Embed Size (px)
Citation preview

Vibrational energy transport in acetylbenzonitrile
described by an ab initio-based quantum tier model
Hiroshi Fujisaki,1, ∗ Kiyoshi Yagi,2, † Hiroto Kikuchi,1, ‡ Toshiya Takami,3, § and Gerhard Stock4, ¶
1Department of Physics, Nippon Medical School,
1-7-1 Kyonan-cho, Musashino, Tokyo 180-0023, Japan2Theoretical Molecular Science Laboratory, Advanced Science Institute, RIKEN, 2-1 Hirosawa, Wako 351-0198, Japan
3Department of Computer Science and Intelligent Systems,
Oita University, 700 Dannoharu, Oita 870-1192, Japan4Biomolecular Dynamics, Institute of Physics, Albert Ludwigs University, D-79104 Freiburg, Germany
(Dated: August 26, 2016)
Performing comprehensive quantum-chemical calculations, a vibrational Hamiltonian of acetyl-benzonitrile is constructed, on the basis of which a quantum-mechanical “tier model” is developedthat describes the vibrational dynamics following excitation of the CN stretch mode. Taking intoaccount 36 vibrational modes and cubic and quartic anharmonic couplings between up to threedifferent modes, the tier model calculations are shown to qualitatively reproduce the main findingsof the experiments of Rubtsov and coworkers (J. Phys. Chem. B 115 (2011) 11063), including theenergy relaxation of the initially excited CN mode and the structure-dependent vibrational trans-port. Moreover, the calculations suggest that the experimentally measured cross-peak among theCN and CO modes does not correspond to direct excitation of the CO normal mode but ratherreflects excited low-frequency vibrations that anharmonically couple to the CO mode. Comple-mentary quasiclassical trajectory calculations are found to be in good overall agreement with thequantum calculations.
I. INTRODUCTION
Recent multidimensional spectroscopy experiments al-low us to monitor the pathways and mechanisms of re-distribution and transport of vibrational energy in con-densed phase molecular systems [1–9]. For example,Dlott and coworkers observed ballistic energy transportalong polymer chains [2], Rubtsov and coworkers mea-sured structure-dependent energy transport in acetylben-zonitrile in solution [4], Hamm and coworkers reportedon diffusive energy transfer along a peptide helix [3],and Mizutani et al. demonstrated anisotropic energy flowin proteins [8]. To model the vibrational dynamics ob-served in these experiments, so far mostly perturbativeGolden Rule-type formulations or classical trajectory cal-culations have been employed [10–19]. In an effort toextend these works towards a quantum-dynamical treat-ment beyond the lowest order of perturbation theory, inthis work we adopt the so-called tier model originally pro-posed by Sibert, Reinhardt, and Hynes [20], and comparethe outcome of these calculations to classical nonequilib-rium trajectory simulations.
To briefly introduce the main idea, we consider the
∗Electronic address: [email protected]
†Electronic address: [email protected]
‡Electronic address: [email protected]
§Electronic address: [email protected]
¶Electronic address: [email protected]
Hamiltonian H = H0 + V , where the harmonic part
H0 =
M∑
k=1
(
p2k2
+1
2ω2kq
2k
)
(1)
is characterized by harmonic frequencies ωk, mass-weighted normal mode coordinates qk and correspondingmomenta pk. Vibrational energy redistribution is causedby anharmonic interactions which are described by
V =1
3!
∑
ijk
C(3)ijkqiqjqk +
1
4!
∑
ijkl
C(4)ijklqiqjqkql + · · · ,(2)
where C(3)ijk and C
(4)ijkl denote third- and forth-order
coupling coefficients, respectively. Adopting a time-dependent formulation, we expand the wave functionas |Ψ(t)〉 =
∑
α Cα(t)|α〉, where basis states |α〉 =|n1, n2, · · · , nM 〉 are build from harmonic oscillator eigen-functions |n〉. The resulting time-dependent Schrodingerequation
ihd
dtCα =
∑
β
HαβCβ (3)
with Hamiltonian matrix elements Hαβ ≡ 〈α|H|β〉 de-scribe the time evolution of the system with respect tosome initial condition |Ψ(0)〉. For example, in a typi-cal laser experiment a high-frequency mode is excited attime t = 0 (e.g., |Ψ(0)〉 = |01, 02, · · · , 1M 〉) and the sub-sequent vibrational energy redistribution is measured bymonitoring the time-dependent energy content
Ek(t) =1
2〈Ψ(t)|
(
p2k + ω2kq
2k
)
|Ψ(t)〉 (4)
© C
opyr
ight
Els
evie
r. Th
is is
the
acce
pted
ver
sion
of a
n ar
ticle
to b
e pu
blis
hed
in C
hem
. Phy
s.
http
://dx
.doi
.org
/10.
1016
/j.ch
emph
ys.2
016.
09.0
10

2
of the normal modes of the system. Alternatively, itmay be instructive to consider some local modes of themolecule, which directly reflect the spatial energy distri-bution of the system. For example, one may calculatethe kinetic energy of, say a local CO vibration, whichcan approximately be obtained from (see Appendix)
EkinCO(t) =
1
2
∑
i,k
U2ik hωknk(t), (5)
where index i runs over the x, y and z components ofthe Cartesian coordinates of the C and O atoms, indexk labels the normal modes with eigenvector matrix U ,nk(t) represents the occupation number of these modes,and the zero-point energy was subtracted.
