Embed Size (px)
Citation preview

The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
1
UV Index 3 as a cut-off
Michael Kimlin
CRE Sun and Health
USC, AUSTRALIA
UV Index - History
The UV index was first introduced in Canada in 1992 in
response to growing concerns about the potential increase
of ultraviolet radiation due to ozone depletion(Fioletov et al., 2010).
In 1995, the World Health Organization (WHO) with the
World Meteorological Organization (WMO), the United
Nations Environment Program (UNEP), the International
Agency for Cancer Research (IARC) and the International
Commission on Non-Ionizing Radiation Protection (ICNIRP)
launched the Global Solar UV Index (UVI) as a vehicle to
visualize the amount of UV radiation reaching the Earth’s
surface (Italia and Rehfuess, 2012, Meves et al., 2003, Azooz and Jallo, 2015) .
UV Index
The UV Index is a simple and informative way of describing the daily danger of solar UV
radiation intensity (WHO, 2015).
The UV index range is expressed as a numeric value and as bands of color representing the
risk level of skin damage due to UV exposure from Low (0-2), Green, to Extreme (11+),
Purple. The higher the index value, the greater the potential for damage to the skin and eye,
and the less time it takes for harm to occur.
Low (0-2): Green
Moderate (3-5): Yellow
High (6-7): Orange
Very High (8-10): Red
Extreme (11+): Purple
In reporting the UVI, most emphasis is placed on the maximum UV level on a given day. This
occurs around solar noon. Depending on geographical location and whether daylight saving
time is applied, solar noon takes place between noon and 2 p.m.
Various Ultraviolet Exposure categories
UV Index Calculation
The calculation of UVI involves both
effect of different wavelengths, and
human susceptibility to them.
The calculations are weighted to those
UV wavelengths that human skin is most
sensitive to according to the McKinlay-
Diffey erythema action spectrum
curve (WHO, 2002). The UVI is a
unitless quantity defined by the
formula:
1.00E-04
1.00E-03
1.00E-02
1.00E-01
1.00E+00
250 270 290 310 330 350 370 390 410
Wavelength (nm)
Re
lativ
e R
esp
on
se
(W
n)
The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
2
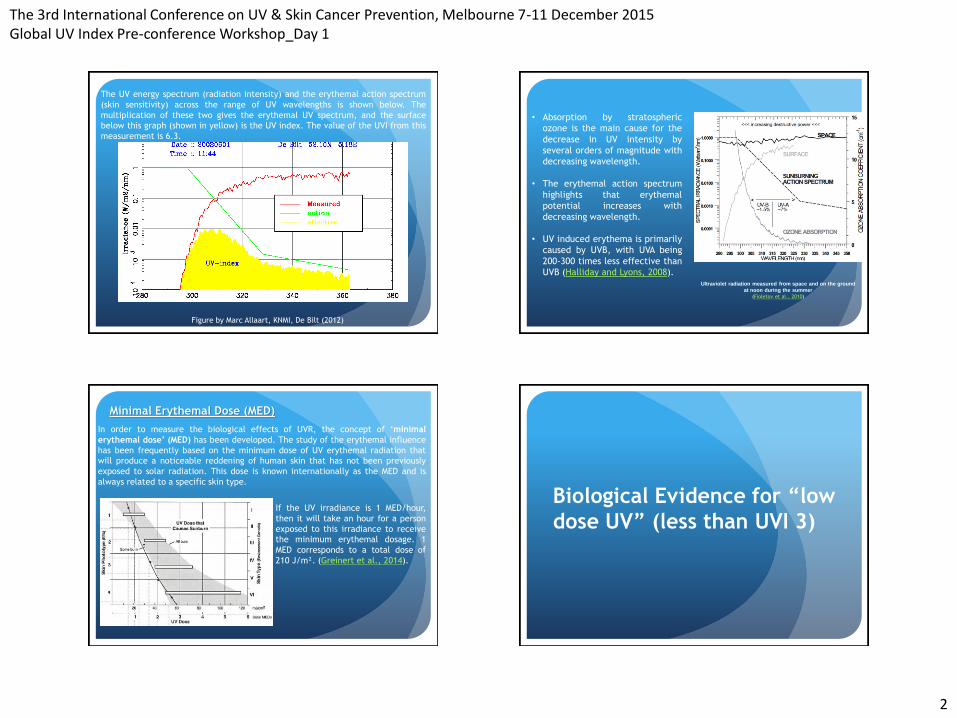
The UV energy spectrum (radiation intensity) and the erythemal action spectrum
(skin sensitivity) across the range of UV wavelengths is shown below. The
multiplication of these two gives the erythemal UV spectrum, and the surface
below this graph (shown in yellow) is the UV index. The value of the UVI from this
measurement is 6.3.
