Embed Size (px)
Citation preview

Chemistry 231Work, Heat and Internal
Energy: The First Law

System – the specific part of the universe of interest to us
Surroundings – the part of the universe not contained in the system
Systems and Surroundings

3 types of Systems• open system – exchanges mass and energy• closed system – exchanges energy but no
mass• isolated system – no exchange of either
mass or energy
State of a System (Continued)
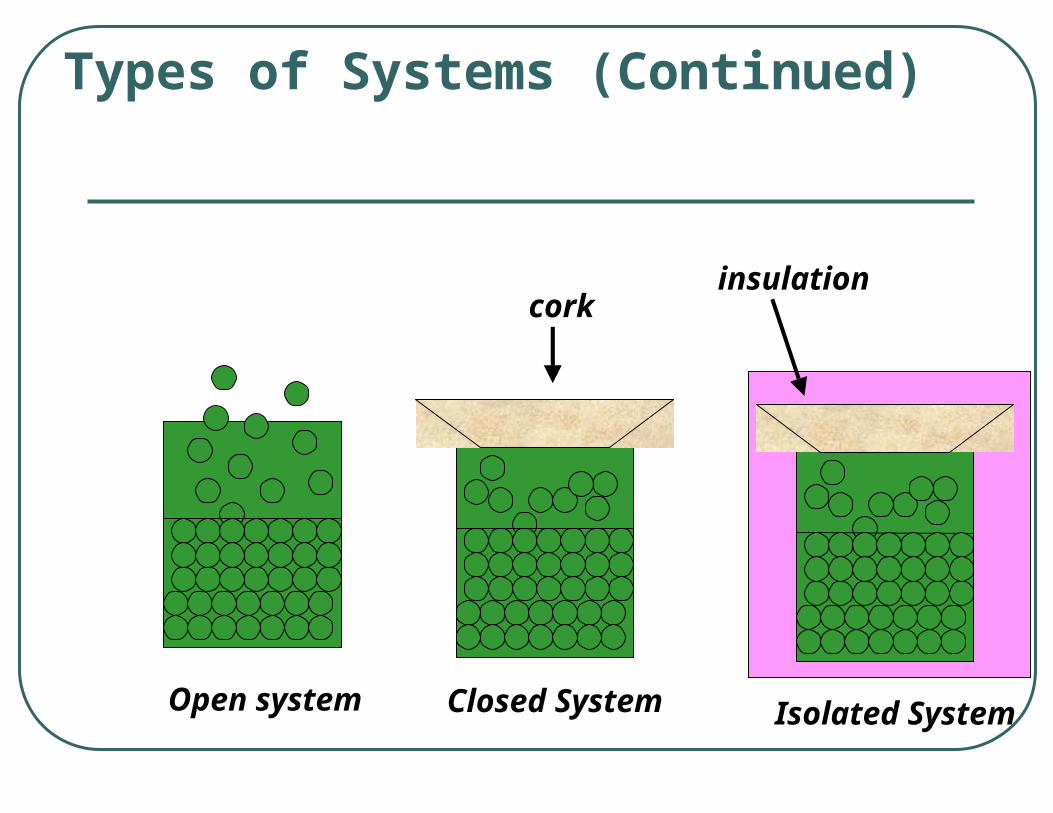
Types of Systems (Continued)
Open system Closed System
corkinsulation
Isolated System

State of a system• the system is in a definite state when each of its
properties has a definite value. Change in state
• initial state• final state
Path• initial and final states• intermediate states
Some Definitions

Process• reversible or irreversible transformation
Cyclic transformation• begins and ends at the same state variables.
Some Definitions (Continued)

Isothermal• dT = 0
Isochoric• dV = 0
Isobaric• dP = 0
Some Definitions (Continued)

