Embed Size (px)
Citation preview

UNIVERSITÄTSKLINIKUM HAMBURG-EPPENDORF
Institut für Pathologie
Prof. Dr. med. Guido Sauter
Intratumorale Heterogenität der EGFR Genvermehrung im nicht-kleinzelligen Bronchialkarzinom
Dissertation
zur Erlangung des Grades eines Doktors der Medizinan der Medizinischen Fakultät der Universität Hamburg.
vorgelegt von:
Tobias Hoenig aus Hamburg
Hamburg 2015
1

Angenommen von der Medizinischen Fakultät der Universität Hamburg am: 27.05.2015
Veröffentlicht mit Genehmigung der Medizinischen Fakultät der Universität Hamburg.
Prüfungsausschuss, der/die Vorsitzende: Prof. Dr. Guido Sauter
Prüfungsausschuss, zweite/r Gutachter/in: Prof. Dr. Heinz-Eckard Laack
Prüfungsausschuss, dritte/r Gutachter/in: PD Dr. Ronald Simon
2

Danksagung
Zunächst möchte ich mich herzlich bei Prof. Dr. med. Guido Sauter bedanken, der mir das Thema dieser Doktorarbeit in seinem Institut für Pathologie angeboten hat.
Dr. Dr. Tobias Grob gilt mein besonderer Dank für seine engagierte Zusammenarbeitmit mir. Dank seiner tatkräftigen Unterstützung war die Publikation unseres Papers im Journal „Lung Cancer“ erst möglich.
Frau Christina Koop danke ich für ihre stetige Betreuung im Labor und ihre große Hilfe beim Erstellen eines Tissue Microarrays.
Zuletzt möchte ich mich bei meinen Eltern bedanken, die mir zu jeder Zeit Rückhalt und Zuversicht gegeben haben.
3

Inhaltsverzeichnis1 Einleitung....................................................................................................................5
1.1 Tyrosinkinaserezeptoren (TKR)..........................................................................51.1.1 Epidermal-Growth-Factor-Rezeptor (EGFR)..............................................6
1.2 Molekular zielgerichtete Therapie.......................................................................71.2.1 Monoklonale Antikörper...............................................................................71.2.2 Tyrosinkinaseinhibitoren (TKI)....................................................................71.2.3 EGFR- Aberrationen als prädiktiver Marker................................................8
1.3 Heterogenität von Tumoren................................................................................91.4 Ziel der Doktorarbeit...........................................................................................9
2 Material und Methoden.............................................................................................102.1 Erstellen eines Tissue Microarrays (TMA)........................................................102.2 Zusammenstellen des Gewebekollektivs.........................................................112.3 EGFR Fluoreszenz-in-situ-Hybridisierung (FISH)............................................112.4 EGFR Immunhistochemie (IHC).......................................................................122.5 EGFR Sequenzierung ......................................................................................12
3 Ergebnisse................................................................................................................133.1 EGFR FISH ......................................................................................................13
3.1.1 Lymphknotenmetastasen..........................................................................143.2 EGFR Immunhistochemie ................................................................................153.3 EGFR Mutationsanalyse amplifizierter Tumore................................................15
4 Diskussion................................................................................................................164.1 Molekulare Diagnostik bei Heterogenität..........................................................164.2 EGFR Aberrationen..........................................................................................164.3 Hohe Polysomie................................................................................................164.5 EGFR Amplifikationen.......................................................................................174.6 Vergleich Primärtumor vs Metastase ...............................................................184.7 Prädiktiver Wert................................................................................................194.8 EGFR Mutationen.............................................................................................19
5 Fazit..........................................................................................................................216 Literaturverzeichnis..................................................................................................227. Paper.......................................................................................................................288. Lebenslauf...............................................................................................................369. Eidesstattliche Versicherung ................................................................................38
4

1 Einleitung
Das nicht-kleinzellige Bronchialkarzinom (NSCLC) ist ein maligner epithelialer Tumor
der Lunge. Es ist eines der weltweit am häufigsten zum Tode führenden Karzinome
bei Männern und Frauen (61).
Je nach Tumorstadium unterscheidet sich das therapeutische Vorgehen. Während
nicht disseminierte NSCLC in frühem Stadium primär kurativ reseziert werden, wird
bei fortgeschrittenem Tumorstadium oder Inoperabilität eine Chemo- und/oder
Radiotherapie durchgeführt (62).
Ein weiterer, seit einigen Jahren etablierter Behandlungsansatz ist eine molekular
gezielte Therapie mit Tyrosinkinaseinhibitoren gegen den Epidermal-Growth-Factor-
Receptor (EGFR). Diese Therapien werden abhängig von molekularen Analysen von
EGFR-Aberrationen in einer (in der Regel durch eine Biopsie gewonnenen)
Tumorgewebeprobe eingeleitet (s. Abschnitt 1.2).
Studien über prädiktive Marker für das Ansprechen dieser molekular zielgerichteten
Therapien ergaben unterschiedliche Ergebnisse (s. Abschnitt 1.2.3). Dies könnte
durch eine Heterogenität von EGFR-Aberrationen in einem Tumor begründet sein (s.
Abschnitt 1.3).
Die folgenden Abschnitte beschreiben die Grundlagen des Epidermal-Growth-Factor-
Receptor und dessen molekular zielgerichteter Therapie bei nicht-kleinzelligen
Bronchialkarzinomen und liefern damit die Basis zur Zieldefinition dieser Arbeit (s.
Abschnitt 1.4).
1.1 Tyrosinkinaserezeptoren (TKR)
Wachstum, Morphogenese und Stoffwechselprozesse einer Körperzelle werden
maßgeblich von Wachstumsfaktoren/-hormonen beeinflusst, welche an spezifische
Rezeptoren binden, diese aktivieren und damit eine intrazelluläre Signalkaskade
initiieren. Viele dieser Prozesse werden durch Phosphorylierung und
Dephosphorylierung von Proteinen reguliert, wobei die Phosphatgruppen meist auf
die Aminosäuren Serin und Threonin aber auch auf Tyrosin übertragen werden (43).
Eine wichtige Familie von membranständigen Wachstumshormonrezeptoren, welche
die Phosphorylierung von Tyrosin katalysieren, sind die sogenannten
Tyrosinkinaserezeptoren, zu denen u.a. die Insulin-, PDGF-, VEGF- und ErbB-
Rezeptor-Familien gehören (s. Abschnitt 1.1.1).
5

Mit der Aktivierung der Tyrosinkinase durch Phosphorylierung der Tyrosinreste
werden weitere Adapterproteine aktiviert. Hierdurch werden verschiedene
Signalkaskaden, wie z.B. der RAS/RAF-MAP-Kinase-Pathway oder der PI3K/AKT-
Pathway aktiviert, die das Zellwachstum stimulieren und den apoptotischen Zelltod
hemmen (41, 42, 44).
Mutationen im Gen von Tyrosinkinaserezeptoren können onkogene Wirkungen
haben, indem sie über verschiedene Mechanismen eine konstitutive Aktivierung der
Rezeptorkinasen bewirken und so zu einem unkontrollierten Wachstum und maligner
Entartung der Zelle führen.
1.1.1 Epidermal-Growth-Factor-Rezeptor (EGFR)
Auf der Basis struktureller Homologien lassen sich die Rezeptor-Tyrosinkinasen in
verschiedene Familien einteilen. Eine von ihnen ist die ErbB- oder auch HER-
(Human Epidermal Growth Factor Receptor) Familie, zu der insgesamt vier
Rezeptoren gezählt werden: HER1/EGFR (ErbB1), HER2/c-neu (ErbB2), HER3
(ErbB3) und HER4 (ErbB4). Diese 170-200 kDa großen Glykoproteine haben trotz
großer Gemeinsamkeiten alle eine unterschiedliche, spezifische Funktion (41, 42).
Der EGF-Rezeptor kann parakrin, juxtakrin und autokrin durch verschiedene
Liganden (EGF, TGF-α, Amphiregulin, Betacellulin und Epiregulin) aktiviert werden
und mehrere, für das Zellwachstum essentielle, Signalkaskaden einleiten. Zu diesen
gehören vor allem der bereits genannte RAS/RAF-MAP-Kinase-Pathway sowie der
PI3K/AKT-Pathway. Des Weiteren findet eine Aktivierung der Proteine STAT3/5 und
Proteinkinase C statt, welche ebenfalls eine proliferative Wirkung besitzen.
In vielen soliden Tumoren konnte eine verstärkte EGFR-Expression (u.a. aufgrund
einer Amplifikation des auf Chromosom 7p12 kodierten EGFR-Gens) nachgewiesen
werden (51, 52). EGFR-Überexpression wurde bei vielen Tumorentitäten mit
fortgeschrittener Krankheit, aggressiveren Phenotyp und generell schlechter
Prognose in Verbindung gebracht (1). Zusätzlich konnte nachgewiesen werden, dass
von diesen Tumoren häufig auch ein oder mehrere EGFR-Liganden produziert
werden, wodurch es zu einer autokrinen Überstimulation kommt (44).
Die proliferativen Eigenschaften und das damit verbundene onkogene Potential des
EGF-Rezeptors machen diesen zu einem stark beforschten Therapieziel in der
Krebsbekämpfung.
6

1.2 Molekular zielgerichtete Therapie
Die zytostatische Wirksamkeit klassischer Chemotherapeutika und die damit
verbundene mittlere Überlebenszeit/-rate ist bei fortgeschrittenen nicht-kleinzelligen
Bronchialkarzinomen relativ gering, die systemischen Nebenwirkungen sind aber
teilweise erheblich (53, 54). Daher ist es von großem Interesse, neue Medikamente
zu entwickeln, die einen spezifischen Prozess der Signaltransduktion, der in den
Tumorzellen verändert ist, hemmen und so molekular gezielt und nebenwirkungsarm
wirken.
1.2.1 Monoklonale Antikörper
Ein möglicher Therapieansatz ist die Gabe von monoklonalen Antikörpern, die an
Oberflächenmoleküle binden, die von den Tumorzellen vermehrt exprimiert werden.
Die Bindung dieses Antikörpers an einen Rezeptor kann die Ligandenbindung und
damit die Initiierung der Signaltransduktion verhindern, oder sie führt zur Zelllyse
durch Aktivierung zytotoxischer Zellen des Immunsystems.
Ein klinisch etabliertes Beispiel bei Mamma- und Magenkarzinomen ist Trastuzumab,
ein monoklonaler Antikörper gegen den HER2/Neu Rezeptor, der von ca 15-20%
aller Mammakarzinome auf Grund einer HER2-Gen-Amplifikation vermehrt exprimiert
wird. Bei Patienten mit HER2/Neu positiven Tumoren konnte unter der Behandlung
mit Trastuzumab eine Überlebenszeitverlängerung, sowie eine Verringerung des
Rezidivrisikos festgestellt werden (55).
Ein weiteres Beispiel ist Bevacizumab, ein Antikörper, der gegen den vaskulären
epithelialen Wachstumsfaktor (VEGF) gerichtet ist und somit die Angioneogenese
von Tumoren hemmt. Dieser Antikörper wird besonders für die Behandlung von
fortgeschrittenen kolorektalen Karzinomen eingesetzt.
Weitere Präparate, welche für die Behandlung von Kolonkarzinomen Verwendung
finden sind Cetuximab und Panitumumab. Sie sind gegen den EGF-Rezeptor
gerichtete Antikörper.
1.2.2 Tyrosinkinaseinhibitoren (TKI)
Neben den extrazellulär wirkenden Antikörpern gibt es auch Medikamente, die
intrazellulär Moleküle inhibieren, die für die Signaltransduktion und Zellproliferation
essentiell sind. Das wohl bekannteste Präparat aus dieser Reihe ist Imatinib, ein
Tyrosinkinaseinhibitor. Imatinib inaktiviert gezielt die Kinase des durch Translokation7

entstehenden BCR-ABL Fusionsproteins, welches für die Entstehung der chronisch-
myeloischen Leukämie verantwortlich ist und führt bei fast allen Patienten zu einer
hämatologischen und in ca. 85% der Fälle zu einer molekulargenetischen Remission
(60).
Auch in der Behandlung des nicht-kleinzelligen Bronchialkarzinoms sind Inhibitoren
der EGF-Rezeptor-Tyrosinkinasen wie Erlotinib und Gefitinib als molekular
zielgerichtete Therapie bereits etabliert. Der Einsatz dieser Pharmaka ist allerdings
nur in einigen Fällen indiziert (s. Abschnitt 1.2.3). In den meisten Fällen konnten
keine oder nur sehr geringe Überlebensvorteile gegenüber der Chemotherapie
beobachtet werden (56).
1.2.3 EGFR- Aberrationen als prädiktiver Marker
EGFR-Mutationen, EGFR-Genkopienanzahl und EGFR-Proteinexpression in NSCLC
wurden in klinischen Studien als prädiktive Marker für das Ansprechen auf molekular
zielgerichtete Therapien untersucht (2, 9, 13, 35, 58). Hierbei traten EGFR-
Mutationen stets als stark prädiktive Marker für ein Ansprechen auf eine Therapie mit
TKI, nicht jedoch mit Cetuximab hervor.
Es konnte gezeigt werden, dass in den Tumoren von Patienten, die besonders gut
auf die Therapie ansprechen, eine Mutation des EGFR-Gens im Bereich der
katalytischen Domäne vorhanden ist (Exon 18 - 21), was zu einer Etablierung der
TKI-Erstlinientherapie bei Patienten mit EGFR-Mutationen führte (5-7, 45, 46).
Verschiedene Studien zur Untersuchung der EGFR-Genkopienanzahl als prädiktiver
Marker für ein Ansprechen auf TKI und Cetuximab ergaben sehr unterschiedliche
Resultate (8, 9, 13-18). Es wurde vorgeschlagen, dass durch Fluoreszenz-in-situ-
Hybridisierung (FISH) ermittelte EGFR-Genkopieerhöhungen als prädiktiver Marker
für eine TKI Therapie nicht nur auf EGFR-Genamplifikationen beschränkt sind,
sondern auch auf hohe Polysomien von Chromosom 7 (≥40% der Tumorzellen mit
mindestens vier Kopien) (10, 13-16, 49).
Die EGFR-Proteinexpression erwies sich in einigen Studien als unbrauchbarer
prädiktiver Maker für TKI-Therapien (2, 3), jedoch wurde sie nach Ergebnissen der
FLEX Studie als prädiktiver Marker für eine Cetuximab-Therapie vorgeschlagen (4).
Eine Validierung dieser Resultate steht jedoch noch aus.
8

