Embed Size (px)
Citation preview

Visualization of electronic density
Bastien Grossoa,b, Valentino R. Cooperc, Polina Pined, Adham Hashibone,Yuval Yaisha, Joan Adlera,∗
aTechnion-IIT, Haifa, IsraelbEPFL, Lausanne, Switzerland
cOakridge National Laboratory, TN, USAdLoyola University, IL, USA
eFraunhofer IWM, Freiburg, Germany
Abstract
The spatial volume occupied by an atom depends on its electronic density.
Although this density can only be evaluated exactly for hydrogen-like atoms,
there are many excellent algorithms and packages to calculate it numerically
for other materials. Three-dimensional visualization of charge density is chal-
lenging, especially when several levels are intertwined in space. In this paper,
we explore several approaches to this, including the extension of an analglyphic
stereo visualization application based on the AViz package for hydrogen atoms
and simple molecules to larger structures such as nanotubes. We will describe
motivations and potential applications of these tools for answering interesting
physical questions about nanotube properties.
Keywords: visualization, electronic charge density, nanotube
2010 MSC: 00-01, 99-00
THIS is a DRAFT for internal circulation to authors and their supervisors!!
Some technical parts have been moved to appendices.
∗Corresponding authorEmail address: [email protected] (Joan Adler)URL: http://phycomp.technion.ac.il (Joan Adler)
Preprint submitted to Journal of LATEX Templates November 4, 2014

1. Introduction and educational applications
The Computational Physics group at the Technion developed a desktop vi-
sualization code for their needs in Atomistic Vizualization, called AViz, [1, 2, 3].5
It is based on Mesa/OpenGL and Qt. Initially we modelled atoms as balls, spins
as cones or vectors and quadrupolar molecules orliquid crystals or pores as cylin-
ders. In a project motivated by educational use we invoked an “off-label” AViz
implementation to illustrate the electric probability density as calulated from
the H atom analytic solution in a smoke rendering form [4], using dots to enable10
semi-transparency. The dot representation of AViz, originally created to enable
quick selection of viewing angle etc for atomistic samples, creates a translucent
effect whereby the sample’s interiors are visible. Combined with color and rota-
tion it gives excellent insight into the nature of the different electronic states[5].
In order to draw the electronic density we (obviously) first need to calculate15
it. In brief, for the H case one calculates the electronic density on a grid, and
defines a box around each grid point. Dots are then drawn at randomly chosen
points within each box at an average density equal to the local electronic density
at the center of the box. Each of these points is given x, y and z coordinates and
is drawn using the dot feature of AViz. The .xyz format is common to many20
molecular visualization packages, but its normally used to indicate atoms, not
density points. For the hydrogen 2s case the datafiles contain some 50,000
points, rather larger than those typically used in atomic visualization, although
since they are not solid spheres, the rendering time is reasonable. In the left
frames of Figure 1 we show the AViz visualization of the electronic density of25
the 2s state of the H atom in both greyscale and color.
Three dimensional visualizations of hydrogen atom wavefunctions are very
helpful for teaching Modern Physics or Quantum Mechanics classes. The con-
cept of electronic density is hard to grasp. Animated gifs of these samples in
rotation are found at [5] using binned color and have been found to be helpful30
to students [6].
2

Figure 1: AViz dot visualizations of electronic density of the 2s state of an H atom in greyscale,
and in color, and an ethylene molecule in color. The atoms in the ethylene are indicated by
small squares, and the color scale of the desity ranges from orange (most dense) through red
and pink to blue (least dense) in all cases.
2. Molecules and solids, especially nanostructures
A natural extension of electronic density visualization for single atoms is to
molecules and simple nanostructures. In this area, experiment has advanced
more quickly than simulation. We note that there are many quite standard35
implementations of smoke density approaches to surface electronic density em-
ulating STM images, but most do not use color as well as concentration to
indicate the density of their “smoke”. Nor do they generally publish 3D im-
ages which can be rotated and sliced as ours can to peer inside the sample.
In less transparent visualization some unique aspects of nanostructures may be40
overlooked.
Our extension of AViz applications aimed to visualize the electronic density
resulting from simulations of larger molecules and solids in the same way. The
first studies [7] concentrated on simple molecules, where there is, of course,
no analytic solution. We used GAMESS [8] to carry out a Density Functional45
theory (DFT) approach with Slater type orbitals (STO-3G) but the rest of
the procedure is similar. In the right frame of Figure 1 we show the colored
electronic density of ethylene. Note that because the density is not shown with
solid curves we can peer nicely into the sample. We have also been able to show
methane molecules both with all orbitals and stripped of the lower densities,50
3

and have also explored specific orbitals [7, 9, 10]
Our next attempt at electronic density visualization was to periodically
bounded samples, employed DFT calculations as implemented in the Vienna
Ab Initio Simulation package VASP[11, 12]. In preparation for the larger sam-
ples, we returned to some of the simple molecules with the VASP code. At that55
time we used slice visualization with VESTA [13], since dot visualization for
many atom samples was limited by the very large datafile size issues. Despite
the VESTA solid visualization rather than the 3D dot-smoke type, we confirmed
that the main features agree. Note that VESTA images also include green in
the color range, the early AViz ones only used a red-blue scale for better depth60
perception.
3. Stereo, binned color smoke rendering
The next stage in our visualization development was to move to 3D stereo.
We selected an approach that has a long history, even predating GL. This old
concept of anaglyphic stereo relies on two images, slightly displaced, and viewed65
on a regular screen/projector or poster [14] through colored glasses, or two
squares of cellophane. Stereo Vision (SV) works by showing a different image
to each eye, thus creating the illusion of a 3D image.
AViz 6.1 [15, 16, 17, 18] has incorporated the possibility of SV, and although
more than two colors are possible there remains some color washout, depending70
on color selection. The SV images generated by AViz, such as those in this paper,
are best viewed using red-cyan anaglyphic glasses. The images in Figures 2 show
nanotube atoms at two different viewing angles in stereo.
In Figure 3 we show (on the left) the electron density of a hydrogen atom in
stereo. Improved colors for stereo for the H atom, as well as clearer instructions75
were given by Meital Kreif in [20]. Two examples are given in Figure 3 (center)
and 3 (right), the former of the n = 3, l = 1, m = 1 orbital and the latter of
the n = 4, l = 3, m = 2 orbital of the H atom. On the website all images can
be rotated to further aid in depth perception.
4

