Embed Size (px)
Citation preview

www.wjpr.net Vol 3, Issue 4, 2014.
1244
Kathpalia et al. World Journal of Pharmaceutical Research
DEVELOPMENT AND EVALUATION OF A READY TO USE
ANTIMALARIAL ORAL SUSPENSION
Harsha Kathpalia*, Chetan Phadke
Department of Pharmaceutics, Vivekanand Education Society‟s College of Pharmacy,
HashuAdvani Memorial Complex, Behind Collector Colony, Chembur (E), Mumbai-
400074, India.
ABSTRACT
Some physiologically active agents such as antibiotics, anti malarial
are chemically unstable in water-based oral suspensions. Non-aqueous
suspensions can be produced which are extremely palatable, are ready-
to-use, without any reconstitution and need not be refrigerated to
maintain the potency of the active ingredient and have a longer shelf
life relative to conventional dry syrup formulations. A ready-to-use
anhydrous suspension of a moisture sensitive antimalarial which can
be stored at room temperature was prepared and evaluated for its
organoleptic properties, sedimentation behavior, redispersibility,
moisture content, viscosity, accelerated physical and chemical stability.
The formulations showed improved redispersibility and no caking
tendency. The products exhibited longer shelf-life at ambient
conditions.
KEYWORDS: Anhydrous vehicle, anti malarial, ready-to-use, stability, sweetener,
thickener.
INTRODUCTION
Marketed moisture sensitive antimalarial suspensions are generally available as dry syrups
and before dispensing it to patient, need to be reconstitution with a diluent. After
reconstitution, suspension requires refrigeration to maintain the potency of the medicament(s)
over the recommended duration of treatment. In dry syrup formulation, if powder is diluted
with an inaccurate volume of diluent or if the liquid and dry components are not properly
mixed, then it leads to formation of suspension having non-uniform dosing due to clumping
World Journal of Pharmaceutical Research SJIF Impact Factor 5.045
Volume 3, Issue 4, 1244-1259. Research Article ISSN 2277 – 7105
Article Received on
18 April 2014,
Revised on 14 May 2014,
Accepted on 07 June 2014
*Author for Correspondence
Harsha Kathpalia
Department of Pharmaceutics,
Vivekanand Education
Society‟s College of
Pharmacy, HashuAdvani
Memorial Complex, Behind
Collector Colony, Chembur
(E), Mumbai-400074, India.