1.3 Heterogenität von Tumoren
Zellpopulationen ein und desselben Tumors können verschiedene Merkmale bzw.
Mutationen tragen. Ursache dafür scheint ein Selektionsvorgang während des
Tumorwachstums zu sein, der es ermöglicht, weitere Mutationen und chromosomale
Aberrationen in den ohnehin schon unkontrolliert wachsenden Tumorzellen
entstehen zu lassen. So kann es sein, dass zwei Biopsien aus demselben Karzinom
verschiedene Ergebnisse der immunhistochemischen Untersuchungen, Mutations-
oder FISH-Analysen haben (27, 34, 35, 47, 48).
1.4 Ziel der Doktorarbeit
Trotz beachtlicher Bemühungen hat sich kein klares Bild vom prädiktiven Wert der
EGFR-Genkopienanzahl für eine Therapie mit TKI oder Antikörpern ergeben. Eine
intratumorale genetische Heterogenität könnte eine mögliche Ursache für die
unterschiedlichen Resultate sein. Solche Tumorheterogenitäten können besonders
bei Bronchialkarzinomen von großer Bedeutung sein, da die Diagnose üblicher
Weise anhand einer kleinen Biopsie (oder sogar weniger Zellen in Fällen von
Zytologieproben) gestellt wird. Beim Vorliegen heterogener Tumore bestünde die
Gefahr der Fehldiagnose und Fehlbehandlung. Bei entsprechender Heterogenität
könnte der Großteil eines vermeintlich „EGFR-negativen“ Tumors durchaus mit TKIs
wirksam in seiner Progression gehemmt werden. Auf der anderen Seite hätte ein
vermeintlich „EGFR-positiver“ Befund bei Heterogenität zur Folge, dass nur ein Teil
der Karzinomzellen durch diese Therapie gehemmt werden und der Rest nicht zur
Remission gebracht werden kann.
Ziel dieser Doktorarbeit ist es, das Ausmaß von intratumoraler Heterogenität der
EGFR-Genkopienanzahl und EGFR-Proteinexpression in nicht-kleinzelligen
Bronchialkarzinome, mittels immunhistochemischer Färbung und Fluoreszenz-in-situ-
Hybridisierung von Tissue Micro Arrays, zu untersuchen.
9

2 Material und Methoden
2.1 Erstellen eines Tissue Microarrays (TMA)
Der Tissue Microarray (TMA) stellt eine seit mehreren Jahren etablierte Methode dar,
mit Hilfe derer man eine große Anzahl von Geweben schnell und kostengünstig
untersuchen kann. Hierzu werden aus den zu untersuchenden, in Paraffin
eingebetteten Tumorgeweben (FFPE) Stanzen entnommen und diese anschließend
in einen neuen Empfänger-Paraffinblock gebettet (Abb. 1). Auf diese Weise ist es
möglich, mehrere hundert verschiedene Tumorgewebe auf einem solchen
Empfängerblock zu sammeln und anschließend das gesamte Kollektiv auf einem
Schnitt mit einer Färbung auf DNA-, RNA- und Proteinebene zu untersuchen (11).
Abbildung 1: Schematische Darstellung der TMA-Herstellung a) Kopf des Arrayers mit Bohrer und
Hohlnadel, HE-Schnitt als Schablone und Tumorparaffinblock; b) Hohlnadel mit Tumorgewebe, Array;
c) und d) Übertragung eines TMA-Schnittes auf einen Objektträger (Sauter et al. 2003)
10

2.2 Zusammenstellen des Gewebekollektivs
Um die Heterogenität von Biomarkerabweichungen in NSCLC zu analysieren,
wurden in der Datenbank des Instituts für Pathologie der Universität Hamburg-
Eppendorf 373 Patienten auf ausreichend verfügbares Tumorgewebe untersucht. In
144 Fällen waren mindestens vier in Paraffinblöcken eingebettetes, formalinfixiertes
Tumorgewebe in den Archiven des Instituts gelagert. Von diesen Tumoren wurden
jeweils acht verschiedene Gewebestanzen für die TMA-Konstruktion entnommen.
Hierbei wurde darauf geachtet, dass die Stanzen von Arealen entnommen wurden,
die weitest möglich von einander entfernt lagen (nach Möglichkeit von verschiedenen
Blöcken; für genauere Angaben siehe Publikation). Außerdem wurden bis zu vier
verschiedene Lymphknotenmetastasen von 62 (von den 144) Patienten mit positivem
Lymphknotenbefall untersucht. Es wurde eine Stanze pro Metastase entnommen.
Alle originalen Schnitte des für den TMA benutzen Gewebes wurden erneut gemäß
den WHO 2004 Kriterien auf ihren histologischen Typ und ihr histologisches Grading
untersucht (12).
2.3 EGFR Fluoreszenz-in-situ-Hybridisierung (FISH)
Die TMA-Schnitte wurden mittels eines kommerziellen Kits proteolytisch
vorbehandelt (Paraffin pre-treatment reagent kit, Abbott). Das Vysis LSI EGFR
Spectrum Orange/CEP7 Spectrum Green Probe Kit (Abbott) wurde nach
Herstelleranweisungen für die Detektion von EGFR-Genen benutzt. Das
Vorhandensein von Tumorzellen wurde in jeder Tumorprobe durch Vergleich mit
einem Hämatoxylin-Eosin (HE) gefärbten Referenz-TMA-Schnitt verifiziert. Auf jedem
Spot wurden 20 Tumorzellen nach vorgeschlagenen Richtlinien zur Evaluation der
EGFR-Genkopienanzahl ausgewertet (19).
FISH-Kriterien für die Wertung „EGFR positiv“:
1.) ≥40% der Tumorzellen weisen ≥4 EGFR Gensignale auf
2.) Tumorproben mit einer EGFR-Genamplifikation entsprechend folgender Kriterien:
a) Das EGFR zu CEP7 Verhältnis aller Zellen ist >2 (bei ≥2 CEP7 Kopien pro Zelle)
b) Gen-Cluster (≥4 EGFR Gensignale mit Clusterverteilung) in ≥10% der Zellen
c) ≥15 EGFR Gensignale in ≥10% der Tumorzellen
11

2.4 EGFR Immunhistochemie (IHC)
Für die immunhistochemische Färbung der TMA-Schnitte wurde das EGFR pharmDx
IHC-Kitsystem (Dako) nach Anweisung des Herstellers angewandt. Im Anschluss an
die Inkubation mit dem primären monoklonalen Antikörper, Klon 2-18C9, gegen
humanes EGFR-Protein verwendet dieses Kit ein gebrauchsfertiges
Visualisierungsreagenz auf der Basis der Dextrantechnologie.
Auswertung:
Die Auswertung wurde von einem geschulten Pathologen mithilfe eines
Lichtmikroskops durchgeführt. Dabei wurde die Intensität der Membranfärbung auf
den einzelnen Gewebespots (in Analogie zum Scoringsystem von HER2 in Brust-
und Magenkrebs (20)) in eine vierstufige Skala unterteilt (0, 1+, 2+, 3+).
2.5 EGFR Sequenzierung
Alle EGFR-amplifizierten Fälle wurden zusätzlich sequenziert, um etwaige
Mutationen in der EGFR-Tyrosinkinasedomäne (Exons 18-21) festzustellen.
Nachdem per Makrodissektion oder Laser-Capture Mikrodissektion (PALM Micro
Beam Laser System, Zeiss Microlmaging) ein Tumorzellgehalt von mindestens 70%
gewährleistet war, wurde die Tumor DNA vom FFPE Gewebe nach Standardprotokoll
extrahiert (QiAmp DNA Mini Kit, Qiagen). Quantität und Qualität der DNA wurde
mittels Nanodrop Spektrophotometer ND-1000 (Thermo-Scientific) evaluiert. 50 ng
der DNA wurden in der PCR Reaktion eingesetzt, wobei der AmpliTaq Gold PCR
Mastermix (Applied Biosystem) unter den empfohlenen Bedingungen des Herstellers
verwendet wurde. DNA-Fragmente der Exons 18-21 wurden mittels spezifischer
Primer amplifiziert. Nach Verifizierung per QlAxcel System (Qiagen) wurden die PCR
Produkte mit einem enzymatischen Verfahren gereinigt (ExoSAP-IT; USB Products)
und eine Sequenzierreaktion mit einem BidDye Terminater Cycle Sequencing Kit
(Applied Biosystems) durchgeführt. Die Sequenzierprodukte wurden mit einem ABI
PRISM 3100 Genetic Analyzer (Applied Biosystems) analysiert.
12

3 Ergebnisse
3.1 EGFR FISH
Die EGFR-Genkopienanzahl war mittels Fluoreszenz-in-situ-Hybridisierung in
insgesamt 1055 Spots (durchschnittlich 7,3 Spots pro Tumor) der TMAs
interpretierbar. In den 261 anderen Spots war die Analyse entweder aufgrund
mangelnder Qualität der Hybridisierung, unzureichender Anzahl der analysierbaren
Tumorzellen, oder komplett fehlender Tumorgewebeprobe auf dem TMA-
Schnittpräparat nicht möglich.
In 37 (25,7%) Tumoren wurde eine hohe Polysomie (40% der Tumorzellen mit ≥4
Signalen des Chromosoms 7, gemäß (19)) nachgewiesen. 36 dieser Tumore waren
heterogen für hohe Polysomie und wiesen andere Proben ohne EGFR-
Genkopieerhöhung auf. In zwei von diesen Fällen konnte in verschiedenen Proben
zusätzlich eine EGFR-Genamplifikation nachgewiesen werden. Ein Tumor konnte
nicht auf seine Heterogenität evaluiert werden, da nur eine Gewebeprobe in der
EGFR FISH Analyse analysierbar war, so dass in keinem der Tumore mit mehreren
interpretierbaren Gewebeproben eine homogene hohe Polysomie aufgefunden
werden konnte (Fig 1B der Publikation). Von den 35 Tumoren ohne gleichzeitige
EGFR-Amplifikation wies ein Durchschnitt von 26,1% (1,8/6,8) Spots eine hohe
Polysomie auf.
In dreizehn Tumoren konnte in ein bis elf Spots eine EGFR-Genamplifikation
festgestellt werden, wobei sechs Fälle eine homogene Amplifikation und sieben Fälle
eine intratumorale Heterogenität aufwiesen. Bei den heterogenen Fällen konnten im
Durchschnitt in 34% (2,4/7,1) der Spots eine Genamplifikation nachgewiesen
werden. Zwei dieser sieben Fälle beinhalteten, wie oben beschrieben, Spots mit
hoher Polysomie des Chromosoms 7 neben Spots mit Amplifikation und negativen
Spots.
Heterogene Tumore wurden separat histologisch untersucht, wobei keine
morphologischen Unterschiede zwischen amplifizierten und nicht amplifizierten
Tumorarealen im Grenzgebiet festgestellt wurden (Abb. 2).
13
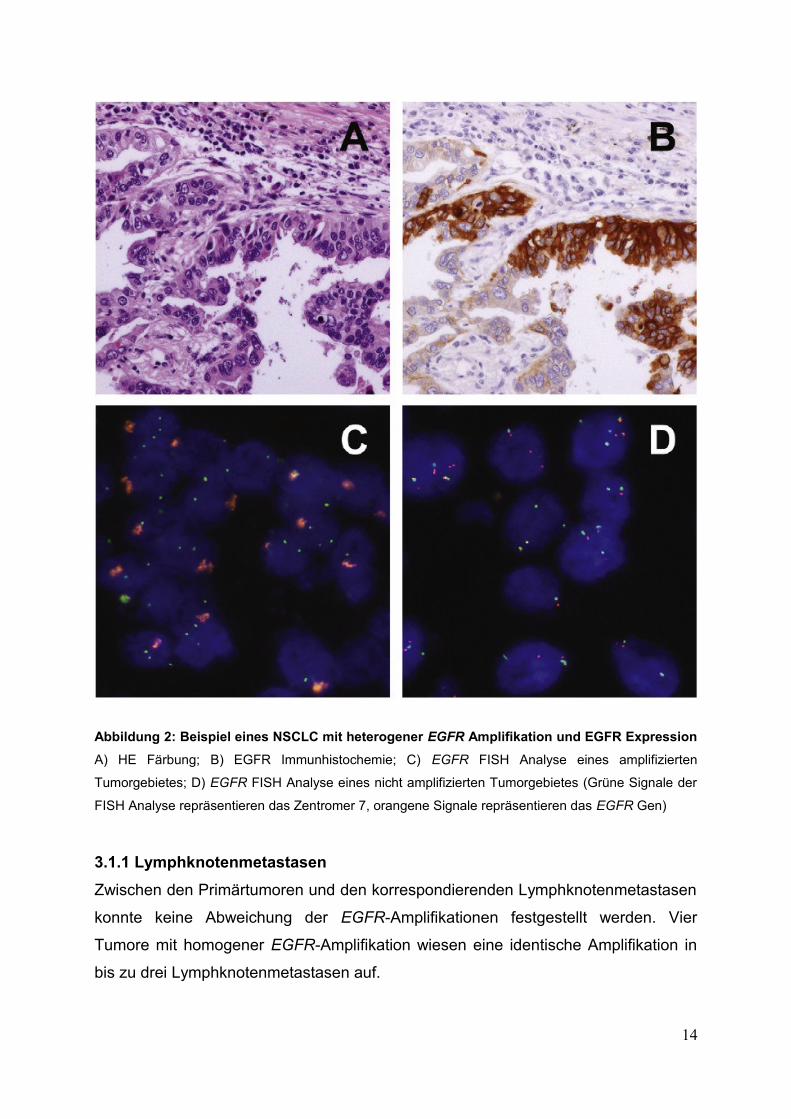
Abbildung 2: Beispiel eines NSCLC mit heterogener EGFR Amplifikation und EGFR Expression
A) HE Färbung; B) EGFR Immunhistochemie; C) EGFR FISH Analyse eines amplifizierten
Tumorgebietes; D) EGFR FISH Analyse eines nicht amplifizierten Tumorgebietes (Grüne Signale der
FISH Analyse repräsentieren das Zentromer 7, orangene Signale repräsentieren das EGFR Gen)
3.1.1 Lymphknotenmetastasen
Zwischen den Primärtumoren und den korrespondierenden Lymphknotenmetastasen
konnte keine Abweichung der EGFR-Amplifikationen festgestellt werden. Vier
Tumore mit homogener EGFR-Amplifikation wiesen eine identische Amplifikation in
bis zu drei Lymphknotenmetastasen auf.
14

