Embed Size (px)
Citation preview

- 1 2 3
WHO/BS/2011.2157 4
ENGLISH ONLY 5 6 7
EXPERT COMMITTEE ON BIOLOGICAL STANDARDIZATION 8
Geneva, 17 to 21 October 2011 9
10
11
Recommendations to Assure the Quality, Safety and Efficacy of BCG Vaccines 12
13
14
Proposed replacement of: TRS 745, Annex 2 and 15
Amendment to TRS 771, Annex 12 16
17 18
This document has been prepared for the purpose of inviting comments and suggestions on the 19 proposals contained therein, which will then be considered by the Expert Committee on Biological 20 Standardization (ECBS). The text in its present form does not necessarily represent an agreed 21 formulation of the Expert Committee. Comments proposing modifications to this text MUST 22 be received by 23 September 2011 and should be addressed to the World Health Organization, 23 1211 Geneva 27, Switzerland, attention: Quality Safety and Standards (QSS). Comments may also 24 be submitted electronically to the Responsible Officer: Dr HyeNa Kang at email: [email protected]. 25 26 The outcome of the deliberations of the Expert Committee will be published in the WHO 27 Technical Report Series. The final agreed formulation of the document will be edited to be in 28 conformity with the "WHO style guide" (WHO/IMD/PUB/04.1). 29
30
© World Health Organization 2011 31 All rights reserved. Publications of the World Health Organization are available on the WHO web site (www.who.int) or 32 can be purchased from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 33 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). 34 Requests for permission to reproduce or translate WHO publications – whether for sale or for noncommercial distribution 35 – should be addressed to WHO Press through the WHO web site 36 (http://www.who.int/about/licensing/copyright_form/en/index.html). 37
The designations employed and the presentation of the material in this publication do not imply the expression of any 38 opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city 39 or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent 40 approximate border lines for which there may not yet be full agreement. 41

WHO/BS/2011.2157
Page 2
42 The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or 43 recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors 44 and omissions excepted, the names of proprietary products are distinguished by initial capital letters. 45 46 All reasonable precautions have been taken by the World Health Organization to verify the information contained in this 47 publication. However, the published material is being distributed without warranty of any kind, either expressed or 48 implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the 49 The named authors/editors alone are responsible for the views expressed in this publication. 50

WHO/BS/2011.2157
Page 3
Recommendations published by the WHO are intended to be scientific and advisory. Each of the following sections constitutes guidance for national regulatory authorities (NRAs) and for manufacturers of biological products. If a NRA so desires, these Recommendations may be adopted as definitive national requirements, or modifications may be justified and made by the NRA. It is recommended that modifications to these Recommendations be made only on condition that modifications ensure that the vaccine is at least as safe and efficacious as that prepared in accordance with the recommendations set out below. The parts of each section printed in small type are comments for additional guidance intended for manufacturers and NRAs, which may benefit from those details.
51
Table of contents 52 53 Introduction ........................................................................................................................5 54 General considerations ...................................................................................................................5 55 Special considerations ..................................................................................................................6 56 57 Part A. Manufacturing recommendations ..............................................................................11 58 A.1 Definitions .................................................................................................................................11 59 A.2 General manufacturing requirements .................................................................................13 60 A.3 Control of source materials ...................................................................................................15 61 A.4 Control of vaccine production ...............................................................................................18 62 A.5 Filling and containers .............................................................................................................20 63 A.6 Control tests on final lot ................................................................................................20 64 A.7 Records ....................................................................................................................................24 65 A.8 Retained samples ...................................................................................................................24 66 A.9 Labeling ....................................................................................................................................25 67 A.10 Distribution and transport ......................................................................................................25 68 A.11 Stability, storage and expiry date ........................................................................................26 69
70 Part B. Preclinical evaluation of BCG vaccines ....................................................................28 71
72 Part C. Clinical evaluation of BCG vaccines .........................................................................30 73 C.1 General considerations ............................................................................................................30 74 C.2 Special considerations.............................................................................................................32 75 C.3 Post-marketing surveillance ...................................................................................................34 76
77 Part D. Recommendations for national regulatory authorities .........................................36 78 D.1 General .....................................................................................................................36 79 D.2 Release and certification .......................................................................................................37 80
81 Authors and Acknowledgements .............................................................................................39 82 83 References ......................................................................................................................................42 84
85 Appendix 1 .....................................................................................................................................49 86 History and genealogy of BCG sub-strains 87 Appendix 2 ......................................................................................................................................50 88

WHO/BS/2011.2157
Page 4
Model summary protocol for manufacturing and control of BCG vaccine 89 Appendix 3 ......................................................................................................................................59 90 Model certificate for the release of BCG vaccine by national regulatory authorities 91

WHO/BS/2011.2157
Page 5
Introduction 92
The last revision of the requirements for dried bacillus Calmette Guérin (BCG) vaccine for human 93
use was in 1985, and an amendment which updated the section on the expiry date was published in 94
1988 (1, 2). Recent WHO consultation meetings (3, 4, 5, 6) have addressed the issues on 95
improvement of vaccine characterization and quality control assays of BCG vaccine to reflect 96
current state-of-the-art technology. In addition, a recommendation to replace the international 97
reference preparation for BCG vaccine by sub-strain specific reference reagents evaluated by 98
collaborative studies has been proposed. This guideline provides recommendations for the 99
production and control of BCG vaccines in Part A, for preclinical evaluation in Part B, and for the 100
content of the clinical development program applicable to BCG vaccines in Part C. The term of 101
'preclinical' evaluation applies for classical BCG vaccine products still in need of such evaluation, 102
including the newly manufactured products requiring clinical trial studies. The clinical part of this 103
document intends to provide a basis for assessment of efficacy and safety of BCG vaccines in pre-104
licensing clinical trials as well as in post-marketing surveillance, monitoring consistency of 105
production and clinical testing of new classical BCG vaccine products. If important changes have 106
been introduced to an authorized production process, the need for preclinical and clinical testing 107
should be considered on a case-by-case basis in consultation with the NRA(s) concerned. 108
109
General considerations 110
Tuberculosis (TB) was declared a global emergency by the WHO in 1993, and Mycobacterium 111
tuberculosis (M. tuberculosis) is now considered to be responsible for more adult deaths than any 112
other pathogens. Vaccination with BCG still remains the standard for TB prevention in most 113
countries because of its efficacy in preventing life-threatening forms of TB in infants and young 114
children. It is inexpensive and usually requires only one administration in either newborn or 115
adolescents (7, 8). As there is currently no suitable alternative, BCG will remain in use in the 116
foreseeable future and may continue to be used as a prime vaccine in a Prime-Boost immunization 117
schedule in conjunction with new TB vaccines (4). 118
119
BCG vaccine is a live attenuated vaccine originated from culturing M. bovis isolated from cattle and 120
cultured for a period of 13 years and a total of 231 passages (7). The BCG vaccine was first used to 121
immunize humans in 1921. Following its introduction into the WHO Expanded Programme on 122

WHO/BS/2011.2157
Page 6
Immunization (EPI) in 1974, the vaccine soon reached global coverage rates exceeding 80% in 123
countries endemic for TB (9). 124
125
Over the years, different BCG vaccine seed strains have evolved from the original vaccine strain for 126
production. A number of BCG vaccine strains that are used worldwide differ in terms of their 127
genetic and phenotypic properties, and their reactogenicity and immunogenicity profile when given 128
to infants and children. With this background of a diversity of sub-strains, manufacturing processes, 129
immunization schedules and levels of exposure to environmental mycobacteria and virulent M. 130
tuberculosis infection, different levels of protective efficacy of BCG vaccines in adult populations 131
have been reported (10). However, the data are insufficient to make recommendations on whether 132
one strain should be preferred over the other (11). The United Nations agencies are the largest 133
supplier of BCG vaccines, distributing more than 120 million doses each year to more than 100 134
countries. Worldwide, the most commonly used vaccine strains are currently Danish 1331, Tokyo 135
172-1 and Russian BCG-I because they are supplied by United Nations Children’s Fund (UNICEF) 136
who purchases the vaccines through a published prequalification process which determines their 137
eligibility for use in national immunization programmes (12). 138
139
There has been particular concern over the safety of BCG vaccination in human immunodeficiency 140
virus (HIV)-infected subjects (8). WHO had previously recommended that in countries with a high 141
burden of TB, a single administration of BCG vaccine should be given to all healthy infants as soon 142
as possible after birth, unless the child presented a symptomatic HIV infection (9). However, recent 143
evidence shows that children who were HIV-infected when vaccinated with BCG at birth, and who 144
later developed AIDS, were at increased risk of developing disseminated BCG disease. Among 145
these children, the benefits of potentially preventing severe TB are outweighed by the risks 146
associated with the use of BCG vaccine; and the use of BCG vaccines at birth should follow the 147
recommendations from WHO Strategic Advisory Group of Experts (SAGE) on immunization and 148
position papers (13, 14). 149
150
Special considerations 151
The formulation of international requirements for freeze-dried BCG vaccine is complicated by the 152
following: (a) a number of different sub-strains derived from the original strain of BCG are used in 153
vaccine manufacture; (b) a number of different manufacturing and testing procedures are employed; 154

WHO/BS/2011.2157
Page 7
(c) difficulties of translation from significant differences in vitro and in vivo between different BCG 155
vaccine strains to any possible differences in protective efficacy against TB in humans; (d) vaccines 156
with different total bacterial content and number of culturable particles are produced; and (e) 157
vaccines intended for administration by different routes are prepared. 158
159
Scope of the Recommendations 160
These revised recommendations refer to freeze-dried BCG vaccines prepared from sub- strains 161
derived from original BCG for use in the prevention of TB. Where BCG vaccine is issued in liquid 162
form, the application of these recommendations is entirely under the responsibility of the national 163
regulatory authority (NRA). In that case, only the relevant parts of these requirements apply because 164
limited stability of liquid BCG limits the possibility of completing the entire recommended control 165
test schedule. Although many of the principles expressed in this document (e.g. manufacturing, 166
quality control) are expected to apply also to new recombinant BCG and other live attenuated 167
mycobacterial vaccines modified by molecular biology techniques, these novel vaccines are outside 168
the scope of this guideline. The same pertains to the use of BCG for immunotherapy (e.g. treatment 169
of bladder cancer). However, applicability of issues on preclinical and clinical evaluations should be 170
considered on a case-by-case basis. These recommendations have been formulated primarily to 171
cover vaccines intended for intradermal and percutaneous administration. Although WHO 172
recommends intradermal administration of the vaccine, preferably in the deltoid region of the arm 173
using syringe and needle, other administration methods such as percutaneous application by the 174
multiple puncture technique are practiced in some countries (9, 15, 16, 17). 175
176
BCG vaccine strains 177
The original BCG vaccine strain was formerly distributed by the Pasteur Institute of Paris and sub-178
cultured in different countries using different culture conditions which were not standardized. Over 179
the years, more than 14 sub-strains of BCG have evolved and been used as BCG vaccine strains in 180
different parts of the world (see Appendix 1). Recently, the various sub-strains have been studied by 181
comparative genomics (18, 19). BCG vaccine strains were thus divided into the “early” strains, in 182
which the original characteristics of 'authentic Pasteur' were conserved with less deletions, insertions 183
and mutation in the genome of the bacilli than the “late” strains. Such strains are represented by 184
BCGs Russia BCG-I, Moreau-RJ, Tokyo 172-1, Sweden, and Birkhaug; and the “late” strains, such 185
as BCGs Pasteur 1173P2, Danish 1331, Glaxo (Copenhagen 1077) and Prague. The genomic 186

WHO/BS/2011.2157
Page 8
sequences of BCG Pasteur 1173P2 as a “late” strain and BCG Tokyo 172-1 as an “early” strain were 187
determined in 2007 and 2009, respectively (18, 19). There is insufficient direct evidence to suggest 188
that various BCG sub-strains differ significantly in their efficacy to protect against TB in humans. 189
However, evidence from animal and human studies indicates differences in the immune responses 190
induced by different BCG vaccine strains (12). Although the “early” strains may confer better 191
protection against TB in some animal studies (18, 20), commonly administered BCG vaccine strains 192
including both evolutionary “early” and “late” strains induce comparable protective immunity 193
against TB (21). 194
195
Only master seed lots that have been shown to be acceptable by laboratory and clinical tests on 196
batches derived from them should be used for production of working seed lots and/ or final product. 197
A suitable seed lot of BCG should yield vaccines that give protection in experimental animals, 198
produce a relatively high level of immunological responses to M. tuberculosis antigens including 199
tuberculin sensitivity in humans, and have an acceptably low frequency of adverse reactions (see 200
A3.1). 201
202
Some manufacturers of freeze-dried BCG vaccine have modified their master seed lot strain to make 203
it more suitable for their particular production procedure. The seed lots prepared in this way may not 204
retain the same immunogenic properties, and should be used only with the approval of the NRA. 205
206
In practice, a product prepared from BCG seed lots may generally be investigated in humans only 207
for their properties of producing tuberculin sensitivity and vaccination lesions. The former should be 208
measured by the distribution of tuberculin reactions according to size in persons vaccinated with a 209
given dose of BCG vaccine. A low dose of tuberculin should be employed (e.g. equivalent to 5 IU of 210
the 1st International Standard for Purified Protein Derivative (PPD) of M. tuberculosis, or 2 211
tuberculin units (TU) of a batch of PPD RT23 with Tween 80). 212
213
Currently three sub-strains specific Reference Reagents for BCG vaccines are available and they are 214
the BCG Danish 1331, Tokyo 172-1 and Russian BCG-I. 215
216
Potency-related tests 217

