Embed Size (px)
Citation preview

Vol. 46 • N. 183luglio-settembre 2016
Perio
dico
trim
estra
le P
OSTE
ITAL
IANE
SPA
- Sp
edizi
one
in A
bbon
amen
to P
osta
le -
D.L.
353
/200
3 co
nv.in
L.2
7/02
/200
4 n°
46 a
rt.1,
com
ma
1, D
CB P
ISA
Aut.
Trib
. di M
ilano
n. 1
30 d
el 1
7/03
/197
1 - S
tam
pa a
tarif
fa ri
dotta
- ta
ssa
paga
ta -
Aut.
Dirp
oste
l Pisa
n. 1
/361
31/4
/1 d
el 1
0/09
/199
3 - T
axe
perç
ue -
Italia
- Fi
nito
di s
tam
pare
pre
sso
IGP,
Pisa
, otto
bre
2016
- IS
SN 0
301-
3642
(prin
t)
Genetica clinica pediatrica (a cura di Angelo Selicorni)
Genetica clinica pediatrica: due focus tematici dalla letteratura più recente
Le problematiche del sonno nelle sindromi malformative: un problema spesso sottostimato
Terapia farmacologica e sindromi genetiche: quali novità?
Oftalmologia pediatrica (a cura di Roberto Caputo)
Novità in Oftalmologia pediatrica
Nuove frontiere in terapia genica: prospettive terapeutiche per le patologie oculari a insorgenza pediatrica geneticamente determinate
Anomalie della motilità oculare e posizioni anomale del capo
Frontiere (a cura di Andrea Biondi, Achille Iolascon, Luigi D. Notarangelo, Massimo Zeviani)
Nuovi approcci nella terapia delle malattie mitocondriali
Focus (a cura di Generoso Andria)
La metabolomica in pediatria: una scienza in continua evoluzione

Vol. 46 • N. 183luglio-settembre 2016
Redazione EditorialeValentina BàrberiTel. 050 3130376 [email protected]
AmministrazionePacini Editore SrlVia Gherardesca, 156121 PisaTel. 050 313011 - Fax 050 [email protected]
StampaIndustrie Grafiche Pacini, Pisa
AbbonamentiProspettive in Pediatria è una rivista trimestrale. I prezzi dell’abbonamento annuo sono i seguenti:
PREZZO SPECIALE RISERVATO A SOCI SIP: € 20,00.Contattare: fax +39 02 45498199 E-mail: [email protected]
Italia € 60,00; estero € 70,00; istituzionale € 60,00; specializzandi € 35,00; fascicolo singolo € 30,00Le richieste di abbonamento vanno indirizzate a: Prospettive in Pediatria, Pacini Editore Srl, Via Gherardesca 1, 56121 Pisa – tel. +39 050 313011 – fax +39 050 3130300 – E-mail: [email protected] dati relativi agli abbonati sono trattati nel rispetto delle disposizioni conte-nute nel D.Lgs. del 30 giugno 2003 n. 196 a mezzo di elaboratori elettronici ad opera di soggetti appositamente incaricati. I dati sono utilizzati dall’editore per la spedizione della presente pubblicazione. Ai sensi dell’articolo 7 del D.Lgs. 196/2003, in qualsiasi momento è possibile consultare, modificare o cancellare i dati o opporsi al loro utilizzo scrivendo al Titolare del Trattamento: Pacini Editore Srl, Via Gherardesca 1, 56121 Pisa.
Le fotocopie per uso personale del lettore possono essere effettuate nei limiti del 15% di ciascun fascicolo di periodico dietro pagamento alla SIAE del compenso previsto dall’art. 68, commi 4 e 5, della legge 22 aprile 1941 n. 633. Le riproduzioni effettuate per finalità di carattere professionale, economico o commerciale o comunque per uso diverso da quello personale possono essere effettuate a seguito di specifica autorizzazione rilasciata da AIDRO, Corso di Porta Romana n. 108, Milano 20122, E-mail: [email protected] e sito web: www.aidro.org.
© Copyright by Pacini Editore Srl
Direttore Responsabile: Patrizia Alma Pacini
Rivista stampata su carta TCF (Total Chlorine Free) e verniciata idro.
DirettoreGeneroso Andria, Napoli
Comitato di DirezioneAndrea Biondi, MonzaFranco Chiarelli, ChietiGiovanni Cioni, PisaGiovanni Corsello, PalermoAchille Iolascon, NapoliAlberto Martini, GenovaLuigi Daniele Notarangelo, BostonFabio Sereni, MilanoRiccardo Troncone, Napoli
Comitato EditorialeSalvatore Auricchio, NapoliSergio Bernasconi, ParmaSilvano Bertelloni, PisaMauro Calvani, RomaLiviana Da Dalt, PadovaMario De Curtis, RomaMaurizio de Martino, FirenzePasquale Di Pietro, GenovaAlberto Edefonti, MilanoCiro Esposito, NapoliCarlo Gelmetti, MilanoGiuseppe Maggiore, PisaGianantonio Manzoni, MilanoBruno Marino, RomaPierpaolo Mastroiacovo, RomaEugenio Mercuri, RomaPaolo Paolucci, ModenaLuca Ramenghi, GenovaDaria Riva, MilanoMartino Ruggieri, CataniaFranca Rusconi, FirenzeFrancesca Santamaria, NapoliLuigi Titomanlio, ParigiPietro Vajro, SalernoMassimo Zeviani, Cambridge, UKGianvincenzo Zuccotti, Milano
Redazione ScientificaRoberto Della Casa (Redattore Capo)Simona FecarottaGiusy Ranucci

Genetica clinica pediatrica (a cura di Angelo Selicorni)
Presentazione ............................................................................................................................................. 189
Genetica clinica pediatrica: due focus tematici dalla letteratura più recenteAlessandra Di Cesare Merlone, Silvia Maitz, Angelo Selicorni ........................................................ 190
Le problematiche del sonno nelle sindromi malformative: un problema spesso sottostimatoGiuseppe Zampino, Roberta Onesimo, Chiara Leoni, Silvia Tajè, Giacomo della Marca .......... 198
Terapia farmacologica e sindromi genetiche: quali novità?Paolo Curatolo, Romina Moavero, Angelo Domenico, Silvia Maitz, Martino Ruggeri, Gioacchino Scarano, Angelo Selicorni .................................................................. 206
Oftalmologia pediatrica (a cura di Roberto Caputo)
Presentazione ............................................................................................................................................. 215
Novità in Oftalmologia pediatricaRoberto Caputo, Gabriele Simonini, Alice Brambilla, Neri Pucci, Cinzia de Libero ................... 216
Nuove frontiere in terapia genica: prospettive terapeutiche per le patologie oculari a insorgenza pediatrica geneticamente determinateMaria Concetta Ferraro, Giacomo Maria Bacci, Roberto Caputo, Francesca Simonelli ........... 227
Anomalie della motilità oculare e posizioni anomale del capoElisabetta Santangelo, Massimiliano Serafino, Paolo Nucci ........................................................... 235
Frontiere (a cura di Andrea Biondi, Achille Iolascon, Luigi D. Notarangelo, Massimo Zeviani)
Nuovi approcci nella terapia delle malattie mitocondrialiMassimo Zeviani, Emanuela Bottani, Carlo Viscomi .......................................................................... 241
Focus (a cura di Generoso Andria)
La metabolomica in pediatria: una scienza in continua evoluzioneAngelica Dessì, Antonio Noto, Roberta Pintus, Vassilios Fanos .................................................... 250
Prospettive in Pediatria
INDICE N. 183luglio-settembre 2016

189
Prospettive in PediatriaLuglio-Settembre 2016 • Vol. 46 • N. 183 • P. 189
Genetica clinica pediatrica
L’invito rivolto a curare una sezione dedicata alla Genetica clinica pediatrica ci ha fortemente entusiasmato. Dopo il primo momento di euforia si è però posto il serio problema di quali argomenti mettere a fuoco, nel ten-tativo di sintetizzare un mondo in estrema evoluzione dal punto di vista tecnologico, assistenziale e di ricerca. Mondo che, tra l’altro, mette in stretta comunicazione branche specialistiche differenti nell’area della medicina e della biologia.La scelta, non facile, è caduta su tre articoli di taglio assai differente e, pensiamo, complementare. Nel primo articolo abbiamo cercato di fare un punto della situazione su due tematiche di particolare interesse sia specu-lativo che pratico inerenti: a) uso nella pratica clinica delle nuove tecnologie genomiche e del whole exome se-quencing (WES) in particolare; b) relazioni sempre più strette tra sindromi genetiche e tumori in età pediatrica. Il secondo articolo, curato dal gruppo del prof. Zampino, è di taglio squisitamente assistenziale, perché nel mondo della Genetica clinica pediatrica la diagnosi clinico-biologica deve sempre essere seguita da una presa in carico a 360°. Abbiamo quindi chiesto ai colleghi del Policlinico Gemelli di Roma di scrivere una revisione sul problema dei disturbi del sonno nelle patologie sindromiche, per comprenderne gli aspetti patogenetici, diagnostici e di trattamento per la pratica clinica.Da ultimo, ma non per ultimo, non potevamo non accendere una luce su nuovi sviluppi terapeutici/farmacologici e sindromi genetiche. È ben noto infatti che si stanno pian piano affacciando, anche nell’ambito delle sindromi genetiche, protocolli più o meno sperimentali in ambito di terapia farmacologica. A questo scopo abbiamo fatto il punto della situazione su sclerosi tuberosa, acondroplasia e neurofibromatosi, chiedendo un sintetico ma stimolante contributo agli esperti italiani più impegnati su queste tematiche.Ringraziamo sin d’ora i colleghi che con pazienza e disponibilità hanno collaborato a costruire questa sezione, nella speranza di essere riusciti a fornire ai lettori informazioni e riflessioni utili, sia per la pratica clinica quo-tidiana che per la comprensione dell’evoluzione potenziale di questa branca della pediatria nei prossimi anni.
Angelo SelicorniUnità Operativa Complessa di Pediatria, ASST Lariana, Como

190
Luglio-Settembre 2016 • Vol. 46 • N. 183 • Pp. 190-197 Prospettive in Pediatria
Genetica clinica pediatrica
Genetica clinica pediatrica: due focus tematici dalla letteratura più recente
Alessandra Di Cesare Merlone1
Silvia Maitz2 Angelo Selicorni1 2
1 Unità Operativa Complessa di Pediatria, ASST Lariana, Como;
2 UOS Genetica clinica pediatrica, Fondazione MBBM, ASST Monza
New technologies for genomic analysis (particularly whole exome sequencing – WES) are becoming used in routine diagnostic tests. The detection rates reported in various studies, the rapidly available results and the possibility of identifying atypical or genetically hetero-geneous phenotypes have led to considering them first or second-line tests for diagnosis of children with suspected monogenic disorders. In this review, we summarise the avail-able genomic studies and critically evaluate the benefits (e.g. use of WES in critically-ill neonates) and challenges, which are mostly related to data interpretation. We also focus our attention on the relationships between germline and somatic mutations on the risk of developing specific forms of cancer, which is an emerging field in pediatric oncology. Be-sides well-known cancer predisposition syndromes, recent studies have highlighted the need for more in-depth clinical and genomic analyses in patients with “isolated tumours”. Patients carrying germline mutations, such as those affected with specific birth defects, may indeed have an increased risk of developing cancer.
Summary
Le nuove tecnologie di studio in ambito genomico (whole exome sequencing – WES in particolare) stanno entrando in modo importante nella routine diagnostica. Le detection rate riportate dai lavori pubblicati, la tempistica di risposta e la loro capacità di risolu-zione di fenotipi atipici o estremamente eterogenei sul piano genetico, impongono una loro considerazione come esami di primo o secondo livello nella diagnostica del bambino con sospetta malattia monogenica. La letteratura a riguardo, che viene qui sinteticamente valutata, ne descrive ampiamente le potenzialità (pensiamo anche all’approccio quasi in urgenza nel neonato critico) e le difficoltà interpretative che ne derivano.Anche in ambito oncologico risulta sempre più evidente un legame assai stretto tra assetto genomico costituzionale (mutazioni germinali) e sviluppo di neoplasia (mutazioni somati-che). Al di là delle ben note condizioni cancer predisposing, i dati della letteratura recente in ambito sia oncologico che clinico genetico sottolineano concordemente la necessità di approfondire la ricerca in ambito clinico genetico, anche di fronte al paziente con “neopla-sia isolata”, e di analizzare con attenzione ogni paziente con patologia dovuta a mutazioni nella linea germinale (a partire dai soggetti con specifici difetti isolati dello sviluppo), quale potenziale soggetto a rischio aumentato di sviluppo di neoplasia.
Riassunto
AbbreviazioniWES: whole exome sequencingWGS: whole genome sequencingNGS: next generation sequencing

191
Focus tematici per la genetica clinica pediatrica
Metodologia della ricerca bibliografica effettuataLa ricerca degli articoli rilevanti su sindromi genetiche e predisposizione al cancro è stata effettuata nella banca bibliografica Medline, utilizzando come moto-re di ricerca PubMed e come parole chiave: “WES”, “WGS”, “NGS”, “genetic syndrome and cancer”. I limiti utilizzati sono stati: “ages: all child 0-18 years”; “dates: last 5 years”. Sono inoltre stati selezionati articoli origi-nali, revisioni, studi epidemiologici anche antecedenti agli ultimi 5 anni, se ritenuti utili alla stesura della re-visione.
ObiettiviNell’ambito delle diverse tematiche afferenti al capi-tolo della genetica clinica pediatrica abbiamo scelto di focalizzare sinteticamente la nostra attenzione su due problematiche di attualità e interesse: punto della situazione sull’utilizzo del whole exome sequencing (WES) nella pratica clinica pediatrica, interrelazioni cliniche e genomiche tra sindromi genetiche/difetti dello sviluppo e neoplasie pediatriche.
Whole Exome Sequencing in pediatria: a che punto siamo?Nel numero di Prospettive in Pediatria di aprile-giugno 2015, Vincenzo Nigro pubblicò un efficace articolo sull’utilizzo delle nuove tecniche di sequenziamento (Next generation sequencing – NGS) in ambito pe-diatrico, con una disamina molto attenta e precisa, focalizzando la sua attenzione sulla descrizione della tecnologia e delle sue possibilità. Il tema è di attualità crescente, tanto che sono stati recentemente pubbli-cati numerosi studi nel tentativo di definire in termini operativi e di pratica clinica vantaggi e svantaggi, limi-ti, preoccupazioni e benefici dell’uso di questa tecno-logia nella routine diagnostica.I dati della letteratura ci dicono che una condizione genetica rara colpisce 1/50 bambini, che sono note almeno 5000-6000 diverse malattie rare e che quasi nel 50% dei pazienti non viene raggiunta una diagno-si eziologica (Sawyer et al., 2016). Secondo le voci
dei pazienti stessi questo percorso viene definito odissea diagnostica ed è costellato di ripetute visite, indagini, analisi molecolari le più differenti con costi, anche sanitari, molto alti. Le analisi eseguite attraver-so le segnalazioni dei pazienti ci dicono che nel 25% dei casi la diagnosi viene raggiunta in un tempo com-preso tra i 5 e i 30 anni e che in questo lasso tempo-rale il 40% dei pazienti riceve diagnosi e trattamenti scorretti, se non interventi chirurgici non necessari (Eurordis, 2007). Mediante il WES vengono contem-poraneamente analizzate il 95% delle regioni codifi-canti dei circa 20.000 geni ipotizzati costituire il no-stro genoma (Patwardhan et al., 2015). Appare quindi chiaro quanto sia attraente un approccio diagnostico di questo tipo che in un breve lasso di tempo potreb-be fornire una diagnosi molecolare precisa, che può meglio guidare il successivo percorso assistenziale e permettere un preciso counseling genetico.
Quali risultati?Negli ultimi anni sono moltissimi i lavori che hanno cercato di dare risposta a questa domanda. Neveling et al. (2013) hanno confrontato i risultati ottenuti con approccio tradizionale (sequenziamento mediante metodo di Sanger di una serie di geni malattia noti) versus quelli ottenuti con WES in 5 categorie di pro-blematiche cliniche (sordità, cecità, disturbi del movi-mento, sospetta patologia mitocondriale e tumori del colon). A esclusione della patologia neoplastica, in tutte le altre categorie la detection rate di WES è stata significativamente maggiore del metodo tradizionale, con un aumento delle diagnosi di ben 4 volte nella categoria ipoacusia (Tab. I).Un’ampia casistica del Baylor College of Medicine, in cui 2000 pazienti consecutivi sono stati sottoposti a WES segnala il raggiungimento di una diagnosi nel 25% circa dei soggetti, con percentuali variabili tra il 20% per sintomi di base non neurologici e il 36% per specifiche categorie di problemi neurologici (atassia, disturbi del movimento) (Yang et al., 2014). In un pro-getto nazionale canadese (progetto FORGE) l’ana-lisi mediante WES di 362 famiglie ha portato a una caratterizzazione molecolare in 188 di esse (51,7%), di cui 105 (pari al 29%) con mutazione dimostrata in geni malattia noti e 83 in nuovi geni; inoltre in 28 fa-
Tabella I. Confronto tra detection rate di un approccio tradizionale (Sanger) e mediante WES.
Problema clinico Detection rate Sanger
Detection rate WES
N. di geni processati con Sanger
N. di geni processati con WES
Sordità 10% 44% 39 104
Cecità 25% 52% 46 164
Disturbi del movimento 5% 20% 32 150
Sospetta patologia mitocondriale 11% 16% 64 211
Tumore del colon 0% 3% 10 115

192
A. Di Cesare Merlone et al.
miglie vi era stata la definizione di una mutazione in un nuovo potenziale gene-malattia, per il quale i dati della letteratura non avevano consentito ancora una conferma assoluta del nesso di patogenicità (Sawyer et al., 2016). Sawyer et al. ben descrivono le ragioni di questo incremento diagnostico dovuto, nella maggio-ranza dei casi, a estrema eterogeneità genetica della condizione/problema di base, presentazione atipica della condizione stessa, estrema rarità della condi-zione, mancata diagnosi molecolare con metodologia tradizionale. Lo studio del trio (genitori + probando) permette di incrementare la detection rate rispetto allo studio del singolo paziente.Savarese et al. (2016) hanno recentemente pubblica-to i risultati di un’analisi, mediante piattaforma NGS (MotorPlex), in 504 pazienti affetti da miopatia o di-strofia muscolare non classificati, raggiungendo la definizione diagnostica in 218 (43,3%). In ulteriori 160 pazienti (31,7%) è stata rilevata una variante di si-gnificato patogenetico ancora incerto. È interessante segnalare come nel 30% dei casi la presentazione fenotipica fosse atipica, allargando quindi lo spettro di presentazione clinica noto per le mutazioni di quel-lo specifico gene. La Tabella II riassume una serie di ulteriori risultati di recenti pubblicazioni in riferimento ad approccio WES per l’inquadramento diagnostico di pazienti con specifiche problematiche pediatriche, soprattutto in ambito neurologico.A fronte di questi incoraggianti dati è bene ricordare che la stessa tecnica presenta anche dei limiti intrin-seci definiti. La copertura del genoma e/o della sua parte codificante (esoma) non è completa e omoge-nea. In un interessante lavoro di comparazione del grado di copertura di diverse piattaforme genomiche commerciali, Patwardham et al. (2015) sottolineano correttamente la necessità, da parte del clinico, di essere consapevole anche dei limiti delle tecniche applicate. Il livello di copertura insufficiente di alcune regioni genomiche contenenti informazioni genetiche rilevanti potrebbe essere infatti alla base di un risulta-to falsamente negativo del test diagnostico richiesto.
Quali ulteriori conseguenze?Nel loro report Valencia et al. (2015) sottolineano un aspetto importante del problema: quello del risparmio economico conseguente all’uso del WES. Riportando
infatti la loro esperienza sui primi 40 casi studiati con WES (con una detection rate del 30%) vanno ad ana-lizzare il numero di test genetici eseguiti dai soggetti diagnosticati prima di accedere a WES (chromosomal microarray analysis, sequenziamento di singoli geni o pannelli di geni). La numerosità di questi test permet-te di sostenere che il costo legato alla loro esecuzione supera di gran lunga il costo del WES stesso. In linea con questo filone di pensiero Shashi et al. (2014) han-no dimostrato come, nell’ambito delle malattie rare, una diagnosi corretta sul piano clinico viene solita-mente posta al massimo alla prima o alla seconda valutazione clinica. La probabilità di diagnosi crolla invece con le successive valutazioni. Ciò consente agli autori di suggerire un algoritmo diagnostico, in cui array CGH e WES diventano o test di prima scelta, in assenza di diagnosi cliniche convincenti, o imme-diatamente successivi a un primo tentativo di confer-ma molecolare, qualora una diagnosi clinica venisse formulata con sufficiente convinzione. Ovviamente il campo di utilizzo delle nuove tecnologie può spaziare ampiamente. Petrikin et al. (2015) riportano ad esem-pio l’esperienza dell’utilizzo di un approccio mediante WGS in una terapia intensiva neonatale di IV livello, su 35 neonati con presentazione clinica variabile, in cui l’ipotesi di una condizione geneticamente deter-minata era fortemente sospettata, pur in assenza di ipotesi diagnostiche convincenti. Grazie alla WGS, il 57% dei pazienti ha ottenuto una diagnosi definitiva permettendo di prendere decisioni terapeutiche mi-rate sul piano medico e/o chirurgico (precision neo-natology) o fornendo, alternativamente, una diagnosi che permettesse di indirizzare il neonato stesso verso un percorso di cure palliative. Il dato ancor più rilevan-te è che quasi la metà delle diagnosi raggiunte (45%) non erano state prese in considerazione nella diagno-si differenziale clinica dei neonati stessi.
Quali nuovi scenari si aprono?Più autori propongono fortemente l’utilizzo delle tecni-che genomiche come primo livello di diagnosi di fron-te al bambino con sospetta patologia monogenica. D. Levenson (2016), all’interno della sessione testing update dell’American Journal of Medical Genetics, cita a sostegno di questa proposta un report di prossi-ma pubblicazione su Genetics in Medicine, da parte
Tabella II. Detection rate di WES in casistiche di pazienti con problematica clinica di base similare.
Problema studiato Numero di pazienti analizzati
Detection rate
Microcefalia e disabilità intellettiva 38 29% Rump et al., 2016
Artrogriposi 17 58,3% Bayram et al., 2016
Patologia cerebellare e disabilità intellettiva 18 50% Megahed et al., 2016
Disabilità intellettiva ed encefalopatia epilettica 43 32,5% Thevenon et al., 2016

193
Focus tematici per la genetica clinica pediatrica
di Stark et al., che giungono a una diagnosi eziologi-ca nel 57% di 80 bambini con sospetta patologia ge-netica (Stark et al., 2016). Sulla scia di queste ipotesi, Douzgou et al. (2016) sostengono che nel prossimo futuro i cambiamenti tecnologici nell’area della ge-nomica modificheranno notevolmente anche il ruolo dei servizi di dismorfologia pur senza sminuirne l’im-portanza. In futuro infatti i genetisti clinici avranno un nuovo decisivo ruolo, all’interno di team multidiscipli-nari, volto a comparare e combinare i risultati dei test genetici in relazione al fenotipo del paziente oltre a mantenere un compito importante nel management e nel possibile futuro trattamento. Di sicuro interes-se a questo proposito l’analisi di Bowdin et al. (2016), che discutono nel dettaglio sfide, opportunità e limiti dell’integrazione della medicina genomica nella prati-ca clinica pediatrica, proponendo un percorso di rife-rimento denominato SickKids Genome Clinic, con lo scopo di analizzarne sicurezza, efficacia e possibilità concreta di implementazione. Il percorso parte dal medico referente (genetista clinico o specialista d’or-gano) e a lui ritorna, dopo un lungo e complesso iter genomico e bio-informatico, per l’interpretazione, la validazione e il successivo utilizzo dei risultati ottenuti.
È sempre e solo tutto semplice e lineare?Tutti i lavori che approfondiscono il tema in oggetto non dimenticano di citare i due elementi problemati-ci cruciali legati all’avvento della nuova tecnologia: i cosiddetti incidental findings (riscontri accidentali di mutazioni in geni responsabili di condizioni non corre-late al fenotipo di partenza e spesso legate a malattie oncologiche e/o degenerative a esordio in età adulta) e il riscontro di varianti di significato incerto, per le quali non sia possibile avere un’interpretazione pre-cisa, sia perché il gene stesso non è stato preceden-temente associato a sindromi genetiche, sia perché vi è incertezza sul significato patogenetico della va-riante identificata. Il primo problema apre un dibattito molto ampio sul consenso informato e sulla necessità di un’accurata informazione pre-test; il secondo, oltre a riprendere l’importanza di un’accurata spiegazione delle possibili risultanze dell’analisi molecolare, con-ferma la potenziale non definitività dell’esito dell’ana-lisi genomica, suscettibile di variazioni interpretative nel tempo, legate all’acquisizione di nuove conoscen-ze.Riteniamo di interesse però citare in conclusione il lavoro di Krabbengorg et al. (2016), che analizza gli effetti psico-sociali dei risultati dell’applicazione del WES su genitori di bambini con sospetta malattia genetica. Gli autori hanno eseguito interviste semi-strutturate a famiglie di 15 pazienti sottoposti a WES con esiti differenti (diagnosi posta, diagnosi dubbia, nessuna diagnosi). Essi dimostrano come le ricadu-te psico-sociali degli esiti del test non siano sempre e necessariamente positive. In caso di mancata dia-gnosi o di interpretazione non conclusiva, infatti, il
quadro di incertezza per il futuro resta inalterato, au-mentando semmai il senso di frustrazione. Ma anche in caso di raggiungimento di una definizione alla fine dell’odissea diagnostica segue l’inizio di una nuova e altrettanto estenuante odissea per la ricerca di in-formazioni sulla rara/rarissima patologia del figlio, di nuovi riferimenti assistenziali e di contatto, spesso estremamente difficile, con genitori che stanno viven-do la medesima esperienza. Secondo gli autori tutte queste istanze devono essere ampiamente discus-se in fase di pre-test, al fine di preparare la famiglia anche da questo punto di vista ai risultati dell’analisi genomica.
Sindromi genetiche e tumoriQuale background biologico?Dall’inizio degli anni Settanta gli studi sull’oncogene-si hanno condotto a formulare ipotesi a supporto di una origine genetica del cancro (two hits hypothesis di Knudson). La cellula, nel processo di oncogene-si, accumula difetti che modificano la sua capacità di differenziazione, di riparo dei danni subiti, di andare incontro ad apoptosi, di moltiplicarsi in risposta a uno stimolo. Secondo la revisione di Bellacosa (2013) l’oncogenesi segue un percorso biologico opposto rispetto al pro-cesso di sviluppo di un organismo, in cui a partire da un piccolo numero di precursori indifferenziati, attraver-so l’attivazione di specifici geni, si arriva alla formazio-ne di diversi tessuti e organi con funzioni specializzate. I difetti dello sviluppo di un organismo che portano a sindromi genetiche e i difetti che si generano durante l’oncogenesi, da condizioni apparentemente distanti vengono così a intersecarsi e talvolta a sovrapporsi. Nell’ambito di condizioni secondarie a mutazioni ger-minali potremo quindi trovarci di fronte a:- difetti genomici che si esprimono quasi esclusiva-
mente (es. coesinopatie) o prevalentemente (RA-Spatie) con anomalie dello sviluppo e con lieve incremento del rischio oncologico;
- difetti che comportano sia significative alterazioni dello sviluppo che altrettanto significativo aumento del rischio oncologico (facomatosi, sindrome Pro-teus, sindromi con iperaccrescimento);
- difetti genomici che hanno una espressione pre-valente in termini di oncogenesi e incidono meno significativamente sullo sviluppo (sindromi auto-somiche recessive con fragilità cromosomica, sin-dromi con predisposizione al cancro autosomiche dominanti) (Fig. 1).
Diversi studi più o meno recenti hanno analizzato perché una sindrome possa esprimersi anche attra-verso una “predisposizione al cancro” (Marshall et al., 2014). È infatti ipotizzabile che la presenza di una mu-tazione genomica germinale possa alterare l’omeo-stasi e la stabilità genomica cellulare, aumentando la suscettibilità allo sviluppo di tumori. D’altra parte sono

194
A. Di Cesare Merlone et al.
note patologie in cui geni e vie metaboliche, le cui anomalie causano sindromi (con mutazione germina-le) e tumore (con mutazione somatica), coincidono. In questi casi la mutazione germinale ha spesso un effetto meno “potente” rispetto all’analoga mutazione somatica, che è caratteristica delle forme sporadiche di neoplasia (es. mutazione somatica e germinale di PTPN11, rispettivamente nella leucemia mielomono-citica giovanile e nella sindrome di Noonan) (Kratz et al., 2005).
Quali dati epidemiologici?Anche al di là di specifiche sindromi cancer predi-sposing, uno sguardo ai dati epidemiologici mostra come le relazioni tra difetti congeniti e tumori siano più che strette. Zhang et al. (2015) hanno stimato che la prevalenza di mutazioni germinali in bambini affetti da cancro è intorno all’8-10%, mentre Schiffman et al. (2013) hanno dimostrato che circa il 25% delle neo-plasie “sporadiche” dell’infanzia può essere legato a un difetto genetico congenito. Già da qualche anno sono noti studi che hanno evidenziato una maggio-re prevalenza di malformazioni congenite in pazienti pediatrici affetti da patologia tumorale rispetto ai con-trolli. Dati più recenti hanno confermato questa asso-ciazione, identificando sia categorie di difetti conge-niti a maggior rischio di associazione con neoplasia (microcefalia, anomalie del sistema nervoso centrale, cardiopatie congenite, schisi palatine, anomalie ocu-lari), sia tipologie di neoplasie maggiormente osser-vate in questi pazienti (tumori del sistema nervoso centrale, neuroblastoma, linfoma e tumori a cellule germinali) (Fisher et al., 2012; Botto et al., 2013). È stata anche evidenziata una correlazione positiva tra maggior rischio oncologico e patologia cromosomica (Botto et al., 2013), anche al di là del noto legame tra neoplasie e sindrome di Down. Altri recenti stu-di hanno ulteriormente dettagliato l’associazione tra specifiche sindromi genetiche e tumori in età pediatri-ca (Zimmerman et al., 2015; Kratz et al., 2015; Varan et al., 2016).
Quali ricadute per la pratica clinica quotidiana?Da tempo è nota l’associazione tra specifiche con-dizioni sindromiche e specifiche tipologie di neopla-sia. Prototipo di questa categoria di sindromi è certa-mente la sindrome di Beckwith-Wiedemann (BWS). In questo senso da tempo sono disponibili protocolli di follow-up che definiscono tipologia e tempistica di accertamenti necessari a giungere a una diagnosi precoce dell’eventuale neoplasia insorta. La pubbli-cazione di Mussa et al. (2016), del Comitato Scienti-fico dell’Associazione Italiana della sindrome stessa, elenca analiticamente queste raccomandazioni. Ciò non sempre è però possibile, in quanto esistono altre condizioni, in cui l’associazione tra sindrome e for-me tumorali è nota ma più eterogenea, impedendo la definizione di protocolli validati (es. sindrome di Rubinstein-Taybi).La BWS rappresenta un interessantissimo modello della relazione tra rischio oncologico e dati genetici (o epigenetici). Negli ultimi anni alcuni lavori hanno infatti dimostrato un’asimmetrica distribuzione del ri-schio oncologico in funzione del difetto molecolare di base (Brioude et al., 2013; Ibrahim et al., 2014; Mussa et al., 2015). Assai recentemente due di questi gruppi hanno quindi proposto due nuovi approcci al follow-up per rischio oncologico dei pazienti affetti da questa condizione (Mussa et al., 2016; Maas et al., 2016). Le Tabelle III e IV li descrivono.Alla luce di questo stretto legame bidirezionale tra pa-tologia tumorale e genetica, da più parti è stato sot-tolineato come ogni bambino affetto da neoplasia do-vrebbe teoricamente essere valutato da un genetista clinico. Poiché ciò può non essere sempre possibile, Hopman et al. (2013) hanno sviluppato uno strumen-to di screening, basato sul lavoro di più esperti, che dovrebbe aiutare, il clinico che segue il bambino af-fetto da tumore, a identificare alcuni indicatori clinici che potrebbero far ipotizzare che la neoplasia possa essere insorta in un quadro più ampio di sindrome genetica. In questa direzione si sviluppa anche il la-
Figura 1. Difetti genomici: continuum tra anomalia dello sviluppo e rischio oncologico.
Anomalia dello sviluppo
Coesinopatie
Ras-patie
pRoteus, FaComatosi, sD Con ipeRaCCResCimento
sinDRomi Con FRagilità CRomosomiCa e autosomiChe Dominanti cancer predisposing
Rischio oncologico

195
Focus tematici per la genetica clinica pediatrica
voro di Jongmans et al. (2016), che suggerisce di va-lorizzare segni clinici quali la presenza di discromie cutanee, emipertrofia e/o iperaccrescimento-scarsa crescita, dismorfismi facciali, disabilità intellettiva, malformazioni congenite, magari associate a patolo-gie ematologiche e/o deficit immunitario, come segni di allarme per la presenza di una sindrome cancer predisposing.
Quale supporto dalle nuove tecnologie di studio del genoma?Nel novembre 2015 Zhang et al. hanno pubblicato sul New England Journal of Medicine un ampio e inte-ressante studio, in cui è stata eseguita analisi di WES, WGS o entrambe alla ricerca di mutazioni germinali in 1120 bambini affetti da neoplasia. Nell’8,5% dei casi è stata ritrovata una mutazione patogenetica o vero-
similmente patogenetica, nella maggioranza dei casi, insorta de novo. Sebbene molti dei riscontri si siano rivelati ben noti, le analisi genomiche hanno consen-tito di rilevare possibili nuove associazioni eziopato-genetiche tra mutazioni di specifici geni e sviluppo di specifiche neoplasie, da confermare con successivi studi. L’indagine ha evidenziato, in un ulteriore 3,4% di pazienti, mutazioni eterozigoti verosimilmente pa-togenetiche in geni malattia predisponenti al cancro a espressione autosomica recessiva e, nel 9,7% dei casi, mutazioni in altri geni di suscettibilità al cancro, oltre a un numero elevato di varianti di incerto signifi-cato patogenetico. Lo studio ha infine evidenziato che l’anamnesi familiare risulta uno strumento di limitato valore nell’identificare la presenza di una condizione genetica “predisponente al cancro”. La percentuale di positività della storia familiare era infatti sovrapponi-
Tabella III. Follow-up del rischio oncologico in rapporto al difetto molecolare di base in pazienti affetti da BWS (da Maas et al., 2016, mod.).
(Epi)genotipo BWS Screening oncologico proposto
Ipometilazione ICR2 (50% dei casi) Nessuno
Ipermetilazione ICR1 (5-10% dei casi) Tumore di Wilms: ecografia renale ogni 3 mesi fino a 5 anni
Mutazioni CDKN1C (5% dei casi) Tumore di Wilms: ecografia renale ogni 3 mesi fino a 5 anni (facoltativo)Epatoblastoma: ecografia addominale ogni 3 mesi fino a 4 anni (facoltativo)Neuroblastoma: catecolamine urinarie ed ecografia addominale ogni 3 mesi fino a 3 anni (facoltativo)
UPD (20-25% dei casi) o difetto molecolare non identificato (15% dei casi)
Tumore di Wilms: ecografia renale ogni 3 mesi fino a 5 anni Epatoblastoma: ecografia addominale ogni 3 mesi fino a 4 anni
ICR1: Imprinting control region 1 a livello della regione cromosomica 11p15.5ICR2: Imprinting control region 2 a livello della regione cromosomica 11p15.5UPD: Disomia Uniparentale Paterna del cromosoma 11p15.5CDKN1C: gene dell’inibitore 1C della chinasi ciclino-dipendente
Tabella IV. Follow-up del rischio oncologico in rapporto al difetto molecolare di base in pazienti affetti da BWS (da Mus-sa et al., 2016 mod.).
(Epi)genotipo BWS Screening oncologico proposto
Ipometilazione ICR2 (> 50% dei casi) Visita medica ogni 4-6 mesi fino all’adolescenza, valutando indicazione a esami supplementari in rapporto a eventuale sospetto specifico
Ipermetilazione ICR1 (5-10% dei casi) Tumore di Wilms: ecografia renale ogni 3 mesi fino a 8-10 anni
Mutazioni CDKN1C (5% dei casi) Neuroblastoma: catecolamine urinarie ed ecografia addominale ogni 4-6 mesi fino a età da definire
UPD (20% dei casi) Tumore di Wilms: ecografia renale ogni 3 mesi fino a 8-10 anni Epatoblastoma: ecografia addominale e dosaggio plasmatico alfa-feto proteina ogni 3 mesi fino a 5 anniCarcinoma surrenalico: ecografia dei surreni ogni 3 mesi fino a età da definire, dosaggio plasmatico DHEAS ogni 3 mesi fino a 5 anni
ICR1: Imprinting control region 1 a livello della regione cromosomica 11p15.5ICR2: Imprinting control region 2 a livello della regione cromosomica 11p15.5UPD: Disomia Uniparentale Paterna del cromosoma 11p15.5CDKN1C: gene dell’inibitore 1C della chinasi ciclino-dipendenteDHEAS: deidroepiandrosterone solfato

196
A. Di Cesare Merlone et al.
bile, intorno al 40%, sia nei soggetti con mutazioni in geni predisponenti al cancro a trasmissione autoso-mica dominante, sia nei soggetti che non presentava-no mutazioni. Questo ampio studio riassume in sé tutte le potenzia-lità e i limiti dell’applicazione della nuova tecnologia genomica in ambito oncologico. Se da un lato infat-ti queste possibilità di caratterizzazione molecolare offrono informazioni utili in termini di approccio tera-peutico, di applicazione di protocolli di follow-up pre-ventivo e di counseling familiare dall’altro le lacune interpretative restano ancora numerose e ampie.In quest’ottica si pongono le indicazioni dell’American Society of Clinical Oncology, che sottolinea come a oggi, nel proporre l’esecuzione di un approfondimen-to genomico sia a livello somatico che germinale, è bene considerare i potenziali esiti anche incidentali del test stesso, al fine di ottenere informazioni real-mente interpretabili e fruibili sia per il paziente che per la sua famiglia; viene inoltre sottolineata con forza l’assoluta importanza di un adeguato counseling pre- e post-test al fine di discutere rischi, potenzialità, limiti
e benefici di indagini di questo genere (Kesserwan et al., 2016; Robson et al., 2015).
Take home message Alla luce di quanto noto e più di recente pubblicato è sin troppo chiaro che le interrelazioni tra genetica clinica pediatrica e oncologia sono profonde. La con-divisone di informazioni tra le due branche ha già per-messo, in parte, di chiarire meccanismi eziopatoge-netici che sono alla base sia di diversi tipi di neoplasia che di condizioni sindromiche. Ci si aspetta che que-sto fecondo travaso di informazioni possa aumentare nel prossimo futuro. Da quanto si può intravedere da questo confronto potrebbero emergere anche fonda-mentali spunti per implementare strategie terapeuti-che mirate alla cura di diverse forme sindromiche, a partire da esperienze già note in ambito oncologico, come per esempio nel caso dell’utilizzo dell’everoli-mus nel trattamento dell’epilessia refrattaria o dell’a-strocitoma subependimale nell’ambito della sclerosi tuberosa (Franz et al., 2014; Krueger et al., 2013).
Bibliografia
Bayram Y, Karaca E, Akdemir ZC, et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J Clin Invest 2016;126:762-78.
Bellacosa A. Developmental disease and cancer: biological and clinical overlaps. Am J Med Genet 2013;161A:2788-96.
** Review che esplicita la continuità esistente tra difetti dello sviluppo e cancro e in quest’ottica delinea possibili futuri ap-procci terapeutici.
Botto LD, Flood T, Little J, et al. Cancer risk in children and adolescents with birth defects: a population-based cohort study. Plos one 2013;8:e69077.
** Ampio studio epidemiologico su inciden-za di neoplasia in pazienti con difetti congeniti.
Bowdin SC, Hayeems RZ, Monfared N, et al. The SickKids genome clinic: developing and evaluating a pediatric model for individualized genomic medicine. Clin Genet 2016;89:10-9.
** Ipotesi analitica di un approccio multidisciplinare all’utilizzo della medicina genomica in pediatria.
Brioude F, Lacoste A, Netchine I, et al. Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to mo-lecular mechanism, and guidelines for tumor surveillance. Horm Res Pediatr 2013;80:457-65.
Douzgou S, Chervinsky E, Gyftodimou Y, et al. Dysmorphology services: a snapshot of current practices and a vision for the fu-ture. Clin Genet 2016;89:27-33.
Eurordis. Survey of the delay in diagno-sis for 8 rare diseases in Europe. Euror-disCare2, 2007. http://archive. eurordis.
Box di orientamento
• Cosa sapevamo primaIl sequenziamento dell’esoma consente di indagare simultaneamente una notevole quantità di informa-zioni genetiche. È nota da tempo l’esistenza di sindromi genetiche con spicccata e definita predisposi-zione allo sviluppo di neoplasie.
• Cosa sappiamo adessoIl calo dei costi relativi all’esecuzione del WES, le sue importanti potenzialità e i risultati derivanti da vari studi in ambito clinico permettono di considerare questa tecnica come prioritaria nello studio di pazienti con sospetta malattia genetica, con un potenziale risparmio di tempo e costi. I rapporti tra mutazioni germinali e patologia tumorale, in aggiunta ai dati più recenti in ambito epidemiologico, giustificano una ricerca attenta in ambito clinico-genetico di fronte a un numero maggiore di bambini affetti da “neoplasia isolata”.
• Quali ricadute sulla pratica clinicaIl WES potrà entrare a breve nel paniere delle disponibilità tecnologiche del genetista clinico di fronte al pa-ziente con sospetta condizione sindromica, tanto più quanto eterogenee saranno le possibilità interpretative cliniche. Il bambino con “neoplasia isolata” dovrà essere maggiormente oggetto di approfondimento gene-tico (clinico e/o molecolare), alla ricerca di una potenziale condizione costituzionale cancer predisposing.

