cual debería corresponder al carbonilo generado
en la posición C-6 y el segundo en 1672,48 cm-1 con una frecuencia de
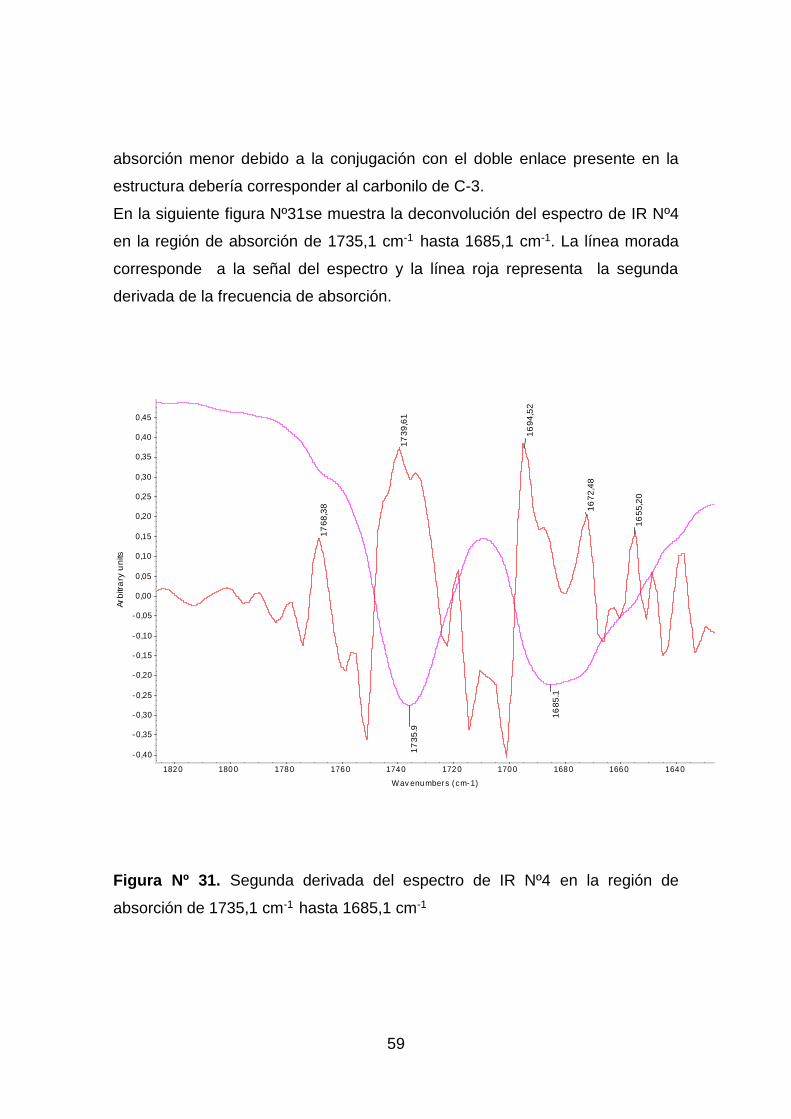
59
absorción menor debido a la conjugación con el doble enlace presente en la
estructura debería corresponder al carbonilo de C-3.
En la siguiente figura Nº31se muestra la deconvolución del espectro de IR Nº4
en la región de absorción de 1735,1 cm-1 hasta 1685,1 cm-1. La línea morada
corresponde a la señal del espectro y la línea roja representa la segunda
derivada de la frecuencia de absorción.
Figura Nº 31. Segunda derivada del espectro de IR Nº4 en la región de
absorción de 1735,1 cm-1 hasta 1685,1 cm-1
16
85
.1
17
35
.9
16
55
,20
16
72
,48
16
94
,52
17
39
,61
17
68
,38
-0,40
-0,35
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
Arb
itra
ry u
nits
1640 1660 1680 1700 1720 1740 1760 1780 1800 1820
Wav enumbers (cm-1)

60
Al producto obtenido se le tomó un espectro UV (Espectro Nº5) utilizando
diclorometano como solvente, en éste se observa una banda de absorbancia a
una λmax. de 242nm. De acuerdo a lo reportado por la literatura [35] los
sistemas Δ4-3,6- dionas de esteroides presentan una banda de absorbancia a
una λmax. de 253 nm cuando el espectro se toma en metanol, al comparar
dicho valor con el resultado obtenido se encuentra cierta similitud en los
resultados obtenidos y la diferencia en la longitud de onda puede atribuirse a la
influencia del solvente empleado durante la realización de los espectros. Es
importante destacar que se utilizó diclorometano como solvente para la
realización del espectro.
En el espectro de RMN-1H (Espectro N°6, CDCl3) se puede observar un
singlete centrado en 0,88 ppm y otro en 1,15 ppm; ambas señales atribuibles a
los protones metílicos localizados en los carbonos 18 y 19 respectivamente. En
6,13 ppm se aprecia un singlete, el cual corresponde al protón olefínico de la
estructura sintetizada, al comparar dicho desplazamiento químico, con el
protón olefínico ubicado en C-6 del colesterol, el cual presenta un
desplazamiento químico de δ=5,35 ppm[36] y cuyo espectro fue tomado en
CDCl3, se logra notar ligeras diferencias en ambos desplazamientos químicos,
ubicándose el protón olefínico para el colesterol a un campo más alto, cuando
el doble enlace se encuentra en el anillo B, con respecto al desplazamiento
químico del producto obtenido. Es importante destacar que la reacción de
oxidación es exotérmica y se desarrolla en un medio fuertemente ácido, hecho
que genera la posibilidad de isomerización del doble enlace del material de
partida (dehidroepiandrosterona) ubicado en la posición 5, a la posición 4,
razón por la cual también se comparó el resultado obtenido con el
desplazamiento del protón perteneciente a la olefina ubicada en C-4 de la
testosterona [37] donde este protón posee un desplazamiento químico de
δ=5,73 ppm, siendo este valor mucho más cercano al desplazamiento
obtenido. La cercanía de ambos valores sugiere la posibilidad de la
isomerización del doble enlace durante la reacción oxidación.

61
La determinación de la posición del doble enlace de la estructura, representa
un aspecto fundamental durante la caracterización del producto. Para
establecer dicha posición también se compararon los datos espectroscópicos
del RMN-13C (Espectro N°7, CDCl3) del producto sintetizado, con los
desplazamientos químicos reportados por la literatura para el doble enlace C-4
de la testosterona y el doble enlace de C-5 del colesterol, de sus respectivos
espectros de RMN-13C, tomados en CDCl3.
En la siguiente tabla se pueden observar algunas de las señales de los
espectros de RMN- 13C, CDCl3, para el colesterol y la testosterona [35][36]
Tabla N°6. Datos espectroscópicos de RMN-13C, CDCl3, para la testosterona y
el colesterol.
Al comparar los desplazamientos químicos de C-4 y C-5 para el colesterol, la
testosterona y el producto obtenido los cuales fueron tomados en CDCl3, se
Asignación
Testosterona
δ(ppm)
Colesterol
δ(ppm)
C-3 199,60 71,3
C-4 123,85 42,4
C-5 171,35 141,2
C-6 32,80 121,3

62
observa que en éste C-4 aparece a 125,88ppm. Este valor está más próximo al
desplazamiento en la testosterona (C-4:123,85ppm) que para C-5 del colesterol
(C-5:141,2ppm); la señal del doble enlace ubicado en C-5 del colesterol se
ubica a un campo más bajo en comparación con la señal del producto
obtenido. Esto indica ocurrió la isomerización del doble enlace de C-5 a C-4
del material de partida durante la reacción de oxidación
En la siguiente figura se puede observar las estructuras del colesterol y la
testosterona
1
5
1
4
2
3
8
7
9
6
13
12
12
1117
16
15
19
18
OH
20
21
22
23
2425
26
2710
5
1
4
2
3
8
7
9
6
13
14
12
1117
16
15
19
18 OH
O
Testosterona Colesterol
Figura Nº 32. Estructuras de la testosterona y el colesterol
Por otra parte el espectro de RMN-13C (Espectro N°7, CDCl3), mostró una
serie de señales, de las cuales se destacan los valores de 201,10 ppm y
198,98 ppm, esto sugiere la formación de dos nuevos grupos carbonilos, es
importante señalar que el carbonilo ubicado en C-17 mostró un desplazamiento
químico alrededor de 219,10 ppm. Vale la pena recordar que la deconvolución
del IR (EspectroNº4) confirmó la existencia de tres carbonilos.

63
En el 2005Hunter y col. [38] reportaron la síntesis de esteroides 4-en-3,6-dionas
a partir de esteroides 5-en-3β-ol. Dentro de los sustratos que emplearon se
encuentra la dehidroepiandrosterona para obtener la Δ4-androst-3,6,17-triona,
al comparar los datos espectroscópicos para RMN-13C, CDCl3, con los
obtenidos` para este producto se encontró total coincidencia, entre los
desplazamientos químicos.
Figura Nº 33. Síntesis de esteroides 4-en-3,6-dionas a partir de esteroides 5-
en-3β-ol, utilizando el Reactivo de Jones
La asignación de los carbonos se realizó tomando en cuenta el trabajo de
Hunter y col. [38]
OH O
O
CrO3/ H
2SO
4/ H
2O
T = 0ºC
Acetona

64
Tabla Nº 7. Datos espectroscópicos de RMN-13C (Espectro N° 7, CDCl3), del
producto de oxidación, empleando el reactivo de Jones
*Datos obtenidos a través de la simulación de la molécula con el programa ACD ChemSketch
versión 3.5
Señal Experimental
δ(ppm)
Señal Teórica
δ(ppm)*
Asignación
219,10 219,91 C-17
201,10 197,06 C-3
198,98 195,76 C-6
160,14 154,90 C-5
125,88 125,16 C-4
51,45 55,57 C-9
50,87 51,65 C-14
47,55 50,31 C-13
45,36 42,11 C-7
39,67 39,35 C-10
35,56 35,70 C-16
35,45 35,54 C-1
33,88 33,91 C-2
33,67 33,23 C-8
30,90 30,60 C-12
21,56 21,95 C-15
20,19 20,81 C-11
17,57 18,20 C-19
13,65 13,74 C-18