Figure by Marc Allaart, KNMI, De Bilt (2012)
• Absorption by stratospheric
ozone is the main cause for the
decrease in UV intensity by
several orders of magnitude with
decreasing wavelength.
• The erythemal action spectrum
highlights that erythemal
potential increases with
decreasing wavelength.
• UV induced erythema is primarily
caused by UVB, with UVA being
200-300 times less effective than
UVB (Halliday and Lyons, 2008). Ultraviolet radiation measured from space and on the ground
at noon during the summer
(Fioletov et al., 2010)
Minimal Erythemal Dose (MED)
In order to measure the biological effects of UVR, the concept of ‘minimal
erythemal dose’ (MED) has been developed. The study of the erythemal influence
has been frequently based on the minimum dose of UV erythemal radiation that
will produce a noticeable reddening of human skin that has not been previously
exposed to solar radiation. This dose is known internationally as the MED and is
always related to a specific skin type.
If the UV irradiance is 1 MED/hour,
then it will take an hour for a person
exposed to this irradiance to receive
the minimum erythemal dosage. 1
MED corresponds to a total dose of
210 J/m². (Greinert et al., 2014).
Biological Evidence for “low
dose UV” (less than UVI 3)
The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
3
Low Dose UV and Skin
Cancer
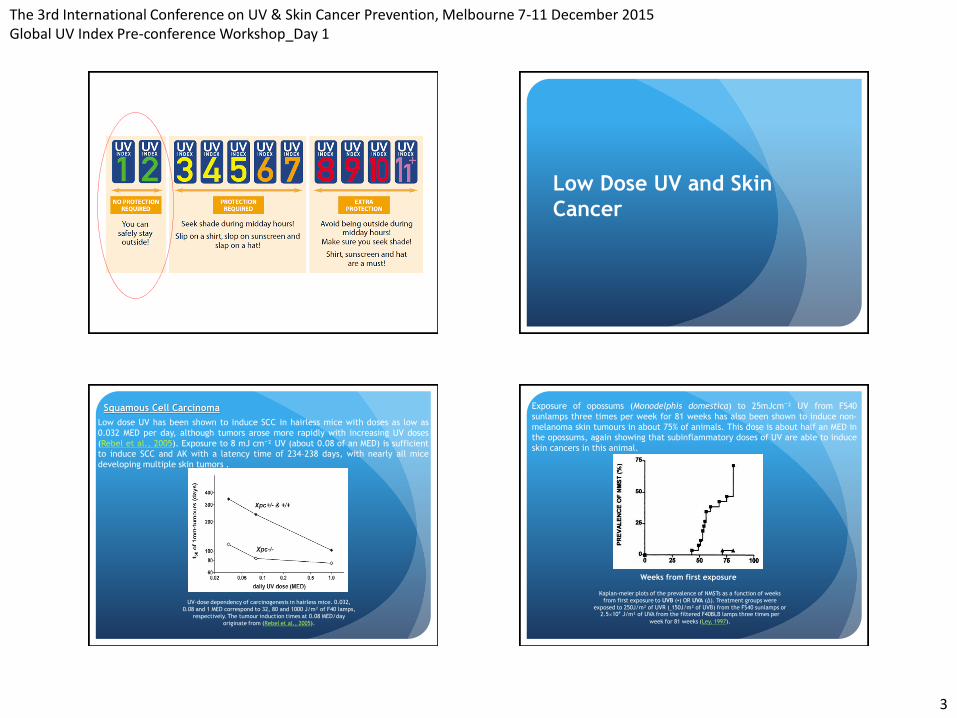
Squamous Cell Carcinoma
Low dose UV has been shown to induce SCC in hairless mice with doses as low as
0.032 MED per day, although tumors arose more rapidly with increasing UV doses
(Rebel et al., 2005). Exposure to 8 mJ cm⁻² UV (about 0.08 of an MED) is sufficient to induce SCC and AK with a latency time of 234–238 days, with nearly all mice
developing multiple skin tumors .