197
Focus tematici per la genetica clinica pediatrica
org/article.php3?id_article=454 (access February 1, 2012).
Fisher PG, Reinolds P, Von Behren J, et al. Cancer in children with nonchromosomal birth defects. J Pediatr 2012;160:978-83.
Franz DN, Belousova E, Sparagana S, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous scle-rosis complex: 2-year open-label extension of the randomized EXIST-1 study. Lancet Oncol 2014;15:1513-20.
Hopman SMJ, Merks JHM, de Borgie CAJM, et al. The development of a clinical screening instrument for tumor predispo-sition syndromes in childhood cancer pa-tients. EJC 2013;49:3247-54.
Ibrahim A, Kirby G, Hardy C, et al. Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics 2014;6:11-20.
Jongmans MCJ, Loeffen JLCM, Waanders E, et al. Recognition of genetic predisposition in pediatric cancer patients: an easy-to-use selection tool. Eur J Med Genet 2016;59:116-25.
Kesserwan C, Friedman Ross L, Brad-bury AR, et al. The advantages and chal-lenges of testing children for heritable pre-disposition to cancer. Am Soc Clin Oncol Educ Book 2016;35:251-69.
Krabbenborg L, Schieving J, Kleefstra T, et al. Evaluating a counseling strategy for diagnostic WES in paediatric neurol-ogy: an exploration of parents’ informa-tion and communication needs. Clin Genet 2016;89:244-50.
** Lavoro interessante che analizza in modo pratico le ricadute, anche problemat-iche, di un approccio di WES sulla stabilità emotiva dei genitori.
Kratz CP, Niemeyer CM, Castelberry RP, et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative dis-ease. Blood 2005;106:2183-5.
Kratz CP, Franke L, Peters H, et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cu-taneous syndromes. BJC 2015;112:1392-7.
Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in Tuberous sclerosis complex. Ann Neurol 2013;74:679-87.
Levenson D. Whole-exome sequencing strategy proposed as first-line test. Am J Med Genet 2016;Testing update:1387-8.
Maas SM, Vansenne F, Kadouch DJM, et al. Phenotype, cancer risk, and survillance in Beckwith-Wiedemann Syndrome de-pending on molecular genetic subgroups. Am J Med Genet 2016;170A:2248-60.
Marshall GM, Carter DR, Cheung BB, et al. The prenatal origins of cancer. Nat Rev Cancer 2014;14:277-89.
Megahed H, Nicouleau M, Barcia G, et al. Utility of whole exome sequencing for the early diagnosis of pediatric-onset cerebel-lar atrophy associated with developmental delay in an inbred population. Orph J Rare Dis 2016;11:57.
Mussa A, Di Candia S, Russo S, et al. Recommendations of the Scientific Com-mittee of the Italian Beckwith-Wiedemann Syndrome Association on the diagnosis, management and follow-up of the syn-drome. Eur J Med Genet 2016;59:52-64.
Mussa A, Molinatto C, Baldassarre G, et al. Cancer risk in Beckwith-Wiedemann Syndrome: a systematic review and meta-analysis outlining a novel (epi)genotype specific histotype targeted screening pro-tocol. J Pediatr 2016;176:142-9.
Neveling K, Feenstra I, Gilissen C, et al. A post-hoc comparison of the utility of Sanger Sequencing and exome sequencing for the diagnosis of heterogeneous diseas-es. Human Mutat 2013;34:1721-6.
Patwardhan A, Harris J, Leng N, et al. Achieving high-sensitivity for clinical appli-cations using augmented exome sequenc-ing. Genome Medicine 2015;7:71-85.
Petrikin JE, Laurel KW, Smith LD, et al. Rap-id whole genome sequencing and precision neonatology. Semin Perinat 2015;623-31.
Robson ME, Bradbury AR, Arun B, et al. American society of clinical oncology poli-cy statement update: genetic and genomic testing for cancer susceptibility. J Clin On-col 2015;33:3660-7.
** Analisi accurata e preziosa di un ap-proccio genomico allo studio germinale e somatico del paziente con neoplasia.
Rump P, Jazayeri O, van Dijk-Bos KK, et al. Whole-exome sequencing is a power-ful approach for establishing the etiologi-cal diagnosis in patients with intellectual disability and microcephaly. BMC Medical Genomics 2016;9:7.
Savarese M, Di Fruscio G, Torella A, et al. The genetic basis of undiagnosed muscu-lar dystrophies and myopathies. Neurology 2016;87:71-6.
Schiffman JD, Geller JI, Mundt E, et al. Update on pediatric cancer predisposi-tion Syndromes. Pediatr Blood Cancer 2013;60:1247-52.
** Efficace revisione sulle condizioni sindromiche cancer predisposing.
Sawyer SL, Hartley T, Dyment DA, et al. Utility of wholw-exome sequencing for those near the end of the diagnostic od-yssey: time to address gaps in care. Clin Genet 2016;89:275-84.
Shashi V, McConckie-Rosell A, Rosell B, et al. The utility of the traditional medi-cal genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet Med 2014;16:176-82.
** Interessante proposta metodologica che integra le nuove tecnologie nel percor-so di diagnosi del bambino con sospetta malattia genetica.
Thevenon J, Duffourd Y, Masurel-Paulet A. Diagnostic odyssey in severe neurode-velopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin Genet 2016;89:700-7.
Valencia CA, Husami A, Holle J, et al. Clinical impact and cost-effectiveness of whole-exome sequencing as a diagnostic tool: a pediatric center’s experience. Front Pediatr 2015;3:67.
** Preziosa analisi della ricaduta anche economica di un’applicazione di WES nella diagnostica pediatrica.
Varan A, Sen H, Aydin B, et al. Neurofi-bromatosis type 1 and malignancy in child-hood. Clin Genet 2016;89:341-5.
Yang Y, Muzny DM, Xia F, et al. Molecu-lar findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312:1870-9.
** Casistica di grande rilevanza numeri-ca per la valutazione della detection rate di WES nella diagnostica routinaria.
Zhang J, Walsh MF, Wu G, et al. Germline mutation in predisposition genes in pediat-ric cancer. N Engl J Med 2015;2336-46.
** Ampio studio che, pur con alcuni lim-iti metodologici, analizza la predisposizione costituzionale in bambini con patologia on-cologica “isolata”.
Zimmerman R, Schimmenti L, Spec-tor L. A catalog of genetic syndromes in childhood cancer. Pediatr Blood Cancer 2015;62:2071-5.
Corrispondenza
Angelo SelicorniUnità Operativa Complessa di Pediatria, ASST Lariana, via Ravona 20, 22020 San Fermo della Battaglia (CO) E-mail: [email protected]

198
Luglio-Settembre 2016 • Vol. 46 • N. 183 • Pp. 198-205 Prospettive in Pediatria
Genetica clinica pediatrica
Le problematiche del sonno nelle sindromi malformative: un problema spesso sottostimato
Giuseppe Zampino1 Roberta Onesimo1
Chiara Leoni1 Silvia Tajè2
Giacomo della Marca3
1UOSA, Malattie Rare e Difetti Congeniti, Polo per la Salute della Donna e del Bambino,
Fondazione Policlinico Universitario “A. Gemelli”, Roma;
2UOC Pediatria ASST Lariana, Como; 3Laboratorio Medicina del
Sonno, Istituto di Neurologia, Fondazione Policlinico
Universitario “A. Gemelli”, Roma
Sleep disorders frequently complicate the life of children with malformation syndromes and their families. The problem is often underestimated. Numerous pathogenetic mecha-nisms are responsible for sleep disorders in patients with these conditions. Such genetic defects can modify neurotransmitters or hormones involved in circadian rhythm, ventila-tor control pattern, craniofacial development and upper airway neuromuscular or con-nective functions with a reduction in the respiratory airway. The comprehension of these mechanisms will allow better management of sleep disorders to avoid variables degrees of cognitive and performance deficits and to reduce behavioural and cardiac problems.
Summary
Le patologie del sonno complicano frequentemente la vita dei bambini con sindrome mal-formativa e delle loro famiglie. Spesso il problema non è diagnosticato. Diversi meccanismi patogenetici sono alla base delle problematiche di sonno nelle numerose sindromi mal-formative. Il difetto genico può determinare modificazioni di neurotrasmettitori, di ormoni coinvolti nel processo del sonno, indurre anomala crescita craniofacciale e dei tessuti molli faringei, che generano riduzione del flusso di aria, o alterare il tessuto connettivo rendendo le pareti più lasse. La comprensione dei meccanismi permette di affrontare il problema con le strategie di trattamento più adeguate, al fine di ridurre le complicanze legate alla patolo-gia del sonno, quali le difficoltà di apprendimento, i disturbi comportamentali e cardiologici.
Riassunto
Metodologia della ricerca bibliografica effettuataLa ricerca degli articoli rilevanti su sindromi genetiche e patologia del sonno è stata effettuata nella banca bi-bliografica Medline, utilizzando come motore di ricer-ca PubMed e come parole chiave: “sleep and genetic syndrome” per la parte generale, e poi incrociando “sleep” con le seguenti sindromi: “Angelman”, “Fragile-X”, “Smith-Magenis”, “Down”, “Marfan”, “Ehlers-Danlos”, “Craniosynostosis”, “Craniofacial anomalies”, “Achon-droplasia”, “Treacher-Collins”, “Beckwith-Wiedemann”. Non sono stati usati limiti di età, ma nella scelta degli articoli sono stati esaminati quelli di carattere gene-
rale o che si riferivano alla popolazione pediatrica, preferibilmente negli ultimi 5 anni. Sono stati selezio-nati articoli originali, revisioni, clinical trials, studi epi-demiologici anche antecedenti agli ultimi 5 anni, se ritenuti utili alla stesura della revisione.
Obiettivo• Effettuare una revisione delle problematiche di
sonno nelle sindromi malformative, scegliendo quelle che per frequenza e intensità sono mag-giormente coinvolte.
• Dare indicazioni di trattamento farmacologico inrelazione alla tipologia di disturbo di sonno.

199
Sonno e sindromi malformative
• Dareuno spaccatodei diversimeccanismipato-genetici che sottendono i disturbi respiratori nel sonno e delle condizioni maggiormente coinvolte, con un orientamento sulle possibilità di trattamen-to conservativo o chirurgico.
IntroduzioneLa patologia del sonno, in particolare i disturbi respi-ratori, è frequente nella popolazione pediatrica. Nei bambini con sindromi malformative la portata del problema è particolarmente importante, sia in termini di frequenza sia in termini di gravità. Però in questi bambini il peso del processo diagnostico da affronta-re, delle molteplici problematiche cliniche da gestire e delle diverse ripercussioni sociali che la condizione determina, fa sottostimare la percezione dei problemi connessi al sonno (Agriman et al., 2015). La patologia del sonno ha un enorme impatto sulla salu-te non solo del bambino, inducendo un’ulteriore variabile compromissione delle abilità cognitive, ponendo un’ulte-riore spina irritativa nel comportamento o aumentando il rischio di cor polmonare, ma anche della famiglia, che soffre di un’importante peggioramento della qualità della vita anche per la riduzione delle ore di sonno.
ClassificazioneLa classificazione dei disturbi del sonno è necessaria per discriminare le diverse patologie e facilitare la com-prensione dei sintomi, dell’eziologia e del trattamento.L’ipersonnia è definita come incapacità a stare in allerta e svegli durante il giorno. Il risultato sono fre-quenti colpi di sonno.Le insonnie sono caratterizzate da difficoltà a iniziare e/o mantenere il sonno, con numerosi risvegli e una durata insufficiente di sonno. In genere i pazienti pre-sentano un sonno di scarsa qualità e non ristorativo.Le parasonnie sono eventi fisici ed esperienziali non desiderati, che avvengono durante il sonno. Consisto-no in anomalie che riguardano movimenti, comporta-menti e funzioni del sistema autonomico. Le discronosi sono disordini del ritmo circadiano del sonno e consistono in un disallineamento tra le abi-tudini di sonno del paziente e quelle della società. I pazienti non riescono a dormire quando il sonno è desiderato, necessario o aspettato. I disturbi respiratori del sonno sono un gruppo che include apnee centrali, dovute a disfunzioni che non fanno partire lo stimolo del respiro, e apnee ostruttive dovute a una resistenza al flusso d’aria nelle vie ae-ree superiori con incremento dello sforzo inspiratorio e ventilazione alterata (Fig. 1, Box 1 e 2).
Sonno e sindromiNelle diverse patologie sindromiche differenti mec-canismi patofisiologici sono alla base della patologia
del sonno. In seguito vengono presentate le sindromi, che per la portata del problema e per le caratteristi-che, sono le più rappresentative delle differenti ano-malie del sonno.
Sindrome di AngelmanLa sindrome di Angelman, causata dalla perdita di espressione del gene UBE3A, è caratterizzata da ri-tardo mentale, epilessia, atassia nel cammino, defi-cit di linguaggio e dismorfismi facciali. Per la elevata frequenza, i problemi di sonno sono inclusi nei criteri diagnostici. Diversi studi evidenziano una prevalenza del problema, che va dal 50% al 90% dei casi. Le problematiche descritte riguardano la riduzione del tempo totale di sonno, dovuta a difficoltà a iniziare il sonno, disorganizzazione dell’architettura del sonno con frequenti risvegli notturni, riduzione del sonno REM. Sono descritte diverse parasonnie quali perio-dici movimenti delle gambe, bruxismo, incubi. Le problematiche di sonno diventano più marcate tra i 2 e i 6 anni di età, si riducono tra i 6-8 anni, fino alla quasi completa scomparsa nell’età adulta. Questo potrebbe essere dovuto a una maturazione neurofi-siologica o alla comparsa nel tempo di fattori protettivi. Si considera che i problemi di sonno nella SA riflet-tono la disregolazione della influenza dei mediatori inibitori GABA nell’interazione talamocorticale, anche se le ricerche che definiscono il meccanismo patoge-netico nei problemi di sonno dei bambini con SA non sono ancora conclusive.Buona parte dei bambini con AS è in trattamento con valproato di sodio, farmaco che determina soppres-sione nella produzione di melatonina. Ma alcuni stu-di hanno notato che bassi livelli di melatonina erano presenti anche nei bambini non trattati con valproato e che la somministrazione di melatonina ha portato un miglioramento del sonno. Al momento l’evidenza dell’efficacia della melatonina non è ancora robusta (Pelc et al., 2008).
Sindrome dell’X-FragileLa sindrome dell’X-Fragile, è la più comune forma di ritardo mentale ereditario. Dovuta a una espansione delle triplette CGG che rende inattivo il gene FMR1, con perdita della proteina FMRP particolarmen-te espressa nel cervello e nei testicoli. La perdita di FMRP causa anomalie di segnale di neurotrasmet-titori che potrebbero essere responsabili del ritardo mentale. Studi molecolari ed elettrofisiologici nel topo hanno dimostrato come la presenza di tale proteina è importante nel regolare il ritmo circadiano del sonno e di altri comportamenti (Zhang et al., 2008). I disturbi del sonno sembrano avere una prevalenza dal 32-50% dei casi e sono caratterizzati essenzialmente da difficoltà a prendere sonno e frequenti risvegli duran-te la notte. Uno studio neurofisiologico ha indentificato in nove pazienti con Fra-X una riduzione del tempo di sonno totale, riduzione della percentuale di sonno REM

200
G. Zampino et al.
Figura 1. Definizione apnee (vedi Box 1 e 2).
Box 1. Classificazione degli eventi respiratori durante il sonno.
Ostruttivi
Apnea Assenza di flusso aereo oronasale di ogni durata con sforzo respiratorio persistente
Ipopnea Distinguibile riduzione del flusso oronasale per due o più respiri, con sforzo respiratorio persistente spesso accompagnato da desaturazione di O2 o risveglio
Sforzi respiratori che determinano risveglio
Incremento dello sforzo respiratorio con riduzione del flusso che determina risveglio, seguito da scomparsa dello sforzo e normalizzazione del flusso
Limitazioni di flusso Appiattimento del segnale inspiratorio sul rilevatore del flusso nasale
Russamento Rumori inspiratori grossolani
Ipoventilazione pCO2 > 50 mmHg per più del 10% del tempo totale di sonno o pCO2 > 53 mmHg e accompagnato da respiro paradosso o eventi ostruttivi
Centrali
Apnea Assenza di flusso aereo oronasale per almeno 20 secondi senza sforzo respiratorio; eventi più brevi sono considerati se associati a risveglio, desaturazione o bradicardia
Ipoventilazione pCO2 > 50 mmHg per più del 10% del tempo totale di sonno o pCO2 > 53 mmHg e accompagnato da riduzione dello sforzo respiratorio
Respiro periodico Successione di almento 3 apnee centrali di diversi secondi separati da un periodo di 20 secondi di respiro normale
Misti
Apnee miste Cessazione di flusso con componente centrale e ostruttiva
Apnea espiratoria prolungata con cianosi
Prolungamento dell’espirazione oltre la capacità funzionale residua
Respiro “apneustico” Marcato prolungamento dell’inspirazione

201
Sonno e sindromi malformative
e un incremento nella prima latenza REM e un aumento della percentuale dello stadio 3-4 NREM.
Sindrome di Smith-MagenisLa sindrome di Smith-Magenis (SMS) è una condi-zione caratterizzata da dismorfismi cranio-facciali, bassa statura e brachidattilia, ritardo mentale, ridot-ta sensibilità al dolore, disturbi del comportamento e disturbi del sonno. È dovuta alla perdita di funzione del gene RAI1, che può avvenire o con mutazioni che inattivano il gene (aploinsufficienza) o per una piccola delezione del cromosoma 17p11.2, dove è localizzato tale gene. La SMS rappresenta la sindrome del son-no, poiché è un criterio fondamentale nella diagnosi. I problemi di sonno possono configurarsi nelle di-scronosi, ovvero nei disturbi del ritmo sonno/veglia. I bambini hanno difficoltà a prendere sonno, hanno frequenti risvegli notturni, si svegliano presto e hanno necessità di fare pisolini durante il giorno. Anomalie del sonno REM sono state individuate nella metà dei casi e l’actigrafia ha indicato che i problemi di sonno iniziano intorno ai 6 mesi di età, con frammentazione del sonno e riduzione delle ore di sonno notturno di 1-2 ore rispetto alla popolazione di controllo, con un compenso fatto di pisolini diurni. La melatonina è un ormone prodotto dalla ghiandola pineale nel cervello e serve per regolare il ritmo son-no veglia, mediante una produzione che è alta la notte e si abbassa durante il giorno. Alcuni studi hanno rile-vato che la quasi totalità dei bambini con SMS hanno inversione del ritmo della melatonina, che rimane alta durante il giorno con un picco diurno; questo dato sia per la frequenza che per la particolarità è un segno della sindrome. Poiché la somministrazione di acebu-tololo sopprime la produzione di melatonina, se dato la mattina e integrato con la somministrazione di me-latonina la sera ristabilisce l’andamento ottimale della melatonina e migliora il ritmo sonno/veglia.La SMS è la dimostrazione delle basi biologiche dei disturbi del sonno, in quanto il gene RAI1 riveste un
ruolo nella regolazione della trascrizione del Circa-dian Locomotor Output Cycles Kaput (CLOCK) che regola molti geni circadiani (Williams et al., 2012). I geni clock non solo mediano la produzione della me-latonina secondo un ritmo circadiano, ma anche rego-lano le funzioni dei nuclei soprachiasmatici e del tratto retinoipotalamico, importanti per la regolazione delle fasi sonno/veglia (De Leersnyder, 2013).
Sindrome di DownLe caratteristiche fenotipiche dei bambini con SD spiegano l’alta frequenza di OSAS, che viene riporta-ta tra il 50-80% dei casi. In particolare l’ipotonia gene-ralizzata, la macroglossia, la tendenza alla glossop-tosi, l’ipoplasia della porzione mediana della faccia, l’ipoplasia mandibolare, l’ipertrofia adenotonsillare, e in alcuni casi l’obesità, contribuiscono a generare il problema. Alcuni studi ipotizzano che anche una disfunzione del midollo allungato determina uno sbi-lanciamento tra sistema simpatico e vagale, che può determinare apnee centrali. Vengono descritti anche quadri di frammentazione e di mantenimento del son-no, risvegli precoci e sonnolenza durante la giornata.Alcuni studi hanno evidenziato che tale problema è sottostimato dalla famiglia. Pertanto è utile informare la famiglia, già in epoca precoce, sui segni e sintomi che caratterizzano le problematiche di sonno in parti-colare sulle OSAS (Bassell et al., 2015).
Sindrome di MarfanAnomalie craniofacciali e aumento della collassabilità delle vie aere superiori, dovuta ad anomalia del tes-suto connettivo, sono alla base di un’aumentata pre-valenza di OSA nei pazienti con sindrome di Marfan. La condizione è reversibile e il trattamento consiste in un trattamento ortodontico con rapida espansione mascellare e avanzamento mandibolare o l’utilizzo di CPAP. Poiché il maggior rischio per la sindrome di Marfan è la dilatazione dell’aorta e la sua dissecazio-ne, alcuni studi hanno rilevato una correlazione tra
Box 2. Classificazione e gravità delle patologie ostruttive nel sonno.
Diagnosi Apnea index Nadir O2 arterioso
End tindalpCO2 peak
End tindal pCO2 > 50 torr
(% del totale del sonno)
Arousal(eventi per ora)
Russamento < 1 evento ora > 92% < 53 torr < 10% EEG < 11
S. Resistenza vie aeree superiori
< 1 evento ora > 92% < 53 torr < 10% RERA > 1;EEG >11
OSAS lievi 1-4 eventi ora 86-91% > 53 torr 10-24% EEG > 11
OSAS moderate 5-10 eventi ora 76-85% > 53 torr 25-49% EEG > 11
OSAS grave > 10 eventi ora < 75% > 53 torr > 50% EEG > 11
EEG: elettroencefalografia; OSAS: obstructive sleep apnea syndrome (sindrome delle apnee ostruttive del sonno) RERA: sforzo respira-torio correlato all’arousal

202
G. Zampino et al.
OSAS e dilatazione dell’arco aortico (Kohler, 2009). Questo rende necessario il riconoscimento del pro-blema e il suo precoce trattamento.
Sindrome di Ehlers-DanlosI pazienti con EDS presentano problematiche respi-ratorie nel sonno che spesso sono misconosciute e hanno un ruolo, in aggiunta ad altri fattori, nella per-cezione del senso di fatica e di sonnolenza durante il giorno. Probabilmente il problema risiede nel coinvol-gimento della cartilagine, che determina la crescita del volto. In uno studio fatto mediante polisonnografia e rinomanometria, si è evidenziato che il problema re-spiratorio nel sonno era presente in tutti i pazienti e si è dimostrato connesso a un aumento delle resistenze nasali (Guilleminault et al., 2013).
Sindrome di Prader-WilliA volte è l’obesità con accumulo di grasso nella fa-ringe che determina le OSAS. La sindrome di Prader-Willi è una condizione caratterizzata da ipotonia ne-onatale, ipogonadismo, iperfagia che associata con riduzione del metabolismo basale determina obesità, ritardo mentale e fragilità emozionale. Si pensa che una disfunzione dell’asse ipotalamico-ipofisario sia alla base delle principali caratteristiche quali l’iper-fagia, la riduzione del metabolismo energetico, l’ipo-gonadismo, la ridotta produzione di GH e l’anomala regolazione della temperatura. S’ipotizza anche che la disfunzione ipotalamica possa essere responsabile dell’anomala risposta ventilatoria all’ipossia e all’iper-capnia (Beauloye et al., 2015).Le problematiche di sonno frequentemente riportate nei bambini con PW variano nelle diverse età: nella prima infanzia sono essenzialmente apnee centrali, poi diventano apnee di tipo ostruttivo e sembrano dovute a obesità, ipotonia, anomalie craniofacciali e ipertrofia adeno-tonsillare (Cohen et al., 2015).Quando l’ipertrofia adenotonsillare è presente, rap-presenta un elemento molto rilevante nel determinare OSA ed è necessario l’intervento di adenotonsillecto-mia. Poiché l’ostruzione respiratoria sembra determi-nare morte improvvisa durante il trattamento con GH, prima di iniziare il trattamento ormonale è raccoman-dato lo studio polisonnografico e una valutazione ORL.
Sindromi con macroglossiaLa riduzione dello spazio aereo posteriore può esse-re connesso a un incremento di volume dei tessuti molli. La macroglossia è uno dei segni che caratte-rizza diverse condizioni, in particolare la sindrome di Beckwith-Wiedemann, la sindrome di Simpson-Gola-bi-Behemel o della sindrome di Costello. In uno studio su una numerosa popolazione di bambini con BWS, nel 48% erano presenti difficoltà respiratorie, ma nella maggior parte erano riconducibili più a una eziologia multifattoriale, dove l’ipertrofia adenotonsillare aveva
un ruolo maggiore rispetto alla macroglossia (Follmar et al., 2014). Nella nostra esperienza la macroglossia nella BW è essenzialmente anteriore determinando prognatismo; quando però la crescita riguarda la base della lingua, allora il problema respiratorio è impor-tante e va gestito con la resezione linguale.
Sindromi con difetti craniofaccialiNei bambini con anomalie craniofacciali, l’ostruzione delle vie respiratorie superiori è una delle cause prin-cipali di morbilità e mortalità, specialmente nel perio-do neonatale. Attraverso l’utilizzo di un questionario, somministrato ai genitori di 575 bambini con malfor-mazioni craniofacciali, il rischio di disturbi respiratori nel sonno è risultato 9 volte superiore, rispetto a bam-bini sani della stessa età, indipendentemente dalla presenza o meno di obesità e con una prevalenza variabile in base all’alterazione morfologica o alla sin-drome (Moraleda-Cibrián et al., 2014).
Anomalie della mandibolaDurante il periodo neonatale, la mandibola è relati-vamente piatta con un ramo corto e un’articolazione scarsamente definita con la base del cranio. Durante l’infanzia la mandibola tende alla retroposizione. Se è presente una riduzione della lunghezza della mandibola, la base della lingua, ancorata alla man-dibola, si sposta postero-inferiormente dislocandosi nell’ipofaringe e determinando ostruzione. Le conse-guenze dell’OSA in questi bambini possono essere importanti, determinando danno cerebrale, deficit di crescita e cor polmonare. La micrognatia è un segno che si ritrova in diverse condizioni sindromiche, ma in particolare nella sequenza di Pierre Robin, nella sindrome di Treacher Collins, nello spettro facio-auri-colo-vertebrale, dove però l’ipoplasia mandibolare in genere è monolaterale (Cielo et al., 2016).
Ipoplasia della MidfaceL’ipoplasia della porzione mediana della faccia rap-presenta il problema principale nell’ostruzione delle vie aeree nelle sindromi craniostenotiche e in alcune displasie scheletriche, quali l’acondroplasia.
CraniostenosiC’è un’alta prevalenza di OSA nei bambini con cranio-stenosi sindromiche e, sebbene vi sia una multifatto-rialità, l’elemento determinante della gravità delle ap-nee è l’ipoplasia della porzione mediana della faccia e questo spiega come la sindrome di Crouzon, Pfeiffer e Apert rappresenta quella in cui l’OSAS è più frequen-te e grave. In questa sindrome la presenza di stenosi coanale è un ulteriore elemento di gravità. L’OSA, de-terminando aumento di CO2, potente vasodilatatore, genera incremento della pressione di perfusione ce-rebrale, che contribuisce all’ipertensione intracranica. Inoltre la possibilità di apnee centrali potrebbe essere

203
Sonno e sindromi malformative
spiegata dall’anomalia di Chiari tipo 1 e il suo tratta-mento sembra migliorare il quadro (Tan et al., 2016).
AcondroplasiaL’acondroplasia determina ipoplasia craniofacciale, con ipoplasia mascellare e della sella nasale. Rus-samento (snoring) occorre frequentemente nei bam-bini con acondroplasia e studi suggeriscono che la prevalenza di OSA è tra 10-35%. Radiograficamente si apprezza restringimento delle vie respiratorie e re-trognatia.
Trattamento L’identificazione precisa dei disturbi del sonno è ne-cessaria per adottare un’adeguata strategia di trat-tamento. Questo include anche l’esclusione e il con-trollo di attività epilettica che può disturbare il sonno. Solitamente, una raccolta attenta di dati del sonno è sufficiente a documentare le caratteristiche del ciclo sonno/veglia, dell’induzione del sonno e del suo man-tenimento, ma è lo studio EEG e/o polisonnografico che permette di rilevare se vi è epilessia.I problemi di sonno nei pazienti con sindromi malfor-mative con ritardo mentale, sono spesso difficili da trattare; l’obiettivo, nella maggior parte dei casi, non è quello di risolvere completamente il problema, quanto un raggiungimento di un accettabile compromesso.
La gestione delle abitudini del sonno è l’approccio più ampiamente applicato nella medicina del sonno pedia-trica. La gestione del comportamento è primariamente basata sulla caratterizzazione dei fattori che sottendono le problematiche di sonno. In alcuni casi, la revisione di questi fattori, ovvero i semplici rituali prima dell’addor-mentamento, possono portare a cambiamenti o stabilire routine che determinano un importante miglioramento del sonno. I ritardi nell’induzione del sonno possono es-sere migliorati da programmi di cronoterapia.A volte sono richiesti farmaci sedativi o ipnoinduttori, sia per brevi periodi sia per lunga durata.C’è un’adeguata esperienza con differenti farmaci che hanno spesso una combinazione di effetti seda-tivi, ipnotici e ansiolitici. La maggior limitazione è il rischio di effetti avversi, che è aumentato con alte dosi o per somministrazioni di lunga durata. Nella pratica clinica, antistaminici, neurolettici, benzodiazepine e antideppressivi sono quelli maggiormente usati, in re-lazione alle coomorbilità e all’esperienza del medico (Beck Blackmer et al., 2016) (Fig. 2).Specialmente nei disturbi del ritmo, la melatonina è il farmaco di prima scelta. Spesso la sua efficacia è discussa in particolare in quelle condizioni dove la melatonina non riesce a svolgere la sua funzione per mancanza di recettori o per differente metabolizzazio-ne. In alcune condizioni, come la sindrome di Smith-Magenis, l’aggiunta di clonidina ne potenzia l’effetto. Il trattamento dei disturbi respiratori nel sonno prevede
Figura 2. Trattamento farmacologico della patologia del sonno.

204
G. Zampino et al.
diverse misure (Fig. 3). Nel caso che l’ostruzione sia lie-ve e che la correzione chirurgica non sia possibile, è ne-cessario un trattamento conservativo (Tan et al., 2016). L’approccio posizionale può essere utile quando l’O-SA è presente nella posizione supina, ad esempio nel caso del bambino con micrognatia, e per evitare la glossoptosi si cerca di far dormire il bambino in una posizione alternativa (prona o laterale). Studi polison-nografici non hanno evidenziato particolare efficacia.In alcuni casi un tubicino nasofaringeo può fungere da stent delle vie aeree superiore e impedire il collas-so. Questo intervento sembra efficace in alcuni quadri di craniostenosi o di Pierre Robin.Continuous positive airway pressure (CPAP) usa
l’aria pressurizzata attraverso il naso per creare uno stent pneumatico e impedire il collabimento delle vie respiratorie.L’adenotonsillectomia è una terapia di prima linea, an-che se la sua efficacia nella popolazione dei bambini con difetti craniofacciali può essere limitata.Interventi chirurgici che prevedono distrazione ossea mandibolare nelle micrognatie, avanzamento mascel-lare nelle craniostenosi complesse, resezione lingua-le nelle macroglossie, disostruzione coanale nelle stenosi/atresie delle coane, correzione della Chiari tipo 1 fino alla tracheostomia dovrebbero essere con-siderati, se l’ostruzione è grave e persistente e non risponde a trattamenti conservativi.
Figura 3. Trattamento della patologia del sonno nel bambino con sindromi.
Box di orientamento
La patologia del sonno è molto frequente nelle sindromi malformative, ma spesso è sottodiagnosticata.È necessaria un’attiva ricerca delle problematiche di sonno, perché la loro precoce individuazione può permettere un trattamento che, oltre a migliorare la qualità della vita del bambino e della sua famiglia, può ridurre una serie di problemi, quali deficit attentivi, problemi di comportamento, ritardo di apprendimento e nei quadri di disturbi respiratorio notturni anche il cor polmonare.A volte la strategia di trattamento non è risolutiva, ma l’obiettivo è il raggiungimento del miglior risultato possibile. Questo può essere ottenuto con modificazioni di abitudini comportamentali, utilizzo di farmaci o miglioramento del flusso di aria nelle vie respiratorie.

205
Sonno e sindromi malformative
Bibliografia
Angriman M, Caravale B, Novelli L, et al. Sleep in children with neurodevel-opmental disabilities. Neuropediatrics 2015;46:199-210.
*** Ottima revisione delle problemat-iche di sonno nel bambino con disabilità su base sindromica.
Bassell JL, Phan H, Leu R, et al. Sleep profiles in children with Down syndrome. Am J Med Genet Part A 2015;167A:1830-5.
Beauloye V, Dhondt K, Buysse W, et al. Evaluation of the hypothalamic-pituitary-adrenal axis and its relationship with cen-tral respiratory dysfunction in children with Prader-Willi syndrome. Orphanet J Rare Dis 2015;10:106.
Beck Blackmer A, James A. Feinstein JA. Management of sleep disorders in children with neurodevelopmental disorders: a re-view. Pharmacotherapy 2016;36:84-98.
*** Ottima revisione sui farmaci da us-are nella patologia del sonno nel bambino.
Cielo CM, Montalva FM, Taylor JA. Cra-niofacial disorders associated with airway obstruction in the neonate. Semin Fetal Neonatal Med 2016 Mar 17. pii: S1744-165X(16)00037-8.
*** Ottima revisione dei disturbi crani-ofacciali e del loro trattamento.
Cohen M, Hamilton J, Narang I. Clinically important age-related differences in sleep related disordered breathing in infants and children with Prader-Willi Syndrome. PLoS One 2014;9:e101012.
De Leersnyder H. Smith-Magenis syn-drome. Handb Clin Neurol 2013;111:295-6.
Follmar A, Dentino K, Abramowicz S, et al. Prevalence of sleep-disordered breathing in patients with Beckwith-Wiedemann syn-drome. J Craniofac Surg 2014;25:1814-7.
Guilleminault C, Primeau M, Chiu HY, et al. Sleep-disordered breathing in Ehlers-Danlos syndrome: a genetic model of OSA. Chest 2013;144:1503-11.
Kohler M, Blair E, Risby P, et al. The prevalence of obstructive sleep apnoea and
its association with aortic dilatation in Mar-fan’s syndrome. Thorax 2009;64:162-6.
Moraleda-Cibrián M, Edwards SP, Kas-ten SJ, et al. Symptoms of sleep disor-dered breathing in children with cranio-facial malformations. J Clin Sleep Med 2014;10:307-12.
Pelc K, Cheron G, Boyd SG, et al. Are there distinctive sleep problems in Angel-man syndrome? Sleep Med 2008;9:434-41.
Tan HL, Kheirandish-Gozal L, Abel F, et al. Craniofacial syndromes and sleep-re-lated breathing disorders. Sleep Med Rev 2016;27:74-88.
Williams SR, Zies D, Mullegama SV, et al. Smith-Magenis syndrome results in disruption of CLOCK gene transcription and reveals an integral role for RAI1 in the maintenance of circadian rhythmicity. Am J Hum Genet 2012;90:941-9.
Zhang J, Fang Z, Jud C, et al. Fragile X-related proteins regulate mammalian circa-dian behavioral rhythms. Am J Hum Genet 2008;83:43-52.
Corrispondenza
Giuseppe ZampinoUOSA, Malattie Rare e Difetti Congeniti, Polo per la Salute della Donna e del Bambino, Fondazione Policlinico Universitario “A. Gemelli”, largo Agostino Gemelli 8, 00168 Roma - E-mail: [email protected]

206
Luglio-Settembre 2016 • Vol. 46 • N. 183 • Pp. 206-214 Prospettive in Pediatria
Genetica clinica pediatrica
Terapia farmacologica e sindromi genetiche: quali novità?
Paolo Curatolo1 Romina Moavero1 2
Angelo Domenico3 5 Silvia Maitz4
Martino Ruggeri5 Gioacchino Scarano6
Angelo Selicorni4 7
1 UOC Neuropsichiatria Infantile, Policlinico Tor Vergata, Roma;
2 UOC Neurologia, Ospedale Pediatrico Bambino Gesù, IRCCS, Roma; 3 Dipartimento di Scienze
Biomediche e Biotecnologiche, Università degli Studi di
Catania; 4 UOS di Genetica Clinica Pediatrica, Fondazione
MBBM, Monza; 5 Dipartimento di Medicina Clinica e Sperimentale,
Sezione di Pediatria e Neuropsichiatria Infantile,
Università degli Studi di Catania; 6 UO di Genetica Medica, Azienda
Ospedaliera Gaetano Rummo, Benevento; 7 UOC Pediatria ASST
Lariana, Como
Recently, the increase in knowledge of the pathogenetic mechanisms underlying some genetic diseases has led to the identification of possible drug treatments that target the most important and disabling clinical manifestations of these diseases. In particular, we present the therapeutic advances and future prospects for four genetic disorders: tuber-ous sclerosis, achondroplasia and neurofibromatosis types 1 and 2. These approaches, still partly on a research basis, represent an important hope for the treatment of genetic diseases.
Summary
Recentemente l’aumento delle conoscenze dei meccanismi patogenetici alla base di alcu-ne patologie genetiche ha portato all’identificazione di possibili trattamenti farmacologici in grado di agire sulle più importanti e invalidanti manifestazioni cliniche delle patologie. In particolare verranno illustrate le novità terapeutiche e le prospettive future per quattro pa-tologie genetiche: sclerosi tuberosa, acondroplasia, neurofibromatosi di tipo 1 e di tipo 2. Questi approcci, ancora in parte sperimentali, rappresentano un’importante speranza per la cura delle patologie genetiche.
Riassunto
Metodologia della ricerca bibliografica effettuataI dati per la realizzazione di questo articolo sono stati selezionati nella banca bibliografica Medline e Sco-pus con le parole chiave “tuberous sclerosis complex”, “neurofibromatosis type 1”, “NF1”, “neurofibromatosis type 2”, “NF2”, “plexiform neurofibroma”, “optic pathway glioma”, “tumors”, “schwannomas”, “ependymomas”,
“astrocytomas”, “biologiocally targeted therapies”, “bio-logical”, “new therapies”, “achondroplasia”. I limiti utiliz-zati sono stati: “all ages”, “children”, “all child = 0-18 ye-ars”, “all years”. Sono stati selezionati articoli originali, revisioni, clinical trials e studi epidemiologici senza limiti di anno di pubblicazione, se ritenuti utili alla ste-sura della revisione. Sono stati considerati solamente gli articoli in lingua inglese.