Figure 2: Left: Nanotube side-view. Right: View along the axis of a nanotube.
Figure 3: Left: Orbital electronic density of the (3,1,0) state of the hydrogen atom. Center:
Image of the n = 3, l = 1, m = 1 orbital. Right: Image of the the n = 4, l = 3, m = 2 orbital.
4. Motivation and preliminary studies of electron density of nan-80
otubes
A nanotube vibrates at a frequency that is a function of its width, length,
tension, boundary conditions and for certain boundary conditions also of its
type. A molecule placed on such a vibrating tube will change this frequency,
enabling elucidation of the mass of the adsorbed molecule. The description of85
these systems with analytic models is limited in cases when both ends are not
completely clamped, as occurs in the laboratory. The essential parameter for
model analysis is the width of the nanotube wall, and it is the electronic cloud
5

around the atomic nuclei that determines this.
In a series of papers and a thesis Pine and coworkers [21, 22, 23, 24, 25,90
26, 27] reviewed the literature and carried out extensive classical molecular
dynamics simulations at the atomistic scale to carefully determine the limits
of applicability of the analytic theory. While values for the wall width were
deduced indirectly by us and many others, direct estimation is of course more
desirable. It would be even more desirable to automate this. We are interested95
in the effect of nanotube local distortions (bending, stretching) on the width,
and also in the effect of total strain.
Of course any study to estimate the width directly has to be quantum me-
chanical. Given the limitations of computer resources, and the need for relatively
long tubes, simulated for long times, with a range of parameters it is natural100
that such width estimates should be deduced with a multiscale approach. Sim-
ulations in tandem at an atomistic scale and at an electronic scale are desirable.
The question of multiscale simulations in an efficient manner that minimizes
the difficulty of using diverse codes for different scales, inputs and outputs is
currently an important research issue. For example, in the European Union FP7105
program several collaborations under the banner of “NMP (Nanosciences, Nan-
otechnologies, materials and new production technologies) multiscale modelling”
are researching this issue. In particular the project “Simulation framework for
multiscale phenomena in nano and microscaled systems, or SimPhoNy” [28]
aims towards a uniform environment for scales from electronic to macroscopic,110
with visualization at all scales. The present study, in addition to its intrinsic in-
terest provides a prototype test bed for SimPhoNy. Together with the atomistic
scale and analytic continuum models developed previously, the present study ex-
plores the electronic scale and its visualization development and transfer from
atomistic scale. There is the caveat that the wrapper codes described below are115
C-based and will have to be moved to python for the SimPhoNy environment.
6

4.1. Molecular dynamics as the starting point
The nanotube simulations at the atomistic scale that form the basis for the
present study are described in [22, 23, 24, 25, 26] with codes given in [27].
In brief the tube is equilibrated with periodic boundary conditions, then “cut120
open” and clamped with the appropriate boundary conditions. When strain is
needed the tube is carefully stretched before clamping. It is then allowed to
vibrate for a long time and the vibration spectrum analysed with MATLAB
codes given in [27].
We used the Brenner [30] potential, and for the present study we selected125
four nanotubes with different strains from [26] and used rings near the center
of the tubes so as to minimize boundary effects. We show one of the nanotubes
with 10 percent strain from [26] in Figure 4. Note the slight extension at the
ends to which we will return below. Thoughout this study we refer to the axis
along the tube as the y direction and the two perpendicular dirctions as x and130
z.
Figure 4: Stretched nanotube, the y axis is along the axial direction and xand y directions
are perpendicular; observe extension near the ends.
4.2. Atomistic visualization from molecular dynamics output
For the nanotube simulations we carried out still and animated visualiza-
tion with AViz, creating .xyz files in the simulations and drawing them post-
processing. In addition to valuable help in the debugging phase, we could show,135
for example regions where bonds become streched (or compressed), with an
example in Figure 5. We note that code files that enable direct processing of
LAMMPS output into AViz input have also been prepared at the Technion [31].
7

