Embed Size (px)
Citation preview

26 G . 0. Schenck, 0 . - A . Neumuller, G. Ohloff und S. Schroeter Bd. 687
Zur Autoxydation des (+)-Limonens
von Giinther 0. Schenck, 0.-Albrecht Neumiiller, Giinther Ohloff und Siegfried Schroeter 1)
Aus dem Max-Planck-Institut fur Kohlenforschung, Abteilung Strahlenchemie, Miilheim/Ruhr
Eingegangen am 23. Marz 1965
Die Autoxydation des (+)-Limonens bei 50" im Dunkeln bzw. bei Belichtung in Abwesenheit von Sensibilisatoren fiihrt zu einem anders zusammengesetzten Substanzgemisch als die photosensibilisierte 02-Ubertragung auf das gleiche Substrat. Der Bildung der Hydroper- oxyde ist wahrscheinlich bei der Autoxydation nach dem a-Methylen-Mechanismus eine Allylumlagerung der primar anzunehmenden Radikale obligatorisch vorgelagert : Die Car- veole entstehen in racemischer Form, Isopiperitenylhydroperoxyd tritt nicht auf. - Das Verhalten der 6 bei der photosensibilisierten 02-Ubertragung auf Limonen erhaltenen Allylhydroperoxyde gegeniiber Cu(1)-chlorid und Oxalsaure wird beschrieben.
Neben a-Pinen gehort Limonen zu denjenigen cyclischen Olefinen, deren Autoxy- dation am eingehendsten untersucht wurdel-24). Die Autoxydation des Kohlenwasser-
1) S. Schroeter, Dissertation Univ. Gottingen 1962. 2) A. Blumann und 0. Zeitschel, Ber. dtsch. chem. Ges. 47, 2623 (1914). 3) G. Dupont und J . Crouzet, Chim. et ind. 23, Sond.-Nr. B, S. 421 (1930) [C. 1931 I, 13681. 4) R . Dupont, Ind. chim. Belge 11, 3 (1940) [C. A. 34, 2353 (1940)l. 5 ) H. J. Strausz, Perfum. essent. Oil Rec. 38, 260 (1947) [C. A. 42, 1025 (1948)l. 6 ) B. E. Proctor und E. M . Kenyon, Food Technol. 3, 387 (1949) [C. A. 44, 10954 (1950)l. 7) H . E . Eschinazi, Bull. Res. Council Israel, Sect. A 1, 109 (1951) [C. A. 46, 4177 (1952)l;
A 2, 73 (1952) [C. A. 47, 11007 (1953)l; Perfum. essent. Oil Rec. 44, 242 (1953) {C. A. 48, 333 (1954)].
8) G. S. Fisher, L. A . Goldblatt, I . Knielund A . D . Snyder, Ind. Engng. Chem. 43, 671 (1951). 9) H . Flores und R. E. Morse, Food Technol. 6, 6 (1952) [C. A. 46, 7674 (1952)l.
10) R. H. Reitsema und F. J . Cramer, Ind. Engng. Chem. 44, 176 (1952). 11) L.-E. Fryklb'' Farmac. Revy 53, 317 (1954) [C. A. 48, 8486 (1954)l. 12) L.-E. FryklC;' Farmac. Revy 54, 244 (1955) [C. A. 49, 14276 (1955)l. 13) N . F. Ermolenko, E. N . Novikova und B. Filonov, Uchenye Zapiski, Beloruss. Gosudarst.
Univ. im. V. I. Lenina, Ser. Khim. 1954, Nr. 20, 106 [C. A. 51, 18672 (1957)l. 14) B. V. Erojeev und A . I. Chirko, Uchenye Zapiski, Beloruss. Gosudarst. Univ. im. V. I.
Lenina, Ser. Khim. 1955, Nr. 24, 16, 31 [C.A.51, 5730, 5731 (1957)l. 15) E. E. Royals und S. E. Hornejr., J. Amer. chem. SOC. 77, 187 (1955); S. E. Hornejr.,
Dissertat. Abstr. 19, 2471 (1959) [C. A.53, 15113 (1960)). l 6 ) G. Widmark und S.-G. Blohm, Acta cbem. scand. 11, 392 (1957). 17) G . Widmark, Ark. Kemi 11, 21 1 (1957) [C. A. 52, 1107 (1958)l. l 8 ) H. Schmidt, Suomen Kemistilehti B31, 61 (1958) [C. A. 52, 20226 (1958)l. 19) E. V. Erofeev, Voprosy khim. Terpenov i Terpenoidov Akad. Nauk. Litovsk. SSR,
Trudy Vsesoyuz. Sovesh. Vil'nyus 1959, 103 [C. A. 55, 17681 (1961)l.

1965 Zur Autoxydation des (+)-Limonens 27
stoffs verlauft als autokatalysierte Kettenreaktion, deren Induktionsperiode durch Zusatz von Peroxyden oder Schwermetallen verkurzt und deren Reaktionsgeschwindig- keit durch Radikalfanger, wie Phenole, gemindert wird 23-25). Als Autoxydations- produkte des (+)-Limonens (1, absol. Konfiguration 26)) bzw. als deren Sekundar- produkte wurden ( &)-Carvon4) (13) und ( ~)-trans-Carveol2.16) (9b) sowie (+)-Li- monenoxyd-(1.2) 18) (14) gefunden. Bain und Mitarbeiter 21,22) erhielten als weitere Autoxydationsprodukte ( i)-cis-Carveol (9a), (+)-Limonenoxyd-(8.9) (15) sowie die beiden tertiaren (+)-p-Menthadien-(2.8)-ole-(l) (Sc bzw. Sd).
Die unter AusschluD von Sauren bzw. Schwermetallkatalysatoren bei 50" im Dun- keln bis zur Aufnahme von 0.6 Moll. 0 2 bzw. unter Belichtung bei 50" in Abwesenheit von Sensibilisatoren durchgefiihrte Autoxydation von 1 lieferte in unseren Versuchen nach Reduktion der Hydroperoxyde mit Na2SO3 neben unveranderteni Ausgangs- material und einem undestillierbaren Ruckstand die in Tabelle 1 aufgefiihrten Pro-
Tabelle 1. Produkte der Autoxydation von (+)-Limonen (nach Reduktion) unter verschiedenen Bedingungen: 1) 12 Stdn. bei 50" im Dunkeln (vgl. Abb. I ) . 2) Vgl. Lit.22); 7 % Verluste, 20 % Ruckstand. 3) 43 Stdn. bei 50" unter Belichtung in GlasgefaB. 4) 20 Stdn.
bei 15" in Aceton unter Belichtung (Quarzgefaf3)
Reduktionsprodukt Ausbeute in % unter Versuchsbedingung 1) 2) 3) 4)
(+)-Limonenoxyd-(1.2) (14) 13 (+)-Limonenoxyd-(8.9) (15) ~
Unbekannte Substanz -
(+)-trans-p-Menthadien-(2.8)-01-( 1) a) (8 c) 11 (+)-cis-p- Menth adien-(2.8)-01-( 1) a) (8d) 12 Unbekannte Substanzb) 10 (f)-Carvon (13) 13 p-Menthadien-[I (7).8]-trans-o1-(2) (1 1 b) (rt)-tvans-Carveol(9 b) 22 (+)-cis-Carveol (9a) 19 p-Menthadien-[I (7).8]-cis-o1-(2) (11 a) - Hohersiedende Anteile -
-
a) trans- bzw. &-Stellung von Methyl- zur Isopropenylgruppe. b) Von Isopiperitenol verschieden.
