Embed Size (px)
Citation preview

Henry Ford Hospital Medical Journal Henry Ford Hospital Medical Journal
Volume 40 Number 3 Article 24
9-1992
Unusual Features of Multiple Endocrine Neoplasia Unusual Features of Multiple Endocrine Neoplasia
Andrea Frilling
Heinz Becker
Hans-Dietrich Roeher
Follow this and additional works at: https://scholarlycommons.henryford.com/hfhmedjournal
Part of the Life Sciences Commons, Medical Specialties Commons, and the Public Health Commons
Recommended Citation Recommended Citation Frilling, Andrea; Becker, Heinz; and Roeher, Hans-Dietrich (1992) "Unusual Features of Multiple Endocrine Neoplasia," Henry Ford Hospital Medical Journal : Vol. 40 : No. 3 , 253-255. Available at: https://scholarlycommons.henryford.com/hfhmedjournal/vol40/iss3/24
This Article is brought to you for free and open access by Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Henry Ford Hospital Medical Journal by an authorized editor of Henry Ford Health System Scholarly Commons.

Unusual Features of Multiple Endocrine Neoplasia
Andrea Frilling,* Heinz Becker," and Hans-Dietrich Roeher̂
In addition to the common presentations of the multiple endocrine neoplasia (MEN) syndromes, unusual organ involvement as rare manifestations ofa single disease may occur. Among our patients we have identified four cases in which unu.sual features of MEN were present. In the first patient, bilateral adrenal cortical adenoma, parathyroid adenoma, multiple pancreatic tumors, andfollicidar thyroid carcinoma were observed. The second patient suffered from thymic carcinoid, parathyroid hyperplasia, gastrinoma, and pituitary adenoma. Addifionally. one family was discovered in which medullary thyroid carcinoma (MTC). Hirschsprung's disease, and pheochromocytoma occurred and another family had MTC and ovarian cancer. Based on these observations, we stress the importance of screening for MEN syndromes in all patients with pathologic findings in any endocrine organ. (Henry Ford Hosp MedJ 1992;40:253-5)
Depending on the different endocrine organs involved, multiple endocrine neoplasia (MEN) type 1 and type 2 (A and
B) are the two major forms of MEN that have been characterized. The two typical MEN syndromes may present incomplete phenotypic expression or unusually overlapping features. In addition, rare organ involvement or unique manifestations of a single disease can occur. Most of the reported cases with uncommon MEN manifestation have no family hi,story of clinicaUy overt endocrine diseases. Only a few families are known in whom the classical MEN appears to cosegregate with other lesions (1-3). The following cases present further rare features of MEN.
Case Reports Since 1986, 27 patients have had surgery for MEN in our de
partment. Of these patients, eight had MEN 1,13 had MEN 2A, and two had MEN 2B. Of the other four patients, three had other family meinbers who presented with unusual features of MEN.
Casel A 16-year-old boy was operated on for medullary thyroid carcinoma
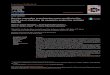
(MTC) following family screening studies (4). In his family, MTC, pheochromocytoma, and Hirschsprung's disease were observed (Fig 1). In the second generation. MTC was found in four members. One other member of this generation presented with MTC and pheochromocytoma. In the third generation, Hirschsprung's di.sease and MTC were found in three of four male individuals. An infant of the fourth generation has Hirschsprung's disea.se. All those with Hirschsprung's disease had severe constipation and underwent resection of the aganglionic colonic segments, two of them neonatally.
Case 2 MTC and ovarian cancer occurred in this family (Fig 2). In the first
and second generations, two women had died of diffuse metastases from ovarian carcinomas. MTC was diagnosed inilially in the third generation in a 48-year-old female with ovarian carcinoma. After surgery basal and pentagastrin stimulated calcitonin levels were normal.
At present, she is the only one in this family presenting both lesions but both of her affected sisters with ovarian carcinoma refused screening for MTC. Three more members, all younger than 25 years, were found by calcitonin screening to have MTC, None of the individuals has presented with pheochromocytomas or hyperparathyroidism.
Case 3 The 47-year-old male seems to be an apparently sporadic MEN case.
No members of his family are known to be affected. In 1979 he underwent gastric resection for muldple ulcers. Serum calcium elevation (2.9 mmol/L) was noted for the first time in 1980. In 1989 a huge calcified mediastinal mass was found to be due to a carcinoid tumor of the thymus. ACTH positivity was demonstrated by immunohistological examination. One year later the serum calcium level was increa.sed at 3 mmol/L. parathormone (FTH) was at the upper normal limit (50 pg/ mL), and the patient underwent a subtotal parathyroidectomy for parathyroid hyperplasia. In 1991 diarrhea and marked elevadon of gastrin, glucagon, pancreatic polypeptide, and prolactin suggested further endocrine tumors. An enlarged sella was found and surgery for gastrinoma is planned.