www.wjpr.net Vol 3, Issue 4, 2014.
1245
Kathpalia et al. World Journal of Pharmaceutical Research
and/or doses which are either super-potent or sub-potent depending upon the amount of
diluent added. Reconstituted suspension is stable only upto 14 days under refrigeration
condition [1, 2]
.
Non-aqueous suspensions can be administered without reconstitution i.e. “ready-to-use”,
pleasant to taste and need not be refrigerated to preserve the strength of the active
ingredient.A ready-to-use suspension has an acceptable shelf life at normal and elevated
temperatures without refrigeration and in addition, has less inter-subject variability[3]
. This is
a viable option for products that may have to be reconstituted where the water supply and
storage facilities are poor. Furthermore, the composition of these suspensions is such that no
special production techniques or packaging components are required. Instead standard filling
equipment and containers such as plastic or glass bottles may be utilized [4]
.
To acquire physical stability is major challenge encountered in developing a good aqueous
suspension [5]
. "The three major issues associated with aqueous suspensions are i) inadequate
dispersion of the particles in the vehicle, ii) settling of the dispersed particles and iii) caking
of these particles in the sediment so as to resist redispersion." These problems can be easily
solved by choosing an edible oil as dispersion vehicle.
The aim of the present study is to furnish a ready-to-use oil based, liquid antimalarial
suspension for oral administration having good shelf life and taste characteristics relative to
conventional dry syrup formulation.
MATERIALS
Lumefantrine was procured from IPCA Labs and Cipla Ltd. Mumbai. Colloidal silicon
dioxide (AEROSIL 200, SYLOID-244FP) was obtained from W. R. Grace &Co. India Pvt
Ltd. Butylatedhydroxylanisole (BHA), Butylatedhydroxyltoluene (BHT), Methyl paraben,
Propyl paraben, Anhydrous citric acid, Menthol were of LR grade quality. Aspartame from
Rubicon Research Pvt Ltd, Acesulfame potassium (SUNETT)from SBS sugar free agency
