Embed Size (px)
Citation preview

Xác định lượng vết thủy ngân bằng phương
pháp chiết pha rắn - quang học
Phùng Thị Thu Huyền
Trường Đại học Khoa học Tự nhiên
Luận văn ThS. ngành: Hóa phân tích; Mã số: 60 44 29
Người hướng dẫn: PGS.TS. Nguyễn Xuân Trung
Năm bảo vệ: 2012
Abstract. Nghiên cứu các điều kiện xác định Hg(II) bằng phương pháp trắc quang
với thuốc thử Dithizone trong môi trường mixen. Nghiên cứu chế tạo vật liệu chiết
pha rắn để làm giàu lượng vết Hg(II) trong môi trường nước. Nghiên cứu ảnh hưởng
của pH, thời gian, nồng độ Hg(II) ban đầu đến dung lượng hấp phụ Hg(II) của vật
liệu vỏ trấu đã biến tính (ERH) và vật liệu vỏ trấu chưa biến tính (NRH) theo
phương pháp tĩnh. Nghiên cứu khả năng làm giàu Hg(II) theo phương pháp động:
khảo sát dung lượng hấp phụ Hg(II), nồng độ và loại axit rửa giải, tốc độ rửa giải,
tốc độ nạp mẫu, thể tích dung môi rửa giải và ảnh hưởng của một số kim loại. Ứng
dụng phân tích hàm lượng thủy ngân vô cơ trong mẫu nước mặt ở hồ Hoàn Kiếm, hồ
Tây, hồ Bảy Mẫu.
Keywords. Hóa phân tích; Quang học; Thủy ngân; Phương pháp chiết pha rắn
Content
MỞ ĐẦU
Nước là nguồn tài nguyên thiên nhiên quý giá, là yếu tố không thể thiếu cho sự sống,
ở đâu có nước ở đó có sự sống. Tuy nhiên, cùng với sự phát triển của xã hội, quá trình đô thị
hóa, công nghiệp hóa, và thâm canh nông nghiệp ngày càng phát triển đã có nhiều ảnh hưởng
xấu đến nguồn tài nguyên này. Nhiều nơi, các nguồn nước bề mặt, thậm chí cả nguồn nước
ngầm đã bị ô nhiễm nghiêm trọng, gây ảnh hưởng xấu tới chất lượng của nước, và ảnh hưởng
đến sức khỏe của con người và động vật, làm giảm năng suất và chất lượng cây trồng. Một
trong những chất gây ô nhiễm là các kim loại nặng (Hg, Pb, Cd, As…) khi ở nồng độ cao
chúng là những chất độc mạnh gây ra tác hại xấu đối với con người đặc biệt là Hg. Khi bị
nhiễm độc thủy ngân sẽ gây ra các tổn thương cho não bộ và gây tử vong. Ngoài ra, nó có thể
gây ra rủi cho và khuyết tật đối với thai nhi.
Do vậy, xác định lượng vết thủy ngân trong nước là một trong những vấn đề thời sự
của hóa học phân tích, nhằm đáp ứng nhu cầu phát triển kinh tế, khoa học kỹ thuật và bảo vệ
môi trường. Tuy nhiên, hàm lượng thủy ngân trong nước là rất nhỏ, vì vậy để phân tích được
thì trước hết ta cần phải làm giàu.
Xuất phát từ những mục tiêu trên chúng tôi đã chọn đề tài: “Xác định lượng vết thủy
ngân bằng phương pháp chiết pha rắn – quang học’’.

CHƢƠNG 1: TỔNG QUAN
1.1. Giới thiệu chung về nguyên tố thủy ngân Thủy ngân được ký hiệu là Hg, có tên La tinh là Hydragyrum, và tên Hy Lạp
Hydrargyros là tổ hợp của 2 từ “nước” và “bạc” – vì nó lỏng giống nước, và có ánh kim
giống như bạc. Trong ngôn ngữ Châu Âu, nguyên tố này được đặt tên là Mercury, lấy theo
tên của thần Mercury của người La Mã.
1.2. Độc tính của thủy ngân
Khi xâm nhập vào cơ thể thuỷ ngân có thể liên kết với những phân tử tạo nên tế bào
sống (axít nucleic, protein.... ) làm biến đổi cấu trúc của chúng và làm ức chế hoạt tính sinh
học của chúng. Sự nhiễm độc thuỷ ngân gây nên những thương tổn trung tâm thần kinh tạo
nên sự run rẩy, sự khó khăn trong cách diễn đạt và nặng hơn nữa có thể gây chết người.
1.3. Các phƣơng pháp xác định thủy ngân
Để xác định thủy ngân với hàm lượng khác nhau có rất nhiều phương pháp, trong các
đối tượng mẫu khác nhau như: phương pháp phân tích trọng lượng và phân tích thể tích dùng
để xác định thủy ngân với hàm lượng lớn, các phương pháp điện hóa và các phương pháp
quang được dùng để xác định lượng vết thủy ngân. Ngoài ra còn có các phương pháp như sắc
ký khí, sắc ký lỏng… cũng được sử dụng nhằm làm tăng độ nhạy của phép phân tích.
CHƢƠNG 2: THỰC NGHIỆM
2.1. Nội dung, đối tƣợng và phƣơng pháp nghiên cứu
Để xác định lượng vết thủy ngân trong đối tượng là nước mặt, chúng tôi đã tập trung
nghiên cứu những vấn đề sau:
- Nghiên cứu các điều kiện xác định Hg(II) bằng phương pháp trắc quang với thuốc
thử Dithizone trong môi trường mixen.
- Nghiên cứu chế tạo vật liệu chiết pha rắn để làm giàu lượng vết Hg(II) trong môi
trường nước.
- Nghiên cứu ảnh hưởng của pH, thời gian, nồng độ Hg(II) ban đầu đến dung lượng
hấp phụ Hg(II) của vật liệu vỏ trấu đã biến tính (ERH) và vật liệu vỏ trấu chưa biến tính
(NRH) theo phương pháp tĩnh.
- Nghiên cứu khả năng làm giàu Hg(II) theo phương pháp động:
- Ứng dụng phân tích hàm lượng thủy ngân vô cơ trong mẫu nước mặt ở hồ Hoàn
Kiếm, hồ Tây, hồ Bảy Mẫu.