Work (w)• any quantity that flows across the system’s
boundary and is completely convertible into the lifting of a mass in the surroundings.
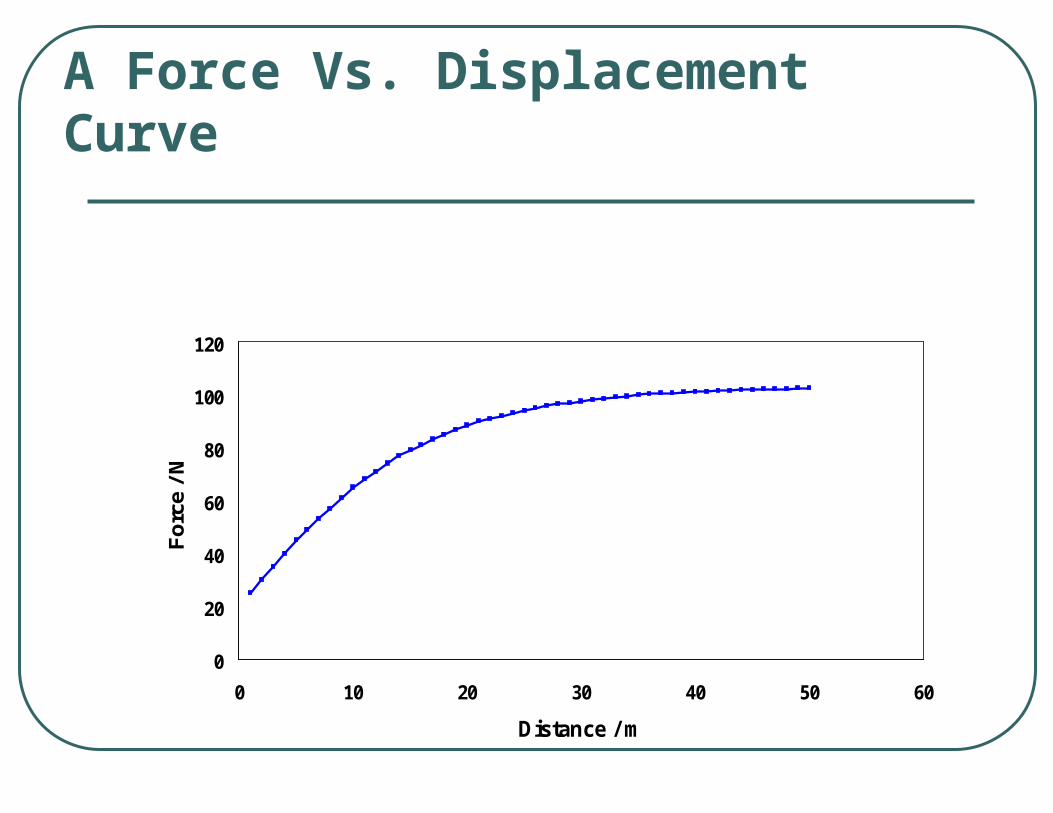
How much work was done?
Work
Unit of work = J = 1 kg m/s2
dzFdw z
0
20
40
60
80
100
120
0 10 20 30 40 50 60
Distance / m
Fo
rce
/ N
A Force Vs. Displacement Curve
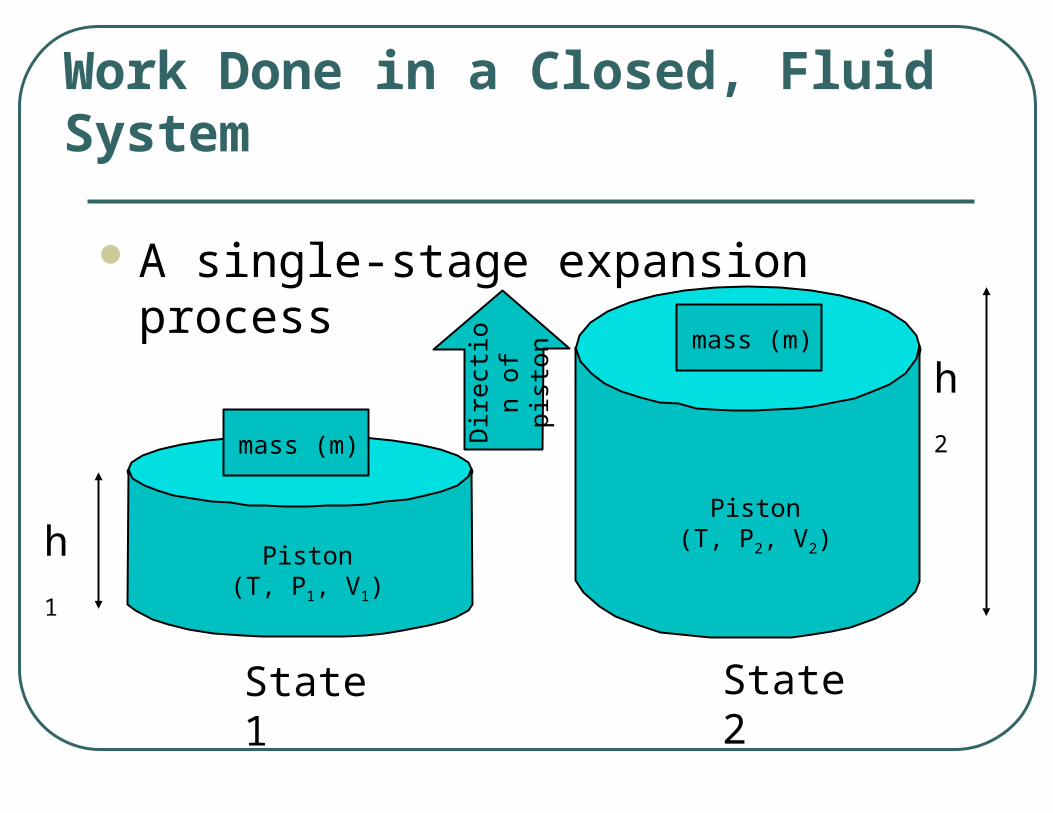
A single-stage expansion process
Work Done in a Closed, Fluid System
State 1 State 2
Piston(T, P1, V1)
mass (m)
Piston(T, P2, V2)
mass (m)
Dir
ecti
on
of p
isto
n
h2
h1

The work done in the surroundings• wsurr= Pext DV
The work done by the system• wsys = - wsurr = - Pext DV
For an infinitesimal volume change• dwsys = - Pext dV
System and Surroundings