65
En 1998 Sheng-Hui y col. [39 ] reportaron la síntesis de 4-en-3,6-dionas a partir
esteroides 5-en-3βol empleando piridin cloro cromato y piridindiclorocromato
(PCC y PDC, respectivamente), estos sugieren que el mecanismo de reacción
se fundamenta en la formación de 5-en-3-ona como intermediario para la
transformación a la 4-en-3,6-dionas a partir de 5-en-3βol. En base a los
resultados obtenidos proponen el siguiente mecanismos de reacción, en el cual
ocurre la oxidación inicial del OH ubicado en la posición 3, para generar la 5-
en-3-ona, posteriormente la isomerización del doble enlace proviene de la
oxidación del mismo, para generar el carbonilo en la posición 6 de la estructura,
tal y como se observa a continuación en la Figura Nº 34
Cr
O
O
O
OH2
CH3
O
HH
OCH3
CH3
O
H
CrOH
O
O
OCH3
H2CrO
3
HCrO3
-
+
O
CH3
O
CH3
CH3
CH3O
O
HCr
O
O
O
C+
O
Cr-
O
H
O
OH
H
CH3
CH3
O
OH2
O
O
CH3
CH3 O
H2CrO
2+
Figura Nº 34.Esquemade síntesis de 4-en-3,6-dionas a partir de 5-en-3β-ol

66
La oxidación directa de la dehidroepiandrosterona también se realizó
empleando atmósfera inerte de nitrógeno y a una temperatura de 0ºC. Bajo
estas condiciones se obtuvo el mismo resultado la Δ4-androst-3,6,17-triona,
por lo que el cambio a una atmósfera inerte (N2) no ejerce ningún efecto
durante la oxidación, pues no evita la epimerización del doble enlace, hecho
que sugiere que la elevada acidez del medio así como la gran cantidad de
agua, podrían ser las condiciones que promueven la epimerización, así como la
formación del nuevo carbonilo en el carbono 6. Por tal razón sería importante
estudiar a futuro el efecto del medio acuoso y su acidez durante este tipo de
oxidaciones empleando el reactivo de Jones en sustratos que poseen otros
grupos funcionales sensibles a la oxidación, así como el efecto de la
temperatura durante la reacción de oxidación.
El siguiente esquema ilustra el producto obtenido durante la reacción de
oxidación:
10
5
1
4
2
3
8
7
9
6
13
14
12
1117
16
15
19
18
OH
O
10
5
1
4
2
3
8
7
9
6
13
14
12
1117
16
15
19
18
O
O
O
CrO3/H2SO4/H2O
Acetona
T= 0 ºC
Figura Nº 35. Síntesis de la Δ4-androst-3,6,17-triona
T=0ºC

67
6.2.2 Reducción Huang-Minlon de la Δ4-androst-en-3,6,17-triona
6.2.2.1 Síntesis de la hidrazona de la Δ4-androst-3,6,17-triona
La síntesis de la hidrazona como primer paso de la reducción, se realizó con la
finalidad de promover la reducción selectiva de los carbonilos ubicados en C-3,
y C-6, ya que el material de partida posee más de un grupo carbonilo en su
estructura. En este primer paso se emplearon cantidades equimolares de
hidrato de hidracina, para promover la reacción de los carbonilos en C-3 y C-6.
Al comparar la reactividad entre los carbonilos ubicados en C-3, C-6 y C-17 se
esperaba que la hidrazona se formara en C-3 y C-6, debido al impedimento
estérico generado por el metilo angular C-18 sobre el carbonilo en C-17, el cual
lo hace menos susceptible para un ataque nucleofílico.
Una vez finalizada la reacción se obtuvo un sólido amarillo, el cual es
parcialmente soluble en diclorometano, cloroformo, acetato de etilo, benceno,
metanol y acetona mostrando un mayor comportamiento de solubilidad en
etanol e insoluble en agua, dicho compuesto era extremadamente polar y
siempre se mantuvo en la línea de sembrado en las distintas cromatografías
de capa fina, realizadas con los siguientes solventes y mezclas de solventes:
metanol, acetona, metanol-agua (50:50 v/v), ácido acético-agua (90:10 v/v),
ácido acético y agua. Además la hidrazona sintetizada mostró un
comportamiento inusual, ya que la parte que se encontraba disuelta en los
distintos solventes orgánicos comenzaba a descomponerse con el pasar de los
días.
Debido a la baja solubilidad del producto obtenido su caracterización se realizó
mediante Espectroscopia de Infrarrojo (IR), ya que al no lograrse la solubilidad
completa del compuesto las señales obtenidas para los espectros de RMN-1H y

68
RMN-13C no se lograron apreciar debido a la poca cantidad de muestra
obtenida en solución.
El espectro de IR (Espectro N°8, tomado en pastilla de KBr) mostró una banda
ancha e intensa con dos picos en 3393,3 cm-1 y 3311,8 cm-1, siendo esta banda
característica de aminas primarias, por otra parte se observan dos señales
intensas en 2940 cm-1 y 2859,9 cm-1 correspondientes a vibraciones de tensión
de enlaces C-H sp3. En 1736,1 cm-1 se aprecia una banda estrecha e intensa,
correspondiente a la vibración de tensión de un enlace carbonílico (C=O), esta
banda es característica del carbonilo ubicado en C-17, hecho que nos indica
que la Hidrazona no se formó en este carbonilo, tal y como se esperaba antes
de comenzar la reacción; por otra parte se observa en 1666,9 cm-1 una señal
intensa la cual puede atribuirse a las vibraciones de tensión C=C, centrada en
1577,3 cm-1 se observa una banda de intensidad media la cual es
características de las vibraciones de tensión de los enlaces del tipo C=N en esa
misma región se nota una banda centrada en 1548 cm-1 también de intensidad
media, la cual podría atribuirse a las vibraciones de tensión de los enlaces de
tipo N-H, para aminas primarias (–NH2). Centrada en 1455,1cm-1 se encuentra
una banda de intensidad media la cual es característica de las vibraciones de
tensión de enlaces C-N.
En vista que el material de partida posee tres grupos carbonilos en su
estructura, se procedió a realizar la deconvolución del espectro IR en la región
de absorbancia de 1736,1 cm-1 hasta 1666,9cm-1 con la finalidad de determinar
la presencia de otro grupo carbonilo en la estructura, ya que como se demostró
en el espectro IR de la Δ4-androst-3,6,17-triona, las señales de los carbonilos
de C-3 y C-6 se encuentran solapadas alrededor de 1685,1cm-1. De acuerdo
con los resultados obtenidos la segunda derivada de la señal en 1736,1 mostró
un solo máximo el cual corresponde a la presencia del carbonilo ubicado en
C-17, es decir que se sintetizo la doble hidrazona en C-3 y C-6, la señal de
1666,9 también mostró un solo máximo, correspondiendo esta señal a la

69
absorbancia C=C, debido a que esta aparece a una frecuencia de absorción
más baja que la obtenida para los carbonilos ubicados en C-3 y C-6, se
descarta la posibilidad que la misma corresponda a algunos de estos
carbonilos.
La siguiente figura Nº36 muestra la deconvolución del espectro de IR Nº7 en la
región de absorción de 1736,1 cm-1 hasta 1666,9 cm-1.La línea turquesa
corresponde a la señal del espectro y la línea roja representa la segunda
derivada de la frecuencia de absorción.
Figura Nº 36. Deconvolución del espectro de IR Nº7 en la región de absorción
de 1736,1 cm-1 hasta 1666,9 cm-1.
.
16
66
.9
17
36
.1
16
74
,04
17
00
,24
17
37
,69
17
74
,44
17
98
,62
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
Arb
itra
ry u
nits
1640 1660 1680 1700 1720 1740 1760 1780 1800 1820 1840
Wav enumbers (cm-1)

70
Como consecuencia de la baja solubilidad del compuesto lamentablemente no
se logran apreciar las señales del mismo en los espectros de RMN hecho que
no permite la asignación del doble enlace en la estructura, el cual se sabe está
presente por las señales obtenidas en el espectro IR, razón por la cual se
proponen dos posibles estructuras como resultado de la síntesis de la
hidrazona.
10
5
1
4
2
3
8
7
9
6
13
14
12
1117
16
15
19
18
N
O
N
NH2
NH2
10
5
1
4
2
3
7
7
9
6
13
14
12
1117
16
15
19
18 O
NH
NH2
NNH2
(19) (20)
Figura Nº 37. Posibles Estructura de la Hidrazona sintetizada.
6.2.2.2 Reducción de la hidrazona de la Δ4-androst-3,6,17-triona

71
La reducción de la hidrazona de la Δ4-androst-3,6,17-triona, generó un
producto aceitoso de color amarillo, el cual posee un Rf=0,44, utilizando una
mezcla de solventes CH2Cl2-CH3OH (90:10 v/v), desarrollado en una placa de
sílice. El producto obtenido es soluble en diclorometano, cloroformo, metanol,
etanol e insoluble en agua. La caracterización del compuesto se realizó
empleando espectroscopia de RMN- 1Hy RMN-13C
En el espectro de RMN-1H (Espectro N°9, CDCl3) se observa una serie de
multipletes cuyas señales son de difícil asignación, por lo tanto para este
espectro sólo fueron consideradas las señales de mayor
intensidad.Centrados0,75 ppm y 1,02 ppm se aprecia un par de singletes,
ambas señales atribuibles a los protones metílicos localizados en los carbonos
18 y 19 respectivamente.
Debido a la poca cantidad de muestra y a la baja resolución de los espectros se
hizo imposible identificar plenamente el compuesto sintetizado. Sin embargo
después de hacer una extensa revisión bibliográfica sobre la reducción de
Wolff-kishner y Huang Minlon de cetonas esteroidales, se proponen un par de
estructuras de los posibles compuestos. Es importante destacar una vez más
que aunque la ausencia de algunas señales en el espectrode RMN-13C no
permite identificar con certeza el producto obtenido, existen señalesen el
mismo que si proporcionan información para su caracterización. Se trata de un
alcohol esteroidal, en el que se han reducido tres grupos carbonilos,se
observan los desplazamientos químicos de los metilos angulares C-18 y C-19 a
11,16 ppm y 15,18 ppm respectivamente, también se aprecian
desplazamientos químicos a 43,05 ppm y a 50,76 ppm, dichos valores se
asignaron a los carbonos C-13 y C-14, siendo estos desplazamientos químicos
característicos del anillo D, de androstanos, en los cuales se ubica un grupo
OH el C-17. De acuerdo con las señales obtenidas en el espectro se logra la