However for the present project potential problems of explosion of Brenner po-
tential samples in LAMMPS meant that we continued to work with our own140
older Molecular Dynamics code. The group has also prepared wrappers for out-
putting Monte Carlo simulations into AViz with both simulations and wrappers
in python [32].
4.3. Earlier visualization from electronic density simulations
In addition to our VASP-VESTA studies mentioned above in which we did145
not succeed to create smoke rendering input, we also carried out exploratory
simulations [34] with Quantum Espresso (QE) [33] DFT code and FORTRAN
wrappers for transfer to AViz, which led to the development of the present more
substantial study.
Figure 5: Vibrating nanotube; upper image shows all nearest neighbour bonds, lower image
shows only shorter bonds to emphasise compression regions.
5. General aspects of our calculations and visualizations150
For this calculation we selected the Quantum Espresso DFT code, [33]. The
selection was based partly on a preference for public domain codes with clear
documentation of their format for the charge density and partly on the possi-
bility of more local support.
We illustrate the complete protocol in Figure 6. From the initial molecular155
dynamics simulation we select one or several rings and place their coordinates in
an .xyz file. We confirm their validity with an atomistic AViz vizualization, for
8
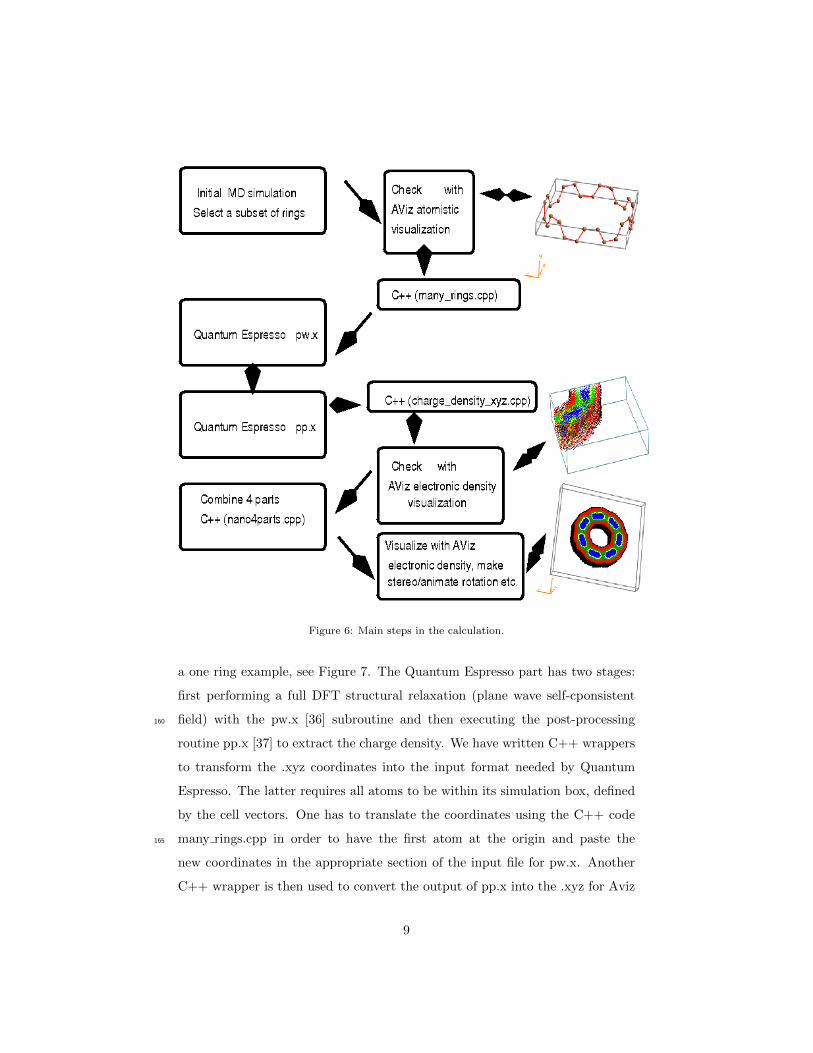
Figure 6: Main steps in the calculation.
a one ring example, see Figure 7. The Quantum Espresso part has two stages:
first performing a full DFT structural relaxation (plane wave self-cponsistent
field) with the pw.x [36] subroutine and then executing the post-processing160
routine pp.x [37] to extract the charge density. We have written C++ wrappers
to transform the .xyz coordinates into the input format needed by Quantum
Espresso. The latter requires all atoms to be within its simulation box, defined
by the cell vectors. One has to translate the coordinates using the C++ code
many rings.cpp in order to have the first atom at the origin and paste the165
new coordinates in the appropriate section of the input file for pw.x. Another
C++ wrapper is then used to convert the output of pp.x into the .xyz for Aviz
9

electronic density visualization. AViz requires an input file in the .xyz format
with two initial rows, the first being the integer number of atoms or dots and
the second a comment line. All other rows have a letter to indicate atomic type170
or dot color and at least three real number spatial coordinates.
All files that we use can be found in a single tar file on the website [35] in
QE charge density/ddl charge density.tar. In addition the input and additional
wrapper routines, currently written in C++ perform the tasks listed below. As
well as being part of the total download file they are also provided as separate175
tar files in the directory QE charge density with the name of the file in brackets
in this list:
• charge density xyz.cpp [35] extracts the charge from pp.x in the correct
input format for AViz (in ddl example input QE.tar)
• many rings.cpp provides the input coordinates for pw.x (in ddl many rings.tar)180
• nano4parts.cpp recombines the 4 quarters into a single ring (in ddl charge density.tar)
(In the current setup QE initially calculates the density in four quadrants
which have to be recombined prior to visualization).
It is also recommended to dilute the points prior to visualization and this is
currently done in this implementation as part of nano4parts.cpp. (This step185
was not needed in the H atom and simple molecule cases because the approach
used to generate points led naturally to a far more dilute concentration.)
We first describe the calculation for a single ring without strain. We then
describe the additional stages needed for systems of several rings and for systems
under strain. The details of the simulations are given in Appendix A, and details190
of the transformations on the output to create AViz input files in Appendix B
and recombination of the 4 parts in Appendix C.
6. Visualization of the charge density in 3D
Once all the steps described above are carried out, we reach the most in-
teresting aspect of this procedure - the visualization of the charge density in195
10