2.2. Hóa chất, thiết bị và dụng cụ thí nghiệm
2.2.1. Hóa chất
Tất cả hóa chất sử dụng đều là hóa chất tinh khiết dùng cho phân tích các nguyên tố
lượng vết, loại P.A của Merck.
- Dung dịch gốc chuẩn Hg2+
1000ppm của Merck
2.2.2. Thiết bị
- Máy trắc quang UV-VIS 1601 PC – Shimazu (Nhật Bản), dải bước sóng đo 190 ÷
900 nm.
2.2.3. Dụng cụ
- Các bình định mức, cốc chịu nhiệt, pipet các loại có dung tích, bình nón.
2.3. Chuẩn bị vật liệu hấp phụ
2.3.1. Giới thiệu thành phần, tính chất của vật liệu vỏ trấu dùng chế tạo pha tĩnh
Trấu là lớp vỏ ngoài cùng của hạt lúa và được tách ra trong quá trình xay xát. Trong
vỏ trấu chứa khoảng 75% chất hữu cơ dễ bay hơi sẽ cháy trong quá trình đốt và khoảng 25%
còn lại chuyển thành tro.
2.3.2. Chuẩn bị nguyên vật liệu
2.3.2.1. Chuẩn bị vỏ trấu
Vỏ trấu được sấy khô ở 100o C trong khoảng 24 giờ, sau đó nghiền nhỏ với kích
thước hạt nhỏ hơn 0,9 mm. Vỏ trấu nghiền nhỏ này được rửa sạch bằng nước cất nóng (có
khuấy) khoảng 650C trong thời gian 1 giờ, rồi sấy khô ở 100
0C. Cuối cùng nó được rửa sạch
lại bằng hỗn hợp n-hexan/etanol (tỉ lệ 1:1) trong hệ chiết soxhlet trong 4 giờ, sau đó phơi khô.
2.3.2.2. Làm sạch vỏ trấu
Cân 10 gam vỏ trấu (chuẩn bị ở mục 2.3.2.1), thêm 270 ml dung dịch NaOH 5M, điều
chỉnh nhiệt độ ở 250C (có khuấy) ngâm trong 24 giờ. Sau đó lọc, rửa sạch với nước cất đến
pH = 7, rửa tiếp bằng etanol và sau đó rửa tiếp bằng axeton, sau đó vỏ trấu được sấy khô ở
1050C trong thời gian 1 giờ và để nguội trong bình hút ẩm.
2.3.2.3. Chuẩn bị EDTAD Cân 50 gam muối của EDTA hòa tan trong nước cất (500 ml). Sau đó nhỏ từng giọt
HCl đặc. Chất rắn thu được được đem lọc, rửa sạch với cồn 95%, rửa tiếp bằng đietylete và
sau đó sấy khô trong thời gian 2 giờ ở 1050C, để nguội trong bình hút ẩm.
Cân 18 gam EDTA vừa để nguội trên cho vào bình kín, thêm 31 ml pyridin, thêm 24
ml anhiđrit axetic, hỗn hợp này được khuấy ở 650 C trong thời gian 24 giờ. Sau đó chất rắn
thu được (EDTAD) đem lọc, rửa sạch với anhiđrit axetic, rửa tiếp bằng đietylete rồi sấy khô
trong tủ sấy chân không và được lưu trữ trong một bình khô.
2.3.3. Biến tính vỏ trấu bằng EDTAD
Cân 5 gam vở trấu (đã làm ở mục 2.3.2.2) thêm 15 gam EDTAD (đã làm ở mục
2.4.1.3), thêm 210 ml đimetyl fomamit, ngâm hỗn hợp trong 20 giờ ở 750C (có khuấy) thu
được vật liệu tương ứng. Sau đó rửa bằng đimetyl fomamit, rửa bằng nước cất, rửa bằng natri
cacbonat bão hòa, rửa bằng nước cất, rửa bằng cồn 95% và cuối cùng rửa bằng axeton rồi
đem sấy khô trong thời gian 1 giờ ở 800C, để nguồi trong bình hút ẩm.
CHƢƠNG 3: KẾT QUẢ VÀ THẢO LUẬN
3.1. Khảo sát các điều kiện đo quang xác định Hg(II)
3.1.1. Khảo sát phổ hấp thụ của phức Hg(II)-đithizon trong môi trƣờng các chất hoạt
động bề mặt khác nhau
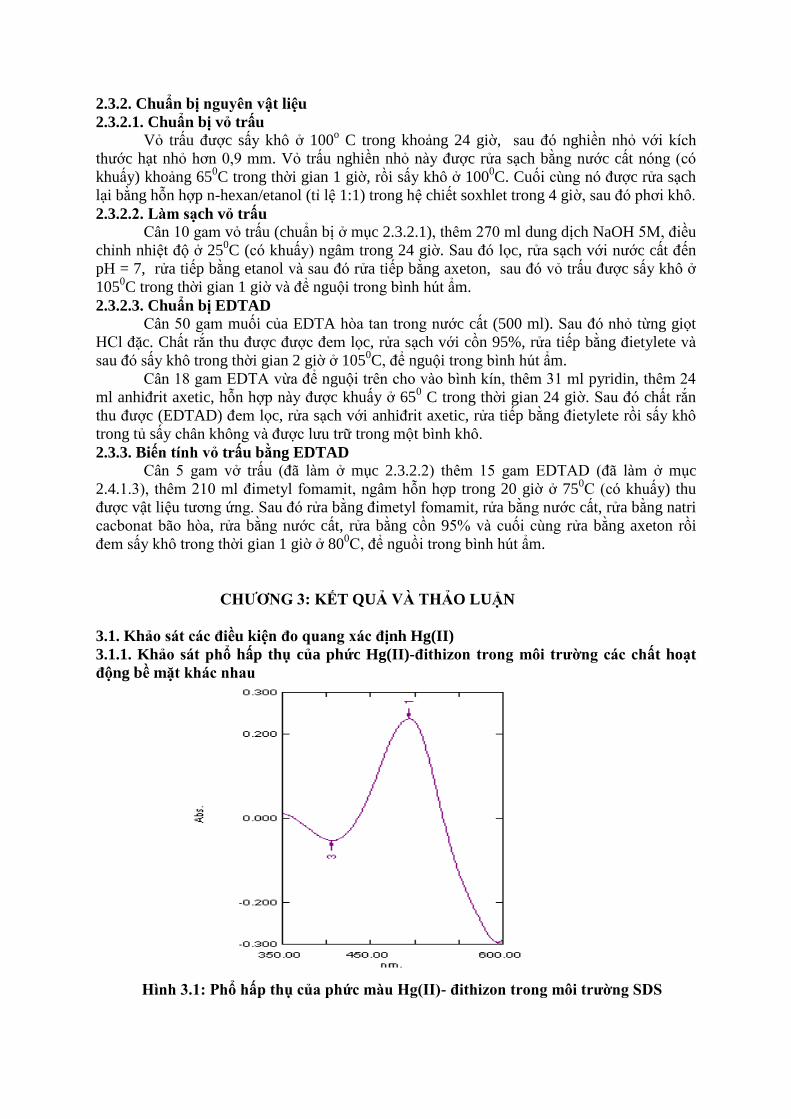
Hình 3.1: Phổ hấp thụ của phức màu Hg(II)- đithizon trong môi trƣờng SDS
Ta thấy phức có cực đại hấp thụ ở 494 nm (λmax = 494nm), độ hấp thụ quang A = 0,24. Như
vậy ta chọn λmax = 494 nm cho các khảo sát tiếp theo.