72
asignación de los carbonos que conforman los anillos D C y B. Se puede
destacar para C-8 δ=31,49 ppm, C-11 δ=20,66 ppm, C-10 δ=35,11ppm, C-9
δ=47,45ppm y C-12 δ=36,68ppm.
En el espectro de RMN-13C(Espectro N°10, CDCl3) se logra apreciar una serie
de señales con desplazamientos químicos característicos de carbonos
pertenecientes a la estructura de esteroides sustituidos con grupos hidroxilos,
estos fueron comparados con los desplazamientos químicos obtenidos para
compuestos androstanos donde los carbonos C-3, C-4, C-6 y C-17 se
encuentran sustituidos con grupos hidroxilos[40]. En la siguiente tabla se
muestran los desplazamientos químicos para los carbonos sustituidos con
grupos hidroxilos en androstanos.
Tabla Nº8.Datos espectroscópicos del espectro de RMN-13C, CDCl3, para
algunos carbonos de la 4ξ,17β-dihidroxi5ξ-androstano y la 3β,6ξ-dihidroxi-5ξ-
androst-17-ona[40].
Asignación
δexp. (ppm)**
4ξ,17β-dihidroxi-5ξ-androstano
Asignación
δexp. (ppm)*
3β,6ξ-dihidroxi-5ξ-
androst-17-ona
C-4 71,22 C-3 66,66
C-17 83,46 C-6 75,98
*δ=Desplazamiento químico.**El espectro de RMN-13C de la 4ξ,17β-dihidroxi5ξ-androstano y la 3β,6ξ-
dihidroxi-5ξ-androst-17-ona, fue tomado en CDCl3
En el espectro de RMN-13C del compuesto sintetizado se observan señalescon
desplazamientos químicos a81,94ppm; 71,85 ppm y 71,64ppm al comparar
dichos valores con los mostrados en la tabla Nº8 se puede presumirque los

73
carbonos C-17, C-3 y C-6 del producto obtenido se encuentran unidos a
grupos hidroxilos, hecho que indica que bajo las condiciones experimentales
utilizadas no se logró obtener la reducción del grupo carbonilo al grupo
metileno, ocurriendo entonces una reducción poco común durante el
procedimiento experimental realizado.
Es imponte destacar que este tipo de reducciones poco comunes fueron
reportadas por primera vez en 1939 por James Dutcher y col. [32] en su trabajo
sobre la reducción de cetonas esteroidales, empleando la reducción de Wolff-
kishner. Uno de los sustratos empleados por ellos fue la colestanona. Su
procedimiento experimental se basó en la síntesis inicial de la hidrazona de la
colestanona, para posteriormente someterla a las condiciones básicas y a la
elevada temperatura característicade la reducción de Wolff-kishner, como base
utilizaron etóxido de sodio en etanol, Dutcher y colaboradores en lugar de
obtener el correspondiente grupo metilenico en C-3, generaron lamezcla de
alcoholes (α y β)- colestanol con un 78% de rendimiento.

74
NNH2
NH2 NH2
EtanolO
NNH2 OHOH
+EtONa/Etanol
Calor
Figura Nº 38. Reducción de la hidrazona de la colestanona a la mezcla de
alcoholes (α y β)- colestanol
Estos sugieren que la obtención del alcohol se debe a que la hidrazona es
hidrolizada en una primera etapa, generando nuevamente la cetona libre, la
cual se reduce con el etoxido de sodio en etanol durante el periodo de
calentamiento, obteniéndose el alcohol secundario y el acetaldehído. En la
siguiente figura se muestran dichas reacciones
Figura Nº 39. Esquema de reducción del carbonilo al alcohol, empleando el
método de Wolff-Kishner.
N
CH3
CH3
NH2OH2 O
CH3
CH3
NH2 NH2
O
CH3
CH3
CH3 OH
H
H
OH
CH3
CH3H
CH3H
O
+
+
+
+
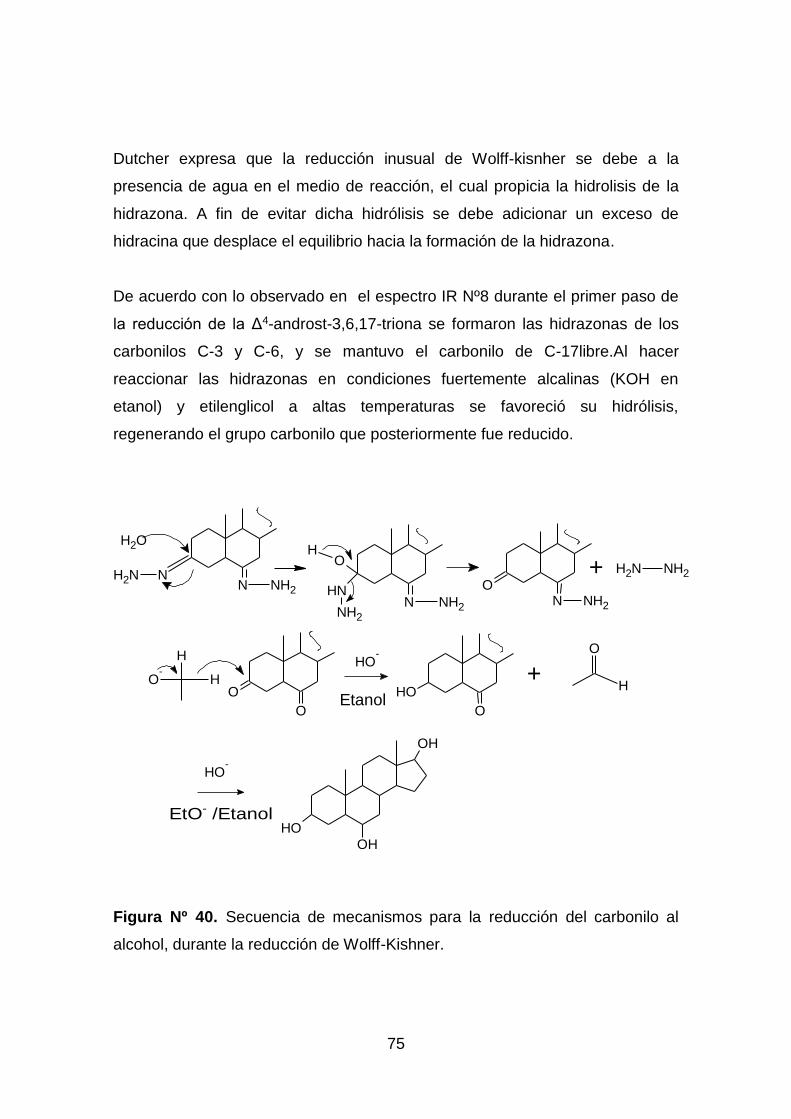
75
Dutcher expresa que la reducción inusual de Wolff-kisnher se debe a la
presencia de agua en el medio de reacción, el cual propicia la hidrolisis de la
hidrazona. A fin de evitar dicha hidrólisis se debe adicionar un exceso de
hidracina que desplace el equilibrio hacia la formación de la hidrazona.
De acuerdo con lo observado en el espectro IR Nº8 durante el primer paso de
la reducción de la Δ4-androst-3,6,17-triona se formaron las hidrazonas de los
carbonilos C-3 y C-6, y se mantuvo el carbonilo de C-17libre.Al hacer
reaccionar las hidrazonas en condiciones fuertemente alcalinas (KOH en
etanol) y etilenglicol a altas temperaturas se favoreció su hidrólisis,
regenerando el grupo carbonilo que posteriormente fue reducido.
NNH2 N NH2 ON NH2
O
O
+
OH2
HO-
H
NH2 NH2
OH
O
HO
NHN NH2NH2
OH
OH
OH
Etanol
OH-
OH-
EtO- /Etanol
+H
O
Figura Nº 40. Secuencia de mecanismos para la reducción del carbonilo al
alcohol, durante la reducción de Wolff-Kishner.

76
En vista de que la reducción del carbonilo involucra la trasferencia de un
hidruro, el carbonilo ubicado en C-17 logró ser reducido a su correspondiente
alcohol. Este hecho puede ser explicado debido a que la transferencia del
hidruro no se ve afectada por el impedimento estérico generado por el metilo
angular C-18.Este resultado concuerda con el encontrado por Dutcher y col.
quienes al hacer reaccionar algunas cetonas esteroidales con etóxido de sodio
a temperatura de reflujo obtuvieron el alcohol correspondiente.
Debido a la poca resolución de los espectros (RMN-13C y RMN-1H) no se logra
detectar ninguna señal característica de los protones y carbonos olefínicos,
razón por la cual no se puede descartar por completo la existencia del doble
enlace en la estructura. Lamentablemente no se cuentó con un espectro IR
para confirmar las señales características producto de las vibraciones de
tensión de enlaces dobles y sencillos, C=C y C(sp2)-H.
De acuerdo a lo antes expuesto una de las posibles estructuras del producto
sintetizado debería ser elΔ4-3ξ,6ξ,17ξ-trihidroxi-androstano, la siguiente figura
muestra su estructura.
10
5
1
4
2
3
8
7
9
6
13
14
12
1117
16
15
19
18
OH
O
OH
(21)
Figura Nº 41.Posible estructura (21) para el producto de reducción de la
hidrazona de la Δ4-androst,3,6,17-triona.