Pvt Ltd and Monoammoniumglycyrrhizinate (MAGNASWEET-MM100)from Mafco
worldwide corporation were obtained. Medium chain triglyceride (MIGLYOL-812) was
obtained from Subhash chemicals, Pune.
www.wjpr.net Vol 3, Issue 4, 2014.
1246
Kathpalia et al. World Journal of Pharmaceutical Research
METHOD
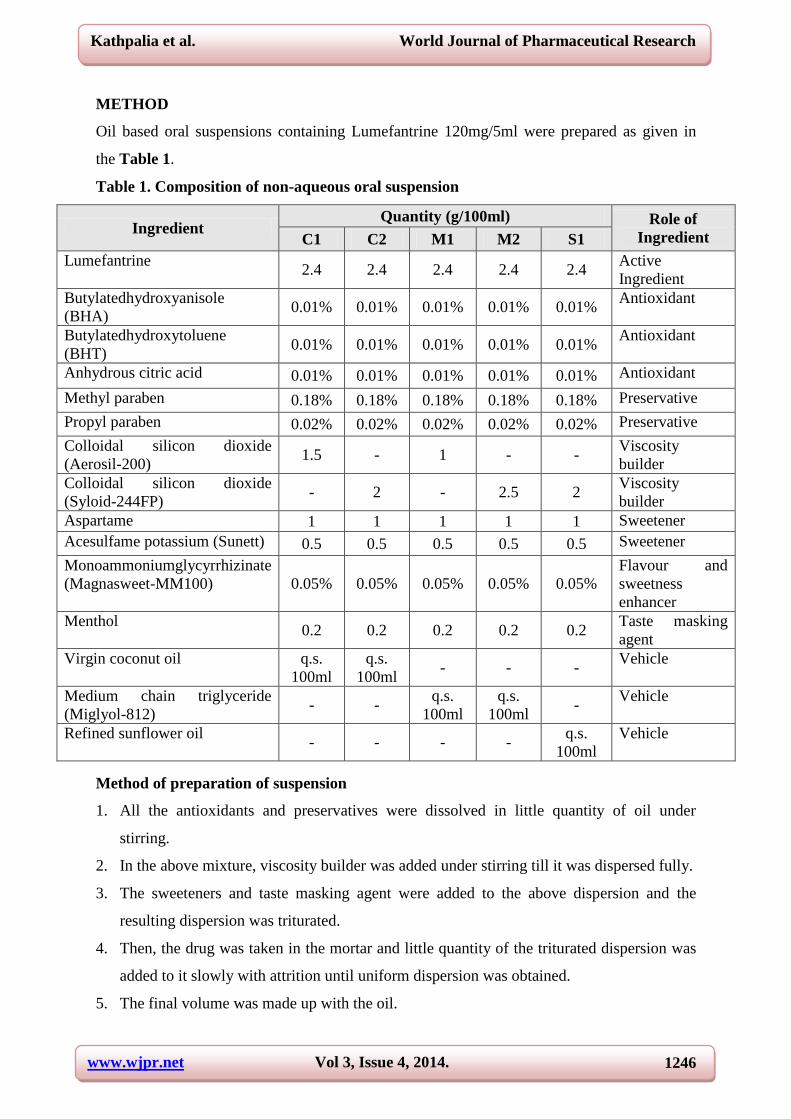
Oil based oral suspensions containing Lumefantrine 120mg/5ml were prepared as given in
the Table 1.
Table 1. Composition of non-aqueous oral suspension
Ingredient Quantity (g/100ml) Role of
Ingredient C1 C2 M1 M2 S1
Lumefantrine 2.4 2.4 2.4 2.4 2.4
Active
Ingredient
Butylatedhydroxyanisole
(BHA) 0.01% 0.01% 0.01% 0.01% 0.01%
Antioxidant
Butylatedhydroxytoluene
(BHT) 0.01% 0.01% 0.01% 0.01% 0.01%
Antioxidant
Anhydrous citric acid 0.01% 0.01% 0.01% 0.01% 0.01% Antioxidant
Methyl paraben 0.18% 0.18% 0.18% 0.18% 0.18% Preservative
Propyl paraben 0.02% 0.02% 0.02% 0.02% 0.02% Preservative
Colloidal silicon dioxide
(Aerosil-200) 1.5 - 1 - -
Viscosity
builder
Colloidal silicon dioxide
(Syloid-244FP) - 2 - 2.5 2
Viscosity
builder
Aspartame 1 1 1 1 1 Sweetener
Acesulfame potassium (Sunett) 0.5 0.5 0.5 0.5 0.5 Sweetener
Monoammoniumglycyrrhizinate
(Magnasweet-MM100) 0.05% 0.05% 0.05% 0.05% 0.05%
Flavour and
sweetness
enhancer
Menthol 0.2 0.2 0.2 0.2 0.2
Taste masking
agent
Virgin coconut oil q.s.
100ml
q.s.
100ml - - -
Vehicle
Medium chain triglyceride
(Miglyol-812) - -
q.s.
100ml
q.s.
100ml -
Vehicle
Refined sunflower oil - - - -
q.s.
100ml
Vehicle
Method of preparation of suspension
1. All the antioxidants and preservatives were dissolved in little quantity of oil under
stirring.
2. In the above mixture, viscosity builder was added under stirring till it was dispersed fully.
3. The sweeteners and taste masking agent were added to the above dispersion and the
resulting dispersion was triturated.
4. Then, the drug was taken in the mortar and little quantity of the triturated dispersion was
added to it slowly with attrition until uniform dispersion was obtained.
5. The final volume was made up with the oil.

