Embed Size (px)
Citation preview

Molecular Cell, Vol. 6, 1461–1472, December, 2000, Copyright 2000 by Cell Press
Crystal Structure of the Hexameric TrafficATPase of the Helicobacter pylori Type IVSecretion System
Type IV secretion systems are used, for example, byHelicobacter pylori, which causes peptic ulcer diseases;Legionella pneumophila, which causes pneumonia; orBordetella pertussis, which causes whooping cough(Vogel et al., 1998; Burns, 1999; Covacci et al., 1999;
Hye-Jeong Yeo,*§ Savvas N. Savvides,*§
Andrew B. Herr,*‖ Erich Lanka,†and Gabriel Waksman*‡
*Department of Biochemistry and Molecular BiophysicsWashington University School of Medicine
Segal et al., 1999a).St. Louis, Missouri 63110Type IV secretion systems are used not only to trans-†Max Planck Institut fur Molekulare Genetik
port molecules toxic to host cells, but also are used toIhnestrasse 73, Dahlemtransport DNA or protein–DNA complexes (Winans etD-14195 Berlinal., 1996). One such process is bacterial conjugationGermanywhereby two mating bacteria exchange genetic material(Lanka and Wilkins, 1995; Pansegrau and Lanka, 1996).By facilitating conjugative transfer, type IV secretion ma-Summarychineries play crucial roles in the spread of antibioticresistance genes among bacteria. Another process uti-The type IV secretion system of Helicobacter pylorilizing type IV secretion systems for nucleoprotein trans-consists of 10–15 proteins responsible for transportport is the transformation of plants by Agrobacteriumof the transforming protein CagA into target epithelialtumefaciens (de la Cruz and Lanka, 1998; Kado, 1998;cells. Secretion of CagA crucially depends on the hex-Rossi et al., 1998). In that process, a fragment of the Tiameric ATPase, HP0525, a member of the VirB11-PulE(tumor-inducing) plasmid, the T-DNA, is transferred intofamily. We present the crystal structure of a binarythe plant cell (de la Cruz and Lanka, 1998; Rossi et al.,complex of HP0525 bound to ADP. Each monomer1998). This transfer system has been exploited in plantconsists of two domains formed by the N- and C-ter-biotechnology to introduce new genes conferring herbi-minal halves of the sequence. ADP is bound at thecide resistance or resistance to pathogens of industrialinterface between the two domains. In the hexamer,crops (D’Halluin and Botterman, 1998; Chua and Sun-the N- and C-terminal domains form two rings, whichdaresan, 2000).together form a chamber open on one side and closed
Type IV secretion systems require a subclass of PulE-on the other. A model is proposed in which HP0525type ATPases, which are known under the generic namefunctions as an inner membrane pore, the closure andof VirB11 ATPases (Motallebi-Veshareh et al., 1992).opening of which is regulated by ATP binding and ADPVirB11 ATPases are essential for T-DNA transfer in A.
release.tumefaciens, conjugative transfer of DNA in most sys-tems studied to date, and protein transfer in H. pylori,
Introduction L. pneumophila, and B. pertussis (Haase et al., 1995;Stephens et al., 1995; Vogel et al., 1998). Despite the
Pathogenicity in gram-negative bacteria is critically de- fact that VirB11 ATPases are known to be essential forpendent upon secretion machineries that mediate the type IV secretion and pathogenicity, little is known abouttransport and injection of toxic molecules into target the function that these proteins fulfill in these machiner-cells (Finlay and Falkow, 1997; Thanassi and Hultgren, ies (Christie and Vogel, 2000). Their role may vary from2000). These secretion systems are classified into four one organism to another depending on the type of trans-types (I to IV) and share a common requirement for port they help mediating. For example, in the conjugativeproteins that utilize ATP as an energy source to drive system encoded by the RP4 plasmid, the VirB11 ATPase,transport of macromolecules (Salmond, 1994). One such TrbB, is known to be involved in pilus biogenesis (Haaseclass of ATPase is that of the PulE-like proteins that et al., 1995). Indeed, many type IV secretion systems,comprise ATPases involved in both type II and type notably those involved in conjugative transfer and
T-DNA transfer, function in conjunction with a fibrousIV secretion (Possot and Pugsley, 1994; Russel, 1998;cell surface organelle called “pilus,” which is thoughtChristie and Vogel, 2000). Pathogenic bacteria that uti-to be important for adhesion between bacteria duringlize type II secretion systems include human pathogens,conjugative transfer or between bacteria and host euk-such as enteropathogenic Escherichia coli (EPEC) caus-aryotic cells during infection (Eisenbrandt et al., 1999;ing infant diarrhea; Vibrio cholerae causing cholera;Soto and Hultgren, 1999; Lai and Kado, 2000). In H.Pseudomona aeruginosa, a leading cause of mortality inpylori, however, there is no known pilus associated withcompromised patients with cystic fibrosis; or Neisseriathe type IV secretion system. As a result, the VirB11gonorrhoea, which causes gonorrhea; as well as plantATPase of H. pylori may be directly involved in transportpathogens, such as Erwinia chrysanthemi or Xanthomo-of proteins through the inner membrane. In either case,nas campestris (reviewed in Finlay and Falkow, 1997).the potential roles of VirB11 proteins are consistent withevidence that these proteins are at least in part associ-‡ To whom correspondence should be addressed (e-mail: waksman@ated with the inner membrane (IM) and localized to thebiochem.wustl.edu).cytoplasmic side of the IM (Thorstenson et al., 1993;§ These authors contributed equally to this work.Rashkova et al., 1997; Grahn et al., 2000).‖Present address: Division of Biology 156-29, California Institute of
Technology, Pasadena, California 91125. Recently, some of the biochemical properties of three

Molecular Cell1462
Figure 1. Structure of the HP0525–ADP Complex
(A) Representative region (the b6 strand) of the experimental electron density at 3 A resolution. The electron density results from a mapcalculated using SAD phases after NCS averaging and solvent flipping and is contoured at a 2s level. The final refined model is shown instick representation color-coded in yellow for carbon, blue for nitrogen, and red for oxygen.(B) Stereo ribbon diagram of the structure of HP0525 bound to ADP and a 9-mer of PEG (Carson, 1997). a helices, 310 helices, strands, andloops are indicated in blue, dark blue, green, and amber, respectively. The ADP is in magenta, and the PEG molecule is in gray. Secondarystructural elements (except for the 310 helices) are labeled from b1 to b13, while helices are labeled from aA to aI. N and C indicate the N andC termini of the protein, respectively.
VirB11 ATPases have been reported, those of the RP4 tion of HP0525 and provides the basis for designing anti-ulcer drugs. HP0525 resembles a six-clawed grappleand R388 plasmid conjugative systems, TrbB and TrwD,
respectively, and that of the type IV secretion system mounted onto a hexameric ring structure. The state cap-tured in the crystal structure is a closed form wherefrom H. pylori, HP0525 (Krause et al., 2000a, 2000b).
The nucleotide requirements vary from one protein to the six grapple elements are clawed together to form achamber that is open on the hexameric ring side andanother. However, ATPase activity is in all cases stimu-
lated by lipid binding, consistent with the localization of closed on the grapple side. The chamber is large enoughto accommodate a large protein, such as CagA. A modelVirB11 ATPases to the membrane. More importantly,
these proteins form hexameric ring structures (z12 nm is proposed as to how proteins could be transportedthrough the ring structure by concerted opening of thein diameter) of identical subunits reminiscent of struc-
tures of ATP-dependent molecular motors, such as heli- six-clawed grapple upon release of nucleotide.cases or F1-ATPases (Krause et al., 2000a; Patel andPicha, 2000). These structures suggest that VirB11 pro- Result and Discussionteins may actively engage in translocation of substrates.