3.1.2. Khảo sát ảnh hƣởng của thời gian đến sự tạo phức
Trong khoảng thời gian khảo sát là 150 phút, cho thấy phức của Hg2+
và đithizon hình
thành khá nhanh ở điều kiện thường và tương đối bền. Vì vậy chúng tôi chọn khoảng thời
gian tốt nhất để tiến hành đo độ hấp thụ quang là 10 – 20 phút sau khi pha dung dịch.
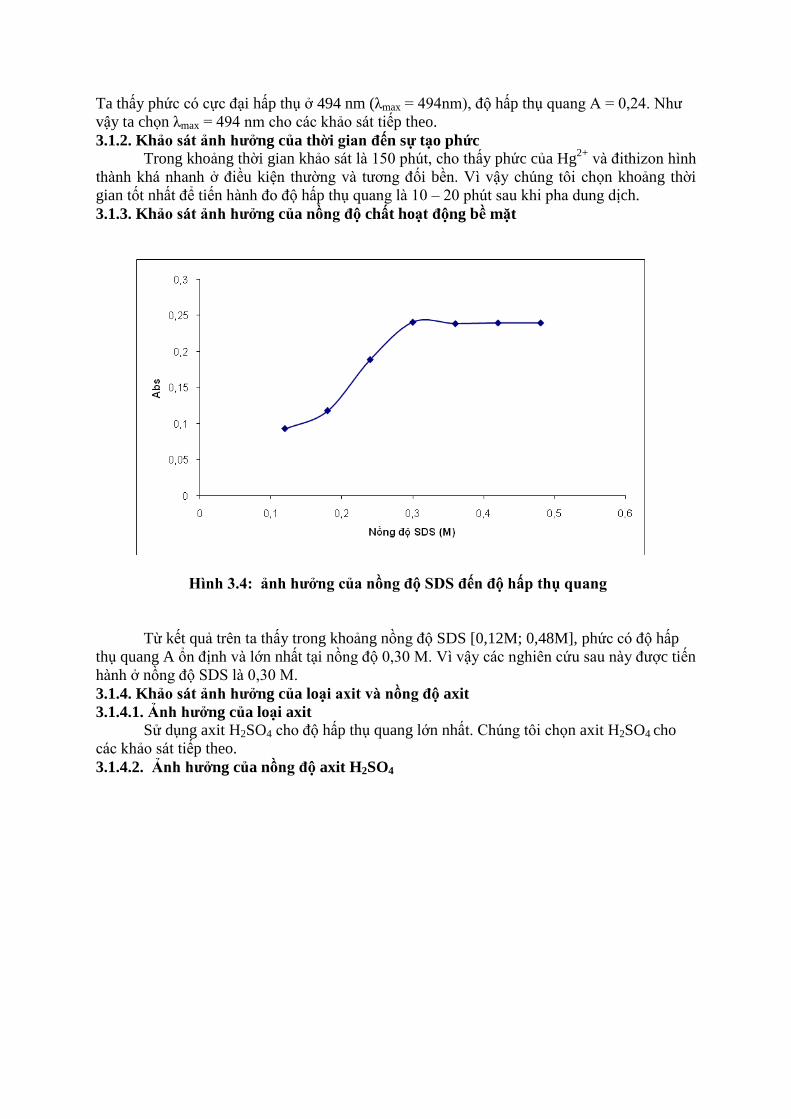
3.1.3. Khảo sát ảnh hƣởng của nồng độ chất hoạt động bề mặt
Hình 3.4: ảnh hƣởng của nồng độ SDS đến độ hấp thụ quang
Từ kết quả trên ta thấy trong khoảng nồng độ SDS [0,12M; 0,48M], phức có độ hấp
thụ quang A ổn định và lớn nhất tại nồng độ 0,30 M. Vì vậy các nghiên cứu sau này được tiến
hành ở nồng độ SDS là 0,30 M.
3.1.4. Khảo sát ảnh hƣởng của loại axit và nồng độ axit
3.1.4.1. Ảnh hƣởng của loại axit
Sử dụng axit H2SO4 cho độ hấp thụ quang lớn nhất. Chúng tôi chọn axit H2SO4 cho
các khảo sát tiếp theo.
3.1.4.2. Ảnh hƣởng của nồng độ axit H2SO4

Hình 3.5: Ảnh hƣởng của nồng độ axit đến độ hấp thụ quang
Tại nồng độ 0,10 M phức có độ hấp thụ quang lớn nhất. Như vậy các thí nghiệm về
sau chúng tôi chọn nồng độ của axit sunfuric là 0,10 M để khảo sát.
3.1.5. Khảo sát ảnh hƣởng của nồng độ thuốc thử đithizon
Hình 3.6: Ảnh hƣởng của nồng độ đithizon đến độ hấp thụ quang của phức
Chúng tôi chọn nồng độ thuốc thử 5,0.10-5
M để tiến hành các khảo sát tiếp theo.