Figure 7: The one ring system with its long axis, in the y direction pointing thru its center.
3D. We make an initial coarse graining of the density in order to use the binned
color approach using dots. The actual color selection is done interactively within
AViz, and can be adapted to the user’s preference, optimal selection if there is
to be greyscale printing or the user’s requirements in the case of a colorblind
person. It would also be different if analglyphic stereo is to be implemented,200
because due to the analyglyphic “washout” some color palettes are better than
others. At this point we have not varied the number of dots in accordance with
their local density as was done for the analytic solutions. The color binning
is done at the same time as the extraction from the QE output format and
all details are in Appendix B. It helps to determine which region of space has205
a higher probability of electron localization. In Figure 8 we see a straight on
view, with colors respectively indicating successively lower densities. The lowest
density is not colored for viewing ease, and with colors black, red, green, yellow
and blue we show successively higher densities. These very bright colors were
selected for their distinctiveness; better palettes for 3D viewing are displayed210
below. This image is not diluted, the diluted version (details in Appendices) is
given in Figure 9.
We observe that there are bad moire effects here. An improved palette for
non-stereo viewing and better angular selection is given in two examples of the
11

Figure 8: Final result
extended systems, a 3 ring system in Figure 10 and for a 6 ring one in Figure215
11.
7. Stereo visualization of electronic density around a nanotube
Further insight into the structure can be found by invoking several advanced
AViz features. The fovy (field of view in the y direction) can be tweaked [18] in
the panel from the viewpoint button on the AViz main panel (Figure 12) via the220
explicit option (Figure 13) so that the moire effects of a straight-on cartesian
12

Figure 9: Final result with dilution
view are minimised.
The analglyphic stereo, as presented in [16] and adjusted by the buttons on
the left with the glasses images (Figure 14 gives an image (Figure 15 which when
viewed with cyan-red glasses appears to come out of the page. In this figure225
one can see the higher density (red color) semitransparently behind the green
grey lower density even without the glasses. A rotating version of this image is
shown on [39].
8. Further analysis
Having carefully described the computation and visualization of the charge230
density, we now turn to physical aspects; thus returning to our initial moti-
vations. The first is research into the deformation of nanotube walls under
distortions. The present project has demonstrated that we can calculate and
13

Figure 10: Visualization of charge density for a 3-rings system without any strain
visualize the electronic density surrounding the tube. We can observe local vari-
ations in its thickness at different axial locations, see for example Figures 10235
and 11. Further investigations with larger samples and a careful numerical data
analysis that falls beyond the scope of the present paper have begun and will
be extended in the near future to larger and more distorted tubes.
The second motivation concerned the integration of simulations on electronic
and atomistic scales as part of SimPhoNy.240
**Adham, I await the promised ref.**
The present computations provide one stage of efforts towards this goal. We
have successfully with intial molecular dynamics input transferred to electronic
density functional theory calculations in a smooth, automatic and reproducible
14
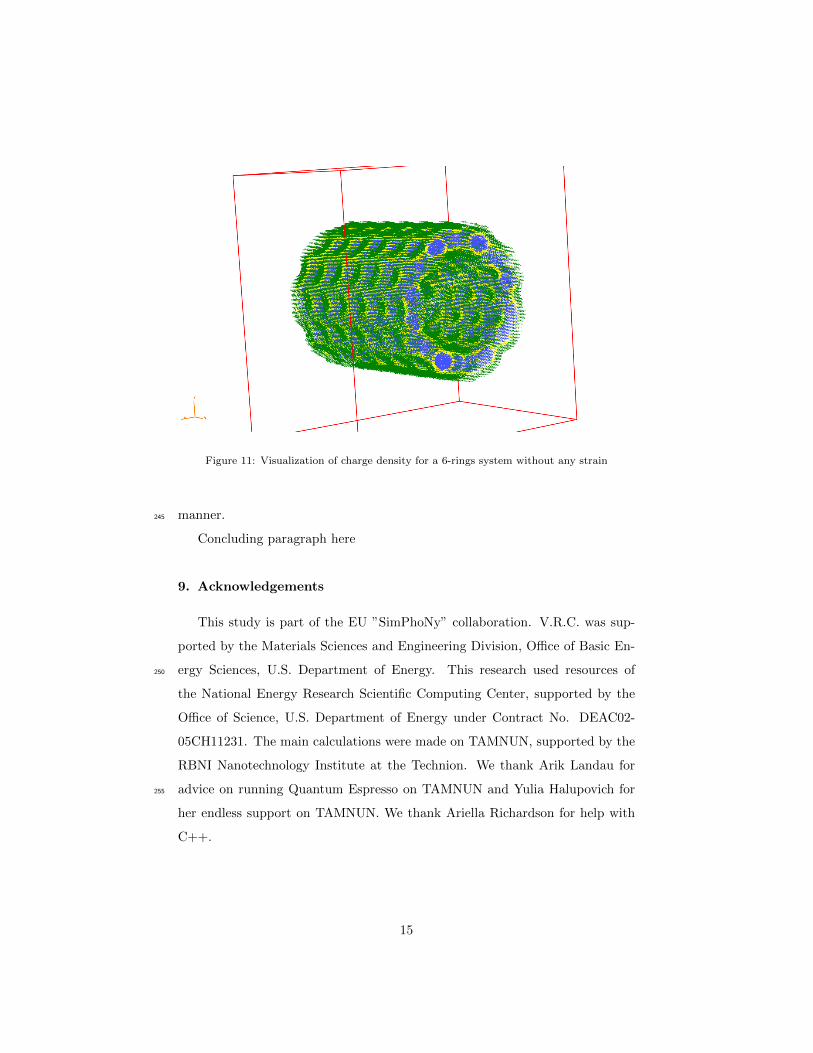
Figure 11: Visualization of charge density for a 6-rings system without any strain
manner.245
Concluding paragraph here
9. Acknowledgements
This study is part of the EU ”SimPhoNy” collaboration. V.R.C. was sup-
ported by the Materials Sciences and Engineering Division, Office of Basic En-
ergy Sciences, U.S. Department of Energy. This research used resources of250
the National Energy Research Scientific Computing Center, supported by the
Office of Science, U.S. Department of Energy under Contract No. DEAC02-
05CH11231. The main calculations were made on TAMNUN, supported by the
RBNI Nanotechnology Institute at the Technion. We thank Arik Landau for
advice on running Quantum Espresso on TAMNUN and Yulia Halupovich for255
her endless support on TAMNUN. We thank Ariella Richardson for help with
C++.
15