In this report, we describe the crystal structure of the Structure of the HP0525 MonomerVirB11 ATPase, HP0525, from H. pylori bound to ADP. The structure of the HP0525 monomer bound to ADP isH. pylori is the causative agent of gastric ulcers, a wide- shown in Figure 1. The structure contains two domainsspread disease in both the developed and developing that are formed by two contiguous parts of the aminoworlds. H. pylori is probably the most common chronic acid sequence (Figure 2). The N-terminal part from resi-bacterial infection in humans, present in almost half of due 6 (the first residue for which interpretable electronthe world population (Covacci et al., 1999). The presence density was observed) to residue 136 forms the N-termi-of the bacterium in the gastric mucosa is associated nal domain, while residues 137 to 328 (the last residuewith chronic active gastritis and is implicated in more for which interpretable electron density was observed)severe gastric diseases, including chronic atrophic and form the C-terminal domain. Topologically, the foldmucosa-associated lymphoid tissue (MALT) lymphomas adopted by the C-terminal domain is that of the RecA(Wotherspoon, 1998; Graham, 2000). One substrate for protein (Story et al., 1992). In contrast, submission ofsecretion is a 145 kDa protein, CagA (Segal et al., 1999b; the coordinates of the N-terminal domain to the DALIAsahi et al., 2000; Backert et al., 2000; Odenbreit et al., server did not return a known fold, suggesting that the2000; Stein et al., 2000). CagA is injected into gastric or fold adopted by the N-terminal domain is novel (Holmduodenal epithelial cells by the type IV secretion sys- and Sander, 1998). Each domain is composed of antem encoded by the Cag pathogenicity island. CagA, extended central b sheet containing six b strands in theonce injected, becomes tyrosine-phosphorylated and is N-terminal domain and seven b strands in the C-terminalthought to interfere with normal signaling in host cells. domain. While the central b sheet of the C-terminal do-
main is flanked on both sides by a helices (three on oneThe crystal structure of HP0525 sheds light on the func-

Structure of the VirB11 ATPase of H. pylori1463
Figure 2. Sequence Alignment of the VirB11 ATPases and Location of Secondary Structures in H. pylori HP0525
The following sequences were used in the alignment: HP0525 from H. pylori, LvhB11 from L. pneumophila, VirB11 from A. tumefaciens, TrbBfrom plasmid RP4, and TraG from plasmid pKM101. Strictly conserved residues are shown in green boxes, while residues that are conservativelysubstituted are shown in pink boxes. Amino acid numbering at the top refers to HP0525. Numbering at the end of the lines refers to thenumber of the last amino acid shown at the end of the line in the corresponding sequence. Secondary structural elements of HP0525 areshown at the bottom with helices and strands indicated as cylinders and arrows, respectively. 310 helices are not shown for clarity. Residuesinvolved in subunit–subunit interactions are shown in blue boxes. Residues involved in ADP binding and proposed to be involved in ATPbinding and hydrolysis are boxed in red.
side [aC, aD, and aE] and four on the other [aF, aG, aH, strand b7 to strand b13 (Story et al., 1992). Only a helicesaH and aI have no equivalent in the RecA fold. Theseand aI]), that of the N-terminal domain is packed against
two a helices on only one side (aA and aB). One of these two helices protrude from the core structure of the do-main, and together with the same regions of the othertwo a helices (helix aA) extends far out away from the
core of the protein. The side of the six-stranded central subunits form the narrow end of the hexameric structure(see below for details). The region between strands b8b sheet opposite the two flanking a helices in the N-ter-
minal domain forms a polar surface that is extensively and b9 also differs significantly from the RecA structure:while these two strands are connected by an extendedused as a subunit-subunit interface in the hexamer.
The two domains are linked by a short loop between loop in HP0525, the equivalent region in RecA containsthree a helices (Story et al., 1992).residues 134 and 141. This linker region emanates from
the b6 strand in the N-terminal domain, which is in anantiparallel arrangement with the two strands preceding Structure of the HP0525 Hexamer
The HP0525 hexamer can be readily assembled usingit (strands b5 and b4; Figure 1B). On the other side, theb6 strand runs antiparallel to the b1 strand, which itself one of the two 3-fold crystallographic symmetry axes
intrinsic to the hexagonal space group into which theforms an antiparallel b sheet with the two strands suc-ceeding it (strand b2 and b3; Figure 1B). A long helix, protein–ADP complex crystallizes. A ribbon diagram
representation of the resulting hexameric structure isaB, spans the length of the entire six-stranded b sheetin the N-terminal domain such that its N terminus con- shown in Figure 3 while a surface diagram onto which
the surface electrostatic potential of the hexameric as-nects to the edge strand b3 on one side, and its Cterminus connects to the other edge-strand b4 on the sembly has been mapped is shown in Figure 4.
The six HP0525 subunits assemble in an intertwinedother side. The N-terminal helix, from residue 6 to resi-due 40, is bent at two positions, at residue 12 and at propeller shape whereby residues in both domains of
individual subunits participate in the subunit–subunitresidue 35. The aA helix is followed by a short loopconnecting to the b1 strand. interface. The entire interface amounts to 2260 A2 of
buried surface area.The first secondary structures in the C-terminal do-main are helices aC and aD. Helix aD is the first second- The overall shape of the hexamer is that of a six-
clawed grapple mounted on a hexameric ring. Indeed,ary structural element that aligns with the RecA protein,as do most of the subsequent secondary structures from when each domain is color-coded differently as is shown
Molecular Cell1464
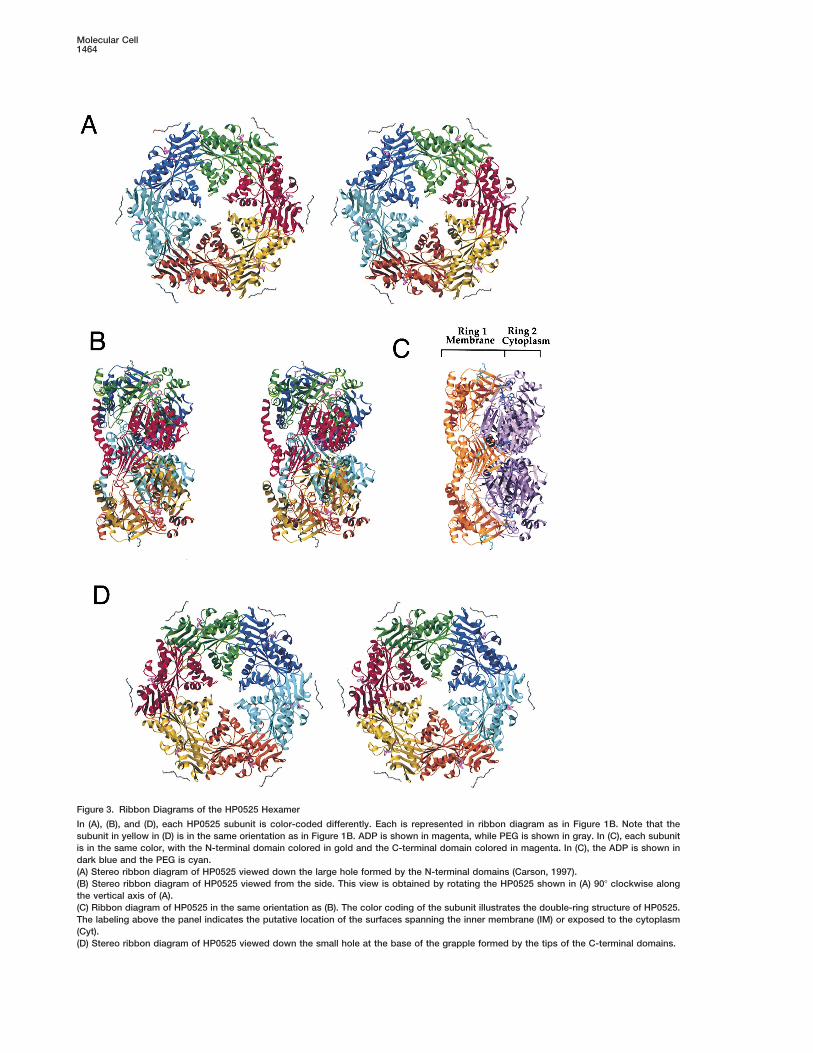
Figure 3. Ribbon Diagrams of the HP0525 Hexamer
In (A), (B), and (D), each HP0525 subunit is color-coded differently. Each is represented in ribbon diagram as in Figure 1B. Note that thesubunit in yellow in (D) is in the same orientation as in Figure 1B. ADP is shown in magenta, while PEG is shown in gray. In (C), each subunitis in the same color, with the N-terminal domain colored in gold and the C-terminal domain colored in magenta. In (C), the ADP is shown indark blue and the PEG is cyan.(A) Stereo ribbon diagram of HP0525 viewed down the large hole formed by the N-terminal domains (Carson, 1997).(B) Stereo ribbon diagram of HP0525 viewed from the side. This view is obtained by rotating the HP0525 shown in (A) 908 clockwise alongthe vertical axis of (A).(C) Ribbon diagram of HP0525 in the same orientation as (B). The color coding of the subunit illustrates the double-ring structure of HP0525.The labeling above the panel indicates the putative location of the surfaces spanning the inner membrane (IM) or exposed to the cytoplasm(Cyt).(D) Stereo ribbon diagram of HP0525 viewed down the small hole at the base of the grapple formed by the tips of the C-terminal domains.