77
Otra posible estructura sería aquella en la que C-4 también estuviese
hidroxilado. En este caso tendríamos 4 grupos hidroxilo en la estructura. El
desplazamiento químico de los carbonos C-4 y C-5 de acuerdo a esta
posibilidad serían72,05 ppm y 54,59 ppm, respectivamente. Sin embargo, es
importante mencionar que son señales con poca intensidad.
Los datos espectroscópicos obtenidos indican que se trata nuevamente de un
alcohol esteroidal en el que se han reducido tres grupos carbonilos, se
observan los desplazamientos químicos característicos de los carbonos C-3, C-
4, C-6 y C-17 cuando estos se encuentran unidos a un grupo –OH. Al igual que
en la estructura anterior los metilos angulares deC-18 y C-19 presentan los
siguientes desplazamientos químicos 11,16ppmy15,18ppm respectivamente,
también se aprecia desplazamientos químicos a 43,05 ppm y a 50,76 ppm,
para los carbonos C-13 y C-14 respectivamente. La caracterización de esta
nueva estructura involucra señales de poca intensidad en el espectro, que
permiten asignar completamente los 19 átomos de carbono que conforman el
esqueleto general de los androstanos.
De acuerdo a lo antes expuesto se propone la estructura (22) como posible
producto de la reacción de reducción, la misma seria llamada 3ξ,4ξ6ξ,17ξ-
tetrahidroxi-androstano y se muestra en la siguiente figura Nº42
10
5
1
4
2
3
8
7
9
6
13
14
12
1117
16
15
19
18
OH
O
OHOH
(22)
Figura Nº 42. Estructura del 3ξ,4ξ6ξ,17ξ-tetrahidroxi-androstano
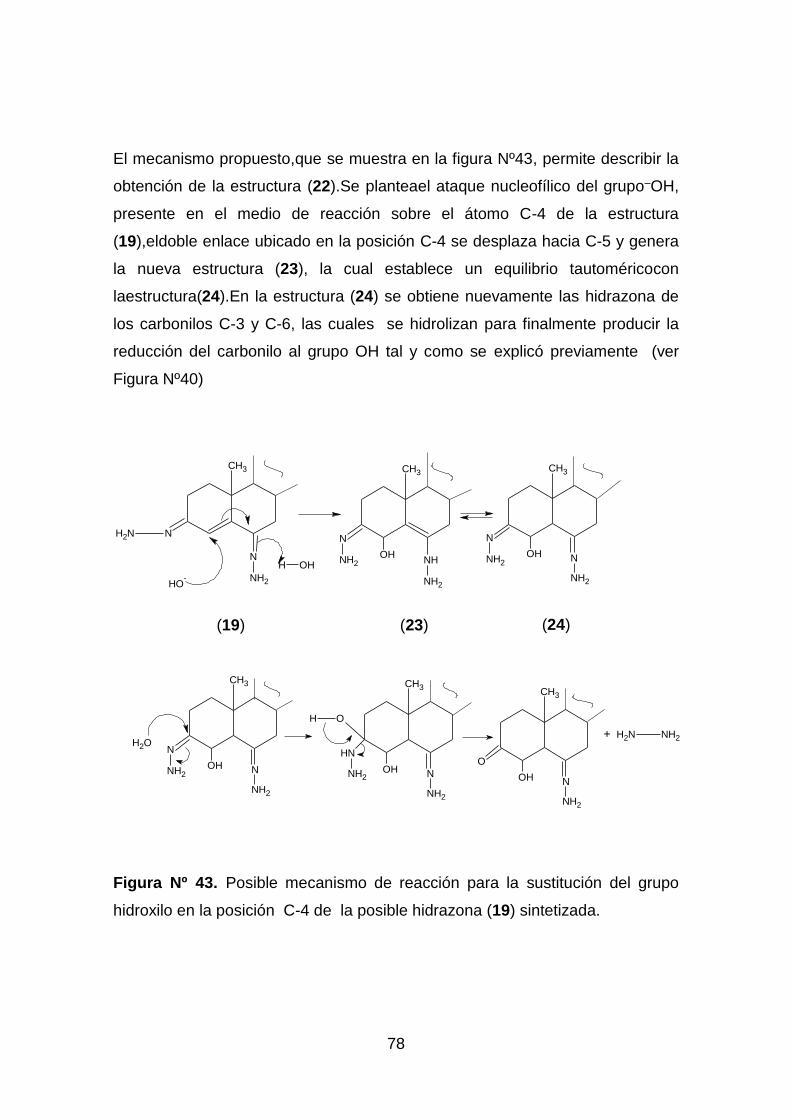
78
El mecanismo propuesto,que se muestra en la figura Nº43, permite describir la
obtención de la estructura (22).Se planteael ataque nucleofílico del grupo–OH,
presente en el medio de reacción sobre el átomo C-4 de la estructura
(19),eldoble enlace ubicado en la posición C-4 se desplaza hacia C-5 y genera
la nueva estructura (23), la cual establece un equilibrio tautoméricocon
laestructura(24).En la estructura (24) se obtiene nuevamente las hidrazona de
los carbonilos C-3 y C-6, las cuales se hidrolizan para finalmente producir la
reducción del carbonilo al grupo OH tal y como se explicó previamente (ver
Figura Nº40)
Figura Nº 43. Posible mecanismo de reacción para la sustitución del grupo
hidroxilo en la posición C-4 de la posible hidrazona (19) sintetizada.
CH3
N
N
NH2
NH2OH-
H OH
CH3
NH
NH2
OH
N
NH2
CH3
OH
N
NH2N
NH2
CH3
OH
N
NH2N
NH2
OH2
H O
CH3
OH N
NH2
NH
NH2
CH3
OH
O
N
NH2
+ NH2 NH2
(19) (23) (24)
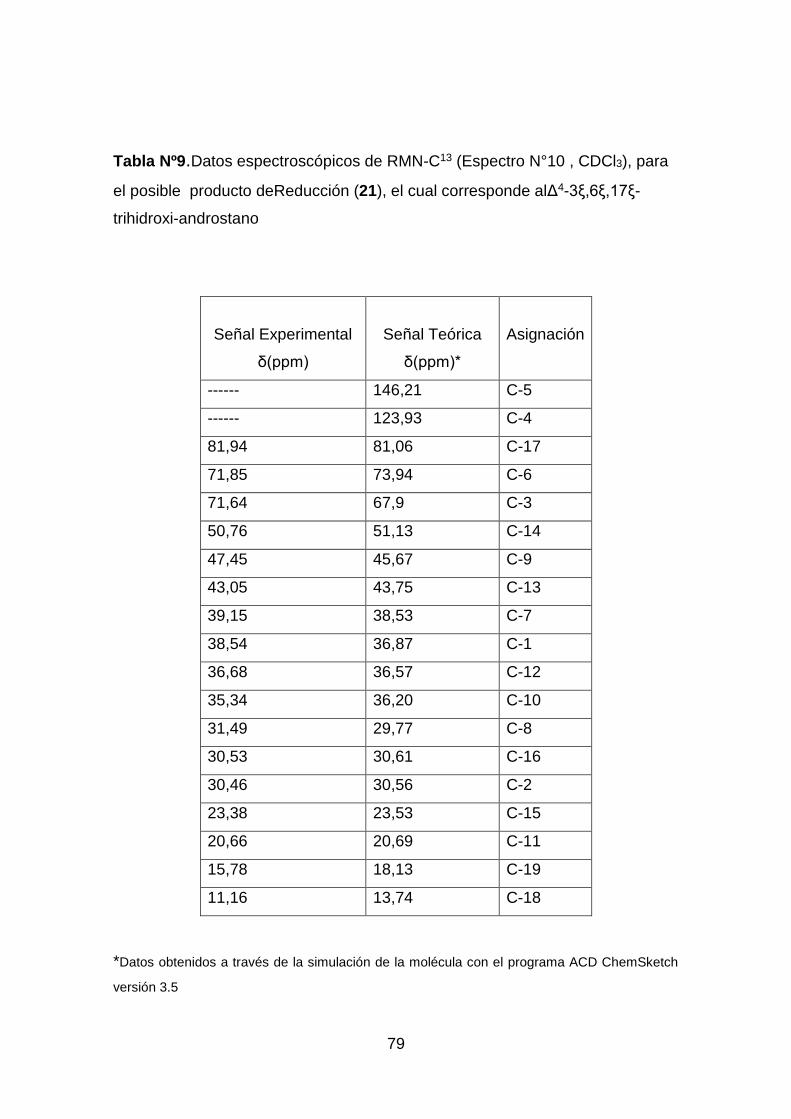
79
Tabla Nº9.Datos espectroscópicos de RMN-C13 (Espectro N°10 , CDCl3), para
el posible producto deReducción (21), el cual corresponde alΔ4-3ξ,6ξ,17ξ-
trihidroxi-androstano
*Datos obtenidos a través de la simulación de la molécula con el programa ACD ChemSketch
versión 3.5
Señal Experimental
δ(ppm)
Señal Teórica
δ(ppm)*
Asignación
------ 146,21 C-5
------ 123,93 C-4
81,94 81,06 C-17
71,85 73,94 C-6
71,64 67,9 C-3
50,76 51,13 C-14
47,45 45,67 C-9
43,05 43,75 C-13
39,15 38,53 C-7
38,54 36,87 C-1
36,68 36,57 C-12
35,34 36,20 C-10
31,49 29,77 C-8
30,53 30,61 C-16
30,46 30,56 C-2
23,38 23,53 C-15
20,66 20,69 C-11
15,78 18,13 C-19
11,16 13,74 C-18

80
Tabla Nº 10.Datos espectroscópicos de RMN-C13 (Espectro N°10, CDCl3), para
el posible producto de Reducción (22), el cual corresponde al 3ξ4ξ,6ξ,17ξ-
tetrahidroxi-androstano.
*Datos obtenidos a través de la simulación de la molécula con el programa ACD ChemSketch
versión 3.5
Señal Experimental
δ(ppm)
Señal Teórica
δ(ppm)*
Asignación
81,94 81,06 C-17
72,05 73,39 C-4
71,85 71,83 C-3
71,64 69,45 C-6
54,39 53,03 C-5
50,76 52,67 C-9
47,45 50,84 C-14
43,05 43,75 C-13
39,15 38,53 C-7
38,53 36,87 C-1
36,68 36,57 C-12
35,34 36,20 C-10
31,49 29,77 C-8
30,53 30,61 C-16
30,46 30,56 C-2
23,38 23,53 C-15
20,66 20,69 C-11
15,78 18,13 C-19
11,16 13,74 C-18

81
6.2.2.3 Reducción directa del producto oxidado empleando en método de
Huang –Minlon.
En vista que la hidrazona sintetizada presentó una baja solubilidad en los
distintos solventes orgánicos utilizados y a la poca estabilidad de la misma se
procedió a realizar la reducción directa de la Δ4-androst-3,6,17-triona, siguiendo
el procedimiento original de Hunag-Minlon, con la finalidad de obtener la
reducción de los carbonilos ubicados en C-3 y C6.
Una vez finalizada la reacción se obtuvo un acetite amarillo el cual era soluble
en metanol, cloroformo, diclorometano y acetona, la caracterización del mismo
se realizó empleando Espectroscopia de Infrarrojo y UV-visible.
El espectro UV (Espectro Nº11) para este compuesto fue tomado en
diclorometano, en el mismo se observa la desaparición de la banda
característica del sistema Δ4-3,6-diona, ubicada a una λmax=242 nm, en este
espectro se nota una banda de absorción a λmax=318 nm, de acuerdo con lo
expresado en trabajos previos sobre el estudio de absorbancia de cetonas en
esteroides[35 ]el carbonilo ubicado en C-17, posee una banda de absorción
ubicada a 293 nm utilizando como solvente metanol y la misma se ve
influenciada por la presencia de otros grupos cromóforos en la estructura. Es
importante destacar que los grupos carbonilos presentan un coeficiente de
extinción pequeño, hecho que genera que sus bandas de absorción sean de
poca intensidad, en el espectro obtenido para el producto de la reducción
directa se observa una banda de poca intensidad a un λmax= 318 nm, esta
puede atribuirse a la presencia de un grupo carbonilo y se ubica a una longitud
de onda mayor debido al efecto del doble enlace en la estructura, sobre el
carbonilo, el cual desplaza la banda de absorción a longitudes de onda
mayores.