Figure 12: The viewpoint
panel of AViz, the ”view”
button was bressed and
the explicit option se-
lected to bring up the ex-
plicit panel.
Figure 13: The explicit
button in the viewpoint
panel of AVizFigure 14: AViz frame
with stereo options on the
right
Appendix A. Details and code links for the simulation part
All tar files are opened with tar -xvf name of file. The instructions assume
use of a LINUX system for the compilation and execution (tested on Red Hat),260
and terminal software that enables ssh -X access if the desktop is a different
system to where the simulations and visualizations are carried out. For com-
pleteness we also include comments related to batch (PBS) submission on our
local SGI RedHat Linux parallel cluster called TAMNUN; with the exception
of commands related to MPI we have also checked their validity on a shared265
memory computer. TAMNUN has Quantum Espresso installed with Intel com-
pilers and AViz 6.1 in the /usr/local partition. Initial versions of the single ring
system were run at NERSC on a Cray XT system (hopper).
The following sections contain explanations for sample input files and pa-
rameter choices; more details are in the full set of downloadable files at [35],270
and non-site specific aspects are in QE documentation [33]. Three cases - single
ring, multiple rings and strained nanotubes are considered. For the single ring
case one downloads the example input tar file.
16

Figure 15: A stereo image of the 10 percent strained, fovy adjusted 3 ring nanotube showing
the semi-transparent outline of the inner cloud.
Appendix A.1. Input file for pw.x
Figure A.16 shows a typical input file for pw.x. We now present a brief275
discussion of the different lines of the input file; a full description of the doc-
umentation is linked above. The path to an user directory is /u/username/,
where username has to be replaced by the login name that is used to connect
on TAMNUN or the appropriate value for another system. In detail:
Appendix A.1.1. &control280
• calculation : defines the type of calculation that one wants to do, ’scf’ is
for self-consistent field
17

Figure A.16: Inupt file for pw.x
• pseudo dir : the path where the potential file(s) are located. This is a
user specific choice.
• prefix : ’NANO’ is an example, one can change it and write whatever285
he/she wants but it is important that all pw.x and pp.x input files use the
same prefix.
• outdir : this path is to the place where all the files will be saved during
and after the computation
18

Appendix A.1.2. &system290
• ibrav : defines the type of lattice that is used. For example 8 is orthorhom-
bic and depending on the lattice the following lines can change. In the
case of ’ibrav=8’ the basis vectors are given by : v1 = (a, 0, 0), v2 =
(0, b, 0), v3 = (0, 0, c).
• celldm : this part defines the crystallographic constants. An important295
remark is that these values are given in Bohr and not in Angstrom (1
Bohr = 0.529177249 Angstrom). In the orthorhombic case, celldm(1) =
a, celldm(2) = b/a and celldm(3) = c/a. Here an explanation about the
values shown in the example (fig. A.16) is necessary. The vector along
the length of the nanotube is given by celldm(2). The two others are set300
to a big value in order to ”insulate” the system from boundary conditions
effects (celldm(1) = 40, means that a = 40, and celldm(3) = 1, means
that c = 40). To define the value that one has to put for celldm(2), the
way is to measure with a visualization program, the distance between
two consecutive rings, convert this value to Bohr (1 nm = 18.8971 Bohr)305
and divide it by celldm(1), because the measured value will give ’b’, but
celldm(2) = b/a.
• nat : is the number of atoms in the simulation. It has to be equal to the
number of lines under the ”ATOMIC POSITIONS” section.
• ntyp : defines the number of different types of atoms present in the simu-310
lation.
• ecutwfc : is the cutoff for the kinetic energy of the wavefunctions.
• ecutrho : is the cutoff for the kinetic energy of the charge density and
potential, the default value (if not specified) is 4 · ecutwfc.
• :input dft:defines the exchange and correlation functionals employed in the315
calculation. here we cose the vdw-DF non-local correlation functional with
19

the C09x exchange functional in order o account for London dispersion
interactions (or van der Waals forces) within our calculations.
Appendix A.1.3. ATOMIC SPECIES
Under this header one has to specify the type of atom, the mass of the320
atom and the exact name of the pseudopotential that will be used for this atom
(located in ’pseudo dir’).
Appendix A.1.4. ATOMIC POSITIONS
These are the coordinates of the atoms present in the system. There are
many ways to do this; we just specified the type of atom (Carbon in the example325
Figure, A.16) and the coordinates using a .xyz file without the two first lines.
After ATOMIC POSITIONS, we specified the unit (see documentation for more
details). For the structures here we emplyed Cartesion coordinates in angstroms.
Appendix A.2. Run pw.x
Now that the input file is created, one can run pw.x using this input file.330
Using MPI the line to execute the code would be :
mpirun -np n /usr/local/espresso/bin/pw.x < name.in > name.out
where n has to be replaced by the number of processors used. The path before
pw.x is the right one for TAMNUN but could differ on another system. The
result of this first part should be a folder with the name ’prefix’.save, where335
’prefix’ is the name chosen for the prefix, (NANO in our example) and a file
name.out.
Appendix A.2.1. vdw-DF calculations
We have included dispersion(van der Waals) interactions within our DFT
calculations using the van der Walls density function (vdw-DF) [29]. To include340
these interactions in QE it is neccesary to first generate the vdW-DF kernel
table. This can be done using the following procedure:
In this case, the solution is :
20