While the above outlined approach in principle isstraightforward, in practice it suffers from the exponen-tial scaling of the dimension of the state space {|α〉}with the number M of vibrational degrees of freedomconsidered. To overcome this problem, a large numberof methods have been proposed, ranging from numeri-cally exact treatments such as the multi-configurationaltime-dependent Hartree (MCTDH) approach [21–23] tosimple classical trajectory calculations [24]. The maybemost popular approach used to calculate transition rateskβα in condensed-phase systems is the Golden Rule ex-pression [25]
kβα ∝ |〈β|V |α〉|2δ(Eβ − Eα), (6)
which is based on first-order time-dependent perturba-tion theory. Starting from some (e.g., experimentallydefined) initial state |α〉, the Golden Rule only requiresstates |β〉 that can be reached by a first-order transition.In practice, one estimates the strength of the transitionby the Fermi resonance parameter [16, 26]
fβα =
∣
∣
∣
∣
〈β|V |α〉Eβ − Eα
∣
∣
∣
∣
, (7)
and includes only transition with
Eβ − Eα ≤ Eth and fβα ≥ fmin, (8)
where Eth and fmin are suitably chosen cut-off parame-ters. In this way, the Golden Rule approach only con-siders the few states of interest for first-order transitions(instead of the total, typically huge dimension of the un-derlying state space).
The so-called tier model represents a generalization ofthis idea to higher orders of time-dependent perturba-tion theory [20, 26–31]. Starting from an initial state|α〉 = |α(0)〉 (zeroth order), all first-order transitionswith sufficient strength fαβ ≥ fmin yield first-order states
|β(1)〉 (first tier, corresponding to a Golden Rule descrip-tion), which subsequently give rise to second-order states|γ(2)〉 (second tier), and so on. The procedure generatesa network in state space shown in Fig. 1, which only in-cludes states that are of importance up to a given order
FIG. 1: Scheme of the network in state space build up by thetier model.
and therefore avoids to directly deal with the exponen-tial size of the underlying state space. In this way, aproblem-adapted basis set is constructed, for which wesolve the time-dependent Schrodinger equation (3). Theefficiency of the method obviously depends on how manytiers with how many states need to be used in order tomodel a given system to some desired accuracy.
In this work, we wish to apply the tier model ansatzto describe the vibrational dynamics of acetylbenzoni-trile (AcPhCN) in chloroform. Upon excitation of theCN stretch mode and by probing a localized CO mode,Rubtsov and coworkers [4] showed that the vibrationalenergy relaxation and energy transport varies for the or-tho, meta and para isomers of AcPhCN (Fig. 2). In par-ticular, they found a clear correlation between the num-ber of bonds connecting the CN mode and the CO modeand the occurrence time of the corresponding cross peakin the two-dimensional infrared spectrum. By performingcomprehensive quantum-chemical calculations to param-eterize the vibrational Hamiltonian [Eqs. (1) and (2)], weconstruct a tier model that accounts for the vibrationaldynamics of AcPhCN following excitation of the CNstretching mode. The quantum dynamics study is com-plemented by quasiclassical trajectory simulations usingthe same model Hamiltonian, which allows us to identifyquantum-mechanical effects (as described by the tier cal-culations) and nonperturbative effects (as described bythe trajectory calculations) of the vibrational dynamics.
II. THEORY AND METHODS
A. Quantum-chemical calculations
All quantum-chemical calculations were performed atthe B3LYP/6-31+G(d) theoretical level using Gaus-sian09 [32]. The effect of the chloroform solvent was im-plicitly treated through the polarizable continuum model(PCM) [33]. After minimizing the structures of the three
© C
opyr
ight
Els
evie
r. Th
is is
the
acce
pted
ver
sion
of a
n ar
ticle
to b
e pu
blis
hed
in C
hem
. Phy
s.
http
://dx
.doi
.org
/10.
1016
/j.ch
emph
ys.2
016.
09.0
10

3
FIG. 2: Molecular structure of the three isomers of acetylben-zonitrile (AcPhCN).
different isomers of AcPhCN, standard normal modeanalysis was performed, which yields the mass-weightedHessian matrix K, the corresponding eigenvector ma-trix U and the diagonal eigenvalue matrix Λ = {ω2
i },i.e., KU = ΛU . Based on the resulting normal modes{qi}, we introduced corresponding harmonic basis states|α〉 = |n1, n2, · · · , nM 〉.
To calculate the anharmonic potential V in Eq. (2),we employed the n-mode coupling representation (nMR)[34–38]. Writing V = V 1MR + V 2MR + V 3MR + . . ., weobtain
V 1MR =∑
i
1
6C
(3)iii q
3i +
1
24C
(4)iiiiq
4i ,
V 2MR =∑
i6=j
1
2C
(3)ijj qiq
2j +
1
6C
(4)ijjjqiq
3j +
∑
i<j
1
4C
(4)iijjq
2i q
2j ,
V 3MR =∑
i<j<k
C(3)ijkqiqjqk +
∑
i6=j<k
1
2C
(4)iijkq
2i qjqk, (9)
that is, the term V nMR accounts for the interactions be-tween n different modes. In the present case of AcPhCN,we included terms containing up to n = 3 modes. Acalculation of the resulting anharmonic frequencies ofAcPhCN using second-order vibrational quasi-degenerateperturbation theory [36] yields in general good agree-ment with the experimental frequencies. An exceptionis the CN mode, where (e.g., for the meta isomer) thecalculated harmonic (2329 cm−1) and anharmonic (2296cm−1) frequencies are well above the experimental fre-quency (2236 cm−1). To better account for the experi-mental situation, we therefore red-shifted the CN modeof all isomers by 60 cm−1.
The 3rd and 4th order coupling coefficients are calcu-lated by numerical differentiation of the Hessian matrix[37]
Kab(x) =1√
mamb
∂2V
∂xa∂xb
, (10)
where xa is the a-th component of the Cartesian coor-dinate x and ma is the associated mass. Configurationsslightly shifted from the equilibrium geometry x0 are gen-erated via
(x±i )a = (x0)a ±
δiUai√ma
, (11)
where δi denotes the step-size of the numerical differen-tiation, which is taken as δi =
√
h/ωi δ0 with δ0 = 0.5[38]. The 3rd and 4th order coupling coefficients are thencalculated as
C(3)ijk =
∑
ab
Kab(x+i )−Kab(x
−i )
2δiUaj Ubk,
(12)
C(4)iijk =
∑
ab
Kab(x+i ) +Kab(x
−i )− 2Kab(x0)
δ2iUaj Ubk.
