Embed Size (px)
Citation preview

2.1 - CHRONIC MYELOID LEUKAEMIA (CML)
1. The clinical features of CMLD - CML is a myeloproliferative disease/neoplasm where there is a proliferation of mature myeloid cells in the BM & PB. Although the causative mutations occurs in the pluripotent HSC and hence is present in all cells, the disease primarily manifests as a neutrophilia
I - i = 1/100k/year (rare); accounts for 15-20% of all leukaemiasA - 50-60yS - M> F (just)GA - UNKNOWN; ionising radiation is a known risk factorP - [see below]C - s/s/e
20-40% of patients are asymptomatic (chronic phase of the disease) Increased metabolism = B symptoms (fatigue, weight loss, sweating, normocytic
anaemia) Acute leuakaemic symptoms manifest in the blastic phase Hepatosplenomegaly (due to extrameduallry haemopoeises)
Investigastions Blood film - mature leucocytosis with blasts <2% of nucleated cells in BM
o The majority is neutrophilia; neutrophil may have decrease nuclear lobules and thus appear as band cells
o Basophilia may also be present; it is otherwise rare in other causes of leucocytosis
Blood count - anaemia. Platelet count may increase as disease progresses (it is very rarely low in the chronic phase)
BM aspirate - HYPOCELLULARo Megakaryocytes can increase in number and appear hypo-lobularo The percentage of erythroid cells falls (increased myeloid:erythroid
ratio)o Increased deposition of reticulin fibreso Pesudo Gaucher cells (macrophaged that have phagocytosed other cells)
can be present Low Neutrophil ALP used to be another diagnostic tool Diagnosis is based on cytogenetics
Managementc - monitor white cell count, cytogenetics and molecular genetics.m - imatinib is a useful treatment (interferons used to be used).S - BM transplant is the only cure but it can not be used in all patients
P - Imatinib gives a good prognosis on haematological, cytogenetic and molecular grounds
2. CML as a 3 phase disease Phase 1 - Chronic; most patients are in this phase Phase 2 - accelerated
o Progression occurs because of additional mutations on top of the background BCR/ABL
o Very variable presentation that can take months or years

o Suspect progression into accelerated phase if patient: Displays decreased response to treatment % of blasts increases to 10-20% % of basophils increases to 20% Platelet count deranges/ splenomegaly increases
Phase 3 - Blastic Phaseo Similar to AML, although some develop into a lymphoid leukaemiao % of blasts >20%o Can be fatal
3. The genetic Pathogenesis of CMLThe hallmark of CML is the Philadelphia chromosome, which is present in 95% of patients. In the remainder, the BCR-ABL fusion gene forms via cryptic translocations involving other chromosomes apart from 9 & 22
The Philadelphia chromosome Translocation of BCR (22q) to ABL (9q)
o The result is an elongated tyrosine kinase product which is constiutively expressed 15% of primary ALL patients have the same mutation, but the splice point on the BCR gene is
upstream - this produces a smaller gene product BCR/ABL expression in the HSC leads to:
o Proliferation & resistance to apoptosis of myeloid lines, particularly neutrophilso Decreased development of lymphoid lineageso Development into accelerated/blastic CML in the context of additional mutations
ABL is usually a nuclear protein, but BCR-ABL is cytoplasmic - has the potential to effect a lot of downstream processes directly

2.2 - ACUTE MYELOID LEUKAEMIA (AML)
1. Clinical features of AMLD - A clonal expansion of myeloid blasts in the BM or other tissues, where the percentage of blasts reaches >20% of nucleated cells in the BM
I - 4/100k/year; accounts for 70% of adult acute leaukaemiasA - adults; median 60yearsS - M>F (just)GA - Known risk factors include ionising radiation, benzene, smoking, chemotherapy and
possibly virusesPC s/s/e
Features relating to BM failure/ replacement of normal cells in BM by leaukaemic cloneso Normocytic Anaemia (fatigue etc)o Neutropenia (infections, mostly bacterial, watch out for sepsis)o Thrombocytopenia (spontaneous bleeds from mucous membranes)
BM expansion/ bone impinging on other tissues - Bone pain Features relating to infiltrates of leukaemic clones into other tissues
o Organomegaly, gyum hypertrophy, sarcoma Symptoms due to increased metabolism - B symptoms Symptoms relating to electrolyte imbalance following release of intraceullar contents
after cell deatho Hyperuricaemia, hyperkalaemiao Hypocalcaemia
Investigations [see below]
ManagementC - supportive treatments; antibiotics (infections), blood transfusions (Hb), platelet transfusions (bleeding), resuscitate, rehydrate, allopurinolM - Chemotherapyto induce remission, differentiating agents (eg ATRA, arsenic trioxide) can be used in M3S - BMT
P - depends on classification but ‘normal’ prognosis AML is roughly 42% 5 year survival
2. Investigations and classifications in AML4 main methods in investigation of AML
Morphology (blood film) Immunophenotyping (flow cytometry) Cytogenetic analysis (FISH) Molecular genetics
WHO classification of AML AML with recurrent genetic abnormalities (translocations; 35% of cases) AML with multi-lineage dysplasia (i.e. following MDS/MPD); worse prognosis Therapy-related AML (secondary to chemo);
o alkylating agents - develops 5-10years after drug use after an MDSo topoisomerase II inhibitors - develops 1-5 years after drug use without an MDS

AML not otherwise categorized - this has been revised in most recent classification - get it
3. Morphological classification in AML - the FAB system is not enough
Classification DescriptionM0 No maturation; immunophneotyping needed to diagnoseM1 Minimal maturationM2 Mature cells with upto 90% non-erythroid; M3 Acute promyelocytic leukaemia; t15,17; DIC; good prognosis if treated earlyM4 MyelomonocyticM5 Monocytic/monoblastic; gum/organ ilfiltrates; hypokalaemiaM6 Erythroid; bad prognosisM7 Megakaryoblastic; immunophenotyping needed to diagnose; bad prognosis
4. Immunophenotyping in AML Is needed for the diagnosis of M0/M7 AML
o Picks up CD41 and CD61 in M7 Has a role in all laeukaemias as a general point because it can pick up CD33/ CD13/ other
myeloid markers Useful in ruling out lymphoid leukaemia if it is negative for CD19 as an example
5. Genetic basis of AMLThere are 4 main translocations in AML; these account for 35% of the cases Good Prognosis
o T8,21 [AML1/ETO] M2 morphology Accounts for 5-12% of cases Presence of auer rods is sufficient to diagnose it on blood film
o Inv16 or inter 16 translocation [CBFb/MYH11] M4 morphology Accounts for 10-12% of cases Associated with abnormal marrow eosinophils and large basophil granules
o T15,17 [PML/RARa] M3 morphology (promyelocytic leuakaemia) Accounts for 5-8% of cases
Bad Prognosiso 11q23 [MLL] mutation
Accounts for 5-6% of cases
The majority of AML cases are caused by genetic mutations These can occur with or without translocations
o FLT3 mutation FLT3 is a gene that is involved in HSC differentiation/proliferation Accounts for 20-40% of cases ITD mutation has worse prognosis than TKD mutation
o KIT mutation [poor prognosis]o Nucleophosmin mutation
Accounts for 1/3 of cases Mutation leads to nucelophosmin, which usually binds RNA in the nucleus, being
expressed in the cytoplasm Better prognosis

2.3 – LAB DIAGNOSIS OF ACUTE LEUKAEMIA: MORPHOLOGY & CYTOCHEMISTRY
1. The initial suspicion of leukaemia comes from signs/symptoms AML
o Anaemia, neutropenia, thrombocytopeniao hepatosplenomegalyo Gum hypertrophy, skin infiltrates (M4)o gum haemorrhage, DIC (M3)o Hypopyon (leukaemic cells in anterior chamber of eye)o Recurrent priapism in men
ALLo Anaemia, neutropenia, thrombocytopeniao hepatosplenomegalyo lymphadenopathyo thymic enlargemento involvement of testes in meno involvement of CNS
2. Blood film, count and bone marrow aspirate are important in Acute leukaemia is diagnosis Things to look for on blood count:
o anaemia +/- MCV (usually normocytic, but anaemia can cause rouleaux = high mcv)o leucocytosis (sometimes leucopenia)o neutropenia & thrombocytopenia
things to look for on blood film:o anaemia + aniso- & polikilocytosiso leucocytosis (sometimes leucopenia)o presence of leukaemic cells (blasts, auer rods)o neutropenia +/- dysplastic neutrophils
dysplastic neutrophil are more characteristic in myeloid leukaemia dysplastic neutrophils are more diffuse and have ?pseudo-pelvis nuclear shape
in MDS the appearance of (white) nucleoli may suggest leukaemia
o thrombocytopenia bone marrow aspirate
o helps to count percentage of blasts in bone marrow (>20% of nucleated cells = acute leukaemia)
o appearance of onky one type of cell (as opposed to all cells in a lineage development) are indicative of clonal expansion and hence acute leukaemia
o gains material for cytogenetic analysis
3. The role of cytochemistry in diagnosis of acute leukaemia is dying out because immunophenotyping is more specificThere are 4 types of stain used in cytochemistry
Myeloperoxidase – stains myeloblastso only identifies M1/2/3 AMLo helps to differentiate M0 & M7 AML from ALL (ALL will not stain +ve)
Sudan Black B – same as myeloperoxidase Chloroacetate esterase – stains myeloblasts
o useful in variant M3 non-specific esterase – stains monoblasts and megakaryocytes

4. the role of morphology in diagnosis of acute leukaemia (FAB)Morphology lineage notesM0 minimally differentiated need immunophenotyping for CD34, CD33,
CD117 to diagnoseM1 slightly matureM2 mature smaller cells, more cytoplasm, nucleus a bit
more condensedM3 APML DIC following degrnaulation of leukaemics
variant M3 has a cleaved nucleus apperanceM4 myelomonocyticM5 monocytic/monoblasticM6 erythroidM7 megakaryoblastic need immunophenotyping to diagnose
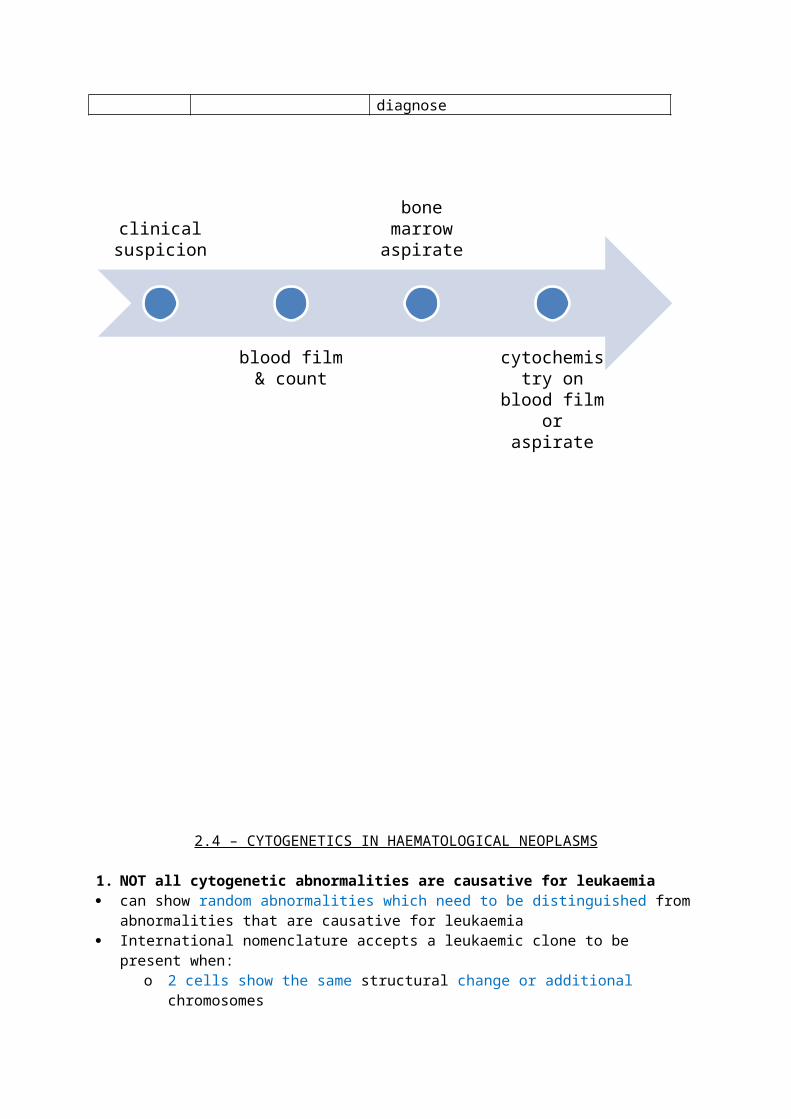
clinical suspicion
blood film & count
bone marrow aspirate
cytochemistry on blood film
or aspirate

2.4 – CYTOGENETICS IN HAEMATOLOGICAL NEOPLASMS
1. NOT all cytogenetic abnormalities are causative for leukaemia can show random abnormalities which need to be distinguished from abnormalities that are
causative for leukaemia International nomenclature accepts a leukaemic clone to be present when:
o 2 cells show the same structural change or additional chromosomeso 3 cells show the same missing chromosome
2. Translocations: Inversions & deletions Reciprocal translocations involve exchange. Non-receiprocal translocations involve one way
transfer Balanced translocations involve no change in amount of DNA. unblanaced translocations give a
gain/ loss of DNAo International nomenclature lists chromosomes in numerical order. if the chromosome is
unbalanced, that which has had DNA inserted into it, is listed first Pericentric inversions involve the centromere
o this involves one chromosome and means one chromosome has 2 fusion genes paracentric inversions do not involve the centromere
o this involves both chromosomes and means both chromosomes have 1 fusion gene terminal deletions involve loss of the telomere Interstitial deletions keep the telomere but intervening material between the centromere and
the telomere is lost
3. Two of the main Cytogenetic techniques are karyoptyping and FISHKaryotyping arrests cells in metaphase and uses a Giemsa/fluoresecent stain to induce a banding pattern on each chromosome so they can be visualised
Single probe single colour FISH Types of probes that can be used in FISH – centromeric, arm or ‘whole chromosome’ Single colour FISH labels a DNA sequence on a pair of chromosomes with one colour
o hence 3 signals = trisomyo signals of different sizes = translocationo 1 signal = partial deletion/ monosomy – not sensitive to this
Double colour FISH Labels targeted sequences of DNA with a particular colour
o possible to label centromere one colour and label an arm a different colour. o hence loss of a chromosome gives loss of both signals whereas a partial deletion gives
loss of one fusion probes target DNA sequences at breakpoints and hence give a colour change when two
targeted sequences come into proximity following translocation breakapart probes target DNA sequences that span across the breakpoint, hence giving a colour
loss following a translocation
Advantages of FISH can give information about an amplified signal (oncogene) as well as a decreased signal (tumour
suppresso gene) can be used with cells in metaphase or interphase – useful for slowly dividing cells (CLL)
Whole paint FISH/ multi colour FISH is useful when the translocation is complexSummary: FISH
Type of FISH Definition Applications(normal) FISH labels DNA sequences with fluorgenic
probedetects numerical abnormalities, translocations, presence of fusion genes, amplifications/loss of gene sequences
M-FISH (multicolour) labels chromsooems with 5 fluogenic probes
can help to make complex karyotopes a bit clearer
SKY (spectral karyotyping)
similar to M-FISH but cominbations of colours are recognised by spectral signature
4. PCR can be adopted if karyoptyping is complex/ cytogenetic analysis has failed
Type of PCR definition Applications(normal) PCR Amplifies DNA in vitro detection of gene rearrangement and hence
leukaemia classification; more senstitive than southern blotting
RT-PCR (reverse transcriptase)
uses RT to generate and subsequently amplify cDNA
analysis of genes that are too long for standard PCR
Multiplex PCR simultaneous identification of a number of mutationsRQ-PCR (real-time quantitative)
fluorogenic probe is broken down over course of reaction
quantification of DNA = quantification of disease burden AND monitoring of minimal residual disease
3. WHO classification of AML
Therapy-related AML has a worse prognosis which is why it is classified separately.
alkylating agents cause worse AML – they are associated with abnormalities in c7, c5, inv(3), t (3;3), (6;9) and (8;16)
topoisomerase inhibitors cause balanced translocations and account for ~10% of APML. this is on the up as treatment of other cancers is improving so that survival is long enough to experience an AML.
importantly, mutations caused by therapy have a worse prognosis than if the same mutation occurred de novo.
The next step in the WHO classification is classifying recurrent genetic abnormalities seen in AML, because they correlate closely to prognosisThe important ones are:
t(8;21) (q22;q22) [RUNX/RUNX1T1]

o good prognosis APML - t(15;17) (q22; q21) [PML/RARA]
o variant M3 is very (but not 100%) similar to true M3 on a genetic and clinical basis. it is ‘variant’ because of morphological differences – granules cannot be seen on LM but they can on EM
o the diagnosis between classic and variant M3 lies with using a fluorescent Ab against PML, which appears microparticulate in variant M3
o survival rates plummet initially because of DICo RARA is involved in a number of leukaemias
inv (16) (p13.1q22) or t(16;16) (p13;q22) [CBFB/MYH11]o eosinophilia and increased basophil granuleso good prognosis
Some others are: t(9;11) (p22;q23) [MLLT3/MLL] – bad
prognosis t(6;9) (p23;q23) [DEK/NUP214] inv (3) (q21q26.2) or t(3;3) (q21;q26.2)
[RPN1/EVI1] t (1;22) (p13;q13) [RBM15-MKL1] NPM1/ CEBPA mutations, these tend to have
a good prognosis.
There are many subtypes of AML that can not be classified on a genetic basis some of these can be classified morphologically by the FAB system M0-M7 the others can not:
o transient myeloproliferative disorder in Downs Syndromeo Acute basophilic leukaemiao acute panmyelosis with myelofibrosiso myeloid sarcoma
Summary – roles of cytogenetics in haematological neoplasms
1. Prognostic rolesa. Demonstrates monoclonality of leukaemia b. Shows which lineages are involved in the neoplasmc. Provides evidence of mechanisms in leukaemia (over-expressed oncogene/ suppressed
tumour suppressor gene)2. Diagnostic roles
a. Provides evidence of aetiology of leukaemia (because certain aetiological agents cause distinctive genetic changes; finding these changes can reveal the cause)
b. Very specific at identifying subtypes of AML (eg M3/APML)c. Distinguishes therapy-related and recurrent AMLd. Can provide evidence for underlying/predisposing disease to a leukaemia
3. Monitoring rolesa. Can provide evidence of regression in response to treatmentb. Can provide evidence for engraftment in response to BMT