22
-
7 11 8
19
19 10
A
-
-
12
5 16 12 6 8 4
21 8 8
-
20) R. A. Bernhurd und A. G. Murr, Food Res. 25, 517 (1960) [C. A. 55, 26011 (1961)l. 21) 21a) Glidden Co., Engl. Pat. 761686 v. 21.4. 1954 IC. 1960. 33821. - 21b) J . P. Bnin.
A. B. Booth und E. A . Klein, Amer. Pat. 2863882 v.-9. 12. 1958, Glidden Co. [C. A. 53, 8194 (1959)l.
22) J. P. Buin, W. Y. Gary und E. A . Klein, Amer. Pat. 3014047 v. 19. 12. 1961, Glidden Co. [C. A. 57, 12557 (1962)l.
23) A. G . Davies, Organic Peroxides, Butterworths & Co., London 1961 ; zur Autoxydation
24) E. G . E. Hawkins, Organic Peroxides: Their Formation and Reactions, Spon, Ltd.,
25) W. 0. Lundberg, Autoxidation and Antioxidants, Bd. 1, Interscience Publ., New York
26) A . J . Birch, Annu. Rep. Progr. Chem. 47, 191 (1950).
bes. S. 18f.
London 1961; zur Autoxydation bes. S. 3731.
1961.
28 G. 0. Schenck, 0 . - A . Neumiiller. G. Ohloff und S. Schroeter Bd. 687
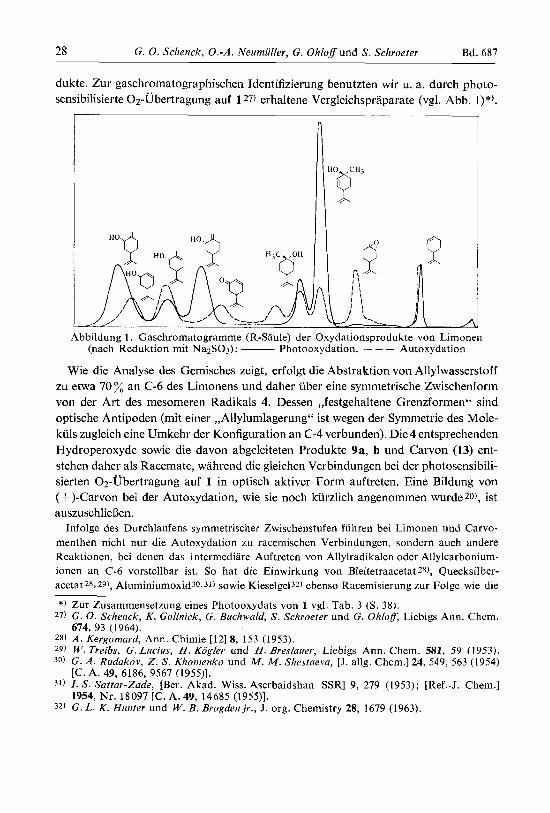
dukte. Zur gaschromatographischen Identifizierung benutzten wir u. a. durch photo- sensibilisierte 02-Ubertragung auf 1 27) erhaltene Vergleichspraparate (vgl. Abb. l)*).
A Abbildung 1 . Gaschromatogramme (R-Saule) der Oxydationsprodukte von Limonen
(nach Reduktion mit NazSOS): - Photooxydation, - - - Autoxydation
Wie die Analyse des Gemisches zeigt, erfolgt die Abstraktion von Allylwasserstoff zu etwa 70 % an C-6 des Limonens und daher iiber eine symmetrische Zwischenform von der Art des mesomeren Radikals 4. Dessen ,,festgehaltene Grenzformen" sind optische Antipoden (mit einer ,,Allylumlagerung" ist wegen der Symmetrie des Mole- kuls zugleich eine Umkehr der Konfiguration an C-4 verbunden). Die 4 entsprechenden Hydroperoxyde sowie die davon abgeleiteten Produkte 9a, b und Carvon (13) ent- stehen daher als Racemate, wahrend die gleichen Verbindungen bei der photosensibili- sierten 02-Ubertragung auf 1 in optisch aktiver Form auftreten. Eine Bildung von (+)-Carvon bei der Autoxydation, wie sie noch kurzlich angenommen wurdezo), ist auszuschlieflen.
Infolge des Durchlaufens symmetrischer Zwischenstufen fuhren bei Limonen und Carvo- menthen nicht nur die Autoxydation zu racemischen Verbindungen, sondern auch andere Reaktionen, bei denen das intermediare Auftreten von Allylradikalen oder Allylcarbonium- ionen an C-6 vorstellbar ist. So hat die Einwirkung von Bleitetraacetat 28), Quecksilber- acetat 28,291, Aluminiumoxid30.31) sowie Kieselgel32) ebenso Racemisierung zur Folge wie die
* ) Zur Zusammensetzung eines Photooxydats von 1 vgl. Tab. 3 (S. 38). 27) G. 0. Schenck, K . Gollnick, G. Buchwald, S . Schroeter und G . Ohlofi Liebigs Ann. Chem.
28) A . Kergomard, Ann. Chimie [12] 8, 153 (1953). 29) W . Treibs, G. Lucius, N. Kogler und H. Breslaucr, Liebigs Ann. Chem. 581, 59 (1953). 30) G. A. Rudakov, Z . S . Khomenko und M . M . Shestaeva, [J . allg. Chem.] 24, 549, 563 (1954)
31) I . S . Sattar-Zade, [Ber. Akad. Wiss. Aserbaidshan SSR] 9, 279 (1953); [Ref.-J. Chem.]
32) G. L. K . Hunter und W . B. Brogden j r . , J. org. Chemistry 28, 1679 (1963).
674, 93 (1964).
[C. A . 49, 6186, 9567 (1955)l.
1954, Nr. 18097 [C. A. 49, 14685 (1955)l.

