
1
STUDIES IN HETEROCYCLIC SYNTHESIS
By
LONGCHUAN HUANG
A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY
UNIVERSITY OF FLORIDA
2010

2
© 2010 Longchuan Huang

3
To my parents Fayun Huang and Miaorong Zhu, to my brother Jiajia Huang, and to my dear friends for their unconditional love and support

4
ACKNOWLEDGMENTS
I would like to express my gratitude to my advisor, Professor Alan R. Katritzky, for
his consistant support and guidance, which were essential for me to complete my
studies. His overall knowledge of science, not just chemistry, and his strong devotions
to science and education is extremely impressive. His mentorship has guided me
through many challenges as a graduate student, and I will always remain appreciative
and thankful for the opportunity working with him. I would especially like to thank Dr. C.
Dennis Hall for his constructive and helpful suggestions for my research and for his
kindness and patience with reading and correcting my writing over and over again. Also,
I want to thank Dr. John Reynolds, Dr. Ion Ghiviriga, Dr. Weihong Tan and Dr. Fazil
Najafi for their time as members of my committee. Their knowledge, advice, and support
have been a valuable and cherished resource during my graduate career.
This work would not have been possible without the hard work of my coworkers
with whom I have interacted: Dr. Rajeev Sakhuja for his expertise in both chemistry and
as a group leader; Dr. Prahbu Mohapatra for the teamwork on the synthesis of 1,3,4-
oxadiazoles in Chapter 3. My thanks must go to Dr. Yuming Song, Ms. Reena Gyanda
and Ms. Ling Wang who all have contributed to the triazole-polymer project described in
Appendix. I would like to thank all of the present and past members of the Katritzky
research group. I have made some great friends and enjoyed their company during the
past four years. Their friendship and support have made this period of my life more
pleasant and memorable.

5
TABLE OF CONTENTS
page
ACKNOWLEDGMENTS .................................................................................................. 4
TABLE OF CONTENTS .................................................................................................. 5
LIST OF TABLES ............................................................................................................ 9
LIST OF FIGURES ........................................................................................................ 10
LIST OF SCHEMES ...................................................................................................... 12
LIST OF ABBREVIATIONS ........................................................................................... 16
ABSTRACT ................................................................................................................... 20
CHAPTER
1 INTRODUCTION TO BENZOTRIAZOLE CHEMISTRY .......................................... 21
1.1 Benzotriazole .................................................................................................... 21
1.1.1 Structure and Isomerization ..................................................................... 21
1.1.2 Synthesis of Benzotriazoles .................................................................... 23
1.2 Activation Ability of the Benzotriazole Ring ....................................................... 24
1.2.1 As a Proton Activator or an Anion Stabilizer ............................................ 24
1.2.2 As a Leaving Group ................................................................................. 25
1.2.3 As an Ambient Anion-Directing Group ..................................................... 26
1.2.4 As a Radical Stabilizer or a Radical Precursor ........................................ 26
1.2.5 As an Anion Precursor ............................................................................. 27
1.3 N-Acylbenzotriazoles in Heterocyclic Synthesis ............................................... 27
1.3.1 Preparation of N-Acylbenzotriazoles ....................................................... 27
1.3.2 N-Acylbenzotriazoles for N-, S- , C- and O- Acylation ............................. 28
1.3.2.1 Selective synthesis of S-acyl and N-acylcysteines ......................... 29
1.3.2.2 Selective synthesis of S-acylglutathiones and N-acylglutathiones ...................................................................................... 29
1.3.2.3 Synthesis of N-Cbz-protected (α-aminoacyl)methylenepyridines and -quinolines ....................................................................................... 30
1.3.2.4 Synthesis of S-acylisotripeptides ................................................... 30
1.3.2.5 Synthesis of azo-dye labeled amino acids and amines .................. 31
1.3.2.6 Synthesis of chiral O-(α-protected-aminoacyl)steroids ................... 31
1.3.2.7 Synthesis of pyridin-2-ylmethyl ketones ......................................... 32
1.3.2.8 Synthesis of 1-(benzotriazol-1-yl)alkyl- ethers and esters .............. 33
1.3.2.9 Bt-mediated C-acylation ................................................................. 33
1.3.3 Expansion of the Scope for N-Acylbenzotriazole Applications in Heterocyclic Synthesis .................................................................................. 34

6
2 EFFICIENT SYNTHESES OF NAPHTHOQUINONE DIPEPTIDES ....................... 35
2.1 Introduction ....................................................................................................... 35
2.1.1 Background ............................................................................................. 35
2.1.2 Interaction of Quinones and Amino Acids in Nature ................................ 39
2.1.3 Application of Quinone-Amino Acid Conjugates ...................................... 39
2.1.4 Literature Preparative Methods for Quinone-Amino Acid Conjugates ..... 40
2.2 Results and Discussion ..................................................................................... 43
2.2.1 Reaction of Naphthoquinone-Amino Acid Conjugates ............................. 43
2.2.2 Reaction of Thio-substituted Benzoquinone with Amino Acids ................ 47
2.2.3 Preparation of Benzotriazole Activated Benzoquinone-Amino Acid Conjugates .................................................................................................... 49
2.3 Conclusion ........................................................................................................ 49
2.4 Experimental Section ........................................................................................ 49
3 1,3,4-OXADIAZOLES FROM FUCTIONALIZED N-ACYLBENZOTRIAZOLES AND ACYLHYDRAZIDES ....................................................................................... 66
3.1 Introduction ....................................................................................................... 66
3.1.1 Oxadiazoles ............................................................................................. 66
3.1.2 Biologically Active 1,3,4-Oxadiazoles ...................................................... 66
3.1.3 Polymeric 1,3,4-Oxadiazoles ................................................................... 67
3.1.4 Luminescent Compounds, Dyes and Photosensitive Materials ............... 68
3.1.5 Other Miscellaneous Applications ........................................................... 69
3.1.6 Literature Preparative Methods for 1,3,4-Oxadiazoles ............................ 70
3.2 Results and Discussion ..................................................................................... 74
3.3 Conclusion ........................................................................................................ 75
3.4 Experimental Section ........................................................................................ 76
3.4.1 General Procedure for the Preparation of 1,3,4-Oxadiazole .................... 77
4 OVERVIEW OF N-HYDROXYAMIDOXIMES, N-AMINOAMIDOXIMES AND HYDRAZIDINES ..................................................................................................... 81
4.1 Introduction ....................................................................................................... 81
4.2 Structure and Configuration .............................................................................. 83
4.2.1 N-Hydroxyamidoximes ............................................................................ 83
4.2.2 N-Aminoamidoxime ................................................................................. 85
4.2.3 Hydrazidines ............................................................................................ 85
4.3 Preparative Methods ......................................................................................... 86
4.3.1 N-Hydroxyamidoximes and Their Derivatives .......................................... 86
4.3.1.1 From oximidoyl chlorides and hydroxyamines ............................... 86
4.3.1.2 From amidoximes and hydroxyamine ............................................ 87
4.3.1.3 From nitrile oxides and hydroxyamines .......................................... 87
4.3.1.4 Miscellaneous preparative methods for di-O-alkyl derivatives of N-hydroxyamidoximes ............................................................................ 88
4.3.2 N-Aminoamidoximes and Their Derivatives ............................................. 89
4.3.2.1 From oxime chlorides or amidoximes ............................................. 89

7
4.3.2.2 From oximebenzotriazoles and hydrazines .................................... 89
4.3.2.3 From N-hydroxyimidates and hydrazides ....................................... 90
4.3.2.4 From oxyimidoylchlorides and hydrazines ..................................... 90
4.3.2.5 From hydrazide imidate and hydroxyamine ................................... 91
4.3.3 Hydrazidines ............................................................................................ 91
4.3.3.1 From imidate salts and hydrazines................................................. 91
4.3.3.2 From amidoximes and hydrazines ................................................. 92
4.3.3.3 From amidrazones and hydrazines ................................................ 92
4.3.3.4 From diethoxy-N,N-dimethylethanamine and hydrazides ............... 93
4.3.3.5 From hydrazonyl bromides and hydrazines ................................... 93
4.3.3.6 From triazines ................................................................................ 94
4.4 Chemistry and Reactions .................................................................................. 94
4.4.1 N-Hydroxyamidoximes ............................................................................ 94
4.4.1.1 Reduction of N-hydroxyamidoximes .............................................. 94
4.4.1.2 Oxidation of N-hydroxyamidoximes................................................ 95
4.4.1.3 Reaction with aldehydes ................................................................ 96
4.4.1.4 Reaction with ketones .................................................................... 97
4.4.2 N-Aminoamidoximes ............................................................................... 97
4.4.2.1 Reaction with aldehydes ................................................................ 97
4.4.2.2 Cyclization in basic media to hydroxytriazoles ............................... 98
4.4.3 Hydrazidines ............................................................................................ 99
4.4.3.1 Reaction with aldehydes ................................................................ 99
4.4.3.2 Reaction with anhydrides ............................................................. 100
4.4.3.3 Reaction with diketones ............................................................... 102
4.3.3.4 Reaction with alpha-keto- acids or esters .................................... 103
4.4.3.5 Reaction with acylnitriles .............................................................. 104
4.4.3.6 Reaction with cyclopentadiene derivatives ................................... 104
4.4.3.7 Reaction with diketoesters ........................................................... 105
4.4.3.8 Reaction with formic acid ............................................................. 106
4.3.3.9 Reaction with thioesters ............................................................... 107
4.3.3.10 Reaction with hydrazine ............................................................. 108
4.4.3.11 Reduction of hydrazidines .......................................................... 108
4.4.3.12 Condensation with α-halo ketones ............................................. 109
4.4.3.13 Miscellaneous reactions ............................................................. 110
4.5 Applications .................................................................................................... 111
4.5.1 N-Aminoamidoximes ............................................................................. 111
4.5.1.1 As a prodrug model ...................................................................... 111
4.5.1.2 Applications in inorganic chemistry .............................................. 111
4.5.2 N-Aminoamidoximes ............................................................................. 112
4.5.2.1 As metal ligands for important coordination compounds .............. 112
4.5.3 Hydrazidines .......................................................................................... 114
4.5.3.1 As new fibrous adsorbents ................................................................. 114
4.5.3.2 As anti-tuberculosis agents .......................................................... 115
4.5.3.3 As environmentally friendly dyes .................................................. 115
4.6 Conclusions .................................................................................................... 116
5 SUMMARY OF ACHIEVEMENTS ........................................................................ 117

8
APPENDIX
A HIGHLY FILLED CROSSLINKED 1,2,3-TRIAZOLE POLYMERS AS NOVEL ROCKET PROPELLANT BINDERS ..................................................................... 118
A-1 Introduction .................................................................................................... 118
A-1-1 Rocket Propellant Binders .................................................................... 118
A-1-2 Triazole Polymers as Novel Rocket Propellant Binders ........................ 119
A-2 Results and Discussion .................................................................................. 124
A-2-1 Selection of Model Polymer System ..................................................... 124
A-2-2 Preparation of Monomers ..................................................................... 124
A-2-3 Preparation of Dogbone Samples ......................................................... 125
A-2-4 Filler Loading Effect .............................................................................. 126
A-3 Conclusions .................................................................................................... 135
A-4 Experimental Section ..................................................................................... 136
LIST OF REFERENCES ............................................................................................. 142
BIOGRAPHICAL SKETCH .......................................................................................... 166