Case 4 An 18-year-old woman was adi-nitted because of a right adrenal lu
mor. In her family an increased frequency of pituitary, adrenal, and thyroid tumors was known although no hospital files concerning the relatives were available. She presented with a two-year history of hirsutism, amenorrhea, facial acne, and diffuse thyroid enlargement. Testosterone was increased to 3 ng/mL (normal 0.2 to 0.8 ng/mL) and serum calcium was 3 mmol/L (normal 2 to 2.5 mmol/L). Basal Cortisol was within normal range at 100 ng/mL but increased only to 148 ng/mL after ACTH stimulation. All other laboralory findings were within normal range. Abdominal computed tomography showed a cystic mass in
Submitted for publication: November 8, 1991. Accepted for publication: January 27, 1992. ^Department of Surgery, University Clinic, Hamburg, Germany. tDepartment of Surgery, Heinrich-Heine-University, Dusseldorf, Germany. Address correspondence to Dr. Frilling, Depanment of Surgei-y, University Clinic.
Mailinistr. 52. 2000 Hamburg 20. Germany.
Henry Ford Hosp Med J—Vol 40, Nos 3 & 4, 1992 Unusual Features of MEN—Frilling et al 253

op
] O " ° symptoms
• O "^TC
H A MTC and Hirschsprung's disease
4in 4a
•
MTC and pheochromocytoma
Hirschsprung's disease
III 6 6p 6p 6p
IV o n n
] ( 3 no symptoms
• O I^TC
ovarian cancer
MTG and ovarian cancer
Fig I—Pedigree ofthe family presenting with MTC, pheochromocytoma, and Hirshsprung's disease. All of the family members have heen .screenedfor MTC.
Fig 2—Pedigree ofthe family presenting with MTC and ovarian cancer. The patients not yet screened for MTC are indicated hy black dots.
the right abdomen. The surgically excised tumor revealed an adrenal cortical adenoma. Eight years later a solitary solid nodule suspicious for malignancy was detected by sonography in the right thyroid lobe which was cold on isotope scanning. All laboratory investigations except the serum calcium (2.7 mmol/L) were within normal range. The padent underwent right hemithyroidectomy which confirmed a 12 mm diameter follicular carcinoma and total thyroidectomy with dissection of the central lymph nodes was performed. No parathyroid enlargement was evident grossly. A subsequent radioacrive scan .showed no distant metastases. In order to exclude pathologic findings in other endocrine organs, sonography of the abdomen and neck was performed demonstrating a 5 cm diameter mass in the head of the pancreas, mulliple < 3 cm diameter tumors within pancreatic body, a 2.7 cm diameter tumor of the left adrenal, but no detectable parathyroid tumor. Computed tomography of the sella showed no abnormalities, PTH was 62 pg/mL (normal range 35 to 55 pg/mL) and calcium was 2,9 mmol/L, but adrenal, pancreatic, and pituitary hormone assays were normal. All pancreatic tumors and a left adrenal tumor were excised followed by resection of an enlarged parathyroid gland (1 cm diameter) localized in the upper thymus. All were histologically adenomata. Immunostained seciions of pancreatic tumors showed chromogranin A positive cells and one tumor was positive for somatostatin. Postoperatively the patient was normocalcemic.
Discussion These unusual cases of MEN support the observation that the
MEN syndromes may present a wide variety of features. Cosegregation of MEN 2 A and aganglionic colon, as seen in our case 1, was reported in 1982 by Verdy et al (3) who documented a family with MEN 2 in which 15 members had MTC and eight had Hirschspmng's disease. Four infants died because of the colonic involvement as early as the neonatal period and a coincidence of MTC and Hirschsprung's disease was seen in one individual. The association of MTC and colonic agangliono.sis in place of ganglioneuromatosis, as seen in MEN 2B, appears to be an unusual variant of MEN 2. The observation of Verdy et al (3) and the present study imply that Hirschsprung's disease associated with the MEN 2A syndrome may be a new entity. The manifestation in one patient of aganglionic megacolon, neurofi
broma, megaloureter, and unilateral pheochromocytomas as described by Shocket and Teloh (5) could be explained as a coincidence of independent lesions.
Cutaneous lichen amyloidosis as another new entity of MEN 2 has been reported previously (1,2). As an explanation for the simultaneous occurrence of these two autosomally dominant hereditary diseases, a contiguous gene syndrome (6) was suggested. Recent results suggest that cutaneous lichen amyloidosis is linked to the MEN 2 locus (7).
The unusual association of ovarian cancer and MTC in the family ofca.se 2 lacks plausible explanation, but it may have occurred by chance in this one family.