207
Terapia delle sindromi genetiche
ObiettivoFornire dati aggiornati rispetto ai nuovi farmaci appro-vati o in corso di sperimentazione per quattro patolo-gie genetiche frequenti: la sclerosi tuberosa, l’acon-droplasia, la neurofibromatosi di tipo 1 e di tipo 2.
IntroduzioneNell’ambito delle sindromi genetiche, le terapie finora disponibili sono basate sulla cura di singoli segni o sintomi correlati alla patologia di base, con un approc-cio chirurgico per le malformazioni maggiori (es. car-diopatie congenite) o con un trattamento farmacolo-gico sintomatico per singole problematiche mediche (reflusso gastroesofageo, epilessia ecc.). Per alcune patologie, in cui è noto un rischio tumorale aumen-tato, vengono proposti dei protocolli di monitoraggio specifici, in relazione alla tipologia di tumore a mag-gior rischio di insorgenza. Negli ultimi anni, l’aumento della conoscenza della patogenesi molecolare di alcune condizioni, ha per-messo di avviare dei protocolli sperimentali farma-cologici per alcune di esse. In questa revisione vie-ne fatto un aggiornato punto della situazione delle terapie farmacologiche in uso o in sperimentazione per quattro importanti patologie genetiche: la sclero-si tuberosa, l’acondroplasia e la neurofibromatosi (di tipo 1 e 2).
Sclerosi tuberosaLa sclerosi tuberosa è una patologia genetica a tra-smissione autosomica dominante, caratterizzata dalla presenza di lesioni istologicamente benigne e circo-scritte. Colpisce circa un bambino ogni 5800 nati, con una prevalenza stimata di 6,8-12,4/100.000 (Curatolo et al., 2015). Il sistema nervoso centrale è interessato in più del 90% dei soggetti con sclerosi tuberosa, con la pre-senza di lesioni tra cui i tuberi cortico-sottocorticali, i noduli subependimali, gli astrocitomi subependimali a cellule giganti e le strie di migrazione della sostan-za bianca (cioè delle strie di displasia della sostanza bianca identificabili alla RM encefalo che vanno dalla regione periventricolare alla corteccia) (Curatolo et al., 2015). Oltre a queste lesioni sono presenti anche segni e sintomi neurologici, tra cui epilessia e disturbi neuropsichiatrici. Le localizzazioni non neurologiche di sclerosi tuberosa includono un interessamento cu-taneo, renale, polmonare, cardiaco e oftalmologico. Le manifestazioni legate alla sclerosi tuberosa pre-sentano un’importante età-dipendenza. Infatti alcune lesioni, come ad esempio gli angiomiolipomi renali o la linfangioleiomiomatosi polmonare, non si presen-tano prima di una certa età, mentre altre, come ad esempio i rabdomiomi cardiaci e i tuberi corticali, ap-paiono già in epoca fetale (Curatolo et al., 2008).La sclerosi tuberosa è causata da mutazioni nel gene
TSC1 sul cromosoma 9 o sul gene TSC2 sul cromo-soma 16, rispettivamente codificanti per le proteine amartina e tuberina, formanti un complesso etero-dimerico.
Le mutazioni in uno dei due geni portano
all’iperattivazione del complesso mTOR (mammalian Target Of Rapamycin), coinvolto in diversi aspetti del funzionamento intracellulare, comprese la crescita e la proliferazione cellulare, la sintesi proteica e il meta-bolismo (Curatolo et al., 2008).
TrattamentoL’aumento delle conoscenze del meccanismo pato-genetico alla base della sclerosi tuberosa ha portato all’identificazione dei farmaci inibitori mTOR come possibili trattamenti farmacologici in grado di agire su diverse manifestazioni cliniche e lesioni patologiche della sclerosi tuberosa (Fig. 1).
Astrocitomi subependimali a cellule giganti (SEGA)Gli inibitori mTOR rappresentano una valida opzione terapeutica per tutti quei pazienti in cui sono presenti SEGA multipli o infiltranti, non suscettibili di resezio-ne chirurgica, o quando la chirurgia non è un’opzione per la presenza di complicanze sistemiche (Jozwiak et al., 2013).L’efficacia degli inibitori mTOR nei SEGA secondari a sclerosi tuberosa è stata descritta per la prima volta nel 2006 (Franz et al., 2006), e il primo studio rando-mizzato in doppio cieco, l’EXIST-1, è stato pubblica-to nel 2013 (Franz et al., 2013). In questo trial clinico di fase 3 il trattamento con everolimus ha portato a una riduzione delle dimensioni dei SEGA di almeno il 50% in più del 35% dei soggetti trattati dopo 6 mesi di trattamento, bloccando la progressione di malattia in tutti i soggetti trattati. Nello studio di estensione, dopo una durata media di trattamento di 29 mesi, il 49% dei pazienti ha presentato una riduzione del volume dei SEGA superiore al 50% (Franz et al., 2014). Tuttavia, la sospensione di everolimus è associata a una ricre-scita delle lesioni, suggerendo così che per tenere i SEGA sotto controllo sia necessario un trattamento continuativo. Le stomatiti e le afte orali sono stati gli eventi avversi più frequentemente riportati nell’E-XIST-1, e le infezioni, osservate nel 14% dei pazienti, sono state il più comune evento avverso serio (Franz et al., 2013). I dati a lungo termine sembrano comun-que mostrare una riduzione degli eventi avversi nel tempo. I risultati di questi studi di fase III hanno porta-to all’approvazione dell’everolimus da parte di EMA e FDA per il trattamento dei SEGA secondari alla scle-rosi tuberosa, non suscettibili di terapia chirurgica.
Angiomiolipomi renali (AML)L’everolimus è attualmente disponibile sia in Europa che negli Stati Uniti anche per il trattamento degli an-giomiolipomi renali in soggetti adulti con sclerosi tu-

208
P. Curatolo et al.
berosa. Lo studio in doppio cieco di fase 3. che ha portato all’approvazione di tale farmaco per le mani-festazioni renali. è lo studio EXIST-2 (Bissler et al., 2013). Il 42% dei pazienti trattati con everolimus ha presentato una risposta significativa; nessuno dei soggetti inseriti nel braccio placebo ha mostrato una riduzione degli angiomiolipomi. La progressione degli angiomiolipomi è stata osservata nel 4% dei pazienti in trattamento con everolimus e nel 21% nel gruppo placebo. I risultati di questo studio hanno confermato che, sebbene eventi avversi siano osservati in quasi tutti i pazienti, questi raramente sono severi, e il trat-tamento è nel complesso ben tollerato. Anche a livello renale la maggior parte delle lesioni torna alle dimen-sioni iniziali in caso di sospensione del trattamento (Bissler et al., 2008).
Epilessia Nonostante i trattamenti farmacologici e non farma-cologici attualmente disponibili, le crisi associate alla sclerosi tuberosa persistono in più del 60% dei pa-zienti, suggerendo la necessità di nuove opzioni tera-peutiche. Studi preclinici suggeriscono che un tratta-mento precoce con inibitori mTOR possono migliora-re la disorganizzazione neuronale e sembrano eserci-tare un’azione antiepilettogenica nel modello animale (Zeng et al., 2008). Esistono dati clinici preliminari di potenziale efficacia dell’everolimus sull’epilessia as-sociata alla sclerosi tuberosa, ma questi sono preva-lentemente casi clinici o piccole serie (Krueger et al.,
2013; Wiegand et al., 2013; Cardamone et al., 2014). Uno studio di fase III, l’EXIST-3 per la valutazione di efficacia e tollerabilità di due diversi dosaggi di evero-limus paragonati al placebo, ha fornito una risposta significativa in termini di riduzione delle crisi epilet-tiche in pazienti con epilessia parziale farmacoresi-stente. La prima analisi ad interim ha mostrato una riduzione di crisi nel 29,3% dei pazienti in trattamento con basse dosi di everolimus vs 39,6% di riduzione dei pazienti in trattamento con alte dosi vs 14,9% dei pazienti in placebo.
Altre manifestazioniGli studi effettuati sulle diverse manifestazioni del-la sclerosi tuberosa hanno mostrato che gli inibitori mTOR riducono in maniera significativa anche gli an-giofibromi del volto. Essi sono inoltre in grado di tene-re sotto controllo l’evoluzione della linfangioleiomio-matosi polmonare. Inoltre in letteratura sono presenti numerosi report clinici che dimostrano un’efficacia anche sui rabdomiomi cardiaci (Moavero et al., 2013).In conclusione, negli ultimi anni l’approccio terapeuti-co alla sclerosi tuberosa si è modificato in maniera si-gnificativa. La disponibilità degli inibitori mTOR infatti mette a disposizione del clinico, che si trova ad affron-tare una malattia sistemica, un’opzione terapeutica in grado di agire contemporaneamente sulle diverse manifestazioni della malattia.
Figura 1. Rappresentazione schematica della cascata mTOR e del meccanismo di azione di everolimus.

209
Terapia delle sindromi genetiche
AcondroplasiaL’acondroplasia (ACH) è la displasia scheletrica più comune causa di bassa statura disarmonica con arti corti. La prevalenza alla nascita è stimata di 1 caso ogni 20.000-25.000 nati circa. La diagnosi di sospetto è clinica: il neonato/piccolo lattante presenta sproporzione tronco/arti per ridu-zione della lunghezza degli arti, macrocrania con la fronte ampia e prominente, ipoplasia del mascellare superiore, naso insellato, torace stretto, addome pro-minente e arti corti con prevalente coinvolgimento dei segmenti prossimali (rizomelici) e dita delle mani cor-te e larghe. È sempre presente una discreta/marcata ipotonia generalizzata e un’iperlassità articolare. Lo studio radiologico dello scheletro consente di rileva-re degli aspetti peculiari anche se non specifici: ossa tubulari corte e robuste, riduzione della distanza in-terpeduncolare fra i corpi delle vertebre lombari, ossa iliache piccole e di forma quadrangolare con caratteri-stico aspetto a tridente dell’acetabolo e ridotte dimen-sioni del forame sacro-ischiatico, parte prossimale del femore ovoidale e traslucida, lievi alterazioni di tutte le metafisi delle ossa lunghe.
Basi biologicheL’acondroplasia è una malattia genetica (MIM #100800) dovuta ad anomalie di un unico gene, FGFR3, mappato per linkage sul braccio corto del cromosoma 4p16.3 nel 1994 (Le Merrer et al., 1994; Velinov et al., 1994) e il cui difetto molecolare è stato identificato dopo appena ulteriori sei mesi (Rousse-au et al., 1994; Shiang et al., 1994). FGFR3 codifica per un recettore del fattore di crescita dei fibroblasti (FGFR3) espresso nella cartilagine di accrescimento che ha un’attività di tipo regolatorio negativo sul pro-cesso di ossificazione encondrale (cartilagineo) del tessuto scheletrico e in parte anche sul processo di ossificazione membranoso. Il recettore FGFR3, nor-malmente attivato da uno dei fattori di crescita dei fibroblasti (FGF), inibisce la proliferazione dei condro-citi e ne induce la maturazione, favorendo uno svi-luppo armonico della crescita delle ossa lunghe. Nei pazienti con acondroplasia questa funzione risulta ampiamente disregolata, causando un’eccesiva ridu-zione della proliferazione e differenziazione dei con-drociti. In oltre il 99% dei casi l’indagine molecolare identifica una sola mutazione nota come G380R: so-stituzione di un residuo di glicina (G) con uno di argi-nina (R) in posizione 380 nel dominio transmembrana della proteina. La mutazione è una mutazione Gain of Function attivante in maniera costitutiva il pathway specifico FGFR3, e quindi l’attività di tipo inibitorio di-venta permanente.La condizione si trasmette in maniera autosomica do-minante e penetranza completa: ogni soggetto porta-tore della mutazione presenta il fenotipo clinico e può
trasmettere la stessa mutazione al 50% della propria progenie. Tuttavia, data la frequenza tra le più alte con cui il gene FGFR3 può subire mutazioni spontanee precocemente nella vita embrionale, nella maggior parte dei casi (circa il 90-95%) i pazienti hanno muta-zioni de novo e genitori non affetti.
Strategie di trattamentoIn passato numerosi studi hanno analizzato l’efficacia della terapia con ormone della crescita (GH). Attual-mente è un dato accertato che, dopo un’iniziale acce-lerazione della crescita, si assiste a un rallentamento. La prosecuzione del trattamento, quindi, comporta risultati molto modesti sulla statura finale dei pazienti e pertanto non ne sussiste l’indicazione.
Drug repositioning strategyNegli ultimi anni la ricerca nel campo farmaceutico ha prestato grande interesse alla cosiddetta drug re-positioning strategy, strategia che valuta l’uso di un farmaco, normalmente assunto per altre indicazioni, in pazienti con una altra diversa condizione. Questa strategia ha un ovvio vantaggio rappresentato dal fat-to che i farmaci identificati possono essere usati fa-cilmente nella pratica clinica, in quanto sono note sia la posologia giusta che gli eventuali effetti collaterali.In questo ambito sono stati prodotti dati sperimentali molto interessanti sull’efficacia di alcuni farmaci, già in uso con altre indicazioni, nell’indurre un incremento della crescita scheletrica. Yangli et al. (2012) hanno trattato ceppi di topi ACH con paratormone (PTH) umano ricombinante per 4 settimane dopo la nascita e hanno dimostrato che questo trattamento nei topi ACH, rispetto ai controlli, è in grado di migliorare la crescita in lunghezza, cor-reggere almeno parzialmente la fusione prematura di alcune suture craniche e migliorare anche la qualità della struttura ossea in termini di mineralizzazione, ipotizzando che l’effetto combinato dell’incremento del peptide correlato al PTH (PTHrP) e la down-re-gulation di FGFR3 possa essere responsabile dell’ef-ficacia del PTH.Matsushita et al. (2013) hanno effettuato uno scree-ning di un ampio gruppo di sostanze (1186), con l’ap-provazione della Federation Drug Amnistration, allo scopo di identificare farmaci utili per la terapia dell’a-condroplasia e delle altre displasie scheletriche della famiglia FGFR3. Hanno scoperto che la meclozina cloridrato (codice ATC R06AE05), farmaco ad azione antistaminica (antagonista recettore H1), efficace nel trattamento della nausea, vomito e vertigini e asso-ciati a chinetosi, è in grado di sopprimere efficace-mente il signaling FGFR3 in tre diverse linee cellulari di cellule condrocitarie e in colture di tessuto osseo embrionale.Gli stessi autori (2015) successivamente hanno te-stato gli effetti della somministrazione di meclozina in embrioni di topi eterozigoti knock-out (KO) per

210
P. Curatolo et al.
FGFR3, dimostrando che la meclozina incrementa la lunghezza totale dell’abbozzo embrionale delle tibie dei topi KO. Hanno poi analizzato la crescita ossea di topi KO e topi wild-type, trattati o meno con me-clozina a dosi nel range utilizzato per disturbi da chi-netosi. Si è verificato un incremento significativo della lunghezza dopo 2 settimane di terapia nei topo KO, incremento evidente anche nei topi wild-type tratta-ti. Questi dati suggeriscono che l’assunzione orale di meclozina nel periodo della crescita è efficace nel mi-gliorare la crescita staturale delle persone con acon-droplasia. La meclozina agirebbe sul pathway di MPK down-regolando la fosforilazione di ERK. Un approccio molto diverso, ma nell’ambito del-lo stesso tipo di strategia, è stato usato da un altro gruppo giapponese. Yamashita et al. (2014) hanno ottenuto, da fibroblasti da biopsia cutanea di pazienti con acondroplasia e controlli, cellule staminali pluri-potenti (iPSC) e su queste hanno testato l’efficacia di alcuni farmaci nel recupero di una normale funzione del processo di ossificazione encondrale del tessuto scheletrico.Le statine, farmaci ipocolesterolemizzanti, hanno mo-strato un significativo miglioramento del processo di condrogenesi delle cellule iPSC derivate dai pazienti. Inoltre hanno dimostrato un’efficacia notevole nel cor-reggere lo sviluppo scheletrico dei topi KO per acon-droplasia. Le statine sarebbero in grado di stimolare sia la differenziazione dei condrociti che la loro ma-turazione, favorendo un incremento dell’espressione
dei geni master dei processi di ossificazione encon-drale e membranoso, rispettivamente Sox9 e Runx2.Gli autori correlano questi risultati, non evidenti però nei controlli, alla riduzione della sintesi e quindi dei livelli di colesterolo che consente un incremento della crescita del tessuto osseo, grazie alla riduzione dei livelli del recettore proteico FGFR3, attivato costitu-zionalmente, mediante degradazione proteolitica da proteasoma.
Il ruolo del peptide natriuretico C (CNP)Oltre la drug repositioning strategy, è in atto un per-corso di ricerca estremamente promettente che ha avuto inizio circa 20 anni fa. Yasoda et al. (1998) han-no identificato il ruolo importante del peptide natriure-tico C (CNP), mediante attivazione di un recettore di membrana Natriuretic Receptor Peptide-B (NRP-B) ad attività guanil-ciclasi, nel controllo dell’ossificazio-ne encondrale, e dimostrato (2004) che la iperespres-sione di CNP nei condrociti della cartilagine di co-niugazione, agendo sul pathway Mitogen-Activated Protein Kinase (MAPK), è in grado di incrementare la crescita scheletrica nei topo KO per acondroplasia (Fig. 2).La dimostrazione (Yasoda et al., 2004) che l’iperpro-duzione di CNP nella cartilagine o la sua continua assunzione per via endovenosa normalizza la statura dei topi KO per acondroplasia ha prospettato la possi-bilità di una terapia specifica per l’acondroplasia, me-diante la somministrazione di CNP a livelli superiori
Figura 2. Struttura e segnale di trasduzione di FGFR3 (da Expert Reviews in Molecular Medicine. Cambridge University Press 2012, mod.).

211
Terapia delle sindromi genetiche
a quelli fisiologici. Poiché l’emivita del CNP in circolo è brevissima (2 minuti dopo la somministrazione per via venosa) il suo utilizzo, come agente terapeutico mediante infusione continua, è sostanzialmente im-praticabile. Lorget et al. (2012) hanno disegnato e pro-dotto un analogo farmacologico di CNP denominato BMN111, costituito da 39 aminoacidi, che ha un’emi-vita che ne consente una unica somministrazione al giorno per via sottocutanea. BMN111 ha dimostrato di agire in maniera simile a CNP, attivando lo specifico recettore NRP-B, ma non i recettori per gli altri ormoni natriuretici. Gli studi pre-clinici hanno rilevato un notevole miglioramento dei parametri di crescita nel topo acondroplasico FGFR3 Y367C/wild-type. Questi risultati hanno consentito di programmare un trial clinico, partito nel 2014, che ha concluso anche la fase 2 di sperimentazione ar-rivando a ottimizzare la dose ottimale. Attualmente siamo all’inizio della fase 3, reclutamento dei pazienti (https://clinicaltrials.gov/).
ConclusioniLe prospettive di identificare una terapia farmacolo-gica della bassa statura disarmonica delle persone con acondroplasia sono da un punto di vista esclusi-vamente sperimentale molto promettenti. Sono stati ottenuti finora dati scientifici sperimentali oggettiva-mente molto interessanti mediante l’assunzione di paratormone o meclozina o statine. Questi risultati hanno bisogno ancora di conferme per poter pensare a un trial clinico.Quindi non c’è molto da stupirsi per le grandi aspetta-tive sia da parte di pazienti e famiglie che dei medici che ha creato l’avvio della sperimentazione clinica da parte delle multinazionale Biomarin mediante il trial clinico, che valuta gli effetti della somministrazione di BMN111. Senza alcun dubbio un grande primo passo nella giusta direzione (Legeai-Mallet, 2016). L’auspicio è che i promettenti primi dati del trial siano confermati e che si giunga in tempi brevi alla sua con-clusione che consentirà finalmente di poter disporre di un farmaco efficace per le persone con acondro-plasia.
Neurofibromatosi tipo 1La neurofibromatosi tipo 1 (NF1), patologia a trasmis-sione autosomica dominante, ha una prevalenza nel-la popolazione generale di circa 1:2500-3000 indivi-dui affetti. Le persone con NF1 presentano macchie caffellatte di grandi dimensioni (> 1,5 cm, distribuite sulla cute) e di piccole dimensioni (pochi mm, localiz-zate nelle regioni ascellari, inguinali e al collo); piccoli noduli rilevati dell’iride (istologicamente dei neurofi-bromi e amartomi) detti di Lisch e neurofibromi cuta-nei e sottocutanei. Alcuni individui con NF1 possono presentare statura ai limiti inferiori della norma (< 25° percentile), note dismorfiche e macrocrania. Un nu-
mero limitato di persone con NF1 presenta anche grave interessamento del sistema nervoso periferico (neurofibromi plessiformi e tumori maligni delle guai-ne nervose periferiche) o coinvolgimento del sistema nervoso centrale (gliomi delle vie ottiche, aree di di-splasia della sostanza bianca cerebrale, malforma-zioni dei vasi cerebrali, disturbi dell’apprendimento), delle ossa (displasie delle ossa lunghe, malformazio-ni di alcuni segmenti ossei, scoliosi) e dell’apparato cardiovascolare (malformazioni cardiache, displasie dei grossi vasi). Vi è, in generale, un più elevato ri-schio oncologico.
Meccanismi molecolariLa NF1 è causata da anomalie della sintesi o della funzione della neurofibromina, proteina che ha il com-pito di inibire il complesso sistema del proto-oncoge-ne Ras-GTP, inattivandolo a Ras-GDP: il Ras-GDP inibisce a sua volta alcuni sistemi proteici complessi “a valle” – RAF/MEK/ERK e PI3K/AKT/mTOR – cau-sando così modulazione (negativa) della crescita e della moltiplicazione cellulare e modulazione di alcuni circuiti neuronali implicati in processi di apprendimen-to e di memoria.
Terapie biologicheI principali bersagli dei protocolli terapeutici nella NF1 sono stati finora i neurofibromi plessiformi, i gliomi delle vie ottiche, e i tumori maligni delle guaine nervo-se periferiche (Blakeley e Plotkin, 2016).
Neurofibromi plessiformiIl trattamento di questi tumori ha comportato lo svi-luppo di più di 20 trials clinici (alcuni tutt’ora in cor-so): sebbene molti di essi abbiano conseguito risultati modesti, in alcuni casi si è ottenuto un rallentamento della crescita tumorale o una riduzione volumetrica delle lesioni.Tra le prime molecole a essere impiegate, il sora-fenib (inibitore del sistema Raf), il tipafernib (attivo sulla farnesil-transferasi), il pirfenidone (attivo sui fi-broblasti) e la talidomide (un anti-angiogenetico) non si sono rivelati utili nell’inibizione della progressione cellulare. Il sirolimus (antagonista del sistema mTOR) ha dimostrato qualche effetto nel rallentamento della crescita tumorale, seppur non mostrando attività on-co-soppressiva. I primi incoraggianti risultati (seppur parziali: 17% di successo nei casi trattati – età 3-65 anni) di riduzione volumetrica dei neurofibromi plessi-formi sono stati ottenuti con l’impiego dell’imatinib, ini-bitore del sistema c-kit e del Platelet derived Growth Factor Receptor Beta (PDGFRbeta) (Robertson et al., 2012). L’interferone alfa-2B peghilato ha garantito la riduzione radiografica dei neurofibromi plessiformi nel 29% dei casi (età 2-35 anni) (Jakacki et al., 2012). L’impiego del selumetinib (inibitore del sistema MEK), ha portato – in uno studio di fase 1 – alla riduzione

212
P. Curatolo et al.
volumetrica nel 100% degli 11 pazienti trattati, di età compresa tra 3 e 18 anni. I più frequenti effetti colla-terali registrati sono stati rash acneiforme, elevazione asintomatica del CPK, nausea, vomito, dolore addo-minale, diarrea e affaticamento (Blakeley e Plotkin, 2016). Lo studio di fase 2, iniziato nel 2015, è tutt’ora in corso. Ulteriori studi, tutt’ora in corso, riguardano l’everolimus (inibitore del sistema mTOR), il nilotinib (a molteplice azione sul sistema c-Kit, BCR-ABL, PDGFRbeta), l’associazione vinblastina/metotrexato (citotossici) e il celecoxib (inibitore dell’angiogene-si, oltre che potente anti-infiammatorio). Oltre al già citato selumetinib, sono in corso di sperimentazione altri due inibitori del sistema MEK, il trametinib e il PD0325901.
Gliomi delle vie otticheIl trattamento di prima linea è ancora affidato ai che-mioterapici, che dimostrano risultati apprezzabili in termini di regressione tumorale e discreta tollerabili-tà clinica. Vengono impiegati (con somministrazioni settimanali) associazioni di carboplatino e vincristina (specie in età infantile) (Moreno et al., 2010). Oltre a una notevole riduzione della massa tumorale, si assi-ste sin dalle prime fasi a un miglioramento generale dell’acuità visiva. Quando il trattamento non risulta efficace, o qualora si verifichi una sensibilizzazione ai farmaci (in particolare al carboplatino), si passa generalmente a un trattamento di seconda linea, che si avvale dell’impiego di temozolamide, bevacizu-mab, cisplatino-etoposide, e vinblastina. Per i gliomi ricorrenti delle vie ottiche è attualmente in sperimen-tazione l’impiego dell’erlotinib, un inibitore del recet-tore tirosin-kinasico del fattore di crescita epidermico (EGF), e dell’everolimus.
Tumori maligni delle guaine nervose perifericheNon sono stati finora raggiunti risultati apprezzabili nella terapia con farmaci biologici dei tumori maligni delle guaine nervose periferiche. Modelli preclinici hanno evidenziato discreti effetti terapeutici degli ini-bitori dell’EGFR, mentre sono tutt’ora in corso studi sull’impiego di everolimus e ranibizumab (inibitore del VEGF).
Neurofibromatosi tipo 2La neurofibromatosi tipo 2 (NF2), anch’essa a tra-smissione autosomica dominante, ha una prevalenza nella popolazione di 1:330.000. È caratterizzata dallo sviluppo di schwannomi dei nervi cranici (principal-mente del nervo VIII – acustico/vestibolare) e dei nervi periferici; tumori multipli del sistema nervoso centrale (es. astrocitomi, ependimomi, meningiomi); poche macchie caffellatte (< 6); cataratta giovanile. L’età d’esordio è quella adulta (25-30 anni), ma ven-gono descritte con frequenza sempre maggiore for-me a esordio in età pediatrica o forme congenite (che
sono più gravi delle forme dell’adulto). A tutt’oggi il trattamento di prima linea è costituito dalla resezione chirurgica, dalla radiochirurgia (gamma-knife) e dalla radioterapia: tali trattamenti sono efficaci solo nel 50% dei casi e gravati da un’elevata incidenza di perdita dell’udito (oltre il 60%) e/o della funzioni delle regioni del sistema nervoso interessate dal tumore.
Meccanismi molecolariLa malattia è causata da anomalie che alterano la sintesi o la funzione della schwannomina [conosciuta anche come Merlina: un membro della famiglia delle proteine ERM (Ezrin-Radixin-Moesin), che regolano la stabilità di membrana e diverse vie di crescita cellu-lare], proteina che favorisce l’endocitosi e l’eliminazio-ne e inibisce il complesso sistema Ras-dipendente. Il mancato controllo della Merlina sul sistema Ras (come nel caso della NF1) causa un’attivazione in-controllata dei sistemi mTOR, RAC1, e FAK, e per-tanto un’eccessiva proliferazione cellulare; la merlina agisce anche sul controllo delle attività delle semafo-rine, molecole ad azione angiogenetica: in sua man-canza si assiste quindi alla proliferazione incontrollata della crescita vascolare (Ruggieri et al., 2015).
Terapie biologicheSono stati testati di recente piccoli gruppi di persone con NF2 con alcuni antagonisti dei membri Her1-2 della famiglia ErbB dei recettori tirosin-kinasi (lapati-nib), con una buona risposta in termini di regressione volumetrica e miglioramento dell’udito (4/17 pazienti); risultati minori si sono ottenuti con l’erlotinib (nessuna riduzione di volume, ma stabilizzazione della malat-tia nel 27% dei pazienti trattati). Gli inibitori del siste-ma mTOR (everolimus) non hanno portato a risultati apprezzabili: nessuno dei 9 pazienti trattati in uno studio di fase 1 ha mostrato una risposta clinica né la stabilizzazione della malattia. Diversi farmaci che agiscono sui sistemi dell’IGF1 e del PDGF/Akt/MEK hanno mostrato discreti risultati in vitro: tra questi in particolare la picropodofillina, inibitore del recettore dell’IGF1, l’OSU-03012, inibitore Akt, e l’imatinib e il nilotinib, che agiscono sul recettore del PDGF e sul sistema c-Kit. L’imatinib è stato anche impiegato in un singolo caso di NF2, mostrando una stabilizzazione della malattia, ma diversi effetti collaterali (nausea, dolore addominale e cefalea grave) che hanno reso necessaria la sospensione del trattamento. Il sorafe-nib, che agisce sugli stessi target di nilotinib e imati-nib e anche sul sistema MEK1-2, è stato impiegato in un singolo paziente, ma anche in questo caso si è osservata un’importante presenza di eventi avversi e il trattamento è stato sospeso.Risultati molto incoraggianti sono stati ottenuti con l’impiego del bevacizumab, un potente anticorpo mo-noclonale diretto contro il fattore di crescita vascolare (VEGF) che al dosaggio di 5 mg/kg ogni 2 settimane ha dimostrato una spiccata inibizione della vascola-

213
Terapia delle sindromi genetiche
Bibliografia
Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, dou-ble-blind, placebo-controlled trial. Lancet 2013;38:817-24.
** Questo lavoro è il primo trial clinico randomizzato sull’efficacia e la tollerabilità di everolimus negli angiomiolipomi renali, e i suoi risultati hanno dato il via all’appro-vazione dell’everolimus per questa indica-zione.
Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lym-
phangioleiomyomatosis. N Engl J Med 2008;358:140-51.
Blakeley JO, Plotkin SR. Therapeu-tic advances for the tumors associated with neurofibromatosis type 1, type 2, and schwannomatosis. Neuro Oncol 2016;18:624-38.
Blakeley JO, Ye X, Duda DG, et al. Efficacy and biomarker study of bevacizumab for he-aring loss resulting from neurofibromatosis type 2-associated vestibular schwannomas. J Clin Oncol 2016;34:1669-75.
** Articolo che segue i primi due lavori dello stesso gruppo che ha per primo ini-ziato la terapia con bevacizumab nella NF2 e che riassume tutta la letteratura, indican-do anche i livelli di alcuni biomarcatori nelle
persone con NF2 trattate con bevacizumab.
Cardamone M, Flanagan D, Mowat D, et al. Mammalian target of rapamycin inhibitors for intractable epilepsy and su-bependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr 2014;164:1195-200.
Curatolo P, Bombardieri R, Jozwiak S. Tu-berous sclerosis. Lancet 2008;372:657-68.
** Revisione completa di tutti gli aspetti della patologia.
Curatolo P, Moavero R, de Vries PJ. Neu-rological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol 2015;14:733-45.
** Revisione completa e aggiornata
rizzazione dei tumori con riduzione volumetrica degli schwannomi del nervo VIII, stabilizzazione della ma-lattia e recupero funzionale: il 90% delle persone trat-tate ha mostrato udito stabilizzato o migliorato dopo 1 anno di trattamento e il 61% dopo 3 anni; l’88% ha avuto stabilizzazione o riduzione delle dimensioni dei tumori dopo 1 anno e il 54% dopo 3 anni. I più comuni eventi avversi sono state le emorragie, ritardata guari-gione delle ferite, proteinuria e ipertensione (Hochart et al., 2015). Nello stesso gruppo di persone con NF2 si è dimostrato (29% dei casi) riduzione volumetrica dei meningiomi, seppure non duratura (3,7-15 mesi). Successivi studi hanno dimostrato l’efficacia del be-vacizumab nella NF2, con riduzione volumetrica degli schwannomi e miglioramento dell’udito osservati in una percentuale tra il 33 e il 100% dei soggetti trattati. L’impiego di terapie combinate (temsirolimus, inibitore del sistema mTOR, e bevacizumab), seppure anco-ra praticato in pochissimi casi, ha portato a riduzioni volumetriche ancora più marcate (fino al 33% delle dimensioni). Il bevacizumab è stato impiegato anche in bambini e adolescenti (età 6-17 anni): i dati della
letteratura e la nostra esperienza personale dimostra-no regressione del volume tra il 5 e oltre il 20% in circa la metà dei pazienti e stabilizzazione della ma-lattia nell’altra metà dei casi. Il trattamento si è inoltre dimostrato efficace anche nel tempo, con effetti bene-fici anche a distanza di 6 anni dall’inizio della terapia.
Conclusioni e prospettive per il futuroLe sindromi genetiche sono malattie rare e a oggi in gran parte prive di terapie efficaci. Negli ultimi anni con l’aumentato interesse, l’avanzamento tecnico e la co-noscenza dei meccanismi patogenetici sono stati fatti passi avanti nell’approccio terapeutico di alcune pato-logie. Sono infatti in corso numerosi trial terapeutici che mirano all’introduzione nella pratica clinica dei farmaci sopradescritti, nella speranza che confermino la pro-mettente efficacia su alcuni degli aspetti più disabilitanti delle patologie che abbiamo preso in considerazione.
Box di orientamento
• Cosa sappiamo primaLa terapia delle patologie genetiche prese in considerazione (sclerosi tuberosa, acondroplasia, neuro-fibromatosi di tipo 1 e 2) si basava unicamente su terapie sintomatiche e su approcci di monitoraggio clinico per la diagnosi precoce di eventuali complicanze.
• Cosa sappiamo adessoNumerosi trial terapeutici hanno mostrato la potenziale efficacia di farmaci che agiscono sulle vie meta-boliche responsabili della patogenesi molecolare di queste patologie, ottenendo un trattamento di molte manifestazioni cliniche e/o complicanze delle stesse.
• Quali ricadute sulla pratica clinicaGli sviluppi attuali della ricerca permettono di individuare, già nel presente e più chiaramente nel pros-simo futuro, i nuovi approcci terapeutici di queste condizioni in grado di modificarne in modo importante la storia naturale.

214
P. Curatolo et al.
delle manifestazione neurologiche e neu-ropsichiatriche della sclerosi tuberosa, con nozioni sulla gestione e il trattamento.
Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis com-plex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013;381:116.
** Questo lavoro è il primo trial clinico randomizzato sull’efficacia e la tollerabilità di everolimus negli astrocitomi subependi-mali a cellule giganti, e i suoi risultati han-no dato il via all’approvazione dell’everoli-mus per questa indicazione.
Franz DN, Leonard J, Tudor C, et al. Ra-pamycin causes regression of astrocyto-mas in tuberous sclerosis complex. Ann Neurol 2006;59:490-8.
Hochart A, Gaillard V, Baroncini M, et al. Bevacizumab decreases vestibular schwannomas growth rate in children and teenagers with neurofibromatosis type 2. J Neurooncol 2015;124:229-36.
* Articolo che indica i principali risultati della terapia con bevacizumab nei bambini e adolescenti con NF2.
Jakacki RI, Dombi E, Potter DM, et al. Phase I trial of pegylated interferon-alpha-2b in young patients with plexiform neuro-fibromas. Neurology 2011;76:265-72.
Jóźwiak S, Nabbout R, Curatolo P, et al. Management of subependymal giant cell astrocytoma (SEGA) associated with tu-berous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol 2013;17:348-52.
Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refracto-ry epilepsy in tuberous sclerosis complex. Ann Neurol 2013;74:679-87.
Le Merrer M, Rousseau F, Legeai-Mal-let L, et al. A gene for achondroplasia-hypochondroplasia maps to chromosome 4p. Nature Genet 1994;6:314-7.
* Il gruppo francese dell’ospedale Ne-cker di Parigi pubblica, in contemporanea al gruppo americano di Tsipouras (Veli-nov et al., 1994), il locus dove mappa il gene per l’acondroplasia e ipocondropla-
sia. Questo risultato è la prima conferma del concetto di famiglia, nell’ambito delle displasie scheletriche, cioè del fatto che forme di gravità clinica diversa possano essere causate da mutazioni in uno stesso gene.
Legeai-Mallet L. C-Type natriuretic pep-tide analog as therapy for achondroplasia. Endocr Dev 2016;30:98-105.
Lorget F, Kaci N, Peng J, et al. Evaluation of the therapeutic potential of a CNP analog in a Fgfr3 mouse model recapitulating achondro-plasia. Am J Hum Genet 2012;91:1108-14.
** È il contributo scientifico fondamen-tale che ha consentito di programmare e procedere all’avvio dell’unico trial clinico in corso. Si tratta della conferma speri-mentale su animali da esperimento che il BMN111, analogo farmacologico del CNP, è in grado di produrre gli stessi suoi effetti, ma ha il vantaggio di avere un’emivita tale da consentire un’unica somministrazione al giorno.
Matsushita M, Hasegawa S, Kitoh H, et al. Meclozine promotes longitudinal skele-tal growth in transgenic mice with achon-droplasia carrying a Gain of Function mu-tation in the FGFR3 gene. Endocrinology 2015;156:548-54.
Moavero R, Coniglio A, Garaci F, et al. Is mTOR inhibition a systemic treatment for tuberous sclerosis? Ital J Pediatr 2013;39:57.
Moreno L, Bautista F, Ashley S, et al. Does chemotherapy affect the visual out-come in children with optic pathway glio-ma? A systematic review of the evidence. Eur J Cancer 2010;46:2253-9.
Robertson KA, Nalepa G, Yang FC, et al. Imatinib mesylate for plexiform neuro-fibromas in patients with neurofibroma-tosis type 1: a phase 2 trial. Lancet Oncol 2012;13:1218-24.
Rousseau F, Bonaventure J, Legeai-Mallet L, et al. Mutations in the gene en-coding fibroblast growth factor receptor-3 in achondroplasia. Nature 1994;371:252-4.
* Lo stesso gruppo francese del Necker di Parigi, che in pochi mesi è riuscito prima a mappare il locus malattia e poi a identifi-care la mutazione causa dell’acondroplasia
nel gene FGFR3 che codifica per il recetto-re 3 per i fattori di crescita dei fibroblasti. Anche in questo caso nello stesso periodo il gruppo americano è giunto allo stesso risultato (Shiang et al., 1994).
Ruggieri M, Praticò AD, Evans DG. Dia-gnosis, management, and new therapeutic options in childhood neurofibromatosis type 2 and related forms. Semin Pediatr Neurol 2015;22:240-58.
Shiang R, Thompson LM, Zhu YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common gene-tic form of dwarfism, achondroplasia. Cell 1994;78:335-42.
Velinov M, Slaugenhaupt SA, Stoilov I, et al. The gene for achondroplasia maps to the telomeric region of chromosome 4p. Nature Genet 1994;6:318-21.
Yamashita A, Morioka M, Kishi H, et al. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature 2014;513:507-11.
Yasoda A, Komatsu Y, Chusho H, et al. Overexpression of CNP in chondrocytes re-scues achondroplasia through a MAPK-de-pendent pathway. Nat Med 2004;10:80-6.
** Il secondo e fondamentale contributo sperimentale che ha dimostrato la possibi-lità CNP di inibire il pathway FGFR3.
Yasoda A, Ogawa Y, Suda M, et al. Na-triuretic peptide regulation of endochon-dral ossification. Evidence for possible roles of the C-type natriuretic peptide/guanylyl cyclase-B pathway. J Biol Chem 1998;273:11695-700.
* Il primo contributo sperimentale che ha consentito di identificare la funzione del pathway CNP-NRP-B sulla regolazione dell’ossificazione encondrale.
Xie Y, Su N, Jin M, et al. Intermittent PTH (1-34) injection rescues the retarded ske-letal development and postnatal lethality of mice mimicking human achondroplasia and thanatophoric dysplasia. Hum Mol Ge-net 2012;21:3941-55.
Zeng LH, Xu L, Gutmann DH, et al. Ra-pamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 2008;63:444-53.
Corrispondenza
Angelo SelicorniUnità Operativa Complessa di Pediatria, ASST Lariana, via Ravona 20, 22020 San Fermo della Battaglia (CO) E-mail: [email protected]

215
Prospettive in PediatriaLuglio-Settembre 2016 • Vol. 46 • N. 183 • P. 215
Oftalmologia pediatrica
Abbiamo accolto con entusiasmo l’iniziativa di Prospettive in Pediatria di trattare una serie di capitoli dedicati al mondo dell’oftalmologia pediatrica, in modo da poter approfondire alcuni temi di largo interesse per le famiglie dei nostri piccoli pazienti. Nell’articolo principale abbiamo analizzato l’incremento di prevalenza della miopia nella popolazione giovanile, evidenziando gli articoli che studiavano le possibili cause alla base di questo au-mento. Abbiamo cercato inoltre di mettere in evidenza quali sono i trattamenti che possono essere utilizzati per il controllo della progressione miopica. Sempre nell’articolo principale sono stati analizzati i nuovi trattamenti locali e sistemici in corso di uveiti pediatriche e congiuntiviti allergiche. Per quanto riguarda le uveiti, abbiamo trattato il ruolo che rivestono i nuovi farmaci biologici e le iniezioni intraoculari di corticosteroidi, mentre nel paragrafo delle congiuntiviti allergiche una particolare attenzione è stata posta nella descrizione del trattamen-to locale della cheratocongiuntivite vernal con preparati locali a base di ciclosporina e tacrolimus, in modo da ridurre gli effetti collaterali dell’ampio uso di corticosteroidi.Nel secondo articolo è stato affrontato il ruolo sempre più ampio che la genetica sta rivestendo nell’aumentare la comprensione dei meccanismi molecolari legati alle manifestazioni dei quadri clinici più vari, soprattutto quelli più complessi e in cui la necessità di un approccio multidisciplinare si rende necessario.Fino a non molti anni fa, infatti, la presenza di una sospetta patologia ereditaria, specie in ambito oculistico, poneva le basi per una situazione clinica incurabile e nei cui confronti anche la scienza medica era in grado di fornire, nel migliore dei casi, solo una definizione diagnostica senza possibilità di avere una prospettiva di terapia auspicabile per i pazienti.Negli ultimi anni, l’evoluzione tecnologica globale ha permesso anche l’applicazione in campo medico di stru-menti rivoluzionari che hanno cambiato l’approccio e l’interesse verso tale gruppo di patologie. Solo per citare la più recente acquisizione, il next generation sequencing (NGS) è in grado di analizzare segmenti di DNA di qualunque grandezza, fino anche a un esame completo delle strutture codificanti (il cosiddetto esoma) e non codificanti (o whole genome sequencing). Il risvolto pratico di tutto questo fiorire di novità e comprensione di meccanismi patogenetici, oscuri fino a non molti anni fa, ha ulteriormente spinto anche la ricerca a identificare i vari target per l’applicazione di terapie che hanno la pretesa di correggere il primo responsabile dell’alterazione patologica e di cui oggi siamo a conoscenza risiedere in punti precisi del nostro genoma.Nel nostro terzo articolo, abbiamo quindi preso in esame una problematica di frequente riscontro nella pratica clinica quotidiana, quali sono le anomalie della motilità oculare e i loro risvolti sulla presenza di torcicolli com-pensativi. Quante volte accade che uno dei principali dubbi dei genitori sia relativo a una problematica postu-rale o di sospetto disallineamento oculare? Abbiamo cercato, in questo caso, di fornire una revisione dei quadri clinici più frequenti che sono di estrema complessità in alcune situazioni, data l’età dei bambini e la difficoltà di eseguire alcuni test diagnostici, che nell’adulto non pongono problematiche di diagnosi differenziale.Comprendiamo bene le difficoltà di una materia che, se non esperita quotidianamente nella pratica clinica, presenta notevoli tecnicismi che possono allontanare l’interesse, data la complessità dei temi e dei meccanismi affrontati, in special modo la parte coinvolgente il settore della genetica oculare. Nel nostro intento speriamo di aver reso un’idea, non eccessivamente semplificata, di quello che significa oggi affrontare una patologia oculare pediatrica e la necessità di collaborare sempre, con approccio multidisciplinare, alla sua definizione clinica e al suo idoneo trattamento.
Roberto CaputoUnità Operativa Oftalmologia Pediatrica, AOU Meyer, Firenze