www.wjpr.net Vol 3, Issue 4, 2014.
1247
Kathpalia et al. World Journal of Pharmaceutical Research
Evaluation[1, 2]
Organoleptic properties
The suspension was evaluated for the general appearance, odor and taste.
Sedimentation Volume
Sedimentation volume (Vs) of the formulation was determined using following formula.
Where,
Hu = ultimate height of the sediment
Ho = initial height of the total suspension.
The height of the sediment was noted at particular time intervals. The Hu/Ho ratios were
obtained and plotted on Y-axis with time on X-axis. Formulations with different grades of
colloidal silicon dioxide at different concentrations were evaluated for the sedimentation
behavior.
Particle Size
Particle size determination was carried out using optical microscopy.
The particle size of the suspended active substance can greatly in fluences properties of oral
suspension.
Ease of redispersibility as such and after freeze-thaw cycling [6]
Twenty milliliter suspension samples were subjected to 3 cycles of 40C & 30
0C each of 24
hours and studied for their physical instability like caking.
The suspension was allowed to settle in a stoppered measuring cylinder. The cylinder was
inverted through 1800 and number of inversions necessary to restore a homogeneous
suspension was determined. If the homogeneity of the suspension was attained in one
inversion, then the suspension was considered 100% easily redispersible. Every additional
inversion decreases the percentage of ease of redispersibility by 5%.
Rheological studies [7]
A Brookfield digital rotational viscometer was used to measure the viscosity of the
suspension in centipoise.Viscosities were determined in triplicate at ambient temperature.
Speed ramp test: The spindle (spindle no. LV 63) was rotated in the suspension at increasing
shear rates i.e. 10, 20, 30, 50, 60 and 100rpm. At each speed, the corresponding dial reading

www.wjpr.net Vol 3, Issue 4, 2014.
1248
Kathpalia et al. World Journal of Pharmaceutical Research
was noted. The speed was then lowered gradually and viscosities were again noted and the
conclusion was drawn from the graph.
Time sensitivity test: The suspensions were studied for change in viscosity with time at a
fixed rpm. The spindle (spindle no. LV 63) was rotated at a fixed rpm in the suspension for a
period of 30 minutes. At each specific time interval, the corresponding dial reading was
noted. Graph of log viscosity Vs time was plotted and conclusion was drawn from the graph.
Moisture content determination by Karl Fischer titrimetry
The water content of the suspension was determined by Karl Fischer titrimetry (Automatic
Potentiometric Titrator, AT-38C, Spectralab).
Assay[8]
An assay was performed on HPLC (Shimadzu-DGU/20A5/Proximal degassor, HIQSIL C8
column 150 x 4.6 mm, 5µm) as per authorized USP salmous standard version 1 for
determining the content of Lumefantrine.
5gms of suspension was weighed in 200 ml volumetric flask; to it 20 ml of 10%w/v
orthophosphoric acid was added and sonicated for 25 minutes. Then, 120 ml of acetonitrile
was added and sonicated for further 20 minutes. It was allowed to cool at room temperature
and diluted up to 200 ml with acetonitrile. Solution was filtered through membrane filter
(GF/C) and analyzed by HPLC method.
Chromatographic conditions
Parameters Specifications
Column Waters symmetry C18 (150mm x 4.6 mm)
Mobile phase composition Buffer: Acetonitrile (30:70)
Flow rate 1.0 ml/min
Injection volume 20µl
Detector wavelength 380 nm
Run time 8 minutes
In-vitro dissolution test[8]
The dissolution studies were performed using a US Pharmacopeia XXXIII type II dissolution
test apparatus. The sample equivalent to 120mg Lumefantrine was placed in each of the
dissolution vessels containing 1000 ml of dissolution media maintained at 37.0±0.5°C and
stirred at 100 rpm. A 5ml aliquot was withdrawn from the dissolution medium at
predetermined intervals of 15, 30 and 45 minutes period. Every time the sample withdrawn