UV-dose dependency of carcinogenesis in hairless mice. 0.032,
0.08 and 1 MED correspond to 32, 80 and 1000 J/m² of F40 lamps,
respectively. The tumour induction times at 0.08 MED/day
originate from (Rebel et al., 2005).
Exposure of opossums (Monodelphis domestica) to 25mJcm⁻² UV from FS40
sunlamps three times per week for 81 weeks has also been shown to induce non-
melanoma skin tumours in about 75% of animals. This dose is about half an MED in
the opossums, again showing that subinflammatory doses of UV are able to induce
skin cancers in this animal.
Weeks from first exposure
Kaplan-meier plots of the prevalence of NMSTs as a function of weeks
from first exposure to UVB (▪) OR UVA (∆). Treatment groups were
exposed to 250J/m² of UVR (˷150J/m² of UVB) from the FS40 sunlamps or 2.5×10⁴ J/m² of UVA from the filtered F40BLB lamps three times per
week for 81 weeks (Ley, 1997).
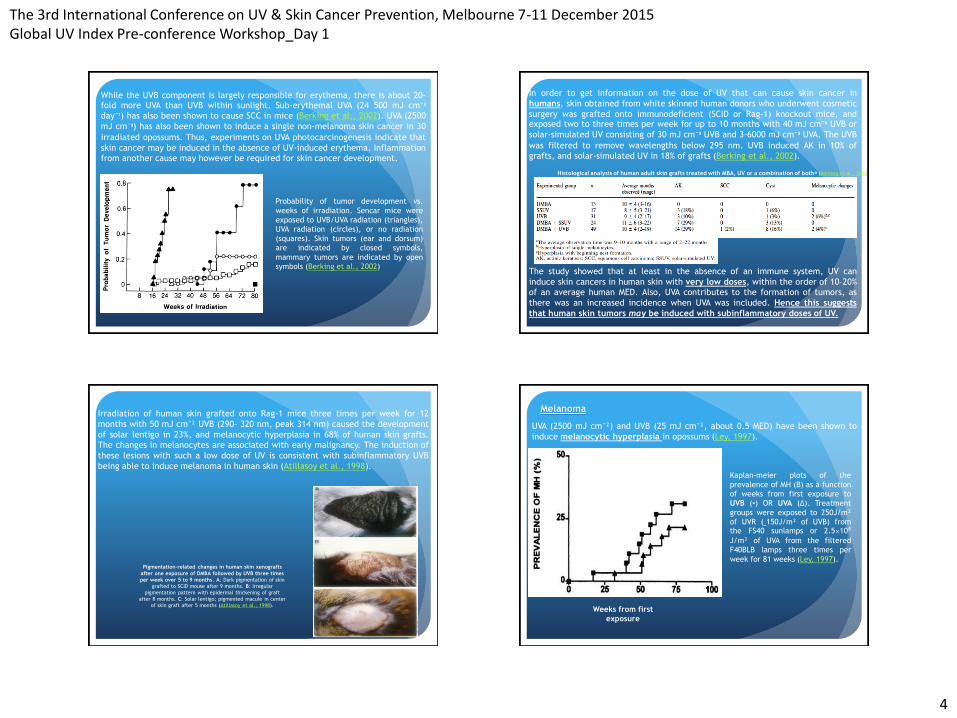
The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
4
While the UVB component is largely responsible for erythema, there is about 20-fold more UVA than UVB within sunlight. Sub-erythemal UVA (24 500 mJ cm⁻² day⁻¹) has also been shown to cause SCC in mice (Berking et al., 2002). UVA (2500
mJ cm⁻²) has also been shown to induce a single non-melanoma skin cancer in 30
irradiated opossums. Thus, experiments on UVA photocarcinogenesis indicate that
skin cancer may be induced in the absence of UV-induced erythema. Inflammation
from another cause may however be required for skin cancer development.