9
LIST OF TABLES
Table page 2-1 Naphthoquinone-amino acid/ester conjugates ................................................... 44
2-2 Naphthoquinone-aminoacylbenzotriazoles ......................................................... 45
2-3 Synthesis of Naphthoquinone-dipeptides ........................................................... 46
2-4 Thiol-substituted benzoquinone-amino acid congjugates ................................... 48
3-1 Reaction of N-acylbenzotriazoles with benzoic acid hydrazide .......................... 76
A-1 Strain and modulus of unfilled and filled crosslinked triazole polymers ............ 127
A-2 Effect of filler loading (Al: 10-14 micron) on strain and modulus of crosslinked triazole polymers .............................................................................................. 128
A-3 Effect of filler loading (Al: < 75 micron) on strain and modulus of crosslinked triazole polymers .............................................................................................. 128
A-4 Effect of filler loading (NaCl: 45-50 micron) on strain and modulus of mechanical properties of crosslinked triazole polymers .................................... 132
A-5 Effect of filler loading (NaCl: 83-105 micron) on strain and modulus of crosslinked triazole polymers ............................................................................ 132
A-6 Effect of mixed filler loading (mixture of two different particle sized Aluminum) on strain and modulus of crosslinked triazole polymers ................................... 133
A-7 Effect of mixed filler loading (mixture of Aluminum and NaCl) on strain and modulus of crosslinked triazole polymers ......................................................... 133
A-8 Effect of mixed filler loading (mixture of Aluminum and NaCl) on strain and modulus of crosslinked triazole polymers ......................................................... 133

10
LIST OF FIGURES
Figure page 1-1 Isomerization of Benzotriazoles .......................................................................... 21
1-2 1H-Benzotriazole functions as an excellent synthetic auxiliary ........................... 22
1-3 Compounds with the Bt-C-O functionality ........................................................... 33
2-1 Important drugs containing quinone moities ....................................................... 36
2-2 Doxorubicin molecules intercalating DNA ........................................................... 37
2-3 Naturally occurring quinones .............................................................................. 39
2-4 Classes of quinones participating in biological redox processes ........................ 39
3-1 Four types of oxadiazoles ................................................................................... 66
3-2 Biologically important oxadiazoles ...................................................................... 67
3-3 Polymers containing 1,3,4-oxadiazoles .............................................................. 68
3-4 1,3,4-Oxdiazoles with interesting optical properties ............................................ 69
3-5 Other applications of 1,3,4-oxidazoles ................................................................ 70
4-1 Structure of N-hydroxyamidoximes, N-aminoamidoxime & hydrazidine ............. 82
4-2 N-Hydroxyamidoximes and their derivatives in the literature .............................. 82
4-3 Known N-aminoamidoximes and their derivatives .............................................. 82
4-4 Hydrazidines and their derivatives ...................................................................... 83
4-5 Tautomerization, conformation and configuration of N-hydroxyamidoxime ........ 85
4-6 Configuration of N-aminoamidoximes ................................................................. 85
4-7 Configuration of hydrazidines ............................................................................. 85
4-8 N-Hydroxybenzamidoxime derivatives ............................................................... 87
4-9 Acetohydroximic oxime and ethylnitrosolic acid ................................................ 112
4-10 N-Aminobenzamidxoime cobalt(II) perchlorate complex .................................. 114
4-11 Environmental friendly dye ligands ................................................................... 116

11
A-1 Common rocket propellant binders ................................................................... 118
A-2 Dogbone mold containing filled and unfilled triazole polymers ......................... 126
A-3. nstron universal tensile testing machine ........................................................... 126
A-4 Effect of filler loading on modulus of crosslinked triazole polymers .................. 130
A-5 Effect of filler loading on strain of crosslinked triazole polymers ....................... 131
A-6 Effect of mixed filler loading on modulus of crosslinked triazole polymers ....... 134
A-7 Effect of mixed filler loading on strain of crosslinked triazole polymers ............ 135
A-8 Dimensions of dogbone mold and dogbone sample ......................................... 137

12
LIST OF SCHEMES
Scheme page 1-1 Alkylation of 1H-benzotriazole ............................................................................ 23
1-2 Synthesis of benzotriazole .................................................................................. 23
1-3 Synthesis of 5,7-dinitro-1-phenylbenzotriazole ................................................... 23
1-4 Reactions of benzotriazolyl-stabilized carbanions with electrophiles .................. 25
1-5 Reaction with Grignard reagent .......................................................................... 26
1-6 Benzotriazole acts as an anion-directing group .................................................. 26
1-7 Benzotriazole acts as an radical stabilizer or precursor ...................................... 27
1-8 Reductive elimination of benzotriazole ............................................................... 27
1-9 Methods for preparation of N-acylbenzotriazoles ............................................... 28
1-10 Selective synthesis of S-acyl and N-acylcysteines ............................................. 29
1-11 Selective synthesis of S-acylglutathiones and N-acylglutathiones ...................... 29
1-12 Synthesis of N-Cbz-protected (α-aminoacyl)methylenepyridines and -quinolines ........................................................................................................... 30
1-13 Preparation of S-acylisotripeptides ..................................................................... 30
1-14 Synthesis of azo-dye labeled amino acids and amines ...................................... 31
1-15 Microwave assisted synthesis of chiral O-(α-protected-aminoacl)steroids and O-(α-protected-dipeptidoyl)steroids .................................................................... 32
1-16 Synthesis of pyridin-2-ylmethyl ketones mediated via N-acylbenzotriazoles ...... 32
1-17 Synthesis of 1-(benzotriazol-1-yl)alkyl esters by N-acylbenzotriazoles .............. 33
1-18 Enaminones via C-acylation of ketimines with N-acylbenzotriazoles .................. 34
2-1 Quinone-amino acid conjugates linked via a vinylic spacer ................................ 41
2-2 Synthesis of quinone-amino acid hybrids via Cross-Enyne Metathesis and Diels-Alder reactions .......................................................................................... 41
2-3 N-Quinonyl amino acids obtained with chloro-substituted quinones ................... 41

13
2-4 Synthesis of N-quinonyl amino acids by addition to S-substituted benzoquinone ..................................................................................................... 42
2-5 Preparation of naphthoquinone-dipeptides ......................................................... 42
2-6 Synthesis of naphthoquinone-amino acid/ester conjugates ................................ 43
2-7 Synthesis of naphthoquinone-aminoacylbenzotriazole conjugates..................... 44
2-8 Preparation of naphthoquinone dipeptide conjugates ......................................... 45
2-9 Synthesis of thiol-substituted benzoquinone-amino acid conjugates .................. 48
2-10 Synthesis of benzoquinone-amino acid benzotriazole derivative ....................... 49
3-1 Cycloaddition reactions of 1,3,4-oxadiazoles in total synthesis of natural product ............................................................................................................... 69
3-2 Preparation of 2,5-disubstituted 1,3,4-oxadiazoles from 1,2-diacylhydrazines ... 70
3-3 Preparation of 2,5-disubstituted 1,3,4-oxadiazoles from hydrazones ................. 71
3-4 Preparation of 1,3,4-oxadiazolinones ................................................................. 71
3-5 1,3,4-Oxadiazole ring synthesis from acyclic precursors .................................... 72
3-6 Preparation of 2-amino-1,3,4-oxadiazoles .......................................................... 72
3-7 One-pot syntheses of unsymmetrical 2,5-disubstituted 1,3,4-oxadiazoles ......... 73
3-8 1,3,4-Oxadiazoles from N-acylbenzotriazoles .................................................... 75
4-1 Preparation of N-hydroxybenzamidoxime ........................................................... 86
4-2 Preparation of N-hydroxypyridylamidoximes ...................................................... 86
4-3 Preparation of 2,6-dichloro-N-hydroxybenzaldoxime hydrochloride salt ............. 87
4-4 Preparation of formic hydroxyamidoxime hydrochloride salt .............................. 87
4-5 Synthesis of N-hydroxyamidoximes from nitrile oxides ....................................... 88
4-6 Preparation of di-O-benzyl derivative of N-hydroxymethylamidoxime ................ 88
4-7 Synthesis of di-O-methylsubstituted p-sulfamido-N-hydroxybenzamidoximes ... 89
4-8 General route to N-aminoamidoximes ................................................................ 89
4-9 Synthesis of N-amino-N´-nitrophenyl benzamidoxime ........................................ 89

14
4-10 Preparation of N-(ethoxycarbonyl)amide benzamidoxime .................................. 90
4-11 Preparation of 3-(3-arylsydnon-4-yl)triazole derivatives ..................................... 90
4-12 Preparation of hydroxamic acid ethoxycarbonylhydrazides ................................ 91
4-13 Synthesis of aliphatic hydrazidines ..................................................................... 91
4-14 Synthesis of substituted formazans .................................................................... 92
4-15 Synthesis of triphenylformazan ........................................................................... 92
4-16 Synthesis of hydrazidine hydrochlorides ............................................................ 92
4-17 Synthesis of diaminoguanidine / amino-hydrazidine ........................................... 93
4-18 Synthesis of hydrazidine derivatives ................................................................... 93
4-19 Synthesis of hydrazidines from hydrazonyl bromide ........................................... 94
4-20 From triazine to hydrazidines.............................................................................. 94
4-21 Conversion of N-hydroxybenamidoxime into benzamidoxime ............................ 95
4-22 Conversion of formic hydroxyamidoxime to its nitrosolic acid ............................. 95
4-23 Synthesis of 3 ,5-diphenyl-1,2,4-oxadiazole ....................................................... 96
4-24 Reaction of nitrosolic acid salts with dinitrogen tetraoxide .................................. 96
4-25 Synthesis of 4-hydroxyoxadiazolines .................................................................. 97
4-26 Reaction of N-hydroxyamidoxime with benzophenone ....................................... 97
4-27 Preparation of 3,5-disustitued 1H-[1,2,4]triazoles ............................................... 98
4-28 Synthesis of 3-benzyl-5-(p-tolyl)-4H-1,2,4-triazol-4-ol ........................................ 98
4-29 Synthesis of 3-phenyl-4-hydroxy-4,5-dihydro-1,2,4-triazol-5-one ....................... 99
4-30 Synthesis of dibenzylidene hydrazidine 4-amino-1,2,4-triazole hydrochloride .... 99
4-31 Reaction of hydrazidines with aldehydes .......................................................... 100
4-32 Synthesis of pyrrolo[1,2-b][1,2,4,5]tetrazines ................................................... 101
4-33 Reaction with diketones .................................................................................... 103
4-34 Syntheses of triazinones .................................................................................. 104

15
4-35 Reaction of hydrazidines with acylnitriles ......................................................... 104
4-36 Synthesis of 4-aminocyclopenta[e]-1,2,4-triazines ........................................... 105
4-37 Reaction of hydrazidines with diketoesters ....................................................... 106
4-38 Reaction hydrazidines with formic acid ............................................................. 107
4-39 Synthesis of unsymmetrically substituted 1,2,4,5-tetrazines ............................ 107
4-40 Synthesis of 3-methyl-6-pyridyl-1,2,4,5-tetrazine .............................................. 108
4-41 Reduction of formazans .................................................................................... 108
4-42 Reaction of α-halo ketones with hydrazidine amine ......................................... 109
4-43 Hydrazidine radical ........................................................................................... 110
4-44 Reaction of hydrazine hydrazidine with acetylacetone ..................................... 110
4-45 In vitro biotransformation of N-hydroxybenzamidoxime .................................... 111
4-46 Synthesis of dinitrosomethanide (DNM) salt ..................................................... 112
4-47 Synthesis of novel vic-dioxime derivatives of hydrazones ................................ 113
4-48 Synthesis of vic-dioxime derivatives and their metal complexes ...................... 114
A-1 Triazole polymer model system ........................................................................ 124
A-2 Preparation of monomers ................................................................................. 125
A-3 General route to crosslinked 1,2,3-triazole polymers with fillers ....................... 141

16
LIST OF ABBREVIATIONS
Ac acetyl (CH3C=O)
Al aluminum
Ala alanine
Ar aryl
Boc t-butyloxycarbonyl
Bn benzyl
br broad (spectral)
brs broad singlet (spectral)
Bt benzotriazoyl
BtH 1H-benzotriazole
BTNO benzotriazole-N-aminoxyl radical (>N−O•)
Bz benzoyl
C carbon
Cu copper
oC degree Celcius
Calcd calculated
CAN ceriumIV ammonium nitrate
Cbz carbobenzyloxy (BnOC=O)
CDCl3 deuterated chloroform
CH3CN acetonitrile
d doublet (spectral)
DCC dicyclohexyl carbodiimide