1965 Zur Autoxydation des (+)-Limonens 29
Gegenwart von Protonen bei der Allylumlagerung des aktiven Carveols33-35). Die unter Ausbildung eines mesomeriefahigen Carbeniat- Anions an C-6 verlaufende Reduktion des (+)-cis-Carvotanacetol-methylathers mit Lithium in Athylamin fuhrt zu racem.-Carvo- menthen36). In gleicher Weise bewirkt Natrium die Racemisierung von 137) .
Die gleichen Betrachtungen gelten analog fur die entsprechenden Reaktionen am A3- Menthen: Autoxydation38), Umsetzung mit N-Brom-succinimid39) oder Quecksilberacetat 29)
und Saurebehandlung40) sowie wahrscheinlich auch die Einwirkung von Maleinsaure- anhydrid41) liefern die racem. Verbindungen. ErwartungsgemaD bleibt die optische Aktivitat erhalten, wenn unsymmetrische Kohlenwasserstoffe, wie Sylvestren34), A3-Caren und A4-Ca- ren42,43). in der gleichen Art behandelt werden.
Eine symmetrische Zwischenstufe der autoxydativen Bildung des racem-Carveols (9a, b) wurde bereits fruher1731*) diskutiert. DaB das bei der Autoxydation erhaltene Hydroperoxydgemisch dennoch optisch aktiv ist, findet seine Erklarung durch den Nachweis der beiden tertiaren Hydroperoxyde 1,*1,22).
Wird Limonen aus 4 zuruckgebildet, so liegt es racemisiert vor. Bei der UV-Belich- tung von (+)-Limonen in Aceton unter Stickstoff, bei der 4 ebenfalls entsteht, erfolgt deshalb auch eine betrachtliche Racemisierung des eingesetzten Kohlenwasserstoffes44~. Gleiches geschieht bei (+)-Carvomenthen45). Das aus dem Autoxydationsgemisch zuruckgewonnene Limonen besitzt jedoch seine volle optische Aktivitat (vgl. Lit. 21,22)). Wir schlieljen daraus, dalj in Gegenwart von 0 2 samtliche einmal gebildeten Radikale 4 zu den ( *)-Carveylhydroperoxyden weiterreagieren. Diese Erklarung gilt auch fur die Belichtung von 1 in 02-gesattigtem Aceton, bei der ebenfalls keine Racemisierung des unverbrauchten Limonens gefunden wird.
Das Auftreten der tertiaren Alkohole 8c und 8d sowie das Fehlen von Isopiperitenol (7a und 7b) unter den Reaktionsprodukten (s. auch Lit. 22,469 zeigen weiter, dalj das
33) A. Blumann und W. R . Wood, J . chem. SOC. [London] 1952, 4420. 34) H. Schmidt, Chem. Ber. 86, 1437 (1953). 35) 0. Wulluch, Liebigs Ann. Chem. 281, 127 (1894). 36) A. S. Hullsworth, H. B. Henbest und T. I . Wrigley, J. chem. SOC. [London] 1957, 1969. 37) H. Pines und H. E . Eschinuzi, J . Amer. chem. SOC. 77, 6314 (1955). 38) K . A. Pecherskuya und K . A. Krasnik, Uchenye Zapiski, Beloruss. Gosudarst. Univ. im.
V. I. Lenina, Ser. Khim. 1954, Nr. 20, 173 [C. A. 51, 11 292 (1957)l. 39) A. K. Macbeth, B. Milligan und J . S . Shannon, J . chem. SOC. [London] 1953, 2574. 40) H . Kwart und L. B. Weisfeld, J . Amer. cheni. SOC. 80, 4670 (1958); W . Hiickel, W . Tuppe
und G. Legutke, Liebigs Ann. Chem. 543, 191 (1940). 41) K . A . Pecherskuya und R . I . Belyakova, Zhidkofaznoe Okislenie Nepredel'nykh Organ.
42) L. M . Yeddanupalli und P. Desikan, J. sci. ind. Res. [New Delhi], Sect. B 18, 392 (1959) Soedin. Sb. 1961, Nr. 1, 145 [C. A. 58, 553 (1963)l.
rc. A. 54. 7770 (1 9 6 0 ~ . 43) 'c. A . Rudukov und 2. T. Murchevskii, [Samml. Aufsatzen allg. Chem.] 2, 1432 (1953)
44) G. 0. Schenck, R . Steinmetz und S. Schroeter, unveroffentlicht. 45) P . De Mayo, J . B. Stothers und W . Templeton, Canad. J. Chem. 39, 488 (1961). 46) R. Lalande und J. Moulines, Bull. SOC. chim. France 1959, 1442.
[C. A. 49, 5390 (195511.

30 G. 0. Schenck, 0 . - A . Neumiiller, G . Ohloff und S. Schroeter Bd. 687
durch Dehydrierung an C-3 entstehende Radikal 2 nur in seiner ,,festgehaltenen" allylmesomeren Form 3 weiterreagiert. ,,Festgehaltene Grenzformen" konnen vorge- tauscht werden, wenn die Moglichkeiten der Ausbildung allyltautomerer Radikal- solvate, etwa durch Addition der Radikalzentren an ein Solvens, nicht beriicksichtigt werden. Im thermodynamischen Gleichgewicht solcher allyltautomerer Radikalsolvate geniigt bei Raumtemperatur bereits eine Energiedifferenz von nur 2 -3 kcal/Mol, um die energetisch begiinstigte Form praktisch allein das Reaktionsbild bestimmen zu lassen. Die unterschiedliche Tendenz zur Verschiebung von Doppelbindungen bei der Autoxydation23-2S347) diirfte hier eine ihrer wesentlichen Ursachen haben.
Aus a-Pinen entsteht neben dem tertiaren Hydroperoxyd das Verbenylhydroperoxyd wahr- scheinlich direkt 21a). Allerdings ist ein strenger Vergleich der Reaktivitaten der Doppel- bindungen bzw. der Allylwasserstoffe nicht moglich, wie auch aus dem beim a-Pinen, nicht aber beim Limonen beobachteten Auftreten von Autoxydationsprodukten mit semicyclischer Doppelbindung hervorgeht. Offensichtlich spielen beim a-Pinen sterische Faktoren eine Rolle, die ein Ausweichen der Dehydrierung auf sonst benachteiligte Wasserstoffe erzwingen.
Die fruher als direkte Autoxydationsprodukte von 1 beschriebenen Isopiperitenole (7a, b) entstehen also erst sekundar durch Allylumlagerung aus trans- bzw. cis-p-Menthadien-(2.8)- 01-(1) (8c bzw. 8d)1,21,22,48). Diese Umwandlungen erfolgen sehr leicht unter dem EinfluB von Sauren, also auch unter den Bedingungen der Chromsaureoxydation. Aus diesem Grund bedeutet hier die Bildung von Isopiperitenon (12) keinen Beweis fur das Vorliegen der Iso- piperitenole. (+)-Isopiperitenon entstand auch, neben (&)-Cawon, Piperitenon und anderen Produkten, bei der Oxydation von Limonen mit tert.-Butylchromat in Gegenwart von Sauerstoff49).
