Embed Size (px)
Citation preview

Listen to this manuscript’s
audio summary by
JACC Editor-in-Chief
Dr. Valentin Fuster.
J O U R N A L O F T H E AM E R I C A N C O L L E G E O F C A R D I O L O G Y VO L . 7 0 , N O . 1 1 , 2 0 1 7
P U B L I S H E D B Y E L S E V I E R O N B E H A L F O F T H E AM E R I C A N
C O L L E G E O F C A R D I O L O G Y F O U N D A T I O N
I S S N 0 7 3 5 - 1 0 9 7 / $ 3 6 . 0 0
h t t p : / / d x . d o i . o r g / 1 0 . 1 0 1 6 / j . j a c c . 2 0 1 7 . 0 7 . 7 5 0
REVIEW TOPIC OF THE WEEK
Unraveling Vascular InflammationFrom Immunology to Imaging
Heather L. Teague, PHD,a Mark A. Ahlman, MD,b Abass Alavi, MD, PHD,c Denisa D. Wagner, PHD,d
Andrew H. Lichtman, MD, PHD,e Matthias Nahrendorf, MD, PHD,f Filip K. Swirski, PHD,f Frank Nestle, MD,g
Joel M. Gelfand, MD, MSCE,h Mariana J. Kaplan, MD,i Steven Grinspoon, MD,j Paul M. Ridker, MD,e
David E. Newby, DM, PHD,k Ahmed Tawakol, MD, PHD,l Zahi A. Fayad, PHD,m Nehal N. Mehta, MD, MSCEl
ABSTRACT
Fro
Sci
syl
MagK
Ar
Me
Ce
Ma
Sin
mo
La
fro
wo
tre
Ph
Ph
res
coi
va
Ac
Dr
All
Ah
Ma
Inflammation is a critical factor in early atherosclerosis and its progression to myocardial infarction. The search for
valid surrogate markers of arterial vascular inflammation led to the increasing use of positron emission tomography/
computed tomography. Indeed, vascular inflammation is associated with future risk for myocardial infarction and can
be modulated with short-term therapies, such as statins, that mitigate cardiovascular risk. However, to better
understand vascular inflammation and its mechanisms, a panel was recently convened of world experts in
immunology, human translational research, and positron emission tomographic vascular imaging. This contemporary
review first strives to understand the diverse roles of immune cells implicated in atherogenesis. Next, the authors
describe human chronic inflammatory disease models that can help elucidate the pathophysiology of vascular
inflammation. Finally, the authors review positron emission tomography–based imaging techniques to characterize
the vessel wall in vivo. (J Am Coll Cardiol 2017;70:1403–12) Published by Elsevier on behalf of the American College
of Cardiology Foundation.
m the aNational Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Maryland; bRadiology and Imaging
ences, Clinical Center, National Institutes of Health, Bethesda, Maryland; cUniversity of Pennsylvania, Philadelphia, Penn-
vania; dHarvard Medical School, Boston, Massachusetts; eBrigham and Women’s Hospital and Harvard Medical School, Boston,
ssachusetts; fCenter for Systems Biology, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts;
ings College, London, United Kingdom; hPerelman School of Medicine, Philadelphia, Pennsylvania; iNational Institute of
thritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, Maryland; jProgram in Nutritional
tabolism, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts; kBritish Heart Foundation
ntre for Cardiovascular Science, University of Edinburgh, Edinburgh, United Kingdom; lCardiovascular Research Center,
ssachusetts General Hospital and Harvard Medical School, Boston, Massachusetts; and the mIcahn School of Medicine at Mount
ai, New York, New York. Dr. Nestle is an employee of Sanofi. Dr. Gelfand has served as a consultant for Coherus (data and safety
nitoring board), Dermira, Janssen Biologics, Merck (data and safety monitoring board), Novartis, Regeneron, Dr. Reddy’s
boratories, Sanofi, and Pfizer; has received honoraria and research grants (to the Trustees of the University of Pennsylvania)
m Abbvie, Janssen, Novartis, Regeneron, Sanofi, Celgene, and Pfizer; has received payment for continuing medical education
rk related to psoriasis that was supported indirectly by Eli Lilly and Abbvie; and coholds a patent on resiquimod for the
atment of cutaneous T cell lymphoma. Dr. Grinspoon has served as a consultant for and received honoraria from Navidea
armaceuticals and Theratechnologies; and has received research grants (to the Massachusetts General Hospital) from Navidea
armaceuticals, KOWA Pharmaceuticals, Gilead Sciences, and Theratechnologies. Dr. Ridker has received investigator-initiated
earch support from the National Heart, Lung, and Blood Institute, Pfizer, AstraZenenca, Novartis, and Kowa; and is listed as a
nventor on patents held by the Brigham and Women’s Hospital that relate to the use of inflammatory biomarkers in cardio-
scular disease and diabetes that have been licensed to AstraZeneca and Siemens. Dr. Tawakol has served as a consultant for
telion Pharmaceuticals; and has received research grants (to the Massachusetts General Hospital) from Actelion and Genentech.
. Mehta is a full-time U.S. government employee; and has received research grants from Abbvie, Janssen, Novartis, and Celgene.
other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Drs. Teague and
lman contributed equally to this work. James Rudd, PhD, served as Guest Editor for this paper.
nuscript received June 16, 2017; revised manuscript received July 20, 2017, accepted July 20, 2017.

ABBR EV I A T I ON S
AND ACRONYMS
CT = computed tomographic
CV = cardiovascular
CVD = cardiovascular disease
FDG = 18F-fluorodeoxyglucose
HIV = human immunodeficiency
virus
IFN = interferon
MI = myocardial infarction
MR = magnetic resonance
NaF = 18F–sodium fluoride
NET = neutrophil extracellular
trap
PAD4 = peptidylarginine
deiminase–4
PET = positron emission
tomographic
SLE = systemic lupus
erythematosus
VI = vascular inflammation
Teague et al. J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7
Immunology of Vascular Inflammation S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2
1404
C ardiovascular disease (CVD) remainsthe leading cause of death world-wide, highlighting the need to eluci-
date its pathogenesis. Once considered apassive biological process, CVD is now recog-nized as an active, immune-driven processthat may begin in childhood (1). Currentresearch into the natural history of atheromadevelopment has implicated many immunecells, including phagocytes, lymphocytes,dendritic cells, and neutrophils (2). Becausethese cells play a major role in initiating pla-que development and complication, leuko-cytes are promising targets for acute andchronic atherosclerosis therapy. However,the complexity of the immune system andits role as a defensive force against infectionrequires novel tools to precisely identify andtreat only the inflammatory cells or processesthat promote atherosclerosis. Biomedicalengineering, specifically in human imaging,offers unique possibilities for diagnosing
and treating atherosclerotic plaque inflammationbefore cardiovascular (CV) events. Thus, interfacingnovel engineering to enhance human imaging withimmunology will be essential to accelerate advancesin management of this disease.