Structure of the VirB11 ATPase of H. pylori1465
Figure 4. Surface of the HP0525–ADP Complex
The surfaces were contoured and displayed using the program GRASP (Nicholls et al., 1991). Color coding is according to charge, blue forthe most positive regions and red for most negative regions with linear interpolation in between.(A) Surface of the HP0525–ADP complex viewed down the large hole. This view corresponds to that of Figure 3A. The inside and outsidedimensions of the HP0525 chamber are indicated in yellow.(B) Surface of the HP0525–ADP complex viewed from the side. Top, the entire HP0525 hexamer. This view corresponds to that shown inFigures 3B and 3C. The putative location of the membrane-spanning and cytoplasm-exposed regions of the HP0525 hexamer are indicated.Also three 9-mer PEG molecules and two ADP molecules are clearly visible and shown as ball-and-stick representation (white for carbon, redfor oxygen, yellow for phosphorus, and blue for nitrogen) and labeled accordingly as PEG and ADP. Bottom, cut-away of the HP0525 hexamer.The orientation is the same as in the top. Four subunits are shown, which allow to visualize the surface potential inside the HP0525 chamber.The inside and outside dimensions of the HP0525 chamber are indicated in yellow.(C) Surface of the HP0525–ADP complex viewed down the small hole of the chamber.
in Figure 3C, it is apparent that the N-terminal domains ring formed by the C-terminal domains). Such a chamberis large enough to accommodate a globular protein orand the C-terminal domains form two separate ring
structures. The N-terminal domains form a hexameric protein domain of about 50 kDa.A 9-mer of polyethylene glycol (PEG) was found boundring that is cylindrical inside (Figures 3A and 3B). In
contrast, the C-terminal domains form a hexameric ring to each of the N-terminal domains at the periphery ofthe hexameric ring (Figure 1B). The presence of PEGthat is conical inside and where each of the C-terminal
domains looks like a grapple claw (Figures 3A and 3D; bound to these surfaces suggests that the N-terminaldomain ring may be embedded into the membrane.Figures 4A and 4C). This comparison facilitates the un-
derstanding of our proposal that HP0525 may act as a VirB11 ATPases are known to be associated to the mem-brane. We propose here that membrane association isprotein translocator.
The HP0525 hexamer has an external diameter of mediated by contacts with the mostly hydrophobic ringformed by the N-terminal domains.100 A and an internal diameter of 50 A (Figure 4A). The
outside cross section (outside, from top to bottom) of While the outside surfaces of the cylindrical hexamericN-terminal domain ring and part of the C-terminal do-the hexamer is 50 A, while its inside cross section (inside,
from top to bottom) is 30 A (Figure 4B). The small hole main ring juxtaposed to it are relatively hydrophobic,the tip of the C-terminal domain ring facing the outsideformed by the closed grapple claws is 10 A in diameter.
Hence, a chamber of about 60,000 A3 is formed, open (the cytoplasmic face) is characterized by a strong nega-tive electrostatic potential (Figure 4C). Hence in theon one side (the side of the ring formed by the N-terminal
domains) and closed on the other side (the side of the closed configuration of the grapple reported here, nega-
Molecular Cell1466
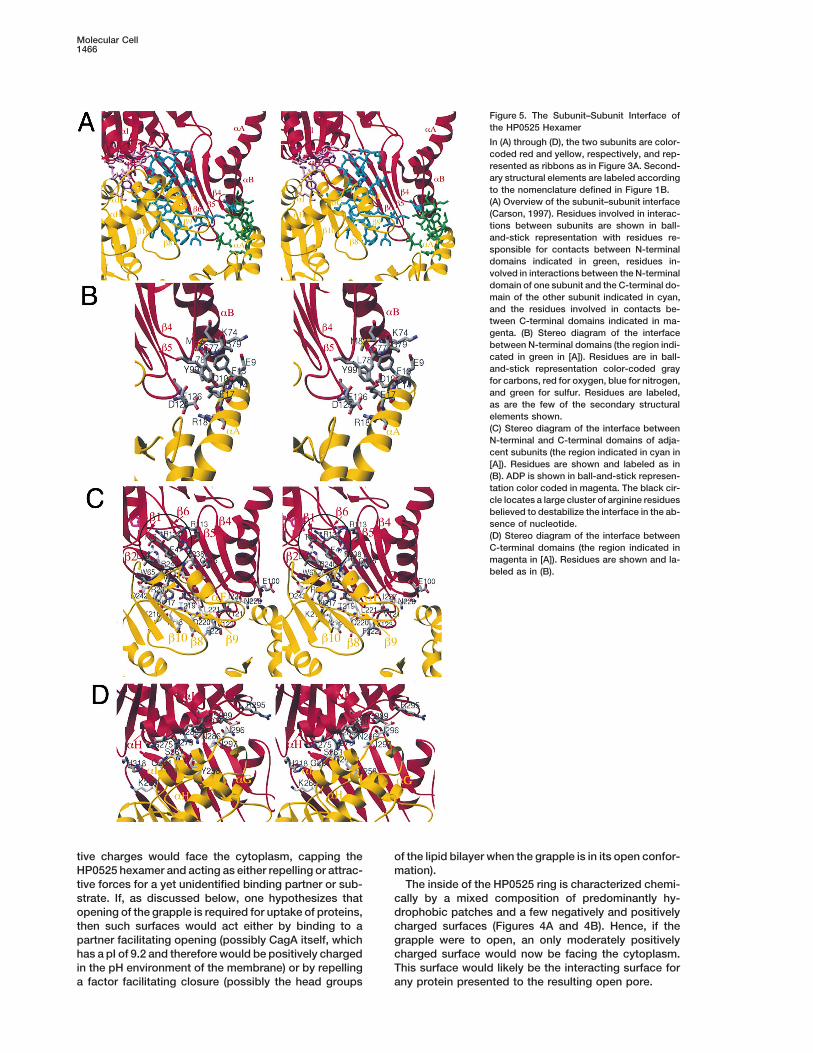
Figure 5. The Subunit–Subunit Interface ofthe HP0525 Hexamer
In (A) through (D), the two subunits are color-coded red and yellow, respectively, and rep-resented as ribbons as in Figure 3A. Second-ary structural elements are labeled accordingto the nomenclature defined in Figure 1B.(A) Overview of the subunit–subunit interface(Carson, 1997). Residues involved in interac-tions between subunits are shown in ball-and-stick representation with residues re-sponsible for contacts between N-terminaldomains indicated in green, residues in-volved in interactions between the N-terminaldomain of one subunit and the C-terminal do-main of the other subunit indicated in cyan,and the residues involved in contacts be-tween C-terminal domains indicated in ma-genta. (B) Stereo diagram of the interfacebetween N-terminal domains (the region indi-cated in green in [A]). Residues are in ball-and-stick representation color-coded grayfor carbons, red for oxygen, blue for nitrogen,and green for sulfur. Residues are labeled,as are the few of the secondary structuralelements shown.(C) Stereo diagram of the interface betweenN-terminal and C-terminal domains of adja-cent subunits (the region indicated in cyan in[A]). Residues are shown and labeled as in(B). ADP is shown in ball-and-stick represen-tation color coded in magenta. The black cir-cle locates a large cluster of arginine residuesbelieved to destabilize the interface in the ab-sence of nucleotide.(D) Stereo diagram of the interface betweenC-terminal domains (the region indicated inmagenta in [A]). Residues are shown and la-beled as in (B).