Ebenso lLRt die Bildung von Menthon bzw. Menthol aus den Autoxydationsprodukten des Limonens 7) nicht auf den direkten Angriff des Sauerstoffs auf die 3-Stellung schlieRen. Diese Produkte sollten vielmehr in der Reaktionsfolge [2 H 31 + 8 -+ 12 mit anschlie- Render Hydrierung entstehen. Entsprechendes gilt fur die bisherige Deutung de<Autoxydation des Carvomenthens50). Hier wurde nach der Chromsaureoxydation der reduzierten Autoxy- dationsprodukte Piperiton nachgewiesen und als dessen Vorstufe Piperitylhydroperoxyd bzw. Piperitol angenommen.
Ein sicherer Nachweis der direkten Bildung sekundarer Allylhydroperoxyde der hier untersuchten Art ist nur moglich, wenn die von uns51) beschriebene Allylumlage- rung tertiarer Allylhydroperoxyde experimentell ausgeschlossen wird, und wenn durch Aufarbeitung der Alkohole unter AusschluD von Sauren Allylumlagerungen der tertiaren Allylalkoholel. 48,50,52) vermieden werden.
47) L. Bateman, Quart. Rev. (chem. SOC., London) 8, 147 (1954). 48) J. P. Bain, A. B. Booth und W. Y. Gary, Amer. Pat. 2894040 v. 7. 7. 1959, Glidden Co.
[C. A. 53, 22067 (1959)l; J . P. Bain, A . B. Booth und E. A. Klein, Amer. Pat. 2827499 v. 18. 3 . 1958, Glidden Co. [C.A. 52, 14719 (1958)l; J. P. Bain und W. Y. Gary, Amer. Pat. 2831 028 v. 15.4. 1958, Glidden Co. [C. A. 52, 14720 (1958)J.
49) K. Fujita, J. Sci. Hiroshima Univ., Ser. A 24, 691 (1960) [C. A. 56, 4799 (1962)J. 5 0 ) V . E. Veijolu und A . 0. Ilvespaa, Acta chem. scand. 13, 301 (1959). 51) G . 0. Schenck, 0 . - A . Neumiiller und W. Eisfeld, Liebigs Ann. Chem. 618, 202 (1958). 52) G. H. Whifham, J. chem. SOC. [London] 1961, 2232.
1965 Zur Autoxydation des (+)-Limonens 31
Wie die Analyse zeigt, erfolgte die Dehydrierung des Limonens ausschlieBlich in u-Stellung zur Al-Doppelbindung an C-3 und an C-6. Die bei einem Angriff an C-7 zu erwartenden Reaktionsprodukte 10 und 11 a, b entstanden unter unseren Bedin- gungen nicht.
15 1 14
-H
1 7
2 3
I 7 8 9 10
6
I R29 11
0 Q 12
7a, 9a, 1la: R' = OH, R~ = H b: R' = H, R2 = OH
8 ~ : R3 = OH, R' = CH3 d: R3 = CH3, R4 = OH
13
Die Ablosung von primaren Wasserstoff-Atomen ist jedoch mit kraftigeren Dehydrierungs- mitteln moglich. So vermag photochemisch angeregtes Aceton derartige Verbindungen auch an der C-7-Methylgruppe zu [5 t--f 61 zu dehydrieren: Wird 1 in Aceton unter Stickstoff UV-bestrahlt, so bildet sichp-Menthadien-[l(7).8] (11, R1 = R2 = H)u). Analog entsteht aus Carvomenthen das p-Menthen-[I (7)]45). 1st bei der UV-Bestrahlung in Aceton ausreichend

32 G. 0. Schenck, 0 . - A . Neumiiller, G . Ohloff und S. Schroeter Bd. 687
Sauerstoff vorhanden, so werden die Allylradikale rnit semicyclischer Doppelbindung abge- fangen. Die aus der Reduktion so gebildeter Hydroperoxyde hervorgehenden Alkohole 11 a und 11 b sind dann in dem Gemisch zu 8 bzw. 4 % enthalten (vgl. Tab. 1, S. 27).
Eine Dehydrierung an C-4 in Allylstellung zur A*-Doppelbindung findet, falls iiberhaupt, nur in untergeordnetem MaBe statt *). Eine Ursache hierfiir ist moglicher- weise in der axialen Stellung des Wasserstoffs an C-4 zu suchen.
Nesmeiunov und Mitarbeite~-53) stellten an bicyclischen Terpenkohlenwasserstoffen fest, dal3 solche quasi-axialen Wasserstoffe selbst dann nicht autoxydativ abgelost werden, wenn sie durch eine Allyldoppelbindung aktiviert sind. cis-p-Menthan rnit aquatorialem Wasserstoff an C-1 bildete bei der Autoxydation wesentlich rascher ein 1-Hydroperoxyd als trans-p-Menthan rnit axialem C-l-HS4). Ein Angriff an C-4-H wurde dabei ebensowenig wie bei Carvomenthen und a-Terpineol beobachtet 21322.54).
Daher liefern Carvomenthen und a-Terpineol bei der Autoxydation gleichartige Produkte, die sich von denen des Limonens nur in der Seitenkette unterscheiden. Dies zeigt Tab. 2 anhand der von Buin und Mitarbeitern22) bei Carvomenthen und a-Ter- pineol destillativ, von uns bei Limonen gaschromatographisch durchgefiihrten Anal ysen.
In der Carvomenthen-Reihe wurden die ersten 4 Oxydationsprodukte in optisch aktiver Form erhalten22). trans-Carvotanacetol sollte jedoch nach dem Radikalmechanismus der Autoxydation in racem.-Form anfallen. Nach der angegebenen Drehung von - 5.8" [reines (-)-trans-Carvotanacetol C(D = - 1 87.3"]55) vermuten wir, daR das Produkt von dem negativ drehenden tertiaren Alkohol (Sc, rnit gesattigter Seitenkette) begleitet war. Angaben iiber die optische Aktivitat der Autoxydationsprodukte des cc-Terpineols liegen in der Patentschrift 22)
nicht vor.