216
Luglio-Settembre 2016 • Vol. 46 • N. 183 • Pp. 216-226 Prospettive in Pediatria
Oftalmologia pediatrica
Novità in Oftalmologia pediatrica
Roberto Caputo1 Gabriele Simonini2
Alice Brambilla2 Neri Pucci3
Cinzia de Libero1
1 Unità Operativa Oftalmologia Pediatrica, AOU Meyer, Firenze; 2 Unità Operativa Reumatologia Pediatrica, AOU Meyer, Firenze;
3 Servizio di Allergologia Pediatrica, AOU Meyer, Firenze
In the last few decades, myopia has increased in prevalence worldwide and has emerged as a public health issue. In this review, we analyse the pathogenesis and treatment of myopic patients. Myopia is a result of the combination of genetic and environmental factors. Looking at treatment strategies, administration of atropine eye drops has been shown to be effective in controlling progression of myopia. We also analyse the treatment strategy of paediatric uveitis, focusing on systemic immune-modulatory therapy and tumour necrosis factor α (TNF-α) an-tagonists. Lastly, we examine new treatments used for ocular allergic diseases.
Summary
La miopia ha avuto un forte aumento di incidenza nella popolazione giovanile negli ultimi 30 anni in tutto il mondo, al punto da diventare un importante problema di salute pubblica. In questo articolo di revisione verranno trattate le cause genetiche e ambientali che sono alla base di questo fenomeno e le possibili metodiche di trattamento, tra cui un ruolo rile-vante gioca la somministrazione locale di atropina. Il secondo argomento che verrà affron-tato è quello che riguarda le novità nel trattamento delle uveiti pediatriche, in particolare delle forme autoimmuni. Saranno analizzati i nuovi farmaci, come le terapie immunomodu-lanti e gli antagonisti del Tumor Necrosi Factor (TNF-α). Nella parte finale viene fatta una revisione delle prospettive terapeutiche in caso di congiuntiviti allergiche, in particolare sull’uso dei nuovi farmaci utilizzati nella cheratocongiuntivite vernalis, come la ciclosporina e il tacrolimus in somministrazione topica.
Riassunto
AbbreviazioniAIG: artrite idiopatica giovanileTNF: Tumor Necrosis FactorMTX: methotrexateSAC: congiuntivite allergica stagionalePAC: congiuntivite allergica perenneVKC: cheratocongiuntivite vernalAKC: cheratocongiuntivite atopicaGPC: congiuntivite gicantopapillare
Obiettivi e metodologia della ricerca bibliografica effettuataL’articolo è una revisione della letteratura inerente al-cuni argomenti oculistici di particolare interesse pedia-trico. La revisione prende in considerazione le pubbli-cazioni prodotte nel periodo 2008-2016, utilizzando il
motore di ricerca PubMed. Sono state utilizzate per la ricerca le seguenti parole chiave: “myopia”, “genetic”, “environment”, “treatment”, “pediatric”, “children”, “atropi-ne”, “uveitis”, “anti tumor necrosis factor”, “allergic con-junctivitis”, “vernal ketatoconjunctivitis”, “cyclosporine”, “tacrolimus”. Particolare attenzione è stata posta ai la-vori di revisione scientifica che comparavano la validità statistica delle precedenti pubblicazioni. Gli argomenti di questo articolo vertono sull’analisi del marcato au-mento di incidenza della miopia avvenuto negli ultimi anni e sulle prospettive di trattamento. Saranno poi evi-denziate le nuove strategie terapeutiche in pazienti af-fetti da uveiti o gravi malattie oculari di origine allergica.
La miopiaPerché parlare di miopia giovanile in un articolo di re-visione pediatrica? Basti pensare che la miopia è di-

217
Novità in Oftalmologia pediatrica
ventato un problema così importante di salute pubbli-ca, al punto che è stato inserito tra le prime 5 priorità in ambito oculistico dall’OMS nella Iniziativa Globale per la Eliminazione della Cecità Evitabile (Huang et al., 2016). La miopia infatti, soprattutto nelle forme più gravi, ha un’aumentata incidenza di complicanze ocu-lari tra cui le degenerazioni maculari, il distacco retini-co e il glaucoma. Non bisogna inoltre dimenticare gli elevati costi che devono sostenere i pazienti miopi e le loro famiglie con visite oculistiche e ricorrenti cam-bi di occhiali o lenti a contatto per il progressivo au-mento del difetto nell’età adolescenziale. Inoltre non possiamo dimenticare che l’accesso alla correzione ottica non è sempre possibile, al punto che si stimano 108 milioni di persone che non possono correggere il loro difetto refrattivo, diventando questa la seconda causa di cecità nel mondo (Bourne et al., 2013).
La miopia: definizioneMa cosa è la miopia? Per miopia s’intende un difetto re-frattivo, per cui le immagini che provengono da lontano invece di cadere sul piano retinico, vengono focalizzate davanti a esso. In genere questo è dovuto a un’aumen-tata lunghezza assiale del bulbo oculare in rapporto al potere delle lenti che compongono l’occhio (Fig. 1). Nei primi anni di vita i bambini sono di solito mode-ratamente ipermetropi. Durante lo sviluppo, la crescita del bulbo oculare e le modifiche del potere delle len-ti che formano l’occhio (cornea e cristallino) portano a una riduzione dell’ipermetropia, in quello che viene definito processo di emmetropizzazione. Un’eccessiva crescita del bulbo oculare provoca quindi un passaggio verso la miopia. La miopia viene generalmente clas-sificata in base all’entità del difetto refrattivo in lieve o grave. Nel primo caso, di solito nelle forme inferiori alle 6 diottrie, l’occhio non presenta alterazioni patologiche e il rischio di complicanze è sovrapponibile a quello della popolazione non miope. Nelle forme gravi (oltre le 6-8 diottrie, a seconda delle classificazioni), il bul-bo oculare si presenta particolarmente allungato, con alterazioni patologiche, tra cui lo sfiancamento della parte posteriore dell’occhio (stafiloma miopico) e con-seguenti alterazioni retiniche (retinocoroidosi miopica) che aumentano il rischio di maculopatie, rotture retini-che e distacco di retina. In questi pazienti risulta anche aumentata l’incidenza di glaucoma. A eccezione delle forme congenite o molto precoci, la miopia insorge in età scolare e tende a progredire nel periodo adole-scenziale. I sintomi manifestati dal paziente consistono in una riduzione dell’acuità visiva da lontano, tendenza a stringere le palpebre nel tentativo di focalizzare le immagini e spesso cefalea. Per la diagnosi è neces-saria una visita oculistica con cicloplegia, in modo da escludere le false miopie da sforzo accomodativo. Il trattamento standard consiste poi nella prescrizione di occhiali o lenti a contatto divergenti, che spostano le immagini posteriormente in modo che vadano a fuoco sul piano retinico.
La miopia: epidemiologiaNumerosi studi hanno dimostrato un importante au-mento dell’incidenza della miopia in tutto il mondo, so-prattutto nei paesi dove è cresciuto in modo evidente il livello di scolarizzazione. In Europa la prevalenza del-la miopia è stimata intorno al 30% degli individui ma con picchi del 47,2% nella popolazione giovanile, una percentuale quasi doppia rispetto ai pazienti anziani (Williams et al., 2015). Uno studio epidemiologico del 2009, condotto negli Stati Uniti, ha analizzato la pre-valenza della miopia nel periodo 1971-1972 e lo ha confrontato con il periodo 1999-2004. I risultati hanno dimostrato una prevalenza media che è passata dal 25% al 41,6%, con una variazione dal 27,1% al 45,8% nella popolazione femminile. Il problema assume pro-porzioni critiche nelle popolazioni asiatiche, nelle quali, probabilmente per particolare background ge-netico, la miopia è più frequente, con un significativo aumento negli ultimi anni. A Singapore, per esempio, la prevalenza della miopia nella popolazione di etnia cinese è passata dal 47% della fine degli anni ’80 a quasi il 90% nel 2010. Un simile aumento è stato ri-scontrato anche nelle popolazioni di etnia indiana e malese (Morgan et al., 2012).Un recentissimo studio ha inoltre valutato che, con questo trend di crescita, nel 2050 i miopi potreb-
Figura 1. Schema rappresentante un occhio miope: A) si nota come le immagini vadano a fuoco davanti la retina; B) l’utilizzo di una lente divergente sposta il fuoco sul piano retinico.
A
B

218
R. Caputo et al.
bero essere circa quasi 5 miliardi (Bourne et al., 2013).
La miopia: cause ambientali e geneticheI dati epidemiologici che abbiamo precedentemente esposto fanno quindi sorgere numerose domande sul perché si sia assistito a questo importante aumento nella prevalenza della miopia. Il dato sicuramente più evidente è quello di un rapporto abbastanza stretto tra l’aumento della scolarizzazione e l’incidenza della miopia. Viene quindi facile ipotizzare che l’attività sco-lastica, e quindi le aumentate attività visive per vicino possano giocare un ruolo importante nel favorire lo sviluppo della miopia. Non si può però non notare che razze diverse hanno incidenze diverse di miopia, così come famiglie con soggetti miopi hanno più frequen-temente figli affetti dal medesimo difetto. Da questi dati si può quindi ipotizzare che anche fattori genetici abbiano il loro ruolo nel giustificare questo crescente aumento della miopia. La letteratura degli ultimi anni ha quindi cercato di valutare in modo chiaro quale tra questi fattori sia maggiormente determinante. Per quanto riguarda l’aspetto genetico, sono stati ese-guiti studi su gemelli o gruppi familiari (Ramessuer et al. 2015; Chua et al., 2015) che hanno dimostrato una marcata influenza della componente genetica sullo sviluppo dei vari difetti refrattivi tra cui la miopia. Inol-tre la miopia è un difetto che si riscontra in molti di-sordini a trasmissione genetica, al punto che nel 2011 nel database Online Mendelian Inheritance in Man (OMIMwww.ncbi.nlm.nih.gov/omim) ne erano elencati ben 261. Tra queste ricordiamo le patologie del tessuto connettivale (Marfan, Stickler) e malattie interessanti i recettori retinici (Cecità Notturna Stazionaria Congenita X linked). Se poi andiamo a valutare soltanto le miopie elevate, questo rapporto con la componente genetica si fa ancora più stretto. Studi GWAS (Genome-Wide Association Studies) hanno identificato oltre 30 pro-babili loci correlati allo sviluppo della miopia (Simpson et al., 2014), che si differenziano a seconda delle po-polazioni studiate ma che nonostante tutto giustificano meno del 12% delle varianti fenotipiche. Alla fine degli anni ’70, studi effettuati su animali, in-clusi i primati, hanno dimostrato che modificando l’in-put visivo, con annebbiamento o lenti, si modificava di conseguenza anche la crescita del bulbo oculare con miopizzazione o ipermetropizzazione, a seconda della tecnica usata. Questi dati, associati a eviden-ze epidemiologiche, hanno cominciato a sottolinea-re il ruolo che può avere l’ambiente nella variazione dello sviluppo refrattivo dell’occhio. È risultato presto evidente che l’aumento della scolarizzazione in molte aree si è affiancato a un progressivo aumento dell’in-cidenza della miopia. Numerosi studi sono stati quindi fatti cercando di correlare il livello di scolarizzazione, il tempo dedicato alle attività visive per vicino e il tempo speso all’aperto, oppure la residenza in aree urbane o rurali con la prevalenza della miopia.
Per quanto riguarda il rapporto tra miopia e scola-rizzazione, esiste sicuramente un legame in tutte le popolazioni studiate (Williams et al., 2015; Fan et al., 2016). Sfortunatamente a un’analisi statistica multifat-toriale, non sembra che il livello di scolarizzazione per sé sia un dato sufficiente a spiegare l’aumento della miopia nelle popolazioni studiate. Gli altri fattori chia-mati in causa quindi a supporto di questi dati sono stati: il tempo trascorso nelle attività da vicino o il tem-po trascorso all’aperto. Per quanto riguarda le attività da vicino, non tutti gli studi concordano sull’esistenza di rapporto diretto fra tempo trascorso nella lettura e miopizzazione (Huang et al., 2015); in alcuni studi tale rapporto appare presente (Fernandez-Montero et al., 2015), mentre in altri non viene confermato (Chua et al., 2015; Rose et al., 2008; Lu et al., 2009). Viceversa la maggior parte degli studi concorda sul rapporto tra il tempo trascorso all’aperto e la minor incidenza di miopia (Rose et al., 2008).Un’attenta revisione di tutti questi studi, porta a con-cludere che la genesi della miopia e del suo impor-tante aumento di prevalenza, siano legati a una stret-ta interazione tra predisposizione genetica e influen-za ambientale (Fan et al., 2016), oppure a un’alterata regolazione epigenetica dell’espressione dei vari geni interessati (Ramessur et al., 2015).
La miopia: il trattamentoLa correzione della miopia avviene classicamente con l’uso di occhiali e lenti a contatto. Queste ultime possono essere utilizzate anche in ambito pediatrico. A fronte di un generale miglioramento delle perfor-mance visive, le lenti a contatto pongono alcuni limi-ti legati prevalentemente a un aumentato rischio di infezioni oculari, per cui la supervisione dei genitori sull’uso corretto e sulle procedure di pulizia e steri-lizzazione diventa fondamentale. Negli ultimi anni si sta facendo ricorso a una nuova tecnica di correzio-ne ottica per la miopia: l’ortocheratologia. Si tratta di una metodica che prevede l’utilizzo di lenti a contatto rigide che vengono indossate di notte, e tramite una calcolata deformazione della cornea, portano a una transitoria riduzione della miopia, in modo da rendere il paziente emmetrope nelle ore diurne. Per quanto riguarda la chirurgia refrattiva, a eccezio-ne di casi particolari, deve essere riservata a pazienti adulti con situazione refrattiva stabile.Negli ultimi anni i ricercatori hanno cercato soluzioni per ridurre la progressione della miopia nel periodo adolescenziale, con metodiche ottiche e farmacologi-che, che andremo ad analizzare:• lenti bifocali: basandosi sull’ipotesi che l’attività da
vicino aumentasse la progressione della miopia e che i miopi avessero un deficit di accomodazione, molti autori hanno sperimentato l’uso di lenti bifo-cali o progressive, che permettessero una messa a fuoco da vicino senza sforzo accomodativo. Pur-troppo questa metodica non ha retto alle evidenze

219
Novità in Oftalmologia pediatrica
scientifiche (Walline et al., 2011) a eccezione di alcuni lavori in cui venivano studiati pazienti miopi con esoforia (tendenza allo strabismo convergen-te) nei quali, la riduzione dello sforzo accomodati-vo con lenti bifocali permetteva una minor progres-sione miopica;
• lenti a contatto: l’uso di lenti a contatto non ha mostrato significativi risultati nella riduzione della progressione della miopia. Uno studio del 2004 ha confrontato l’uso di lenti a contatto morbide ver-so lenti a contatto gas permeabili e ha evidenzia-to una riduzione della progressione miopica nel gruppo con lenti gas permeabili nel primo anno, che però non è stato riconfermato al terzo anno di follow-up. Attualmente sono in corso studi con l’uso di lenti a contatto morbide con curvatura pe-riferica modificata che, riducendo lo sfocamento periferico, potrebbero avere effetto sulla riduzio-ne della progressione miopica. Su tale metodica mancano però ancora conferme certe;
• ortocheratologia: una revisione degli ultimi studi effettuati in pazienti miopi trattati con la tecnica dell’ortocheratologia ha ipotizzato una possibile efficacia nella riduzione della progressione miopi-ca (Sun et al., 2015) utilizzando questa metodica;
• sottocorrezione ottica: per molti anni era consue-tudine prescrivere una correzione miopica lieve-mente inferiore al difetto refrattivo totale. Questa pratica, non solo non si è dimostrata efficace nel mantenere un controllo della progressione miopi-ca, ma sembra addirittura peggiorarla;
• atropina collirio: l’atropina è un agente antimusca-rinico che ha dimostrato una notevole efficacia nel controllo della progressione miopica (Chia et al., 2016). Purtroppo il collirio a base di atropina at-tualmente in commercio (1% e 0,5%) provoca una midriasi e un blocco dell’accomodazione, con con-seguente disturbo visivo, che ne impedisce l’uso quotidiano. Gli studi più recenti hanno però dimo-strato che, preparazioni in collirio alla concentra-zione di 0,01%, mantengono una buona efficacia nel controllo della miopia, con pochi effetti collate-rali come midriasi e cicloplegia;
• occhiali forati: sono occhiali con lenti nere e picco-li fori che “promettono il blocco e addirittura il mi-glioramento della miopia”. Attualmente NON esiste alcun supporto scientifico a favore di questi stru-menti;
• metodo Bates: si basa su un concetto di utilizzo del-la mente e degli occhi in modo naturale con la con-sapevolezza delle interferenze esterne, ma anche per questa metodica NON esistono dati scientifici e non è stata valutata tramite trials clinici.
Possiamo quindi concludere che il paziente miope beneficia di una correzione ottica totale che deve por-tare a permanenza. Nella fase adolescenziale, l’unica metodica che sta dimostrando una buona efficacia nel controllo della progressione della miopia è l’utilizzo di
atropina in collirio alla concentrazione di 0,01%, men-tre per l’ortocheratologia aspettiamo ulteriori conferme.
Novità in tema di trattamento delle uveiti pediatricheL’uveite è una patologia infiammatoria, a eziologia variabile, che coinvolge parzialmente o totalmente la tunica vascolare dell’occhio (uvea). Il processo flogi-stico può essere acuto, con esordio clinico eclatante, o cronico e paucisintomatico. Classicamente le uveiti sono suddivise in base alla porzione interessata in an-teriori (iridocicliti), intermedie, posteriori (corioretiniti) e diffuse (panuveiti). Il 5-10% dei casi esordiscono du-rante l’infanzia, con un’incidenza di circa 4-7/100.000 bambini per anno e una prevalenza di 28/100.000 bambini. La corretta diagnosi e l’adeguata terapia sono necessari al fine di prevenire l’insorgenza di se-vere complicanze oculari e deficit visivi permanenti. All’uveite pediatrica sono infatti imputabili fino al 10% dei casi di cecità infantile nei paesi a elevato sviluppo socio-economico.L’Artrite Idiopatica Giovanile (AIG) è la principale causa di uveite cronica anteriore in età pediatrica, essendo responsabile del 1,8-47% dei casi diagno-sticati. Più raramente la flogosi oculare si sviluppa nel contesto di malattie autoimmuni o infiammatorie siste-miche, patologie tumorali o malattie infettive. I casi a eziologia non determinabile rientrano nel quadro di uveite idiopatica, a verosimile patogenesi autoimmu-ne (Simonini et al., 2010).
Terapia “step by step”La scelta di un appropriato percorso terapeutico è di-rimente al fine di controllare la flogosi intraoculare, li-mitare il ricorso ai corticosteroidi e garantire una buo-na prognosi visiva a lungo termine. L’infiammazione cronica può determinare l’insorgenza di complicanze quali cataratta, glaucoma, cheratopatia a bandelletta, sinechie, edema maculare cistoide, ipotonia oculare, distacco retinico, ischemia retinica e atrofia ottica. Fino al 30% dei pazienti affetti presenta ridotta acu-ità visiva, mentre il 10% dei casi va incontro a cecità. Complicanze oculari possono poi essere legate al fre-quente uso di corticosteroidi e tra queste ricordiamo la cataratta e il glaucoma.Con l’esclusione delle forme a eziologia infettiva, per le quali è essenziale un’appropriata terapia antimicro-bica, il trattamento dell’uveite si basa attualmente su un percorso step by step (Fig. 2). In caso di flogo-si lieve-moderata con prevalente coinvolgimento del segmento anteriore, la somministrazione di steroidi topici associati a midriatici può essere sufficiente a risolvere il quadro infiammatorio. Se la flogosi è più marcata diventa invece necessario un trattamento steroideo per via sistemica. I pazienti che non rispondono al trattamento di prima linea o che sviluppano dipendenza dagli steroidi sono
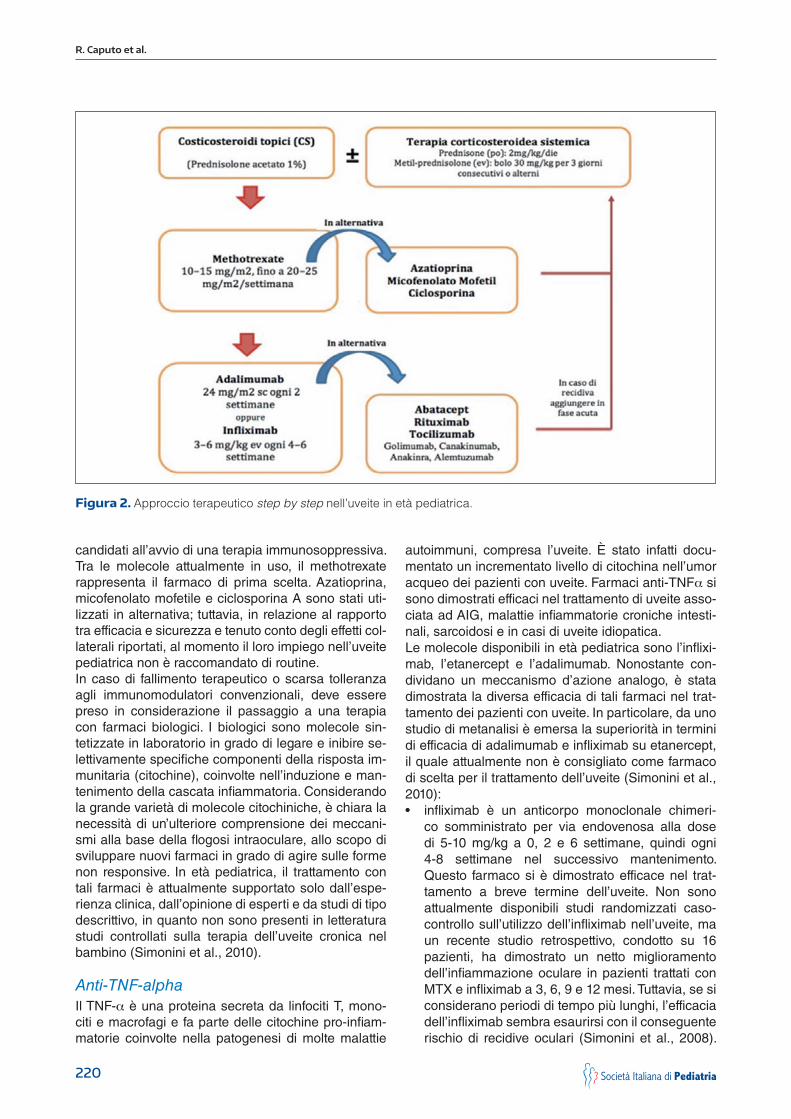
220
R. Caputo et al.
candidati all’avvio di una terapia immunosoppressiva. Tra le molecole attualmente in uso, il methotrexate rappresenta il farmaco di prima scelta. Azatioprina, micofenolato mofetile e ciclosporina A sono stati uti-lizzati in alternativa; tuttavia, in relazione al rapporto tra efficacia e sicurezza e tenuto conto degli effetti col-laterali riportati, al momento il loro impiego nell’uveite pediatrica non è raccomandato di routine. In caso di fallimento terapeutico o scarsa tolleranza agli immunomodulatori convenzionali, deve essere preso in considerazione il passaggio a una terapia con farmaci biologici. I biologici sono molecole sin-tetizzate in laboratorio in grado di legare e inibire se-lettivamente specifiche componenti della risposta im-munitaria (citochine), coinvolte nell’induzione e man-tenimento della cascata infiammatoria. Considerando la grande varietà di molecole citochiniche, è chiara la necessità di un’ulteriore comprensione dei meccani-smi alla base della flogosi intraoculare, allo scopo di sviluppare nuovi farmaci in grado di agire sulle forme non responsive. In età pediatrica, il trattamento con tali farmaci è attualmente supportato solo dall’espe-rienza clinica, dall’opinione di esperti e da studi di tipo descrittivo, in quanto non sono presenti in letteratura studi controllati sulla terapia dell’uveite cronica nel bambino (Simonini et al., 2010).
Anti-TNF-alphaIl TNF-α è una proteina secreta da linfociti T, mono-citi e macrofagi e fa parte delle citochine pro-infiam-matorie coinvolte nella patogenesi di molte malattie
autoimmuni, compresa l’uveite. È stato infatti docu-mentato un incrementato livello di citochina nell’umor acqueo dei pazienti con uveite. Farmaci anti-TNFα si sono dimostrati efficaci nel trattamento di uveite asso-ciata ad AIG, malattie infiammatorie croniche intesti-nali, sarcoidosi e in casi di uveite idiopatica. Le molecole disponibili in età pediatrica sono l’inflixi-mab, l’etanercept e l’adalimumab. Nonostante con-dividano un meccanismo d’azione analogo, è stata dimostrata la diversa efficacia di tali farmaci nel trat-tamento dei pazienti con uveite. In particolare, da uno studio di metanalisi è emersa la superiorità in termini di efficacia di adalimumab e infliximab su etanercept, il quale attualmente non è consigliato come farmaco di scelta per il trattamento dell’uveite (Simonini et al., 2010):• infliximab è un anticorpo monoclonale chimeri-
co somministrato per via endovenosa alla dose di 5-10 mg/kg a 0, 2 e 6 settimane, quindi ogni 4-8 settimane nel successivo mantenimento. Questo farmaco si è dimostrato efficace nel trat-tamento a breve termine dell’uveite. Non sono attualmente disponibili studi randomizzati caso-controllo sull’utilizzo dell’infliximab nell’uveite, ma un recente studio retrospettivo, condotto su 16 pazienti, ha dimostrato un netto miglioramento dell’infiammazione oculare in pazienti trattati con MTX e infliximab a 3, 6, 9 e 12 mesi. Tuttavia, se si considerano periodi di tempo più lunghi, l’efficacia dell’infliximab sembra esaurirsi con il conseguente rischio di recidive oculari (Simonini et al., 2008).
Figura 2. Approccio terapeutico step by step nell’uveite in età pediatrica.

221
Novità in Oftalmologia pediatrica
Inoltre, la maggioranza degli studi presenti in let-teratura considera l’efficacia separata della terapia con MTX e infliximab, pertanto non è chiaro quale sia lo specifico e reale contributo di ciascuno di questi farmaci al miglioramento clinico (Simonini et al., 2014);
• etanerceptèunaproteinadifusionetrailrecetto-re del TNF-α e il dominio Fc dell’IgG1. È sommini-strato per via sottocutanea alla dose di 0,4 mg/kg per 2 volte a settimana. Pur mostrando una buo-na efficacia nel trattamento dell’artrite, non è sta-ta dimostrata la sua effettiva utilità nella terapia dell’uveite. L’unico studio randomizzato controlla-to, fino a ora condotto sulla terapia delle uveiti in età pediatrica, non ha dimostrato un effettivo vantaggio della somministrazione di etanercept rispetto a placebo, mentre è stata riportata una netta superiorità dell’infliximab sull’etanercept nel controllo della malattia oculare associata ad AIG. Esso pertanto non è consigliato come farmaco di prima scelta per il trattamento dell’uveite (Simo-nini et al., 2014);
• adalimumabèunanticorpoumanizzato,sommini-strato per via sottocutanea ogni 7 giorni alla dose di 24 mg/m2. Numerosi studi riportano l’efficacia di questo farmaco nell’uveite (Simonini et al., 2014). In particolare, il nostro gruppo, in uno studio di co-orte che includeva 33 pazienti con uveite cronica, ha concluso che adalimumab è più efficace di in-fliximab nel mantenimento della remissione clini-ca a lungo termine (Simonini et al., 2011). Questo dato è stato poi confermato anche in uno studio multicentrico italiano su 108 pazienti con uveite associata ad AIG e trattati con anti-TNF-α. In tale studio sia infliximab che adalimumab sembrano essere sicuri ed efficaci nel trattamento dell’uveite, ma quest’ultimo presenta una maggiore efficacia nel controllo dell’infiammazione oculare a medio termine (Zannin et al., 2013). Inoltre, la sua effi-cacia appare maggiore qualora esso sia utilizzato come primo anti-TNF-α, altrimenti perde la capa-cità di mantenere una remissione prolungata nel tempo (Simonini et al., 2013).
Circa il 25% dei pazienti trattati con anti-TNF sviluppa intolleranza al farmaco o presenta una risposta clini-ca insufficiente; tali pazienti possono beneficiare del passaggio a un’altra categoria di modificatori dell’atti-vità biologica (Simonini et al., 2015). Altri modificatori dell’attività biologica:• abatacept è una proteina di fusione formata dal
dominio extracellulare dell’antigene 4 dei linfociti T citotossici e dal dominio Fc dell’IgG1; esso agi-sce inibendo l’attivazione dei T linfociti attraverso il blocco di segnali di co-stimolazione. Il dosaggio raccomandato è 10 mg/kg a 0, 2 e poi ogni 4 set-timane per via endovenosa. L’esperienza legata all’impego di abatacept nel trattamento delle uvei-ti risulta ancora limitata. In un ristretto numero di
pazienti affetti da uveite AIG-correlata, l’utilizzo di abatacept ha portato al miglioramento della flogosi oculare, oltre alla riduzione della frequenza delle recidive e del ricorso agli steroidi. Non è stata de-scritta l’insorgenza di nuove complicanze, né l’ag-gravamento di complicanze preesistenti durante la terapia, se pur non confermato in altre casistiche messe a confronto (Simonini et al., 2015);
• rituximab è un anticorpomonoclonale chimericodiretto verso l’antigene CD20 dei linfociti B. Esso viene utilizzato nel trattamento di linfomi, malattie autoimmuni e malattia infiammatorie oculari, e re-centemente si sta studiando il suo possibile ruo-lo di alternativa terapeutica in pazienti con uveite associata ad AIG scarsamente responsiva ad altri trattamenti. In uno studio retrospettivo multicentri-co che includeva 10 pazienti con uveite complicata e refrattaria al trattamento con CS topici, sistemici e anti-TNF-α, il rituximab si è dimostrato efficace in 7 su 10 pazienti nel controllo della malattia ocu-lare; la sua efficacia sembra più marcata in sog-getti affetti da AIG oligoarticolare ANA positiva e HLAB27 negativa. Ulteriori studi sono comunque necessari per provarne l’efficacia e la sicurezza (Simonini et al., 2015);
• tocilizumabèunanticorpomonoclonaleumanizzatodiretto contro IL-6, citochina pro-infiammatoria coin-volta nella proliferazione e differenziazione di linfoci-ti T e B. La somministrazione di tocilizumab per via endovenosa, al dosaggio di 8 mg/kg ogni 4 settima-ne, si è dimostrata efficace nell’indurre l’inattivazione di malattia in una piccola coorte di pazienti affetti da uveite AIG-correlata, associata a complicanze ocula-ri, non responsiva ad altre terapie. Alcuni casi clinici sembrano suggerire il possibile promettente ruolo di tocilizumab nel trattamento di pazienti adulti affetti da uveite severa e refrattaria. L’impiego di tale farmaco in pediatria è ancora limitato per la mancanza di dati relativi a efficacia e sicurezza in questa fascia d’età (Simonini et al., 2015).
Relativamente ai farmaci diretti contro interleuchina 1, nota citochina pro-infiammatoria apparentemente coinvolta anche nell’insorgenza e mantenimento del-la flogosi intra-oculare, sono stati descritti pochi casi clinici che hanno visto l’impiego di anakinra e cana-kinumab in 4 pazienti affetti da sindrome di Blau con coinvolgimento oculare, malattia di Behçet giovanile e sindrome Cronica, Infantile, Neurologica, Cutanea, Articolare (CINCA) (Simonini et al., 2015). Dalla nostra recente revisione sistematica, volta a va-lutare l’efficacia e sicurezza della terapia con biologi-ci non anti-TNF nel trattamento delle uveiti croniche idiopatiche severe e refrattarie, è emerso che il ricor-so a tali farmaci è in grado di portare a una buona risposta clinica in circa due terzi dei pazienti affetti. Tale tentativo terapeutico è pertanto consigliabile in questa categoria di pazienti, che presenta tra l’altro un elevato rischio di sequele visive invalidanti e per-

222
R. Caputo et al.
manenti. I pochi dati a disposizione in letteratura non consentono invece di dimostrare un chiaro vantaggio nell’impiego dei farmaci biologici come prima linea te-rapeutica e pertanto è attualmente ancora consiglia-bile l’approccio step by step in tutti i pazienti (Simoni-ni et al., 2010) (Fig. 2).
I trattamenti intravitrealiLe uveiti posteriori, intermedie, e panuveiti non infet-tive raramente rispondono alla sola terapia topica e spesso causano importanti deficit visivi, che possono derivare dall’insorgenza di edema maculare, infiam-mazione del nervo ottico e del vitreo. La sola terapia topica con colliri cortisonici risulta inef-ficace, per scarsa capacità del farmaco a raggiungere il segmento posteriore dell’occhio per cui è necessa-rio l’utilizzo di farmaci sistemici come descritto nella parte precedenti di questo lavoro. Per ridurre gli effetti collaterali di una terapia sistemica, si è cercato di tro-vare vie di somministrazione locali che permettessero una disponibilità del farmaco a dosaggi efficaci nella parte posteriore del bulbo oculare.Una via di somministrazione per i corticosteroidi usata da molti anni è quella dell’iniezione perioculare, che permette una discreta penetrazione del farmaco a livel-lo bulbare posteriore, ma che per ovvi motivi è difficil-mente praticabile in modo ripetuto in ambito pediatrico.Negli ultimi anni i ricercatori hanno valutato l’efficacia e sicurezza dell’iniezione intravitreale di vari farmaci, tra cui i corticosteroidi. Iniezioni intravitreali di triamci-nolone acetonide vengono utilizzate nel trattamento dell’edema maculare cistoide, secondario al processo uveitico refrattario al trattamento topico e/o sistemico. Questa metodica consente di ottenere in camera vitrea una rapida e maggior concentrazione del far-maco, che esplica la sua massima biodisponibilità direttamente a contatto con il tessuto retinico, supe-rando la barriera emato-oculare. Il triamcinolone, che essendo un farmaco solo in parte idrosolubile ha una durata in camera vitrea di circa 3 mesi. L’effetto è co-munque limitato nel tempo e si ha quindi la necessità di ripetute iniezioni con aumento delle complicanze oculari rappresentate dall’insorgenza di glaucoma e cataratta. Per aumentare la permanenza di farmaco e quindi ridurre la frequenza di iniezioni, negli ultimi anni sono stati immessi sul mercato Impianti di desa-metasone o fluorcinolone intravitreale a lento rilascio (slow – release device) (Tomkins Netzer et al., 2016). Alcuni di questi farmaci sono stati approvati dalla FDA per l’utilizzo delle uveiti dell’adulto. Le principali complicanze riscontrate nell’utilizzo di questi impianti sono l’aumento della pressione endoculare (glauco-ma), l’insorgenza di cataratta, emorragie vitreali e ra-ramente infezioni intraoculari (endoftalmiti).Bisogna ricordare che, nonostante la provata effica-cia, questi trattamenti sono considerati off label in ambito pediatrico.
Novità in tema di trattamento delle congiuntiviti allergicheLe congiuntiviti allergiche sono un vasto gruppo di condizioni allergiche caratterizzate da infiammazione della congiuntiva. Si riconoscono 5 forme principali: la congiuntivite allergica stagionale (SAC), la congiuntivite allergica perenne (PAC), la cheratocongiuntivite Vernal (VKC), la cheratocongiuntivite atopica (AKC), la congiuntivi-te gigantopapillare (GPC). I quadri clinici delle varie forme presentano similitudini e ciò può comportare problemi di diagnosi differenziale. Alcune di queste sono notevolmente frequenti (SAC e PAC) e non comportano particolari problemi di gestione tera-peutica; le altre sono abbastanza rare (VKC, AKC) e il trattamento con i comuni farmaci antistaminici, sia locali che sistemici, risulta spesso inefficace. In questi casi si rende necessario il ricorso agli steroidi topici che, se usati per periodi prolungati, espongo-no il paziente al rischio di effetti collaterali avversi, anche gravi come infezioni herpetiche, cataratta e glaucoma.Di particolare interesse pediatrico sono le congiuntivi-ti allergiche (SAC e PAC) e la VKC, in quanto la che-ratocongiuntivite atopica è un’infiammazione cronica che interessa prevalentemente pazienti di età tra i 16 e i 19 anni, spesso associata a eczema (95%) o asma e la cheratocongiuntivite gigantopapillare (GPC) soli-tamente è associata all’uso prolungato di lenti a con-tatto (LAC) o protesi oculari.
La congiuntivite allergica stagionale (SAC) e la congiuntivite allergica perenne (PAC)SAC è la forma più comune di congiuntivite allergica e rappresenta più del 50% delle congiuntiviti allergi-che. È una congiuntivite stagionale e i sintomi oculari, spesso associati a riniti, sono strettamente legati a pollini delle graminacee, parietaria e pollini di piante arboree nelle forme più precoci. I sintomi più impor-tanti sono prurito, bruciore e lacrimazione.In casi più impegnativi si possono riscontrare sintomi corneali quali fotofobia; eccezionali sono comunque gli esiti permanenti. I segni clinici, usualmente bila-terali, sono rappresentati da iperemia congiuntivale, ipertrofia papillare a livello della congiuntiva tarsale e infine produzione di essudato, inizialmente chiaro/trasparente (fase acuta) e successivamente denso/fibroso (fase cronica). Può interessare l’età pediatrica anche se il picco d’incidenza si colloca negli ultimi anni della seconda decade. La diagnosi è relativa-mente facile: pressoché costante è la contemporanea presenza di rinite allergica la quale, anzi, è spesso prevalente come intensità. Un’anamnesi familiare per allergie IgE-mediate e una personale per dermatite atopica sono frequenti. La PAC è considerata una variante della SAC che persiste per tutto l’anno ed è per lo più associata a

223
Novità in Oftalmologia pediatrica
rinite pluristagionale; circa il 79% dei pazienti presen-ta esacerbazioni stagionali. Acari della polvere, epi-teli di animali, spore fungine presenti negli ambienti domestici o lavorativi sembrano essere gli allergeni più comuni. La prevalenza è più bassa della SAC. Nelle forme perenni la sintomatologia è meno pro-nunciata; è comunque caratterizzata da prurito, lieve iperemia congiuntivale, bruciore, sensazione di cor-po estraneo. Sia la SAC che la PAC hanno all’incirca lo stesso esordio e si associano entrambe a sintomi, quali asma/eczema. Il sospetto diagnostico di SAC e PAC si basa sull’evidenza clinica (eritema, iperemia congiuntivale, ipertrofia papillare, edema palpebrale), sulla sintomatologia soggettiva (in particolare il pru-rito). La diagnosi sarà confermata con reperti di la-boratorio, come l’aumento di eosinofili e neutrofili nel sangue, presenza elevata di IgE specifiche sieriche e cutanee (Skin prick Test).
La cheratocongiuntivite Vernal (VKC)La cheratocongiuntivite Vernal (letteralmente “pri-maverile”, VKC) è un’affezione oculare ritenuta rara, classificata come allergica, se pure a eziologia sco-nosciuta. La VKC ha un forte impatto negativo sulla qualità di vita del bambino, condizionando le attività quotidiane sia scolastiche che di vita relazionale. Si tratta di un’affezione cronica bilaterale, spesso se-vera, a rischio di esiti permanenti se non adeguata-mente trattata. Il termine cheratocongiuntivite implica un possibile coinvolgimento della cornea (sofferenza corneale, abrasioni, ulcere a scudo) con conseguente compromissione del visus nel 5-6% dei casi. I sintomi si manifestano in primavera (febbraio-aprile) e peg-giorano progressivamente in estate, per poi regre-dire nel periodo autunno-inverno (forma stagionale 86,5%); in casi gravi la sintomatologia persiste per tutto l’anno (forma perenne 13,5%).La VKC inizia a manifestarsi nella prima decade di vita, solitamente non prima dei 3 anni e tende a risol-versi spontaneamente (anche se non sempre) dopo la pubertà, alla fine della seconda decade.Geograficamente è più diffusa nelle aree a clima cal-do e temperato, come il bacino del Mediterraneo, il Medio Oriente, la penisola dell’Anatolia, la penisola arabica, l’India, il Pakistan, il Giappone. Segni oculari patognomonici sono rappresentati dalla presenza delle papille giganti tarsali superiori (Fig. 3), e/o da papille limbari (Fig. 4).Possiamo distinguere 3 forme cliniche di VKC:• formabulbare:presenzadipapillelimbari;• formatarsale:presenzadipapilletarsali;• formamista: presenza di papille sia limbari che
tarsali.La diagnosi differenziale con la classica congiuntivite allergica (SAC e PAC) si basa essenzialmente sulla presenza delle papille giganti e tarsali, sul peggiora-mento nel periodo estivo, sulla differente gravità dei sintomi e sulla scarsa risposta agli antistaminici.
La morbilità di quest’affezione dipende soprattutto dalla scarsa efficacia dei farmaci tradizionali impiega-ti per il trattamento delle congiuntiviti allergiche (an-tistaminici, cromoni, NSAIDs), utili solo nelle forme più lievi, e dalla conseguente necessità di impiegare cortisonici, unici farmaci efficaci, per periodi prolun-gati, con i ben noti rischi di effetti secondari sull’oc-chio, come glaucoma, cataratta e aumentato rischio di infezioni.Nelle ultime decadi si sono utilizzati farmaci off label, principalmente ciclosporina e tacrolimus, per via to-pica per ridurre l’utilizzo di cortisonici locali. Studi cli-nici in doppio cieco contro placebo hanno dimostrato un’efficacia notevole della ciclosporina per via oculare (alla concentrazione dell’1 o 2%) nelle forme modera-te e severe di VKC in assenza di reazioni avverse si-gnificative. La ciclosporina per via oculare, infatti, non provoca alterazioni del cristallino e non aumenta la pressione oculare; inoltre, dato il basso dosaggio, i li-velli ematici sono sostanzialmente indosabili in questi pazienti. Le concentrazioni di ciclosporina che si sono dimostrate efficaci per uso topico, negli studi citati va-
Figura 3. Papille giganti tarsali nella VKC.
Figura 4. Papille limbari nella VKC.