2.5 – IMMUNOPHENOTYPING IN ACUTE LEUKAEMIA
1. When do we need immunophenotyping? In EVERY case of acute leukaemia that is not obviously myeloid Particularly in M0 & M7 AML
o lymphoblasts cannot be distinguished from M0 myelobasts because M0 do not have granules (so myeloperoxidase staining will fail) and do not have auer rods (so morphological tests will fail)
2. Principles of Immunophenotyping the process identifies CSM antigens, and, if the cell can be permeabilised, cytoplasmic and
nuclear antigens too Most immunophenotyping is achieved by use of a flow cytometer
o forward light scatter can count and size cells in a streamo sideways light scatter can characterize cells based on granularity/ complexityo gating (to increase expression and hence scatter) of a particular antigen can increase
specificity in flow cytometric analyses gating fails with some blast cells, particularly monoblasts and M3 AML blasts
the principle is using a flurosecent monoclonal antibody against the antigen. o monoclonal antibodies are stable and specifico coexpression of different antigens can be measures if >1 Ab is usedo Monoclonal antibodies are expensive and it would not be practical to check for every
surface antigen instead, panels of antibodies that are appropriate for subtypes of leukaemia are
used to make a diagnosis supplementary panels are used to identify markers that correlate with stages of
cell maturation and hence is of prognostic value rarely, antibopdies are labeleed to an enzyme which releases a colour when it catalyses a
reaction. enzyme-labelled antibodies require the sample to be on a fixed medium
3. Applications of Immunophenotyping in haematological malignancy differentiation AML from ALL and determining the subtype of AML/ ALL distinguish acute leukaemia from a lymphoproliferative disorder/ other tumour revealing the abnormal phenotype which is useful in monitoring minimal residual disease after
remission is achieved identify antigens for therapeutic targets
o you rarely need immunophenotyping to diagnose CML because usually blood film/ count/ cytochemistry will give you the diagnosis accurately
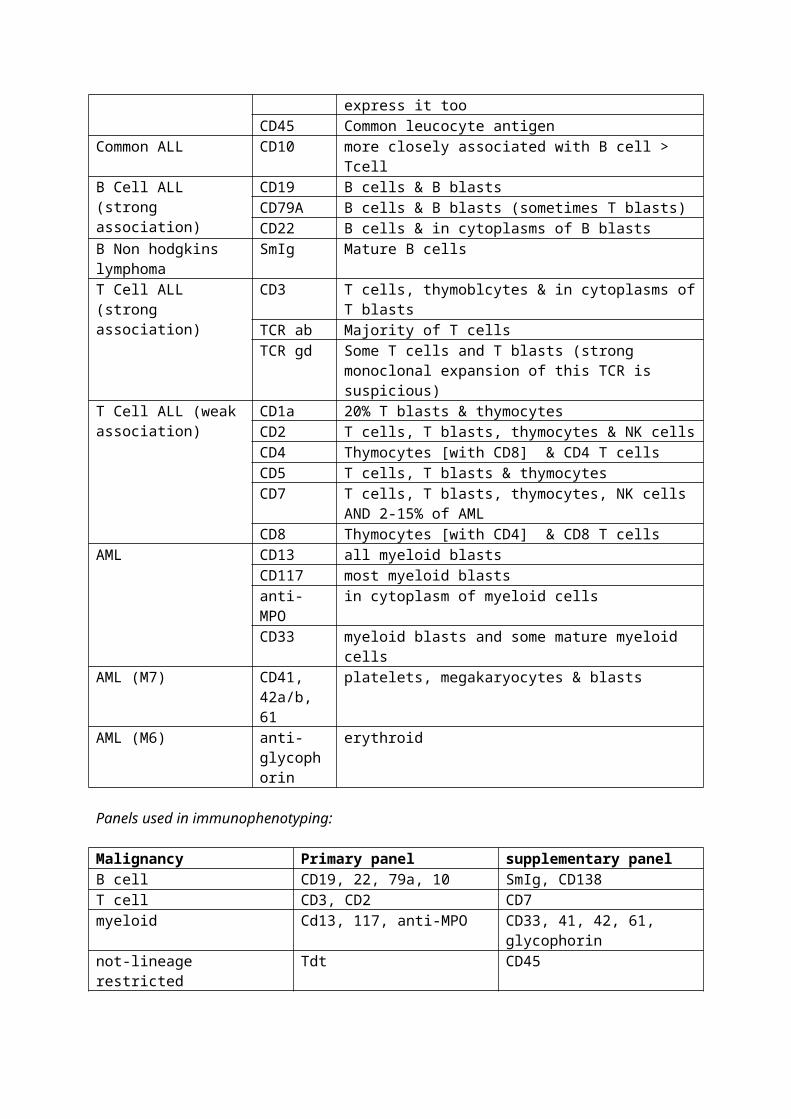
4. Common CD markers
Diagnostic use CD Distribution of CDBoth AML & ALL CD34 HSCs, immature cells
TdT ALL blasts and 10-20% of AML blasts express it tooCD45 Common leucocyte antigen
Common ALL CD10 more closely associated with B cell > TcellB Cell ALL (strong association)
CD19 B cells & B blastsCD79A B cells & B blasts (sometimes T blasts)CD22 B cells & in cytoplasms of B blasts
B Non hodgkins SmIg Mature B cells

lymphomaT Cell ALL (strong association)
CD3 T cells, thymoblcytes & in cytoplasms of T blastsTCR ab Majority of T cellsTCR gd Some T cells and T blasts (strong monoclonal expansion of
this TCR is suspicious)T Cell ALL (weak association)
CD1a 20% T blasts & thymocytesCD2 T cells, T blasts, thymocytes & NK cellsCD4 Thymocytes [with CD8] & CD4 T cellsCD5 T cells, T blasts & thymocytesCD7 T cells, T blasts, thymocytes, NK cells AND 2-15% of AMLCD8 Thymocytes [with CD4] & CD8 T cells
AML CD13 all myeloid blastsCD117 most myeloid blastsanti-MPO in cytoplasm of myeloid cellsCD33 myeloid blasts and some mature myeloid cells
AML (M7) CD41, 42a/b, 61
platelets, megakaryocytes & blasts
AML (M6) anti-glycophorin
erythroid
Panels used in immunophenotyping:
Malignancy Primary panel supplementary panelB cell CD19, 22, 79a, 10 SmIg, CD138T cell CD3, CD2 CD7myeloid Cd13, 117, anti-MPO CD33, 41, 42, 61, glycophorin not-lineage restricted Tdt CD45
Supplementary Panel outcomes in B-cell ALL
Type of B-ALL MarkersPro-B or early B CD34+, TDT+, CD10-, SmIg-Common CD34+, TDT+, CD10+ SMIg-Pre-B CD34+, TDT-, mu Ig (cytoplasmic only)Mature B (non Hodgkin lymphoma) CD34-, TDT-, kappa/lamda Ig (cytoplasmic or surface)
3. Minimal Residual Disease MRD can not be detected morphologically and cytogenetic analysis is also too insensitive Immunophenotyping is useful in monitoring minimal residual disease after remission is
achieved But PCR is the best method of monitoring MRD Another method is multiparameter flow cytometric immunophenotyping – this uses a
number of antibodies simultaneously to try and detect leukaemic characteristics. such characteristics include:
o aberrant antigen expression loss/weak expression of a (tumour suppressor) antigen over expression of a (oncogenic) antigen simultaneous expression of markers of immature cells (CD34, tdt)

2.6 – MANAGEMENT OF CML
1. Recap of the CML disease processWithout Treatment, CML patients undergo the following process:
5-6years in the chronic phase (anaemia, B symptoms, early satiety and abdominal discomfort because of splenomegaly)
~1year in the accelerated phase 3-6months in a blastic crisis – followed by death
2. The genetic pathophysiology of CML is the key to treatment design22q- is the Philadelphia chromosome This is because BCR is upstream of ABL in 22 whereas it is downstream of ABL in 9q+ The breakpoint in 9 is anywhere before the 2nd exon of ABL The breakpoint on 33 is at either exon 13 or 14 of BCR
o Hence the breakpoint region in 9 is much larger than in 22
The role of BCR/ABL Normally, a tyrosine kinase domain on ABL, called SH1, keeps it regulated BCR ‘unlocks’ and hence activates SH1, and thus ABL
o Since BCR is a housekeeping gene, it is always on and unregulated. o Hence the fusion gene leads to constitutive activation of SH1
The active BCR-ABL gene product uses ATP to phosphorylate side chains on tyrosine residueso Cytoskeletal protein phosphorlyation = increased cell migrations and decreased
adherence to BM matrixo Nuclear protein phosphorylation = induction of c-myc = proliferationo Mitochondrial protein phosphorylation = escape from apaoptosis
RT-PCR in disease monitoring Because the breakpoint region in 9 is large – traditional PCR cannot detect the gene sequences
involved at the BCR/ABL border following translocationo Hence, RT-PCR has to be used to replicate the mRNA transcript and analyse cDNA
At diagnosis, you usually have 10e13 leukaemic cells. PCR is only sensitive to the point that where it doesn’t pick up anymore BCR/ABL transcript, you
probably have less than 10e7 leukaemic cellso It is thought that the host immune system can manage this leukaemic burden
3. Imatinib and other monoclonal antibodies can ‘cure’ CML
Imatinib is a tyrosine kinase inhibitor The mechanism of action is that the drug mimics ATP so BCR-ABL uptakes it instead of ATP, and
thus cannot start phosphorylation cascades 1/3 of patients fail to responf to Imatinib. The reasons for this are:
o Lack of complianceo Drug interactions and CYP3A4 polymorphisms giving reduced drug titre in plasmao Plasma binding (AGP) to give reduced drug titreo Resistance
15 mutations account for 90% of cases of resistance to Imatinibo One of these is a point mutation in BCR/ABL [t315i] which means Imatinib can not bind
the gene product Newer agents dasatanib and nilotinib give a response in 40% of resistant patients

o Subsequently, they have been show to be more powerful and have a decreased risk of developing blastic crises compared to imatinib, when given to patients in the chronic phase
With treatment, about 85-90% of CML patients now have a normal life expectancy The main issue is that Imatinib costs about £500million/yr; newer agents are about 1/3 more
expensive
Measuring the response to Imatinib [clinical endpoints from the IRIS study] Haematological response
o WCC <10e9o Plt <450 x10e9o <5% myelo- & metamyelocyteso No blasts, promyelocytes, no extramedullar haemopoeisis
Cytogenetic response in marrow cellso No Philadelphia chromosome in metaphase arrested cells
Molecular responseo PCR to measure ‘log reduction’ of ratio of BCR/ABL:BCR transcripts
2.7 – ACUTE LYMPHOBLASTIC LEUKAEMIA (ALL)
1. Clinical features of ALLD - haematological neoplasm characterized by clonal expansion of lymphoblasts in BM and PB. I - commonest childhood malignancy; 35% of all childhood cancersA - peak incidence 2-5 yearsS - M>FG - White > BlackA - UnknownP - [see classification]C - s.s.e
Bone marrow failure = anaemia, neutropenia (chest infections in kids), thrombocytopeniaOrgan infiltration =
tender bones (limping children) organomegaly and lymphadenopathy meningeal syndrome (rare)
o confusion, hemiplegia testicular swelling in boys, mediastinal compression in T-ALL
P cure rate in children ~90%; worse in adults (~50%)5 year disease free survival is now about 90% in kids who get ALL aged 2-5
2. Differential diagnosis of ALL AML Aplastic anaemia, neuroblastoma, rhabdomyosarcoma Infectious disease
o Pertussis – look for ‘clefted lymphocyte’ on morphology – more indicative of reactive lymphocytosis
o EBV – look for scalloping (cupping) of RBCs by lymphctic cytoplasm
Diagnosis of ALL depends on immunophenotyping Tdt is the most important antigen – present on lymphoblasts and lost on mature cells B/T markers can subsequently diagnose the sub-type
o 75% of adult ALL is B-ALLo 85% of childhood ALL is B-ALL
3. Classification of ALL The old FAB classification was based on morphology
L1 – uniform, small blast cells L2 – large blast cells with prominent nucleoli and more cytoplasm L3 – blasts with perinuclear and cytoplasmic vacuoles
o L3 is now known as Burkitt’s lymphoma (which is associated with HIV)
Blasts – increased nuclear: cytoplasmic ratioAgranular cytoplasmVisible nucleoliLarge cells

The new classification given by WHO is based on cytogenetic & molecular subtypes of ALL ALL
o B- cell Good prognosis
T(12;21) (p12,q22) [TEL/AML1] – this is the commonest genetic abnormality in B-ALL
T(1;19) (q23;p13) [PBX/E2A] Hyperdiploidy – trisomy 4,10,17 have decent prognoses
Bad prognosis T(9;22)(q34;q11) [BCR/ABL] T(11q23) [MLL] – AL diagnosed in babies <1yr old usually have this hypodiploidy
o T-cell Burkitt’s lymphoma Biphenotypic acute leukaemia ALL not otherwise classified
o Aplastic variant of ALLo Down syndrome ALLo Secondary ALL
4. Pathways affected by mutations in B-ALL Block in B-cell maturation
o PAX5 is common mutation; IKZF1 and EBF1 mutations are others Downregulation of tumour suppressor genes = dysregulated cell cycle Abnormal lymphoid signaling Activation of transcription factors Modification of epigenomes and histone proteins
5. Prognostic factors in ALL At diagnosis
o Age – unless <1yr old (MLL) having ALL young is better prognostically then getting it oldo WCC – higher WCC = worse prognosis (T-ALL tends to present with high WCC than B)o Cytogenetics [see above]o Immunophenotype – not of importance in prognosis of B-ALL. T-ALL which is CD1a+ has
a favourable prognosis At Induction of treatment
o Remission/MRD = <5% blastso To avoid relapse, you aim for no blasts/100k cells o MRD+ve = worse prognosis
Long-term effects of ALL Chemotherapy related – infertility Radiotherapy related
o Skin cancer (SCC/BCC)o Meningiomao endocrinopathies
6. Management of ALL depends on chemotherapy Step 1- Induction Phase – 2x28days
o Aim is to induce remissiono Common drugs are vincristine, dexamethasone, daunorubicin, asparaginase

Asparaginase can cause thrombosis/bleeding, pancreatitis, hepatoxicity Step 2 – intensification
o Aim is to offer prohylaxis against CNS malignancyo Methotrexate and radiotherapy
Step 3 – either HSC transplant in bad prognosis groups or consolidation in good prognosis groupso Indication of transplant include MLL, Ph+, MRD+/relapsing diseaseo Stem cells can be obtained from sibling, Anthony Nolan register or unbilical cord
transplant Step 4 –(post consolidation) – maintenance phase; can take years
o Methotrexate and mercaptopurine
Supportive measures in management Hickman line (central venous access) Blood/platelet transfusions and antibiotics to prevent infections Antiemetics Allopurinol to prevent tumour lysis syndrome Ph+ ALL needs Imatinib
New options in management – the BITE trial used anti-CD19 in B-ALL; it stimulates host T cells to kill B-blasts; great!

2.8 – ONCOGENES AND TUMOUR SUPPRESSOR GENES IN ACUTE LEUKAEMIA; M2 & M3 AML
1. It is thought that most cases of leukaemia are caused by the 2 hit hypothesis of genetic mutation
Acute leukaemias are clonal expansions of abnormal cells, commonly as a result of a somatic mutation in a proto-oncogene, resulting in oncogenesis
o Mutations in oncogenes often affect transcription factors Mutations in acute leukaemia often demonstrate the dominant negative effect
o The mutant inhibits the gene-product of the normally functioning allele, as well as making a dysfunctional gene product itself
One mutation is rarely enough – you need one affecting survival and one affecting differentiation ALL follows the 2 hit hypothesis
o You get one mutation in utero (eg t12;21)o Another mutation after birth triggers ALL
2. The nature of mutations in oncogenesis (5) Point mutations Interstitial deletions, bringing proto-oncogenes into contact with the wrong gene promoters Internal tandem duplication (MLL, FLT3) Amplification of an oncogene (rare) Translocations, which, again, bring a proto-oncogene into contact with a wrong promoter Insertional oncogenesis occurs after gene therapy, where the gene is inserted into the wrong
place
Inherited genetic abnormalities in oncogenesis can be the first hit Haploinsufficiency (loss of 1 allele) of a gene that is critical for normal differentiation
o Eg loss of 1 copy of RUNX1 in familial thrombocytopenia can predispose to AML Loss of function of tumour suppressor genes
o Neuroblastomas (NF1), retinoblastomas (RB1) Chromosomal fragility syndromes increase likelihood of somatic mutations occurring
o Fanconi anaemia Abnormalities of genes that repair DNA mean that random errors are not corrected Abnormalities of genes that degrade mutagens can lead to environmental mutagenesis
3. Abnormalities in tumour suppressor genes can cause a variety of leukaemias Whole/partial chromosome deletion as well as small mutations can affect normal tumour
suppressor function Tumour suppressor loss can contribute to:
o Familial leukaemiaso Therapy-related leukaemiao AML in elderlyo Transformation of chronic leukaemia to an acute/blastic phase
4. The genetic basis of M3 AML; APMLNormal action of RARa and RA
RARa uses Zinc to bind to the retinoid X receptor [RXR]; this dimer can bind to retinoic acid response elements [RARE] on target genes
This complex recruits NCoR/HDAC and Sin3; this is a repressor complex. It also interacts with another repressor; SMRT
o Hence normally, RARa supresses transcription – tumour suppressor gene In the presence of physiological levels of retinoic acid:

o the repressor complex cannot assembleo RARa binds to transcriptional coactivators eg CBPo Histone acetylase is recruited; forming an activator complex
Hence, normally, retinoic acid activates transcription – proto-oncogene
Normal functions of PML [tumour suppressor]PML has 2 isoforms: Normal nuclear PML makes the outer shell of nuclear bodies (NBs)
o NBs contain other proteins eg pRb, p53 and BLM hence normally functioning NBs play key roles in apoptosis, senescence, growth suppression and genome stability
Cytoplasmic isoform enhances TGFb – this also achieves tumour suppressor activity
Overall actions of PML-RARa fusion geneSince all patients express PML-RARa but only 70% express the reciprocal (?relevance), the former is likely to be the causative fusion gene
Resistance to retinoic acid = decreased transcription = block in differentiation at the promyelocyte stage
Continued proliferation Failure of apoptosis
Mechanism of PML-RARa; suppression of transcription leads to block of differentation Oncogenic - Dominant negative inhibition of the normal PML allele [both isoforms] and the
normal RARa alleleo Abnormal PML = microparticualte distribution of proteins usually encased within Nbso Cytoplasmic isoform is sequestered = decreased TGFb
Suppressive - PML-RARa dimerizes with RXR and binds to RARE as normal, BUT oligomerization leads to binding of nuclear PML
o Nuclear PML and RXR are subsequently sequesteredo The resulting complex binds the repressor complex more stronglyo The resulting complex has resistance to retinoic acid
Suppressive – PML-RARa can bind to RARE without the need for RXR – this has a stronger repressive activity as there are more binding sites for repressor complex proteins
Suppressive – PML-RARa recruits DNA methyltransferase; methylation of DNA leads to decreased transcription
5. The genetic basis of variant APMLNot all acute leukaemias involving the RARa gene also involve PML. These variants may or may not respond to ATRA/Arsenic trioxide – and knowing the molecular basis of disease helps us to make new therapies. Some examples are:
T(11;17)(q23;q21) PLZF/RARao ATRA can not disrupt binding of some repressor molecules to PLZF; adding HDACi
may help T(11;17)(q13;q21) NUMA1/RARa T(5;17)(q35;q21) NPM1/RARa T(5;17)(q25;q21) NPM1/RARa
o All of these variants have oligomerizaton domains, meaning they can bind to repressor complexes more strongly. They also all display the dominant negative effect.