Die Ubereinstimmung in den Ausbeuten einander entsprechender Verbindungen aus Limonen, Carvomenthen und a-Terpineol ist bis auf die der sekundaren Alkohole mit cis-standiger OH-Gruppe befriedigend. Die Abweichungen lassen sich leicht auf unterschiedliche experimentelle Bedingungen zuriickfiihren :
Der Epoxydgehalt steigt rnit zunehmender Dauer der Autoxydation an, wahrend gleich- zeitig die Peroxydzahl sinkt 12922). Die Ausbeute vom zuletzt destillierenden cis-Carve01 bzw. cis-Carvotanacetol ist stets dann zu gering, wenn der Ruckstand nicht geniigend erhitzt wird. cis-Sobrerol geht jedoch bei der Destillation bereits in Pinol iiber22).
Ob bei der Autoxydation des Limonens zwei 1.2-Epoxyde (14) gebildet werden, konnte mit der von uns zur Gaschromatographie der Autoxydationsprodukte verwendeten R-Saule nicht
*) Moglicherweise handelt es sich bei der zu etwa 10% entstehenden, nicht identifizierten Verbindung um das Produkt eines Angriffs in Allylstellung zur exocyclischen Doppel- bindung.
53) A . N . Nesmeianov, K . A . Pecherskaia, A . N . Akhramovich und L. M. Minakova, Proc. Acad. Sci. USSR, Chem. Sect. 121, 601 (1958).
54) J . S. Stinson, G . S. Fisher und J . E. Hawkins, J. org. Chemistry 24, 1084 (1959); s. aber Y. Mafsubara, Y. Heya und H. Aizawu, J. chem. SOC. Japan, pure Chem. Sect. [Nippon Kagaku Zasshi] 81, 479 (1960) [C. A. 56, 1484 (1962)l.
5 5 ) S. Schroeter, Liebigs Ann. Chem. 674, 118 (19643.

1965 Zur Autoxydation des (+)-Limonens 33
entschieden werden; aus 1 mit Peressigsaure hergestelltes 14 erwies sich nur auf der K-Saule als in zwei Komponenten spaltbarl,s6). Lediglich im Fall des a-Terpineols ist es gelungen, die bei der Autoxydation entstandenen beiden 1.2-Epoxyde destillativ zu trennenzz).
Der Vergleich der stereoselektiv verlaufenden photosensibilisierten 02-Ubertragung auf Limonen 27) mit der Autoxydation zeigt, daB letztere keine Steveoselektivitat
Tabelle 2. Autoxydationsprodukte von Limonen, Carvomenthen und a-Terpineol (nach der Reduktion)
Autoxvdations- X-Anteil im Reaktionsgemisch aus
Limo n e n Carvomenthen a-Terpineol produkt
(R=/,) ( R = /I\ ) OH (R =A)
0 a: : /\i
14
k HO CH3 0 8c
R
13
R Unbekannt
13
11
16.8 19
1 3 . 7 a ) 12
12 1 0 . 0 s ) 15
22 23.4 36
19 8.5 4 b)
1 3 9.0 7
10 3 . 1 - 15.5 Riickstand -
a) Konfigurative Zuordnung bei Lit. 27.55). - b) AlsYPinol. I
56) H. Kuczyriski und K . Piatkowski, Roczniki Chem. [Ann. SOC. chim. Polonorum] 33, 311 (1959) [C. A. 53, 18083 (1959)l.
Liebigs Ann. Chem. Bd. 687 3

34 G . 0. Schenck, O. -A. Neumiiller, G . Ohloff und S . Schroeter Bd. 687
erkennen la&: Die entstehenden cis-trans-Paare der tertiaren Alkohole 8 wie der Carveole 9 werden jeweils im Verhaltnis 1 : 1 gebildet *). Die Unspezifitat der Autoxy- dation ergibt sich als einfache Folge des cc-Methylen-Mechanismus57) : Die Addition des Sauerstoffs an die durch primare Dehydrierung in Allylstellung entstandenen allylmesomeren Radikale 2/3 bzw. 4 vermag statistisch von beiden Seiten des Molekiils unter Bildung gleicher Mengen cis- und trans-isomerer Hydroperoxyde zu erfolgen.
Aus der Gegenuberstellung 1,27,58) von Autoxydation und photosensibilisierter 0 2 -
Ubertragung an den Beispielen des cr-Pinens 2 1 ~ 5 9 ) und des A3-Carens6oI61) geht ebenfalls hervor, daR die Autoxydation nicht stereoselektiv verlauft.
Katalytische Zersetzung der Limonenhydroperoxyde Crab. 3, S 38, und Abb. 2)
Da die tertiaren p-Menthadien-(2.8)-ole-(l) (Sc, d) unter den Autoxydationspro- dukten lange unentdeckt geblieben waren, nahmen wir an, daB die entsprechenden tertiaren Hydroperoxyde sich katalytisch zersetzt hatten. Zur Prufung dieser Annahme untersuchten wir qualitativ das Verhalten des Gemisches der aus der photosensibili- sierten 02-Ubertragung erhaltlichen Limonenhydroperoxyde27) gegeniiber Schwer- metallkatalysatoren und Sauren. Das Photooxydat enthielt aufgrund seiner Bildungs- weise weder Carvon noch Isopiperitenylhydroperoxyd.
Da wir nur an der Untersuchung der Stabilitat der Hydroperoxyde gegen die genannten Agenzien interessiert waren, begnugten wir uns damit, das aus der Umsetzung resultierende Gemisch nach seiner Reduktion qualitativ auf das Vorhandensein oder die Abwesenheit der bekannten27) Produkte zu prufen.
Verwendeten wir CuCl als Katalysator, so verhielten sich die Reaktionsgemische nur noch schwach bzw. nicht mehr peroxydisch. Nach Reduktion mit Na2S03 verblieb in so behandelten Losungen neben den tertiaren Alkoholen 8c, d haupt- sachlich Carvon (vgl. Abb. 2 ) : Die tertiaren Hydroperoxyde reagieren also mit CuCl unter Sauerstoffabspaltung zu den tertiaren Alkoholen, die sekundaren
*) Streng gilt dies nur unter der Annahme, daI3 (+)-Carvon (13) aus den Carveylhydro- peroxyden (9, R1 bzw. R2 = OOH, R2 bzw. R1 = H) zu gleichen Teilen entsteht.
57) Die Entdeckung des cc-Methylen-Mechanismus wird im allgemeinen E. H. Farmer, Trans. Faraday SOC. 38, 340 (1942); 42, 228 (1946), sowie E . H. Farmer, C. F. Bloomfield, A . Sundralingam und D . A . Sutton, ebenda 38, 348 (1942), zugeschrieben. Der Angriff des Sauerstoffs in Allylstellung bei der Autoxydation wurde jedoch bereits von H . Schmidt, Dissertation Univ. Gottingen 1925; Ber. dtsch. chem. Ges. 63, 1129 (1930), und H. Wienhaus, Angew. Chem. 41, 617 (1928), formuliert. Noch friihere Hinweise auf die Allyloxydation finden sich bei A . Blumann und 0. Zeitschel, Ber. dtsch. chem. Ges. 46, 1178 (1913), bes. S. 1187.