tive charges would face the cytoplasm, capping the of the lipid bilayer when the grapple is in its open confor-mation).HP0525 hexamer and acting as either repelling or attrac-
tive forces for a yet unidentified binding partner or sub- The inside of the HP0525 ring is characterized chemi-cally by a mixed composition of predominantly hy-strate. If, as discussed below, one hypothesizes that
opening of the grapple is required for uptake of proteins, drophobic patches and a few negatively and positivelycharged surfaces (Figures 4A and 4B). Hence, if thethen such surfaces would act either by binding to a
partner facilitating opening (possibly CagA itself, which grapple were to open, an only moderately positivelycharged surface would now be facing the cytoplasm.has a pI of 9.2 and therefore would be positively charged
in the pH environment of the membrane) or by repelling This surface would likely be the interacting surface forany protein presented to the resulting open pore.a factor facilitating closure (possibly the head groups
Structure of the VirB11 ATPase of H. pylori1467
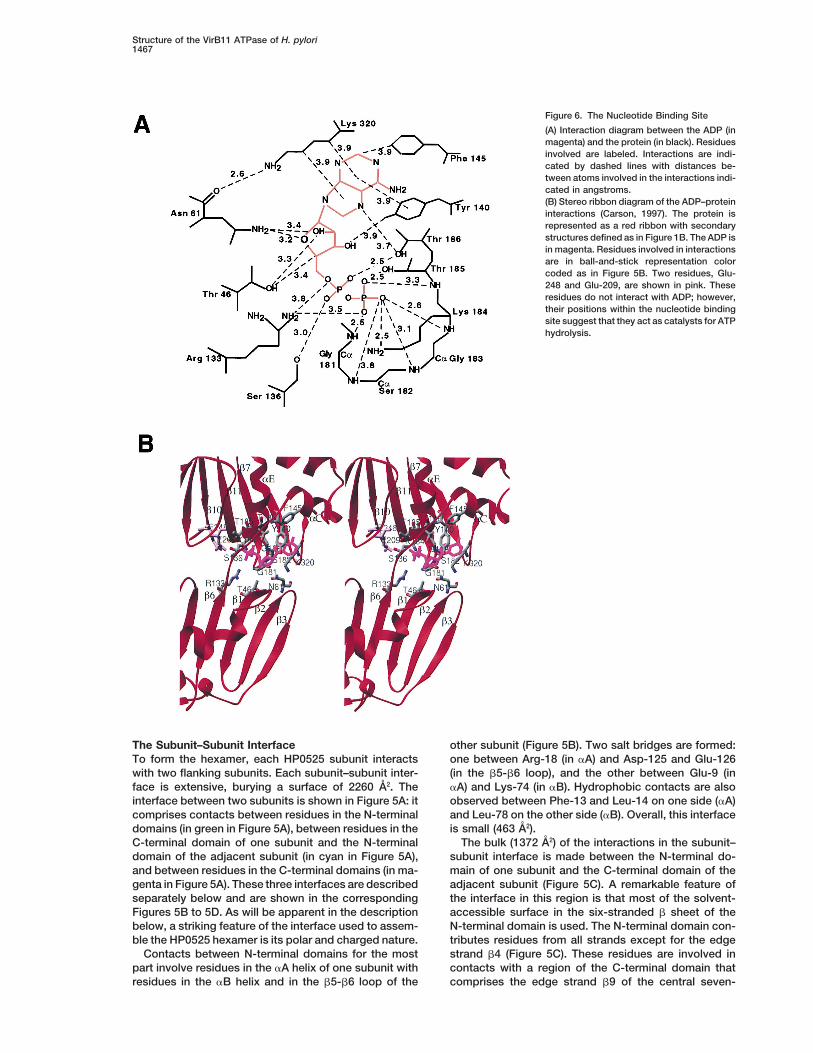
Figure 6. The Nucleotide Binding Site
(A) Interaction diagram between the ADP (inmagenta) and the protein (in black). Residuesinvolved are labeled. Interactions are indi-cated by dashed lines with distances be-tween atoms involved in the interactions indi-cated in angstroms.(B) Stereo ribbon diagram of the ADP–proteininteractions (Carson, 1997). The protein isrepresented as a red ribbon with secondarystructures defined as in Figure 1B. The ADP isin magenta. Residues involved in interactionsare in ball-and-stick representation colorcoded as in Figure 5B. Two residues, Glu-248 and Glu-209, are shown in pink. Theseresidues do not interact with ADP; however,their positions within the nucleotide bindingsite suggest that they act as catalysts for ATPhydrolysis.
The Subunit–Subunit Interface other subunit (Figure 5B). Two salt bridges are formed:one between Arg-18 (in aA) and Asp-125 and Glu-126To form the hexamer, each HP0525 subunit interacts
with two flanking subunits. Each subunit–subunit inter- (in the b5-b6 loop), and the other between Glu-9 (inaA) and Lys-74 (in aB). Hydrophobic contacts are alsoface is extensive, burying a surface of 2260 A2. The
interface between two subunits is shown in Figure 5A: it observed between Phe-13 and Leu-14 on one side (aA)and Leu-78 on the other side (aB). Overall, this interfacecomprises contacts between residues in the N-terminal
domains (in green in Figure 5A), between residues in the is small (463 A2).The bulk (1372 A2) of the interactions in the subunit–C-terminal domain of one subunit and the N-terminal
domain of the adjacent subunit (in cyan in Figure 5A), subunit interface is made between the N-terminal do-main of one subunit and the C-terminal domain of theand between residues in the C-terminal domains (in ma-
genta in Figure 5A). These three interfaces are described adjacent subunit (Figure 5C). A remarkable feature ofthe interface in this region is that most of the solvent-separately below and are shown in the corresponding
Figures 5B to 5D. As will be apparent in the description accessible surface in the six-stranded b sheet of theN-terminal domain is used. The N-terminal domain con-below, a striking feature of the interface used to assem-
ble the HP0525 hexamer is its polar and charged nature. tributes residues from all strands except for the edgestrand b4 (Figure 5C). These residues are involved inContacts between N-terminal domains for the most
part involve residues in the aA helix of one subunit with contacts with a region of the C-terminal domain thatcomprises the edge strand b9 of the central seven-residues in the aB helix and in the b5-b6 loop of the

Molecular Cell1468
stranded b sheet, helix aF, and the loops between helixaF and strand b10 and between helix aE and strand b8.Here again, polar and ionic interactions are numerous.Two tryptophans, Trp-57 (in b2) and Trp-65 (in b3), areinvolved in extensive contacts with residues in the b8-b9 loop and in the aE-b8 loop. Also involved in theinterface is a continuous stretch of residues from Tyr-218 to Phe-222 (i.e., residues in the entire b9 strand ofthe C-terminal domain are implicated in subunit–subunitassembly).