B. Tier model
As explained in the Introduction, the applicability andefficiency of the tier model crucially depends on howmany tiers with how many states are required to modelthe vibrational energy redistribution of AcPhCN to somedesired accuracy. In a first step, we therefore determinedthe normal modes to be included in the model. Sinceinitially the CN stretching mode (ω ≈ 2330 cm−1) is ex-cited, we can safely neglect all modes of higher frequency(such as CH stretches). Moreover, we did not include thefive lowest normal modes (ω <∼ 230 cm−1, reflecting, e.g.,methyl group rotations), since standard expansions suchas in Eq. (9) are known to lead to artifacts due to thelarge anharmonicity of these modes. In this way, M = 36normal modes are taken into account.
Next, we need to select the contributing states of thevarious tiers. Including interaction terms with qnk up ton = 3 with 〈l|q3k|m〉 ∝ δl,m±3, maximal three quantaare exchanged in a single transition. Starting from ini-
tial state |α(0)〉 = |n(0)1 , n
(0)2 , · · · , n(0)
M 〉 (zeroth order),
we consider all possible states of the first tier |β(1)〉 =
|n(1)1 , n
(1)2 , · · · , n(1)
M 〉 with n(1)i = n
(0)i ± p (p = 0, 1, 2, 3).
By calculating the Fermi parameter fαβ [Eq. (7)] forthese states, we use condition (8) to further restrict thestate space. In this way, all first-tier states are deter-mined. For each first-tier state, we then repeat the pro-cedure to select the second-tier states |γ(2)〉 (from whichthe third-tier states are determined). States identical tostates already found for a previous tier are discarded as
© C
opyr
ight
Els
evie
r. Th
is is
the
acce
pted
ver
sion
of a
n ar
ticle
to b
e pu
blis
hed
in C
hem
. Phy
s.
http
://dx
.doi
.org
/10.
1016
/j.ch
emph
ys.2
016.
09.0
10

4
well as states with ni > nmax = 10. Using thresholdsEth = 200 cm−1 as well as fmin = 0.006, 0.06 and 0.2for the 1st, 2nd, and 3rd tier, respectively, the resultingdimensions of the relevant Hilbert space for the three iso-mers of AcPhCN (given as sum of the number of statesof the three tiers) are 54+825+3229 = 4108 (ortho),51+806+3146 = 4003 (meta), and 47+670+2176 = 2893(para). Employing the resulting Hamiltonian matrixHαβ = 〈α|H|β〉, the time-dependent Schrodinger equa-tion (3) is solved using a leap-frog algorithm [39] with atime step ∆t = 0.1 fs.
C. Classical trajectory calculations
Using the anharmonic model Hamiltonian describedabove, we also performed nonequilibrium classical tra-jectory simulations of the vibrational dynamics. Tosolve Newton’s equation, we employed Yoshida’s 6th-order symplectic integrator [40] with a time step of 0.05fs. Using classical action-angle variables as in [11, 14], weinitially assigned a thermal energy of kBT to all normalmodes, except for the CN mode which was excited by onequantum hω. We decided against including zero-pointenergy into the CN mode (see, e.g., the discussion inRefs. 11, 41), as test calculations yielded somewhat bet-ter results in this case. In this way we prepared 300 initialstates, ran classical trajectories and calculated ensembleaveraged normal mode energies [Eq. (4)]. Trajectoriesthat escaped from the energy basin (a few %) due tothe approximate description of the anharmonic Hamilto-nian were excluded in the analysis. For easy comparisonto the quantum results, all classical vibrational energieswere shifted such that Ek(t=0) = 0.
III. RESULTS AND DISCUSSION
A. Convergence of tier model
We first want to study the convergence of the tier-model with respect to the number of tiers and the numberof states per tier. To address the latter point, we con-sider the time evolution of the normal mode energy ofthe initially excited CN mode for different cut-off valuesof fmin and Eth [see Eq. (8)]. As a representative ex-ample, Fig. 3a shows the CN mode energy for the orthoisomer of AcPhCN, which is seen to decay on a picosec-ond time scale. Compared are three-tier calculations us-ing our standard thresholds given above and by 50 %reduced thresholds, for which the dimension of the statespace doubles. Up to about 4 ps, the agreement betweenthe results for the two thresholds is excellent, for longertimes we find deviations up to ≈ 10 %. For our purposes,it seems sufficient to use the smaller basis set in all re-maining calculations. Moreover, we consider the effectsof the number of tiers on the relaxation dynamics of theCN mode energy. Figure 3b reveals that the Golden Rule
result (corresponding to tier 1) is only sufficient for a fewhundreds of femtoseconds. Including two tiers appearsappropriate for a few picoseconds, where the results arequalitatively similar to our 3-tiers calculations. At longertimes, we may also expect contributions from higher tierswhich, however, is beyond the scope of this paper.
0
500
1000
1500
2000
2500
3000
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
(a) high thresholdlow threshold
0
500
1000
1500
2000
2500
3000
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
(b) 1st2nd3rd
FIG. 3: Convergence of the tier model for the ortho isomer ofAcPhCN with respect to (a) the number of states per tier and(b) the number of tiers. Shown is the normal mode energy ofthe initially excited CN mode for (a) our standard basis (red)and for a basis with doubled number of states (green), as wellas (b) for one (red), two (green) and three (blue) tiers.