www.wjpr.net Vol 3, Issue 4, 2014.
1249
Kathpalia et al. World Journal of Pharmaceutical Research
was replaced by fresh dissolution medium maintained at the same temperature. The sample
removed was filtered, diluted and analyzed by measuring the absorbance using a
spectrophotometer (UV 150-02, Shimadzu Corporation, Kyoto, Japan) at wavelength 342nm
after suitable dilution. Amount of drug dissolved at various time intervals was calculated for
all the formulations. According to the Authorized USP SALMOUS standard version 1 of
Artemether and Lumefantrine for oral suspension, 60% of the amount of Lumefantrine
should dissolve in 40 minutes.
i) Dissolution rate study in official dissolution media
Dissolution study was carried out in 1%w/v Benzalkonium chloride (BKC) in 1000ml of
0.1N hydrochloric acid (HCl).
ii) Dissolution rate study in unofficial dissolution media[9]
Dissolution study was carried out in 2%w/v Myrj-52 (Polyethyeneglycol-40-stearate) in
1000ml of 0.005M HCl.
iii) Dissolution rate study in Bio-relevant media
1 dose (53.6 gms) of Ensure nutritional powder (Abbott Laboratories) was dissolved in
1000ml of 1%w/v Benzalkonium chloride solution containing 0.1N HCl. The drug was
extracted in chloroform and then analyzed at 342nm using UV-Vis spectrophotometer.
Stability Studies
Stability of the selected formulation was monitored at 400C/75%RH and 25
0C/60%RH for a
period of three months in amber colored glass bottles with screw cap. The formulation was
visually inspected for appearance, sedimentation volume, redispersibility and evaluated for
viscosity, particle size, moisture content, drug content and in-vitro dissolution study.
RESULTS AND DISCUSSION
Results of evaluation tests of Lumefantrine oil based suspensions are shown in Table2.
Table2. Evaluation results of physico-chemical studies
Parameters
Evaluated M1 M2 C1 C2 S1
Appearance Greenish
yellow
viscous
liquid
Greenish
yellow
viscous
liquid
Greenish
yellow
viscous
liquid
Greenish
yellow
viscous
liquid
Slightly
greenish
yellow viscous
liquid
Odor No odor No odor
Smell of
coconut oil
Smell of
coconut oil
Smell of
sunflower oil
www.wjpr.net Vol 3, Issue 4, 2014.
1250
Kathpalia et al. World Journal of Pharmaceutical Research
Taste and
Palatability
Sweet,
palatable
Sweet,
palatable
Sweet,
palatable
Sweet,
palatable
Sweet,
palatable
Sedimentation
Volume (After
24hrs)
0.85 0.9 0.9 0.85 0.8
Median Particle
size (μm) 37.32 21.58 29.62 27.70 26.13
Redispersibility
(%) as such 100 100 90 95 100
Redispersibility
after freeze-thaw
cycling
95 95 80 90 90
Viscosity in cP
(Spindle no. 63,
at 100rpm)
199 238 431 175 189
Moisture content
(%) 0.195 0.24 0.356 0.296 0.358
The prepared formulations of Lumefantrine oil based suspension were found to possess an
excellent redispersibility property with optimum particle size. Sedimentation studies showed
that the sedimentation volume of all formulations is close to 1, which indicated that the
formulations were optimum and acceptable. The viscosity of all the formulations was such
that it would be easily pourable from the container and also showed a shear thinning effect.
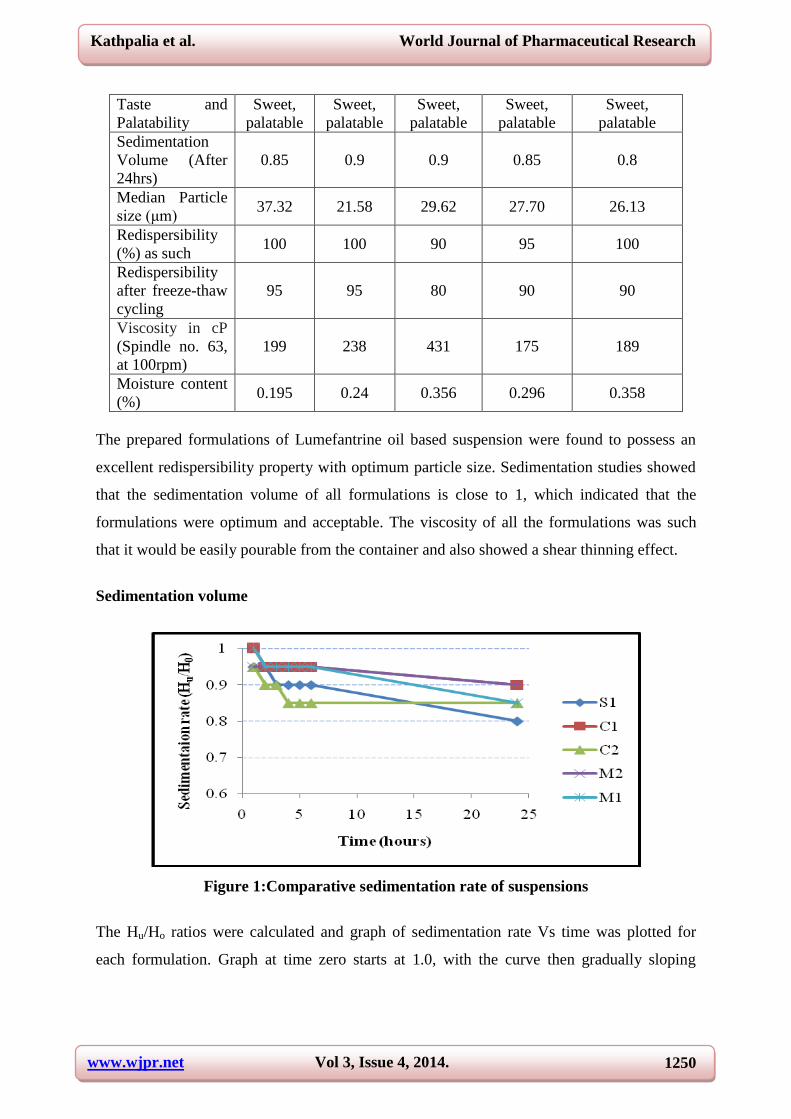
Sedimentation volume
Figure 1:Comparative sedimentation rate of suspensions
The Hu/Ho ratios were calculated and graph of sedimentation rate Vs time was plotted for
each formulation. Graph at time zero starts at 1.0, with the curve then gradually sloping