224
R. Caputo et al.
riano dall’1 al 2%; in uno studio più recente della dura-ta di 2 anni, è stata utilizzata una preparazione a con-centrazione molto più bassa (0,05%) di ciclosporina, che ha dimostrato di essere più efficace del ketotifene in 31 pazienti per la prevenzione delle riacutizzazioni stagionali (Lambiase et al., 2011). Nonostante ciò, in accordo con la nostra personale esperienza, circa il 7-10% dei pazienti con VKC, non risponde al tratta-mento, transitoriamente o permanentemente, con col-lirio alla ciclosporina e in essi vi è un’alta prevalenza di casi con sintomatologia perenne. Nei pazienti resi-stenti alla terapia con ciclosporina, può trovare indica-zione il tacrolimus per via oculare. Il tacrolimus, come la ciclosporina, è un inibitore della calcineurina che blocca l’attivazione dei linfociti T, è indicato nella profi-lassi del rigetto d’organo nei pazienti trapiantati e vie-ne utilizzato come seconda linea di trattamento della dermatite atopica nei pazienti immunocompetenti di età maggiore di 2 anni. Il collirio al tacrolimus inol-tre, è stato usato per il trattamento di patologie oculari severe negli adulti e al meeting della Committee for Orphan Medicinal Products nel gennaio 2004 e stato designato dalla European Medicines Agency (EMEA) come un farmaco orfano per il trattamento della VKC. Alla prima descrizione di Vichyanond et al. sono se-guiti studi che mettono in luce sia l’efficacia che la sicurezza del collirio al tacrolimus allo 0,1% come unguento oftalmico nella VKC (Labcharoenwongs et al., 2012). Uno studio di Ebihara et al. effettuato su 52 pazienti trattati con collirio al tacrolimus allo 0,1% per 12 settimane, 15 dei quali erano affetti da VKC e 37 da cheratocongiuntivite allergica ha dimostrato una buona sicurezza del tacrolimus. La massima con-centrazione di tacrolimus determinata nel sangue è stata < a 2 mg/ml e circa il 75% dei pazienti trattati avevano concentrazioni ematiche di tacrolimus infe-riori allo 0,5 mg/ml. In un nostro recente studio cli-nico in doppio cieco, in cui sono stati arruolati i soli pazienti con VKC severa e resistenti alla terapia con ciclosprina, comparandone l’efficacia del tacrolimus, è emerso come il collirio al tacrolimus allo 0,1% sia efficace e sicuro nell’uso a breve termine per il tratta-mento della VKC severa anche resistente alla terapia con ciclosporina (Pucci et al., 2015). I valori ematici di transaminasi e creatinina monitorizzati durante il trattamento si sono mantenuti nei limiti della norma e
il tacrolimus è risultato indosabile nel sangue durante il trattamento. Da un punto di vista pratico, i pazienti che utilizzano il collirio al tacrolimus allo 0,1%, lamen-tano transitorio bruciore dopo instillazione del collirio. In circa 2/3 dei pazienti tale sintomatologia si riduce in circa 1-2 settimane. Un ultimo studio policentrico in aperto ha mostrato un’elevata efficacia del collirio con tacrolimus 0,1%, utilizzato su oltre 1.400 pazienti affetti da cheratocongiuntiviti croniche, tra cui la VKC, con un follow-up a 6 mesi (Fukushima et al., 2014). Nel 2005 la Pediatric Advisory Commitee of the US FDA ha aggiunto il tacrolimus unguento e il pime-crolimus in crema per il trattamento della dermatite atopica fra i farmaci potenzialmente a rischio per ne-oplasie cutanee a causa della assenza di dati certi sulla sicurezza di questi farmaci per l’utilizzo a lungo termine. Ciò nonostante alcune autorevoli società scientifiche hanno criticato questa posizione. A oggi non ci sono però, in letteratura, sufficienti studi epi-demiologici per concludere che gli inibitori della cal-cineurina possano essere causa di tumori, al con-trario, recenti studi indicano comunque un elevato livello di sicurezza nella somministrazione di questi farmaci (Car, 2013).Nell’ultima decade sono stati pubblicati case report di pazienti con VKC trattati con omalizumab, anticorpo monoclonale anti-IgE (de Clerck, 2013). In tutto 4 pa-zienti insensibili ad altri trattamenti sono stati trattati con successo. Nello studio di Heffler e coll. (Heffler et al., 2016) si è dimostrata una significativa riduzione della conta degli eosinofili nello scraping congiuntiva-le in corso di terapia. Si tratta peraltro di una casistica limitata e sono pertanto necessari ulteriori studi com-parativi e su casistiche più ampie. In conclusione la ciclosporina in collirio rappresen-ta, a oggi, la terapia di elezione nel trattamento della VKC moderata-severa. Ciononostante circa il 7-10% dei pazienti è transitoriamente o permanentemente non responsivo. In questi casi, il tacrolimus collirio allo 0,1% è risultato efficace e sicuro per il trattamento a breve-medio termine, mentre studi ulteriori sono ne-cessari per la valutazione della efficacia e sicurezza a lungo termine. L’anticorpo monoclonale anti-IgE oma-lizumab potrebbe, nel prossimo futuro, rappresentare una valida alternativa nei casi severi.

225
Novità in Oftalmologia pediatrica
Bibliografia
Bourne RRA, Stevens GA, White RA, et al. Causes of vision loss worldwide, 1990-2010: a systematic analysis. Lancet Glob Health 2013;1:e339-49.
Car WW. Topical calcineurin inhibitor for atopic dermatitis:review and treatment rac-comendations. Pediatr Drug 2013;15:303-10.
Chia A, Lu QS, Tan D. Five-Year clinical trial on atropine for the treatment of myo-pia 2: myopia control with atropine 0.01% eyedrops. Ophthalmol 2016;123:391-9.
** Follow-up a 5 anni sull’efficacia dell’atropina nel controllo della progres-sione miopica.
Chua SY, Ikram MK, Tan CS, et al. Rela-tive contribution of risk factors for early-onset myopia in young asian children. IOVS 2015;56:8101-7.
de Clerk TA, Sharma V, Arkwright PD, et al. Severe vernal keratoconjunctivitis suc-cessfully treated with subcutaneous omali-zumab. JAAPOS 2013;17:305-6.
Fan Q, Verhoeven VJ, Wojciechowski R, et al. Meta-analysis of gene-environment-wide association scans accounting for edu-cation level identifies additional loci for re-fractive error. Nat Commun 2016;7:11008.
Fernandez-Montero A, Olmo-Jimenez JM, Olmo N, et al. The impact of computer use in myopia progression: a cohort study in Spain. Prev Med 2015;71:67-71.
Fukushima A, Ohashi Y, Ebihara N, et al.
Therapeutic effects of 0.1% tacrolimus eye drops for refractory allergic ocular diseases with proliferative lesion or corneal involve-ment. Br J Ophthalmol 2014;98:1023-7.
* In questo studio si dimostra efficacia e sicurezza del tacrolimus per via oculare.
Heffler E, Picardi G, Liuzzo MT, et al. Omalizumab treatment of vernal kerato-conjunctivitis. JAMA Ophthalmol 2016:1-3.
Huang HM, Chang DS, Wu PC. The association between near work activi-ties and myopia in children-a system-atic review and meta-analysis. PloS one 2015;10:e0140419.
* Review sul rapporto tra attività da vi-cino e progressione miopica.
Huang J, Wen D, Wang Q, et al. Efficacy comparison of 16 interventions for myopia control in children: a network meta-analy-sis. Ophthalmol 2016;123:697-708.
* Review sulle varie metodiche utilizzate per il controllo della miopia.
Labcharoenwongs P, Jirapongsana-nuruk O, Visitsunthorn N, et al. A double masked comparison of 01% tacrolimus ointment and 2% cyclosporine eyedrops in the treatment of Vernal keratoconjunctivitis in children. Asian Pac J Allergy Immunol 2012;30:177-84.
Lambiase A, Leonardi A, Sacchetti M, et al. Topical cyclosporine prevents seasonal recurrences of Vernal keratoconjunctivitis in a randomized, double-masked, con-trolled 2-years study. J Allergy Clin Immu-nol 2011;128:896-7.
** Il lavoro, molto ben condotto, che rib-adisce l’efficacia della ciclosporina topica e ne evidenzia anche il ruolo preventivo con l’utilizzo di un basso dosaggio.
Lu B, Congdon N, Liu X, et al. Associa-tions between near work, outdoor activity, and myopia among adolescent students in rural China: the Xichang Pediatric Refrac-tive Error Study report no. Arch Ophthal-mol 2009;127:769-75.
Morgan IG, Ohno-Matsui K, Saw S-M. Myopia. Lancet 2012;379:1739-48.
** Review sulla miopia.
Pucci N, Caputo R, Di Grande L, et al. Tacrolimus vs cyclosporine eyedrops in se-vere cyclosporine –resistant Vernal kerato-conjunctivitis: a randomized, comparative, double-blind crossover study. Ped Allergy Immunol 2015;26:256-61.
** Il lavoro dimostra che il tacrolimus per uso oculare è efficace nei pazienti re-sistenti al trattamento con ciclosporina.
Ramessur R, Williams KM, Hammond CJ. Risk factors for myopia in a discord-ant monozygotic twin study. Ophthalmic Physiol Opt 2015;35:643-51.
Rose KA, Morgan IG, Ip J, et al. Outdoor activity reduces the prevalence of myopia in children. Ophthalmol 2008;115:1279-85.
* Primo studio che ha correlato il tempo trascorso all’aria aperta e la progressione miopica.
Simonini G, Cantarini L, Bresci C, et al. Current therapeutic approaches to autoim-
Box di orientamento
• Lamiopiahamostratounenormeaumentodiprevalenzanegliultimianni,soprattuttonellepopolazionimaggiormente scolarizzate al punto da essere inserita nelle 5 priorità dell’OMS per la prevenzione della cecità evitabile.
• Questoaumentodiprevalenzasembralegatoaun’interazionetraelementigeneticieambientali(aumen-tata attività scolare, riduzione attività all’aperto).
• Perridurrelaprogressionedellamiopiainetàadolescenzialesonostatepropostenumerosemetodiche,ma attualmente soltanto l’utilizzo di atropina in collirio alla concentrazione di 0,01% e il trattamento con ortocheratologia hanno dimostrato un’efficacia scientificamente dimostrabile.
• Leuveitipediatrichesonocausadel10%dicecitàpediatricaneipaesisviluppati.• Iltrattamentodelleuveitiprevedeunapprocciostep by step, iniziando con cortisonici locali per poi pas-
sare a farmaci sistemici di prima linea come il methotrexate, e successivamente all’impiego di farmaci biologici (Anti TNF – alpha o altri modificatori dell’attività biologica).
• Incasiselezionatidiuveiticoninteressamentovitrealeocorioretinicopossonoessereutilizzati tratta-menti corticosteroidei iniettati direttamente nella cavità vitreale.
• Lacheratocongiuntivitevernalisèunapatologiacheoriginadaunarispostaimmunologicacellulo-media-ta di tipo IV e che può assumere caratteri di particolare gravità, andando a coinvolgere anche la cornea. Presenta lesioni tipiche quali le papille giganti (Fig. 3) e le papille limbari (Fig. 4) e tende a peggiorare nel periodo estivo.
• Negliultimianniiltrattamentodelleformepiùgraviprevedel’affiancamentoaicorticosteroidilocalidialtriimmunosoppressori, come la ciclosporina e il tacrolimus.

226
R. Caputo et al.
Corrispondenza
Roberto CaputoUnità Operativa Oftalmologia Pediatrica, AOU Meyer, viale Pieraccini 24, 50139 Firenze - E-mail: [email protected]
mune chronic uveitis in children. Autoim-mun Rev 2010;9:674-83.
** Revisione sistematica delle strategie terapeutiche attualmente disponibili per la cura delle uveite infiammatorie croniche pediatriche e proposta di flow-chart tera-peutico.
Simonini G, Cimaz R, Jones GT, et al. Non anti TNF biologic modifier drugs in non-infectious refractory chronic uveitis: the current evidence from a sys-tematic review. Semin Arthritis Rheum 2015;45:238-50.
** Revisione sistematica e confronto meta-analitico dei farmaci non anti-TNFa attualmente utilizzati per la cura delle uveiti infiammatorie croniche dell’infanzia.
Simonini G, Druce K, Cimaz R, et al. Cur-rent evidence of anti-tumor necrosis factor α treatment efficacy in childhood chronic uveitis: a systematic review and meta-anal-ysis approach of individual drugs. Arthritis Care Res 2014;66:1073-84.
** L’unico studio con approccio meta-analitico attualmente disponibile in lettera-tura, che misura il confronto di efficacia dell’utilizzo degli anti-TNFα per la cura
delle uveiti infiammatorie croniche pedi-atriche.
Simonini G, Taddio A, Cattalini M, et al. Superior efficacy of adalimumab in treating childhood refractory chronic uveitis when used as first biologic modifier drug: adali-mumab as starting anti-TNF-α therapy in childhood chronic uveitis. Pediatric Rheu-matol Online J 2013;15:11-6.
Simonini G, Taddio A, Cattalini M, et al. Prevention off flare recurrences in childhood-refractory chronic uveitis: an open-label comparative study of adali-mumab versus infliximab. Arthritis Care Res 2011;63:612-8.
** L’unico studio pediatrico di tipo prospettico comparativo di efficacia fra infliximab e adalimumab per la cura delle uveiti infiammatorie pediatriche. Livello di evidenza IIb.
Simonini G, Zannin ME, Caputo R, et al. Loss of efficacy during long-term infliximab therapy for sight-threatening childhood uveitis. Rheumatology 2008;47:1510-4.
Simpson CL, Wojciechowski R, Oexle K, et al. Genome-wide meta-analysis of myopia and hyperopia provides evi-
dence for replication of 11 loci. PloS one 2014;9:e107110.
Sun Y, Xu F, Zhang T, et al. Orthokeratol-ogy to control myopia progression: a meta-analysis. PloS one 2015;10:e0124535.
Tomkins-Netzer O, Talat L, Seguin-Greenstein S, et al. Outcome of treating pediatric uveitis with dexamethasone im-plants. Am J Ophthalmol 2016;161:110-5 e111-2.
Walline JJ, Lindsley K, Vedula SS, et al. Interventions to slow progression of myo-pia in children. Cochrane Database Syst Rev 2011(12):CD004916.
** Review della bibliografia sulle meto-diche di intervento per frenare la progres-sione miopica.
Williams KM, Bertelsen G, Cumberland P, et al. Increasing prevalence of myopia in europe and the impact of education. Oph-thalmol 2015;122:1489-97.
Zannin ME, Birolo C, Gerloni VM, et al. Safety and effecacy of infliximab and adali-mumab for refractory uveitis in juvenile idiopathic arthritis: 1 year follow up data from the Italian Registry. J Rheumatol 2013;40:74-9.

227
Luglio-Settembre 2016 • Vol. 46 • N. 183 • Pp. 227-234 Prospettive in Pediatria
Oftalmologia pediatrica
Nuove frontiere in terapia genica: prospettive terapeutiche per le patologie oculari a insorgenza pediatrica geneticamente determinate
Maria Concetta Ferraro1 Giacomo Maria Bacci2
Roberto Caputo2 Francesca Simonelli1
1 UOC Oculistica, Dipartimento Multidisciplinare di Specialità
Medico-Chirurgiche e Odontoiatriche, Seconda
Università degli Studi di Napoli; 2 Unità Operativa Oftalmologia Pediatrica, AOU Meyer, Firenze
Herein we examine the current state of knowledge about the potential of gene therapy for paediatric onset inherited retinal dystrophies. These diseases are a leading cause of major visual loss and are caused by genetic mutations that alter normal processes of retinal phototransduction and lead to anatomic degeneration that is associated with loss of normal visual function. Specifically, we focused on the main clinical trials of ocular gene therapy and assessed future prospects in the context of paediatric onset inherited retinal degenerations.
Summary
L’articolo si propone di approfondire lo stato attuale della conoscenza sulla potenzialità della terapia genica applicata alle distrofie retiniche ereditarie a insorgenza pediatrica. Tali malattie rappresentano una delle principali cause di importante deficit visivo e risultano da mutazioni genetiche esitanti nell’alterazione dei normali meccanismi di elaborazione dello stimolo luminoso a livello retinico e, più in generale, in degenerazioni anatomiche incom-patibili con una normale funzione visiva. Nello specifico abbiamo focalizzato l’attenzione sui principali trial clinici di terapia genica oculare, e abbiamo analizzato le prospettive futu-re nell’ambito delle degenerazioni retiniche ereditarie a insorgenza pediatrica.
Riassunto
AbbreviazioniAAV: virus adeno-associatoAd: adenovirus BVMD: degenerazione maculare vitelliforme di Best CHM: coroideremiaCSNB: cecità notturna congenita stazionariaIRD: degenerazioni retiniche ereditarieLCA: amaurosi congenita di LeberLV: lentivirusNP: nanoparticellePR: fotorecettori retiniciRP: retinite pigmentosaRPE: epitelio pigmentato retinico
RS: retinoschisi STGD: malattia di Stargardt USH1B: sindrome di Usher tipo 1B
GlossarioAmaurosi: grave deficit visivo fino alla cecità, transi-toria o permanenteColoboma: assenza di parte di una struttura oculare come risultato di un’incompleta chiusura della fessura embrionaleElettroretinogramma: indagine diagnostica in grado di misurare graficamente i potenziali di azione dei fo-torecettori retinici, una volta stimolati da un impulso luminoso

228
M.C. Ferraro et al.
Emeralopia-nictalopia: condizione di difficoltà nella visione crepuscolareFotofobia: condizione di intensa sensibilità alla lucePseudo-fovea: nuova area retinica di fissazione che permette di compensare la perdita di visione fovealeRoving: deviazioni oculari orizzontali lente, coniuga-te, casuali ed “erranti”
Metodologia della ricerca bibliografica effettuataPer la revisione della letteratura sulla terapia genica oculare è stata utilizzata la banca bibliografica Medli-ne, utilizzando come motore di ricerca PubMed. Le parole chiave utilizzate sono “ocular gene therapy”, “pediatric retinal dystrophies”, “AAV”, “LCA”, “RPE65”, “inherited retinal degenerations”, con selezione di arti-coli pubblicati dal 2010 al 2016.
IntroduzioneCon il termine distrofie retiniche a insorgenza pedia-trica geneticamente determinate, più comunemente chiamate degenerazioni retiniche ereditarie (Inherited retinal degenerations – IRD), s’identifica un gruppo eterogeneo di disfunzioni retiniche ereditarie che, sebbene piuttosto rare qualora considerate come singole entità, rappresentano nell’insieme una delle principali cause di importante deficit visivo a esordio precoce ed evoluzione progressiva, colpendo circa 2 milioni di persone nel mondo (Sahel et al., 2014). Si tratta di malattie mono o multigeniche, isolate o sin-dromiche, causate dalla mutazione di uno o più dei circa 200 geni maggiormente espressi nei fotorecet-tori retinici (PR), coni e bastoncelli, e in misura mi-nore nell’epitelio pigmentato retinico (retinal pigment epithelium – RPE). (Dalkara e Sahel, 2014; Trapani et al., 2015) (Fig. 1).
ClassificazioneLa classificazione delle degenerazioni retiniche ere-ditarie si basa sul tipo di difetto genetico, sul pattern di trasmissione ereditaria che può essere autosomica dominante (AD), autosomica recessiva (AR), X-linked e mitocondriale; sul tipo di cellule prevalentemen-te coinvolte (PR o RPE); sull’epoca di insorgenza e sull’entità del deficit visivo, nonché sulle caratteristi-che del fundus oculi. Per i modelli AD e AR non sem-bra esserci un’evidente correlazione con il sesso. Le manifestazioni fenotipiche possono variare tra la reti-nite pigmentosa (RP) nelle sue varie forme, l’amau-rosi congenita di Leber (LCA), la malattia di Stargardt (STGD), le distrofie dei coni, la distrofia maculare vitelliforme di Best (BVMD), la retinoschisi giovanile (RS), la cecità notturna congenita stazionaria (CSNB) e la coroideremia (CHM). Tali condizioni si caratteriz-zano per la comparsa di una sintomatologia eteroge-
nea, che si accompagna a un deficit visivo più o meno severo e/o progressivo. È pertanto di fondamentale importanza la collaborazione tra il pediatra e l’oculista per l’identificazione precoce della malattia e la piani-ficazione di un corretto iter diagnostico-terapeutico, al fine di poter migliorare la qualità visiva del piccolo pa-ziente e porre le basi per una prospettiva terapeutica.
Eziopatogenesi e aspetti cliniciDa un punto di vista patogenetico, le distrofie retini-che ereditarie sono accomunate dalla ridotta o as-sente funzione di proteine che svolgono ruoli chiave nel processo di fototrasduzione o che sono coinvolte nel metabolismo delle cellule dell’epitelio pigmenta-to retinico, condizione che porta a una progressiva degenerazione delle strutture retiniche e a un inevi-tabile decadimento delle funzioni visive, talvolta fin dai primi anni di vita. Tra i sintomi e segni clinici più frequentemente associati alle IRD, sono da ricordare la comparsa di movimenti oculari anomali (nistagmo; roving); fotofobia d’intensità variabile e non sempre presente, emeralopia (scarsa capacità visiva in am-bienti poco illuminati); difficoltà nella distinzione dei colori (discromatopsia-acromatopsia); riduzione pro-gressiva della capacità visiva con difficoltà del piccolo paziente a muoversi negli ambienti o comparsa di dif-ficoltà nella lettura in età scolare; atteggiamenti parti-colari, come la digitopressione degli occhi nelle for-me più gravi (segno di Franceschetti); associazione a vizi di refrazione potenzialmente elevati; eventuale
Figura 1. Disposizione topografica dell’RPE rispetto ai fotorecettori (Cone, Rod) e la precisa localizzazione dei principali geni coinvolti nelle IRD a livello dell’epitelio pig-mentato retinico e del segmento esterno/interno dei foto-recettori.

229
Nuove frontiere in terapia genica
associazione con altre manifestazioni oculari quali strabismo, cataratta, nonché ad alterazioni retiniche più o meno peculiari. In alcuni casi, infine, le distrofie retiniche ereditarie possono presentarsi come facenti parte di quadri sindromici.
Strategie terapeuticheData l’importanza dell’impatto clinico e sociale di tali patologie, la terapia mirata al ripristino visivo, o all’ar-resto dell’evoluzione della malattia, nelle IRD rappre-senta a oggi un’importante esigenza medica. Attual-mente, tuttavia, non sono ancora disponibili terapie efficaci: nel tempo si sono succeduti diversi approcci terapeutici, quali una supplementazione vitaminica, la somministrazione di fattori di crescita, l’utilizzo delle cellule staminali e la realizzazione di impianti protesici retinici. Al momento i risultati ottenuti si sono rivelati estremamente variabili. L’approccio mediante terapia genica è quello che, negli ultimi anni, è apparso offrire una concreta prospettiva di raggiungimento dell’obiet-tivo terapeutico. Infatti, nelle ultime tre decadi, grazie all’identificazione di specifici geni coinvolti nell’ezio-patogenesi delle IRD, si sono poste le basi per lo sviluppo di diverse strategie basate sulla terapia ge-nica. Sembrerebbe che le forme monogeniche di tali affezioni, e in particolare quelle causate da mutazioni recessive di tipo null, rappresentino le condizioni cli-niche maggiormente sensibili alla terapia genica: in tali forme, infatti, è possibile la correzione del difet-to ereditario, causato da un’alterazione di un singolo gene, tramite l’identificazione di un target terapeutico gene-specifico. Tali mutazioni sono localizzate nel-la maggior parte dei casi nella retina esterna (PR o RPE). In particolare, le forme candidabili alla terapia genica sono rappresentate dall’amaurosi congenita di Leber tipo 2 (LCA2) (gene RPE65), la LCA1 (gene
GUCY2D), la retinoschisi X-linked (gene RS1), la acromatopsia (geni CNGA3 e CNGB3), la sindrome di Usher 1B (gene MYO7A), la malattia di Stargardt (gene ABCA4), la coroideremia (gene CHM) e la reti-nite pigmentosa X-linked RP3 (gene RPGR) (Fig. 2).
Terapia genica: strategie e target L’occhio, in virtù delle sue caratteristiche peculiari, ri-sulta un target ideale per tale approccio: • sitrattadiunorganoincui,grazieallatrasparenza
delle strutture oculari (cornea, cristallino, vitreo), è possibile verificare in vivo gli effetti della terapia ge-nica attraverso le più recenti tecniche di imaging;
• èuncompartimentochiuso,graziealqualepicco-le quantità di vettore possono essere utilizzate con minimo rischio di tossicità sistemica;
• èunsitoprivilegiatoperlesuecaratteristicheana-tomiche, che consentono allo strumento terapeutico di svolgere la sua azione in assenza di significati-ve reazioni immunitarie. Tali peculiarità permettono che non si verifichi né una diffusione sistemica del vettore virale, né una risposta immune nei confronti del vettore o dei prodotti transgenici;
• èunorganopariesimmetrico:leIRD,hannounamanifestazione bilaterale, più o meno simmetrica, consentendo di poter comparare gli effetti della somministrazione del complesso gene-vettore in un occhio e la progressione della malattia nell’oc-chio controlaterale (Trapani et al., 2014);
• accessibilitàchirurgicadiretta:leprocedureperviaintravitreale o sottoretinica permettono di collocare direttamente il vettore in prossimità del sito di azio-ne e di minimizzare i problemi di biodisponibilità.
Quali pazienti e quali condizioni?Poiché l’efficacia del trasferimento genico si basa
Figura 2. Distribuzione percentuale delle IRD a esordio in età pediatrica.
Altre distrofie retiniche ereditarie 10%
Coroideremia 2%
Retinoschisi 3%
Distrofia dei coni-bastoncelli 10% Retinite pigmentosa 40%
Sindrome di Usher 10%Cecità notturna
congenita stazionaria 5%
Amaurosi congenita di Leber 5%
Degenerazione corioretinica 5%
Degenerazione maculare ereditaria 10%

230
M.C. Ferraro et al.
sulla presenza di cellule bersaglio vitali, risulta fonda-mentale l’identificazione precoce della patologia per ottenere la migliore efficacia della terapia genica. Con-dizioni come la LCA tipo 1 e tipo 2, l’acromatopsia o la retinoschisi giovanile X-linked, mostrano una struttura retinica conservata per decenni dopo la diagnosi, no-nostante il deficit visivo grave e congenito; per tale motivo questi pazienti sono candidati ideali per una terapia genica. Per molte condizioni che presenta-no una degenerazione retinica rapida dal momento dell’esordio, in combinazione con un difetto funziona-le (p.e. la malattia di Stargardt tipo 1 (STGD1), in cui il trasporto di retinale è compromesso o la LCA tipo 4 (LCA4), in cui enzimi chiave nella cascata della fo-totrasduzione sono destabilizzati, una terapia genica precoce potrebbe essere impiegata per prevenire la degenerazione della retina e ottenere il ripristino della funzione visiva dei fotorecettori (Trapani et al., 2015).
Quali strategie?Differenti strategie possono essere applicate nella te-rapia genica per le IRD: la sostituzione genica, l’inibi-zione genica, strategie mutazione-indipendente (neu-roprotezione). La sostituzione genica è impiegata nei disordini causati da mutazioni null esitanti nella per-dita della funzione specifica di una proteina e si basa sul trasferimento di una corretta copia del gene muta-to, senza la rimozione dello stesso. L’inibizione genica consiste nell’inibire l’espressione del gene mutato at-traverso modifiche a carico dell’RNA messaggero ed è impiegato per quelle forme causate da una mutazio-ne con “acquisizione di funzione”, nelle quali si ha la produzione di una proteina con funzione anomala che interferisce con il funzionamento del peptide normale prodotto dall’allele sano. La neuroprotezione, invece, si propone quale possibile strategia per prolungare la sopravvivenza delle cellule retiniche, rallentando la degenerazione mediante la riduzione degli effetti secondari alla disfunzione proteica.
I vettori: fulcro della terapia genicaA prescindere dalla strategia “mutazione-dipendente” o “indipendente”, i vettori svolgono un ruolo cardine nella terapia genica oculare. Esistono due tipi di vet-tori: 1 non virali (DNA nudo): sicuri per la bassa immuno-
genicità, ma che tuttavia presentano maggiori diffi-coltà nel raggiungimento della cellula target, per la presenza di barriere fisiche intraoculari. Pertanto sono stati introdotti metodi chimici quali l’associa-zione ad altre molecole (liposomi, polipeptidi, na-noparticelle compattate) e metodiche fisiche, quali la iontoforesi e l’elettropolarizzazione, che ne mi-gliorano la penetrazione;
2 virali: in grado di trasferire gli acidi nucleici nel nu-cleo delle cellule ospite attraverso specifiche in-terazioni con recettori di membrana e successiva internalizzazione. A livello retinico sono stati testati
differenti vettori virali, tra i quali i lentivirus (LV), gli adenovirus (AV) e i virus adeno-associati (AAV), sono quelli maggiormente impiegati. In particola-re gli AAV sono i vettori virali preferiti nella terapia genica delle IRD grazie alla loro piccola dimen-sione, l’abilità nel raggiungere la cellula target, la presenza di vari sierotipi legati alla variabilità del capside, l’eccellente profilo di sicurezza e la bassa immunogenicità, caratteristiche fondamentali per il trattamento delle malattie croniche, come le IRD, per le quali si può prevedere più di una sommini-strazione sottoretinica.
Procedure chirurgiche: quale scegliere? Il raggiungimento dall’esterno del bersaglio intraocu-lare, rappresentato dai fotorecettori retinici e dall’epi-telio pigmentato retinico, può essere attuato mediante due differenti procedure chirurgiche: l’iniezione intra-vitreale e quella sottoretinica. Quest’ultima consiste nella somministrazione sottoretinica di una quantità variabile di vettore virale in un volume di soluzione salina tamponata con fosfato, che crea un tempora-neo distacco retinico localizzato, che generalmente si riassorbe rapidamente.Una procedura alternativa, meno invasiva, è l’iniezione intravitreale in grado di distribuire il vettore su tutta la retina senza causarne un rischioso distacco. Tuttavia, quando iniettati per via intravitreale, la maggior parte dei vettori virali, tra cui la maggior parte dei sierotipi AAV, non raggiunge la retina a eccezione dei sierotipi AAV2/2, e in qualche misura di AAV2/6 e di AAV2/8, la cui trasduzione è tuttavia limitata principalmente alle cellule ganglionari della retina e alle cellule Muller nel-lo strato retinico interno. Il fallimento dei vettori iniettati per via intravitreale nel raggiungere i PR e RPE nella retina esterna sembra essere dovuto alla presenza di barriere fisiche, quali la membrana limitante interna, particolarmente spessa nei grandi animali, nonché la relativa abbondanza di recettori AAV che catturano i vettori dopo la somministrazione intravitreale. Un’ulteriore comprensione delle barriere retiniche ini-benti la trasduzione, così come la valutazione in vivo eseguita direttamente su grandi animali, potrebbe identificare la strada per la diffusione dei vettori dal vitreo alla retina dei primati. Fino a quando ciò non sarà possibile, l’iniezione sottoretinica rimane la pro-cedura standard più efficace per raggiungere l’epitelio pigmentato retinico e i fotorecettori (Fig. 3).
Trial clinici: primi risultati e prospettive futureTre indipendenti trial clinici nell’uomo, iniziati contempo-raneamente nel 2007 (NCT00481546, NCT00516477, NCT00643747, clinicaltrials.gov) di cui uno effettuato su 5 pazienti italiani, sono stati eseguiti al fine di va-lutare la sicurezza e l’efficacia della terapia genica per l’amaurosi congenita di Leber tipo 2 (Tab. I).

231
Nuove frontiere in terapia genica
L’amaurosi congenita di Leber tipo 2 rappresenta un candidato ideale per la terapia genica per due distinte ragioni, in primis in quanto il deficit di RPE65 causa difetti nel ciclo visivo e una scarsa funzione visiva già nella 1ª decade di vita, e poi perché la struttura reti-nica risulta conservata fino alla 2ª-3ª decade di vita, periodo in cui la progressiva degenerazione fotorecet-toriale risulta evidente (Simonelli et al., 2007).La LCA è dunque il primo esempio di IRD per la quale sia stato effettuato un trial clinico sulla terapia genica in fase I/II e i risultati ottenuti rappresentano, a oggi, il maggior successo per ciò che concerne la terapia genica oculare (Trapani et al., 2015). I pa-zienti affetti da tale patologia mostrano una severa compromissione visiva già nella 1ª decade di vita associata a nistagmo, intensa fotofobia, nictalopia, riduzione dei riflessi pupillari e risposte elettroreti-nografiche ipovoltate o estinte. L’aspetto oftalmosco-pico è estremamente variabile, potendo oscillare da alterazioni dell’EPR con aspetto tipo “sale e pepe”, fino allo pseudo coloboma maculare. Tuttavia nella maggior parte dei casi, nelle fasi iniziali di malattia non vi è una correlazione direttamente proporzionale tra il decadimento delle funzioni visive e la degene-razione istologica delle cellule retiniche. Tale condi-
zione evolve poi progressivamente verso la cecità entro la 3°-4° decade di vita (Simonelli et al., 2010; Maguire et al., 2009). L’amaurosi congenita di tipo 2 (LCA2) è una forma a trasmissione AR associata alla mutazione del gene RPE65 che codifica per una proteina (RPE65, con at-tività di isomeroidrolasi) espressa nell’EPR e coinvol-ta nel ciclo della fototrasduzione. Conseguentemente all’assenza o alla mancata funzione di tale proteina, si verifica una riduzione dell’11-cis-retinale necessario alla rigenerazione del pigmento visivo dopo l’esposi-zione alla luce con conseguente degenerazione foto-recettoriale (Maguire et al., 2009; Bennett et al., 2012). Nel trial clinico del Children Hospital di Philadelphia e della Seconda Università di Napoli, i pazienti sono stati trattati nell’occhio con visione peggiore, me-diante una singola iniezione sottoretinica, effettuata in anestesia generale, di una dose bassa (1,5×1010 particelle/occhio), media (4,8x1010 particelle/occhio) o alta (1,5x1011 particelle/occhio) di virus adeno-asso-ciato 2 (AAV2.hRPE65v2) contenente RPE65 cDNA.Durante i follow-up i pazienti sono stati sottoposti a un esame fisico generale, test clinici e di laboratorio, tra cui una valutazione della biodistribuzione del vet-tore e valutazione della risposta immunitaria, visita
Figura 3. Localizzazione del sito di iniezione sottoretinica, a livello dell’epitelio pigmentato retinico e rapporti anatomici con le altre strutture intraoculari.
A B
Tabella I. Numeri di riferimento dei trials clinici (NCT), titolo e strutture partecipanti.
NCT Titolo Partecipanti
NCT00481546 Phase I Trial of Gene Vector to Patients with Retinal Disease due to RPE65 Mutations
University of Pennsylvania University of Florida
NCT00516477 Safety Study in Subjects With Leber Congenital Amaurosis Children Hospital Philadelphia Seconda Università Studi Napoli
NCT00643747 Safety Study of RPE65 gene Therapy to Treat Leber Congenital Amaurosis
University College London Moorfields Eye Hospital NHC Foundation Trust

232
M.C. Ferraro et al.
oculistica, nonché test di mobilità per valutare la ca-pacità del paziente di muoversi lungo un percorso a ostacoli. Non sono stati evidenziati importanti eventi avversi e solo in un paziente è stata osservata una minima risposta immunitaria sistemica. Dall’analisi dei dati longitudinali, la singola iniezione sottoretinica di AAV2 sembrerebbe responsabile di un incremento dell’acuità visiva, riduzione del nistagmo con incre-mento della stabilità di fissazione e miglioramento del campo visivo. Inoltre, a un’analisi dei test di mobilità è stato evidenziato un andamento più sicuro dei sog-getti durante il percorso a ostacoli, con una migliore percezione del percorso e dei suoi ostacoli. I risultati ottenuti sono rimasti stabili in tutti i pazienti a 3 anni di follow-up. È stato inoltre osservato che il tasso di successo nel miglioramento delle funzioni visive è le-gato all’epoca del trattamento, con i migliori risultati ottenuti nei pazienti più giovani, che presentano una struttura retinica meglio conservata. Tali risultati di sicurezza ed efficacia hanno consentito di includere anche bambini nei trial clinici e hanno fornito le basi per l’approccio della terapia genica al trattamento di altre forme di degenerazione retinica ereditaria.
Lezioni e criticitàAnche se i dati preliminari di questi primi studi clinici risultano estremamente promettenti, lo sviluppo della terapia genica della retina ha permesso di trarre delle lezioni molto importanti e ha sollevato alcune criticità. Essa infatti ha delineato la finestra terapeutica tra il primo tempo di intervento e il punto in cui i processi degenerativi non possono essere più ripristinati con cellule bersaglio ormai atrofiche e degenerate. Sono inoltre stati messi in evidenza eventuali rischi chirur-gici: la somministrazione sottoretinica della soluzione, contenente il vettore nella regione parafoveale, provo-ca un transitorio distacco della retina, che in alcuni casi è esitato in danni permanenti a livello retinico (Simo-nelli et al., 2007; Maguire et al., 2008). Da tutti gli studi è comunque emerso che la terapia genica è sufficien-temente sicura, e sostanzialmente efficace, in partico-lare per la regione retinica extrafoveale. Infatti, sembra esserci qualche rischio nel trattamento della fovea, probabilmente per la particolare natura del legame tra i fotorecettori foveali e il sottostante epitelio EPR (An-derson e Fisher, 1979) e ciò potrebbe spiegare perché il riaccollamento della fovea, dopo il piccolo distacco indotto dalla somministrazione del vettore virale, è più complesso rispetto a quello di altre zone retiniche. Inol-tre, a differenza di quanto osservato in cani affetti da LCA2, nessun miglioramento nell’elettroretinogramma è stato finora documentato nei pazienti LCA2 trattati con AAV (Annear et al., 2011). Questi dati possono in-dicare un inadeguato apporto di RPE65 e un ripristino solo parziale del ciclo visivo nei pazienti trattati. Inoltre, più recentemente, è stato riportato un beneficio solo transitorio dell’efficacia della terapia genica in pazienti con LCA RPE65-correlata, seguita da un progressivo
declino funzionale (Bainbridge et al., 2015). Sulla base di tali evidenze, sarebbe quindi auspicabile lo sviluppo di vettori più efficienti, al fine di ottenere un completo restauro del ciclo visivo e un trattamento più precoce nei pazienti.
Nuove sfide e prospettive future Attualmente la società di biotecnologie Spark Thera-peutics sta testando il vettore AAV2/2-RPE65 per la LCA tipo 2 in un trial clinico avanzato di fase III (Lok, 2014), coinvolgente piccoli pazienti come bambini di 3 anni (NCT00999609). Inoltre, un approccio alterna-tivo per il trattamento della LCA2 è ora in fase di valu-tazione in uno studio clinico che utilizza il promotore RPE65 in combinazione con l’AAV2/4 (NCT01496040): tale strategia potrebbe aumentare la specificità e l’ef-ficacia della terapia. È importante sottolineare che il successo degli studi clinici sulla LCA2 ha favorito una più ampia applicazione della terapia genica per le IRD a causa di mutazioni in geni espressi in differenti strati retinici, e che provocano altre forme di IRD.Visto l’importante sviluppo della terapia genica ocu-lare viene da sé l’importanza di porsi nuove sfide e traguardi. La prima sfida è quella di trasferire geni di grandi dimensioni a livello retinico. Infatti, nonostante la popolarità acquisita dai vettori AAV, uno dei prin-cipali ostacoli alla loro applicazione diffusa è la loro capacità di confezionamento di circa 5 kb, precluden-done il loro utilizzo per il trattamento di IRD come la malattia di Stargardt e la sindrome di Usher tipo 1B (STGD e l’USH1B), che sono causate da mutazioni in geni di grandi dimensioni. Così, molti ricercatori stanno esplorando vettori alternativi con capacità di clonaggio maggiori rispetto agli AAV, quali adenovirus (Ad), lentivirus (LV) e nanoparticelle di DNA (NP). Tut-tavia, l’esperienza limitata con questi vettori, insieme con la necessità di chiarire le loro caratteristiche di trasduzione in modelli animali di grandi dimensioni, inclusi i primati non umani, rende necessaria l’ese-cuzione di ulteriori test, prima che il DNA compatta-to con le NP possa essere utilizzato per trasferire ai fotorecettori retinici umani geni di grosse dimensioni.
ConclusioniIn sintesi, l’intervento tempestivo, che richiede una precoce diagnosi clinica e molecolare, in combina-zione con una storia naturale ben caratterizzata della malattia, sarà necessario per massimizzare l’efficacia della terapia genica per le IRD. È fondamentale inoltre la standardizzazione dei parametri clinici per la sele-zione dei pazienti, e infine lo sviluppo e validazione di endpoint clinici (es. modalità di imaging e nuovi bio-marcatori di malattia), adatti per quantificare l’effettivo vantaggio del trattamento.In conclusione, trasformare la terapia genica della re-tina da ricerca sugli animali in studi clinici è ancora un