Another remarkable feature of the N-terminal/C-termi-Figure 7. Schematic Model for the Opening and Closure of the
nal domains interface region is the involvement of a HP0525 Chambercluster of basic and acidic residues located near the The N-terminal and C-terminal domains are represented in dark andATP binding site (indicated in a circle in Figure 5C; also light blue, respectively. The view is down the big hole as in Figurenote the location of the ADP molecule in magenta in 3A. ATP molecules are indicated as yellow balls.Figure 5C). Residues in contact are on one side Arg-238 and Arg-240, and in the other side Glu-47, Arg-113,and Arg-133. While Glu-47 makes ionic interactions with In the C-terminal domain, a predominant region in-Arg-238, the side chains of Arg-113, Arg-240, and Arg- volved in contact with the ADP is the P loop, which is133 would clash electrostatically if it were not for the located between strand b7 and helix aE. Interactionspresence of the bound ADP molecule. Indeed, Arg-133 involve main chain amide groups of Lys-84, Gly-183,interacts directly with the a- and b-phosphate groups Ser-182, and Gly-181. The charged tip of Lys-184 coordi-of the ADP (Figure 6), and the b-phosphate group is nates the b-phosphate, while two threonines immedi-5.5 A away from the guanido group moiety of Arg-240. ately C-terminal to the P loop (Thr-185 and Thr-186)A modeled g-phosphate (not shown) would interact di- make hydrogen bonding interactions with both therectly with Arg-240. Only the charge emanating from a- and b-phosphates. The C-terminal domain is alsoArg-113 would not apparently be neutralized: its guanido involved in contacts with the adenine moiety with thegroup is 7.3 A away from the b-phosphate of ADP. How- adenine ring lodged between Tyr-140, Phe-145, and theever, this side chain is solvent exposed and could readily aliphatic moiety of Lys-320. Coordination of the riboseundergo a local conformational change that would bring moiety of the ADP molecule is mostly carried out byit within contact with the modeled g-phosphate of ATP. residues in the N-terminal domain (Thr-46 and Asn-61).Thus, ADP or ATP would appear to be crucial to neutral- However, the N-terminal domain is also involved in coor-ize the like-charge clashes in this region. We propose dinating the phosphate moiety of ADP through Arg-133that in the absence of the nucleotide, such clashes and Ser-136.would have a destabilizing effect on the whole interface In RecA-like proteins, ATP hydrolysis is catalyzed byleading to the release of the C-terminal domain from the acidic residues located within the ATP binding site. Ininterface and its swiveling to an open conformation. RecA, two acidic residues have been implicated in catal-
Finally, contacts are made between the C-terminal ysis: Glu-96 has been proposed to activate the attackingdomains of two adjacent subunits (Figure 5D). The inter- water during hydrolysis of ATP, while Asp-144 is thoughtface in this region buries 425 A2 of surface area. Interac- to coordinate the magnesium ion (electron density fortions are between the claws of the grapple and possibly magnesium was not observed in the HP0525-ADP com-help maintaining the grapple in the closed conformation plex) (Story and Steitz, 1992). Both Glu-96 and Asp-144seen in the crystal. More specifically, interactions in- have their equivalents in HP0525: those are Glu-209 andvolve primarily residues in the C-terminal end of the aG Glu-248, respectively. Thus, we propose that these twohelix of one subunit with the residues of the aH helix of
residues are involved in ATP hydrolysis in HP0525.the adjacent subunit. Here also, the contacts made are
As noted before, the ADP binding site is close toprimarily polar.
the subunit–subunit interface and is contiguous with acluster of arginine residues contributed by both sub-The Nucleotide Binding Siteunits. We postulate that this observation has functionalThe nucleotide binding site is described in Figure 6. Thesignificance since, in the absence of ADP or ATP, desta-ADP is located between the N-terminal and C-terminalbilization of the interface between the C-terminal domaindomains, and residues in both domains contribute toof one subunit and the N-terminal domain of the adjacentthe binding site. It is important to note that, without ADPsubunit may occur leading to a possible release of thebound, contacts between the two domains within oneC-terminal domain from the interface.subunit would be very scarce. Indeed, there are only a
few direct contacts between the two domains, and theA Model for HP0525 Functioncontact area directly involving the two domains is aVirB11 ATPases are known to be essential in severalmere 90 A2. This implies that the nucleotide-mediatedimportant processes, such as T-DNA transfer, conjuga-contacts between the two domains are the driving forcetive transfer of many plasmids, and type IV secretion.maintaining the two domains together. One would there-VirB11 proteins are also homologous to PulE ATPases,fore predict that, in the absence of nucleotide, the do-which play crucial roles in type II secretion and typemain–domain interface within a subunit would not beIV pilus biogenesis (not to be mistaken with type IVsufficient to maintain the two domains in a specific rela-
tive orientation. secretion, since the assembly machinery of type IV pili
Structure of the VirB11 ATPase of H. pylori1469
Table 1. Data Collection, Phasing, and Refinement
Data Collectiona
Data Set Resolution Reflections (Total/Unique) Completeness (%) Rsym (%)b I/s(I)
SeMet-peak (all data) 30–2.5 A 163,385/29,176 94.7 (84.2) 10.8 (25.9) 10.8 (5.7)SeMet-peak (I/s(I) . 0) 30–2.5 A 137,723/27,352 88.8 (71.4) 10.2 (15.8) 10.7 (5.7)
Refinement
Resolution 30–2.5 A (2.59–2.50 A)uFu /s(uFu) .0No. of reflections (working/test) 26,424 (1354)Completeness (overall/last shell) 89% (71%)Total No. of atoms 5,321
Protein atoms 5,104 (646 residues)ADP molecules 2PEG (n 5 9) 1Water molecules 300
R factor 0.223R-free factor 0.289Averaged B factors (A2)
Main chain 36.0Side chain 36.3
Wilson plot B (A2) 38.0Rms deviations
NCS (A) 0.09Bonds (A) 0.010Angles (8) 1.50B values (A2) 1.6/2.5
a Numbers in parentheses indicate values in the highest resolution shell (2.59–2.50 A).b Rsym 5 ouI 2 ,I. u/oI, where I equals observed intensity, and ,I. equals average intensity for symmetry-related reflections.
is related to type II secretion systems). However, very domains and its occupancy is similar in all subunits.Hence, subunits of HP0525 may operate simultaneouslylittle is known as to how they are implicated in these
biological processes. upon binding of ATP or release of ADP.The opening of the HP0525 chamber would create aAlthough, in the absence of biochemical data, any
derivation of function from structure is speculative, one large cylinder, which could accommodate passage oflarge proteins or protein domains. The CagA protein iscan infer from the structure of HP0525 bound to ADP a
working model for the function of this protein in the type certainly a potential candidate for transport. Anotherpossibility, however, would be that HP0525 is utilizedIV secretion system encoded by the Cag pathogenicity
island of H. pylori. We propose here that HP0525 acts as an inner membrane usher to ferry the protein subunitsrequired to build the parts of the type IV secretion ma-as a hexameric pore or portal, the closure and opening
of which is regulated by concerted binding of ATP and chinery bridging the inner and outer membranes. Sucha role is conceivable, since HP0525 is a homolog of therelease of ADP, respectively (Figure 7). We further pro-
pose that this ATP/ADP-controlled pore or portal is in- PulE class of ATPases, some of which, such as the PilFATPase of N. gonorrhea, is required for macromolecularvolved in translocating proteins, either the CagA protein
itself or components of the type IV secretion machinery, assembly of type IV pili (Freitag et al., 1995; Watson etal., 1996; Sauvonnet et al., 2000). In either case, thesethrough the inner membrane.