WHO/BS/2011.2157
Page 9
There is some evidence that BCG seed lots that have been shown to produce vaccines with 218
protective potency in laboratory animals and tuberculin sensitivity in humans will give effective 219
protection against TB in humans. It should be noted that tuberculin sensitivity is a marker for cell-220
mediated immune responses to mycobacteria and not a direct indicator of protective immunity. A 221
number of alternative laboratory tests have been developed primarily for research purposes, but to 222
date, none have been proven to be reliable indicators of protective immune-conversion following 223
administration of different vaccines. 224
225
Field observations should be made in conjunction with laboratory studies in animals. The latter 226
should include protection tests, tests of vaccination lesions, and tests for tuberculin conversion. 227
Immunizing efficacy should be measured in terms of degree of protection afforded to the test 228
animals against a challenge with virulent M. tuberculosis. Sensitizing efficacy should be measured 229
by the average dose of vaccine that will convert a negative tuberculin reaction in guinea-pigs to a 230
positive one, as well as by the reaction time that such conversion is effected. In these animal tests, 231
the inclusion, for comparative purposes, of an in-house reference BCG vaccine prepared from a seed 232
lot known to be effective in animals and humans is recommended. 233
234
As currently there is no biomarker, which directly correlates to clinical efficacy of BCG vaccine, the 235
laboratory tests at present in use and included in these requirements are designed to ensure that new 236
vaccine lots do not differ appreciably from those that have already been shown to be safe and 237
effective in humans as regards their ability to induce adequate sensitivity to tuberculin, or from an 238
in-house reference vaccine prepared from a seed lot shown to be safe and effective in humans. At 239
present, for batch control purposes, much reliance is placed on tests for the estimation of the total 240
bacterial content and for the number of culturable particles. It is not possible to specify single 241
requirements for the total bacterial content and for the number of culturable particles for all vaccines 242
(22), since different sub-strains and methods of manufacture may yield different specifications for 243
these parameters. For example, although the number of culturable bacteria in a single human dose 244
may differ for different vaccines, these vaccines may show satisfactory properties as regards their 245
ability to induce adequate sensitivity to tuberculin and their safety in humans. It is therefore 246
essential that clinical studies for dose optimisation in humans be carried out to estimate suitable total 247
bacterial contents and the number of culturable particles for a particular manufacturer’s product. For 248
a particular vaccine, the difference between the lower and upper specification for the number of 249

WHO/BS/2011.2157
Page 10
culturable particles should not be larger than 4-fold. In addition, it is necessary to perform animal 250
experiments that give an indication of the safety and efficacy of the vaccines to the satisfaction of 251
the NRA. 252
253

WHO/BS/2011.2157
Page 11
Part A. Manufacturing recommendations 254
255
A.1 Definitions 256
A.1.1 International name and proper name 257
The international name should be "Freeze-dried BCG vaccine". The proper name should be the 258
equivalent of the international name in the language of the country of origin. The use of the 259
international name should be limited to vaccines that satisfy the recommendations formulated below. 260
261
A.1.2 Descriptive definition 262
Freeze-dried BCG vaccine is a freeze-dried preparation containing live bacteria derived from a 263
culture of the bacillus of Calmette and Guérin, known as BCG, intended for intradermal injection. 264
The name of the freeze-dried vaccine intended for percutaneous vaccination, should be “Freeze-265
dried BCG vaccine, Percutaneous”. The preparation should satisfy all the recommendations 266
formulated below. 267
268
A.1.3 International reference preparation/ reagents 269
The 1st International Reference Preparation for BCG vaccine was established in 1965 and the 1st 270
International Standard for PPD of M. tuberculosis, in 1951. Because of the age of these preparations, 271
the need for replacements has been recognized, especially for the International Reference 272
Preparation for BCG vaccine which is a live bacterial preparation. WHO has initiated the 273
development of replacement for the BCG reference preparation. These have been presented to the 274
ECBS in 2009 and 2010 as candidates for the 1st WHO Reference Reagents for BCG vaccines of 275
sub-strain Danish 1331, Tokyo 172-1 and Russian BCG-I (23). These reference reagents cover the 276
major proportion of BCG vaccine strains currently used in production. The establishment of sub-277
strain Moreau-RJ as the WHO Reference Reagent for BCG vaccine is currently in progress and 278
scheduled to submit to the ECBS in 2012 for adoption. These preparations are intended as reference 279
reagents if required for: 280
− periodical consistency monitoring of quantitative assays such as viability estimates (such as 281
culturable particle count and modified ATP assays); 282
− residual virulence/ local reactogenicity assays and protection assays in animal models for 283
nonclinical evaluation; and/ or 284

WHO/BS/2011.2157
Page 12
− as reference BCG sub-strains for identity tests using multiplex PCR as included in the 285
collaborative study or in other molecular biology techniques. 286
The NIBSC-HPA, Potters Bar, UK distributes the WHO Reference Reagents for BCG vaccines. 287
288
A.1.4 Terminology (alphabetical order) 289
The definitions given below apply to the terms as used in these recommendations. They may have 290
different meanings in other contexts. 291
292
Final bulk: The homogeneous finished liquid vaccine present in a single container from which the 293
final containers are filled, either directly or through one or more intermediate containers derived 294
from the initial single container. 295
Final lot: A number of sealed, final containers that are equivalent with respect to the risk of 296
contamination during filling and, when it is performed, freeze-drying. A final lot should therefore 297
have been filled from a single container and freeze-dried in one continuous working session. 298
In-house reference: A batch of vaccine prepared from the same BCG strain as the tested vaccine 299
and used in parallel to the vaccine tested in: 300
− quantitative assays such as viability estimates (such as culturable particle count and modified 301
ATP assays); and 302
− residual virulence assays. 303
Master seed lot: A bacterial suspension of a single sub-strain originated from the bacillus of 304
Calmette and Guérin that has been processed as a single lot and is of uniform composition. A seed 305
lot should be maintained in the freeze-dried form stored at -20°C or below (in the liquid form stored 306
at -80°C or below) in order to maintain viability. In each manufacturing establishment, a master 307
seed lot is that from which material is drawn for inoculating media for the preparation of working 308
seed lots or single harvests. 309
Single harvests: The material obtained from one batch of cultures that have been inoculated with the 310
working seed lot (or with the inoculum derived from it), harvested and processed together. Single 311
harvests should be prepared from cultures originating from a seed lot by as few cultural passages as 312
possible, and by not more than 12 passages from the master seed lot. 313
Working seed lot: A quantity of bacterial organisms of a single sub-strain derived from the master 314
seed lot by growing the organisms and maintaining them in aliquots in the freeze-dried form stored 315
at -20°C or below (in the liquid form stored at -80°C or below). The working seed lot should be 316

WHO/BS/2011.2157
Page 13
prepared from the master seed lot by as few cultural passages as possible, e.g. 3-6 passages from the 317
master seed lot, having the same characteristics as the master seed lot and intended for inoculating 318
media for the preparation of single harvests. 319
320
A.2 General manufacturing recommendations 321
The general manufacturing recommendations for manufacturing establishments contained in the 322
Good Manufacturing Practices for Pharmaceuticals Products: main principles (24) and the Good 323
Manufacturing Practices for Biological Products (25) should apply to establishments manufacturing 324
BCG vaccine. Also, the compliance with current good manufacturing practices should apply with 325
the addition of the following: 326
327
Details of standard operating procedures for the preparation and testing of BCG vaccines adopted by 328
the manufacturer together with evidence of appropriate validation of each production step should be 329
submitted for the approval of the NRA. As may be required, proposals for the modification of 330
manufacturing and control methods should also be submitted for approval to the NRA before they 331
are implemented. 332
333
The NRA should satisfy itself that adequate control of the manufacturing, shipping, and storage of 334
the BCG vaccine has been achieved. NRAs may consider that a formal clinical lot-to-lot consistency 335
study is not necessary if there are adequate and satisfactory data provided to support consistency of 336
manufacture. However, several different lots of the product should be used in randomized studies 337
and should elicit comparable immune responses in similar populations. 338
339 The degree of consistency in producing satisfactory final lots is an important 340 factor in judging the efficacy and safety of a particular manufacturer's product. 341
342
The data that should be considered in determining the consistency of production should include the 343
results obtained with consecutive vaccine lots when tested as described in Part A, section 6, for 344
example, the test for viability (Part A, section 6.7), and thermal stability test (Part A, section 6.8). 345
346
More than two consecutive vaccine lots should have been satisfactorily prepared before any vaccine 347
from a given manufacturer, or resulting from a new method of manufacture, is released. In 348
subsequent routine production, if a specified proportion of vaccine lots or a specified number of 349

WHO/BS/2011.2157
Page 14
consecutive vaccine lots fails to meet the requirements, the manufacture of BCG vaccine should be 350
discontinued and not be resumed until a thorough investigation has been made and the cause or 351
causes of the failures determined to the satisfaction of the NRA. 352
353
Conventionally, production of BCG vaccine should take place in dedicated area, completely 354
separate from areas used for production of other medicines or vaccines, and using dedicated separate 355
equipment. Such areas should be so situated and ventilated that the hazard of contamination is 356
reduced to a minimum. No animals should be permitted in the vaccine production areas. Tests for 357
the control of vaccine that require cultures to be made of contaminating microorganisms should be 358
carried out in a completely separate area. Tests in which animals are used should also be carried out 359
in a completely separate area. 360
For the purposes of these requirements, the processes of vaccine production that should take place in 361
dedicated facilities are all operations up to and including the sealing of the vaccine in the final 362
containers. 363
364 In some countries, the production of BCG vaccine - although isolated - is carried 365 out in a building in which other work takes place. This should be done only after 366 consultation with, and with the approval of, the NRA. If production takes place 367 in part of a building, the work carried on in other parts of the building should be 368 of such a nature that there is no possibility of cross-contamination to the BCG 369 vaccine. 370
371
No cultures of microorganisms other than the BCG vaccine strain approved by the NRA for vaccine 372
production should be introduced into the manufacturing areas. In particular, no strains of other 373
mycobacterial species, whether pathogenic or not, should be permitted in the BCG vaccine 374
production area. 375
376
BCG is susceptible to sunlight. Therefore, the procedures for the preparation of the vaccine should 377
be so designed that all cultures and vaccines are protected from direct sunlight and ultraviolet light 378
at all stages of manufacture, testing, and storage, until the vaccine is issued. 379
380
BCG vaccine should be produced by a staff consisting of healthy persons who do not work with 381
other infectious agents; in particular, they should not work with virulent strains of M. tuberculosis, 382
nor should they be exposed to a known risk of tuberculosis infection. Precautions should be taken 383
also to ensure that no worker should be employed in the preparation of BCG vaccine unless he or 384

WHO/BS/2011.2157
Page 15
she has been shown by medical examination to be free from TB. The scope and nature of the 385
medical examination should be at the discretion of the NRA, but it should include a radiological 386
examination and should be repeated at intervals or when there is reason to suspect illness. 387
388 The frequency with which the radiological examination should be carried out is 389 at the discretion of the NRA. It is advisable to keep radiation exposure to a 390 minimum, but the examination should be of sufficient frequency to detect the 391 appearance of early active TB. It is estimated that, if workers in BCG vaccine 392 laboratories were given one or two conventional X-ray examinations of the chest 393 each year, not using fluoroscopic methods, and if the best available techniques 394 were employed to minimize the radiation dose, the doses received would be 395 considerably lower than the maximum permissible doses for workers 396 occupationally exposed to radiation that have been set by the International 397 Commission on Radiological Protection (26, 27). 398
399
Should an examination reveal signs of TB or suspected TB in a worker, he or she should no longer 400
be allowed to work in the production areas and the rest of the staff should be examined for possible 401
TB infection. In addition, all cultures should be discarded and the production areas decontaminated. 402
If it is confirmed that the worker has TB, all vaccine made while he or she was in the production 403
areas should be discarded. In addition, distributed batches should be recalled. 404
405
Persons not normally employed in the production areas should be excluded from them unless, after a 406
medical examination, including radiological examination, they are shown to be free from TB. In 407
particular, persons working with mycobacteria other than the BCG seed strain should be excluded at 408
all times. 409
410
Written descriptions of procedures for the preparation of BCG vaccine should be submitted for 411
approval to the NRA. Proposals for modification should be submitted for approval to the NRA 412
before their implementation. 413
414
A.3 Control of source materials 415
A.3.1 Seed lot system 416
The production of vaccine should be based on the seed lot system. A seed lot prepared from a strain 417
approved by the NRA (see Part D, section 1.1) should be prepared under conditions satisfying the 418
requirements of Part A, sections 2, 3 and 4. 419
420

WHO/BS/2011.2157
Page 16
The BCG vaccine strain used should be identified by historical records that include information on 421
its origin and subsequent manipulation. It would be preferable for the master seed lot to have 422
protection proven clinically through clinical studies with a batch derived from it by a production 423
process that is representative of the commercial process; also it is recommended to use a batch 424
derived from such a clinically ‘validated’ seed lot as in-house reference in the laboratory to help 425
ensure consistency in production. 426
427
If a working seed lot is being used, the total number of passages for a single production harvest 428
should not exceed 12 including the passages necessary for preparing the working seed lot. 429
430
Clinically relevant antimicrobial sensitivity testing should be carried out as a part of the ongoing 431
characterization of BCG sub-strains. It would be appropriate to test this property at the level of both 432
master and working seed lots for licensing purposes and to monitor this in final lot. 433
434
A.3.2 Tests on seed lot 435
When a new working seed lot is established, a suitable test for delayed hypersensitivity in guinea-436
pigs is carried out; the vaccine is show to be not significantly different in activity from the in-house 437
reference. 438
439
A.3.2.1. Identity test 440
The bacteria in the master and working seed lots are identified as M. bovis BCG using 441
microbiological techniques, for example morphological appearance of the bacilli in stained smears 442
and by the characteristic appearance of the colonies grown on solid media. Molecular biology 443
techniques, for example PCR test can supplement to identify the specific sub-strain of BCG. The 444
techniques will also ensure genetic consistency in production, from master seed through working 445
seed and to final product (4). 446
447
A.3.2.2. Test for bacterial and fungal contamination 448
Each master and working seed lot should be tested for bacterial and fungal contamination by 449
appropriate tests as specified in Part A, section 5.2 (28) of the General Requirements for the 450
Sterility of Biological Substances, or by the validated methods approved by the NRA. 451
452