• copy the file from usr/local/espresso/PW/src generate vdW kernel table.x
into the user’s directory - go into the destination folder and use the com-345
mand (note the . at the end, it is important):
cp usr/local/espresso/PW/src/generate vdW kernel table.x .
• run it with : generate vdW kernel table.x
• move the resulting table file (vdW kermel table) into the directory where350
the pseudopotential is located.
Remark : it can take a moment to execute the generate vdW kernel table.x
file but it only needs to be generated once. (If this is not done properly you
may get:
Error in routine read kernel table (1) :355
No \ ”vdW kernel table \ ” file could be found
If you see this error try again to follow the above directions carefully.)
)
Appendix A.3. Systems of multiple rings
Adding more rings is not totally trivial and should be omitted in a first360
calculation. As explained in section Appendix A.1.2, it is clear that if the
number of rings changes, the number of atoms will change (nat) and the unit
cell length along the c-axis, celldm(2) also change. For example, if the system
is now a 6-rings system the value of celldm(2) has to be multiplied by 6.
The position of the atoms is another aspect that requires care. Indeed, the365
celldm vectors define the simulation box. We recall the values are given in Bohr.
On the other hand, the values of the positions are given in Angstrom. The fact
that the positions are in Angstroms is a special case, it could be defined in
Bohrs if we wanted to. So an important thing to check is that all the atoms
are contained in the volume defined by the celldm vectors. For simplicity our370
box starts from (0,0,0) and has the size of the ’celldm vectors’ values. (Remark
21

: 1 Bohr = 0.5291 Angstrom). So if the atoms are not contained in these
boundaries they have to be translated. To check it, the y-coordinate of the
atom (in Angstrom) has to be in the interval [0, celldm(2) · 0.5291 · celldm(1)].
The atoms just need to be within one unit cell of each other and could be defined375
starting at say (-1/4, -1/4, -1/4).
The translation is done by a script that needs as input file an .xyz file with
the coordinates. After downloading and opening the many rings tar file,
• compile the code with : g++ many rings.cpp -o exec many rings
• put the xyz file in the same folder and execute the code with : ./exec many rings380
• enter the exact name of the file (.xyz included)
• select to do a manual translation or automatic one
A file with the same name plus ” translated.xyz” will be created in the current
directory. These are the new coordinates that will be used.
In the case one chooses to translate automatically, the program will find the385
minimum value in the first and last ring and translate every atom by this value
along the y direction in order to have one atom at the origin and all the others
contained inside of the simulation box.
——————————Valentino and Bastien can you please sort out this
next part—————————————————————390
Appendix A.4. System under strain
The third case that has to be considered with care is the case where there is a
strain in the nanotube. This can also be omitted in a first trial. The consequence
of the strain will be to change the distance between the atoms. When one adds a
strain in the nanotube the distance along y is increased and the distance along395
x and z (direction of the radius) is decreased. As the constraint is constant
the distance between two neighboor-cells along one given direction should be
the same. Nevertheless, as one can probably notice in Figure 4 the slice of
the nanotube does not fall along a perfectly straight line, which means that the
22

distance is not constant everywhere between the consecutive cells. This problem400
is due to the way that the nanotube was built numerically and more specifically
due to the boundary conditions. A way to minimise this problem is to take the
atoms in the middle of the nanotube in order to be as far as possible from the
edges. Every ring has 28 atoms. For the nanotube presented in Figure 4, if
one wants to select a system with 3 rings, the way to select them in the middle405
is to remove the 18 first rings removing the 504 first coordinates, then jump
84 coordinates and remove the rest. One can check that the distance is now
constant between all the atoms. This has been tested for a system of 3 rings
under 2.5, 5 and 10 percent strain. For a larger number of rings this could be
problematic if the distance is not constant.410
Appendix A.5. Input file for pp.x
After the first step, one should have the two files mentioned above. The
next step is to extract the charge density from the output file. To do so, one
will create an input file for pp.x and obtain an output file that will contain the
charge density for each point of the grid, as shown in our later figures. In detail:415
Figure A.17: Inupt file for pp.x
23

Appendix A.5.1. &inputpp
• prefix : as mentioned before it is really important that the name of the
prefix is exactly the same as it was for pw.x
• filplot : this is the name of the outputfile and can be changed
• plot num : defines the quantity that one is interested in during the post-420
processing, 0 is for the charge density (see the QE documentation for other
values)
• outdir : same as before
Appendix A.5.2. &plot
This section may not be needed for other systems but from our experience425
on TAMNUN it is better to specify it.
• filepp(1) : is the name of the output file that will contain the quantity to
be plotted and saved in fileout, here it is the charge density for example.
• output format : the integer defines the format of the output file (see doc-
umentation).430
• fileout : name of the file that contains the data to do the plot. This is not
used for AViz but it can be useful if one wants to use other software such
as xcrysden [38].
Appendix A.5.3. Remark
On TAMNUN it is important to put the outdir at the end of the section435
&inputpp. An other observation is that it is possible to specify the amount of
data saved during the computation using ’disk io’. If this is not specified the
default value is ’low’ but the less data are saved the more RAM is used and it
can be a problem if the available RAM is not sufficient. The default value is
advised.440
24