3.1.6. Khảo sát sự phụ thuộc của độ hấp thụ quang A vào nồng độ Hg(II)
0 1 2 3 4 5 6
0,0
0,2
0,4
0,6
0,8
Ab
s
[Hg2+]ppm
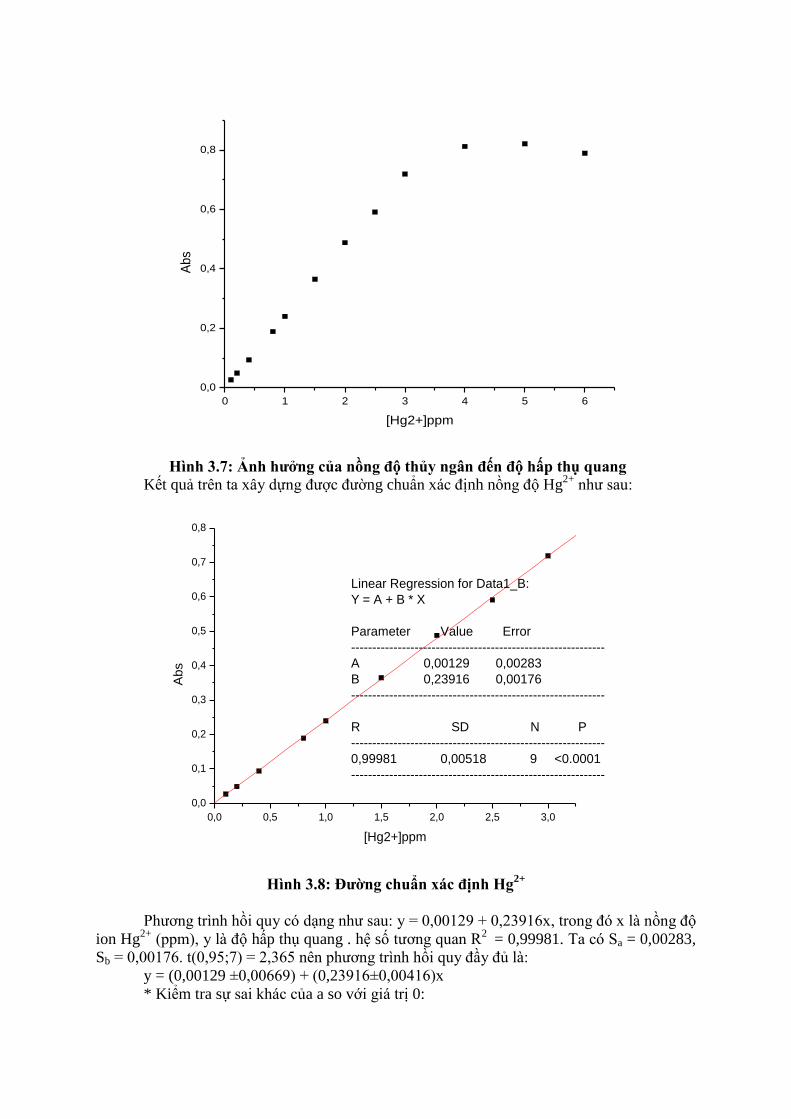
Hình 3.7: Ảnh hƣởng của nồng độ thủy ngân đến độ hấp thụ quang
Kết quả trên ta xây dựng được đường chuẩn xác định nồng độ Hg2+
như sau:
0,0 0,5 1,0 1,5 2,0 2,5 3,0
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
Linear Regression for Data1_B:
Y = A + B * X
Parameter Value Error
------------------------------------------------------------
A 0,00129 0,00283
B 0,23916 0,00176
------------------------------------------------------------
R SD N P
------------------------------------------------------------
0,99981 0,00518 9 <0.0001
------------------------------------------------------------
Ab
s
[Hg2+]ppm
Hình 3.8: Đƣờng chuẩn xác định Hg
2+
Phương trình hồi quy có dạng như sau: y = 0,00129 + 0,23916x, trong đó x là nồng độ
ion Hg2+
(ppm), y là độ hấp thụ quang . hệ số tương quan R2
= 0,99981. Ta có Sa = 0,00283,
Sb = 0,00176. t(0,95;7) = 2,365 nên phương trình hồi quy đầy đủ là:
y = (0,00129 ±0,00669) + (0,23916±0,00416)x
* Kiểm tra sự sai khác của a so với giá trị 0:

Sự khác nhau giữa a và giá trị 0 là không có ý nghĩa thống kê, nên ta có thể coi a = 0.
Vậy phương pháp nghiên cứu trên không mắc sai số hệ thống.
3.1.7. Giới hạn phát hiện và giới hạn định lƣợng
Độ lệch chuẩn Sy = 1
1
2
)(
n
i
n
i
xx = 0,00247
Từ đó suy ra: 3 3 0,00247
LOD = 0,03( )0,23916
Syppm
b
10 10 0,00247
LOQ = 0,10( )0,23916
Syppm
b
Như vậy, khoảng tuyến tính xác định Hg2+
là: [0,1ppm; 3,0ppm].
3.1.8. Độ lặp lại của phép đo
Trong khoảng nồng độ [0,1ppm; 3,0ppm], RSD < 5% tức là phép phân tích có độ lặp
lại cao.
3.2. Nghiên cứu ảnh hƣởng của các ion kim loại đến sự tạo phức
3.2.1. Ảnh hƣởng của các ion kim loại đến phép xác định Hg(II)
Các ion kim loại Mg2+
, Al3+
, Ca2+
, K+, Na
+ gây ảnh hưởng không nhiều đến giá trị độ
hấp thụ quang , nên chúng tôi chỉ tiếp tục nghiên cứu cách loại trừ các ion Cu2+
, Pb2+
, Zn2+
,
Mn2+
,Fe3+
. Một trong các phương pháp phổ biến để loại trừ ảnh hưởng của các ion kim loại
cản trở là dùng các chất tạo phức để loại trừ ảnh hưởng của chúng. Để khảo sát, chúng tôi
chọn EDTA làm thuốc thử để che ảnh hưởng của các ion kim loại Cu2+
, Pb2+
, Zn2+
, Mn2+
,
Fe3+
.
3.2.2. Ảnh hƣởng của EDTA
EDTA ảnh hưởng không nhiều đến độ hấp thụ quang của phức màu.
3.2.3. Loại trừ ảnh hƣởng của ion kim loại
EDTA thích hợp để che được các ion kim loại gây cản trở đến phép xác định thủy
ngân bằng phương pháp đo quang.