6. The molecular basis of the management of classic APML (and some variants)Actions of ATRA
Induces terminal differentiation Facilitates apoptosis of neutrophils Leads to loss of leukaemic clone and replacement by normal cells
Mechanisms of ATRA Degrades PML-RARa
o Caspase mediated cleavage The degradation releases RXR to bind with normal RARa The degradation also redistributes nuclear PML back into nuclear bodies The net result is return to normal transcription
ATRA doesn’t cure, you will get relapse. Arsenic trioxide and HDAC inhibitors (which interferes with the repressor complex), in synergy with ATRA, may cure APML without the need for chemotherapy.
Actions of Arsenic trioxide: Degrades PML-RARa without degrading RARa – this induces partial differentiation which may be
the keyo It Activates caspases and induces apoptosis
Nuclear bodies are reformed Unlike ATRA, it also effects leukaemia-initiating cells. These cells are quiescent but have the
potential to cause relapse; this is why arsenic trioxide has lower relapse rates in APML.
7. The genetic basis of M2 AMLNormal RUNX1 function Usually encodes a transcription factor CBFa in myeloid cells
o CBFa usually activates p14ARF CBFa binds to CBFb via its ‘runt’ domain; this complex allows another protion of the runt domain
to bind DNAo In the complex, CBFb protects CBFa from ubiquitization
After binding to DNA, CBFb recruits histone-acetyl-transferase – this leads to transcription of many genes including G-CSF, GM-CSF, M-CSF, CD13, IL3, IL5 and MPO
o CBFb also leads to repressed transcription of other genes by interacting with TLE
Normal RUNX1T1 is a tumour repressor that is usually expressed in the brain
Actions of the RUNX1/RUNX1T1 fusion gene Dominant negative inhibition of normal RUNX1
o Hence p14ARF is suppressed The fusion product binds CBFb with greater affinity so DNA binding is enhanced Similar to PML-RARa, there is oligomerization of domains in the RUNX1t1 protion of the
gene producto Oligomerization leads to binding of the repressor complex (NCoR, HDAC, Sin3)o Repression of DNA repair genes increases likelihood of further mutation
The fusion gene also inhibits PLZF; which leads to excessive proliferation of myeloid cells Its inhibits the TGFb pathway and this also leads to proliferation

T(16;21)(q24;q22) involves a different translocation (CBFA2T3 instead of RUNX1T1) but mechanism of action is similar because oligomerization domains are similar
8. Other mutations in RUNX1 (apart from translocations) can also give AMLFamilial thrombocytopenia predisposes to AML
A mutation in the runt domain leads to failure of recruitment of histone-acetyl-transferase This leads to the inability to activate transcription(haplo-insufficiency of the mutated allele)
– which is necessary but not sufficient (other mutations needed) for the development of AML
Sometimes, this mutation also displays a dominant negative effect A similar mutation is seen in acquired cases of M0 AML and MDS
9. The genetic basis of M4 AMLAction of CBFb/MYH11 fusion gene
MYH11 forms multimers and this leads to sequestration of RUNX1/CBFa from the CBFa/CBFb complex
MYH11 recruits co-repressorso Hence, all leukaemias involving CBF genes tend to involve oligomerization of the
fusion product leading to sequestration of CBFa/RUNx1
10. Genetic basis of AML without evident translocations This still tends to follow a two hit hypothesis First hit:
o NPM1 mutations are common in adultso CEBPA mutations are implicated in familial leukaemias
Second hit:o FLT3 (ITD) mutationo Others
Key summary points for M2/M3 leukaemias: Both involve excessive transcriptional repression due to oligomerization Both involve fusion genes with dominant negative effects To reverse the leukaemic phenotype, you need to:
o Inhibit HDACo Recruit HATo Recruit transcriptional activators
2.9 - MYELODYSPLASTIC SYNDROMES (MDS)
1. Clinical features of MDS

D - A group of clonal haematopoetic stem cell diseases characterised by abnormalities in one or more myelod cell lines and manifested by:
Cytopenia - particularly manifesting as anaemia The cycle between hyperactive, ineffective haemopoeiss which aims to compensate
for abnormal cells, but fails to because all the BM can make is more abnormal cells Increased risk of transforming into AML
I - 3-5/100kA - elderly; median 70yearsS - M>FGA - UNKNOWN - environemtnal exposure to benzene, cigarette smoking may be relevant
Secondary to Prior chemo/radiotherapy accounts for 15% of caseso Cyclophosphamide (used in breast and prostate ca)o Cisplatin (used in breast, testicular and ovarian ca)
In the differential diagnosis, you must rule out other causes of acquired dysplasis eg infectious (EBV), drugs (azathioprine, cyclosporine) and B12 deficiency
P - [see classification]C - s/s/e
Cytopenia - anaemia [commonest finding], neutropenia, thrombocytopenia
ManagementC - supportive treatment (transfusions, antibiotics). Especially for people who are old and
have comorbitiesM - EPO may help with anaemia; lenalidomide improves cytopenia [watch major SFX is VTE]. Danozol can help in those with severe thrombocytopenia and pyrimidine analogues (azacytidine, decitabine) can be used in people with evident chromosomal abnormalitesS - BMT for young patients only
P - The international prognostic scoring system is adopted [see below]. Age and underlying disease also affects prognosis
Score 0 0.5 1 1.5 2% BM blasts <5% 5-10% 11-19% >20% (AML)Karyotype good intermediate PoorCytopenia 0.1 2-3
Characteristics of cytopenia: Neutrophils <1.8e9;plt <100e9; Hb<10g/dl
Low risk = 0 = good prognosiso Morphology - RARSo Cytogenetics - normal, del 5q, del20q, del Y
Intermediate risk = 0.5-2 o Morphology = RAEB1, RCMDo Cytogenetics - all that do not fall into good/bad risk
High risk = >2 = poor prognosiso Morphology - RAEB2o Cytogenetics - monsomy 7/ del7q, complex karyotypes
2. WHO classification of MDS
Disease PB BM

Refractory unilineage cytopenia (anaemia/neutropenia or thrombocytopenia)
No blasts unilineage dysplasia<5% blasts<15% Ring sideroblasts (RS)
Refractory anaemia with RS (RARS)
No blasts <5% blasts>15% RS
Refractory cytopenia with multilineage dysplasia (RCMD)
No blasts Dysplasia >10% in two or more lineages<5% blasts
Refractory anaemia with excess of blasts-1 (RAEB1)
<5% blasts, no auer rods Uni/mulit-lineage dysplasia5-9% blasts, no auer rods
Refractory anaemia with excess of blasts-2 (RAEB2)
5-19% blasts with auer rods Uni/multi-lineage dysplasia10-19% blasts with auer rods
MDS associated with del (5q) [5q syndrome]
<5% blasts, macrocytosis Hypolobulated megakaryocytes<5% blasts, no auer rods
Unclassified MDS No blasts Dysplasia; 5% blasts
3. Morphological appearances of dysplastic lineages Granulocyte lineage:
o Hypolobualted nuclei (pseudo-pelger/dumbbell shaped nuclei)o Hypo/agranular blue cytoplasmso >1 nucleus per cello Auer rods may or may not be present in MDS
Erythroid lineage:o Cytoplasm
Poor Hb, vacuole appearances Heavy perinuuclear siderotic granules (ring sideroblasts) Cytoplasmic bridge formation PAS positivity
o Nuclear Incomplete division, budding, fragmentation >1 nucleus per cell with Internuclear bridges
o Megakaryoblast lineage: Smaller cells with hypolobulated nuclei [normal megakaryocyte nucleus looks
like a bunch of grapes] >1 nucleys per cell
4. Cytogenetic changes in MDS Only 50% of MDS have a cytogenetic abnormality evident. Cytogenetic changes are more common in t-MDS The majority of t-MDS are monosomies of 5/7 or deletions of 5q/7q
o Two other translocations found in t-MDS are balanced - t (11;16) and t(3;21) Some other cytogenetic changes in de novo MDS are:
o Unbalanced translocations - trisomy 8, monsomy 5/7, del 20q, del y, abnormal 17 Del 5q (5q syndrome) is more common in women and presents with a good prognosis; it has
a low AML transformation rate can some patients respond to lanalidomide
5. Clinical features of mixed MDS/MPDD - Clonal haematopoetic neoplasms that at the time of presentation, have findings that support both MDS and MPD including:

Hypercellular BM with <20% blasts Proliferation in one cell lineage and dysplasia (without proliferation) in another Hepatosplenomegaly is common
I/A - 3/100k >age 60; develops in 5% of people with MDS
6. Sub- Classification of MDS/MPD Chronic myelomonocytic leukaemia (CMML)
o This is a persistent monocytosis in the absence of other causes including infection, BCR/ABL, PDGFRA rearrangement
o Presents with B symptoms, splenomegaly and infiltrates into other organs/skino Median survival is <3years and 15-30% progress to AMLo CMML1 has <5% PB blasts and <10% BM blastso CMML2 has 5-19% PB blasts and 10-19% BM blastso Cytogenetic abnormalities only seen in 20-40% of cases; about 40% have a RAS
mutation; n-ras mutations are relatively frequent in MDS/MPD Atypical CML (BCR-ABL negative)
o Presents very similarly to CML but fusion gene is absent on PCR and/or Px is not responding to imatinib
o Multilineage dysplasia is what differentiates it from CMLo Cytogenetic abnormalities are seen in 80% of caseso Upto 40% progress to AML
Juvenile myelomonocytic leukaemia (JMML)o Diagnosis includes high HbF, hypersensitive to Gm-CSF in vitroo Usually present in boys younger than 14 with thrombocytopenia and neutropenia,
hepatosplenomegaly is almost always thereo Associated with neurofibromatosis and monosomy 7 is seen in 40% of cases
Unclassifiable MDS/MPD ?RARS with thrombocytosis
2.10 - CASE PRESENTATION/TUTORIAL - MDS/AML/BMT TRANSPLANTS
MDS/AML

Key points in the history of someone presenting with ‘B symptoms’o Things that could mean it is not a haematological disease - recent drugs, quick onset,
alcohol/IVDUo Thigns that could mean it is a haematological disease - Hx of cancer and chemo/radio,
HIV (causes MDS) Key points in investigations:
o Hb on ABG is not reliableo 1 set of abnormal results always need to be redoneo Blasts & dysplastic cells in the BM occur in MDS and AML
Girls given chemo are probably going to be infertile, boys should be sperm-banked 50% AML patients relapse after induction chemo
Autologous BMT transplants The principle with autologoug transplants is to wipe out the clone (or monoclonal antibody in AI
disease) with high dose chemotyherapy and then re-itnroduce the normal host stem cells Uses in malignancy:
o Haem - Lymphoma, myeloma, some ALLo Solid - Testicular, placental, ovariano Other - neuroblastoma
Uses in AI disease - MS/ RA/ scleroderma
Allogenic BMT transplants Sibling/ matched allogenic - of use in all tumours? Unrelated/ unmatched allogenic
o Can be used in AML/ ALL
Problems with BMT transplantation Infection
o High dose chemo knocks out fast dividing cells - including gut cells Gut is full of G-ve bacteria that can then enter bloodstream to cause sepsis -
BADo Hickman lines are often needed to deliver chemo - source of infectiono Immunosuppresion leads to susceptibility to all sorts of oppurtunisitics including:
CMV - retinitis, pneumonitis, gastroenteritis Aspergillus in the lungs
Hepatic/ renal failureo Both of these are sensitive to damage by chemotherapy.o The kidney is sensitive to antiviral drugs that are used to treat CMV
GVHDo Usually occurs 2 weeks after chemotherapy; lymphocytes are the 1st cell lineage to
recovero ‘foreign’ lymphocytes proceed to attack, in particular, the skin, gut and liver. But they
also attack the leukaemic cloneo Steroids need to be given to supress the response - the problem here is reintroduction
of infection and relapse of leukaemia Transplant mortality is roughly 15%. You need to nurse patients through the 2 week barrier and
hope for a sub-clinical level of GVHD.
2.11 - CHRONIC LYMPHOCYTIC LEUKAEMIA (CLL)
1. Clinical Features of CLL

D - proliferation of mature (B) lymphocytesI - accounts for ~20% of all haematological malignancies; incidence 3/100kA - increase incidence with age; median age between 65-70yearsS - male>female 2:1G - Commonest leukaemia in western world (20x more than eastern)A - Unknown; deletions of microRNA may have a roleP - [See below]C - s/s/e
disease course is very variable. It tends to be a slow, indolent progression of mature B cells; the time from diagnosis by which symptoms of BM overcrowding is seen is variable
70% diagnosed incidentallyo Family hx (you can find monoclonal B cells in relatives of CLL patients)o comorbididiteso Infections (bacterial/ fungal)o anaemiao Peripheral lymphadenopathy - syymetrical enlargement of superficial odeso Hepatosplenomegaly
Progressive phase/other symptoms relating to management AI diseases in CLL
o Haem - AIHA, AI thrombocytopenia, neutropenia, pure red cell aplasia AIHA is stimulated by treatment (chlorambucil)
o Non-haem - nephrotic syndrome, angio-oedema, paraneoplastic pemphigus Progression
o Richter syndrome - transformation toa high grade lymphoma that effects 3% of patients, associated with fludarabine usage
Investigations Blood count/film
o Count - Lymphocytosis between 5-300e9, Normochromic anaemia, thrombocytopenia
o Film - Smear cells (artefact cause by spreading blood on film) Immunophenotyping
o The main thing with mature CLL B cells is that they express CD5, which is usually exclusively espressed on T cells - normal mature B cells do not express it
The CLL score gives 1 point to CD5+. CD23+, FMC7- and weak expression of Cd22 and SmIg. A score of 5/5 = CLL likely
Other tests include antigolubulin test (coombs test), reticulocyte count, serum Ig, bone marrow aspirate/ LN biopsy
o Serum Ig - redyuced concentrations/ immuneparesiso BM aspirate - lymphocytic replacement of normal cells
Differential diagnosis Mantle cell lymphoma - non hodkins lymphoma associated with t(11;14) (q13;q32)
where cyclin D1 is overexpressed - very aggressive cancer.o Cannot differentiate on immunophenotyping because they also co-express CD5
2. Prognostic factors in CLLClinical Stage

Binet Staging score:Stage Features Prevalence (%)A <3 lymphoproliferative areas 60B >3 lymphoproliferative areas 30C B + Hb<10g/dl + Plt < 100e9 10
Rai staging score:Stage Prognosis Features Prevalence (%)0 Good Lymphocytosis 301 lymphadenopathy 252 Intermediate Hepatosplenomegaly +/- [stage1] 253 bad 2+ Hb <11g/dl 104 2+ Plt <100e9 10
CD38/ZAP70Lower ZAP70 expression is a good prognosis
Ig gene mutation statusCLL with Mutated Ig heavy chain genes has a better survival than non-mutated. The principle behind this is that Ig somatic hyper-mutation is a normal process by which Ig’s transform to bind antigens better. Lack of mutation = worsenes B cell function
Cytogenetics - Karyotpyping is impossible in CLL because cells are too slow diving - you need FISH Good prognosis - normal karyotype, del(13q), trisomy 12 Worse prognosis - del 11q, del 17p
o Del 17p confers p53 loss on one allele and mutation of the other = genomic instability, accumulation of mutations and clonal expansion/ resistance to chemo (p53 usually coordinates responses to DNA damage)
Anti CD52 is first line treatment for del (17p) CLL - watch out for reduced t cell response, increased risk of fever and CMV infections
o Del 11q gives an ATM mutation which also leads to p53 dysfunctiono P53 deletions have a very bad prognosis
3. Management of CLL Watch and wait for all patients until they show an indication for intervention:
o Progressive BM failureo Massive/progressive lymphadenopathyo Progressive lymphocytosis defined as 50% increase of 2months and/or a doubling
time less than 6 monthso >10% weight loss in 6months, fever >38 for >2weeks, fatigue, night sweatso Autoimmune cytopenias
If an intervention is indicated - the following treatment strategies are adopted:o First line treatments - steroids, alkylating agent, purine analogues, anti CD20o Second line - purine analogues, anthracyclines, allogeneic BMTo Treatment of patients who are refractory - high dose steroids, anti-CD52
Young patients may be cured by allogeneic SCT Supportive management for people who would not benefit from intensive chemo
2.12 Prophylaxis/treatment of infections (infections account for 50% CLL deaths)2.12- MANAGEMENT OF AML

New points Early infections in AMl are largely from commensal bacteria - watch for G-ve sepsis AML has decreased associated with lymphadenopathy but increased association with leukostasis
as compared to lymphoid leukaemias. Choice of treatment is influenced by
o Type of amlo Ageo Curative versus palliative treatment
Pricniples of treatmento Induction of remission
1 cycle of Combination chemo with cytarabine and daunorubicin 60-80% achieve remission after induction therapy
o Consolidation 3 cycles of the same Chemo Autologous/allogeneic SCT
o At all times - supportive care, psychosocial support Well established prognostic factors in AML:
o Patient performance (can they handle chemo? Did they achieve induction-remission?)o Ageo Primary/secondary AMLo Cytogenetic features
Recap of old points: Clonal proliferation of precursor cells AND maturation arrest Biphenotypic leukaemias (HSC mutations) are treates as AML Commonest leukaemia in adults, median age 63 Main differentials are MPN/MDS and aplastic anaemia De novo AML occurs due to risk factors eg
o Genetic disorders (downs, NFM, kilnefelters, fanconi anaemia)o Mutagen exposure (benzene, cigarettes)
Secondary AML occurs because ofo Progression of previous MDS/MPNo Previous chemo/radiation therapy
Symtoms occur due to marrow failure (anaemia, neutropenia, thrombocytopenia) and tissue infiltration (hepatospleno- & lymphadenopathy, gum infiltration[m4/5], granulocytic sarcoma in skin, bone pain)
Can usualy diagnose with cirscropy (>20% in BM, auer rods), cytochemsitry, cytogenetics and immunophenotypic is needed for M0/M7 subtypes
2.13- CONSTITUTIONAL SYNDROMES PREDISPOSING TO ACUTE LEUKAEMIA

1. Pathways in leukaemogenesis Common oncogenesis involves changes in gene expression implicated in networks/ circuits
includingo Cellular motility circuitso Proliferationo Differentiationo Viability
In leukamogenesis there are 3 main groups of chnges in gene expressiono Upregulated oncogenes = excessive proliferationo Down regulated tumour suppressor genes = evasion of apoptosiso Aberrant DNA repair = defective differentiation; oncogene stimulation and tumour
repressor gene inhibition
2. Genetic defects predisposing to leukaemia Tumour suppressor genes
o Li fraumeni syndrome (p53)o Neurofibromatosis (NF1)
DNA repair geneso Fanconi anaemiao Ataxia-telangiectasia syndrome (ATM)o Also bloom syndrome and Nijmegen breakage syndrome
Ribosomopathieso Blackfan-diamond anaemia (rps19/24)o Schwachmann-diamond syndrome (Sdo1)o Dyskeratosis congentia (cbf5)
In utero mutationso TEL-RUNX1
Aneuploidyo Down syndrome
3. Fanconi Anaemia is a bone marrow failure syndrome associated with defective recombinational DNA repair Normally there are three main pathways in response to DNA damage:
o Repair Single strand breaks fixed by base excision repair Adducts fixed by nucleotide excision repair Double strand breaks fixed by recombinational repair or end-joining Mistahces/inertions/deletions fixed by mismatch repair
o Transcriptional respsonseo Apoptosis
Fanconi anaemia is an AR disease associated with congenital malformations and progressive BM failure with propensity to suffer malignancy, leukaemia and MDS
o FANCA is the commonest of 13 associated mutationso Underlying mechanism is defective homologous recombination DNA repair and
hence increased sensitivity to DNA damage by cross linking agentso Most patients present with sympttoms of pancytopenia at 7years of ageo 50% develop MDs/AML by age 40
4. Ribosomopathies

Blakcfan diamond anaemia is associated with increased activity of RBC adenosine deaminase (ADA) giving red cell aplasia in early infancy (sometimes aplastic anaemia), growth failure with an increased risk of developing AML and osteosarcomas
o Pathogenesis is defective ribosome 40S synthesis which is toxic to RBCso The undlerying mutation commonly involves ribosomal proteins (rps) 19 & 24 and
there seems to be a block in differentiation of red cellso ?treat with steroidso Some patients spontaneously enter remission
Schwachman diamond syndrome is an AD disease with SBDS mutations presenting with:o BM failure (neutropenia is common)o Exocrine pancreatic insufficiency (improves with age)o Increased risk od developing aplastic anaemia, MDs, AMLo Proposed mehcnaism is decreased levels of mature 80S ribosomes [defective
binding of 40S/60S subunits]
5. Dyskeratosis congenital is a mixed ribosomopathy/ Telomeropathy Normally, telomeres have repeated TTAGGG sequences which stabilize the chromosome by
preventing loss of genetic information and end-to-end fusions. Repeats are lost with each cell divison
The telomerase complex replaces thje repeats. It is comprised of Dyskerin, TERC, shelterin, TERT, NOP10, NHP2, GAR1 and probably more proteins
o The telomerase complex is also involved in pseudouridylation of rRNA - which is necessary for normal ribosome synthesis
Dyskeratosis congentia commonly presents with a mucocutaneous triad (skin pigmentation, nail dystrophy, mucosal leukoplakia) and bone marrow failure
o DC is due to mutations in any of the proteins in the telomerase complex - commonly Dyskerin which is inherited in an X-linked fashion. This leads to a mixed ribosome and telomerase-related disease
o DC demonstrates the idea of anticipation in genetics - this is the idea that a disease has a worse phenotype and earlier onset in successive geenrations. DC correlates because telomere length seems to shorten with generations.
6. In utero mutations T12;21 in childhood B-ALL has a TEL-RUNX1 fusion gene that forms in utero in 1% of
suffererso There is hyperdiploidy and production of a pre-leukaemic clone, which is selected
for following ?an infection after birth to produce ALL
2.14- STEM CELL TRANSPLANTATION