5 8 ) G. 0. Schenck, H. Eggert und W. Denk, Liebigs Ann. Chem. 584, 177 (1953). 5 9 ) G . Helms, Dissertation, Univ. Gottingen 1961. 60) B. A . Arbuzov, Z . G. Isaeva und V. V. Ratner, Proc. Acad. Sci. USSR, Chem. Sect. 134,
1031 (1960). 61) G . 0. Schenck, S . Schroeter und G . Ohloff, Chem. and Ind. 1962, 459; K. Gollnick,
S. Schroeter, G . Ohloff, G . Schade und G . 0. Schenck, Liebigs Ann. Chem. 687, 14 (1965).
1965 Zur Autoxydation des (+)-Limonens 35
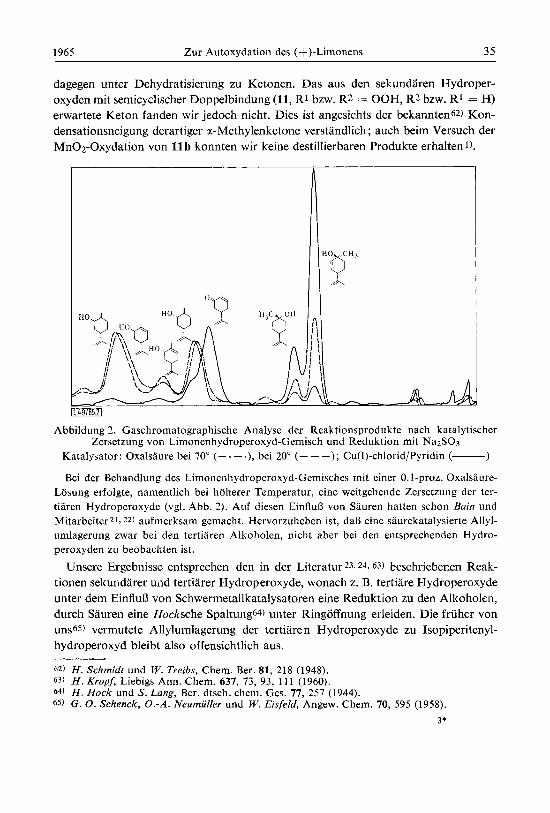
dagegen unter Dehydratisierung zu Ketonen. Das a m den sekundaren Hydroper- oxyden mit seniicyclischer Doppelbindung (11, R1 bzw. R2 = OOH, R2 bzw. R1 = H) erwartete Keton fanden wir jedoch nicht. Dies ist angesichts der bekannten62) Kon- densationsneigung derartiger a-Methylenketone verstandlich; auch beim Versuch der Mn02-Oxydation von 11 b konnten wir keine destillierbaren Produkte erhaltenl).
un U " 0 K i-
Abbildung 2. Gaschromatographische Analyse der Reaktionsprodukte nach katalytischer Zersetzung von Limonenhydroperoxyd-Gemisch und Reduktion mit Na2SO3
Katalysator: Oxalsaure bei 70" (- . - .), bei 20" (- - -); Cu(1)-chlorid/Pyridin (- 1 Bei der Behandlung des Limonenhydroperoxyd-Gemisches mit einer 0. I-proz. Oxalsaure-
Losung erfolgte, namentlich bei hoherer Temperatur, eine weitgehende Zersetzung der ter- tiaren Hydroperoxyde (vgl. Abb. 2). Auf diesen EinfluB von Sauren hatten schon &in und Mitarbeiter 21,22) aufmerksam gemacht. Hervorzuheben ist, daB eine saurekatalysierte Allyl- umlagerung zwar bei den tertiaren Alkoholen, nicht aber bei den entsprechenden Hydro- peroxyden zu beobachten ist.
Unsere Ergebnisse entsprechen den in der Literatur 23,24,63) beschriebenen Reak- tionen sekundarer uild tertiarer Hydroperoxyde, wonach z. B. tertiare Hydroperoxyde unter dem EinfluB von Schwermetallkatalysatoren eine Reduktion zu den Alkoholen, durch Sauren eine Hocksche Spaltung64) unter Ringoffnung erleiden. Die fruher von uns65) vermutete Allylumlagerung der tertiaren Hydroperoxyde zu Isopiperitenyl- hydroperoxyd bleibt also offensichtlich aus.
6 2 ) H . Schmidt und W. Treibs, Chem. Ber. 81, 218 (1948). 63) H . Kropf, Liebigs Ann. Chem. 637, 73, 93, 11 1 (1960). 64) H . Hock und S. Lang, Ber. dtsch. chem. Ges. 77, 257 (1944). 65) G . 0. Schenck, 0 . - A . Neumiiller und W. Eisfeld, Angew. Chem. 70, 595 (1958).
3'

36 G. 0. Schenck, 0 . -A . Neumiiller, G. Ohloff und S. Schroeter Bd. 687
Nachtrag bei der Korrektur (28. 6. 65): Nach AbschluR der vorliegenden Arbeit haben A . Blumann, H. Farnow und F. Porsch, J. chem. SOC. [London] 1965, 2990, aus Limonen, das wahrend der Sommermonate in Australien in offenen StahlgefaRen der Luft ausgesetzt war, auch Perillaalkohol (10) in 0.04-proz. Ausbeute isoliert.
Beschreibung der Versuche
Die Schmelzpunkte wurden nach Kofler bestimmt und sind korrigiert. - NIR-Spektren von CC4-Losungen wurden rnit einem Gerat Beckman.DK-2, IR-Spektren von Filmen zwischen KBr-Platten bzw. von KBr-PreRlingen rnit einem Gerat Beckman-IR-4 aufgenom- men. - Zur Gaschromatographie (GC) diente ein Perkin-Elmer-Fraktometer 11 6 mit C-, K- und R-Saule; letztere wurde bei 170-180", 1.5-1.55 atu und 106ccm He/Min. betrieben. - Die Bestimmung der optischen Drehung von Flussigkeiten erfolgte in Substanz, von Festsubstanzen in Chloroform bei c = 1-5. - Die Analysen wurdenvon A . Bernhardt, Mulheim/Ruhr, ausge- fuhrt. - Die Belichtungen mittels Hg-Hochdruckbrenners nahmen wir bei 15-60" in Belich- tungsgefaRen.66) mit Tauchlampenschacht aus Weichglas bzw. Quarz unter kontinuierlicher Registrierung des 02-Verbrauches vor.
