Embed Size (px)
Citation preview

What You Should Know When You Make Manufacturing Changes to Biotechnology Products
May 16-18, 2011 | Beijing, China
Mark Rosolowsky, Ph.D.Vice President,
Global Regulatory Sciences-CMCBristol-Myers Squibb

Disclaimers
– The information within this presentation represents the views of the presenter and is based on the presenter’s expertise and experience
2

3
Overview
– Comparability - key points for consideration
– Discuss changes to biotechnology products:• During development• Post-marketing

4
Why are Changes to Biotechnology Products so Complex?
Small Molecules– Usually synthetic, organic
compounds having well defined structures and chemical characteristics
– Typically produced through chemical synthesis
– Usually micromolecules having molecular weights of less than 500 Daltons
– Generally very stable, and not extremely sensitive to heat
Biotechnology Products– Usually a protein- or
carbohydrate-based product with complex structure
– Either composed of / or extracted from a living organism or produced via cell culture
– Macromolecular by nature, and usually have a molecular weight greater than 500 Daltons
– Tend to be rather labile, and are usually very heat- and sheer-sensitive
– Tend to be immunogenic

What is Comparability?
• ICHQ5E Definitions• Comparable:
– A conclusion that products have highly similar quality attributes before and after manufacturing process changes and that no adverse impact on the safety or efficacy, including immunogenicity, of the drug product occurred. This conclusion can be based on an analysis of product quality attributes. In some cases, nonclinical or clinical data might contribute to the conclusion.
• Comparability Exercise: – The activities, including study design, conduct of studies,
and evaluation of data, that are designed to investigate whether the products are comparable.
5

Comparability Exercise Considerations
– No one right answer when deciding a comparability package
– Decision must be made with consideration of:• Product clinical development plan • Complexity of the product • Stage of development of the product • Robustness of analytical methods • Existence of relevant animal models • Previous health authority interactions
6

What is “Highly Similar”?
– Comparability does not require quality attributes of pre-change and post-change product to be identical
– “Highly similar” depends upon whether:• Existing knowledge can adequately support that
differences in quality attributes have no adverse impact upon safety or efficacy
– Side-by-side analysis of “post-change” vs. “pre-change” product is useful for contemporaneous evaluation
7

8
Comparability is a Sequential Process
Non-clinicalNon-clinical Clinical and/or Pharmacovigilance
Clinical and/or PharmacovigilanceQualityQuality
Confirmatory clinical testing may be necessaryConfirmatory clinical testing may be necessary
Non-clinical studies may be necessary, if non-clinical studies cannot discern relevant differences, then…
Non-clinical studies may be necessary, if non-clinical studies cannot discern relevant differences, then…
If the analytical procedures used are not sufficient to discern relevant differences, then…
If the analytical procedures used are not sufficient to discern relevant differences, then…
OVERALL GOAL: Assess potential impact to safety and efficacy of the product

Parameters to Consider
– Production step where changes are introduced.– Potential impact of changes to: purity , physicochemical and
biological properties • considering complexity and degree of knowledge (e.g., impurities,
product related substances).
– Availability of suitable analytical techniques to detect potential modifications
– Understanding of relationship between quality attributes and safety and efficacy, based on overall nonclinical and clinical experience.
– Relevant physicochemical and biological characterization data regarding quality attributes;
– Need for stability data, including accelerated or stress conditions, • to provide insight into potential product differences in the degradation
pathways
9

Parameters to Consider (continued)
– Batches used for demonstration of manufacturing consistency;– Historical data that provide insight into potential “drift” of quality
attributes– Critical control points in the manufacturing process that affect
product characteristics, – Impact of the process change on the quality of in-process materials
& ability of downstream steps to accommodate material from a changed cell culture process;
– Adequacy of the in-process controls (critical control points & in-process testing:
• In-process controls for post-change process should be confirmed, modified, or created, as appropriate, to maintain product quality
– Nonclinical or clinical characteristics of the drug product and its therapeutic indications
10

Risk Assessment
– Utilize prior knowledge and development studies– Categorize risk change of process parameters
based upon potential to impact product quality– Examples:
• High: Change to Master Cell Bank (MCB)• Moderate: Media composition change using
established raw materials, <50% output • Low: Step optimization (e.g. flow rate, wash volumes,
elution collection criteria)
– Provides an effective tool for internal discussion of change & subsequent communications to regulators regarding the assessment
11

Risk Assessment Model
Phase of clinical program
Ch
ang
e T
ype
Low risk
High risk
High risk
Red = Stop & ReconsiderHigh likelihood toImpact program
Yellow = Proceed with CautionModerate likelihood to impact program
Green = GoUnlikely to impactprogram