Insgesamt konnten in 16 Fällen mit heterogener EGFR-Genkopieerhöhung
zusätzliche Lymphknotenmetastasen evaluiert werden. Hierbei zeigten zwei Tumore
eine fokale Amplifikation und 14 Tumore eine fokale hohe Polysomie. Die EGFR-
Genkopienanzahl der Lymphknotenmetastasen war stets mit einem Teil des
Primärtumors identisch. Unterschiedliche EGFR-Genkopieerhöhungen zwischen
einzelnen Lymphknotenmetastasen eines Primärtumors konnten in vier von 11 Fällen
nachgewiesen werden.
3.2 EGFR Immunhistochemie
Die immunhistochemische Analyse des EGFR-Proteins konnte in 1203 Gewebespots
erfolgreich durchgeführt werden. Die anderen 113 Gewebespots wurden aus der
Analyse ausgeschlossen, da die Spots auf dem TMA-Schnitt fehlten, oder zu wenige
Tumorzellen für eine Evaluation enthielten. Im Durchschnitt waren 8,4 Spots eines
jeden Tumors analysierbar. Eine starke Immunfärbung mindestens eines Spots kam
in 84 (58,3%) und eine komplett negative Immunfärbung aller untersuchten Spots in
8 (5,6%) Tumoren vor. Die meisten Tumore zeigten ein gewisses Ausmaß an
Heterogenität der EGFR-Expression. Nur 20 (13,9 %) Tumore wiesen eine komplette
Homogenität in allen untersuchten Spots auf (8 Tumore negativ, 1 Tumor schwach
positiv, 11 Tumore stark positiv). Alle 6 homogen amplifizierten Fälle wiesen hierbei
eine homogen stark positive IHC-Färbung auf. 31 (21,5%) Tumore zeigten Areale mit
starker Expression neben vollständig negativen Tumoranteilen.
3.3 EGFR Mutationsanalyse amplifizierter Tumore
In vier der dreizehn amplifizierten Tumore konnte per Sequenzanalyse eine typische
Mutation des EGFR-Gens nachgewiesen werden. Zwei dieser Tumore zeigten eine
homogene EGFR-Amplifikation, die zwei anderen eine heterogene EGFR-
Amplifikation.
15

4 Diskussion
4.1 Molekulare Diagnostik bei Heterogenität
Für molekular zielgerichtete Krebstherapien stellt Tumorheterogenität ein großes
Problem dar. Der zu untersuchende Biomarker könnte lediglich in einer Fraktion
eines Tumors vorhanden sein. Die molekulare Diagnostik von durch Biopsien
gewonnenen Gewebeproben wäre dadurch in ihrer Aussagekraft limitiert. Unter einer
zunächst effektiven molekular zielgerichteten Krebstherapie könnte es daher nach
einiger Zeit zu Resistenzen kommen, nachdem zielstrukturpositive Tumorzellen
abgestorben sind und zielstrukturnegative Zellen die Überhand gewinnen.
Gemessen an ihrer Wichtigkeit wurde die Heterogenität von Tumoren bisher in relativ
wenigen Studien untersucht. In den meisten dieser Studien wurde die Heterogenität
durch Analyse eines oder zweier Gewebeblöcke pro Tumor/Patient quantifiziert (28,
29, 37). Die gesamte Biologie eines großen Tumors kann jedoch nicht durch lediglich
eine kleine Gewebesektion repräsentiert werden.
Unser Verfahren, in dem wir mittels TMA Proben von acht so weit wie möglich von
einander lokalisierten Arealen pro Tumor analysiert haben, ermöglicht eine deutlich
umfassendere Analyse molekularer Eigenschaften in einer großen Anzahl von
Tumoren.
4.2 EGFR Aberrationen
In unserer Studie fanden wir in 48 (33,3%) von 144 NSCLC eine Erhöhung der
EGFR-Genkopienanzahl. Hierbei waren keine signifikanten Unterschiede zwischen
den verschiedenen histologischen Subtypen erkennbar. Wir konnten auch keine
signifikante Assoziation einer erhöhten EGFR-Genkopienanzahl mit dem
histologischen Grad, oder mit dem Staging des Tumors feststellen. Vorherige
Studien, die mittels FISH die EGFR-Genkopienanzahl in Bronchialkarzinomen
untersucht haben, haben, mit einer Erhöhung bei 31-45% aller Tumore,
vergleichbare Ergebnisse erzielt (15, 22, 23).
4.3 Hohe Polysomie
Hohe Polysomie wurde in der Vergangenheit als prädiktiver Marker für die
Wirksamkeit einer Anti-EGFR-Therapie vorgeschlagen (10). Dieses Vorgehen hat
16

sich jedoch nicht bewährt. Eine ausgeprägte Heterogenität könnte Grund für den
niedrigen prädiktiven Wert sein.
Die Daten unserer Studie zeigen eine sehr hohe Rate von Bronchialkarzinomen mit
einer Heterogenität der Aberrationen der EGFR-Genkopienanzahl. Dies trifft
insbesondere zu, wenn die EGFR-Aberration durch hohe Polysomie bedingt ist. Alle
36 Bronchialkarzinome (mit mindestens zwei auswertbaren Gewebespots), in denen
eine hohe Polysomie nachgewiesen wurde, hatten Tumoranteile mit und ohne hohe
Polysomie. Da die Evaluation von hoher Polysomie durch FISH schwierig und
anfällig für Fehler ist, sind weitere Studien zur Validierung unserer Resultate sinnvoll
(19).
Technische Limitationen können zu der Variabilität unserer Daten beigetragen
haben. So ist z.B. bei der Determinierung, ob 40% der Zellen 4 oder mehr Signale
aufweisen, oder nicht, die Dicke des Paraffinschnittes von entscheidender
Bedeutung. Mit Zunahme der Schnittdicke werden FISH Analysen immer mehr
Signale hervorbringen, da der Durchmesser von Tumorzellkernen stets größer ist, als
die Tiefe des Schnittes.
Die Validität unserer FISH Daten wird durch die signifikante Assoziation zwischen
hoher Polysomie und hohem Level von EGFR-Proteinexpression unterstützt. TMAs
ermöglichen eine größtmögliche Standardisierung. Dennoch ist anzunehmen, dass
sich in unseren Daten Fehler befinden können. Die lange Erfahrung unseres Labors
für FISH Analyse bestärkt wiederum, dass eine wesentlich bessere Quantifikation
von hoher Polysomie schwer zu erzielen ist (25-27).
Unabhängig davon zeigt unsere Studie, dass Analysen der EGFR-Genkopienanzahl
durch eine einzelne Gewebeproben nur sehr unwahrscheinlich ein gesamtes
Bronchialkarzinom repräsentieren können. Die Tumorgewebeproben auf unserem
TMA simulieren bis zu einem gewissen Grad die Vorgehensweise in der Klinik, in
der Biomarkeranalysen bei Bronchialkarzinomen mit sehr kleinen Biopsien
durchgeführt werden.
4.4 EGFR Amplifikationen
Wir konnten in 13 (9,0%) Bronchialkarzinomen hochgradige EGFR-
Genamplifikationen, ohne signifikante Unterschiede zwischen den verschiedenen
histologischen Subtypen nachweisen. Dies ist mit Ergebnissen anderer Studien17

vereinbar, die eine Genamplifikation in 6-13% der NSCLC festgestellt haben (10, 13,
24).
Die Diagnose von Genamplifikationen durch FISH ist gegenüber der von hoher
Polysomie einfach und mit sehr geringer Variabilität zu stellen (26). In 7 der 13
EGFR-amplifizierten NSCLC unserer Studie konnte ein heterogenes
Amplifikationsmuster nachgewiesen werden. Dies demonstriert noch klarer die hohe
Frequenz einer intratumoralen Heterogenität der EGFR-Genkopienanzahl.
Nur in 2 von 37 Tumoren mit hoher Polysomie konnte eine gleichzeitige EGFR-
Genamplifikation in anderen Tumorarealen nachgewiesen werden. Dies spricht eher
gegen die Hypothese, dass sich eine Amplifikation aus einer hohen Polysomie
entwickelt.
4.5 Vergleich Primärtumor vs Metastase
Daniele et al. und Sun et al. führten zwei Studien durch, welche mittels FISH die
Anzahl der EGFR-Genkopien in primären NSCLC und ihren Fernmetastasen
verglichen (28, 29). Sun et al. untersuchten 55 Primärtumore und ihre
korrespondierenden Gehirnmetastasen. Hierbei stimmten die Ergebnisse für hohe
Polysomie in 50% und die für Genamplifikation in 67% der Fälle nicht überein. Die
Autoren stellten eine deutlich höhere Frequenz von EGFR-Amplifikationen in
Gehirnmetastasen im Vergleich zu ihren korrespondierenden Primärtumoren fest.
Daniele et al. verglichen 38 NSCLC mit ihren korrespondierenden Gehirn- und
Nebennierenmetastasen. Zwei der Fälle wiesen eine EGFR-Amplifikation auf. Ein
Unterschied zwischen Primärtumor und Metastasen wurde hierbei nicht beschrieben.
Die Ergebnisse für hohe Polysomie waren in 53% der Fälle nicht übereinstimmend.
Es lag eine Tendenz höherer Polysomie in Metastasen verglichen mit den jeweiligen
Primärtumoren vor.
In unserer Studie konnten wir keinen signifikanten Anstieg von Genamplifikationen
oder hoher Polysomie in Lymphknotenmetastasen verglichen mit ihren
Primärtumoren beobachten.
18