Probability of tumor development vs.
weeks of irradiation. Sencar mice were
exposed to UVB/UVA radiation (triangles),
UVA radiation (circles), or no radiation
(squares). Skin tumors (ear and dorsum)
are indicated by closed symbols,
mammary tumors are indicated by open
symbols (Berking et al., 2002)
In order to get information on the dose of UV that can cause skin cancer in
humans, skin obtained from white skinned human donors who underwent cosmetic
surgery was grafted onto immunodeficient (SCID or Rag-1) knockout mice, and exposed two to three times per week for up to 10 months with 40 mJ cm⁻² UVB or
solar-simulated UV consisting of 30 mJ cm⁻² UVB and 3–6000 mJ cm⁻² UVA. The UVB
was filtered to remove wavelengths below 295 nm. UVB induced AK in 10% of
grafts, and solar-simulated UV in 18% of grafts (Berking et al., 2002).
The study showed that at least in the absence of an immune system, UV can
induce skin cancers in human skin with very low doses, within the order of 10–20%
of an average human MED. Also, UVA contributes to the formation of tumors, as
there was an increased incidence when UVA was included. Hence this suggests
that human skin tumors may be induced with subinflammatory doses of UV.
Histological analysis of human adult skin grafts treated with MBA, UV or a combination of bothᵃ (Berking et al., 2002)
Pigmentation-related changes in human skin xenografts
after one exposure of DMBA followed by UVB three times
per week over 5 to 9 months. A: Dark pigmentation of skin
grafted to SCID mouse after 9 months. B: Irregular
pigmentation pattern with epidermal thickening of graft
after 8 months. C: Solar lentigo; pigmented macule in center
of skin graft after 5 months (Atillasoy et al., 1998).
Irradiation of human skin grafted onto Rag-1 mice three times per week for 12
months with 50 mJ cm⁻² UVB (290– 320 nm, peak 314 nm) caused the development
of solar lentigo in 23%, and melanocytic hyperplasia in 68% of human skin grafts.
The changes in melanocytes are associated with early malignancy. The induction of
these lesions with such a low dose of UV is consistent with subinflammatory UVB
being able to induce melanoma in human skin (Atillasoy et al., 1998).
Melanoma
UVA (2500 mJ cm⁻²) and UVB (25 mJ cm⁻², about 0.5 MED) have been shown to
induce melanocytic hyperplasia in opossums (Ley, 1997).
Weeks from first
exposure
Kaplan-meier plots of the
prevalence of MH (B) as a function
of weeks from first exposure to
UVB (▪) OR UVA (∆). Treatment
groups were exposed to 250J/m²
of UVR (˷150J/m² of UVB) from the FS40 sunlamps or 2.5×10⁴ J/m² of UVA from the filtered
F40BLB lamps three times per
week for 81 weeks (Ley, 1997).

The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
5
Summary – Animal Models
Doses of UVB more than 10-fold lower to produce erythema can induce SCC in
animal models.
UVA at doses several hundred fold lower to produce erythema can induce skin
cancers in animal models and at least contribute to photocarcinogenesis in
human skin grafted onto immunodeficient mice.
Therefore, it can be suggested that sub-erythemal UVB or UVA may cause
melanocytic lesions in a variety of animal models and in human skin grafted onto
immunoincompetent mice.
This shows that it is possible for sub-erythemal doses of UV to cause skin cancer.
However, the relative importance of subinflammatory compared to erythemal
doses of sunlight in causing skin cancer in humans remains unknown.
Low Dose UV and
immunosuppression
It is now known that in order for skin cancers to develop, both
genetic damage and immunosuppression is required.
While it cannot be directly experimentally determined
whether or not erythemal doses of UV are required for
photocarcinogenesis in humans, photoimmunosuppression and
gene mutations can be studied as surrogate markers because
these two biologic events are essential for skin cancer
induction.