1. Introduction to transplantsAutologous transplants
Give GCSF 5-16microg/kg every day for 4days; 3 hours in total 4 days later, Collect HSCs (CD34+ve) from PB & freeze Thaw HSCs and reinfuse back into host after high dose chemo
Allogeneic transplants Give cancer sufferer high dose chemo +/- radio Transplant BM cells from a different donor Donor stem cells can be obtained from BM/ PB or umbilical cord
o BM - procedure is done under GA and involves taking a lot of BM samples (can only take 5ml at a time and you need about a litre)
o PB - like autologous, needs GCSF prior to HSC extractiono Umbilical cord - can only get a little sample - not useful for adults because you need
roughly 2mill CD34+ cells/kg body weight Donor is ideally matched for HLA-type aka sibling/identical twin
o Increasing the specificity requirement of matching decreases the chance that a donor will be available
Mortality rate is upto 50% and it costs roughly £150k per transplant done
2. Indications for transplantAutologous transplants
Indicated for any patient where there is no allogeneic match and if the patient is too old to survive (aggressive) allogeneic therapy
Useful in the management of diseases that are not based in the bone marrowo Mianly for acute leukaemias, lymphomas and solid tumourso Can also be used for myeloma and CLL
Allogeneic transplants All leukaemia, myeloma, other causes of Bm failure and congenital immune deficiencies Can also be used for lymphoma
3. Outcomes/ complications or transplantsStandard parameters
Overall survival Disease-free survival Transplant related mortality Relapse incidence
Factors that affect transplant outcome in allogeneic transplants
Factor 0 1 2Age <20 20-40 >40Disease phase Early Intermediate lateGenders of receipient/donor Female male*Time from diagnosis to BMT <1yr >1yrDonor matching sibling unmatched

*female to male is a worse outcome because of ?lack of Y chromosome; outcome is even worse if she is pregnant with a male child and has formed antibodies against him?
Higher score = worse prognosis
Complications of BMT Graft failure
o Prevented by suppressing the host immune response with chemotherapyo Host-versus graft disease may manifest as an engraftment syndrome
WCC, neutrophils and platelets all go up Infections
o Post-transplant (chemo) pancytopenia can last for upto 1 montho Contributing factors to infection are neutropenia, loss of barrier function, suppressed
cellular and antibody-mediate immunityo In the first few weeks of allogeneic BMT you are aplastic - bacterial infections because of
loss of innate immunity Weeks 3 - 6 months you are at risk for GVHD - viral and fungal infections
because of impaired adaptive immunity Beyond 6 months is the late phase - latent infections?
o Aspergillosis occurs in 6% of BMTs and has a mortality rate of 92%o CMV is also a common problem
This is primarily because at any given time, 20% of a person’s T-cell reserve is dedicated to controlling CMV - when you are T-cell depleted, shit hits the fan
Risk factors for CMV = negative patients/donors serological stuaus, type of donor, type of transplant and total viral load
Graft versus host diseaseo The cut off between acute and chronic forms is 100days. Chornic GvHD presents like
sclerodermao Donors T cells and receipient t cells attack each other
You can prevent this by depleting the donor T cells using monoclonal antibodies - but this leads to relapse
This suggests that donor T cells are playing a key role in attacking the host leukaemic cells
Hence in relapse, you can reinfuse donor t cells to control ito Donor t cells attack host skin, liver, bowelo Treated with steroid and cyclosporine amongst other drugso Prophylaxis regimens include steroids, cyclosporine and methotrexate
Relapse of original disease

2.15- DESIGNED DRUGS
1. The ideal drug target in leukaemia is… Crucial to the malignant phenotype and definably correlated with clinical outcome Not expressed significantly in vital organs or tissues Reproducibly measured in readily available clinical samples When interrupted, interfered with, or inhibited a clinical response is yielded in a significant
proportion of patients For antibodies: once bound to the surface the antibody is internalised or endocytosed Minimal response in patients whose tumours do not express the target
2. There are 4 categories of drug targets in leukaemia
1. CSM markers/ receptors and secreted proteinsa. Normally a reaction usually leads to dimerization & signal transuctionb. Eg FLT3 (intercellular communication), CD markers, VEGF
2. Intracellular kinasesa. Normally phosphorylate amino acid residues and tyrosine kinases (BCR/ABL) or
serine/threonine kinases (aurora)b. Can be receptor bound (FLT3) or intracellular (BCR/ABL)
3. Gene fusion products a. Confer dysregulated gene function which is usually unique to the tumour cellb. Ege BCR/ABL; PML/RARa
4. Other targets eg proteasomea. The 26S proteasome is involved in intracellular protein degradation via ubiquitinationb. It has 3 subunits (1x20S, 2x19S)
3. There are 3 categories of targeted therapy in leukaemia
1. Antibody therapies - specificity and high affinitya. Naked - confer cell mediated cytotoxicity (via attraction of macrophages and NK cells)
and complement activationi. Eg Rituximab (anti-CD20 [IgG]) in B-cell leukaemia and NHL (also used in RA and
management of graft rejection post-transplantii. Eg alemtuzumab (anti-CD-52) in CLL and t-cell lymphoma in patients who failed
treatment with alkylating agentsb. Conjugated - focussed delivery radiation/ cellular toxin
i. Eg gemtuzumab ozogamicin (anti-Cd33 tagged with a cytotoxin) used for elderly relapsed AML. The toxin is released following intraceullar lysosomal cleavage - loss of specificity makes is undesirable as it has a wide range of side effects eg hepatotoxicity
ii. Eg yttrium90-anti-CD66 - beta emitter used in mice studies for multiple myeloma
2. Anti-secreted protein antibodiesa. Eg bevacizumab (anti-VEGF) inhibits angiogenesis and hence tumour growth in
colorectal, breast and lung ca
3. Small molecule inhibitorsa. TK inhibitors

i. Imatinib - blocks proliferation, induces apoptosis and low toxicity. Mimics ATP binding site for BCR-ABL in CML (t9;22)
1. T315I (cytosine to thymine at 944) makes isoleucine instead of threonine and is the most clinically relevant of 50+ point mutations in BCR-ABL that confer imatinib resistance
2. Others - 2nd gen = nilotinib, dasatinib, bosutinib are useful in all resistant patients except for T315I
3. 3rd gen agents eg ponatinib show some effect in T315I resistant patientsii. [under development] tandutinib, lestaurtinib, midostaurin target FLT3 ITD in
AML. The ITD mutations makes the FLT3 ligand independent and is associated with a more aggressive disease
b. Differentation agentsi. ATRA - targets retinoic acid receptor in APML (t15;17; PML-RARa)
c. Proteasome inhibitorsi. Bortezomib inhibits 26S proteasome and thus blocks degradation of IkB = less
NfkB activation = antiprolioferative and pro-apoptotic in multiple myeloma and ?MCL
1. It appears myeloma cells more sensitive to the drug, but it can effect other cells to so bortezomib does have side effects
Bortezomib inhibits proteasome 26S
Proteasome usually degrades IkB
IkB usually inhibits Nfkb
Nfkb usually promotes proliferation & anti-apoptotic pathways
hence the net result of the drug is anti-proliferative & pro-apoptotic

2.16 - ACUTE PROMYELOCYTIC LEUKAEMIA (APML)
1. Clinical features of Acute Promyelocytic Leukaemia Patients usually present with haemorrhage resulting from DIC
o Bleeding from venupuncture sitreo Mucosal bleedingo Low platelet count (platelets used up)
The mean age of onset is about 40years old, DIC occurs within the first few weeks and can be fatal
o Watch out for ganagrene, pulmonary and cerebral haemorrhageso The pathophysiology is due to increased TF rexpression/release by the
promyelocytes and increased fibrinolysis (because of annexin 2) Patient may present with a low white cell count, and a higher white cell count is a bad
prognostic marker
2. There is no role for immunophenotyping in the diagnosis of APML Morphology - Blood film/ bone marrow aspirate
o M3 FAB AML - granular blasts where you can’t really see the nucleuso Faggott cells may appear - they have an abundance of auer rodso M3 variant has less granular/darkly staining cytoplasms - the nucleus may also appear
clefted Cytogenetics - FISH
o Karyotyping takes about a week - FISH is better because DIC might kill them before thiso Looking for characteristic t(15;17)(q22;q21) - PML-RARa
Anti-PML antibody?
3. The roles of PML-RARa and outlines of treatment for APML Usually retinoic acid binds to RARa to unwind chromatin and activate transcription PML-RARa binds strongly to the repressor-complex and is resistant to physiological levels of RA.
o Less transcription = Block in differentiation at promyelocyte stageo Continued prolioferation and failure of apoptosis
ATRA is essentially a super-charged RA doseo It induces differentiation into neutrophils
Blast PM M metaM band cell neutrophilo It facilitates apoptosis o The leukaemic clone is replaces by normal cells
ATRA is usually used in combination with one cycle of anthracycline-based-chemotherapy. This reduces DIC and thus haemorrhage-related-early-mortality
Arsenic is emerging as another treatmento it releases caspase III from mitochondria to facilitate apoptosiso it degrades PMl-RARa without degrading normal RARao it may be of use in ATRA-resistant patients
4. the ATRA (/differentiation) syndrome is a side effect of ATRA treatment clinical features - SOB, oedema, weight gain, pulmonary infilitrates and hypoBP
o (can also occur with arsenic treatment) Believed to be related to rising numbers of differentiating myeloid cells Treat by temporarily stopping ATRA and administering dexamethasone

2.17- PATHOBIOLOGY OF MULTIPLE MYELOMA
1. Origin and phenotype of the myeloma cell and the cellular compartment where oncogenesis develops
Functional Ig rearrangement (VDJ), somatic hypermutation and Ig heavy-chain class (from IgM to IgG/A) switching following antigen exposure converts B cells into plasma cells, which reside in the marrow
o All these three processes involves double strand breaks = propensity to mutateo Somatic hypermutation and class switching occurs in germinal centres of LNs
Plasma cells are dependent on interactions with Bm stroma for survival and proliferation hence it is not normally found in the peripheral blood
o They have a low labelling index and make Ig inefficientlyo Plasma cell leukaemia is a progression where plasma cells become independent of
stroma and hence begin to appear in peripheral blood 2 compartments of dividing cells in myeloma
o End stage cell (plasma cell)o Feeder cell (plasmablast)
Immunophenotype of myeloma cellso Plasma cells are marked by CD38 & CD138 (syndican1)
Myleomatous plasma cells lack CD19 and 50% lose CD27; loss continues with disease progression
o Expression of CD56; this is absent in normal plasma cels and in plasma-cell leukaemiao ?CD28 expression in relapse patientso Under-expression of CD45 and over-expression of Cd221 are associated with a worse
prognosis
2. Genetic events that lead to myeloma Karyotyping is difficult because cells have slow turnover - you need FISH Hyperdiploidy seen in 60%; pseudo/hypodiploidy seen in 15-20%
o Monsomy 13/del (13q) common; upto 50% patients with MM -aasspcoted with worse prognosis
o Trisomy 3,5,7,9,11,15,19 common Diploid cytogenetic commonly involve IgH gene locus (14q23)
o Primary MM present early with B-cell specific mutations involving switch of J region of the IgH locus (also present in MGUS).
o Secondary MM presents late with more complex abnormalitieso Juxtaposition of IgH with a gene promoter is found in 48% MGUS, 73% of all MM
and 84% PCL 4 categories : t(11;14)(q13;q32) 15-20% Bcl -1 Cyclin D1
NB a different breakpoint in the same 14q32 locus gives mantle cell lymphoma
t(4;14)(p16.3;q32) 12% FGFR-1 and MMSET worst prognosis
t(14;16)(q32;q23) 5-10% c-maf t(6;14)(p21;q32) 5% Cyclin D3
o Other translocations involving the IgH locus include 8q24 (c-myc), 18q21 (BCL2), 11q23 (MLL1) 20q12 (MAFB). These are rarer
Secondary genetic events become commoner with advanced disease. These include further mutatuons in IgH as well as mutations in lamda light chain, del (Rb), P18/ras mutations
o Ras mutations accumulate and correlate with disease stage. Mutant ras has less dependence on stroma (IL6) for activation

3. Emerging role of cyclin D activation Cyclins are usually not expressed in lymphoid cells D cyclins (1,2,3) are important or cell cycle progression from G0 G1
o They interact with CDK4 = release of E2F from Rb = transcription & progression T(11;14)(q13;q32) involved with overexpression of cyclin D1 - seen predominantly in light
chain myeloma. Associated with a good prognosis T(6,14)(p21;q32) involved with overexpression of cyclin d3 Maf/MAFb mutations may increase levels of Cyclin D2 and trisomy 11 may contribute to
increase in cyclin D1
4. Role of stroma cytokines and adhesion molecules in myeloma Normal plasma cell development depends of stroma via interactions through adhesion
molecules, receptors and cytokines These interaction regulate proliferation , differentiation and apoptosis of plasma cells Growth/pro-proliferative/anti-apoptosis factors:
o IL6, IGF1, VEGF, SDF1ao BM stroma produces IL6. Myeloma cells may also make it when in contact with stroma
Inhibitory factors:o IFN-a,b,g, TGF-1b,a

2.18- TREATMENT OF MULTIPLE MYELOMA
1. Spectrum of disease caused by myeloma Proliferation of plasma cells within BM
o Symptoms of BM overcrowding/failure; anaemia, neutropenia, thrombocytopenia Cytokine production by plasma cells
o Bone resorption = pain. Fractures, hypercalcaemia Paraprotein production by plasma cells can lead to Hyperviscous blood Excess light chain production may lead to amyloidosis RENAL FAILURE caused by recurrent infections (neutropenia), hypercalcaemia, paraprotein
and light chain deposits
2. The choice of treatment depends on the ‘stage’ of the disease MGUS may precede active myeloma directly, an intervening period of ‘indolent’ myeloma may
also occur There is no treatment benefit by starting early in MGUS/ indolent patients - watch and wait
unless there is signs of active /progression of disease:o Presence of M component in serum (as kappa or lamda light chains) and/or urine (as
bence jones proteins) Note, M proteins correlate weakly with disease progress
o Intact Ig, BJPo Presence of plasma cells in BMo Raised serum light chains in abscence of >10% plasma cells in BMo Ca>2.65, bone diseaseo Creatinine >177o Hb <10
Active MM is incurable so aims of treatment are to prolong life/ reduce symptomso Give Bisphosponates because they reverse bone disease and block mevalonic acid
pathway which may trigger an anti-myeloma response Ultimately, the only treatment for active myeloma patients is autologous BM transplant
o If patients are not candidates for autografts - melphalan Lastly, if patients relapse after chemo/ auto-SCT - new drugs eg Bortezomib may be used
3. The choice of chemotherapy regimen for autologous stem-cell transplant Standard therapy - Melphalan +/- prednisolone
o Low CR [complete response] rate but oral therapy and produces response in 50-60% Px High dose therapy - Cyclophosphamide, thalidomide and dexamethasone has now replaced VAD
as high dose regimen in the UKo VAD - vincristine, Adriamycin, dexamethasone + autologous SCT
High response rate 65%, CR achieved in 10% Bm suppression, high dose steroids, hickman line and unsustainability of
response are weaknesses (median survival is about the same)o Other regimens with similar survival outcomes - COP, CVAMP, VBMCP, VMCP, BVAP
Median survival between either regimen is roughly similar at about 4-5 years , relapses also seem to continue at a constant rate between either
Allogeneic SCT only applicable to a minority of patients [because most people get myeloma when they are old]. Does offer a cure but has a high relapse rate and high treatment-related-mortality
o Should be considered for anyone <age 55 with matched sibiling

4. Treatment options in relapsed myeloma/ refractory myeloma THALIDOMIDE
o Low renal excretion; particularly useful in MM because renal failure is a problemo Supresses TNfa and angiogenesis
Morphology of advanced disease shows microvessels - hence anti-angiogenesis may be its most important mechanism
o Stimulates IL10, Tcells, B cells (cell mediated immunity)o Teratogenicity, constipation, somnolence, peripheral neuropathy, and
thromboembolism are side effects Lenalidomide is more potent than thalidomide in enhancing Nk cell activity, has less incidence of
side effects but may produce cytopenias. It is also very expensive. Bortezomib - proteasome inhibitor that leads to decreased activity of NfKB
o Proteasomes degrades proteins that are ‘polubiquinated’ [tagged in many sites by ubiquitin]
o Tumour cells are highly dependent on proteasome activity because they are high turnover cells
o Proteasome inhibiton leads to growth retardation and apoptosis of tumour cells Inhibition of NfKB = decreased proliferation, adhesion molecule and IL6
production, and increased spoptosis through ?caspase mediated pathwayso Bortezomib is potent, reversible and selective; it binds to proteasomes with a high
affinity and a low turnover rate

2.19 - MOLECULAR TECHNIQUES IN HAEMATOLOGY
1. Descriptions of some molecular techniques in haematologyThe f irst step is ALWAYS PCR
You have to extract DNA/RNa from cellular (usually WCC) suspensions Then you do PCR - you can use upto 70 cycles if you are looking for a rare molecular target Primer design is important
o 18-30bp longo Avoid repeats & complimentary sequences to prevent self-annealingo Limit C/G because they require more heat to denature (4degs as opposed to 2degs
with A/T)
Then you analyse your product Agarose gel analysis/ Qiaxel segregate fragments within the product according to size/molecular
weight - qualitative information onlyo Qiaxel is preferred because it avoids the need for the bromide (carcinogen) and is much
quicker Gene scanning is more sensitive/ specific/qualitative
o It can determine sequences and hence is useful in segregating fragments that vary by only a few bases, which would otherwise have a similar weight and be missed on agarose gel/Qiaxel
Heteroduplex gel analysis is useful when one allele is mutated and the other is noto It uses slow re-annealing which can generate mismatches in such caseso However this is laborious and you need a lot of sample material
Real time PCR uses 2 primers and 1 probeo The probe is degraded by TAQ-polymerase after about 15 cycles, and then produces lighto Concentration of your molecular target at the beginning of the reaction effects the
number of cycles needed to produce light (cycle threshold)o Hence you can use RQ-PCR to tell how much of your target was in a particular sampleo All samples reach the same plateau, but after a different number of cycles
Sanger Sequencing is similar to heteroduplex but it will tell you what nucleotides are mutated, hence it is quantitivate, whereas heteroduplex can only tell you if a mutation is present or not (qualitative)
2. Clinical applications of some molecular techniques in haematology Establish clonality in T/B cell proliferations
o Sanger sequencing is used to sequence immunoglobulin genes in B cell leukamiaso Recap: you have 125V, 3D and 4J segments, any of which can combine for the
antigen cinding component of an antibody, then nucleotides are added at V/D and D/J interfaces to get more variety
No 2 individuals will ever develop the same sequence on the Ig gene in leukaemia - if you see this, your sample is contaminated
Demonstrate chimerism/ monitor treatment post BM transplanto After a transplant, You use PCR with primers designed to flank short tandem repeats
and then use gene scanning to see whether the pre-transplant genotype of the host returns - this would indicate relapsed disease/ failure of treatment
o Real time PCR can identify very small events - so this is also useful in monitoring treatment response
PCr detection of chromosomal abnormalities/ fusion geneso Eg in BCR-ABL, cooperation of primers would suggest the translocation

Lastly - microarray analysis is a useful research tool by matching disease to gene expression - but you need a lot of patient samples for it.

