Embed Size (px)
Citation preview

UNIVERSITA’ DEGLI STUDI DI PERUGIA
Facoltà di Medicina e Chirurgia
Corso di Laurea Specialistica in Biotecnologie Mediche
Anno accademico 2010-2011
TESI DI LAUREA
IDENTIFICAZIONE E CARATTERIZZAZIONE FUNZIONALE DI NUOVE
MUTAZIONI GENETICHE NEI CANALI DEL POTASSIO Kir 4.1 (KCNJ10)
IN PAZIENTI AFFETTI DA AUTISMO, EPILESSIA E RITARDO MENTA-
LE.
Laureanda Relatore
Francesca Rollo Mauro Pessia

1
INDICE
SINTESI…………………………………………………………………………..4
INTRODUZIONE………………………………………………………………..5
Cenni di Storia della Neurofisiologia……………………………………………5
Canali del potassio………………………………………………………………..6
Canali Kir………………………………………………………………………....9
Struttura molecolare…………………………………………………………….10
Dominio transmembrana……………………………………………………….12
Dominio citoplasmatico………………………………………………………...12
Rettificazione Inward e funzione dei canali Kir……………………………...14
Regolazione del poro dei canali Kir…………………………………………..15
Fisiologia dei canali Kir4.x e Kir5.1…………………………………………..20
Localizzazione intracellulare…………………………………………………..24
Localizzazione nei microdomini di membrana……………………………….26
Funzioni fisiologiche…………………………………………………………...27
Farmacologia…………………………………………………………………..32
Canalopatie associate ai Kir4.1 e Kir5.1……………………………………..32
EPILESSIA……………………………………………………………………...35
Cause dell’epilessia…………………………………………………………….36
Patofisiologia dell’epilessia…………………………………………………….37
AUTISMO……………………………………………………………………….40

2
Cause dell’autismo……………………………………………………………..41
Manifestazioni cliniche dell’autismo………………………………………….42
Il fenotipo “autismo-epilessia”………………………………………………..44
RAZIONALE DELLO STUDIO………………………………………………45
MATERIALI E METODI……………………………………………………...47
PAZIENTI……………………………………………………………………….47
Reclutamento dei pazienti e indagini cliniche………………………………...47
ANALISI DI MUTAZIONI……………………………………………………..48
BIOLOGIA MOLECOLARE…………………………………………………..49
ANALISI STRUTTURA E FUNZIONE………………………………………50
ESPRESSIONE ETEROLOGA DEI CANALI IONICI……………………...51
Prelievo e selezione degli ovociti………………………………………………...53
Microiniezione di mRNA in ovociti……………………………………………..54
ELETTROFISIOLOGIA………………………………………………………..56
Two-electrode voltage-clamp…………………………………………………...56
Tecnica del patch-clamp………………………………………………………..59
RISULTATI……………………………………………………………………..61

3
Caratteristiche cliniche dei pazienti…………………………………………..61
Risultati delle analisi di mutazioni……………………………………………62
Caratterizzazione funzionale dei canali omomerici che presentano le mutazioni
R18Q e V84M……………………………………………………………………64
Caratterizzazione funzionale dei canali eteromerici che presentano le mutazio-
ni R18Q e V84M …………………………………………………………………...69
Modeling molecolare……………………………………………………………...72
Conferma dei risultati……………………………………………………………73
DISCUSSIONE……………………………………………………………………75
BIBLIOGRAFIA…………………………………………………………………80
RINGRAZIAMENTI………………………………………………………......111

4
SINTESI
Lo scopo di questa tesi è quello di verificare se mutazioni nel gene, che codifica per
il canale del potassio Kir4.1 (KCNJ10), inducono modificazioni nell’attività della
proteina, in pazienti appartenenti ad un particolare sottogruppo dello spettro autisti-
co, che è stato denominato come "fenotipo autismo-epilessia".
Per verificare se i canali Kir fossero responsabili del fenotipo oggetto di studio, il ge-
ne KCNJ10 è stato analizzato alla ricerca di mutazioni da DNA genomico estratto da
leucociti del sangue periferico dei pazienti e utilizzando tecniche standard di genetica
molecolare.
Successivamente attraverso studi funzionali, utilizzando la tecnica elettrofisiologica
del TEVC (two-electrode voltage-clamp) e del patch clamp, si è analizzato l’effetto
delle mutazioni identificate sull’attività del canale Kir4.1. I risultati ottenuti mostra-
no che queste mutazioni inducono una gain-of-function dell’attività del canale sia at-
traverso un aumento di espressione in superficie o della conduttanza della proteina.
I canali Kir 4.1 sono abbondantemente espressi negli astrociti in cui assieme al
Kir5.1 contribuiscono al buffering di K+. La disfunzione dell’omeostasi del K
+ indot-
ta dalle mutazioni del canale Kir4.1, in pazienti con DSA e suscettibilità all’epilessia,
potrebbe contribuire allo sviluppo del fenotipo autistico e degli attacchi epilettici, al-
terando l’eccitabilità e la funzione sinaptica, e potrebbe rappresentare un target per
nuovi approcci terapeutici.

5
INTRODUZIONE
Cenni di Storia della Neurofisiologia
Il potenziale d'azione (PA) è il fenomeno particolare che si manifesta nei neuroni, e
che prevede un rapido cambiamento di carica tra l'interno e l'esterno della membrana
cellulare. Nel corso dei secoli il lavoro pionieristico di alcuni scienziati ha permesso
di chiarire i fenomeni che sono alla base della generazione del PA e delle proprietà
elettriche delle cellule nervose.
Luigi Galvani, tra il 1791 ed il 1797, pubblicò il suo più famoso esperimento metten-
do le basi di una nuova scienza e proponendo la teoria sull’elettricità animale. Infatti
su un preparato di rana eseguì i primi esperimenti di elettrofisiologia. Successiva-
mente Alessandro Volta illuminato dai risultati di Galvani, inventò la prima batteria.
Gli effetti elettrici e fisiologici ottenuti da questo strumento su preparati di tessuto sia
umano che di rana, inaugurarono definitivamente l’era moderna della neurofisiologia
(Piccolino 1997 e 2000).
Cento anni dopo, Julius Bernstein ipotizzò l’esistenza di potenziali bioelettrici nelle
membrane eccitabili e propose che la permeabilità di queste strutture dovesse neces-
sariamente cambiare in seguito ad una stimolazione. Questa teoria venne confermata
alla fine del 1930 da Kenneth Cole e Howard Curtis. Con degli esperimenti che pre-
vedevano l’uso di un assone gigante di calamaro, i due ricercatori mostrarono che la
conduttanza della membrana aumentava durante un PA (Cole and Curtis 1939). Suc-
cessivamente Alan Hodgkin e Andrei Huxley osservarono che sorprendentemente il
potenziale della membrana superava il valore di 0mV e cambiava di segno durante
un impulso elettrico. Un decennio dopo Hodgkin e Katz dimostrarono inequivoca-
bilmente che l’entrata degli ioni sodio attraverso la membrana determinava la fase di
ascesa ed la propagazione del PA, così come il superamento dello zero (Hodgkin e
Katz 1949). Immediatamente dopo, utilizzando l’innovativa tecnica del Voltage-
Clamp, riuscirono a dissociare le correnti ioniche di sodio e di potassio negli assoni
stabilendo la loro voltaggio dipendenza, le loro proprietà cinetiche ed il ruolo nella
generazione e nella propagazione del PA (Hodgkin e Huxley 1952).
Hodgkin e Huxley dedussero il coinvolgimento di proteine contenenti dei pori nella
generazione del segnale elettrico nei neuroni. Sebbene l’esistenza dei pori fosse stata
originariamente proposta da Ernst Brücke nel 1843, solo nel 1970 vennero misurate
le correnti dei singoli canali quando Ervin Neher e Bert Sakmann svilupparono la
tecnica del patch-clamp (Neher e Sakmann 1976). Ed è grazie a queste affascinanti

6
scoperte che noi oggi sappiamo che le aperture e chiusure concertate di canali ionici
del sodio, del potassio e del calcio svolgono un ruolo di assoluta importanza nella
neurotrasmissione. In particolare loro generano e stabiliscono la forma dei PA, mo-
dulano il rilascio dei neurotrasmettitori, controllano l’eccitabilità, le proprietà elettri-
che e la frequenza di scarica dei neuroni sia centrali che periferici (Hille B. 2001).
Molti di questi canali sono stati clonati negli ultimi vent’anni. La ricchezza di infor-
mazioni ottenute hanno inaugurato un intenso periodo di analisi della struttura-
funzione di questi canali ionici. Inoltre, la dettagliata delucidazione della struttura dei
geni ha permesso di individuare delle mutazioni responsabili di canalopatie eredita-
rie. A coronamento di quest’era, studi di cristallografia hanno recentemente chiarito
la struttura di molti canali ionici, fornendoci informazioni preziosissime sul funzio-
namento di queste proteine.
Canali del potassio
I canali del potassio rappresentano la più antica famiglia di canali di membrana e so-
no espressi in molti tipi di cellule, sia nel regno animale che in quello vegetale oltre
che in lieviti e batteri (Hille B. 2001). Questi canali hanno la funzione fondamentale
di stabilizzare il potenziale di riposo di una membrana; intervengono nella fase di ri-
polarizzazione dopo un potenziale d'azione e sono importanti perché condizionano
l'intervallo tra un PA ed il successivo in caso di scariche ripetitive; hanno poi la fun-
zione di trasporto dei soluti negli epiteli e rimuovono gli eccessi di potassio dagli
spazi intercellulari, che è una funzione tipica delle cellule gliali.
Questi canali possono essere regolati da cambiamenti del potenziale di membrana e
dello stato metabolico della cellula o tramite trasmettitori e ormoni. Tale regolazione
è importante per le funzioni di signaling tra i neuroni e nei meccanismi di protezione
durante eventi stressanti quali anossia e ischemia. Questo giustifica il fatto che agenti
farmacologici che incrementano o diminuiscono l’attività dei canali al potassio pos-
sano essere utilizzati come potenziali anti-ischemici, antiaritmici, antipertensivi o an-
tianginosi.
L’importanza fisiologica della funzione dei canali al potassio è evidente dalle conse-
guenze che mutazioni possono generare. Mutazioni dei geni che codificano per tali
canali sono associati a malattie neurologiche come l’atassia episodica (Browne et al.
1994; Adelman et al. 1995) e l’aritmia cardiaca (Curran et al. 1995, Sanguinetti et al.
1995). Mutazioni nel gene che codifica per il recettore della sulfanilurea, una β subu-

7
nità del canale per il potassio ATP-dipendente nel pancreas, induce ipoglicemia ipe-
rinsulinemica infantile (Inagaki et al. 1995, Thomas et al. 1995).
Questi canali ionici conducono selettivamente lo ione K+ secondo gradiente elettro-
chimico ad una velocità di 106-10
8 ioni al secondo. Nello stato di riposo la concen-
trazione degli ioni potassio fuori dalla cellula è di circa 35 volte inferiore rispetto alla
concentrazione nei fluidi intracellulari ([K+]e=4mEq/l, [K
+]i=140mEq/l) e in seguito
all’apertura dei canali si genera un flusso di cariche positive verso l’esterno che per-
mettono la ripolarizzazione e/o l’iperpolarizzazione della membrana cellulare.
I canali del potassio mostrano una straordinaria eterogeneità dovuta in parte alla mol-
titudine di geni che codificano per i canali stessi, ma anche ad altri processi come lo
splicing alternativo, che da luogo alla formazione di mRNA multipli trascritti da un
singolo gene, o l’assemblamento eteromerico di differenti subunità principali come
anche possibili modificazioni post-traduzionali. Grazie ai progressi nel campo della
biologia molecolare ed alla conoscenza del genoma umano, negli ultimi anni sono
stati identificati più di 80 canali del potassio con i relativi geni anche se in ogni caso
è possibile identificare alcuni elementi strutturali comuni. I canali del potassio, infat-
ti, sono tutti complessi generati dal coassemblaggio di quattro subunità che possono
essere identiche o simili (isoforme) e che sono chiamate principali o α-subunità.
Queste subunità costituiscono il canale ionico. Tali proteine possono essere formate
da un numero variabile di segmenti transmembrana (TMs) uniti da loops intra ed e-
xtracellulari con il dominio amminico e quello carbossi-terminale localizzati nel lato
citoplasmatico.
I canali del potassio, quindi, sono tutti tetrameri e a seconda del numero di α-eliche
che costituiscono ogni dominio del tetramero si distinguono in tre categorie come si
può osservare nella figura seguente:

8
Fig. 1 Classificazione dei canali del K+
Canali del potassio a 6 α-eliche transmembrana (6 STM), che comprendo-
no:
o Canali rettificanti che si aprono con ritardo (o delayed rectifier). So-
no canali del potassio attivati dalla depolarizzazione della membrana.
Hanno la funzione di regolare la durata del potenziale d’azione.
o Canali che generano correnti transienti (KA). Sono canali che si apro-
no in modo transiente quando una cellula si depolarizza in seguito ad
un’iperpolarizzazione. La loro funzione è quella di stabilizzare il po-
tenziale di riposo delle cellule nervose e ridurre la frequenza dei treni
di potenziali d’azione.
o Canali del K+ calcio-dipendenti. Si distinguono in canali ad alta
(BKCa) e bassa conduttanza (SKCa). Nella cellula a riposo questi canali
sono chiusi a causa della bassa concentrazione di calcio. In seguito a
depolarizzazione si aprono i canali del calcio voltaggio-dipendenti.
Per cui, la concentrazione intracellulare di Ca2+
aumenta. Ciò provoca
l’apertura di questi canali del potassio che così può uscire dalla cellu-
la. Tipicamente, hanno un tempo di apertura prolungato ed il fusso di
ioni potassio verso l’esterno iperpolarizza la cellula e quindi riduce la
frequenza dei potenziali d’azione generati da un neurone.
Canali del potassio a 4 α-eliche transmembrana (4 TMs), ancora non ben
caratterizzati.

9
Canali del potassio a 2 α-eliche transmembrana (2 TMs). Sono canali che
permangono aperti in condizioni di iperpolarizzazione della membrana e si
chiudono quando questa si depolarizza. Tipici delle cellule muscolari cardia-
che, stabilizzano il potenziale quando la membrana è nello stato di riposo, ma
quando uno stimolo sovrasoglia induce un potenziale d’azione, i canali si
chiudono.
Fanno parte di questa famiglia i canali del potassio attivati da proteine G
(GIRK = G protein coupled inward rectifier K+ channels).
L’attività di questi canali è stimolata da numerosi recettori inibitori presenti a livello
del sistema nervoso centrale, del cuore e delle ghiandole endocrine, in cui svolgono
un ruolo di iperpolarizzazione con blocco dell’attività elettrica di membrana.
La loro attivazione richiede l’interazione della subunità βγ di proteine Gi con struttu-
re citosoliche del canale che determinano l’apertura del poro mediante una modifica-
zione conformazionale delle eliche che lo delimitano.
La caratteristica principale di questi canali è il filtro di selettività che permette il pas-
saggio degli ioni K+ solo se deidratati. Al centro del canale è presente una cavità pie-
na d’acqua.
Canali Kir
I canali del potassio a 2 α-eliche transmembrana inward rectifier (Kir) sono stati i-
dentificati in un’ampia varietà di cellule oltre ai miociti cardiaci (Beeler e Reuter
1970; Kurachi Y. 1985; McAllister e Noble 1966; Noble D. 1965; Rougier et al.
1968) ed ai neuroni (Brown et al. 1990; Gahwiler e Brown 1985; Lacey et al. 1988;
North et al. 1987; Takahashi T. 1990; Williams et al. 1988).
Infatti sono stati individuanti anche nelle cellule del sangue (Lewis et al. 1991;
McKinney e Gallin 1988), cellule endoteliali (Silver e DeCoursey 1990), osteoclasti
(Sims e Dixon 1989), e cellule gliali (Kuffler e Nicholls 1966; Newman EA. 1984) e
sono molto importanti per la regolazione del potenziale di membrana a riposo e per l’
attività elettrica cellulare.
Questi canali sono anche regolatori essenziali dei processi di trasporto del K+
nell’organismo e/o di secrezione di ormoni (KATP e insulina) (Nichols e Lopatin
1997; Reimann, e Ashcroft 1999).
I canali Kir, quindi, non solo controllano le proprietà elettriche attive e passive delle
cellule, ma intervengono anche nelle pathway intracellulari di recettori accoppiati a

10
proteine G (GPCR), rappresentando un punto di collegamento tra stato metabolico
cellulare ed eccitabilità di membrana.
A tutt’oggi sono stati identificati 15 geni codificanti per le subunità Kir, classificati
in sette famiglie (Kir 1.x-Kir 7.x). A loro volta queste famiglie possono essere orga-
nizzate in quattro gruppi funzionali come mostrato nella figura seguente:
Fig. 2 Famiglie dei canali Kir
1) Canali Kir classici (Kir2.x)
2) Canali Kir associati a proteine G (Kir3.x)
3) Canali K+ sensibili all’ATP (Kir6.x)
4) Canali di trasporto del K+ (Kir1.x, Kir4.x, Kir5.x e Kir7.x)
Struttura molecolare
I primi ad essere descritti già a partire dal 1993 sono stati i canali Kir 1.1 e i Kir 2.1.
La struttura primaria di questi canali è costituita da due domini transmembrana (TM1
e TM2) uniti da una sequenza aminoacidica che costituisce la porzione extracellula-
re del poro (H5), e da due domini citoplasmatici amino (NH2)- e carbossi (COOH)-
terminale.
Rettifica-
zioo-
ne,spiega
bene ora o
dopo

11
Fig. 3 Struttura primaria delle subunità dei canali Kir
Ogni subunità Kir contiene due domini transmembrana (TM1 e TM2), una regione
formante il poro del canale (H5) e le estremità citosoliche NH2- e COOH- terminali.
Il segmento H5 costituisce il filtro di selettività (Heginbotham et al. 1994) e presenta
la classica sequenza aminoacidica presente in tutti gli altri canali selettivi agli ioni
K+, T-X-G-Y(F)- G (Bichet et al. 2003). Le estremità NH2 e COOH esposte nel cito-
plasma, sono associate l’una all’altra in modo da costituire il dominio citoplasmatico
che è legato, ma comunque distinto, dal dominio transmembrana. Si pensa che pro-
prio il dominio citoplasmatico sia responsabile del meccanismo di gating del canale
(meccanismo per cui l’azione di un ligando in una regione del canale o una variazio-
ne del potenziale di membrana, inducono un cambiamento di conformazione del ca-
nale). Mediante cristallografia, si è osservato che questa struttura molecolare è co-
mune a tutti i canali Kir. Rispetto ai canali Na+, Ca
2+ (formati da 4 subunità, ognuna
a sua volta costituita da 6 eliche transmembrana) e K+ voltaggio dipendenti (che pos-
siedono 6 domini transmembrana), i canali Kir non possiedono il sensore del voltag-
gio. Di conseguenza lo stato di attivazione dei canali Kir non dipende dal potenziale
di membrana ma da meccanismi specifici per ogni tipo di canale (es. ATP determina
chiusura canali K-ATP dipendenti o proteina G causa attivazione canali KG). Dal
momento che la struttura primaria dei due domini transmembrana è insufficiente a
formare un canale ionico completo, i canali Kir funzionali sono costituiti da quattro
di queste subunità che si uniscono a formare un complesso tetramerico (Glowatzki et
al. 1995; Yang et al. 1995). In virtù della forte omologia e semplicità delle subunità, i
canali Kir possono essere sia omomerici, quindi costituiti dall’unione di 4 subunità
H2N+ COO-

12
identiche, che eteromerici. In quest’ultimo caso, il canale è costituito da 4 subunità
differenti, normalmente appartenenti alla stessa famiglia (per esempio la subunità Kir
2.1 si può associare con un altro membro della famiglia Kir 2.x (Preisig-Muller et al.
2002; Schram et al. 2002), anche se possono verificarsi delle eccezioni come nel caso
del canale Kir4.1-5.1.
Dominio transmembrana
Come è stato descritto precedentemente, questo dominio è composto da due strutture
ad elica che attraversano la membrana, una esterna (TM1) e una interna (TM2), e da
due piccole eliche addizionali (―pore‖ e ―side‖ helix). Il poro del canale è delimitato
dall’elica TM2 di ognuna delle quattro subunità Kir. Il poro attraverso il quale passa-
no gli ioni può essere suddiviso, dal punto di vista funzionale, in tre regioni distinte:
il filtro di selettività, la cavità centrale piena d’acqua e la superficie interna del poro
costituita dalle basi delle quattro eliche TM2. La sequenza T-X-G-Y(F)-G, che rap-
presenta il filtro di selettività, separa la cavità centrale del canale dalla soluzione e-
xtracellulare.
Fig. 4 Dominio trans membrana
Nella figura sono mostrate alcune strutture secondarie del dominio transmembrana,
importanti per il funzionamento dei canali Kir. Il dominio transmembrana comprende
tre eliche: TM1, ―pore helix‖ e TM2. Sul lato citoplasmatico della membrana c’è an-
che l’elica anfipatica ―side helix‖. I residui mostrati in giallo sono responsabili
dell’interazione con Mg2+
e poliammine e, quindi, coinvolti nei meccanismi di retti-
ficazione. Nei canali Kir ci sono due diversi meccanismi di gating, lento e veloce.
Filtro di selettività
“pore helix”
TM2
TM1
“side helix”
Residui che intera-
giscono con Mg2+
e
poliammine

13
Nelle registrazioni delle correnti di singolo canale, il gating lento corrisponde a rapi-
de aperture (bursts) intervallate da lunghi periodi di chiusura, mentre il gating veloce
corrisponde a rapide aperture e chiusure del canale.Diverse analisi svolte sui canali
Kir hanno mostrato che entrambe le eliche TM1 e TM2 sono coinvolte in questi
meccanismi di gating.
Dominio citoplasmatico
Il dominio citoplasmatico dei canali Kir è costituito dalle estremità NH2 e COOH
delle quattro subunità Kir. Ogni terminazione è ricca di filamenti β, i quali si unisco-
no a formare tre foglietti β. In realtà il dominio citoplasmatico è costituito principal-
mente dalle terminazioni COOH, mentre le terminazioni NH2 contribuiscono alla
formazione dell’interfaccia tra le subunità posizionandosi tra le estremità COOH a-
diacenti. Ogni terminazione NH2 contiene un singolo filamento β (βA) che si unisce
a due filamenti β (βL e βM) nell’estremità COOH per formare un foglietto β. Tutto
ciò è importante per conferire stabilità all’intera struttura citoplasmatica del canale.I
quattro gruppi formati dall’associazione delle terminazioni NH2 e COOH costitui-
scono una struttura cilindrica che circonda il cosiddetto poro citoplasmatico. Questa
architettura è caratteristica dei canali Kir e determina un prolungamento di ≈30 Å
della pathway di conduzione (Nishida e MacKinnon 2002). Quindi in questi canali, il
potassio deve percorrere più di 60 Å attraverso il poro composto dal dominio tran-
smembrana e citoplasmatico.
Fig. 5 Dominio citoplasmatico
membrana
N217D
S225E
E300
D260
S256D
H222
R52 R190
R219
L262
G336
L333
H57

14
In BLU sono mostrati i residui aminoacidici coinvolti nel legame del PtdIns(4,5)P2,
necessario per l’apertura del canale. Questi sono distribuiti sulla superficie del domi-
nio citoplasmatico, a contatto con la membrana. La parte centrale del dominio cito-
plasmatico è costituita da una cavità acquosa che contribuisce alla permeazione degli
ioni. A livello di tale cavità sono localizzati alcuni residui coinvolti nella rettificazio-
ne inward (mostrati in ROSSO). I canali Kir3.x sono attivati dall’interazione diretta
con la subunità βγ delle proteine G e i canali contenenti le subunità Kir3.2 e Kir3.4
possono essere attivati anche dal Na+. Gli aminoacidi responsabili di queste intera-
zioni sono rispettivamente indicati in VERDE e in ROSA.
La regione citoplasmatica dei canali Kir è importante anche perché permette il lega-
me del fosfatidilinositolo 4,5 bifosfato (PtdIns(4,5)P2), necessario per il corretto fun-
zionamento del canale (Bendahhou et al. 2003; Hilgemann e Ball 1996; Huang et al.
1998; Lopes et al. 2002; Shyng e Nichols 1998; Zhang et al. 1999), così come di altre
proteine di trasporto degli ioni come lo scambiatore Na+/Ca
2+ e i canali del Ca
2+ vol-
taggio-dipendenti (Hilgemann e Ball 1996; Hilgemann et al. 2001; Suh e Hille
2005). E’ stato proposto che l’azione del PtdIns(4,5)P2 coinvolga numerosi residui
basici e privi di carica nella regione citoplasmatica del canale, in particolare a livello
della superficie rivolta verso la membrana (Du X et al. 2004; Fan e Makielski 1997;
Huang et al. 1998; Schulze et al. 2003; Shyng et al. 2000; Zhang et al. 1999). Anche
nella regione transmembrana sono presenti residui che partecipano all’associazione
tra il PtdIns(4,5)P2 ed il canale, si tratta di residui basici raggruppati nella parte più
bassa dell’elica interna TM2 e residui privi di carica localizzati, invece, sulla ―slide
helix‖ (Lopes et al. 2002; Roha´cs et al. 2002). Tuttavia il meccanismo d’azione del
PtdIns(4,5)P2 non è ancora del tutto compreso.
La modulazione della conformazione delle terminazioni NH2 e COOH e la loro inte-
razione gioca chiaramente un ruolo fondamentale nella regolazione del gating dei ca-
nali Kir.
Rettificazione Inward e funzione dei canali Kir
Inizialmente le correnti Kir sono state definite correnti rettificanti ―anomale‖ del po-
tassio. Infatti la relazione tra queste correnti e il potenziale di membrana non segue la
cinetica descritta da Hodgkin-Huxley (Hagiwara S. 1983) e il loro comportamento
sembra dipendere principalmente dal gradiente elettrochimico del potassio (potenzia-
le di membrana (Em) meno potenziale di equilibrio del K+ (EK)). Infatti, la rettifica-

15
zione Inward delle correnti dei canali Kir è dovuta al blocco del poro del canale da
parte di Mg2+
e poliammine intracellulari quando il potenziale di membrana (Em) è
maggiormente depolarizzato rispetto al potenziale di equilibrio del potassio (EK)
(Hagiwara e Takahashi 1974). Molti dei residui aminoacidici responsabili di questo
blocco sono stati individuati a livello dell’elica interna TM2, ma ne sono stati identi-
ficati anche altri a livello della parete del poro nel dominio citoplasmatico. Nelle cel-
lule che esprimono elevate quantità di questi canali, quindi, ci si aspetta un potenzia-
le di membrana prossimo al potenziale di equilibrio del potassio e mancanza di attivi-
tà elettrica spontanea. Proprio queste caratteristiche, insieme alla voltaggio indipen-
denza, rendono i canali Kir molto importanti per la regolazione del potenziale di
membrana a riposo.
L’attività fisiologica dei canali Kir dipende da diversi fattori tra i quali l’apertura del
poro e il flusso ionico. Anche la localizzazione del canale in particolari regioni della
cellula (per esempio a livello della membrana pre- o post-sinaptica per quanto ri-
guarda i neuroni), o in microdomini della membrana cellulare (ad esempio in prossi-
mità di altre molecole trasportatrici), da un contributo importante al ruolo funzionale
dei canali Kir nei diversi organi o tessuti.
Regolazione del poro dei canali Kir
Ci sono diversi fattori che regolano il flusso ionico e le cinetiche di apertura del poro,
i principali sono ioni, poliammine, nucleotidi, lipidi e diverse proteine intracellulari
che nella maggior parte dei casi interagiscono direttamente con componenti fonda-
mentali del canale.
Mg2+
intracellulare e poliammine.
La capacità rettificante dei canali Kir è il risultato dell’interazione tra due so-
stanze intracellulari, Mg2+
e poliammine (in particolare spermidina e spermi-
na), e la regione citoplasmatica del poro del canale (Lopatin et al. 1994; Ma-
tsuda et al. 1987). Depolarizzazioni di membrana fanno si che queste sostanze
blocchino la fuoriuscita del potassio legandosi a residui localizzati a livello
dei domini transmembrana o citoplasmatici dei canali. Diversi studi hanno
dimostrato che la rettificazione in realtà è il risultato dell’azione combinata di
questo blocco mediato dal Mg2+
intracellulare e di un processo di attivazione
intrinseco, il meccanismo sterico del gating (Ishihara et al. 1989; Kurachi
Y.1985; Matsuda H. 1988; Matsuda et al. 1987). Ad esempio, nel caso delle

16
correnti dei canali Kir cardiaci, gli ioni Mg2+
determinano una rettificazione
inward istantanea, seguita dall’aumento tempo-dipendente della rettificazio-
ne dovuto al meccanismo intrinseco di gating. Mediante registrazioni di
patch-clamp nella configurazione ―inside-out‖, si è osservato che, nel caso in
cui venga meno, questo meccanismo intrinseco di gating può essere ripristi-
nato mediante l’applicazione, a livello della porzione intracellulare del cana-
le, di poliammine (spermidina e spermina) in concentrazioni sub-micromolari
(Fakler et al. 1995; Ficker et al. 1994; Lopatin et al. 1994; Yamada e Kurachi
1995). Grazie a questa osservazione e dal momento che le poliammine sono
presenti in concentrazioni sub-millimolari nella cellula, si può affermare che
il meccanismo di gating è il risultato della capacità delle poliammine di bloc-
care e sbloccare lentamente i canali Kir ( in modo tempo-dipendente). Quindi,
in condizioni di depolarizzazione (precedentemente chiamata ―deattivazio-
ne‖) si ha inibizione tempo-dipendente delle correnti outward (correnti in u-
scita dalla cellula) dovuta all’azione bloccante delle poliammine. In caso di
iperpolarizzazione invece, le correnti inward del potassio inizialmente au-
mentano in modo indipendente dal tempo a causa del rapido sblocco del ca-
nale per rimozione degli ioni Mg2+
e continuano poi ad aumentare in modo
tempo-dipendente per lo sblocco dovuto alle poliammine. Quest’ultima fase
viene definita ―attivazione‖(Lopatin et al. 1995).
Non tutti i tipi di canali Kir comunque presentano lo stesso grado di rettifica-
zione inward, esistono infatti ―forti rettificatori‖ (canali Kir 2.1 e Kir3.x),
―rettificatori intermedi‖ (Kir 4.x) e ―deboli rettificatori‖ (Kir 1.1 e Kir 6.x).
Diversi studi di questi canali hanno permesso di identificare la posizione 172
del secondo dominio transmembrana (TM2) come sito critico nel determinare
la capacità rettificante dei canali Kir. Si è giunti a questa conclusione osser-
vando che in questa posizione nei forti rettificatori Kir 2. 1 è presente un re-
siduo di Aspartato (D) carico negativamente, mentre nei rettificatori deboli
Kir 1.1 c’è un residuo di Asparagina (N) privo di carica (Lu Z e MacKinnon
1994; Stanfield et al. 1994; Wible et al. 1994; Yang et al. 1995). Per questo
motivo il sito 172 del dominio TM2 è stato definito ―D/N site‖ ed è stato di-
mostrato che sostituendo questo residuo di Asparagina con l’Aspartato si ot-
tiene un forte aumento dell’affinità dei Kir 1.1 per il Mg2+
e di conseguenza
un aumento della rettificazione.