82
El espectro de IR (Espectro N°12, tomado en pastilla de KBr) mostró una señal
poco intensa en 3008,4 cm-1 la cual corresponde a vibraciones de tensión de
enlaces C (sp2)-H. Centrada en 2925,8 cm-1 y 2854,7 cm-1se notan un par de
señales agudas e intensas correspondientes a las vibraciones de tensión de
enlaces C (sp3)-H. Por otra parte centrada en 1745,1 cm-1 una banda intensa,
corresponde a la frecuencia de absorción, para las vibraciones de tensión de
un enlace carbonilo (C=O). Esta frecuencia de absorción es característica del
grupo carbonilo ubicado en C-17 de androstanos. Centrada en 1462,2 cm-1 se
encuentra una banda de intensidad media, la cual se atribuye a las vibraciones
de tensión de los grupos metilenos –CH2.
Al comparar los espectros IR de la Δ4-androst-3,6,17-trionacon el producto
obtenido durante su reducción empleando el procedimiento de Huang Minlon
se observa la desaparición de una banda ubicada en 1681,5cm-1 la cual
corresponde al solapamiento de las bandas correspondientes a las vibraciones
de tensión de los enlaces C=O para los carbonilo C-3 y C-6, además que solo
se observa en 1745,1 cm-1 la banda correspondiente al carbonilo ubicado en
C-17, hecho que sugiere que pudo haber ocurrido la reducción de los
carbonilos C-3 y C-6 o de uno de ellos, por tal razón se procedió a tomar la
segunda derivada de la frecuencia de absorción del espectro IR , ya que en
este caso el material de partida posee más de un grupo carbonilo que absorben
a la misma frecuencia de vibración o una frecuencia muy cercana por lo que se
observa una sola banda de absorción en el espectro.
De acuerdo con los resultados obtenidos la segunda derivada generó un solo
máximo de absorción, en la región de absorción de los grupos carbonilos, es
decir, que el producto obtenido posee un solo carbonilo. Con este
procedimiento experimental se lograron reducir los carbonilos de C-3 y C-6 del
sustrato.
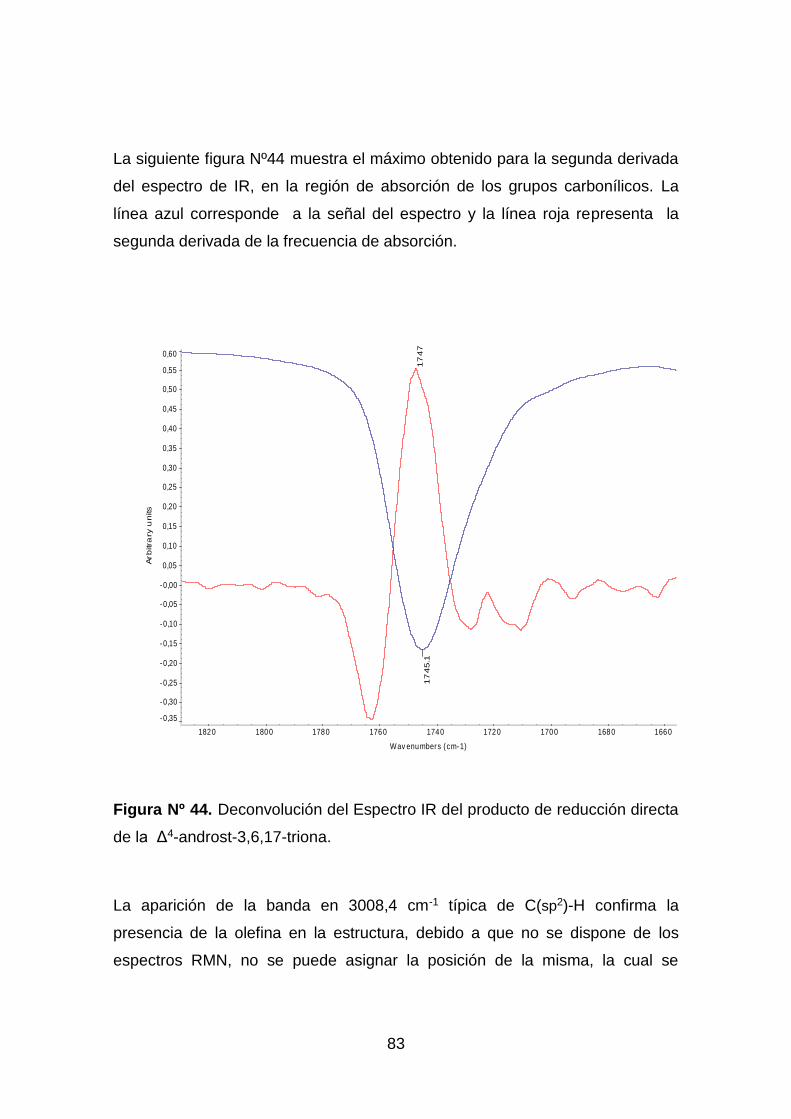
83
La siguiente figura Nº44 muestra el máximo obtenido para la segunda derivada
del espectro de IR, en la región de absorción de los grupos carbonílicos. La
línea azul corresponde a la señal del espectro y la línea roja representa la
segunda derivada de la frecuencia de absorción.
Figura Nº 44. Deconvolución del Espectro IR del producto de reducción directa
de la Δ4-androst-3,6,17-triona.
La aparición de la banda en 3008,4 cm-1 típica de C(sp2)-H confirma la
presencia de la olefina en la estructura, debido a que no se dispone de los
espectros RMN, no se puede asignar la posición de la misma, la cual se
17
45
.11
74
7,5
0
-0,35
-0,30
-0,25
-0,20
-0,15
-0,10
-0,05
-0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
Arb
itra
ry u
nits
1660 1680 1700 1720 1740 1760 1780 1800 1820
Wav enumbers (cm-1)

84
encuentra en los carbonos C4-C5 del material de partida utilizado para la
reducción.
A continuación se muestran las posibles estructuras del compuesto sintetizado
10
5
1
4
2
3
8
7
9
6
13
14
12
1117
16
15
19
18 O
(25)
10
5
1
4
2
3
8
7
9
6
13
14
12
1117
16
15
19
18 O
(26)
Δ4-androst-17-ona o Δ5-androst-17-ona
Figura Nº 45. Posibles producto de la reducción directa de la Δ4-androst-
3,6,17-triona.
Este procedimiento experimental fue mucho más efectivo para la reducción de
los carbonilos a sus correspondientes grupos metilenos debido a que se
adicionó un exceso de hidracina, el cual favoreció la formación de la hidrazona
además que la colocación de una trampa de agua durante la reacción ayudó a
eliminar el agua presente en el medio, evitando así la hidrolisis de la
hidrazona. Por otra parte, no se observó la reducción del carbonilo C-17 a su
respectivo alcohol, una posible causa fue que se disminuyó el tiempo de
reacción.

85
7. CONCLUSIONES
Se logró de manera efectiva la síntesis la Δ4-androst-3,6,17-triona a partir de
la oxidación directa de la dehidroepiandrosterona, a bajas temperaturas,
utilizando el Reactivo de Jones.
La disminución de la temperatura durante el procedimiento de oxidación
empleando el Reactivo de Jones, no logra evitar que grupos sensibles como
dobles enlaces se vean afectados frente a las condiciones ácidas de la
reacción. Por lo tanto se presume que el medio acuoso en la reacción sería el
encargado de promover la isomerización del doble enlace.
El reemplazo del aire atmosférico por nitrógeno (N2), durante la oxidación de la
dehidroepiandrosterona a bajas temperaturas no logra evitar la formación del
sistema Δ4-3,6-diona en el sustrato, por lo que la atmósfera utilizada durante la
síntesis no es un parámetro que modifique la reacción del doble enlace frente
al Reactivo de Jones.
La bromación con ácido acético de la dehidroepiandrosterona generó la
síntesis de la 3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona, hecho que no
permitió la síntesis de la Δ5androst-3,17-diona, siendo esto una consecuencia
de la acetilación del grupo hidroxilo ubicado en C-3.
La protección de dobles enlaces ubicados en el anillo B de androstanos
mediante bromaciones con ácido acético, debe llevarse a cabo cuando dentro
de la estructura no se encuentren grupos propensos a reaccionar con el ácido

86
acético, ya que éste es capaz de promover la bromación de cetonas cíclicas así
como generar esterificaciones cuando se tienen grupos OH ubicados en la
posición 3 del anillo A.
El control de la relación estequiométrica de hidracina / carbonilo, la cantidad de
agua en el medio de reacción y la temperatura a la que se lleve a la cabo la
reducción de Huang Minlon son los parámetros fundamentales que deben ser
controlados para la reducción efectiva de los carbonilos de C-3 y C-6 de la Δ4-
androst-3,6,17-triona. Ya que se puede generar una reducción poco común de
los carbonilos generando grupos hidroxilos en lugar de grupos metilenos
La elevada temperatura y el medio fuertemente básico puede generar la
reducción del carbonilo ubicado en la posición C-17 de la Δ4-androst-3,6,17-
triona, a través de un proceso de óxido-reducción, el cual involucre la
transferencia de un hidruro al carbonilo para generar el respectivo grupo
hidroxilo.