Low Dose UV and Immunosuppression Sub-erythemal Doses of UV are Immunosuppressive in
Humans
Ultraviolet-induced immunosuppression is a key contributor to the development of
skin cancer. There are different lab models to describe UV-induced
immunosuppression:
The local model is when the antigen is applied to UV-irradiated skin of
unimmunized individuals.
Systemic immunosuppression is when the antigen is applied to an unirradiated
skin site of an unimmunized individual who has been exposed to UV at a
different skin site.
An established or memory immune response can also be suppressed by UV
irradiation.

The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
6
Daily irradiations with a suberythemal UV dose of 144 mJ cm⁻² UVB can cause
increasing levels of immunosuppression (local immunosuppression) with
increasing numbers of daily irradiations (Halliday and Lyons, 2008).
Increasing doses of single exposures to UVB also cause greater levels of
immunosuppression in humans with the suppression being discernible within 48h
of irradiation with 300 mJ cm⁻² UVB, which is subinflammatory (Poon et al.,
2005).
Single-exposure ultraviolet (UV) immunosuppression
time course and dose response (Poon et al., 2005)
UVB and Immunosuppression UVA and Immunosuppression
UVA is immunosuppressive at doses much lower than are required for this waveband
to cause erythema (Poon et al., 2005).
Irradiation with UVA suppresses reactivation of memory immunity to nickel in
humans. UVA also causes systemic suppression of recall immunity in humans
(Fourtanier et al., 2005).
In mice, UVA suppresses the induction of local immunosuppression and the
reactivation of memory immunity. Systemic suppression of the induction of primary
CHS can occur with three daily exposures to the relatively low UVA dose of 1680 mJ cm⁻² (Byrne et al., 2002).
• It has to be noted that unlike UVB, UVA is
immunosuppression but not until 48h following
exposure.
Single-exposure ultraviolet (UV)
immunosuppression time course
and dose response (Poon et al.,
2005)
UV and Immunosuppression
Humans are not exposed to pure UVB or UVA, but to sunlight, which consists of a
mixture of these two wavebands.
A single exposure to solar-simulated UV, less than half of what is required to cause
erythema has been shown to suppress the reactivation of memory immunity to
nickel in humans (Poon et al., 2005)
In mice a dose of 1820 mJ cm⁻² solar-simulated UV which is only half that required
to cause barely detectable inflammation can cause systemic immunosuppression
(Byrne et al., 2002).
Single-exposure ultraviolet (UV) immunosuppression time course and
dose response (Poon et al., 2005)
Summary
• Sub-erythemal doses of UVB can cause local immunosuppression.
• Erythemal doses + may be required for systemic immunosuppression in humans
• UV dose as low as 12.5% of an erythemal dose can cause systemic
immunosuppression in mice (Byrne et al., 2002)
• UVA at doses far too low to be inflammatory can cause local and systemic
immunosuppression in humans and mice.
• UV that mimics the UV portion of the solar spectrum is immunosuppressive in
humans and mice at subinflammatory doses.

The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
7
Low Dose UV and genetic
mutations
Low Dose (sub –erythemal) of UV are Mutagenic
Melanomas have been shown to contain significantly elevated numbers of UVR
signature mutations compared with internal cancers;
Mutations that give cells a growth advantage—inhibit cell death by apoptosis or
other mechanisms, establish genomic instability or overcome senescence—can all
contribute to the development of cancer. Such mutations are an essential step in
the formation of skin cancer;
UV can cause gene mutations in normal keratinocytes that result in the formation
of benign skin lesions, while inflammatory responses, developing for unknown
reasons as a post-UV exposure event, drive progression beyond an initial lesion;
UV induced mutations can cause epidermal cells to make pro-inflammatory
factors or to induce them in the surrounding stroma, creating an oxidizing
environment in which additional oncogenic mutations are likely to take place,
even in the absence of UV.
The high level of mutations in melanomas that have the signature of UVR-induced
damage indicates that the normal mechanisms that detect and repair this damage
must be defective in this system.
UVR causes DNA damage in the form of cyclobutane pyrimidine dimers (CPD) and
6-4 photoproducts (6-4PP), which are repaired by nucleotide excision repair
(NER) and if it not repaired, can lead to mutation. The 6–4PP are less abundant and
efficiently repaired, whereas the more abundant CPDs repair is slower and
misrepair of these lesions produce the UVR signature C>T and CC>TT mutations.