4.6 Prädiktiver Wert
Zwar liegen uns keine klinischen Verlaufsdaten der Patienten vor, dennoch rufen
unsere Ergebnisse Zweifel an einem relevanten prädiktiven Wert von EGFR FISH
Untersuchungen hervor, welche an Biopsien durchgeführt werden.
In unserer Studie wurden 6 von 144 (4,2%) Tumoren mit homogenen EGFR-
Amplifikationen im Primärtumor und dessen Metastasen identifiziert. Angenommen
dass EGFR, mit massiver Gen-Amplifikation, eine entscheidende biologische Rolle in
diesen Krebszellen spielt, ist zu erwarten, dass zielgerichtetes Therapieren mit
Tyrosinkinaseinhibitoren oder monoklonalen Antikörpern eine effektive Strategie bei
solchen Tumoren darstellen könnte.
4.7 EGFR Mutationen
Mehrere klinische Studien haben eine positive Korrelation zwischen einer durch FISH
bewerteten, erhöhten Anzahl der EGFR-Genkopien und dem Ansprechen auf
EGFR-TKI gezeigt (13-16). In der großen Phase III INTEREST-Studie war eine hohe
EGFR-Genkopienanzahl jedoch nicht prädiktiv für ein, durch eine Gefitinibtherapie
erzieltes, progressionsfreies Überleben bei Patienten mit fortgeschrittenem NSCLC
(17, 18). Dementsprechend haben Ergebnisse der IPASS Studie nahegelegt, dass
der anscheinende Erfolg von Gefitinib, der bei Patienten mit hoher EGFR-
Genkopienanzahl beobachtet wurde, auf eine gleichzeitig vorhandene EGFR-
Mutation zurückzuführen ist (5). Es fand sich bei einem hohen Anteil (77,6%) der
Tumore der Patienten eine EGFR-Mutation. Patienten ohne diese gleichzeitige
Mutation haben nicht von einer Gefitinibtherapie profitiert.
Eine Mutation in der Tyrosinkinasedomäne vom EGFR-Gen ist der bisher am
stärksten prädiktive Wert für das Ansprechen auf eine Anti-EGFR-Therapie. Hierbei
ist zu beachten, dass möglicherweise auch Mutationen heterogen in Tumoren
ausgeprägt sein können (35, 47, 48).
Taniguchi et al. entnahm aus 21 NSCLC mit bekannter EGFR-Mutation jeweils 50-60
separate Gewebeproben, die er einzeln auf das Vorhandensein einer Mutation
untersuchte. Es wurden 15 homogene und 6 heterogene Fälle beschrieben. Bai et al.
untersuchte in einer Studie 45 EGFR-mutierte NSCLC auf eine solche Heterogenität.
Er analysierte jeweils 28-34 Foki pro Tumor. In ca 30% der Tumore zeigte sich eine
intratumorale Heterogenität für EGFR-Mutationen. Heterogene Fälle waren dabei im
19

Vergleich zu homogenen Fällen mit einer niedrigeren EGFR-Genkopienanzahl
assoziiert.
In unserer Studie wurde zur Mutationsanalyse der EGFR-amplifizierten NSCLC die
DNA von jeweils nur einem großen Gewebefokus, anstatt mehrerer kleiner
sequenziert. Eine Aussage zur Heterogenität dieser Mutationen lässt sich somit nicht
machen.
Die Heterogenität von EGFR-Mutationen ist seltener, als die von Änderungen der
EGFR-Genkopienanzahl (34, 36). Yatabe et al. legen daher nahe, dass EGFR-
Amplifikationen während der Tumorprogression in EGFR-mutierten
Adenokarzinomen erworben werden (37). Erhöhungen der EGFR-Genkopienanzahl
in Tumoren mit gleichzeitiger Mutation sind mit einem ansteigenden Verhältnis von
mutiertem zu Wildtypallel assoziiert, was impliziert, dass das mutierte Allel selektiv
amplifiziert wird (38).
Mehrere Studienergebnisse lassen vermuten, dass EGFR-Mutationen mit höherer
Frequenz in EGFR-amplifizierten NSCLC auftreten (30-32). Die berichtete Frequenz
von gleichzeitigen EGFR-Mutationen in EGFR-amplifizierten NSCLC hängt jedoch
stark vom untersuchten Patientenkollektiv ab. EGFR-Mutationen sind in spezifischen
Subgruppen wesentlich häufiger vorhanden. So z.B. bei Asiaten, Patienten mit
Adenokarzinomen, Nichtraucher und Frauen (57, 59). Auf der anderen Seite ist die
berichtete Frequenz der EGFR-Genkopienanzahl nicht abhängig vom ethnischen
Ursprung (33), oder histologischem Subtyp (13, 24). Wir fanden gleichzeitig
vorhandene EGFR-Mutationen in 4 von 13 amplifizierten Tumoren (30,8%). Dieses
Ergebnis ist mit anderen Berichten über unselektierte europäische Kohorten
vergleichbar. Capuzzo et al. fand in 6 von 9 amplifizierten NSCLC EGFR-Mutationen,
während die gesamtdurchschnittliche Mutationsfrequenz der untersuchten Kohorte
bei 17% lag (13).
EGFR-Mutationen kommen fast ausschließlich in Adenokarzinomen vor (39). Einige
Autoren schlagen deshalb eine Limitierung von EGFR-Testungen auf
Adenokarzinome vor (40). Es ist erwähnenswert, dass ein EGFR-mutiertes
Plattenepithelkarzinom in unserer Studie gefunden wurde. Diese Beobachtung
könnte ein Hinweis dafür sein, dass zumindest einige Patienten mit
Plattenepithelkarzinomen von einer TKI-Therapie profitieren könnten.
20

5 Fazit
Der EGF-Rezeptor ist ein etabliertes therapeutisches Ziel bei fortgeschrittenen nicht-
kleinzelligen Bronchialkarzinomen. EGFR-Mutationen sind der bisher stärkste
prädiktive Marker für ein positives Ansprechen auf eine Therapie mit
Tyrosinkinaseinhibitoren. Der prädiktive Wert einer erhöhten EGFR-
Genkopienanzahl ist bis heute nicht einheitlich geklärt.
Unsere Daten zeigen, dass Aberrationen der EGFR-Genkopienanzahl in nicht-
kleinzelligen Bronchialkarzinomen in den meisten Fällen heterogen sind. Dies trifft
insbesondere zu, wenn die EGFR-Aberration auf eine hohe Polysomie
zurückzuführen ist. In unserer Studie zeigten 100% der Tumore mit hoher Polysomie
(36 von 36) und 54% der Tumore mit einer EGFR-Amplifikation (7 von 13) eine
heterogene Ausprägung. Diese Beobachtung stellt das Konzept in Frage, dass
therapeutische Entscheidungen auf EGFR-FISH-Analysen basieren, welche durch
kleine Biopsien gewonnen wurden.
Diskrepante Studienergebnisse über den prädiktiven Wert von EGFR-Aberrationen
für molekular zielgerichtete Therapien mit Tyrosinkinaseinhibitoren (wie Erlotinib und
Gefitinib) oder monoklonalen Antikörpern (z.B. Cetuximab) können durchaus durch
eine solche intratumorale Heterogenität begründet sein.
21

6 Literaturverzeichnis
1. Nicholson RI, Gee JM, Harper ME: EGFR and cancer prognosis. Eur J
Cancer 37 Suppl 2001, 4:S9-15
2. Clark GM, Zborowski DM, Culbertson JL, et al: Clinical utility of
epidermal growth factor receptor expression for selecting patients with advanced
non-small cell lung cancer for treatment with erlotinib. J Thorac Oncol 2006, 1:837-46
3. Keedy VL, Temin S, Somerfield MR, et al: American Society of Clinical
Oncology provisional clinical opinion: epidermal growth factor receptor (EGFR)
Mutation testing for patients with advanced non-small-cell lung cancer considering
first-line EGFR tyrosine kinase inhibitor therapy. J Clin Oncol 2011, 29:2121-7
4. Pirker R, Pereira JR, von Pawel J, et al: EGFR expression as a
predictor of survival for first-line chemotherapy plus cetuximab in patients with
advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study.
Lancet Oncol 2011, 13:33–42
5. Fukuoka M, Wu YL, Thongprasert S, et al: Biomarker Analyses and
Final Overall Survival Results From a Phase III, Randomized, Open-Label, First-Line
Study of Gefitinib Versus Carboplatin/Paclitaxel in Clinically Selected Patients With
Advanced NSCLC in Asia (IPASS). J Clin Oncol 2011, 29:2866-74
6. Mitsudomi T, Morita S, Yatabe Y, et al: Gefitinib versus cisplatin plus
docetaxel in patients with non-small-cell lung cancer harbouring mutations of the
epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3
trial. Lancet Oncol 2010, 11:121-8
7. Zhou C, Wu YL, Chen G, et al: Erlotinib versus chemotherapy as first-
line treatment for patients with advanced EGFR mutation-positive non-small-cell lung
cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3
study. Lancet Oncol 2011, 12:735–42
8. Carlson JJ, Garrison LP, Ramsey SD, et al: Epidermal growth factor
receptor genomic variation in NSCLC patients receiving tyrosine kinase inhibitor
therapy: a systematic review and meta-analysis. J Cancer Res Clin Oncol 2009,
135:1483-93
9. Dahabreh IJ, Linardou H, Siannis F, et al: Somatic EGFR mutation and
gene copy gain as predictive biomarkers for response to tyrosine kinase inhibitors in
non-small cell lung cancer. Clin Cancer Res 2010, 16:291-303
22

10. Hirsch FR, Varella-Garcia M, Bunn PA, Jr., et al: Epidermal growth
factor receptor in non-small-cell lung carcinomas: correlation between gene copy
number and protein expression and impact on prognosis. J Clin Oncol 2003,
21:3798-807
11. Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton
S, et al.: Tissue microarrays for high-throughput molecular profiling of tumor
specimens. Nat Med 1998, 4:844–7
12. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, editors. World
Health Organization classification of tumors. Lyon: IARC Press; 2004. p. 9–124.
13. Cappuzzo F, Hirsch FR, Rossi E, et al: Epidermal growth factor
receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J
Natl Cancer Inst 2005, 97:643-55
14. Goss G, Ferry D, Wierzbicki R, et al: Randomized phase II study of
gefitinib compared with placebo in chemotherapy-naive patients with advanced non-
small-cell lung cancer and poor performance status. J Clin Oncol 2009, 27:2253-60
15. Hirsch FR, Varella-Garcia M, Bunn PA, Jr., et al: Molecular predictors of
outcome with gefitinib in a phase III placebo-controlled study in advanced non-small-
cell lung cancer. J Clin Oncol 2006, 24:5034-42
16. Hirsch FR, Varella-Garcia M, McCoy J, et al: Increased epidermal
growth factor receptor gene copy number detected by fluorescence in situ
hybridization associates with increased sensitivity to gefitinib in patients with
bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group Study. J Clin
Oncol 2005, 23:6838-45
17. Douillard JY, Shepherd FA, Hirsh V, et al: Molecular predictors of
outcome with gefitinib and docetaxel in previously treated non-small-cell lung cancer:
data from the randomized phase III INTEREST trial. J Clin Oncol 2010, 28:744-52
18. Kim ES, Hirsh V, Mok T, et al: Gefitinib versus docetaxel in previously
treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet
2008, 372:1809-18
19. Varella-Garcia M, Diebold J, Eberhard DA, et al: EGFR fluorescence in
situ hybridisation assay: guidelines for application to non-small-cell lung cancer. J
Clin Pathol 2009, 62:970-7
20. Wolff AC, Hammond ME, Schwartz JN, et al: American Society of
Clinical Oncology/College of American Pathologists guideline recommendations for
23

human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol
2007, 25:118-45
21. Varella-Garcia M: Stratification of non-small cell lung cancer patients for
therapy with epidermal growth factor receptor inhibitors: the EGFR fluorescence in
situ hybridization assay. Diagn Pathol 2006, 1:19
22. Hirsch FR, Varella-Garcia M, Dziadziuszko R, et al: Fluorescence in situ
hybridization subgroup analysis of TRIBUTE, a phase III trial of erlotinib plus
carboplatin and paclitaxel in non-small cell lung cancer. Clin Cancer Res 2008,
14:6317-23
23. Tsao MS, Sakurada A, Cutz JC, et al: Erlotinib in lung cancer -
molecular and clinical predictors of outcome. N Engl J Med 353:133-44, 2005
24. Morinaga R, Okamoto I, Fujita Y, et al: Association of epidermal growth
factor receptor (EGFR) gene mutations with EGFR amplification in advanced non-
small cell lung cancer. Cancer Sci 2008, 99:2455-60
25. Press MF, Sauter G, Buyse M, et al: Alteration of topoisomerase II-
alpha gene in human breast cancer: association with responsiveness to
anthracycline-based chemotherapy. J Clin Oncol 2011, 29:859-67
26. Sauter G, Lee J, Bartlett JM, et al: Guidelines for human epidermal
growth factor receptor 2 testing: biologic and methodologic considerations. J Clin
Oncol 2009, 27:1323-33
27. Sauter G, Moch H, Moore D, et al: Heterogeneity of erbB-2 gene
amplification in bladder cancer. Cancer Res 1993, 53:2199-203
28. Daniele L, Cassoni P, Bacillo E, et al: Epidermal growth factor receptor
gene in primary tumor and metastatic sites from non-small cell lung cancer. J Thorac
Oncol 2009, 4:684-8
29. Sun M, Behrens C, Feng L, et al: HER family receptor abnormalities in
lung cancer brain metastases and corresponding primary tumors. Clin Cancer Res
2009, 15:4829-37
30. Hirsch FR, Varella-Garcia M, Cappuzzo F, et al: Combination of EGFR
gene copy number and protein expression predicts outcome for advanced non-small-
cell lung cancer patients treated with gefitinib. Ann Oncol 2007, 18:752-60
31. Pinter F, Papay J, Almasi A, et al: Epidermal growth factor receptor
(EGFR) high gene copy number and activating mutations in lung adenocarcinomas
24