B. Vibrational energy redistribution
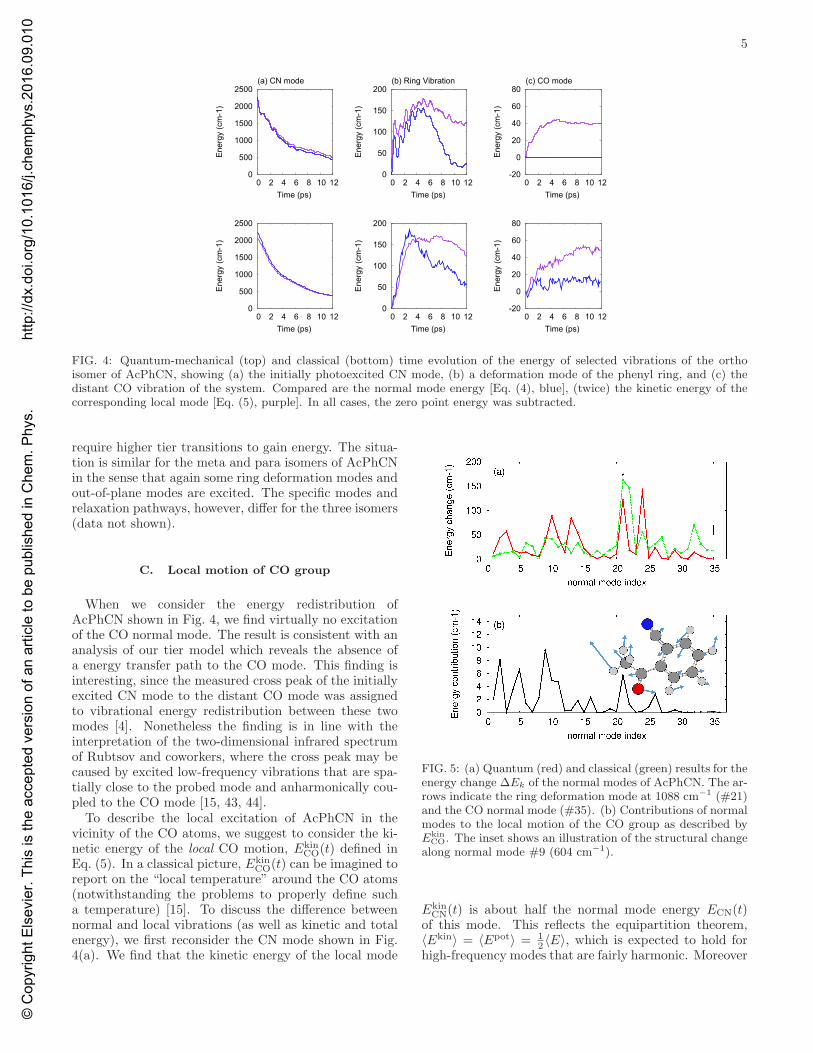
We now wish to employ the above developed tiermodel to discuss the photoinduced vibrational dynamicsof AcPhCN. Focusing first on the ortho isomer, Fig. 4(a)shows the time evolution of the normal mode energy ofthe initially excited CN mode. To facilitate the compar-ison to experiment, here and in all following figures thevibrational energies [Eqs. (4) and (5)] are multiplied witha phenomenological damping factor exp(−t/τ) with τ =14 ps, which accounts for the dissipation of the molecule’svibrational energy into chloroform solvent [4, 42]. An ex-ponential fit to the resulting CN energy ECN(t) gives 5.6ps, which is in qualitative agreement with the experimen-tal result of 3.4 ps [4].
The energy of the initially excited CN mode is re-distributed into a number of weakly excited vibrations,most prominently deformation modes of the phenyl ring.As a representative example, Fig. 4(b) shows the timeevolution of a ring deformation mode at 1088 cm−1,which picks up ≈ 150 cm−1 of vibrational energy withina few picoseconds. To provide an overview of the vi-brational energy redistribution following CN excitation,Fig. 5 presents the integrated energy change ∆Ek =
1/(12ps)∫ 12ps
0Ek(t)dt − Ek(0) of the included normal
modes of AcPhCN. Apart from the ring deformation vi-brations at 1088 cm−1 (mode #21) and 1211 cm−1 (mode#24), the out-of-plane modes at 393 cm−1 (mode #3),617 cm−1 (mode #10) and 786 cm−1 (mode #13) arefound to receive significant energy. An analysis of thedominant Fermi resonance parameters [Eq. (7)] of the tiermodel reveals that the ring deformation modes are ex-cited by tier-1 transitions, while the out-of-plane modes
© C
opyr
ight
Els
evie
r. Th
is is
the
acce
pted
ver
sion
of a
n ar
ticle
to b
e pu
blis
hed
in C
hem
. Phy
s.
http
://dx
.doi
.org
/10.
1016
/j.ch
emph
ys.2
016.
09.0
10
5
0
500
1000
1500
2000
2500
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
(a) CN mode
0
50
100
150
200
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
(b) Ring Vibration
-20
0
20
40
60
80
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
(c) CO mode
0
500
1000
1500
2000
2500
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
0
50
100
150
200
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
-20
0
20
40
60
80
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
FIG. 4: Quantum-mechanical (top) and classical (bottom) time evolution of the energy of selected vibrations of the orthoisomer of AcPhCN, showing (a) the initially photoexcited CN mode, (b) a deformation mode of the phenyl ring, and (c) thedistant CO vibration of the system. Compared are the normal mode energy [Eq. (4), blue], (twice) the kinetic energy of thecorresponding local mode [Eq. (5), purple]. In all cases, the zero point energy was subtracted.
require higher tier transitions to gain energy. The situa-tion is similar for the meta and para isomers of AcPhCNin the sense that again some ring deformation modes andout-of-plane modes are excited. The specific modes andrelaxation pathways, however, differ for the three isomers(data not shown).