233
Nuove frontiere in terapia genica
processo lungo, ma si sta lavorando attivamente per definire i punti mancanti che porteranno all’introduzio-ne di molti nuovi approcci promettenti in questo cam-po ancora non ben compreso e conosciuto. Ne deriva la necessità di porsi nuove sfide che si basano su una
più vasta conoscenza dei meccanismi fisiopatologici della malattia, promozione di studi longitudinali sulla storia naturale, per predire la progressione della ma-lattia in relazione allo specifico genotipo.
Box di orientamento
• Ledistrofieretinicheereditarierappresentanounadelleprincipalicausedigraveeprogressivodeficitvisivo.
• Lemutazioniallabaseditalidisfunzionicoinvolgonoproteinefondamentalineiprocessidifototrasduzio-ne retinica, con riduzione delle funzioni visive fin dai primi anni di vita.
• Nonesistonoaoggiterapieefficaci,sebbenesianostativalutatidifferentiapprocciterapeutici.• Laterapiagenicaoculare,chesembrerebbeoffrireun’opzioneterapeuticamoltopromettente,necessita,
per poter essere efficace, della presenza di cellule bersaglio vitali, condizione caratterizzante la LCA2, che presenta una struttura retinica conservata per decenni nonostante il calo visivo precoce.
• Iprimi3trialclinicisull’impiegodeivettoriviraliadeno-associatialivellooculare,nell’uomo,sonostatieffettuati su pazienti affetti da LCA tipo 2 e iniziati contemporaneamente nel 2007.
• Irisultatipromettentideiprimitrialclinicihannopostolebasiperlosviluppodiulterioristudi.• Aoggisonoinattotrialclinici,sudifferentiformedidistrofieretinicheereditarie,alcunideiqualiestesi
anche alla popolazione pediatrica.
Bibliografia
Adijanto J, Naash MI. Nanoparticle-based technologies for retinal gene thera-py. Eur J Pharm Biopharm 2015;95:353-67
Anderson DH, Fisher SK. The rela-tionship of primate foveal cones to the pigment epithelium. J Ultrastruct Res 1979;67:23-32.
* Articolo che esplora le caratteristiche di connessione tra coni foveali ed epitelio pigmentato retinico nei primati.
Annear MJ, Bartoe JT, Barker SE, et al. Gene therapy in the second eye of RPE65-deficient dogs improves retinal function. Gene Ther 2011;18:53-61.
** Articolo che evidenzia la bassa rispo-sta immunogenica indotta dalla terapia ge-nica con vettori virali.
Bainbridge JW, Mehat MS, Sundaram V, et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med 2015;372:1887-97.
** L’articolo evidenzia la possibile dura-ta temporanea del beneficio indotto dalla terapia genica per amaurosi congenita di Leber.
Bennett J, Ashtari M, Wellman J, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci-ence Transl Med 2012;4:120ra15.
** L’articolo indica la sicurezza anche nella somministrazione ripetuta di vettore adenovirale per terapia genica della amau-rosi congenita di Leber.
Binley K, Widdowson P, Loader J, et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: safety and biodistribution of Star-Gen for Stargardt disease. Invest Ophthal-mol Vis Sci 2013;54:4061-71.
Cideciyan AV, Aguirre GK, Jacobson SG, et al. Pseudofovea formation after gene therapy for RPE65-LCA. Invest Ophthalmol Vis Sci 2015;56:526-37.
Cideciyan AV, Hauswirth WW, Aleman TS, et al. Vision 1 year after gene therapy for Leber’s congenital amaurosis. N Engl J Med 2009;361:725-7.
Colella P, Trapani I, Cesi G, et al. Effi-cient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther 2014;21:450-6.
Conlon TJ, Deng WT, Erger K, et al. Pre-clinical potency and safety studies of an AAV2-mediated gene therapy vector for the treatment of MERTK associated retini-tis pigmentosa. Hum Gene Ther Clin Dev 2013;24:23-8.
Dalkara D, Sahel JA. Gene therapy for inherited retinal degenerations. C R Biol 2014;337:185-92.
* Articolo di revisione sui recenti svilup-pi nella terapia genica delle distrofie retini-che ereditarie.
Dong B, Nakai H, Xiao W. Characterization of genome integrity for oversized recombi-nant AAV vector. Mol Ther 2010;18:87-92.
Han Z, Conley SM, Makkia RS, et al. DNA nanoparticlemediated ABCA4 delivery res-
cues Stargardt dystrophy in mice. J Clin Invest 2012;122:3221-6.
Hirsch ML, Li C, Bellon I, et al. Oversized AAV transduction is mediated via a DNA-PKcs-independent, Rad51C dependent re-pair pathway. Mol Ther 2013;21:2205-16.
Koilkonda RD, Yu H, Chou TH, et al. Safety and effects of the vector for the Leber hereditary optic neuropathy gene therapy clinical trial. JAMA Ophthalmol 2014;132:409-20.
Lok C. Curing blindness: vision quest. Nature 2014;513:160-2.
* Prospettive di terapia genica in pazienti pediatrici.
Lopes VS, Boye SE, Louie CM, et al. Retinal gene therapy with a large MYO7A cDNA using adeno-associated virus. Gene Ther 2013;20:824-33.
Maclaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 2014;383:1129-37.
Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 2009;374:1597-605.
* Articolo che identifica la sicurezza e la stabilità del trattamento per amaurosi con-genita di Leber determinate da mutazioni su gene RPE65.
Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for

234
M.C. Ferraro et al.
Leber’s congenital amaurosis. N Engl J Med 2008;358:2240-8.
** Articolo di revisione sulla sicurezza ed efficacia della terapia genica nell’amau-rosi congenita di Leber.
Sahel JA, Marazova K, Audo I. Clinical characteristics and current therapies for in-herited retinal degenerations. Cold Spring Harb Perspect Med 2014;5:pii: a017111.
* Articolo di revisione sulle terapie attua-li delle distrofie retiniche e correlazione con i quadri clinici.
Sahel JA, Roska B. Gene therapy for blind-ness. Annu Rev Neurosci 2013;36:467-88.
Simonelli F, Maguire AM, Testa F, et al. Gene therapy for leber’s congenital am-aurosis is safe and effective through 1.5 years after vector administration. Molecu-lar Therapy 2010;18:643-50.
** Articolo di revisione sulla sicurezza e durata del risultato ottenuto con terapia genica nell’amaurosi congenita di Leber.
Simonelli F, Ziviello C, Testa F, et al. Clini-cal and molecular genetics of Leber’s con-genital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci 2007;48:4284-90.
* Articolo che esplora le caratteristiche di popolazione per identificare quadri di amaurosi congenita di Leber.
Trapani I, Banfi S, Simonelli F, et al. Gene therapy of inherited retinal degenerations: prospects and challenges. Hum Gene Ther 2015;26:193-200.
** Articolo di revisione sulle strategie terapeutiche per migliorare e ampliare le prospettive di terapia genica delle distrofie retiniche ereditarie.
Trapani I, Colella P, Sommella A, et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med 2014;6:194-211.
** Articolo che esplora la possibilità di terapia genica con vettori virali idonei per geni di grandi dimensioni.
Trapani I, Puppo A, Auricchio A. Vec-tor platforms for gene therapy of inher-ited retinopathies. Prog Retin Eye Res 2014;43:108-28.
** Articolo che esplora i possibili vettori per la terapia genica delle distrofie retini-che ereditarie.
Zallocchi M, Binley K, Lad Y, et al. EIAV-based retinal gene therapy in the shaker1 mouse model for usher syndrome type 1B: development of UshStat. PLoS One 2014;9:e94272.
Corrispondenza
Giacomo Maria BacciDipartimento di Oftalmologia Pediatrica, AOU Meyer, viale Pieraccini 24, 50139 Firenze - Tel. +39 055 5662526 - E-mail: [email protected]

235
Luglio-Settembre 2016 • Vol. 46 • N. 183 • Pp. 235-240 Prospettive in Pediatria
Oftalmologia pediatrica
Anomalie della motilità oculare e posizioni anomale del capo
Elisabetta Santangelo Massimiliano Serafino
Paolo Nucci
Ospedale San Giuseppe, Milano
Ocular motility disorders are still a controversial issue for the role of the pediatrician in screening for visual defects, yet its relationship with the ophthalmologist is undoubtedly important, as such defects may interfere with the development of normal vision. This re-view examines clinical, diagnostic and therapeutic approaches to ocular motility disor-ders, with specific attention on anomalous head posture.
Summary
Attualmente non esiste un consenso univoco relativamente alle modalità e al ruolo del pediatra nell’effettuazione dello screening dei difetti visivi. È tuttavia indubbio che il pe-diatra svolga un ruolo fondamentale nell’interazione con lo specialista per l’individuazione precoce di quelle patologie, che potrebbero interferire in maniera significativa sul norma-le sviluppo dell’apparato visivo. Quest’articolo di approfondimento mira ad analizzare gli aspetti clinico-diagnostici, nonché l’iter terapeutico da attuarsi in presenza di anomalie della motilità oculare, con particolare attenzione all’identificazione e al significato delle posizioni anomale del capo.
Riassunto
Metodologia della ricerca bibliografica effettuataLa revisione della letteratura scientifica oftalmologi-ca è stata effettuata utilizzando la banca bibliografica Medline, tramite il motore di ricerca PubMed. Gli arti-coli sono stati valutati in base al loro valore scientifi-co per il medico non oftalmologo, al fine poter offrire la migliore panoramica possibile, relativamente alle anomalie della motilità oculare e in particolare alle posizioni anomale del capo.
Anomalie della motilità oculare: elementi generaliLo strabismo è una condizione caratterizzata da un mancato allineamento degli assi visivi, che si può evidenziare con un angolo di deviazione che varia al variare della posizione dello sguardo (strabismo in-comitante) o che invece rimane costante (strabismo concomitante). Tale deviazione può essere presente costantemente, e viene in questo caso definita etero-tropia, oppure può comparire occasionalmente, venen-do svelata da specifiche procedure diagnostiche quali l’occlusione di uno dei due occhi; si parla in questo caso di eteroforia.
Una deviazione degli assi visivi sul piano orizzontale viene definita esotropia o exotropia, a seconda che la deviazione sia nasale o temporale, una deviazione verticale viene definita ipertropia o ipotropia, mentre in presenza di un disallineamento oculare di tipo tor-sionale si parla di ciclotropia (Wright, 2003.).L’ispezione consente di identificare gli elementi che possono ragionevolmente far sospettare un’anomalia della motilità oculare. A tal fine, è importante ricordare che ci sono alcune varianti morfologiche palpebrali e orbitarie che possono dare una falsa impressione di deviazione oculare; ad esempio, l’ipertelorismo e l’e-picanto possono indurre erroneamente a ipotizzare, rispettivamente, un quadro di exotropia e di esotropia (Hwang, 2012).
Valutazione eziopatogeneticaL’iter diagnostico in presenza di anomalie della moti-lità oculare deve mirare a valutare l’integrità del siste-ma oculomotorio, dalle vie sopranucleari ai muscoli oculari, considerando quelle che possono essere le diverse noxae patogenae determinanti una deviazio-ne oculare (Tab. I) (Peragallo, 2015). A tal fine è indi-spensabile non trascurare un’approfondita anamnesi perinatale, che deve sempre prendere in considera-

236
E. Santangelo et al.
zione la probabilità che diverse condizioni patologi-che possano determinare la presenza di anomalie della motilità oculare (Tab. II).
Quadri clinici EsodeviazioniTra le varie forme di strabismo, l’esotropia è sicura-mente la più frequente, e tra queste in particolare la forma essenziale infantile, la forma accomodativa e quella sensoriale.L’esotropia essenziale infantile è un quadro conge-nito a esordio tipicamente entro il sesto mese di vita, caratterizzata da un angolo di deviazione solitamente stabile compreso tra le 30 e le 70 diottrie prismatiche. In circa il 50% di questi bambini, si può evidenziare, in presenza di occlusione di un occhio, la comparsa di nistagmo latente. Il trattamento è sostanzialmente chirurgico, oltre che anti-ambliopico.Questo quadro va sempre distinto da eventuali forme di esotropia sensoriale, nelle quali lo strabismo insor-ge come conseguenza di una ridotta acutezza visiva monolaterale, dovuta ad esempio a opacità corneali, cataratta congenita (Merino, 2007) o ad altre patolo-gie neuroretiniche.L’esotropia di tipo accomodativo, invece, compare più tardivamente, in genere intorno ai due-tre anni di vita, in associazione a una ipermetropia non corretta
che porta il bambino a un eccesso di convergenza; il trattamento principale consiste nella correzione ottica (Campos, 2008).
ExodeviazioniLe exodeviazioni sono molto frequenti, ma non sono tutte da considerarsi patologiche, dato che circa il 70% dei neonati presenta piccole deviazioni che nor-malmente scompaiono entro i primi 2-4 mesi di vita. L’exotropia intermittente è un quadro che si caratte-rizza per periodi in cui la deviazione è perfettamen-te controllata, alternata a momenti in cui l’exoforia si slatentizza, in particolare a seguito di occlusione o in presenza di stanchezza e stati febbrili. Raramente va incontro a miglioramento spontaneo, ed è consiglia-bile attuare un trattamento chirurgico, preferibilmente oltre i 6 anni di età (Joyce, 2015). A differenza dell’esotropia, le forme di exotropia con-genita sono estremamente rare, e solitamente asso-ciate a quadri malformativi cranio-faciali. Analogamente a quanto visto per l’esotropia, è pos-sibile che si realizzino quadri di exotropia sensoriale, conseguenti a ridotta acutezza visiva monolaterale.
Deviazioni oculari verticaliLe deviazioni oculari verticali possono essere isolate o associate a deviazioni orizzontali. Lo strabismo ver-ticale è più frequentemente incomitante, a differenza delle deviazioni orizzontali. Le deviazioni verticali incomitanti possono essere restrittive, associate ad esempio a fratture del pavimento dell’orbita (Lueder, 2015) o a monocular elevation deficiency (caratteriz-zata da un deficit di entrambi i muscoli elevatori di un occhio, cioè del retto superiore e dell’obliquo in-feriore), oppure, più raramente, non restrittive, con-seguenti a patologie cerebellari e tronco-encefaliche. Esiste un quadro, noto come dissociated strabismus complex, che si caratterizza per la presenza di mo-vimenti dissociati variabili lungo i diversi piani, verti-cale, orizzontale o torsionale. La deviazione verticale dissociata è la forma di gran lunga più frequente in questo ambito (Christoff, 2014).
Strabismo paraliticoÈ importante distinguere questi quadri da forme di strabismo paralitico, nelle quali una lesione può lo-calizzarsi a qualsiasi livello dall’origine nucleare del nervo fino alla terminazione neuromuscolare. Il nervo oculomotore (III nervo cranico) ha il suo nu-cleo somatomotore nella sostanza grigia ventrale all’acquedotto mesencefalico di Silvio, in corrispon-denza del collicolo superiore; le sue fibre, dopo aver attraversato il fascicolo longitudinale mediale e il nucleo rosso, emergono a livello della fossa interpe-duncolare e decorrono nel seno cavernoso insieme alle fibre del IV e del VI nervo cranico, penetrando nell’orbita a livello della fessura orbitaria superiore.
Tabella I. Cause di deviazioni oculari.
Patologie muscolari
Infiammazione, trauma e incarceramento, patologie infiltrative
Patologie della giunzione neuromuscolare
Miastenia gravis, botulismo
Patologie dei nervi cranici e centrali
Patologie ischemico/emorragiche, traumatiche, neoplastiche, degenerativeIpertensione endocranicaPatologia del seno cavernoso
Tabella II. Valutazione anamnestica delle condizioni as-sociate ad anomalie della motilità oculare.
Infezioni congeniteToxoplasma gondii, CMV, HSV,
Prematurità
Patologie cerebrali acquisiteEncefalopatia ipossico-ischemica, stroke arterioso, leucomalacia periventricolare, emorragia della matrice germinativa
Traumi pre- e post-natali
Anomalie malformative

237
Anomalie della motilità oculare e posizioni anomale del capo
Innerva il muscolo retto superiore, retto inferiore, retto mediale, obliquo inferiore ed elevatore della palpebra superiore. Il nucleo del nervo trocleare (IV nervo cranico) si trova nella sostanza grigia mesencefalica in corrisponden-za del collicolo inferiore, caudalmente al nucleo del III nervo cranico. Emerge dalla superficie dorsale del tronco e circonda il peduncolo cerebrale portandosi ventralmente, per decorrere insieme al III e al VI nel seno cavernoso, penetrando quindi a livello orbitario. Innerva il muscolo obliquo superiore. Il nervo abducente (VI nervo cranico) ha il nucleo localizzato a livello pontino, in corrispondenza del pavimento del IV ventricolo, e le sue fibre emergono ventralmente tra bulbo e ponte. Decorre insieme al III e al IV nervo cranico e, a livello orbitario, si porta a innervare il muscolo retto laterale.
Valutazione delle posizioni anomale del capoÈ sempre importante verificare il corretto allineamen-to del capo, che deve essere mantenuto eretto e in asse rispetto al corpo. Ci sono diverse condizioni patologiche che possono comportare una posizione anomala del capo (PAC), e in particolare le forme a eziologia oculare sono state descritte per la prima vol-ta da Cuignet nel 1873, come una posizione obbliga-ta della testa causata da anomalie del sistema visivo (Cuignet, 1873). Si tratta di una condizione di frequente riscontro nella pratica pediatrica, con un’incidenza stimata intorno al 3% (Mitchell, 1999).Una volta valutata la presenza di una PAC, ancora pri-ma di definirne l’eziologia, è importante descriverne le caratteristiche. Bisogna quindi definire se si tratta di una rotazione del capo rispetto all’asse verticale (head turn), se si ha una posizione con mento eleva-to (chin-up) o depresso (chin-down) rispetto all’asse orizzontale, o se si ha un’inclinazione (head tilt) su l’una o l’altra spalla; tali posizioni possono essere pre-senti in associazione. Non sempre una PAC è manifesta in modo evidente a una prima analisi, e quindi l’ispezione deve affidarsi a criteri clinici ben precisi. Per identificare un’inclinazione è necessario valutare se le orecchie e gli occhi sono allineati, per identificare una rotazione bisogna valu-tare se il naso è centrato e se entrambi gli occhi sono ugualmente visibili, mentre una posizione in chin-up è identificata dalla possibilità di visualizzare le narici e quella in chin-down si caratterizzata per una maggiore visualizzazione delle sclere (Davitt, 2001).Gli elementi fisiologicamente implicati nel corretto mantenimento della postura di capo e collo sono in-formazioni afferenti dalla retina, dai propriocettori pre-senti a livello cervicale, e dal labirinto, e per questo motivo una PAC è da attribuire a una di queste tre componenti.
Dal punto di vista eziologico, una posizione anomala del capo può essere imputabile principalmente a cau-se oculari, ortopediche e neurologiche, le altre sono presenti più raramente (Nucci, 2005).
Posizione anomala del capo di origine extraoculareNell’ambito delle forme extraoculari (Tab. III), le prin-cipali sono quelle di tipo ortopedico; in particolare, queste includono il torcicollo muscolare congenito, associato a rigidità del muscolo sternocleidomastoi-deo, l’anomalia di Klippel-Feil, caratterizzata da fu-sione delle vertebre cervicali, ed eventuali lesioni del plesso brachiale. Più raramente, può trattarsi di for-me di torcicollo di tipo funzionale o correlate a quadri sindromici rari quali la sindrome di Sandifer (Deskin, 1995), in cui si ha l’associazione con ernia iatale e reflusso gastroesofageo. Le forme ortopediche, insieme a quelle neurologiche, sono più frequentemente congenite, e si distinguono in questo senso dalle forme a eziologia oculare, che sono più frequentemente acquisite. Nello specifico, al di sotto dei 3 anni di età tendono a essere più fre-quenti le forme di tipo extraoculare, mentre in non più del 30% dei pazienti si riconosce un’eziologia ocula-re; viceversa, nei bambini al di sopra dei 6 anni, l’e-ziologia di una PAC è in circa il 90% dei casi oculare. Dal punto di vista clinico, le forme extraoculari sono molto accentuate rispetto a quelle oculari, sono di so-lito associate ad altre asimmetrie facciali e danno una difficile mobilizzazione del capo.
Posizione anomala del capo di origine oculareL’adozione di una particolare postura in presenza di patologie oculari rappresenta un tentativo di ottene-re un miglioramento della visione, motivo per cui si parla anche di posizione compensatoria del capo. I
Tabella III. PAC di origine extraoculare.
Cause ortopedicheTorcicollo muscolare congenitoTorcicollo spasticoTorcicollo da alterazioni del tratto cervicale
Cause neurologicheNeoplasieLesioni post-infiammatorie del SNCRitardo psicomotorioQuadri malformativi siringomielici
Cause otorinolaringoiatricheIpoacusiaPatologie vestibolari
RareSindrome di SandiferTorcicollo funzionale

238
E. Santangelo et al.
meccanismi coinvolti sono rappresentati dal miglio-ramento dell’acuità visiva e della visione binoculare (Kraft, 2005), mentre in presenza di aree di cecità o di visione monoculare si sposta il volto dal lato di cecità nel tentativo di centrare il residuo del campo visivo con l’asse corporeo.Dal punto di vista epidemiologico, lo strabismo inco-mitante è la causa più comune, rappresentando circa il 63-70% di tutte le cause di PAC (Nucci, 2006). Il nistagmo rappresenta la seconda causa più comune, rappresentando il 17-20% di tutti i casi di PAC (Hertle, 2000).È molto importante che la diagnosi e il trattamento vengano realizzate il più precocemente possibile, in quanto il persistere di una PAC, associata a un pro-blema oculare, determina anomalie muscolo-schele-triche secondarie che rendono ancora più complessa la diagnosi differenziale e le possibilità terapeutiche successive. L’iter diagnostico, in presenza di una PAC, prevede innanzitutto di osservare la PAC, e in seguito si andrà a posizionare il capo nella direzione opposta, valutando un peggioramento del nistagmo o della de-viazione e lo stato sensoriale del paziente.Di seguito, sono presentate gli aspetti caratterizzanti i principali quadri oculari, responsabili dello sviluppo di una posizione compensatoria del capo (Tab. IV).
PAC associata a strabismo incomitanteNei soggetti con strabismo incomitante, la deviazione non è la stessa in tutte le posizioni di sguardo, ma ne esiste una in cui il disallineamento è massimo. Questi pazienti utilizzano la PAC per orientare gli assi visivi in tale direzione; in linea generale, la posizione com-pensatoria della testa è nella direzione di azione del muscolo deficitario, nella quale la diplopia è assente. Il trattamento si basa sulla correzione dello strabismo che consenta di ottenere l’ortotropia in posizione pri-maria. In presenza di un deficit non ben evidente, l’occlusio-ne dell’occhio paretico può determinare la scomparsa della PAC.Il deficit del muscolo obliquo superiore, da paralisi del IV nervo cranico o da lassità tendinea congenita, è la causa più frequente, sia come forma congenita
che acquisita (Davitt, 2001); il paziente in questo caso si presenta tipicamente con un’ipertropia dell’occhio paretico che aumenta in adduzione, essendo l’obli-quo superiore un depressore soprattutto in adduzio-ne. Un deficit congenito può associarsi a dismorfismi facciali, con un caratteristico maggiore sviluppo del volto dell’emilato paretico rispetto al controlaterale (Greenberg, 2000). Questi soggetti presentano tipica-mente un’inclinazione della testa dal lato non affet-to, con un’eventuale componente di tipo rotatorio. La conferma diagnostica viene dall’head tilt test di Biel-schowsky, che viene definito positivo nel caso in cui, inclinando la testa dal lato opposto a quello in cui si trova il paziente, si osservi un incremento della devia-zione verticale dell’occhio. Il trattamento prevede un indebolimento del muscolo piccolo obliquo. Un deficit dell’elevazione in adduzione può essere inoltre correlato a un ostacolato scorrimento del ten-dine del muscolo obliquo superiore all’interno della sua troclea, quadro che prende il nome di sindrome di Brown; in tal caso si renderà necessaria una teno-tomia. Il deficit del muscolo retto laterale, associato a para-lisi del VI cranico, è la seconda causa per frequen-za tra quelle acquisite. Determina un’esodeviazione compensata dai pazienti con una rotazione della te-sta verso il lato dell’occhio affetto per ridurre o elimi-nare la diplopia. Essendo questo quadro tipicamente acquisito, l’obiettivo è risolvere la causa sottostante; la chirurgia oculare, con indebolimento del muscolo retto mediale, si rende necessaria nel momento in cui non si ha una risoluzione spontanea dopo 6 mesi e dopo aver escluso altre lesioni intracraniche (Nucci, 2015).La sindrome di Duane è una condizione caratteriz-zata da ipoplasia/aplasia congenita del nucleo del VI nervo cranico, in cui si realizza un’innervazione anomala del muscolo retto laterale da parte di una branca del III nervo cranico. È un quadro che presen-ta, in circa il 10% dei casi, un pattern ereditario di tipo autosomico dominante. I quadri clinici conseguenti a una sindrome di Duane sono rappresentati in circa il 50-80% da un deficit dell’abduzione con esotropia, mentre più raramente si ha il riscontro di exotropia con deficit dell’adduzione. Nel caso in cui vi sia una deviazione in posizione primaria, il paziente avrà una PAC rotatoria dal lato affetto se si ha esotropia, o dal lato opposto se si ha exotropia (Zanin, 2010). La pre-senza di una PAC deve essere sempre seguita con attenzione nel tempo, in quanto rappresenta una tra le principali ragioni di correzione chirurgica di questo quadro (Nucci, 2005).Le sindromi alfabetiche con pattern “A” o “V” sono del-le forme di strabismo orizzontale caratterizzato da un incremento o da un decremento della deviazione ri-spetto alla posizione primaria in modo variabile nello sguardo verso l’alto e verso il basso. Nel caso in cui la deviazione sia presente in posizione primaria, il bam-
Tabella IV. PAC di origine oculare
Strabismo incomitante
Esotropia essenziale infantile
Nistagmo
Errori refrattiviIpermetropia AstigmatismoMiopia
Ptosi palpebrale
Difetti perimetrici

239
Anomalie della motilità oculare e posizioni anomale del capo
bino potrebbe assumere una posizione in chin-up o in chin-down per mantenere la visione binoculare (Da-vitt, 2001). Numerose altre patologie muscolari possono determi-nare una limitazione meccanica nei movimenti oculari, tali da determinare il crearsi di una PAC. Tra queste in particolare si ricorda la frattura blow-out del pavimen-to orbitario, frequente in età pediatrica, caratterizzata da un “intrappolamento” del muscolo retto inferiore, che determina l’assunzione di una PAC in chin-up, per sopperire al deficit dell’elevazione (Soo, 2009).
PAC associata a esotropia essenziale infantileL’esotropia essenziale infantile si caratterizza per la presenza, nella maggior parte dei pazienti, di una ti-pica rotazione della testa verso l’occhio fissante con una lieve inclinazione sulla spalla dello stesso lato della rotazione. Tipicamente, la PAC aumenta quan-do il bambino guarda la tv o quando a scuola legge alla lavagna; la PAC fa sì che il bambino assuma un atteggiamento definito anche hunter position, nel ten-tativo di avere una buona fissazione. A oggi, è ancora discussa la reale prevalenza di PAC in corso di eso-tropia congenita (Brodsky, 2004).
PAC associata a nistagmoIl nistagmo si caratterizza per movimenti oculari in-volontari di tipo oscillatorio; è possibile descrivere un null-point, un punto dello spazio nel quale l’intensità delle oscillazioni è minima, con la migliore acuità vi-siva possibile. Nel caso in cui il null-point non sia in posizione primaria, la PAC tende a portare lo sguardo verso il null-point. Il paziente assumerà una posizione della testa compensatoria variabile in base al tipo di nistagmo: se torsionale, il movimento sarà di inclina-zione, se orizzontale sarà di rotazione, se verticale sarà in chin-up o in chin-down; nella maggior parte dei soggetti, tuttavia, si hanno posizioni combinate. Si noti però che non sempre in presenza di nistagmo il soggetto ricerca il null-point; infatti, è possibile che il paziente ruoti il capo massimalmente per bloccare, in versione estrema, gli occhi; quest’ultima eventualità rende inutile un trattamento chirurgico, da realizzarsi invece nel caso in cui questo possa migliorare la fun-zionalità visiva e la PAC (Boricean, 2011).
PAC associata a errori refrattiviSono stati descritti alcuni casi di PAC compensatori secondari a errori refrattivi, ma i meccanismi che ne determinano l’instaurarsi sono poco conosciuti. Più
frequentemente, una PAC come conseguenza di un errore refrattivo è presente in modo incostante, poten-dosi ridurre in condizioni di riposo visivo.Il quadro più frequentemente descritto è una posizio-ne in chin-down associato a ipermetropia superiore a +5.00 diottrie, risolta con l’utilizzo di lenti correttive (Havertape, 1998); una PAC con inclinazione laterale della testa è presente in corso di astigmatismo, men-tre in presenza di miopia è più frequente il chin-up, probabilmente nel tentativo di sfruttare l’effetto steno-peico di tale posizione.
PAC associata a ptosi palpebraleIn presenza di ptosi palpebrale tale da coinvolgere l’asse ottico interferendo così con la funzione visiva, il paziente assume una PAC in chin-up. La presenza di PAC è comunque prognosticamente favorevole in questi pazienti, in quanto indicativo di un tentativo di sviluppare le funzioni visive (Fogagnolo, 2008).
PAC associata a difetti perimetriciIn presenza di difetti del campo visivo associati a pa-tologie neuro-oftalmologiche, è possibile che i pazien-ti assumano una PAC tale da poter sfruttare la por-zione di retina visivamente indenne e massimizzare l’ampiezza del campo visivo residuo (Paysse, 1997).
ConclusioniL’importanza di una diagnosi e di un trattamento pre-coce delle anomalie della motilità oculare è spesso dirimente, sia per il riconoscimento di eventuali qua-dri patologici sottostanti, sia per garantire il raggiungi-mento della migliore funzionalità visiva possibile, sia, non da ultimo, per evitare disagi sociali legati all’este-tica del disturbo. Ciò assume particolare rilevanza per la risoluzione di una posizione anomala del capo, in cui la diagno-si precoce è molto significativa, in quanto impedisce che il quadro determini anomalie muscolo-scheletri-che secondarie, che renderebbero la condizione di più difficile risoluzione. Da questo punto di vista, il ruolo dell’interazione tra il pediatra e gli oftalmologi e ortopedici che si occu-pano di sviluppo posturale è fondamentale. L’obiettivo è cercare di trattare la condizione oculare di base ed evitare così che s’instaurino conseguenze significati-ve dal punto di vista dello sviluppo muscolo-schele-trico. Anche in questo, al di là degli aspetti funzionali legati al persistere della PAC, non bisogna dimentica-re il disagio legato agli aspetti estetici dell’anomalia posturale.

240
E. Santangelo et al.
Box di orientamento
• Leanomaliedellamotilitàocularesonounquadroestremamentefrequenteinetàpediatrica,eilpediatraè lo specialista che ha la maggiore possibilità di intercettarle precocemente, in particolare per quanto riguarda la presenza di posizioni anomale del capo.
• Lostrabismoeleposizionianomaledelcapopossonoessereassociateaconseguenzenegative,siadalpunto di vista della funzionalità visiva, che dello sviluppo muscolo-scheletrico.
• Ilpediatrasvolgeunruolofondamentalenell’identificazioneprecocedelleanomaliedellamotilitàoculare;l’interazione con lo specialista è fondamentale per garantire il migliore trattamento possibile e accompa-gnare la gestione del follow-up.
Bibliografia
Boricean ID, Bărar A. Understanding ocular torticollis in children. Oftalmologia 2011;55:10-26.
Brodsky MC, Jenkins R, Nucci P. Unex-plained head tilt following surgical treat-ment of congenital esotropia: a postural manifestation of dissociated vertical diver-gence. Br J Ophthalmol 2004;88:268-72.
Campos EC. Why do the eyes cross? A review and discussion of the nature and origin of essential infantile esotropia, mi-crostrabismus, accommodative esotropia, and acute comitant esotropia. J AAPOS 2008;12:326-31.
Christoff A, Raab EL, Guyton DL, et al. DVD--a conceptual, clinical, and surgical overview. J AAPOS 2014;18:378-84.
Cuignet FLU. Torticollis oculaire. Re-ceuil Ophtalmol 1873. In: Bielschowsky A (Ed.). Lectures on MotorAnomalies. Ha-nover, NH: Dartmouth College Publications 1943/1956.
Davitt BV. Anomalous head postures: a review. Am Orthop J 2001;51:137-43.
** Questa review presenta in modo com-pleto e dettagliato le caratteristiche fisiopa-tologiche delle posizioni anomale del capo.
Deskin RW. Sandifer syndrome: a case of torticollis in infancy. Int J Pediatr Otorhi-nolaryngol 1995;32:183-5.
Fogagnolo P, Serafino M, Nucci P. Sta-bility of silicone band frontalis suspension for the treatment of severe unilateral upper
eyelid ptosis in infants. Eur J Ophthalmol 2008;18:723-7.
Greenberg MF, Pollard ZF, Schnall BM. Ocular plagiocephaly: ocular torticollis with skull and facial asymmetry. Ophthalmology 2000;107:173-9.
Havertape SA, Cruz OA. Abnormal head posture associated with high hyperopia. J AAPOS 1998;2:12-6.
Hertle RW, Zhu X. Oculographic and clinical characterization of thirty-seven children with anomalous head postures, nystagmus, and strabismus: the basis of a clinical algorithm. JAAPOS 2000;4:25-32.
Hwang JM, Baek RM, Lee SW. Ocu-lar findings in children with orbital hypertelorism. Plast Reconstr Surg 2012;130:624e-627e.
Joyce KE, Beyer F, Thomson RG, et al. A systematic review of the effectiveness of treatments in altering the natural history of intermittent exotropia. Br J Ophthalmol 2015;99:440-50.
Kraft SP. Abnormal head postures in children. In: Taylor D, Hoyt CS (Eds.). Pe-diatric Ophthalmology and Strabismus. Philadelphia: Elsevier Saunders 2005, pp. 987-1000.
Lueder GT. Orbital causes of incomitant strabismus. Middle East Afr J Ophthalmol 2015;22:286-91.
Merino P, Gómez-de-Liaño P, Gil MR, et al. Strabismus and congenital cataracts. Arch Soc Esp Oftalmol 2007;82:623-8.
Mitchell PR. Ocular torticollis. Tr Am Ophth Soc 1999;XCVII:697-769.
Nucci P, Curiel B, Lembo A, et al. Anom-alous head posture related to visual prob-lems. Int Ophthalmol 2015;35:241-8.
Nucci P, Kushner BJ, Serafino M, et al. A multi-disciplinary study of the ocu-lar, orthopedic, and neurologic causes of anomalous head postures in children. Am J Ophthalmol 2005;140:65-8.
Nucci P. Superior oblique palsy: promot-ing a simpler approach. Eur J Ophthalmol 2006;16:1-2.
Paysse EA, Coats DK. Anomalous head posture with early-onset homonymous hemianopia. JAAPOS 1997;1:209-13.
Peragallo JH, Pineles SL, Demer JL. Recent advances clarifying the etiolo-gies of strabismus. J Neuroophthalmol 2015;35:185-93.
Soo K, Taik-Kun K. Clinico-radiologic findings of entrapped inferior oblique mus-cle in a fracture of the orbital floor. Korean J Ophthalmol 2009;23:224-7.
Wright KW, Spiegel PH. Pediatric Oph-thalmology And Strabismus. New York: Springer-Verlag 2003.
* Il testo contiene una descrizione siste-matica dei diversi quadri di strabismo che possono essere alla base delle anomalie della motilità oculare.
Zanin E, Gambarelli N, Denis D. Distinctive clinical features of bilateral Duane retraction syndrome. J AAPOS 2010;14:293-7.
Corrispondenza
Massimiliano SerafinoOspedale San Giuseppe, via San Vittore 12, 20123 Milano - E-mail: [email protected]

241
Luglio-Settembre 2016 • Vol. 46 • N. 183 • Pp. 241-249 Prospettive in Pediatria
Frontiere
Nuovi approcci nella terapia delle malattie mitocondriali
Massimo Zeviani Emanuela Bottani
Carlo Viscomi
MRC Mitochondrial Biology Unit, Cambridge, UK
Le malattie mitocondriali sono un importante gruppo di malattie genetiche caratterizzate dal malfunzionamento della fosforilazione ossidativa (OxPhos), il processo che converte l’energia ottenuta dalle sostanze nutritive in ATP, la molecola ad alta energia utilizzata dalle cellule. Questi disordini sono caratterizzati da un’estrema variabilità di sintomi, coinvolgi-mento di organi e decorso clinico, con un notevole impatto sulla qualità e, spesso, sulla durata della vita. Benché negli ultimi 20 anni si sia assistito a un impressionante aumento delle nostre conoscenze sui meccanismi genetici e biochimici alla base delle malattie mi-tocondriali, lo sviluppo di approcci terapeutici in grado di migliorare in modo significativo il decorso clinico non è stato altrettanto soddisfacente. Al momento, la terapia delle malattie mitocondriali rimane basata su interventi supportivi, mirati ad affrontare le complicanze della malattia. Tuttavia negli ultimi anni sono emerse nuove strategie terapeutiche, la cui efficacia è stata dimostrata almeno a livello preclinico. Questo articolo riassume i risultati principali ottenuti dalla terapia sperimentale e illustra alcuni degli sviluppi futuri in questo campo.
Riassunto
SummaryMitochondrial disorders are an important group of genetic conditions characterised by impaired oxidative phosphorylation (OXPHOS). Mitochondrial disorders come with an impressive variability of symptoms, organ involvement and clinical course, which con-siderably impact the quality of life and often shortens life expectancy. Although in the last 20 years there has been an exponential increase in understanding the genetic and bio-chemical mechanisms leading to the disease, this has not resulted in the development of effective therapeutic approaches that have significantly improved the clinical course and outcome of these conditions. Therapeutic options for mitochondrial diseases still remain focused on supportive interventions aimed at relieving complications. However, new ther-apeutic strategies are recently emerging, some of which have shown potential efficacy at the pre-clinical level. This review will present the state of the art on experimental therapy for mitochondrial disorders.
Introduzione
Concetti di base di biologia e medicina mitocondrialeI mitocondri sono organelli semiautonomi circondati da una doppia membrana lipidica, esterna e interna; quest’ultima si ripiega a formare numerose invagina-zioni chiamate cristae mitocondriali, sulle quali sono posizionati i complessi della catena respiratoria (CR).I mitocondri convertono l’energia liberata dalla de-gradazione ossidativa completa di zuccheri e acidi grassi, un processo noto come respirazione cellula-re e i cui prodotti finali sono CO2 e H2O, in calore e
ATP, la molecola energetica della cellula. Respirazio-ne e sintesi di ATP sono processi accoppiati che de-finiscono la via metabolica nota come fosforilazione ossidativa (OXPHOS). La respirazione è svolta da quattro complessi della CR (complessi I-IV, CI-CIV) che trasferiscono gli elettroni, estratti dai nutrienti in forma di atomi di idrogeno, all’ossigeno molecolare. Gli elettroni sono convogliati alla CR in forma di due intermedi di riduzione, il NADH che cede elettroni al gruppo prostetico FMN, incorporato nel CI, e il FADH2 che fa parte sia del CII (il quale riceve elettroni princi-palmente dal ciclo di Krebs) sia di altre deidrogenasi mitocondriali (es. la Electron-Transfer Factor dehydro-genase – ETF-DH, per quanto riguarda gli intermedi