Example Risk Assessment Tool
13
Each risk factor category has built-in drop-down boxes with potential values
Higher values indicate greater risk Team discussion is critical to document thought process that
drove the scores (“no one right answer”)
Total risk score generated by multiplying individual risk factors

Evaluating Changes During Development
14

Phase-based Approach to Changes
Early Development:– Before nonclinical studies:
• Comparability is not generally not a concern• Subsequent nonclinical and clinical studies using the
post-change product as part of the development process support change
– Early phases of nonclinical and clinical studies:• Comparability testing is generally not as extensive as
for an approved product– As knowledge and information accumulate, the
comparability exercise will generally become more comprehensive

Phase-based Approach to Changes
• During Pivotal Clinical Studies:– Changes are discouraged– Sponsor should seek scientific advice from the relevant
health authorities• If process changes are introduced in late (post-
pivotal studies) stages of development:– Thorough comparability exercise is generally required:
• Physicochemical and biological in vitro studies • Clinical pharmacokinetic and / or pharmacodynamic
comparability studies may also be required– If comparability exercise cannot rule out impact to the
efficacy and safety profile:• Additional clinical studies may be required
16

Guidelines for Acceptance Criteria Setting for Analytical Comparability
Pre Pivotal Post Pivotal
Release Test Development Release Specification 3SD from small number of lots
Development or Commercial Release Specification 3SD from larger number of lotsCQA - closer to manufacture history
Extended Characterization a
Reported as found but summarized by expert analyst as being comparable or not comparable
Quantified acceptance criteria where possible. Visual equivalence should have defined criteria
the analyst uses to make the determination.
Stability Same requirements depending on whether the test is a release test or extended characterization.
Same requirements depending on whether the test is a release test or extended characterization.b
Stability CQA stress changes demonstrate comparable rate changes c
a Orthogonal support for release test or independent attribute assessment for structure-function relationshipb If a stability indicating attribute is identified under recommended storage conditionsc If a forced degradation or stressed condition degradation product attribute is identified and believed relevant to structure-function

Case Study - Development
18

Case Study Process Changes
Overview of Changes– Drug Substance
• New MCB (higher producing subclone of current MCB)• New DS manufacturing site (Site “A” Site “B”)• New DS manufacturing process (cell culture and purification)
– Drug Product• New DP manufacturing site (Site “X” Site “Y”)• Minor change to sterile filtration (0.1 µm0.2 µm)
Reason for Changes– Increase drug substance yield ~4X– Manufacturing control– COG

Comparability Exercise
Goal - to ensure the quality, safety and efficacy of drug product produced by a changed manufacturing process
How - through collection and evaluation of the relevant data based on process and product knowledge– Analytical assays– Biological assays– Nonclinical data– Clinical data

Analytical Comparability
Established at multiple points– In-process– Release
• Release tests• Extended characterization tests
– Stability profile• Recommended storage condition• Accelerated/Stress storage conditions
– Downstream• Drug substance changes may only be seen in the drug product
(release, stability profile)

Analytical Acceptance Criteria
In-process– Comparable process/product related impurity/adventitious
agent clearance Release tests
– Current (pre-change) specification• Additional tests may be needed
Extended characterization– May need to evaluate additional pre-change batches to
establish appropriate acceptance criteria
Evaluation against historical data (i.e. clinical experience)Side-by-side comparison of pre- and post-change product
by various analytical characterization techniques

Binding Kinetics by Surface Plasmon Resonance
-10
0
10
20
30
40
50
60
70
80
0 100 200 300 400 500 600 700 800
Tim e s
Re
sp. D
iff.
RU
Association
Dissociation

Electron Spray Ionization Mass Spectrometry
Reference material (pre-change)
Post-change product
Slight increase in one subtype observed