87
8. ESPECTROS

88
Espectro Nº 1IR de la 3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona
556.0
590.1
651.4
679.6
756.5
838.2
907.7
964.1
1030.1
1085.5
1128.9
1158.1
1240.1
1365.7
1377.1
1435.0
1454.1
1732.9
1759.9
2871.9
2948.3
3012.5
3443.3
3499.8
bromacion de dhea
0
10
20
30
40
50
60
70
80
90
100
%T
500 1000 1500 2000 2500 3000 3500 4000
Wav enumbers (cm-1)

89
Espectro Nº1.RMN-1H de la 3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona
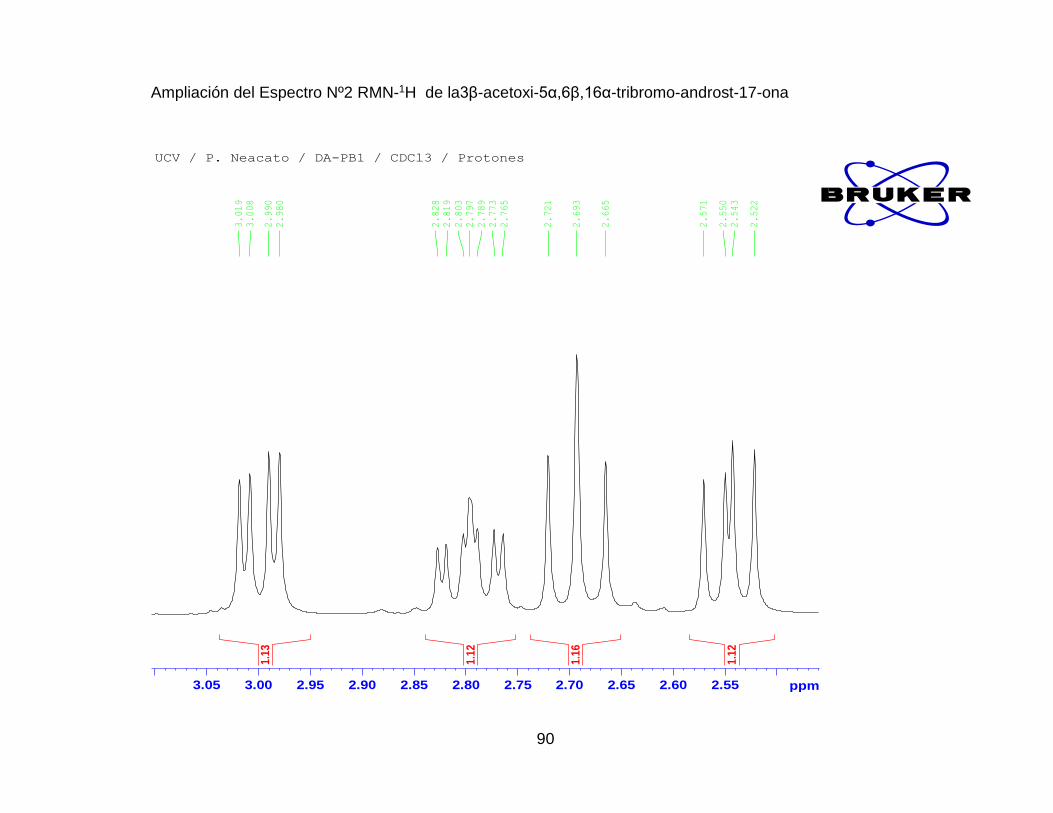
90
Ampliación del Espectro Nº2 RMN-1H de la3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona
UCV / P. Neacato / DA-PB1 / CDCl3 / Protones
2.552.602.652.702.752.802.852.902.953.003.05 ppm
2.522
2.543
2.550
2.571
2.665
2.693
2.721
2.765
2.773
2.789
2.797
2.803
2.819
2.828
2.980
2.990
3.008
3.019
1.1
2
1.1
6
1.1
2
1.1
3

91
Espectro Nº2. RMN- 13 C de la3β-acetoxi-5α,6β,16α-tribromo-androst-17-ona

92
Espectro Nº3. IR de la Δ4-androst-3,6,17-triona.
41
6.8
50
6.0
56
6.8
75
5.7
82
8.0
87
6.6
92
9.4
95
4.6
10
19
.3
10
56
.51
09
2.7
11
16
.91
16
8.6
12
18
.51
23
9.2
12
67
.01
32
8.9
13
46
.01
37
8.7
14
06
.51
45
4.3
14
68
.7
16
00
.0
16
85
.11
73
5.9
28
56
.62
91
6.0
29
54
.1
30
12
,53
33
22
.53
34
9.1
34
45
.4
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100%
T
500 1000 1500 2000 2500 3000 3500 4000
Wav enumbers (cm-1)
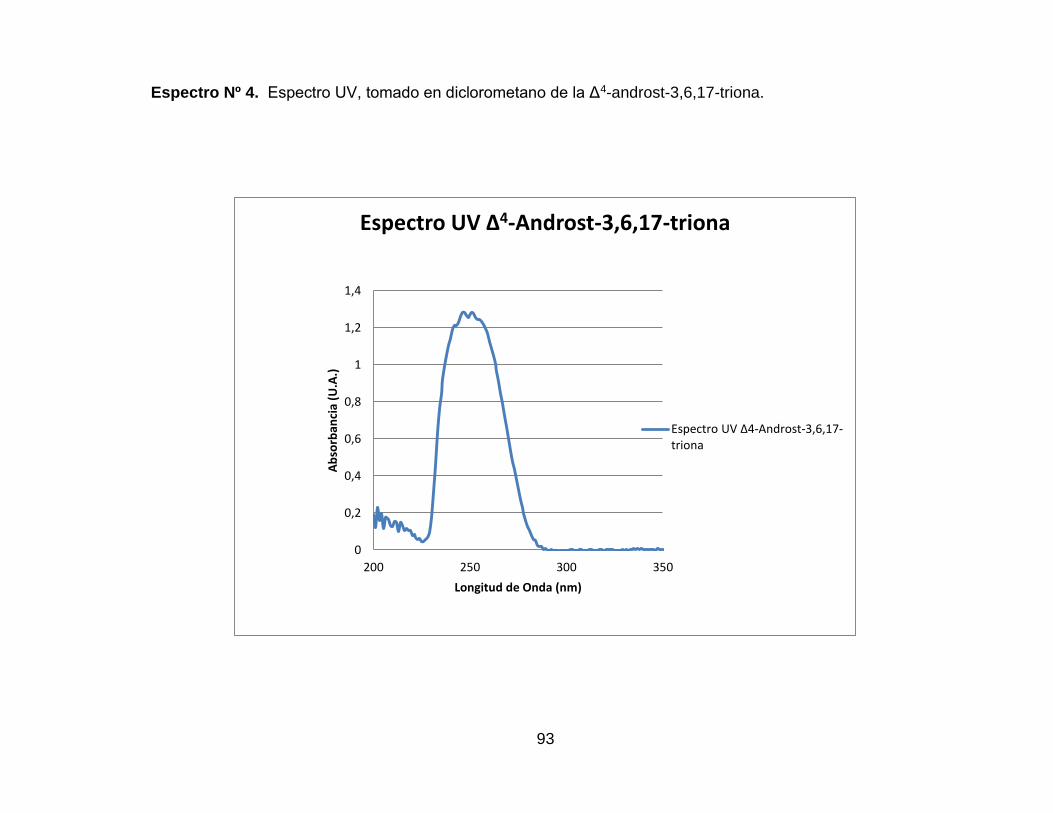
93
Espectro Nº 4. Espectro UV, tomado en diclorometano de la Δ4-androst-3,6,17-triona.
0
0,2
0,4
0,6
0,8
1
1,2
1,4
200 250 300 350
Ab
sorb
anci
a (U
.A.)
Longitud de Onda (nm)
Espectro UV Δ4-Androst-3,6,17-triona
Espectro UV Δ4-Androst-3,6,17-triona