Appendix A.6. Run pp.x
The execution line is similar to the previous one :
mpirun -np n /usr/local/espresso/bin/pp.x < name.in > name.out
Appendix A.7. Example of a 1 ring system
For the one ring system (illustrated in Figure 7), the tar contains an input445
file for pw.x and an other one for pp.x and also two bash scripts that create
these files and submit them through the queuing system on TAMNUN.
Appendix A.8. Creation of the .xyz file for the charge density
The output file from pp.x should now be name.charge. This file has the
structure shown in Figure A.18. The first line has 8 numbers, the three first450
numbers are the size of the grid in the three spatial directions, the three following
are the same repeated. The seventh number corresponds to the number of atoms
and the last one to the number of different type of atoms. About the second
line, the first number is the type of Bravais lattice, and the three following
numbers are the celldm defined in the input file of pw.x. The first lines are not455
of interest to us because what we want is the charge density, which is defined
for each point of the grid starting at line 34.
Our aim is to create a .xyz format file with the charge density. More precisely,
each value of charge density given in the output file corresponds to a point in the
grid. To convert this file into an appropriate format one can download a script460
written in C++ as part of the example input file and carry out the following:
1. compile and execute the code : g++ charge density xyz.cpp -o exec charge density
2. copy the file you wish to process into the directory where the script is
3. run ./exec charge density to open the program
4. enter the name of the file to convert and press enter465
A new file with the right format is now created in the same folder and is ready
to be visualized. An older fortran convert file for QE output is at http://
phycomp.technion.ac.il/~aviz/download/download.html.
25

Figure A.18: Output file from pp.x, 40 first lines.
Appendix B. Further data processing and visualization
Appendix B.1. Initial visualization with AViz470
As it was mentioned previously, the raw charge density output from QE is
displayed in four different parts (see Figure B.19). This is a special case and
could hav ebeen done differently.
Appendix B.1.1. Input file for AViz
The output file after the execution of the C++ routine is a standard .xyz file475
for AViz (see Figure (B.20) and Section 5) having the first line with an integer
total number of dots, then a comment line and then the third and subsequent
lines containing the information. All numbers from line 3 onward have to be
real ones. The different columns in our example are :
1. A letter (a,b,c,d,e,f) that indicates the colors (transparent, black, red,480
green, yellow, blue
2. The x coordinate
26

Figure B.19: Charge density for a single ring of nanotube - initial visualization with AViz
3. The y coordinate
4. The z coordinate
5. The charge density at that point485
Figure B.20: Input .xyz file for AViz, containing a letter for color selection, coordinates and
charge density
In this file the coordinates go from 1 to 200 in each direction. The colors are
in order of increasing density ranging from less than 0.0005 to 0.5 or above.
**Bastien, units please**
27

Appendix C. C++ code to recombine the circle
In this part the different important functions implemented will be presented.490
The general procedure is to read the file mentionned above, find which part
corresponds to which quarter of circle, recombine them and create a new input
file for AViz.
Appendix C.1. Function to read the initial .xyz file
The first step is to read the file in order to work on it after. To do so, it495
is possible to read line after line and store each line in a vector of string (fig.
(C.21)).
Figure C.21: Code to read from a file and create a vector of strings.
Appendix C.2. Separate the four parts
Once the vector of string is created, the second step is to separate the four
different quarters. As mentioned above, the coordinates go from 1 to 200.500
The structure of the .xyz file is the following : taking half of lines in the file
( total number lines2 ) corresponds to take half of the sample (cutting in the z-
direction). The result is a parallelepiped of dimension (x2 , y, z). Indeed, if one
takes the 200 first lines of the file, it corresponds to one single line on the plane
(a one dimensional path). The 200 following lines represent the same line but505
28

translated in the y-direction. That means the second layer of points forming
the plane.
This means that in order to separate the four parts, one has to take the lines
labeled from 1 to 100 for the first half of the file (part1), the lines from 101 to
200 for the first half of the file (part2) and the same for the second part of the510
file (part3 and part4). This work is done by the loop shown in Figure (C.22).
Each part is stored in a vector of strings.
Appendix C.3. Recombine the circle
This is the most difficult part. The elements of the different vectors are
strings but we have to exctract the numbers for the coordinates and also the515
charge density, process the coordinates and recreate the file. To read the num-
bers from a string, one can use the function sscanf(), but the input line to read
has to be a char. So the idea is to read each line of the vector, convert the string
into a const char using .c str(), extract the different variables in the appropriate
format, modify the coordinates and store the line in the same format than it520
was initially. One has just to be carefull to erase the string and char variables
after each iteration. (See fig. (C.23))
Appendix C.4. Dilute points
The sample is quite thick and the points are dense but in order to see the
physics ”inside” the sample one needs to dilute the points randomly. The idea525
is to delete a given number of points randomly in the vector that contains the
information about the coordinates and charge density. (See fig. (C.24)
Appendix C.5. Create the final output
The last step is to write an output file with the .xyz format described above
but with the changed coordinates to have the right circle. This can be done530
using ofstream. The two first lines contain the number of points followed by a
comment line. The rest can be done with a loop reading each element of the
vector of string recombined and writting it in the output file. (See fig. C.25).
29

Figure C.22: Part of the code to separate the quarters of the sample
30

Figure C.23: Code to recombine the four parts of the sample and create a vector containing
the information.
Appendix C.6. Run the code and results
To run the code, one has to compile it first using the command :535
g++ -o nano4parts nano4parts.cpp.
31

Figure C.24: Code to dilute the points and see through the sample.
Figure C.25: Code to create the output file with the changed coordinates.
This will create an executable file named nano4parts which should be placed
in the same folder as the initial input file executed. The name of the input file
will be requested, enter it (include the extension in the name : example.xyz)
and press enter. The code generates five .xyz files : four to visualize each of the540
four parts of the sample and the fifth is the full circle. They will be generated
in the same folder. One can then visualize the .full circle.xyz file with AViz and
obtain the result presented in Figure (8).
An easy way to view the inside of the sample is to make a random dilution.
After this random dilution, one obtains the result shown in fig. (9).545
32