17
DCM methylene chloride
DMAP 4-dimethylaminopyridine (base catalyst)
DMSO dimethyl sulfoxide (solvent)
DMSO-d6 deuterated dimethyl sulfoxide
DMF dimethylformamide (solvent)
E entgegen (opposite, trans)
EDC 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
equiv equivalent(s)
et al. and others
EtOAc ethyl acetate
Fe iron
g gram(s)
Glu glutamic acid
Glu-OMe glutamic methylester
Gly glycine
h hour
H hydrogen
HBT 1-hydroxybenzotriazole
HOBT N-hydroxybenzotriazole
HBTU O-benzotriazolye-N,N,N’,N’-tetramethyluroniumhexafluoro-
phosphate
HCl hydrochloric acid
HDPE high density polyethylene

18
HMDS hexamethyldisilazide
HRMS high resolution mass spectrometry
HTPB hydroxy-terminator polybutadiene
Hz hertz
i-Pr isopropyl
J coupling constant (NMR)
LDA lithium aluminium hydride
LDPE low density polyethylene
Leu leucine
lit literature
Lle isoleucine
Lys lysine
m multiplet (spectral); metre(s); milli
MeCN acetonitrile
MgSO4 magnesium sulfate
m. p. melting point
Ms methanesulfonyl (mesyl, CH3SO2)
m/z mass-to-charge ratio
N nitrogen
NaCl sodium chloride
NMR nuclear magnetic resonance
O oxygen
PBAN polybutadiene acrylic acid acrylonitrile

19
Phe phenylalanine
PMMA polymethylmethacrylate
PU polyurethane
RT room temperature
s singlet (spectral)
S sulfur
SOCl2 thionyl chloride
t triplet (spectral)
t tertiary
TBAF tetrabutylammonium floride
TEA triethylamine (Et3N)
THF tetrahydrofuran (solvent)
TMS tetramethylsilane, also trimethylsilyl
Tryp tryptophan
Ts tosyl (p-CH3C6H4SO2)
UV ultra violet
Val valine
wt% weight percent
Z zusammen (together, cis)

20
Abstract of Dissertation Presented to the Graduate School of the University of Florida in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
STUDIES IN HETEROCYCLIC SYNTHESIS
By
Longchuan Huang
December 2010
Chair: Alan R. Katritzky Major: Chemistry
1H-Benzotriazole and its derivatives are versatile synthetic auxiliaries. My
research studies have further investigated the application of N-acylbenzotriazoles in the
synthesis of heterocyclic compounds. In Chapter 2, an efficient N-acylbenzotriazole
mediated preparation of naphthoquinones-dipeptides from naphthoquinone-α-amino
acid conjugates as potential cytotoxic agents is reported. In Chapter 3, a convenient
preparation of 1,3,4-oxadiazoles from functionalized N-acylbenzotriazoles and acyl
hydrazides is described. Chapter 4 presents a review of the synthesis, reactivity and
utility of N-amino- and N-hydroxy- amidoximes and hydrazidines, which are important
classes of nitrogen-rich heterocycle precursors.

21
CHAPTER 1 INTRODUCTION TO BENZOTRIAZOLE CHEMISTRY
1.1 Benzotriazole
1.1.1 Structure and Isomerization
Benzotriazole (1.1) is classified as a 1,2,3-triazole, i.e. a cyclic compound featuring
two fused rings containing the linkage -N=N-NH- or =N-NH-N=. Benzotriazole is used as
corrosion inhibitor, e.g. for silver protection in dishwashing detergents and an anti-fog
agent in photographic development. [2009JAE269, 2009JMPT1729, 2009ME367]
Benzotriazole derivatives are employed in pharmaceuticals such as antifungal,
antibacterial, anthelmintic drugs, and polymerization catalysts. [2003CEJ4586,
2010CR1564]
Figure 1-1. Isomerization of Benzotriazoles
1H-Benzotriazole exists in solution as an equilibrium mixture of 1-benzotriazole
(1.1) and 2-benzotriazole (1.2) (Figure 1-1). [1975JCS(PT1)1181] Such isomerization is
general for disubstituted N-(aminomethyl)benzotriazoles, such as N-(aminoalkyl)- (1.3)

22
[1987JCS(PT1)2673, 1989H1121, 1990JOC5683], N-(alkoxyalkyl)- [1992JOC4932], N-
(alkylthioalkyl)- (1.5) [1991HCA1936], and N-(diarylmethyl)-benzotriazoles
[1990JCS(PT2)2059], but not for simple N-alkylbenzotriazoles.
1H-Benzotriazole is an excellent synthetic auxiliary. [1991T2683, 1998CR409,
2003CEJ4586] As summarized in Section 1.1.2, it can act as a leaving group, an
electron-withdrawing group and an electron-donating group (Figure 1-2). As another
aspect of a good auxiliary, BtH can act as a weak base (pKa = 1.6) or a weak acid (pKa
= 8.3) [1948JCS2240, 1991T2683], which facilitates the easy removal of benzotriazole
from the reaction mixture by washing with base or acid. Moreover, 1H-benzotriazole is
an inexpensive, stable compound that is soluble in common organic solvents such as
ethanol, benzene, chloroform, and DMF.
Figure 1-2. 1H-Benzotriazole functions as an excellent synthetic auxiliary
Alkylation of 1H-benzotriazole (1.1) with alkyl halides or sulfates in the presence
of a base yield mixtures of 1-alkylbenzotriazoles (1.10) and 2-alkylbenzotriazoles (1.11).
The ratio of product depends on the bulkiness of the alkyl group and varies from 78:22
(R = Et) to 50:50 (R = C6H11CH2) (Scheme 1-1). [1994LAC1]

23
Scheme 1-1. Alkylation of 1H-benzotriazole
1.1.2 Synthesis of Benzotriazoles
Benzotriazole is produced by reaction of o-phenylenediamine (1.12) with sodium
nitrite and acetic acid. The conversion proceeds via diazotization of one of the amino
groups (Scheme 1-2). [2001HYDX350, 1981USP4299965]
Scheme 1-2. Synthesis of benzotriazole
Reduction of compound (1.13) gave 4,6-dinitro-N1-phenylbenzene-1,2-diamine
(1.14), which were further subjected to the reaction with acetic acid and sodium nitrite to
yield 5,7-dinitro-1-phenyl-benzotriazole (1.15). Dinitrobenzotriazole (1.15) may be
further nitrated with nitric or mixed acid, and its derivatives have been examined as
potential energetic materials with particular reference to their densities (Scheme 1-3).
[1992AJC513]
Scheme 1-3. Synthesis of 5,7-dinitro-1-phenylbenzotriazole

24
1.2 Activation Ability of the Benzotriazole Ring
Benzotriazole derivatives are important synthetic auxiliaries that offer versatile
applications in organic chemistry including a vast array of synthetic transformations.
[1998CR409, 2003CEJ4586] Benzotriazole methodology has been applied to alkylation
[1994CSR363], acylation [2003JOC4932, 2003JOC5720, 2005S1656], imine acylation
[2000S2029], and imidoylation [1997TL6771, 1999OL977, 2002JOC4667,
2003CEJ4586]. It has also been utilized in Mannich reactions [1994JHC917], Michael
reactions [2001BCSJ2133] and Grignard reactions [2007S3141].
Many heterocycles are biologically active compounds; therefore, heterocyclic
scaffolds are of major interest to chemists. The application of benzotriazole derivatives
in organic synthesis has been studied meticulously by our group since 1980s, especially
with reference to the synthesis of heterocyclic molecules. A benzotriazole group
commonly activates the carbon atom to which it is attached; hence, benzotriazole
intermediates are widely used to introduce a variety of functional groups into molecules.
Five major applications of benzotriazole group in organic transformations are illustrated
below:
1.2.1 As a Proton Activator or an Anion Stabilizer
Many synthetic applications of benzotriazole derivatives are based on the ability of
the benzotriazolyl substituent to stabilize an adjacent carbanion. [1998CR409,
2003CEJ4586, 2006S3231]
n-BuLi or LDA can convert 1-(n-alkyl)benzotriazoles (1.16) to anions (1.17) (R1 =
H or alkyl), consecutively treating with alkyl halides will give 1-alkylbenzotriazoles (1.18)
bearing secondary alkyl groups. Carbonyl electrophiles can be used to trap the Bt-
stabilized anion (1.17) to form (1.20). Reaction of (1.17) with CO2, or ethyl benzoate
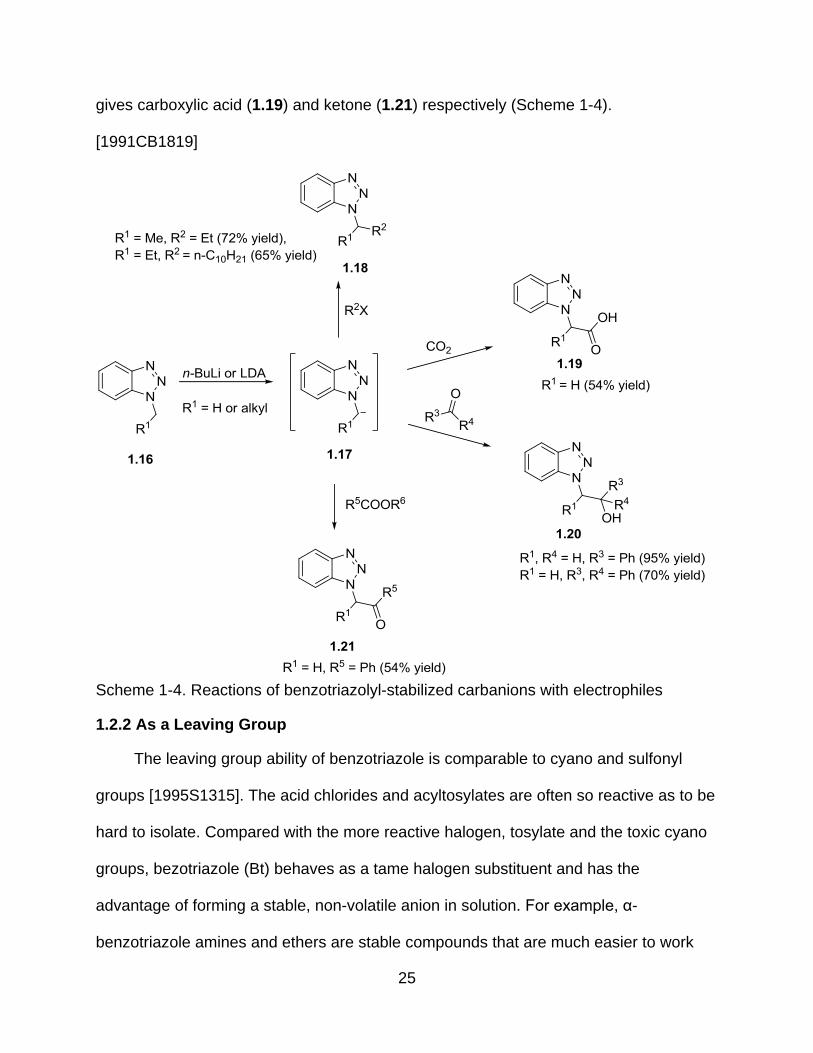
25
gives carboxylic acid (1.19) and ketone (1.21) respectively (Scheme 1-4).
[1991CB1819]
Scheme 1-4. Reactions of benzotriazolyl-stabilized carbanions with electrophiles
1.2.2 As a Leaving Group
The leaving group ability of benzotriazole is comparable to cyano and sulfonyl
groups [1995S1315]. The acid chlorides and acyltosylates are often so reactive as to be
hard to isolate. Compared with the more reactive halogen, tosylate and the toxic cyano
groups, bezotriazole (Bt) behaves as a tame halogen substituent and has the
advantage of forming a stable, non-volatile anion in solution. For example, α-
benzotriazole amines and ethers are stable compounds that are much easier to work

26
with than the corresponding toxic chloro derivatives. The displacement of benzotriazole
group can be easily achieved by nucleophilic attack [1994CSR363], or by different
nucleophilic atoms such as C, S, N, O, or even by Grignard reagents (Scheme 1-5).
[1991T2683, 1996JOC1624]
Scheme 1-5. Reaction with Grignard reagent
1.2.3 As an Ambient Anion-Directing Group
In an allylic system (1.25), the benzotriazolyl moiety acts as an anion-directing
group. Hence, the alpha position to Bt group is favored for attack of various
electrophiles (Scheme 1-6). [1990HC21, 1992LAC843]
Scheme 1-6. Benzotriazole acts as an anion-directing group
1.2.4 As a Radical Stabilizer or a Radical Precursor
The benzotriazolyl moiety can act as a radical precursor (1.30) (Scheme 1-7). The
generation of the aminoxyl radical benzotriazole-N-oxyl (>N−O•) (i.e., BTNO) (1.29a)
from 1-hydroxybenzotriazole (HBT) (1.28a) by monoelectronic oxidation with ceriumIV
ammonium nitrate (i.e., CAN) in MeCN solution is shown in Scheme 1-7. [2004CC2356,
2005JOC9521] BTNO radical (1.29a) can be trapped and used to initiate other radical
reactions via generating a different radical such as (1.30a).