WHO/BS/2011.2157
Page 17
A.3.2.3 Test for absence of virulent mycobacteria 453
The test for absence of virulent mycobacteria, described in Part A, section 4.2.3, should be made in 454
at least ten healthy guinea-pigs injected with a quantity of vaccine not less than 50 single human 455
doses and should be observed for at least 6 weeks. If none of the animals shows signs of progressive 456
TB and at least 90% survive (i.e. should 1 out of 10 animals dies) the observation period, the seed 457
lot should be considered to be free from virulent mycobacteria . 458
459
Should more than 10% of the guinea-pigs die (i.e. should 2 out of 10 animals die) during the 460
observation period and freedom from progressive TB disease is verified, the test should be repeated 461
on at least 10 more guinea-pigs . On the second occasion, the seed lot passes the test if not more 462
than 10% animal die (i.e. should 1 of 10 animals dies) during the observation period and autopsy 463
does not reveal any sign of TB . 464
465
A.3.2.4. Test for excessive dermal reactivity 466
Use 6 healthy guinea-pigs, each weighing not less than 250g and having received no treatment likely 467
to interfere with the test. Inject intradermally into each guinea-pig, according to a randomized plan, 468
0.1 ml of the reconstituted vaccine and of vaccine dilutions 1:10 and 1:100. The same dilutions of 469
the appropriate international reference reagent or in-house reference should be injected into the same 470
guinea-pigs at randomly selected sites. Observe the lesions formed at the sites of injection for at 471
least 4 weeks. The vaccine complies with the test if the reactions it produces are not markedly 472
different from that produced by the appropriate international reference reagent or in-house reference. 473
474
A.3.3 Production culture medium 475
The production culture medium should contain no substances known to cause toxic or allergic 476
reactions in humans. The use of material originated from animals should be discouraged. However, 477
if constituents derived from animal origin are necessary, approval of the NRA should be sought and 478
the materials should comply with current Transmissible Spongiform Encephalopathies (TSE) policy 479
(29, 30, 31, 32, 33, 34). A risk assessment for TSE would need to be included for the materials of 480
culture medium. The revised WHO Guidelines on TSE in relation to biological and pharmaceutical 481
products (29) provide guidance on risk assessments for master and working seeds and should be 482
consulted. Substances used in that medium should meet such specifications as the NRA may 483
prescribe. 484

WHO/BS/2011.2157
Page 18
485
A.4 Control of vaccine production 486
A.4.1 Control of single harvests 487
All cultures should be examined visually, and any that have grown in an uncharacteristic manner 488
should not be used for vaccine production. 489
490
A.4.2 Control of final bulk 491
A.4.2.1 Final bulk 492
The final bulk should be prepared from a single harvest or by pooling a number of single harvests. 493
494
A.4.2.2 Test for bacterial and fungal contamination 495
The final bulk should be tested for bacterial and fungal contamination by appropriate tests as 496
specified in Part A, section 5.2 (28) of the General Requirements for the Sterility of Biological 497
Substances, or by the validated methods approved by the NRA. No vaccine lot should be passed for 498
use unless the final bulk has been shown to be free from such contamination. 499
500
A.4.2.3 Test for absence of virulent mycobacteria 501
The test for absence of virulent mycobacteria should be carried out on each final bulk or final lot. 502
503
At least 6 healthy guinea-pigs, all of the same sex, each weighing 250 - 400 g are used. They have 504
not received any treatment or diet, such as antibiotics, likely to interfere with the test. A sample of 505
the final bulk intended for this test should be stored at 4°C for not more than 72 hours after harvest. 506
507
A dose of BCG organisms corresponding to at least 50 single human doses of vaccine intended for 508
intradermal injection should be injected into each guinea-pig by the subcutaneous or intramuscular 509
route.1 The guinea-pigs should be observed for at least 6 weeks. If, during that time, they remain 510
healthy, gain weight, show no signs of progressive TB and not more than one die, the final bulk 511
should be considered to be free from virulent mycobacteria . 512
513
1 When a more concentrated vaccine, intended for administration by the percutaneous route, is tested, a dilution factor approved by the NRA should be applied so that the mass of BCG injected corresponds to at least 50 human doses of intradermal vaccine.

WHO/BS/2011.2157
Page 19
At the end of the observation period, the animals should be sacrificed and examined post-mortem 514
for macroscopic evidence of progressive TB disease; similarly, any animals that die before the end 515
of the observation period should be subjected to a post-mortem examination. 516
Should one-third of the guinea-pigs die (i.e. should 2 out of 6 animals die) during the observation 517
period (and freedom from progressive TB disease is verified), the test should be repeated on at least 518
6 more guinea-pigs. 519
On the second occasion, the vaccine lot passes the test if not more than one animal dies during the 520
observation period and autopsy does not reveal any sign of TB. 521
522
Should a vaccine lot fail to satisfy the requirements of this test because animals 523 die from causes other than TB, the procedure to be followed by the manufacturer 524 should be determined with the approval of the NRA. 525
526
If signs of TB disease are seen, the vaccine lot should be rejected, all subsequent vaccine lots should 527
be withheld, and all current vaccine stocks should be held pending further investigation. The 528
manufacture of BCG vaccine should be discontinued and it should not be resumed until a thorough 529
investigation has been made and the cause or causes of the failure determined and appropriate 530
actions have been taken. Production should be allowed to resume only upon the approval of the 531
NRA. 532
533
A.4.2.4 Test for bacterial concentration 534
The bacterial concentration of the final bulk should be estimated by a validated method approved by 535
the NRA and should have a value within a range approved by the NRA (see Part D, section 1.2). 536
Based on manufacturers' experience, the opacity method is the method of choice, 537 the International Reference Preparation of Opacity,1 or an equivalent reference 538 preparation approved by the NRA, may be employed in comparative tests. 539
540
A.4.2.5 Test for number of culturable particles 541
The number of culturable particles on a solid medium of each final bulk should be determined by an 542
appropriate method approved by the NRA. Alternatively, a bioluminescense or other biochemical 543
method can be used (35, 36), provided that the method is properly validated against the culturable 544
particle test, for the production step in question. If properly validated, such tests can be used as 545
1The International Reference Preparation of Opacity is in the custody of the National Institute for Biological Standards and Control, Health Protection Agency, Potters Bar, Hertfordshire, England, which supplies samples on request.

WHO/BS/2011.2157
Page 20
equivalent methods. Regular calibration with the reference method as agreed with NRA would be 546
relevant. 547
548 The medium used in this test should be such that the number of culturable 549 particles may be determined at an optimal time point (usually 3-5 weeks) after 550 the medium has been inoculated with dilutions of the vaccine. 551 552 There are various methods of determining the number of culturable particles in 553 BCG vaccine, and it is essential that only one culture method be used for all the 554 vaccine lots produced by a manufacturer (5). It is also desirable for assay 555 validation that the test be carried out in parallel with the appropriate international 556 reference reagent or in-house reference, e.g. the same vaccine that has been used 557 in clinical trials and assured safety (including immunogenicity) and efficacy. 558
559
A.4.2.6 Substances added to the final bulk 560
Substances used in preparing the final bulk should meet such specifications as the NRA may 561
prescribe. In particular, the NRA should approve the source(s) of any animal-derived raw materials 562
that should comply with the guidelines on tissue infectivity distribution of TSEs (30). 563
564
Substances added to improve the efficiency of the freeze-drying process or to aid the stability of the 565
freeze-dried product should be sterile and of high and consistent quality, and should be used at 566
suitable concentrations in the vaccine. 567
568
A.5 Filling and containers 569
The general requirements concerning filling and containers given in Good Manufacturing Practices 570
for Biological Products (25) should apply to vaccine filled in the final form. 571
572 The containers should be in a form that renders the process of reconstitution as 573 simple as possible. Their packaging should be such that the reconstituted 574 vaccine is protected from direct sunlight. 575
576
A.6 Control tests on final lot 577
Tests on the final lot should be performed after reconstitution, except for appearance and residual 578
moisture tests. The fluid supplied or recommended for reconstitution should be used, unless such 579
fluid would interfere with any of the tests, in which case some other suitable fluid should be used. 580
The vaccine should be reconstituted to the concentration at which it is to be used for injection into 581
humans; an exception may be made in the case of the test for absence of virulent mycobacteria (Part 582
A, section 6.4.1), when a higher concentration of reconstituted vaccine may be necessary. 583

WHO/BS/2011.2157
Page 21
584
A.6.1 Inspection of final containers 585
Every container in each final lot should be inspected visually, and those showing abnormalities 586
should be discarded. 587
588
The appearance of the freeze-dried vaccine and the reconstituted vaccine should be described with 589
respect to its form and colour. If reconstitution with the product diluent does not allow for the 590
detection of particulates, an alternative diluent may be used. 591
592
A.6.2 Identity test 593
An identity test should be performed on samples of the vaccine from each final lot. The identity test 594
for final lots should be used to identify the product as BCG as approved by NRA. The identity of 595
each final lot of vaccine should be verified by the morphological appearance of the bacilli in stained 596
smears and by the characteristic appearance of the colonies grown on solid media. Preferably a 597
validated nucleic acid amplification technique (such as PCR) should be used and the morphological 598
technique. 599
600
A.6.3 Test for bacterial and fungal contamination 601
Samples from each final lot should be tested for bacterial and fungal contamination by appropriate 602
tests as specified in Part A, section 5.2 (28) of the General Requirements for the Sterility of 603
Biological Substances, or by the validated methods approved by the NRA. 604
605
A.6.4 Safety tests 606
A.6.4.1 Test for absence of virulent mycobacteria 607
Provided the test for virulent mycobacteria has been carried out with satisfactory results on the final 608
bulk vaccine, it may be omitted on the final lot. 609
610
If the test for the absence of virulent mycobacteria, applied to the final bulk, is unsatisfactory (and 611
freedom from progressive TB disease is verified), it should be repeated with a sample of a final lot 612
(see Part A, section 4.2.3). 613
614
A.6.4.2 Test for excessive dermal reactivity 615

WHO/BS/2011.2157
Page 22
Provided the test has been carried out with satisfactory results on the working seed lot and on 5 616
consecutive final lots produced from it, the test may be omitted on the final lot. 617
618
A.6.5 Test for bacterial concentration 619
The total bacterial content of the reconstituted vaccine should be estimated for each vaccine lot by a 620
validated method approved by the NRA, and should have a value within a range approved by the 621
NRA (see Part D. section 1.2). 622
623 The estimation of tota1 bacterial content may be made either directly, by 624 determining the dry weight of organisms, or indirectly, by an opacity method 625 that has been calibrated in relation to the dry weight of the organisms. 626 627 It is desirable that one method of estimation should be adhered to for all the 628 vaccine lots produced by a manufacturer. 629
630
A.6.6 Test for residual moisture 631
The average moisture content of a freeze-dried vaccine should be determined by a validated method 632
accepted by the NRA. Values should be within limits of the preparations shown to be adequately 633
stable in the stability studies of the vaccine. 634
635
A.6.7 Tests for viability 636
A.6.7.1 Test for number of culturable particles 637
The number of culturable particles of each final lot should be determined by an appropriate method 638
approved by the NRA (see Part A, section 4.2.5). The viable count should have a value within a 639
range approved by the NRA that should not be wider than a 4-fold difference between the lower and 640
upper levels of the specification for numbers of culturable particles (see Part D, section 1.2). By 641
comparison with the results of the test for number of culturable particles carried out on final bulk, as 642
described in Part A. section 4.2.5, the percentage survival on freeze-drying may be calculated and 643
this value should be not less than one approved by the NRA. The appropriate international reference 644
reagent or in-house reference should be used for every test in order to validate the assay. 645
646 The purpose of including the appropriate international reference reagent or in-647 house reference is to have a check on the quality and consistency of the culture 648 medium and the accuracy of the technique used for the determination of the 649 number of culturable particles. It is not intended to adjust the count of the 650 vaccine by comparison with the reference preparation. 651 652 The survival rate after freeze-drying is usually not less than 20%. 653

WHO/BS/2011.2157
Page 23
654
A.6.7.2 Rapid test for viability 655
As an alternative to the colony counting method, a bioluminescense or other biochemical method 656
can be used, provided that the method is properly validated against the culturable particle test, for 657
the production step in question. If properly validated, such tests may be considered by the NRA to 658
replace the culturable particle test. 659
660 The bioluminescence reaction occurring in fireflies depends upon the presence 661 of adenosine triphosphate (ATP), luciferin luciferase, oxygen, and magnesium 662 ions. This reaction can be reproduced in vitro by mixing these components. If all 663 components except ATP are present in excess, the amount of light emitted is 664 proportional to the amount of ATP coming from the vaccine. 665 666 Since ATP is present in all living cells and is immediately destroyed when the 667 cell dies, ATP is a reliable marker for living cells. 668 669 Studies, have shown that if properly validated, measurement of ATP using the 670 bioluminescence reaction can be used to estimate the viable count of freeze-671 dried BCG vaccine within 1- 2 days, as accurately as other, more time-672 consuming methods, once the mean content of ATP per culturable particle has 673 been estimated for a given vaccine production. 674
675
A.6.8 Thermal stability test 676
The thermal stability test is as part of characterization and consistency demonstration of the vaccine 677
production. This requirement of this test should be at discretion of NRA and if required, each final 678
lot should be tested for thermal stability by a validated method approved by the NRA. If the 679
production consistency is demonstrated, this test may be omitted on the final lot and subjected to 680
NRA approval (6). 681
682
If performed, the test should involve the determination of the number of culturable particles before 683
and after the samples have been held at appropriate temperatures and for appropriate periods. 684
685
For example, the thermal stability test may be carried out by taking samples of the 686 vaccine and incubating them at 37°C for 28 days. 687
688
The percentage decrease in the number of culturable particles is then compared with that of samples 689
of the same vaccine lot stored at 2° - 8°C. The number of culturable particles in the vaccine after 690
heating should be not less than 20% of that stored at 2° - 8°C (37). The absolute value should be 691
approved by the NRA. The viability test should also be performed with the appropriate international 692
reference reagent or in-house reference for checking validity of the assay. One method of 693