If the system is in equilibrium• Fsys = -Fext
• P = Pext
For a simple system• d wrev = - P dV
Reversible (Multistage) Expansion
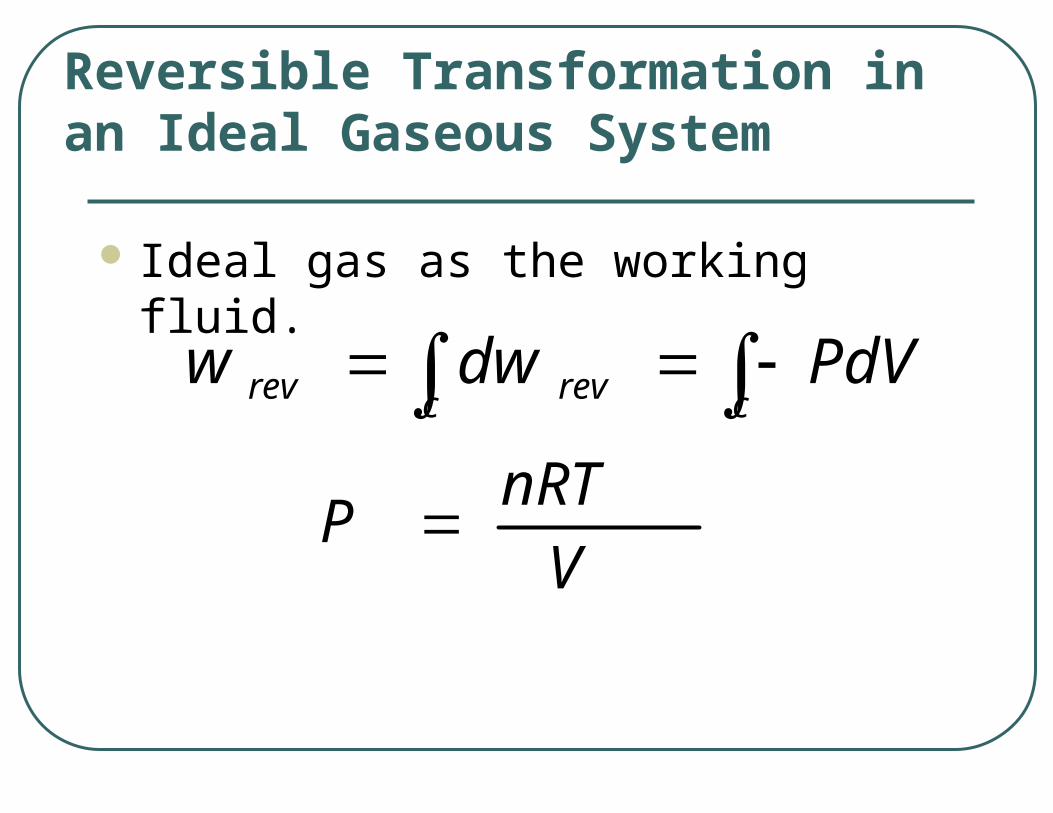
Ideal gas as the working fluid.
Reversible Transformation in an Ideal Gaseous System
cc revrev PdVdww
VnRT
P

For an isothermal process (ideal gas as working fluid)
Reversible Transformation (Continued)
1
2lnVV
nRTdwwc revrev

dwirr = -Pext dV for a constant external pressure
Irreversible Transformations
12
2
1
VVP
dVPdww
ext
extc irrirr

Heat - the quantity that flows across the boundary of the system during a change in state• due to temperature difference between
system and surroundings• HOT to COLD (never the other way
around)!!!
Heat

Measured by determining the temperature change of some known object
'Amount of Heat'
C - the heat capacity of the system.
CdTdq

Integrate the infinitesimal heat flow
Macroscopic Heat Flows
2
1
T
Tc c
CdTCdTdqq
TCq
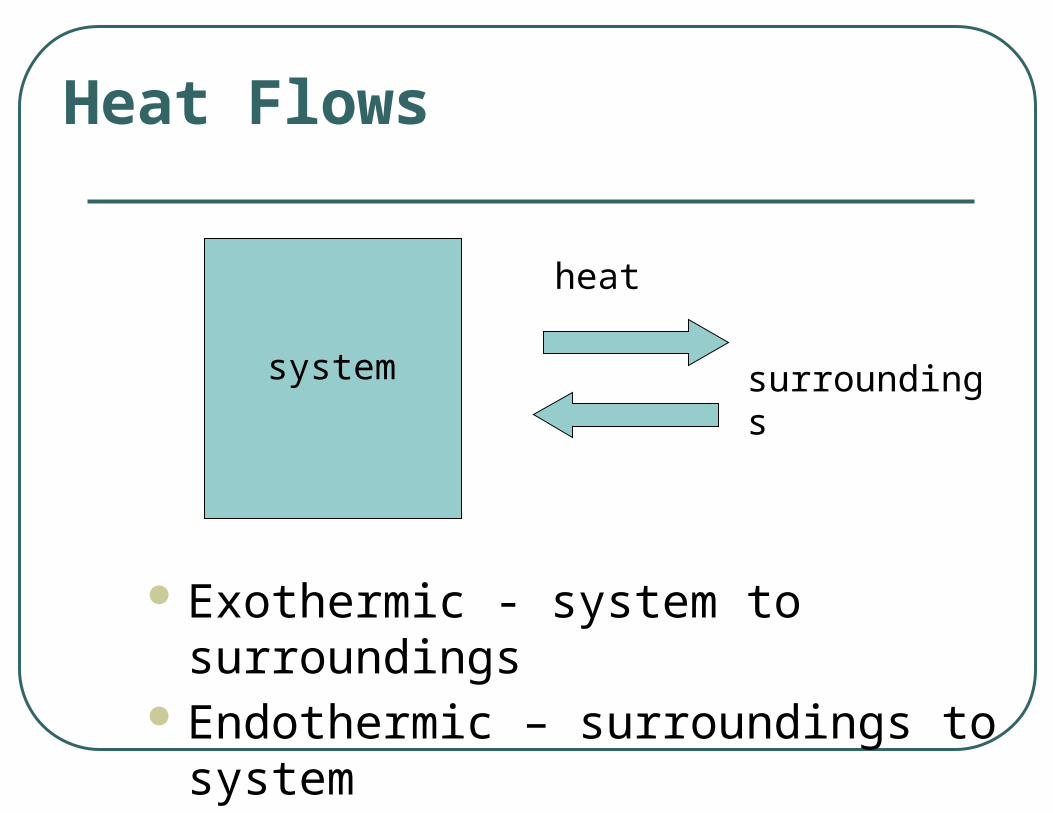
Exothermic - system to surroundings Endothermic – surroundings to system
Heat Flows
surroundingssystem
heat