27
Scheme 1-7. Benzotriazole acts as an radical stabilizer or precursor
1.2.5 As an Anion Precursor
Benzotriazole moieties can act as a carbanion (1.32) precursor via reductive
elimination (Scheme 1-8). [1997JOC4148, 1996LAC745, 1992JCS(PT1)1111] The
carbanion can react further with other electrophiles such as ketones/aldehydes to form
alcohols (1.33).
Scheme 1-8. Reductive elimination of benzotriazole
1.3 N-Acylbenzotriazoles in Heterocyclic Synthesis
1.3.1 Preparation of N-Acylbenzotriazoles
N-Acylbenzotriazoles are stable crystalline compounds that can be easily prepared
and handled in the lab. The classical preparation of acylazoles was from the
corresponding acid chlorides (Scheme 1-9). N-Acylbenzotriazoles can now be prepared
directly from carboxylic acids (1.35), obviating the necessity of isolating acid chlorides.
The second method is reaction of carboxylic acids with thionyl chloride in the presence
of excess benzotriazole, providing N-acylbenzotriazoles (1.35) in high yields (Scheme
1-9). [2003S2795] The third method uses a sulfonylbenzotriazole (1.36) as a “counter

28
attack” reagent; in the presence of Et3N, carboxylic acids (1.35) are directly converted
into the acylbenzotriazoles (1.34) through intermediate formation of the mixed
carboxylic sulfonic anhydride and benzotriazole anion, which are then acylated by the
mixed anhydride. [1992T7817, 2000JOC8210]
A wide range of N-acylbenzotriazoles have been prepared in our group via the
methods mentioned above, including alkyl and aryl carboxylic acids, many heterocyclic
carboxylic acids, unsaturated carboxylic acids, and carboxylic acids with various other
functionalities. [1992T7817, 2000JOC8210, 2003S2795]
Scheme 1-9. Methods for preparation of N-acylbenzotriazoles
1.3.2 N-Acylbenzotriazoles for N-, S- , C- and O- Acylation
N-Acylbenzotriazoles are advantageous for N-, O-, C-, and S-acylation,
[2000JOC8210, 2003JOC5720, 2005SL1656, 2005S397, 2006S411, 2006S3231,
2008OBC2400] especially where the corresponding acid chlorides are unstable or
difficult to prepare [1998AA35, 1999T8263]. Several recent demonstrations of 1-acyl-
1H-benzotriazoles as versatile synthetic auxiliaries in our group include:
29
1.3.2.1 Selective synthesis of S-acyl and N-acylcysteines
Cysteine (1.37) can be exclusively S- or N- acylated to (1.38) or (1.39) with N-
acylbenzotriazoles (1.34) under slightly different reaction conditions (Scheme 1-10).
[2009JOC7165]
Scheme 1-10. Selective synthesis of S-acyl and N-acylcysteines
1.3.2.2 Selective synthesis of S-acylglutathiones and N-acylglutathiones
1-Acyl-1H-benzotriazoles (1.40) were used in the selective syntheses of S-
acylglutathiones (1.42) and N-acylglutathiones (1.43). [2010SL1337] The transformation
is facile and has general applications for S-acylation and N-acylation of biologically
important larger peptides and glycopeptides (Scheme 1-11).
Scheme 1-11. Selective synthesis of S-acylglutathiones and N-acylglutathiones

30
1.3.2.3 Synthesis of N-Cbz-protected (α-aminoacyl)methylenepyridines and -quinolines
Aminoacyl-conjugates of nitrogen heterocycles (1.46) were synthesized as chiral
potential novel pharmacophores from 2-methyl- and 4-methylpyridine and 2-
methylquinoline (1.45) by reacting with benzotriazole-activated (Cbz)-protected amino
acids (1.44) (Scheme 1-12). [2010JOC3938]
Scheme 1-12. Synthesis of N-Cbz-protected (α-aminoacyl)methylenepyridines and -
quinolines
1.3.2.4 Synthesis of S-acylisotripeptides
Cysteine and C-terminal cysteine peptides (1.47) are selectively S-acylated by N-
(Pg-α-aminoacyl)benzotriazoles (1.34) to give N-Pg-S-acylisotripeptides (1.48) (Scheme
1-13), which can undergo chemical ligation after deprotection to give the corresponding
native tetra-peptides via migration of the cysteine S-acyl groups to the N-terminal amino
acids. [2010OBC2316]
Scheme 1-13. Preparation of S-acylisotripeptides

31
1.3.2.5 Synthesis of azo-dye labeled amino acids and amines
Traditional methods to link azo-dye carboxylic acids to bio-moieties have used
coupling reagents such as DCC, EDCI, HOBT, HBTU, or via acyl chloride
intermediates, and usually require complex procedures, harsh reaction conditions
and/or give low yields. By comparison, the new methods for preparing azo-dye labeled
amino acids (1.52) and amines (1.53) were developed by reaction of N-(4-
arylazobenzoyl)-1H-benzotriazole (1.49) with amino acids (1.50) or amines (1.51) under
mild reaction conditions to give high yields with no racemization of chiral compounds
(Scheme 1-14). [2008OBC2400]
Scheme 1-14. Synthesis of azo-dye labeled amino acids and amines
1.3.2.6 Synthesis of chiral O-(α-protected-aminoacyl)steroids
Chiral O-(α-protected-aminoacyl)steroids (1.56) and O-(α-protected-
dipeptidoyl)steroids (1.59, 1.61) were prepared under microwave irradiation from
naturally occurring steroidal alcohols (1.55, 1.58, 1.60) with complete retention of
chirality mediated by N-(Z-α-aminoacyl)-benzotriazoles (1.54) and Z-
dipeptidoylbenzotriazole (1.57). (Scheme 1-15) [2006Steroids660]

32
Scheme 1-15. Microwave assisted synthesis of chiral O-(α-protected-aminoacyl)steroids
and O-(α-protected-dipeptidoyl)steroids
1.3.2.7 Synthesis of pyridin-2-ylmethyl ketones
Katritzky el. al. reported that 2- or 4-picoline (1.62) was lithiated by LDA and then
treated with acylbenzotriazoles (1.63) to afford pyridin-2-ylmethyl ketones (1.64) in good
yields (60-84%) (Scheme 1-16). In comparison with previous methods, this approach
utilizing N-acylbenzotriazole simplifies the procedure and provides generally better
yields. [2005ARKIVOC329, 2010CR1564]
Scheme 1-16. Synthesis of pyridin-2-ylmethyl ketones mediated via N-
acylbenzotriazoles

33
1.3.2.8 Synthesis of 1-(benzotriazol-1-yl)alkyl- ethers and esters
Benzotriazole derivatives containing the Bt-C-O functionality are versatile
intermediates in organic synthesis. [1998CR409] One of the examples is 1-
(benzotriazol-1-yl) alkyl ethers (1.65) (Figure 1-3) which have been widely used for the
preparation of various heterocycles [1995JOC7612, 1995JOC7625], α-functionalized
ketones [1995JOC7619, 1997JOC706], amides [1988JOC5854], and ethers
[1989JOC6022]. Another example is 1-(benzotriazol-1-yl) alkyl esters (1.66) which
should offer similar synthetic functions. The initial route for the preparation of (1.66) was
reported from forming unstable intermediates with high sensitivity to moisture.
[1991S69] A more general and useful synthesis of the 1-(benzotriazol-1-yl) alkyl esters
(1.66) was achieved by the use of N-acylbenzotriazoles (1.67) reacting with aldehydes
(1.68) (Scheme 1-17). [1999JHC777]
Figure 1-3. Compounds with the Bt-C-O functionality
Scheme 1-17. Synthesis of 1-(benzotriazol-1-yl) alkyl esters by N-acylbenzotriazoles
1.3.2.9 Bt-mediated C-acylation
Carbon acylations provide an entry to carbon-carbon bonds. [1973JOC514] 1-
Acylbenzotriazoles-mediated C-acylation was demonstrated in the regioselective
synthesis of β-diketones. [2000JOC3679] Reactions of alkyl and aryl N-

34
acylbenzotriazoles with saturated cyclic ketones, unsaturated cyclic ketones, and
aliphatic ketones in the presence of lithium diisopropylamide (LDA) and tetrahydrofuran
(THF) at -78°C resulted in C-acylated products in excellent yields. Enaminones (1.71)
were obtained by C-acylation of ketimines (1.70) with N-acylbenzotriazoles (1.69)
(Scheme 1-18). [2000S2029]
Scheme 1-18. Enaminones via C-acylation of ketimines with N-acylbenzotriazoles
1.3.3 Expansion of the Scope for N-Acylbenzotriazole Applications in Heterocyclic Synthesis
N-Acylbenzotriazoles are versatile synthetic auxiliaries used as C-, O-, S- and N-
acylating agents as well as precursors to many valuable heterocycles. Part of my
research efforts has been focused on the expansion of the scope of N-
acylbenzotriazoles as activated reagents toward heterocyclic synthesis, specifically,
naphthoquinone-dipeptides and 1,3,4-oxadiazoles. In Chapter 2, an N-acylbenzotriazole
mediated preparation of naphthoquinones-dipeptides from naphthoquinone-α-amino
acid conjugates as potential cytotoxic agents is described, also some investigation of
benzoquinone amino acid conjugates are documented. In Chapter 3, 1,3,4-oxadiazoles
were prepared from functionalized N-acylbenzotriazoles and acylhydrazides.