C. Local motion of CO group
When we consider the energy redistribution ofAcPhCN shown in Fig. 4, we find virtually no excitationof the CO normal mode. The result is consistent with ananalysis of our tier model which reveals the absence ofa energy transfer path to the CO mode. This finding isinteresting, since the measured cross peak of the initiallyexcited CN mode to the distant CO mode was assignedto vibrational energy redistribution between these twomodes [4]. Nonetheless the finding is in line with theinterpretation of the two-dimensional infrared spectrumof Rubtsov and coworkers, where the cross peak may becaused by excited low-frequency vibrations that are spa-tially close to the probed mode and anharmonically cou-pled to the CO mode [15, 43, 44].
To describe the local excitation of AcPhCN in thevicinity of the CO atoms, we suggest to consider the ki-netic energy of the local CO motion, Ekin
CO(t) defined inEq. (5). In a classical picture, Ekin
CO(t) can be imagined toreport on the “local temperature” around the CO atoms(notwithstanding the problems to properly define sucha temperature) [15]. To discuss the difference betweennormal and local vibrations (as well as kinetic and totalenergy), we first reconsider the CN mode shown in Fig.4(a). We find that the kinetic energy of the local mode
FIG. 5: (a) Quantum (red) and classical (green) results for theenergy change ∆Ek of the normal modes of AcPhCN. The ar-rows indicate the ring deformation mode at 1088 cm−1 (#21)and the CO normal mode (#35). (b) Contributions of normalmodes to the local motion of the CO group as described byE
kin
CO. The inset shows an illustration of the structural changealong normal mode #9 (604 cm−1).
EkinCN(t) is about half the normal mode energy ECN(t)
of this mode. This reflects the equipartition theorem,〈Ekin〉 = 〈Epot〉 = 1
2 〈E〉, which is expected to hold forhigh-frequency modes that are fairly harmonic. Moreover
© C
opyr
ight
Els
evie
r. Th
is is
the
acce
pted
ver
sion
of a
n ar
ticle
to b
e pu
blis
hed
in C
hem
. Phy
s.
http
://dx
.doi
.org
/10.
1016
/j.ch
emph
ys.2
016.
09.0
10
6
it suggests that the kinetic energies of normal and localvibrations are roughly the same for a localized excitedvibrational mode.
While we get similar results for the weakly excited ringdeformation mode (Fig. 4(b)), the situation is quite dif-ferent for the distant CO vibration. Although the corre-sponding normal mode is not excited, the C and O atomsnonetheless reveal significant local motion, which is re-flected in a nonzero kinetic energy of the correspondinglocal mode Ekin
CO. This finding is readily explained byEq. (5) which expands the kinetic energy of the CO localmode in terms of the normal modes of the system. Show-ing the resulting (time-integrated) normal mode contri-butions, Fig. 5b reveals that particularly normal modeswith low frequency contribute to Ekin
CO. As an example,the inset of the figure shows an illustration of the struc-tural change along normal mode #9. The vibration re-veals a global motion of the COCH3 group with respectto the phenyl ring, which obviously results in motion ofthe CO and thus contributes to Ekin
CO.
D. Structure dependent energy flow
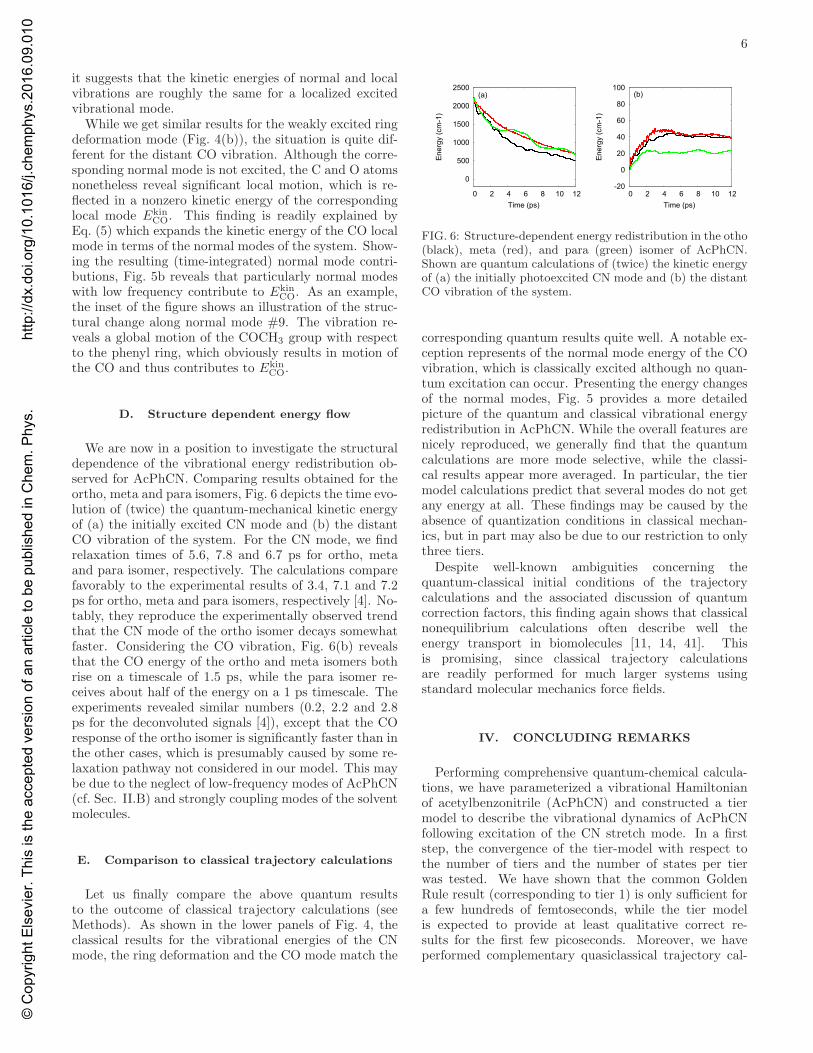
We are now in a position to investigate the structuraldependence of the vibrational energy redistribution ob-served for AcPhCN. Comparing results obtained for theortho, meta and para isomers, Fig. 6 depicts the time evo-lution of (twice) the quantum-mechanical kinetic energyof (a) the initially excited CN mode and (b) the distantCO vibration of the system. For the CN mode, we findrelaxation times of 5.6, 7.8 and 6.7 ps for ortho, metaand para isomer, respectively. The calculations comparefavorably to the experimental results of 3.4, 7.1 and 7.2ps for ortho, meta and para isomers, respectively [4]. No-tably, they reproduce the experimentally observed trendthat the CN mode of the ortho isomer decays somewhatfaster. Considering the CO vibration, Fig. 6(b) revealsthat the CO energy of the ortho and meta isomers bothrise on a timescale of 1.5 ps, while the para isomer re-ceives about half of the energy on a 1 ps timescale. Theexperiments revealed similar numbers (0.2, 2.2 and 2.8ps for the deconvoluted signals [4]), except that the COresponse of the ortho isomer is significantly faster than inthe other cases, which is presumably caused by some re-laxation pathway not considered in our model. This maybe due to the neglect of low-frequency modes of AcPhCN(cf. Sec. II.B) and strongly coupling modes of the solventmolecules.