2.20 - CYTOGENETICS IN LEUKAEMIA - PRACTICAL OVERVIEW
The average gene is about 10-50kb in size, the average chromosome 50-100Mb in sizeo Cytogenetic is low resolution - usally only to 5mb
Acquried cytogenetics looks at mutations in individual cells and is what is used in leukaemia Constitutional cytogenetics looks for mutations in meiosis; inherited diseases like CF, downs
Karyotping The first step is preparing metaphase cells for analysis
o Colchicine arrests mitosis by inhibiting spindle formationo BrdU/U?
Then you stain with giemsa and analyse band formation under a microscope/ using a computero Dark bands are gene poor and light bands are gene richo Single cell abnormalities may be artefacts - you need to analyse several metaphaseso You need to clone 2 or more cells to prove a translocation/chromosome gaino You need to clone 3 or more cells to prove a chromosome losso Red herrings are loss/gain of X/Y
The genome is usually in equilibrium between tumour suppressor genes and proto-oncogeneso Cytogenetic analyses can pick up monosomies/ trisomies/ trnasloactions which obsiously
upset this balanceo This provides diagnostic, prognostic and treat-reponse information
In nomenclature - for trnaslocations - the involved chromosome that contributed the centromere is termed a derivative
It is fast, cheap, screens the whole genome but is low resolution
FISH FISH involves labelling targets within the genome with a fluorescent probe
o It is rapid, requires less analytical skill, detects sub-micrscopic abnormalities and is useful for slowly dividing cells
o Traditional karyotyping is less narrow - this is the major disadvantage of FISH Chronic eosinophilic leukaemia is characterised by CHIC2 (non terminal) deletion which brings
PDGFRA into proximity with FIP1L1, creating a fusion gene on chromosome 4
aCGH Array comparative genomic hybridisation involves co-hybridising the sample to a genomic array
and using a computer to analyse it. Its major problems are:o Require relatively large quantities of purified disease cellso Expenseo Data analysis is complicated by germline copy number variationo Difficult technique to master, prone to unreliable resultso More clinical correlations requiredo Unable to detect balanced rearrangements

2.21 - TECHNIQUES USED IN MONITORING MINIMAL RESIDUAL DISEASE (MRD)
1. Definition of MRD and requirements of monitoring techniques MRD is the lowest level of disease detectable in patients in complete cytogenetic remission by
the methods available, hence these methods need to be:o Specific (to demonsrate malignant from normal cells)o Sensitive (because you want to pick up the few malignant cells left)o Reproducibleo Quantitative
The ideas behind monitoring MRD is that if you can’t completely eliminate the leukaemic clone, you can live with a small number of laukaemic cells because your immune system can keep them under check. Monitoring is needed to make sure the clone does not re-proliferate, so treatment strategies preventing relapse can be employed.
2. Advantages and disadvantages of main techniques used Morphology
o Advantage - easy access to source (PB/BM) and rapid testo Disadvantage - poor sensitivity, high false negative rate (especially post-chemo)
Immunophenotypingo Ads - rapid, cost effectiveo Disads - only applicable for about 60% of cases, requires a lot of analytical expertise, and
can have low sensitivity Cytogenetics (structural changes, deletions, numerical changes)
o Reproducible, cost effectiveo Poor sensitivity and requires cell division for karyotyping - you would have to wait
6months-1year post chemotherapy for cell division to reach ‘normal’ - therefore cytogenetics is of limited use in monitoring MRD
It is useful in diagnostics, prognoses and telling us epidemiology of certain translocations eg young >old
Molecular techniqueso You can look for (clonal) genes
Ig genes for B cells - the ?complimentary dependent region 3 (CDR3) is a locus of added nucleotides that joins [V to D] and [D to J] and is typical of leukaemia - primers are designed against this region
TCR genes for T cells - primers built against gamma/delta chains (immature)o You can look for fusion genes generated by translocations
Fusion genes are specific to leukaemia. Ig/TCR gene rearrangement are done in a polyclonal fashion by normal cells as well, but no normal cell will have a fusion gene
You need PCR to monitor patients who have a number of leukaemic cells that is too low for micrscopes to detect. It can also help to identify the very start of a relapse, which can inform clinical decision making
Eg. BCR/ABL, PML/RARa
3. Strengths, weaknesses of molecular techniquesAds - reproducible, applicable to 90% of cases, sensitive, becoming more cost effective
Disads - requires molecular expertise, can be laborious

2.22 - LYMPHOMA - DLBCL & MCL (this lecture was not actually given)Not sure if this lecture was given or not
1. Pathogenesis in lymphoma Mutations that confer survival advantage/clonal proliferation early in the lymphoid ontogeny
leads to:o ALLo Precursor B/T cell lymphoma
Mutations that occur later in the ontogeny leads to:
o CLLo Mature B/T cell lymphoma
85% of lymphoma is non-hodgkins, i.e. reed-sternberg cells are absent
2. Structure of a lymph node The outer layer (PARACORTICAL) contains T cells The inner layer in the lymphoid follicle:
o the germinal centre is where B cells bind antigen and become activated
o the surrounding mantle zone still contains naïve, unstimulated, virgin b cells
3. Clinical features of lymphoma Presentation
o proliferation of lymphocytes in tissues = lymphadenopathy (+/- local compression/ obstruction) hepatosplenomegaly High WCC Leukostasis
o Proliferation of (immature) blasts in BM = Anaemia, neutropenia, thrombocytopenia
o B symptoms Diagnosis/staging - BM/ LN biopsy, PET scan
4. Diffuse large B cell lymphoma DLBCL Aggressive Mature b cell (non-hodgkin) lymphoma; accounts for 30% of all NHL Occurs in the elderly Morphology - sheets of large B cells seen in (34%) or after germinal centre (49%)
o Within germinal centre is a better prognostic phenotype - associated with ongoing Ig mutations, bcl2 and c-rel amplification
o Post-germinal centre has no ongoing Ig mutations and associated with NfKB activation Moleculr - p53 mutation is a worse prognosis Prognosis determined by IPI [international prognostic index]
o 1 point each for Age >60, raised LDH, ‘performance status’ 2-4, stage III/IV and >1 extranodal site
o 5 year survival is 73% for score 0/1; 26% for score 4/5 Treated with 6-8 cycles of rituximab + CHOP (cyclophos, Adriamycin, vincristine, prednisolone) 50% are cured, consider Autologous SCT for people who relapse - can save 25% of them
5. Mantle cell lymphoma

Aggressive Mature B cell (non-hodgkin) lymphoma Affects adult males more (6:1), is disseminated at presentation (multiple large nodes) median survival is 3-5years is worst of all B cell lymphomas - frequently relapses Morphology - low rate of somatic hypermutation of V genes in small pre-germinal centre B cells Immunophenotyping - - aberrant cd5, cd19, cd20, cd22, delta chain, sIgM/D (cd10 and cd23
negative) Molecular - 50-75% associated with t(11;14)(q13;q32) and cyclin d1 overexpression
o BCL1/IgH fusion gene = upregulated BCL1 = increased cyclin D1 Cyclin d1 not normally expressed in lymphocytes = unchecked progression into S
phaseo The same translocation can be found in large cell lymphoma, CLL and MMo additional cytogenetic abnormalities occur in 50% (+12, 13q-(BCMS), 17p- (p53), 11q-
(ATM), 9p- (p14,15,16) Treat with 3 cycles of R-CHOP, 3 cycles of R-DHAP (platinum-based) and auto-SCT
6. Other non-hodgkins lymphomasBurkitts Aggressive Mature b cell (non-hodgkin) lymphoma Jaw/abdominal sweeling secondary to lymphadenopathy seen in young people Associated with EBV Morphology - proliferation within germinal centre; ‘starry sky appearance’ Cytogenetic/molecular - t(8;14), t(2;8), t(8;22) - all involving c-myc
Follicular lymphoma Mature b cell (non Hodgkin)
lymphoma - accounts for 22% of NHL Affects elderly as an indolent disease
that can transform into an aggressive lymphoma
Morphology - arises in germinal centre in a follicular pattern
Immunophenotyping - cd10 & bcl6 +ve
Molecular - associated with t(14;18) and bcl-2 translocation
7. Outlook in Non hodgkins lymphoma
Phenotype Survival CurabilityAggressive Weeks without treatment Curable - leukaemia protocols~ Months without treatment ~ R-CHOP +/- A-SCTIndolent Years (upto 15) without treatment/
transformationIncurable; wait and watch or R-COP
2.23 - CYTOGENETICS AND MOLECULAR GENETICS OF ALL
1. The roles of cytogenetics in ALL

Determines prognosis, informs treatment decisions and is useful for MRD monitoring o Vary rarely needed for diagnosis in ALL
Cand distinguish and recognise relapses, leukaemogenic mutations and molecular mechanisms in leukaemogenesis
2. Important cytogenetic features of childhood and adult B-ALL Cytogenetic and molecular abnormalities between B & T-ALL differ greatly Children tend to have better prognoses in ALL because ‘good prognosis’ cytogenetic
abnormalities are more common, and they are more resilient and hence can survive more aggressive treatment regimens
o High Hyperdiploidy (25% children; 8% adults)o T(12;21) (10-30% children; 3% adults)o T (1;19) (5% children; 3% adults)
Bad prognosis t (9;22) is occurs in 25% of adult ALL (2% children)
Age, gender (lymphoblasts are protected from chemo in CNS & testes) and country where you are
treated also influence survival from ALL
3. Genetic features of B-ALL2 hit model Initial mutation (eg a translocation) in childhood ALL may occur in utero The second hit leads to emergence of the leukaemic clone
o Exposure to an environmental agent/ pathogen? - a concordant time gap in between ‘hits’ might suggest this
o Deletion of the normal allele in a person where the other copy is already mutated (eg ETV6)
o Other cytogenetic abnormalities
Child - High Hyperdiploidy 10,17,21 are often triplicated You need centromeric probes in FISH to pick it up The same hyperdiplouid karyotype is odten seen in identical twins Morphologically, it is associated with Common B ALL
Child - T(12;21) (p13;q22) Leads to formation of the ETV6-RUNX1 fusion gene
o RUNX1 codes for CBFa, which binds CBFb and HAT. HAT allows it to bind to DNA and activate transciprtion for a number of genes including tyrosine kinases
o The fusion gene leads to a product which: cannot bind HAT, but remains tethered to DNA binds co-repressor molecules like sin3a, ncor, HDAC heterodimerises via ETV6 to give further transcriptional inhibition
Although associated with a good prognosis (95% 7yr EFS), ‘relapses’ can occur - these are thought to be emergence of a sub-clone
The translocation is cryptic; the derivatives look similar to the originals - SO YOU ALWAYS FISH Morphologically, it is associated with Common B ALL
Child - T(1;19)(q23;p13) Leads to formation of the TCF3-PBX1 fusion gene Unbalanced translocations are common, and involve loss of the derivative of 1 and subsequent
duplication on the c1 that was not involved in the translocation Morphologically, it is associated with Pre-B ALL (mu positive)

Infant - t(4;11)(q21; q23) Leads to the MLL-MLLT2 fusion gene
o This leads to activation of transcription of genes like HOX Morphologically, it is associated with Pro-B ALL; mutations occur in early precursors or the
pluripotent stem cell - this can lead to a biphenotypic leukaemia Has the worst prognosis despite more intensive treatment
Adult - T(9;22)(q34;q11.2) Leads to the BCR-ABL1 fusion gene
o Involves a different breakpoint from that seen in CML Not all cases have abnormal cytogenetics - you need FISH & PCR Presents in middle age and is largely responsible for the ‘second peak’ of ALL incidence with age Poor prognosis, often relapses
Another rare cytogenetic abnormality in B-ALL is dic(9;12) where, following translocation, the result is one chromosome with centromeres from both the participants.
A Marker chromosome is an abnormal chromosome that marks the leukaemic clone
4. Overview of genetic & cytogenetic features in T-ALL Important loci are 14q32 and 14q11 as these code for the TCR The most frequent cytogenetic abnormality is an interstitial deletion that leads to the STIL-TAL
fusion gene (33%)o TAL is Not usually expressed in T cellso The deletion is cyrtpic - you need RT-PCR
T(5;14)(q35;q32)is seen in about 20% of T-ALLo Translocation is Cryptic - FISH for ito Leads to HOX11L2 dysregulation
T(10;14)(q24;q11) is the third most common cytogenetic abnormality in T-ALLo Leads to HOX11 dysregulation
5 - RECAP - mutations affecting oncogenes in leukaemia Point mutations (eg RAS) Small deletions (STIL-TAL) Internal tandem duplications (FLT3) Translocations that produce fusion genes (many) either by promoter exchange or bringing an
oncogene under influence of another promoter Direct gene amplification Insertional mutagenesis following gene therapy
2.24 - IMMUNOPHENTYPING PRACTICAL
Morphology- when in doubt, just say you can not tello M0/M7 AML needs immunophenotyping

o Monoblasts may be non-specific esterase +ve Immunophenotyping
o Common white cell antigen - CD45o Myeloid markers - CD117, 13, 33, 15o 1 immature marker (CD34 or Tdt) is enough
Monoblasts can be CD34 negativeo CD56 marks Nk cells but is aberrantly expressed on myeloid and myeloma cellso HLA-DR negative is ALWAYS APML
MDS - 5q syndrome progresses to AML more readily because of proneness to further mutations, lenalidomide to treat
APML - watch for headaches, treat DIC with platelets, plasma and ATRA straight away tAML - t(9;11) [MLL rearrangement] secondary to doxorubicin
2.25 - MULTIPLE MYELOMA (CLINICAL)
1. Epidemiology of Multiple Myeloma
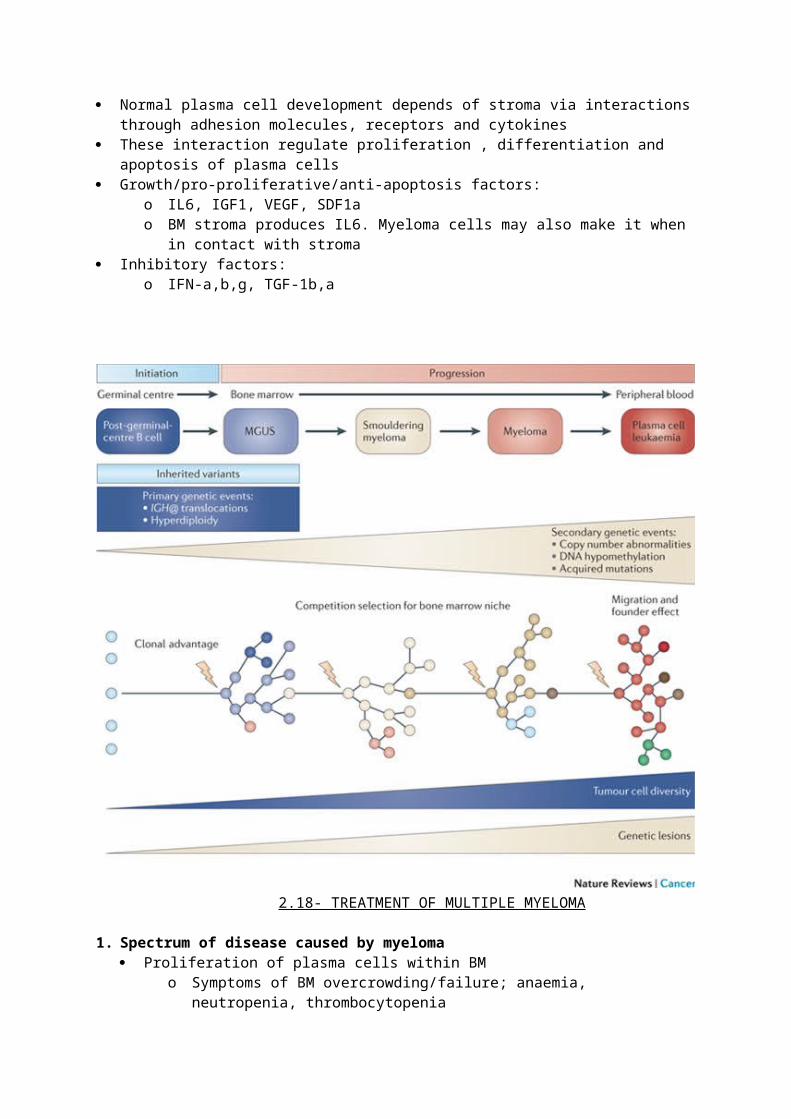
D - clonal proliferation of bone marrow plasma cells, characterised by production of one/both of monoclonal antibodies (termed paraproteins) and free Ig light chains (BJP).I - incidence is 3.5k in UKA -OLD; median age 70S - M>F (just)G - Blacks > whiteA - UNKNOWN
2. Pathophysiology of disease in Multiple Myeloma3 main mechanisms of disease Proliferation of plasma cells
o Anaemia (common presenting symptom)o Neutropenia (recurrent infections +/- sepsis)o Thrombocytopenia
Cytokine productiono TNFa & IL1b stimulate osteoclasts = bone damage, lytic lesions,
hypercalcaemia Osteoclasts make IL6 which stimulates plasma cells,
reduces albumin and raises CRPo Bind to osteoblasts/ BM stromal cells to further stimulate
osteoclasts via RANK/RANKl pathway Monoclonal antibody and/or light chain production
o Most commonly IgG / IgA are produced, there raise ESR and lead to rouleaux formation
o Free light chains circulate as BJP and can contribute to renal failure
o BJPs can bind to plasma protein to lead to amyloidosiso 25% of patients have BJP only (no paraprotein) - hence these may have normal ESR
Pathophysiology of renal failure - Hyeprcalcaemia, recurrent infections, antibody and/or light chain deposits
Pathophysiology of bone disease = lytic lesions, BM growth due to plasma cell proliferation, osteopenia
3. Clinical features of multiple myeloma Symptoms/signs/examination
o Symptomatic patients present with symptoms of anaemia, hypercalcaemia, bone disease and renal disease
o Patients can be aymptomatic in MGUS and early active MM Investigations
o Bloods - Raised ESR (in paraprotein +ve MM), CRP, calcium, low albumino Film - rouleaux in blood film, clonal plasma cells >10% in BM (presence in BM = PCL)o Immunophenotyping - CD56, CD138, clonal expression of either kappa/lamda light chain o Cytochemistry - immunoperoxidase staining for CDs, cyclin D1 and light chainso Serum protein electrophoresis - dominant M bando Beta2macroglobulin = marker of tumour load and renal function
ISS staging system (2005):
Stage B2 macroglobulin Albumin

1 <3.5 >352 Between 3.5 and 5.5 <353 >5.5
4. Treatment options in multiple myelomaSymptomatic management Hypercalcaemia - fluids, steroids bisphosphonates (IV zoledronic acid)
o Bisphosphonates improve bone symptoms and reduce need for radiotherapy to ease bone pain etc
Conventional chemotherapy regimens & high dose chemotherapy + AutoSCT have similar OS Conventional:
o Cyclophosphamide, Thalidomide and Dexamethasone for most patientso Melphalan and prednisolone for the elderly
High dose chemotherapy + AutoSCTo Attal et al 1996 - as compared to conventuional therapy, high-dose therapy achieves
greater CR but has similar OS Hence AutoSCT is the treatment of choice
o Child et al 2003 (Myeloma VII) confirmed these findings
AlloSCT is the only cure but is only applicable to the minority of patients Only applicable for a small group of patients: age <50 and with a matched sibling Transplant related mortality is high Curative in about 20% of patients Females, early disease stage, being in CR are good prognostic factors
How to improve survival in MM? [median survival now is about 6 years] Use different chemo regimens Tandem (>1) transplants - IFM94 study says it improves OS CD34 selection and starting SCT early seem to have no effect
5. New agents in Multiple Myeloma Thalidomide
o Immunomodulatory - anti adhesive, anti TNfa and anti IL10; stimulates host lymphocyteso anti-angiogenic; anti FGF and anti VEGFo Effective in about 60% of patients, oral admin and CHEAPo SFX = constipation, somnolence, peripheral neuropathy, VTE
Bortezomibo Reversibly inhibits chymotryptic active site of proteasome beta subunit = NfkB
inactivation = less transcription of IL4, VEGF, adhesion moleculeso Key study was APEx; Richardson et al 2005o Effective in 40% of patients, fast responseso Main SFX = thrombocytopenia, neutropenia, anaemia, peripheral neuropathy. Very
expensive and requires IV admin Lenalidomide
o Advantages - oral admin, appears to be more potent than thalidomideo Diadvantages - very expensive, VTE. neutropenia
2.26 - COMPLICATIONS OF LEUKAEMIA THERAPY

1. Types of leukaemia and their therapy CML - TKIs ALL - combination chemo (FCR; fludarabine, cyclophosphamide, rituximab) Acute leukaemias - combination chemo
SCT is an option for all leukaemias
2. Principles in chemotherapyChemotherapy is aimed at dividing cells. Tumour cells are more proliferative and they divide inefficiently and hence spend a longer total time undergoing division = proness to chemo
Types of chemo: Antimetabolites Mitosis inhibitors Enzyme inhibitors Alkylating agents Anthracylines Other - TKIs, monoclonal antibodies
3. Systemic effects of chemotherapy Nausea/Vomiting
o Melphalan and anthracycline are bado Prophylactic ally/therapeutically give anti-emetics, particularly 5HT3 inhibitors eg
ondansteron Alopecia
o Can recover but may be patchy. Offer wig Dose-dependent bone marrow suppression
o Alkylating agents are the worsto Treat anaemia with transfusions/ EPOo Treat thrombocytipenioa with plateletso Be careful of neutropenia; <0.2e9 is high risk of infection; <0.1 is high risk of sepsis from
gut commensals Drug extravasation (local inflammation, necrosis etc)
o Rarely a problem because most people are on central lines rather than cannulas TUMOUR LYSIS SYNDROME
o Particularly pertinent in acute leukaemias with a high WCC and Burkitts lymphomao = Hyperuricaemia, kalaemia, phosphataemia and hypocalcaemia
= dehydration, metabolic acidosis, ARF, deatho Prevent with allopurinol or rasburicase for the above two diseases
4. Organ-specific effects of chemotherapy
renal bladder cardio liver GI neuro ReproAntimetabolites √ √Mitosis inhibitors √Alkylating agents √ √ √ √ √ √ √Anthracyclines √ √
Long term effects: Cerebellar ataxia with antimetabolites (cytosine arabinoside)