(A) Different forms of DNA damage are repaired by specific
repair mechanisms. (B) The extent of damage and success of
repair determine the outcome, either cell cycle checkpoint
arrest to allow for repair and return to proliferation or
where damage is too extensive for successful repair,
senescence, or death (Pavey et al., 2013).
Defects in checkpoint mechanisms have been identified in melanomas and are
likely to be responsible for increased mutation load in melanoma.
UVR causes both G1 and G2 phase checkpoint arrest in vitro cultured cells:
• The G1 arrest appears to be through a p53-dependent mechanism
• The G2 arrest is independent of p53, and involves a block in the cdc25-
dependent activation of the mitotic cyclin dependent kinase (CDK) complexes
(p16/CDKN2A)
Cell cycle checkpoints: Cell cycle checkpoints are
triggered in response to specific stresses and utilize
different signaling mechanisms to arrest the cells at
specific points in the cell cycle until the stress has been
resolved. (Pavey et al., 2013)

The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
8
Sub-erythemal UV doses are able to induce mutations (Mouret et al., 2006).
Chronic irradiation with 0.5 MED UV for 59 or 75 days has been shown to cause the
formation of p53 patches in hairless mice. The majority of these patches contained
mutant p53 (Rebel et al., 2005).
UVB and Mutation
Average numbers of mutant p53 patches
in Xpc-deficient mice (open diamonds)
and wild-type littermates (closed
diamonds) (n=4) at 1.0 of a wild-type
MED/day. Wild-type SKH1 data at 1.0
MED (closed circles and regression line)
are derived from (Rebel et al., 2005).
The melanoma susceptibility gene CDKN2A which encodes the cell cycle inhibitor
p16INK4A (p16) is commonly defective in melanoma.
Mutations of p16/CDKN2A have also been identified in non-melanoma skin cancers
(Pavey and Gabrielli, 2002).
P16/CDKN2A, which is functionally inactivated in a high proportion of melanoma
cell lines and in 10 - 30% of tumors, is a direct effectors of cell cycle progression
(Pavey et al., 2001).
Melanoma associated mutations of p16 disrupt its ability to promote senescence
arrest. In addition to this role, increased p16 expression has been correlated with
the G2 phase checkpoint arrest in response to suberythemal UVR (Pavey et al., 2013).
Checkpoint Defects (G2 – p16)
G2 phase checkpoint and repair response to UV lesions detected in S phase (Pavey et al.,
2013)
The studies show that in response to suberythemal dose of UVB, basal and
suprabasal layer cells (keratinocytes and melanocytes 24 - 48 h after exposure ) in
human skin arrest in G2 phase, and this is associated with increased levels of p16
(Pavey et al., 2001).
p16 expression is a delayed response, peaking at 24h after UV exposure. The
proportion of cells expressing p16 dramatically increased at 16h post-irradiation with 250 Jm⁻² UVB, peaked at 24h, and substantially diminished by 72h post-
irradiation. Similar data were obtained with 150 and 875 Jm⁻² UVB.
• Several studies identified p53 mutations in a majority of human non-melanoma
skin cancers (NMSC) and the commonest preneoplastic lesion, actinic keratosis,
from sun-exposed body sites (Roshan and Jones, 2012). Clusters of p53-positive
epidermal keratinocytes (p53 patches) are clonal outgrowths of keratinocytes
that contain mutations in p53 and are related to the formation of carcinomas
(Nicolien et al., 2005).
• Loss of p16 function may not only provide a growth advantage by disabling the
senescence mechanism, thus extending the proliferative capacity of the cells,
but may also result in cells undergoing mitosis with an increased burden of UV-
induced DNA mutations, the result of an inability to delay in G2 phase to permit
complete repair of the UV-induced lesions(Pavey and Gabrielli, 2002)
• It is therefore suggests that low dose UV is able to induce signature UV
mutations.