E. Comparison to classical trajectory calculations
Let us finally compare the above quantum resultsto the outcome of classical trajectory calculations (seeMethods). As shown in the lower panels of Fig. 4, theclassical results for the vibrational energies of the CNmode, the ring deformation and the CO mode match the
0
500
1000
1500
2000
2500
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
(a)
-20
0
20
40
60
80
100
0 2 4 6 8 10 12
Energ
y (
cm
-1)
Time (ps)
(b)
FIG. 6: Structure-dependent energy redistribution in the otho(black), meta (red), and para (green) isomer of AcPhCN.Shown are quantum calculations of (twice) the kinetic energyof (a) the initially photoexcited CN mode and (b) the distantCO vibration of the system.
corresponding quantum results quite well. A notable ex-ception represents of the normal mode energy of the COvibration, which is classically excited although no quan-tum excitation can occur. Presenting the energy changesof the normal modes, Fig. 5 provides a more detailedpicture of the quantum and classical vibrational energyredistribution in AcPhCN. While the overall features arenicely reproduced, we generally find that the quantumcalculations are more mode selective, while the classi-cal results appear more averaged. In particular, the tiermodel calculations predict that several modes do not getany energy at all. These findings may be caused by theabsence of quantization conditions in classical mechan-ics, but in part may also be due to our restriction to onlythree tiers.
Despite well-known ambiguities concerning thequantum-classical initial conditions of the trajectorycalculations and the associated discussion of quantumcorrection factors, this finding again shows that classicalnonequilibrium calculations often describe well theenergy transport in biomolecules [11, 14, 41]. Thisis promising, since classical trajectory calculationsare readily performed for much larger systems usingstandard molecular mechanics force fields.
IV. CONCLUDING REMARKS
Performing comprehensive quantum-chemical calcula-tions, we have parameterized a vibrational Hamiltonianof acetylbenzonitrile (AcPhCN) and constructed a tiermodel to describe the vibrational dynamics of AcPhCNfollowing excitation of the CN stretch mode. In a firststep, the convergence of the tier-model with respect tothe number of tiers and the number of states per tierwas tested. We have shown that the common GoldenRule result (corresponding to tier 1) is only sufficient fora few hundreds of femtoseconds, while the tier modelis expected to provide at least qualitative correct re-sults for the first few picoseconds. Moreover, we haveperformed complementary quasiclassical trajectory cal-
© C
opyr
ight
Els
evie
r. Th
is is
the
acce
pted
ver
sion
of a
n ar
ticle
to b
e pu
blis
hed
in C
hem
. Phy
s.
http
://dx
.doi
.org
/10.
1016
/j.ch
emph
ys.2
016.
09.0
10

7
culations, which in general were found to reproduce theenergy redistribution of the quantum calculations quitewell.
To study the structure dependent energy transport, wehave considered the time evolution of the energy of thevibrational modes of AcPhCN in chloroform. The tiermodel calculations have been shown to qualitatively re-produce the main findings of the experiments of Rubtsovand coworkers [4], including the energy relaxation of theinitially excited CN mode and isomer-dependent vibra-tional redistribution. The general performance of themodel seems remarkable, in particular in the light of thefact that the cooling of photoexcited AcPhCN in the chlo-roform solvent was simply modeled by an overall phe-nomenological relaxation term. We have shown that thecross-peak among the CN and CO modes measured inthe two-dimensional infrared spectrum does not neces-sarily correspond to direct excitation of the CO normalmode but may rather reflect excited low-frequency vi-brations that are spatially close to the probed mode andanharmonically couple to the CO mode [15, 43]. To ac-count for this transient feature of infrared spectra, wehave proposed to consider the kinetic energy of the local
CO mode, which can be imagined to report on the “localtemperature” around the CO atoms.
Acknowledgments
This work is dedicated to Professor Lorenz Cederbaumon the occasion of his 70th birthday. Moreover, we aregrateful to Igor Rubtsov, Peter Hamm, David Leitner,Mikito Toda, Shinji Saito, and John Straub for inspiringand useful discussions. This research was supported bythe Alexander von Humboldt Foundation and Japan So-ciety for the Promotion of Science (KAKEN 22540421).