17
Fig. 6 Caratteristiche elettrofisiologiche dei canali del potassio Kir
Concentrazione extracellulare di K+.
La conduttanza di tutti i canali Kir, ad eccezione del Kir7.1 (Doring et al.
1998; Krapivinsky et al. 1998), aumenta all’aumentare della concentrazione
extracellulare di potassio ([K]o). Ad esempio, la conduttanza del canale
Kir2.1, in assenza di Mg2+
e poliammine, dipende dalla radice quadrata della
[K]o, suggerendo che questa dipendenza possa essere una proprietà del poro
dei canali aperti (Lopatin e Nichols 1996). Tale proprietà implica che il pas-
saggio di ioni attraverso i canali Kir non segue la legge di permeabilità di
Goldman-Hodgkin-Katz (secondo la quale gli ioni influenzano il potenziale
di membrana in base alla loro permeabilità relativa e si muovono attraverso il
poro in maniera del tutto indipendente l’uno dall’altro), ma piuttosto è in ac-
cordo con il modello del ―poro multi-ione‖ secondo il quale il poro del canale
può essere occupato contemporaneamente da più di uno ione (Hille e
Schwarz 1978). A tal proposito, uno studio di Hagiwara e collaboratori ha

18
proposto che il poro dei canali Kir avrebbe almeno 2 siti di legame per il K+
(Ciani et al. 1980; Ciani et al. 1978).
Fosfatidilinositolo 4,5-bifosfato.
Il fosfatidilinositolo 4,5-bifosfato [PI(4,5)P2] è un lipide ancorato alla mem-
brana cellulare che è molto importante per la funzionalità della maggior parte
dei canali Kir (Hilgemann et al. 2001). Le analisi mutazionali di questi canali
hanno mostrato che il PI(4,5)P2 si lega a livello di residui aminoacidici cari-
chi positivamente a livello dell’estremità carbossi-terminale (Huang et al.
1998; Lopes et al. 2002). Molto spesso quando si effettuano registrazioni di
patch-clamp su frammenti di membrana separati dalla cellula, l’attività dei
canali Kir diminuisce progressivamente, ma può essere ripristinata mediante
l’applicazione di ATP sulla superficie intracellulare della membrana poiché
l’ATP ripristina il PI(4,5)P2 mediante l’attivazioni di chinasi lipidiche (Hil-
gemann e Ball 1996). Tutto ciò dimostra quindi l’importanza del PI(4,5)P2
per la funzionalità dei canali Kir.
Ulteriori sostanze
La funzionalità di alcuni tipi di canali Kir, in particolare i Kir 1.1, Kir 2.3 e i
Kir contenenti la subunità 4.1, può essere regolata anche da variazioni del pH
intracellulare e/o extracellulare. Generalmente l’abbassamento di pH (acidifi-
cazione) determina la riduzione dell’attività del canale.
I canali Kir associati a proteine G (KG) che contengono la subunità Kir3.2 o
Kir3.4 possono essere attivati dal sodio intracellulare (Nai+) (Ho e Murrell-
Lagnado 1999; Petit-jacques et al. 1999).
I canali Kir ATP - dipendenti (KATP), che sono costituiti da una subunità 6.x e
dal recettore ausiliario della sulfonilurea (SURx), sono inibiti dall’ATP intra-
cellulare che interagisce direttamente con le regioni citoplasmatiche della su-
bunità Kir 6.2 per regolare la funzione del poro del canale (Anticliff et al.
2005; Cukras et al. 2002; John et al. 2003; Li et al. 2000). Invece i nucleotidi
difosfato intracellulari (NDPs) attivano i KATP legandosi al recettore della sul-
fonilurea il quale a sua volta influenza la funzione del poro in modo tale da
aumentare la probabilità di apertura del canale (Babenko et al. 2000; Shyng et
al. 2000; Ueda et al. 1997).

19
Fosforilazione
L’attività dei canali Kir può essere modulata anche da proteine chinasi, in
particolare proteina chinasi A e C, mediante la fosforilazione delle loro subu-
nità. Alcuni esempi di questa regolazione sono: la fosforilazione della subuni-
tà Kir1.1 da parte della PKC che risulta nella soppressione dell’attività del
canale (Lin et al. 2002); la fosforilazione delle subunità 6.1 e SUR2B dei ca-
nali KATP nel muscolo liscio da parte della PKA , che determina invece un
aumento della loro attività (Quinn et al. 2004; Shi et al. 2007); la fosforila-
zione della subunità Kir1.1 da parte di una chinasi serica regolata da gluco-
corticoidi (SGK) che promuove la sua espressione sulla superficie cellulare
(Yoo et al. 2003).
Interazione proteina-proteina
Le interazioni proteina-proteina coinvolte nella regolazione della funzione
del poro dei canali Kir includono l’interazione tra le subunità βγ delle protei-
ne G (Gβγ) e la subunità Kir 3.x, l’associazione del recettore SUR con Kir 6.x
e il legame di proteine di ancoraggio della membrana a diverse subunità Kir. I
canali KG sono attivati dall’associazione diretta della subunità Kir 3.x e della
subunità Gβγ rilasciata da recettori accoppiati a proteine Gi/o (Chan et al.
1997; He et al. 1999). I canali KATP per essere funzionali necessitano
dell’associazione tra le subunità del recettore per la sulfonilurea e le subunità
6.x per formare strutture etero-ottameriche (Inagaki et al. 1996; Inagaki et al.
1995). I domini citoplasmatici di SURx interagiscono con Kir 6.x per modifi-
care l’attività del canale (Babenko e Bryan 2002; Reimann et al. 1999).
Inoltre, proteine di ancoraggio come PSD-95,SAP97 e Nexina27, giocano un
ruolo chiave nel determinare la localizzazione di alcuni canali Kir sulla super-
ficie cellulare (Alewine et al. 2007; Horio et al. 1997).

20
Fisiologia dei canali Kir4.x e Kir5.1
I Kir4.x e i Kir5.1 fanno parte del gruppo dei canali trasportatori del K+. Inizialmente
diversi gruppi di ricerca hanno identificato il canale Kir4.1 da librerie di cDNA del
cervello, assegnandogli diversi nomi quali BIR10 (Bond et al. 1994), KAB-2 (Takumi
et al. 1995), BIRK1 (Bredt 1995) e Kir1.2 (Shuck et al. 1997). Analisi immunoisto-
chimiche di ibridazione in situ hanno dimostrato che il canale Kir4.1 era espresso so-
prattutto nelle cellule gliali del cervello. Quindi, Kir4.1 è stato considerato il respon-
sabile della conduttanza al K+ di queste cellule che è responsabile del processo chia-
mato ―K+-Buffering‖, fondamentale nel controllo della funzione neuronale (Kuffler
e Nicholls 1996; Newman EA. 1984).
La sequenza aminoacidica del Kir4.1 ha rispettivamente il 53, 43 e 53% di identità
con la sequenza dei Kir1.1, Kir2.1 e Kir3.1. Invece, il Kir5.1 mostra rispettivamente
il 39, 50 e 40% di identità con gli stessi canali.
Quando il canale Kir4.1 è espresso da solo in cellule eterologhe, costituisce un tetra-
mero e suscita una corrente K+ (Pessia et al. 1996), al contrario di quanto accade in-
vece quando è il Kir5.1 ad essere espresso da solo. Infatti, le subunità Kir 5.1 sem-
brano non formare canali omomerici in cellule eterologhe, mentre formano canali e-
teromerici con le subunità Kir4.1 e Kir4.2 (Pessia et al., 1996; Tucker et al., 2000). I
canali eteromerici Kir4.1-5.1 esistono anche in forma tetramerica (Pessia et al. 1996)
e sono stati individuati a livello di vari tessuti, inclusi reni e cervello. Per quanto ri-
guarda i Kir5.1 omomerici, è stato osservato che questi sono espressi nel citoplasma
di cellule come i fibrociti della coclea dove però non sembrano formare canali fun-
zionali sulla superficie della membrana cellulare.
I Kir4.2 sono stati isolati per la prima volta nel rene umano (Shuck et al. 1997), ma
sono espressi anche nel fegato, nei fibrociti embrionali e nelle cellule endoteliali mi-
crovascolari. Mediante il sequenziamento della regione q22.2 del cromosoma 21, re-
sponsabile della sindrome di Down, è stato possibile identificare il gene della subuni-
tà Kir4.2 (Gosset et al. 1997). Il Kir4.2 mostra un’identità del 62% con il Kir4.1.
Un’importante differenza tra le due subunità sta nel fatto che il Kir4.1 possiede un
dominio legante ATP (Walker type A ATP-binding cassette) nell’estremità COOH
(Takumi et al. 1995), mentre il Kir4.2 ne è sprovvisto (Pearson et al. 1999).
Il Kir4.2 è in grado di formare un canale funzionale anche se è espresso da solo, ma
l’eteromero Kir4.2-5.1 presenta una differente conduttanza del singolo canale e una

21
maggiore espressione sulla superficie cellulare che determina un aumento della cor-
rente dell’intera cellula (Pearson et al. 1999; Pessia et al. 2001).
Struttura e funzione del poro
Kir4.1 e Kir5.1
I canali omomerici Kir4.1 e gli eteromerici Kir4.1-5.1 presentano differenze
nella conduttanza del singolo canale, probabilmente perché essi mostrano sta-
ti multipli di subconduttanza. La conduttanza degli omomeri Kir4.1 è com-
presa tra 20 e 40pS (Lourdel et al. 2002; Tanemoto et al. 2000; Yang et al.
2000), mentre quella degli eteromeri Kir4.1-5.1 è compresa tra 40 e 60pS. I
Kir4.1 sono classificati come ―rettificatori intermedi‖, mentre i Kir5.1 sono
dei rettificatori molto più forti. I residui a livello del sito D/N sono il glutam-
mato (Glu) nei Kir4.1 e l’asparagina (Asn) nei Kir5.1. Nelle registrazione di
patch-clamp è stato osservato che nei Kir4.1 anche in presenza di alte concen-
trazioni di spermina, rimane una conduttanza residua del 10-15%. Questa ap-
parente insensibilità potrebbe essere dovuta alla parziale permeazione della
spermina attraverso il poro del canale Kir4.1 (Kucheryavykh et al. 2007), in-
fatti la filantotossina, una poliammina che ha una coda voluminosa, blocca
completamente le correnti outward attraverso questo canale.
Per la formazione del canale eteromerico è necessaria la presenza del residuo
E177 nella regione prossimale dell’estremità COOH proprio al di sotto del
dominio TM2 (Konstas et al. 2003).
Una proprietà fondamentale dei canali eteromerici Kir4.1/Kir5.1 è la loro
sensibilità alle variazioni di pH intracellulare (pHi) negli intervalli fisiologi-
ci. Questa caratteristica li rende particolarmente importanti come regolatori
dei flussi di K+ attraverso le membrane in funzione delle concentrazioni in-
tracellulari di H+ (Yang e Jiang, 1999; Tsai et al., 1995; Tucker et al., 2000;
Pessia et al., 2001; Tanemoto et al., 2000). L’accoppiamento funzionale tra
variazioni di pH e flusso di K+ coordinata da questi canali potrebbe regolare
l’eccitabilità neuronale (Giebisch G. 2001). E’ stato osservato che i canali e-
teromerici Kir4.1/Kir5.1 sono molto più sensibili alle variazioni intracellulari
di pH rispetto ai canali omomerici Kir4.1. Infatti, quando i valori di pH si
mantengono fisiologicamente tra 6.5 e 8.0, l’attività del canale Kir4.1 è inibi-
ta dall’acidificazione con una pKa=6.1, mentre l’attività dei canali eteromerici

22
è drammaticamente soppressa anche da una lieve acidificazione intracellulare
e aumentata da un’alcalinizzazione con pKa di ≈ 7.4 (Pessia et al., 2001; Ta-
nemoto et al., 2000; Xu et al., 2001). Alla luce di queste osservazioni, è pos-
sibile affermare che la eteropolimerizzazione del Kir 4.1 con il Kir5.1, in
qualche modo, modula le proprietà intrinseche del sensore del pH del canale
(Pessia et al., 2001). Sono stati individuati diversi residui aminoacidici della
subunità Kir4.1 sia nei canali omomerici che negli eteromeri Kir4.1-5.1 che
sono responsabili della sensibilità alle variazione di pHi, tra questi ci sono il
residuo E158 nella regione TM2 (Xu et al. 2000), il K67 nell’estremità cito-
plasmatica NH2 (Yang et al. 2000) e l’H190 nella regione COOH-terminale
(Casamassima et al. 2003).
Fig.7 Sensibilità del Kir4.1-Kir5.1 alle variazioni citoplasmatiche di pH.
(In alto) Tracce rappresentative di registrazioni di ovociti di Xenopus Leavis microi-
niettati con mRNA di Kir4.1-Kir5.1, e Kir4.1 da solo. Le correnti son evocate da i-
perpolarizzazioni da un potenziale di mantenimento di -10 mV a -110mV per 4 s in
una soluzione di K+ 90mM. L’acidificazione citoplasmatica determina una riduzione
marcata della corrente nell’eterodimero Kir4.1/Kir5.1. Viceversa, l’effetto
sull’omodimero Kir4.1 è molto minore. (In basso) Curva dose risposta di inibizione
della corrente di Kir4.1-Kir5.1 e Kir4.1 a diversi valori di pHi.

23
Fig. 8 Registrazioni di patch-clamp di Kir4.1-Kir5.1 a diverse concentrazioni di pHi.
Nella figura sono mostrate registrazioni di patch-clamp in configurazione in-out da
ovociti di Xenopus Leavis microiniettati con mRNA di Kir4.1-Kir5.1. Sulla mem-
brana sono presenti diversi canali. Cambiando il pH della soluzione a contatto con la
porzione citoplasmatica del canale l’attività dello stesso viene modificata.
L’acidificazione riduce la probabilità di apertura del canale, mentre
l’alcalinizzazione determina un aumento della probabilità.
Una caratteristica che differenzia ulteriormente i canali eteromerici Kir4.1-
5.1 dai Kir4.1 omomerici è la sensibilità al Na+. Infatti, l’attività dei canali
Kir4.1-5.1 è aumentata dal Na+ (Rosenhouse-Dantsker et al. 2008) e questa
proprietà sembra dipendere dal residuo D205 localizzato a livello della regio-
ne citoplasmatica COOH-terminale. Questo residuo non è presente nella su-
bunità Kir4.1 e per questo motivo i canali omomerci Kir4.1 non sono sensibi-
li al Na+.

24
Localizzazione intracellulare
Kir4.x e Kir5.1
Localizzazione sulla membrana cellulare
Tessuto epiteliale
Quando il canale Kir4.1 è espresso nelle cellule epiteliali renali MDCK (Ma-
din Darby Canine Kidney), esso si trova a livello della membrana basolatera-
le (Tanemoto et al. 2005) dove interagisce con una proteina PDZ associata al-
la membrana, la guanilato kinasi (MAGI-1) (Tanemoto et al. 2008).
L’interazione avviene tra il quinto dominio PDZ della MAGI-1 e il motivo
legante il PDZ dell’estremità COOH del Kir4.1. La fosforilazione di un resi-
duo (S377) di questo motivo legante il PDZ, distrugge l’interazione e deter-
mina lo spostamento del Kir4.1 dalla membrana basolaterale a compartimenti
perinucleari nelle cellule MDCK (Tanemoto et al. 2008). Per quanto riguarda
invece la subunità Kir5.1, quando è espressa da sola nelle cellule MDCK, es-
sa rimane nei compartimenti intracellulari, mentre la coespressione con la su-
bunità Kir4.1 ne determina la localizzazione sulla membrana basolaterale
(Tanemoto et al. 2005). Ciò è stato dimostrato anche da analisi di immunoi-
stochimica e registrazioni patch-clamp che hanno messo in evidenza come i
Kir4.1 e Kir5.1 formino canali eteromerici sulla membrana basolaterale di al-
cune cellule epiteliali renali (Ito et al. 1996; Lourdel et al. 2002; Tanemoto
et al. 2005; Tucker et al. 2000).
A livello dello stomaco, invece, i canali omomerici Kir4.1 sono localizzati
sulla membrana apicale delle cellule parietali (Fujita et al. 2002; Kaufhold et
al. 2008). In particolare, specifiche analisi hanno dimostrato che tali canali
sono espressi sui microvilli della superficie della membrana apicale ma non
nel sistema tubulo-vescicolare (intracellulare). Il meccanismo che determina
questa specifica localizzazione dei Kir4.1 nelle cellule parietali rimane, però,
ancora sconosciuto.
Il pattern di distribuzione dei Kir4.1 e Kir5.1 nella coclea dell’orecchio inter-
no, che contiene una varietà di cellule epiteliali, è unico (Hibino e Kurachi
2006). Entrambe le subunità sono espresse nella parete laterale cocleare, ma
in diversi modi. I Kir4.1 sono localizzati nel tessuto epiteliale nella stria va-
scolare (Hibino et al. 1997); in particolare, tecniche di immunoistochimica ad
alta risoluzione hanno evidenziato l’espressione di questi canali nel polo api-

25
cale delle cellule intermediate (Ando e Takeuchi 1999; Hibino et al. 2004). I
Kir5.1, invece, sono espressi nei fibrociti del legamento spirale e sono loca-
lizzati soprattutto a livello intracellulare piuttosto che sulla superficie della
membrana (Hibino et al. 2004).
Cellule gliali
Nel sistema nervoso centrale (SNC) e nella retina, le subunità Kir4.1 e Kir5.1
sono prevalentemente espresse nelle cellule astrogliali, dove formano canali
omomerici Kir4.1 ed eteromerici Kir4.1-5.1. Entrambi i tipi di canale mostra-
no un pattern di distribuzione unico in diversi tipi di cellule. La subunità
Kir4.1 è maggiormente espresso negli astrociti del SNC, sia a livello cerebra-
le che del midollo spinale (Brady et al. 1996; Kaiser et al. 2006; Li et al.
2001; Neusch et al. 2006; Olsen et al. 2006; Poopalasundaram et al. 2000;
Takumi et al. 1995). Anche la subunità Kir5.1 è espressa negli astrociti del
cervello (Benfenati et al. 2006; Hibino et al. 2004; Lichter-Konecki et al.
2008), inoltre entrambe le subunità sono presenti nelle cellule di Muller della
retina(Ishii et al. 2003; Ishii et al. 1997; Nagelhus et al. 2004). Nella cortec-
cia cerebrale, a livello dei processi astrocitari che circondano le sinapsi (pro-
cessi perisinaptici), si evidenzia la presenza sia dei canali omomerici Kir4.1
che degli eteromerici Kir4.1-5.1, mentre nei processi perivascolari (―end fe-
et‖), sono espressi solo gli eteromeri. Nelle cellule di Muller della retina, so-
no presenti sia i canali omomerici Kir4.1, localizzati negli ―end feet‖, che i
canali etromerici Kir4.1-5.1 nei processi perisinaptici.
Il meccanismo responsabile dell’espressione di ogni canale a livello di diffe-
renti macrodomini di membrana non è stato ancora completamente chiarito.
Numerosi studi suggeriscono che il complesso proteico associato alla distro-
fina ha un ruolo importante nel determinare la localizzazione dei canali
Kir4.1 negli ―end feet‖. Infatti, topi mutanti che non possiedono distrofina
(topi knockout o ―topi mdx3CV‖ ), mostrano una ridotta espressione dei
Kir4.1 a livello degli ―end feet‖ delle cellule di Muller (Connors et al. 2002;
Dalloz et al. 2003), mentre non è influenzata l’espressione di questi canali
sulla membrana perisinaptica, suggerendo che la localizzazione dei canali
Kir4.1 in quest’area è determinata da un meccanismo diverso.
Le cellule satelliti sono un altro sottotipo di cellule gliali che esprimono i ca-
nali Kir4.1. Queste cellule circondano i corpi cellulari di vari neuroni con

26
multipli strati di guaina mielinica. In particolare, i Kir4.1 sono espressi sulla
guaina mielinica che circonda i gangli spirali cocleari, i gangli del trigemino e
i gangli cervicali superiori (Hibino et al. 1999; Vite t al. 2006; Vite t al.
2008; Zou et al. 2009).
Fig. 9 Localizzazione dei canali Kir nelle cellule astrogliali.(a sinistra) Distribuzione
dei canali Kir4.1 omomerici e dei Kir4.1-5.1 sulla membrana degli astrociti. A livello
dei processi perisinaptici sono espressi sia i Kir4.1 che i Kir4.1-5.1, mentre negli
―end feet‖ sono espressi esclusivamente gli eteromeri. (a destra) Cellule di Muller.
Queste cellule esprimono i canali eteromerici nei processi perisinaptici e i Kir4.1 a
livello dei processi perivascolari e degli ―end feet‖. Sia negli astrociti che nelle cellu-
le di Muller i canali Kir sono espressi in associazione all’acquaporina 4 (AQP4).
Localizzazione nei microdomini di membrana
I canali Kir4.1 sono concentrati in particolari microdomini della membrana delle cel-
lule astrogliali, i DRMs (Hibino et al. 2007). Analisi biochimiche e immunistochimi-
che hanno evidenziato che i Kir4.1 sono abbondanti in domini DRMs non-caveolari
del cervello, così come in astrociti messi in coltura e in cellule T HEK293 quando
espressi esogenamente. Nelle cellule T HEK293, la deplezione del colesterolo di
membrana mediante metil-β-ciclodestrina (MβCD) risultava nella perdita di associa-
zione tra il Kir4.1 e i DRMs e nella perdita di funzionalità del canale. Quindi, la lo-

27
calizzazione dei canali Kir4.1 nei domini DRMs non-caveolari sensibili alla MβCD
sembra essere vincolante per la funzionalità del canale. Anche i canali acquosi AQP4
si trovano a livello dei DRMs nelle cellule astrogliali, ma questi microdomini sono
insensibili alla MβCD e questa caratteristica ha suggerito che in queste regioni la
membrana fosse povera di colesterolo. Analisi immunoistochimiche hanno dimostra-
to che questi differenti DRMs si trovano in stretta vicinanza nella membrana degli
astrociti e proprio questa vicinanza potrebbe essere coinvolta nell’accoppiamento del
trasporto di K+ e acqua.
Funzioni fisiologiche
Kir4.x e Kir5.1
Reni e stomaco
Nei reni, l’epitelio del tubulo contorto distale (DCT) gioca un ruolo importante nel
riassorbimento del Na+
(Hebert et al. 2005). Questo trasporto è mediato da canali del
Na+ localizzati a livello della membrana apicale (Costanzo LS. 1984), la cui attività
si associa a quella delle pompe Na+-K
+-ATPasi presenti sulla membrana basolaterale.
I canali del K+ sulla membrana basolaterale sono fondamentali in questo processo di
riassorbimento del Na+ perché mantengono costante la concentrazione del K
+ sul lato
extracellulare delle Na+-K
+-ATPasi in modo da mantenerle attive. Questo meccani-
smo è chiamato ―K+ recycling‖. Infatti, bloccando i canali del K
+ della membrana
basolaterale con Ba2+
si ottiene l’inibizione del trasporto di Na+ stimolato dalla vaso-
pressina (Schafer e Troutman 1987).
Nella membrana basolaterale delle cellule del tubulo contorto distale di topo (Lour-
del et al. 2002) e coniglio (Taniguchi et al. 1989) sono stati individuati canali Kir con
conduttanza unitaria di 37-40pS. Nel modello murino, è stato osservato che i canali
sono inibiti da un abbassamento del pH intracellulare con una pKa di ≈7.6. E’ stato
dimostrato che questi canali sono eteromeri Kir4.1-5.1 e/o Kir4.2-5.1 (Lourdel et al.
2002; Tanemoto et al. 2000; Tucker et al. 2000), la cui sensibilità alle variazioni del
pHi suggerisce che essi fungono da sensori del pHi stesso e che sono coinvolti nel
trasporto di ioni pHi-dipendente nelle cellule renali.
Nel DCT, sia i Kir4.1 che i Kir4.2 possono interagire direttamente con un recettore
sensore del Ca2+
a livello della membrana basolaterale (Huang et al. 2007). Questa
interazione riduce significativamente la corrente che attraversa entrambe le subunità.

28
Sebbene il ruolo fisiologico di questa interazione sia ancora sconosciuto, si pensa che
possa essere coinvolta nel controllo dell’omeostasi salina a livello renale.
Recentemente è stato dimostrato che a livello della membrana basolaterale delle cel-
lule principali nel dotto collettore (CCD) sono presenti dei canali eteromerici Kir4.1-
5.1 (Lachheb et al. 2008) con una conduttanza unitaria di ≈40 pS che potrebbero ave-
re la stessa funzione dei canali Kir presenti nel DCT.
Nelle cellule parietali dello stomaco, i canali omomerici Kir4.1 sono localizzati sulla
membrana apicale insieme alle H+-K
+-ATPasi (Fujita et al. 2002; Kaufholf et al.
2004). Queste pompe secernono H+ all’esterno scambiandoli con il K
+ che entra co-
sì nella cellula. Questi canali Kir4.1, potrebbero essere coinvolti nel mantenimento
dell’attività delle pompe H+-K
+-ATPasi rifornendo di K
+ il compartimento extracel-
lulare, probabilmente in modo simile a quanto si verifica nelle cellule epiteliali renali
per il mantenimento dell’attività delle pompe Na+-K
+-ATPasi. Tale ipotesi è confer-
mata dall’evidenza che applicando Ba2+
alle cellule parietali, che induce il blocco
dei canali Kir4.1, si ottiene una ridotta secrezione protonica da parte di queste cellule
(Fujita et al. 2002).
Coclea
La coclea dell’orecchio interno contiene due tipi di fluido extracellulare: la perilinfa
e l’endolinfa. La composizione ionica della perilinfa è pressoché identica al comune
fluido extracellulare. L’endolinfa, invece, contiene K+ (≈150mM) e possiede un po-
tenziale altamente positivo di ≈+80mV (potenziale endococleare) (Hibino et al. 2006;
Von Bekesy G. 1951; Von Bekesy G. 1952) rispetto al sangue o alla perilinfa. Que-
sto ambiente eccezionale dal punto di vista del potenziale e della composizione ioni-
ca è essenziale per una corretta capacità uditiva.
Il potenziale endococleare si pensa sia mantenuto per mezzo del passaggio di K+ dal-
la perilinfa all’endolinfa attraverso la parete cocleare laterale. Questa è costituita da
due componenti, la stria vascolare e il legamento spirale. E’ stato osservato che
l’applicazione di Ba2+
a livello della stria vascolare sopprime il potenziale endoco-
cleare (Marcus et al. 1985). L’unico tipo di subunità Kir presente a livello della stria
vascolare è il Kir4.1 (Hibino et al. 1997) ed è probabile che questi canali, espressi
specificatamente sulla membrana apicale delle cellule intermediate della stria (Ando
e Takeuchi 1999), determinino il passaggio di un’ampia corrente di ioni K+ attraver-
so la membrana e che quindi siano responsabili di una importante frazione del poten-
ziale endococleare (Hibino e Kurachi 2006; Nin et al. 2008; Takeuchi et al. 2000).