WHO/BS/2011.2157
Page 24
determining the number of culturable particles should be adhered to, as suggested in Part A, section 694
4.2.5. 695
696 The purpose of including the appropriate international reference reagent or in-697 house reference is to have a check on the quality and consistency of the medium 698 used for the determination of the number of culturable particles. It is not 699 intended to adjust the count of the vaccine by comparison with the reference 700 preparation. 701
702
All manufacturers should keep their product for the approved storage period and should determine 703
the number of culturable particles from time to time to demonstrate that the number is being 704
maintained at an adequate level. 705
706 In some countries, the thermal stability test is carried out only after the vaccine 707 has been stored for 3-4 weeks after freeze-drying, since it is considered that the 708 degree of stability during the first 3 weeks may not be related to the long-term 709 stability of the product. 710 711 As a guide to stability, some manufacturers of freeze-dried BCG vaccine 712 determine the residual moisture content of the final vaccine, since failure to 713 achieve a certain degree of desiccation results in an unstable product. However, 714 such a test cannot be regarded as an alternative to tests involving the 715 determination of the number of culturable particles. 716
717
A.7 Records 718
The recommendations in Section 8 of Good Manufacturing Practices for Biological Products 719
should apply (25) 720
721
Written records should be kept of all seed lots, all cultures intended for vaccine production, all 722
single harvests, all final bulk vaccines, and all vaccine in the final containers produced by the 723
manufacturing establishments, including all tests irrespective of their results. 724
725
The records should be of a type approved by the NRA. An example of a suitable protocol is given in 726
Appendix 2. 727
728
A.8 Retained samples 729
The recommendations in Section 9.5 of Good Manufacturing Practices for Biological Products 730
should apply (25). 731
732

WHO/BS/2011.2157
Page 25
It is desirable that samples should be retained for at least one year after the 733 expiry date for the final lot. 734
735
A.9 Labeling 736
The recommendations in Section 7 of Good Manufacturing Practices for Biological Products (25) 737
should apply including the following. 738
739
The label, and/ or the packaging insert in some countries, printed on or affixed to each container 740
should show the volume and nature of the reconstituting fluid. Also, this label, or the label on the 741
carton enclosing several final containers, or the leaflet accompanying the containers, should contain 742
the following additional information: 743
- the fact that the vaccine fulfils the requirements of this document; 744
- instructions for use of the vaccine and information concerning contraindications and the 745
reactions that may follow vaccination; 746
- the conditions recommended during storage and transport, with information on the reduced 747
stability of the vaccine if exposed to temperatures higher than that stated on the label; 748
- warnings that the vaccine should be protected from direct sunlight; 749
- a statement that, after a final container of freeze-dried BCG has been reconstituted, the vaccine 750
should be kept on ice or otherwise refrigerated until used, should be used as soon as possible, 751
and that any reconstituted container remaining at the end of the immunization session 752
(maximum six hours) should be discarded (38), and 753
- information on clinically relevant antimicrobial sensitivity. 754
755
The label for the reconstituting fluid should state 'Reconstituting fluid for BCG vaccine Proprietary 756
name'. 757
758
A.10 Distribution and transport 759
The recommendations given in Section 8 of Good Manufacturing Practices for Biological Products 760
(25) should apply. Also, the document for Safe Vaccine Handling, Cold Chain and Immunizations 761
(39) should apply. Further guidance is provided in the WHO Model Guidance for the Storage and 762
Transport of Time and Temperature–sensitive Pharmaceutical Products (40). 763
764 Diluent used in reconstitution should be shipped and distributed together with 765 the vaccine in immediate container, i.e. vial or ampoules (41). This ensures that 766

WHO/BS/2011.2157
Page 26
the correct diluent will be used for the vaccine. The freeze-dried vaccine is not 767 damaged by freezing and can be frozen and thawed. However, repeated freeze-768 thawing is not recommended. The diluent should never be frozen. 769 770
A.11 Stability, storage and expiry date 771
A.11.1 Stability testing 772
Adequate stability studies form an essential part of vaccine development. Current guidance on 773
evaluation of vaccine stability is provided in the recommendations given in WHO guidelines on 774
stability evaluation of vaccines should be applied (42). Stability testing should be performed at 775
different stages of production if stored for a given time period, namely as appropriate on single 776
harvests or pool of single harvests, final bulk, final lot. In addition, such studies should be 777
undertaken on reconstituted vaccine. Stability-indicating parameters should be defined or selected 778
appropriately according to the stage of production. It is advisable to assign a storage period to all in-779
process materials during vaccine production, in particular intermediates such as single harvests and 780
final bulk; and a shelf-life period to the final lots. 781
782
BCG vaccines require special precautions to ensure sufficient stability. In this connection the most 783
important measures are lyophilization, the use of an effective stabilizer, and proper sealing of 784
vaccine containers. 785
786 Historically the use of ampoules sealed under vacuum was the most common 787 practice for increasing stability. However, vacuum-sealing is difficult 788 compared to sealing in the presence of inert gas. There were no significant 789 differences between BCG vaccines sealed under vacuum and under nitrogen 790 or carbon dioxide at either 4° or 37°C (19). Manufacturers now prepare BCG 791 vaccines in vials/ ampoules, and under well-validated conditions, the product 792 is adequately stable. 793
794
A.11.2 Storage conditions 795
The Guideline for Establishing or Improving Primary and Intermediate Vaccine Stores (41) should 796
apply. 797
798
Storage conditions should be based on stability studies and approved by the NRA. Before being 799
distributed by the manufacturing establishment, or before being issued from a depot for the storage 800
of vaccine, all vaccines in their final containers should be stored constantly at 2°-8°C (37, 44) and 801
vaccine diluents should be stored as recommended by manufacturer. Freeze-dried BCG vaccines, 802

WHO/BS/2011.2157
Page 27
regardless of their sub-strain, are sensitive to ultraviolet and fluorescent light. They should be 803
protected from direct sunlight (37). 804
805 BCG vaccines are sensitive to light as well as to heat. Normally, these vaccines 806 are supplied in vials/ ampoules made from dark brown glass, which gives them 807 some protection against light damage, but care should still be taken to keep them 808 covered and protected from strong light at all times (44). 809 810 Freeze-dried BCG vaccines may be kept frozen at -15°C to -25°C if cold chain 811 space permits, but this is neither essential nor recommended (37). 812 813 Precautions should also be taken to maintain the vaccine, during transport and 814 up to the time of use, at the temperature and under the storage conditions 815 recommended by the manufacturer. 816
817
A.11.3 Expiry date 818
The expiry date should be approved by the NRA and based on the stability of the final product as 819
well as the results of the stability tests referred to in section 11.1. It is established for each batch by 820
adding the shelf-life period to the date of manufacture. Most freeze-dried BCG vaccines are stable at 821
temperatures of 2°-8°C for at least two years (37) from the date of manufacture. The storage of final 822
product at -20°C to extend the shelf-life should be validated. 823
824 Freeze-dried BCG vaccines become much more heat sensitive after they have 825 been reconstituted with diluent. Reconstituted BCG vaccine is very unstable and 826 at risk of contamination (37, 39, 44). Once reconstituted, BCG vaccine should 827 be stored on ice or at 2°-8°C and use within 6 hours (4, 41). 828

WHO/BS/2011.2157
Page 28
Part B. Preclinical evaluation of BCG vaccines 829
830
Details on the design, conduct, analysis and evaluation of preclinical studies are available in WHO 831
Guidelines for Nonclinical Evaluation of Vaccines (45). 832
833
Preclinical testing of a new strain (i.e. derived by selection from existing BCG strains in Appendix 1) 834
or for a new manufacturer of a BCG vaccine is a prerequisite for initiation of clinical studies in 835
humans, and includes immunogenicity, protection studies (proof of concept) and safety testing in 836
animals. The vaccine lots used in preclinical studies should be adequately representative of the 837
formulation intended for clinical investigation and, ideally, should be the same current Good 838
Manufacturing Practice (cGMP) manufactured lots used in clinical studies. If this is not feasible, 839
then the lots used clinically should be comparable to those used in the preclinical studies with 840
respect to potency, stability and other characteristics of quality, often the technical manufacturing 841
consistency lots may be used for these purposes. 842
843
New manufacturers of BCG vaccine for human use will need to refer to the range of preclinical 844
safety and characterisation tests that are recommended for existing, licensed BCG vaccines. 845
Although there is currently no requirement for additional preclinical testing beyond that already 846
described for licensed BCG vaccines, the development of new variants of BCG, the potential for 847
new fermentation technologies and the possibility of novel live vaccines against TB have shown that 848
additional preclinical studies beyond that required of licensed BCG vaccine can be helpful in 849
demonstrating that a new BCG product has satisfactory preclinical efficacy, safety and stability. 850
851 Guideline example on protective potency testing: Hartley Guinea-pigs are used for 852 potency testing. Guinea-pigs are vaccinated with a small amount of BCG (~103 853 CFU). Eight weeks after the vaccination, guinea-pigs are challenged with virulent 854 M. tuberculosis H37Rv (ATCC 27294) by the pulmonary route with a low dose (10 855 – 15 CFU) per animal. Five weeks after the infection, guinea pigs are euthanized, 856 the spleen and the lung lobes are removed. Then these organs are homogenized 857 separately. Appropriate dilutions are inoculated onto duplicate solid medium and 858 incubated at 37°C for 3 weeks. The number of M. tuberculosis H37Rv colonies is 859 counted, and expressed as mean log10 CFU per tissue. The CFU results are compared 860 between the vaccinated and non-vaccinated groups (46). 861
862
If there are two pharmacologically relevant species for the clinical candidate (one rodent and one 863
non-rodent), then both species should be used for short-term (up to 1 month duration) toxicology 864
studies. If the toxicological findings from these studies are similar in both species, then longer-term 865

WHO/BS/2011.2157
Page 29
studies in one species are usually considered sufficient; the rodent species should be considered 866
unless there is a rationale for using non-rodents. Studies in two nonrodent species are not 867
appropriate. Other in vivo studies should address both potency (such as tuberculin sensitivity and 868
immunological tests) and safety (such as tests for excessive dermal reactivity and absence of 869
virulent mycobacteria) issues of the classical BCG vaccines. 870
871
It may be of benefit for new BCG vaccine developers to consider the points raised in the recent 872
meetings establishing recommendations for new live vaccines against TB (47, 48). 873

WHO/BS/2011.2157
Page 30
Part C. Clinical evaluation of BCG vaccines 874
875
Clinical trials should adhere to the principles described in the WHO Guidelines for Good Clinical 876
Practice (GCP) for Trials on Pharmaceutical Products (49) and the general principles described in 877
the WHO Guidelines on Clinical Evaluation of Vaccines: Regulatory Expectations (50). All clinical 878
trials should be approved by the relevant NRAs and local Ethics Committees. Continued licence of 879
BCG vaccines should be viewed in the light of on-going post-marketing data on the safety, 880
immunogenicity and effectiveness of BCG vaccines in the target population. 881
882
The section considers the provision of clinical data required a) when a new candidate "classical" 883
BCG vaccine derived from (the same master seed of) one of the strains recognized (see Appendix 1) 884
is developed; b) when there have been major changes to the manufacturing process of an established 885
vaccine, including preparation of new master seed lot of an established strain; c) when technology 886
transfer of existing vaccine is planned to a new manufacturer; and d) when revalidation of existing 887
vaccines used in national immunization program is considered. 888
889
Vaccines manufactured using a "new strain (i.e. derived by selection from existing BCG strains in 890
Appendix 1)" should require a full clinical development program that provides evidence of safety, 891
efficacy, and the reactogenicity profile in all age target age groups. 892
893
Other vaccines against M tuberculosis derived from M. bovis or other mycobacterial strains cannot 894
be considered as “BCG” and would require a full clinical development program and are not included 895
here. 896
897
C.1 General considerations 898
C.1.1 Comparative or Placebo-controlled clinical trials 899
It would not be considered ethical to conduct a placebo-controlled trial of protective efficacy of a 900
BCG vaccine in a TB endemic area, particularly in infants. A comparative trial with a licensed, or 901
internationally accepted (WHO pre-qualified) BCG vaccine could be accepted. 902
903
C.1.2 Value of PPD response 904

WHO/BS/2011.2157
Page 31
It is recognized that the response to PPD is not an indicator of a protective immune response. 905
Nonetheless this has been used for over 50 years to indicate a cellular immune response to an 906
infection with M. tuberculosis or as evidence of “successful” BCG vaccination. At best a PPD 907
reaction is an indicator of exposure to antigens of TB, and the generation of a cellular immune 908
response. Thus, it can be used in a PPD naïve population as an indicator of an immune response to 909
the BCG vaccine (51). Other immunological measures may be more closely related to M. 910
tuberculosis infection or vaccination, but currently none has been agreed as a correlate of protection 911
from infection or disease. 912
913
C.1.3 BCG in HIV-infected infants 914
A very important safety consideration with regard to vaccination policy is establishing, during 915
clinical trials, the potential for disseminated BCG disease in immunocompromised children that may 916
be more pronounced. The use of BCG vaccines at birth should follow the recommendations from 917
WHO Strategic Advisory Group of Experts (SAGE) on immunization and position papers (13, 14). 918
These consider the policies for immunization exclusion for infants known to be infected with HIV, 919
infants symptomatic for HIV infection, and those infants, born to mothers known to be HIV infected, 920
and who may be infected. 921
922
C.1.4 Post-vaccination reactions and complications 923
Vaccines intended for intradermal or percutaneous injection should be given strictly intradermally or 924
percutaneously, and vaccinators should be trained accordingly. Incorrect vaccination technique can 925
result in adverse reactions, including discharging ulcers, abscesses and keloid scars. 926
927
Current BCG vaccines have a known reactogenicity profile after intradermal inoculation (52). Local 928
reaction at the vaccination site is normal after a BCG vaccination. It may take the form of a nodule 929
that, in many cases, will break down and suppurate. The reaction developing at the vaccination site 930
usually subsides within 2 - 5 months and in practically all children leaves a superficial scar of 2 - 10 931
mm in diameter. The nodule may persist and ulcerate. Swelling of regional lymph nodes may also 932
be seen, and this may be regarded as a normal reaction, but the size should be limited. 933
934
Keloid and lupoid reactions may occur at the site of the vaccination. Children with such reactions 935
should not be revaccinated. Inadvertent subcutaneous injections produce abscess formations and 936