3.3. Nghiên cứu khả năng làm giàu Hg(II)
3.3.1. Xác định hình dạng và nhóm chức của vật liệu
3.3.1.1. Xác định nhóm chức bằng phổ hồng ngoại
Theo kết quả từ phổ hồng ngoại của vật liệu ta thấy vật liệu đã biến tính xuất hiện các
pic 1661,38 cm-1
; 1512,92 cm-1
; 1463,43 cm-1
; 1375,45 cm-1
; 1048,28 cm-1
;
976,80 cm-1
có thể giả thuyết rằng có sự tham gia của hai nhóm cacbonyl, một nhóm liên
quan đến phản ứng este hóa, một nhóm là thuộc nhóm cacboxylat của phân tử EDTAD.
Chứng tỏ phân tử EDTAD tham gia vào mạng lưới phân tử của cellulose của vỏ trấu.
3.3.1.2. Hình dạng SEM của vật liệu

Hình 3.11: Ảnh chụp bề mặt vỏ trấu trƣớc khi biến tính
Hình 3.12: Bề mặt vật liệu sau khi biến tính
Ta thấy ở vỏ trấu sau khi biến tính có bề mặt xốp hơn rất nhiều so với vỏ trấu khi
chưa biến tính. Do đó đã làm tăng diện tích bề mặt của vật liệu lên rất nhiều, tạo điều kiện
cho việc hấp phụ của vật liệu tốt hơn.
3.3.2. Ứng dụng vật liệu hấp phụ để tách, làm giàu và xác định lƣợng vết Hg(II)
3.3.2.1. Nghiên cứu khả năng làm giàu lƣợng vết Hg(II) theo phƣơng pháp tĩnh
a. Khảo sát ảnh hƣởng pH đến khả năng hấp phụ Hg(II) lên vật liệu
Kết quả thu được cho thấy, trong khoảng giá trị pH từ 1÷7, dung lượng hấp phụ
Hg(II) của vật liệu ERH tăng và gần như không đổi từ pH=4÷7 và có dung lượng hấp phụ lớn
nhất ở pH=5. Như vậy, các nghiên cứu tiếp theo đối với vật liệu ERH chúng tôi điều chỉnh
giá trị pH cả dung dịch mẫu bằng 5. Còn vật liệu NRH dung lượng hấp phụ Hg(II) tăng trong
khoảng pH từ 1 đến 3 và đặt giá trị cực đại ở pH=3. Như vậy đối với vật liệu NRH nghiên
cứu tiếp theo chúng tôi điều chỉnh giá trị pH cả dung dịch mẫu bằng 3.
b. Khảo sát thời gian đạt cân bằng hấp phụ
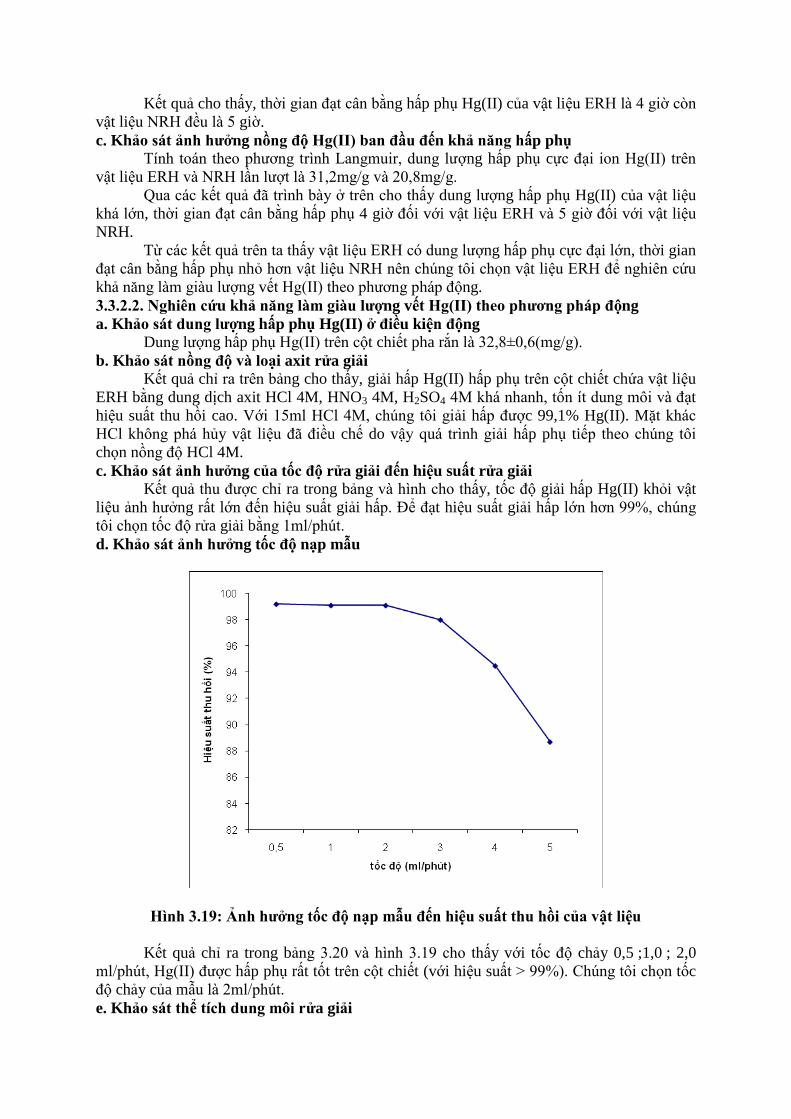
Kết quả cho thấy, thời gian đạt cân bằng hấp phụ Hg(II) của vật liệu ERH là 4 giờ còn
vật liệu NRH đều là 5 giờ.
c. Khảo sát ảnh hƣởng nồng độ Hg(II) ban đầu đến khả năng hấp phụ
Tính toán theo phương trình Langmuir, dung lượng hấp phụ cực đại ion Hg(II) trên
vật liệu ERH và NRH lần lượt là 31,2mg/g và 20,8mg/g.
Qua các kết quả đã trình bày ở trên cho thấy dung lượng hấp phụ Hg(II) của vật liệu
khá lớn, thời gian đạt cân bằng hấp phụ 4 giờ đối với vật liệu ERH và 5 giờ đối với vật liệu
NRH.
Từ các kết quả trên ta thấy vật liệu ERH có dung lượng hấp phụ cực đại lớn, thời gian
đạt cân bằng hấp phụ nhỏ hơn vật liệu NRH nên chúng tôi chọn vật liệu ERH để nghiên cứu
khả năng làm giàu lượng vết Hg(II) theo phương pháp động.