Appendix C.7. Download : Recombine circle
The C++ script is downloadable here : http://phycomp.technion.ac.il/
~sbgrosso/QE_charge_density/ddl_nano4parts.tar.
In order to execute the program, one has to :
1. type g++ -o name of executable nano4parts.cpp550
2. be sure that the file with the data is in the same folder
3. run the program using : ./name of executable
—for submission a pdf is good enough, afterwards –i think this journal wants
a .bib file and if so i will pay Liz to do this—
[1] J.Adler, “Visualization in Atomistic and Spin Simulations” Computers in555
Science and Engineering, 5 pp 61-65
[2] http://phycomp.technion.ac.il/∼aviz
[3] Adler J, Hashibon A, . Schreiber N, Sorkin A, Sorkin S and Wagner G
2002 “Visualization of MD and MC Simulations for Atomistic Modeling”
Computer Physics Communications, 147 pp 665-9560
[4] S. Johnson, W. Potter, K. Malkin, Proceedings of the International Con-
ference on Simulation in Engineering Education, (1994), 26, p. 199-202.
[5] http://phjoan23.technion.ac.il/∼phr76ja/joeyfox/Hydrogen.html
[6] Adler J, Fox J, Kalish R, Mutat T, Sorkin A and Warszawski E 2007
”The essential role of visualization for modeling nanotubes and nanodia-565
mond”Computer Physics Communications 177 pp 19-20
[7] http://phelafel.technion.ac.il/∼orcohen/DFTVisualize.html
[8] http://classic.chem.msu.su/gran/gamess/index.html
[9] J. Adler, J. Zaffran, A. Silverman, A. Sorkin, O. Cohen and R. Kalish,
“Simulation and visualization of nanodiamond and nanographite”, Com-570
puter Physics Communications, (2011), 182, 2009.
33

[10] http://phycomp.technion.ac.il/∼jeremie
[11] G. Kresse and J. Furthmuller, (1996), Phys. Rev. B 54, 11 169.
[12] G. Kresse and D. Joubert, (1999), Phys. Rev. B. 59, 1758.
[13] http://www.geocities.jp/kmo mma/crystal/en/vesta.html575
[14] Sprott J C “Simple programs create 3D Images” 1992 Computers in Physics
6 pp 132-8
[15] http://phelafel.technion.ac.il/$\sim$peledan
[16] D. Peled, A. Silverman and J. Adler, “3D visualization for atomistic simu-
lations on every desktop”, IOP Conference Series, (2013) 454, p. 012076.580
[17] J. Adler, Y. Koenka and A. Silverman, 2011 “Adventures in carbon visu-
alization with AViz” Physics Procedia 15 pp 7-16
[18] http://phycomp.technion.ac.il/$\sim$newaviz
[19] http://phelafel.technion.ac.il/~meytal
[20] Joan Adler, Yaron Artzi, Liron ben Bashat, Tzipora Yael Izraeli, Meital585
Kreif, Ido Lavi, Alexander Leibenzon, Adam Levi, Itai Schlesinger, Elad
Toledano, Uria Peretz, Yonatan Weisler and Alon Yagil, “How do I simulate
problem X?”, IOP Conference Series, (2014) 510, 012003.
[21] ”Visualization Techniques for Modelling Carbon Allotropes”, J. Adler and
P. Pine, Computer Physics Communications, (2009), 180 , 580-2.590
[22] P. Pine, Y. Yaish and J. Adler, “Simulational and vibrational analysis
of thermal oscillations of single-walled carbon nanotubes”, Phys. Rev. B
(2011), 83, 155410.[/3]
[23] P. Pine, Y. Yaish and J. Adler, “Thermal oscillations of structurally distinct
single-walled carbon nanotubes”, Phys. Rev. B (2011).84,245409.595
34

[24] P. Pine, Y. Yaish and J. Adler, “The affect of boundary conditions on
the vibrations of single-walled carbon nanotubes”, J. App. Phys., (2011),
110, 124311, Also featured in Virtual Journal of Nanoscale Science and
Technology, 25, no 2.
[25] P. Pine, Y. Yaish and J. Adler, “Simulation of nanosensors based on single600
walled carbon nanotubes”, IOP Conference Series, (2012) 402, p.012002.
[26] P. Pine, Y. Yaish and J. Adler, “Vibrational analysis of thermal oscillations
of single-walled carbon nanotubes under axial strain”, PRB, (2014) 89,
115405.
[27] P. Pine, Ph. D. thesis, Technion, 2012, http://phycomp.technion.ac.605
il/~newphr76ja/polina.pdf
[28] http://www.simphony-project.eu
[29] http://pwscf.org/mailman/listinfo/pw_forum
[30] D. W. Brenner, Phys. Rev. B 42, 9458 (1990).
[31] http://phycomp.technion.ac.il/~tamnun/easy_lammps.html610
[32] Joan Adler, Hila Glanz, and Nadir Izrael, “A “Rosetta stone” for AViz”,
Physics Procedia, (2014), 57c pp 2-6. (DOI10.1016/j/phpro.2014.08.122)
[33] http://www.quantum-espresso.org
[34] http://phycomp.technion.ac.il/~tamnun/qe/draw.html
[35] http://phycomp.technion.ac.il/~sbgrosso615
[36] http://www.quantum-espresso.org/wp-content/uploads/Doc/INPUT_
PW.html#idm1680
[37] http://www.quantum-espresso.org/wp-content/uploads/Doc/INPUT_
PP.html
[38] http://www.xcrysden.org/XCrySDen.html620
35

[39] http://phycomp.technion.ac.il/~phr76ja/bastiendata/bastien2.
gif
36