Embed Size (px)
Citation preview

FIND MEANING IN COMPLEXITY
© Copyright 2014 by Pacific Biosciences of California, Inc. All rights reserved.
Steve Picton PhD,
Pacific Bioscience - Europe
What could you do with a 10kb read?: SMRT®
sequencing for de novo bacterial assembly and
methylation analysis.

2
Close to home………………2013

Close to home………………2013
3

4
What, where (from) ……………….and why. 2011

Cost implications…………….
5

• We know <1% of the Earth’s microbiome
• Horizontal gene transfer is wide-spread and frequent
• High-quality, finished genomes are the starting point for:
– Functional genomic studies
– Comparative genomics
– Forensics………………
– Metagenetics
The case for de novo, Finished Microbial Genomes
Chain et al. (2009) Science 326: 236-237 Fraser et al. (2002) J Bacteriology 184: 6403-6405

Finished Genomes to Fight Foodborne Outbreaks
• ~76 million illnesses each year
• ~325,000 hospitalizations
• $78 billion economic loss (US)
• High serotype diversity
• Emerging hypervirulence
• ~76 million illnesses each year
• ~325,000 hospitalizations
• $78 billion economic loss (US)
• High serotype diversity
• Emerging hypervirulence

National Collection of Type Cultures (NCTC)
• Collaboration with Public Health England & the Wellcome Trust
Sanger Institute
• Plan to finish 3000 bacterial genomes
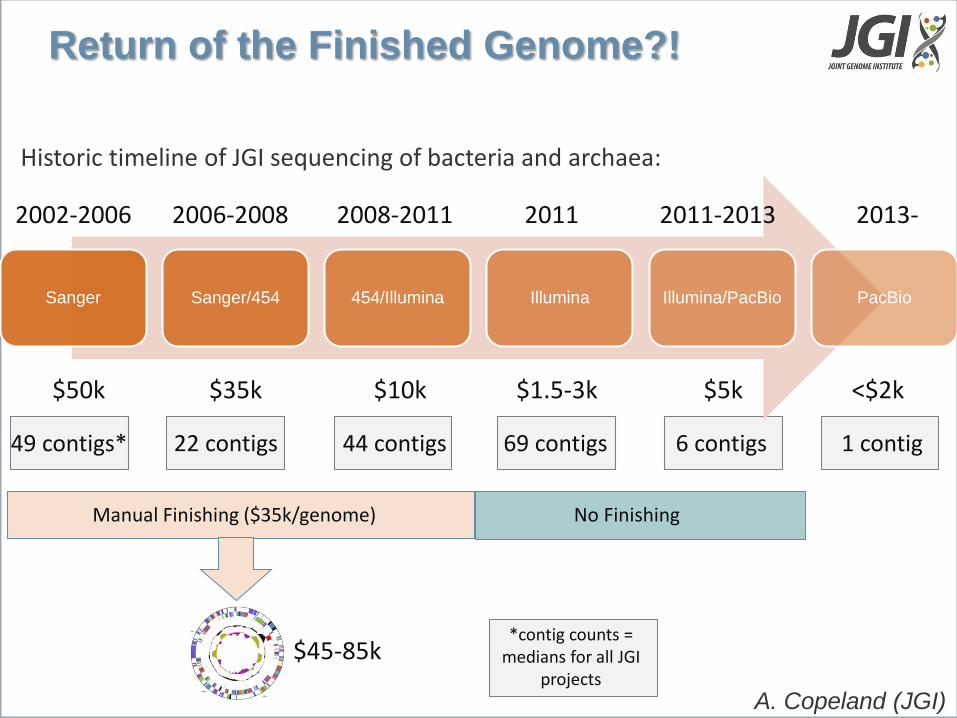
Sanger Sanger/454 454/Illumina Illumina Illumina/PacBio PacBio
2002-2006 2006-2008 2011 2008-2011 2011-2013
Historic timeline of JGI sequencing of bacteria and archaea:
2013-
Return of the Finished Genome?!
$50k $35k $1.5-3k $10k $5k <$2k
No Finishing
49 contigs* 44 contigs 69 contigs 22 contigs 6 contigs 1 contig
*contig counts = medians for all JGI
projects
Manual Finishing ($35k/genome)
$45-85k
A. Copeland (JGI)