3.3.2.2. Nghiên cứu khả năng làm giàu lƣợng vết Hg(II) theo phƣơng pháp động
a. Khảo sát dung lƣợng hấp phụ Hg(II) ở điều kiện động
Dung lượng hấp phụ Hg(II) trên cột chiết pha rắn là 32,8±0,6(mg/g).
b. Khảo sát nồng độ và loại axit rửa giải
Kết quả chỉ ra trên bảng cho thấy, giải hấp Hg(II) hấp phụ trên cột chiết chứa vật liệu
ERH bằng dung dịch axit HCl 4M, HNO3 4M, H2SO4 4M khá nhanh, tốn ít dung môi và đạt
hiệu suất thu hồi cao. Với 15ml HCl 4M, chúng tôi giải hấp được 99,1% Hg(II). Mặt khác
HCl không phá hủy vật liệu đã điều chế do vậy quá trình giải hấp phụ tiếp theo chúng tôi
chọn nồng độ HCl 4M.
c. Khảo sát ảnh hƣởng của tốc độ rửa giải đến hiệu suất rửa giải
Kết quả thu được chỉ ra trong bảng và hình cho thấy, tốc độ giải hấp Hg(II) khỏi vật
liệu ảnh hưởng rất lớn đến hiệu suất giải hấp. Để đạt hiệu suất giải hấp lớn hơn 99%, chúng
tôi chọn tốc độ rửa giải bằng 1ml/phút.
d. Khảo sát ảnh hƣởng tốc độ nạp mẫu
Hình 3.19: Ảnh hƣởng tốc độ nạp mẫu đến hiệu suất thu hồi của vật liệu
Kết quả chỉ ra trong bảng 3.20 và hình 3.19 cho thấy với tốc độ chảy 0,5 ;1,0 ; 2,0
ml/phút, Hg(II) được hấp phụ rất tốt trên cột chiết (với hiệu suất > 99%). Chúng tôi chọn tốc
độ chảy của mẫu là 2ml/phút.
e. Khảo sát thể tích dung môi rửa giải
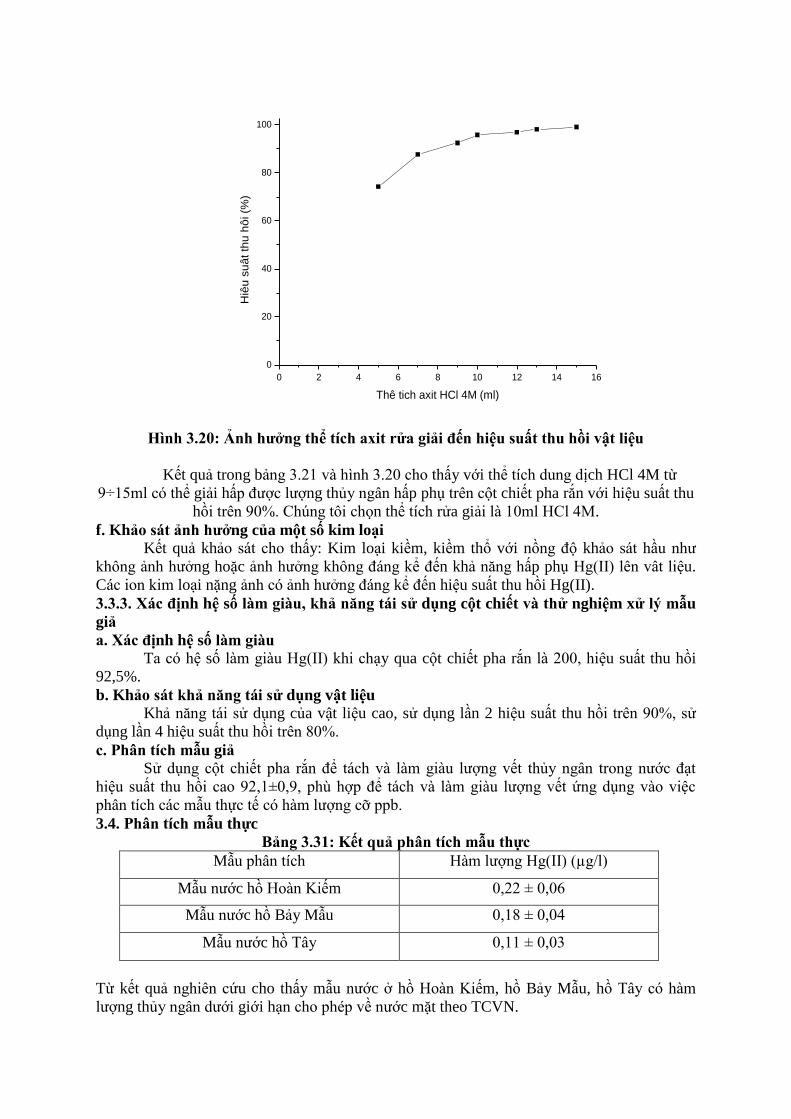
0 2 4 6 8 10 12 14 16
0
20
40
60
80
100
Hiê
u s
uâ
t th
u h
ôi (%
)
Thê tich axit HCl 4M (ml)
Hình 3.20: Ảnh hƣởng thể tích axit rửa giải đến hiệu suất thu hồi vật liệu
Kết quả trong bảng 3.21 và hình 3.20 cho thấy với thể tích dung dịch HCl 4M từ
9÷15ml có thể giải hấp được lượng thủy ngân hấp phụ trên cột chiết pha rắn với hiệu suất thu
hồi trên 90%. Chúng tôi chọn thể tích rửa giải là 10ml HCl 4M.
f. Khảo sát ảnh hƣởng của một số kim loại
Kết quả khảo sát cho thấy: Kim loại kiềm, kiềm thổ với nồng độ khảo sát hầu như
không ảnh hưởng hoặc ảnh hưởng không đáng kể đến khả năng hấp phụ Hg(II) lên vât liệu.
Các ion kim loại nặng ảnh có ảnh hưởng đáng kể đến hiệu suất thu hồi Hg(II).