www.wjpr.net Vol 3, Issue 4, 2014.
1251
Kathpalia et al. World Journal of Pharmaceutical Research
downward to the right as time proceeds. The formulation producing least slope values were
considered to have good suspendibility.
The formulations C1 and M2 were found to have good sedimentation but, viscous in nature
and less pourable. Thus, the formulations M1 and C2 were found to have satisfactory
suspendibility.
Rheological studies
Figure 2: Time sensitivity test of suspensions
Figure 3: Speed ramp test of suspensions
Speed ramp test and Time sensitivity test can help in determining the flow behavior of the
suspension on storage and on shaking.
In speed ramp test, if the upward-curve and downward-curve overlap the fluid is time-
independent. If they don't, the fluid is time dependent. If the upward curve indicates a higher
viscosity than the downward curve, the fluid is thixotropic. If the upward curve indicates

www.wjpr.net Vol 3, Issue 4, 2014.
1252
Kathpalia et al. World Journal of Pharmaceutical Research
lower viscosity than the downward curve, the fluid is rheopectic. Formulations M1, C2 were
showed time dependent behavior and thixotropic nature. Since, there was a decrease in
viscosity with increasing shear rate; it indicated pseudoplastic nature of suspension. In time
sensitivity test, if there is a change in fluid's viscosity over time, it indicates time-dependent
behavior of the suspension. If there is decrease in viscosity, it indicates thixotropy andif there
is increase in viscosity, it indicates rheopexy. Formulations M1, M2 and C2 showed time-
dependent behavior and thixotropic nature.
From graph, it was concluded that formulations M1, M2 and C2 have thixotropic and
pseudoplastic nature which is desirable for a good suspension.
Moisture content analysis
The initial moisture content of all formulations is given in Table2. Since the all suspensions
contains less than about 0.5%w/v moisture, they can be termed as „anhydrous‟.
Assay
Assay results of Lumefantrine in selected formulations were listed in Table 3.
Table 3. Assay of Lumefantrine in selected formulations
Sr.
No. Formulations
Lumefantrine
(%)
1 M1 96.16
2 C2 98.14
3 M2 96.41
The assay values of Lumefantrine in selected formulations complied with the acceptance
criteria for assay given in the USP specification which is 95% to 105% of Lumefantrine.
In-vitro dissolution rate study
i) Dissolution rate profile of Lumefantrine
Figure4:Comparative dissolution profile of Lumefantrine in suspensions

www.wjpr.net Vol 3, Issue 4, 2014.
1253
Kathpalia et al. World Journal of Pharmaceutical Research
Dissolution profile of Lumefantrine in all formulations failed to comply with USP
specifications.
Probable reasons for poor dissolution rate could be-
1) Floating of sample on surface of dissolution media i.e. improper dispersion of sample into
the dissolution media;
2) Drug has poor affinity to aqueous dissolution media.
2%w/v Myrj-52 solution and Biorelevent dissolution media were also tried to check
dissolution rate of selected formulations. Since, formulation M1 showed better dissolution
result compared to others. Hence, this formulation was further studied.
ii) Dissolution rate study in unofficial dissolution media and Bio-relevant media
mimicking fed state
Figure5:Comparative dissolution profile of formulation M1 in 2%w/v Myrj dissolution
media and Biorelevent dissolution media
Dissolution rate of Lumefantrine in 2%w/v Myrj-52 solution failed to comply with USP
specifications and dissolution rate of Lumefantrine was same as official media. Dissolution
rate of formulation M1 for Lumefantrine in Biorelevent media (Ensure dissolution media)
showed significant improvement compared with official dissolution media and 2%w/v Myrj-
52 solution. Also, it complied with USP dissolution specification.
Therefore, on basis of dissolution studies carried out in unofficial dissolution media and
Biorelevent dissolution media, it was concluded that official dissolution conditions could be
inappropriate for dissolution study of Lumefantrine oil based suspensions.

www.wjpr.net Vol 3, Issue 4, 2014.
1254
Kathpalia et al. World Journal of Pharmaceutical Research
Stability Studies
Table 4. Stability study data of selected formulation M1of Lumefantrine suspension at
the end of 3 months interval
Test parameters Initial 25°C ± 60%RH 40°C ± 75%RH
Appearance Yellowish green,
pourable liquid
Yellowish green,
pourable liquid
Yellowish green,
pourable liquid
Taste and Palatability Sweet, palatable Sweet, palatable Sweet, palatable
Odor No smell No smell Slight smell
Sedimentation rate
(After 24 hours) 0.85 0.95 0.95
Particle size (μm) 37.32 53.18 60.29
Redispersibility as such (%) 100 95 90
Redispersibility after freeze-
thaw cycling (%) 95 90 85
Viscosity (cP) 199 234 500
Moisture content (%) 0.195 0.349 0.413
Assay (%) Lumefantrine 96.16 95.44 95.37
In-vitro dissolution rate (%)
in USP dissolution media 52.28 50.17 49.99
There was change in particle size at the end of 3 months but it was within acceptable range. A
significant increase in viscosity at accelerated temperature was observed on storage, but after
shaking it showed shear thinning effect. The drug content and moisture content were found to
be within limits throughout 3 months interval at both the conditions studied.There was no
significant physico-chemical change observed during accelerated stability studies.
FIGURES
Figure 1:Comparative sedimentation rate of suspensions