WHO/BS/2011.2157
Page 32
may lead to ugly retracted scars. Among the major complications, suppurative lymphadenitis has 937
been observed. In the case of certain vaccines, it has been revealed that there is a strong correlation 938
between the incidence of these complications in newborns and the number of culturable particles in 939
the vaccine. 940
941
Thus, a reduction of the dose for the newborn may reduce these complications to acceptable levels. 942
It is recommended that the dose for newborns or infants should be one-half to one-quarter of that for 943
teenage children or adults. The concentration of the vaccine should be shown to be effective and 944
tolerated in the age groups for which the vaccine is intended (53). 945
946
The NRA should issue guidelines for the treatment of complications. 947
948
C.2 Special considerations 949
C.2.1 New “classical” BCG vaccines 950
This section is limited to the clinical development of new “classical” BCG vaccines manufactured 951
following these recommendations and using strains of BCG that are derived from (the same master 952
seed of) one of the strains recognized in Appendix 1. 953
954
The use of comparative studies with a licensed BCG vaccine can provide evidence of the similarity 955
of safety and immune responses to a new classical BCG vaccine product. 956
957
The target population for the vaccine would be newborns or infants according to the current 958
recommendations for use of BCG vaccines. 959
960
The preclinical expectations for a new classical BCG vaccine are outlined in Part B. 961
For such a new classical BCG vaccine, these preclinical studies should be 962 conducted in comparison to an existing licensed BCG vaccine, preferably derived 963 from the same BCG sub-strain. It would be expected that the results of preclinical 964 studies would be similar for the new vaccine product and for the comparator. 965
966
The clinical development program should ideally be designed to show the safety and protective 967
efficacy for the vaccine. However, for such a new classical BCG vaccine product, comparative 968
studies with an existing licensed BCG vaccine, using immunological responses as a marker for 969
efficacy, may be acceptable to the responsible NRA. 970

WHO/BS/2011.2157
Page 33
Comparable PPD response (proportion of PPD converters, intensity of response) 971 may be acceptable. 972
973
Clinical studies should provide evidence of safety in all the potential target populations, including 974
those with a high incidence of diseases that may affect the safety or efficacy of the new vaccine 975
product. 976
977
Phase-I/II: Safety and reactogenicity in healthy adults (comparative) 978
End points 979
Safety and reactogenicity – can include healthy HIV-infected adults 980
Immune responses – non-inferior PPD response and may include other 981
immunological markers. 982
983 These studies are difficult to interpret as adults will most likely have received BCG 984 vaccination at birth. Dose-finding studies may be considered unnecessary for these 985 vaccines. The safety in HIV-infected individuals and infants needs to be considered. 986
987
Dose-finding and age-de-escalation can be included in these studies but review by a 988
suitable Independent Safety Committee at each step should be considered. 989
990
Phase-III: Safety and reactogenicity in infants (comparative) 991
End points 992
Safety and reactogenicity 993
Non-inferior PPD immune response 994
995
Post-marketing risk management: 996
As it may not be practically possible to evaluate protective efficacy for a new classical BCG vaccine, 997
the responsible NRA in the country of manufacture should require post-marketing surveillance 998
activities for safety and effectiveness in a suitable environment. Sentinel surveillance sites in an 999
endemic country may be considered. 1000
1001
C.2.2 Revalidation of existing vaccines within national immunization program 1002
The responsible NRA of a country of manufacture may require a demonstration that adequate 1003
control of BCG vaccine has been achieved, by arranging for studies in children to be made at regular 1004
intervals on some of the final lots prepared. 1005

WHO/BS/2011.2157
Page 34
1006
Such studies on immunological responses to M. tuberculosis antigens should be made, including 1007
sensitivity to tuberculin. In at least 100 tuberculin-negative persons per year, and records should be 1008
obtained of the degree of sensitivity to tuberculin induced (distribution of tuberculin reactions by 1009
size)1 with a defined dose of tuberculin,2 local skin lesions (nature and size of reaction at injection 1010
site), and the occurrence of untoward vaccination reactions. It is desirable that such tests should be 1011
performed in parallel on two or more vaccine lots in the same population group, one of the vaccine 1012
lots being preferably a reference vaccine. 1013
1014
In relation to the tuberculin sensitivity test, different practices have been adopted according to the 1015
country situation. In the United State of America, Germany and Republic of Korea, routine 1016
demonstration of BCG-induced tuberculin conversion in humans is currently not required. This test 1017
is used in the UK as a diagnostic tool for TB disease in high risk children before BCG vaccination; 1018
and tuberculin-positive children are not vaccinated. 1019
1020
The frequency of testing of batches will depend on the number of batches of vaccine produced, but, 1021
in any case, at least one batch each year should be tested. The age groups of children in whom the 1022
vaccine is tested should be the same as those in which the vaccine will be eventually used. 1023
1024
If a batch of vaccine is to be exported, it should be ascertained in which age group it will be used in 1025
the importing country; the vaccine should be then tested accordingly. 1026
1027
C.3 Post-marketing surveillance 1028
The responsible NRA in the country of manufacture may require periodic safety update reports and 1029
periodic revalidation of the BCG safety, and immunogenicity. 1030
1031
C.3.1 BCG vaccine used in a national immunization program 1032
1 In some countries, the proportion of cases showing a negative reaction to tuberculin before BCG vaccination, but giving a positive result after vaccination, is called the “tuberculin conversion rate”. Unless positive and negative reactions are carefully defined, however, such a rate may not include certain cases in which a weak reaction to tuberculin is changed after BCG vaccination into a strong reaction. 2An intradermal test with a dose of tuberculin equivalent to 5 IU of tuberculin PPD is suitable. A description of an appropriate method and a design for a study to assess BCG vaccines in man are available on application to Chief, Tuberculosis and Respiratory Infections, World Health Organization, 1211 Geneva 27, Switzerland.

WHO/BS/2011.2157
Page 35
As in all immunization programmes, the adverse events following immunization with BCG vaccines 1033
should be monitored. 1034
1035
For BCG vaccines the following are important: 1036
- All injection site abscesses; 1037
- All cases of BCG lymphadenitis; 1038
- All deaths that are thought by health workers, or the public, to be related to immunization; 1039
- All cases requiring hospitalization that are thought by health workers, or the public, to be 1040
related to immunization; and 1041
- Other severe or unusual medical incidents that are thought by health workers, or the public, to 1042
be related to immunization. 1043
1044
Appropriate training of health care workers is important as some medical incidents can be related to 1045
immunization even if they have a delayed onset (54). 1046
1047
C.3.2 WHO pre-qualified BCG vaccines 1048
Pre-qualified vaccines may be used in a wide range of countries world-wide. Periodic safety update 1049
reports supplied to WHO should include specific analysis of countries where the vaccine has been 1050
used. 1051

WHO/BS/2011.2157
Page 36
Part D. Recommendations for national regulatory authorities 1052
1053
D.1 General 1054
The general recommendations for NRAs provided in the Guidelines for National Authorities on 1055
Quality Assurance for Biological Products should apply (55). These specify that no new biological 1056
substance should be released until consistency of manufacturing and quality as demonstrated by a 1057
consistent release of batches has been established. The detailed production and control procedures as 1058
well as any significant change in them that may affect the quality, safety or efficacy of BCG vaccine 1059
should be discussed with and approved by the NRA. For control purposes, the NRA should obtain 1060
the WHO Reference Reagents as comparators for potency-related testing and, where necessary, 1061
establish national working reference preparation(s) calibrated against the international reference. 1062
In addition, the NRA should provide a reference vaccine or approve one used by a manufacturer, 1063
and should give directions concerning the use of the reference vaccine in specified tests. The NRA 1064
should also give directions to manufacturers concerning the BCG sub-strain to be used in vaccine 1065
production, the total content of bacteria, the number of culturable particles, and the stability required 1066
of the vaccine, and should specify the requirements to be fulfilled by the manufacturer in accordance 1067
with the provisions of Part A of this document, including those for consistency of quality in respect 1068
of the points referred to in Part A, section 2. 1069
1070
D.1.1 BCG vaccine strain 1071
The sub-strain of BCG (maintained in the form of a seed lot) used in the production of vaccine 1072
should be derived from the original strain maintained by Calmette and Guérin and should be 1073
identified by historical records that include information on its origin and subsequent manipulation. 1074
On the basis of cultures and biochemical and animal tests, the BCG seed lot should show 1075
characteristics that conform to those of BCG and generally differ from those of other mycobacteria. 1076
The identity test should be supplemented by molecular biology techniques to identify the specific 1077
BCG sub-strain used. The seed lot should show consistency in the morphological appearance of 1078
colonies and genetic stability on serial subculture. It should also have been shown to yield vaccines 1079
that, upon administration by intradermal injection to children and adults, induce relevant 1080
immunological responses to M. tuberculosis antigens including sensitivity to tuberculin, and with a 1081

WHO/BS/2011.2157
Page 37
low frequency of untoward effects. In addition, the seed lot should have been shown to give 1082
adequate protection against TB in experimental animals in tests for protective potency. 1083
1084
D.1.2 Concentration of BCG vaccine 1085
The concentration of BCG vaccine varies with different vaccine products and is dependent on a 1086
number of factors, such as the sub-strain of BCG used and the method of manufacture. It is therefore 1087
essential, for each manufacturer as well as for each different method of manufacture, for the 1088
optimum potency of vaccine to be ascertained by trials in tuberculin-negative subjects (newborns, 1089
older children, and adults) to determine the response to vaccination in respect of the induction of 1090
relevant immunological responses to M. tuberculosis antigens including sensitivity to tuberculin, the 1091
production of acceptable local skin lesions, and the occurrence of a low frequency of untoward 1092
reactions. As a result of such trials, the NRA should give directions to the manufacturer concerning 1093
the total bacterial content and the number of culturable particles required for the vaccine. 1094
1095 If a manufacturer changes its procedure of preparing BCG vaccine, and if the 1096 NRA considers that the change might affect the final product, it may be 1097 necessary to conduct further clinical trials in order to determine the optimum 1098 content of BCG organisms in the new product. 1099
1100
D.2 Release and certification1 1101
A vaccine lot should be released only if it fulfils the national requirements and/or Part A of these 1102
Recommendations. Before any vaccine lot is released from a manufacturing establishment, the 1103
recommendations for consistency of production provided in Guidelines for national authorities on 1104
quality assurance for biological products (55) should be met. Also, the general recommendations 1105
for NRAs provided in the Guidelines for Independent Lot Release of Vaccines by Regulatory 1106
Authorities, which has been prepared, should be followed (56). A protocol based on the model given 1107
in Appendix 2, signed by the responsible official of the manufacturing establishment, should be 1108
prepared and submitted to the NRA in support of a request for release of vaccine for use. 1109
A statement signed by the appropriate official of the NRA (or authority as appropriate) should be 1110
provided if requested by a manufacturing establishment and should certify whether or not the lot of 1111
vaccine in question meets all national requirements, as well as Part A of these recommendations. 1112
The certificate should also state the date of manufacture, the lot number, the number under which 1113
1 Where there is no NRA, a manufacturer may request advice and help from: Chief, Biologicals, World Health Organization, 1211 Geneva 27, Switzerland

WHO/BS/2011.2157
Page 38
the lot was released, and the number appearing on the labels of the containers. In addition, the date 1114
of the last satisfactory potency test as well as the expiry date assigned on the basis of shelf-life 1115
should be stated. A copy of the official national release document should be attached. The certificate 1116
should be based on the model given in Appendix 3. The purpose of the certificate is to facilitate the 1117
exchange of vaccines between countries. 1118

WHO/BS/2011.2157
Page 39
Authors and Acknowledgements 1119
1120 The scientific basis for the revision of the Requirements published in WHO TRS 745 and TRS 771 1121 was developed at the meetings from 2003 to 2007 attended by the following people: 1122 1123 Dr Lewellys Barker, Aeras Global Tuberculosis Vaccine Foundation, Rockville, MD, USA; 1124 Professor Marcel Behr, Montreal General Hospital, Quebec, Canada; Dr Tagir Bektimirov, 1125 Tarassevich State Research Institute for Standardization and Control of Medical Biological 1126 Preparations, Moscow, Russian Federation; Dr Timothy Brewer, Brigham and Women’s Hospital, 1127 Boston, USA; Dr Roland Brosch, Institut Pasteur, Paris, France; Dr Luiz Roberto Castello-Branco, 1128 Fundacao Ataulpho de Paiva, Brazilian League Against Tuberculosis, Rio de Janeiro, Brasil; Dr 1129 Miliana Chouchkova, National Center of Infectious and Parasitic Diseases, Sofia, Bulgaria; Dr 1130 Stewart Cole, Institut Pasteur, Paris, France; Dr Michael Corbel, National Institute for Biological 1131 Standards and Control (NIBSC), Potters Bar, UK; Dr Janet Darbyshire, MRC Clinical Trials Unit, 1132 London, UK; Dr Hazel Dockrell, London School of Hygiene and Tropical Medicine, London, UK; 1133 Mr Isao Fujita, Japan BCG Laboratory, Tokyo, Japan; Dr Marina Gheorghiu, Institut Pasteur, Paris, 1134 France; Dr Elwyn Griffiths, Biologics and Genetic Therapies Directorate, Health Canada, Ottawa, 1135 Canada; Dr Cristina Gutierrez Pèrez, Institut Pasteur, Paris, France; Dr Kaare Haslov, Statens Serum 1136 Institut, Copenhagen, Denmark; Dr Mei Mei Ho, NIBSC, Potters Bar, UK; Dr Suresh Jadhav, 1137 Serum Institute of India Ltd., Pune, India; Dr Sten E. Jensen, Statens Serum Institut, Copenhagen, 1138 Denmark; Dr Micheline Lagranderie, Institut Pasteur, Paris, France; Mr François Leguellec, 1139 Aventis Pasteur, Val de Reuil, France; Dr Yolanda Lopez Vidal, Universidad Nacional Autonoma 1140 de Mexico, Facultad de Medicina, Mexico City Mexico; Dr Gilles Marchal, Institut Pasteur, Paris, 1141 France; Dr Harriet Mayanja-Kizza, Makarere Medical School, Kampala, Uganda; Dr Serge 1142 Mostowy, McGill University Health Centre, Montreal General Hospital, Quebec, Canada; Dr Micha 1143 Roumaintzeff, Lyon, France; Dr Masaaki Seki, BCG Laboratory, Tokyo, Japan; Dr Veronique 1144 Vincent, Institut Pasteur, Paris, France; Dr Frank Weichold, Aeras Global Tuberculosis Vaccine 1145 Foundation, Rockville, MD, USA; Dr Saburo Yamamoto, National Institute of Infectious Diseases, 1146 Tokyo, Japan; Dr Ikuya Yano, Japan BCG Laboratory, Tokyo, Japan; Dr Douglas Young, Imperial 1147 College of Science, Technology & Medicine, London, UK; Dr Uli Fruth, IVR/IVB, WHO, Geneva, 1148 Switzerland; Dr Ivana Knezevic, IVB, WHO, Geneva, Switzerland; Dr Joelle Daviaud, ATT/IVB, 1149 WHO Geneva, Switzerland; Dr David Wood, IVB, WHO, Geneva, Switzerland; Dr Tiequn Zhou, 1150 IVB, WHO, Geneva, Switzerland. 1151 1152 The first draft was prepared by Dr Hye-Na Kang, QSS/IVB/FCH, WHO, Geneva, Switzerland, 1153 taking into account comments received from: 1154
Dr Murielle Andre, Agence Française de Sécurité Sanitaire des Produits de Santé, Lyon, France; Dr 1155 Iin Susanti Budiharto, Bio Farma, Bandung, Indonesia; Dr Hyungok Chun, Korea Food and Drug 1156 Administration, Seoul, Republic of Korea; Dr Michael Corbel, National Institute for Biological 1157 Standards and Control (NIBSC), Potters Bar, UK; Dr Roland Dobbelaer, Lokeren, Belgium; Dr 1158 Sunil Gairola, Serum Institute of India, Hadapsar, India; Dr Leander Grode, Vakzine Projekt 1159 Management GmbH, Hannover, Germany; Dr Mei Mei Ho, NIBSC, Potters Bar, UK; Dr Suresh 1160 Jadhav, Serum Institute of India Ltd., Pune, India; Professor Diana Levi, Tarassevich State Research 1161 Institute for Standardization and Control of Medical Biological Preparations, Moscow, Russian 1162 Federation; Dr Sheldon Morris, US Food and Drug Administration, Silver Spring, MD, USA; Dr 1163 Volker Öppling, Paul Ehrlich Institut, Langen, Germany; Dr Micha Roumiantzeff, Lyon, France; Dr 1164