94
Espectro Nº 5.RMN-1Hde la Δ4-androst-3,6,17-triona

95
Ampliación H-RMN de la Δ4-androst-3,6,17-triona
6.2 ppm
6.134
1.0
0
UCV / P. Neacato / DA-OxD1 / CDCl3 / Protones
1.41.51.61.71.81.92.02.12.22.32.42.52.62.72.8 ppm
1.314
1.321
1.340
1.347
1.386
1.409
1.422
1.429
1.443
1.452
1.469
1.477
1.495
1.503
1.529
1.535
1.554
1.572
1.579
1.746
1.752
1.773
1.778
1.852
1.869
1.886
1.895
1.908
1.927
1.937
1.950
1.962
2.050
2.068
2.079
2.087
2.098
2.106
2.123
2.398
2.430
2.448
2.454
2.466
2.486
2.731
2.756
6.6
9
1.3
7
4.0
3
5.1
7
3.9
1
1.2
4
0.91.01.11.2 ppm
0.878
1.145
3.8
8
3.7
8
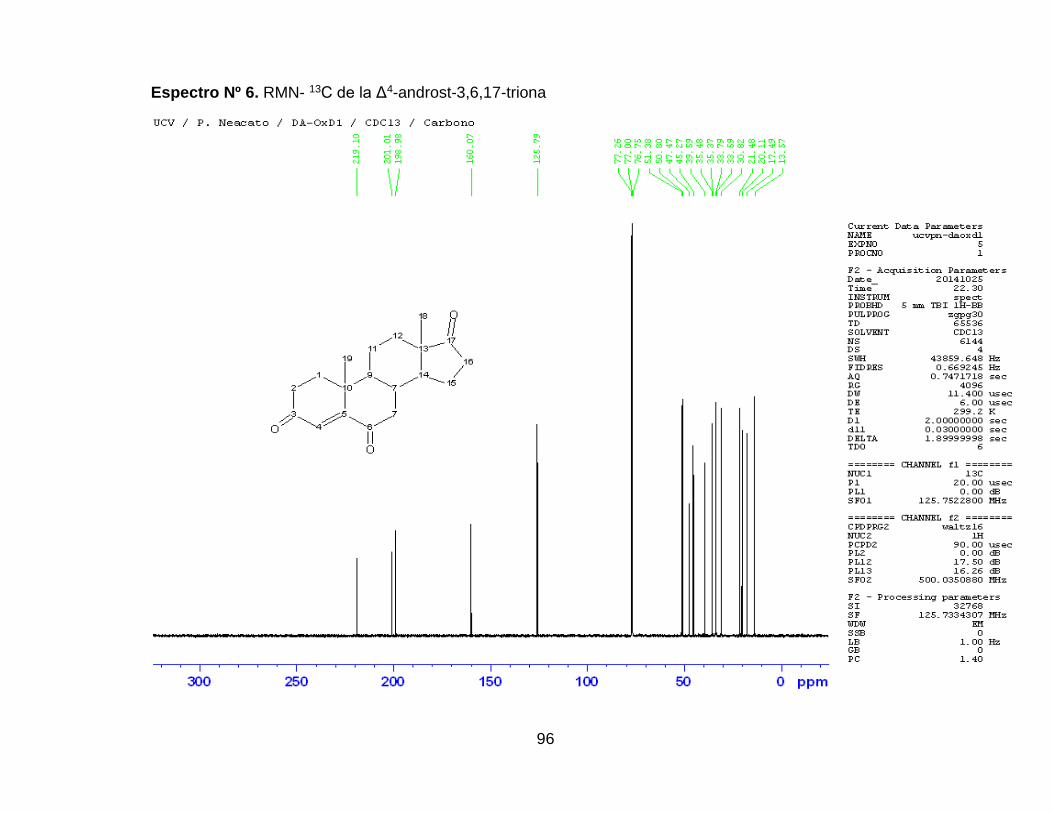
96
Espectro Nº 6. RMN- 13C de la Δ4-androst-3,6,17-triona
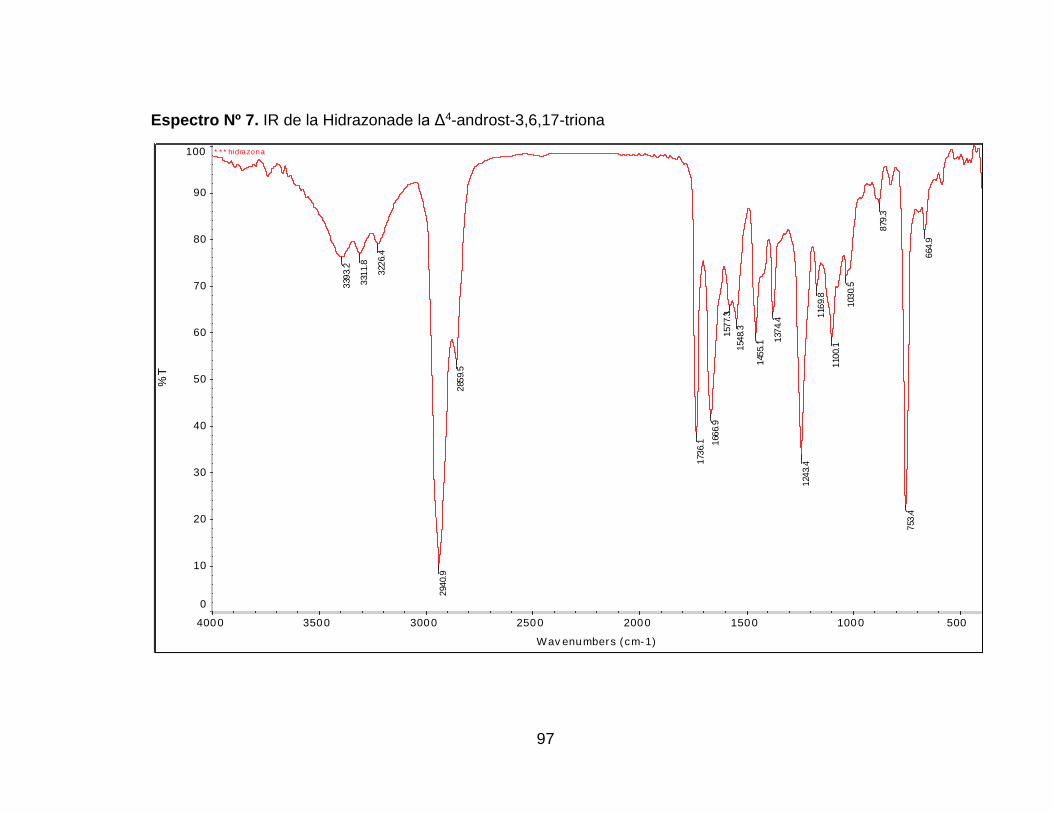
97
Espectro Nº 7. IR de la Hidrazonade la Δ4-androst-3,6,17-triona
664
.9
753
.4
879
.3
103
0.5
110
0.1
116
9.8
124
3.4
137
4.4
145
5.1
154
8.3
157
7.3
166
6.9
173
6.1
285
9.5
294
0.9
322
6.4
331
1.8
339
3.2
* * * hidrazona
0
10
20
30
40
50
60
70
80
90
100%
T
500 1000 1500 2000 2500 3000 3500 4000
Wav enumbers (cm-1)
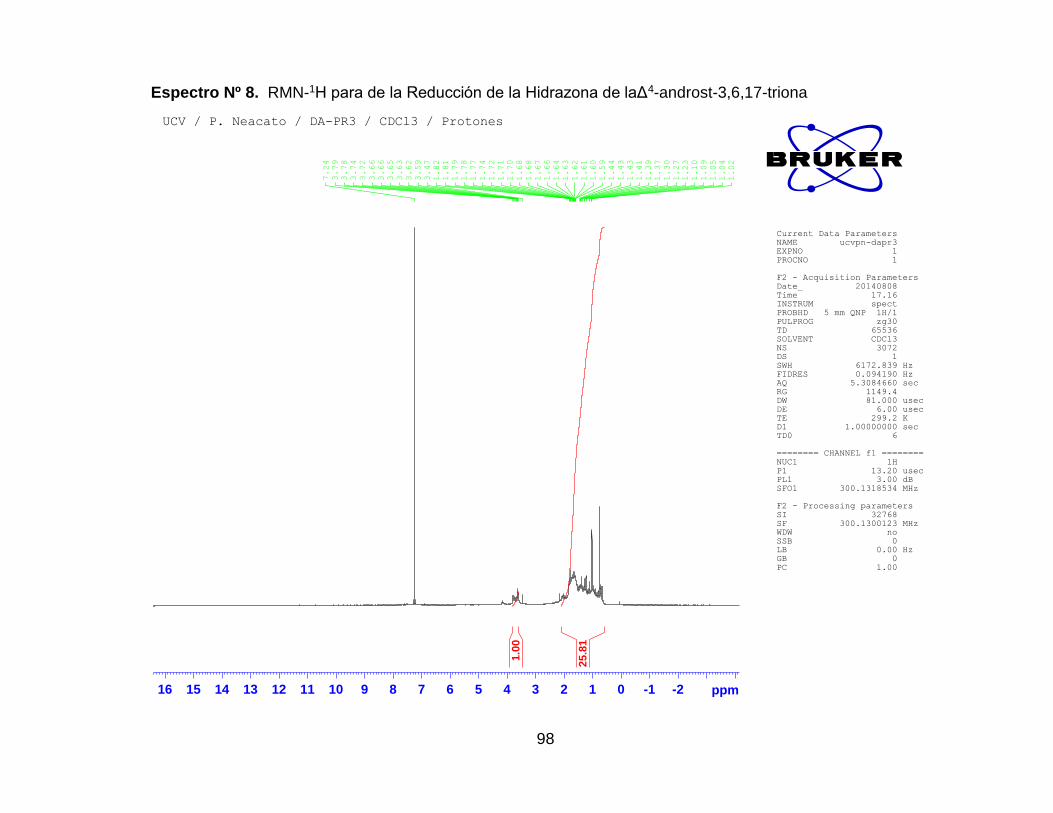
98
Espectro Nº 8. RMN-1H para de la Reducción de la Hidrazona de laΔ4-androst-3,6,17-triona
-2-116 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
1.02
1.04
1.05
1.09
1.10
1.23
1.27
1.30
1.37
1.39
1.41
1.43
1.43
1.44
1.59
1.60
1.61
1.62
1.63
1.64
1.66
1.67
1.68
1.68
1.70
1.71
1.72
1.74
1.77
1.78
1.79
1.81
1.82
3.47
3.59
3.62
3.63
3.65
3.66
3.66
3.72
3.74
3.78
3.79
7.24
25.8
1
1.0
0
UCV / P. Neacato / DA-PR3 / CDCl3 / Protones
Current Data Parameters
NAME ucvpn-dapr3
EXPNO 1
PROCNO 1
F2 - Acquisition Parameters
Date_ 20140808
Time 17.16
INSTRUM spect
PROBHD 5 mm QNP 1H/1
PULPROG zg30
TD 65536
SOLVENT CDCl3
NS 3072
DS 1
SWH 6172.839 Hz
FIDRES 0.094190 Hz
AQ 5.3084660 sec
RG 1149.4
DW 81.000 usec
DE 6.00 usec
TE 299.2 K
D1 1.00000000 sec
TD0 6
======== CHANNEL f1 ========
NUC1 1H
P1 13.20 usec
PL1 3.00 dB
SFO1 300.1318534 MHz
F2 - Processing parameters
SI 32768
SF 300.1300123 MHz
WDW no
SSB 0
LB 0.00 Hz
GB 0
PC 1.00