www.wjpr.net Vol 3, Issue 4, 2014.
1255
Kathpalia et al. World Journal of Pharmaceutical Research
Figure 2: Time sensitivity test of suspensions
Figure 3: Speed ramp test of suspensions
Figure 4:Comparative dissolution profile of Lumefantrine in suspensions

www.wjpr.net Vol 3, Issue 4, 2014.
1256
Kathpalia et al. World Journal of Pharmaceutical Research
Figure 5:Comparative dissolution profile of formulation M1 in 2%w/v Myrj dissolution
media and Biorelevent dissolution media
TABLES
Table 1. Composition of non-aqueous oral suspension
Ingredient Quantity (g/100ml) Role of
Ingredient C1 C2 M1 M2 S1
Lumefantrine 2.4 2.4 2.4 2.4 2.4
Active
Ingredient
Butylatedhydroxyanisole
(BHA) 0.01% 0.01% 0.01% 0.01% 0.01% Antioxidant
Butylatedhydroxytoluene
(BHT) 0.01% 0.01% 0.01% 0.01% 0.01% Antioxidant
Anhydrous citric acid 0.01% 0.01% 0.01% 0.01% 0.01% Antioxidant
Methyl paraben 0.18% 0.18% 0.18% 0.18% 0.18% Preservative
Propyl paraben 0.02% 0.02% 0.02% 0.02% 0.02% Preservative
Colloidal silicon dioxide
(Aerosil-200) 1.5 - 1 - -
Viscosity
builder
Colloidal silicon dioxide
(Syloid-244FP) - 2 - 2.5 2
Viscosity
builder
Aspartame 1 1 1 1 1 Sweetener
Acesulfame potassium (Sunett) 0.5 0.5 0.5 0.5 0.5 Sweetener
Monoammoniumglycyrrhizinate
(Magnasweet-MM100) 0.05% 0.05% 0.05% 0.05% 0.05%
Flavour and
sweetness
enhancer
Menthol
0.2 0.2 0.2 0.2 0.2
Taste
masking
agent
Virgin coconut oil q.s.
100ml
q.s.
100ml - - - Vehicle
Medium chain triglyceride
(Miglyol-812) - -
q.s.
100ml
q.s.
100ml - Vehicle
Refined sunflower oil - - - -
q.s.
100ml Vehicle

www.wjpr.net Vol 3, Issue 4, 2014.
1257
Kathpalia et al. World Journal of Pharmaceutical Research
Table2. Evaluation results of physico-chemical studies
Parameters
Evaluated M1 M2 C1 C2 S1
Appearance Greenish
yellow
viscous
liquid
Greenish
yellow
viscous
liquid
Greenish
yellow
viscous
liquid
Greenish
yellow
viscous
liquid
Slightly
greenish
yellow
viscous liquid
Odor No odour No odour
Smell of
coconut oil
Smell of
coconut oil
Smell of
sunflower oil
Taste and
Palatability
Sweet,
palatable
Sweet,
palatable
Sweet,
palatable
Sweet,
palatable
Sweet,
palatable
Sedimentation
Volume (After
24hrs)
0.85 0.9 0.9 0.85 0.8
Median Particle
size (μm) 37.32 21.58 29.62 27.70 26.13
Redispersibility
(%) as such 100 100 90 95 100
Redispersibility
after freeze-thaw
cycling
95 95 80 90 90
Viscosity
(Spindle no. 63,
at 100rpm)
199 238 431 175 189
Moisture content
(%) 0.195 0.24 0.356 0.296 0.358
Table 3. Assay of Lumefantrine in selected formulations
Sr.
No. Formulations
Lumefantrine
(%)
1 M1 96.16
2 C2 98.14
3 M2 96.41
Table 4. Stability study data of selected formulation M1 of Lumefantrine suspension at
the end of 3 months interval
Test parameters Initial 25°C ± 60%RH 40°C ± 75%RH
Appearance Yellowish green,
pourable liquid
Yellowish green,
pourable liquid
Yellowish green,
pourable liquid
Taste and Palatability Sweet, palatable Sweet, palatable Sweet, palatable
Odor No smell No smell Slight smell
Sedimentation rate
(After 24 hours) 0.85 0.95 0.95
Particle size (μm) 37.32 53.18 60.29
Redispersibility as such (%) 100 95 90
Redispersibility after freeze-
thaw cycling (%) 95 90 85