Our proposal is based on the following observations: observations suggest that VirB11 and PulE ATPasesserve as “traffic” ATPases, facilitating traffic of proteins(1) there is little contact between the N-and C-terminal
domains within a given subunit, and hence, each C-ter- through the inner membrane.VirB11 ATPases are also parts of machineries thatminal domain is only stabilized by the extended polar
interface that it forms with the N-terminal domain of transport nucleic acid (T-DNA transfer and conjugativetransfer). Nucleic acids are in most cases transportedthe adjacent subunit; (2) the nucleotide is an important
component of the subunit–subunit interface in that it as single-stranded (ss) DNAs coated with ssDNA bindingproteins (Rees and Wilkins, 1989, 1990; Gelvin, 1998;neutralizes like charges, which, in its absence, would
clash and possibly destabilize the whole interface in that Wilkins and Thomas, 2000). Hence, the transport of nu-cleic acid may not differ in fundamental ways from theregion. A possible effect of these clashes would be to
release the C-terminal domain from the interface, transport of protein. It would however have to be asequential unidirectional process, since ssDNA bindingallowing it to swivel out to open up the chamber; (3)
many properties of nucleotide binding in HP0525 are proteins form long clusters along the ssDNA, which re-semble beads on strings (Gray, 1989; Griffith et al., 1984).reminiscent of the way nucleotide binding and hydroly-
sis operate the opening and enlargement of the chamber Hence, continuous transport of a single string of severalhundreds of ssDNA binding proteins bound to ssDNAof chaperones, such as GroEL/GroES (Sigler et al., 1998;
Weber et al., 1998). Indeed, in HP0525, the nucleotide may require an active system of translocation actingunidirectionally. In that context, the VirB11 ATPase ap-plays a critical role in the interface within and between

Molecular Cell1470
terson search (program CNS version 1.0; Brunger et al., 1998) usingpears to be well suited to accomplish this task. Its cham-the single wavelength peak data yielded 12 of the 14 possible sites.ber is large enough to accommodate one ssDNA bindingInspection of the three-dimensional arrangement of these sites re-protein/ssDNA complex, such as the one generated byvealed two clusters of six selenium atoms related by a 2-fold non-
E. coli SSB, for example (Raghunathan et al., 2000). crystallographic symmetry (NCS) axis parallel to the crystallographicFurthermore, one complex could be translocated and c axis (hence this 2-fold axis was not observed in the self-rotation
function). The 12 identified sites were then used in phasing usingprevented from stepping back by the partial closure ofthe SAD phasing method as implemented by the program CNS.the HP0525 chamber. In that case, sequential cycles ofDensity modification (solvent flipping as implemented by the pro-ATP binding and ADP release would serve to ratchetgram SOLOMON; Abrahams and Leslie, 1996) resulted in a partlythe ssDNA binding protein/ssDNA complexes through.interpretable electron density map, into which 60% of the secondary
There are to date no drugs targeting specifically type structural elements could be placed as poly-Ala fragments (programII or IV secretion machineries. Such drugs would have O [Jones and Thirup, 1986; Jones et al., 1991]). From this partial
model, a NCS mask was constructed that was used to refine thethe advantage of applying to a narrow spectrum of or-NCS operators previously derived from the relationship betweenganisms. The structure of HP0525 provides the basisselenium atoms using the program PHASES (Furey and Swamina-for designing anti-ulcer drugs and could serve as athan, 1997). This mask and the refined NCS operators were thenstructural model for other ATPases of the PulE family toused in a protocol combining NCS averaging and solvent flipping
help design drugs targeting other important pathogens. (program CNS): the result was a greatly improved electron densitymap. Representative regions of this electron density map are shown
Experimental Procedures in Figure 1. This map was used to place 90% of the secondarystructural elements as poly-Ala. Several cycles of combining calcu-
Purification of Selenomethionyl HP0525 lated phases from the minimized partial model with SAD experimen-HP0525 (in plasmid pWP4760; Krause et al., 2000a) was expressed tal phases followed by NCS averaging and solvent flipping (programin the methionine auxotroph E. coli strain DL41 in the presence of CNS) resulted in electron density maps that allowed building of aselenomethionine using standard procedures. After sonication in a continuous poly-Ala chain for residues 7–328 and the identificationbuffer containing 100 mM HEPES (pH 6.6), 200 mM NaCl, 2 mM of an ADP molecule. As phases continued to improve, side chainsEDTA, 2 mg/ml aprotinin, 2.5 mg/ml leupeptin, 1 mM PMSF, and 2 were gradually introduced. A thin tube of electron density locatedmM DTT, cell lysates were centrifuged at 15,000 rpm for 20 min. near the N-terminal domain became clearly apparent and was inter-Proteins in the supernatant were precipitated by the addition of preted as a 9-mer polyethylene glycol molecule. The resulting atomic(NH4)2SO4 at 60% saturation on ice. After centrifugation, pellets were model was refined against 2.5 A data using conjugate gradient mini-resuspended in 20 ml (per 1 liter culture) of buffer A containing 20 mization (program CNS). NCS restraints were applied to the mainmM HEPES (pH 6.6), 0.1 M NaCl, 0.1 mM EDTA, 1 mM DTT, 10% chain atoms of the two HP0525 molecules, and B factors wereglycerol, and dialyzed against 2 liter of buffer A overnight. The dia- refined individually. Water molecules were added conservatively.lyzed protein sample was applied to a 5 ml Hitrap Q column equili- After bulk solvent correction, the refinement converged to a final Rbrated in buffer B containing 20 mM HEPES (pH 6.6), 0.1 M NaCl, 1 factor of 22. 3% with an R-free factor of 28.9% (30–2.5 A resolutionmM DTT, 10% glycerol. The flowthrough contained selenomethionyl range; |F|/s(|F|) . 0.0) with good stereochemistry (Table 1) (Brunger,HP0525 as the only major protein and was dialyzed against buffer 1992). The NCS restraints applied to the main chain atoms wereC (20 mM HEPES (pH 8.0), 0.1 M NaCl, 1 mM DTT, 10% glycerol) such that the root-mean-square (rms) deviation between main chainfor at least 4 hr and loaded onto a second Hitrap Q column equili- atoms in the final model is 0.09 A. Average B factors were 36 A2
brated in the same buffer. Selenomethionyl HP0525 eluted at 0.3 M and 36.3 A2 for main chain and side chain atoms, respectively, andNaCl. Pooled fractions were dialyzed against buffer D (20 mM MES rms deviations for bonded atoms was 1.6 A2 for main chain atoms[pH 7.0], 0.1 M NaCl, 1 mM DTT, 10% glycerol) and applied to a 2 and 2.5 A2 for side chain atoms. The model includes residues 6–328,ml hydroxyapatite column (BioRad CHT2 column) equilibrated in 1 ADP molecule for each of the HP0525 molecules in the asymmetricbuffer D. Proteins were eluted with a 40 ml linear gradient from 0 unit, 1 PEG molecule (n 5 9), and 300 water molecules. All φ, Cto 0.35 M potassium phosphate (pH 7.0) in buffer D. Selenomethionyl angles lie in the allowed region of the Ramachandran plot with 90%HP0525 eluted at 0.17 M potassium phosphate. HP0525 was further in the most favored regions.purified by gel filtration (Sephacryl S-200 26/60 column, Pharmacia)equilibrated in buffer D. The protein is 99% pure at this stage. Acknowledgments