3.3.3. Xác định hệ số làm giàu, khả năng tái sử dụng cột chiết và thử nghiệm xử lý mẫu
giả
a. Xác định hệ số làm giàu
Ta có hệ số làm giàu Hg(II) khi chạy qua cột chiết pha rắn là 200, hiệu suất thu hồi
92,5%.
b. Khảo sát khả năng tái sử dụng vật liệu
Khả năng tái sử dụng của vật liệu cao, sử dụng lần 2 hiệu suất thu hồi trên 90%, sử
dụng lần 4 hiệu suất thu hồi trên 80%.
c. Phân tích mẫu giả
Sử dụng cột chiết pha rắn để tách và làm giàu lượng vết thủy ngân trong nước đạt
hiệu suất thu hồi cao 92,1±0,9, phù hợp để tách và làm giàu lượng vết ứng dụng vào việc
phân tích các mẫu thực tế có hàm lượng cỡ ppb.
3.4. Phân tích mẫu thực
Bảng 3.31: Kết quả phân tích mẫu thực
Mẫu phân tích Hàm lượng Hg(II) (µg/l)
Mẫu nước hồ Hoàn Kiếm 0,22 ± 0,06
Mẫu nước hồ Bảy Mẫu 0,18 ± 0,04
Mẫu nước hồ Tây 0,11 ± 0,03
Từ kết quả nghiên cứu cho thấy mẫu nước ở hồ Hoàn Kiếm, hồ Bảy Mẫu, hồ Tây có hàm
lượng thủy ngân dưới giới hạn cho phép về nước mặt theo TCVN.

Bảng 3.32: Giá trị giới hạn hàm lƣợng thủy ngân trong nƣớc mặt theo TCVN
STT Thông số Đơn vị
Giá trị giới hạn
A B
A1 A2 B1 B2
1 Thủy ngân mg/l 0,001 0,001 0,001 0,002
KẾT LUẬN
Sau quá trình nghiên cứu hoàn thành luận văn thạc sĩ với nội dung đề tài: “Xác định
lượng vết thủy ngân bằng phương pháp chiết pha rắn – quang học’’, chúng tôi đã thực hiện
được một số công việc sau:
1. Đã nghiên cứu tối ưu hóa các điều kiện xác định Hg(II) bằng phương pháp trắc quang với
thuốc thử đithizon trong môi trường mixen.
2. Đã nghiên cứu các yếu tố ảnh hưởng đến quá trình hấp phụ Hg(II) trên vật liệu ERH và
NRH (vật liệu vỏ trấu biến tính và vật liệu vỏ trấu chưa biến tính):
Đã khảo sát được ảnh hưởng của nồng độ đầu và tìm được dung lượng hấp phụ cực
đại ion Hg2+
trên vật liệu ERH và NRH lần lượt là 31,2mg/g và 20,8mg/g.
3. Đã khảo sát khả năng hấp phụ Hg(II) của vật liệu ở điều kiện động:
Dung lượng hấp phụ cực đại đối với Hg(II) là 32,8±0,6mg/g.
4. Áp dụng xác định Hg(II) trong nước mặt ở một số hồ thuộc thành phố Hà Nội và thu được
kết quả:
- Mẫu nước hồ Hoàn Kiếm: 0,22 ± 0,06 µg/l.
- Mẫu nước hồ Bảy Mẫu: 0,18 ± 0,04 µg/l.
- Mẫu nước hồ Tây: 0,11 ± 0,03 µg/l.
Với những gì đã làm được trong bản luận văn này, chúng tôi hy vọng đây là một đề
tài hữu ích cho việc áp dụng xác định lượng vết Hg(II) trong mẫu nước, ít tốn kém.
References
Tiếng Việt
1. Phạm Tiến Đức (2008), Nghiên cứu khả năng hấp thu kim loại nặng của đá ong biến
tính có gia thêm đất hiếm, Luận án thạc sĩ trường Đại học khoa học Tự Nhiên, Đại học
quốc gia Hà Nội.
2. Trịnh Xuân Giản, Hoàng Bạch Dương, Lê Lan Anh, Nguyễn Thị Huệ, Vũ Đình Lợi, Phạm
Gia Môn (1999), “Phương pháp von-ampe hòa tan xác định lượng vết thủy ngân trong
mẫu nước”, Tạp trí phân tích hóa lý và sinh học, 4(3), tr. 36-38.
3. Trần Từ Hiếu, Từ Vọng Nghi, Nguyễn Văn Ri, Nguyễn Xuân Trung (2003), Hóa học phân
tích phần 2, Đại học Quốc Gia Hà Nội, Hà Nội.
4. Nguyễn Thị Thu Hương (2006), Xác định thủy ngân trong mẫu môi trường bằng phương
pháp phổ hấp thụ nguyên tử hóa hơi lạnh (CV- AAS), Luận án thạc sĩ trường Đại học
khoa học Tự Nhiên, Đại học quốc gia Hà Nội.
5. Hoàng Nhâm (2002), Hóa học vô cơ Tập 3, Nhà xuất bản Giáo dục, Hà Nội.
6. Nguyễn Văn Ri, Tạ Thị Thảo (2006), Thực tập hóa học phân tích, Đại học Quốc
Gia Hà Nội, Hà Nội.
7. Đỗ Quang Trung (2002), Ứng dụng kỹ thuật chiết pha rắn để tách làm giàu và xác định
lượng vết thủy ngân, asen trong nước, Luận án tiến sỹ hóa học trường Đại học khoa học
Tự Nhiên, Đại học quốc gia Hà Nội.
8. Đào Hữu Vinh, Lâm Ngọc Thụ (1979), Chuẩn độ phức chất, NXB khoa học kỹ thuật.

Tiếng Anh
9. A. Krata, E.Bulska(2005), “Critical evaluation of analytical performace of atomic
absorption spectrometry and inductively coupled plasma mass spectrometry for mercury
determition”, Spectrochimica Acta, 60(B), pp. 345-350.
10. Ali Mohammad, Haji Shabani, and Navid Nasirizdel (2006), “Preconcentration,
speciation and determination of ultra trace amounts of mecury by using dithizone
modified dithizone naphthalene membrane disk/electron beam irradiation and cold
vapor atomic absorption spectrometry”, Journal of Hazardous Materials, 35, pp 468-
475.
11. David E. Nixon, Mary F. Burritt, Thomas P. Moyer (1999), “ The determination of
mercury in whole blood and urine by inductively coupled plasma mass spectrometry”,
Spectrochimica Acta, B(54), pp. 1141-1153.
12. Fausun Okc, Hasan Ertasa, F.Nil Erta (2008), “Determination of mercury in table salt
samples by on-line medium exchange anodic stripping voltammetry”, talanta, 75, pp.