29
Questa ipotesi è supportata dal fatto che topi knockout per il Kir4.1 sono sordi e mo-
strano un potenziale endococleare prossimo a 0 mV con una riduzione di ≈ 50% della
[K+] nell’endolinfa (Marcus et al. 2002).
Nel legamento spirale, i fibrociti che sono bagnati dalla perilinfa esprimono canali
omomerici Kir5.1 (Hibino et al. 2004). La maggior parte di questi canali sembra es-
sere localizzata nel compartimento intracellulare, ma dal momento che la perfusione
di Ba2+
attraverso la perilinfa aumenta leggermente il potenziale endococleare, è pos-
sibile che una piccola popolazione di canali Kir5.1 possa essere localizzata sulla
membrana cellulare e che possa regolare negativamente la circolazione di K+ (Hibi-
no et al. 2004; Marcus DC 1984).
Cellule gliali
Gli astrociti cerebrali e le cellule di Muller della retina proiettano i loro processi non
solo sulle sinapsi e sul soma dei neuroni, ma anche verso i vasi sanguigni, e l’umore
vitreo. Queste cellule astrogliali hanno diversi ruoli nel controllo della funzione si-
naptica. Una delle più importanti funzioni degli astrociti è il mantenimento
dell’ambiente ionico e osmotico nello spazio extracellulare. Queste cellule conduco-
no ampie correnti Kir che sono coinvolte nel trasporto del K+ dalle regioni con alta
[K+], dovuta all’eccitazione sinaptica, a quelle con bassa [K
+] (Kuffler e Nicholls
1966; Newman EA 1984). Dal momento che un’eccessiva [K+] provocherebbe uno
stato di depolarizzazione continua nei neuroni, alterando quindi la trasmissione dei
segnali nervosi (Newman EA 1984; Orkand et al. 1966), la rapida clearance del K+
da parte degli astrociti è essenziale per il corretto funzionamento delle sinapsi. Tale
trasporto polarizzato è conosciuto come ―spatial buffering of K+‖ nel cervello e ―K
+-
siphoning‖ nelle cellule di Muller della retina. Per espletare queste funzioni di ―K+-
buffering‖, le cellule astrogliali si servono dei canali Kir localizzati a livello di speci-
fici domini della membrana. I canali Kir4.1 sono abbondantemente espressi nelle cel-
lule astrogliali e sono i principali responsabili della loro conduttanza al K+ nel cervel-
lo, nel midollo spinale e nella retina (Higashi et al 2001; Ishii et al. 2003; Ishii et al.
1997; Kaiser et al. 2006; Li et al. 2001; Naghelus et al. 2004; Neusch et al. 2006;
Olsen et al. 2006; Poopalasundaram et al. 2000; Takumi et al. 1995). Infatti, la dele-
zione o la mutazione del gene Kir4.1 determina un forte aumento della resistenza agli
input, riduzione della conduttanza al K+ e/o depolarizzazione del potenziale di ripo-
so nelle cellule astrogliali (Djukic et al. 2007; Kofuji et al. 2000; Neusch et al.
2006; Olsen et al. 2006). Il fenotipo dei topi knockout per il gene Kir4.1 evidenzia

30
l’importanza di questo gene per la funzione di ―K+-buffering‖ delle cellule astroglia-
li. Con un elettroretinogramma (ERG) è possibile osservare che nella retina di topi
mutanti, la risposta lenta da stimolazione luminosa è assente (Kofuji et al. 2000). Dal
momento che questa risposta si pensa sia generata da una diminuzione, evocata dalla
luce, della [K+] nella porzione distale della retina, essa potrebbe essere attribuita al
flusso di K+ dalla regione distale a quella prossimale della retina in risposta
all’iperpolarizzazione dei fotorecettori nella quale giocano un ruolo i canali Kir4.1.
Anche l’ippocampo nei topi knockout per il gene Kir4.1 mostra un ridotto assorbi-
mento di K+ in risposta all’eccitazione neuronale (Djukic et al. 2007) e ciò suggeri-
sce che i Kir4.1 sono coinvolti nel ―K+-buffering‖ non solo nella retina ma anche nel
cervello. I canali omomerici Kir4.1 e quelli eteromerici Kir4.1-5.1 sono distribuiti in
maniera differente nelle cellule astrogliali di cervello e retina (Hibino et al. 2004; I-
shii et al. 2003). Negli astrociti corticali del cervello, sia i canali omomerici che quel-
li eteromerici sono localizzati nei processi perisinaptici, mentre solo gli eteromeri
sono espressi a livello degli ―end feet‖. Ciò suggerisce che, negli astrociti corticali, il
K+ è assorbito attraverso canali omomerici ed eteromerici ed espulso solo attraverso
canali eteromerici. Per quanto riguarda, invece, gli astrociti del talamo e
dell’ippocampo, che possiedono numerose sinpasi, essi esprimono principalmente i
canali omomerici Kir4.1. Le cellule di Muller della retina, esprimono gli eteromeri
Kir4.1-5.1 a livello dei processi perisinaptici e i Kir4.1 alle loro estremità in contatto
con l’umore vitreo e i piccoli vasi sanguigni. Quindi, in questa regione si verifica
l’espulsione del K+, mentre l’assorbimento avviene a livello dei processi perisinapti-
ci. Come descritto precedentemente, la principale differenza tra Kir4.1 omomerici e
Kir4.1-5.1 e la sensibilità al pHi.
Gli astrociti esprimono diversi sistemi di trasporto degli ioni, incluso il cotrasportato-
re Na+-HCO3
-. L’attivazione di questo trasportatore, determinando l’ingresso nella
cellula di uno ione Na+ e di due o tre ioni HCO3
- (Anderson e Swanson 2000; Blau-
stein et al. 2002), induce l’alcalinizzazione intracellulare e l’iperpolarizzazione di
membrana. L’eccesso di K+ extracellulare, dovuto all’attivazione sinaptica, accelera
l’attività di questo trasportatore tramite la depolarizzazione della membrana degli a-
strociti. La conseguente alcalinizzazione intracellulare potrebbe aumentare l’attività
dei canali eteromerici Kir4.1-5.1 facilitando, quindi, l’assorbimento di K+. Invece,
gli astrociti dell’ippocampo e del talamo, che esprimono principalmente i canali o-
momerici Kir4.1, possiedono solo una piccola quantità dei cotrasportatori Na+-

31
HCO3- (Schmitt et al. 2000). Il ruolo fisiologico dell’eterogenicità dei canali Kir e-
spressi nei processi perisinaptici degli astrociti cerebrali non è ancora conosciuto.
Nei processi perivascolari degli astrociti cerebrali e delle cellule di Muller della reti-
na, i canali Kir sono colocalizzati con i canali dell’acqua AQP4 (Hibino et al. 2004;
Ishii et al. 2003; Nagelhus et al. 1999; Nagelhus et al. 2004). Nei modelli murini, la
delezione della proteina AQP4, rallenta l’assorbimento del K+ da parte degli astrociti
(Padmawar et al. 2005). Nei topi konockout per l’α-Sintrofina, che mantengono i
canali Kir4.1 ma perdono i canali AQP4 nei processi perivascolari delle cellule a-
strogliali, la clearence dell’eccesso di K+, indotta dalla stimolazione neuronale, è ri-
tardata (Amiry-Moghaddam et al. 2003). Questa osservazione evidenzia l’esistenza
di un collegamento tra il trasporto di acqua e di K+.
I canali Kir4.1 posso anche essere funzionalmente accoppiati a trasportatori del glu-
tammato. Nelle cellule astrogliali i Kir4.1 sono espressi insieme ai trasportatori GLT-
1 e GLAST del glutammato (Djukic et al. 2007; Olsen et al. 2007). Infatti, la dele-
zione o la mutazione del gene Kir4.1 determina una riduzione dell’assorbimento del
glutammato negli astrociti (Djukic et al. 2007; Kucheryavykh et al. 2007). Quando è
funzionale, il trasporto di glutammato depolarizza la membrana. L’espressione di ca-
nali Kir4.1 funzionali iperpolarizza la membrana, e potrebbe quindi facilitare
l’influsso di glutammato nella cellula attraverso il trasportatore.
Nei primi stadi postnatali, il midollo spinale esprime i canali Kir4.1 a livello sia della
materia grigia che della materia bianca (Dibaj et al. 2007; Neusch et al. 2001). Dato
che i Kir4.1 non sono espressi nei neuroni, questa osservazione suggerisce che essi
sono localizzati negli oligodendrociti così come negli astrociti. Nel periodo postnata-
le i Kir4.1 vengono espressi solo negli astrociti (materia grigia) del midollo (Dibaj et
al. 2007; Kaiser et al. 2006).
L’espressione dei canali Kir4.1 potrebbe, quindi, essere necessaria per lo sviluppo
precoce degli oligodendrociti (Neusch et al. 2001). Un possibile ruolo dei Kir4.1 du-
rante lo sviluppo potrebbe essere quello di iperpolarizzare la membrana e determina-
re così l’ingresso di Ca2+
attraverso i canali permeabili a questo ione, come nel caso
dell’associazione con il Kir2.1 (Konig et al. 2004).
I Kir4.1 sono stati identificati negli oligodendrociti di animali adulti, nel nervo ottico
e nel cervello dove questi canali sono localizzati a livello dei corpi cellulari degli oli-
gondrociti e più raramente nei loro processi (guaine mieliniche) (Kalsi et al. 2004;
Poopalasundaram et al. 2000). Questo tipo di distribuzione implica che i Kir4.1 negli
oligondendrociti adulti non sono coinvolti nel ―K+-buffering‖.

32
I canali omomerici Kir4.1 espressi nelle guaina mielinica delle cellule satelliti, cir-
condando i neuroni gangliari. In questa regione ci si aspetta che i Kir4.1 si comporti-
no come nelle cellule astrogliali assorbendo l’eccesso di K+ extracellulare indotto
dall’eccitazione dei gangli (―K+-buffering‖) (Hibino et al. 1999). Grazie al silenzia-
mento del Kir4.1 mediante le tecnologia ―RNA-interference‖ (RNAi) è stato possi-
bile chiarire il ruolo fisiologico di questi canali nelle cellule satelliti del ganglio tri-
geminale di ratto (Vit et al. 2008). Infatti, la distruzione del Kir4.1 comporta la ridu-
zione della soglia nocicettiva e porta a comportamenti facciali sia spontanei che evo-
cati meccanicamente simili alle reazioni suscitate da uno stimolo doloroso. Un possi-
bile meccanismo è che l’attenuazione del ―K+-buffering‖ delle cellule satelliti au-
menti la concentrazione di K+
nello spazio extracellulare dei gangli, incrementando la
loro eccitabilità.
Farmacologia
I canali Kir che contengono le subunità Kir4.1 e Kir4.2 sono bloccati dagli ioni Ba2+
e Cs+ (Konstas et al. 2003; Takumi et al. 1995; Tanemoto et al. 2000). Oltre a questi
bloccanti non selettivi dei canali Kir, alcuni antidepressivi triciclici (TCAs) come
nortriptilina, amitriptilina, desiprismina e imipramine, bloccano i canali Kir4.1 (Su et
al. 2007). L’azione inibitoria della nortiptramina dipende dalla differenza di voltag-
gio rispetto all’EK ed ha un maggiore effetto sulle correnti outward. Anche inibitori
selettivi del riassorbimento della serotonina (SSRIs), come fluoxentina, sertralina e
fluvoxamina, bloccano i canali Kir4.1 in modo voltaggio-indipendente (Ohno et al.
2007). Questi farmaci hanno solo un debole effetto sulle correnti del K+ attraverso i
canali Kir2.1 e Kir1.1. Il fatto che i TCAs e dei SSRIs bloccano i canali Kir nelle cel-
lule astrogliali potrebbe contribuire all’azione terapeutica e/o agli effetti collaterali di
questi composti. Studi recenti hanno dimostrato che questi antidepressivi possono in-
teragire con i residui T128 e E158 che sono a contatto con la cavità centrale del poro
dei canali Kir4.1 (Furutani et al. 2009).
Canalopatie associate ai Kir4.1 e Kir5.1
Il fenotipo dei topi knockout per i Kir4.1 suggerisce che questi canali sono responsa-
bili di una varietà di condizioni patofisiologiche. Per prima cosa, i topi knockout per
i Kir4.1 mostrano una perdita della risposta lenta da stimolo luminoso nell’ ERG

33
(Kofuji et al. 2000). Inoltre, la distruzione dei canali Kir4.1 che sono abbondante-
mente espressi negli oligodendrociti durante il primo sviluppo postnatale (Kaiser et
al. 2006), induce un rilevante danneggiamento delle capacità motorie nei topi
(Neusch et al. 2001). Le basi cellulari di questo fenotipo sembrano essere: ipomieli-
nazione a livello del midollo spinale associata a una grave vacuolizzazione spongi-
forme, gonfiore e degenerazione assonale. Questi topi sono anche sordi a causa del-
la mancanza del potenziale endococleare e della perdita del K+ endolinfatico (Marcus
et al. 2002).
Recentemente è stato ipotizzato che l’anormalità del funzionamento dei canali Kir4.1
nel cervello potrebbe indurre l’epilessia. L’analisi delle membrane di cellule gliali ot-
tenute da pazienti affetti da forme intrattabili di epilessia talvolta mostrano una per-
dita quasi completa della conduttanza Kir (Bordey e Sontheimer 1998), riduzione
della rettificazione inward (Hinterkeuser et al. 2000) e della capacità di clearance del
K+ (Gabriel et al. 1998; Jauch et al. 2002). Studi precedenti, hanno dimostrato che il
gene Kir4.1 potrebbe essere coinvolto nelle crisi epilettiche. Analisi di Linkage han-
no permesso di identificare mutazioni nel gene dei canali Kir4.1 umani (R271C) che
potrebbe essere associata con le crisi epilettiche generalizzate (Buono et al. 2004).
Comunque, mutazioni equivalenti del gene Kir4.1, espresso da solo o con il Kir5.1,
suscitano una corrente K+ praticamente identica a quella generata dai canali wild-
type (Shang et al. 2005). Una recente analisi genetica ha permesso di identificare
numerose mutazioni del gene Kir4.1 in pazienti affetti da crisi epilettiche,sordità sen-
so neuronale, atassia, ritardo mentale e squilibrio elettrolitico (Bockenhauer et al.
2009; Scholl et al. 2009). Gli organi interessati da questo tipo di patologie sono gli
stessi in cui sono particolarmente espressi i canali Kir4.1. Grazie ad analisi elettrofi-
siologiche in vitro, si è osservato che alcune mutazioni riscontrate in queste patolo-
gie (R65P e G77R), determinano una rilevante riduzione della corrente attraverso i
Kir4.1 (52). In particolare la mutazioni R65P sembra coinvolgere anche il sito di le-
game del PI (4,5)P2 determinando quindi la perdita di funzionalità del canale (Scholl
et al. 2009).
Topi knockout che non possiedono canali Kir4.1 nelle cellule astrogliali sono affetti
da una severa forma di atassia e crisi indotte dallo stress (Djukic et al. 2007), inoltre
l’ablazione del gene Kir4.1 induce sordità nei topi (Marcus et al. 2002), supportando
l’ipotesi della relazione patofisiologia tra danneggiamento dei canali Kir4.1 ed epi-
lessia e disordini uditvi.

34
Recentemente alcuni esperimenti hanno dimostrato che i Kir4.1 sono potenzialmente
coinvolti nella protezione delle cellule atsrogliali da gravi danneggiamenti in caso di
lesioni cerebrali (Dibaj et al. 2007). L’esperimento consisteva nell’applicazione a li-
vello del midollo spinale di una soluzione ipotonica (al 30%), questa, nelle cellule a-
strogliali, determinava un rigonfiamento che però interessava esclusivamente i corpi
cellulari, mentre non aveva alcun effetto sugli ―end feet‖. Nei topi knockout per il
Kir4.1, invece, si osservava il rigonfiamento ipotonico anche degli ―end feet‖ astro-
citici. Il meccanismo mediante il quale i canali Kir4.1 prevengono il rigonfiamento
dei processi astrociti è però ancora sconosciuto.
In alcuni casi, lesioni del midollo spinale possono determinare condizioni di ipotoni-
cità che causano accumulo di fluido e quindi edema.
Inoltre, in seguito a eventi ischemici della retina, l’espressione dei canali Kir4.1 è
down regolata (Pannicke et al. 2005). La conseguente riduzione della funzione di
―K+-buffering‖ dei canali Kir, potrebbe essere coinvolta nella formazione
dell’edema.
Infine, il gene Kir4.2 è localizzato in stretta vicinanza al locus DCR1 (regione cro-
mosomica 1 della sindrome di Down). Ciò suggerisce un possibile collegamento tra
questa patologia e la disfunzione dei canali Kir4.2 , sebbene l’espressione della pro-
teina Kir4.2 nel cervello dei pazienti sani e di quelli affetti dalla sindrome di Down
sia praticamente la stessa (Ferrando-Miguel et al. 2004).

35
EPILESSIA
L'epilessia è causata dall'abnorme alterazione dell'attività elettrica di alcuni neuroni,
generalmente localizzati a livello della corteccia cerebrale (lo "strato più esterno"
dell'encefalo). Ciò comporta ad un'insieme di manifestazioni caratterizzate da brevi
episodi di perdita di conoscenza (assenze) e da alterazioni sensitive, psichiche o mo-
torie, più o meno accompagnate da spasmi o da contrazioni della muscolatura sche-
letrica di tipo convulsivo. Gli attacchi epilettici muscolari possono essere distinti in:
MIOCLONICI: spasmi di lieve entità;
TONICI: contrazioni più intense;
TONICI/CLONICI: violenti spasmi muscolari seguiti dal rilassamento della
stessa muscolatura.
L'alternanza di questi due stati è responsabile delle tipiche scosse muscolari ritmiche
('convulsioni') associate alla crisi epilettica.
Si definiscono foci epilettogeni i punti in cui originano gli attacchi epilettici; in tale
sede si concentra la popolazione neuronale con attività anomala. Questi foci possono
rimanere silenti per periodi prolungati dal momento che i neuroni sani che li circon-
dano tendono ad inibirne o neutralizzarne le scariche elettriche anomale. Quando
l'attività di questi neuroni viene sopraffatta e la cosiddetta "soglia di convulsività"
superata, insorgono i sintomi tipici della malattia. Da notare che tale soglia varia da
individuo ad individuo ed è particolarmente bassa negli epilettici.
In letteratura sono stati descritti oltre 150 tipi di epilessia, classificabili in parziali e
generalizzati:
EPILESSIE PARZIALI: il focus epilettogeno interessa soltanto un emisfero
cerebrale. Possono essere ulteriormente classificate in semplici o complesse.
Nel primo caso si caratterizzano per attacchi leggeri, che non si traducono
mai in perdite di conoscenza; al contrario, le epilessie complesse comportano
manifestazioni più severe, sempre accompagnate da perdita di conoscenza
(generalmente di breve durata - pochi secondi -) e da contrazioni muscolari
più intense.
EPILESSIE GENERALIZZATE: i neuroni che causano gli attacchi interes-
sano entrambi gli emisferi. Si accompagnano quasi sempre a perdita di cono-
scenza (assenza) associata a manifestazioni contrattili e spasmi di tipo mio-
clonico/tonico e tonico/clonico.

36
Si definisce stato epilettico il succedersi di manifestazioni epilettiche in modo fre-
quente e duraturo (vari episodi si possono notare anche nell'arco di alcune ore). In
questo caso ci troviamo di fronte ad una vera e propria emergenza medica che va
trattata il prima possibile, onde evitare la morte del soggetto per insufficienza respi-
ratoria.
Cause dell'epilessia
Le cause che portano un encefalo normale ad attivarsi in maniera parossistica fino a
provocare una crisi epilettica sono ancora in parte oscure: le ultime ricerche si stanno
rivolgendo verso il ruolo dei canali per gli elettroliti transmembrana voltaggio-
dipendenti. Vi sono evidenze che portano a considerare in difetti a carico dei carriers
per il sodio ed il calcio, dei punti di partenza interessanti per capire la fisiopatologia
del neurone epilettico. Ciò per due ragioni: la scoperta di mutazioni genetiche a cari-
co di canali voltaggio-dipendenti per questi elettroliti come substrato per numerose
sindromi epilettiche giovanili; l'evidenza dell'attività antiepilettica di numerose mo-
lecole che agiscono a questo livello molecolare.Dal punto di vista clinico, invece, si è
soliti suddividere le epilessie dal punto di vista eziologico in tre famiglie: genetiche,
sintomatiche e criptogenetiche. Le epilessie genetiche sono quelle nelle quali si è
trovato una specifica mutazione genetica: la più importante sarebbe quella per l'epi-
lessia mioclonica giovanile, il cui gene mutato mapperebbe nel cromosoma 6 e codi-
ficherebbe per una subunità per un canale del Sodio voltaggio-dipendente.
Le epilessie sintomatiche, sono quelle numericamente più diffuse. Costituiscono
quelle epilessie la cui causa è riscontrabile anatomicamente in una lesione parenchi-
male visibile alle neuroimmagini (principalmente la risonanza magnetica). Le cause
sono veramente tante di cui le principali sono costituite da:
lesioni pre-peri natali, che possono essere traumi da parto oppure complican-
ze come l'anossia perinatale; infezioni perinatali (specialmente da Cytomega-
lovirus - CMV), malformazioni (come la lissencefalia, o l'eterotopia);
malattie cerebrovascolari, che modificano l'architettura cellulare a livello del-
la lesione, con alterazioni anche a carico della rete dei neurotrasmettitori (ad
esempio per il glutammato). A volte, una crisi epilettica può essere indice
premonitore di sofferenza di una determinata regione cerebrale, ed essere un
"campanello d'allarme" per l'insorgenza futura di un accidente cerebrovasco-

37
lare; neoplasie, di cui spesso la crisi epilettica, più frequentemente di tipo
parziale, è il sintomo di esordio;
traumi cranici specialmente quelli aperti rispetto a quelli chiusi;
malattie infiammatorie come encefaliti, meningiti o infezione da virus HIV;
patologie degenerative, come il morbo di Alzheimer.
L'epilessie criptogenetiche sono epilessie che non hanno una causa organica visibile.
Sono un'evenienza statisticamente alta. Nella maggioranza dei casi l'epilessia è cau-
sata da una sofferenza organica del cervello, ad eccezione di una discreta percentuale
di casi idiopatici, di cui peraltro ancora si discute, che può guarire spontaneamente
durante l'età dello sviluppo (quando l'epilessia è di tipo semplice).
Patofisiologia dell’epilessia
L'attività sincrona parossistica di estesi aggregati neuronali (‛aggregati neuronali e-
pilettici'; Ajmone-Marsan, 1961) è espressione tipica della presenza di attività epilet-
tica nel cervello. Ne consegue che le anormalità bioelettriche registrabili con la elet-
troencefalografia rappresentano sintomi importanti dell'epilessia. Negli ultimi cin-
quant'anni sono stati compiuti ripetuti tentativi per ricondurre l'epilessia a un disordi-
ne metabolico primario e per dare una definizione dei caratteri specifici di tale di-
sturbo. Un’ influenza metabolica o biochimica capace di modificare la predisposizio-
ne epilettica è rappresentata dal livello di polarizzazione della membrana del neurone
o, in altri termini, dalla differenza di potenziale tra l'interno e l'esterno della cellula.
Il livello di polarizzazione della membrana neuronale è mantenuto dal trasporto atti-
vo di ioni, specialmente di quelli sodio e potassio. Le mutazioni in geni che codifi-
cano per le subunità proteiche dei canali ionici voltaggio-dipendenti e ligando-
dipendenti sono stati associati con le forme di epilessia generalizzata e sindromi se-
questro infantile. Infatti un meccanismo ipotizzato per alcune forme di epilessia ere-
ditaria sono le mutazioni dei geni che codificano per le proteine del canale del sodio;
questi canali del sodio difettosi rimangono aperti per troppo tempo rendendo il neu-
rone iper-eccitabile. Il glutammato, un neurotrasmettitore eccitatorio, può quindi es-
sere rilasciato da questi neuroni in grande quantità che, legandosi con i neuroni vicini
glumtamanergici attiva un eccessivo rilascio di Ca + + in queste cellule. Tale ecces-
sivo rilascio del calcio può essere neurotossico alla cellula colpita. Un altro possibile
meccanismo coinvolge la ‛pompa del sodio'. La principale sorgente dell'energia ne-
cessaria per assicurare il gradiente di membrana è rappresentata dal processo di ossi-

38
dazione del glucosio. Una depolarizzazione della membrana può essere provocata da
un'insufficienza relativa della pompa del sodio con conseguente accumulo di ioni so-
dio nell'interno del neurone. La membrana può inoltre essere depolarizzata, ripolariz-
zata ed iperpolarizzata dall'azione delle sinapsi eccitatorie e inibitorie. I segnali si-
naptici sono trasmessi da mediatori chimici; tra essi l'aceticolina è impiegata in pre-
valenza dalle sinapsi eccitatorie e l'acido gamma-ammino-butirrico (GABA) è, con
tutta probabilità, il mediatore delle sinapsi inibitorie. Queste inducono una iperpola-
rizzazione della membrana, mentre quelle eccitatorie la depolarizzano. Gli agenti
biochimici epilettogeni possono interferire su ognuno di questi meccanismi fonda-
mentali della polarizzazione della membrana neuronale. Ciò spiega perché, ad esem-
pio, la soglia epilettica può essere abbassata da differenti condizioni patologiche che
diminuiscono l'apporto al tessuto nervoso di ossigeno o di glucosio, sostanze indi-
spensabili per il metabolismo generatore dell'energia necessaria al trasporto attivo di
ioni. In campo clinico si descrivono vari casi nei quali si verificano crisi epilettiche
in conseguenza di insufficienza o arresto di circolazione ematica cerebrale, di ipossia
cerebrale (eccessiva altitudine) o di ipoglicemia (terapia con insulina, adenomi del
pancreas,ecc). La catena metabolica preposta alla sintesi del GABA sembra avere
una speciale importanza nella biochimica dell'epilessia. Tale sostanza, oltre a svolge-
re funzioni di mediatore chimico ad effetto inibitorio, concorre al metabolismo ossi-
dativo del glucosio, garantendo una alternativa al ciclo ossidativo di Krebs o rappre-
sentandone un processo collaterale, e pertanto concorre alla produzione neuronale di
energia. La piridossina (vitamina B6) entra nel ciclo metabolico del GABA. La grave
ipovitaminosi B6 da carenza dietetica, o la cattiva utilizzazione metabolica di tale so-
stanza, sono talora causa di epilessia. Fattori metabolici, farmacologici o tossici pos-
sono inoltre interferire con la sintesi, il deposito, l'attivazione e l'inattivazione dei
mediatori chimici delle sinapsi favorendo di conseguenza i processi epilettici. Per e-
sempio il diisopropilfluorofosfato (DFP) e il tetraetilpirofosfato (TEPP) bloccano l'i-
nattivazione provocando crisi epilettiche. Altri fattori metabolici possono direttamen-
te influenzare il trasporto di ioni attraverso la membrana neuronale. Ciò può realiz-
zarsi ad esempio a seguito di disordini del ricambio idrosalino che s'accompagnano
ad alterazioni ormoniche e metaboliche. In particolare tutte le condizioni cliniche che
inducono una ritenzione di acqua e di NaCl nell'organismo possono favorire o indur-
re attacchi convulsivi. Recentemente sono state offerte prove convincenti sul difetto-
so funzionamento della pompa del sodio nei neuroni epilettici e sulla capacità dei
farmaci anticonvulsivi (specialmente dintoina e suoi derivati) nel migliorare tale fun-

39
zione ( Woodbury, 1969). Ulteriori delucidazioni dei meccanismi biochimici coin-
volti nella genesi dell'epilessia sicuramente contribuiranno a far progredire la terapia
farmacologica di questa malattia.