In this review, we begin with an overview of theemerging understanding of CVD as a systemicinflammatory disorder relating to monocytes, mac-rophages, neutrophils, and T cells. We then discussspecific human conditions with increased CVD risk tostudy the natural history of atherosclerosis, includinghuman immunodeficiency virus (HIV), systemic lupuserythematosus (SLE), and psoriasis as human modelsof vascular disease initiation and progression.Finally, we review current applications of positronemission tomographic (PET) imaging and emergingPET tracer agents used in vascular characterization.
IMMUNOLOGY OF INFLAMMATION AS IT
PERTAINS TO THE VESSEL WALL
MONOCYTES AND MACROPHAGES. The most numerouscells in atherosclerotic plaques are macrophages (3),leukocytes that are central to innate immunity. Inatherosclerosis, macrophage accumulation com-mences as bone marrow–derived, Ly6Chigh mono-cytes infiltrate the lesion. These Ly6Chighinflammatorymonocytes exit the bonemarrow in a C-Cmotif chemokine receptor 2–dependent manner,accumulate in the vessel wall, and differentiate intomacrophages, which are sustained through self-renewal (4). Notably, as atherosclerosis progresses,
local macrophage proliferation, rather than monocyterecruitment, becomes more important in lesiongrowth.
In addition to the accumulation of monocytes inatherosclerotic lesions, these innate immune cellscontribute to the biological response following amyocardial infarction (MI). Monocytes are bothdestructive and protective, in that they give rise toinfarct rupture and contribute to infarct healing.However, an overabundance of monocytes caninterfere with healing, resulting in heart failure. In anacute MI, an oversupply of monocytes to the aorta israpid, concomitant with a reduction in C-X-C motifchemokine ligand 12 expression in the bone marrow.Diminished C-X-C motif chemokine ligand 12 expres-sion enables myeloid cells and their progenitors to exitthe bone marrow and take up residence in the spleen,where they trigger extramedullary hematopoiesis (5).Additionally, differentiated leukocytes, especiallymonocytes and neutrophils, take up residence in end-organ tissues, giving rise to plaques and inflammation.The vascular sympathetic innervation (i.e., nervefibers traveling along the aorta and arterioles) plays arole in the increased emergency supply of leukocytes.In the periphery, sympathetic innervation activatesendothelial cells on the luminal surface of athero-sclerotic plaques, increasing adhesion moleculeexpression, which augments leukocyte recruitment(6). To directly investigate the role of the vascularsympathetic innervation system in atherogenesis, therole of stress in monocyte proliferation is a topic ofongoing investigation. Stress elevates noradrenalinelevels in the bone marrow and activates bone marrowstem cells (7). Hematopoietic stem and progenitor cellproliferation is significantly enhanced, C-X-C motifchemokine ligand 12 is lowered, and monocytes enterthe systemic circulation in increased numbers.Collectively, these mechanisms suggest a multiorgancommunication system that activates the bonemarrow through the sympathetic innervation system,increasing hematopoietic stem and progenitor cellproliferation and thus enhancing leukocytosis.Therefore, current studies aim to unravel the systemicmechanisms that control the production, recruitment,differentiation, and proliferation of monocytes andtheir descendent macrophages in atherosclerosis, todetermine how these processes can be balanced toexert themost benefit, and to elucidate specific controlpoints in atherogenesis.
NEUTROPHILS. Neutrophils are increasingly recog-nized in the initiation of atherosclerotic plaquedevelopment. Neutrophils are the initial immune cellto infiltrate inflammatory sites produced by injury or

FIGURE 1 Formation of Neutrophil Extracellular Traps Induces Vascular Damage
Neutrophil extracellular traps (NETs) in the timeline of deep vein thrombosis (DVT): a model. (A) DVT is initiated by local hypoxia and
activation of endothelial cells (ECs) as a result of flow restriction and disturbances. Activated endothelium releases ultralarge von Willebrand
factor (ULVWF) and P-selectin from Weibel-Palade bodies (WPBs), which mediate platelet and neutrophil adhesion. Activated platelets recruit
tissue factor (TF)–containing microparticles that enhance thrombin generation in the growing thrombus. (B) Activated platelets and
endothelium or other stimulus induce NET formation in adherent neutrophils. NETs provide an additional scaffold for platelet and red blood
cell (RBC) adhesion, promote fibrin formation, and exacerbate platelet and endothelial activation. (C) Plasmin, a disintegrin and
metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13), and deoxyribonuclease (DNase) mediate thrombolysis by
degrading fibrin, ULVWF, and deoxyribonucleic acid, respectively. Monocytes/macrophages (Mø) release an additional source of DNase and
generate plasmin and promote restoration of blood flow (10).
J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7 Teague et al.S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2 Immunology of Vascular Inflammation
1405
infection. Although the antimicrobial action of neu-trophils is indispensable to combat infection, theirmechanisms of action yield significant tissue damageand toxic debris. A newly identified defense mecha-nism is the ability of neutrophils to form neutrophilextracellular traps (NETs) (8); however, NET forma-tion, or “NETosis,” remains poorly understood.Currently, 2 distinct mechanisms have beendescribed, suicidal and vital NETosis. During suicidalNETosis, the internal membranes of the neutrophilsdissolve, followed by cell lysis and decondensedchromatin release (8). During an infectious process,neutrophils directly secrete nuclear contents from thecell without killing the neutrophils (9). Althoughmany questions remain unanswered regarding
NETosis, it is evident that NETs themselves areproinflammatory, induce endothelial and tissuedamage, and are highly prothrombotic (Figure 1) (10).Additionally, NETs provide a communication plat-form between neutrophils and macrophages, primingmacrophages to produce pro–interleukin-1b, culmi-nating in atherosclerotic plaque destabilization (11).