Heat flows during phase changes - latent heats• Latent heat of vapourisation• Latent heat of fusion
Latent Heats
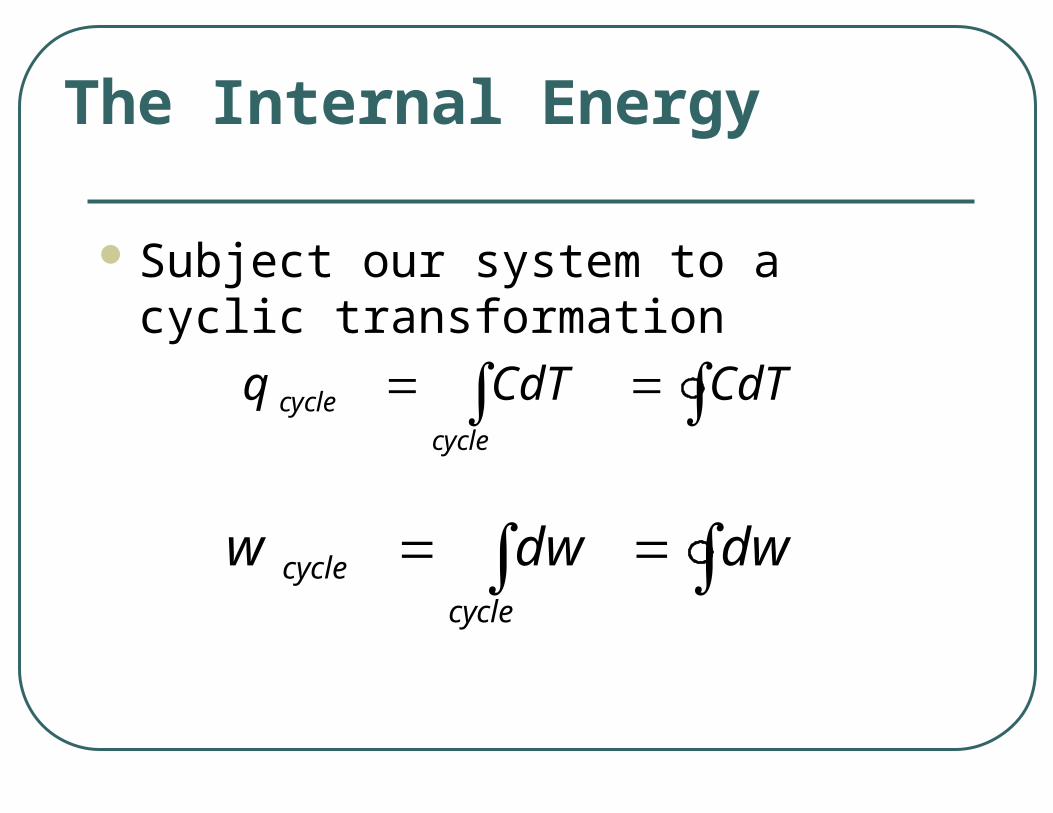
Subject our system to a cyclic transformation
The Internal Energy
CdTCdTqcycle
cycle
dwdwwcycle
cycle

The following would be true for an exact differential
Cyclic Integrals of Exact Differentials
exact is df if 0df

The infinitesimal change in the internal energy
The Internal Energy
dwdqdU
wqdwdqUc
For a general process

In general, we write U as a function of T and V
The Properties of U
dVVU
dTTU
dUTV

Examine the first partial derivative
Isochoric Changes in U
0
dTTU
dUV

Define the constant volume heat capacity, CV
The Constant Volume Heat Capacity
VVV T
UdTdq
C

For a system undergoing an isochoric temperature change
Heat Flows Under Constant Volume Conditions
For a macroscopic system
dTCdU V
2
1
T
TVV dTCqU