99
Espectro Nº9.RMN-13C para de la Reducción de la Hidrazona de laΔ4-androst-3,6,17-triona
200 180 160 140 120 100 80 60 40 20 0 ppm
11.16
14.83
15.78
20.66
23.38
29.11
30.38
30.46
30.53
31.49
33.06
33.98
35.34
35.51
36.22
36.68
38.54
39.15
39.28
41.77
43.05
47.45
50.76
54.06
54.39
66.61
71.64
71.85
72.05
76.58
77.00
77.42
81.94
Current Data Parameters
NAME ucvpn-dapr3
EXPNO 2
PROCNO 1
F2 - Acquisition Parameters
Date_ 20140808
Time 22.53
INSTRUM spect
PROBHD 5 mm QNP 1H/1
PULPROG zgpg30
TD 65536
SOLVENT CDCl3
NS 54758
DS 4
SWH 17985.611 Hz
FIDRES 0.274439 Hz
AQ 1.8219508 sec
RG 5160.6
DW 27.800 usec
DE 6.00 usec
TE 299.2 K
D1 2.00000000 sec
d11 0.03000000 sec
DELTA 1.89999998 sec
TD0 56
======== CHANNEL f1 ========
NUC1 13C
P1 12.50 usec
PL1 2.00 dB
SFO1 75.4752953 MHz
======== CHANNEL f2 ========
CPDPRG2 waltz16
NUC2 1H
PCPD2 100.00 usec
PL2 3.00 dB
PL12 20.59 dB
PL13 19.00 dB
SFO2 300.1312005 MHz
F2 - Processing parameters
SI 32768
SF 75.4677495 MHz
WDW EM
SSB 0
LB 2.00 Hz
GB 0
PC 0.80
UCV / P. Neacato / DA-PR3 / CDCl3 / Carbono

100
Ampliación del espectro RMN-13C para de la Reducción de la Hidrazona de laΔ4-androst-3,6,17-triona

101
Espectro Nº10. Espectro UV (CH2Cl2) del producto de la reducción directa del Δ4-androst-3,6,14-triona
empleando en método de Huang –Minlon
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
200 250 300 350 400 450
Ab
sorb
anci
a (U
.A.)
Longitud de Onda (nm)
Espectro UV del producto de reducción de la Δ4-Androst-3,6,17-triona

102
Espectro Nº 11. Reducción directa del Δ4-androst-3,6,14-trionaempleando en método de Huang –Minlon
72
3.5
10
99
.2
11
62
.8
12
40
.91
27
0.4
13
78
.2
14
62
.2
17
45
.1
28
54
.7
29
25
.8
30
08
.4
-0
10
20
30
40
50
60
70
80
90
100
%T
500 1000 1500 2000 2500 3000 3500
Wav enumbers (cm-1)

103
9. BIBLIOGRAFÍA
[1]Flores, J. Amado, J.A. (1997). Farmacología Humana. España. Editorial Masson,
S.A. Capítulo 1, pág. 14-15. Tercera Edición.
[2]Goodman y Gilman. (2006) Las Bases Farmacológicas de la Terapéutica. Editorial
Mc Graw Hill. Volumen I Capítulo 2, pág. 35-36. Undécima edición
[3] Solomons T.W. Graham. (1999). Química Orgánica. New York Editorial Wiley,
Capítulo 23, pág. 942-951. Segunda Edición
[4] Shulman, S. Phiair, J. Peterson, L. Warren, J. (1997) Estados Unidos. The
biological and clinical basis of Infectious Diseases. W.B Saunders Compañy.
Capítulo 5, pág. 47-48. Quinta Edición.
[5] Antonio Peña. (2004).Bioquímica, México. Editorial Limusa S.A. Capítulo 4,
pag.118-120. Segunda Edición.
[6] Marcano, D. Hasegawa, M. (1991) Venezuela. Fitoquímica Orgánica. Consejo de
Desarrollo Humanístico y Científico – UCV. Capítulo 4, pág 171-250. Primera Edición
[7]Finar, I.L. (1976). Química Orgánica Vol II: Estereoquímica y química de los
Productos Naturales. España Editorial Alhambra S.A. Capítulo VIII, pág. 257-259.
Primera Edición
[8] Cram, D.J. Hammond G.S. (1963) Química Orgánica. Editorial Limusa. Capítulo 24,
pág. 555; 560-566. Primera Edición.
[9]Flores, J. Amado, J.A. (1997) Farmacología Humana. España. Editorial Masson,
S.A. Capítulo 52, pág. 901-908. Tercera Edición.

104
[10] Marcano D. Cortes L. (2010) Química Orgánica Vol. II, Universidad de los Andes,
Ediciones Vicerrectorado Académico, CODEPRE. Mérida-Venezuela. Capítulo11, pág.
752; 755-757; 760-761; 765. 2da Edición.
[11] Levy D. (2008) Arrow Pushing in Organic Chemistry, Estados Unidos. Editorial
John Wiley & Sons, Publication. Capítulo 7, pág.116-117. Cuarta Edición
[12] Pine S. Briggs J. (1970) Química Orgánica. Editorial McGraw Hill Capitulo 11, pág.
525-536
[13] Solomons T.W. Graham. (1999) Química Orgánica. Editorial Wiley, New York
Capítulo 5, pág. 190-192 Segunda Edición
[14] Carey Sundberg. (1977) Química Orgánica Avanzada Editorial McGraw Hill
Capítulo 6 pág 393-394. Cuarta Edición
[15] Fieser L. Williamson K. Organic Experiments, Estados Unidos D.C. HEATH AND
COMPANY,. Capítulo 23, pág. 247-249. 7th Edition.
[16] Marcano D. Cortes L. (2004) Química Orgánica Vol. II, Universidad de los Andes,
Ediciones Vicerrectorado Académico, CODEPRE. Mérida-Venezuela. Capítulo12, pág.
803; 816-818; 830,836-838; 840-842. Tercera Edición.
[17] March, J. y Smith, M. (2007). Advanced Organic Chemistry. Estados Unidos
John Wiley & Sons, Publication. Capítulo 19, pág.1715-1716; 1727; 1835-1836; 1838 6th
Edition.
[18] Fieser L. Williamson K. Organic Experiments. Estados Unidos.D.C. HEATH AND
COMPANY,. Capítulo 225, pág. 261. 7th Edition

105
[19] Allinger N. (1997) Química Orgánica Vol. I, España. Editorial Reverte S.A.
Capítulo 17, pág.632-633. Segunda Edición.
[20] Fieser L. and Fieser M. Reagents for Organic Synthesis Vol.I, Editorial John
Wiley & Sons, pág. 1287
[21]J. C. Buchanan y P. D. Woodgate. (1976) Department of Chemistry, Stanford
University, California J. Am. Chem. Soc.
[22] Goldsmith D. J., Menger F (1977) Química Orgánica. Argentina. Editorial Limusa.
Capítulo 13, pag. 359-360. Segunda Edición
[23] G.I. Poos, G.E. Arth, R.E. Beyler y L.H. Sarett(1953) J. Am. Chem. Soc. 75,425
[24] John R.Holum. (1961). J.Org. Chem.26, 4814,
[25] J. C. Collins, W. W. Hess y F. J. Frank, (1968).TetrahedronLett.3363,
[26] R. Ratcliffe y R. Rodehorst, (1970) J. Org. Chem. 35, 4000,
[27] E. Piers y Paul M.Worster. (1957) .Can J. Chem. 55
[28] LouisF. Fieser (1953).J. Am. Chem. Soc. 75, 5421,
[29] C. Djerassi, R. R. Engle y A. Bowers, (1956) J. Am. Chem. Soc. 21, 1548
[30]Curtis, I. Heilbron, E. R. H. Jones y G. F. Woods, (1953).J. Chem. Soc., 457
[31]Huang Minlon. (1949) J. Am. Chem. Soc. 71, 3301,
[32] Dutcher y Wintersteiner (1939) J. Am. Chem. Soc. 61, 1991,
[33] Erno Prestch and Philippe Buhlmann (2010) .Structure Determination of Organic
Compounds Pág. 198. 4th Edition.

106
[34] Marcano D. Cortes L. (2010) Química Orgánica Vol. II, Universidad de los Andes,
Ediciones Vicerrectorado Académico, CODEPRE. Mérida-Venezuela. Cap. 9
Compuestos carbonílicos Pág. 634. Segunda Edición.
[35] Luis Dorfman, Ultraviolet Absorption of Steroids J. Chem,Rev. (1953), 59 pág.
50-52; 67
[36] Espectros de RMN-13C y RMN-1H para el colesterol consultado el 24-09-2014.
Disponible en http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi
[37] Espectros de RMN-13C y RMN-1H para la testosterona consultado el 24-09-2014.
Disponible en http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi
[38]A. Christy Hunter . (2005). ElsevierScienceInc. Steroids71:30-33
[39]Sheng-Hui. (1998). ElsevierScienceInc. Steroids 63:76-79
[40] Alvarado R.. (2013) Trabajo Especial de Grado. ESTUDIO DE LA REACCIONES
DE HIDROBORACION SOBRE SISTEMAS Δ4-Δ5 EN ANDROSTANOS, Universidad
Central de Venezuela. Págs.52-72




![Study on Georeferencing methods - TN-ITS › docs › emaps › eMaPS-D2.42-Triona... · 20 2012 Triona AB Peo Svensk, Lars Wikström 13-0 2 Version 1.2 [GEOREFERENCING METHODS] A](https://img.pdfslide.net/doc/110x75/5ed8b3336714ca7f476868b8/study-on-georeferencing-methods-tn-its-a-docs-a-emaps-a-emaps-d242-triona.jpg)











![FEDERICO TAVELLA, arXiv:1801.04774v2 [cs.ET] 18 Jan 2018 · FEDERICO TAVELLA, University of Padua, Italy ALBERTO GIARETTA, Örebro University, Sweden TRIONA MARIE DOOLEY-CULLINANE,](https://img.pdfslide.net/doc/110x75/5f7baa7b44f32a295b58e245/federico-tavella-arxiv180104774v2-cset-18-jan-2018-federico-tavella-university.jpg)
![Analytical Method Development and Validation for the ...Chemical name Bis(trifluoromethyl)phenyl]-3-oxo-4-aza-5α-androst-1-ene-17β-carboxamide Molecular formula C27H30F6N2O2 Molecular](https://img.pdfslide.net/doc/110x75/61388fa70ad5d20676495427/analytical-method-development-and-validation-for-the-chemical-name-bistrifluoromethylphenyl-3-oxo-4-aza-5-androst-1-ene-17-carboxamide.jpg)