35
CHAPTER 2 EFFICIENT SYNTHESES OF NAPHTHOQUINONE DIPEPTIDES
2.1 Introduction
2.1.1 Background
Quinones play vital roles in the biochemistry of living cells including respiration,
photosynthesis and cellular defense against bacteria, fungi and parasites.
[2007BMCL2340] Some quinonic derivatives are used as medicines for treating
bacterial and fungal diseases, and others exhibit potent antimalarial capacities.
[2002AA71] Many naturally occurring quinones are antitumor agents, and those
approved for clinical use include: menadione (2-methyl-1,4-naphthoquinone) (2.1),
anthracycline-glycosides (daunorubicin (2.2), doxorubicin (2.3)), benzoquinone
derivatives (mitomycin C (2.4), carbazilquinone (2.5), diaziquone (2.6)), and more
complex quinones (mitoxantrone (2.7), streptonigrin (2.8)) (Figure 2-1) [2005MRMC449,
2008OBC637, 2007MRMC481].
Menadione (2.1) has been used experimentally as a chemotherapeutic agent for
cancer. The combination of vitamin C and Menadione (2.1) has antitumor activities and
ability to prevent and treat breast and prostate cancer. [2001JN158S] Daunorubicin
(2.2) is a chemotherapeutic natural product of the anthracycline family. It has been used
for treatment of some cancers, and also specific types of leukaemia. Doxorubicin (2.3)
is another anthrocycline-type of drug used in cancer chemotherapy. All anthracyclines
have anticancer abilities by intercalating DNA and. inhibiting DNA replication in cancer
cells. The cartoon diagram of two doxorubicin molecules intercalating DNA is shown in
Figure 2-2. [1990B2538]
Reproduced in part with permission from Synthesis, 2010, 12, 2011, Copyright © 2010 Wiley

36
Mitomycin C (2.4) is a type of anti-tumor antibiotic that binds covalently to DNA.
[2008OBC637] Mitomycin-C (2.4) and Carbazilquinone (2.5) both contain quinonyl,
aziridinyl and carbamoyloxy groups, and both have significant effects on plasmacytoma
X5563 in C3H/He mice. [2007MRMC481] Diaziquone (2.6) is a synthetic quinonic
derivative with potential antineoplastic activities. It can damage DNA via initiating radical
reactions with DNA strand breaks. Also, it can disrupt DNA function by alkylating or
crosslinking DNA during all phases of the cell cycle. [1998L139]
Figure 2-1. Important drugs containing quinone moities

37
Mitoxantrone (2.7) is a type II topoisomerase inhibitor which can disrupt DNA
synthesis and repair in both healthy cells and cancer cells by intercalation with DNA. It
has been used in the treatment of several types of cancer. [1979JMC1024]
Streptonigrin (2.8) is an aminoquinone isolated from the bacterium Streptomyces
flocculus. It can act as a reverse transcriptase inhibitor and cause free radical-mediated
cellular damage. It can also complex with DNA and topoisomerase II, resulting in DNA
cleavage and inhibition of DNA replication and RNA synthesis. [1977BBRC387]
(wwwPDB – Worldwide Protein Data Bank) Figure 2-2. Doxorubicin molecules intercalating DNA

38
The cell cytotoxicity of quinonic drugs is due to (i) their ability to undergo a
reversible one electron reduction followed by formation of semiquinone radicals and (ii)
their ability to associate and intercalate with DNA duplexes, thus impairing appropriate
template function and nucleic acid synthesis. [2000AA439]
Varieties of human tumors are hormone-dependent and contain corresponding
hormone receptors. Receptors for peptide hormones such as luteinizing hormone-
releasing hormone (LH-RH, also known as Gonadotropin-releasing hormone (GnRH)
and luliberin which is a tropic peptide hormone responsible for the release of Follicle-
stimulating hormone (FSH) and luteinizing hormone (LH) from the anterior pituitary
[1998LPS421], somatostatin [1990JSBMB1083], bombesin [1983P683], vasoactive
intestinal peptide [1990P1205] and growth factors, including epidermal and insulin-like
[1995JANYAS402], have been detected in the cancers of prostate, breast pancreas,
ovary, lung, colon and in brain tumors [1987CL223]. In view of abundancy of tumors
having LH-RH receptors, related target chemotherapy has gained considerable
attention over the years. Thus, different analogs of LH-RH, agonists and antagonists,
were conjugated to cytotoxic compounds such as alkylated nitrogen mustard, anticancer
antibiotics and quinones derivatives, which exhibited a wide range of specific binding
affinities towards LH-RH receptors. Preliminary finding proved that quinonyl-amino acids
incorporated into a biological active peptide showed cytotoxic and anticancer activity
[1998LPS421], which aroused our interest in synthesis of different quinones-amino
acids dipeptides. Several peptides with quinone moieties attached through the ε-amino
side chain of a D-lysine residue possess cytotoxic activity against human breast and
prostate cancer cell lines. [1992PNAS972, 1996LPS263]

39
2.1.2 Interaction of Quinones and Amino Acids in Nature
Quinones and amino acids both exist in living systems, but usually in separate
organs. Naturally occurring quinones include naphthoquinone (2.9) [2005BMCL5324],
p-benzoquinone (2.10) [2006AA173], o-benzoquinone (2.11) [2007EJOC1244] and
anthraquinones (2.12) [2006AA173] (Figure 2-3). Quinones are involved in mechanisms
of electron and hydrogen transfer. [2005BMCL5324, 2007EJOC1244]
Figure 2-3. Naturally occurring quinones
2.1.3 Application of Quinone-Amino Acid Conjugates
Quinones and amino acids [2003AAPP34] constitute two ubiquitous classes of
naturally occurring compounds with diverse important properties and applications.
Naphthoquinones (2.13), ubiquinone (2.14) and plastoquinones (2.15) examplify many
classes of quinones that can participate in the electron-transporting chains during
diverse biological redox processes, involving cellular respiration and photosynthesis.
[2007EOTP649, 2005BMCL5324]
Figure 2-4. Classes of quinones participating in biological redox processes

40
The efficiency of the quinonic compounds in inhibiting cancer cells growth is
believed to stem from their ability to associate and intercalate with DNA duplexes and
their participation in key cellular redox mechanisms with consequent generation of
highly reactive oxygen species (ROS), which in turn modify and degrade nucleic acids
and proteins within the cancer cells. [2002AA71] In the living cells quinones can
undergo non-enzymatic or enzymatic one-electron reduction to give toxic semiquinone
anion radicals. After additional redox reactions semiquinone anion radicals form
superoxide anion radicals and hydroxyl radical which produces high cytotoxicity. Cell
cytotoxicity is expressed by various mechanisms including redox cycling, arylation,
intercalation, induction of DNA strand breaks, generation of site-specific free radicals
and interference with mitochondrial respiration. [2005BMCL5324] Many biological
peptides and proteins exert their activity following binding to specific cellular receptors
and have therefore been used extensively as vectors for drug targeting.
Quinone-amino acid conjugates [1996S1468, 2000AA439, 2001AA135,
2001AA381, 2001T407, 2002AA71, 2005BMCL5324, 2007EJOC1244] have significant
potential for drug applications, and thus cytotoxic quinone-peptide conjugates
[1996LPS263, 1998LPS421] are attractive synthetic targets. Quinone-amino acid
conjugates are made up of two components and thus offer almost unlimited potential
structural variations, for the reason that the combination of the features of two or more
biologically active natural moieties in a single molecule may result in more pronounced
pharmacological activities. [2002CSR324, 2003ACIE3996]
2.1.4 Literature Preparative Methods for Quinone-Amino Acid Conjugates
Considerable efforts have been devoted to the synthesis of quinone-amino acid
conjugates utilizing diverse routes including (Figure 2-5): (i) transamination, to give
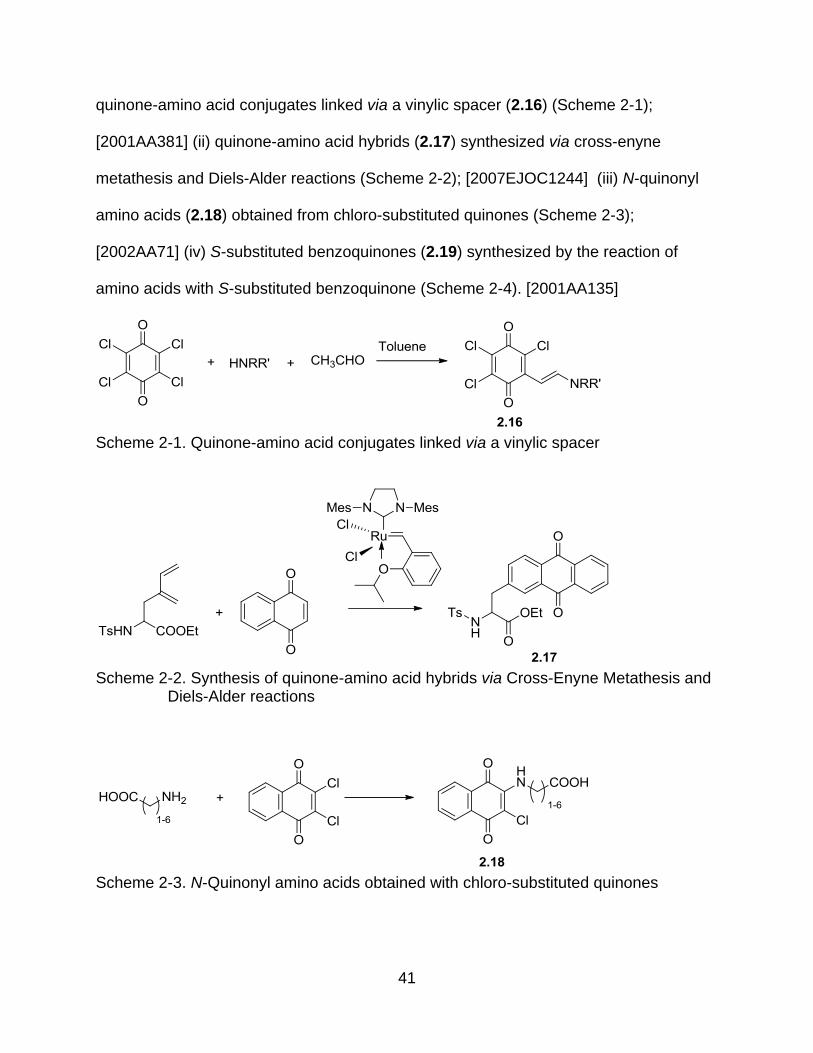
41
quinone-amino acid conjugates linked via a vinylic spacer (2.16) (Scheme 2-1);
[2001AA381] (ii) quinone-amino acid hybrids (2.17) synthesized via cross-enyne
metathesis and Diels-Alder reactions (Scheme 2-2); [2007EJOC1244] (iii) N-quinonyl
amino acids (2.18) obtained from chloro-substituted quinones (Scheme 2-3);
[2002AA71] (iv) S-substituted benzoquinones (2.19) synthesized by the reaction of
amino acids with S-substituted benzoquinone (Scheme 2-4). [2001AA135]
Scheme 2-1. Quinone-amino acid conjugates linked via a vinylic spacer
Scheme 2-2. Synthesis of quinone-amino acid hybrids via Cross-Enyne Metathesis and
Diels-Alder reactions
Scheme 2-3. N-Quinonyl amino acids obtained with chloro-substituted quinones
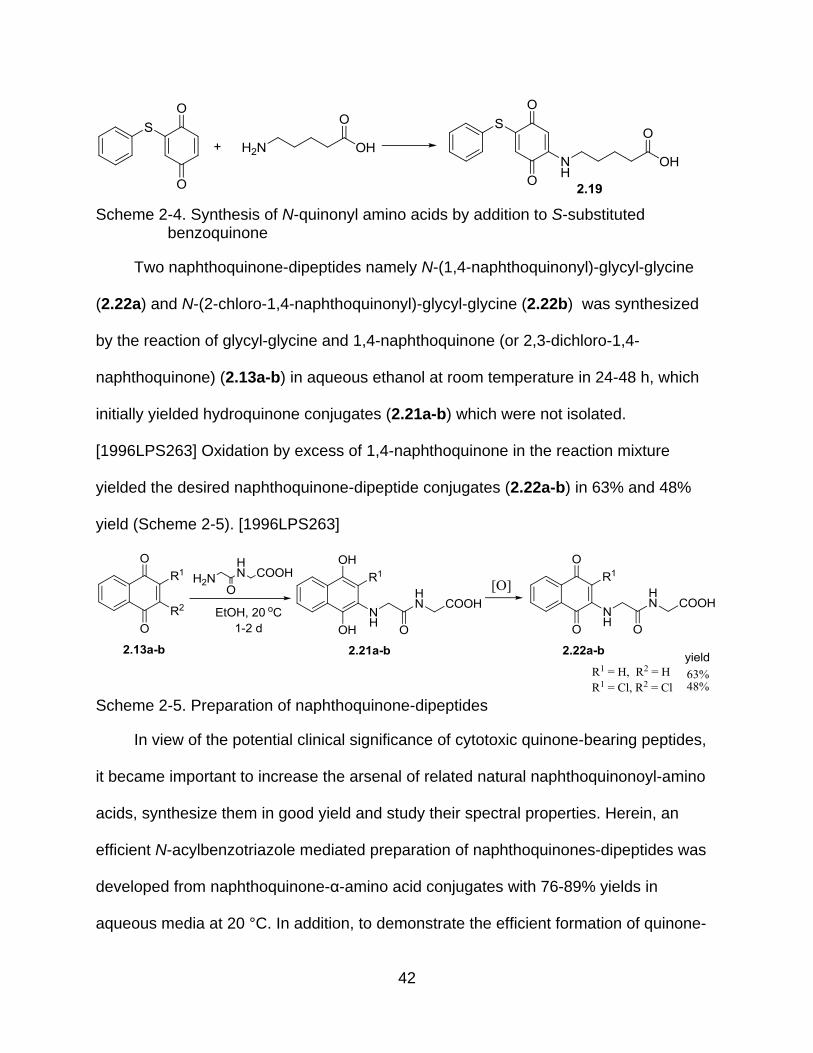
42
Scheme 2-4. Synthesis of N-quinonyl amino acids by addition to S-substituted
benzoquinone
Two naphthoquinone-dipeptides namely N-(1,4-naphthoquinonyl)-glycyl-glycine
(2.22a) and N-(2-chloro-1,4-naphthoquinonyl)-glycyl-glycine (2.22b) was synthesized
by the reaction of glycyl-glycine and 1,4-naphthoquinone (or 2,3-dichloro-1,4-
naphthoquinone) (2.13a-b) in aqueous ethanol at room temperature in 24-48 h, which
initially yielded hydroquinone conjugates (2.21a-b) which were not isolated.
[1996LPS263] Oxidation by excess of 1,4-naphthoquinone in the reaction mixture
yielded the desired naphthoquinone-dipeptide conjugates (2.22a-b) in 63% and 48%
yield (Scheme 2-5). [1996LPS263]
Scheme 2-5. Preparation of naphthoquinone-dipeptides
In view of the potential clinical significance of cytotoxic quinone-bearing peptides,
it became important to increase the arsenal of related natural naphthoquinonoyl-amino
acids, synthesize them in good yield and study their spectral properties. Herein, an
efficient N-acylbenzotriazole mediated preparation of naphthoquinones-dipeptides was
developed from naphthoquinone-α-amino acid conjugates with 76-89% yields in
aqueous media at 20 °C. In addition, to demonstrate the efficient formation of quinone-