NET formation is a peptidylarginine deiminase–4(PAD4)–dependent mechanism induced by extracel-lular stimuli via microbes, activated platelets, cyto-kines, antibodies to neutrophils, hypoxia, andcholesterol crystals. Upon activation, PAD4 convertsarginine residues on histones to citrulline, triggeringchromatin decondensation and NET release (12).Consequently, PAD4 is expelled in conjunction with
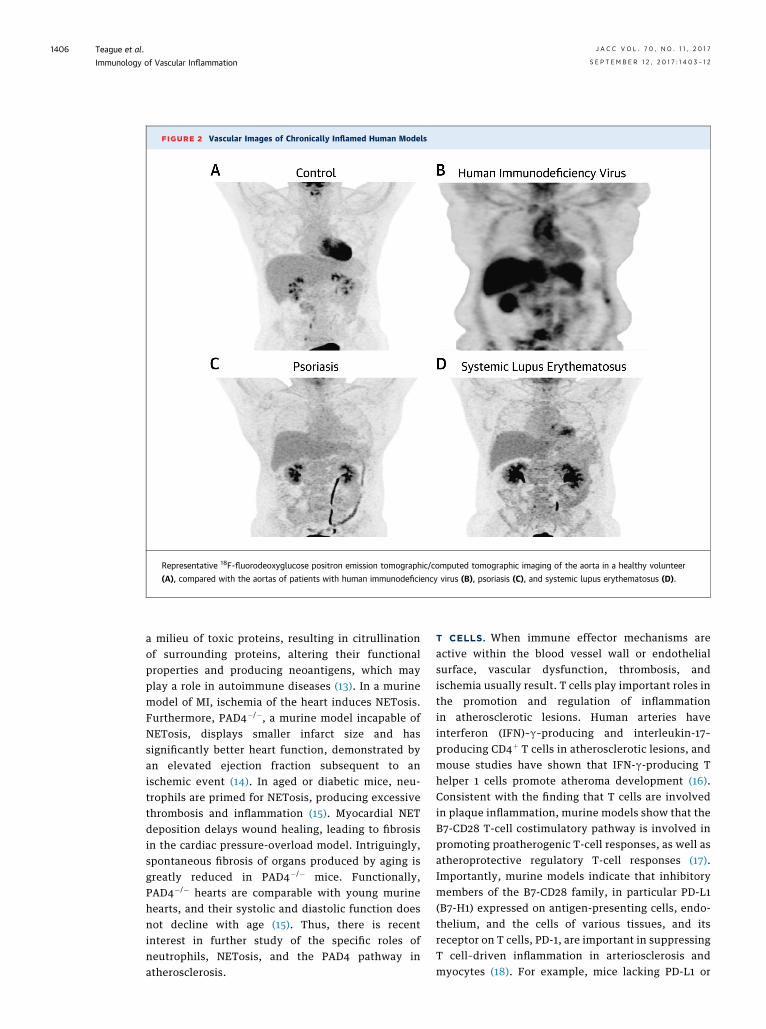
FIGURE 2 Vascular Images of Chronically Inflamed Human Models
Representative 18F-fluorodeoxyglucose positron emission tomographic/computed tomographic imaging of the aorta in a healthy volunteer
(A), compared with the aortas of patients with human immunodeficiency virus (B), psoriasis (C), and systemic lupus erythematosus (D).
Teague et al. J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7
Immunology of Vascular Inflammation S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2
1406
a milieu of toxic proteins, resulting in citrullinationof surrounding proteins, altering their functionalproperties and producing neoantigens, which mayplay a role in autoimmune diseases (13). In a murinemodel of MI, ischemia of the heart induces NETosis.Furthermore, PAD4�/�, a murine model incapable ofNETosis, displays smaller infarct size and hassignificantly better heart function, demonstrated byan elevated ejection fraction subsequent to anischemic event (14). In aged or diabetic mice, neu-trophils are primed for NETosis, producing excessivethrombosis and inflammation (15). Myocardial NETdeposition delays wound healing, leading to fibrosisin the cardiac pressure-overload model. Intriguingly,spontaneous fibrosis of organs produced by aging isgreatly reduced in PAD4�/� mice. Functionally,PAD4�/� hearts are comparable with young murinehearts, and their systolic and diastolic function doesnot decline with age (15). Thus, there is recentinterest in further study of the specific roles ofneutrophils, NETosis, and the PAD4 pathway inatherosclerosis.
T CELLS. When immune effector mechanisms areactive within the blood vessel wall or endothelialsurface, vascular dysfunction, thrombosis, andischemia usually result. T cells play important roles inthe promotion and regulation of inflammationin atherosclerotic lesions. Human arteries haveinterferon (IFN)–g–producing and interleukin-17–producing CD4þ T cells in atherosclerotic lesions, andmouse studies have shown that IFN-g-producing Thelper 1 cells promote atheroma development (16).Consistent with the finding that T cells are involvedin plaque inflammation, murine models show that theB7-CD28 T-cell costimulatory pathway is involved inpromoting proatherogenic T-cell responses, as well asatheroprotective regulatory T-cell responses (17).Importantly, murine models indicate that inhibitorymembers of the B7-CD28 family, in particular PD-L1(B7-H1) expressed on antigen-presenting cells, endo-thelium, and the cells of various tissues, and itsreceptor on T cells, PD-1, are important in suppressingT cell–driven inflammation in arteriosclerosis andmyocytes (18). For example, mice lacking PD-L1 or

FIGURE 3 Fluorine-18–Sodium Fluoride Uptake in the
Coronary Arteries
A discrete focus of 18F–sodium fluoride uptake overlying an
otherwise heavily calcified left anterior descending coronary
artery, suggesting a locus of active calcification with potentially
increased vulnerability for rupture (arrow).
J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7 Teague et al.S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2 Immunology of Vascular Inflammation
1407
PD-1 display a marked increase in CD4þ- and CD8þ-mediated inflammation in arterial lesions. Mice lack-ing PD-1 or PD-L1, or treated with PD-1 blockade, aremore susceptible to T cell–mediated myocardialinjury. Furthermore, IFN-g-induced up-regulation ofPD-L1 by heart endothelial cells in vitro or in vivoprotects against CD8þ cytotoxic T lymphocyte–mediated damage (19).
Mouse studies demonstrating the protective rolesof the PD-1 pathway in arteries and heart highlight thepossibility of increasing CV risks by targeting PD-1 orPD-L1 in cancer immunotherapy. In fact, many casesof acute severe lymphocytic myocarditis are nowbeing reported in the context of checkpoint blockadecancer immunotherapy, including patients treatedwith anti-PD-1 (20). Histopathologic analyses oftissues from some of these cases reveal up-regulationof endothelial human leukocyte antigen–antigen Drelated, as well as myocyte and endothelial PD-L1associated with T-cell infiltrates, consistent with anIFN-mediated effect.