40
AUTISMO
I disturbi dello spettro autistico (DSA) sono caratterizzati da diversi gradi di com-
promissione dell'interazione sociale, della comunicazione verbale e non verbale, e da
caratteristici schemi di interessi e comportamenti. I DSA sono diffusi in tutto il mon-
do. In Italia, non ci sono tassi di prevalenza chiari, ma le stime sono di circa 60 casi
per 10.000 bambini come in molti paesi occidentali, con una prevalenza nei maschi
che sembra essere 3-4 volte superiore a quello delle femmine (American Psychiatric
Association, 2000). In molti bambini, l’inizio di autismo è graduale; comunque circa
il 30% ha un inizio ―regressivo‖. Il 50% dei bambini con autismo sono definiti men-
talmente ritardati dal test del QI. Circa il 25% dei bambini ai quali è diagnosticato
l’autismo all’età di 2-3 anni riesce a raggiungere i vari gradi di comunicazione e a
mescolarsi con la popolazione generale intorno ai 6-7 anni. Il rimante 75% continua
a chiedere supporto parentale, scolastico e sociale. Attualmente i DSA sono conside-
rati disturbi eterogenei rispetto alla eziologia, biologia e fenotipo. Si ritiene infatti
che l'autismo sia una condizione "multifattoriale", data cioè dal coinvolgimento
combinato di diversi fattori genetici, e dalla loro possibile interazione con altri fattori
di rischio non genetici, non ancora conosciuti. Un certo numero di condizioni prena-
tali o postnatali, anomalie cromosomiche (in particolare, delezioni e duplicazioni del
cromosoma 16p e 15q) (Shen Yet al., 2010), e malattie monogeniche (ad esempio, la
Sindrome dell’X Fragile e la Sclerosi tuberosa) sono state associate a DSA. Tuttavia,
con l'eccezione della sindrome di Rett (RS), che è principalmente dovuta a mutazioni
del gene che codifica per la proteina dimetil-CpG-binding 2 (MECP2), l'eziologia dei
DSA rimane in gran parte sconosciuta. Circa il 90% dei casi sono considerati idiopa-
tici e, nonostante la mancanza di ereditarietà mendeliana, si ritiene possano essere
trasmessi geneticamente. Tuttavia, non sono stati identificati i geni associati con l'au-
tismo idiopatico, a conferma della difficoltà di trovare comuni meccanismi genetici e
molecolari che possono spiegare questa patologia eterogenea. Parecchi esperimenti
suggeriscono che la predisposizione a DSA sia dovuta a variabili genetiche. Si consi-
dera un’ elevata concordanza di DSA in gemelli monozigoti (90%) che gemelli dizi-
goti (10%)(Bailey A et al., 1995). Inoltre i genitori e i fratelli dei bambini colpiti mo-
strano spesso la manifestazione di DSA più delicata e subclinica, definita ― vasto fe-
notipo di autismo‖ (Piven J et al., 1997), suggerendo che DSA sia legato a fattori
comuni di suscettibilità genetica. Tuttavia, sebbene vari studi genetici sono stati ese-
guiti, nessun gene è stato associato al fenotipo di autismo idiopatico, confermando la
sua complessità.

41
Cause dell’autismo
La prima ipotesi sviluppata sulle cause dell'autismo, ormai sostanzialmente abbando-
nata nella ricerca scientifica ma ancora frequentemente citata, è quella di Leo Kan-
ner, che per primo, nel 1943, pubblicò uno studio abbastanza esaustivo sulla sindro-
me. Nonostante avesse concluso trattarsi di un disturbo innato, Kanner che aveva in-
dividuato nelle famiglie con figli autistici molti genitori, nonni, e parenti di alto livel-
lo culturale, ipotizzò che l'ossessività fosse una sorta di caratteristica fondamentale di
queste famiglie.
Eisenberg, suo stretto collaboratore, più tardi sottolineò quanto fosse difficile non
considerare la configurazione affettiva del nucleo famigliare, ipotizzando che il com-
portamento dei genitori non aiutasse né stimolasse il bambino a uscire dal suo guscio
di "refrigerazione affettiva".
I primi studi sull'autismo successivi a quello di Kanner si sono poi concentrati preva-
lentemente sul ruolo dei genitori. Molti e diversi sono però i fattori osservati che pos-
sono contribuire allo sviluppo della sindrome, e comprendono sia fattori ereditari che
non ereditari ( Charles JM et al., 2008). Poiché nel 60% dei casi due gemelli omozi-
goti risultano entrambi affetti, con tutta probabilità una componente genetica esiste
(Muhle R et al., 2004) anche se non è il solo fattore scatenante (altrimenti il 100%
dei monozigoti svilupperebbe la sindrome); si ipotizza quindi una causa di tipo mul-
tifattoriale, con elementi genetici e ambientali. L'autismo può inoltre presentarsi in
comorbilità ad altre sindromi già note: sindrome dell'X-fragile (Zingerevich C et al.,
2008), sclerosi tuberosa, fenilchetonuria (non trattata) e rosolia congenita.
È invece ormai completamente discreditata la vecchia ipotesi della causa vaccinale,
che era stata erroneamente e frodatoriamente avanzata, in uno studio poi ritrattato e
basato sulla manipolazione di dati sperimentali, da Andrew Wakefield; il quale, se-
condo il British Medical Journal (Brian Deer, 2011), asserisce scorrettamente di aver
trovato un'ipotetica correlazione fra il disturbo e l'assunzione del vaccino trivalente
(contro morbillo, parotite e rosolia) (Wakefield AJ et al, 1998); questo studio, poi ri-
velatosi basato su dati falsi e completamente manipolati, spinse ad avviare una serie
di altri studi su una più ampia popolazione, per comprendere se realmente esistesse
una correlazione o meno. Nessuno studio confermò i dati, che erano del tutto errati,
di Wakefield.
Ad esempio, un noto studio condotto su tutti i bambini nati
in Danimarca dal 1991 al 1998, ai quali venne iniettato il vaccino (a quasi mezzo mi-
lione di bambini), non trovò alcuna differenza di incidenza dell'autismo con i bambi-

42
ni non vaccinati (Madsen KM et al., 2002). Quella non fu l'unica smentita di tale af-
fermazione: nel corso del tempo si sono avuti molti studi similari. Inoltre è stato an-
che escluso il ruolo negativo del thimerosal (Immunization Safety Review Commit-
tee, 2004) .Infine, l'ipotesi ha subìto un'ulteriore forte smentita a opera d'uno studio
giapponese (Honda et al., 2005), nel quale si è evidenziato che, nonostante la sospen-
sione completa di tale vaccinazione nel 1993, l'incidenza della patologia è continuata
ad aumentare.
Manifestazioni cliniche dell’autismo
Normalmente i sintomi si manifestano come un ritiro autistico (nel senso di compor-
tamenti notevolmente anomali e non sempre comprensibili, a causa dei quali la per-
sona si trova esposta a un alto rischio di isolamento sociale), dovuto a gravi altera-
zioni nelle aree funzionali descritte qui di seguito:
Comunicazione verbale e non verbale
Il 50% dei soggetti autistici non è in grado di comunicare verbalmente. I soggetti che
sono in grado di utilizzare il linguaggio si esprimono in molte occasioni in modo biz-
zarro; spesso ripetono parole, suoni o frasi sentite pronunciare (ecolalia). L'ecolalia
può essere immediata(ripetizione di parole o frasi subito dopo l'ascolto), oppure eco-
lalia differita (ripetizione a distanza di tempo di frasi o parole sentite in precedenza).
Anche se le capacità imitative sono integre, queste persone spesso hanno notevoli
difficoltà a impiegare i nuovi apprendimenti in modo costruttivo a situazioni diverse
da quelle che li hanno generati in prima istanza.
Interazione sociale
Gli autistici mostrano un'apparente carenza di interesse e di reciprocità relazionale
con gli altri; tendenza all'isolamento e alla chiusura sociale; apparente indifferenza
emotiva agli stimoli o, al contrario, ipereccitabilità agli stessi; difficoltà a instaurare
un contatto visivo diretto: il bambino che intorno ai due anni di età continua a evitare
lo sguardo degli altri mostra, secondo diversi studi, una maggiore possibilità di svi-
luppare l'autismo. Gli autistici hanno difficoltà nell'iniziare una conversazione o a ri-
spettarne i "turni", oltre a difficoltà a rispondere alle domande e a partecipare alla vi-
ta o ai giochi di gruppo. Non è infrequente che bambini affetti da autismo vengano
inizialmente sottoposti a controlli per verificare una sospetta sordità, dal momento
che non mostrano apparenti reazioni (proprio come se avessero problemi uditivi)
quando vengono chiamati per nome.

43
Immaginazione o repertorio di interessi
Di solito un limitato repertorio di comportamenti viene ripetuto in modo ossessivo; si
possono osservare posture e sequenze di movimenti stereotipati (per es. torcersi o
mordersi le mani, sventolarle in aria, dondolarsi, compiere complessi movimenti del
capo, ecc.) detti appunto stereotipie. Queste persone possono manifestare eccessivo
interesse per oggetti o parti di essi, in particolare se hanno forme tondeggianti o pos-
sono ruotare (palle ovali, biglie, trottole, eliche, ecc.). Talvolta la persona affetta da
autismo tende ad astrarsi dalla realtà per isolarsi in una sorta di "mondo virtuale", in
cui si sente di vivere a tutti gli effetti (dialogando talora con personaggi inventati).
Pur mantenendo in molti casi la consapevolezza del proprio fantasticare, è con fatica
e solo con delle sollecitazioni esterne (suoni improvvisi, richiami di altre persone)
che riesce a essere in varia misura partecipe nella vita di gruppo.
Importanza dell'ordine
Si riscontra una marcata resistenza al cambiamento, che per alcuni può assumere le
caratteristiche di un vero e proprio terrore fobico. Questo può accadere se viene al-
lontanato dal proprio ambiente (camera, studio, giardino, ecc.), o se nell'ambiente in
cui vive si cambia inavvertitamente la collocazione di oggetti, del mobilio o comun-
que l'aspetto della stanza.
Lo stesso può verificarsi se si lasciano in disordine oggetti (sedie spostate, finestre
aperte, giornali in disordine): la reazione spontanea della persona autistica sarà quella
di riportare immediatamente le cose al loro ordine o, se impossibilitato a farlo, mani-
festare comunque inquietudine. La persona può allora esplodere in crisi di pianto o di
riso, o anche diventare autolesionista e aggressiva verso gli altri o verso gli oggetti.
Altri soggetti, al contrario, mostrano un'eccessiva passività, aprassia motoria
e ipotonia, che sembra renderli impermeabili a qualsiasi stimolo

44
Bambino affetto da autismo che si manifesta nel suo intento continuo di dare un dato ordine
alle cose.
Il fenotipo “autismo-epilessia”
In questo progetto abbiamo deciso di investigare il meccanismo molecolare di un
particolare sottogruppo all'interno dello spettro autistico, che è stato denominato co-
me "fenotipo autismo-epilessia". La forte correlazione tra autismo e epilessia ha
permesso di identificare il fenotipo ―autismo-epilessia‖( Tuchman R et al.; 2009). Il
rischio di attacchi epilettici nell’autismo è fra il 5% (Bryson SE et al.; 1988) e il 46%
(Hughes JR et al.; 2005), superando di gran lunga la percentuale che si associa alla
popolazione generale (0.5-1%). Infatti in letteratura la prevalenza di autismo nella
popolazione epilettica è riportata per essere circa 50 volte superiore rispetto la popo-
lazione generale (Clarke DF et al.; 2005). Sebbene il significato patofisiologico di
questa relazione non è stato ancora del tutto chiarito, esso offre le tracce adeguate per
individuare i possibili meccanismi molecolari e genetici che accomunano gli attacchi
epilettici e le disfunzioni socio-comunicative che caratterizzano l’autismo.
Un locus di suscettibilità per l'autismo è stato mappato in prossimità di un cluster di
geni che codificano per canali del sodio voltaggio-dipendenti (SCN1A e SCN2A),
sul cromosoma 2, che sono anche geni di suscettibilità per l'epilessia (Bryson SE et
al., 1988). Inoltre, uno squilibrio fra i sistemi eccitatori ed inibitori GABAergici e
glutamatergici, e varianti in geni che codificano per le subunità del recettore GABA,
sono stati associati alla suscettibilità per autismo e convulsioni, suggerendo che la
classificazione dei geni per l'epilessia può essere d’aiuto anche nell’identificare geni
candidati per l'autismo (Hughes JR et al., 2005; Clarke DF et al., 2005; Weiss LA et
al., 2003).

45
RAZIONALE DELLO STUDIO
Attualmente suscita enorme interesse come possibile responsabile del fenotipo ―auti-
smo-epilessia‖ il canale del potassio inwardly-rectfying Kir 4.1 localizzato nel cer-
vello.
I canali Kir4.1 e Kir5.1 sono stati clonati inizialmente da cervello (Powell EM et al.,
2003) , in cui sono espressi principalmente nelle cellule gliali (Delahanty RJ et al.,
2011), inclusi sia oligodendrociti che astrociti (Collins AL et al., 2006). In particola-
re i canali Kir4.1, sono convolti nel mantenimento dell’ambiente osmotico e ionico
dello spazio extracellulare promuovendo il trasporto di K+
da regioni ad alta [K+]o,
che risulta dall’eccitazione sinaptica, a regioni con bassa [K+]o. Questo trasporto po-
larizzato di K+
negli astrociti, svolge un ruolo importante nel "buffering spaziale"
della concentrazione extracellulare K+ ([K+]o) che è essenziale per il mantenimento
dell'attività neuronale.
In studi passati è stato dimostrato che la subunità Kir5.1 è in grado di co-assemblare
selettivamente con la subunità Kir4.1, conferendo al canale eteromerico una condut-
tanza di singolo canale molto più grande, una più forte rettificazione, una ―relaxa-
tion‖ tempo-dipendente ed una maggiore sensibilità ai cambiamenti del pHi. Il ruolo
fisiologico dei canali Kir4.1/Kir5.1 negli astrociti è ancora sconosciuto, tuttavia que-
ste differenze funzionali fanno ipotizzare che entrambi i tipi di canali abbiano una
specifica funzione nel buffering di K+ nella glia. I canali eteromerici Kir4.1/Kir5.1,
in particolare, sono abbondantemente espressi nel tronco encefalico, soprattutto nel
locus coeruleus (Fakler B et al., 1996), dove la loro elevata sensibilità ai cambiamen-
ti in pHi contribuisce alla chemiosensibilità di questa regione del cervello. Nella neo-
corteccia di topo, i Kir4.1 ed i Kir5.1 sono espressi esclusivamente negli astrociti, e
non nei neuroni. Sia canali eteromerici Kir4.1/5.1 che canali omomerici Kir4.1 sono
stati identificati in prolungamenti astrocitari che circondano le sinapsi (processi peri-
sinaptici). Al contrario, solo il canale eteromerico è presente in processi astrocitari
connessi alla pia e ai vasi sanguigni (terminazioni periferiche) e potrebbe essere re-
sponsabile dell'estrusione di un eccesso di K+ intracellulare.
Varie evidenze sperimentali suggeriscono che il canale del potassio inward-rectifying
del cervello Kir4.1 (KCNJ10) sia tra i possibili meccanismi fisiopatologici che giusti-
ficano la copresenza di epilessia e di disturbo dello spettro autistico. Inoltre, una serie
di dati in letteratura indicano che la disfunzione delle correnti Kir negli astrociti pro-
voca malattie invalidanti, anche se, il ruolo fisiologico e patologico dei canali Kir in
queste cellule rimane in gran parte sconosciuto. Registrazioni da biopsie di pazienti

46
affetti da epilessie intrattabili hanno mostrato una riduzione significativa della con-
duttanza dei Kir negli astrociti e una ridotta capacità di rimozione del potassio (Wu J
et al., 2004; Bordey A et al, 1998 ). I topi knockout per il Kir4.1 mostrano mortalità
precoce e convulsioni scatenate da stress. Mutazioni nel gene KCNJ10 sono state ri-
scontrate in pazienti affetti da convulsioni, atassia, sordità neurosensoriale, tubulopa-
tia (sindrome EST (Jauch R et al., 2002), sindrome di SESAME (Djukic B et al.,
2007)). In generale, sembra che sia la riduzione o l'assenza di Kir4.1 causi epilessia.
Il rapporto tra i canali Kir e disturbi dello spettro autistico è meno semplice. Tuttavia,
una maggiore espressione di Kir4.1 è stata riscontrata nel locus coeruleus (LC) di to-
pi knock-out per la proteina MECP2, un modello animale di RS (Ferraro TN et al.,
2004). Questo risultato sembra essere alla base dei comportamenti autistici osservati
nei pazienti con sindrome di Rett, dovuto ad una alterata neuromodulazione dei neu-
roni LC. Infine, uno studio recente su un pedigree finlandese ha proposto di estende-
re il gene KCNJ10 come candidato per i DSA (Shang L et al.,2005).
Pertanto cambiamenti nel funzionamento dei canali Kir astrocitari (Kir4.1, Kir5.1)
dovuti a mutazioni nel KCNJ10, potrebbero causare un’alterazione nella regolazione
dell'omeostasi del K+ nel cervello e contribuire alla comparsa di autismo con suscet-
tibilità a crisi epilettiche.

47
MATERIALI E METODI
PAZIENTI
Reclutamento dei pazienti e indagini cliniche
Poiché i disturbi dello spettro autistico sono disturbi eterogenei dal punto di vista cli-
nico è opportuno selezionare accuratamente i pazienti da includere nello studio. In
questo progetto si è deciso di investigare il meccanismo molecolare di un particolare
sottogruppo all'interno dello spettro autistico, che è stato denominato come "fenotipo
autismo-epilessia". E’stato reclutato un totale di almeno 150 pazienti divisi in tre
gruppi: I) DSA idiopatico, con storia di crisi epilettiche; II) DSA idiopatico senza
storia di crisi epilettiche, ma con elettroencefalografia (EEG) parossistico; III) DSA
idiopatico, con nessuna storia di crisi epilettiche, né anomalie EEG. La caratterizza-
zione dei fenotipi è stata effettuata attraverso colloqui clinici, revisione di precedenti
dati clinici, esame clinico generale, dismorfismi, valutazione neurologica, psichiatri-
ca e neuropsicologica, analisi di laboratorio (compresi i livelli di elettroliti nel pla-
sma e nelle urine), esame neurofisiologico e, ove mancante, risonanza magnetica
(MRI) del cervello. I criteri di esclusione erano la presenza di DSA e\o epilessia se-
condari a squilibri cromosomici, mutazioni di geni noti o secondari ad altre patolo-
gie. La diagnosi di DSA è stata effettuata secondo i criteri diagnostici del DSM-IV-
TR e ulteriormente confermata con l'uso di strumenti come ADOS-G e ADI-R. La
valutazione clinica e neuropsicologica comprendeva anche le scale Wechsler o Leiter
International Performance Scale-R e la scala Vineland del comportamento adattivo.
Registrazioni video-EEG di superficie sono effettuate attraverso un sistema di moni-
toraggio video-EEG. Al fine di caratterizzare il fenotipo epilettico, sono stati effet-
tuati lunghi monitoraggi con video-EEG durante il sonno e la veglia, per registrare
episodi critici. Gli elettrodi sono posizionati sul cuoio capelluto secondo il sistema
10-20. I segnali sono acquisiti e analizzati off-line. Il fenotipo epilettico viene classi-
ficato in base ai criteri ILAE (Commission of Classification and Terminology-
International League Against Epilepsy, 1981;1989).
Le analisi di mutazioni nel gene KCNJ10 sono state eseguite su cinquantadue pazien-
ti (trentaquattro ragazzi e diciotto ragazze, età media 10.0±4.9 anni, range 2.3-2.4
anni) con epilessia a ―causa sconosciuta‖ associata a compromissione di una delle a-
bilità comunicative o cognitive o entrambe. Il termine epilessia a ―causa sconosciuta‖
indica che gli attacchi non sono il risultato di un danno cerebrale noto, anomalie
strutturali o altri disordini neurologici; inoltre i risultati clinici e quelli ottenuti
dall’elettroencefalogramma non sono caratteristici delle sindromi epilettiche idiopa-

48
tiche. In questo studio la risonanza magnetica del cervello risultava normale in tren-
tanove pazienti, considerando che tredici soggetti avevano delle anomalie medie
(quattro presentavano demielinizzazione, due manifestavano ipoplasia cerebellare e
in altri due soggetti si era riscontrato la manifestazione di entrambi; in due microce-
falia, ipoplasia del corpo calloso in altri due soggetti, ipoplasia del tronco cerebrale
in un paziente). Tuttavia i risultati ottenuti dalla risonanza magnetica erano insuffi-
cienti per definire l’eziologia specifica dell’epilessia. Al comparire dell’epilessia,
ventitre bambini presentavano spasmi infantili, venti avevano attacchi focali e nove
pazienti manifestavano attacchi generalizzati (tonico, tonico-clonico, mioclonico, as-
senze atipiche) in aggiunta alle crisi tutti i pazienti avevano segni neurologici gravi,
come ad esempio disabilità o handicap mentale di comunicabilità o difficoltà cogni-
tive o entrambe. Si è valutato che la disabilità mentale era presente in quarantacinque
pazienti (lieve-moderata in ventidue, grave-profonda in ventitre), considerando che
tre soggetti avevano un livello cognitivo normale. Questi ultimi casi avevano gravi
problemi comunicativi e relazionali, non giustificati soddisfacentemente dal processo
epilettico. Vari gradi di compromissione delle funzioni motorie sono state osservate
in trentuno pazienti (ritardo motore/goffagine in dicianove, atassia in due, disordine
motorio in un caso, tetraparesi spastica in nove pazienti).
ANALISI DI MUTAZIONI
Per verificare se i canali Kir fossero responsabili del fenotipo oggetto di studio, il ge-
ne KCNJ10 è stato analizzato alla ricerca di mutazioni. Dopo aver purificato il DNA
genomico da leucociti del sangue periferico, la ricerca di mutazioni dei geni KCNJ10
(Kir4.1), KCNJ16 (Kir5.1) è stata realizzata utilizzando tecniche standard di genetica
molecolare, compresa l'analisi dHPLC seguita dal sequenziamento diretto degli esoni
codificanti e delle sequenze a margine utilizzando il sistema Big Dye 3,1. La PCR
quantitativa in tempo reale (q-PCR) o un test MLPA (Multiplex ligation-dependent
Probe Amplification) sono utilizzati per l'individuazione di delezioni o duplicazioni
di esoni multipli nel gene. Linee cellulari (fibroblasti della pelle o linfoblasti) sono
analizzate tramite q-PCR per valutare i livelli di espressione di mRNA o per verifica-
re gli effetti delle mutazioni sullo splicing, ove possibile. Le variazioni nucleotidiche
individuate sono convalidate utilizzando metodologie standard basate sulla PCR, in-
cluso il sequenziamento capillare, effettuato nei parenti affetti e nei parenti sani ed
escluse in almeno 400 cromosomi sani. L’analisi di segregazione nei parenti sarà e-
seguita per verificare se le mutazioni sono ereditate o sono mutazioni de novo.

49
BIOLOGIA MOLECOLARE
I canali di membrana vengono generalmente studiati utilizzando tecniche elettrofisio-
logiche e tecniche di biologia molecolare. Una delle possibilità d’utilizzo di tali tec-
niche è attraverso lo studio di canali mutanti, espressi eterologamente in ovociti di
Xenopus leavis i quali sono particolarmente agevoli poiché ci permettono di lavorare
su cellule di dimensioni notevoli. La tecnologia del DNA ricombinante, più nota col
nome di ingegneria genetica, consente la creazione di combinazioni geniche comple-
tamente nuove, cioè non presenti negli organismi che esistono in natura. Tale costru-
zione si ottiene incorporando una molecola di acido nucleico in un virus, in un pla-
smide batterico o in un altro tipo di sistema vettoriale; il vettore inserisce poi il co-
strutto di DNA in un organismo ospite, all’interno del quale il gene esogeno riesce a
riprodursi. I vettori finora sviluppati per il trasferimento di geni sono o vettori pla-
smidici (basati su piccole unità di DNA, distinte dai cromosomi) o batteriofagi (cioè
derivanti dai batteriofagi, i virus dei batteri) e devono essere in grado di penetrare
nella cellula ospite e di replicarsi al suo interno. I vettori che offrono le maggiori
possibilità di applicazione sono quelli plasmidici. Il DNA codificante per la proteina
viene inserito nel vettore utilizzando endonucleasi di restrizione, che tagliano la mo-
lecola di DNA a livello di specifiche sequenze di basi, dando così origine a frammen-
ti particolari. Alcune endonucleasi di restrizione tagliano i filamenti del DNA in pun-
ti non identici, generando in questo modo delle corte regioni a singola elica
all’estremità della molecola di DNA (estremità coesive); altre tagliano i due filamenti
in punti identici producendo estremità piatte. I frammenti di DNA ottenuti dalla dige-
stione del DNA stesso con gli enzimi di restrizione possono poi essere separati in ba-
se al peso molecolare. L’enzima DNA ligasi salda il frammento utile (gene) con il
vettore, purché trattato con lo stesso enzima di restrizione (ciò dà origine a estremità
complementari, che possono poi unirsi). Una volta costruito, il DNA ricombinante
può essere introdotto nella cellula ospite mediante un processo di trasformazione. Se
è compatibile con l’ospite, il DNA inserito si replicherà, cioè sarà clonato conte-
stualmente al processo di divisione cellulare dell’ospite. La popolazione delle cellule
batteriche contenenti DNA ricombinante è numerosa; alcune di esse ospitano proba-
bilmente al loro interno plasmidi non ricombinanti che riacquistano spontaneamente
una forma circolare dopo la digestione operata dall’endonucleasi. Il problema è dun-
que quello di selezionare un clone cellulare che possieda il gene desiderato. Bisogna,
per prima cosa, selezionare i ricombinanti. Uno dei metodi più semplici e più usati è
quello di adoperare vettori plasmidici resistenti ad uno o più antibiotici. Supponiamo,

50
ad esempio, di poter disporre di un cDNA e di inserirlo in uno di questi vettori. Suc-
cessivamente ai processi di amplificazione del plasmide ibrido mediante il suo trasfe-
rimento in cellule competenti (trasformazione), le cellule trasformate che hanno in-
corporato il plasmide ibrido potranno essere facilmente individuate facendole cresce-
re su piastre contenti l’antibiotico per cui il vettore plasmidico è resistente. Le colo-
nie individuate saranno quindi propagate, si procederà dunque all’estrazione del
DNA, che sarà infine testato per la sua correttezza (analisi di restrizione). Questo
DNA può essere utilizzato come tale o utilizzato come stampo per ottenere RNA
mediante un processo di trascrizione in vitro.
Nei nostri esperimenti, le mutazioni sono state introdotte nel cDNA del canale Kir4.1
usando il kit di mutagenesi QuikChangeTM Site-Directed Mutagenesis Kit (Strata-
gene) secondo le istruzioni riportate (D’Adamo et al., 1998). La sequenza di nucleo-
tidi di tutte le subunità sono state determinate utilizzando un sequenziatore automati-
co. Gli oligonucleotidi sono stati ottenuti da EUROBIO, Milano, Italia. Tutti i cDNA
utilizzati sono stati subclonati nel vettore di espressione per ovociti pBF, che presen-
ta le regioni non tradotte al 5’ e 3’ del gene della beta globina di Xenopus, vicino ad
un polylinker che contiene molti siti di restrizione. L’RNA codificante per il canale
Kir4.1 è stato sintetizzato in vitro usando la RNA polimerasi SP6 .
ANALISI STRUTTURA FUNZIONE
In questo studio la struttura 3D dell’etero-tetramero Kir4.1/5.1 è stata costruita attra-
verso modelli comparativi usando il software Modeller (Sali and Blundell, 1993). La
struttura a raggi X della chimera Kir3.1-canale procariote Kir (PDB id.: 2QKS) è sta-
to usato come modello (Nishida et al., 2007). Lo studio dell’allineamento di sequen-
za del frammento di interesse in contrapposizione con il campione è stato eseguito
usando ClustalX e perfezionato con Muscle (Edgar, 2004); la percentuale di identità
calcolata nelle sequenze allineate era di 36,7% mentre la similarità era 66,3%; solo i
residui 25-349 della struttura primaria Kir4.1 e i residui 31-347 della sequenza
Kir5.1 potevano essere allineati con i segmenti corrispondenti dei campioni analizza-
ti ai raggi X. I modelli omologhi generati sono stati venti, fra loro i cinque modelli
con energia più bassa sono stati selezionati e analizzati usando Procheck (Laskowski
et al., 1993). Il modello finale è stato scelto rispettando la percentuale più alta di re-
sidui nella regione definita dai diagrammi Ramachandran (>90%). Il modello è stato
poi immerso nel palmitoyl-oleoyl-fosfatidilcolina (POPC) lipide del doppio strato e
tutte le molecole di sovrapposizione sono state rimosse. Infine il mutante V84M è

51
stato generato sostituendo la catena laterale Val84 con la metionina usando VMD
(Humphrey et al., 1996). Il modello finale è stato definito mimetizzando con GRO-
MACS4 e GROMOS96 (Hess et al., 2008) l’ingombro sterico degli atomi limitrofi.
ESPRESSIONE ETEROLOGA DEI CANALI IONICI
Il sistema di espressione eterologa in ovociti di Xenopus è stato introdotto nel 1971
da Gurdon, per lo studio del controllo dell’epressione genica (Gurdon et al., 1971). È
comunque nel 1982 che Miledi e coll. hanno dimostrato che vari tipi di recettori e
canali ionici possono essere espressi negli ovociti iniettando al loro interno mRNA
opportunamente isolato a partire da appropriati tessuti.
Alcuni tra i più comuni tipi di studio che utilizzano l’espressione di canali ionici e
recettori esogeni negli ovociti di Xenopus includono:
1. analisi delle proprietà di canali ionici mutati, per comprendere le relazioni
struttura-funzione dei canali ionici
2. studio dei processi post-traduzionali e dell’assemblaggio dei recettori e dei
canali multisubunità
3. comparazione delle proprietà dei canali espressi in differenti tessuti, in un
ambiente comune
4. esame della modulazione delle funzioni di canali e recettori da parte di se-
condi messaggeri
5. screening funzionale dei geni clonati codificanti recettori e canali
La specie Xenopus laevis, anfibio artigliato del Sud Africa, è un membro della fami-
glia dei Pipidae, dell’ordine degli anuri. Esistono sei specie di Xenopus, tutte indi-
gene dell’Africa, ma solo la Xenopus laevis è utilizzata comunemente nei laboratori
di ricerca. Gli Xenopus laevis si distinguono per la presenza di tre dita dotate d’artigli
in ogni zampa dell’animale, da cui il nome improprio di ―rana artigliata‖.