43
α-amino acid conjugates derived from S-substituted benzoquinone, the thiol group is
considered to contribute redox properties to the target conjugates and potentially
increase biological activities. For this purpose S-substituted p-benzoquinones were first
reacted with natural α-amino acids via N- addition (Scheme 2-9), then further activated
with benzotriazole group, which were used for the next step peptide synthesis.
However, the preparation of acylbenzotriazoles from S-substitued quinone-amino acid
conjugates proved difficult, but only one example was obtained after many attempts
(Scheme 2-10).
2.2 Results and Discussion
2.2.1 Reaction of Naphthoquinone-Amino Acid Conjugates
Naphthoquinone-amino acid conjugates (2.25a-g) were synthesized from 2-
naphthalene-1,4-dione (2.23) and amino acid or amino ester (2.24a-g) by modifying a
literature procedure [1996LPS263] in aqueous EtOH at room temperature for 10-12 h in
presence of Et3N. The reaction mixture was subjected to column chromatography to first
yield naphthoquinone-amino acid triethylammonium salt, which upon washing with
aqueous hydrochloric acid solution yielded the expected naphthoquinone-amino acid
conjugates (2.25a-g) in 58-90% yield (Scheme 2-6, Table 2-1). [2010S2011]
Scheme 2-6. Synthesis of naphthoquinone-amino acid/ester conjugates

44
Table 2-1. Naphthoquinone-amino acid/ester conjugates
Entry Amino acid (2.24)
Target Compounds (2.25)
Yield (%)
Mp (ºC)
1 L-Phenylalanine (2.24a) 2.25a 72 200-203 2 L-Leucine (2.24b) 2.25b 79 115-117 3 L-Alanine (2.24c) 2.25c 64 137-139a 4 L-Tryptophan (2.24d) 2.25d 58 208-211b 5 L-Proline OMe (2.24e) 2.25e 90 151-153 6 D-Val-OMe (2.24f) 2.25f 76 255-256 7 β-alanine (2.24g) 2.25g 80 205-207 aLit. m. p. 139-142 ºC [2000AA469];
b lit. m. p. 210-213 ºC [1996S1468]
Activation of the terminal carboxylic acid of naphthoquinone-amino acid
conjugates (2.25a-d) was achieved by reaction with same equivalent of benzotriazole
(2.26) and N, N’-dicyclohexylcarbodimide (DCC) to yield naphthoquinone amino-
acylbenzotriazoles (2.27a-d). The reaction was initially attempted with BtH/SOCl2/THF/
RT, 2-5h or BtSO2Me/THF/Et3N/reflux, 8-12h, but a complex mixture was obtained.
Finally, N-acylbenzotriazole derivatives were obtained in DCM at room temperature in
4h using DCC as the coupling agent. (Scheme 2-7, Table 2-2).
Scheme 2-7. Synthesis of naphthoquinone-aminoacylbenzotriazole conjugates

45
Table 2-2. Naphthoquinone-aminoacylbenzotriazoles
Entry Naphthoquinone Amino Acid Conjugates (2.5)
Target compounds (2.27)
Yield (%)
Mp (ºC)
1 2.25a 2.27a 86 115-117 2 2.25b 2.27b n/a n/a 3 2.25c 2.27c n/a n/a 4 2.25d 2.27d 83 114-115
N-Acylbenzotriazole (2.27a-d) derivatives were coupled with various natural amino
acids (2.24a-f) in aqueous acetonitrile-triethylamine at 20 °C in 4 hours to give
naphthoquinone dipeptides (2.28a-k) in good to excellent yields (76-89%). (Scheme 2-
8, Table 2-3)
Scheme 2-8. Preparation of naphthoquinone dipeptide conjugates

46
Table 2-3. Synthesis of Naphthoquinone-dipeptides (continued on the next page)
Entry
Naphthoquinone - amino N-acylbenzotriazole conjugate
Amino acid (2.24)
Target compound (2.28)
yield (%)
Mp (ºC)
1 2.27a L-Alanine (2.24c)
2.28a
89 166-168
2 2.27a L-Valine (2.24e)
2.28b
81 175-177
3 2.27a L-Tryptophan (2.24d)
2.28c
81 215-217
4 2.27b L-Tryptophan (2.24d)
2.28d
81 223-225
5 2.27b L-Alanine (2.24c)
2.28e
89 172-174
6 2.27b L-Glutamic acid methyl ester (2.24f)
2.28f
81 153-155

47
Table 2-3. Continued
Entry
Naphthoquinone - amino N-acylbenzotriazole conjugate
Amino acid (2.24)
Target compound (2.28)
yield (%)
Mp (ºC)
7 2.27c L-Tryptophan (2.24d)
2.28g
82 243-245
8 2.27d L-Leucine (2.24b)
2.28h
79 114-120
9 2.27d L-Glutamic acid methyl ester (2.24f)
2.28i
76 104-111
10 2.27d L-Phenylalanine (2.24a)
2.28j
81 121-123
11 2.27b L-Phenylalanine (2.24a)
2.28k
78 161-164
2.2.2 Reaction of Thio-substituted Benzoquinone with Amino Acids
2-(Cyclohexylsulfanyl)-p-benzoquinone (2.29) was prepared from cyclohexyl
mercaptan (2.20) by reaction with two equivalents of p-benzoquinone (2.10) at room
temperature for 2 hours. Compound (2.29) was used as the starting material for the
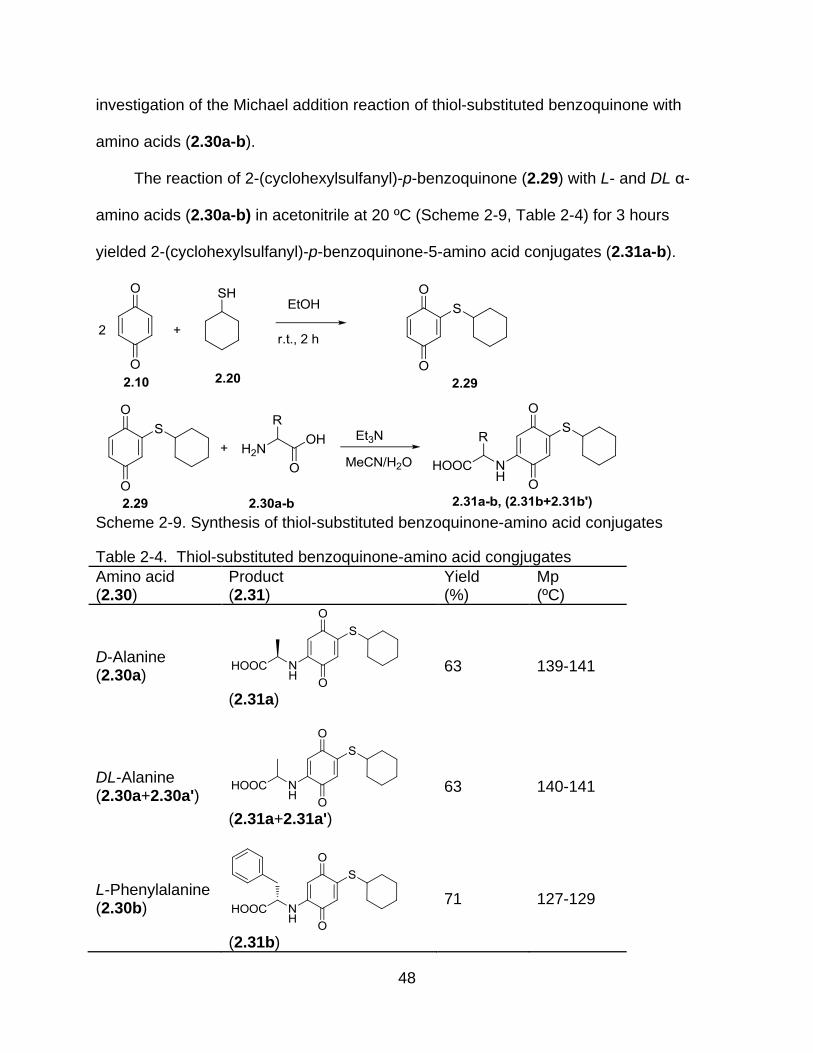
48
investigation of the Michael addition reaction of thiol-substituted benzoquinone with
amino acids (2.30a-b).
The reaction of 2-(cyclohexylsulfanyl)-p-benzoquinone (2.29) with L- and DL α-
amino acids (2.30a-b) in acetonitrile at 20 ºC (Scheme 2-9, Table 2-4) for 3 hours
yielded 2-(cyclohexylsulfanyl)-p-benzoquinone-5-amino acid conjugates (2.31a-b).
Scheme 2-9. Synthesis of thiol-substituted benzoquinone-amino acid conjugates
Table 2-4. Thiol-substituted benzoquinone-amino acid congjugates
Amino acid (2.30)
Product (2.31)
Yield (%)
Mp (ºC)
D-Alanine (2.30a)
(2.31a)
63 139-141
DL-Alanine (2.30a+2.30a')
(2.31a+2.31a')
63 140-141
L-Phenylalanine (2.30b)
(2.31b)
71 127-129