Notably, CVD initiation and progression involvebiological activity from multiple immune cells, bothinnate and adaptive. Future studies deciphering theinterplay among these immune cells in CVD are crit-ical for developing therapies targeting CVD initiationin order to reduce clinical CVD outcomes andultimately decrease CVD prevalence.
HUMAN DISEASE MODELS OF IMAGING TO
STUDY VASCULAR INFLAMMATION
Currently, 2 large ongoing CV trials in patients withprior MI are testing if treatment of inflammation willreduce a second CV event: CIRT (CardiovascularInflammation Reduction Trial) and inhibition ofinterleukin-1B in CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study). There isan initial report that CANTOS met the primaryendpoint for a reduction in recurrent major adverse CVevents. These trials will provide critical data onwhether inhibition of nonspecific T-cell inflammation(methotrexate) or inflammasome activation (canaki-numab) reduces further CV events in high-risk pa-tients. As the results of these trials become available,emerging data from human chronic diseases associ-ated with high CV risk and systemic inflammationprovide models to understand vascular disease initia-tion and progression. Indeed, 18F-fluorodeoxyglucose(FDG) PET/computed tomographic (CT) imaging hasbeen used to characterize vascular inflammation (VI)in HIV infection, psoriasis, and SLE (Figure 2).HUMAN IMMUNODEFICIENCY VIRUS. HIV treatmenthas become very effective over the past decade;however, the rate of MI, stroke, and sudden cardiacdeath remains elevated 50% to 100% in HIV infection(21). Although dyslipidemia is more prevalent in HIVinfection, traditional risks, including dyslipidemia,hypertension, and diabetes, only account for 25% ofthe excess risk. Using coronary CT angiography, astudy demonstrated a high prevalence of noncalcifiedplaque with high-risk morphological features, asso-ciated with increased immune activation indexes (22)and inflammation, on FDG PET imaging thatremained elevated after effective antiretroviraltherapy (23).
Among patients with HIV infection, coronary pla-ques are often inflamed and noncalcified (24), andpatients exhibit increased myocardial fibrosis imagedby magnetic resonance (MR) (25). Imaging with FDGhas demonstrated that HIV-infected patients with anundetectable viral load have increased aortic target-to-background ratios compared with healthy controlsubjects and subjects with known CVD, indicatingsignificant arterial inflammation, even in the contextof immune restoration and viral suppression(Figure 2) (26). Together, coronary CT angiographyand FDG PET imaging have helped identify theunique pathophysiology of arterial inflammation andplaque in HIV infection, proved to be a readout for theefficacy of anti-inflammatory strategies now beingtargeted in HIV infection, including newer statins.Statins effectively decrease noncalcified plaque as

TABLE 1 A Summary of Agents and Their Potential Mechanisms of Uptake Applicable to
Vascular Inflammation Imaging
Agent (Ref. #) Potential Mechanism of Uptake
18F-FLT (62) Structural analogue of thymidine, images DNA synthesis withinatheroma
11C-PK11195 (63) Affinity for translocator protein, upregulated on inflammatorycells
18F-A85380 (64) Binds arterial nicotinic acetylcholine receptors, possibly relatedto vascular damage
18F-choline (65) Images increased cell wall synthesis within atheroma68Ga-DOTA-octreotate (60) Affinity for somatostatin receptors, which are highly expressed
on macrophages64Cu-ATSM (66) Trapped within cells in hypoxic state18F-MISO (67) Trapped within cells in hypoxic state68Ga-NOTA-RGD (68) Images neoangiogenesis as a result of hypoxia or chronic
inflammation64Cu-DOTA-CANF (69) Images neoangiogenesis via natriuretic peptide receptor
affinity18F-FDG A glucose analogue imaging increased metabolic rate in the
presence of inflammation and hypoxia18F–sodium fluoride (59) Images active calcification as a result of necrosis or
inflammation68Ga-CXCR4 (70) Images CXCR4 receptor expressed by inflammatory cells18F-florbetapen (61) Imaging b-amyloid plaque as a component of inflammation
11C-PK11195 ¼ 11C-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinolinecarboxamide; A85380 ¼3-([2S]-azetidinylmethoxy)pyridine dihydrochloride; ATSM ¼ diacetyl-bis(N-methylthiosemicarbazone;CXCR4 ¼ C-X-C chemokine receptor type 4; DOTA-CANF ¼ 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraaceticacid atrial natriuretic factor; FDG ¼ fluorodeoxyglucose; FLT ¼ fluorothymidine; MISO ¼ fluoromisonidazole;NOTA-RGD ¼ 1,4,7-triazacyclononane-N,N0 ,N00-triacetic acid arginine-glycine-aspartate.
Teague et al. J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7
Immunology of Vascular Inflammation S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2
1408
well as low-density lipoprotein in patients with HIVinfection (27). On the basis of these data, a large trialof 6,500 patients with HIV infection and a low tomoderate risk for CVD is now under way to testwhether pitavastatin can prevent CV events byreducing low-density lipoprotein and concomitantlyimprove inflammatory pathways of immune activa-tion (REPRIEVE [Randomized Trial to PreventVascular Events in HIV]; NCT02344290).
PSORIASIS. Psoriasis is a chronic T helper 1 cellT helper 17 cell inflammatory disease that affectsmore than 125 million people worldwide and about2% to 4% of the adult population, with the mostcommon type manifesting as plaque psoriasis.The pathophysiology of the disease is localizedskin inflammation, epidermal hyperproliferation,up-regulated T-cell and neutrophil activation, andincreases in C-reactive protein, serum amyloid A, andintercellular adhesion molecule 1 (28). Consistentwith chronically inflamed pathologies, largepopulation-based epidemiological studies suggestthat patients with psoriasis, particularly moderate tosevere, have an increased risk for MI, stroke, andCV mortality independent of traditional risk factors(29–31). Notably, studies demonstrate strong associ-ations of psoriasis and CVD with increasing severityof skin disease. Patients with moderate to severe
psoriasis experience premature mortality, dyingapproximately 5 years younger than their non-psoriatic counterparts in adjusted analysis (32). Over10-year periods, patients with moderate to severepsoriasis have about 6% excess risk for developing amajor CV event, beyond the conventional Framing-ham risk score calculation (33). Interestingly, trans-lational studies have demonstrated dramaticup-regulation of genes known to adversely affect CVrisk in the skin lesions of psoriasis. Furthermore,transgenic mouse models of psoriasis in which thegenetic construct is confined to the skin demonstrateaortic inflammation and thrombosis (34). Similarly,patients with psoriasis have an increased amount ofaortic FDG PET activity that is positively correlatedwith skin disease severity (35).