are not consistently accompanied by positivity for EGFR protein by standard
immunohistochemistry. J Mol Diagn 2008, 10:160-8
32. Sholl LM, Yeap BY, Iafrate AJ, et al: Lung adenocarcinoma with EGFR
amplification has distinct clinicopathologic and molecular features in never-smokers.
Cancer Res 2009, 69:8341-8
33. Li C, Sun Y, Fang Z, et al: Comprehensive analysis of epidermal growth
factor receptor gene status in lung adenocarcinoma. J Thorac Oncol 2011, 6:1016-21
34. Sakurada A, Lara-Guerra H, Liu N, et al: Tissue heterogeneity of EGFR
mutation in lung adenocarcinoma. J Thorac Oncol 2008, 3:527-9,
35. Taniguchi K, Okami J, Kodama K, et al: Intratumor heterogeneity of
epidermal growth factor receptor mutations in lung cancer and its correlation to the
response to gefitinib. Cancer Sci 2008, 99:929-35
36. Yatabe Y, Matsuo K, Mitsudomi T: Heterogeneous distribution of EGFR
mutations is extremely rare in lung adenocarcinoma. J Clin Oncol 2011, 29:2972-7
37. Yatabe Y, Takahashi T, Mitsudomi T: Epidermal growth factor receptor
gene amplification is acquired in association with tumor progression of EGFR-
mutated lung cancer. Cancer Res 2008, 68:2106-11
38. Takano T, Ohe Y, Sakamoto H, et al: Epidermal growth factor receptor
gene mutations and increased copy numbers predict gefitinib sensitivity in patients
with recurrent non-small-cell lung cancer. J Clin Oncol 2005, 23:6829-37
39. Marchetti A, Martella C, Felicioni L, et al: EGFR mutations in non-small-
cell lung cancer: analysis of a large series of cases and development of a rapid and
sensitive method for diagnostic screening with potential implications on
pharmacologic treatment. J Clin Oncol 2005, 23:857-65
40. Horn L, Pao W: EML4-ALK: honing in on a new target in non-small-cell
lung cancer. J Clin Oncol 2009, 27:4232-5
41. Mohammed Akli Ayoub, Heng B. See, Ruth M. Seeber, et al: Profiling
Epidermal Growth Factor Receptor and Heregulin Receptor 3 Heteromerization
Using Receptor Tyrosine Kinase Heteromer Investigation Technology. PLOS ONE
2013, 8(5):e64672
42. Monilola A.Olayioye, Richard M.Neve, et al: The ErbB signaling
network: receptor heterodimerization in development and cancer. EMBO J 2000,
19:3159-67
25

43. Markus BACH, Mark LARANCE, et al: The serine/threonine kinase
ULK1 is a target of multiple phosphorylation events. Biochem. J 2011, 440:283–291
44. Adi F. Gazdar, John D. Minna: Deregulated EGFR Signaling during
Lung Cancer Progression: Mutations, Amplicons, and Autocrine Loops. Cancer Prev
Res (Phila) 2008, 1(3):156–160
45. William Pao, Vincent Miller, Maureen Zakowski, et al: EGF receptor
gene mutations are common in lung cancers from ‘‘never smokers’’ and are
associated with sensitivity of tumors to gefitinib and erlotinib. PNAS 2004,
101(36):13306–13311
46. Lecia V. Sequist, Victoria A. Joshi, Pasi A. Jänne, et al: Epidermal
Growth Factor Receptor Mutation Testing in the Care of Lung Cancer Patients. Clin
Cancer Res 2006, 12:4403-4408
47. Akira Sakurada, Humberto Lara-Guerra, et al: Tissue Heterogeneity of
EGFR Mutation in Lung Adenocarcinoma. Journal of Thoracic Oncology 2008, 3(5):
527-579
48. Hua Bai, Zhijie Wang, Yuyan Wang, et al: Detection and Clinical
Significance of Intratumoral EGFR Mutational Heterogeneity in Chinese Patients with
Advanced Non-Small Cell Lung Cancer. PLOS ONE 2013, 8(2):e54170
49. Fang Wang, Sha Fu, Qiong Shao, et al: High EGFR copy number
predicts benefits from tyrosine kinase inhibitor treatment for non-small cell lung
cancer patients with wild-type EGFR. Journal of Translational Medicine, 11:90, 2013
50. Soley Bayraktar, Caio M Rocha-Lima: Molecularly targeted therapies for
advanced or metastatic non-small-cell lung carcinoma. World J Clin Oncol 2013, 4(2):
29-42
51. Hiroaki Kato, Tokuzo Arao, Kazuko Matsumoto, et al: Gene
amplification of EGFR, HER2, FGFR2 and MET in esophageal squamous cell
carcinoma. Int. Journal of Oncology 2013, 42:1151-1158
52. Krishna M. Talasila, Anke Soentgerath, Philipp Euskirchen, et al: EGFR
wild-type amplification and activation promote invasion and development of
glioblastoma independent of angiogenesis. Acta Neuropathol 2013, 125:683–698
53. J. Michael Randall, Anjali A. Bharne, Lyudmila A. Bazhenova:
Hypersensitivity reactions to carboplatin and cisplatin in non-small cell lung cancer. J
Thorac Dis 2013, 5(2):E53-E57
26

54. Rajeswaran A, Trojan A, Burnand B, Giannelli M.: Efficacy and side
effects of cisplatin- and carboplatin-based doublet chemotherapeutic regimens
versus non-platinum-based doublet chemotherapeutic regimens as first line
treatment of metastatic non-small cell lung carcinoma: a systematic review of
randomized controlled trials. Lung Cancer 2008, 59(1):1-11
55. Untch M, et al: Pathologic complete response after neoadjuvant
chemotherapy plus trastuzumab predicts favorable survival in human epidermal
growth factor receptor 2-overexpressing breast cancer: results from the TECHNO
trial of the AGO and GBG study groups. J Clin Oncol. 2011, 29(25):3351-7
56. Herbst RS, Prager D, Hermann R, et al: TRIBUTE: a phase III trial of
erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel
chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol. 2005,
23(25):5892-9
57. Bell DW, Brannigan BW, Matsuo K, et al: Increased prevalence of
EGFR-mutant lung cancer in women and in East Asian populations: analysis of
estrogen-related polymorphisms. Clin Cancer Res 2008, 14:4079-4084
58. Mukohara T, Engelman JA, Hanna NH, Yeap BY, et al: Differential
effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal
growth factor receptor mutations. J Natl Cancer Inst 2005, 97:1185-1194
59. Bell DW, Lynch TJ, Haserlat SM, et al: Epidermal growth factor receptor
mutations and gene amplification in non-small-cell lung cancer: molecular analysis of
the IDEAL/INTACT gefitinib trials. J Clin Oncol 2005, 23:8081-92
60. W. Siegenthaler, H. Blum: Klinische Pathophysiologie, Thieme 2006, 9:
1110
61. Ferlay J, Shin HR, Bray F, et al: Estimates of worldwide burden of
cancer in 2008: GLOBOCAN 2008. Int J of Cancer 2010, 127(12):2893-2917
62. Goulart B, Reys C, Fedorenko C, Mummy D, et al: Referral and
Treatment Patterns Among Patients With Stages III and IV Non-Small-Cell Lung
Cancer. J Oncol Pract 2013, 9(1): 42-50
27