49
2.2.3 Preparation of Benzotriazole Activated Benzoquinone-Amino Acid Conjugates
2-(4-Cyclohexylsulfanyl-3,6-dioxocyclohexa-1,4-dienylamino)propionic acid
(2.31a+2.31a’) on treatment with 1H-benzotriazole and thionyl chloride in DCM gave the
corresponding stable, crystalline racemic acylbenzotriazole (2.32a+2.32a’) in 65 % yield
(Scheme 2-10).
Scheme 2-10. Synthesis of benzoquinone-amino acid benzotriazole derivative
2.3 Conclusion
Naphthoquinones-dipeptides (2.28a-j) were synthesized as potential cytotoxic
agents from naphthoquinone-amino acid conjugates (2.25a-d) by N-acylbenzotriazole
methodology in aqueous medium at 20 °C in 76-89% yield. Three examples of thiol-
subsituted benzoquinone-amino acids (2.31a-b) were prepared in moderate yields, but
the preparation of thio-substituted benzoquinone-N-aminoacylbenzotriazoles was
challenging, due to the many side products formed during the reaction. Only one
example (2.32a+2.32a’) was obtained after many attempts. Disubstituted
benzoquinones resist further substitution in the presence of N- or S- nucleophiles.
2.4 Experimental Section
General methods. Melting points were determined on a capillary point apparatus
equipped with a digital thermometer and are uncorrected. NMR spectra were recorded
in CDCl3 or DMSO-d6 with TMS for 1H (300 MHz) and 13C (75 MHz) as internal
reference. Free amino acids were purchased from Fluka (Buchs, Switzerland) and

50
Acros (Suwanee, GA, USA) and used without further purification. Elemental analyses
were performed on a Carlo Erba-1106 instrument.
General method for preparation of naphthoquinone-amino acid conjugates
(2.25a-g). 2-Naphthalene-1,4-dione (20 mmol) and amino acid/ester (10 mmol) were
dissolved in a mixture of EtOH-H2O (50 : 5 mL). Triethylamine (20 mmol) was added to
the reaction mixture and the mixture was stirred at room temperature for 12 h. The
resulting solution was evaporated under reduced pressure, and the residue was
subjected to column chromatography, eluting with EtOAc/Hexane (2:8) first to remove
the nonpolar impurities, and then with 100% EtOAc to yield a solid, which was
characterized as the triethylamine salt of the expected product. The salt was dissolved
in EtOAc (50 mL), and washed with 3N HCl solution (3 x 50 mL). The organic layer was
dried over sodium sulfate anhydrous, filtered and evaporated under vacuum to yield the
required naphthoquinone-amino acid/ester conjugate.
(S)-2-((1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)-3-phenylpropanoic acid (2.25a)
Black microcrystals; yield: 72%; m. p. 200-203 oC. (lit. m. p. 200-202 oC);
[2002AA71] 1H NMR (DMSO-d6) : 3.22 (t, J = 7.5 Hz, 2H), 4.54-4.48 (m, 1H), 5.74 (s,
1H), 7.26-7.01 (m, 6H), 7.74 (t, J = 7.5 Hz, 1H), 7.84 (t, J = 7.5 Hz, 1H), 7.91 (d, J = 7.5
Hz, 1H), 7.96 (d, J = 7.5 Hz, 1H); 13C NMR (DMSO-d6) : 35.9, 55.7, 101.2, 125.6,
126.2, 126.8, 128.4, 129.4, 130.3, 132.6, 132.9, 135.2, 137.0, 147.6, 172.0, 181.4,
182.0.

51
(S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)-4-methylpentanoic acid (2.25b)
Black crystals; yield: 79%; m. p. 115–117 oC; 1H NMR (DMSO-d6) : 0.87 (d, J =
6.0 Hz, 3H). 0.92 (d, J = 6.3 Hz, 3H), 1.71-1.65 (m, 2H), 1.88-1.96 (m, J = 8.4 Hz, 1H),
4.11-4.07 (m, 1H), 5.68 (s, 1H), 7.31 (d, J = 8.1 Hz, 1H), 7.74 (td, J = 7.5 & 1.5 Hz, 1H),
7.83 (td, J = 7.5 & 1.5 Hz, 1H), 7.94 (dd, J = 7.5 & 1.2 Hz, 1H), 8.00 (dd, J = 7.5 & 1.2
Hz, 1H); 13C NMR (DMSO-d6) : 21.6, 22.7, 24.6, 53.4, 100.7, 125.4, 126.0, 130.3,
132.5, 132.8, 134.9, 148.1, 172.9, 181.3, 181.7; HRMS calcd for C16H18NO4: [M+H]+
288.1320, found 288.1233.
(S)-2-(1,4-Dioxo-1,4-dihydronaphthalen-2-ylamino)propanoic acid (2.25c)
Red crystals; yield: 64%; m. p. 137–139 oC. (lit. m. p. 139–142 ºC); [2000AA439]
1H NMR (DMSO-d6) : 1.31 (d, J = 2.7 Hz, 3H), 3.60-3.80 (m, 1H), 5.58 (s, 1H), 7.43 (d,
J = 6.0 Hz, 1H), 7.71 (t, J = 7.5 Hz, 1H), 7.82 (t, J = 7.2 Hz, 1H), 7.92-7.99 (m, 2H); 13C
NMR (DMSO-d6) : 17.1, 51.5, 99.1, 125.3, 125.8, 130.2, 132.0, 133.3, 134.8, 146.5,
173.6, 181.0, 181.6.
(S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)-3-(1H-indol-3-yl)propanoic acid (2.25d)

52
Orange brown crystals; yield: 58%; m. p. 208–211 oC. (lit. m. p. 210–213 ºC);
[1996S1468] 1H NMR (DMSO-d6) : 3.36-3.38 (m, 2H), 4.45-4.52 (m, 1H), 5.74 (s, 1H),
6.91-6.97 (m, 2H), 7.05 (t, J = 7.8 Hz, 1H), 7.18 (d, J = 2.4 Hz, 1H), 7.32 (dd, J = 8.1,
0.6 Hz, 1H), 7.52 (d, J = 8.1 Hz, 1H), 7.74 (t, J = 7.5 Hz, 1H), 7.83 (t, J = 7.5 Hz, 1H),
7.92-8.00 (m, 2H), 10.90 (s, 1H); 13C NMR (DMSO-d6) : 26.1, 55.2, 100.9, 108.8,
111.4, 118.1, 118.4, 120.9, 124.0, 125.3, 125.9, 127.2, 130.0, 132.3, 134.9, 136.0,
147.2, 172.1, 181.1, 181.7.
(S)-Methyl 1-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)pyrrolidine-2-carboxylate (2.25e)
Black crystals; yield: 90%; m. p. 151–153 oC (lit. m. p. 149–150 ºC);
[1977BCSJ2170] 1H NMR (DMSO-d6) : 1.83-2.13 (m, 3H), 2.21-2.34 (m, 1H), 3.38-
3.47 (m, 2H), 3.69 (s, 3H), 4.98 (bs, 1H), 5.77 (s, 1H), 7.71 (t, J = 7.5Hz, 1H), 7.83 (t, J
= 7.5 Hz, 1H), 7.91 (d, J = 7.2 Hz, 2H); 13C NMR (DMSO-d6) : 21.8, 31.0, 50.9, 52.1,
62.4, 105.1, 124.8, 126.3, 131.2, 132.2, 132.3, 134.5, 148.4, 172.6, 181.3, 182.6.
(R)-Methyl 2-((1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)-3-methylbutanoate (2.25f)
Black crystals; yield: 76%; m. p. 255–256 oC (lit. m. p. 256–257 oC);
[1977BCSJ2170] 1H NMR (CDCl3) : 0.98 (d, J = 6.9 Hz, 3H), 1.04 (d, J = 6.6 Hz, 3H),
2.23-2.30 (m, 1), 3.76(s, 3H), 3.84-3.89 (m, 1H), 5.66 (s, 1H), 6.29 (d, J = 8.7 Hz, 1H),

53
7.60 (t, J = 7.5 Hz, 1H), 7.69 (t, J = 7.5 Hz, 1H), 8.01-8.05 (m, 2H); 13C NMR (CDCl3) :
18.6, 19.0, 31.3, 52.6, 60.6, 102.0, 126.3, 126.5, 130.5, 132.3, 133.4, 134.9, 147.3,
171.1, 181.5, 183.3.
3-((1,4-Dioxo-1,4-dihydronaphthalen-2-yl)amino)propanoic acid (2.25g)
Brown crystals; yield: 80%; m. p. 205–207 oC (lit. m. p. 206–207 ºC); [2002AA71]
1H NMR (DMSO-d6) : 2.60 (t, J = 6.9 Hz, 2H), 3.38-3.42 (m, 2H), 5.72 (s, 1H), 7.50 (t, J
= 6.0 Hz, 1H), 7.73 (t, J = 7.5 Hz, 1H), 7.83 (t, J = 7.5 Hz, 1H), 7.93-7.99 (m, 2H); 13C
NMR (DMSO-d6) : 32.3, 38.0, 99.8, 125.5, 126.1, 130.5, 132.4, 133.2, 135.1, 148.5,
172.9, 181.6.
General method for preparation of naphthoquinone-amino acyl benzotriazole
conjugates (2.27a-d). To a solution of naphthoquinone-amino acid conjugate (2.25a-d)
(5 mmol) in anhydrous DCM (30 mL), benzotriazole (0.60 g, 5 mmol) and N,N'-
dicyclohexylcarbodiimide (DCC) (0.95 g, 5 mmol) were added. The reaction mixture was
stirred at room temperature for 4 h, then filtered through celite at least twice. The
organic layer was concentrated under vacuo and the residue was recrystallized from
EtOAc/Hexane to yield 2.27a and 2.27d as pure products. Compounds 2.27b-c were
not isolated in pure form, but used as crude (NMR shows trace amount of DBU
coexisting with the product) for the next coupling reaction.

54
(S)-2-((1-(1H-Benzo[d][1,2,3]triazol-1-yl)-1-oxo-3-phenylpropan-2-yl)amino)naphthalene-1,4-dione (2.27a)
Black crystals; yield: 86%; m. p. 115–117 oC; 1H NMR (CDCl3) : 3.37 (dd, J = 7.5
& 13.8 Hz, 1H), 3.60 (dd, J = 4.8 & 13.8 Hz, 1H), 5.76 (s, 1H), 5.56-5.82 (m, 1H), 6.55
(d, J = 7.8 Hz, NH), 7.33 (m, 5H), 7.72-7.54 (m, 4H), 8.04 (t, J = 6.0 Hz, 2H), 8.20 (t, J =
7.5 Hz, 2H); 13C NMR (CDCl3) : 38.9, 56.6, 103.0, 114.4, 120.8, 126.4, 126.6, 127.1,
127.9, 129.1, 129.3, 130.5, 131.0, 131.3, 132.5, 133.2, 134.8, 134.9, 146.3, 146.6,
169.6, 181.3, 183.3; Anal. Calcd for C25H18N4O3: C, 71.08; H, 4.29; N, 13.26. Found: C,
70.80; H, 5.04; N, 13.49.
2-(((2S)-1-(1H-Benzo[d][1,2,3]triazol-1-yl)-3-(2,7a-dihydro-1H-indol-3-yl)-1-oxopropan-2-yl)amino)naphthalene-1,4-dione (2.27d)
Yellow microcrystals; yield: 83%; m. p. 114.0 – 115.0 oC; 1H NMR (CDCl3) : 3.60
(dd, J = 7.6, 15.0 Hz, 1H), 3.80 (dd, J = 4.8, 14.7 Hz, 1H), 5.72 (s, 1H), 5.92 (q, J = 4.8
Hz, 1H), 6.61 (d, J = 8.1 Hz, 1H), 7.00 (t, J = 7.5 Hz, 1H), 7.10-7.17 (m, 2H), 7.29 (d, J =
8.4 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.54 -7.72 (m, 5H), 8.02 (d, J = 7.8 Hz, 1H), 8.18
(t, J = 6.9 Hz, 2H), 8.26 (br s, 1H); 13C NMR (CDCl3) : 29.3, 56.1, 103.0, 111.7, 114.6,
118.5, 120.3, 120.8, 122.9, 123.7, 126.5, 126.7, 127.1, 131.4, 132.6, 135.0, 147.0,
170.1; HRMS calcd for C27H20N5O3 [M+H]+ 462.1488, found 462.1556.