A series of randomized, placebo-controlled clinicaltrials are evaluating the impact of treatmentssuch as adalimumab compared with phototherapy(NCT01553058), ustekinumab (NCT02187172), andsecukinumab (NCT02690701) on aortic inflammation(measured by FDG PET imaging) and CV biomarkersin psoriasis. These trials will provide greater insightinto the ability of anti-inflammatory therapies todiminish the risk for CVD.SYSTEMIC LUPUS ERYTHEMATOSUS. Patients withSLE have a heightened risk for developing athero-sclerosis, including MI and stroke (36), with youngwomen having up to a 50-fold increased risk fordeveloping vascular events (37). Furthermore, evenpatients with SLE with no prior CVD events havesubclinical vascular dysfunction, endothelialdysfunction, arterial stiffness, myocardial perfusionabnormalities, carotid plaque, and coronary calcifi-cation not fully explained by Framingham risk scoreor by medications used to treat SLE (36).
Altered innate immune responses characteristic ofSLE contribute to the development of lupus vascul-opathy and atherosclerotic plaque formation (38).Type I IFNs, a group of cytokines elevated in manypatients with SLE, particularly during periods of flare,have pleiotropic deleterious roles in the vasculature(39). Type I IFNs independently associated withendothelial function, carotid plaque, and severity ofcoronary calcification in patients without histories ofCVD (40). Specifically, these cytokines promote animbalance of enhanced endothelial cell damage anddecreased vascular repair that may promote initiationof vasculopathy and contribute to the development offoam-cell formation and platelet activation. SLE isalso characterized by the presence of a distinct subsetof pathogenic neutrophils, low-density granulocytes,which have an enhanced capacity to form NETs(38,39). NETs can amplify inflammatory responses in

CENTRAL ILLUSTRATION Progression of Vascular Inflammation in Human Inflammatory Models
Teague, H.L. et al. J Am Coll Cardiol. 2017;70(11):1403–12.
A gross illustration of the aortic arch has been taken in cross section to magnify the vessel wall. Neutrophil activation due to systemic inflammation leads to
the formation of neutrophil extracellular traps and may initiate damage to the endothelium. Monocytes and T cells then infiltrate the lesion. Monocytes
differentiate into macrophages, where they proliferate to sustain their population. Macrophages within the vessel wall have high glycolytic rates and take up the18F-fluorodeoxyglucose (FDG) tracer, which is detectable by FDG positron emission tomography in human models of inflammation. IFN ¼ interferon; IL ¼ interleukin;
TNF ¼ tumor necrosis factor.
J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7 Teague et al.S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2 Immunology of Vascular Inflammation
1409
the plaque and other tissues, including inflamma-some activation in macrophages and induction oftype I IFN synthesis by plasmacytoid dendritic cells(38,41).
Two pathways are of potential interest in targetingvascular risk in SLE: the inhibition of type 1 IFNs andthe blockage of NETosis through PAD-4 inhibition.Type 1 IFNs signal through the JAK/STAT pathway,which is inhibited by tofacitinib. Tofacitinib amelio-rated vascular dysfunction in a murine lupus model(42) and is being tested in humans in an ongoingclinical trial (NCT02535689). Inhibition of pathwaysimplicated in NET formation using chemical in-hibitors of PAD-4 leads to amelioration of endothelialdysfunction, a prothrombotic phenotype, and plaque
formation in murine models of lupus and athero-sclerosis (41). Overall, SLE represents a unique modelfor understanding the role of the innate immunesystem in the development of vascular disease, andmay allow the characterization of novel therapeutictargets in subsets of patients.
PET IMAGING OF THE ARTERY WALL
Currently, FDG PET imaging is a cost-effective clinicalstandard of care for diagnosis, staging, and moni-toring the response of many malignancies, and itsrole is significantly enhanced by the introduction ofPET/CT imaging and PET/MR imaging. Imaginghuman inflammation models with positron emission

Teague et al. J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7
Immunology of Vascular Inflammation S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2
1410
tomography allows investigations to be conductedwithout expensive animal models, a step towardclinical translation.
PET imaging of VI using FDG was first describedmore than 15 years ago (43,44) and has becomeimportant in atherosclerosis research. FDG accumu-lates within living cells in proportion to their glyco-lytic rates (45). Several tissues have particularly highglycolytic rates and hence tend to accumulate FDG,including tumors, brain tissue, myocytes, andinflammatory cells. Phagocytes have particularly highglycolytic rates, especially after proinflammatoryactivation (46), leading to high FDG retention. Apotential advantage of PET imaging in addition toother available anatomic imaging techniques (e.g., CTimaging, MR, and ultrasound) is through the ability toadminister miniscule concentrations of a substancethat can be targeted for a specific molecular process.PET images can be interpreted both qualitatively andquantitatively to evaluate early phases of disease orshort-term changes in VI related to treatment thatmay not have concordant changes in morphology.Several studies show that FDG uptake, whenmeasured in the arterial wall in vivo, reflects the levelof macrophage accumulation within the atheroma(47). Atherosclerotic lesions show higher VI by FDGand tend to experience greater subsequent progres-sion (48). Moreover, the FDG measurement of VI mayprovide an independent index of the risk for subse-quent CV events (48–50), which is the subject oflarger ongoing prospective studies (51) to evaluate thestandardization of ideal imaging procedures (52,53)and subsequent clinical application.
FDG imaging of VI is increasingly used to evaluatetherapeutic compounds targeting atherosclerosis.The best therapies studied in this regard are statins,which show a reduction in the arterial FDG signalconsistent with their impact on CV events in ran-domized clinical trials (54,55). However, a lack ofreduction in arterial FDG uptake associates with aparallel failure to reduce CV events for a number ofexperimental treatments (56–58). Prospectiveoutcome data and response to novel therapies usingFDG in vascular diseases are limited. However, giventhe general concordance between VI imaging andCVD outcomes, relatively small and swift FDG imag-ing studies may facilitate drug discovery.