Crystallization of the HP0525–ADP Binary Complex We thank J. Vogel, D. Berg, S. Beverley, F. Sauer, C. Frieden, andThe selenomethionyl HP0525–ADP complex was formed by mixing S. Hultgren for comments on the manuscript and the staff of theselenomethionyl HP0525 (0.5 mg/ml) with ADP-Mg to a final concen- Structural Biology Center for assistance during data collection. Thistration of ADP-Mg of 5 mM. The complex was then concentrated to work was supported by funds from the Washington Universityz30 mg/ml. Crystals of the selenomethionyl HP0525–ADP complex School of Medicine.were grown at room temperature by vapor diffusion in hanging dropsagainst a reservoir solution containing 100 mM Tris-HCl (pH 7.0–7.5), Received September 13, 2000; revised October 31, 2000.19%–21% PEG1000, 0.2 M calcium acetate, and 15% glycerol (Mc-Pherson, 1990). Hexagonal-shaped plate crystals were formed Referenceswithin 24 hr and continued to grow for 1 week. Single crystals (0.3 3
0.3 3 0.07 mm) were flash-frozen to liquid nitrogen temperature. Abrahams, J.P., and Leslie, A.G.W. (1996). Methods used in theCrystals were in space group P6322 with cell dimensions a 5 b 5 structure determination of bovine mitochondrial F1 ATPase. Acta112.57 A, and c 5 234.65 A with two HP0525 subunits in the asym- Crystallogr. D52, 32–42.metric unit. MAD data were collected to a resolution of 2.5 A at
Asahi, M., Azuma, T., Ito, S., Ito, Y., Suto, H., Nagai, Y., Tsubokawa,three wavelengths using a single crystal (Beamline 19ID, StructuralM., Tohyama, Y., Maeda, S., Omata, M., et al. (2000). HelicobacterBiology Center, Advanced Photon Source). However, due to sharppylori CagA protein can be tyrosine phosphorylated in gastric epi-decay during data collection, only the data collected at the firstthelial cells. J. Exp. Med. 191, 593–602.wavelength (the selenium absorption peak; Table 1) was usable, andBackert, S., Ziska, E., Brinkmann, V., Zimny-Arndt, U., Fauconnier,the structure was solved using phasing based on single-wavelengthA., Jungblut, P.R., Naumann, M., and Meyer, T.M. (2000). Transloca-anomalous diffraction (SAD).tion of the Helicobacter pylori CagA protein in gastric epithelial cellsby a type IV secretion apparatus. Cell. Microbiol. 2, 155–164.Structure Determination and Refinement
The HP0525 monomer contains 7 methionines; hence, 14 seleniums Brunger, A.T. (1992). The free R value: a novel statistical quantity forassessing the accuracy of crystal structures. Nature 355, 472–474.were expected in the asymmetric unit. Anomalous difference Pat-

Structure of the VirB11 ATPase of H. pylori1471
Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Krause, S., Barcena, M., Pansegrau, W., Lurz, R., Carazo, J.M.,and Lanka, E. (2000a). Sequence-related protein export NTPasesGrosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M.,
Pannu, N.S., et al. (1998). Crystallography and NMR system: a new encoded by the conjugative transfer region of RP4 and by the cagpathogenicity island of Helicobacter pylori share similar hexamericsoftware suite for macromolecular structure determination. Acta
Crystallogr. D54, 905–921. ring structures. Proc. Natl. Acad. Sci. USA 97, 3067–3072.
Krause, S., Pansegrau, W., Lurz, R., de la Cruz, F., and Lanka, E.Burns, D.L. (1999). Biochemistry of type IV secretion. Curr. Opin.Microbiol. 2, 25–29. (2000b). Enzymology of type IV macromolecule secretion systems:
the conjugative transfer regions of plasmids RP4 and R388 and theCarson, M. (1997). Ribbons. Methods Enzymol. 277, 493–505.cag pathogenicity island of Helicobacter pylori encode structurallyChristie, P.J., and Vogel, J.P. (2000). Bacterial type IV secretion:and functionally related nucleoside triphosphate hydrolases. J. Bac-conjugation systems adapted to deliver effector molecules to hostteriol. 182, 2761–2770.cells. Trends Microbiol. 8, 354–360.Lai, E.M., and Kado, C.I. (2000). The T-pilus of Agrobacterium tume-Chua, N.H., and Sundaresan, V. (2000). Plant biotechnology. Thefaciens. Trends Microbiol. 8, 361–369.ins and outs of a new green revolution. Curr. Opin. Biotechnol. 11,Lanka, E., and Wilkins, B.M. (1995). DNA processing reactions in117–119.bacterial conjugation. Annu. Rev. Biochem. 64, 141–169.Covacci, A., Telford, J.L., Del Giudice, G., Parsonnet, J., and Rappu-McPherson, A. (1990). Current approaches to macromolecular crys-oli, R. (1999). Helicobacter pylori virulence and genetic geography.tallization. Eur. J. Biochem. 189, 1–23.Science 284, 1328–1333.Motallebi-Veshareh, M., Balzer, D., Lanka, E., Jagura-Burdzy, G.,D’Halluin, K., and Botterman, J. (1998). The use of Agrobacteriumand Thomas, C.M. (1992). Conjugative transfer functions of broad-tumefaciens for plant genetic engineering. In The Rhizobiaceae:host-range plasmid RK2 are coregulated with vegetative replication.Molecular Biology of Model Plant-Associated Bacteria. (Amsterdam:Mol. Microbiol. 6, 907–920.Kluwer Academic Publishers), pp. 339–345.Nicholls, A., Sharp, K.A., and Honig, B. (1991). Protein folding andde la Cruz, F., and Lanka, E. (1998). Function of the Ti-plasmid Virassociation: insights from the interfacial and thermodynamic prop-proteins: T-complex formation and transfer to the plant cell. In Theerties of hydrocarbons. Protein Struct. Funct. Genet. 11, 281–296.Rhizobiaceae: Molecular Biology of Model Plant-Associated Bacte-
ria. (Amsterdam: Kluwer Academic Publishers), pp. 281–301. Odenbreit, S., Puls, J., Sedlmaier, B., Gerland, E., Fischer, W., andHaas, R. (2000). Translocation of Helicobacter pylori CagA into gas-Eisenbrandt, R., Kalkum, M., Lai, E.M., Lurz, R., Kado, C.I., andtric epithelial cells by type IV secretion. Science 287, 1497–1500.Lanka, E. (1999). Conjugative pili of IncP plasmids, and the Ti plas-
mid T pilus are composed of cyclic subunits. J. Biol. Chem. 274, Pansegrau, W., and Lanka, E. (1996). Enzymology of DNA transferby conjugative mechanisms. Prog. Nucleic Acid Res. Mol. Biol. 54,22548–22555.197–251.Finlay, B.B., and Falkow, S. (1997). Common themes in microbial
pathogenicity revisited. Microbiol. Mol. Rev. 61, 136–169. Patel, S.S., and Picha, K.M. (2000). Structure and function of hexa-meric helicases. Annu. Rev. Biochem. 69, 651–697.Freitag, N.E., Seifert, H.S., and Koomey, M. (1995). Characterization
of the pilF-pilD pilus-assembly locus of Neisseria gonorrhoeae. Mol. Possot, O., and Pugsley, A.P. (1994). Molecular characterization ofPulE, a protein required for pullulanase secretion. Mol. Microbiol.Microbiol. 16, 575–586.12, 287–299.Furey, W., and Swaminathan, S. (1997). PHASES 95: a program
package for the processing and analysis of diffraction data from Raghunathan, S., Kozlov, A.G., Lohman, T.M., and Waksman, G.(2000). Structure of the DNA binding domain of E. coli SSB boundmacromolecules. Methods Enzymol. 277, 307–326.to ssDNA. Nature Struct. Biol. 7, 648–652.Gelvin, S.B. (1998). Agrobacterium VirE2 proteins can form a com-
plex with T strands in the plant cytoplasm. J. Bacteriol. 180, 4300– Rashkova, S., Spudich, G.M., and Christie, P.J. (1997). Characteriza-tion of membrane and protein interaction determinants of the Agro-4302.bacterium tumefaciens VirB11 ATPase. J. Bacteriol. 179, 583–591.Graham, D.Y. (2000). Helicobacter pylori infection is the primary
cause of gastric cancer. J. Gastroenterol. 35, 90–97. Rees, C.E., and Wilkins, B.M. (1989). Transfer of tra proteins intothe recipient cell during bacterial conjugation mediated by plasmidGrahn, A.M., Haase, J., Bamford, D.H., and Lanka, E. (2000). Compo-ColIb-P9. J. Bacteriol. 171, 3152–3157.nents of the RP4 conjugative transfer apparatus form an envelope
structure bridging inner and outer membranes of donor cells: impli- Rees, C.E., and Wilkins, B.M. (1990). Protein transfer into the recipi-ent cell during bacterial conjugation: studies with F and RP4. Mol.cations for related macromolecule transport systems. J. Bacteriol.