Examine the second partial derivative
Isothermal Changes in U
dVVU
dUT
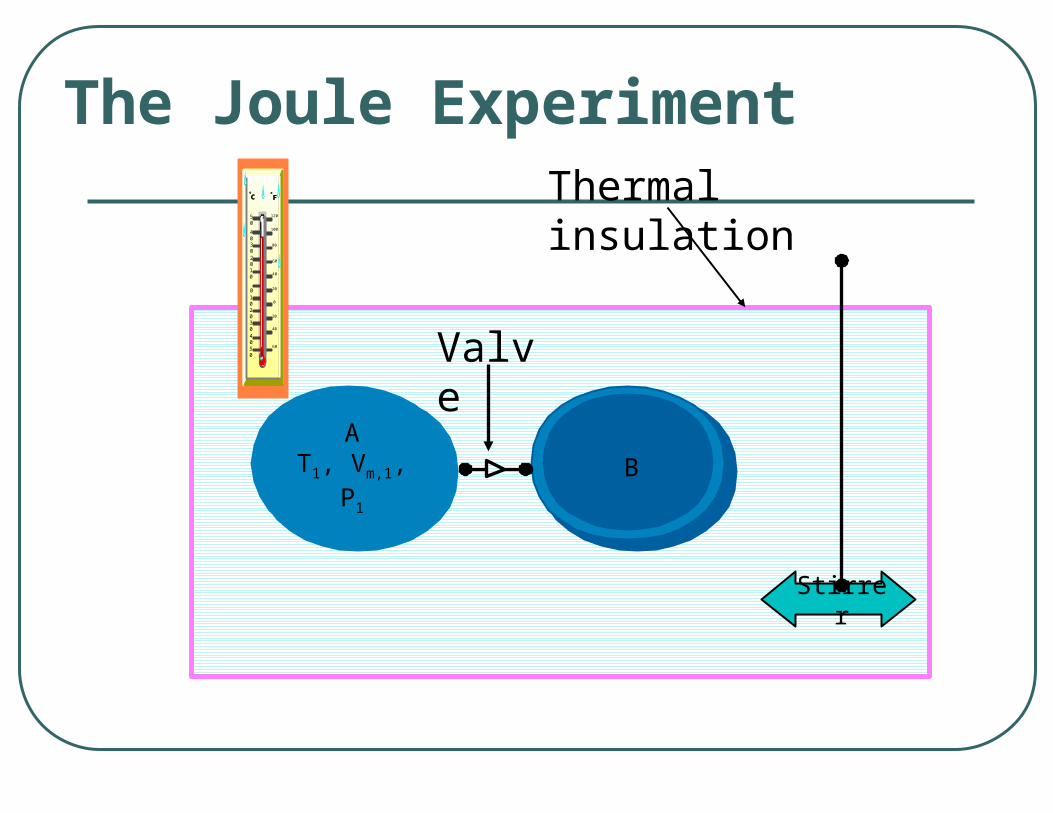
The Joule Experiment
AT1, Vm,1, P1
B
Stirrer
Valve
Thermal insulationFFOO
CO
CO
50
40302010 01020304050
120
100
80
0
20
20
40
60
60
40

The partial derivative
The Joule Coefficient
is known as the Joule coefficient, J. UV
T

The change in the internal energy under isothermal conditions is related to the Joule Coefficient
Internal Energy and the Joule Coefficient
VUT TU
VT
VU
JVT
CVU

For an adiabatic process, q = 0!! The first law becomes
Adiabatic Processes
wU
dTCdwdU V
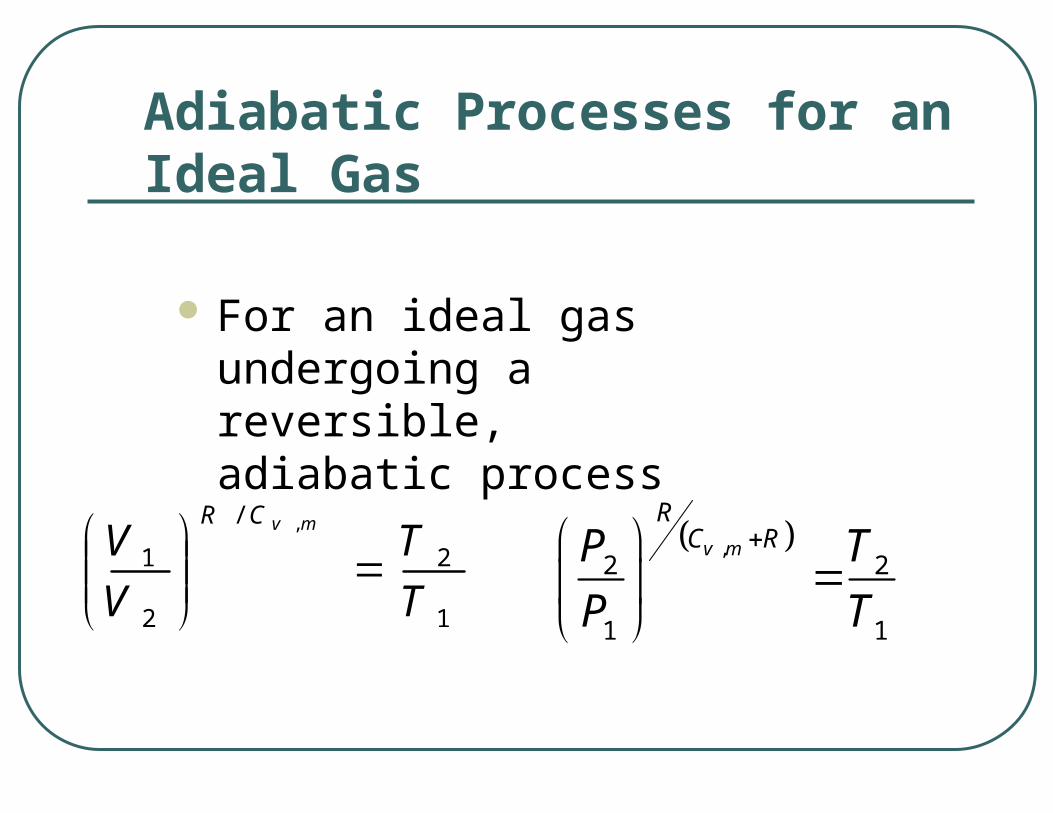
Adiabatic Processes for an Ideal Gas
For an ideal gas undergoing a reversible, adiabatic process
1
2
/
2
1,
TT
VV
mvCR
,2 2
1 1
v m
RC RP T
P T