FIND MEANING IN COMPLEXITY
© Copyright 2014 by Pacific Biosciences of California, Inc. All rights reserved.
SMRT® technology - Overview

Single Molecule, Real-Time (SMRT®) DNA Sequencing
PacBio® RS II Trace
SMRT® Cells Zero-Mode
Waveguides
Phospholinked
Nucleotides

PacBio® Advances in Read Length
12
Early PacBio chemistries
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
453 1012 1734 LPR
FCR ECR2
C2–C2
P4–C2
Read L
ength
(bp)
2008 2009 2010 2011 2012 2013 2014 2015
P5–C3

Long Read Lengths needed to Resolve Repeats
• E. coli:
>1 kb
>2 kb
>5 kb
Repeats
>99% identical
with length: 5.4 kb repeat
Repeat instance 1:
Repeat instance 2:

Fully Resolving the Legionella
pneumophila rtxA Locus • D4174:
• D4174: 13.8 kb
• LR1342: 17.8 kb
Collaboration with A. Ensminger (U Toronto & PHO) & K. Dewar (McGill)
• LR1342:
25 repeats; 14kb region
550bp
12
10
14
18kb region

Resolving the Legionella pneumophila
rtxA Locus • D4174:
Collaboration with A. Ensminger (U Toronto & PHO) & K. Dewar (McGill)
• A single 23 kb PacBio read:
• A single 300bp Illumina read:

The Pertussis Genome is Extremely Repetitive
• Bordetella pertussis: • E.coli:
>1 kb
>2 kb
>5 kb
Repeats
>99% identical
with length:
>8% repeats >500bp
First sequenced 2002, Parkhill

Genome Organization Comparison – Presented AGBT 2013
Collaboration with A. Zeddeman, H. van der Heide, M. Bart & F. Mooi
National Institute for Public Health and the Environment (RIVM), Netherlands
1917
3582
1920
3585
3640
3405
3658
3913
3921
2011, CS
2003, Tohama

Organization of Virulence Genes Differs Between Strains
Collaboration with A. Zeddeman, H. van der Heide, M. Bart & F. Mooi
National Institute for Public Health and the Environment (RIVM), Netherlands

Tracking hospital associated
bacteria with epidemiology and
genomic sequencing
Julie Segre, PhD
Senior Investigator
National Human Genome Research Institute
Presented at the 60th NIH Clinical Center Anniversary, November 6th, 2013

De novo assembly of plasmids from draft genomes is difficult
poorly aligned contig
repeat regions
indel
ind
el
reference sequence
Fold Coverage Alignment to Reference
Fragmented and incomplete information, plasmids unresolved

Targeted sequencing could have given misleading result
Identical
Unrelated
pt #1 pt #53
*
Full genome sequencing rules out patient-patient transmission
Klebsiella pneumoniae Klebsiella pneumoniae pKpQIL
KPC-2
KPC-3

Horizontal gene transfer from patient to environment
Klebsiella pneumoniae Enterobacter cloacae Citrobacter freundii
KPC-2
KPC-3