Tryptic Peptide MapsA
U
0.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
0.20
0.22
0.24
0.26
Minutes
34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00 50.00 52.00 54.00 56.00 58.00 60.00 62.00
H1
- 4
1.5
37
pG
lu-H
1 -
52
.05
8
Overlay of pre- and post-change materials
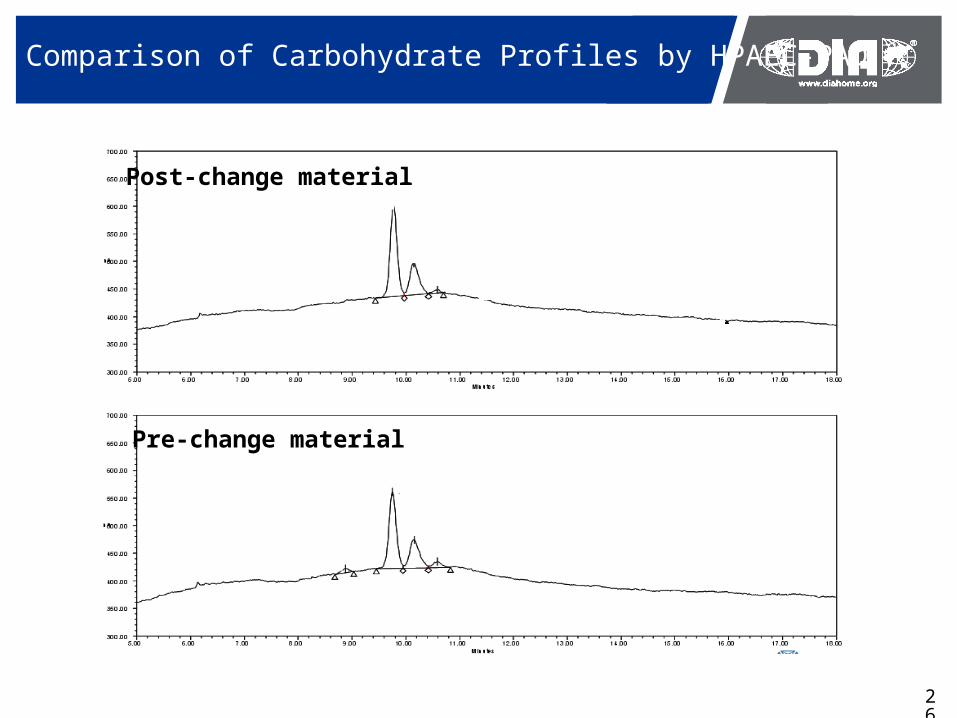
Comparison of Carbohydrate Profiles by HPAEC-PAD
26
Pre-change material
Post-change material

Isoelectrically Focused
Lane 1: Reference material (pre-change)Lanes 2 through 5: Post-change material

Cation Exchange Profiles
Gray line: Pre-change materialBlack line: Post-change material

Evaluating Changes for Marketed Products
29

Changes to Marketed Products
• Changes during life-cycle are inevitable:– Yield increases necessary to meet market
demand / address cost of goods issues– Quality improvements are necessary to
adhere to current GMPs– Unexpected events require corrective
action, such as process parameter changes– Vendor / supply issues necessitate use of
alternate materials
30

Hypothetical Process Evolution
Process Step
Phase II Phase III & Commercial
Process C Process D Process E
Cell Bank XYZ-01 XYZ-01.1 Same Same Same
Media DE CD-CHO, eRDF & Yeastolate
Same, Plus Additional Minerals
Same Same
Production Conditions
No temp. shift
Two phase temp. shift
Same Same Single temp. shift
Downstream Sequence
Six columns
Five columns
Same Change of one resin
Change of one resin
Other Process Changes
N/A N/A Centrifugation & filtration parameters
Filter changes N/A

Case Study – Marketed Product
32

Case Study: Change in Media
– Quality: Media component change resulted in minor differences to quality attributes
– Nonclinical: Study conducted using non-human primate model with previously well-established concordance to human PK for the product• Model had demonstrated sensitivity to
changes in:– Minor glycosylation alterations– Moderate sialic acid profile shifts
33

Non-Human Primate Results
Red line = pre-changeBlue line = post-change
Time in Hours
Ser
um
Co
nce
ntr
atio
n [
ug
/mL
]

FDA Feedback
– Quality attributes evaluated against historic data alone deemed insufficient• “…the Agency strongly encourages the use of
side-by-side analysis as the most rigorous assessment of comparability…”
– Side-by-side comparison required:• “Since the product approved for marketing
authorization was made using the…media..., this product should be directly compared to the product produced from the proposed process…”

Regulatory Experiences with Comparability
– Comparability exercises can be successfully used to support changes
– Most changes can be supported on Quality attributes alone
• Specifications alone are generally NOT sufficient to support comparability
– Additional characterization required– Side-by-side analysis preferred by FDA
– Additional non-clinical & clinical data may be required
– Safety & efficacy data are generally not required
36

In Summary
– Comparability is a key issue for biotech products– There is no single, correct strategy to demonstrate
comparability– Decision must be made with consideration of multiple factors:
• Complexity of the product • Stage of development of the product • Process knowledge & robustness of analytical methods • Existence of relevant animal models • Previous health authority interactions
– Comparability should be approached stepwise:• Quality• Non-clinical• Clinical
37

Acknowledgements
Cheryl Watson Reb RussellDave Peck
Charlene CraigGary Lazarus
38





