WHO/BS/2011.2157
Page 40
Keigo Shibayama, National Institute of Infectious Diseases, Tokyo, Japan; Dr James Southern, 1165 Capetown, South Africa; Mr Apichai Supasansatorn, Ministry of Public Health, Nonthaburi, 1166 Thailand; Dr Sri Wahyuningsih, National Agency of Drug and Food Control (NADFC), Jakarta 1167 Pusat, Indonesia; Dr Kenneth Barry Walker, NIBSC, Potters Bar, UK; Dr Saburo Yamamoto, 1168 National Institute of Infectious Diseases, Tokyo, Japan; Mrs AiHua Zhao, National Institute for the 1169 Control of Pharmaceutial & Biological Products, Beijing, P.R. China. 1170
Following the informal consultation meeting on standardization and evaluation of BCG vaccines in 1171 September 2009, Geneva, Switzerland, draft recommendations were revised by Dr Hye-Na Kang, 1172 QSS/IVB/FCH, WHO, Geneva, Switzerland, taking into account information on the current 1173 manufacturing and regulatory practice provided at the meeting attended by the following 1174 participants: 1175 1176 Dr Murielle Andre, Agence Française de Sécurité Sanitaire des Produits de Santé, Lyon, France; Dr 1177 Lewellys F. Barker, Aeras Global Tuberculosis Vaccine Foundation, Rockville, MD, USA; Dr Arun 1178 Bhardwaj, Central Research Institute, Kasuli, Himachal Pradesh, India; Dr Michael Brennan, Aeras 1179 Global TB Vaccine Foundation, Rockville, MD, USA; Dr Iin Susanti Budiharto, Bio Farma, 1180 Bandung, Indonesia; Dr Luiz Roberto Castello-Branco, Fundacao Ataulpho de Paiva, Brazilian 1181 League Against Tuberculosis, Rio de Janeiro, Brasil; Dr Miliana Chouchkova, National Center of 1182 Infectious and Parasitic Diseases, Sofia, Bulgaria; Dr Hyungok Chun, Korea Food and Drug 1183 Administration, Seoul, Republic of Korea; Dr Mike Corbel, NIBSC, Potters Bar, UK; Dr Bernd 1184 Eisele, Vakzine Projeckt Management GmbH, Hannover, Germany; Dr Sunil Gairola, Serum 1185 Institute of India, Hadapsar, India; Dr Leander Grode, Vakzine Projeckt Management GmbH, 1186 Hannover, Germany; Dr Kaare Haslov, Statens Serum Institut, Copenhagen, Denmark; Dr Mei Mei 1187 Ho, NIBSC, Potters Bar, UK; Dr Peter Hubrechts, Statens Serum Institut, Copenhagen, Denmark; 1188 Dr Greg Hussey, South African TB Vaccine Initiative, Cape Town, South Africa; Professor Diana 1189 Levi, Tarassevich State Research Institute for Standardization and Control of Medical Biological 1190 Preparations, Moscow, Russian Federation; Dr Kevin Markey, NIBSC, Potters Bar, UK; Dr Carlos 1191 Martin, Universidad de Zaragoza, Zaragoza, Spain; Dr Sheldon Morris, FDA/CBER, Bethesda, MD, 1192 USA; Dr Volker Öppling, Paul Ehrlich Institut, Langen, Germany; Dr Micha Roumiantzeff, Lyon, 1193 France; Dr. Masaaki Seki, Japan BCG Laboratory, Tokyo, Japan; Dr Keigo Shibayama, National 1194 Institute of Infectious Diseases, Tokyo, Japan; Dr James Southern, Advisor to Medicines Control 1195 Council in South Africa, Capetown, South Africa; Mr Apichai Supasansatorn, Ministry of Public 1196 Health, Nonthaburi, Thailand; Dr Yolanda Lopez Vidal, Universidad Nacional Autonoma de 1197 Mexico, Copilco-Universidad, Mexico City, Mexico; Dr Sri Wahyuningsih, National Agency of 1198 Drug and Food Control (NADFC), Jakarta Pusat, Indonesia; Dr Kenneth Barry Walker, NIBSC, 1199 Potters Bar, UK; Dr Saburo Yamamoto, Japan BCG Laboratory, Tokyo, Japan; Dr Zihou Yan, 1200 Chengdu Institute of Biological Products, Chengdu, P. R. China; Dr Lei Zhang, Chengdu Institute 1201 of Biological Products, Chengdu, P. R. China; Mrs AiHua Zhao, National Institute for the Control of 1202 Pharmaceutial & Biological Products, Beijing, P.R. China; Dr Hye-Na Kang and Dr Ivana Knezevic, 1203 QSS/IVB/FCH, WHO, Geneva, Switzerland. 1204 1205 Since then, several draft recommendations were prepared by Dr Mei Mei Ho, NIBSC, Potters Bar, 1206 UK with support from the drafting group, Dr Mike Corbel, Milton Keynes, UK; Dr Roland 1207 Dobbelaer, Lokeren, Belgium; Dr James Southern, Capetown, South Africa; Dr Kenneth Barry 1208 Walker, NIBSC, Potters Bar, UK; Dr Hye-Na Kang, QSS/IVB/FCH, WHO, Geneva, Switzerland. 1209 1210

WHO/BS/2011.2157
Page 41
Following the meeting of the drafting group in March 2011, Potters Bar, UK, draft 1211 recommendations were updated taking into account the comments received from: 1212 1213 Dr Murielle Andre, Agence Française de Sécurité Sanitaire des Produits de Santé, Lyon, France; Dr 1214 Miliana Chouchkova, National Center of Infectious and Parasitic Diseases, Sofia, Bulgaria; Dr 1215 Hazel Dockrell, London School of Hygiene and Tropical Medicine, London, UK; Dr Sunil Gairola, 1216 Serum Institute of India, Hadapsar, India; Dr Peter Hubrechts, Statens Serum Institut, Copenhagen, 1217 Denmark; Jeewon Jung, Korea Food and Drug Administration, Chungchengbuk-do, Republic of 1218 Korea; Dr Micheline Lagranderie, Institut Pasteur, Paris, France; Professor Diana Levi, Tarassevich 1219 State Research Institute for Standardization and Control of Medical Biological Preparations, 1220 Moscow, Russian Federation; Dr Volker Öppling, Paul Ehrlich Institut, Langen, Germany; Gandjar 1221 Trisnasari, Bio Farma, Bandung, Indonesia; Dr Yolanda Lopez Vidal, Universidad Nacional 1222 Autonoma de Mexico, Facultad de Medicina, Mexico City Mexico; Dr Veronique Vincent, Institut 1223 Pasteur, Paris, France; Dr Sri Wahyuningsih, National Agency of Drug and Food Control (NADFC), 1224 Jakarta Pusat, Indonesia; Dr Tang Ying, China National Biotec Group, Beijing, China; Mrs AiHua 1225 Zhao, National Institute for the Control of Pharmaceutial & Biological Products, Beijing, P.R. China. 1226 1227 The draft recommendations were posted on the WHO biologicals web site for public consultation 1228 from 1 to 23 June 2011. 1229 1230 The WHO/BS/2011.xxxx document was prepared by Dr Mei Mei Ho, NIBSC, Potters Bar, UK; Dr 1231 Hye-Na Kang, QSS/IVB/FWC, WHO, Geneva, Switzerland; Dr James Southern, Capetown, South 1232 Africa; Dr Kenneth Barry Walker, NIBSC, Potters Bar, UK, taking into account comments 1233 received from the following reviewers: 1234 1235 Chiyoung Ahn, Korea Food and Drug Administration, Chungchengbuk-do, Republic of Korea; Dr 1236 Murielle Andre, Agence Française de Sécurité Sanitaire des Produits de Santé, Lyon, France; Dr 1237 Michael Brennan, Aeras Global TB Vaccine Foundation, Rockville, MD, USA; Dr Iin Susanti 1238 Budiharto, Bio Farma, Bandung, Indonesia; Yejin Choi, Korea Food and Drug Administration, 1239 Chungchengbuk-do, Republic of Korea; Dr Miliana Chouchkova, National Center of Infectious and 1240 Parasitic Diseases, Sofia, Bulgaria; Dr Sunil Gairola, Serum Institute of India, Hadapsar, India; Dr 1241 Kaare Haslov, Statens Serum Institut, Copenhagen, Denmark; Dr Hamidreza Hozouri, Pasteur 1242 Institute of Iran, Tehran, Iran; Dr Peter Hubrechts, Statens Serum Institut, Copenhagen, Denmark; 1243 Jeewon Joung, Korea Food and Drug Administration, Chungchengbuk-do, Republic of Korea; 1244 Hyeonguk Kim, Korea Food and Drug Administration, Chungchengbuk-do, Republic of Korea; Jun-1245 Gyou Kim, Korea Food and Drug Administration, Chungchengbuk-do, Republic of Korea; Young 1246 Lim Kim, Korea Food and Drug Administration, Chungchengbuk-do, Republic of Korea; 1247 Kwangmoon Lee, Korea Food and Drug Administration, Chungchengbuk-do, Republic of Korea; Dr 1248 Volker Öppling, Paul Ehrlich Institut, Langen, Germany; Dr Micha Roumiantzeff, Lyon, France; Dr. 1249 Masaaki Seki, Japan BCG Laboratory, Tokyo, Japan; Mr Sang-Cheol Shin, Green Cross, Yongin, 1250 Republic of Korea; Dr Iin Susanti Budiharto, Bio Farma, Bandung, Indonesia; Dr Yolanda Lopez 1251 Vidal, Universidad Nacional Autonoma de Mexico, Copilco-Universidad, Mexico City, Mexico; Dr 1252 Veronique Vincent, Institut Pasteur, Paris, France; Dr Saburo Yamamoto, Japan BCG Laboratory, 1253 Tokyo, Japan; Dr Jinho Shin, QSS/IVB/FWC, WHO, Geneva, Switzerland. 1254

WHO/BS/2011.2157
Page 42
References 1255 1256 1. Requirements for dried BCG vaccines. In: WHO Experts Committee on Biological 1257
Standardization. Thirty-six Report. Geneva, World Health Organization, 1987, Annex 2 1258
(WHO Technical Report Series, No. 745). 1259
2. Requirements for dried BCG vaccines. In: WHO Experts Committee on Biological 1260
Standardization. Thirty-eight Report. Geneva, World Health Organization, 1988, Annex 12 1261
(WHO Technical Report Series, No. 771). 1262
3. Cobel MJ et al. Report on a WHO consultation on the characterization of BCG strains, 1263
Imperial College, London 15-16 December 2003. Vaccine, 2004, 22:2675-2680. 1264
4. Ho M, Knezevic I, Roumiantzeff M. Report on a WHO consultation on the characterization of 1265
BCG vaccines held at WHO Headquarters, Geneva. 8-9 December 2004. Vaccine, 2005, 1266
23:5700-5704. 1267
5. Knezevic I, Cobel MJ. WHO discussion on the improvement of the quality control of BCG 1268
vaccines held at Pasteur Institute, Paris, France, 7 June 2005. Vaccine, 2006, 24:3874-3877. 1269
6. Ho MM et al. Report on a WHO informal consultation on standardization and evaluation on 1270
BCG vaccines, 22-23 September 2009, WHO, Geneva, Switzerland. Vaccine, 2010, 28: 6945 - 1271
6950. 1272
7. State of the Art of New Vaccines: Research and Development. Geneva, World Health 1273
Organization, 2006 (WHO/IVB/06.01). 1274
8. Issues Relating to the Use of BCG in Immunization Programes. Geneva, World Health 1275
Organization, 1999 (WHO/V&B/99.23). 1276
9. WHO position paper of BCG vaccine. Weekly Epidermiological Record, 2004, 79:27-38. 1277
10. Colditz GA et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of 1278
the published literature. Journal of the American Medical Association, 1994, 271(9):698–702. 1279
11. Global Advisory Committee on Vaccine Safety, 12-13 June 2007. Weekly Epidermiological 1280
Record, 2007, 82: 252-259. 1281
12. Ritz N et al. Influence of BCG vaccine strain on the immune response and protection against 1282
tuberculosis. FEMS Microbiology Review, 2008, 32:821-41. 1283
13. Revised BCG vaccination guidelines for infants at risk for HIV infection. Weekly 1284
Epidermiological Record, 2007, 82:193-196. 1285
14. Use of BCG vaccine in HIV-infected infants. Weekly Epidermiological Record, 2010, 85:32-1286
33. 1287