www.wjpr.net Vol 3, Issue 4, 2014.
1258
Kathpalia et al. World Journal of Pharmaceutical Research
Viscosity (cP) 199 234 500
Moisture content (%) 0.195 0.349 0.413
Assay (%) Lumefantrine 96.16 95.44 95.37
In-vitro dissolution rate (%)
in USP dissolution media 52.28 50.17 49.99
CONCLUSION
Improved suspend ibility and redispers ibility results in an improved product because less
shaking of the suspension is required before dosing and allows the product to have a longer
shelf-life. Since, the moisture sensitive drug in the oily vehicle shows significantly improved
stability in the ready-to-use suspension form as it prevents hydrolysis of the drug. Such
ready-to-use suspension is very suitable in conditions as in remote areas and in the third
world countries where the people suffering from malaria are large in numbers and illiterate
people cannot understand the direction for use of dry syrups requiring reconstitution and
storage at refrigeration condition.
The oil used in the non-aqueous suspension may improve the absorption of Lumefantrine in-
vivo apparently due to digestion of the oil and subsequent solubilization of the drug. The oil
may also facilitate the transport of Lumefantrine across the intestinal membrane10
. Further
study is needed to optimize dissolution media mimicking the in-vivo fed conditions for oil
based formulations, since Lumefantrine is orally absorbed lipophilic antimalarial drug with
less bioavailability and its absorption is enhanced in the presence of food.
ACKNOWLEDGEMENT
We are thankful to principal of VES College of Pharmacy, Chembur, Mumbai for providing
the facilities to carry out this work and also thankful to IPCA Lab. and Cipla Ltd. for
providing Lumefantrine.I am grateful to University Grant Commission for granting me a
Junior Research fellowship for the project.
REFERENCE
1. Kathpalia H, Mittal S, Bhatia V. Development and evaluation of a ready to use
cefpodoximeproxetil suspension. Int J Pharm Sci Res 2011;2(8):2173-7.
2. Kathpalia H, Shidhaye S, Mittal S, Bhatia V, Pillai P. Development and evaluation of a
ready to use paediatric antibiotic suspension. Int J Advances Pharm Nanotech
2011;1(2):71–7.

www.wjpr.net Vol 3, Issue 4, 2014.
1259
Kathpalia et al. World Journal of Pharmaceutical Research
3. Mulligan, S. Liquid suspension for oral administration, U.S. Patent 5,156,842, October 20,
1992.
4. Song-Ling lin.; Pramoda, M. Permanent suspension pharmaceutical dosage form, U.S.
patent 4,080,445, March 21, 1978.
5. Higuchi WI, Swarbrick J, Simonelli AP, Martin A. Particle phenomena and coarse
dispersions. Remington's Pharmaceutical Sciences. 17th
ed., Easton: Mack Publishing
Company; 1985; 301-329.
6. Elkheshen SA. Optimization of a reconstitutable suspension of rifampicin using 24factorial
designs. Drug DevInd Pharm 1996;22(7):623-30.
7. More solutions to sticky problems: A guide to getting more from your Brookfield
viscometer: Brookfield Engineering Laboratories, Inc. pp. 4-12.
8. Google. Authorized USP SALMOUS standard version 1, Lumefantrine and artemether
tablet. [cited2012 October 17]. Available from:
http://www.usp.org/sites/default/files/usp_pdf/EN/nonUSStandards/lumefantrineArtemet
herTablets.pdf
9. Umapathi P, Ayyappan J, DarlinQuine S. Development and validation of a dissolution test
method for artemether and lumefantrine in tablets. Tropical J Pharm Res 2011;10(5):643-
53.
10.Carrigan PJ, Bates TR.Biopharmaceutics of drugs administered in lipid containing dosage
forms: GI absorption of griseofulvin from an oil-in-water emulsion in the rat. J Pharm
Sci1973; 62: 1476-9.

![27 Kathpalia Updates on Esophageal Disorders€¦ · [ADDPRESENTATIONTITLE:INSERTTAB>HEADER&$ FOOTER$>$NOTES$AND$HANDOUTS] 1 5/25/17 Overview$of$Esophageal$Disorders Priya&Kathpalia,&MD](https://img.pdfslide.net/doc/110x75/6013e0cdb9861a111f4ceab4/27-kathpalia-updates-on-esophageal-disorders-addpresentationtitleinserttabheader.jpg)



