Sterility with most (freeze sperm/ eggs/ embryos to prevent) T-AML, Bladder Ca and Pulmonary fibrosis with alkylators
5. Side effects of stem cell transplant
Acute/chronic toxicity of chemo = problems seen with the table above Acute neutropenia = infections (bacterial and fungal) Effects of immunosuppression =
o Chronic/late infections (pneumococcus, PCP, CMV)o Acute GvHD (skin, gut liver)
Prevent/treat with immunosuppressive (cyclosporine, tacrolimus, rapamicin, MMF, CAMPATH) and corticosteroids
o Chronic GvHD (presents like scleroderma; skin, gut, liver lungs) Prevent/treat with immunosuppressive (cyclosporine, tacrolimus, rapamicin,
MMF, CAMPATH) and corticosteroids Long term complications
o Endocrinopathy (thyroid, adrenals, growth retardation)o Infertilityo Cataractso Pulmonary dysfunctiono T-AML
2.27 - LEUKAEMIA - PAST,PRESENT & FUTURECml

1882 - Arthur conan doyle found arsenic to have an effect on CML like syndrome 1950s - nitrogen mustards and bisulphan used to treat 1973- 9,22 translocation established 1980 - BCR/ABL1 fusion gene established 1980s - alpha interferon used to treat IRIS trial 2003 - imatinib Now we can use PCR to monitor MRD with people on imatinib therapy
ALL Used to be universally fatal 1930s - folic acid found to make it worse
o Idea emerged that antifolate (aminopterin) can induce remission 1950s - 6mercaptopurine used but associated with CNS relapse Now combo chemotherapy mean >90% ALL can be cured We can use cytogenetics to monitor MRD
Hodgkin lymphoma Hodgkin recognised the syndrome in 1832 following post mortem analyses Reed and Sternberg characterised the characteristic multinucleate cell between 1898-1902
o Both postulated TB to be associated with it 1940s - role of brucella in aetiology? 1960 - cytogenetics demonstrated aneuploidy and clonal derivation Staging from the 50s onwards has progressed from lymphograms open surgery CT
PET-CT (this is what is now used) Combination radiotherapy (ABVD) and radiotherapy is now used to treat
2.28 - APML - A PARADIGM FOR MOLECULARLY TARGETED THERAPY
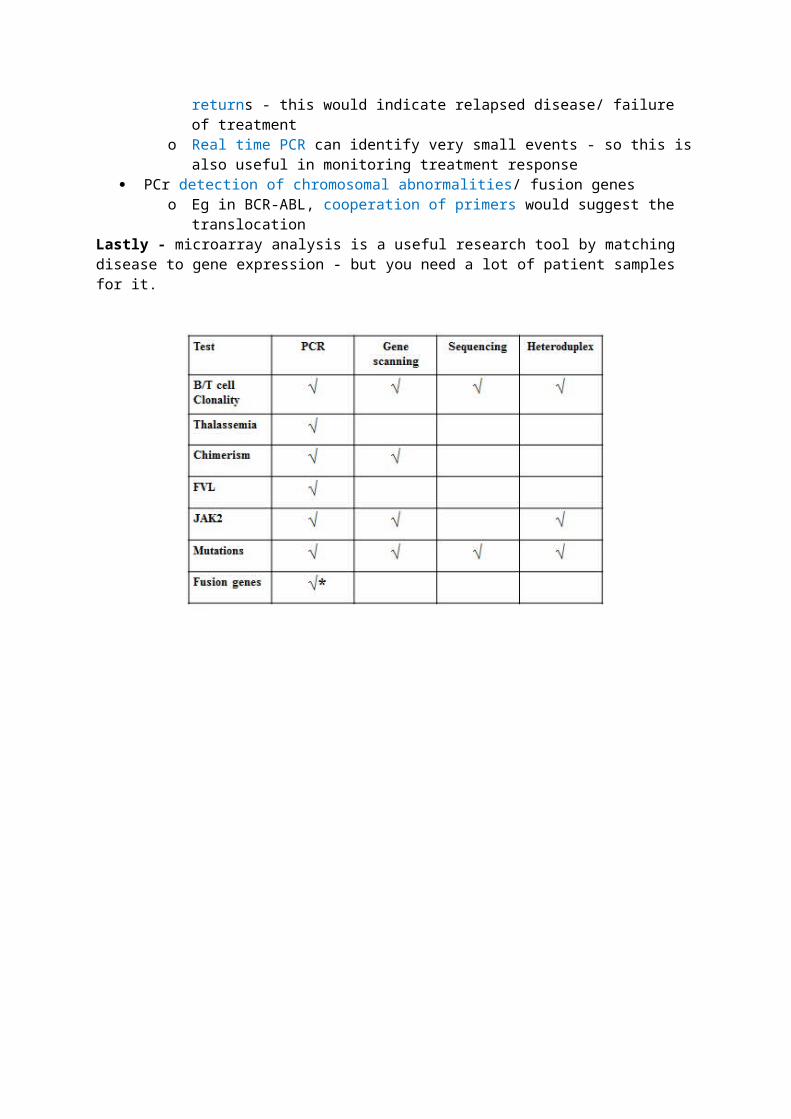
1. Recap of genetic basis of APML APML is all about repressed transcription giving a halt in matuaration at the promyelocyte stage.
Mutations in these committed cells gives them the capacity to self renew - which is a property of stem cells
Disruptuon of PML nuclear bodies by PML-RARa may be a key effect o Normal PML makes the outer shell of nuclear bodies o NBs contain other proteins eg pRb, p53 and BLM hence normally functioning NBs play
key roles in apoptosis, senescence, growth suppression and genome stabilityo Abnormal PML leads to microparticualte distribution of proteins usually encased within
Nbs
2. Mechanisms in t-APML can inform us of ‘weaknesses’ in the genomeIn t-APML, there is clustering of breakpoint s within intron 6 of PML and RARa following prior exposure to mitoxantrone
o mitoxantrone is used for breast cancer and MS 4 breakpoints are clustered within 8 base pairs on PML
o The fact that 1 in 400 MS sufferers treated with mitoxantrone develop t-APML, and experience the same breakpoints, suggests that this area of the genome is a favoured topoisomerase-II cleavage site and thus is prone to sustained damage but topoisomerase-Ii poisons such as mitoxantrone
3. The future of APML management may see the abolishment of chemotherapy ATRA achieves the following by binding to the ligand-binding domain of RARA:
o Degradation of PML-RARao Replaces co-repressor molecules bound to PML-RARa with activators o Reformation of PML in nuclear bodieso Terminal differentiation, upregulation of TRAIL and induction of apoptosis
Arsenic achieves the following by binding to PML:o also induces degradation of PML-RARa and thus achieves differentationo inhibits angiogenesiso induces apoptosis by multiple mechanisms
you can use molecular monitoring (PCR for PML-RARa) to:o tailor treatment regimeso intervene early and monitor continually - this reduces relapse rateo allow admin and monitoring of low dose chemo (which confers reduced toxicity)
studies suggest low dose regimes have better survival current evidence suggests completely withdrawing chemo does not affect
survival - win!o Molecular monitoring is probably cost effective in badly diseased patients
other potentials for molecularly targeted therapy: anti-CD33 + chemo for AML that has core-binding-factor rearrangements (eg t8/21, inv(16)) molecular monitoring may also be useful to predict relapse in NPM1 mutated AML patients
2.29 - LYMPHOMA AETIOLOGY AND BURKITTS LYMPHOMA

1. Overview of aetiology of lymphomaMultifactorial disease aetiology Inherited predisposition may relate to immunodeficiency:
o Abberant immune response to infections (certain HLA groups are prone, SCID)o Defective DNA repair (ataxia telangiectasia/ Nijmegen breakage syndrome)o Polymorphisms in DNA repair geneso Polymorphisms in genes encopding complement]
Acquired immunodeficiencyo Induced by HIVo Iatrogenic - particulary following heart/lung transplant or methotrexate use
Environmental exposure to mutagens (pesticides, herbicides, fertilizers in follilcular lymphoma) INFECTIONS
o Bacteria, viruses, malaria Auto immune diseases (cioeliac, hashimotos thyroiditis, sjogrens, RA) Blood transfusion? NHL
2. Role of bacterial agents, namely H.pylori, in lymphoma aetiologyTypes of bacteria involved in lymphoma
H Pylori (T cell gastric MALT lymphoma) Campylobacter - (immunoproliferative small intestinal disease; IPSID) TB - pyothorax associated lymphoma Maybe borrelia and chlamydia too
H.Pylori in gastric MALT lymphoma Proof of aetiology
o 90-95% of g-MALT lymphoma patients are infectedo Incidence of gastric MAL lymphoma is increased 6 fold in infected peopleo elimination of the infection (antibiotics) leads to remission
The bacteria stimulates T cells, the T cells lead to chronic stimulation of B cells The disease becomes autonomous from infective stimulation (and thus refractory to
antibiotics treatment) following other genetic eventso T(11;18) BIRC3-MALT1 fusion gene = BCL10 dysregulationo T(1;14)= BCL10 dysregulation o T (14;18) = BCL2 dysregulation because a MALT1 fusion gene is formedo Associations with autoimmune diseases and fas mutations can also lead to
refractory gMALT Borrelia may play a similar role in the aetiology of cutaneous MALT lymphoma
3. Role of viral agents, namely EBV, in lymphoma aetiologyTypes of viruses involved in lymphoma
HIV o Both HL & NHL (intracerebral DLFBL, Burkitts)
HIV surpresses T cell function in burkitts and EBV/oppurtunistics stimulate B cells
o May be in part related to EBV co-infection (1/3 patients have this) Herpes viruses
Infection = stimulated CD4 T
cells
polyclonal B cell expansion,
dependent on bacterial antigens
random translocation etc
monoclonal B cell expansion;
independent of bacterial antigens

o EBV - endemic Burkitts, many classical HLo HHV8 - primary effusion lymphoma (EBV may be a cofactor); CD30+ CD138+
HTLV1 - TALL HCV - salivary gland MALT lymphoma
Lymphoma mechanisms in viral infection Expression of viral oncogenes Transactivation of host ptoro-oncogenes Insertional mutagenesis Induction of chronic B cell stimulation Supressed T cell function
EBV in endemic Burkitt Lymphoma Endemic Burkitts is an aggressive lymphoma that causes jaw tumour in children, spread across
the malarial belto Sub aharan Africa, south America, papua new guineao Geographical areas within these countries that confer pro-malaria conditions have
higher sub-incidences EBV and malaria act as a team in the aetiology of endemic burkitts
o EBV causes chronic B cell stimulationo Malaria does the same but it also surpresses t cell function
It is also associated with splenic marginal zone lyphomao A random mutation (eg dysregulation of myc or p53) is what ultimately leads to
monoclonal expansion of (usually) IgM B cells Diagnosis of Burkitts
o Cytogenetics - 3 trasnlocations associated with emergence of endemic Burkitts A breakapart probe in FISH is useful because they all involve myc so it can pick
up any of the threeo Morphology - vacuolated basophilic cytoplasms; appearance of macrophages that have
ingested apoptotic materialo Immunophneotype - CD34-, Tdt-, SmIG+, B markers and BCL6 [mature B]
CD10+ (but this is also seen is normal follicle cells)Translocation Fusion product FrequencyT(8;14)(q24;q32) MYC-Ig heavy chain 90&T (2;8) Myc-kappa light chain 5%T (8;22) Myc-lamda light chain 5%
Other roles of EBV in lymhpoma:o Can cause classical hodkin lymphoma
incidence increases 2-3y after glandular fever is present in neoplastic cells encodes for oncogenic proteins
latent membrane protein 1 [LMP1] induces BCL2 LMP2 downregulates development genes and upregulates proliferation genes
o Can cause DLBCL (latent membrane protein 1 may be important)o implicated in post-transplant lymphoprliferative diseaseso Richters syndrome in CLL (where B cells that are clonally related to CLL invade tissues)o Lymphoma in people who are immunodefiencent follwoinf drug therapy (infliximab,
methotrexate)Sporadic (non-endemic) Burkitts

The same translocations are seen but the exact breakpoints are different from what is seen in endemic burkitts
o EBV might be the key difference in mediating this Soradic is only associated with EBv in 10% of caes Affects abdominal organs (ovaries, caecum0 and breasts
HCV in lymphoma The importance of HCV in lymphoma seems to vary by country It causes monoclonal cryoglobulinaemia which leads to tissue necrosis
o Associated with SLVL, MALt lymphoma of salviares, DLBCL Patients with splenic marginal zone lymphoma appear to regress when HCV infection is
treated
4. Role of auto-immune and other non-infective diseases in lymphoma etiology Principle is the same with inefctions - antigenic stimulation of host lymphocytes = chronic
proliferation Autoimmune diseases seem to give an increased incidence of Hodgkin lymphoma
o Sjogrens increases risk 6 fold (marginal zone, DLBCL, FL)o SLE increases risk of DLBCL and Marginal Zone Lymphoma 3 foldo Psoriasis and coeliac assoacited with T cell lymphoma
Coeliac gives a high grade lymphoma of T mucosal lymphocyteso i.e. the lymphoma arises in a T cell subset which is different to that which mediates the
autoimmune diseaseo Incidence is increased 80x in people are left undiagnosed/untreated
The autoimmune lymphoproliferative syndrome (ALPS)is a chronic polyclonal disorder resulting from mutations that lead to failure of apoptosis
o Increased incidence of B NHL & HL as well as autoimmune diseases
2.30 - HTLV1 AND ATLL

1. Spectrum of disease caused by HTLV1 infection is somewhat dependent of proviral-loadBrief Epidemiology of HTLV1
The infection is almost as prevalent as HIV worldwide Particularly common in sub Saharan Africa, peru, japan and amongst aboriginals Disease of the elderly
Proviral load and disease spectrum Importantly, the proviral load does not vary over an individuals’ disease course, but there is
varability between different infectees Low proviral load correlates to asymptomatic disease features
o Recurrent shingleso TBo Helminth infections e.g. strongyloides (watch for G-ve sepsis following bowel
inflammation)o Infective dermatitis (chronic infection in children)
Higher proviral loads correlate with symptomatic disease features after a long latent periodo Presentation is at ago 60+ usuallyo Adult T cell leukaemia/lymphoma (4 subtypes)o HTLV1-associated- myopathy
Weak & stiff legs, lumbar pain, leg pain, bladder dysfunction, erectile dysfunction, constipation
Strongly resembles MS The presence of CSF HTLV1 antibodies distinguishes it from MS
o Chronic multisystem inflammatory diseases (alveolitis, uveitis, polymyositis)
The STLV1 virus produces a similar disease phenotype hence the immune response to the pathogen, rather than the pathogen itself, might be key.
2. Features of adult T cell leukaemia/lymphoma
Signs/symptomso Bm failure (anaemia, thrombocytopenia)o Hypercalcaemia (mehcnaisms similar to what is seen in myeloma) o Generalised lymphadenopathy - watch for SVC obstructiono Hepatosplenomegalyo Skin lesions - always biopsy - skin lesion appearance varies gretly between patients.
Nodular lesions are the ‘classic’ appearanceo Lytic bone lesions
Investigations
Type Lymphocytes
Abnormal T cells
Ca LDH Other Survival
indolent Smouldering (5%)
<4e9 >5% Normal <1.5x Skin/lung lesions
24mon
Chronic Leuk (15%)
>4e9 >5% Normal <2x Liver,spleen, skin, lung
24mon
Aggressive Lymphoma (20%)
<4e9 <1% Can be high
Can be high
LNs 10mon
Acute Leuk (60%) >4e9 >5% Can be high
Can be high
Tumour lesions
6mon

o Bloods - Bm failure, high WCC, high Ca, high LDH (marker of cell turnover)o Film - flower cells (large cells with indented/lobulated nuclei)o Cytogenetics - no classic hallmarko Immunophenotpye - CD4+ CD25+o Always do an Lp because, like burkitt lymphoma, it commonly spreads to CNS and
crytpoccal meningitis is also common in ATLL so rule this out too Management
o Aggressive T-ALL - ½ cycles of CHOP + AZT/IFN but we don’t know when to start AZTo Indolent t-ALL - AZT/IFNo Relapsed t-all - DHAP/ new drugs (anti CD25, HDACi’s, bortexomib, ATO)/ alloSCT
Difficulty with alloSCT is that most sbilings are infected too so you have to look for unmatched donors
3. Virology in T-ALLHTLV1 is essential to cause T-ALL
Vertical transmission is via breastmilk Horizontal transmission is similar to that of other blood borne viruses
Mechanism of infection It is a retrovirus - its uses reverse transcriptase before incorporating into host T-cells In early stages of disease - cell-cell contact is how it spreads between cells
o This is why you can’t find anything in the plasma From then on, the virus replicates by conferring the clonal expansion of the infected cells
How does HTLV1 cause cancer? Two ways in which the provirus affects host genome
o Acute - Transcription of an oncogene that happens to have existed within the provirus o Chronic - the provirus acts as a gene promoter itself and leads to transcription of proto-
oncogenes within the host genome e.g. myc Important proteins
o TAX - expressed in 40% of T-ALL Involved in a lot of cellular pathways but generally it immortalises the CD4 cell,
drives tumour formation and is immunodominant Tax activates NfKb, AP1, SRF, NFAT, CREB/ATF It downregulates beta-polymerase Cell cycle progression (expression of cyclin G2) Inactivation of p53 (ATLL with wild type p53 responds to zidovudine) Induction of apoptosis (through Fas/FasL and IL-1b converting enzyme
(ICE)). Inhibition of apoptosis (Tax inhibits anti-APO-1 induced T-cell death,
represses transcription of Bax gene and up regulates BCL2). Decreased Genomic Stability Telomerase
o HBZ - its mRNA in seen in all cases of TALL and it seems to drive tumour formationo These proteins are not oncogenes - only 5% of HTLV1 infected people actually get TALL
The basic leuice zipper domain promotes t cell proliferation and represses taxo This suggests a complex regulatory mechanism of viral load etc
2.31 - LYMPHOPROLIFERATIVE DISEASES
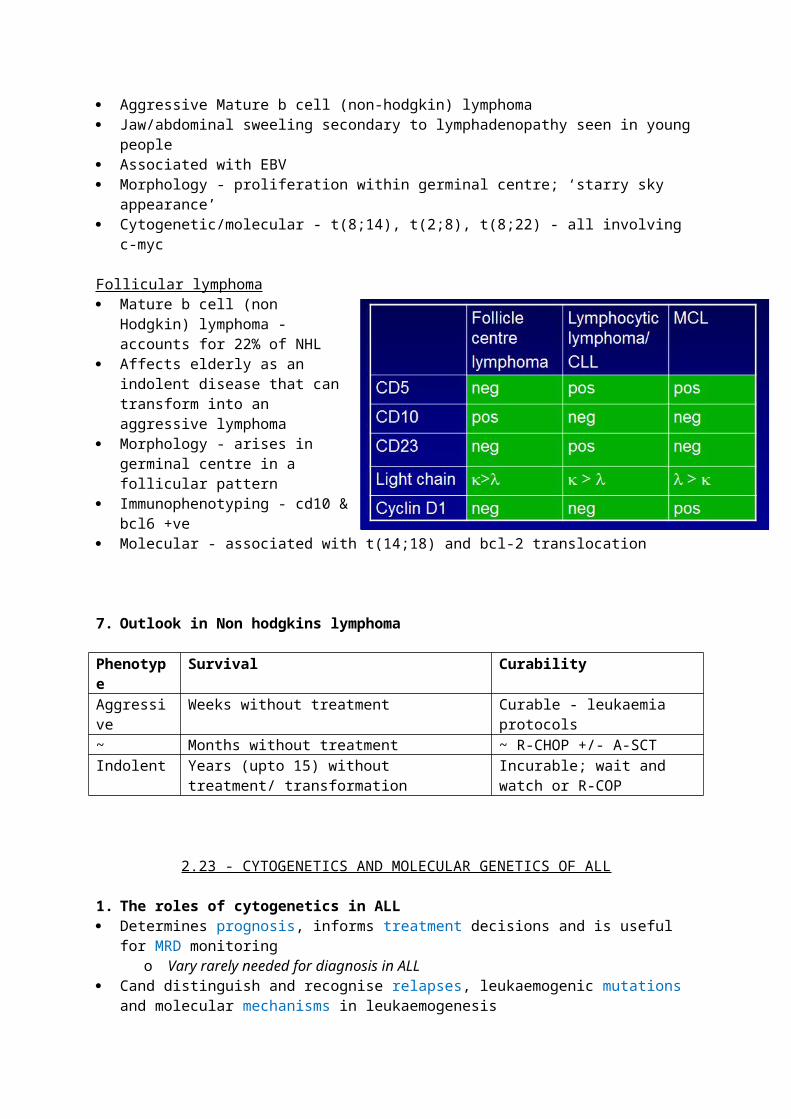
1. MALT lymphoma Epidemiology
o Defined as extranodal marginal zone lymphoma of MALTo Comprises 8% of all NHLo Presents at 55-60years oldo Commonly arises in stomach, but also in many other organs
Disease courseo Indolent in most patients; 5yr survival 80-95%o Nodes usually only involved after local spread; <15% involve BMo A minority transform to aggressive DLBCL
Diagnosiso Cytogenetics - t (11;18)(q21;q21)o Immunophenotype - CD20+ CD10- CD5-o Molecular - BIRC3-MALT1
Aetiology - H pylori Treatment - 2 weeks triple therapy (PPI, amoxicillin, clari/metro) works in 85% of cases
o 50% of those who don’t respond/relapse after MRD, respond to chlorambucilo Rituximab may also be useful
2. Primary CNS Lymphoma Epidemiology
o Comprises <2% of all NHLo Presents at median 61 years oldo Incidence of upto 10% in AIDS patients
Disease courseo >95% are B cell tumourso 2/3 present as solitary CNS lesionso Median overall surviala about 5 years following combination therapy; radiation is
bad? Outlook for PCNSL has improved significantly due to intensified chemotherapy Removing DXT from therapy of patients may reduce toxicity without compromising outcome Some patients with refractory/relapsed disease may be salvaged by HDT
3. Lymphoma in Pregnancy Epidemiology
o Occurs in 1/3k pregnancies, usually as a HLo No evidence that HL influences outcome of pregnancy or vice versao If NHL – particularly bad prognsosis if it presents aggressively (90% of px)
Difficulties:o Accurate staging is difficult because you can’t use CT in pregnancy - dangerous for
babyo Is it safe to do chemo?
Treatmento Do nothing - probably safe for HLo Terminate pregnancy? Probably only for aggressive NHL presenting early in
pregnancyo Avoid radioo ABV/ABVD in second half of pregnancy for HL; CHOP for aggressive NHLo vinblastine until pregnancy is completed and then revert back to ABV/ABVD