7 Paper
28

Lung Cancer 79 (2013) 221– 227
Contents lists available at SciVerse ScienceDirect
Lung Cancer
j our na l ho me p age: www.elsev ier .com/ locate / lungcan
Frequent intratumoral heterogeneity of EGFR gene copy gain in non-small cell
lung cancer
Tobias J. Grob a,∗,1, Tobias Hoenig a,1, Till S. Clauditz a, Djordje Atanackovicb, Alexandra M. Koenigd,Yogesh K. Vashistd, Hans Kloseb, Ronald Simon a, Klaus Pantel c, Jakob R. Izbickid, Carsten Bokemeyerb,Guido Sauter a, Waldemar Wilczak a
a Department of Pathology, University Medical Center Hamburg-Eppendorf, Hamburg, Germanyb Department of Oncology, Hematology with Section Pneumology, Hubertus-Wald Tumorzentrum (University Cancer Center Hamburg), University Medical Center Hamburg-Eppendorf,
Hamburg, Germanyc Institute of Tumour Biology, University Medical Center Hamburg-Eppendorf, Hamburg, Germanyd Department of General, Visceral and Thoracic Surgery, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
a r t i c l e i n f o
Article history:
Received 23 April 2012
Received in revised form 8 November 2012
Accepted 11 November 2012
Keywords:
Non-small cell lung cancer
EGFR
Fluorescence in situ hybridization
Immunohistochemistry
Mutation
Heterogeneity
a b s t r a c t
Next to EGFR mutation, EGFR gene copy number evaluated by fluorescence in situ hybridization (FISH)
emerged as a potential predictive marker for sensitivity to EGFR tyrosine kinase inhibitors, although
controversial data exist. As the diagnostic accuracy of predictive biomarkers can be substantially limited
by regional differences within tumors, heterogeneity of EGFR gene copy gain in NSCLC was assessed in
this study. For this purpose, a novel tissue microarray (TMA) based analysis platform was developed.
TMAs were constructed containing 8 different tissue cylinders from 144 primary NSCLCs. From 62 of
these patients additional nodal metastases were sampled. EGFR gene copy number and EGFR expression
was analyzed by FISH and immunohistochemistry according to the suggested guidelines. 13 (9.0%) of the
144 evaluated tumors showed EGFR amplification and 37 (25.7%) tumors high polysomy in at least one
tumor area. In 7 (53.8%) of 13 amplified cases the analysis of different tumor areas revealed subclones
without EGFR gene copy gain next to subclones with amplification. All of the 36 evaluable tumors with
high polysomy showed heterogeneity of EGFR gene copy number with areas negative for gene copy
gain within the individual tumors. Heterogeneity of EGFR gene copy gain in lung cancer challenges the
concept of using small biopsies for the analysis of EGFR FISH status. EGFR gene copy number is highly
heterogeneous within individual NSCLCs and this finding might well be a reason for the controversial
clinical data existing regarding responsiveness to anti-EGFR therapy.
© 2012 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
The epidermal growth factor receptor (EGFR) is a receptor tyro-
sine kinase of the ErbB family and has been implicated in the
proliferation and survival of cancer cells. Aberrant expression of
EGFR has been detected in many human epithelial malignancies
including non-small cell lung cancer (NSCLC). Moreover EGFR over-
expression has been linked to advanced disease, a more aggressive
phenotype and overall poor prognosis in many tumor entities [1].
EGFR is the target for tyrosine kinase inhibitors (TKIs) such as
gefitinib or erlotinib and monoclonal antibodies such as cetuximab
∗ Corresponding author at: Institute of Pathology, University Medical Center
Hamburg-Eppendorf, Martinistr. 52, 20246 Hamburg, Germany.
Tel.: +49 40 7410 53173; fax: +49 40 7410 56815.
E-mail address: [email protected] (T.J. Grob).1 These authors contributed equally.
or panitumumab. The identification of the patients who are most
likely to derive clinical benefit from these drugs is of paramount
importance.
EGFR mutations, EGFR gene copy number and EGFR protein
expression have been studied as predictive biomarkers in major
clinical trials in NSCLC. EGFR protein expression yielded largely
disappointing results predicting response to TKI [2,3]. However,
recently published results from the FLEX study suggest EGFR
expression as a predictor for cetuximab therapy [4]. EGFR mutation
analysis has consistently emerged as strong predictor for response
to EGFR TKI therapy in NSCLC and lead to the introduction of first-
line EGFR TKI treatment for patients with activating EGFR mutations
[5–7]. Findings regarding EGFR gene copy number as a predictive
marker of response to TKIs, have been inconsistent [8,9]. In con-
trast to HER2 testing for trastuzumab therapy in breast cancer, it
is suggested that EGFR gene copy gain as a predictive marker is
not restricted to gene amplification but also to an increased EGFR
copy number due to balanced polysomy of chromosome 7 [10].
0169-5002/$ – see front matter © 2012 Elsevier Ireland Ltd. All rights reserved.
http://dx.doi.org/10.1016/j.lungcan.2012.11.009
222 T.J. Grob et al. / Lung Cancer 79 (2013) 221– 227
The criteria for EGFR positivity assessed by fluorescence in situ
hybridization (FISH) are either gene amplification or a minimum
subset of tumor cells (≥40%) with at least four copies of the EGFR
gene (high polysomy). This definition led to considerable obstacles
[11]. Especially the reproducibility using different methods such
as quantitative real-time PCR is limited [12]. Nevertheless, sev-
eral clinical trials showed positive correlation between increased
EGFR gene copy number as assessed by FISH and response to EGFR
TKI [13–16]. However, in the large phase III INTEREST study high
EGFR gene copy number was not predictive for progression-free
survival achieved by gefitinib treatment in patients with advanced
NSCLC [17,18]. Accordingly, results from the IPASS study suggest
that the apparent benefit of gefitinib seen in patients with high
EGFR gene copy number was driven by overlap with a coexisting
EGFR mutation [5].
Despite these considerable efforts a clear picture of the predic-
tive value of EGFR gene copy number has not yet emerged. Genetic
tumor heterogeneity represents a possible source for the controver-
sial results. Genetic heterogeneity might be particularly essential in
lung cancer because diagnosis is usually made on small biopsies or
even on a few cells in cases of cytology specimens. To better under-
stand the extent of such heterogeneity we analyzed 144 surgical
specimens of non-small cell lung cancers extensively on the EGFR
protein and gene copy number level using a novel tissue microarray
(TMA) based platform for heterogeneity analysis. Our results sug-
gest that substantial heterogeneity exists for EGFR amplification in
lung cancer and that high polysomy of chromosome 7 as assessed
by FISH is an unreliable predictive marker without consistency in
NSCLC.
2. Materials and methods
2.1. Tissues and construction of tissue micro arrays
Patients with extensive retained tumor material (at least 4 tis-
sue blocks per patient) were identified to analyze heterogeneity of
biomarker alterations in NSCLC. Among 373 patients reviewed for
availability of large tissue quantities, there were 144 patients for
whom at least 4 blocks of paraffin embedded formalin fixed tis-
sue had been stored at the archives of the Institute of Pathology
of the University Medical Center, Hamburg-Eppendorf. The num-
ber of available tissue blocks was 8 in 24, 7 in 12, 6 in 26, 5 in
32 and 4 in 50 patients. Eight different tissue cores were taken
per patient for TMA construction. Accordingly no more than two
cores were removed from one tissue block. Emphasis was placed
on taking these samples from areas that were as distant from each
other as possible. Up to 4 different lymph node metastases were
also examined from 62 (of the 144) patients with nodal positive
disease. One tissue core was analyzed per metastasis. The number
of analyzed metastases was 4 in 18, 3 in 11, 2 in 18, and 1 in 15
patients. TMAs were constructed as previously described [19]. The
usage of TMAs for research purposes has been approved by the local
ethics committee. Consecutive 4 �m sections of the resulting multi-
tumor TMA blocks were transferred to an adhesive coated slide
system (Instrumedics, Hackensack, USA) for fluorescence in situ
hybridization, immunohistochemical analysis and H&E stained ref-
erence. All original slides of the tissues included in these TMAs
had been reviewed determining histological type based on mor-
phology and histological grade according to WHO 2004 [20]. The
pathologic stage was obtained from the primary report of the Insti-
tute of Pathology. For four cases the pathologic stage is missing
due to incomplete lymph node sampling. The median patient age
at surgery was 65 years (range 38–82 years). An overview of the
clinical and pathological parameters of the studied cohort is given
in Table 1.
Table 1
Clinical and pathological characteristics of the study cohort. EGFR gene copy gain
(high polysomy or EGFR amplification) found in at least one tumor area.
High polysomy EGFR amplification
Total cases 144 37 (25.7%) 13 (9.0%)
Sex
Female 36 7 (19.4%) 5 (13.9%)
Male 108 30 (27.8%) 8 (7.4%)
Histologic type
Adenocarcinoma 79 22 (27.8%) 6 (7.6%)
Squamous cell
carcinoma
48 12 (25.0%) 5 (10.4%)
Adenosquamous
carcinoma
2 1 (50.0%) 1 (50.0%)
Large cell
undifferentated
carcinoma
15 2 (13.3%) 1 (6.7%)
Histologic grade
1 2 0 0
2 64 19 (29.7%) 4 (6.3%)
3 63 16 (25.4%) 8 (12.7%)
4 15 2 (13.3%) 1 (6.7%)
Stage (UICC)
Ia 12 3 (25.0%) 1 (8.3%)
Ib 29 6 (20.7%) 0
IIa 6 1 (16.7%) 0
IIb 37 11 (29.7%) 4 (10.8%)
IIIa 30 6 (20.0%) 3 (10.0%)
IIIb 19 6 (31.6%) 3 (15.8%)
IV 7 3 (42.9%) 1 (14.3%)
2.2. EGFR fluorescence in situ hybridization
FISH was performed on TMA sections using a commercial kit for
proteolytic slide pre-treatment (paraffin pre-treatment reagent kit,
Abbott Molecular, Wiesbaden, Germany). The Vysis LSI EGFR Spec-
trum Orange/CEP7 Spectrum Green Probe (Abbott Molecular) was
used for EGFR copy number detection according to manufacturer’s
instructions. Presence of tumor cells was verified in each spot by
comparison of an H&E stained consecutively cut reference section
of the TMA. Tumor cells of each spot were evaluated according to
the suggested guidelines for evaluation of EGFR gene copy gain [21].
2.3. EGFR immunohistochemistry
For EGFR protein detection, the EGFR pharmDX kit (Dako,
Glostrup, Denmark) was used according to the manufacturer’s
instructions. Membrane staining intensity in tumor cells was
scored in four categories (0–3+): no staining (0), weak staining (1+,
light brown membrane staining, visible only with high magnifica-
tion), intermediate staining (2+, between 1+ and 3+), and strong
staining (3+, dark-brown linear membrane staining).
2.4. EGFR sequencing
All EGFR amplified cases were sequenced for mutations in
the EGFR tyrosine-kinase domain (exons 18–21). Tumor DNA
was extracted from FFPE tissue after macrodissection or laser-
capture microdissection (PALM Micro Beam Laser System, Zeiss
MicroImaging, Munich, Germany) to ensure tumor cell content of
at least 70%. Extraction was performed as per standard protocols
(QiAmp DNA Mini Kit, Qiagen, Hilden, Germany). 100 ng of DNA
were subjected to PCR reactions using the AmpliTaq Gold PCR
mastermix (Applied Biosystems, Darmstadt, Germany) under the
conditions recommended by the manufacturer. Specific DNA frag-
ments of the individual exons were amplified using the following
primers: EGFR exon 18 forward 5′-GCTGAGGTGACCCTTGTCTC-
3′ and reverse 5′-TGGAGTTTCCCAAACACTCAG-3′; exon 19
T.J. Grob et al. / Lung Cancer 79 (2013) 221– 227 223
diffe
rent are
as
prim
ary
tum
or
corr
espondin
gm
eta
sta
se
s
#026
#028
#059
#021
#097
#091
#116
#038
#080
#089
#139
#023
#107
EGFR ampli fication
high po lysomy
EGFR FISH ne gative
test fail ure
A
diffe
rent are
as
prim
ary
tum
or
corr
espondin
gm
eta
sta
se
s
#100
#065
#085
#089
#033
#067
#101
#035
#020
#041
#052
#066
#082
#087
#088
#122
#132
#045
#139
#008
#012
#032
#040
#048
#054
#060
#072
#075
#102
#112
#115
#118
#125
#133
#009
#046
#144
B
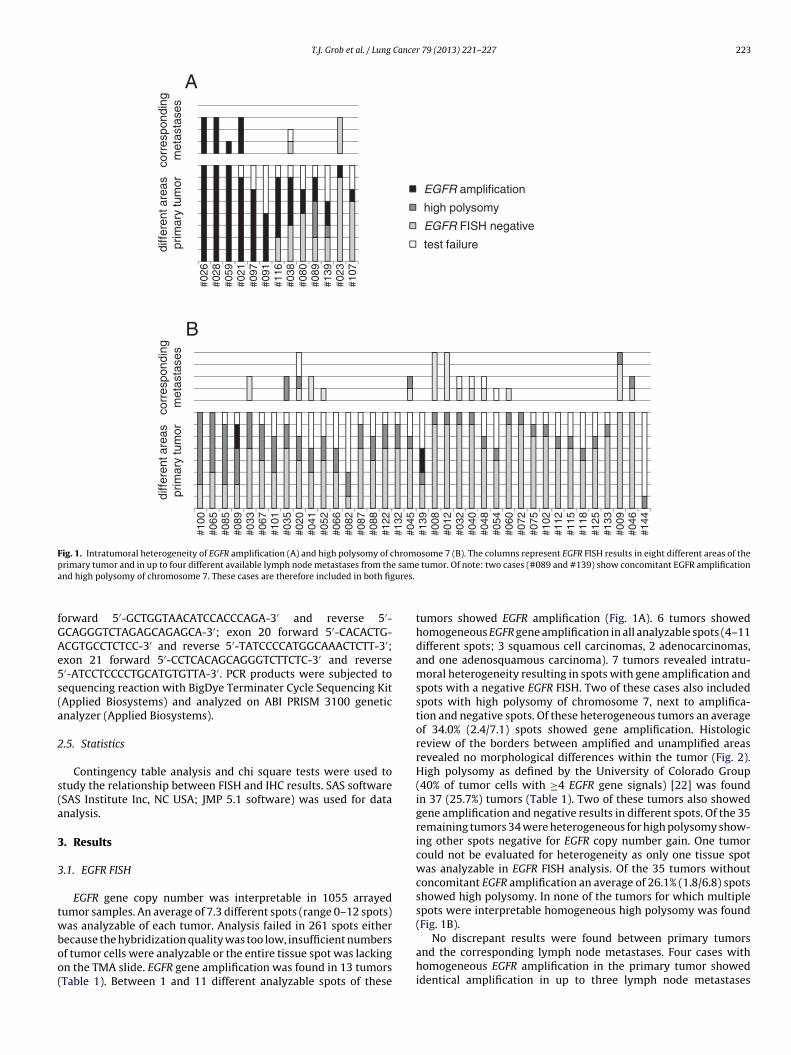
Fig. 1. Intratumoral heterogeneity of EGFR amplification (A) and high polysomy of chromosome 7 (B). The columns represent EGFR FISH results in eight different areas of the
primary tumor and in up to four different available lymph node metastases from the same tumor. Of note: two cases (#089 and #139) show concomitant EGFR amplification
and high polysomy of chromosome 7. These cases are therefore included in both figures.
forward 5′-GCTGGTAACATCCACCCAGA-3′ and reverse 5′-
GCAGGGTCTAGAGCAGAGCA-3′; exon 20 forward 5′-CACACTG-
ACGTGCCTCTCC-3′ and reverse 5′-TATCCCCATGGCAAACTCTT-3′;
exon 21 forward 5′-CCTCACAGCAGGGTCTTCTC-3′ and reverse
5′-ATCCTCCCCTGCATGTGTTA-3′. PCR products were subjected to
sequencing reaction with BigDye Terminater Cycle Sequencing Kit
(Applied Biosystems) and analyzed on ABI PRISM 3100 genetic
analyzer (Applied Biosystems).
2.5. Statistics
Contingency table analysis and chi square tests were used to
study the relationship between FISH and IHC results. SAS software
(SAS Institute Inc, NC USA; JMP 5.1 software) was used for data
analysis.
3. Results
3.1. EGFR FISH
EGFR gene copy number was interpretable in 1055 arrayed
tumor samples. An average of 7.3 different spots (range 0–12 spots)
was analyzable of each tumor. Analysis failed in 261 spots either
because the hybridization quality was too low, insufficient numbers
of tumor cells were analyzable or the entire tissue spot was lacking
on the TMA slide. EGFR gene amplification was found in 13 tumors
(Table 1). Between 1 and 11 different analyzable spots of these
tumors showed EGFR amplification (Fig. 1A). 6 tumors showed
homogeneous EGFR gene amplification in all analyzable spots (4–11
different spots; 3 squamous cell carcinomas, 2 adenocarcinomas,
and one adenosquamous carcinoma). 7 tumors revealed intratu-
moral heterogeneity resulting in spots with gene amplification and
spots with a negative EGFR FISH. Two of these cases also included
spots with high polysomy of chromosome 7, next to amplifica-
tion and negative spots. Of these heterogeneous tumors an average
of 34.0% (2.4/7.1) spots showed gene amplification. Histologic
review of the borders between amplified and unamplified areas
revealed no morphological differences within the tumor (Fig. 2).
High polysomy as defined by the University of Colorado Group
(40% of tumor cells with ≥4 EGFR gene signals) [22] was found
in 37 (25.7%) tumors (Table 1). Two of these tumors also showed
gene amplification and negative results in different spots. Of the 35
remaining tumors 34 were heterogeneous for high polysomy show-
ing other spots negative for EGFR copy number gain. One tumor
could not be evaluated for heterogeneity as only one tissue spot
was analyzable in EGFR FISH analysis. Of the 35 tumors without
concomitant EGFR amplification an average of 26.1% (1.8/6.8) spots
showed high polysomy. In none of the tumors for which multiple
spots were interpretable homogeneous high polysomy was found
(Fig. 1B).
No discrepant results were found between primary tumors
and the corresponding lymph node metastases. Four cases with
homogeneous EGFR amplification in the primary tumor showed
identical amplification in up to three lymph node metastases
224 T.J. Grob et al. / Lung Cancer 79 (2013) 221– 227
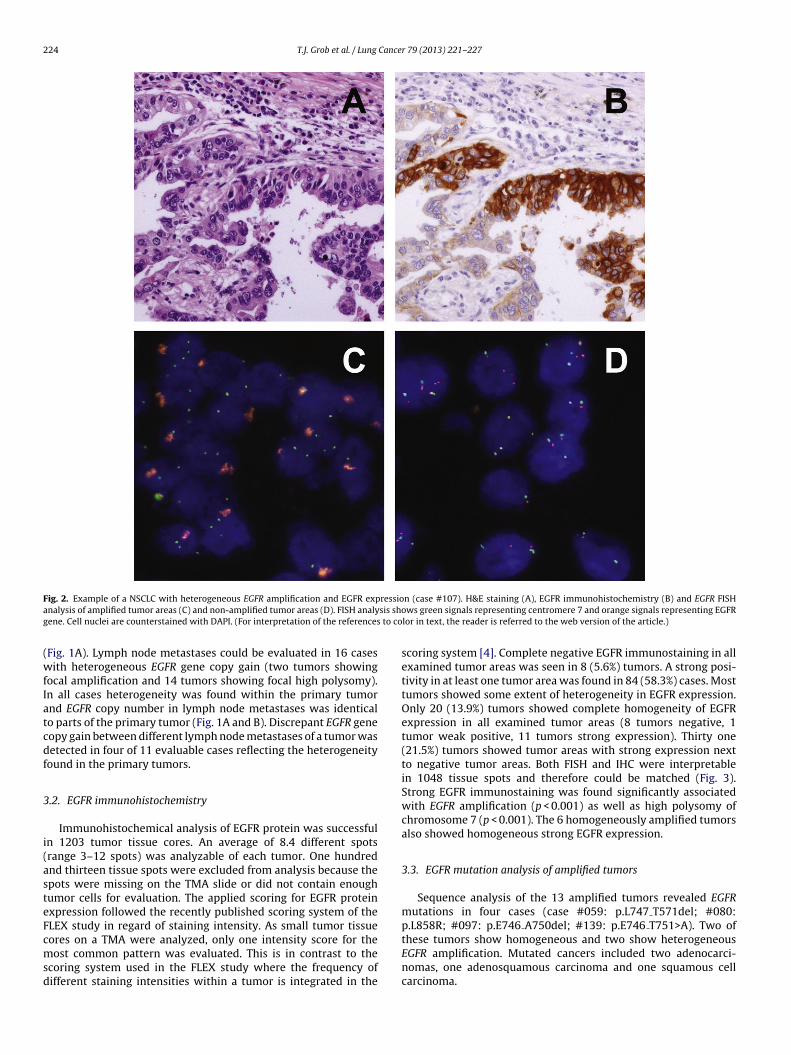
Fig. 2. Example of a NSCLC with heterogeneous EGFR amplification and EGFR expression (case #107). H&E staining (A), EGFR immunohistochemistry (B) and EGFR FISH
analysis of amplified tumor areas (C) and non-amplified tumor areas (D). FISH analysis shows green signals representing centromere 7 and orange signals representing EGFR
gene. Cell nuclei are counterstained with DAPI. (For interpretation of the references to color in text, the reader is referred to the web version of the article.)
(Fig. 1A). Lymph node metastases could be evaluated in 16 cases
with heterogeneous EGFR gene copy gain (two tumors showing
focal amplification and 14 tumors showing focal high polysomy).
In all cases heterogeneity was found within the primary tumor
and EGFR copy number in lymph node metastases was identical
to parts of the primary tumor (Fig. 1A and B). Discrepant EGFR gene
copy gain between different lymph node metastases of a tumor was
detected in four of 11 evaluable cases reflecting the heterogeneity
found in the primary tumors.
3.2. EGFR immunohistochemistry
Immunohistochemical analysis of EGFR protein was successful
in 1203 tumor tissue cores. An average of 8.4 different spots
(range 3–12 spots) was analyzable of each tumor. One hundred
and thirteen tissue spots were excluded from analysis because the
spots were missing on the TMA slide or did not contain enough
tumor cells for evaluation. The applied scoring for EGFR protein
expression followed the recently published scoring system of the
FLEX study in regard of staining intensity. As small tumor tissue
cores on a TMA were analyzed, only one intensity score for the
most common pattern was evaluated. This is in contrast to the
scoring system used in the FLEX study where the frequency of
different staining intensities within a tumor is integrated in the
scoring system [4]. Complete negative EGFR immunostaining in all
examined tumor areas was seen in 8 (5.6%) tumors. A strong posi-
tivity in at least one tumor area was found in 84 (58.3%) cases. Most
tumors showed some extent of heterogeneity in EGFR expression.
Only 20 (13.9%) tumors showed complete homogeneity of EGFR
expression in all examined tumor areas (8 tumors negative, 1
tumor weak positive, 11 tumors strong expression). Thirty one
(21.5%) tumors showed tumor areas with strong expression next
to negative tumor areas. Both FISH and IHC were interpretable
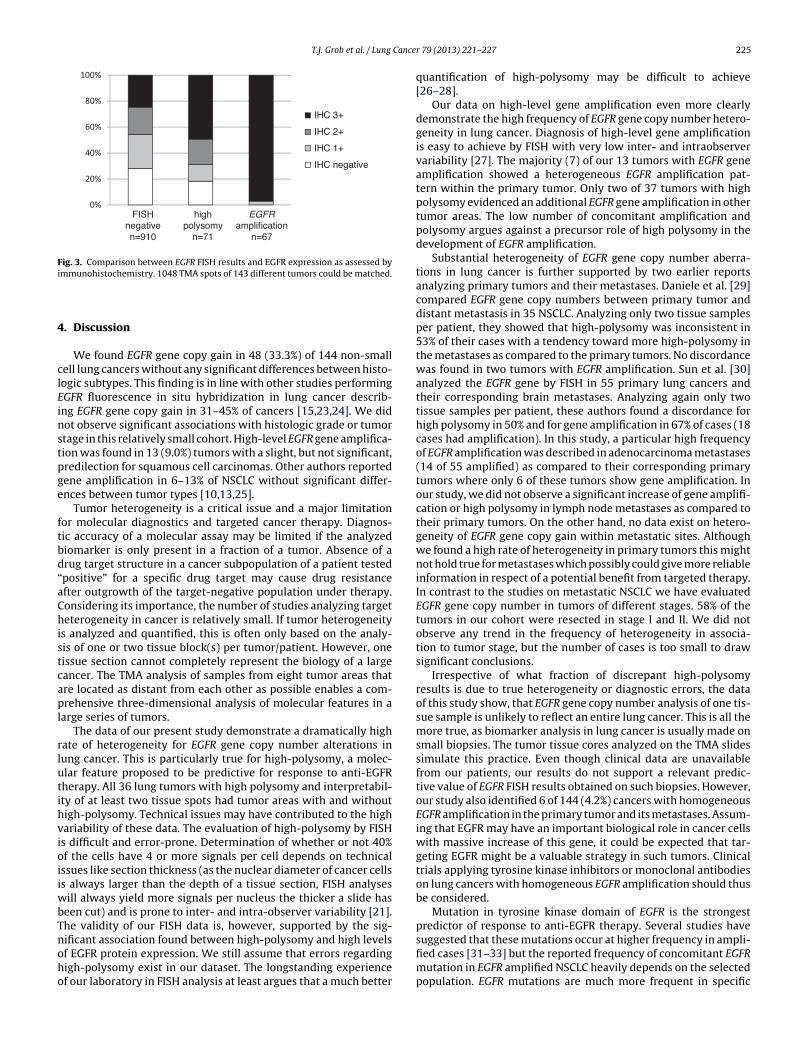
in 1048 tissue spots and therefore could be matched (Fig. 3).
Strong EGFR immunostaining was found significantly associated
with EGFR amplification (p < 0.001) as well as high polysomy of
chromosome 7 (p < 0.001). The 6 homogeneously amplified tumors
also showed homogeneous strong EGFR expression.
3.3. EGFR mutation analysis of amplified tumors
Sequence analysis of the 13 amplified tumors revealed EGFR
mutations in four cases (case #059: p.L747 T571del; #080:
p.L858R; #097: p.E746 A750del; #139: p.E746 T751>A). Two of
these tumors show homogeneous and two show heterogeneous
EGFR amplification. Mutated cancers included two adenocarci-
nomas, one adenosquamous carcinoma and one squamous cell
carcinoma.
T.J. Grob et al. / Lung Cancer 79 (2013) 221– 227 225
FISHnegativen=910
highpolysomy
n=71
EGFR
amplificatio nn=67
IHC 3+
IHC 2+
IHC 1+
IHC negative
Fig. 3. Comparison between EGFR FISH results and EGFR expression as assessed by
immunohistochemistry. 1048 TMA spots of 143 different tumors could be matched.
4. Discussion
We found EGFR gene copy gain in 48 (33.3%) of 144 non-small
cell lung cancers without any significant differences between histo-
logic subtypes. This finding is in line with other studies performing
EGFR fluorescence in situ hybridization in lung cancer describ-
ing EGFR gene copy gain in 31–45% of cancers [15,23,24]. We did
not observe significant associations with histologic grade or tumor
stage in this relatively small cohort. High-level EGFR gene amplifica-
tion was found in 13 (9.0%) tumors with a slight, but not significant,
predilection for squamous cell carcinomas. Other authors reported
gene amplification in 6–13% of NSCLC without significant differ-
ences between tumor types [10,13,25].
Tumor heterogeneity is a critical issue and a major limitation
for molecular diagnostics and targeted cancer therapy. Diagnos-
tic accuracy of a molecular assay may be limited if the analyzed
biomarker is only present in a fraction of a tumor. Absence of a
drug target structure in a cancer subpopulation of a patient tested
“positive” for a specific drug target may cause drug resistance
after outgrowth of the target-negative population under therapy.
Considering its importance, the number of studies analyzing target
heterogeneity in cancer is relatively small. If tumor heterogeneity
is analyzed and quantified, this is often only based on the analy-
sis of one or two tissue block(s) per tumor/patient. However, one
tissue section cannot completely represent the biology of a large
cancer. The TMA analysis of samples from eight tumor areas that
are located as distant from each other as possible enables a com-
prehensive three-dimensional analysis of molecular features in a
large series of tumors.
The data of our present study demonstrate a dramatically high
rate of heterogeneity for EGFR gene copy number alterations in
lung cancer. This is particularly true for high-polysomy, a molec-
ular feature proposed to be predictive for response to anti-EGFR
therapy. All 36 lung tumors with high polysomy and interpretabil-
ity of at least two tissue spots had tumor areas with and without
high-polysomy. Technical issues may have contributed to the high
variability of these data. The evaluation of high-polysomy by FISH
is difficult and error-prone. Determination of whether or not 40%
of the cells have 4 or more signals per cell depends on technical
issues like section thickness (as the nuclear diameter of cancer cells
is always larger than the depth of a tissue section, FISH analyses
will always yield more signals per nucleus the thicker a slide has
been cut) and is prone to inter- and intra-observer variability [21].
The validity of our FISH data is, however, supported by the sig-
nificant association found between high-polysomy and high levels
of EGFR protein expression. We still assume that errors regarding
high-polysomy exist in our dataset. The longstanding experience
of our laboratory in FISH analysis at least argues that a much better
quantification of high-polysomy may be difficult to achieve
[26–28].
Our data on high-level gene amplification even more clearly
demonstrate the high frequency of EGFR gene copy number hetero-
geneity in lung cancer. Diagnosis of high-level gene amplification
is easy to achieve by FISH with very low inter- and intraobserver
variability [27]. The majority (7) of our 13 tumors with EGFR gene
amplification showed a heterogeneous EGFR amplification pat-
tern within the primary tumor. Only two of 37 tumors with high
polysomy evidenced an additional EGFR gene amplification in other
tumor areas. The low number of concomitant amplification and
polysomy argues against a precursor role of high polysomy in the
development of EGFR amplification.
Substantial heterogeneity of EGFR gene copy number aberra-
tions in lung cancer is further supported by two earlier reports
analyzing primary tumors and their metastases. Daniele et al. [29]
compared EGFR gene copy numbers between primary tumor and
distant metastasis in 35 NSCLC. Analyzing only two tissue samples
per patient, they showed that high-polysomy was inconsistent in
53% of their cases with a tendency toward more high-polysomy in
the metastases as compared to the primary tumors. No discordance
was found in two tumors with EGFR amplification. Sun et al. [30]
analyzed the EGFR gene by FISH in 55 primary lung cancers and
their corresponding brain metastases. Analyzing again only two
tissue samples per patient, these authors found a discordance for
high polysomy in 50% and for gene amplification in 67% of cases (18
cases had amplification). In this study, a particular high frequency
of EGFR amplification was described in adenocarcinoma metastases
(14 of 55 amplified) as compared to their corresponding primary
tumors where only 6 of these tumors show gene amplification. In
our study, we did not observe a significant increase of gene amplifi-
cation or high polysomy in lymph node metastases as compared to
their primary tumors. On the other hand, no data exist on hetero-
geneity of EGFR gene copy gain within metastatic sites. Although
we found a high rate of heterogeneity in primary tumors this might
not hold true for metastases which possibly could give more reliable
information in respect of a potential benefit from targeted therapy.
In contrast to the studies on metastatic NSCLC we have evaluated
EGFR gene copy number in tumors of different stages. 58% of the
tumors in our cohort were resected in stage I and II. We did not
observe any trend in the frequency of heterogeneity in associa-
tion to tumor stage, but the number of cases is too small to draw
significant conclusions.
Irrespective of what fraction of discrepant high-polysomy
results is due to true heterogeneity or diagnostic errors, the data
of this study show, that EGFR gene copy number analysis of one tis-
sue sample is unlikely to reflect an entire lung cancer. This is all the
more true, as biomarker analysis in lung cancer is usually made on
small biopsies. The tumor tissue cores analyzed on the TMA slides
simulate this practice. Even though clinical data are unavailable
from our patients, our results do not support a relevant predic-
tive value of EGFR FISH results obtained on such biopsies. However,
our study also identified 6 of 144 (4.2%) cancers with homogeneous
EGFR amplification in the primary tumor and its metastases. Assum-
ing that EGFR may have an important biological role in cancer cells
with massive increase of this gene, it could be expected that tar-
geting EGFR might be a valuable strategy in such tumors. Clinical
trials applying tyrosine kinase inhibitors or monoclonal antibodies
on lung cancers with homogeneous EGFR amplification should thus
be considered.
Mutation in tyrosine kinase domain of EGFR is the strongest
predictor of response to anti-EGFR therapy. Several studies have
suggested that these mutations occur at higher frequency in ampli-
fied cases [31–33] but the reported frequency of concomitant EGFR
mutation in EGFR amplified NSCLC heavily depends on the selected
population. EGFR mutations are much more frequent in specific