Defining the enthalpy of the system Re-examine the piston with the weight
on top
State Changes Under Constant Pressure Conditions
Piston(T, P, V)
mass (m)

The first law
A Constant Pressure Process
Integrating
PdVdqdU P
2
1
2
1
V
VcP dVPdqdU

Define the enthalpy of the system, H
Enthalpy
PVUH

In general, we write H as a function of T and P
The Properties of H
dPPH
dTTH
dHTP

Examine the first partial derivative
Isobaric Changes in H
0
dTTH
dHP

Define the constant pressure heat capacity, CP
The Constant Pressure Heat Capacity
PPP T
HdTdq
C

For a system undergoing an isobaric temperature change
Heat Flows Under Constant Pressure Conditions
For a macroscopic system
dTCdH P
2
1
T
TPP dTCqH

For an ideal gas
Relating CP and CV
nRCC VP
In general
TVP
TVCC
2

Examine the second partial derivative
Isothermal Changes in H
dPPH
dHT
0
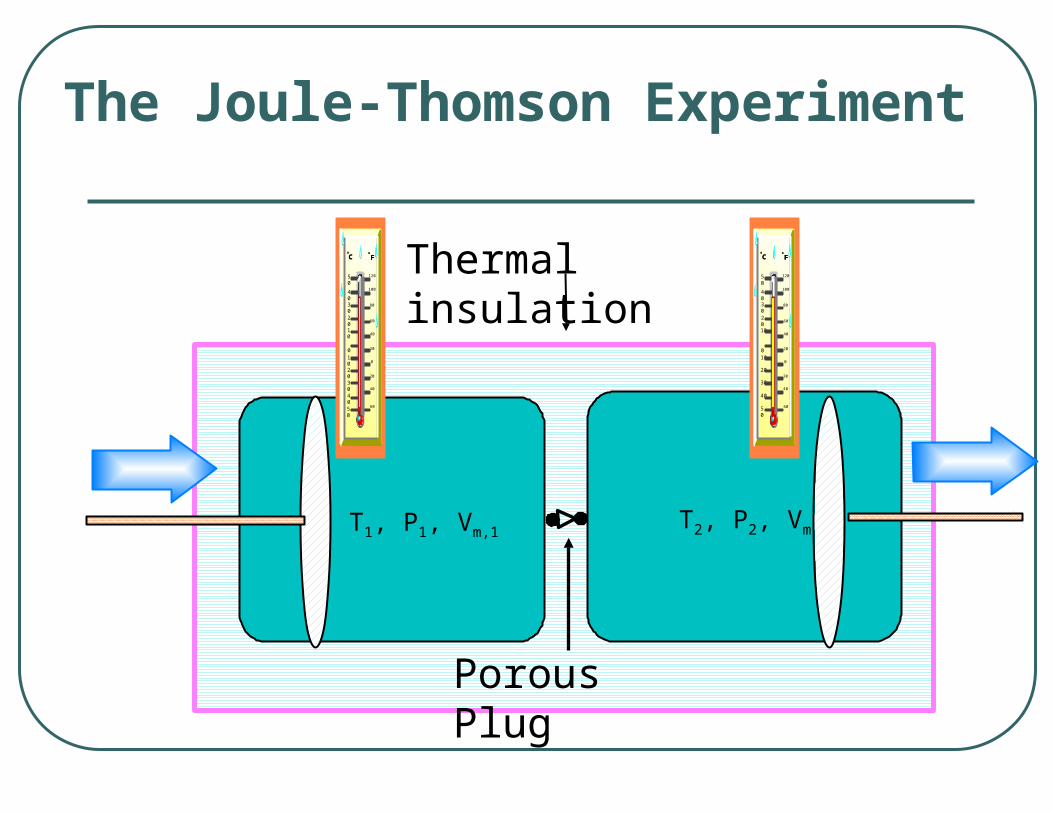
The Joule-Thomson Experiment
Porous Plug
Thermal insulation
T1, P1, Vm,1T2, P2, Vm,2
FFOO
CO
CO
50
40302010 01020304050
120
100
80
0
20
20
40
60
60
40
FFOO
CO
CO
50
40302010 01020304050
120
100
80
0
20
20
40
60
60
40

The partial derivative
The Joule-Thomson Coefficient
is known as the Joule-Thomson coefficient, JT.
HPT

The change in the enthalpy under constant pressure conditions is related to the Joule-Thomson Coefficient
Relating H to the Joule-Thompson Coefficient
PHT TH
PT
PH
JTPT
CPH

Enthalpy Changes for Reactions
The shorthand form for a chemical reaction
J
JJ0
J = chemical formula for substance JJ = stoichiometric coefficient for J

Reaction Enthalpy Changes
The enthalpy change for a chemical reaction
JHnHJ
mJr
Hm [J] = molar enthalpies of substance J
nJ = number of moles of J in the reaction

The Enthalpy Change
Reaction beginning and ending with equilibrium or metastable states
JHn
HHH
JmJ
initialfinalr
Note – Initial and final states have the same temperature and pressure!