V. APPENDIX: DERIVATION OF EQ. (5)
Given mass-weighted Cartesian coordinates Qi andmomenta Pi (i = 1, . . . , 3N), the corresponding normalmode coordinates qk and momenta pk (k = 1, . . . , 3N−6)
are related via
Pi =∑
k
Uikpk, Qi −Qeqi =
∑
k
Uikqk, (13)
where Uik represents the eigenvector matrix of the trans-formation. The kinetic energy associated with a compo-nent (x, y or z) of an atom is given by
Ekini =
P 2i
2=
1
2
∑
k,l
UikUil〈pkpl〉, (14)
〈pkpl〉 =∑
α,β
C∗α(t)Cβ(t)〈α|pkpl|β〉. (15)
The somewhat cumbersome calculations simplify con-siderably if we assume that 〈pkpl 6=k〉 ≪ 〈pkpk〉 (which,though, may not be well fulfilled by low-frequencymodes). Assuming furthermore that |〈α|p2k|β〉| ≪|〈α|p2k|α〉| (which is typically well fulfilled), we obtain
Ekini =
1
2
∑
k
U2ik
∑
α
|C∗α(t)|2〈α|p2k|α〉
=1
2
∑
k
U2ikhωk (nk(t)+1/2), (16)
where nk(t) is the occupation probability of normal modek. The kinetic energy Ekin
CO(t) of the diatomic CO vibra-tions in Eq. (5) is obtained by sumation of the i = x, yand z components of the Cartesian coordinates of thetwo atoms. In the case of the ring deformation mode,we included the kinetic energy of all six C atoms of thephenyl ring, and subsequently scaled the energy by a fac-tor 0.5 in order to roughly account for the fact that onlyhalf of the ring atoms contribute to the correspondingnormal mode. We note that the kinetic energy Ekin
CO(t)includes the relative motion of the two atoms to eachother (the local mode) as well as the relative motion ofthe two atoms with respect to the rest of the molecule.Owing to its approximate nature, Eq. (16) allows us toexpress the atomic kinetic energy in terms of a simplesum over its normal mode contributions.
[1] D. M. Leitner and J. E. Straub, Proteins: Energy, Heat
and Signal Flow, Taylor and Francis/CRC Press, Lon-don, 2009.
[2] Z. Wang, J. A. Carter, A. Lagutchev, Y. K. Koh, N.-H.Seong, D. G. Cahill, and D. D. Dlott, Ultrafast flashthermal conductance of molecular chains, Science 317,787 (2007).
[3] V. Botan, E. Backus, R. Pfister, A. Moretto, M. Crisma,C. Toniolo, P. H. Nguyen, G. Stock, and P. Hamm, En-ergy transport in peptide helices, Proc. Natl. Acad. Sci.USA 104, 12749 (2007).
[4] V. Kasyanenko, S. Tesar, G. Rubtsov, A. Burin, and
I. Rubtsov, Structure dependent energy transport:Relaxation-asissted 2DIR measurement and theoreticalstudies, J. Phys. Chem. B 115, 11063 (2011).
[5] W. J. Schreier, T. Aumller, K. Haiser, F. O. Koller,M. Lweneck, H.-J. Musiol, T. E. Schrader, T. Kiefhaber,L. Moroder, and W. Zinth, Following the energy transferin and out of a polyprolinepeptide, Peptide Science 100,38 (2013).
[6] H. M. Muller-Werkmeister, Y.-L. Li, E. W. Lerch,D. Bigourd, and J. Bredenbeck, Ultrafast hopping fromband to band: Assigning infrared spectra based on vi-brational energy transfer, Angew. Chem. Int. Ed. Engl.
© C
opyr
ight
Els
evie
r. Th
is is
the
acce
pted
ver
sion
of a
n ar
ticle
to b
e pu
blis
hed
in C
hem
. Phy
s.
http
://dx
.doi
.org
/10.
1016
/j.ch
emph
ys.2
016.
09.0
10

8
52, 6214 (2013).[7] G. Li, D. Magana, and R. B. Dyer, Anisotropic energy
flow and allosteric ligand binding in albumin, Nat. Com-mun. 5, 3100 (2014).
[8] N. Fujii, M. Mizuno, H. Ishikawa, and Y. Mizutani, Ob-serving vibrational energy flow in a protein with the spa-tial resolution of a single amino acid residue, J. Phys.Chem. Lett. 5, 3269 (2014).
[9] N. I. Rubtsova and I. V. Rubtsov, Vibrational en-ergy transport in molecules studied by relaxation-assistedtwo-dimensional infrared spectroscopy, Ann. Rev. Phys.Chem. 66, 717 (2015).
[10] D. E. Sagnella and J. E. Straub, Directed energy ”funnel-ing” mechanism for heme cooling following ligand photol-ysis or direct excitation in solvated carbonmonoxy myo-globin, J. Phys. Chem. B 105, 7057 (2001).
[11] P. H. Nguyen and G. Stock, Nonequilibrium molecular-dynamics study of the vibrational energy relaxation ofpeptides in water, J. Chem. Phys. 119, 11350 (2003).
[12] D. Schwarzer, P. Kutne, C. Schroder, and J. Troe, In-tramolecular vibrational energy redistribution in bridgedazulene-anthracene compounds: Ballistic energy trans-port through molecular chains, J. Chem. Phys. 121, 1754(2004).
[13] D. M. Leitner, Frequency resolved communication mapsfor proteins and other Nanoscale materials, J. Chem.Phys. 130, 195101 (2009).
[14] H. Fujisaki, K. Yagi, J. E. Straub, and G. Stock, Quan-tum and classical vibrational relaxation dynamics of n-methylacetamide on ab initio potential energy surfaces,Int. J. Quant. Chem. 109, 2047 (2009).
[15] P. H. Nguyen, S. M. Park, and G. Stock, Nonequilibriummolecular dynamics simulation of the energy transportthrough a peptide helix, J. Chem. Phys. 132, 025102(2010).
[16] H. Fujisaki, Y. Zhang, and J. E. Straub, Non-Markoviantheory of vibrational energy relaxation and its applica-tions to biomolecular systems, Adv. Chem. Phys. 145, 1(2011).
[17] S. Tesar, V. Kasyanenko, I. Rubtsov, G. Rubtsov, andA. Burin, Theoretical study of internal vibrational relax-ation and energy transport in polyatomic molecules, J.Phys. Chem. A 117, 315 (2012).