WHO/BS/2011.2157
Page 43
15. Birkhaug K. Protective value of the intracutaneous and percutaneous methods of BCG 1288
vaccination, Acta Media Scandinavica, 1944, CXVII, fasc. III-IV:274-312. 1289
16. Mande R, Fillastre C, et Pinelli L. A propos des complications de la vaccination B.C.G. Ann 1290
Pediatr, 1976, 23:219-225. 1291
17. Mori T, Yamauchi Y, Shiozawa K. Lymph node swelling due to bacilli Calmette-Guerin 1292
vaccination with multipuncture method. Tubercle and Lung Disease, 1996, 77:269-273. 1293
18. Brosch R et al. Genome plasticity of BCG and impact on vaccine efficacy. Proceedings of the 1294
National Academy of Sciences, 2007, 104 (13): 5596-5601. 1295
19. Seki M et al. Whole genome sequence análisis of Mycobacterium bovis Bacillus Calmette-1296
Guerin (BCG) Tokyo 172: A comparative study of BCG vaccine substrains. Vaccine, 2009, 1297
27:1710-1716. 1298
20. Hayashi D et al. Comparable Studies of immunostimulating activities in vitro among 1299
Mycobacterium bovis bacillus Calmette-Guerin (BCG) substrains. FEMS Immunology and 1300
Medical Microbiology, 2009, 56:116-123. 1301
21. Horwitz MA et al. Commonly administered BCG strains including an evolutionarily early 1302
strain and evolutionarily late strains of disparate genealogy induce comparable protective 1303
immunity against tuberculosis. Vaccine 2009, 27(3): 441-5 1304
22. In vitro assays of BCG products. Unpublished working document, Geneva, World Health 1305
Organization, 1977 (WHO/TB/Technical guide/77.9). 1306
23. International collaborative study to evaluate and establish the 2nd reference reagent for BCG 1307
vaccine of three different substrains (unpublished data, 2009). 1308
24. Good Manufacturing Practices For Pharmaceutical Products: Main Principles. In: WHO Expert 1309
Committee On Specifications For Pharmaceutical Preparations. Thirty-Seventh Report. 1310
Geneva, World Health Organization, 2003, Annex 4 (WHO Technical Report Series, No. 908). 1311
http://Whqlibdoc.Who.Int/Trs/Who_Trs_908.Pdf#Page=46. Accessed on 11 April 2011. 1312
25. Good manufacturing practices for biological products. In: WHO Expert Committee on 1313
Biological Standardization. Forty-second Report. Geneva, World Health Organization, 1992, 1314
Annex 1 (WHO Technical Report Series, No. 822). 1315
http://www.who.int/biologicals/publications/trs/areas/vaccines/gmp/WHO_TRS_822_A1.pdf. 1316
Accessed on 11 April 2011. 1317

WHO/BS/2011.2157
Page 44
26. INTERNATIONAL COMMISSION ON RADIOLOGICAL PROMOTION. Report of 1318
Committee II on permissible dose for internal radiation. London, Pergamon Press, 1959 (ICRP 1319
Publication 2). 1320
27. UNITED NATIONS SCIENTIFIC COMMITTEE ON THE EFFECT OF ATOMIC 1321
RADIATION. Ionizing radiation: levels and effects. Report. New York, United Nations, 1972. 1322
28. General requirements for the sterility of biological substances ((Requirements for Biological 1323
Substances No. 6, revised 1973). In: WHO Expert Committee on Biological Standardization. 1324
Twenty-fifth report. Geneva, World Health Organization, 1973, Annex 4 (WHO Technical 1325
Report Series, No. 530). 1326
http://www.who.int/biologicals/publications/trs/areas/vaccines/sterility/WHO_TRS_530_A4.p1327
df. Accessed on 11 April 2011. 1328
29. Guidelines on transmissible spongiform encephalopathies in relation to biological and 1329
pharmaceutical products. Geneva, World Health Organization, 2003 1330
(WHO/BCT/QSD/2003.01). 1331
30. Recommendations for the evaluation of animal cell cultures as substrates for the manufacture 1332
of biological medicinal products and for the characterization of cell banks. In: WHO Expert 1333
Committee on Biological Standardization. 2010. 1334
31. WHO guidelines on tissue infectivity distribution in transmissible spongiform 1335
encephalopathies. WHO 2006. 1336
http://www.who.int/bloodproducts/tse/WHO%20TSE%20Guidelines%20FINAL-1337
22%20JuneupdatedNL.pdf. Accessed on 11 April 2011. 1338
32. WHO tables on guidelines on tissue infectivity distribution in transmissible spongiform 1339
encephalopathies. Geneva, World Health Organization, 2010. 1340
http://www.who.int/bloodproducts/tablestissueinfectivity.pdf. Accessed on 11 April 2011. 1341
33. Update of the opinion on TSE infectivity distribution in ruminant tissues. European 1342
Commission, 2002. http://ec.europa.eu/food/fs/sc/ssc/out296_en.pdf. Accessed on 11 April 1343
2011. 1344
34. Note for guidance on minimizing the risk of transmitting animal spongiform encephalopathy 1345
agents via human and veterinary medicinal products (EMEA/410/01 Rev. 2 - October 2003) 1346
adopted by the Committee for Proprietary Medicinal Products (CPMP) and by the Committee 1347
for Veterinary Medicinal Products (CVMP). Official Journal of the European Union, 2004. 1348

WHO/BS/2011.2157
Page 45
http://www.ema.europa.eu/pdfs/human/bwp/TSE%20NFG%20410-rev2.pdf. Accessed on 11 1349
April 2011. 1350
35. Ho MM et al. Report of an international collaborative study to establish the suitability of using 1351
modified ATP assay for viable count of BCG vaccine. Vaccine, 2008, 26(36):4754-4757. 1352
36. Jensen SE et al. Development and validation of an ATP method for rapid estimation of viable 1353
units in lyophilised BCG Danish 1331 vaccine. Biologicals, 2008, 36(5):308-314. 1354
37. Temperature Sensitivity of Vaccines. Geneva, World Health Organization, 2006 1355
(WHO/IVB/06.10). 1356
38. WHO Policy Statement: the use of opened multi-dose vials of vaccine in subsequent 1357
immunization sessions. 2000. WHO/V&B/00.09. http://www.who.int/vaccines-1358
documents/DocsPDF99/www9924.pdf. Accessed on 11 April 2011. 1359
39. Safe Vaccine Handling, Cold Chain and Immunizations. Geneva, World Health Organization, 1360
1998 (WHO/EPI/LHIS/98.02). 1361
40. Model guidance for the storage and transport of time and temperature-sensitive pharmaceutical 1362
products. In: WHO Expert Committee on Biological Standardization. 2010 (in press). 1363
41. Proper handling and reconstitution of vaccines avoids programme errors. Vaccines and 1364
Biologicals Update. Volume 34, World Health Organization, 2000. 1365
http://www.who.int/vaccines-documents/DoxNews/updates/updat34e.pdf. Accessed on 11 1366
April 2011. 1367
42. WHO guidelines on stability evaluation of vaccines. In: WHO Expert Committee on Biological 1368
Standardization. Fifty-Seventh report. Geneva, World Health Organization, 2006 (WHO 1369
Technical Report Series, in press). WHO/BS/06.2049 - Final: 1370
http://www.who.int/biologicals/publications/trs/areas/vaccines/stability/Microsoft%20Word%1371
20-%20BS%202049.Stability.final.09_Nov_06.pdf. Accessed on 11 April 2011. 1372
43. Guideline for Establishing or Improving Primary and Intermediate Vaccine Stores. Geneva, 1373
World Health Organization, 2002 (WHO/V&B/02.34). http://www.who.int/vaccines-1374
documents/DocsPDF02/www715.pdf. Accessed on 11 April 2011. 1375
44. Immunization in Practice: A practical resource guide for health staffs, 2004 update. Geneva, 1376
World Health Organization, 2004 (WHO/IVB/04.06). 1377
45. WHO guidelines on nonclinical evaluation of vaccines. In: WHO Expert Committee on 1378
Biological Standardization. Fifty-fourth report. Geneva, World Health Organization, 2005, 1379
Annex 1 (WHO Technical Report Series, No. 927). 1380

WHO/BS/2011.2157
Page 46
http://www.who.int/biologicals/publications/trs/areas/vaccines/nonclinical_evaluation/ANNE1381
X%201Nonclinical.P31-63.pdf. Accessed on 11 April 2011. 1382
46. Yamamoto T et al. Mycobacterium bovis BCG vaccination modulates TNF-a production after 1383
pulmonary challenge with virulent Mycobacterium tuberculosis in guinea pigs. Tuberculosis, 1384
2007, 87:155-165. 1385
47. Walker KB et al. The second Geneva Consensus: Recommendations for novel live TB 1386
vaccines. Vaccine, 2010, 28(11):2259-2270. 1387
48. Kamath AT et al. New live mycobacterial vaccines: the Geneva consensus on essential steps 1388
towards clinical development. Vaccine, 2005, 23(29):3753-3761. 1389
49. Guidelines for good clinical practice (GCP) for trials on pharmaceutical products. In: WHO 1390
Expert Committee on the Use of Essential Drugs. Sixth report. Geneva, World Health 1391
Organization, 1995, Annex 3 (WHO Technical Report Series, No. 850). 1392
http://www.who.int/medicinedocs/collect/edmweb/pdf/whozip13e/whozip13e.pdf. Accessed 1393
on 11 April 2011. 1394
50. WHO guidelines on clinical evaluation of vaccines: regulatory expectations. In: WHO Expert 1395
Committee on Biological Standardization. Fifty-second report. Geneva, World Health 1396
Organization, 2004, Annex 1 (WHO Technical Report Series, No. 924). 1397
http://www.who.int/biologicals/publications/trs/areas/vaccines/clinical_evaluation/035-1398
101.pdf. Accessed on 11 April 2011. 1399
51. Roth A et al. Vaccination technique, PPD reaction and BCG scarring in a cohort of children 1400
born in Guinea-Bissau 2000-2002. Vaccine, 2005, 23(30):3991-3998. 1401
52. WHO Expert Committee on Tuberculosis. Ninth report. Geneva, World Health Organization, 1402
1974 (WHO Technical Report Series, No. 552). 1403
53. Hoft DF et al. Clinical reactogenicity of intradermal bacille Calmette-Guerin vaccination. 1404
Clinical Infectious Disease, 1999, 28(4):785-790. 1405
54. Surveillance of Adverse Events Following Immunization. Geneva, World Health Organization, 1406
1997 (WHO/EPI/TRAM/93.02. Rev 1). 1407
55. Guidelines for national authorities on quality assurance for biological products. In: WHO 1408
Expert Committee on Biological Standardization. Forty-second Report. Geneva, World Health 1409
Organization, 1992, Annex 2 (WHO Technical Report Series, No. 822). 1410
http://whqlibdoc.who.int/trs/WHO_TRS_822.pdf. Accessed on 11 April 2011. 1411

WHO/BS/2011.2157
Page 47
56. Guidelines for independent lot release of vaccines by regulatory authorities. In: WHO Expert 1412
Committee on Biological Standardization. 2010 (in press). 1413

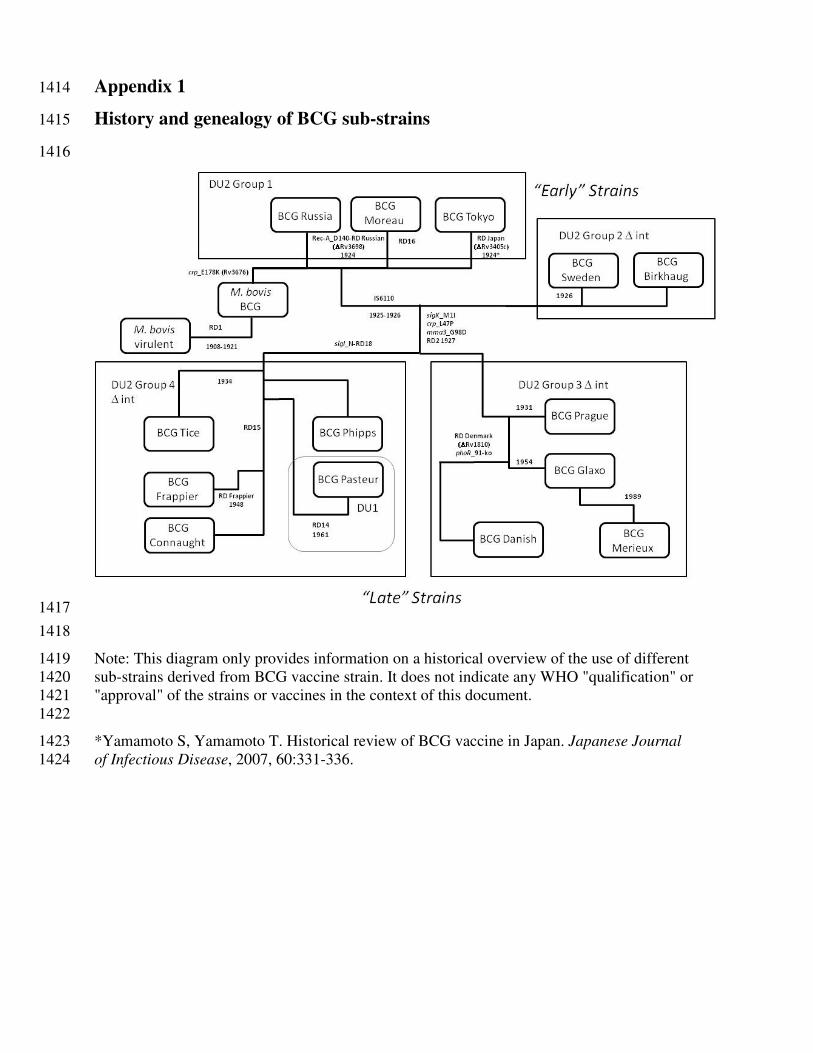
Appendix 1 1414
History and genealogy of BCG sub-strains 1415
1416
1417
1418
Note: This diagram only provides information on a historical overview of the use of different 1419 sub-strains derived from BCG vaccine strain. It does not indicate any WHO "qualification" or 1420 "approval" of the strains or vaccines in the context of this document. 1421 1422
*Yamamoto S, Yamamoto T. Historical review of BCG vaccine in Japan. Japanese Journal 1423 of Infectious Disease, 2007, 60:331-336. 1424