52
Figura 10. Esemplari di Xenopus laevis adulti.
Sono note quattro sottospecie di Xenopus laevis, denominate Xenopus laevis laevis,
Xenopus laevis petersi, Xenopus laevis victorianus e Xenopus laevis borealis. Tutti
gli Xenopus sono aerobici, ma la loro vita è completamente acquatica, hanno quindi
necessità di respirare aria, ma non necessitano di una qualsiasi vita terrestre, sono
molto suscettibili alla disidratazione, e possono morire se poste al di fuori
dell’ambiente acquatico anche per poche ore.
Normalmente presentano pigmentazione scura sul lato dorsale e sono dotate di cellu-
le cromatofore che permettono di variare rapidamente colore in risposta all’ambiente
in cui si trovano. Questi anfibi secernono sia un liquido muco-simile sia una sostanza
tossica di proprietà affini all’epinefrina, ma questi strumenti di difesa non sono peri-
colosi ai fini del maneggio da parte dell’uomo. La dieta di mantenimento è costituita
da polmoni, reni e cuore di bue, anche se per gli animali provenienti da allevamenti è
possibile utilizzare mangimi sintetici specifici. La stabulazione degli Xenopus avvie-
ne in vasche o acquari di polipropilene con acqua priva di cloro, ad una temperatura
di circa 20°C, esponendole ad un ciclo luce-buio di 12h. Generalmente per la scelta
degli esemplari per la produzione degli ovociti si tende a prediligere soggetti di sesso
femminile, di grandi dimensioni e sessualmente maturi, che sono maggiormente in-
dicati nella produzione degli ovociti allo stadio V, ma anche d’ovociti in altri stadi.
Per sincronizzare lo sviluppo degli ovociti è eventualmente possibile iniettare gona-
dotropina corionica umana che stimola l’ovulazione e la deposizione delle uova, ma
giacché è necessario attendere due mesi dopo la somministrazione e che in ogni caso
è possibile ottenere ovociti anche da esemplari non iniettati, è preferibile evitare tale
operazione.

53
Prelievo e selezione degli ovociti
Gurdon et al. hanno per primi dimostrato che gli ovociti di Xenopus possono essere
utilizzati per l’espressione di mRNA esogeno, quando è microiniettato nel citopla-
sma. Molti ricercatori hanno utilizzato, da allora, questa proprietà per studiare
l’espressione di un vasto numero di molecole. L’asportazione degli ovociti avviene
in un ambiente adeguatamente trattato ed utilizzando strumenti sterili. Si estraggono
le ovaie e si preleva la quantità di ovociti desiderati.
Figura 11. Xenopus laevis durante la fase di prelievo degli ovociti
Gli ovociti prelevati vengono inseriti in una capsula Petri, lavati con una soluzione di
OR-2 (NaCl 82,5 mM, KCl 2 mM, MgCl2 1 mM, Hepes 5 mM; pH 7,5-7,55 con
NaOH) aiutandosi con un pasteur senza punta ed avendo cura di evitare che gli ovo-
citi vengano a contatto con l’aria che potrebbe danneggiarli. Mediante l’uso di un a-
gitatore si digerisce la membrana follicolare con collagenasi alla concentrazione fina-
le di 2 mg/ml per 20 minuti. Gli ovociti vengono lavati tre volte con ND-96 (NaCl
96 mM, KCl 2 mM, CaCl2 1,8 mM, MgCl2 1 mM, Hepes 5 mM, Na-piruvato 2,5
mM, teofillina 0.5 mM, gentamicina 50μg/ml) e selezionati, al IV stadio di matura-
zione, utilizzando uno stereo microscopio. La selezione degli ovociti avviene sce-
gliendo quelli che si presentano nelle condizioni migliori e allo stato di maturazione
ottimale per poter essere iniettati. Se sono in buona salute, essi appaiono di forma ro-
tondeggiante, piuttosto turgidi e con una netta separazione tra il polo vegetale e quel-
lo animale, che deve avere una pigmentazione scura ed omogenea.

54
Alcuni ovociti possono essere ancora ricoperti dalla membrana follicolare che deve
essere rimossa in modo da evitare disagi al momento dell’iniezione e durante le regi-
strazioni elettrofisiologiche. Infatti, la presenza di canali ionici endogeni localizzati a
livello del follicolo è fonte di correnti che possono disturbare la rilevazione di quelle
studiate. Una volta selezionati, gli ovociti sono conservati in capsule di Petri a 18° C
in attesa di essere iniettati.
Figura 12. Ovociti di Xenopus
Microiniezione di mRNA in ovociti
Un ovocita isolato è in grado di tradurre efficientemente mRNA esogeno iniettato nel
citoplasma. Generalmente gli ovociti sono capaci di modificare (tramite fosforilazio-
ni, glicosilazioni, ecc.) correttamente i polipeptidi tradotti da mRNA iniettato. Inoltre
è stato dimostrato che gli ovociti sembrano in genere convogliare correttamente
all’appropriato compartimento subcellulare proteine espresse eterologamente. La ca-
pacità degli ovociti di tradurre efficientemente e fedelmente informazioni genetiche
estranee associata all’abilità di assemblare i complessi oligomerici recettore/canale e
di inserirli nella membrana plasmatica per generare risposte rilevabili elettrofisiolo-
gicamente, fanno di essi un potente mezzo per le indagini molecolari neurobiologi-
che. Due possono essere gli approcci utilizzati per l’espressione di proteine di tra-
sporto di membrana: il primo comporta l’iniezione di mRNA poli(A+) isolato da cel-
lule che esprimono la proteina d’interesse; in alternativa, quando è reperibile un
cDNA della proteina in questione, questo può essere utilizzato per generare un
mRNA sintetico da adoperare per la successiva iniezione. L’RNA è facilmente de-
gradato da RNAsi ambientali quindi è necessario prendere una serie di precauzioni
per evitare che questo avvenga. Gli elettrodi per l’iniezione vengono prodotti tirando
capillari in vetro con un puller, la punta viene poi tagliata a becco di clarino e
l’elettrodo viene riempito di paraffina e successivamente di mRNA.

55
Figura 13. Iniezione di RNA e formazione della proteina in ovociti di Xenopus
La quantità di RNA da iniettare è regolata tramite un codice a quattro numeri posto
sull’iniettore. Utilizzando una Petri, sul cui fondo è stata applicata una reticella con
maglie di 1 mm di larghezza, utile a tenere fermi e ordinati gli ovociti durante
l’iniezione, si precede alla microiniezione. Nei nostri esperimenti gli ovociti sono
stati iniettati ciascuno con 50nl di RNA codificante per le subunità da iniettare. Negli
esperimenti di co-iniezione uguali quantità di RNA codificante per le diverse subuni-
tà sono iniettate in rapporto 1:1. Gli ovociti iniettati sono conservati in gruppi di 20
in capsule di Petri contenenti ND-96 ed incubati a 18°C per un periodo di tempo va-
riabile (1-6 giorni) necessario affinchè la proteina sia espressa.

56
ELETTROFISIOLOGIA
Two-electrode voltage-clamp
Il blocco del voltaggio (voltage-clamp) permette allo sperimentatore di fissare o
bloccare il valore del potenziale di membrana mantenendolo fisso a livelli stabiliti.
L’apparato consta di un generatore di corrente connesso a una coppia di elettrodi (Fi-
gura 14). Misurando la quantità di corrente che è stato necessario erogare per mante-
nere il potenziale di membrana al suo nuovo valore, si misura automaticamente an-
che la corrente di membrana che passa attraverso i canali ionici. Il voltaggio di mem-
brana è registrato mediante un elettrodo di potenziale relativamente ad un valore di
riferimento mentre l’elettrodo di corrente inietta corrente nella cellula. Lo sperimen-
tatore stabilisce un potenziale imposto (holding voltage, Vh o command potential) e il
voltage clamp utilizza un sistema a feedback negativo per mantenere la cellula a que-
sto valore. Gli elettrodi sono connessi ad un amplificatore che misura il potenziale di
membrana e invia il segnale all’amplificatore a feedback. Questo amplificatore rice-
ve anche un input dal generatore di segnali che decide il potenziale imposto, e sottrae
il potenziale di membrana dal potenziale imposto (Vh-Vm), amplifica questa differen-
za ed invia un output all’elettrodo di corrente. Ogni volta che la cellula devia dal po-
tenziale imposto, l’amplificatore genera un segnale di errore, che è la differenza tra il
potenziale imposto e il potenziale della cellula, il circuito a feedback invia corrente
nella cellula per ridurre a zero il segnale di errore. In questo modo il circuito del vol-
tage clamp produce una corrente che è uguale ed opposta alla corrente che passa at-
traverso i canali ionici nella membrana. Il metodo del blocco del voltaggio è ancora
oggi ampiamente utilizzato per studiare le correnti ioniche nei neuroni e in altre cel-
lule. La versione attualmente più diffusa di questa tecnica è il metodo del potenziale
imposto ad un frammento di membrana (patch-clamp), una tecnica che si può appli-
care praticamente a qualunque tipo di cellula e che offre un potere di risoluzione suf-
ficientemente elevato da permettere di misurare le microcorrenti elettriche che attra-
versano singoli canali ionici. Per effettuare registrazioni di corrente macroscopica
sugli ovociti abbiamo utilizzato la tecnica elettrofisiologica del two-electrode volta-
ge-clamp, con un elettrodo intracellulare per registrare il potenziale intracellulare ed
un secondo elettrodo per il passaggio della corrente necessaria a mantenere il poten-
ziale desiderato. La corrente necessaria per mantenere un potenziale dato è il parame-
tro misurato.

57
Figura 14. Two-electrode voltage-clamp
Figura 15. Two-electrode voltage-clamp
La maggior parte dei set-up di voltage-clamp utilizzano anche un elettrodo per la re-
gistrazione del potenziale della soluzione esterna (elettrodo di riferimento). Questo
evita errori di polarizzazione derivanti dalla corrente che passa attraverso l’elettrodo
di terra. In questa configurazione, il potenziale transmembrana è ricavato dalla diffe-
renza tra il potenziale dell’elettrodo intracellulare e l’elettrodo di riferimento. In que-
sto modo è possibile correggere i piccoli errori derivanti dalla non esatta corrispon-
denza del potenziale della soluzione esterna con il potenziale di terra. Nel nostro ca-
so, gli elettrodi sono connessi ad un amplificatore GeneClamp 500 (Axon Instru-
ments) collegato ad un computer tramite un’interfaccia ITC-16 (Instrutech). Le cor-
renti sono registrate e analizzate con il software Pulse+PulseFit (HEKA Elektronic).
Gli elettrodi intracellulari standard contengono una soluzione interna di KCl 3M o di
solfato di potassio (o aspartato) 0.5M e KCl 30mM. La soluzione di KCl 3M può es-
ser utilizzata per svariate applicazioni; le soluzione di aspartato/solfato sono utili
quando si vuole ridurre il carico di Cl- all’interno degli ovociti, in questo caso la pre-
senza di KCl è comunque necessaria per l’elettrodo Ag/AgCl. Tipicamente il diame-
tro della punta dell’elettrodo desiderato per questo tipo di esperimento dovrebbe tro-

58
varsi in un range da 1 a 5μm, con resistenza da 0.4 a 2MΩ. Il contatto elettrico con la
soluzione di riempimento è assicurata da un elettrodo di cloruro d’argento. Tale elet-
trodo può essere fabbricato a partire da un filo di cloruro d’argento, da un filo
d’argento immerso in cloruro d’argento fuso o da un pellet composto da argento e
cloruro d’argento. La durata degli elettrodi menzionati è in funzione del diametro del
filo utilizzato. Molti amplificatori per il voltage clamp incorporano uno strumento di
misurazione per la resistenza degli elettrodi, comunque in mancanza di tale strumen-
tazione è possibile misurare tali valori applicando un impulso di corrente (ΔI) attra-
verso l’elettrodo Ag/AgCl e misurando il salto di potenziale indotto. La resistenza
dell’elettrodo sarà ΔV/ΔI. Finché gli elettrodi mantengono valori di bassa resistenza
possono essere utilizzati per diversi ovociti. Data la notevole dimensione degli ovoci-
ti, gli elettrodi possono essere posizionati semplicemente con un comune micromani-
polatore. E’ comunque importante che il set-up sia privo di vibrazioni, poiché le o-
scillazioni possono produrre lacerazioni nella membrana, nel punto di penetrazione
degli elettrodi. Un tavolo isolato dalle vibrazioni non è solitamente necessario se il
terreno è sufficientemente tranquillo. Una gabbia di Faraday non è essenziale per
questa tecnica, ma può comunque limitare il rumore di fondo presente nel laborato-
rio. Il microscopio non necessita ingrandimenti eccessivi, può quindi essere semplice
(obiettivo 5-10X), sia invertito che non. Una volta che gli elettrodi sono immersi nel-
la soluzione, bisogna compensare il potenziale degli elettrodi a valori compresi tra
±1mV. È consigliabile effettuare questa regolazione dopo ogni esperimento, per assi-
curare che non ci siano scostamenti dal valore di potenziale imposto agli ovociti. Gli
ovociti vengono a tal punto impalati prima con l’elettrodo della corrente, poi con
quello del potenziale. L’avvenuta penetrazione da parte dell’elettrodo del potenziale
può essere verificata tramite i valori di potenziale registrati compresi in un range di -
30/-60 mV. Più complesso è il monitoraggio della penetrazione dell’elettrodo della
corrente. Si valuta l’ampiezza delle variazioni di potenziale in risposta a stimoli da
parte dell’elettrodo della corrente. Ampiezze minime lasciano intendere che l’ovocita
non contiene molti canali. Negli esperimenti riportati in seguito, la corrente che passa
attraverso la membrana degli ovociti è stata registrata 1-6 giorni dopo le iniezioni di
mRNA e le correnti di ―leak‖ e capacitive sono state sottratte applicando un protocol-
lo P/4. Questo metodo sviluppato da Armtrong e Bezanilla nel 1977, prevede la regi-
strazione di quattro correnti evocate da uno step iperpolarizzante di ampiezza pari ad
un quarto dell’ampiezza dello step depolarizzante di registrazione. La media di que-

59
ste quattro correnti viene quindi sottratta alla corrente evocata dello step depolariz-
zante di registrazione:
Ii=It+∑4
i=1 Is
La soluzione di registrazione utlizzata contiene (mM): NaCl 96, KCl 2, MgCl2 1,
CaCl2 1.8, HEPES 5, pH=7.4.
Il software di registrazione Pulse+Pulsefit ci consente di creare una serie di protocolli
sperimentali in cui il voltaggio è bloccato al valore di volta in volta desiderato, e, di
seguito, di misurare il flusso di ioni potassio che passa attraverso i canali espressi
sulla membrana dell’ovocita. Variando il protocollo e registrando la corrente otte-
niamo informazioni su diverse caratteristiche del canale. Il parametro che è stato ana-
lizzato nei nostri esperimenti è il livello di espressione o ampiezza della corrente in
risposta ad una depolarizzazione della membrana.
L’analisi delle correnti e la statistica è stata fatta con il software KaleidaGraph,
Synergy Software, USA.
Tecnica del patch-clamp
Lo studio delle correnti elettriche originate dai flussi ionici nelle cellule eccitabili ha
avuto una svolta epocale quando, negli anni settanta, grazie alla tecnica del patch-
clamp è stato possibile analizzare l’attività dei singoli canali ionici da frammenti di
membrana. Gli ideatori di questa metodica, Ervin Neher e Bert Sakmann, sono stati
insigniti del premio Nobel per la fisiologia e la medicina nel 1991, per la messa a
punto di questa tecnica. Il patch-clamp, concettualmente semplice, ha però richiesto
anni d’elaborazione e perfezionamento. L’estremità di una sottile pipetta di vetro
(1μm) è fatta aderire perfettamente ad una membrana cellulare, permettendo così
d’isolare meccanicamente ed elettricamente una piccola area della membrana stessa e
i canali ionici in essa presenti. Come nelle altre tecniche di voltage-clamp, il proble-
ma fondamentale, nelle misurazioni su di un singolo canale, è dato dal rumore di
fondo, ben superiore all’esile corrente da misurare. Tale problema fu risolto dagli au-
tori praticando una lieve aspirazione attraverso la pipetta e portando, così, la resi-
stenza della saldatura (seal) a più di un miliardo di Ohm, il che rappresentò un mi-
glioramento di parecchi ordini di grandezza. È proprio la scoperta del gigaseal, ovve-
ro la creazione, attraverso l’aspirazione, di una zona sigillata tra la pipetta e la mem-
brana cellulare, con elevatissima resistenza, che ha permesso alla registrazione di
singoli canali ionici di divenire una tecnica ad alta risoluzione. Attualmente la tecni-
ca del patch-clamp è utilizzata per studiare cellule eccitabili come neuroni, fibre mu-

60
scolari e cellule endocrine. Inoltre è stata usata per caratterizzare canali ionici batte-
rici in particolari sistemi cellulari artificiali. A differenza di altre tecniche di voltage-
clamp, questa tecnica utilizza un singolo elettrodo per bloccare il potenziale della
cellula. Questo consente di mantenere il voltaggio costante e misurare i cambiamenti
della corrente o, in alternativa, di bloccare la corrente e osservare le variazioni di po-
tenziale della cellula. Esistono diverse possibili configurazioni per le registrazioni di
corrente in seguito alla formazione di un gigaseal ed è possibile cambiare la compo-
sizione della soluzione di riempimento dell’elettrodo per studiare i canali in differenti
condizioni sperimentali (Hamil et al., 1981; Fig.16). Nella configurazione cell-
attached, l’elettrodo rimane legato al frammento di membrana. Questa configurazio-
ne consente di registrare correnti attraverso singoli canali nel patch di membrana o di
verificare sui canali l’effetto di farmaci o ligandi presenti nella soluzione
dell’elettrodo. Applicando ulteriore suzione in modo da rompere la porzione di
membrana interna all’elettrodo, si passa nella configurazione whole-cell, in cui
l’elettrodo ha accesso al contenuto intracellulare della cellula. Le configurazioni
inside-out ed outside-out sono anche chiamate tecniche di excised-patch, perché il
frammento di membrana è rimosso dal corpo cellulare. In questo modo è possibile
studiare la modulazione dei canali dal lato intra- ed extracellulare.
Figura 16. Configurazioni della tecnica del patch-cla

61
RISULTATI
Caratteristiche cliniche dei pazienti
I disturbi dello spettro autistico sono disturbi eterogenei dal punto di vista clinico ed
è quindi opportuno selezionare accuratamente i pazienti da includere nello studio.
Famiglia A:
I pazienti A-III:1 e A-III:2 (Fig.17A) sono gemelli monozigoti di otto anni. A cinque
mesi si era notata la presenza di ipotonia, difficoltà durante il sonno, disabilità
nell’interazione sociale per entrambi. Durante il settimo mese per entrambi, nell’arco
delle ventiquattro ore si verificavano spasmi epilettici curati con somministrazione di
ACTH. A dieci mesi, il paziente A-III:2 non manifestava attacchi epilettici, mentre il
fratello presentava crisi persistenti che sono state attenuate verso il dodicesimo mese
di età, sottoponendolo a terapia con valproato e topimarato. I bambini mostravano
deficit posturale, ipotonia, nistagmo e assenza di parola. Gli elettroencefalogrammi
erano normali mentre erano svegli; le registrazioni durante il sonno mostravano atti-
vità ritardata, normalizzata successivamente in entrambi all’età di tre anni. Le analisi
di laboratorio (inclusi gli esami del siero e delle urine), il cariotipo ad alta definizio-
ne, il CGH-array e le analisi mutazionali ARX erano normali. La rappresentazione
somatosensoriale dei potenziali uditivi evocati nel tronco cerebrale era irrilevante. Il
paziente A-III:1 presentava deficit motorio persistente, confusione e ha pronunciato
le sue prime parole all’età di cinque anni; il soggetto ha anche manifestato un severo
deficit nell’interazione sociale, stereotipi, e atteggiamenti ripetitivi. Questi sintomi
rientrano nella diagnosi di DSM-IV-TR caratteristiche del DSA. Un’ ultima valuta-
zione, all’età di otto anni ha permesso di rilevare deficit negli atteggiamenti comuni-
cativi e sociali, movimenti ripetitivi, confusione e moderato ritardo mentale (IQ: 43).
La maggior parte dei sintomi, anche se in maniera meno severa, si è verificata anche
nel paziente A-III:2, il quale mostrava anche comportamenti rituali. Abilità sociali al-
terate, ansia, depressione, disordini ossessivi-compulsivi e ritardo mentale moderato
(IQ:58). La madre di quarantotto anni (soggetto A-II:1) aveva un tic multiplo, sinto-
mi ossessivi-compulsivi, cambiamenti d’umore con impulsività e confusione moto-
ria. Però non presentava attacchi epilettici.
Famiglia B:
Il soggetto di quattordici anni (B-III:1; Fig.17A ) è il primo bambino sano di genitori
non consanguinei. Lo sviluppo psicomotorio era normale fino all’età di 12 mesi, in
seguito l’alterazione del sonno, l’assenza dello sviluppo del linguaggio, la non rispo-

62
sta al nome e la manifestazione di comportamenti rituali sono diventati evidenti. A
diciotto mesi gli è stato diagnosticato DSA. In quel periodo l’elettroencefalogramma,
la tac al cervello, la risposta del tronco cerebrale, il fondo oculare, il cariotipo e il test
del X fragile erano normali. All’età di sei anni, ha avvertito gli attacchi epilettici par-
ziali che parzialmente rispondevano al valproato. Gli elettroencefalogrammi hanno
mostrato i parossismi frontali bilaterali sia da sveglio sia durante il sonno, mentre la
risonanza magnetica del cervello risultava normale. Le analisi del sangue e valori
chimici (inclusi elettroliti e test dell’urine) erano considerati nella norma. Le caratte-
ristiche autistiche peggioravano nei giorni, con severo deficit nell’abilità comunicati-
va e sociale, atteggiamenti ripetitivi e stereotipati, severo ritardo mentale, e compor-
tamenti nocivi per se stesso e per gli altri. La sorella più giovane (B-III:2) e il nonno
paterno (B-I:1) sono sani ma non non stati esaminati.
Fig.17. (A) Albero genealogico della famiglia A e della famiglia B e analisi di se-
quenza. albero genealogico di due famiglie portatrici delle nuove mutazioni in
KCNJ10. I quadrati rappresentano i maschi e i cerchi le femmine. I simboli neri rap-
presentano i soggetti che hanno la mutazione; i tagli denotano un individuo deceduto.
(B) Sequenziamento della mutazione. Elettroferogramma dell’esone 2 fiancheggiante
la mutazione in KCNJ10 in individui non affetti (WT) e soggetti affetti (A-III:1, B-
III:1). La posizione della mutazione eterozigote è indicata dalla freccia e la conse-
guenza della mutazione è mostrata sotto. Paziente A-III:1 manifesta una mutazione
eterozigote c.53G>A, prevedendo una nuova mutazione V84M.
Risultati delle analisi di mutazioni
Per verificare se i canali Kir fossero responsabili del fenotipo oggetto di studio, il ge-
ne KCNJ10 è stato analizzato alla ricerca di mutazioni. Dopo aver purificato il DNA

63
genomico da leucociti del sangue periferico, la ricerca di mutazioni dei geni KCNJ10
(Kir4.1), KCNJ16 (Kir5.1) è stata realizzata utilizzando le tecniche di genetica mo-
lecolare già descritte.
Nei pazienti A-III:1 e A-III:2 (Fig.17A) si è individuata una mutazione eterozigote
c.53G>A in KCNJ10, prevedendo una variante p. R18Q nell’estremità-N-terminale
citoplasmatica del canale Kir4.1 (Fig. 17B, D). Il paziente B-III:1 presentava una
mutazione eterozigote c250G>a (V84M) nel primo dominio di transmembrana di
KCNJ10 (Fig. 17B, D). Le due mutazioni non sono state individuate in 528 cromo-
somi sani. Entrambe le mutazioni sono state ereditate dalla madre. Nella famiglia A,
la nonna materna (A-I:2) era wild-type. Nella famiglia B, la mutazione era anche in-
dividuata in B-I:1 e B-III:2. Le due mutazioni identificate erano nuove e non erano
presenti in dbSNP. Queste mutazioni colpiscono i residui altamente conservati nei
mammiferi e in Xenopus tropicalis (Fig 17C). In questo studio, si è anche identificato
una mutazione eterozigote p.R271C in due bambini. Questa variante si riscontra ge-
neralmente nei bambini sani (Buono et al., 2004) e sono state studiate le conseguenze
funzionali di questa mutazione sulla funzione del canale e sulla sua struttura (Shang
et al.2005). Inoltre sono state individuate due altre mutazioni in KCNJ10 (g.250T>C
e g.83_88dup6). L’analisi di mutazione di KCNJ16 ha individuato una variante poli-
morfica eterozigote (rs34408089) in quattro pazienti, incluso il caso B-III:1.
Fig.17. (C) Analisi bioinformatiche. Sequenza proteica ortologa e paraloga del gene
KCNJ10 di vertebrato sono state allineate usando ClustalW algoritmo. I numeri di
accesso in Gene Bank per le sequenze ortologhe e paraloghe sono: NP_002232.2
(human KCNJ10), XP_001115293.2 (macaca), XP_513920.2 (pantroglodytes),
NP_001034573.1 (mouse), NP_113790.1 (rattus), NP_001075070.1 (bos taurus),
AAW30192.1 (sus scrofa), NP_001072312.1 (Xenopus tropicalis). (D) Rappresenta-
zione schematica della sub unità umana Kir4.1, composta da un poro delimitante la

64
regione (S1-H5 loop-S2), con le estremità sia C-terminale sia N-terminale poste
all’interno della cellula. La variante R18Q è situata nel dominio citoplasmatico N-
terminale, mentre la variante V84M è localizzata nel dominio di transmembrana M1.
Caratterizzazione funzionale dei canali omomerici che presentano le mutazioni
R18Q o V84M.
Per verificare l’effetto delle mutazioni identificate sull’attività del canale Kir4.1 sono
state effettuate registrazioni in voltage-clamp con due elettrodi (TEVC) da ovociti di
Xenopus che esprimono il canale Kir4.1 wild-type e mutato o co-espresso con Kir5.1.
Il cDNA dei canali Kir è stato clonato in un vettore di espressione specifico per ovo-
citi. Il cDNA che codifica per il canale mutato è stato generato mediante mutagenesi
sito-diretta.
Per confrontare le differenze nelle proprietà intrinseche e nel livello di espressione
del canale conseguenti alle mutazioni, uguali quantità di RNA codificante per il ca-
nale wild-type e per i mutanti è stato iniettato nel sistema di espressione eterologo di
ovociti di Xenopus e le correnti macroscopiche sono state registrate con la tecnica
del voltage-clamp. L’espressione di entrambi i canali R18Q o V84M presentava cor-
renti con cinetiche evidenti simili al WT (Fig.18A-C). Tuttavia, il rapporto corren-
te/voltaggio per entrambi i canali mutati presentava ampiezze più grandi rispetto a
quelle del WT quando ugual quantità di WT o RNAs mutati erano espressi sulla
membrana (n= 129; Fig.18D, F). Per mimare lo stato eterozigote della malattia,
RNAs WT e R18Q o V84M sono stati co-iniettati (1:1 ratio) e presentavano ampiez-
ze di corrente intermedia tra l’omomerico WT e R18Q o V84M (Fig. 18E, G). Il gra-
fico a barre riportato (Fig. 18E) rappresenta 4 esperimenti indipendenti eseguiti fa-
cendo uso di serie differenti di ovociti. Il numero totale di ovociti del gruppo esami-
nato è approssimativamente 120. Si noti che l’ampiezza delle correnti è strettamente
legata alla quantità di cRNA iniettata. Le mutazioni R18Q e V84M aumentano no-
tevolmente le ampiezze di corrente sia dei canali omomerici sia eteromerici. I dati
sono la media di 80-120 cellule ± SEM. Il significato statistico delle differenze os-
servate è stato valutato paragonando ogni risultato con il controllo e usando il test di
Student. valori P<0.005, valori P<0.01. Le correnti di singolo canale sono state e-
saminate per mezzo di registrazioni di patch-clamp in configurazione cell-attached o
in-out, per determinare differenze funzionali come conduttanza, probabilità di apertu-
ra, tempi medi di apertura e chiusura. Le registrazioni pacth clamp di un singolo ca-
nale rilevavano che la mutazione V84M incrementava la conduttanza di circa 1.5

65
volte rispetto al WT (Fig.19; n=14). Non sono state osservate differenze evidenti nel-
la probabilità e nel tempo di apertura tra il canale WT e il canale V84M (risultati non
mostrati). La mutazione R18Q non alterava la conduttanza unitaria (Fig.19; n=8) o i
parametri di altri singoli canali (risultati non mostrati). Considerando i risultati, si e-
vince che le mutazioni R18Q e V84M colpiscono le ampiezze delle correnti princi-
palmente aumentando l’espressione del canale in superficie e la sua conduttanza.