242
M. Zeviani et al.
della beta ossidazione degli acidi grassi). Sia il NADH sia il FADH2 donano elettroni al coenzima Q (CoQ), un trasportatore di elettroni idrofobico mobile della membrana mitocondriale interna. Il flusso elettronico che determina la riduzione del CoQ avviene median-te centri ossidoriduttivi ferro-zolfo (Fe-S) contenuti sia nel CI (8 centri Fe-S), sia nel CII (3 centri Fe-S). Il CoQH2 (ridotto) dona elettroni al CIII, che a sua volta li trasferisce a un’altra “navetta” ossidoriduttiva mobile, il citocromo c, mediante l’azione di due citocromi, c1 e b, e un centro Fe-S incorporato in una proteina catali-tica, la proteina di Rieske. Il citocromo c è un elemen-to redox mobile che cede un elettrone per volta al CIV (citocromo c ossidasi, COX), il quale infine opera la riduzione di O2 molecolare a H2O. Nei complessi I, III, e IV, ma non in CII, il flusso elettronico libera energia, che sostiene l’attività di pompe protoniche incorpora-te nei complessi stessi per effettuare la traslocazione di protoni (H+) dall’interno del mitocondrio (matrice) allo spazio intermembrana. Si genera in questo modo un potenziale elettrochimico di membrana (ΔP), for-mato da una componente elettrostatica (ΔY) e una chimica (ΔpH), che è infine utilizzato dal complesso V della CR (ATP sintasi) per condensare ADP e fosfato inorganico in ATP (Wallace et al., 2010). I mitocondri hanno un proprio genoma (DNA mito-condriale – mtDNA), ereditato per via matrilineare e costituito nei mammiferi da una molecola circolare di 16,5 kb che codifica 13 subunità dei complessi I, III, IV e V della CR (il complesso II è invece composto da 4 subunità tutte codificate dal DNA nucleare). Inoltre, il mtDNA contiene geni codificanti 22 RNA transfer (mt-tRNA) e 2 RNA ribosomali (mt-rRNA), necessari per la traduzione in situ delle 13 subunità della CR co-dificate dal mtDNA. Il mtDNA è presente in centinaia o migliaia di copie nei vari tipi cellulari di ogni individuo. Negli individui sani, tutte le molecole di mtDNA sono uguali tra loro, una condizione definita come omopla-smia. Per contro, nei pazienti con mitocondriopatia, genomi mitocondriali mutati spesso coesistono con genomi normali (eteroplasmia). Il resto del proteoma mitocondriale, stimato intorno a 1500 polipeptidi, è codificato da geni nucleari, che vengono tradotti nel citosol e quindi trasportati nei mitocondri per mezzo di un processo attivo che richiede energia. Il CI (NADH-ubiquinone ossidoreduttasi) contiene set-te subunità codificate dal mtDNA (ND1-ND6 e ND4L) e almeno 37 subunità codificate nel nucleo. Il CII (succinato-ubiquinone ossidoreduttasi) è com-posto da quattro subunità tutte codificate dal geno-ma nucleare, contenenti 3 centri Fe-S, e trasferisce elettroni dal FADH2, prevalentemente generato dalla beta-ossidazione degli acidi grassi, al CoQ.Il CIII (ubiquone-ferricitocromo c ossidoreduttasi) è composto da una singola subunità codificata dal mtDNA (l’apocitocromo b) e 10 subunità codificate dal genoma nucleare, di cui una contenente un cen-tro Fe-S.
CIV (citocromo c ossidasi, COX), che è composto da 3 subunità codificate dal mtDNA e 11 dal genoma nu-cleare, contiene due molecole di eme A e due centri catalitici contenenti rame (centri Cu++A e Cu++B). Il complesso V (CV – ATPasi oligomicina sensibile) utilizza l’energia potenziale del gradiente elettrochimi-co per sintetizzare ATP. Il CV è composto da due su-bunità codificate dal mtDNA (ATPasi 6 e 8) e da alme-no 13 subunità codificate nel nucleo. Queste subunità formano le due porzioni del complesso: la porzione F0 è immersa nella membrana mitocondriale interna e costituisce il rotore attivato dal flusso di protoni at-traverso di essa, mentre la porzione F1 catalizza la biosintesi dell’ATP (Walker, 2013). Numerosi fattori di assemblaggio e chaperone sono necessari per assemblare la struttura portante dei complessi, inserire i gruppi prostetici e i centri reattivi contenenti ioni metallici, assemblare gli olocomples-si (Fernandez-Vizarra et al., 2009) ed eventualmente produrre strutture quaternarie costituite da più com-plessi interagenti fra loro, chiamati supercomplessi (Lapuente-Brun et al, 2013).Altre componenti del proteoma mitocondriale sono richieste per numerosissimi processi biologici, come la replicazione, trascrizione e traduzione del mtDNA, la fusione e fissione della rete mitocondriale, vie di segnalazione ed esecuzione (come la produzione di specie reattive dell’ossigeno, ROS, e l’apoptosi), la biosintesi di componenti lipidiche essenziali delle membrane mitocondriali, la formazione delle cristae, l’eliminazione di prodotti tossici ecc.
Introduzione alle malattie mitocondrialiLe malattie mitocondriali primarie possono essere classificate in due principali categorie, a seconda che la mutazione responsabile sia nel genoma mitocon-driale o nucleare. Le mutazioni nel mtDNA compren-dono mutazioni puntiformi omo- o eteroplasmiche, e riarrangiamenti su larga scala, che sono sempre eteroplasmici. Mutazioni puntiformi eteroplasmiche sono state trovate in tutti i geni mitocondriali e danno origine a diversi fenotipi clinici, incluse alcune sindro-mi classiche come l’encefalomiopatia mitochondriale con acidosi lattica ed episodi di ictus (MELAS) (Goto et al., 1990), l’epilessia mioclonica con accumulo di fibre “rosse stracciate” ragged red fibers – RRF (MERRF) (Shoffner et al., 1990), la sindrome da debolezza neurogenica, atassia e retinite pigmento-sa (NARP) (Holt et al., 1990), la sindrome di Leigh (LS). La presenza di RRF è un segno di proliferazione mitocondriale spesso osservato nelle miopatie mito-condriali, specie nel paziente adulto. Esistono anche mutazioni omoplasmiche patogene, principalmente quelle che causano la neuropatia ottica ereditaria di Leber (LHON) (Wallace et al., 1988). I riarrangiamenti singoli del mtDNA (singole delezioni o duplicazioni) danno origine all’oftalmoplegia esterna progressiva

243
Nuovi approcci nella terapia delle malattie mitocondriali
(PEO) (Moraes et al., 1989), alla sindrome di Kearns-Sayre (KSS) (Moraes et al., 1989) e alla sindrome di Pearson (Rotig et al., 1989). Le delezioni singole, a differenza delle mutazioni puntiformi, sono di solito sporadiche.Sono state trovate mutazioni in un vastissimo numero di geni nucleari direttamente o indirettamente legati alla catena respiratoria e codificanti, per esempio, proteine coinvolte nel mantenimento e/o replicazione del mtDNA; subunità strutturali dei complessi della catena respiratoria; fattori di assemblaggio dei com-plessi respiratori; proteine coinvolte nella struttura di-namica dei mitocondri, come la fissione/fusione dei mitocondri o in processi di segnale/esecuzione come l’apoptosi (si veda [Koopman et al., 2012] per una lista completa).L’estrema complessità biochimica e genetica del me-tabolismo mitocondriale fa sì che le malattie mitocon-driali siano caratterizzate da una notevole eteroge-neità clinica, che rende difficile la raccolta di gruppi omogenei di pazienti per stabilire l’efficacia di un trat-tamento. Un’area nosologica esemplificativa di tale eterogeneità sono le deficienze primarie di CoQ, che possono presentarsi come encefalomiopatia, disordi-ni multisistemici, atassia cerebellare, miopatia isola-ta o sindrome nefrosica (Lopez et al., 2014). Inoltre, per ragioni ignote, solo il 20% dei pazienti risponde al trattamento con CoQ, l’unica terapia al momento disponibile (Lopez et al., 2014).
Strategie terapeuticheNotevoli progressi sono stati fatti negli ultimi anni nella comprensione dei processi patogenetici alla base delle malattie mitocondriali, dei meccanismi che controllano la biogenesi mitocondriale e delle vie di trasduzione del segnale. Partendo da queste conoscenze, sono state recentemente proposte alcune strategie tera-peutiche per le malattie mitocondriali, e le prove spe-rimentali della loro efficacia si stanno accumulando in modelli cellulari e animali. Queste terapie si possono suddividere in strategie “generaliste” che possono, in linea di principio, essere utilizzate in un ampio numero di patologie, e strategie “su misura”, applicabili a una singola malattia. Il primo gruppo include: (i) la regolazione/attivazione della biogenesi mitocon-
driale; (ii) la regolazione/attivazione dell’autofagia/mitofagia; (iii) l’inibizione dell’apoptosi; (iv) l’eliminazione di composti tossici, quali le specie
reattive dell’ossigeno (ROS) generati dalla disfun-zione della CR;
(v) il bypass dei difetti di trasferimento elettronico sul-la CR;
(vi) il trasferimento nucleare. Il secondo gruppo include: (i) l’eliminazione di metaboliti tossici in specifiche
condizioni;
(ii) l’integrazione di desossinucleotidi precursori del mtDNA;
(iii) la terapia di sostituzione genica/cellulare. Ciascuna di queste strategie può essere perseguita attraverso diversi approcci, quali il trattamento far-macologico, il trasferimento genico (per esprimere il gene mancante o una proteina terapeutica), il trapian-to di cellule staminali o d’organo. In questo articolo ci concentreremo sulle terapie sperimentali emergenti (cioè in fase preclinica) per le malattie mitocondriali, e, in questo ambito, sugli approcci più prossimi all’ap-plicazione clinica (Fig. 1).
Interventi farmacologici e metabolici
Aumento della biogenesi mitocondrialeLe malattie mitocondriali sono caratterizzate da difetti bioenergetici, che causano una ridotta sintesi di ATP. È quindi possibile che interventi terapeutici mirati ad aumentare la quantità di ATP disponibile all’interno delle cellule siano efficaci nell’arrestare, mitigare o risolvere alcune mitocondriopatie. È importante con-siderare che le malattie mitocondriali si manifestano quando l’attività residua di una proteina scende sotto una soglia critica, e perciò un recupero anche par-ziale dell’attività può essere di beneficio dal punto di vista clinico. Ad esempio, è stato evidenziato che i portatori sani di mutazioni LHON hanno un contenuto di mtDNA maggiore dei loro parenti affetti e della po-polazione generale (Giordano et al., 2014).L’aumento della biogenesi mitocondriale è una rispo-sta omeostatica a condizioni di stress (es. freddo, esercizio fisico, stato nutrizionale) (Scarpulla, 2008).Le vie che controllano la biogenesi mitocondria-le sono state particolarmente studiate nel muscolo scheletrico e nel grasso bruno, dove risultano esse-re sotto il controllo dei coattivatori trascrizionali della famiglia PGC (Peroxisome proliferator-activated re-ceptor Gamma (PPARγ) Coactivators), in particolare di PGC-1α e β. Le proteine PGC interagiscono con numerosi fattori trascrizionali, attivandoli. Tra questi ricordiamo i Nuclear Respiratory Factors (NRF1 and 2), e i Peroxisomal Proliferator Activator Receptors (PPAR α, β, and γ), che stimolano la trascrizione di geni collegati all’OXPHOS e all’ossidazione degli aci-di grassi (FAO), rispettivamente.PGC-1α è la più nota tra le proteine PGC. La sua attivi-tà è controllata dai livelli di acetilazione, che a loro volta dipendono dall’attività dell’acetilasi GNC5 e della dea-cetilasi sirtuina 1 (SIRT1), e di fosforilazione, control-lata da varie chinasi (Bastin et al., 2008; Fernandez-Marcos e Auwerx, 2011). Ridotti livelli di acetilazione e aumentati livelli di fosforilazione stimolano l’attività di PGC-1α e inducono la biogenesi mitocondriale.L’attivazione della biogenesi mitocondriale è stata sperimentalmente ottenuta utilizzando varie strategie

244
M. Zeviani et al.
farmacologiche. Un primo approccio consiste nell’u-tilizzo del bezafibrato, un farmaco largamente impie-gato nel trattamento delle dislipidemie. Il bezafibrato è un agonista generico dei recettori PPAR, che a loro volta agirebbero come attivatori trascrizionali di PGC-1α . L’utilizzo del bezafibrato in un topo privo del gene COX10, un enzima coinvolto nella biosintesi del gruppo prostetico eme A presente nella citocromo c ossidasi, e caratterizzato da una grave miopatia con deficit di COX, ha portato a un netto miglioramento della performance motoria e della sopravvivenza, ri-spetto a topi non trattati (Wenz, 2009). Questi risulta-ti non sono però stati confermati da studi successivi (Viscomi et al., 2011; Yatsuga e Suomalainen, 2012). Ciononostante un trial clinico è stato recentemente avviato su pazienti con miopatia mitocondriale (www.clinicaltrial.gov: NCT02398201). Vie alternative per l’attivazione della biogenesi mito-condriale dipendente da PGC-1α sono basate sulla stimolazione della chinasi AMP-dipendente (AMPK) o della deacetilasi nucleare Sirt1. AICAR, un agonista di AMPK, causa una robusta induzione di geni collegati all’OXPHOS e l’aumento delle attività dei complessi della catena respiratoria in modelli murini di deficit di COX (Viscomi et al., 2011). Sirt1 utilizza il NAD+ per deacetilare i residui di lisina acetilata delle proteine. L’aumento della concentrazione di NAD+ ne stimola l’attività. La somministrazione del precursore di NAD+ nicotinammide riboside (NR) o l’inibizione, con siste-
mi genetici o farmacologici, della poli(ADP)-ribosil po-limerasi 1 (Parp1), un enzima nucleare che compete con Sirt1 per l’utilizzo del NAD+ come substrato, de-terminano l’attivazione di Sirt1 e aumentano la respi-razione mitocondriale inducendo i geni dell’OXPHOS attraverso la stimolazione di PGC1α (Cerutti et al., 2014; Khan et al., 2014). Nonostante questi approc-ci siano basati su sostanze potenzialmente innocue (NR) o già utilizzate in clinica (inibitori di PARP), non sono al momento presenti sperimentazioni cliniche per confermarne l’efficacia nei pazienti.A ogni modo, questi risultati aprono la nuova ed ecci-tante prospettiva di una terapia in grado di correggere un ampio spettro di malattie mitocondriali dovuto a dif-ferenti cause genetiche.
Eliminazione di metaboliti tossiciAlcuni interventi terapeutici sono stati utilizzati in malattie mitocondriali caratterizzate dall’accumulo di sostanze tossiche dovuto al blocco di specifiche vie metaboliche. Un primo esempio è l’uso di N-acetilci-steina (NAC) e metronidazolo per ridurre l’accumulo di acido solfidrico (H2S) associato all’encefalopatia etilmalonica (EE) (Viscomi et al., 2010). EE è una ma-lattia infantile multisistemica causata da mutazioni in ETHE1, un gene che codifica una sulfur-diossigenasi mitocondriale coinvolta nello smaltimento di H2S (Ti-ranti et al., 2009). H2S è un composto altamente tossi-
Figura 1. Schema riassuntivo delle principali strategie terapeutiche per le malattie mitocondriali.

245
Nuovi approcci nella terapia delle malattie mitocondriali
co prodotto dal catabolismo degli aminoacidi solforati nei tessuti e dalla flora batterica anaerobica presente in grandi quantità nell’intestino crasso.L’N-acetil-cisteina (NAC) è un composto permeabile alle cellule che agisce come precursore del glutatione, necessario per lo smaltimento di H2S. Il metronidazo-lo è un antibiotico specifico per i batteri anerobici, che producono H2S. La somministrazione di NAC e me-tronidazolo determina un significativo prolungamento della durata della vita e delle condizioni cliniche di un modello murino Ethe1-/-. Gli stessi composti si sono dimostrati efficaci nel mitigare i sintomi dei pazienti EE (Viscomi et al., 2010).Altri composti potenzialmente tossici sono le specie re-attive dell’ossigeno (ROS), generate come prodotti se-condari della respirazione mitocondriale. Nei mitocondri l’anione superossido (O2
-•), una molecola altamente re-attiva, viene prodotta in diversi siti della matrice e dello spazio intermembrana. O2
-• è rapidamente convertito in perossido di idrogeno (H2O2) dalle superossido dismu-tasi mitocondriale e citosolica; l’H2O2 è un composto molto meno reattivo ed è rapidamente convertibile ad acqua da numerosi e abbondanti enzimi presenti negli stessi mitocondri, nei perossisomi e nel citosol (Sena e Chandel, 2012). In presenza di ferro ridotto, Fe2+, l’a-nione superossido può invece generare, mediante la reazione di Fenton, il radicale ossidrilico (•OH), un com-posto altamente reattivo che può danneggiare le struttu-re cellulari, perossidando proteine, lipidi e acidi nuclei-ci. Un’aumentata produzione di ROS è una frequente conseguenza di difetti di OXPHOS (Raimundo et al., 2012), e può determinare danni importanti a proteine, lipidi e acidi nucleici. Queste osservazioni costituiscono la base razionale per l’utilizzo di agenti antiossidanti nel-la terapia delle malattie mitocondriali. Miscele di antios-sidanti, quali acido lipoico, vitamine C ed E, CoQ ecc., sono state a lungo usate nella terapia delle malattie mi-tocondriali, ma di fatto la loro efficacia non è mai stata validata da studi controllati e rigorosi in modelli animali o nei pazienti. Alcuni trial clinici sono in corso per testare i potenziali benefici di due nuovi antiossidanti in varie malattie mitocondriali. Il primo composto è EPI-743, un derivato della vitamina E, il secondo, KH176, derivato dall’antiossidante Trolox. Entrambi sono stati sviluppati a partire da screening di librerie di composti, e portati alla sperimentazione clinica saltando lo stadio dei test su modelli animali della malattia.
Sommistrazione di nucleotidiLa somministrazione di desossiribonucleotidi è stata utilizzata per corregge la deplezione del mtDNA in fi-broblasti e/o modelli murini di pazienti con mutazioni in enzimi coinvolti nel controllo dei pool mitocondriali di deossinucleotidi precursori del mtDNA (per esem-pio la deossiguanosina chinasi, dGK, e la timidina chinasi mitocondriale, TK, codificate rispettavamente dai geni DGUOK e TK2).
In particolare, risultati promettenti sono stati ottenu-ti in un modello murino che riproduce la mutazione umana H126N nella timidina chinasi 2 (TK2) (Garone et al., 2014), che fosforila nei mitocondri la timidina e la deossicitidina monofosfato (dCMP). La mancanza di TK2 causa deplezione severa del mtDNA nel mu-scolo e nel cervello. Il trattamento con 200 o 400 mg/kg/die di nucleotidi determina un aumento nei livelli di dNTP e di mtDNA, un recupero delle attività della RC e un significativo prolungamento della sopravvivenza da 13 a 34 giorni.
Terapie nutrizionali Vari approcci basati sulla manipolazione della dieta sono stati tentati con risultati controversi. Una dieta chetogenica (KD), cioè con alto contenuto di grassi e basso contenuto di carboidrati, è stata proposta con lo scopo di stimolare la β-ossidazione mitocondriale e produrre chetoni, che costituiscono una fonte alterna-tiva di energia per cervello, cuore e muscolo scheletri-co. I corpi chetonici sono metabolizzati ad acetil-CoA, che entra nel ciclo di Krebs, e sono ossidati cedendo elettroni alla catena respiratoria, così generando ATP attraverso l’OXPHOS. Questa via catabolica esclude parzialmente il complesso I, perché aumenta la sintesi di succinato, che dona elettroni al complesso II della catena respiratoria. Questa osservazione suggerisce un potenziale beneficio della dieta chetogenica nei difetti di complesso I. Inoltre, l’aumento di corpi che-tonici determina una maggiore espressione di geni OXPHOS, probabilmente per mezzo di una risposta simile a quella del digiuno (Nunnari e Suomalainen, 2012), una condizione di stress cellulare che induce la biogenesi mitocondriale. La KD è stata utilizzata per ri-durre l’eteroplasmia associata a delezione del mtDNA in modelli cellulari (Santra et al., 2004), e per indurre l’espressione di proteine disaccoppianti (uncoupling proteins, – UCP) e di geni coinvolti nella biogenesi mi-tocondriale nell’ippocampo di modelli animali (Jarrett et al., 2008) e per tentare di arrestare la progressione del-la miopatia in un modello murino di delezioni multiple del mtDNA (Ahola-Erkkila et al., 2010). Tuttavia, altri la-vori hanno dimostrato che KD può avere effetti opposti e peggiorare il difetto mitocondriale in vivo, ad esempio nel modello murino Mpv17-/- (Bottani et al., 2013).Come KD, anche una dieta high-fat, che ha un con-tenuto più elevato di carboidrati rispetto alla dieta KD, ha dimostrato avere effetti protettivi in modelli di ma-lattie mitocondriali in vivo e in vitro (Schiff et al., 2011).Risultati simili potrebbero in linea di principio esse-re ottenuti con composti che rilasciano succinato nei mitocondri. Ad esempio, la trieptaoina è un compo-sto che induce un rapido aumento di succinil-CoA, e migliora significativamente sia la cardiomiopatia in pazienti con deficit di VLCAD sia i sintomi miopatici in pazienti con deficit di CPT2 (Roe et al., 2002; Wat-mough et al., 1990).

246
M. Zeviani et al.
Un trial clinico è stato recentemente concluso sull’uso di un dieta ricca di trigliceridi a media catena in pa-zienti con mutazione MELAS 3243A>G, ma i risultati non sono ancora noti.
Approcci molecolari al trattamento delle malattie mitocondriali
Terapia genicaLa correzione di una mutazione attraverso la reintro-duzione del gene funzionante in organi critici è stata a lungo considerata come la cura definitiva per le ma-lattie genetiche. In particolare, vettori virali adenoasso-ciati (AAV) non sono patogeni per l’uomo, e il DNA da essi veicolato rimane episomico nelle cellule dei tessu-ti post-mitotici (ad es. cuore, cervello, muscolo schele-trico, e in parte fegato e rene) per molto tempo, ridu-cendo il rischio di mutagenesi inserzionale (Mingozzi e High, 2011). Sono stati sviluppati vari serotipi AAV con diversa specificità cellulare, che consentono di effet-tuare terapie mirate a singoli organi (Gao et al., 2002). Il serotipo AAV2/8 in particolare è già stato utilizzato con successo sull’uomo per il trattamento dell’emofi-lia B (Nathwani et al., 2011). Lo stesso serotipo è stato utilizzato per riesprimere selettivamente nel fegato il gene Ethe1, responsabile dell’encefalopatia etilmalo-nica, determinando un’efficace clearance di H2S dal sangue, la normalizzazione dei biomarcatori della ma-lattia, e un evidente miglioramento sia delle condizioni cliniche che della sopravvivenza (Di Meo et al., 2012). Questi risultati hanno fornito un’importante prova di principio che la riattivazione del filtro epatico è suffi-ciente a migliorare decisamente le malattie mitocon-driali da accumulo di sostanze tossiche, e costituisco-no quindi il razionale per proporre il trapianto di fegato nell’encefalopatia etilmalonica (Dionisi-Vici et al., 2016) e nella MNGIE (mitochondrial neuro-gastro-intestinal encephalomyopathy). Quest’ultima è una grave ma-lattia autosomica recessiva causata da mutazioni nel gene TYMP, codificante per la timidina fosforilasi (TP), un enzima citosolico che catalizza il primo passaggio del catabolismo della timidina (dThd) e della deossiuri-dina (dUrd) (Nishino et al., 1999). Nella MNGIE il mal-funzionamento di TP causa un accumulo sistemico di dThd e dUrd, che determina uno sbilanciamento dei pool di deossinucleotidi trifosfati (dNTP), con effetti mutageni sul mtDNA. Alterazioni deleterie del mtDNA (deplezione, delezioni multiple e mutazioni puntiformi) colpiscono soprattutto organi postmitotici, in particolare la muscolatura liscia intestinale, il muscolo scheletrico e il sistema nervoso centrale e periferico, causando un progressivo deficit mitocondriale e disfunzione d’orga-no. I sintomi si manifestano in età giovanile adulta e comprendono grave dismotilità intestinale con diarrea cronica, dolori addominali importanti ed eventualmente perdita di peso fino alla cachessia, oftalmoplegia ester-na progressiva con miopatia, e grave neuropatia peri-
ferica sensori-motoria. I pazienti di solito muoiono per le complicazioni dovute al loro stato nutrizionale molto compromesso, con una vita media di 37 anni (Nishi-no et al., 2001). In topi TYMP-/-, la somministrazione sistemica ad alte dosi di AAV2/8 esprimente la forma nativa di TYMP determina la normalizzazione dei livelli plasmatici di dCTP e dTTP fino a 8 mesi dalla som-ministrazione (Torres-Torronteras et al., 2014). Il tratta-mento correntemente usato per la MNGIE è basato sul trapianto di midollo osseo, che ha però una mortalità post-operatoria superiore al 50% a causa delle reazio-ni da rigetto, incluse quelle del trapianto contro l’ospite (graft-versus-host disease) e la necessità di elimina-re il tessuto emopoietico endogeno (haematopoietic conditioning), che possono compromettere definitiva-mente le condizioni già molto critiche dei pazienti al momento del trapianto (Hirano et al., 2006; Rahman e Hargreaves, 2007). La somministrazione di AAV in-gegnerizzati è una procedura molto meno invasiva e quindi probabilmente più facilmente accettabile e por-terebbe a un sostanziale miglioramento della prognosi di questa malattia invariabilmente mortale.L’epatotropismo dei ceppi AAV2/8 può anche essere sfruttato per correggere il difetto alla base di malattie mitocondriali che colpiscono selettivamente il fega-to. Noi abbiamo applicato questo principio al modello murino Mpv17-/-. Mpv17 è una proteina mitocondriale a funzione ignota, mutata in pazienti affetti da forme epato-cerebrali di sindrome da deplezione del mtDNA (Karadimas et al., 2006; Spinazzola et al., 2006). Come nella malattia umana, il modello murino mostra una pro-fonda riduzione del mtDNA nel fegato ma, in contrasto con quanto avviene nell’uomo, nessun fenotipo clinico, almeno in condizioni standard. Tuttavia i topi Mpv17-
/- sviluppano una steatosi con evoluzione cirrotica e di-sfunzione epatica rapidamente progressiva se esposti a dieta chetogenica (Bottani et al., 2013). Un vettore virale AAV2/8 esprimente Mpv17 umano è in grado di cor-reggere completamente la deplezione del mtDNA e di prevenire la cirrosi indotta da KD in topi Mpv17-/-, soprat-tutto quando il trattamento genico precede l’inizio della KD, mentre la progressione della malattia è ritardata ma non abolita, quando la terapia genica è effettuata dopo l’inizio del regime KD. La recente introduzione di nuovi serotipi epatotropi, come AAV5, apre la possibilità di una seconda somministrazione di virus, sfuggendo così alla neutralizzazione da parte del sistema immunitario (Pa-neda et al., 2009; Unzu et al., 2011).Complessivamente questi risultati preclinici dimostra-no il grande potenziale della terapia genica basata su AAV per combattere specifiche malattie mitocondriali. Un certo numero di problemi devono tuttavia essere ancora risolti, quali lo sviluppo di strategie per colpire organi cri-tici come cervello, cuore e muscolo scheletrico. Benché qualche successo sia stato ottenuto su miopatie e distro-fie non mitocondriali in modelli preclinici (Childers et al., 2014; Greelish et al., 1999; Gregorevic et al., 2004), la loro efficacia nell’uomo deve ancora essere dimostrata.

247
Nuovi approcci nella terapia delle malattie mitocondriali
Una strategia ripetutamente proposta per la correzio-ne di mutazioni in geni del mtDNA è basata sull’e-spressione allotopica, secondo la quale il gene, rico-dificato secondo il codice genetico universale, viene fuso con una sequenza di localizzazione mitocon-driale (MTS) e trasfettato nel nucleo. Il gene verrebbe quindi trascritto nel nucleo, tradotto nel citoplasma, e indirizzato ai mitocondri dove verrebbe trasloca-to all’interno della matrice/membrana mitocondriale interna. Questo approccio è stato tentato in modelli cellulari (Bonnet et al., 2008; Bonnet et al., 2007; Kal-timbacher et al., 2006) e animali (Ellouze et al., 2008). Complessivamente, i risultati di questi esperimenti sono molto controversi, poiché dati conflittuali sono stati ottenuti da diversi gruppi sulla capacità dei geni ricodificati e indirizzati ai mitocondri di essere effetti-vamente importati nell’organello e di essere corretta-mente integrati nei complessi della catena respiratoria (Perales-Clemente et al., 2011). Tuttavia tre trial clinici open label basati sull’espressione allotopica di geni mitocondriali della LHON sono al momento in corso (NCT02161380, NCT01267422, NCT02652767).
Modulazione dei processi di fissione-fusione e dell’ultrastruttura mitocondrialeI mitocondri sono organelli molto dinamici la cui forma e massa sono finemente regolati dall’attività di protei-ne pro-fusione, quali mitofusina 1 (MFN1), MNF2 e la proteina associata ad atrofia ottica 1 (OPA1), e pro-fissione, quali la dynamin-related protein 1 (DRP1) e la mitochondrial fission 1 FIS1 (Friedman e Nunnari, 2014; Mishra e Chan, 2014). Mutazioni in geni che codificano per questi complessi macchinari causano malattie nell’uomo. Ad esempio, mutazioni in OPA1 sono associate ad atrofia ottica dominante (Alexander et al., 2000) e mutazioni in MFN2 causano la malat-tia di Charcot-Marie-Tooth di tipo 2A (Zuchner et al., 2004). Alterazioni della dinamica e dell’ultrastruttura dei mitocondri sono osservate in numerose patologie e la loro correzione può portare a un miglioramento della patologia. La prova genetica di questo principio è stata ottenuta dimostrando che l’overespressione di Opa1, una GTPasi coinvolta nella biogenesi del-le cristae mitocondriali e nel promuovere la fissione della membrana mitocondriale interna, aumenta l’ef-ficienza respiratoria (Cogliati et al., 2013). Inoltre, la (moderata) sovra-espressione di OPA1 migliora il fe-notipo clinico di topi con deficit di complesso I e IV e protegge da una serie di insulti, quali il danno da ischemia-riperfusione, l’atrofia muscolare indotta da denervazione, o il danno miopatico (Civiletto et al., 2015; Varanita et al., 2015). Alcuni composti speri-mentali, quali l’inibitore di Drp1 MDIVI-1, e l’idrazone M1 che si ritiene promuova la fusione mitocondriale agendo su MNF o OPA1, sono in grado di interferire con i processi di fissione-fusione, ma il potenziale te-
rapeutico di questi composti deve comunque essere verificato. Tuttavia, i cosiddetti peptidi di Szeto-Schiller (SS) sono stati utilizzati per correggere i difetti dell’ul-trastruttura mitocondriale in varie condizioni, quali l’a-trofia muscolare, l’insufficienza cardiaca, l’ischemia-riperfusione e il diabete (Szeto e Birk, 2014). I peptidi SS sono tripeptidi in grado di penetrare in cellula e di accumularsi nei mitocondri, dove legano la cardiolipi-na, un componente lipidico della membrana mitocon-driale interna con attività di modulazione della catena respiratoria e di strutturazione delle cristae. Inoltre, la cardiolipina modula l’attività di Opa1 ed è possibile che questo possa spiegare almeno in parte gli effetti dei peptidi SS sulla struttura della cristae. I peptidi SS sono stati ampiamente caratterizzati dal punto di vista farmacologico e uno di essi (MTP131 o Bendavia) è al momento in trial clinico per le miopatie mitocondria-li (NCT02367014).
Trasferimento somatico nucleareData la difficoltà nella manipolazione del mtDNA e le incertezze nella consulenza genetica per le mutazioni nel mtDNA, al momento la migliore opzione per le don-ne con mutazioni patogene nel mtDNA è la diagnosi preimpianto. Recenti progressi tecnici in primati non umani (Tachibana et al., 2009) ed embrioni umani non vitali (Craven et al., 2010) hanno aperto la strada alla sostituzione del mtDNA materno mutato con quello di un donatore sano. Questo può essere ottenuto attra-verso il trasferimento del fuso cromosomico di oociti maturi della donna affetta in un oocita sano o dei pro-nuclei durante la fase pre-zigotica dell’uovo fecondato (Craven et al., 2010; Tachibana et al., 2013). Entrambe le tecniche sono state rifinite per ridurre al minimo il trasferimento della minor quantità possibile di mtDNA mutato dell’ooplasma accettore. Un bambino nato da queste procedure avrà quindi i geni nucleari della ma-dre affetta (e del padre sano) ma i geni mitocondriali sani del donatore (si veda (Chinnery et al., 2014) per un sommario recente su questi argomenti).
ConclusioniLe malattie mitocondriali sono straordinariamente complesse e la loro biologia ha finora reso impossi-bile sviluppare terapie efficaci per la maggior parte di esse. Tuttavia, negli ultimi anni si sono osservati numerosi tentativi di modificare in modo significativo il fenotipo di modelli cellulari o animali, utilizzando strategie specifiche per una malattia o strategie ad ampio spettro applicabili a vari disordini. Il patrimonio di conoscenze accumulato in oltre 25 anni di intensi studi sulle cause e i meccanismi delle malattie mi-tocondriali hanno aperto la strada all’acquisizione di importanti “prove di principio” sperimentali in fase pre-clinica, che ora aspettano di essere traslate e testate sui pazienti.

248
M. Zeviani et al.
Bibliografia
Ahola-Erkkila S, Carroll CJ, Peltola-Mjosund K, et al. Ketogenic diet slows down mitochon-drial myopathy progression in mice. Hum Mol Genet 2010;19:1974-84.
Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin-related GT-Pase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 2000;26:211-5.
Bastin J, Aubey F, Rotig A, et al. Activa-tion of peroxisome proliferator-activated receptor pathway stimulates the mito-chondrial respiratory chain and can cor-rect deficiencies in patients’ cells lacking its components. J Clin Endocrinol Metabol 2008;93:1433-41.
Bonnet C, Augustin S, Ellouze S, et al. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim Biophys Acta 2008;1783;1707-17.
Bonnet C, Kaltimbacher V, Ellouze S, et al. Allotopic mRNA localization to the mitochondrial surface rescues respira-tory chain defects in fibroblasts harboring mitochondrial DNA mutations affecting complex I or v subunits. Rejuvenation Res 2007;10:127-44.
Bottani E, Giordano C, Civiletto G, et al. AAV-mediated liver-specific MPV17 ex-pression restores mtDNA levels and pre-vents diet-induced liver failure. Mol Ther 2014;22:10-7.
Cerutti R, Pirinen E, Lamperti C, et al. NAD-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochon-drial disease. Cell Metab 2014;19:1042-9.
** Un importante studio sulla possibilità di modulare la biogenesi mitocondriale at-traverso la regolazione dei livelli endogeni di NAD+.
Childers MK, Joubert R, Poulard K, et al. Gene therapy prolongs survival and re-stores function in murine and canine mod-els of myotubular myopathy. Sci Transl Med 2014;6:220ra10.
* Uno dei primi esempi di terapia genica nel muscolo di un mammifero di medie di-mensioni.
Chinnery PF, Craven L, Mitalipov S, et al. The challenges of mitochondrial replace-ment. PLoS Gen 2014;10:e1004315.
Civiletto G, Varanita T, Cerutti R, et al. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Met 2015;21:845-54.
* La prima dimostrazione che la mod-ulazione dell’ultrastruttura mitocondri-ale può essere efficace nella terapia delle malattie mitocondriali.
Cogliati S, Frezza C, Soriano ME, et al. Mitochondrial cristae shape determines res-piratory chain supercomplexes assembly and respiratory efficiency. Cell 2013;155:160-71.
Craven L, Tuppen HA, Greggains GD, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010;465:82-5.
** La prima dimostrazione di fattibilità del trasferimento dei pronuclei in embrioni umani.
Di Meo I, Auricchio A, Lamperti C, et al. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encepha-lopathy. EMBO 2012;4:1008-14.
** Il primo studio in cui la terapia genica epatica viene utilizzata per correggere un difetto mitocondriale dovuto all’accumulo di sostanze tossiche.
Dionisi-Vici C, Diodato D, Torre G, et al. Liver transplant in ethylmalonic encepha-lopathy: a new treatment for an otherwise fatal disease. Brain 2016;139:1045-51.
* Il primo esempio di trapianto di fe-gato utilizzato per correggere l’accumulo di sostanze tossiche dovuto a disfunzione mitocondriale.
Ellouze S, Augustin S, Bouaita A, et al. Optimized allotopic expression of the hu-man mitochondrial ND4 prevents blind-ness in a rat model of mitochondrial dys-function. Am J Hum Gen 2008;83:373-87.
Fernandez-Marcos PJ, Auwerx J. Regu-lation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr
2011;93:884S-90.Fernandez-Vizarra E, Tiranti V, Zeviani M.
Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta 2009;1793:200-11.
Friedman JR, Nunnari J. Mitochondrial form and function. Nature 2014;505:335-43.
Gao GP, Alvira MR, Wang L, et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A 2002;99:11854-9.
Garone C, Garcia-Diaz B, Emmanuele V, et al. Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 defi-ciency. EMBO 2014;6:1016-27.
Giordano C, Iommarini L, Giordano L, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 2014;137:335-53.
Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial en-cephalomyopathies. Nature 1990;348:651-3.
Greelish JP, Su LT, Lankford EB, et al. Sta-ble restoration of the sarcoglycan complex in dystrophic muscle perfused with hista-mine and a recombinant adeno-associated viral vector. Nature Med 1999;5:439-43.
Gregorevic P, Blankinship MJ, Allen JM, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vec-tors. Nature Med 2004;10:828-34.
Hirano M, Marti R, Casali C, et al. Alloge-neic stem cell transplantation corrects bio-chemical derangements in MNGIE. Neurol-ogy 2006;67:1458-60.
Holt IJ, Harding AE, Petty RK, et al. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Gen 1990;46:428-33.
Jarrett SG, Milder JB, Liang LP, et al. The ke-togenic diet increases mitochondrial glutathione levels. J Neurochemistry 2008;106:1044-51.
Kaltimbacher V, Bonnet C, Lecoeuvre G, et al. mRNA localization to the mitochon-drial surface allows the efficient transloca-
Box di orientamento
• Cosa sapevamo prima Le malattie mitocondriali sono al momento incurabili. La variabilità clinica , biochimica e genetica rende estremamente difficile lo sviluppo di nuove terapie.
• Cosa sappiamo adessoNegli ultimi anni sono stati fatti i primi passi verso la messa a punto di nuovi interventi terapeutici, alcuni dei quali si sono mostrati efficaci in modelli animali. Questi comprendono l’uso di farmaci per aumentare la biogenesi mitocondriale, eliminare composti tossici e correggere l’ultrastruttura dei mitocondri e l’uso di vettori virali adenoassociati in alcune malattie specifiche.
• Quali ricadute sulla pratica clinicaSupportati dai risultati raccolti in modelli cellulari e animali, i primi trial clinici sui pazienti sono stati sviluppati e sono attualmente in corso. L’impatto di questi approcci nella pratica clinica sarà verificato nei prossimi anni.

249
Nuovi approcci nella terapia delle malattie mitocondriali
tion inside the organelle of a nuclear recod-ed ATP6 protein. Rna 2006;12:1408-17.
Karadimas CL, Vu TH, Holve SA, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Gen 2006;79:544-8.
Khan NA, Auranen M, Paetau I, et al. Ef-fective treatment of mitochondrial myopa-thy by nicotinamide riboside, a vitamin B3. EMBO Mol Med 2014;6:721-31.
Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med 2012;366:1132-41.
* Un’ampia e recente revisione sulle malattie mitocondriali.
Lapuente-Brun E, Moreno-Loshuertos R, Acín-Pérez R, et al. Supercomplex as-sembly determines electron flux in the mi-tochondrial electron transport chain. Sci-ence 2013;340:1567-70.
Lopez LC, Luna-Sanchez M, Garcia-Cor-zo L, et al. Pathomechanisms in coenzyme q10-deficient human fibroblasts. Molec Syndromol 2014;5:163-9.
Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nature Rev Genet 2011;12:341-55.
Mishra P, Chan DC. Mitochondrial dy-namics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 2014;15:634-46.
Moraes CT, DiMauro S, Zeviani M, et al. Mitochondrial DNA deletions in pro-gressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med 1989;320:1293-9.
Nathwani AC, Tuddenham EG, Ranga-rajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemo-philia B. N Engl J Med 2011;365:2357-65.
** La prima applicazione all’uomo di una terapia genica basata su AAV2/8.
Nishino I, Spinazzola A, Hirano M. Thy-midine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999;283:689-92.
Nishino I, Spinazzola A, Hirano M. MNGIE: from nuclear DNA to mitochondrial DNA. Neuromuscul Disord 2001;11:7-10.
Nunnari J, Suomalainen A. Mito-chondria: in sickness and in health. Cell 2012;148:1145-59.
* Un’ampia analisi della biologia dei mi-tocondri.
Paneda A, Vanrell L, Mauleon I, et al. Ef-fect of adeno-associated virus serotype and genomic structure on liver transduction and biodistribution in mice of both genders. Hu-man Gene Ther 2009;20:908-17.
Perales-Clemente E, Fernandez-Silva P, Acin-Perez R, et al. Allotopic expres-
sion of mitochondrial-encoded genes in mammals: achieved goal, undemonstrated mechanism or impossible task? Nucleic Acids Res 2011;39:225-34.
Rahman S, Hargreaves IP. Allogeneic stem cell transplantation corrects bio-chemical derangements in MNGIE. Neurol-ogy 2007;68:1872-3.
Raimundo N, Song L, Shutt TE, et al. Mitochondrial stress engages E2F1 ap-optotic signaling to cause deafness. Cell 2012;148:716-26.
Roe CR, Sweetman L, Roe DS, et al. Treatment of cardiomyopathy and rhabdo-myolysis in long-chain fat oxidation dis-orders using an anaplerotic odd-chain tri-glyceride. J Clin Invest 2002;110:259-69.
Rotig A, Colonna M, Bonnefont JP, et al. Mitochondrial DNA deletion in Pear-son’s marrow/pancreas syndrome. Lancet 1989;1:902-3.
Santra S, Gilkerson RW, Davidson M, et al. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Annals Neurol 2004;56:662-9.
Scarpulla RC. Transcriptional para-digms in mammalian mitochondrial bio-genesis and function. Physiological Rev 2008;88:611-38.
Schiff M, Benit P, El-Khoury R, et al. Mouse studies to shape clinical trials for mitochondrial diseases: high fat diet in Harlequin mice. PloS One 2011;6:e28823.
Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen spe-cies. Mol Cell 2012;48:158-67.
Shoffner JM, Lott MT, Lezza AMet al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mi-tochondrial DNA tRNA(Lys) mutation. Cell 1990;61:931-7.
Spinazzola A, Viscomi C, Fernandez-Vizarra E, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet 2006;38:570-5.
Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther 2014;96:672-83.
Tachibana M, Amato P, Sparman M, et al. Towards germline gene therapy of in-herited mitochondrial diseases. Nature 2013;493:627-31.
Tachibana M, Sparman M, Sritanaudom-chai H, et al. Mitochondrial gene replace-ment in primate offspring and embryonic stem cells. Nature 2009;461:367-72.
Tiranti V, Viscomi C, Hildebrandt T, et al. Loss of ETHE1, a mitochondrial di-oxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nature Med 2009;15:200-5.
* Uno studio sul meccanismo patoge-netico dell’encefaloptia etilmalonica.
Torres-Torronteras J, Viscomi C, Ca-brera-Perez R, et al. Gene therapy using a liver-targeted AAV vector restores nu-cleoside and nucleotide homeostasis in a murine model of MNGIE. Molecular Ther 2014;22:901-7.
Unzu C, Sampedro A, Mauleon I, et al. Sustained enzymatic correction by rAAV-mediated liver gene therapy pro-tects against induced motor neuropathy in acute porphyria mice. Molecular Ther 2011;19:243-50.
Varanita T, Soriano ME, Romanello V, et al. The OPA1-dependent mitochon-drial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Met 2015;21:834-44.
Viscomi C, Bottani E, Civiletto G, et al. In vivo correction of COX deficiency by acti-vation of the AMPK/PGC-1alpha axis. Cell Met 201;14:80-90.
** La prima dimostrazione in vivo della possibilità di utilizzare la biogenesi mitocon-driale nella terapia delle malattie mitocondriali.
Viscomi C, Burlina AB, Dweikat I, et al. Combined treatment with oral metronida-zole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nature Med 2010;16:869-71.
* Uno studio in cui si dimostra l’efficacia di una terapia farmacologica nell’encefaloptia etilmalonica.
Walker JE. The ATP synthase: the un-derstood, the uncertain and the unknown. Biochem Soc Trans 2013;41:1-16.
Wallace DC, Fan W, Procaccio V. Mito-chondrial energetics and therapeutics. An-nual Rev Pathol 2010;5:297-348.
Wallace DC, Singh G, Lott MT, et al. Mi-tochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988;242:1427-30.
Watmough NJ, Bindoff LA, Birch-Machin MA, et al. Impaired mitochondrial beta-oxidation in a patient with an abnormality of the respiratory chain. Studies in skel-etal muscle mitochondria. J Clin Invest 1990;85:177-84.
Wenz T. PGC-1alpha activation as a ther-apeutic approach in mitochondrial disease. IUBMB Life 2009;61:1051-62.
Yatsuga S, Suomalainen A. Effect of bezafibrate treatment on late-onset mito-chondrial myopathy in mice. Human Mol Gen 2012;21:526-35.
Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GT-Pase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nature Gen 2004;36:449-51.
Corrispondenza
Massimo ZevianiMRC Mitochondrial Biology Unit, Wellcome Trust/MRC, Building Hills Road, Cambridge, CB2 0XY, UK Tel. +44 1223252704 - E-mail: [email protected]

250
Luglio-Settembre 2016 • Vol. 46 • N. 183 • Pp. 250-258 Prospettive in Pediatria
Focus
La metabolomica in pediatria: una scienza in continua evoluzione
Angelica Dessì Antonio Noto
Roberta Pintus Vassilios Fanos
Unità di Terapia Intensiva Neonatale, Sezione di Patologia
Neonatale, Azienda Ospedaliero-Universitaria di Cagliari
Le scienze ‘omiche’ comprendono un ampio spettro di discipline che studiano la relazione che intercorre tra genotipo e fenotipo. La metabolomica è una nuova tecnologia d’indagine che permette di studiare le modificazioni del metaboloma di un individuo in tempo reale, sia in condizioni fisiologiche che patologiche e/o a seguito dell’esposizione individuale a fattori epigenetici quali ambiente, dieta e uso di farmaci. Il metaboloma, ovverosia l’insieme delle variazioni dei metaboliti nel tempo, può essere analizzato con metodi non invasivi, poiché ci si avvale della raccolta di fluidi biologici quali sangue, saliva, urina e feci. Le tec-niche di analisi dei campioni possono essere di tipo qualitativo o quantitativo, come la spet-troscopia di risonanza magnetica e la cromatografia in fase gassosa o liquida, accoppiata con la spettrometria di massa. Le potenziali applicazioni di questa tecnica in campo medi-co sono molteplici e negli ultimi anni c’è stato un incremento esponenziale degli studi per quanto riguarda l’applicazione della metabolomica in pediatria. In questa review sono stati analizzati alcuni tra i più recenti studi di metabolomica in campo pediatrico, in particolare sulla sepsi, le malattie renali, il ritardo di crescita intrauterino e il neonato grosso per età gestazionale, l’enterocolite necrotizzante e l’autismo. La metabolomica sembra essere una tecnica promettente e i nuovi biomarcatori potrebbero essere utilizzati nella pratica clinica come strumento diagnostico predittivo o per valutare l’efficacia e la tossicità di un farmaco, in modo tale da poter fornire al bambino una medicina personalizzata a sua misura.
Riassunto
SummaryThe ‘omics’ sciences include a wide range of disciplines that investigate the interplay between genotype and phenotype. Metabolomics is a new investigation technology that enables to study the changes in the metabolome of a human being in real time, in physi-ological or pathological conditions and/or due to individual exposition to epigenetic fac-tors such as environment, diet and pharmacological therapies. The metabolome, namely the variations of metabolites that occur over time, can be analyzed through non-invasive methods using the collection of biological fluids such as blood, saliva, urine, feces. The analysis techniques of samples can be qualitative or quantitative including the nuclear magnetic resonance spectroscopy and the liquid or gas phase chromatography, cou-pled with mass spectroscopy. The potential applications of this technique in medicine are manifold and in the past few years there was an exponential increment of the studies concerning the application of metabolomics in pediatrics. In this review, the most recent studies concerning metabolomics in pediatrics have been analyzed, in particular those addressing sepsis, renal diseases, intrauterine growth restriction and large for gestational age, necrotizing enterocolitis and autism. Metabolomics seems to be a promising tech-nique and the new biomarkers could be used in clinical practice as predictive diagnostic tools or to evaluate the efficacy or toxicity of a certain drug, in order to provide children with a personalized medicine.