Reaction Enthalpies (cont’d)
We note that 1 mole of a reaction occurs if
JJn
JHHJ
mJr

A Standard State Reaction
A reaction that begins and ends with all substances in their standard states
The degree sign, either or
• P = 1.00 bar• [aqueous species] = 1.00 mol/ kg• T = temperature of interest (in data tables -
25C or 298 K).

Standard Reaction Enthalpies
We note that for 1 mole of a reaction under standard conditions
JHHJ
mJr

The Formation Reaction
A "chemical thermodynamic reference point."
For CO and CO2
C (s) + O2 (g) CO2 (g)
C (s) + ½ O2 (g) CO (g)

The Formation Reaction
The formation reaction• 1 mole of a compound • constituent elements • stable state of aggregation at that temperature.
Formation of 1.00 mole of Na2SO3(s)
2 Na(s) + S(s) + 3/2 O2 (g) Na2SO3 (s)
‘Formation enthalpy of Na2SO3(s)’, fH°[Na2SO3 (s)]

The Significance of the Formation Enthalpy
fH° is a measurable quantity!
Compare CO (g) with CO2 (g)
C (s) + 1/2 O2 (g) CO (g)
fH° [CO(g)] = -110.5 kJ/mole
C (s) + O2 (g) CO2 (g)
fH° [CO2(g)] = - 393.5 kJ/mole

Formation Enthalpies
Formation enthalpies - thermodynamic reference point! • Ho
m [J] = fH [J]
• Hm [elements] = 0 kJ / mole.
Use the tabulated values of the formation enthalpies

The General Equation
The enthalpy change for a given reaction is calculated from the formation enthalpies as
Notes Reverse a reaction Multiply a reaction by an integer
JHHJ
fJr

The Calorimeter
A calorimeter - device containing water and/or another substance with a known heat capacity
Calorimeters – either truly or approximately adiabatic systems
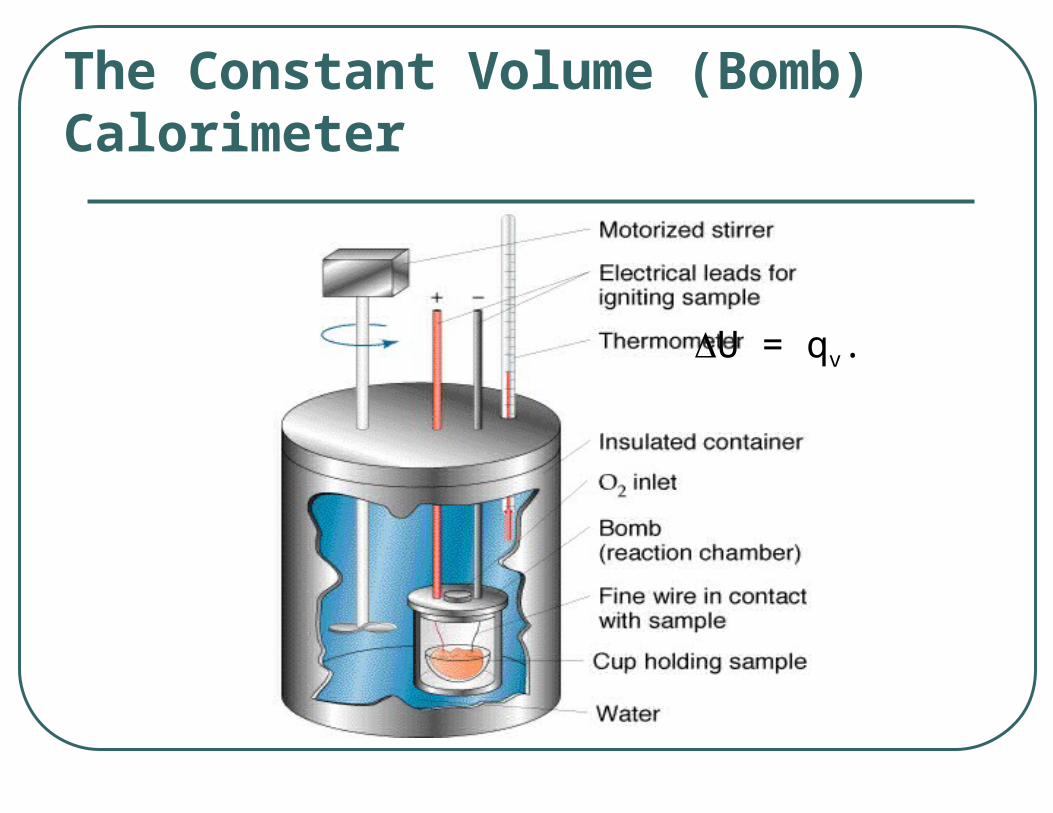
The Constant Volume (Bomb) Calorimeter
U = qv.
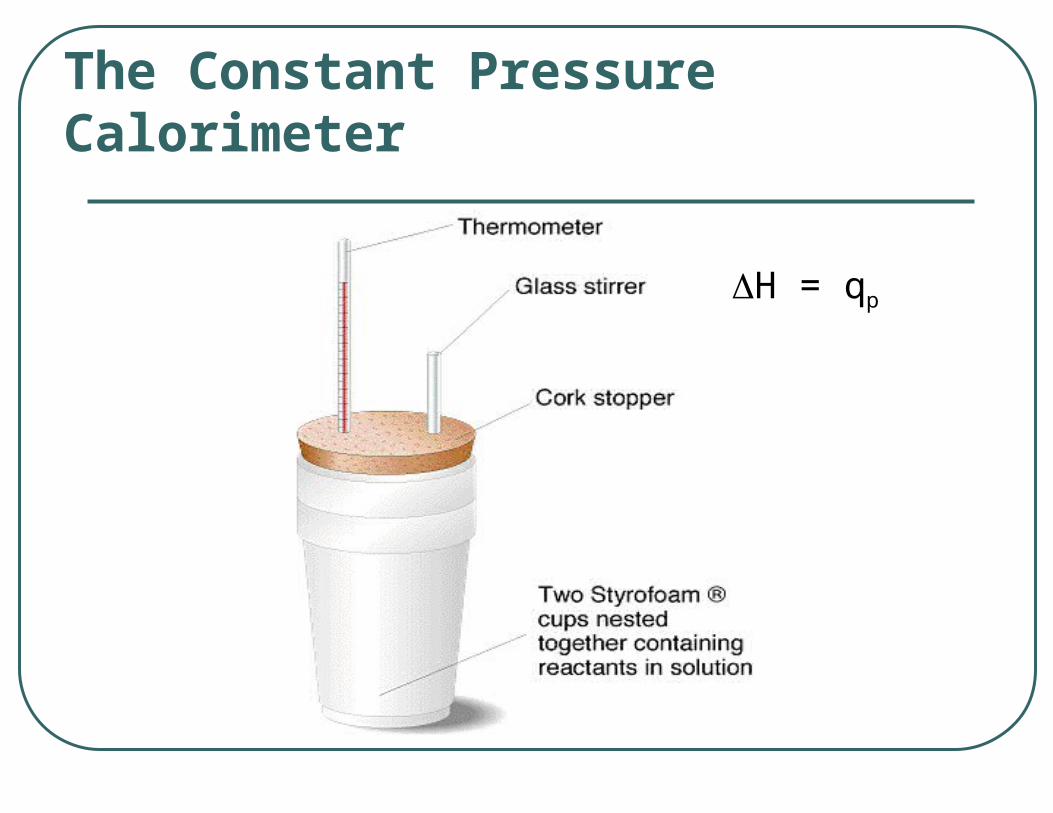
The Constant Pressure Calorimeter
H = qp