Salmonella De Novo Assemblies
Strain Genome size Additional genomic elements
S. Bareilly (SAL2881) 4,730,611 bp 78,193 bp
S. Heidelberg (318_04) 4,793,478 bp 117,929 bp; 35,296 bp; 3969 bp
S. Heidelberg (2069) 4,783,941 bp 110,345 bp; 37,704 bp
S. Typhimurium (2048) 4,967,892 bp 142,804 bp; 48,532 bp
S. Javiana (1992_73) 4,629,444 bp 24,013 bp; 17,094 bp
S. Cubana (2050) 4,977,480 bp 166,668 bp; 122,863 bp
S. St.Paul SP3 4,730,130 bp none
S. St.Paul SP48 4,940,224 bp 44,606 bp; 40,801 bp
Collaboration with M. Allard, E. Brown, E. Strain, M. Hoffman, T. Muruvanda,
S. Musser (FDA), R. Roberts (NEB), B. Weimer (UC Davis)
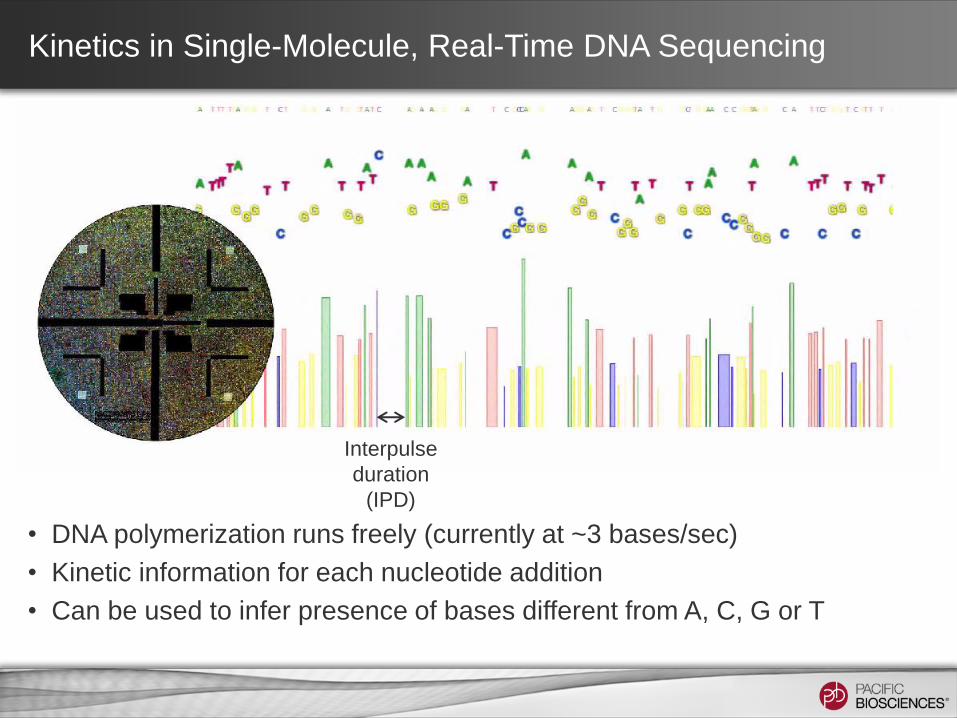
• DNA polymerization runs freely (currently at ~3 bases/sec)
• Kinetic information for each nucleotide addition
• Can be used to infer presence of bases different from A, C, G or T
Kinetics in Single-Molecule, Real-Time DNA Sequencing
Interpulse
duration
(IPD)

Epigenome Information

Comprehensive Understanding of Pathogens

The E. coli Outbreak Strain Methylome
E. Schadt (Mt. Sinai), M. Waldor (Harvard) & Rich Roberts (NEB)

Lambda-like phage element specific to
outbreak strain
• Contains stxAB
• Contains putative methyltransferase
and restriction enzyme for CTGCAG
motif 55989
C227-11
C734-09
C35-10
C682-09
C760-09
C754-09
C777-09
Novel sequence motif: CTGCm6AG

Effects of Horizontal Gene Transfer on Pathogenicity
M.EcoGIII

Summary -
32
• ‘Whole genome sequencing’ does not always deliver whole genome assembly
• High accuracy , long contiguous reads enable
• Complete genome assembly
• Identification of complex genome rearrangements
• Precise location of phage insertions
• Identity of extra chromosomal elements
• Kinetic data enables identification of methylation events
• Strain specific methylation may provide addition identifiers

Thank you

34

35





