As in case 3, Rosai and Higa (8) described carcinoid tumor of the thymus in three patients with MEN 1, each without a positive famdy history although presenting with additional features of MEN 1, such as parathyroid, pituitary, thyroid, and/or adrenal cortex lesions. As in our case 4, the initial manifestation of MEN 1 was the Zollinger-Ellison syndrome in one patient (8). An aggressive course of the thymic carcinoids was observed, with ad three patients dying of diffuse metastatic spread or local invasive tumor recurrence. Other cases of patients with thymic carcinoid and hyperparathyroidism have been reported (9,10). Shepherd (11) reported that of 110 Tasmanian patients with MEN 1, three had thymic carcinoids. Thymic carcinoids occur most often in male MEN I patients older than 40 years. Only a few MEN 2A cases presenting with this tumor are known (12). Possibly due to ectopic ACTH secretion of the tumor, Cushing's syndrome has been frequently observed in MEN 2. In contrast to most ofthe midgut carcinoids, carcinoid syndrome has not been seen in patients with thymic carcinoids (10).
The coexistence of fodicular carcinoma and MEN 1, as in case 4, appears to be a rare event. Unfortunately, we have not been able to obtain detailed data on the family of the patient. Apparently MEN was not recognized initially, since a semm calcium elevation was evident in 1982 at the time of the first adrenal surgery. An awareness of MEN at the time of the thyroid surgery may have allowed detection of the parathyroid adenoma earlier.
254 Henry Ford Hosp Med J—Vol 40. Nos 3 & 4,1992 Unusual Features of MEN—Frilling et al

A good review of endocrine tumor combinations and possible overlap syndromes is provided by Schimke (13). To avoid overlooking MEN syndromes, each patient with endocrine organ pathology needs careful evaluation for other endocrine tumor involvement and probably careful family investigation.
Addendum One other MEN 2 family with Hirschspmng's disease was
reported at the 1991 Workshop (14).
References 1. Donovan DT. Levy ML, Furst EJ, et al. Familial cutaneous lichen amy
loidosis in association with multiple endocrine neoplasia type 2A: A new variant. Henry Ford Hosp Med J 1989;37:147-50.
2. Nunziata V, di Giovanni G, Lettera AM, D'Armiento M, Mancini M. Cutaneous lichen amyloidosis associated with multiple endocrine neoplasia type 2A. Henry Ford Hosp Med J 1989;37:144-6.
3. Verdy M, 'Weber AM, Roy CC, Morin CL, Cadotte M, Brochu P. Hirschsprung's disease in a family with multiple endocrine neoplasia type 2. J Pediatr Gastroenterol Nutr 1982;1:603-7.
4. Frilling A, Goretzki PE, Ba.stian L, Roeher HD. The importance of screening for medullary thyroid carcinoma in families of patients with MEN 2. Henry FordHospMedJ 1989;37:122-3.
5. Shocket E, Teloh HA. Aganglionic megacolon, pheochromocytoma, megaloureter, and neurofibroma; co-occurrence of several neural abnormalities. Am J Dis Child 1957;94:185-91.
6. Schmickel RD. Contiguous gene syndromes: A component of recognizable syndromes. J Pediatr 1986; 109:231 -41.
7. Robinson MF, Furst EiJ, Nunziata V, et al. Characterization of the clinical features of five families with hereditary primary cutaneous lichen amyloidosis and multiple endocrine neoplasia type 2. Henry Ford Hosp Med J 1992;40:249-52.
8. Rosai J, Higa E. Mediastinal endocrine neoplasm, of probable thymic origin, related to carcinoid tumor: Clinicopathologic study of 8 cases. Cancer 1972;29:1061-74.
9. Manes JL. Taylor HB. Thymic carcinoid in familial multiple endocrine adenomatosis. Arch Pathol 1973;95:252-5.
10. Vener JD, Zuckerbraun L, Goodman D. Carcinoid tumor of the thymus associated with a parathyroid adenoma. Arch Otolaryngol 1982;108:324-6.
11. Shepherd JJ. The natural history of multiple endocrine neoplasia type 1: Highly uncommon or highly unrecognized? Arch Surg 1991:126:935-52.
12. Marchevsky AM, Dikman SH. Mediastinal carcinoid with an incomplete Sipple's syndrome. Cancer 1979;43:2497-501.
13. Schimke RN. Multiple endocrine neoplasia: How many syndromes? Am J Med Genet 1990;37:375-83.
14. Ward JL. Hyland VJ. Andrew DS, Marsh DJ, Robinson BG. Medullary thyroid carcinoma: Australian experience with genetic testing, Henry Ford Hosp MedJ 1992;40:220-3.
Henry Ford Hosp Med J—Vol 40, Nos 3 & 4, 1992 Unusual Features of MEN—Frilling et al 255