442-446.
13. Humaira Khan, M. Jamaluddin Ahmed, and M. Iqbal Bhanger (2005), “A simple
Spectrophotometric determination of trace level mecury using 1,5-
Diphenylthiocabazone solubilized in micelle”, Analytical sciences may 2005, 21, pp.
507-512.
14. Japan public heath association (2001), Preventive measures against environmental
mecury pollution and its health effects, pp.10-4.
15. Kalameqham R, Ash KO (1992), “ A simple ICP-MS procedure for the determination of
total mercury in whole blood and urine”, J Clin Lab Anal, 6(4), pp. 190-193.
16. L. Aduna de Paz, A. Alegria, R. Barber & R. Far & M.J. Lagarda (1997), “Determination
of mercury in dry-fish samples by microwave digestion and flow injection analysis
system cold vapor atomic absorption spectrometry”, Food Chemistry, 58, pp. 169-172.
17. Masatoshi Morita, Jun Yoshinaga, John S.Edmonds (1998), “The determination of
mercury Species in environmental and biological samples”, International union of pure
and applied chemistry, 70(8), pp. 1585-1615.
18. M. Horvat vm LupSina (1991), ’’Determination of total mercury in coal fly ash by goal
amalgamation cold vapour atomic absorption spectrometry’’, Analytica Chimica Acta
(243), pp.71-79.
19. M.Narsiruddin Khan, B.Se (Iions), M.Sc, M.Phil (2006), Kinetic Determination of iron
and spetrophotometric Determination of mercury By exploiting The reactions of
Neutral Red, University of Karachi, Karachi.
20. Moon-Sook Jeoung and Hee-Seon Choi (2004), “Spectrophotometric Determination of
Trace Hg(II) in Cetyltrimethylammonium Bromide Media”, Bull. Korean Chem. Soc,
25(12), pp. 1877-1880.
21. Nguyen Trong Ngo, Nguyen Thanh Binh, Truong Y, Nguyen Van Phuc, Le Nhu Sieu, Le
Ngoc Chung (2000), “Methol for simultaneous determination of cadmium, mercury and
selenniun in the marine envieronment by neutron activation analysis”, Tap chi phan tich
Hoa, Ly va Sinh hoc, 5(1), pp. 52-55.
22. P.Shetty, A.A.Moosavy- Movahedi, K. Regan(1994), “Determination of trace level
mercury in biological and enviromental samples by neutron activation anlysis, Journal
of Radioanalytical and Nuclear Chemistry”, Articles, 1182(2), pp. 205-211.
23. Qiufen Hu, Guangyu Yang, Jiayuan Yin, Yun Yao (2002), “Determination of trace lead,
cadmium and mercury by on-line column enrichment followed by RP-HPLC as metal-
tetra-(4-bromophenyl)-porphyryl chelates”, Talanta, 57, pp. 751-756.
24. Ralph G.Smith (1993), “ Determination of Mercury in Environmental Samples by Isotope
Dilution/ICPMS”, Anal. Chem, 65(18), pp. 2485-2488.

25. Rakesh Kumar Mahajan, Ravneet Kaur, Inderpreet Kaur, Vandana Sharma, Manoj Kumar
(2004), “Mercury (II) ion – Selective Electrodes Based on p-tert-Butyl Calix [4] crowns
with Imine Units”, Analytical Sciences, 4, pp. 811-814.
26. R.Falter, H.F.Scholer (1994), “Determination of Methyl-Ethyl-phenyl, and total mercury
in Neckar river fish”, Chemosphere, 29(6), pp. 1333-1338.
27. Rita Giovannetti, Vito Bartocci (1998), “Kinetic and equilibrium studies on mercury(II)-
Coprorphyrin-I. Metalion Exchange reaction with cobalt(II) and application to
dertermination of trace mercury (II)”, Talanta, 46, pp. 997-984.
28. R. Saran, T.S.Basu Baul (1994), “Determination of submicrogram amounts of mercury
(II) with 5-(2-carborbomethoxyphenyl) azo-8-quynolinol in presence of anionic
surfactant by derivative spectrophotometry”, Talanta, 41(9), pp. 1537-1544.
29. Shayessteh Dadfarnia, Ali Mohammed Salmanzadeh, and Ali Mohammed Haji Shabani
(2002), “Preconcentration and Deternation of Mercury(II) and Methylmercury in
Waters by Immobilized 1,5-Diphenylcarbazone and Cold Vapor Atomic Absorption
spectrometry”, Talanta, 23(12), pp. 1719-1723.
30. S. Mishra, R.M. Tripathi, S. Bhalke, V.K. Shukla, V.D. Puranik (2005),’’Determination
of methylmercury and mercury(II) in a marine ecosystem using solid-phase
microextraction gas choromatography-mass spectrometry’’, Analytica Chimica Acta,
551, pp. 192-198.
31. Surkumar Chatterjee, Ajay pillai, V.K. Gupta (2002), “Spectrophotometric determination
of mecury in environmental sample and fungicides based on its complex with o-
carboxyphenyl diazoamino p-azobenzen”, Talanta, 57, pp. 461-465.
32. Susan C.Hight, John Cheng (2006), “Determination of methylmercury and estimation of
total mercury in seafood using high performance liquid chromatoghraphy (HPLC) and
inductively coupled plasma-mass spectrometry (ICP-MS) metrod development and
validation”, Analytica Chimica Acta, 567, pp. 160-172.
33. T.V.Ramakrisna, G.Aravamudan and M.Vijayakumar (1976), “Spectrophotometric
determination of mecury (II) as the ternary complex with Rhodamine 6G and Iodide”,
Analytica Chimica Acta , 84( 2), pp. 369-375.
34. Weizhu Yang, Qun Hu, Jing Ma, Liming Wang, Guangyu Yang and Gang Xie (2006),
“Solid phase extraction and spectrophotometric Determination of mecury in Tobacco
and Tobacco Additivies with 5-(p-aminobenzyllidene)-thiorhodanine (ABTR)”,
Talanta, 17(5), pp. 1039-1044.
35. Yong Cal, Rudolf Jaffe, Ronald Jones (1997), “Ethylmercury in the soil and sediments of
the Florida Everglades”, Environmental science & technology, 31(1), pp. 302-305.





