226 T.J. Grob et al. / Lung Cancer 79 (2013) 221– 227
sub-groups such as the Asian population, patients with adeno-
carcinomas, non-smokers and females. On the other hand, the
reported frequency of EGFR gene copy gain is not dependent on
ethnic origin [34] or histologic subtype [13,25]. We found con-
comitant EGFR mutations in 4 of the 13 amplified tumors (30.8%).
This result is comparable with other reports on unselected cohorts
of Caucasian origin. Cappuzzo et al. found EGFR mutation in 6 of
9 amplified NSCLC, while the overall mutation frequency of the
examined cohort was 17% [13]. Evidence suggests that heterogene-
ity for EGFR mutation is less extensive than for EGFR gene copy
number changes [35–37]. Yatabe et al. therefore suggested that
EGFR gene amplification is acquired upon tumor progression in
EGFR mutated adenocarcinomas [38]. EGFR copy gains in tumors
with concomitant mutations are associated with an increasing ratio
of mutant to wild-type allele, implying that the mutant allele is
selectively gained [39].
5. Conclusion
In summary, our data demonstrate that EGFR gene copy number
alterations are mostly heterogeneous in lung cancer. This observa-
tion challenges the concept of using small biopsies for the analysis
of EGFR FISH status. Studies evaluating EGFR targeted therapy in
the small subgroup of homogeneously EGFR amplified lung cancers
appear to be warranted.
Conflict of interest statement
None declared.
Acknowledgement
We appreciate the excellent technical support of Silvia Schnöger
and Sascha Eghtessadi.
References
[1] Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer2001;37(Suppl 4):S9–15.
[2] Clark GM, Zborowski DM, Culbertson JL, Whitehead M, Savoie M, Seymour L,et al. Clinical utility of epidermal growth factor receptor expression for selectingpatients with advanced non-small cell lung cancer for treatment with erlotinib.J Thorac Oncol 2006;1:837–46.
[3] Keedy VL, Temin S, Somerfield MR, Beasley MB, Johnson DH, McShane LM, et al.American Society of Clinical Oncology provisional clinical opinion: epidermalgrowth factor receptor (EGFR) mutation testing for patients with advancednon-small-cell lung cancer considering first-line EGFR tyrosine kinase inhibitortherapy. J Clin Oncol 2011;29:2121–7.
[4] Pirker R, Pereira JR, von Pawel J, Krzakowski M, Ramlau R, Park K, et al. EGFRexpression as a predictor of survival for first-line chemotherapy plus cetuximabin patients with advanced non-small-cell lung cancer: analysis of data from thephase 3 FLEX study. Lancet Oncol 2012;13:33–42.
[5] Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V,et al. Biomarker analyses and final overall survival results from a phase III, ran-domized, open-label, first-line study of gefitinib versus carboplatin/paclitaxelin clinically selected patients with advanced non-small-cell lung cancer in Asia(IPASS). J Clin Oncol 2011;29:2866–74.
[6] Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefiti-nib versus cisplatin plus docetaxel in patients with non-small-cell lung cancerharbouring mutations of the epidermal growth factor receptor (WJTOG3405):an open label, randomised phase 3 trial. Lancet Oncol 2010;11:121–8.
[7] Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemother-apy as first-line treatment for patients with advanced EGFR mutation-positivenon-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011;12:735–42.
[8] Carlson JJ, Garrison LP, Ramsey SD, Veenstra DL. Epidermal growth factor recep-tor genomic variation in NSCLC patients receiving tyrosine kinase inhibitortherapy: a systematic review and meta-analysis. J Cancer Res Clin Oncol2009;135:1483–93.
[9] Dahabreh IJ, Linardou H, Siannis F, Kosmidis P, Bafaloukos D, Murray S. SomaticEGFR mutation and gene copy gain as predictive biomarkers for responseto tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res2010;16:291–303.
[10] Hirsch FR, Varella-Garcia M, Bunn Jr PA, Di Maria MV, Veve R, Bremmes RM,et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: cor-relation between gene copy number and protein expression and impact onprognosis. J Clin Oncol 2003;21:3798–807.
[11] Eberhard DA, Giaccone G, Johnson BE. Biomarkers of response to epidermalgrowth factor receptor inhibitors in non-small-cell lung cancer working group:standardization for use in the clinical trial setting. J Clin Oncol 2008;26:983–94.
[12] Bell DW, Lynch TJ, Haserlat SM, Harris PL, Okimoto RA, Brannigan BW, et al.Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials.J Clin Oncol 2005;23:8081–92.
[13] Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, et al. Epidermalgrowth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst 2005;97:643–55.
[14] Goss G, Ferry D, Wierzbicki R, Laurie SA, Thompson J, Biesma B, et al. Random-ized phase II study of gefitinib compared with placebo in chemotherapy-naivepatients with advanced non-small-cell lung cancer and poor performance sta-tus. J Clin Oncol 2009;27:2253–60.
[15] Hirsch FR, Varella-Garcia M, Bunn Jr PA, Franklin WA, Dziadziuszko R, ThatcherN, et al. Molecular predictors of outcome with gefitinib in a phase IIIplacebo-controlled study in advanced non-small-cell lung cancer. J Clin Oncol2006;24:5034–42.
[16] Hirsch FR, Varella-Garcia M, McCoy J, West H, Xavier AC, Gumerlock P, et al.Increased epidermal growth factor receptor gene copy number detected by flu-orescence in situ hybridization associates with increased sensitivity to gefitinibin patients with bronchioloalveolar carcinoma subtypes: a Southwest oncologygroup study. J Clin Oncol 2005;23:6838–45.
[17] Douillard JY, Shepherd FA, Hirsh V, Mok T, Socinski MA, Gervais R, et al. Molec-ular predictors of outcome with gefitinib and docetaxel in previously treatednon-small-cell lung cancer: data from the randomized phase III INTEREST trial.J Clin Oncol 2010;28:744–52.
[18] Kim ES, Hirsh V, Mok T, Socinski MA, Gervais R, Wu YL, et al. Gefitinib versusdocetaxel in previously treated non-small-cell lung cancer (INTEREST): a ran-domised phase III trial. Lancet 2008;372:1809–18.
[19] Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, et al.Tissue microarrays for high-throughput molecular profiling of tumor speci-mens. Nat Med 1998;4:844–7.
[20] Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, editors. World HealthOrganization classification of tumors. Lyon: IARC Press; 2004. p. 9–124.
[21] Varella-Garcia M, Diebold J, Eberhard DA, Geenen K, Hirschmann A, Kockx M,et al. EGFR fluorescence in situ hybridisation assay: guidelines for applicationto non-small-cell lung cancer. J Clin Pathol 2009;62:970–7.
[22] Varella-Garcia M. Stratification of non-small cell lung cancer patients for ther-apy with epidermal growth factor receptor inhibitors: the EGFR fluorescencein situ hybridization assay. Diagn Pathol 2006;1:19.
[23] Hirsch FR, Varella-Garcia M, Dziadziuszko R, Xiao Y, Gajapathy S, Skokan M,et al. Fluorescence in situ hybridization subgroup analysis of TRIBUTE, a phaseIII trial of erlotinib plus carboplatin and paclitaxel in non-small cell lung cancer.Clin Cancer Res 2008;14:6317–23.
[24] Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, et al. Erlotinibin lung cancer – molecular and clinical predictors of outcome. N Engl J Med2005;353:133–44.
[25] Morinaga R, Okamoto I, Fujita Y, Arao T, Sekijima M, Nishio K, et al. Associationof epidermal growth factor receptor (EGFR) gene mutations with EGFR ampli-fication in advanced non-small cell lung cancer. Cancer Sci 2008;99:2455–60.
[26] Press MF, Sauter G, Buyse M, Bernstein L, Guzman R, Santiago A, et al.Alteration of topoisomerase II-alpha gene in human breast cancer: associa-tion with responsiveness to anthracycline-based chemotherapy. J Clin Oncol2011;29:859–67.
[27] Sauter G, Lee J, Bartlett JM, Slamon DJ, Press MF. Guidelines for humanepidermal growth factor receptor 2 testing: biologic and methodologic con-siderations. J Clin Oncol 2009;27:1323–33.
[28] Sauter G, Moch H, Moore D, Carroll P, Kerschmann R, Chew K, et al.Heterogeneity of erbB-2 gene amplification in bladder cancer. Cancer Res1993;53:2199–203.
[29] Daniele L, Cassoni P, Bacillo E, Cappia S, Righi L, Volante M, et al. Epidermalgrowth factor receptor gene in primary tumor and metastatic sites from non-small cell lung cancer. J Thorac Oncol 2009;4:684–8.
[30] Sun M, Behrens C, Feng L, Ozburn N, Tang X, Yin G, et al. HER family recep-tor abnormalities in lung cancer brain metastases and corresponding primarytumors. Clin Cancer Res 2009;15:4829–37.
[31] Hirsch FR, Varella-Garcia M, Cappuzzo F, McCoy J, Bemis L, Xavier AC, et al.Combination of EGFR gene copy number and protein expression predicts out-come for advanced non-small-cell lung cancer patients treated with gefitinib.Ann Oncol 2007;18:752–60.
[32] Pinter F, Papay J, Almasi A, Sapi Z, Szabo E, Kanya M, et al. Epidermalgrowth factor receptor (EGFR) high gene copy number and activating muta-tions in lung adenocarcinomas are not consistently accompanied by positivityfor EGFR protein by standard immunohistochemistry. J Mol Diagn 2008;10:160–8.
[33] Sholl LM, Yeap BY, Iafrate AJ, Holmes-Tisch AJ, Chou YP, Wu MT, et al. Lungadenocarcinoma with EGFR amplification has distinct clinicopathologic andmolecular features in never-smokers. Cancer Res 2009;69:8341–8.
[34] Li C, Sun Y, Fang Z, Han X, Fang R, Zhang Y, et al. Comprehensive analysis ofepidermal growth factor receptor gene status in lung adenocarcinoma. J ThoracOncol 2011;6:1016–21.