Although FDG imaging adequately addressescertain pathophysiological and treatment-relatedquestions, the specificity for inflammation with thisagent is not clearly defined, because of the variableaffinity for glucose of all cells in the body. Addition-ally, high FDG activity in the blood pool and tissuesnear the vessel wall complicates quantification. Thus,
there are opportunities for other imaging agents foratherosclerosis, such as vascular calcification imagedwith 18F–sodium fluoride (NaF). Originally approvedby the U.S. Food and Drug Administration for boneimaging in oncology, NaF PET imaging is an emergingtechnique reputed to capture a different aspect of thedevelopment of atheroma compared with FDG (59),related to calcification and microcalcification withinan atherosclerotic plaque (Figure 3). In patients withMI and stable angina, Joshi et al. (59) demonstratedconspicuous uptake of NaF in plaque, with histolog-ical evidence of macrophage infiltration, calcification,necrosis, and apoptosis, as well as high-risk plaquefeatures imaged by intravascular ultrasound. Advan-tages of NaF over FDG include rapid backgroundblood and tissue clearance and absence of activity inthe myocardium, improving the potential for imagingpathological NaF uptake in the coronary arteries (53).As with FDG, elements of the cell/process specificityof NaF in atherosclerosis have been described, andimaging technique standardization is an active realmof research.
In parallel with further FDG and NaF work, inves-tigation continues with other PET, CT, and MR tech-niques to study varied mechanisms and applicationsfor CVD imaging. An abbreviated list of PET agentscurrently under investigation is shown in Table 1.Agents from the list that already have Food and DrugAdministration approval for human imaging ofnonvascular processes include 68Ga-DOTA-octreotateand 18F-florbetapen. 68Ga-DOTA-octreotate has highaffinity for somatostatin receptors that are present inhigh concentration on inflammatory leukocytes (60).Its low background activity and high signal make it apromising candidate for plaque imaging, and itsfeasibility in humans has been demonstrated incomparison with FDG (60). 18F-florbetapen is a PETagent that is used clinically to characterize b-amyloidplaque in neurodegenerative processes, such as Alz-heimer’s disease. Bucerius et al. (61) demonstratedthat higher carotid artery uptake of 18F-florbetapen isassociated with male sex, independent of centralnervous system uptake, which potentially implicatesthe presence of higher concentrations of inflamma-tory b-amyloid within the vessel wall. Further in-vestigations of the performance characteristics ofthese new agents are ongoing.
In summary, with a new understanding of immu-nologic processes applied to the established feasibilityof promising PET agents for imaging atherosclerosisin humans, we now can administer these techniquesin newly identified human disease models to betterunderstand atherogenesis (Central Illustration).Acknowledging the great potential for pathology-

J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7 Teague et al.S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2 Immunology of Vascular Inflammation
1411
specific and quantitative information that PET imag-ing has for atherosclerosis, an outcome-based multi-disciplinary approach for further investigation,integrating biology, engineering, imaging physics, andeconomics, will continue to help us understand howthese techniques ultimately fit into clinical care andtranslational research.
CONCLUSIONS
Inflammation has emerged as a critical factor in earlyatherosclerosis. An understanding of the immunecells involved in the initiation and progression ofVI may facilitate the identification of important
pathways to target for future CVD therapeutics.Furthermore, the use of PET imaging to measure VImay improve our ability to identify the most prom-ising therapies to take to phase III clinical trials. Assuch, advances in immunology and vascular imaginghave the potential to accelerate discovery of newtreatments to reduce the burden of CVD.
ADDRESS FOR CORRESPONDENCE: Dr. Nehal N.Mehta, Cardiovascular and Pulmonary Branch,National Heart, Lung, and Blood Institute, 10 CenterDrive, CRC, Room 5-5140, Bethesda, Maryland, 20892.E-mail: [email protected].
RE F E RENCE S
1. Libby P. Inflammation in atherosclerosis. Nature2002;420:868–74.
2. Galkina E, Ley K. Immune and inflammatorymechanisms of atherosclerosis. Annu Rev Immunol2009;27:165–97.
3. Moore KJ, Sheedy FJ, Fisher EA. Macrophages inatherosclerosis: a dynamic balance. Nat RevImmunol 2013;13:709–21.
4. Robbins CS, Hilgendorf I, Weber GF, et al. Localproliferation dominates lesional macrophageaccumulation in atherosclerosis. Nat Med 2013;19:1166–72.
5. Dutta P, Courties G, Wei Y, et al. Myocardialinfarction accelerates atherosclerosis. Nature2012;487:325–9.
6. Sager HB, Dutta P, Dahlman JE, et al. RNAitargeting multiple cell adhesion moleculesreduces immune cell recruitment and vascularinflammation after myocardial infarction. SciTransl Med 2016;8:342ra80.
7. Heidt T, Sager HB, Courties G, et al. Chronicvariable stress activates hematopoietic stem cells.Nat Med 2014;20:754–8.
8. Brinkmann V, Reichard U, Goosmann C, et al.Neutrophil extracellular traps kill bacteria. Science2004;303:1532–5.
9. Yipp BG, Petri B, Salina D, et al. Infection-induced NETosis is a dynamic process involvingneutrophil multitasking in vivo. Nat Med 2012;18:1386–93.
10. Fuchs TA, Brill A, Wagner DD. Neutrophilextracellular trap (NET) impact on deep veinthrombosis. Arterioscler Thromb Vasc Biol 2012;32:1777–83.
11. Warnatsch A, Ioannou M, Wang Q,Papayannopoulos V. Inflammation. Neutrophilextracellular traps license macrophages for cyto-kine production in atherosclerosis. Science 2015;349:316–20.
12. Wang Y, Li M, Stadler S, et al. Histone hyper-citrullination mediates chromatin decondensationand neutrophil extracellular trap formation. J CellBiol 2009;184:205–13.
13. Dwivedi N, Radic M. Citrullination ofautoantigens implicates NETosis in the
induction of autoimmunity. Ann Rheum Dis2014;73:483–91.
14. Savchenko AS, Borissoff JI, Martinod K, et al.VWF-mediated leukocyte recruitment with chro-matin decondensation by PAD4 increasesmyocardial ischemia/reperfusion injury in mice.Blood 2014;123:141–8.
15. Martinod K, Witsch T, Erpenbeck L, et al.Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med 2017;214:439–58.
16. Witztum JL, Lichtman AH. The influence ofinnate and adaptive immune responses onatherosclerosis. Annu Rev Pathol 2014;9:73–102.