Autoxydation von (+)-Limonen
a) 350 g reines (+)-Lirnonen (l), [cA]&O = + 123", wurden im Dunkeln im geschlossenen Sauer- stof-Kreislauf bei 50" autoxydiert; Sauerstoffaufnahme wahrend 12 Stdn. 0.62 Moll. Nach Reduktion rnit 250 g NuzS03 + 5 g KOH in 900 ccm Wasser (15 Stdn. bei 20", 2 Stdn. bei 60") und Trocknen der ausgeatherten Phase rnit N a ~ S 0 4 und K2CO3 wurde das nicht umge- setzte 1 unter N2 uber eine 40 cm lange, rnit V2A-Wendeln gefullte Kolonne uber Na2CO3 destilliert: [a]D = +123" bis +120". Destillation der angereicherten Fraktionen bei 35 bis 75"/0.1 Torr lieferte, neben 9 g undestillierbarem Ruckstand, 55 g Gemisch (nL0 = 1.431, a23 - - +50.8"), das noch 12% Limonen enthielt (GC).
b) In einem weiteren Ansatz belichteten wir 1 kg 1 bei 50-60" wahrend 43 Stdn. rnit einer Quecksilberdampflampe HgH 2000 (Osram) in einer Glasapparatur im geschlossenen 0 2 -
Kreislauf. Nach Reduktion rnit 400 g N a ~ S 0 3 + 6 g KOH und Abdestillieren des unver- brauchten Limonens wurden 221 g Oxydationsgemisch neben 130 g Ruckstand erhalten.
Die gaschromatographischen Analysen der beiden Gemische (vgl. Tab. I , S. 27) wurden auf einer 2-m-R-SSule durchgefuhrt. Eine Trennung von 7a, b und 13 sowie von 9a und 11 a war unter diesen Bedingungen nicht moglich. Nach Reduktion des Gemisches rnit LiAIH4 konnten 7a, b und 9a, b (aus 13) unterschieden werden.
Trennung der Autoxydationsprodukte: 50 g des im Dunkelansatz erhaltenen Gemisches wurden uber Na2CO3 fraktioniert destilliert (Drehbandkolonne, Haage, Mulheim) :
I ) (+)-Limonenoxyd-(I.2) (14). - Die ersten Fraktionen bestanden aus Limonen. Diesem folgte eine Fraktion, die reines 14 (GC) enthielt: nL0 = 1.4661, cc',' = +54.5"; Lit.18) nh0 =
6 6 ) G. 0. Schenck, Dechema-Monogr. 24, 105 (1955).

1965 Zur Autoxydation des (+)-Limonens 37
1.4693, CLD = + 60.4". Beim Schutteln dieser Fraktion mit verd. Schwefelsaure kristallisierte unter Temperaturerhohung Limonenglykol-(1.2) vom Schmp. 69-70" (Lit. 18,671 72-73"). Das durch Autoxydation erhaltene 14 wurde mit authent. (+)-Limonenoxyd [am (+)-Li- monen mit Peressigsaure: nkO = 1.4761, d: = 0.9305, t ~ ; ~ = +55"] identifiziert (GC, IR). Das Vergleichspraparat bestand aus 2 Verbindungen im Verhaltnis 1 : 1 (GC, K-Siule, 130").
2) (+)-trans- und (+)-cis-p-Menthadien-(2.8)-oZe-(I) (8c bzw. 8d). - Die in den folgenden Fraktionen befindlichen tertiaren Alkohole wurden gaschromatographisch durch Zumischen der bei der photosensibilisierten 02-Ubertragung auf 1 2 7 ) erhaltenen Verbindungen identi- fiziert. Die an 8 c und 8d angereicherten Fraktionen zeigten die IR-Maxima der cis-Doppel- bindung bei 738 und 746 cm-1.
3) (&)-Carvon (13). - In den anschliel3end destillierenden Fraktionen wurde das Keton gas- chromatographisch durch Zumischen von authent. Material sowie als 2.4-Dinitro-phenyl- hydrazon vom Schmp. und Misch-Schmp. 185" (Lit.37) 190") identifiziert.
4) (&)-trans-Carveol (9b). - Die folgenden Fraktionen enthielten als Hauptbestandteil9 b, wie gaschromatographisch durch Zufiigen von reinem (-)-trans-Carveol27) gezeigt wurde. Schwer losliches 3.5-Dinitro-benzoat von 9b vom Schmp. 121" (Lit.68) 1!9O), [a]$' = *O".
5 ) (*)-&-Carveal (9a). - Die Identitat von 9 a ergab sich durch Vergleich (GC, IR) mit authent. (-)-cis-Carveol27). Die zuletzt iibergehenden Fraktionen ergaben das 3.5-Dinitro- benzoat von 9 a vom Schmp. 93" (Lit.68) 91.5'), [a]&' = *O" .
Isomerisierung yon 8 c zu 8d und Isopiperitenol (7a, b)
21 g gaschromatographisch reines 8 c (aus 1 durch photosensibilisierte 02-Ubertragung dargestellt27)) wurden mit 150 ccm Wasser unter intensivem Riihren 4 Stdn. bei 150" (Olbad) erhitzt. Das nach Aufnehmen in Ather, Trocknen mit K2C03 und Na2S04 und einfacher Destillation erhaltene Gemisch bestand (GC, R-Siule) aus 8a, 8d und 7a,b im Ver- haltnis 1 : 1 : 2. Durch Drehbanddestillation iiber Na2C03 erhielten wir weitgehend reines (GC, IR) 8d: nhO = 1.4890, d: = 0.942, cck2 = Jr146.8"; Lit.27) M D = +180.2". Daneben wurden Fraktionen erhalten, die zu mehr als 95% aus 7a,b bestanden: nk' = 1.4930, d,20 =
0.953, CL$~ = t35.97". Das Isopiperitenol-Gemisch setzte sich aus 23% 7 a und 77% 7 b zusammen (GC, C-Saule, 160", 1 atu). Oxydation mit aktivem Braunstein und Umsetzen mit 2.4-Dinitro-phenylhydrazin gab das bekannte49) 2.4-Dinitro-phenylhydrazon von 12 vom Schmp. 163". Das gleiche 1R-Spektrum wie das aus der Allylumlagerung hervorgegangene Isopiperitenol zeigle durch LiAlH4-Reduktion von (+)-Tsopiperitenon erhaltenes 7a, b, das durch folgende Daten charakterisiert war: Sdp.9 98", nkO = 1.4947, d p = 0.947, = + 114.3".
p-Nitro-benzoat von 7b (?): Schmp. 106.5- 107" (aus Petrolather), [a12 = -174.5".
C17H19N04 (301.3) Ber. C 67.76 H 6.36 N 4.65 Gef. C 67.74 H 6.41 N 4.73
67) H. Schmidt, Chem. Ber. 82, 11 (1949). 6 8 ) R . C. Johnston und J . Read, J. chem. SOC. [London] 1934, 233.