WHO/BS/2011.2157
Page 50
Appendix 2 1425
Model summary protocol for manufacturing and control of BCG vaccine 1426
1427 The following protocol is intended for guidance, and indicates the information that should be 1428 provided as a minimum by the manufacturer to the NRA. 1429 Information and tests may be added or deleted as required by the NRA, if applicable. 1430 1431 It is thus possible that a protocol for a specific product may differ in detail from the model 1432 provided. The essential point is that all relevant details demonstrating compliance with the 1433 license and with the relevant WHO recommendations of a particular product should be given 1434 in the protocol submitted. 1435 1436 The section concerning the final product must be accompanied by a sample of the label and a 1437 copy of the leaflet that accompanies the vaccine container. If the protocol is being submitted 1438 in support of a request to permit importation, it must also be accompanied by a lot release 1439 certificate from the NRA or national control laboratory of the country in which the vaccine 1440 was produced stating that the product meets national requirements as well as Part A 1441 recommendations of this document published by WHO. 1442 1443
Summary information on the finished product (final lot)
International name _______________________________________
Trade name _______________________________________
Product licence (marketing authorization)
number
_______________________________________
Country _______________________________________
Name and address of manufacturer _______________________________________
Site of manufacture of final lot _______________________________________
Name and address of licence holder if
different
_______________________________________
BCG sub-strain _______________________________________
Authority that approved BCG sub-strain _______________________________________
Date approved _______________________________________
Final bulk number _______________________________________
Volume of final bulk _______________________________________
Final product
Type of vaccine Intradermal/ Percutaneous/ Other
Final lot number _______________________________________
Type of container _______________________________________
Number of doses per container: _______________________________________
Number of filled containers in this final lot _______________________________________

WHO/BS/2011.2157
Page 51
Date of manufacture of final lot _______________________________________
Date on which last determination of
bacterial count was started or date of start
of period of validity
_______________________________________
Shelf-life approved (months) _______________________________________
Expiry date _______________________________________
Diluent _______________________________________
Storage conditions _______________________________________
Volume of single human dose _______________________________________
Volume of vaccine per container _______________________________________
Number of doses per container _______________________________________
Summary of the composition (Include a
summary of the qualitative and quantitative
composition of the vaccine per human dose
_______________________________________
Release date _______________________________________
Production information
A genealogy of the lot numbers of all vaccine components used in the formulation of the final product will be informative. The following sections are intended for the reporting of the results of the tests performed during the production of the vaccine, so that the complete document will provide evidence of consistency of production; thus if any test has to be repeated, this must be indicated. Any abnormal results should be recorded on a separate sheet.
1444
Control of source materials (A.3) 1445
The information requested below is to be presented on each submission. Full details on master and 1446 working seed-lots upon first submission only and whenever a change has been introduced. 1447
Master seed lot
Origin of seed lot _______________________________________
Master seed lot number. _______________________________________
Name and address of manufacturer _______________________________________
Passage level _______________________________________
Date of preparation of seed lot _______________________________________
Date of receipt of seed lot (if applicable) _______________________________________
Date of reconstitution of seed lot ampoule _______________________________________
Date approved by the National Regulatory Authority:
_______________________________________
Working seed lot
Working seed lot number. _______________________________________

WHO/BS/2011.2157
Page 52
Name and address of manufacturer _______________________________________
Passage level _______________________________________
Date reconstitution of seed lot ampoule _______________________________________
Date approved by the National Regulatory Authority
_______________________________________
Tests on working seed lot production (A.3.2)
Identity test (A.3.2.1)
Method used _______________________________________
Date test start _______________________________________
Date test complete ______________________________________
Results _______________________________________
Test for bacterial and fungal contamination (A.3.2.2)
Method used _______________________________________
Number of containers tested _______________________________________
Volume of inoculum per container _______________________________________
Volume of medium per container _______________________________________
Observation period (specification) _______________________________________
Incubation Media used Inoculum Date test start Date test complete Results
20–25 °C __________ __________ ___________ ___________ ________
30–36 °C __________ __________ ___________ ___________ ________
Negative control
__________ __________ ___________ ___________ ________
Test for absence of virulent mycobacteria (A.3.2.3)
Method used _______________________________________
No. of human dose injected per guinea-pig _______________________________________
Inoculation route _______________________________________
No. of guinea-pigs given injection _______________________________________
Weight range of guinea-pigs _______________________________________
Observation period (specification) _______________________________________
Date test start _______________________________________
Data test complete _______________________________________
Health of animals during test _______________________________________
Weight gains (losses) _______________________________________
Result _______________________________________
Test for excessive dermal reactivity (A.3.2.4)
vaccine reference vaccine
Method used __________________ __________________

WHO/BS/2011.2157
Page 53
Dilutions injected __________________ __________________
Inoculation route __________________ __________________
No. of guinea-pigs given injection __________________ __________________
Observation period (specification) __________________ __________________
Date test start __________________ __________________
Data test complete __________________ __________________
Mean diameter of lesions (for each dilution) __________________ __________________
Result __________________ __________________
Production of culture medium (A.3.3)
Any components of animal origin _______________________________________
Certificate for BSE/TSE-free _______________________________________
Control of vaccine production (A.4)
Control of single harvests (A.4.1)
Derived from master seed lot number. _______________________________________
Working seed lot number _______________________________________
Passage level from master seed ______________________________________
Culture medium
Number and volume of containers inoculated
Date of inoculation ______________________________________
Temperature of incubation ______________________________________
Date of harvest ______________________________________
Results of visual inspection ______________________________________
Control of final bulk (A.4.2)
Tests for bacterial and fungal contamination (A.4.2.2)
Method used _______________________________________
Number of containers tested _______________________________________
Volume of inoculum per container _______________________________________
Volume of medium per container _______________________________________
Observation period (specification) _______________________________________
Incubation Media used Inoculum Date test start Date test complete Result
20°–25°C __________ ___________ ___________ ___________ ________
30°–36°C __________ ___________ ___________ ___________ ________
Negative control
__________ ___________ ___________ ___________ ________
Test for absence of virulent mycobacteria (A.4.2.3) (if test not performed on final lot)

WHO/BS/2011.2157
Page 54
Method used _______________________________________
No. of human dose injected per guinea-pig _______________________________________
Inoculation route _______________________________________
No. of guinea-pigs given injection _______________________________________
Weight range of guinea-pigs _______________________________________
Observation period (specification) _______________________________________
Date test start _______________________________________
Data test complete _______________________________________
Health of animals during test _______________________________________
Weight gains (losses) _______________________________________
Result _______________________________________
Test for bacterial concentration (A.4.2.4)
Method used _______________________________________
Date test start _______________________________________
Data test complete _______________________________________
Specification _______________________________________
Result _______________________________________
Test for number of culturable particles (A.4.2.5)
Method used _______________________________________
Date test start _______________________________________
Data test complete _______________________________________
Specification _______________________________________
Result _______________________________________
Information of working reference preparation _______________________________________
Substances added (A.4.2.6)
Any components of animal origin _______________________________________
Certificate for BSE/TSE-free _______________________________________
Filling and containers (A.5)
Lot number _______________________________________
Date of filling _______________________________________
Volume of final bulk filled _______________________________________
Filling volume per container _______________________________________
Number of containers filled (gross) _______________________________________
Date of freeze-drying _______________________________________
Number of containers rejected during inspection
_______________________________________

WHO/BS/2011.2157
Page 55
Number of containers sampled _______________________________________
Total number of containers (net) _______________________________________
Maximum period of storage approved _______________________________________
Storage temperature and period _______________________________________
Control tests on final lot (A6)
Inspection of final containers (A.6.1)
Appearance _______________________________________
Date of test _______________________________________
Specification _______________________________________
Result _______________________________________
Recommended reconstitution fluid _______________________________________
Volume of reconstitution fluid per final container
_______________________________________
Identity test (A.6.2)
Method used _______________________________________
Date test start _______________________________________
Date test complete ______________________________________
Specification ______________________________________
Result _______________________________________
Tests for bacterial and fungal contamination (A.6.3)
Method used _______________________________________
Number of containers tested _______________________________________
Volume of inoculum per container _______________________________________
Volume of medium per container _______________________________________
Observation period (specification) _______________________________________
Specification ______________________________________
Incubation Media used Inoculum Date test start Date test complete Result
20°–25°C __________ ___________ ___________ ___________ ________
30°–36°C __________ ___________ ___________ ___________ ________
Negative control
__________ ___________ ___________ ___________ ________
Safety tests (A.6.4)
Test for absence of virulent mycobacteria (A.6.4.1) (if test not performed on final bulk)
Method used _______________________________________
No. of human dose injected per guinea-pig _______________________________________

WHO/BS/2011.2157
Page 56
Inoculation route _______________________________________
No. of guinea-pigs given injection _______________________________________
Weight range of guinea-pigs _______________________________________
Observation period (specification) _______________________________________
Date test start _______________________________________
Data test complete _______________________________________
Health of animals during test _______________________________________
Weight gains (losses) _______________________________________
Specification ______________________________________
Result _______________________________________
Test for excessive dermal reactivity (A.6.4.2) if applicable
vaccine reference vaccine
Method used __________________ __________________
Dilutions injected __________________ __________________
Inoculation route __________________ __________________
No. of guinea-pigs given injection __________________ __________________
Observation period (specification) __________________ __________________
Date test start __________________ __________________
Data test complete __________________ __________________
Mean diameter of lesions (for each dilution) __________________ __________________
Specification __________________ __________________
Result __________________ __________________
Test for bacterial concentration (A.6.5)
Method used _______________________________________
Date test start _______________________________________
Data test complete _______________________________________
Specification _______________________________________
Result _______________________________________
Test for residual moisture (A.6.6)
Method ______________________________________
Date ______________________________________
Specification ______________________________________
Result ______________________________________
Tests for viability (A.6.7)
Test for number of culturable particles (A.6.7.1)
Method used ______________________________________
Medium ______________________________________

WHO/BS/2011.2157
Page 57
Date test start ______________________________________
Data test complete ______________________________________
Before lyophilization After lyophilization
No. of containers tested __________________ __________________
Mean count of culturable particles per mL __________________ __________________
Mean survival rate (%) __________________ __________________
Specification ______________________________________
Result ______________________________________
Information of working reference preparation ______________________________________
Rapid test for viability (A.6.7.2) if applicable
Method ______________________________________
Mean survival rate (%) ______________________________________
Date ______________________________________
Specification ______________________________________
Result ______________________________________
Thermal stability test (A.6.8)
Method used ______________________________________
Date test start ______________________________________
Data test complete ______________________________________
Unheated containers Heated containers
No. of containers tested _______________ ________________
Culturable particles in each container per mL _______________ ________________
Mean survival rate (%) _______________ ________________
Specification ______________________________________
Result ______________________________________
Information of working reference preparation ______________________________________
1448 Submission addressed to national regulatory authority 1449 1450 1451 Name of responsible person (typed) _________________________ 1452 1453 Certification by the person from the control laboratory of the manufacturing company taking 1454 over responsibility for the production and control of the vaccine: 1455 1456 I certify that lot no. ______________ of BCG vaccine, whose number appears on the label of 1457 the final container, meets all national requirements and/or satisfies Part A of the 1458 Recommendations for Biological Substances No. 3 (Recommendations for BCG vaccine, 1459 revised 2011) 1460 1461

WHO/BS/2011.2157
Page 58
Signature: __________________________________________________ 1462 1463 Name (typed): ______________________________________________ 1464 1465 Date: ____ 1466

WHO/BS/2011.2157
Page 59
Appendix 3 1467
Model certificate for the release of BCG vaccine by national regulatory 1468
authorities 1469 1470 LOT RELEASE CERTIFICATE 1471 1472 1473 The following lot(s) of BCG vaccine produced by ____________________________(1) in 1474 _______________(2), whose numbers appear on the labels of the final containers, meet all 1475 national requirements(3) and Part A(4) of the WHO recommendations to assure the quality, 1476 safety and efficacy of freeze-dried BCG vaccines (_____)(5), and comply with Good 1477 Manufacturing Practices for Pharmaceutical Products: Main Principles(6) and Good 1478 Manufacturing Practices for Biological Products(7). 1479 As a minimum, this certificate is based on examination of the summary protocol of 1480 manufacturing and control. 1481 1482 The certificate may include the following information: 1483
• Name and address of manufacturer; 1484 • Site(s) of manufacturing; 1485 • Trade name and/common name of product; 1486 • Marketing authorization number; 1487 • Lot number(s) (including sub-lot numbers, packaging lot numbers if necessary); 1488 • Type of container; 1489 • Number of doses per container; 1490 • Number of containers/lot size; 1491 • Date of start of period of validity (e.g. manufacturing date) and/or expiry date; 1492 • Storage condition; 1493 • Signature and function of the authorized person and authorized agent to issue the 1494
certificate; 1495 • Date of issue of certificate; and 1496 • Certificate number. 1497
1498 1499 1500 1501 The Director of the National Regulatory Authority (or Authority as appropriate): 1502 1503 Name (Typed) 1504 Signature 1505 Date 1506 1507 1508 1Name of manufacturer 1509 2Country of origin 1510 3If any national requirements are not met, specify which one(s) and indicate why release of the 1511 lot(s) has nevertheless been authorized by the NRA. 1512

WHO/BS/2011.2157
Page 60
4With the exception of provisions on distribution and shipping, which the NRA may not be in a 1513 position to assess. 1514 5WHO Technical Report Series, No. ___, YYYY, Annex __. 1515 6WHO Technical Report Series, No. 908, 2003, Annex 4. 1516 7WHO Technical Report Series, No. 822, 1992, Annex 1. 1517 1518 1519
= = = 1520 1521





