182, 1564–1574. Microbiol. 4, 1199–1205.
Rossi, L., Tinland, B., and Hohn, B. (1998). Role of virulence proteinsGray, C.W. (1989). Three-dimensional structure of complexes of sin-gle-stranded DNA-binding proteins with DNA. J. Mol. Biol. 208, of Agrobacterium tumefaciens in the plant. In The Rhizobiaceae:
Molecular Biology of Model Plant-Associated Bacteria (Amsterdam:57–64.Kluwer Academic Publishers), pp. 303–320.Griffith, J.D., Harris, L.D., and Register, J. (1984). SSB protein forms
several different complexes with single stranded DNA, one of which Russel, M. (1998). Macromolecular assembly and secretion acrossthe bacterial cell envelope: type II protein secretion systems. J. Mol.is active in the assembly of stable RecA protein-DNA filaments. Cold
Spring Harb. Symp. Quant. Biol. 49, 553–559. Biol. 279, 485–499.
Salmond, G.P.C. (1994). Secretion of extracellular virulence factorsHaase, J., Lurz, R., Grahn, A.M., Bamford, D.H., and Lanka, E. (1995).Bacterial conjugation mediated by plasmid RP4: RSF1010 mobiliza- by plant pathogenic bacteria. Annu. Rev. Phytopathol. 32, 181–200.tion, donor-specific phage propagation, and pilus production re- Sauvonnet, N., Vignon, G., Pugsley, A.P., and Gounon, P. (2000).quire the same Tra2 core components of a proposed DNA transport Pilus formation and protein secretion by the same machinery incomplex. J. Bacteriol. 177, 4779–4791. Escherichia coli. EMBO J. 19, 2221–2228.Holm, L., and Sander, C. (1998). Touring protein fold space with Segal, G., Russo, J.J., and Shuman, H.A. (1999a). RelationshipsDali/FSSP. Nucleic Acids Res. 26, 316–319. between a new type IV secretion system and the icm/dot virulence
system of Legionella pneumophila. Mol. Microbiol. 34, 799–809.Jones, T.A., and Thirup, S. (1986). Using known substructures inprotein model building and crystallography. EMBO J. 5, 819–822. Segal, E.D., Cha, J., Lo, J., Falkow, S., and Tompkins, L.S. (1999b).
Altered states: involvement of phosphorylated CagA in the inductionJones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. (1991).Improved methods for building protein models in electron density of host cellular growth changes by Helicobacter pylori. Proc. Natl.
Acad. Sci. USA 96, 14559–14564.maps and the location of errors in these models. Acta Crystallogr.A47, 110–119. Sigler, P.B., Xu, Z., Rye, H.S., Burston, S.G., Fenton, W.A., and
Horwich, A.L. (1998). Structure and function in GroEL-mediated pro-Kado, C.I. (1998). Agrobacterium-mediated horizontal gene transfer.Genet. Eng. 20, 1–24. tein folding. Annu. Rev. Biochem. 67, 581–608.

Molecular Cell1472
Soto, G.E., and Hultgren, S.J. (1999). Bacterial adhesins: commonthemes and variations in architecture and assembly. J. Bacteriol.181, 1059–1071.
Stein, M., Rappuoli, R., and Covacci, A. (2000). Tyrosine phosphory-lation of the Helicobacter pylori CagA antigen after cag-driven hostcell translocation. Proc. Natl. Acad. Sci. USA 97, 1263–1268.
Stephens, K.M., Roush, C., and Nester, E. (1995). Agrobacteriumtumefaciens VirB11 protein requires a consensus nucleotide-bind-ing site for function in virulence. J. Bacteriol. 177, 27–36.
Story, R.M., and Steitz, T.A. (1992). Structure of the recA protein-ADP complex. Nature 355, 374–376.
Story, R.M., Weber, I.T., and Steitz, T.A. (1992). The structure of theE. coli recA protein monomer and polymer. Nature 355, 318–325.
Thanassi, D.G., and Hultgren, S.J. (2000). Multiple pathways allowprotein secretion across the bacterial outer membrane. Curr. Opin.Cell Biol. 12, 420–430.
Thorstenson, Y.R., Kuldau, G.A., and Zambryski, P.C. (1993). Subcel-lular localization of seven VirB proteins of Agrobacterium tumefa-ciens: implications for the formation of a T-DNA transport structure.J. Bacteriol. 175, 5233–5241.
Vogel, J.P., Andrews, H.L., Wong, S.K., and Isberg, R.R. (1998).Conjugative transfer by the virulence system of Legionella pneu-mophila. Science 279, 873–876.
Watson, A.A., Alm, R.A., and Mattick, J.S. (1996). Identification of agene, pilF, required for type 4 fimbrial biogenesis and twitchingmotility in Pseudomonas aeruginosa. Gene 180, 49–56.
Weber, F., Keppel, F., Georgopoulos, C., Hayer-Hartl, M.K., andHartl, F.U. (1998). The oligomeric structure of GroEL/GroES is re-quired for biologically significant chaperonin function in protein fold-ing. Nat. Struct. Biol. 5, 977–985.
Wilkins, B.M., and Thomas, A.T. (2000). DNA-independent transportof plasmid primase protein between bacteria by the I1 conjugationsystem. Mol. Microbiol., in press.
Winans, S.C., Burns, D.L., and Christie, P.J. (1996). Adaptation of aconjugal transfer system for the export of pathogenic macromole-cules. Trends Microbiol. 4, 64–68.
Wotherspoon, A.C. (1998). Gastric lymphoma of mucosa-associatedlymphoid tissue and Helicobacter pylori. Annu. Rev. Med. 49,289–299.
Protein Data Bank ID Code
Coordinates have been deposited with the ID code 1g6o.





