2.32 - INFECTION & LYMPHOMA
1. How infectious agents contribute to lymphoma Direct infection & neoplastic transformation of lymphocytes
o EBV - germinal centre/post germinal centre B lymphomas and almost all extranodal NK/T cell lymphomas
o HHV8 - primary effusion lymphoma (with EBV as a cofactor) & LBCL arising from multicentric castleman disease
o HTLV1 - ATLLo HIV - primary CNS DLBCL, DLBCL and Burkitts
Immune suppression leading to increased risk of lymphomao HIVo EBV - causes >2/3 of B cell related post-transplant lymphoprolfierative disorders
(within 5 years of transplant)o Immunosuppressive medication, increasing age, SCID
Chronic (antigen-mediated) stimulation of the immune systemo i.e. the lymphoma cell is not infected itselfo H pylori (gMALT) and other bacterial infections; tend to be associated with low
grade, indolent, usually marginal zone lymphomas
2. Methods of detecting infectious agents in lymphoma Detection of EBV
o FISH for EBER1o Cytochemistry using anti- EBNA1, anti-EBNA2 or anti LPM1
Detection of HHV8o Cytochemistry using anti-LANA1 or anti-vIL6
Detection of HTLV1o Serology
3. Role of EBV in molecular pathogenesis of Burkitt & classical Hodgkin lymphomaClassical HL - EBV confers autonomous signalling pathways
EBV infects >90% of cHL patients, and these patients have a higher EBV Ab titre The rise in EBV Ab development precedes the lymphoma There is an increased risk of developing cHL following glandular fever There appears to be a need for CD4 cells for EBV to mediate disease - incidence increases as
you start treatment for HIV in co-infected patients Molecular roles of EBV
o EBV rescues B cells with crippling IgH mutations from apoptosis before they leave the germinal centre
o LMP1 induces B cell activation markers, adhesion molecules and upregulates anti-apoptosis genes
o LMP1 also activates JAK/STAT NfKb pathways so they become ligand independento LMP2A allows B cells to survive without signalling from Ig
Endemic Burkitt lymphoma - EBV generally cofners an escape from apoptosis Malaria infects RBCs and presents the molecule PfEMP1 on their CSMs. A region on this
molecule, called C1DR1a can bind to and activate B cells In Burkitt lymphoma each follicle expands roughly 10x The translocations in Burkitt lymphoma involve upregulation of myc
o Subsequent p53 mutations are seen in 1/3 patients - aberrant apoptosiso Mutant myc fails to upregulate BIM1, which is usually essential for apoptosis

o Upregulated PIM1 further counteracts p53 Molecular roles of EBV
o EBNA3A & -3C hypermethylate BIM1 = downregulation = less apoptosiso EBNA1 is also anti-apoptotico EBER1 promotes proliferation and secretion of the b cell growth factor IL10
4. Molecular roles of HHV8 in primary effusion lymphoma
LANA1 inhibits p53 and thus impairs apoptosis Viral cyclins resemble host cyclins so this leads to continuous proliferation FLICE protein blocks caspase activation and apoptosis mediated by Nfkb Viral IL6 stimulates B cells

2.33 - FOLLICULAR LYMPHOMA1. Introduction to lymphoma Lymphoma is a clonal expansion of lymphocytes, usually B lineage, into tissues of the body Lymphomas can be classified based on the characteristics of the B cell they arise from,
particularly the stage of maturity in reference to lymph node anatomy:o Naïve/pre germinal - CLL/SLLo Pre-germinal/manle zone - MCLo Germinal - Burkitt, Follicularo Post-germinal - MZL/ extranodal MALT lymphomaso Plasma cell myeloma
2. Clinical features of follicular lymphomaD - low grade germinal centre, non Hodgkin, lymphomaI - accouints for 22% of all lymphomas; second most common after DLBCLA - adultsS - F>M G - western world/industrialised countriesA - almost nothing is known; folate deficiency may be a risk factor
P - growth of LN follicles; lack of macrophages from follicles helps differentiate it from reactive hyperplasia; (which requires macrophages as it is an antigen driven process)
C - usually spreads slowly so that it is a disseminated disease at presentation - LNpathy, splenomegaly and BM invasion my occur. Can transform to a high grade DLBCL or Burkitts. When it relpases, the time intervals between relapses progressively decline.
3. Grading/ Staging of follicular lymphomaGrade Grade 1 & 2 are low grade. 3 a can also behave indolently Grade 3b behaves more like a high grade lymphoma
Stage Determined by examination, bloods, biopsies, CXR, CT and PET (radioactively labelled
glucose - picks up high metabolism tissues inc. heart, liver, bowel) 1 - single LN/ structure affected 2 - >2 structures on same side of diaphragm 3 - involvement of both sides of diaphragm 4 - invasion of non lymphoid tissues
o All - A/B depending on whether ‘B’ symptoms are present or not
4. Morphology of follicular lymphoma Nuclear cleft presence is indicative of germinal cell lymphomas Chromatin is evenly condensed - this helps differentiate it from CLL, where chromatin is patchy Trephine biopsy shows paratrabecular infiltration
5. Immunophenotype of follicular lymphoma Flow:
o CD10+ (germinal cell), CD19, 20,22 + (B), CD 45+ (WCC)o CD5 - (identifies it from CLL), CD3-, CD15,30- (these are HL markers), CD34, Tdt-
(lymphoma cells are mature) NB CLL will also display CD23

o One of kappa or lamda light chain Immunohisto:
o BCL2+ - this is key to diagnosis
6. Cytogenetics & molecular features of follicular lymphoma T (14;18)(q32;q21) is the hallmark - brings BCL2 under the influence of IgH@ enhancer
o There are 4 cluster regions of breakpoints on IgH - 2 major & 2 minor; each major is nearer the 3’ end of the gene locus
o BCL2 is juxtraposed against the J region of IgH. It normally prevents efflux of cytochrome C from the mitochondria and thus confers apoptosis
o Normally, unstimulated B cells die - this control is lost T(2;18)(p12;q21) kappa and t(18;22)(q21;q11) are also possibilities Hence follicular lymphoma could be an error of normal class switching
You can detect this with FISH & PCRo Use a breakapart probe for 14q32 and this will detect any of the 3 translocations
RQ-PCR is useful for MRD monitoring
7. Treatment of follicular lymphoma Most options are aimed at palliation. AlloSCT + local radio offers the only hope for cure but very
few patients can survive the treatment Involved field radiotherapy - stages 1 & 2 Chemo-immunotherapy is now considered the optimum treatment for symptomatic stages 3/4
o CVP + rituximab We still don’t know what to do for advanced stage, asymptomatic patients - the rationale for
delaying treatment until the patient experiences symptoms is to avoid drug resistance

2.34 - IMMUNOPHENOTYPING IN LYMPHOMA
1. The roles of immunophenotyping in lymphoma Distinguiosh lymphoma from reactive conditions Distinguish lymphoma from CLL/ALL Help make a diagnosis Helps to reveal specific aetiologies Helps to give prognostic information Can allow monitoring or MRD
2. Techniques of immunophenotyping Immunocytochemistry Immunohistochemistry Flow Cytometry
All of the methods can detect CSM antigens, cytoplasmic antigens and nuclear antigens. However, flow cytometry needs cells to be permealised to detect the latter two.
3. Distinguishing reactive lymphocytosis from lymphoma by immunophenotyping In reactive conditions, about 2/3 of lympohcytes display kappa whereas 1/3 display lamda In neoplasms, all cells express one or the other For t cells, uniform expression of CD4/CD8 points to lymphoma
o Also look out for abnormal expression on T-antigens for example a reduced expression of TCR a/b as compared to g/d [normally, it is the other way around]
4. Distinguishing lymphoma for ALL/CLL B-lymphoma
o ALL will usually express TdT, maybe CD34 and are usually SmIg and FMC7 negative T-lymphoma
o ALL will usually present TdT and maybe CD34o Thymic t cells make CD1o Early T cells have cytoplasmic CD3, whereas mature lymphomatous cells will express
surface CD3o Early t cells co-express CD4/CD8 before they are selected for
5. Panels for diagnosing lymphomas Diagnosing B cell lymphoma
o Exclude T cell lymphoma - CD2 negativeo Confirm B cell lymphoma - CD19, 22, 23, 79b kappa/lamda positive
Diagnosing T cell lymphomao CD3, CD7, CD4/8, CD25
6. Classic immunophenotypes lymphomaB-cell lymphomas CLL - CD5+, CD23+, CD19+ weak SmIg, 22, 79b FMC7
o [CLL score >4 is very likely; CD23 is not included in this scoring system] o ZAP70 and CD38 in CLL confer a worse prognosis
NHL – opposite to CLL - CD5-ve (except MCL), CD23-ve, SmIG+ve, CD22,79b, FMC7 +veo Mantle cell - cyclin D1+, CD5+, CD19+o Follicular CD10+ve

o Hairy cell - CD11c, CD103, CD25+22, CD123 [Most patients have atleast 3 of these four markers]
Myeloma - CD19-ve CD56+ve, CD138+, CD38+ve, cytIg, CD79a [normal plasma cells are CD19+ve, CD56-ve i.e. the opposite to myeloma cells]
T-cell lymphomas Large granular lymphocyte leukaemia - CD11b, CD16, CD56, CD57, TIA1
o NB this can be T or NK lineage ATLL - CD4+, CD25+ve, HLADR, CD38 and CD71 +ve -
o increased sideways scatter because of flower nucleus (more complex = more scatter) NK lymphoma - CD5-ve, CD79a-ve
7. Monitoring lymphoma MRD Needs multi-parameter flow cytometry
o This allows different cell populations to be identifiedo It uses more antibodies to detect subtle changes i.e. strength of antigen expression,
clonality/ emergence of a sub-clone/ aberrant antigen expression

2.35 - PATHOLOGY OF NON-HODGKIN LYMPHOMAS
1. WHO classification of lymphoma
B NHL T NHLLow Grade SLL (small lymphocytic lymphoma)
MCLFollicularMALT
Mycosis fungoides
High grade DLBCLBurkittsLymphoblastic lymphoma
AnaplasticATLLenteropathic
2. Clinical features of NHL (largely a repetition from previous Barbara bain lecture)D - a lymphoma without the presence of reed-sternberg cellsI -on the up due to increasing AIDS & more frequent SCTs (immunosupression)A - mean = 42S - M>FG
A - Immunodeficiency (AIDS, SCT)AI disease (RA, SLE)Chromosomal abnormalities
T14/18 = follicular T11/14 = mantle T8/14 = burkitts
Infections (EBV, HIV, HTLV1, H.Pylori)Chemicals (AAs, radiation)Inherited factors/ familial and racial trends
P -excised lymph nodes usually display diffuse whiteness and increased size
C - insidious (especially with low grade) lymphadenopathy with or without B symptoms- tumour bulk can impact local structures- lymphomas can invade BM (anaemia, neutropenia, thrombocytopenia)- you can get autoimmune disease with some lymphomas
Diagnosis - FNA cytology can be useful in those that have spread to blood, but otherwise you need LN excision biopsy under GA
P - depends on age, LDH, staging, extranodal involvement(s). some molecular and histological markers can also determine prognosis
3. Low grade lymphoma vs high grade lymphoma
LOW GRADE HIGH GRADEIndolent/ slow progression Aggressive/rapid progressionPresents late, disseminated Presents earlyDifficult to treat; impossible to cure May be curable with chemotherapy (40-50%)Occurs in adults Can occur at any age

4. B cell NHLCLL/SLL
Accounts for 5% of NHL; presents in elderly (60years) Indolent lymphoma that is usually disseminated at presentation, maybe with BM
involvement Chemo can not cure it - median survival is still 10years (indolent) 3% cases can transform to DLBCL Pathology
o Small round lymphocytes with little cytoplasmo Cant see follicles in LNso Lymphocytes may invade nearby adipose tissueo Immunophenotype - CD5+, CD23+
MCL Indolent lymphoma that presents in the elderly but disseminates early and thus only has
median survival of 3-4years Pathology - cells appear similar to CLL/SLL Immunophenotype - nuclear cyclin D1, CD5+, CD23- Cytogenetics - t11/14 = upregulated cyclin D1
Follicular Indolent lymphoma that presents in the elderly of developed countries Pathology - follicles expand and extened into LN medulla Immunophenotype - CD10+ve, Bcl2+vem CD10+ve Cytogenetics - most have T14/18 = dysregulated BCL2 = dysregualted apoptosis
MALT An extranodal marginal zone lymphoma associated with autoimmune disease (sjogrens in
salivary gland lymphoma) and infection (h pylori, campylobacter) One of the only haematological malignancies that is more common in women (AI Dx) Pathology - lymphatic destruction of glands Immuniphenotype is of no use in gastric MALT - you need an endoscopy
Lymphoplasmacytic lymphoma/ Waldenstroms Can progress to Waldenstroms macroglobinaemia if IgM expression is large enough:
o Hyperviscous blood, high ESR (small thrombosis can = visual problems, VTE) 50% have t9/14 which leads to PAX-mediated p53 dysregulation IgM (surface and cytoplasmic) is what you look for in immunophenotyping
DLBCL Aggressive disease that invades extranodal sites in 60% of cases De novo cases arise in follicles, transformations of MCL/SLL/follicular arise in other areas Pathology - cells have more cytoplasm, more nucleoli and more apoptotic bodies Immunophenotype and cytogenetics are varied because the disease is so heterogeneous
Burkitts

Endemic/ sporadic disease that responds well to chemo EBV immortalises cells and malaria stiomultes their proliferation in endemic forms. The
subsequent 8/14 trasnlocation leads to emergence of the clone Pathology - starry sky (macrophages) Immunophenotype - CD10+, mib1/ki67+ve
5. T cell NHLMycosis fungoides
Cutaneous disease with nodular infiltration of skin May be limited to skin for years before dissemination Pathology - ‘pouches’ that contain malignant cells underneath the surface of the skin
Anaplastic LCL Heterogenous group of nodal T lymphomas with a good prognosis Pathology - expansion of paracortex. Large cells Immunophenotype - CD30+ve, alk1+ve, EMA+ve Cytogenetics - t2/5 = NPM;Alk fusion product = altered growth receptor function
ATLL Aggressive disease secondary to HTLV1 infection, presenting with LNpathy, skin rashes,
organ dissemination and hypercalcaemia Virus activataes IL2 and CD25 via TAX as well as other genes involves in apoptosis (p16) CD25+ is the immunological hallmark
2.36 - FLOW CYTOMETRY (AGAIN!)

Advantages of Flow cytometry
Can identify and quantify a cell sample (qualitive and qwuantitaive) Multi-paramteric Can analyse a large number of cells (10e6)
Principles of Flow Uses flourochrome conjugated monoclonal antibodies against known cellular markers
o Usually CSM, but can be internal if cells are permalised A stream of cells is passed through a laser Cells sctter the light so that fluorochromes are excited to different energy levels; this change
in energy is emitted as a light photon when it returns to its original energy level, and this can be measured by a light detector
Detectors for different wavelengths of light match the light emitted to the antoibody it was matched to and hence the antigen presence
Forward light scatter is an indicator of how large a cell is Sideways light scatter is an indicator of how granular the cell is/ how complex the nucleus is
Applications of Flow Diagnosis of leukaemia, lymphoma, PNH, immunodeficiency, platelet disorders Monitoring MRD Measurement of absolute cell numbers (CD4 in HIV, CD34 after SCTs)
Important markers
Important Diagnostic Hallmarks APL - CD34-; HLADR -ve
B-ALL - CD34+, CD10+, B markers + HCL - Cd11c +ve, CD103+ve SLVL - CD103-ve, CD123-ve MCL - cyclin D1 Myeloma - CD19-ve, CD56+ve, CD138 +ve, CD38+ve PNH- CD59 is lost (as with other GPi anchored CD markers)
2.37 - MOLECULAR GENETICS OF LYMPHOMAS
1. General features of lymphoma (think this covers both HL and NHL) EPIDEMIOLOGY :

o Disease of men, the elderly, can be predisposed to by infections, genetics, enivormental mutagens, autoimmune diseases
SYMPTOMS:o 1st symptoms may, but not always, emerge at the lymph nodes involvedo Indolent lymphomas may present with b symptoms without overt/evident LN size
change DIAGNOSIS:
o LN biopsy, bloods, BM aspirate (to exclude metastases), XR, CT, PETo Staging depends on invovlement either side of the diaphragm and presence/absence
of B symptoms CLASSIFICATION:
o Hl/NHL depends of the presence of reed Sternberg cellso Diffuse NHLs are associated with complete loss of LN architecture, whereas follicular
ones concur follicular expansion into other, still defined, areaso HLs are generally easier to treat
2. Ig rearrangement in lymphomas 85-90% of translocations underlying B lymphoma involve Ig genes:
o 14q32 (IgH@)o 2p11 (Igk)o 22q11 (Iglamda)
Ig genes are very active transcriptionally in B cells because they are key to function, hence genes brought under their influence following translocation become constitutively expressed
o The partner gene doesn’t have to be downstream either, because a promoter region on IgH@ (switch mew?) can transcribe in both directions
Ig genes give rise to lymphoma because of rearrangement AND somatic mutations Translocations lead to one of two outcomes:
o Transcriptional dysregulation - 8/14 burkitt = cmyc activation = proliferation, growth 14/18 follicular = bcl2 activation = anti-apoptosis
Other cases involve BCL6 - this is implicated in lymphoma after it is subjected to a number of somatic mutations
11/18 MCL = cyclin d1 activation = unchecked proliferationo Fusion gene production
T cell lymphomas involve TCR loci translocations HENCE most are diagnosed with FISH, PCR, karyoptying. New techniqwues include microarrays
and array competitce genomic hydribisation (CGH)
3. Other mutations in lymphomas BCL10 - MALT (under influence of 14q32) MALT1 - MALT (under influence of 14q32) - believed to be due to nfkb activation PAX5 - waldenstroms (under influence of 14q32) Point/somatic mutations in c-myc and bcl6 C-myb translocations; ink mutations Tp53 deletions after 17q- or monosomy 17; often an acquired event in lymphoma; bad
prognosis Epigenetic changes - methylation = gene inactivation
2.38 - THERAPY OF NHL
1. Prognostic factors in NHL; the Internation Prognostic IndexCure rate varies from 25-75% depending on these factors:

Age >60 LDH>normal Performance status 2-4 Disease stage 3/4 more than one extranodal site
IPI score 5 year survival0-1 73%2 51%3 43%4-5 26%
2. Therapeutic options in NHL
Indolent b lymphomas usually have small cells. o The most common is follicular lymphoma. o Median survival is about 8 years. o 80-85% present with stage ¾ disease. o Watchful waiting is best until they present with progressive disease/transformation,
watch for B symptoms or BM failure symptoms o Give them CHOP (cyclophos, doxo, vincristine, pred)o Those who relapse may need high dose chemo (LACE; lomustine, cytarabine, cyclo,
etop) + autoSCTo Randomised data is needed to assess the efficancy of rituximab
Aggressive lymphomas usually have large cellso the most common is DLBCLo CHOP +R is first line
2.39 - MANAGEMENT OF MDS (includes overview of whole disease because lecturer was good)
1. Clinical features of MDS

D - heterogenous group of clonal disorders characterised by progressive cytopenia and qualitative abnormalities in myeloid cells, leading to ineffective haematopoiesis. It is now recognised as a cancer. 50% die because of cytopenia, 30% progress to AML.I - 5/100k in UKA - median = 67S - M>FGA - unknownP - Bone Marrow features:
hypercellular bone marrow and peripheral (pan)cytopenia abnormal microenvironment due to ineffective erythropoiesis, increased TNFa
production = apoptosis and an angiogenic response, CD4 lymphopenia and emergence of auto-reactive T cells
Cellular features:o nuclear:cytoplasmic size asynchronyo disorganised chromatin and cytoplasmo loss of lobules in neutrophils and agranular cytoplasms
C - anaemia may be the only presenting symptom
2. Prognosis of MDS + the IPSS0 0.5 1.0 1.5 2.0
Blast % <5 5-10 11-20 >20 [AML]Karyotype good intermediate PoorCytopenia 0-1 2-3
Karyotypes: Good - normal, del5q, del 20q Intermediate - anything that isn’t good or bad Bad - complex or c7 abnoramlities
Cytopenia; 1 point each for: Hb<10 Neutrophils <1.5e9 Plt <100e9
Prognostic risk Score Prevalence Median survival Years AMLLow 0 31% 5.7y 9.4Int-1 0.5-1 39% 3.5y 3.3Int-2 1.5-2 22% 1.2y 1.1High >2.5 8% 0.4y 0.2
3. WHO Classification of MDS RA RARS - with only erythroid dysplasia
o RA & RARS are the most indolent forms of MDS and account for the majority of paitents; very few MDS [patients present with worst prognostic subtypes
o Mean survival is about 4-5 years RCMD - refractory cytopenia with multilineage dysplasia RCMD + ring sideroblasts

RAEB 1 (5-9% blasts) & RAEB 2 (10-19% blasts) MDS-U (unclassified) 5q syndrome
5q syndrome Interstitial deletion between q31 and q33 = macrocytic anaemia (MCV usually normal is other cases of MDS) = thrombocytosis + increase in mononuclear megakaryocytes
o Those with high Plt counts may have the JAK2 V617F mutation, which is also seen in other myeloproliferative diseases
Presents is women, splenomegaly may be present Accounts for only 15% of patients with MDS
4. The use of EPO+GCSF in MDS Supportive care is employed in many patients because MDS is a disease of the elderly and its
heterogenous nature makes it difficult to cure 80% of patients MDS patients are anaemia, 40% are trabnsufsion dependent Endogenous EPO varies greatly within patients and does not correlate to disease sub-type of
transfusion dependenceo Other factors such as ring sideroblasts, transfusion dependence, cytogenetics, seem to
predict response EPO+GCSF seems to only prvoke a response in those who are less transfusion dependent
o RA, RARS and RAEB1o The response wears off after 2 years though, so it isn’t completely effectiveo Additionally, GCSF may contribute to progression to AML as a SFXo Current recommendations are giving a 6 week trial of EPO+GCSF in Ra/RAEB patients
with low transfusion reqyuirement and a basal EPO of <200
5. The use of targeted therapy in MDS Farnesyl transferase inhibitors were designed to inhibit ras but only gave responses in 29% of
patients Thalidomide achieves responses in 51% of patients (but you need high doses)
o Anti-angiogenesis (anti-VEGF) - angiogenesis is important in tumour growth, vascularity and metastasis
o TNFa synthesis inhibitiono Immunomodulatoryo Because of high dose requirement - SFX - somnolence, peripheral neuropathy, VTE
Lenalidomide is really good for 5q- - removes transfusion dependence in 2/3 of patientso Anti-inflammtory, enchance t/nk cells, anti-angiogenic - pretty much returns bone
marrow microenvironment to normal
6. The use of non-intensive chemo in MDS The idea behind immunosuppression is getting rid of the clonal t cells and letting the others grow
o Antithymocyte globulin (ATG) is effective in hypoplastic MDS patientso Cyclosporin blocks IL2 production but is of limited benefit outside hypoplastic RA
patientso Cytarabine & 5-aza-cytidine hypomethylate DNA (i.e. p15, which is hypermethylated in
many high risk MDS cases) and gives a 49% response rate
7. The use of intensive chemo in MDS

There is a reluctance to treat people with high dose chemo + SCT until they actually develop AML because these patients are already profoundly cytopenic and most cannot tolerate >2 chemo cycles
FLAG/Ida is what is used now for high risk MDS because it has minimal toxicity and gives rapid haemoatopoietic recovery
o Younger patients and those with favourable karyotypes have more favourable responses High dose chemo +AlloSCT is the only cure but most patients are elderly so you can’t give the full
whack chemo dose because transplant related mortality increases with ageo Less ability to deal with toxicityo Increased incidence and severity of GVHDo Increased non-malignant causes of death
Predictors for outcome in AlloSCTo Older Age is poorer outcomeo Morphology (RAEB onwards is poorer outcome; 3year survival 30%)o High risk cytogenetics is poorer outcomeo Prolonged disease duration, marrow fibrosis, t-MDS are all poor outcome
There is a huge difference in survival amongst MDS patients who have a sibling and a matched-stranger AlloSCT
Hence, patients who are young enough and have a sibling donor should go for high dose chemo + alloSCT early in the disease course
2.40 - IMMUNE COMPLICATIONS IN CLL
1. Incidence of infections in CLL 50% patients have recurrent infections
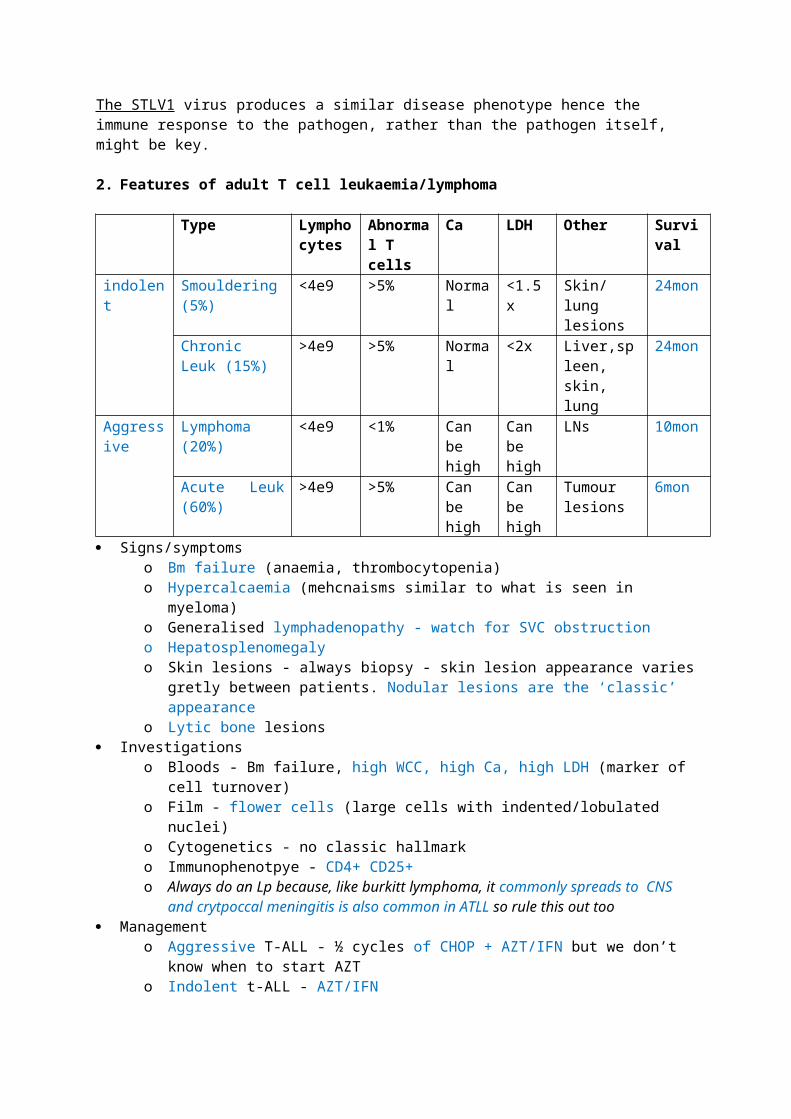
Incidence of infections increases with:o Ageo Disease progression – hypogammaglobulinaemia, AI diseaseo Repeated therapies – neutropenia, AI disease??
Prevent infections with:o IVIG - useful for peventing bacterial infectionso Fluconazole for candida (but this increases incidence of non-albicans candida
infections)o septrin/ co-trimoxazole to prevent PCPo GCSF may improve neutropeniao Immunizations against strep pneumo, haemophilus, influenza
2. Hypogammaglobulinaemia (low IgG?) & impaired immunity in CLL Hypogammaglobulinaemia may be due to defective cell mediated immunity due to
abnormally functioning T cells and thus dysregulated B cell functiono Reduced Cd4:Cd8 ratio is seeno Leads to increased risk of encapsulated organism infection
Skin shows decreased tuberculin response VZV- shingles
o Related to disease progression Complement system is also defective in CLL - reduced opsoniosation of bacteria by C3b
3. Neutropenia in CLL Neutropenia = bacterial & fungal infections
o Strep Pneumonia is the most common and severeo Staph Aureus, Haemophilus influenza, legionella and slamonella also commono Fungal - PCP, candida, aspergillus
Mostly arises because of therapy, not diseaseo Steroids = defective chemotaxis + lymphopenia
PCP, candidao Fludarabine (purine analogue) = lymphopenia + myelosuppression
Strep pneumo, CMV/HSV/VZV, PCP, mycobacterium fortuitumo Anti-CD52 (alemtuzumab)
Bacterial, CMV, PCP infections CMV reactivation is at highest risk when CD4<50
4. Tumours in CLL Skin cancer Kaposi Sarcoma (HHV8)
5. AI complications in CLL AIHA
o Incidence = 4-40%o Due to polyclonal IgG autoantibodies production against RBCs secondary to immune
dysregulation (not Ab production by the malignant cells)o = anaemia, jaundice, splenomegaly, reticulocytes, +ve coombs testo Treat with steroids, rituximab, may need transfusion
Auto-Immune thrombocytopenic purpurao Incidence 1-2%

o IgG antibodies against platelets + premature destruction by the spleeno Treat with steroids, IVIg
Pure red cell aplasiao Incidence <1%, occurs early in diseaseo = anaemia, erythroid hypoplasia in BMo Treat with steroid and ciclosporin
Risk factors for AI disease:o Disease stage, age, high WCCo Maleo CD38, ZAP70, porr risk cytogenetics, beta2m
2.41 - HODGKIN LYMPHOMA
1. Clinical features of Hodgkin lymphoma

D - any lymphoma with the presence of Reed-sternberg and mononuclear hodkin cells on morphology. Rarer than NHL and more likely to present with localised disease.I - rare 4-5/100kA - 2 peaks; late adolescence and >60S/G/A/P
C - (cervical) lymphadenopathy is usually the presenting feature. Hepatosplenomegaly may occur in advanced disease. B symptoms (fever may be episodic) are also present
Investigations- Bloods; High WCC, anaemia, high ESR & LDH- LN aspirate/ biopsy is diagnostic; perhaps with CT guiding for central nodes
2. Classification of Hodgkin lymphoma 2 broad categories
o Nodular, lymphocyte dominant HL - behaves more like a NHLo Classical HL
Nodular sclerosis Mixed cellularity Lymphocyte rich Lymphocyte depleted
Nodular ClassicalIncidence 5% 95%Age Anyone Normal 2 peaksGender 70%male equalaetiology Possibly EBVAffected organs Peripheral LNs Mediastinal/cervical LNsAffected cell Germinal B HeterogeneousRelapse Can DLBCL rareImmunophenotype CD20+, 45+, 79a+, 15-, 30-, 138-
BCL6+, SmIg weakBSAP+, Oct2 +, BOB1+
CD20-, 45-, 79a-, SmIg-, 15+, 60+,138+, IRF4/MUM1 +BSAP+, Oct2-, BOB1-
Cytogenetics 3q27 translocations (BCL6) REL/ FAS mutations
3. Aetiology of HL Familal factors (HLA polymorphisms implicated in classical HL) EBV and HIV shows strong links in classical HL patients
o EBV is present in reed-sternberg cells in 40-50% of patientso HL incidence increases 4x after glandular fevero HL incidence rises are CD4 count rises (i.e. after you start treating someone for HIV)
ALPS (very rare) can predispose to nodular LDHL
4. Mechanisms of HL Constitutive activation of Nfkb = proliferation and survival of HL B cells
o Defective Ikb may be involved in HL cases not secondary to infection In EBV related HL, LMP1 mimics Cd40 = nfkb activation and LMP2 confers proliferation and
anti-apoptosis Impaired cell mediated immunity in HIV/old age means that reduced immunosurveillance is
another problem in controlling proliferating b cells

5. Staging of HL Staging is based on
o Examinationo CXR
mediastinal widening, hilar LNpathyo CTo PET (more useful for monitoring disease after follow up)
Staging is 1,2,3,4 depending on structures involved either side of diaphragm (+A/B)
6. Treatment of HL Combo involved field radio and chemo
(ABVD/BEACOPP) Use PET to monitor disease after 2 cycles ?Anti CD20 in nodular HL ?Anti CD30 in classical HL Treatment SFX = toxicity, malignancies, CVD
7. Prognosis of HL Median survival untreated in less than a year 5 year survival of early stage HL is now >90% Prognostic factors are:
a. Disease stageb. Agec. Albumind. Lymphocyte counte. LDHf. ESR
2.42 - MYELOMA BONE DISEASE
1. Bone disease in myeloma
Is the most frequent presentation of MM; 70% of caseso 60% present with osteolytic lesionso 10% present with diffuse osteopeniao Very rarely patients present with increased bone formation; POEMS syndrome
The consequences of increased bone resorption in MM are:o Pathological fractures (37%)o Osteoporosiso Hypercalcemia (9%)o Bone paino Spinal cord compression (3%)
2. Recap of normal bone-cell functions Osteoblasts are derived from mesencymal stem cells. Osteoclasts come from the
myeloid precursor Osteoblasts produce osteoid matrix, the main components of which are type 1 collagen
and hydroxyapatite.o The matrix is calcified extracellularlyo Osteoblasts are stimulated by growth factors eg IGF, EGF, BMP/TGFb, GPCRo The GF Wnt requires LRP5/6 as a cofactor to activate transcription. Sclerostin, a
molecule produced by osteoclasts, prevents this interaction and thus inhibits osteoblast growth via this pathway.
Osteoclasts resorp boneo RANKl is a key stimulator of osteoclasts. It is made by osteoblastso OPG is a decoy RANKl receptor
Humoral Local+ve osteoclasts PTH, T4, VitD IL6, RANKl, M-CSF, PDGF, IGFs-ve osteoclasts Cortisol, T cell [IL4,18,IFNg] OPG
3. Pathogenesis of bone disease in MM Myeloma cells stimulate osteoclasts directly via MIP1a, IL3, HGF Myeloma cells stimulate bone marrow stromal cells via adhesion
o Stromal cells produce sclerostin, dickkopf1, activin A and other molecules that inhibit osteoblasts
o Stromal cells display RANKl to stimulate osteoclasts Osteoclasts produce IL6 which stimulates plasma cells - vicious circle The RANKl/OPG ratio is usually increased in MM- it may have prognostic value Serum MIP1a, dickkopf1, activin A is also increased and may have prognostic value
4. Treatment of myeloma bone disease Prevent the first skeletal-related event/ prevent recurrence after a first event
o Bisphosphonates - induce osteoclast apoptosis and inhibit migration into boneso The mechanism is by inhibition of FFP synthase o Zoledronic acid is the best one but needs to be given IV. Clodronate is second
choice.

o Side effects: Oral therapy - GI intolerance; ulcers in upto 1/3 patients IV - flu/fevers are common, renal disease and jaw osteonecrosis are rare
o Current guidelines are tio give them for two years for all symptomatic px, have regular dental monitoring and then after 2 years its at the prescribing doctors discretion
Palliate and control bone paino Analgesiao Palliative radio
5. Other drugs in MM Proteasome inhibitors
o Proteasomes degrade Ikb = free Nfkb = transcription o The end point of the RANKl pathway of osteoclast activation is Nfkbo Bortezomib bind to the chymotryptic site in the core of the proteasome to inhibit
ito Bortezomib increases bone marrow density
Monoclonal Antibodieso Denosumab (anti-RANKl) leads to osteoclast apoptosis and prevents skeletal-
related-events but increases risk of death compared to zoledronic acido Anti-Dkk1 antibodies lead to bone formation in mice
Novel anabolic agents eg BHq880, sotatercept
2.43 - MYELODYSPLASTIC SYNDROMES

1. Recap of clinical features of MDSD - heterogeneous haematological disorders characterised by ineffective clonal haematopoiesis, peripheral cytopenias and hypercellular BM.IA - 70-75yearsSGA - previous chemo/radio are risk factorsPC - cytopenias = anaemia, thrombocytopenia, neutropenia
2. WHO Classification of MDS
Name Feature %of patients Refractory cytopenia with unilineage dysplasia
One of anaemia/neutropenia/ thrombocytopenia
10 (usually anaemia)
Refractory anaemia with ring sideroblasts >15% ring sideroblasts 55q- syndrome Hypolobulated megaKcytes 5Refractory cytopenia with multilineage dysplasia
Pelger-huet anomaly 20
Refractory anaemia with excess blasts 1 5-9% blasts 20Refractory anaemia with excess blasts 2 10-19% blasts 20unclassifiable 10
Survival worsens and potential to transform into leukaemia increases as you move down the table
Has features of the IPSS which were covered in a previous lecture [2.39]
3. Pathogenesis of MDS BM features:
o Hypercellularo Dysplastic/megaloblastic erythropoiesis (RA)
Ringed sideroblasts - formed when iron accumulates in mitochondria of developing erythroblasts. Identify with Prussian blue stain
o Dysgranulopoiesis; hyporgranular neutrophilso Micromegakaryocyteso Increased blast number in RAEB
Pathogenesiso Reduced proliferation and differentiation of erythroid progenitorso Excess presence of apoptosed progenitors
High caspase 1/3 activity Escape from apoptosis and transformation to AML may be due to bcl2
expression seen in RAEBo VEGF is increased in MDSo Immune dysfunction in MDS:

Autoantibody production, monoclonal gammopathy, hypo- or hyper-gammaglobulinaemia
Presence of antinuclear antibodies, rheumatoid factor, anti-DNA antibodies, positive Coombs test
Increased frequency of auto-immune disorders (~10%-15% of MDS patients): vasculitis, arthritis, pleuritis, pericarditis, myositis
Decreased NK-cell activity, decreased antibody-dependent killing and diminished CD4 numbers
T-cells inhibit MDS CFU-E ; CD8+ cells inhibit CFU-GM
4. Treatment of MDS





