17. Foks AC, Lichtman AH, Kuiper J. Treatingatherosclerosis with regulatory T cells. Arterio-scleros Thrombos Vasc Biol 2015;35:280–7.
18. Gotsman I, Sharpe AH, Lichtman AH. T-cellcostimulation and coinhibition in atherosclerosis.Circ Res 2008;103:1220–31.
19. Lichtman AH. The heart of the matter: pro-tection of the myocardium from T cells.J Autoimmun 2013;45:90–6.
20. Johnson DB, Balko JM, Compton ML, et al.Fulminant myocarditis with combination immunecheckpoint blockade. N Engl J Med 2016;375:1749–55.
21. Triant VA, Lee H, Hadigan C, Grinspoon SK.Increased acute myocardial infarction rates andcardiovascular risk factors among patients withhuman immunodeficiency virus disease. J ClinEndocrinol Metab 2007;92:2506–12.
22. Zanni MV, Abbara S, Lo J, et al. Increasedcoronary atherosclerotic plaque vulnerability bycoronary computed tomography angiography inHIV-infected men. AIDS 2013;27:1263–72.
23. Zanni MV, Toribio M, Robbins GK, et al. Effectsof antiretroviral therapy on immune function andarterial inflammation in treatment-naive patientswith human immunodeficiency virus infection.JAMA Cardiol 2016;1:474–80.
24. Lo J, Abbara S, Shturman L, et al. Increasedprevalence of subclinical coronary atherosclerosisdetected by coronary computed tomography
angiography in HIV-infected men. AIDS 2010;24:243–53.
25. Thiara DK, Liu CY, Raman F, et al. Abnormalmyocardial function is related to myocardialsteatosis and diffuse myocardial fibrosis in HIV-infected adults. J Infect Dis 2015;212:1544–51.
26. Subramanian S, Tawakol A, Burdo TH, et al.Arterial inflammation in patients with HIV. JAMA2012;308:379–86.
27. Lo J, Lu MT, Ihenachor EJ, et al. Effects ofstatin therapy on coronary artery plaque volumeand high-risk plaque morphology in HIV-infectedpatients with subclinical atherosclerosis: a rando-mised, double-blind, placebo-controlled trial.Lancet HIV 2015;2:e52–63.
28. Lowes MA, Suárez-Fariñas M, Krueger JG.Immunology of psoriasis. Annu Rev Immunol2014;32:227–55.
29. Gelfand JM, Dommasch ED, Shin DB, et al. Therisk of stroke in patients with psoriasis. J InvestDermatol 2009;129:2411–8.
30. Gelfand JM, Neimann AL, Shin DB, Wang X,Margolis DJ, Troxel AB. Risk ofmyocardial infarctionin patientswith psoriasis. JAMA 2006;296:1735–41.
31. Mehta NN, Azfar RS, Shin DB, Neimann AL,Troxel AB, Gelfand JM. Patients with severepsoriasis are at increased risk of cardiovascularmortality: cohort study using the General PracticeResearch Database. Eur Heart J 2010;31:1000–6.
32. Yeung H, Takeshita J, Mehta NN, et al. Psori-asis severity and the prevalence of major medicalcomorbidity: a population-based study. JAMADermatol 2013;149:1173–9.
33. Mehta NN, Yu Y, Pinnelas R, et al. Attributablerisk estimate of severe psoriasis on major cardio-vascular events. Am J Med 2011;124:775.e1–6.
34. Wang Y, Gao H, Loyd CM, et al. Chronic skin-specific inflammation promotes vascular inflam-mation and thrombosis. J Invest Dermatol 2012;132:2067–75.
35. Naik HB, Natarajan B, Stansky E, et al. Severityof psoriasis associates with aortic vascular inflam-mation detected by FDG PET/CT and neutrophilactivation in a prospective observational study.Arterioscler Thromb Vasc Biol 2015;35:2667–76.

Teague et al. J A C C V O L . 7 0 , N O . 1 1 , 2 0 1 7
Immunology of Vascular Inflammation S E P T E M B E R 1 2 , 2 0 1 7 : 1 4 0 3 – 1 2
1412
36. Roman MJ, Shanker BA, Davis A, et al. Preva-lence and correlates of accelerated atherosclerosisin systemic lupus erythematosus. N Engl J Med2003;349:2399–406.
37. Manzi S, Meilahn EN, Rairie JE, et al. Age-specific incidence rates of myocardial infarctionand angina in women with systemic lupuserythematosus: comparison with the FraminghamStudy. Am J Epidemiol 1997;145:408–15.
38. Villanueva E, Yalavarthi S, Berthier CC, et al.Netting neutrophils induce endothelial damage,infiltrate tissues, and expose immunostimulatorymolecules in systemic lupus erythematosus.J Immunol 2011;187:538–52.
39. Denny MF, Yalavarthi S, Zhao W, et al.A distinct subset of proinflammatory neutrophilsisolated from patients with systemic lupus ery-thematosus induces vascular damage and synthe-sizes type I IFNs. J Immunol 2010;184:3284–97.
40. Somers EC, Zhao W, Lewis EE, et al. Type Iinterferons are associated with subclinical markersof cardiovascular disease in a cohort of systemiclupus erythematosus patients. PloS ONE 2012;7:e37000.
41. Knight JS, Luo W, O’Dell AA, et al. Peptidy-larginine deiminase inhibition reduces vasculardamage and modulates innate immune responsesin murine models of atherosclerosis. Circ Res 2014;114:947–56.
42. Furumoto Y, Smith CK, Blanco L, et al. Tofa-citinib ameliorates murine lupus and its associatedvascular dysfunction. Arthritis Rheumatol 2017;69:148–60.
43. Yun M, Yeh D, Araujo LI, Jang S, Newberg A,Alavi A. F-18 FDG uptake in the large arteries: anew observation. Clin Nucl Med 2001;26:314–9.
44. Rudd JH, Warburton EA, Fryer TD, et al. Im-aging atherosclerotic plaque inflammation with[18F]-fluorodeoxyglucose positron emissiontomography. Circulation 2002;105:2708–11.
45. Joseph P, Tawakol A. Imaging atherosclerosiswith positron emission tomography. Eur Heart J2016;37:2974–80.
46. Tawakol A, Singh P, Mojena M, et al. HIF-1band PFKFB3 mediate a tight relationship betweenproinflammatory activation and anerobic meta-bolism in atherosclerotic macrophages. Arte-rioscler Thromb Vasc Biol 2015;35:1463–71.