• Photocarcinogenesis in humans could only occur in response to UV doses too low
to cause erythema if these low doses are able to suppress immunity and mutate
genes in humans
Summary

The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
9
Summary
Skin Cancer may be induced with Sub-erythemal Doses of UV
With low dose UV (less than an MED):
Studies have indicated that sub-erythemal doses of UV
may cause benign skin tumors.
UV induced immunosuppression and gene mutations
may be sufficient to cause skin carcinogenesis.
Other considerations:
Most studies in this area (low dose UV) have been conducted in animal models – large area of research;
Limited human studies and epidemiological data
Dose rate of UV
UVI based on erythema as an end point
Other possible factors to consider – eye, Vitamin D
(Prof Lucas this afternoon will discuss balance)
Skin cancer may occur in response to lower doses of UV than are required to cause
erythema. Skin cancer, immunosuppression and gene mutations can all occur in
response to lower doses of UV radiation than are required to cause erythema (Halliday and Lyons, 2008).
MED
Thank you

The 3rd International Conference on UV & Skin Cancer Prevention, Melbourne 7-11 December 2015 Global UV Index Pre-conference Workshop_Day 1
10
References
ATILLASOY, E. S., SEYKORA, J. T., SOBALLE, P. W., ELENITSAS, R., NESBIT, M., ELDER, D. E., MONTONE, K. T., SAUTER, E. &
HERLYN, M. 1998. UVB induces atypical melanocytic lesions and melanoma in human skin. The American journal of
pathology, 152, 1179.
AZOOZ, A. & JALLO, N. 2015. Estimation of UV Index from Integrated Detector Type Measurements. American Institute of
science 1, 5. BERKING, C., TAKEMOTO, R., BINDER, R. L., HARTMAN, S. M., RUITER, D. J., GALLAGHER, P. M., LESSIN, S. R. & HERLYN,
M. 2002. Photocarcinogenesis in human adult skin grafts. Carcinogenesis, 23, 181.
BYRNE, S. N., SPINKS, N. & HALLIDAY, G. M. 2002. Ultraviolet A Irradiation of C57BL/6 Mice Suppresses Systemic Contact
Hypersensitivity or Enhances Secondary Immunity Depending on Dose. Journal of Investigative Dermatology, 119, 858-
864. FIOLETOV, V., KERR, J. B. & FERGUSSON, A. 2010. The UV Index: Definition, Distribution and Factors Affecting It.
Canadian Journal of Public Health / Revue Canadienne de Sante'e Publique, 101, I5-I9.
FOURTANIER, A., MOYAL, D., MACCARIO, J., COMPAN, D., WOLF, P., QUEHENBERGER, F., COOPER, K., BARON, E.,
HALLIDAY, G., POON, T., SEED, P., WALKER, S. L. & YOUNG, A. R. 2005. Measurement of Sunscreen Immune Protection
Factors in Humans: A Consensus Paper. Journal of Investigative Dermatology, 125, 403-409. GREINERT, R., DE VRIES, E., ERDMANN, F., ESPINA, C., AUVINEN, A., KESMINIENE, A. & SCHÜZ, J. 2014. European Code
against Cancer 4th Edition: Ultraviolet radiation and cancer. Cancer Epidemiology.
HALLIDAY, G. M. & LYONS, J. G. 2008. Inflammatory Doses of UV May Not Be Necessary for Skin Carcinogenesis.
Photochemistry & Photobiology, 84, 272-283.
ITALIA, N. & REHFUESS, E. A. 2012. Is the Global Solar UV Index an Effective Instrument for Promoting Sun Protection? A Systematic Review. Health Education Research, 27, 200-213.
KAPPES, U. P., LUO, D., POTTER, M., SCHULMEISTER, K. & RÜNGER, T. M. 2006. Short- and Long-Wave UV Light (UVB and
UVA) Induce Similar Mutations in Human Skin Cells. Journal of Investigative Dermatology, 126, 667-675.
KELLY, D. A., YOUNG, A. R., MCGREGOR, J. M., SEED, P. T., POTTEN, C. S. & WALKER, S. L. 2000. Sensitivity to sunburn is
associated with susceptibility to ultraviolet radiation-induced suppression of cutaneous cell-mediated immunity. The Journal of Experimental Medicine, 191, 561.