66

67
Fig. 18 (A, B, C) Espressione dei canali Kir4.1 wild type, canali R18Q e V84M.
Correnti registrate in configurazione TEVC da ovociti di Xenopus che esprimono u-
gual quantità di mRNA Kir4.1 WT, R18Q o V84M. Le correnti di K+
rettificano
fortemente, si attivano rapidamente e si riducono nel tempo a potenziali iperpolariz-
zati. Le correnti sono evocate da un potenziale di holding -10mV variando il poten-
ziale di membrana con incrementi di -10mV da +50 a -150mV. Il protocollo speri-
mentale è mostrato come inserzione in A. Le linee orizzontali tratteggiate indicano il
livello di corrente uguale a 0. (D, F) Curva corrente-voltaggio (I/V) per canali WT
(●) e R18Q (○) (D) o WT (●) e V84M (○) (F), quando uguale quantità di cRNA per
ogni tipo di canale è stata espressa. Si noti che la relazione I/V sia per R18Q sia per
V84M presenta ampiezze più grandi rispetto ai canali WT.(E, G) Grafico a barre che
mostra la quantità di corrente media registrata a -100mV negli ovociti che esprimo-
no i canali indicati. La quantità di cRNA iniettata in ogni gruppo è indicata in paren-
tesi.

68

69
Fig. 19. (A, B, C)Registrazioni si patch-clamp dei canali Kir4.1 WT, R18Q e
V84M. Tracce rappresentative ottenute da registrazioni dei canali WT (A), R18Q (B)
e V84M (C) a -80mV in configurazione cell-attached. Le tracce in basso sono mo-
strate su una scala di tempo ampliata. Le aperture del canale sono indicate con de-
flessioni verso il basso e le frecce indicano il livello di chiusura (C) e apertura (O).
Le tracce delle correnti sono state filtrate a 0.5kHz.Analisi della conduttanza dei ca-
nali Kir4.1 WT, R18Q e V84M. (D, E, F) Istogramma delle correnti unitarie registra-
te a -80mV da ovociti che esprimono i canali WT, (D) R18Q (E) e V84M (F). Gli i-
stogrammi sono costruiti utilizzando le registrazioni in cui è presente un solo canale.
(G) Le correnti registrate a vari voltaggi, sono state analizzate e plottate per calcolare
la conduttanza del canale.Non ci sono differenze nella conduttanza del canale WT
(25.8pS; nero) e R18Q (26.1pS; blu), mentre è significativamente aumentata la con-
duttanza della mutazione V84M (39.5 pS; arancione).
Caratterizzazione funzionale di canali eteromerici Kir4.1-Kir5.1 che presentano
le mutazioni R18Q e V84M
La subunità Kir5.1 co-assembla con la subunità Kir4.1, conferendo importanti pro-
prietà al canale eteromerico Kir4.1/Kir5.1 rispetto al canale omomerico Kir4, quali
ad esempio una più piccola corrente istantanea, una ―relaxation‖ tempo-dipendente a
potenziali iperpolarizzati e un drammatico aumento della sensibilità alla inibizione
tramite H+
intracellulare (pHi) (Pessia et al.1996) (confrontare Figs. 18A e 20A). Per
verificare se entrambe le mutazioni influenzano le proprietà biofisiche e la sensibilità
al pHi dei canali eteromerici Kir4.1/Kir5.1, RNA codificante per canali wild-type e
mutanti è stato iniettato in ovociti di Xenopus Laevis e le correnti relative sono state
registrate con la tecnica del TEVC. Entrambe le mutazioni incrementavano la corren-
te istantanea e annullavano la componente lenta della ―relaxation‖ dei canali
Kir4.1/Kir5.1. Per contro, la loro sensibilità al pHi non mostrava cambiamenti
(Fig.20A-I).
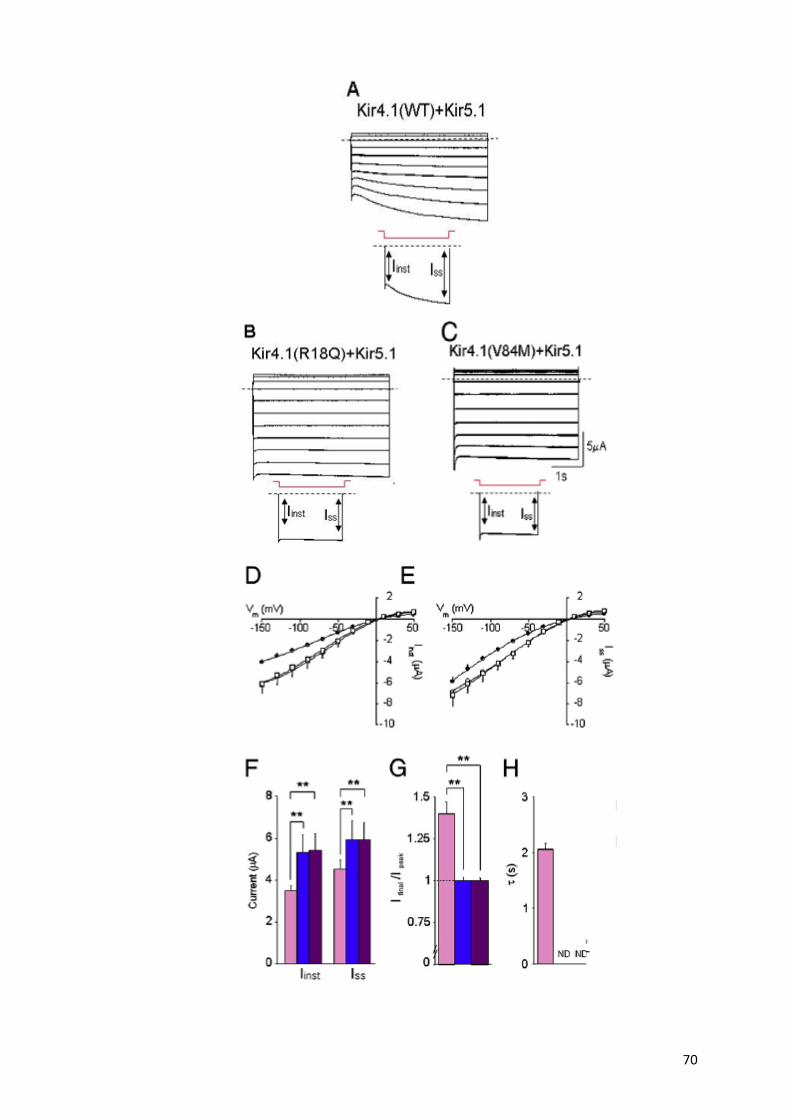
70

71
Fig. 20. (A,B, C) Effetti delle mutazioni R18Q e V84M sulla funzione del canale ete-
romerico Kir4.1/Kir5.1. (A) Tracce rappresentative di correnti registrate da ovociti
che esprimono i canali Kir4.1 (WT)/Kir5.1, (B) Kir4.1(R18Q)/Kir5.1, e (C) Kir4.1
(V84M)/ Kir 5.1. Le correnti sono evocate secondo la descrizione in Fig. 2. Le linee
orizzontali tratteggiate indicano il livello di corrente 0. Si nota la tipica ―relaxation"
tempo-dipendente (componente lenta) della corrente del canale Kir4.1 (WT)/Kir 5.1
(A). Le tracce di corrente mostrano una corrente istantanea (Iinst) e una corrente allo
stato stazionario (Iss) (inserzione in A). Invece, in entrambe le correnti di
Kir4.1(R18Q)/Kir5.1 (B) e Kir4.1 (V84M)/Kir5.1 (C) le componenti Iinst e Iss hanno
una ampiezza simile (inserzione in B e C), denotando un incremento di Iinst e comple-
ta assenza della ―relaxation‖ lento tempo-dipendente. Curva corrente-voltaggio (D,
E).Rappresentazione della media dei rapporti Iinst/V (D) e Iss/V (E) per i canali
Kir4.1(WT)/ Kir5.1(●), Kir4.1 (R18Q)/Kir5.1 (○) e Kir4.1(V84M)/Kir5.1 (□).I dati
sono la media di 80-120 cellule ± SEM. Il significato statistico delle differenze os-
servate è stato valutato paragonando ogni risultato con il controllo e usando il test di
Student. valori P<0.005, valori P<0.01.(F) Il grafico a barre mostra che a -100mV
sia Iinst sia Iss per Kir4.1 (R18Q)/Kir5.1 (blu) e Kir4.1(V84M)/Kir5.1(viola) sono più
ampie rispetto a quelle dei canali Kir4.1(WT)/Kir5.1 (rosa), quando un ugual quanti-
tà di cRNA per ogni tipo di canale è stata espressa. (G) Iss Iinst / calcolato a -100mV
per i canali Kir4.1(WT)/Kir5.1 (rosa), Kir4.1(R18Q)/Kir5.1 (blu), o
Kir4.1(V84M)/Kir5.1 (viola). Questa analisi denota che la componente lenta rappre-
senta il 40% della corrente totale per WT, mentre è quasi assente nei canali muta-
ti.(H) La componente lenta dell'attivazione dei canali Kir4.1 (WT)/Kir5.1 è fittata
con una funzione esponenziale e la costante di tempo media è rappresentata in un
grafico a barre; questo valore non può essere calcolato per i canali mutati.I grafici a
barre riportati rappresentano quattro esperimenti indipendenti usando serie differenti
di ovociti. Il numero totale di ovociti esaminati per ogni gruppo è approssimativa-

72
mente 100. Questi risultati indicano che le mutazioni R18Q e V84M aumentano le
ampiezze della corrente del canale eteromerico e che le correnti inwardly-rectifying
K+
hanno cinetiche di attivazione più veloce rispetto al WT. (I) Inibizione della cor-
rente in funzione del pH intracellulare dei canali omomerici Kir4.1 (WT) (●),
Kir4.1(R18Q) (▲), Kir4.1(V84M) (■), e dei i canali Kir4.1(WT)/Kir5.1 (○),
Kir4.1(R18Q)/Kir5.1( ),e Kir4.1(V84M)/Kir5.1 (□). I risultati sono stati ottenuti da
correnti registrate a -100mV con la tecnica del TEVC durante la perfusione con una
soluzione di acetato di K+ che riduce il pH intracellulare degli ovociti al valore in-
dicato (mean±S.E.M. di 6–8 ovociti). La linea continua mostra il ―fit‖ con
l’equazione 1/[1+([H+]i/K)
n] dalla quale il valore di pKa è stato calcolato. Si noti che
le mutazioni R18Q e V84M non cambiano la sensibilità al pHi del canale Kir4.1 o
del canale Kir4.1/Kir5.1. I dati sono la media di 80-120 cellule ± SEM. Il significato
statistico delle differenze osservate è stato valutato paragonando ogni risultato con il
controllo e usando il test di Student. valori P<0.005, valori P<0.01.
Modeling molecolare
Basandosi sui risultati ottenuti dalla struttura cristallina, si è generato un modello 3D-
omologo del canale eteromerico Kir4.1/Kir5.1. L’analisi di questo modello indicava
che la mutazione V84M era localizzata vicino all’interfaccia membrana/acqua del
primo segmento trans membrana (TM1) e rivelava che gli atomi della catena laterale
Met-84 erano posizionati verso le molecole lipidiche del doppio strato di membrana
(Fig.21). Tuttavia la posizione del residuo Arg-18 nella regione N-terminale non è
stata determinata per mancanza di risultati strutturali.

73
Fig.21. Modello 3D-omologo del canale Kir4.1/Kir5.1 in cui è indicata la posizione
di Val-84. (A)La struttura cristallina mostra la regione a forma di cono del canale
immerso nel doppio strato fosfolipidico di membrana (rappresentati a forma di ba-
stoncini di colore blu chiaro). Gli elementi della struttura secondaria sono indicati
come nastri. Le catene blu e rosse si riferiscono ai monomeri Kir4.1 e Kir5.1. (B)
Immagine in primo piano del canale in cui è indicata la posizione di Val-84 nel pri-
mo dominio transmembrana che è stata mutata in metionina. Si nota
che gli atomi della catena laterale di Met 84 sono rivolti verso le molecole del dop-
pio strato che sono state posizionate a una distanza di ~3,0 Å. La posizione del resi-
duo Arg-18 nella regione N-terminale non è stata rappresentata a causa della man-
canza di dati strutturali. Il primo residuo disponibile nel sistema cristallino è Arg-
27(indicato come un bastone).
Conferma dei risultati
In collaborazione con l’ Istituto di ricovero e cura a carattere scientifico (IRCCS)
fondazione Stella Maris di Pisa, si sono eseguiti esperimenti di conferma usando sia
la tecnica del western blot sia le tecniche di immufluorescenza. Il western blot è stato
fatto con anticorpi anti-Kir e anti-istidina su estratti di citosol e membrane delle linee
cellulari U251, mentre per le immunofluorescenze si è usato sempre l’anti-istidina e
un marcatore per l’actina. In questo modo è stato possibile identificare dove vengono
espresse le proteine ricombinanti e valutare se ci sono differenze tra mutanti e WT.

74
Da una prima analisi (Fig.22.) si è dedotto che la mutazione R/Q del Kir4.1 è mag-
giormente espressa sia nel citosl che nelle membrane mentre la mutazione V/M è e-
spressa a livelli più bassi rispetto al WT. C’ è poi un effetto anche sulla morfologia
delle cellule che potrebbe essere dovuto all’espressione dei canali Kir4.1 WT e muta-
ti indifferentemente. Quindi ciò permette di confermare la deduzione del nostro stu-
dio: le mutazioni R18Q e V84M colpiscono le ampiezze delle correnti rispettivamen-
te aumentando l’espressione del canale in superficie e la sua conduttanza.
U251overesprimenti:
HIS_Kir4.1 WT – R/Q – V/M
IF HIS/ACTIN WB: antiHIS
WB: antiKir4.1
51 -
64 -
39 -
CITOSOLMEMBRANE
U251
CITOSOLMEMBRANE
51 -
64 -
39 -
HIS_Kir4.1 WTHIS_Kir4.1 WT
HIS_Kir4.1 R/Q
HIS_Kir4.1 V/M
Fig.22. Caratterizzazione mediante western blot e immunofluorescenza delle linee
cellulari U251 overesprimenti i canali Kir4.1 WT e mutanti.

75
DISCUSSIONE
In questo studio abbiamo individuato per la prima volta mutazioni nel gene che codi-
fica per il canale del potassio Kir4.1 associate ad attacchi epilettici, DSA e deficit in-
tellettivo. Inoltre abbiamo dimostrato che queste mutazioni, che cambiano residui al-
tamente conservati nella struttura, alterano l’attività del canale sia attraverso un au-
mento di espressione della proteina in membrana sia attraverso un aumento di con-
duttanza del canale.
E’ noto che epilessia e DSA sono fortemente associati. La prevalenza degli attacchi
epilettici è infatti maggiore nei malati con DSA (5-6%) (Bryson et al., 1988; Hughes
and Melyn, 2005), rispetto alla popolazione generale (0.5-1%). La presenza di auti-
smo nei pazienti epilettici è approssimativamente del 32%, un valore circa cinquanta
volte superiore rispetto a quello riscontrato nella popolazione generale (Clarke et al.,
2005). Pertanto, è stato definito il fenotipo ―autismo-epilessia‖ e i meccanismi pato-
genetici comuni sono statti ipotizzati (Tuchman et al., 2009). Nel nostro studio sono
state identificate due famiglie portatrici delle nuove mutazioni in KCNJ10.
Una penetranza ridotta è stata osservata in tre portatori apparentemente asintomatici
delle mutazioni (B-I:1, B-II: 1 e B-III: 2, Fig.17A) ed in uno che era leggermente
colpito dalla mutazione (A-II:1,Fig.17A). La penetranza incompleta è possibile nelle
― canalopatie‖ (Camerino et al., 2008; Li and Lester, 2001), in particolare quando
colpiscono il sistema nervoso centrale (Gargus, 2009). Alternativamente, dovrebbe
essere considerata la presenza di mutazioni del gene KCNJ10 in uno scenaro multi-
genico più complesso (Abrahams and Geschwind, 2008; Toro et al., 2010). Tuttavia
non si può escludere che altri meccanismi, quali metilazione di un allele paterno o ef-
fetto genitore di origine o altri fattori genetici o epigenetici possano contribuire
all’espressione clinica delle variabili indotte dalle mutazioni del gene KCNJ10. Per
esempio, i fattori epigenetici sono considerati possibili cause della penetranza ridotta
delle mutazioni in KCNJ11 (Pinney et al., 2009). D’altra parte, i DSA non seguono le
classiche leggi di Mendel (Gupta and State, 2007) e il tasso di concordanza tra ge-
melli monozigoti può essere basso (40%) (Rosenberg et al., 2008). Le manifestazioni
psichiatriche osservate nel caso A-II:1 indicano la diagnosi della sindrome di Touret-
te e disordini ossessivi-compulsivi. I risultati suggeriscono che questi disordini psi-
chiatrici e DSA condividono delle similarità in fenomenologia, struttura del cervello,
caratteristiche famigliari e genetiche (Hollander et al., 2009). I genitori e i fratelli dei
pazienti autistci mostrano spesso manifestazioni di autismo subcliniche, il così detto
―fenotipo di ampio autismo‖(Pive net al., 1997), suggerendo che DSA è associato

76
debolmente con le varianti preesistenti nei genitori. La possibilità che KCNJ10 possa
essere non correlato con il fenotipo, non può essere esclusa. I DSA non erano fra le
caratteristiche cliniche associate con EAST/sindrome SeSAME e la mutazione auto-
somica recessiva del gene KCNJ10 e la disabilità intellettuale non erano obbligato-
riamente una manifestazione della malattia. Mutazioni bialleliche in EAST/sindrome
SeSAME compromettevano la funzione del canale attraverso differenti meccanismi
di loss-of-function, quali una riduzione dell’espressione in superficie del canale, ri-
duzione del tempo di apertura medio e uno spostamento della sensibilità al pH ad un
range alcalino (Reichold et al., 2010; Sala-Rabanal et al., 2010; Tange t al., 2010).
Questi difetti funzionali sono stati proposti per essere alla base delle manifestazioni
renali della malattia, includendo alcalosi metabolica ipokaliemica e attivazione del
sistema renina-angiotensina-aldosterone.
I nostri pazienti esibiscono un fenotipo neuropsichiatrico puro, senza manifestazioni
di danni renali o inibizione dell’udito. I meccanismi molecolari sono diversi, grazie
ai quali le nuove mutazioni del gene KCNJ10 contribuiscono alla malattia. In partico-
lare, la mutazione V84M che è localizzata in TM1, vicino alla superficie membra-
na/acqua, incrementava la conduttanza del canale producendo un effetto di gain-of-
function. Questo risultato strutturale e funzionale suggerisce che questa mutazione al-
tera il gating del canale o l’interazione tra il canale e i lipidi del doppio strato di
membrana. Interessante notare che la mutazione dei canali Kir3.2 in TM1 (N94H) ri-
sulta nei canali costitutivamente attivi e le correnti di base sono circa 30 volte più
ampie rispetto WT (Yi et al., 2001). D’altra parte, la mutazione R18Q, localizzata
nella parte N-terminale dei canali Kir4.1, genera una gain-of-function del canale pro-
vocando un aumento dell’ampiezze della corrente whole-cell, senza cambiare la con-
duttanza del canale, la probabilità di apertura e il tempo medio di apertura. Tutto ciò
suggerisce che la mutazione R18Q aumenta l’espressione della proteina sulla super-
ficie cellulare. Questa conclusione è supportata dai risultati di struttura funzione ot-
tenuti dai canali Kir3.2. La mutazione S9A in Kir3.2 incrementava l’ espressione in
superficie di circa 3 volte rispetto al WT in cellule HEK293 senza cambiamento della
conduttanza del canale (Chung et al., 2009). Gli allineamenti di sequenza hanno rile-
vato che sia la mutazione S9A in Kir3.2 e la mutazione R18Q in Kir4.1 risiedono in
un dominio simile in posizione N-terminale del canale e probabilmente condividono
meccanismi di azione comuni. Le mutazioni R18Q e V84M causavano un drammati-
co incremento della correnti istantanee del canale eteromerico Kir4.1/Kir5.1 e aboli-
vano quasi completamente la componente lenta della corrente, denotando una rispo-

77
sta più veloce e più efficiente dei canali mutati. Il fatto che la sensibilità al pHi dei
canali mutati Kir4.1/Kir5.1 è simile al WT dimostra che l’assemblaggio delle subuni-
tà mutate Kir4.1 ha luogo normalmente, indipendentemente dalla mutazione. Una se-
rie di evidenze convalidano che le mutazioni in KCNJ10 dovrebbero contribuire alla
disfunzione del cervello, suscettibilità agli attacchi e DSA. Kir4.1 svolge un ruolo
importante nel "buffering spaziale" della concentrazione extracellulare K+
([K+]o) che
è essenziale per il mantenimento dell'attività neuronale (Chever et al., 2010). Gli a-
strociti sono il 90% delle cellule che compongono il cervello umano e ogni astrocita
controlla l’attività di molte sinapsi (circa 140000 nell’ippocampo) (Benarroch, 2009).
La manifestazione sia di epilessia sia di ASD in pazienti portatori di mutazioni gain-
of-function del gene KCNJ10 suggerisce che la disfunzione del buffering di K+ a-
strociti-dipendenti da K+
dovrebbe essere un meccanismo comune che contribuisce
sia agli attacchi sia alle caratteristiche nei ASD. Inoltre le registrazioni su campioni
di pazienti con epilessie intrattabili hanno dimostrato una riduzione della conduttanza
dei canali Kir negli astrociti (Bordey and Sontheimer, 1998) e della rimozione del
potassio (Jauch et al., 2002). Topi knockout per Kir4.1 mostrano attacchi indotti da
stress (Djukic et al., 2007). Gli attacchi e il ritardo mentale sono parte dello spettro
clinico delle mutazioni loss-of-function in Kir4.1. (Bockenhauer et al., 2009; Scholl
et al., 2009). La relazione tra KCNJ10 e ASD sembra poco lineare. Tuttavia, uno stu-
dio recente di linkage su una famiglia Finlandese ha proposto KCNJ10 come gene
candidato per ASD (Kilpinen et al., 2009). KCNJ10 è stato considerato un possibile
responsabile sia di ASD e sia dei topi privi di MECP-2, in modelli animali della sin-
drome di Rett (Zhang et al., 2010). E’ stato proposto che la mutazione loss-of-
function dovrebbe favorire l’accumulo extracellulare di K+, contribuendo
all’ipereccitabilità neuronale e epilessia (Chever et al., 2010; Orkand et al.,1966).
Nelle cellule gliali knock-out per i canali Kir4.1, non sono state osservate delle va-
riazioni del potenziale di membrana durante l’incremento di [K+]o indotto da stimo-
lazione nervosa (Chever et al., 2010). Recentemente, è stato dimostrato che un epi-
sodio isolato di iperattività locale neuronale scatena un grande aumento di calcio ne-
gli astrociti vicini. Gli astrociti attivati rieccitano i neuroni generando la scarica sin-
crona tipica dell’attacco epilettico. L’analisi funzionale effettuata in questo studio
suggerisce, come un meccanismo patogenetico alternativo, che l’incremento e il più
veloce flusso di K+ negli astrociti attraverso i canali Kir4.1 durante l’intensa attività
neuronale, dovrebbe aumentare la depolarizzazione di membrana e aumentare la
concentrazione di calcio in queste cellule. Le elevate concentrazione di calcio negli

78
astrociti sono connesse con il rilascio di gliotrasmettitori, come glutammato e D-
serina che attivano la scarica dei neuroni, promuovendo un’ attività epilettica e la
sincronia neuronale locale (Angulo et al., 2004; Bezzi et al., 1998; Felli net al., 2004;
Mothet et al., 2005; Parpura et al., 1994; Pasti et al., 2001; Tian et al., 2005). Quindi
il ciclo eccitatorio ricorrente neurone-astrocita-neurone dovrebbe svilupparsi in un si-
to ristretto del cervello, come conseguenza della gain-of-function dei canali Kir4.1 e
contribuire allo sviluppo degli attacchi. L’alto livello di espressione dei canali Kir4.1
e Kir5.1, canali eteromerici presenti sui neuroni del Locus Coeruleus (LC)
(D’Adamoet al., 2010), suggerisce che queste mutazioni dovrebbero alterare il siste-
ma noradrenergico (NA) del cervello. (LC-NA) (Samuels and Szabadi, 2008). Il LC
produce e trasporta tutta la noradrenalina (NA) nella corteccia cerebrale e nell'ippo-
campo e la maggior parte delle fibre NA in altre parti del nevrasse, compreso il cer-
velletto. Una alterazione nello sviluppo di questa rete potrebbe causare comporta-
menti autistici negli uomo (MeHler and Purpura, 2009). Inoltre, l’ upregolazione di
Kir4.1 è stata individuata nei neuroni LC di topi knock-out per MECP-2. Questa so-
vraespressione di Kir4.1 altera la neuromodulazione dei neuroni LC, inducendo
comportamenti autistici che si verificano nella sindrome di Rett (Zhang et al., 2010).
Sia le caratteristiche autistiche sia quelle conoscitive possono essere legate a eventi
di sviluppo postnatali, dovuti a cambiamenti nella attività sinaptica coinvolti nei
meccanismi di plasticità sinaptica per l’apprendimento e la memoria (Hong et al.,
2005; Zoghbi, 2003). L’attività dei canali Kir4.1 mostra una regolazione durante lo
sviluppo, che correla sia con la differenziazione delle cellule e sia con la regolazione
durante lo sviluppo delle dinamiche extracellulare di K+
(Connors Et al., 1982; Ma-
cFarlane and Sontheimer, 2000; Neusch et al., 2001). Inoltre le sostanze neuroattive
rilasciate dagli astrociti regolano varie funzioni tra le quali l’eccitabilità neuronale
(D’Adamo et al., 2010; Tian et al., 2005), la trasmissione sinaptica eccitatoria e inibi-
toria e la plasticità (Beattie et al., 2002; Fiacco and McCarthy, 2004; Kang et al.,
1998; Pascual et al., 2005; Yang et al., 2003), come anche la sinaptogenesi e i colle-
gamenti neuronali (Collazos-Castro and NietoSampedro, 2001; Elmariah et al., 2005;
Fase net al., 2003; Ullian et al., 2004).
Quindi i possibili effetti delle mutazioni R18Q sull’aumento di espressione e V84M
sulla conduttanza del singolo canale, alterano i diversi meccanismi correlati
all’omeostasi del K+
, alla differenziazione cellulare e alla plasticità sinaptica.

79
I risultati suggeriscono che il meccanismo molecolare che contribuisce all’ auti-
smo/epilessia con disabilità mentale si correla a un incremento di espressione in su-
perficie o della conduttanza dei canali Kir4.1, o entrambi gli aumenti.
Gli astrociti dovrebbero rappresentare un target cruciale del controllo farmacologico
per alterazioni sinaptiche e scariche elettriche anormali.