38 G. 0. Schenck, 0 . - A . Neumiiller, G . Ohloff und S. Schroeter Bd. 687
Isopiperitenon (12). - 158 g 81227) in 480 ccm Benzol wurden unter Riihren langsam zu einer Mischung von 308 g K2Cr207, 800 ccm Wasser und 204 g konz. Schwefelsaure getropft. AnschlieRend wurde 1 Stde. bei 60" erwarmt. Nach Destillation iiber eine rnit V2A-Wendeln gefiillte Kolonne wurden 117 g 12 erhalten: nk0 = 1.5037, d,zO = 0.9611, E D = 4 83.7"; Lit.22) a'," = +68.3". Das Gaschromatogramm (C-Saule, 160", 1.5 atii) zeigte 2 Peaks im Verhaltnis 9 : 1. - 1R-Spektrum: VC=CH~ : 893, 6190 cm-1 (E = 0.3)27,69).
2.4-Dinitro-phenylhydrazon: Schmp. 162- 163" (Lit.49) 159- 160"). A,,, (in Methanol) =
282.4 m p (log E = 4.48), 311 mp (log E = 4.25).
Zersetzungsreaktionen der Limonenhydroperoxyde (vgl Tab. 3 und Abb. 2, S 35)
Reaktion mit CnCIIPyridin: Zu etwa 125 g Gemisch der Limonen-Photohydroperoxyde 27)
in 350 ccm Pyridin wurde eine Spatelspitze CuCl gegeben. Unter Aufsieden des Pyridins trat heftige Reaktion ein. Nach Reduktion der Mischung mit wanriger Na2SO3-Losung wurde ausgeathert, rnit Na2S04 getrocknet und nach Verdunsten des Athers von dem hochviskosen Ruckstand abdestilliert. Das Destillat (38 g), das stark nach Carvon roch, wurde gaschromato- graphisch analysiert (vgl. Tab. 3, Versuch 1). Da Isopiperitenol (7a,b) und Carvon (13) nicht zu unterscheiden waren, wurde das Gemisch rnit LiAlH4 reduziert, wobei Carvon in 9004 cis- und 10 % trans-Carveol iiberging, die beide gaschromatographisch von Isopiperitenol unter- schieden werden konnten (Tab. 3, Versuch 2).
Reaktion mit CuClIPyridinlMethanoI: 84 g Hydroperoxyd-Gemisch in 600 ccm Methanol wurden unter Eiskuhlung und Riihren zu 40 ccm Pyridin in 300 ccm Methanol und einer Spatelspitze CuCl getropft. Nach 12stdg. Riihren bei 20" wurde die Losung mit wasriger Na~SO3-Losung versetzt und aufgearbeitet. Das erhaltene Gemisch (21.7 g; n'," = 1.4919, CLD = i-64.4") wurde gaschromatographisch analysiert (Tab. 3, Versuch 3).
Tabelle 3. Gaschromatographische Analyse der Reduktionsprodukte nach katalytischer Zer- setzung der Limonenhydroperoxyde (GC : 2-m-R-Saule)
Reduktionsprodukte Anteil in % bei Versuch 1 2 3 4 5 6 7
trans-p-Menthadien-(2.8)-01-(1) (812) 46 44 50 37 19 5 34 cis-p-Menthadien-(2.8)-01-(1) (8 d) 15 11 18 11 7 4 10 Unbekannte Substanz - 3 - - - 4 -
p-Menthadien-[1(7).8]-truns-o1-(2) ( l lb) 13 8 14 20 26 21 20 trans-Carveol (9 b) 2 5 3 11 14 10 10 cis-Carveol (9a) + p-Menthadien-[1(7).8]- 1 25 6 15 25 32 26
Isopiperitenol (7a, b) + Carvon (13)~) 1 8 4 9 6 - - -
cis-01-(2) (11 a)b) Hohersiedende Anteile 5 - - - 9 2 4 -
a) Keine Trennung; bei der Aufarbeitung (alkal. NazSO3-Losung) ging der Keton-Anteil z. T. verloren. b) Keine Trennung.
69) R. F. Goddu, Analytic. Chem. 29, 1790 (1957).

1965 Zur Autoxydation des (+)-Limonens 39
Reaktion rnit CuCIIMethanol: Wir lieBen 84 g Hydroperoxyd-Gemisch in 600 ccm Methanol mit CuCl3 Tage bei 20" stehen. Danach wurde die Losung, die rnit Triphenylphosphin noch peroxydisch reagierte, mit Na2SO3 reduziert, destilliert (30.1 g) und gaschromatographisch analysiert (Tab. 3, Versuch 4).
Reaktion mit Oxalsaure: 168 g Hydroperoxyd-Gemisch in 1.2 1 Methanol wurden rnit 5 g Oxalsaure + 30 ccm Wasser versetzt und 3 Tage bei Raumtemperatur stehen gelassen. Die Halfte der Losung wurde mit waRriger alkalischer NazSO3-Losung reduziert (35.7 g). Die Reduktionsprodukte wurden gaschromatographisch bestimmt (Tab. 3, Versuch 5). Die andere Halfte des Reaktionsgemisches wurde 2 Stdn. auf 70", dann 7 Stdn. auf 40" erwarmt und anschlieRend rnit alkalischer NazS03-Losung reduziert. Wir erhielten 49.4 g Gemisch (n'," = 1.4744, CCD = +17.3"), dessen Zusammensetzung aus Tab. 3, Versuch 6, hervorgeht.
Tab. 3, Versuch 7, gibt die Zusammensetzung eines Reduktionsgemisches ohne vorher- gehende katalytische Zersetzung wieder (vgl. Lit. 27)).
Aceton-sensibilisierte Photooxydation
150 ccm (+)-Lirnonen (c&l = + 101.9") wurden in Gegenwart von 20 ccm Aceton im Sauerstof-Kreislauf rnit einem Quecksilberbrenner HPK 125 W (Quarzschacht, 15") belichtet. Nachdem die Losung in 20 Stdn. 9 I Sauerstoff aufgenommen hatte, wurde sie mit Na2S03- Losung 1 Stde. auf 50" erwarmt und nach dem Erkalten ausgeathert. Destillation iiber eine kurze Vigreux-Kolonne gab neben unverandertem Limonen (ag = +lO1.2") 10 g eines Ge- misches, das die in Tab. l (S. 27) angegebene Zusanfmensetzung hatte (GC, K-Saule, 160"). Die Identifizierung der einzelnen Komponenten erfolgte durch Zumischen von authen- tischen 27) Substanzen. 149/651





