KIM, S.-I., PFEIFER, G. P. & BESARATINIA, A. 2007. Mutagenicity of ultraviolet A radiation in the lacI transgene in Big Blue
mouse embryonic fibroblasts. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 617, 71.
LEY, R. D. 1997. Ultraviolet radiation A-induced precursors of cutaneous melanoma in Monodelphis domestica. Cancer
research, 57, 3682-3684. MEVES, A., REPACHOLI, M. H. & REHFUESS, E. A. 2003. Commentary Global Solar UV Index: a physician's tool for fighting
the skin cancer epidemic.International Journal of Dermatology.
References
MOURET, S., BAUDOUIN, C., CHARVERON, M., FAVIER, A., CADET, J. & DOUKI, T. 2006. Cyclobutane Pyrimidine Dimers Are
Predominant DNA Lesions in Whole Human Skin Exposed to UVA Radiation. National Academy of Sciences.
NICOLIEN, K., ANJA, W., SANDER, B., HENK, J. V. K. & FRANK, R. D. G. 2005. Relationship between UV-induced mutant p53
patches and skin tumours, analysed by mutation spectra and by induction kinetics in various DNA-repair-deficient mice. Carcinogenesis, 26, 2123.
PAVEY, S., CONROY, S., RUSSELL, T. & GABRIELLI, B. 1999. Ultraviolet radiation induces p16CDKN2A expression in human
skin. Cancer research, 59, 4185-4189.
PAVEY, S. & GABRIELLI, B. 2002. α-melanocyte stimulating hormone potentiates p16/CDKN2A expression in human skin after ultraviolet irradiation. Cancer research, 62, 875-880.
PAVEY, S., RUSSELL, T. & GABRIELLI, B. 2001. G 2 phase cell cycle arrest in human skin following UV irradiation. Oncogene,
20, 6103-6110.
PAVEY, S., SPOERRI, L., HAASS, N. K. & GABRIELLI, B. 2013. DNA repair and cell cycle checkpoint defects as drivers and
therapeutic targets in melanoma. Pigment cell & melanoma research, 26, 805-816.
PERSSON, A. E., EDSTROM, D. W., BACKVALL, H., LUNDEBERG, J., PONTEN, F., ROS, A. M. & WILLIAMS, C. 2002. The
mutagenic effect of ultraviolet-A1 on human skin demonstrated by sequencing the p53 gene in single keratinocytes. Photodermatology, Photoimmunology & Photomedicine, 18, 287.
POON, T. S. C., BARNETSON, R. S. C. & HALLIDAY, G. M. 2005. Sunlight-Induced Immunosuppression in Humans Is Initially
Because of UVB, Then UVA, Followed by Interactive Effects. Journal of Investigative Dermatology, 125, 840-846.
REBEL, H., KRAM, N., WESTERMAN, A., BANUS, S., VAN KRANEN, H. J. & DE GRUIJL, F. R. 2005. Relationship between UV-
induced mutant p53 patches and skin tumours, analysed by mutation spectra and by induction kinetics in various DNA-
repair-deficient mice. Carcinogenesis, 26, 2123-2130.
RECIO, J. A., NOONAN, F. P., TAKAYAMA, H., ANVER, M. R., DURAY, P., RUSH, W. L., LINDNER, G., DE FABO, E. C., DEPINHO,
R. A. & MERLINO, G. 2002. Ink4a/arf deficiency promotes ultraviolet radiation-induced melanomagenesis. Cancer research, 62, 6724-6730.
ROSHAN, A. & JONES, P. H. 2012. Chronic low dose UV exposure and p53 mutation: Tilting the odds in early epidermal
preneoplasia? International Journal of Radiation Biology, 88, 682-687.
WIGAN, M., PINDER, A., GILES, N., PAVEY, S., BURGESS, A., WONG, S., STURM, R. A. & GABRIELLI, B. 2012. A UVR-induced
G2-phase checkpoint response to ssDNA gaps produced by replication fork bypass of unrepaired lesions is defective in
melanoma. Journal of Investigative Dermatology, 132, 1681-1688.
TEMIS. 2012. UV radiation monitoring: UV index and UV dose, http://www.temis.nl/uvradiation/info/uvindex.html