[18] T. Ishikura, Y. Iwata, T. Hatano, and T. Yamato, Energyexchange network of inter-residue interactions within athermally fluctuating protein molecule: A computationalstudy, J. Comput. Chem. 36, 1709 (2015).
[19] S. Buchenberg, D. Leitner, and G. Stock, Scaling rulesfor vibrational energy transport in globular proteins, J.Phys. Chem. Lett. 7, 25 (2016).
[20] E.L. Sibert III, W. Reinhardt, and J. Haynes, Intramolec-ular vibrational relaxation and spectra of ch and cd over-tones in benzene and perdeuterobenzene, J. Chem. Phys.81, 1115 (1984).
[21] H.-D. Meyer, U. Manthe, and L. S. Cederbaum, Themulti-configurational time-dependent Hartree approach,Chem. Phys. Lett. 165, 73 (1990).
[22] H. D. Meyer, F. Gatti, and G. A. Worth, editors, Multidi-
mensional Quantum Dynamics, Willey-VCH, Weinheim,2009.
[23] K. Giese, M. Petkovic, H. Naundorf, and O. Kuhn,Multidimensional quantum dynamics and infrared spec-troscopy of hydrogen bonds, Phys. Rep. 430, 211 (2006).
[24] W. L. Hase, Advances in Classical Trajectory Methods,
volume 1, Jai Press, London, 1992.[25] D. J. Tannor, Introduction to Quantum Mechanics- A
Time-Dependent Perspective, University Science Books,Sausalito, 2007.
[26] R. Wyatt and J. Zhang, editors, Dynamics of Molecules
and Chemical Reactions, Dekker, 1996.[27] S. Schofield and P. Wolynes, A scaling perspective on
quantum energy flow in molecules, J. Chem. Phys. 98,1123 (1993).
[28] A. Stuchebrukhov and R. Marcus, Theoretical study ofintramolecular vibrational relaxation of acetylenic CHvibration for v=1 and 2 in large polyatomic molecules(CX3)3YCCH, where X=H or D and Y=C or Si,, J.Chem. Phys. 98, 6044 (1993).
[29] M. Gruebele and R. Bigwood, Molecular vibrational en-ergy flow: beyond the golden rule, Int. Rev. Phys. Chem.17, 91 (1998).
[30] S. Keshavamurthy, Scaling perspective on intramolecu-lar vibrational energy flow: analogies, insights, and chal-lenges, Adv. Chem. Phys. 153, 43 (2013).
[31] D. M. Leitner, Quantum ergodicity and energy flow inmolecules, Adv. Phys. 64, 445 517 (2015).
[32] M. J. Frisch et al., Gaussian 09 Revision A.1, GaussianInc. Wallingford CT 2009.
[33] J. Tomasi, B. Mennucci, and R. Cammi, Quantum me-chanical continuum solvation models, Chem. Rev. 105,2999 (2005).
[34] S. Carter, S. Culik, and J. Bowman, Vibrational self-consistent field method for many-mode systems: A newapproach and application to the vibrations of CO ad-sorbed on Cu(100), J. Chem. Phys. 107, 10458 (1997).
[35] K. Yagi, S. Hirata, and K. Hirao, Efficient configurationselection scheme for vibrational second-order perturba-tion theory, J. Chem. Phys. 127, 034111 (2007).
[36] K. Yagi, S. Hirata, and K. Hirao, Vibrational quasi-degenerate perturbation theory: Applications to Fermiresonance in CO2, H2CO, and C6H6, Phys. Chem. Chem.Phys. 10, 1781 (2008).
[37] K. Yagi, T. Taketsugu, K. Hirao, and M. S. Gordon,Direct vibrational self-consistent field method: Appli-cations to H2O and H2CO, J. Chem. Phys. 113, 1005(2000).
[38] K. Yagi, K. Hirao, T. Taketsugu, M. W. Schmidt, andM. S. Gordon, Ab initio vibrational state calculationswith a quartic force field: Applications to H2CO, C2H4,CH3OH, CH3CCH, and C6H6, J. Chem. Phys. 121, 1383(2004).
[39] T. Iitaka, Solving the time-dependent schrdinger equa-tion numerically, Phys. Rev. E 49, 4684 (1994).
[40] H. Yoshida, Construction of higher order symplectic in-tegrators, Phys. Lett. A 150, 262 (1990).
[41] G. Stock, Classical simulation of quantum energy flow inbiomolecules, Phys. Rev. Lett. 102, 118301 (2009).
[42] S. M. Park, P. H. Nguyen, and G. Stock, Molecular dy-namics simulation of cooling: Heat transfer from a pho-toexcited peptide to the solvent, J. Chem. Phys. 131,184503 (2009).
[43] P. Hamm, S. M. Ohline, and W. Zinth, Vibrational cool-ing after ultrafast photoisomerization of azobenzene mea-sured by femtosecond infrared spectroscopy, J. Chem.Phys. 106, 519 (1997).
[44] V. M. Kasyanenko, Z. Lin, G. I. Rubtsov, J. P. Donahue,and I. V. Rubtsov, Energy transport via coordinationbonds, J. Chem. Phys. 131 (2009).
© C
opyr
ight
Els
evie
r. Th
is is
the
acce
pted
ver
sion
of a
n ar
ticle
to b
e pu
blis
hed
in C
hem
. Phy
s.
http
://dx
.doi
.org
/10.
1016
/j.ch
emph
ys.2
016.
09.0
10













![Japan IPv6 Measurement, by Tomohiro Fujisaki [APNIC 38 / IPv6 Readiness Measurement BoF]](https://img.pdfslide.net/doc/110x75/549215a9b4795956138b5624/japan-ipv6-measurement-by-tomohiro-fujisaki-apnic-38-ipv6-readiness-measurement-bof.jpg)