251
La metabolomica in pediatria: una scienza in continua evoluzione
Metodologia della ricerca bibliografica effettuataGli autori hanno selezionato dalla letteratura più re-cente gli studi più rilevanti per ciò che riguarda le ap-plicazioni della metabolomica in pediatria, utilizzando come motore di ricerca PubMed e le seguenti parole chiave per i diversi temi trattati: “metabolomics in pe-diatrics”, “metabolomics and sepsis”, “metabolomics and renal diseases”, “metabolomics and IUGR”, “me-tabolomics and necrotizing enterocolitis”, “metabolo-mics and autism”.
Introduzione: la metabolomicaLe scienze definite “omiche” comprendono un ampio spettro di discipline che studiano la relazione che in-tercorre tra genotipo e fenotipo. La genomica, ossia lo studio del genoma inteso come l’insieme dei geni di un organismo, è nata con l’intento di svelare il ruo-lo chiave della genetica nei processi vitali e ha con-dotto al sequenziamento dell’intero genoma umano. La genomica è risultata essere limitata, però, dalla scarsità delle associazioni lineari tra le varianti ge-netiche e la complessità fenotipica (Jansen et al., 2004). Questo ha determinato un passaggio dallo studio del genotipo umano, alla valutazione del suo fenotipo, e quindi allo studio degli effetti che scatu-riscono dall’interazione tra questi due elementi. Il cambio di prospettiva ha generato lo sviluppo di altre scienze “omiche”, tra cui la trascrittomica (lo studio dei trascritti del RNA prodotti dal genoma), la prote-omica (lo studio delle proteine prodotte da una spe-cie) e infine la metabolomica. Quest’ultima rappre-senta una recente branca scientifica il cui oggetto di interesse è il metaboloma, inteso come l’insieme dei metaboliti costituenti un organismo vivente. Secon-do la definizione di Nicholson et al. la metabolomica è una misurazione quantitativa della risposta dina-mica di un organismo vivente a stimoli esterni, sia patologici che fisiologici, o a modificazioni interne, quindi di natura genetica. Tale risposta è valutabile mediante l’analisi delle variazioni dei metaboliti nel sistema biologico considerato. Risulta evidente, per-tanto, come la metabolomica sia in grado di studiare la complessità biologica dell’interazione tra genetica e ambiente, mostrando un profilo metabolico speci-fico per ogni evento generato da questa relazione (Fig. 1). Queste informazioni sono inoltre ricavabili in modo minimamente invasivo, dato che per l’analisi dei me-taboliti ci si può avvalere della raccolta di fluidi biologi-ci quali il sangue, la saliva, le urine, le feci. L’approccio metabolomico consiste nell’impiego sequenziale di una tecnica di rilevamento, che si avvale della spettro-scopia di risonanza magnetica nucleare (1H-NMR) e la cromatografia in fase gassosa o liquida, accoppiate con la spettrometria di massa (LC-MS e GC-MS), con
le quali si possono misurare simultaneamente, nel campione esaminato, la concentrazione di un gran numero di metaboliti (Ellis et al., 2007).Le applicazioni della metabolomica in campo medi-co possono essere numerose (Tab. I). Sin dallo svi-luppo embrionale la vita umana è un sistema biolo-gico in continua evoluzione, con stimoli differenti che possono lasciare un’impronta nel processo di cresci-ta. Per questo motivo un obiettivo specifico, su cui si sta concentrando la recente ricerca medica, è la definizione dei profili metabolici che si susseguono nel corso delle diverse fasi della vita, individuando la traiettoria metabolica caratterizzante la vita umana dal suo principio fino al suo termine (Dessì et al., 2013). Infatti la ricerca di biomarcatori, correlati con uno stato fisiopatologico e con uno specifico stile di vita, permettono di valutare lo stato di benessere o meno dell’organismo, così come l’efficacia o la tossi-cità di un farmaco.
Figura 1. Le frecce in grigio rappresentano 4 vie metabo-liche individuali (biochimica tradizionale). Le frecce nere mostrano le opportunità offerte dalla metabolomica: esiste una via metabolica che connette queste 4 vie tramite gli intermediari di ciascuna via (da Fanos et al., 2016, mod.).
Tabella I. Alcune delle più importanti applicazioni della metabolomica in medicina (da Fanos et al., 2016, mod.).
• Fotografia dello stato fisiologico• Diagnosi di malattia• Scoperta di biomarcatori• Farmacometabolomica: misura della risposta ai
farmaci• Nutrimetabolomica: misura degli effetti della
nutrizione• Classificazione dei fenotipi• Identificazione di vie metaboliche alterate da malattie
o terapie• Genomica funzionale• Caratterizzazione dei prodotti naturali

252
A. Dessì et al.
La metabolomica in pediatria: alcuni esempiLa metabolomica sembra essere una tecnica molto promettente anche in campo pediatrico: nell’ultimo decennio c’è stato un incremento esponenziale di studi pubblicati in letteratura. L’esigenza di trovare nuove soluzioni in questo ambito nasce dal fatto che nonostante le numerose indagini nel corso degli ultimi anni, non esiste ancora un singolo test che soddisfi i criteri di marcatore ideale per la diagnosi precoce di diverse malattie pediatriche. Attualmente nella pra-tica clinica pediatrica vengono dosati un numero li-mitato di biomarcatori nei liquidi biologici, mediante delle tecniche analitiche convenzionali. Inoltre, que-sti vengono applicati in neonatologia e in pediatria, senza considerare che la patogenesi e la risposta alla terapia di molte malattie nei bambini è differen-te rispetto all’adulto. L’analisi dell’intero metaboloma sembra rappresentare quindi un buon metodo per la determinazione di variazioni metaboliche correlate a patologie che determinano notevoli cambiamenti nei metaboliti dell’organismo (Fanos et al., 2011), come ad esempio in caso di sepsi, malattie renali, problemi nutrizionali come il ritardo di crescita (IUGR) e il neo-nato grosso per età gestazionale (GEG), enterocolite necrotizzante (NEC) e autismo, di cui parleremo di seguito.
SepsiLa sepsi è una sindrome caratterizzata da profonde modificazioni immunologiche, metaboliche, emodina-miche e respiratorie, dovute a un processo infettivo che colpisce l’organismo (Vergnano et al., 2005). A oggi sono stati pubblicati diversi studi di metabolo-mica e sepsi, sia su modelli animali sia sull’uomo e di questi alcuni su pazienti pediatrici (Mickiewicz et al., 2013; Fanos et al., 2014; Mickiewicz et al., 2013) (Tab. II). È interessante notare come vi siano delle analogie nei metaboliti riscontrati, caratterizzanti i
soggetti settici: un aumento dei metaboliti extracellu-lari (sangue e urine) del metabolismo ossidativo degli acidi grassi (idrossibutirrato, acilcarnitine e acetoace-tato) e un aumento del glucosio e del lattato. Questo potrebbe essere spiegato dal fatto che in condizioni di forte stress, come nella sepsi, si ha un aumento della lipolisi e del catabolismo proteico per soddisfare un surplus del fabbisogno energetico: questo si traduce in un aumento dell’escrezione renale della carnitina e di altri metaboliti coinvolti nel metabolismo ossidativo degli acidi grassi. Inoltre è noto come nei pazienti con shock settico vi è un maggiore uso di glucosio e un aumento della produzione di lattato a livello anerobico (Dessì et al., 2014). Dai risultati di questi studi sembra che l’approccio metabolomico possa rivelarsi utile per implementare la diagnosi precoce e per una migliore individuazione della popolazione a rischio.
Malattie renaliAttualmente i lavori di metabolomica e nefrologia pe-diatrica pubblicati in letteratura sono pochi (Tab. III) (Beger et al., 2016; Atzori et al., 2010; Blydt-Hansen et al., 2014; Hanna et al., 2013; Klepacki et al., 2015; Sedic et al., 2015). Da uno studio di Atzori e coll., si evince come l’analisi metabolomica sembra essere un metodo innovativo e non invasivo per studiare e monitorare anche le malattie nefro-urologiche pedia-triche. In questo lavoro è stata valutata la possibilità che le malattie nefrologiche pediatriche potessero es-sere associate con un modello metabolico ben defi-nito. A tal fine, con metodi di metabolomica basati su 1H-NMR, sono state analizzate le urine di 21 bambini affetti da nefropatie (displasia renale, reflusso vesci-co-ureterale, infezioni del tratto urinario e insufficien-za renale acuta) e sono state confrontate con quelle di 19 controlli sani. Da questo studio sono emerse alterazioni degli spettri di aminoacidi, di purine e pi-rimidine, nonché alterazioni del ciclo dell’acido citrico che caratterizzavano i soggetti patologici dai controlli. Alcuni studi hanno invece applicato la metabolomica
Tabella II. Studi di metabolomica che hanno analizzato i profili metabolici di pazienti pediatrici con sepsi.
Autore Tipo di paziente
Tipo di campione
Analisi metabolomica
Metaboliti
Mickiewicz et al., 2013
60 bambini con shock settico vs 40 SIRS vs 40
controlli
Siero 1H-NMR 2-idrossibutirrato, lattato, glucosio, creatina, creatinina,
istidina, fenilalanina
Fanos et al., 2014
9 neonati settici vs 16 controlli
Urine 1H-NMR e GC-MS Acetato, glicina, lattato, glucosio, ribitolo
Mickiewicz et al., 2015
181 bambini settici vs 63
controlli
Siero e plasma 1H-NMR Acetato, mannosio, taurina, dimetilamina, 2-idrossibutirrato,
alanina
SIRS: sindrome da risposta infiammatoria sistemica.

253
La metabolomica in pediatria: una scienza in continua evoluzione
per la valutazione della nefrotossicità da parte di alcuni farmaci (Hanna et al., 2013; Klepacki et al., 2015). Ad esempio, nello studio sperimentale di Hanna e coll., condotto su ratti neonati, la nefrotossicità indotta da gentamicina è stata associata con un profilo di meta-boliti urinari. In particolare, 14 parametri erano signi-ficativamente alterati dopo somministrazione di gen-tamicina: glucosio, galattosio, N-acetil-glucosammina, mio-inositolo, acido butanoico e 3-idrossibutirrato sono aumentati di tre volte rispetto ai valori normali, seguiti da citrullina e pseudo-uridina che sono comunque au-mentati, ma a livelli più bassi (Hanna et al., 2013). Be-ger et al. hanno applicato un’analisi metabolomica per lo studio di danno renale come complicazione dopo intervento chirurgico cardiopolmonare in età pediatri-ca. In questo studio, 40 campioni di urine sono stati prelevati da bambini che avevano subito un intervento chirurgico per correggere difetti cardiaci congeniti. Il danno renale acuto, definito come un elevato aumen-to di valori sierici basali di creatinina, è stato riportato in 21 neonati 48-72 h successivamente all’intervento chirurgico cardiaco. L’analisi mediante spettrometria di massa delle urine dei campioni prelevati a 4 e 12 ore dopo l’intervento ha permesso di identificare l’acido omovanillico, un metabolita della dopamina, come un nuovo, sensibile e predittivo biomarcatore precoce di danno renale, dopo intervento chirurgico cardiaco pe-diatrico (Beger et al., 2008). La metabolomica trova spazio anche nell’analisi del rapporto tra la nutrizione e lo stato di salute o ma-lattia dell’uomo (Watkins et al., 2013). La gravidanza, la nascita, l’allattamento e la composizione del latte
materno sono influenzate dalla nutrizione e dai suoi costituenti, sia macroscopici che microscopici (Moco et al., 2013). La metabolomica, e più nello specifico la nutrimetabolomica, può rivelarsi uno strumento utile nell’analisi di questi elementi, e può fornire preziose informazioni sui loro effetti biologici nell’organismo.Allo stesso tempo in letteratura stanno emergendo nuove ipotesi relative ai meccanismi biologici alla base dell’obesità infantile, dello sviluppo del diabete e della sindrome metabolica in età adulta. Studi sperimentali dimostrano come anche la malnutrizione fetale, sia in eccesso che in difetto, possa predisporre allo sviluppo di patologie croniche dell’adulto. Il primo autore a ipo-tizzare la programmazione fetale (teoria del fetal pro-gramming) di patologie come la cardiopatia ischemica, l’obesità o il diabete di tipo 2, è stato David Barker. In condizioni di rifornimento non ottimale di ossigeno e nutrienti, si altera l’equilibrio fra vie anaboliche e ca-taboliche e si può innescare una modificazione nelle traiettorie metaboliche della programmazione fetale verso malattie croniche, come la sindrome metabolica. La teoria del fetal programming fa riflettere sul ruolo della malnutrizione fetale e neonatale, e su quali stra-tegie possano essere adottate per prevenire gli scom-pensi metabolici che possono condurre da un lato a un ritardo di crescita intrauterino (IUGR) e dall’altro a macrosomia fetale (GEG). Le condizioni di IUGR e GEG sono caratterizzate da biomarcatori metabolici specifici, e l’interesse per la loro individuazione è testi-moniato dal crescente numero di studi presenti in lette-ratura (Nissen et al., 2011; Alexandre-Gouabau et al., 2011; Dessì et al., 2011; Horgan et al., 2011; Lin et al.,
Tabella III. Studi sulla metabolomica in nefrologia pediatrica.
Autore Tipo di pazienti e campione
Nefrouropatia Analisi metabolomica
Biomarcatori identificati
Beger et al. (2008)
Bambini(urine)
Insufficienza renale acuta dopo intervento cardiaco
LC-MS Acido omovanillico solfato
Atzori et al. (2010)
Bambini(urine)
Nefrouropatie varie 1H-NMR Ippurato, triptofano, fenilalanina, malato, tirosina, idrossibutirrato, N-acetil glutammato, triptofano,
prolina
Hanna et al. (2013)
Rattineonati(urine)
Nefrotossicità da gentamicina
GC-MS Glucosio, galattosio, N-acetil-glucosammina, mioinositolo,
acido butanoico, 3-idrossibutirrato, citrullina, pseudouridina
Sedic M et al. (2014)
Bambini(urine)
Sindrome nefrosica idiopatica
LC-MS Idrossifenilacetato, uridina, glutammina, fenilalanina
Blydt-Hansen TD et al. (2014)
Bambini(urine)
Rigetto T cellulo-mediato di trapianto di rene
LC-MS Prolina, chinurenina, fosfatidilcolina
Klepacki J et al. (2015)
Bambini(urine)
Trapianto renale e nefrotossicità da
Tacrolimus
HPLC-MS/MS Glucosio, sorbitolo, ossido di trimetilammina
Nutrimetabolomica: IUGR e GEG

254
A. Dessì et al.
2012; Favretto et al. 2012; van Vliet et al., 2013; Sanz-Cortés et al., 2013; Logan et al., 2012; Dani et al., 2014; Dessì et al., 2014; Barberini et al., 2014; Marincola et al., 2015; Dessì et al., 2016). Diversi studi metabolo-mici riguardanti i neonati IUGR e GEG sono stati con-dotti col fine di portare alla luce nuove evidenze che potessero confermare quanto ipotizzato inizialmente da Barker (Tab. IV). Dalle evidenze scientifiche emerse proprio con la metabolomica sembra che, seppur ap-parentemente così diversi, IUGR e GEG, condividano un pattern metabolico comune, caratterizzato da uno stato di resistenza glucidica e di ipoglicemia alla na-scita che li distingue dai neonati di peso adeguato per età gestazionale (AGA). Le anomalie energetiche che si ripercuotono nello sviluppo fetale dei neonati IUGR e GEG sono legate a dei deficit in una comune via bio-chimica, ossia il ciclo di Krebs. Questo elemento man-tenuto costante durante il processo di crescita, sembra predisporre sia gli IUGR che i GEG a un’aumentata suscettibilità a sviluppare diabete di tipo 2 e obesità nell’età adulta (Dessì et al., 2013).Recentemente un’area di crescente interesse è anche la correlazione tra microbiota intestinale e le molecole bio-logiche (microbiomica): l’applicazione della metabolomi-ca dimostra infatti un grande effetto della microflora in-testinale sui metaboliti riscontrati nel nostro organismo. Bisogna tenere conto non solo di come la genetica insieme a ciò che mangiamo e i farmaci che assumia-mo condizionino il nostro metabolismo, ma anche di come il cibo e i farmaci vengano metabolizzati dal no-stro microbiota e quali effetti questa interazione possa avere nei diversi organi (Fig. 2).Lo studio dell’interazione tra i metaboliti dell’organismo e dei microrganismi della flora intestinale (microbiomi-ca) sembra avere un ruolo importante anche in campo pediatrico e neonatologico (Fanos et al., 2014).
NEC – Enterocolite necrotizzanteAnche la NEC, che rappresenta ancora oggi l’emer-genza intestinale più frequente nelle terapie intensive neonatali, è stata recentemente oggetto di alcuni stu-di di metabolomica (Dessì et al., 2016). Diversi gruppi di studiosi, attraverso l’analisi di campioni di urine o di sangue, hanno cercato di determinare se esistessero dei metaboliti caratterizzanti che potessero in qualche modo correlarsi con la patologia. Poiché la patogene-si della NEC è ancora sconosciuta, la metabolomica sembra ricoprire un ruolo fondamentale nella com-prensione delle cause scatenanti di questa patologia. Morrow et al. hanno ipotizzato un ruolo nella disbiosi precoce nella patogenesi di questa malattia, analiz-zando campioni di urine provenienti da 32 neonati di età gestazionale inferiore alle 29 settimane, raccolti prima dell’insorgenza della patologia. Di questi neonati 11 hanno contratto la NEC, mentre 21 sono sopravvis-suti. Nei loro campioni è stata riscontrata una disbiosi precoce, in particolare una modifica della composizio-
ne della specie dei Firmucutes ha preceduto la necro-si, mentre una modifica in quella dei Proteobatteri è stata associata a un esordio più tardivo della malattia. È stata inoltre osservata l’assenza di Propionilbatteri in tutti i casi di NEC. I metaboliti discriminanti riscontrati sono stati l’alanina (associata positivamente alla NEC) e l’istidina (ha mostrato una correlazione inversa con i casi di NEC tardiva). Invece Garner et al. hanno analizzato la presenza di composti organici volatili (COV) nelle feci di 6 neona-ti che hanno sviluppato la NEC, raccolte prima e dopo l’insorgenza della malattia confrontandole con quelle di 7 neonati pretermine sani per un periodo di 8 mesi (Gar-ner et al., 2009). Nei neonati affetti da NEC, la quantità di COV è risultata essere nettamente inferiore rispetto al gruppo di controllo, pertanto gli autori concludono che questo genere di composti potrebbe essere usato in fu-turo come biomarker diagnostico per la NEC.Nel 2015 è stato condotto un lavoro da De Magistris et al., che ha esaminato le urine di neonati affetti da NEC in un periodo di 60 giorni dalla nascita. In questo studio gli autori hanno evidenziato una distri-buzione dei profili metabolici correlata al momento dell’insorgenza della patologia. I pazienti affetti da NEC mostravano infatti un aumento dell’acido gluco-nico urinario immediatamente dopo la comparsa del-la patologia che li caratterizzava rispetto ai controlli. Anche in questo caso vi è una stretta correlazione tra metabolita e microrganismi della flora intestinale: l’a-cido gluconico può essere prodotto in diversi modi dal nostro organismo, sia attraverso l’ossidazione spon-tanea del glucosio, sia dal microbiota intestinale. Le specie produttrici di acido gluconico, o che comunque lo utilizzano all’interno del proprio metabolismo, sono contraddistinte da una maggiore capacità prolifera-tiva, da una maggiore virulenza e da una spiccata farmacoresistenza. Sembra quindi che la presenza di
Figura 2. Rappresentazione schematica dell’influenza dell’interazione tra genetica, farmaci, dieta e microbiota sui diversi organi umani.

255
La metabolomica in pediatria: una scienza in continua evoluzione
questo metabolita sia in grado di perturbare molteplici vie metaboliche e favorire la colonizzazione da parte di batteri aggressivi.
AutismoL’autismo, così come i disturbi dello spettro autistico (ASD), sono un grave disturbo psichiatrico caratteriz-zato da un insieme di sintomi come comportamenti ri-
Tabella IV. Studi di metabolomica su IUGR e GEG.
Classe antropometrica
Autori Anno Pazienti Campioni Metodica Metaboliti
IUGR
(* studi condotti su animali)
Nissen et al. 2011 12 IUGR vs 12 AGA*
Plasma 1H-NMR Mioinositolo, D-chiro-inositolo
Alexandre-Gouabau et
al.
2011 8 IUGR vs 8 AGA*
Plasma LC-MS Prolina, arginina, istidina, tirosina, carnitina
Dessì et al. 2011 26 IUGR vs 30 AGA
Urine 1H-NMR Mioinositolo, sarcosina, carnitina, creatinina
Horgan et al. 2011 8 IUGR vs 6 AGA
Sangue venoso
Cordone ombelicale
UPLC-MS Fenilacetilglutammina, carnitina, idrossibutirrato
Lin et al. 2012 18 IUGR vs 18 AGA*
Sangue venoso
Cordone ombelicale
Q-TOF-MS Acido piroglutammico, carnitina, creatinina
Favretto et al. 2012 22 IUGR vs 21 AGA
Sangue Cordone
ombelicale
LC-MS Fenilalanina, triptofano, metionina
Van Vliet et al.
2013 10 IUGR vs 6 AGA*
Cervello LC-QTOF-MS Asparagina, ornitina, acido N-acetilaspartilglutammico,
acido N-acetilaspartato, acido palmitoleico
Sanz-Cortés et al.
2013 23 IUGR vs 23 AGA
Sangue venoso
Cordone ombelicale
1H-NMR Colina, fenilalanina, tirosina, creatina, glutammina,
valina, leucina
GEG
Logan et al. 2012 18 GEG vs 12 AGA
Urine 1H-NMR Glucosio, formato, fumarato, succinato, citrato
Dani et al. 2014 30 GEG vs 40 AGA
Siero 1H-NMR Glucosio, piruvato, istidina, alanina, valina, metionina,
arginina, lisina
IUGR, AGA, GEG
Dessì et al. 2014 12 IUGR e 9 GEG vs 17
AGA
Urine 1H-NMR Mioinositolo, creatinina, creatina, citrato, urea,
glicina
Barberini et al.
2014 23 IUGR e GEG vs 10
AGA
Urine GC-MS Mioinositolo
Marincola et al.
2015 8 IUGR e 8 GEG vs 11
AGA
Urine 1H-NMR Mio-inositolo, creatinina, citrato
Dessì et al. 2016 11 IUGR e 12 GEG vs 14
AGA
Urine GC-MS Acido treonico, pseudouridina, ribosio,
mioinositolo, glicina

256
A. Dessì et al.
petitivi, scarsa comunicazione orale, ridotta empatia, e incapacità di risposta sociale (American Psychiatric Association, 2013).Un recente sondaggio ha calcolato che l’incidenza di tali patologie è aumentata notevolmente negli ulti-mi anni, basti pensare che nel 1980 veniva eseguita diagnosi di autismo/ASD su un soggetto ogni 2.500, mentre a oggi viene eseguita diagnosi su uno ogni 68 soggetti (Center for Disease Control and Prevention, 2014; Baron-Cohen et al., 2015).È stato inoltre calcolato il costo economico di tali pa-tologie, tenendo in considerazione il numero di perso-ne malate, le spese per le cure mediche, e la perdi-ta economica per la società in termini di produttività. Sulla base di questi tre paramenti è stato stimato un costo che si attesta tra i 161 e 367 miliardi di dollari nel 2015, che aumenteranno fino a circa 1.010,6 mi-liardi di dollari nel 2025 (Leigh e Du, 2015; Tarricone, 2006).Le ricerche più recenti pongono un notevole interesse sul ruolo della metabolomica per l’identificazione di nuovi marcatori diagnostici, prognostici e terapeutici. Tra i nuovi biomarcatori più interessanti vi sono i de-rivati del microbiota intestinale, che si è visto essere presenti nei soggetti affetti da autismo/ASD. L’ipotesi è che possano modulare il fenotipo comportamentale dell’ospite, influenzando notevolmente le vie meta-boliche dell’ospite e il sistema immunitario, determi-nando la suscettibilità individuale alla malattia. Ana-lizzando i risultati emersi dallo studio di Noto et al. è interessante notare come i metaboliti, caratterizzanti i soggetti autistici siano strettamente correlati con il mi-crobiota intestinale. Ad esempio nei bambini autistici è stata ritrovata una concentrazione urinaria più ele-vata di acido ippurico e di acido 3-idrossipropanoico rispetto ai controlli. L’acido ippurico è noto essere un co-metabolita della flora microbica mammifera men-tre l’acido 3-idrossipropanoico è stato già riscontra-to elevato nelle urine in precedenti studi sull’autismo ed è considerato un metabolita anomalo derivante da batteri anaerobi dell’intestino, come ad esempio i Clostridi. Questi risultati sembrano avere un risvolto clinico importante dal momento che in bambini con disturbi dello spettro autistico è stata osservata una colonizzazione abnorme di Clostridi, a discapito di
altri batteri comunemente riscontrati nella flora intesti-nale di bambini sani.In accordo con lo studio precedente vi sono diversi studi pubblicati in letteratura che identificano, nel me-taboloma urinario dei pazienti autistici, cicli metabolici alterati come il metabolismo intestinale batterico, de-gli aminoacidi e il metabolismo che riguarda lo stress ossidativo (Mussap et al., 2016).
ConclusioniLe nuove tecnologie come la metabolomica e la mi-crobiomica rappresentano degli strumenti in rapida evoluzione potenzialmente applicabili in tutti i campi della medicina. Per quanto riguarda i pazienti neonatali e pediatrici, l’approccio metabolomico si rivela particolarmente utile, in quanto trattandosi di soggetti che hanno subi-to delle influenze dall’ambiente esterno in misura infe-riore rispetto ai pazienti adulti, è possibile identificare tempestivamente le alterazioni patologiche del profilo metabolico e intervenire precocemente. La metabo-lomica, nonostante le potenzialità di analisi applicate a gruppi di soggetti, non è ancora utilizzabile se rife-rita al singolo paziente, in un approccio di medicina di precisione, a causa della variabilità dei dati che si possono incontrare in una determinata patologia pe-diatrica. Pertanto, a nostro avviso, si tratta di tecniche molto promettenti, ma che al momento trovano anco-ra difficoltà a essere applicate al letto del paziente.Non è difficile ipotizzare entro i prossimi 5 anni un progresso tecnologico verso questa direzione, con l’utilizzo di strumenti semplici ed efficaci, quali i dip-stick delle urine.Le informazioni ottenute dall’analisi di metaboliti pre-senti nei biofluidi è fondamentale per la comprensione delle influenze esercitate sull’organismo sia da fattori genetici, sia da fattori ambientali come ad esempio la dieta, il microbiota intestinale o l’eventuale assunzio-ne di farmaci.Tutto questo si traduce in una migliore comprensione dei meccanismi fisiopatologici nelle diverse patologie, permettendo lo sviluppo di terapie adeguate, efficaci e sartoriali per il paziente.

257
La metabolomica in pediatria: una scienza in continua evoluzione
Box di orientamento
• Cosa sapevamo primaLe conoscenze sullo stato metabolico globale nel neonato e nel bambino sono, a tutt’oggi, ancora scarse. Nella pratica clinica pediatrica vengono dosati comunemente gli stessi biomarcatori dell’adulto, mediante delle metodiche analitiche convenzionali.
• Cosa sappiamo adessoGli studi recenti sull’applicazione della metabolomica in pediatria hanno messo in luce le differenze di concentrazione dei metaboliti nei bambini sani rispetto a quelli affetti da diverse patologie quali sepsi, malattie renali, problemi nutrizionali, enterocolite necrotizzante e autismo.
• Quali ricadute sulla pratica clinicaI biomarcatori scoperti grazie alla metabolomica potrebbero essere utili per comprendere la fisiopatolo-gia ancora poco chiara di diverse malattie pediatriche. La metabolomica sembra essere uno strumento diagnostico predittivo, che ci permetterà di trovare delle strategie terapeutiche più efficaci per quel deter-minato paziente.
Bibliografia
Alexandre-Gouabau MC, Courant F, Le Gall G, et al. Offspring metabolomic re-sponse to maternal protein restriction in a rat model of intrauterine growth restriction (IUGR). J Proteome Res 2011;10:3292-302.
American Psychiatric Association. Di-agnostic and Statistical Manual of Mental Disorders, 5th edn. Arlington, VA: Ameri-can Psychiatric Publishing 2013.
Atzori L, Antonucci R, Barberini L, et al. 1H NMR-based metabolic profiling of urine from children with nephrouropathies. Front Biosci (Elite Ed) 2010;2:725-32.
Barberini L, Noto A, Fattuoni C, et al. Uri-nary metabolomics (GC-MS) reveals that low and high birth weight infants share elevated inositol concentrations at birth. J Matern Fetal Neonatal Med 2014;2:20-6.
Barker DJ. Fetal origins of coronary heart disease. BMJ 1995;311:171-4.
Baron-Cohen S, Lombardo MV, Auyeung B, et al. Why are autism spectrum condi-tions more prevalent in males? PLoS Biol 2011;9:e1001081.
Beger RD, Holland RD, Sun J, et al. Me-tabonomics of acute kidney injury in chil-dren after cardiac surgery. Pediatr Nephrol 2008;23:977-84.
Blydt-Hansen TD, Sharma A, Gibson IW, et al. Urinary metabolomics for non-invasive detection of borderline and acute T cell-mediated rejection in children after kidney transplantation. Am J Transplant 2014;14:2339-49.
Centers for Disease Control and Preven-tion (CDC). Prevalence of autism spectrum disorder among children aged 8 years – au-tism and developmental disabilities moni-toring network, 11 sites, United States, 2010. MMWR 2014;63:1-21.
Dani C, Bresci C, Berti E, et al. Metabo-lomic profile of term infants of gestational diabetic mothers. J Matern Fetal Neonatal Med 2014;27:537e42.
De Magistris A, Corbu S, Cesare Flamin-cola F et al. NMR-based metabolomics analysis of urinary changes in neonatal en-terocolitis. JPNIM 2015;4:37-8.
Dessì A, Atzori L, Noto A, et al. Me-tabolomics in newborns with intrauterine growth retardation (IUGR): urine reveals markers of metabolic syndrome. J Matern Fetal Neonatal Med 2011;2:35-9.
** Primo studio sulla metabolomica e il ritardo di crescita intrauterino nel neonato.
Dessì A, Corsello G, Stronati M, et al. New diagnostic possibilities in systemic neonatal infections: metabolomics. Early Hum Dev 2014;1:S19-21.
Dessì A, Marincola FC, Pattumelli MG, et al. Investigation of the ¹H-NMR based urine metabolomic profiles of IUGR, LGA and AGA newborns on the first day of life. J Matern Fetal Neonatal Med 2014;2:13-9.
Dessì A, Murgia A, Agostino R, et al. Ex-ploring the role of different neonatal nutri-tion regimens during the first week of life by urinary GC-MS metabolomics. Int J Mol Sci 2016;17:265.
Dessì A, Pintus R, Marras S, et al. Me-tabolomics in necrotizing enterocolitis: the state of the art. Expert Rev Mol Diagn 2016;22:1-6.
Dessì A, Puddu M, Ottonello G, et al. Metabolomics and fetal-neonatal nutrition: between “not enough” and “too much”. Molecules 2013;18:11724-32.
* Aggiornata review sulla nutrimetabo-lomica feto-neonatale.
Ellis DI, Dunn WB, Griffin JL, et al. Meta-bolic fingerprinting as a diagnostic tool. Pharmacogenomics 2007;8:1243-66.
Fanos V, Barberini L, Antonucci R, et al. Metabolomics in neonatology and pediat-rics. Clin Biochem 2011;44:452-4.
Fanos V, Caboni P, Corsello G, et al. Uri-nary (1)H-NMR and GC-MS metabolomics predicts early and late onset neonatal sep-sis. Early Hum Dev 2014;1:S78-83.
Fanos V, Mussap M, Faa G, et al. The next ten years in neonatology: new directions in research. J Pediatr Neonat Individual Med 2014;3:e030239.
Favretto D, Cosmi E, Ragazzi E, et al. Cord blood metabolomic profiling in in-trauterine growth restriction. Anal Bioanal Chem 2012;402:1109-21.
Garner CE, Ewer AK, Elasouad K. Analy-sis of faecal volatile organic compounds in preterm infants who develop necrotising enterocolitis: a pilot study. J Pediatr Gas-troenterol Nutr 2009;49:559-65.
Hanna MH, Segar JL, Teesch LM, et al. Urinary metabolomic markers of amino-glycoside nephrotoxicity in newborn rats. Pediatr Res 2013;73:585-91.
Horgan RP, Broadhurst DI, Walsh SK, et al. Metabolic profiling uncovers a phe-notypic signature of small for gestational age in early pregnancy. J Proteome Res 2011;10:3660-73.
Jansen JJ, Hoefsloot HC, Boelens HF, et al. Analysis of longitudinal metabolomics data. Bioinformatics 2004;20:2438-46.
Klepacki J, Klawitter J, Klawitter J, et al. A high-performance liquid chromatog-raphy-tandem mass spectrometry-based targeted metabolomics kidney dysfunction marker panel in human urine. Clin Chim Acta 2015;446:43-53.
Leigh JP, Du J. Forecasting the Eco-nomic Burden of Autism in 2015 and 2025 in the United States. J Autism Dev Disord 2015;45:4135-9.

258
A. Dessì et al.
Lin G, Liu C, Feng C, et al. Metabolomic analysis reveals differences in umbilical vein plasma metabolites between normal and growth-restricted fetal pigs during late gestation. J Nutr 2012;142: 990-8.
Logan KM, Wijeyesekera AD, Perez IG, et al. Infants of mothers with diabetes have altered urinary metabolic profile at birth. Available at: www.neonatalsociety.ac.uk/abstracts/logank_2012_diabeticurinary-profile.shtml
Marincola FC, Dessì A, Pattumelli MG, et al. 1H NMR-based urine metabolic pro-file of IUGR, LGA, and AGA newborns in the first week of life. Clin Chim Acta 2015; 451:28-34.
Mickiewicz B, Vogel HJ, Wong HR, et al. Metabolomics as a novel approach for ear-ly diagnosis of pediatric septic shock and its mortality. Am J Respir Crit Care Med 2013;187:967-76.
Mickiewicz B, Thompson GC, Blackwood J, et al. Development of metabolic and in-flammatory mediator biomarker phenotyp-ing for early diagnosis and triage of pediat-ric sepsis. Crit Care 2015;19:320.
Moco S, Collino S, Rezzi S, et al. Metab-olomics perspectives in pediatric research. Pediatr Res 2013;73:570-6.
Morrow AL, Lagomarcino AJ, Schibler KR. Early microbial and metabolomic sig-natures predict later onset of necrotizing enterocolitis in preterm infants. Microbi-ome 2013;1:13.
Mussap M, Noto A, Fanos V. Metabo-lomics of autism spectrum disorders: early insights regarding mammalian-microbial cometabolites. Expert Rev Mol Diagn 2016;16:8.
Nicholson JK, Lindon JC, Holmes E. Metabonomics: understanding the metabolic responses of living systems to pathophysiological stimuli via multi-variate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999;29:1181-9.
Nissen PM, Nebel C, Oksbjerg N, et al. Metabolomics reveals relationship be-tween plasma inositols and birth weight: possible markers for fetal programming of type 2 diabetes. J Biomed Biotechnol 2011;2011:378268.
* Primo lavoro sperimentale di metabo-lomica e ritardo di crescita intrauterino su maialini.
Noto A, Fanos V, Barberini L, et al. The urinary metabolomics profile of an Italian autistic children population and their un-
affected siblings. J Matern Fetal Neonatal Med 2014;2:46-52.
Sanz-Cortés M, Carbajo RJ, Crispi F, et al. Metabolomic profile of umbilical cord blood plasma from early and late intrauter-ine growth restricted (IUGR) neonates with and without signs of brain vasodilation. PLoS One 2013;8:e80121.
Sedic M, Gethings LA, Vissers JP, et al. Label-free mass spectrometric profiling of urinary proteins and metabolites from pae-diatric idiopathic nephrotic syndrome. Bio-chem Biophys Res Commun 2014;452:21-6.
Tarricone R. Cost-of-illness analysis. What room in health economics? Health Policy 2006;77:51-63.
van Vliet E, Eixarch E, Illa M, et al. Me-tabolomics reveals metabolic alterations by intrauterine growth restriction in the fetal rabbit brain. PLoS One 2013;8:e64545.
Vergnano S, Sharland M, Kazembe P, et al. Neonatal sepsis: an international perspective. Arch Dis Child Fetal Neonatal 2005;220-4.
Watkins SM, Hammock BD, Newman JW, et al. Individual metabolism should guide agriculture toward foods for im-proved health and nutrition. Am J Clin Nutr 2001;74:283-6.
Corrispondenza
Vassilios FanosUnità di Terapia Intensiva Neonatale, Sezione di Patologia Neonatale, Azienda Ospedaliero-Universitaria di Cagliari, Policlinico Monserrato, Blocco Q - S.S 554 (bivio Sestu), 09042 Monserrato (CA) - E-mail: [email protected].










![Rene. Frank Zampino - Quebec · 2013. 4. 25. · Frank Zampino[780178].html .Joge conjointe hockey-ville Montréal . chard Deschamp-LaSalle ichel Prescott ank Zampino assimo lezzoni](https://img.pdfslide.net/doc/110x75/60c9f3f7c2c84b0d6e07eb38/rene-frank-zampino-quebec-2013-4-25-frank-zampino780178html-joge-conjointe.jpg)


