80
BIBLIOGRAFIA
Abrahams, B.S., Geschwind, D.H., 2008. Advances in autism genetics: on the thre-
shold of a new neurobiology. Nat. Rev. Genet. 9, 341–355.
Alewine C, Kim BY, Hegde V, Welling PA. (2007). Lin-7 targets the Kir 2.3 chan-
nel on the basolateral membrane via a L27 domain interaction with CASK. Am J
Physiol Cell Physiol 293: C1733–C1741.
American Psychiatric Association (2000): Diagnostic and Statistical Manual of Men-
tal Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric
Press.
Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagel-
hus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. (2003). Delayed K+ clear-
ance associated with aquaporin-4 mislocalization: phenotypic defects in brains of -
syntrophin-null mice. Proc Natl Acad Sci USA 100: 13615–13620.
Anderson CM, Swanson RA. (2000). Astrocyte glutamate transport: review of prop-
erties, regulation, and physiological functions. Glia 32:1–14.
Ando M, Takeuchi S. (1999). Immunological identification of an inward rectifier K+
channel (Kir4.1) in the intermediate cell (melanocyte) of the cochlear stria vascularis
of gerbils and rats. Cell Tissue Res 298: 179–183.
Angulo, M.C., Kozlov, A.S., Charpak, S., Audinat, E., 2004. Glutamate released
from glial cells synchronizes neuronal activity in the hippocampus. J. Neurosci. 24,
6920–6927.
Auranen M, Vanhala R, Varilo T, Ayers K, Kempas E, Ylisaukko-Oja T, Sinsheimer
JS,Peltonen L, Jarvela I. (2002): A genomewide screen for autism-spectrum disord-
ers: evidence for a major susceptibility locus on chromosome 3q25-27. American
Journal of HumanGenetics 71:777-790.

81
Babenko A, Gonzalez G, Bryan J. (2000). Pharmaco-topology of sulfonylurea recep-
tors. Separate domains of the regulatory subunits of KATP channel isoforms are re-
quired for selective interaction with K+ channel openers. J Biol Chem 275: 717–720
Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M.
(1995):Autism as a strongly genetic disorder: evidence from a British twin study.
PsychologicalMedicine 25:63-77.
Beattie, E.C., Stellwagen, D., Morishita, W., Bresnahan, J.C., Ha, B.K., Von Za-
strow, M., et al., 2002. Control of synaptic strength by glial TNFalpha. Science 295,
2282–2285.
Beeler GW Jr, Reuter H. (1970). Voltage clamp experiments on ventricular myocar-
dial fibres. J Physiol 207: 165–190.
Benarroch, E.E., 2009. Astrocyte–neuron interactions: implications for epilepsy.
Neurology 73, 1323–1327
Bendahhou S, Donaldson MR, Plaster NM, Tristani-Firouzi M, Fu YH, Ptacek LJ.
(2003). Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil
syndrome. J Biol Chem 278: 51779–51785
Benfenati V, Caprini M, Nobile M, Rapisarda C, Ferroni S. (2006). Guanosine pro-
motes the up-regulation of inward rectifier potassium current mediated by Kir4.1 in
cultured rat cortical astrocytes. J Neurochem 98: 430–445.
Bezzi, P., Carmignoto, G., Pasti, L., Vesce, S., Rossi, D., Rizzini, B.L., et al., 1998.
Prostaglandinsstimulate calcium-dependent glutamate release in astrocytes. Nature
391, 281–285.
Bichet D, Haass FA, Jan LY. (2003). Merging functional studies with structures of
inward-rectifier K_ channels. Nat Rev Neurosci 4:957–967.

82
Blaustein MP, Juhaszova M, Golovina VA, Church PJ, Stanley EF. (2002).Na/Ca
exchanger and PMCA localization in neurons and astrocytes: functional implications.
Ann NY Acad Sci 976: 356–366.
Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, To-
bin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross
JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank
M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA,
Warth R, Sheridan E, Kleta R. (2009). Epilepsy, ataxia, sensorineural deafness, tubu-
lopathy, and KCNJ10 mutations. N Engl J Med 360: 1960–1970
Bond CT, Pessia M, Xia XM, Lagrutta A, Kavanaugh MP, Adelman JP. (1994).
Cloning and expression of a family of inward rectifier potassium channels. Receptors
Channels 2: 183–191
Bordey A, Sontheimer H. (1998). Properties of human glial cells associated with epi-
leptic seizure foci. Epilepsy Res 32: 286–303.
Brady PA, Alekseev AE, Aleksandrova LA, Gomez LA, Terzic A. (1996). A disrup-
ter of actin microfilaments impairs sulfonylurea-inhibitory gating of cardiac KATP
channels. Am J Physiol Heart Circ Physiol 271: H2710–H2716.
Bredt DS, Wang TL, Cohen NA, Guggino WB, Snyder SH. (1995). Cloning and ex-
pression of two brain-specific inwardly rectifying potassium channels. Proc Natl
Acad Sci USA 92: 6753–6757
Brian Deer, How the vaccine crisis was meant to make money, British Medical Jour-
nal, 2011; 342:c5258 doi: 10.1136/bmj.c5258 (Published 11 January 2011) BMJ
Brown DA, Gahwiler BH, Griffith WH, Halliwell JV. (1990). Membrane currents in
hippocampal neurons. Prog Brain Res 83: 141–160.
Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P & Litt M (1994).
Episodic ataxia/myokymia syndrome is associated with point mutations in the human
potassium channel gene, KCNA1. Nat Genet 8, 136-40.

83
Bryson SE, Clark BS, Smith IM. (1988): First report of a Canadian epidemiological
study of autistic syndromes. Journal of Child Psychology and Psychiatry 29:433-445.
Buono RJ, Lohoff FW, Sander T, Sperling MR, O’Connor MJ, Dlugos DJ, Ryan SG,
Golden GT, Zhao H, Scattergood TM, Berrettini WH, Ferraro TN. (2004). Associa-
tion between variation in the human KCNJ10 potassium ion channel gene and sei-
zure susceptibility. Epilepsy Res 58: 175–183.
Camerino, D.C., Desaphy, J.F., Tricarico, D., Pierno, S., Liantonio, A., 2008. Thera-
peutic approaches to ion channel diseases. Adv. Genet. 64, 81–145.
Casamassima M, D’Adamo MC, Pessia M, Tucker SJ. (2003). Identification of a he-
teromeric interaction that influences the rectification, gating, and pH sensitivity of
Kir4.1/Kir5 1 potassium channels. J Biol Chem 278: 43533–43540.
Chan KW, Sui JL, Vivaudou M, Logothetis DE. (1997). Specific regions of hetero-
meric subunits involved in enhancement of G proteingated K+ channel activity. J Bi-
ol Chem 272: 6548–6555
Charles JM, Carpenter LA, Jenner W, Nicholas JS. (2008). Recent advances in aut-
ism spectrum disorders.. Int J Psychiatry Med. 38: 133-140..
Chever, O., Djukic, B., McCarthy, K.D., Amzica, F., 2010. Implication of k(ir)4.1
channel inexcess potassium clearance: an in vivo study on anesthetized glial-
conditional k(ir)4.1 knock-out mice. J. Neurosci. 30, 15769–15777.
Chung, H.J., Qian, X., Ehlers, M., Jan, Y.N., Jan, L.Y., 2009. Neuronal activity regu-
latesphosphorylation-dependent surface delivery of G protein-activated inwardly rec-
tifying potassium channels. Proc. Natl. Acad. Sci. U.S.A. 106, 629–634.
Ciani S, Krasne S, Hagiwara S. (1980). A model for the effects of potential and ex-
ternal K+ concentration on the Cs+ blocking of inward rectification. Biophys J 30:
199–204

84
Ciani S, Krasne S, Miyazaki S, Hagiwara S. (1978). A model for anomalous rectifi-
cation: electrochemical-potential-dependent gating of membrane channels. J Membr
Biol 44: 103–134.
Clarke DF, Roberts W, Daraksan M, Dupuis A, McCabe J, Wood H, Snead OC 3rd,
Weiss SK. (2005): . The prevalence of autistic spectrum disorder in children sur-
veyed in a tertiary care epilepsy clinic. Epilepsia 46:1970-1977.
Cole K.S. and Curtis H.J. (1939) Electrical impedance of the squid giant axon during
activity. J.Gen. Physiol. 22,649-70.
Collazos-Castro, J.E., Nieto-Sampedro, M., 2001. Developmental and reactive
growth of dentate gyrus afferents: cellular and molecular interactions. Restor. Neu-
rol. Neurosci.19, 169–187.
Collins AL, Ma D, Whitehead PL, Martin ER, Wright HH, Abramson RK, Hussman
JP,Haines JL, Cuccaro ML, Gilbert JR, Pericak-Vance MA. (2006): Investigation of
autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics
7:167-174.
Connors, B.W., Ransom, B.R., Kunis, D.M., Gutnick, M.J., 1982. Activity-
dependent K +accumulation in the developing rat optic nerve. Science 216, 1341–
1343.
Curran ME, Splawski I, Timoth KW, Vincent GM, Green ED, Keating MT (1995). A
molecular basis for cardiac arrhythmia: HERG mutation cause long QT syndrome.
Cell 80, 795-03.
D'Adamo, M.C., Liu, Z., Adelman, J.P., Maylie, J., Pessia, M., 1998. Episodic ataxia
type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of
the channel. EMBO J. 17, 1200–1207.
D'Adamo, M.C., Shang, L., Imbrici, P., Brown, S.D., Pessia, M., Tucker, S.J., 2010.
Genetic inactivation of KCNJ16 identifies Kir5.1 as an important determinant of neu-
ronal PCO2/pH sensitivity. J. Biol. Chem. Nov 3 [Epub ahead of print].

85
Dalloz C, Sarig R, Fort P, Yaffe D, Bordais A, Pannicke T, Grosche J, Mornet D,
Reichenbach A, Sahel J, Nudel U, Rendon A. (2003). Targeted inactivation of dy-
strophin gene product Dp71: phenotypic impact in mouse retina. Hum Mol Genet 12:
1543–1554.
DeHart GW, Jin T, McCloskey DE, Pegg AE, Sheppard D. (2008). The 9 1 integrin
enhances cell migration by polyamine-mediated mod-ulation of an inward-rectifier
potassium channel. Proc Natl Acad Sci USA 105: 7188–7193.
Delahanty RJ, Kang JQ, Brune CW, Kistner EO, Courchesne E, Cox NJ, et al.
(2011):Maternal transmission of a rare GABRB3 signal peptide variant is associated
with autism.Molecular Psychiatry 16:86-96.
Derst C, Wischmeyer E, Preisig-Muller R, Spauschus A, Konrad M, Hensen P, Jeck
N, Seyberth HW, Daut J, Karschin A. (1998). A hyperprostaglandin E syndrome mu-
tation in Kir1.1 (renal outer medullary potassium) channels reveals a crucial residue
for channel function in Kir1 3 channels. J Biol Chem 273: 23884–23891
Dibaj P, Kaiser M, Hirrlinger J, Kirchhoff F, Neusch C. (2007). Kir4.1 channels re-
gulate swelling of astroglial processes in experimental spinal cord edema. J Neuro-
chem 103: 2620–2628.
Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. (2007). Conditional
knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium
and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27:
11354–11365.
Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. (2007). Conditional
knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium
and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27:
11354–11365

86
Doring F, Derst C, Wischmeyer E, Karschin C, Schneggenburger R, Daut J, Kar-
schin A. (1998). The epithelial inward rectifier channel Kir7.1 displays unusual K+
permeation properties. J Neurosci 18: 8625–8636.
Du X, Zhang H, Lopes C, Mirshahi T, Rohacs T, Logothetis DE. (2004). Characte-
ristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of
Kir channels by diverse modulators. J Biol Chem 279: 37271–37281.
Edgar, R.C., 2004. MUSCLE: multiple sequence alignment with high accuracy and
high throughput. Nucleic Acids Res. 32, 1792–1797.
Elmariah, S.B., Oh, E.J., Hughes, E.G., Balice-Gordon, R.J., 2005. Astrocytes regu-
late inhibitory synapse formation via Trk-mediated modulation of postsynaptic GA-
BAA receptors. J. Neurosci. 25, 3638–3650.
Fakler B, Bond CT, Adelman JP, Ruppersberg JP. (1996): Heterooligomeric assem-
bly of inward-rectifier K+ channels from subunits of different subfamilies: Kir2.1
(IRK1) and Kir4.1(BIR10). Pflugers Archives 433:77-83.
Fakler B, Brandle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP.
(1995). Strong voltage-dependent inward rectification of inward rectifier K+ chan-
nels is caused by intracellular spermine. Cell 80: 149–154
Fan Z, Makielski JC. (1997). Anionic phospholipids activate ATP-sensitive potas-
sium channels. J Biol Chem 272: 5388–5395.
Fasen, K., Elger, C.E., Lie, A.A., 2003. Distribution of alpha and beta integrin sub-
units in the adult rat hippocampus after pilocarpine-induced neuronal cell loss, axon-
al reorganization and reactive astrogliosis. Acta Neuropathol. (Berl.) 106,319–322.
Fellin, T., Pascual, O., Gobbo, S., Pozzan, T., Haydon, P.G., Carmignoto, G., 2004.
Neuronal synchrony mediated by astrocytic glutamate through activation of extrasy-
naptic NMDA receptors. Neuron 43, 729–743.

87
Ferrando-Miguel R, Cheon MS, Lubec G. (2004). Protein levels of genes encoded on
chromosome 21 in fetal Down Syndrome brain (Part V): overexpression of phospha-
tidyl-inositol-glycan class P protein (DSCR5). Amino Acids 26: 255–261.
Ferraro TN, Golden GT, Smith GG, Martin JF, Lohoff FW, Gieringer TA, Zamboni
D, Schwebel CL, Press DM, Kratzer SO, Zhao H, Berrettini WH, Buono RJ. (2004):
Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination
of Kcnj10 as a causative gene. Mammalian Genome 15:239-251.
Fiacco, T.A., McCarthy, K.D., 2004. Intracellular astrocyte calcium waves in situ in-
crease the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neu-
rons.J. Neurosci. 24, 722–732.
Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. (1994). Spermine and
spermidine as gating molecules for inward rectifier K+ channels. Science 266: 1068–
1072
Fujita A, Horio Y, Higashi K, Mouri T, Hata F, Takeguchi N, Kurachi Y. (2002).
Specific localization of an inwardly rectifying K+ channel, Kir4.1, at the apical
membrane of rat gastric parietal cells: its possible involvement in K recycling for the
H -K -pump. J Physiol 540: 85–92.
Funke L, Dakoji S, Bredt DS. (2005). Membrane-associated guanylate kinases regu-
late adhesion and plasticity at cell junctions. Annu Rev Biochem 74: 219–245.
Furutani K, Ohno Y, Inanobe A, Hibino H, Kurachi Y. (2009). Mutational and in si-
lico analyses for antidepressant block of astroglial inward-rectifier kir4.1 channel.
Mol Pharmacol 75: 1287–1295
Gabriel S, Kivi A, Kovacs R, Lehmann TN, Lanksch WR, Meencke HJ, Heinemann
U. (1998). Effects of barium on stimulus-induced changes in [K ]o and field poten-
tials in dentate gyrus and area CA1 of human epileptic hippocampus. Neurosci Lett
249:91–94.

88
Gahwiler BH, Brown DA. (1985). GABAB-receptor-activated K+ current in voltage-
clamped CA3 pyramidal cells in hippocampal cultures. Proc Natl Acad Sci USA 82:
1558–1562.
Ganz ML. (2007): The lifetime distribution of the incremental societal costs of aut-
ism. Archives of Pediatrics and Adolescent Medicine 161:343-349.
Gargus, J.J., 2009. Genetic calcium signaling abnormalities in the central nervous
system: seizures, migraine, and autism. Ann. N.Y. Acad. Sci. 1151, 133–156.
Glowatzki E, Fakler G, Brandle U, Rexhausen U, Zenner HP, Ruppersberg JP, Fak-
ler B. (1995). Subunit-dependent assembly of inward-rectifier K_ channels. Proc Bi-
ol Sci 261: 251–261.
Gómez-Gonzalo, M., Losi, G., Chiavegato, A., Zonta, M., Cammarota, M., Brondi,
M., et al.,2010. An excitatory loop with astrocytes contributes to drive neurons to
seizure threshold. PLoS Biol. 8,e1000352.
Gosset P, Ghezala GA, Korn B, Yaspo ML, Poutska A, Lehrach H, Sinet PM, Creau
N. (1997) A new inward rectifier potassium channel gene (KCNJ15) localized on
chromosome 21 in the Down syndrome chromosome region 1 (DCR1). Genomics
44: 237–241.
Gupta, A.R., State, M.W., 2007. Recent advances in the genetics of autism. Biol.
Psychiatry 61, 429 437.
Hagiwara S, Takahashi K. (1974). The anomalous rectification and cation selectivity
of the membrane of a starfish egg cell. J Membr Biol 18: 61–80.
He C, Zhang H, Mirshahi T, Logothetis DE. (1999). Identification of a potassium
channel site that interacts with G protein βγ subunits to mediate agonist-induced sig-
naling. J Biol Chem 274: 12517–12524
Hebert SC, Desir G, Giebisch G, Wang W. (2005). Molecular diversity and regula-
tion of renal potassium channels. Physiol Rev 85: 319–371.

89
Heginbotham L, Lu Z, Abramson T, MacKinnon R. (1994). Mutations in the K+
channel signature sequence. Biophys J 66: 1061–1067.
Helbig, I., Scheffer, I.E., Mulley, J.C., Berkovic, S.F., 2008. Navigating the channels
and beyond: unravelling the genetics of the epilepsies. Lancet Neurol. 7, 231–245.
Hess, B., Kutzner, C., van der Spoel, D., Lindahl, E., 2008. GROMACS 4: algo-
rithms for highly efficient, load-balanced, and scalable molecular simulation. J.
Chem. Theor. Comp. 4, 435–447.
Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. (2004). Differential assembly of
inwardly rectifying K channel subunits, Kir4.1 and Kir5 1, in brain astrocytes. J Biol
Chem 279: 44065–44073.
Hibino H, Higashi-Shingai K, Fujita A, Iwai K, Ishii M, Kurachi Y. (2004). Expres-
sion of an inwardly rectifying K+ channel, Kir5.1, in specific types of fibrocytes in
the cochlear lateral wall suggests its functional importance in the establishment of
endocochlear potential. Eur J Neurosci 19: 76–84.
Hibino H, Horio Y, Fujita A, Inanobe A, Doi K, Gotow T, Uchiyama Y, Kubo T,
Kurachi Y. (1999). Expression of an inwardly rectifying K+ channel, Kir4.1, in satel-
lite cells of rat cochlear ganglia. Am J Physiol Cell Physiol 277: C638–C644.
Hibino H, Horio Y, Inanobe A, Doi K, Ito M, Yamada M, Gotow T, Uchiyama Y,
Kawamura M, Kubo T, Kurachi Y. (1997). An ATPdependent inwardly rectifying
potassium channel, KAB-2 (Kir4.1), in cochlear stria vascularis of inner ear: its spe-
cific subcellular localization and correlation with the formation of endocochlear po-
tential. J Neurosci 17: 4711–4721.
Hibino H, Kurachi Y. (2006). Molecular and physiological bases of the K+ circula-
tion in the mammalian inner ear. Physiology 21: 336–345
Hibino H, Kurachi Y. (2007). Distinct detergent-resistant membrane microdomains
(lipid rafts) respectively harvest K+ and water transport systems in brain astroglia.

90
Eur J Neurosci 26: 2539–2555
Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. (2001).
An inwardly rectifying K+ channel, Kir4.1, expressed in astrocytes surrounds syn-
apses and blood vessels in brain. Am J Physiol Cell Physiol 281: C922–C931
Higashi, K., Fujita, A., Inanobe, A., Tanemoto, M., Doi, K., Kubo, T., et al., 2001.
An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds syn-
apses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 281, C922–C931.
Hilgemann DW, Ball R. (1996). Regulation of cardiac Na+,Ca2+ exchange and
KATP potassium channels by PIP2. Science 273: 956–959
Hilgemann DW, Feng S, Nasuhoglu C. (2001). The complex and intriguing lives of
PIP2 with ion channels and transporters. Sci STKE 2001:RE19.
Hille B (2001). Ionic channels of excitable membranes, 3nd edn. Sinauer, Sunder-
land, Mass.
Hille B, Schwarz W. (1978). Potassium channels as multi-ion single-file pores. J Gen
Physiol 72: 409–442
Hinterkeuser S, Schroder W, Hager G, Seifert G, Blumcke I, Elger CE, Schramm J,
Steinhauser C. (2000). Astrocytes in the hippocampus of patients with temporal lobe
epilepsy display changes in potassium conductances. Eur J Neurosci 12: 2087–2096.
Ho IH, Murrell-Lagnado RD. (1999). Molecular determinants for sodiumdependent
activation of G protein-gated K+ channels. J Biol Chem 274: 8639–8648
Hodgkin AL and Katz B. (1949) The effect of sodium ions on the electrical activity
of the giant axon of the squid. J.Physiol. 108, 37-77.
Hodgkin AL, Huxley AF (1952) Propagation of electrical signals along giant nerve
fibers. Proc R Soc Lond B Biol Sci. Oct 16;140(899):177-83.

91
Honda et al., 2005
Horio Y, Hibino H, Inanobe A, Yamada M, Ishii M, Tada Y, Satoh E, Hata Y, Takai
Y, Kurachi Y. (1997). Clustering and enhanced activity of an inwardly rectifying po-
tassium channel, Kir4.1, by an anchoring protein, PSD-95/SAP90. J Biol Chem 272:
12885–12888.
Huang C, Sindic A, Hill CE, Hujer KM, Chan KW, Sassen M, Wu Z, Kurachi Y,
Nielsen S, Romero MF, Miller RT. (2007).Interaction of the Ca2 -sensing receptor
with the inwardly rectifying potassium channels Kir4.1 and Kir4.2 results in inhibi-
tion of channel function. Am J Physiol Renal Physiol 292: F1073–F1081
Huang CL, Feng S, Hilgemann DW. (1998). Direct activation of inward rectifier po-
tassium channels by PIP2 and its stabilization by Gβγ. Nature 391: 803–806
Hughes JR, Melyn M. (2005): EEG and seizures in autistic children and adolescents:
further findings with therapeutic implications. Clinical EEG and Neuroscience
36:15-20.
Immunization Safety Review Committee. 2004. Immunization safety review: vac-
cines and autism. National Academies Press. Washington, DC, USA. pag 214.
Inagaki N, Gonoi T, Clement JP IV, Namba N, Inazawa J, Gonzalez G, Aguilar-
Bryan L, Seino S, Bryan J (1995). Reconstitution of IKATP: an inward rectifier sub-
unit plus the sulfonylurea receptor. Science 270, 1166-69
Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S.
(1996). A family of sulfonylurea receptors determines the pharmacological proper-
ties of ATP-sensitive K+ channels. Neuron 16: 1011–1017.
Inanobe A, Yoshimoto Y, Horio Y, Morishige KI, Hibino H, Matsumoto S, Tokuna-
ga Y, Maeda T, Hata Y, Takai Y, Kurachi Y. (1999). Characterization of G-protein-
gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the
substantia nigra. J Neurosci 19: 1006–1017

92
Ishihara K, Mitsuiye T, Noma A, Takano M. (1989). The Mg2+ block and intrinsic
gating underlying inward rectification of the K+ current in guinea-pig cardiac myo-
cytes. J Physiol 419: 297–320
Ishii M, Fujita A, Iwai K, Kusaka S, Higashi K, Inanobe A, Hibino H, Kurachi Y.
(2003). Differential expression and distribution of Kir5.1 and Kir4 1 inwardly recti-
fying K channels in retina. Am J Physiol Cell Physiol 285: C260–C267.
Ishii M, Horio Y, Tada Y, Hibino H, Inanobe A, Ito M, Yamada M, Gotow T,
Uchiyama Y, Kurachi Y. (1997). Expression and clustered distribution of an inward-
ly rectifying potassium channel, KAB-2/Kir4.1, on mammalian retinal Muller cell
membrane: their regulation by insulin and laminin signals. J Neurosci 17: 7725–
7735.
Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonosaki A, To-
moike H, Kurachi Y. (1996). Immunolocalization of an inwardly rectifying K+
channel, KAB-2 (Kir4.1), in the basolateral membrane of renal distal tubular epithe-
lia. FEBS Lett 388: 11–15.
Jauch R, Windmüller O, Lehmann TN, Heinemann U, Gabriel S. (2002): Effects
of barium, furosemide, ouabaine and 4,4'-diisothiocyanatostilbene-2,2'-disulfonic ac-
id (DIDS) on ionophoretically-induced changes in extracellular potassium concentra-
tion in hippocampal slices from rats and from patients with epilepsy. Brain Research
925:18-27.
Jauch, R., Windmüller, O., Lehmann, T.N., Heinemann, U., Gabriel, S., 2002. Ef-
fects of barium, furosemide, ouabaine and 4,4'-diisothiocyanatostilbene-2,2'-
disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potas-
sium concentration in hippocampal slices from rats and from patients with epilep-
sy.Brain Res. 925, 18–27.
Kaiser M, Maletzki I, Hulsmann S, Holtmann B, Schulz-Schaeffer W, Kirchhoff F,
Bahr M, Neusch C. (2006). Progressive loss of a glial potassium channel (KCNJ10)
in the spinal cord of the SOD1 (G93A) transgenic mouse model of amyotrophic lat-

93
eral sclerosis. J Neurochem 99: 900–912
Kaiser M, Maletzki I, Hulsmann S, Holtmann B, Schulz-Schaeffer W, Kirchhoff F,
Bahr M, Neusch C. (2006). Progressive loss of a glial potassium channel (KCNJ10)
in the spinal cord of the SOD1 (G93A) transgenic mouse model of amyotrophic lat-
eral sclerosis. J Neurochem 99: 900–912
Kalsi AS, Greenwood K, Wilkin G, Butt AM. (2004). Kir4.1 expression by astro-
cytes and oligodendrocytes in CNS white matter: a developmental study in the rat
optic nerve. J Anat 204: 475–485.
Kang, J., Jiang, L., Goldman, S.A., Nedergaard, M., 1998. Astrocyte-mediated po-
tentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692.
Kaufhold MA, Krabbenhoft A, Song P, Engelhardt R, Riederer B, Fahrmann M,
Klocker N, Beil W, Manns M, Hagen SJ, Seidler U. (2008). Localization, trafficking,
and significance for acid secretion of parietal cell Kir4.1 and KCNQ1 K channels.
Gastroenterology 134: 1058–1069.
Kilpinen H, Ylisaukko-oja T, Rehnström K, Gaál E, Turunen JA, Kempas E, von
Wendt L,Varilo T, Peltonen L. (2009): Linkage and linkage disequilibrium scan for
autism loci in an extended pedigree from Finland. Human Molecular Genetics
18:2912-2921.
Kilpinen, H., Ylisaukko-oja, T., Rehnström, K., Gaál, E., Turunen, J.A., Kempas, E.,
et al.,2009.Linkage and linkage disequilibrium scan for autism loci in an extended
pedigree from Finland. Hum. Mol. Genet. 18, 2912–2921.
Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. (2000). Genet-
ic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice:
phenotypic impact in retina. J Neurosci 20: 5733–5740
Kohda Y, Ding W, Phan E, Housini I, Wang J, Star RA, Huang CL. (1998). Locali-
zation of the ROMK potassium channel to the apical membrane of distal nephron in

94
rat kidney. Kidney Int 54: 1214–1223.
Konig S, Hinard V, Arnaudeau S, Holzer N, Potter G, Bader CR, Bernheim L.
(2004). Membrane hyperpolarization triggers myogenin and myocyte enhancer fac-
tor-2 expression during human myoblast differentiation. J Biol Chem 279: 28187–
28196.
Konstas AA, Korbmacher C, Tucker SJ. (2003). Identification of domains that con-
trol the heteromeric assembly of Kir5.1/Kir4.0 potassium channels. Am J Physiol
Cell Physiol 284: C910–C917.
Koyrakh L, Lujan R, Colon J, Karschin C, Kurachi Y, Karschin A, Wickman K.
(200). Molecular and cellular diversity of neuronal Gprotein-gated potassium chan-
nels. J Neurosci 25: 11468–11478.
Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE. (1998). A
novel inward rectifier K+ channel with unique pore properties. Neuron 20: 995–1005
Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Rei-
chenbach A, Skatchkov SN, Eaton MJ. (2007). Downregulation of Kir4.1 inward
rectifying potassium channel subunits by RNAi impairs potassium transfer and glu-
tamate uptake by cultured cortical astrocytes. Glia 55: 274–281.
Kucheryavykh YV, Pearson WL, Kurata HT, Eaton MJ, Skatchkov SN, Nichols CG.
(2007). Polyamine permeation and rectification of Kir4.1 channels. Channels 1: 172–
178
Kuffler SW, Nicholls JG. (1966). The physiology of neuroglial cells. Ergeb Physiol
57: 1–90.
Kurachi Y. (1985). Voltage-dependent activation of the inward-rectifier potassium
channel in the ventricular cell membrane of guinea-pig heart. J Physiol 366: 365–385

95
Kurachi Y. (1997). Clustering and enhanced activity of an inwardly rectifying potas-
sium channel, Kir4.1, by an anchoring protein, PSD-95/SAP90. J Biol Chem 272:
12885–12888.
Lacey MG, Mercuri NB, North RA. (1988). On the potassium conductance increase
activated by GABAB and dopamine D2 receptors in rat substantia nigra neurones. J
Physiol 401: 437–453.
Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Van-
dewalle A, Teulon J, Paulais M. (2005). Kir4.1/Kir5.1 channel forms the major K
channel in the basolateral membrane of mouse renal collecting duct principal cells.
Am J Physiol Renal Physiol 294: F1398–F1407
Laskowski, R.A., MacArthur, M.W., Moss, D.S., Thornton, J.M., 1993. PRO-
CHECK—aprogram to check the stereochemical quality of protein structures. J. App.
Cryst. 26, 283–291.Li, M., Lester, H.A.,2001. Ion channel diseases of the central
nervous system. CNS DrugRev. 7, 214–240.
Levy SE, Mandell DS, Schultz RT. (2009): Autism. Lancet 374 :1627-
1638.Stefanatos GA. (2008): Regression in autistic spectrum disorders. Neuropsy-
chology Review18:305-319.
Lewis DL, Ikeda SR, Aryee D, Joho RH. (1991). Expression of an inwardly rectify-
ing K+ channel from rat basophilic leukemia cell mRNA in Xenopus oocytes. FEBS
Lett 290: 17–21.
Li L, Head V, Timpe LC. (2001). Identification of an inward rectifier potassium
channel gene expressed in mouse cortical astrocytes. Glia 33: 57–71
Liao YJ, Jan YN, Jan LY. (1996). Heteromultimerization of G-proteingated inwardly
rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in
weaver brain. J Neurosci 16: 7137–7150
Lichter-Konecki U, Mangin JM, Gordish-Dressman H, Hoffman EP, Gallo V.
(2008). Gene expression profiling of astrocytes from hyperammonemic mice reveals

96
altered pathways for water and potassium homeostasis in vivo. Glia 56: 365–377.
Lin D, Sterling H, Lerea KM, Giebisch G, Wang WH. (2002). Protein kinase C
(PKC)-induced phosphorylation of ROMK1 is essential for the surface expression of
ROMK1 channels. J Biol Chem 277: 44278–44284
Livak, K.J., Schmittgen, T.D., 2001. Analysis of relative gene expression data using
real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408.
Lopatin AN, Makhina EN, Nichols CG. (1994). Potassium channel block by cytop-
lasmic polyamines as the mechanism of intrinsic rectification. Nature 372: 366–369
Lopatin AN, Makhina EN, Nichols CG. (1995). The mechanism of inward rectifica-
tion of potassium channels: ―long-pore plugging‖ by cytoplasmic polyamines. J Gen
Physiol 106: 923–955
Lopatin AN, Nichols CG. (1996). [K+] dependence of open-channel conductance in
cloned inward rectifier potassium channels (IRK1, Kir2.1). Biophys J 71: 682–694
Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE. (2002). Alterations in
conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 34: 933–
944.
Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A,
Teulon J. (2002). An inward rectifier K+ channel at the basolateral membrane of the
mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J
Physiol 538: 391–404
Lu Z, MacKinnon R. (1994). Electrostatic tuning of Mg2+ affinity in an inward-
rectifier K+ channel. Nature 371: 243–246
MacFarlane, S.N., Sontheimer, H., 2000. Changes in ion channel expression accom-
pany cell cycle progression of spinal cord astrocytes. Glia 30, 39–48.