Relating H and U
The enthalpy and the internal energy both represent quantities of heat.
U = qv.
H = qp.
Relate the two state functions using the following relationship
U = H - PV

Other Important Enthalpy Changes
Enthalpy of solution Enthalpy of dilution Enthalpy of fusion Enthalpy of vapourisation

The Solution Enthalpy
solH - heat absorbed or released when a quantity of solute is dissolved in fixed amount of solvent
solH = Hm(sol’n) – Hm(component)• H(component) = Hm(solid) + Hm(solvent)
Two definitions• Standard• Limiting

The Dilution Enthalpy
For the process,
HCl (aq, 6 M) HCl (aq, 1 M). The Enthalpy of dilution of the acid.
dilH = Hm(sol’n 2) – Hm(sol’n ,1)

Reaction Enthalpy Changes With Temperature
Differentiate the reaction enthalpy with temperature
JHHJ
mJr
JHdTd
dTHd
JmJ
r

The Result
rCp
- the heat capacity change for the reaction
TCKHTH prrr 298
J
pJpr JCC

Internal Energy Changes in Chemical Reactions
Examine a chemical reaction.
C (s) + O2 (g) CO2 (g)
U = U[CO2 (g)] – U[C(s)] – U[O2(g)]
Note - rH = -393.5 kJ/mole
RTnUH
JUU
grr
JfJr

Enthalpies and Hess’s Law
Use tabulated values of formation enthalpies to obtain rH°.
May also estimate reaction enthalpies using an indirect method.

Hess’s Law
Hess’s Law – • the enthalpy change for a given reaction is
the same whether the reaction occurs in a single step or in many steps.

Bond Energies
Examine the following reactions H2 (g) ® H (g) + H (g) DU° = 433.9 kJ
Cl2 (g) ® Cl (g) + Cl (g)DU° = 239.5 kJ
Bond dissociation energies. Enthalpy changes are designated D (H-
H) and D (Cl-Cl).

For Polyatomic Molecules
CO2 (g) ® C (g) + 2 O (g)DU = 740 kJ DH of this reaction D(C=O) What about dissociating methane into C
+ 4 H’s?CH4(g) ® C(g) + 4 H(g) DU° = 1640 kJ
4 C-H bonds in CH4 \ D (C-H) 410 kJ/mol

Make or Break!!
Note: all chemical reactions involve the breaking and reforming of chemical bonds• Bonds break - we add energy. • Bonds form - energy is released.
rU° S D(bonds broken) - S D(bonds formed)

A Word of Caution
These are close but not quite exact. Why?
The bond energies we use are averaged bond energies !
This is a good approximation for reactions involving diatomic species.
Can only use the above procedure for GAS PHASE REACTIONS ONLY!!!





