T.J. Grob et al. / Lung Cancer 79 (2013) 221– 227 227
[35] Sakurada A, Lara-Guerra H, Liu N, Shepherd FA, Tsao MS. Tissue heterogeneityof EGFR mutation in lung adenocarcinoma. J Thorac Oncol 2008;3:527–9.
[36] Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K. Intratumor hetero-geneity of epidermal growth factor receptor mutations in lung cancer and itscorrelation to the response to gefitinib. Cancer Sci 2008;99:929–35.
[37] Yatabe Y, Matsuo K, Mitsudomi T. Heterogeneous distribution of EGFR muta-tions is extremely rare in lung adenocarcinoma. J Clin Oncol 2011;29:2972–7.
[38] Yatabe Y, Takahashi T, Mitsudomi T. Epidermal growth factor receptor geneamplification is acquired in association with tumor progression of EGFR-mutated lung cancer. Cancer Res 2008;68:2106–11.
[39] Takano T, Ohe Y, Sakamoto H, Tsuta K, Matsuno Y, Tateishi U, et al. Epidermalgrowth factor receptor gene mutations and increased copy numbers predictgefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J ClinOncol 2005;23:6829–37.

8 Lebenslauf
entfällt aus datenschutzrechtlichen Gründen
36

entfällt aus datenschutzrechtlichen Gründen
37

9 Eidesstattliche Versicherung
Ich versichere ausdrücklich, dass ich die Arbeit selbständig und ohne fremde Hilfeverfasst, andere als die von mir angegebenen Quellen und Hilfsmittel nicht benutztund die aus den benutzten Werken wörtlich oder inhaltlich entnommenen Stelleneinzeln nach Ausgabe (Auflage und Jahr des Erscheinens), Band und Seite desbenutzten Werkes kenntlich gemacht habe.Ferner versichere ich, dass ich die Dissertation bisher nicht einem Fachvertreter aneiner anderen Hochschule zur Überprüfung vorgelegt oder mich anderweitig umZulassung zur Promotion beworben habe.
Unterschrift: ......................................................................
38





