97
Madsen KM, Hviid A, Vestergaard M, Schendel D, Wohlfahrt J, Thorsen P, Olsen J,
Melbye M. (novembre 2002). A population-based study of measles, mumps, and ru-
bella vaccination and autism. N Engl J Med. . 347: 1477-82.
Marcus DC, Wu T, Wangemann P, Kofuji P. (2002). KCNJ10 (Kir4.1) potassium
channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol 282:
C403–C407.
Marcus DC. (1984). Characterization of potassium permeability of cochlear duct by
perilymphatic perfusion of barium. Am J Physiol Cell Physiol 247: C240–C246.
Matson, J.L., Shoemaker, M., 2009. Intellectual disability and its relationship to aut-
ism spectrum disorders. Res. Dev. Disabil. 30, 1107–1114.
Matsuda H, Saigusa A, Irisawa H. (1987). Ohmic conductance through the inwardly
rectifying K channel and blocking by internal Mg2+. Nature 325: 156–159
Matsuda H. (1988). Open-state substructure of inwardly rectifying potassium chan-
nels revealed by magnesium block in guinea-pig heart cells. J Physiol 397: 237–258.
McAllister RE, Noble D. (1966). The time and voltage dependence of the slow out-
ward current in cardiac Purkinje fibres. J Physiol 186:632–662.
McKinney LC, Gallin EK. (1988). Inwardly rectifying whole-cell and single-channel
K currents in the murine macrophage cell line J774.1. J Membr Biol 103: 41–53.
McNicholas CM, MacGregor GG, Islas LD, Yang Y, Hebert SC, Giebisch G. (1998).
pH-dependent modulation of the cloned renal K+ channel, ROMK. Am J Physiol
Renal Physiol 275: F972–F981
Mehler, M.F., Purpura, D.P., 2009. Autism, fever, epigenetics and the locus coeru-
leus.Brain Res. Rev. 59, 388–392.
Misgeld U, Muller W, Brunner H. (1999). Effects of (-)baclofen on inhibitory neu-
rons in the guinea pig hippocampal slice. Pflu¨ gers Arch 414: 139–144

98
Mothet, J.P., Pollegioni, L., Ouanounou, G., Martineau, M., Fossier, P., Baux, G.,
2005.Glutamate receptor activation triggers a calcium-dependent and SNARE pro-
tein-dependent release of the gliotransmitter D-serine. Proc. Natl. Acad. Sci. U.S.A.
102,5606–5611.
Muhle R, Trentacoste SV, Rapin I (2004). The genetics of autism. Pediatrics 113:
472-486.
Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Otter-
sen OP. (1999). Immunogold evidence suggests that coupling of K+ siphoning and
water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1
and AQP4 in specific membrane domains. Glia 26: 47–54.
Nagelhus EA, Mathiisen TM, Ottersen OP. (2004). Aquaporin-4 in the central nerv-
ous system: cellular and subcellular distribution and coexpression with KIR4.1. Neu-
roscience 129: 905–913.
Neher E. and Sakmann B. (1976) Single-channel currents recorded from membrane
of denervated frog muscle fibres. Nature , 260, 779-802.
Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Hand-
schuh M, Bahr M, Richter DW, Kirchhoff F,Hulsmann S. (2006). Lack of the Kir4.1
channel subunit abolishes K+ buffering properties of astrocytes in the ventral respira-
tory group:impact on extracellular K regulation. J Neurophysiol 95: 1843–1852
Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. (2001). Kir4.1 potassium
channel subunit is crucial for oligodendrocyte development and in vivo myelination.
J Neurosci 21: 5429–5438.
Newman EA. (1984). Regional specialization of retinal glial cell membrane. Nature
309: 155–157.
Nichols, C.G. and Lopatin, A.N. (1997) Ann. Rev. Physiol. 59, 171-91.

99
Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP.
(1997). Specialized membrane domains for water transport in glial cells: high-
resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci 17:
171–180
Nin F, Hibino H, Doi K, Suzuki T, Hisa Y, Kurachi Y. (2008). The endocochlear po-
tential depends on two K diffusion potentials and an electrical barrier in the stria
vascularis of the inner ear. Proc Natl Acad Sci USA 105: 1751–1756
Nishida M, MacKinnon R. (2002). Structural basis of inward rectification: cytoplas-
mic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell 111:
957–965
Nishida, M., Cadene, M., Chait, B.T., MacKinnon, R., 2007. Crystal structure of a
Kir3.1-prokaryotic Kir channel chimera. EMBO J. 26, 4005–4015.
Noble D. (1965). Electrical properties of cardiac muscle attributable to inward going
(anomalous) rectification. J Cell Comp Physiol 66: 127–136.
North RA, Williams JT, Surprenant A, Christie MJ. (1987).Mu and delta receptors
belong to a family of receptors that are coupled to potassium channels. Proc Natl
Acad Sci USA 84: 5487–5491.
Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y.(2007). Inhibition of astroglial
Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178: 44–51.
Olsen ML, Higashimori H, Campbell SL, Hablitz JJ, Sontheimer H. (2006). Func-
tional expression of Kir4.1 channels in spinal cord astrocytes. Glia 53: 516–528.
Orkand RK, Nicholls JG, Kuffler SW. (1996). Effect of nerve impulses on the mem-
brane potential of glial cells in the central nervous system of amphibia. J Neurophy-
siol 29: 788–806.

100
Orkand, R.K., Nicholls, J.G., Kuffler, S.W., 1966. Effect of nerve impulses on the
membrane potential of glial cells in the central nervous system of amphibia.J. Neu-
rophysiol. 29, 788–806.
Padmawar P, Yao X, Bloch O, Manley GT, Verkman AS. (2005). K+ waves in brain
cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nat Me-
thods 2: 825–827.
Pannicke T, Uckermann O, Iandiev I, Biedermann B, Wiedemann P, Perlman I, Rei-
chenbach A, Bringmann A. (2005). Altered membrane physiology in Muller glial
cells after transient ischemia of the rat retina. Glia 50: 1–11.
Parpura, V., Basarsky, T.A., Liu, F., Jeftinija, K., Jeftinija, S., Haydon, P.G., 1994.
Glutamate mediated astrocyte–neuron signalling. Nature 369, 744–747.
Pascual, O., Casper, K.B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J.Y., et
al., 2005.Astrocytic purinergic signaling coordinates synaptic networks. Science 310,
113–116.
Pasti, L., Zonta, M., Pozzan, T., Vicini, S., Carmignoto, G., 2001. Cytosolic calcium
oscillations in astrocytes may regulate exocytotic release of glutamate. J. Neurosci.
21, 477–484.
Pearson WL, Dourado M, Schreiber M, Salkoff L, Nichols CG. (1999). Expression
of a functional Kir4 family inward rectifier K channel from a gene cloned from
mouse liver. J Physiol 514: 639–653
Pessia M, Imbrici P, D’Adamo MC, Salvatore L, Tucker SJ. (2001). Differential pH
sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by hetero-
polymerisation with Kir5.1. J Physiol 532: 359–367.
Pessia M, Tucker SJ, Lee K, Bond CT & Adelman JP (1996). Subunit positional ef-
fects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J 15,
2980-7.

101
Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. (1996). Subunit positional ef-
fects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J 15:
2980–2987.
Pessia, M., Imbrici, P., D'Adamo, M.C., Salvatore, L., Tucker, S.J., 2001. Differen-
tial pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by
heteropolymerisation with Kir5.1. J. Physiol. 532, 359–367.
Pessia,M.,Tucker,S.J.,Lee,K.,Bond,C.T.,Adelman,J.P.,1996.Subunitpositionaleffects
revealed by novel heteromeric inwardly rectifying K+channels. EMBOJ. 15, 2980–
2987.
Piccolino M. (1997) Luigi galvani and animal electricity:Two centuries anfter the
fundation of electrophysiology. Trends Neurosci.,20,443-8
Piccolino M. (2000) The bicentennial of the Voltaic battery (1800-2000): the artifi-
cial electric organ Trends Neurosci.,23,147-151.
Pinney, S.E., MacMullen, C., Becker, S., Lin, Y.W., Hanna, C., Thornton, P., et al.,
2008.Clinical characteristics and biochemical mechanisms of congenital hyperinsu-
linism associated with dominant KATP channel mutations. Clin. Invest. 118, 2877–
2886.
Piven J, Palmer P, Jacobi D, Childress D, Arndt S. (1997): Broader autism pheno-
type:evidence from a family history study of multiple-incidence autism families.
American Journalof Psychiatry 154:185-190.
Ponce A, Bueno E, Kentros C, Vega-Saenz de Miera E, Chow A, Hillman D, Chen
S, Zhu L, Wu MB, Wu X, Rudy B, Thornhill WB. (1996). G-protein-gated inward
rectifier K+ channel proteins (GIRK1) are present in the soma and dendrites as well
as in nerve terminals of specific neurons in the brain. J Neurosci 16: 1990–2001

102
Poopalasundaram S, Knott C, Shamotienko OG, Foran PG, Dolly JO, Ghiani CA,
Gallo V, Wilkin GP. (2000). Glial heterogeneity in expression of the inwardly recti-
fying K+ channel, Kir4.1, in adult rat CNS. Glia 30: 362–372
Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P. (2003):
Genetic disruption of cortical interneuron development causes region- and GABA
cell type-specific deficits, epilepsy, and behavioral dysfunction. Journal of Neuros-
cience 23:622-631.
Preisig-Muller R, Schlichthorl G, Goerge T, Heinen S, Bruggemann A, Rajan S,
Derst C, Veh RW, Daut J. (2002). Heteromerization of Kir2.x potassium channels
contributes to the phenotype of Andersen’s syndrome. Proc Natl Acad Sci USA
99:7774–7779
Quinn KV, Giblin JP, Tinker A. (2004) Multisite phosphorylation mechanism for
protein kinase A activation of the smooth muscle ATPsensitive K+ channel. Circ Res
94: 1359–1366
Reichold, M., Zdebik, A.A., Lieberer, E., Rapedius, M., Schmidt, K., Bandulik, S., et
al.,2010.KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensori-
neural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad.
Sci.U.S.A. 107, 14490–14495.
Reimann F, Ryder T, Tucker S, Ashcroft F. (1999). The role of lysine 185 in the
Kir6.2 subunit of the ATP-sensitive channel in channel inhibition by ATP. J Physiol
520: 661–669.
Reimann, F. and Ashcroft, F.M. (1999) Curr. Opin. Cell Biol. 11, 503-8.
Roha´cs T, Lopes C, Mirshahi T, Jin T, Zhang H, Logothetis DE. (2002). Assaying
phosphatidylinositol bisphosphate regulation of potassium channels. Methods Enzy-
mol 345: 71–92

103
Rosenberg, R.E., Law, J.K., Yenokyan, G., McGrea-
dy,J.,Kaufmann,W.E.,Law,P.A.,2009.Arch.Pediatr. Adolesc. Med. 163, 907–914.
Rosenhouse-Dantsker A, Sui JL, Zhao Q, Rusinova R, Rodriguez-Menchaca AA,
Zhang Z, Logothetis DE. (2008). A sodium-mediated structural switch that controls
the sensitivity of Kir channels to PtdIns(4,5)P2. Nat Chem Biol 4: 624–631
Rougier O, Vassort G, Stampfli R. (1968). Voltage clamp experiments on frog atrial
heart muscle fibres with the sucrose gap technique. Pflu¨ gers Arch 301: 91–108.
Sala-Rabanal, M., Kucheryavykh, L.Y., Skatchkov, S.N., Eaton, M.J., Nichols, C.G.,
2010.Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1
(KCNJ10).J. Biol. Chem. 285, 36040–36048.
Sali, A., Blundell, T.L., 1993. Comparative protein modelling by satisfaction of spa-
tial restraints. J. Mol. Biol. 234, 779–815.
Salvatore L., D'Adamo M. C., Polishchuk R., Salmona M., Pessia M.(1999) Locali-
zation and age-dependent expression of the inward rectifier K+channel subunit Kir
5.1 in a mammalian reproductive system FEBS Letters 449; 146-152
Samuels, E.R., Szabadi, E., 2008. Functional neuroanatomy of the noradrenergic lo-
cuscoeruleus: its roles in the regulation of arousal and autonomic function part
I:principles of functional organisation. Curr. Neuropharmacol. 6, 235–253.
Schafer JA, Troutman SL. (1987). Potassium transport in cortical collecting tubules
from mineralocorticoid-treated rat. Am J Physiol Renal Fluid Electrolyte Physiol
253: F76–F88.

104
Schmitt BM, Berger UV, Douglas RM, Bevensee MO, Hediger MA, Haddad GG,
Boron WF. (2000). Na/HCO3 cotransporters in rat brain: expression in glia, neurons,
and choroid plexus. J Neurosci 20: 6839–6848.
Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW,
Farhi A,Nelson-Williams C, Lifton RP. (2009): Seizures, sensorineural deafness,
ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by
mutations in KCNJ10.PNAS 106:5842-5847.
Schram G, Melnyk P, Pourrier M, Wang Z, Nattel S. (2002). Kir2.4 and Kir2.1 K+
channel subunits co-assemble: a potential new contributor to inward rectifier current
heterogeneity. J Physiol 544: 337–349
Schulte U, Hahn H, Konrad M, Jeck N, Derst C, Wild K, Weidemann S, Ruppers-
berg JP, Fakler B, Ludwig J. (1999). pH gating of ROMK [Kir1.1] channels: control
by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proc Natl Acad Sci
USA 96:15298–15303
Schulze D, Krauter T, Fritzenschaft H, Soom M, Baukrowitz T. (2003). Phosphatidy-
linositol 4,5-bisphosphate (PIP2) modulation of ATP and pH sensitivity in Kir chan-
nels. A tale of an active and a silent PIP2 site in the N terminus. J Biol Chem 278:
10500–10505
Shang L, Lucchese CJ, Haider S, Tucker SJ. (2005): Functional characterisation of
missensevariations in the Kir4.1 potassium channel (KCNJ10) associated with sei-
zure susceptibility.Brain Research Molecular Brain.Research 13;139:178-183.
Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, Miller KJ,
Frazier JA,Silverstein I, Picker J, Weissman L, Raffalli P, Jeste S, Demmer LA, Pe-
ters HK, Brewster SJ,Kowalczyk SJ, Rosen-Sheidley B, McGowan C, Duda AW 3rd,
Lincoln SA, Lowe KR,Schonwald A, Robbins M, Hisama F, Wolff R, Becker R, Na-
sir R, Urion DK, Milunsky JM,Rappaport L, Gusella JF, Walsh CA, Wu BL, Miller
DT; Autism Consortium ClinicalGenetics/DNA Diagnostics Collaboration. (2010):

105
Clinical genetic testing for patients with
Shi Y, Wu Z, Cui N, Shi W, Yang Y, Zhang X, Rojas A, Ha BT, Jiang C. (2007).
PKA phosphorylation of SUR2B subunit underscores vascular KATP channel activa-
tion by β-adrenergic receptors. Am J Physiol Regul Integr Comp Physiol 293:
R1205–R1214
Shuck ME, Piser TM, Bock JH, Slightom JL, Lee KS, Bienkowski MJ. (1997). Clon-
ing and characterization of two K+ inward rectifier (Kir) 1.1 potassium channel ho-
mologs from human kidney (Kir1.2 and Kir1.3). J Biol Chem 272: 586–593.
Shyng S, Ferrigni T, Nichols C. Regulation of KATP channel activity by diazoxide
and MgADP. (1997). Distinct functions of the two nucleotide binding folds of the
sulfonylurea receptor. J Gen Physiol 110: 643–654.
Shyng SL, Cukras CA, Harwood J, Nichols CG. (2000). Structural determinants of
PIP2 regulation of inward rectifier KATP channels. J Gen Physiol 116: 599–608.
Shyng SL, Nichols CG. (1998). Membrane phospholipid control of nucleotide sensi-
tivity of KATP channels. Science 282: 1138–1141
Siegrist SE, Doe CQ. (2007). Microtubule-induced cortical cell polarity. Genes Dev
21: 483–496
Silver MR, DeCoursey TE. (1990). Intrinsic gating of inward rectifier in bovine pul-
monary artery endothelial cells in the presence or absence of internal Mg2+. J Gen
Physiol 96: 109–133.
Simons K, Ikonen E. (1997). Functional rafts in cell membranes. Nature 387: 569–
572.
Sims SM, Dixon SJ. (1989). Inwardly rectifying K_ current in osteoclasts. Am J
Physiol Cell Physiol 256: C1277–C1282.

106
Stanfield PR, Davies NW, Shelton PA, Sutcliffe MJ, Khan IA, Brammar WJ, Conley
EC. 1994. A single aspartate residue is involved in both intrinsic gating and blockage
by Mg2+ of the inward rectifier, IRK1. J Physiol 478: 1–6
Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, Kurachi Y. (2007). Inhibition of as-
troglial inwardly rectifying Kir4.1 channels by atricyclic antidepressant, nortripty-
line. J Pharmacol Exp Ther 320: 573–580
Suh BC, Hille B. (2005). Regulation of ion channels by phosphatidylinositol 4,5-
bisphosphate. Curr Opin Neurobiol 15: 370–378.
Takeuchi S, Ando M, Kakigi A. (2000). Mechanism generating endocochlear poten-
tial: role played by intermediate cells in stria vascularis. Biophys J 79: 2572–2582.
Takumi T, Ishii T, Horio Y, Morishige K, Takahashi N, Yamada M, Yamashita T,
Kiyama H, Sohmiya K, Nakanishi S, Kurachi Y. (1995). A novel ATP-dependent
inward rectifier potassium channel expressed predominantly in glial cells. J Biol
Chem 270: 16339–16346.
Takumi T, Ishii T, Horio Y, Morishige K, Takahashi N, Yamada M, Yamashita T,
Kiyama H, Sohmiya K, Nakanishi S, Kurachi Y. (1995). A novel ATP-dependent
inward rectifier potassium channel expressed predominantly in glial cells. J Biol
Chem 270: 16339–16346.
Takumi, T., Ishii, T., Horio, Y., Morishige, K., Takahashi, N., Yamada, M., et al.,
1995. A novel ATP-dependent inward rectifier potassium channel expressed predo-
minant-ly in glial cells. J. Biol. Chem. 270, 16339–16346.
Tanemoto M, Abe T, Ito S. (2005). PDZ-binding and di-hydrophobic motifs regulate
distribution of Kir4.1 channels in renal cells. J Am Soc Nephrol 16: 2608–2614

107
Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. (2000). In vivo formation of a pro-
ton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J
Physiol 525: 587–592.
Tanemoto M, Toyohara T, Abe T, Ito S. (2008). MAGI-1a functions as a scaffolding
protein for the distal renal tubular basolateral K+ channels. J Biol Chem 283: 12241–
12247
Tang, X., Hang, D., Sand, A., Kofuji, P., 2010. Variable loss of Kir4.1 channel func-
tion in SeSAME syndrome mutations. Biochem. Biophys. Res. Commun. 3399, 537–
541.
Taniguchi J, Yoshitomi K, Imai M. (1989). K+ channel currents in basolateral mem-
brane of distal convoluted tubule of rabbit kidney. Am J Physiol Renal Fluid Electro-
lyte Physiol 256: F246–F254
Thompson SM, Gahwiler BH. (1992).Comparison of the actions of baclofen at pre-
and postsynaptic receptors in the rat hippocampus in vitro. J Physiol 451: 329–345
Tian, G.F., Azmi, H., Takano, T., Xu, Q., Peng, W., Lin, J., et al., 2005. An astrocyt-
ic basis ofepilepsy.Nat. Med. 11, 973–981.
Toro, R., Konyukh, M., Delorme, R., Leblond, C., Chaste, P., Fauchereau, F., et al.,
2010. Keyrole for gene dosage and synaptic homeostasis in autism spectrum disord-
ers.Trends Genet. 26, 363–372.
Tuchman R, Moshé SL, Rapin I. (2009): Convulsing toward the pathophysiology of
autism. Brain and Development 31:95-103.
Tucker SJ, Imbrici P, Salvatore L, D’Adamo MC, Pessia M. (2000). pH dependence
of the inwardly rectifying potassium channel, Kir5.1, localization in renal tubular ep-
ithelia. J Biol Chem 275: 16404–16407.

108
Ueda K, Inagaki N, Seino S. (1997). MgADP antagonism to Mg2+ -independent
ATP binding of the sulfonylurea receptor SUR1. J Biol Chem 272: 22983–22986
Ullian, E.M., Christopherson, K.S., Barres, B.A., 2004. Role for glia in synaptogene-
sis. Glia 47,209–216.
Vit JP, Jasmin L, Bhargava A, Ohara PT. (2006). Satellite glial cells in the trigeminal
ganglion as a determinant of orofacial neuropathic pain. Neuron Glia Biol 2: 247–
257.
Vit JP, Ohara PT, Bhargava A, Kelley K, Jasmin L. (2008). Silencing the Kir4.1 po-
tassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in
pain-like behavior in the absence of nerve injury. J Neurosci 28: 4161–4171
Von Bekesy G. (1951). DC potentials and energy balance of the cochlear partition. J
Acoust Soc Amer 23: 576–582.
Wakefield AJ, Murch SH, Anthony A, Linnell J, Casson DM, Malik M, Berelowitz
M, Dhillon AP, Thomson MA, Harvey P, Valentine A, Davies SE, Walker-Smith JA.
(1998). Ileal-lymphoid-nodular hyperplasia, non-specific colitis, and pervasive deve-
lopmental disorder in children.. Lancet. 351: 637-641.
Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, Reichert J,
BuxbaumJD, Meisler MH. (2003): Sodium channels SCN1A, SCN2A and SCN3A in
familial autism.
Wible BA, Taglialatela M, Ficker E, Brown AM. (1994). Gating of inwardly rectify-
ing K+ channels localized to a single negatively charged residue. Nature 371: 246–
249
Williams J.T., North R.A., Shefner S.A., Nishi S., Egan T.M. (1984) Membrane
properties of rat Locus Coeruleus neurones. Neuroscience 13, 137-156.

109
Williams JT, Colmers WF, Pan ZZ. (1988). Voltage- and ligand-activated inwardly
rectifying currents in dorsal raphe neurons in vitro. J Neurosci 8: 3499–3506.
Woodbury, D. M., Mechanisms of action of anticonvulsants, in Basic mechanisms of
the epilepsies (a cura di H. H. Jasper e altri), Boston 1969, pp. 647-681.
Wu J, Xu H, Shen W, Jiang C. (2004): Expression and Coexpression of CO2-
sensitive Kir Channels in Brainstem Neurons of Rats. J. Membrane Biology 197:179-
191.
Xu H, Yang Z, Cui N, Chanchevalap S, Valesky WW, Jiang C. (2000). A single re-
sidue contributes to the difference between Kir4.1 and Kir1.1 channels in pH sensi-
tivity, rectification and single channel conductance. J Physiol 528: 267–277
Yamada M, Kurachi Y. (1995). Spermine gates inward-rectifying muscarinic but not
ATP-sensitive K_ channels in rabbit atrial myocytes. Intracellular substance-
mediated mechanism of inward rectification. J Biol Chem 270: 9289–9294
Yang J, Jan YN, Jan LY. (1995). Determination of the subunit stoichiometry of an
inwardly rectifying potassium channel. Neuron 15: 1441–1447.
Yang, Y., Ge, W., Chen, Y., Zhang, Z., Shen, W., Wu, C., et al., 2003. Contribution
of astrocytes to hippocampal long-term potentiation through release of D-serine.Proc.
Natl. Acad. Sci. U.S.A. 100, 15194–15199.
Yi, B.A., Lin, Y.F., Jan, Y.N., Jan, L.Y., 2001. Yeast screen for constitutively active
mutant G protein-activated potassium channels. Neuron 29, 657–667.

110
Ylisaukko-Oja T, Nieminen-Von Wendt T, Kempas E, Sarenius S, Varilo T, Wendt
Lv L,Peltonen L, Jarvela II. (2004): Genome-wide scan for loci of Asperger syn-
drome. MolecularPsychiatry. 9:161-168.
Yoo D, Kim BY, Campo C, Nance L, King A, Maouyo D, Welling PA. (2003). Cell
surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-
induced kinase, SGK-1, and protein kinase A. J Biol Chem 278: 23066–23075
Zhang H, He C, Yan X, Mirshahi T, Logothetis DE. (1999). Activation of inwardly
rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat Cell Biol 1: 183–
188.
Zhang X, Cui N, Wu Z, Su J, Tadepalli JS, Sekizar S, Jiang C. (2010): Intrinsic
membrane properties of locus coeruleus neurons in Mecp2-null mice. American
Journal Physiology Cell Physiology 298:C635-C646.
Zhang, J.M., Wang, H.K., Ye, C.Q., Ge, W., Chen, Y., Jiang, Z.L., et al., 2003. ATP
released by astrocytes mediates glutamatergic activity-dependent heterosynaptic sup-
pression.Neuron 40, 971–982.
Zingerevich C, Greiss-Hess L, Lemons-Chitwood K, Harris SW, Hessl D, Cook K,
Hagerman RJ. (2008). Motor abilities of children diagnosed with fragile X syndrome
with and without autism.. J Intellect Disabil Res..
Zoghbi, H.Y., 2003. Postnatal neurodevelopmental disorders: meeting at the syn-
apse?Science 302,826–830.
Zou J, Zhang Y, Yin S, Wu H, Pyykko I. (2009). Mitochondrial dysfunction disrupts
trafficking of Kir4.1 in spiral ganglion satellite cells. J Neurosci Res 87: 141–149.

111
RINGRAZIAMENTI
Ringrazio sentitamente:
Il Prof. Mauro Pessia per i preziosi consigli e per l’importante supporto al
mio studio;
La Dott.ssa Cristina D’Adamo per l’aiuto fornitomi durante l’esperienza di
laboratorio;
La Dott.ssa Paola Imbrici che oltre ad essermi stata vicina con grande dispo-
nibilità e cortesia nella parte pratica della tesi, mi è stata di valido aiuto du-
rante la stesura.





