47. Taqueti VR, Di Carli MF, Jerosch-Herold M,et al. Increased microvascularization and vesselpermeability associate with active inflammation inhuman atheromata. Circ Cardiovasc Imaging 2014;7:920–9.
48. Abdelbaky A, Corsini E, Figueroa AL, et al.Early aortic valve inflammation precedes calcifi-cation: a longitudinal FDG-PET/CT study. Athero-sclerosis 2015;238:165–72.
49. Rominger A, Saam T, Wolpers S, et al. 18F-FDGPET/CT identifies patients at risk for futurevascular events in an otherwise asymptomaticcohort with neoplastic disease. J Nucl Med 2009;50:1611–20.
50. Figueroa AL, Abdelbaky A, Truong QA, et al.Measurement of arterial activity on routine FDGPET/CT images improves prediction of risk offuture CV events. J Am Coll Cardiol Img 2013;6:1250–9.
51. Mehta NN, Torigian DA, Gelfand JM,Saboury B, Alavi A. Quantification of atheroscle-rotic plaque activity and vascular inflammationusing [18-F] fluorodeoxyglucose positron emissiontomography/computed tomography (FDG-PET/CT). J Vis Exp 2012:e3777.
52. Huet P, Burg S, Le Guludec D, Hyafil F, Buvat I.Variability and uncertainty of 18F-FDG PET imagingprotocols for assessing inflammation in athero-sclerosis: suggestions for improvement. J NuclMed 2015;56:552–9.
53. Bucerius J, Hyafil F, Verberne HJ, et al., for theCardiovascular Committee of the European Asso-ciation of Nuclear Medicine (EANM). Positionpaper of the Cardiovascular Committee of theEuropean Association of Nuclear Medicine (EANM)on PET imaging of atherosclerosis. Eur J Nucl MedMol Imaging 2016;43:780–92.
54. Tawakol A, Fayad ZA, Mogg R, et al. Intensi-fication of statin therapy results in a rapid reduc-tion in atherosclerotic inflammation: results of amulticenter fluorodeoxyglucose-positron emissiontomography/computed tomography feasibilitystudy. J Am Coll Cardiol 2013;62:909–17.
55. Cholesterol Treatment Trialists’ (CTT) Collab-oration. Efficacy and safety of more intensivelowering of LDL cholesterol: a meta-analysis ofdata from 170,000 participants in 26 randomisedtrials. Lancet 2010;376:1670–81.
56. Fayad ZA, Mani V, Woodward M, et al., forthe dal-PLAQUE Investigators. Safety and efficacyof dalcetrapib on atherosclerotic diseaseusing novel non-invasive multimodality imaging(dal-PLAQUE): a randomised clinical trial. Lancet2011;378:1547–59.
57. Tawakol A, Singh P, Rudd JH, et al. Effect oftreatment for 12 weeks with rilapladib, alipoprotein-associated phospholipase A2 inhibitor,on arterial inflammation as assessed with 18F-fluorodeoxyglucose-positron emission tomogra-phy imaging. J Am Coll Cardiol 2014;63:86–8.
58. O’Donoghue ML, Glaser R, Cavender MA,et al., for the LATITUDE-TIMI 60 Investigators.Effect of losmapimod on cardiovascular outcomesin patients hospitalized with acute myocardialinfarction: a randomized clinical trial. JAMA 2016;315:1591–9.
59. Joshi NV, Vesey AT, Williams MC, et al.18F-fluoride positron emission tomography foridentification of ruptured and high-risk coronaryatherosclerotic plaques: a prospective clinical trial.Lancet 2014;383:705–13.
60. Tarkin JM, JoshiFR, EvansNR,etal. Detectionofatherosclerotic inflammation by 68Ga-DOTATATEPET compared to [18F]FDG PET Imaging. J Am CollCardiol 2017;69:1774–91.
61. Bucerius J, Barthel H, Tiepolt S, et al. Feasi-bility of in vivo 18F-florbetaben PET/MR imagingof human carotid amyloid-b. Eur J Nucl Med MolImaging 2017;44:1119–28.
62. Ye YX, Calcagno C, Binderup T, et al. Imagingmacrophage and hematopoietic progenitor prolif-eration in atherosclerosis. Circ Res 2015;117:835–45.
63. Pugliese F, Gaemperli O, Kinderlerer AR, et al.Imaging of vascular inflammation with [11C]-PK11195 and positron emission tomography/computed tomography angiography. J Am CollCardiol 2010;56:653–61.
64. Bucerius J, Manka C, Schmaljohann J, et al.Feasibility of [18F]-2-fluoro-A85380-PET imagingof human vascular nicotinic acetylcholinereceptors in vivo. J Am Coll Cardiol Img 2012;5:528–36.
65. Vöö S, Kwee RM, Sluimer JC, et al. Imagingintraplaque inflammation in carotid atheroscle-rosis with 18F-fluorocholine positron emissiontomography-computed tomography: prospectivestudy on vulnerable atheroma with immunohisto-chemical validation. Circ Cardiovasc Imaging 2016;9:e00447.
66. Nie X, Laforest R, Elvington A, et al. PET/MRIof hypoxic atherosclerosis using 64Cu-ATSM in arabbit model. J Nucl Med 2016;57:2006–11.
67. Joshi FR, Manavaki R, Fryer TD, et al. Vascularimaging with 18F-fluorodeoxyglucose positronemission tomography is influenced by hypoxia.J Am Coll Cardiol 2017;69:1873–4.
68. Paeng JC, Lee YS, Lee JS, et al. Feasibility andkinetic characteristics of 68Ga-NOTA-RGD PET forin vivo atherosclerosis imaging. Ann Nucl Med2013;27:847–54.
69. Liu Y, Pressly ED, Abendschein DR, et al.Targeting angiogenesis using a C-type atrialnatriuretic factor-conjugated nanoprobe and PET.J Nucl Med 2011;52:1956–63.
70. Hyafil F, Pelisek J, Laitinen I, et al. Imaging thecytokine receptor CXCR4 in atheroscleroticplaques with the radiotracer 68Ga-pentixafor forPET. J Nucl Med 2017;58:499–506.
KEY WORDS cardiovascular imaging,monocytes, neutrophils, T cells





