55
General method for preparation of Naphthoquinone-dipeptides (2.28a-j). A
solution of L-amino acid (1 mmol) (2.24a-f) and Et3N (1.2 mmol) in water (4 mL) was
added to a solution of N-acyl benzotriazole derivative (1 mmol) (2.25a-d) in MeCN (50
mL). The reaction mixture was stirred at room temperature for 3-4 h, and then quenched
with 4 N aqueous HCl (2 mL). The reaction mixture was concentrated, diluted with
EtOAc (100 mL), and washed with 4N aqueous HCl (30 mL x 3), and brine (30 mL x 2).
The organic layer was concentrated, and cold hexane (30 mL) was added to the
resulting solution. The precipitated solid was filtered and dried under vacuum to yield
naphthoquinone-dipeptides (2.28a-j).
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-3-phenylpropanamido) propanoic acid (2.28a)
Orange crystals; yield = 89%; m. p. 166–168 ºC; 1H NMR (CD3COCD3-d6) : 1.40
(d, J = 7.2 Hz, 3H), 3.21 (dd, J = 14.1 Hz & 7.5 Hz, 1H), 3.37 (dd, J = 13.8 Hz & 4.8 Hz,
1H), 4.45-4.52 (m, 2H), 5.66 (s, 1H), 6.45 (s, 1H), 7.14 (d, J = 6.3 Hz, 1H), 7.19-7.36 (m,
5H), 7.72 (t, J = 7.5 Hz, 1H), 7.82 (t, J = 7.5 Hz, 1H), 8.01 (t, J = 6.0 Hz, 2H); 13C NMR
(CDCl3) : 18.3, 21.9, 22.8, 24.4, 48.8, 54.5, 100.7, 125.4, 126.0, 130.3, 132.4, 132.8,
134.9, 147.8, 169.9, 181.2, 181.8; HRMS calcd for C22H21N2O5: [M+H]+ 393.1445, found
393.1601.

56
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-3-phenylpropanamido)-3-methylbutanoic acid (2.28b)
Red crystals; yield: 81%; m. p. 175–177 oC; 1H NMR (CD3COCD3-d6) : 0.97 (t, J
= 5.4 Hz, 6H), 2.27-2.16 (m, 1H), 3.23 (dd, J = 13.8 & 7.8 Hz, 1H), 3.36 (dd, J = 13.8 &
5.1 Hz, 1H), 4.60-4.47 (m, 2H), 5.72 (s, 1H), 6.68 (d, J = 7.2 Hz, 1H), 7.34-7.16 (m, 5H),
7.81-7.65 (m, 2H), 7.83 (t, J = 13.9 Hz, 1H), 8.02 (t, J = 7.5 Hz, 2H); 13C NMR
(CD3COCD3-d6) : 18.2, 19.5, 31.6, 38.6, 57.77, 57.84, 58.1, 102.6, 126.5, 126.6, 126.8,
127.7, 129.3, 129.5, 130.3, 131.5, 133.1, 134.2, 135.5, 137.7, 147.9, 170.9, 172.8,
182.2, 182.7; HRMS calcd for C24H25N2O5: [M+H]+ 421.1758, found 421.1778.
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-3-phenylpropanamido)-3-(1H-indol-2-yl)propanoic acid (2.28c)
Red crystals; yield: 81%; m. p. 215–217 oC; 1H NMR (DMSO-d6) : 3.14-3.06 (m,
3H), 3.23 (dd, J = 5.4 & 5.1 Hz, 1H), 4.38-4.31 (m, 1H), 4.59-4.52 (m, 1H), 5.57 (s, 1H),
6.93 (t, J = 7.2 Hz, 1H), 7.04-7.00 (m, 2H), 7.24-7.14 (m, 5H), 7.32 (d, J = 7.8 Hz, 1H),
7.53 (d, J = 7.8 Hz, 1H), 7.73 (t, J = 7.5 Hz, 1H), 7.82 (t, J = 7.5 Hz, 1H), 7.91 (d, J = 7.2
Hz, 1H), 7.97 (d, J = 7.5 Hz, 1H), 8.65 (d, J = 8.1 Hz, 1H), 10.87 (s, 1H); 13C NMR
(DMSO-d6) : 27.1, 37.1, 53.0, 56.6, 100.9, 109.5, 111.3,116.4, 118.1, 118.3, 120.8,
123.6, 124.5, 125.3, 125.7, 125.9, 126.0, 126.4, 127.1, 128.1, 128.5, 129.1, 130.0,

57
132.3, 132.6, 134.8, 136.0, 136.9, 147.2, 169.6, 172.8, 180.9, 181.6; HRMS calcd for
C30H26N3O5: [M+H]+ 508.1867, found 508.1886.
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-4-methylpentanamido)-3-(1H-indol-2-yl)propanoic acid (2.28d)
Red crystals; yield: 81%; m. p. 223–225 oC; 1H NMR (DMSO-d6) : 0.82 (d, J = 6.3
Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H), 1.61-1.52 (m, 2H), 1.75-1.64 (m, 1H), 3.06 (dd, J =
14.4 & 5.4 Hz, 1H), 3.19 (dd, J = 14.4 & 5.4 Hz, 1H), 4.08-4.01 (m, 1H), 4.54-4.51 (m,
1H), 5.73 (s, 1H), 7.03-6.90 (m, 3H), 7.14 (d, J = 2.1 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H),
7.51 (td, J = 7.5 & 1.5 Hz,1H), 7.74 (td, J = 7.5 & 1.5 Hz, 1H), 7.84 (td, J = 7.5 &1.2 Hz,
1H), 7.95 (dd, J = 8.1 & 1.2 Hz, 1H), 8.00 (dd, J = 7.8 & 1.2 Hz, 1H), 8.47 (d, J = 8.1 Hz,
1H), 10.81 (s, 1H); 13C NMR (DMSO-d6) : 22.0, 22.7, 24.3, 53.0, 54.4, 100.8, 109.6,
111.4, 118.1, 118.3, 120.8, 123.6, 125.4, 126.0, 127.2, 130.3, 132.4, 132.8, 134.9,
136.1, 147.6, 170.7, 172.9, 181.2, 181.8; Anal. Calcd for C27H27N3O5: C, 68.48; H, 5.75;
N, 8.87. Found: C, 68.58; H, 5.51; N, 8.45.
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-4-methylpentanamido) propanoic acid (2.28e)
Orange crystals; yield: 89%; m. p. 172–174 oC; 1H NMR (DMSO-d6) : 0.85 & 0.92
(dd, J = 6.3 Hz, 6H), 1.23 (d, J = 7.2 Hz, 3H), 1.69-1.55 (m, 2H), 1.80-1.69 (m, 1H),
4.12-4.05 (m, 2H), 5.73 (s, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.73 (t, J = 7.5 Hz, 1H), 7.83

58
(t, J = 7.5 Hz, 1H), 7.93 (d, J = 7.5 Hz, 1H), 7.99 (d, J = 7.5 Hz, 1H), 8.19 (d, J = 6.9 Hz,
1H); 13C NMR (DMSO-d6) : 18.3, 21.9, 22.8, 24.4, 48.8, 54.5, 100.7, 125.4, 126.0,
130.3, 132.4, 132.8, 134.9, 147.8, 169.9, 181.2, 181.8; HRMS calcd for C19H23N2O5:
[M+H]+ 359.1601, found 359.1596.
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-4-methylpentanamido)-5-methoxy-5-oxopentanoic acid (2.28f)
Red crystals; yield: 81%; m. p. 153–155 oC; 1H NMR (DMSO-d6) : 0.86 (d, J = 6.3
Hz, 3H), 0.92 (d, J = 6.0 Hz, 3H), 1.71-1.58 (m, 2H), 1.85-1.76 (m, 2H), 2.03-2.00 (m,
1H), 2.35-2.29 (m, 1H), 3.51 (s, 3H), 4.16-4.00 (m, 2H), 5.75 (s, 1H), 7.12 (d, J = 8.7 Hz,
1H), 7.73 (t, J = 7.5 Hz, 1H), 7.83 (t, J = 7.5 Hz, 1H), 7.93 (d, J = 7.5 Hz, 1H), 7.99 (d, J
= 7.2 Hz, 1H), 8.22 (d, J = 7.5 Hz, 1H); 13C NMR (DMSO-d6) : 13.9, 21.9, 22.1, 22.8,
24.4, 26.9, 29.7, 31.0, 51.2, 52.0, 54.5, 100.9, 125.4, 126.0, 130.3, 132.4, 132.8, 134.9,
147.7, 170.4, 172.9, 181.3, 181.7; HRMS calcd for C22H27N2O7: [M+H]+ 431.1813 found
431.1816.
(S)-2-((S)-2-((1,4-dioxo-1,4-dihydronaphthalen-3-yl)amino)propanamido)-3-(1H-indol-3-yl)propanoic acid (2.28g)

59
Red crystals; yield: 82%; m. p. 243–245 oC; 1H NMR (DMSO-d6) : 1.36 (d, J = 6.6
Hz, 3H), 3.08 (dd, J = 14.7 & 5.1 Hz, 1H), 3.22 (dd, J = 14.7 & 5.1 Hz, 1H), 4.12 (t, J =
7.2 Hz, 1H), 4.57-4.50 (m, 1H), 5.60 (s, 1H), 7.08-6.93 (m, 3H), 7.16 (d, J = 2.1Hz,
1H), 7.31 (d, J = 7.8 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H),7.75 (td, J = 7.5 & 1.2 Hz, 1H),
7.85 (td, J = 7.5 & 1.2 Hz, 1H), 7.95 (d, J = 6.6 Hz, 1H), 8.00 ( d, J = 6.9 Hz, 1H), 8.50
(d, J = 8.1Hz, 1H), 10.85 (s, 1H); 13C NMR (DMSO-d6) : 17.6, 27.0, 50.8, 52.9, 100.7,
109.5, 111.3, 118.0, 118.3, 120.8, 123.5, 125.3, 125.9, 127.1, 130.2, 132.3, 132.8,
134.8, 136.0, 147.0, 171.0, 172.8, 181.1, 181.6; Anal. Calcd for C24H21N3O5: C, 66.81;
H, 4.91; N, 9.74. Found: C, 66.57; H, 4.79; N, 9.50.
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-3-(1H-indol-3-yl)propanamido)-4-methylpentanoic acid (2.28h)
Orange crystals; yield: 79%; m. p. 114–120 ºC; 1H NMR (DMSO-d6) : 0.81 (d, J =
6.0 Hz, 3H), 0.91 (d, J = 6.3 Hz, 3H), 1.22-1.25 (m, 1H), 1.54-1.77 (m, 2H), 3.27-3.30
(m, 2H), 4.28-4.38 (m, 2H), 5.59 (s, 1H), 6.90-6.99 (m, 2H), 7.05 (t, J = 7.8 Hz, 1H),
7.27 (s, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.71 (t, J = 7.2 Hz, 1H),
7.80 (t, J = 7.5 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.94 (d, J = 7.5 Hz, 1H), 8.61 (d, J =
7.8 Hz, 1H), 10.89 (s, 1H); 13C NMR (DMSO-d6) : 21.0, 22.8, 24.3, 27.4, 33.3, 38.6,
50.2, 56.1, 100.9, 109.0, 111.3, 118.2, 118.3, 120.9, 124.2, 125.3, 125.8, 127.2, 130.1,

60
132.3, 132.7, 134.8, 136.1, 147.3, 170.2, 173.7, 181.0, 181.5; Anal. Calcd for
C27H27N3O5: C, 68.48; H, 5.75; N, 8.87. Found: C, 68.20; H, 5.90; N, 8.47.
(S)-2-((S)-2-(1,4-Dioxo-1,4-dihydronaphthalen-2-ylamino)-3-(1H-indol-3-yl)propanamido)-5-methoxy-5-oxopentanoic acid (2.28i)
Yellow crystals; yield: 76%; m. p. 104–111 ºC; 1H NMR (DMSO-d6) : 1.85-1.92
(m, 1H), 1.98-2.15 (m, 1H), 2.37 (t, J = 7.2 Hz, 2H), 3.29-3.38 (m, 2H), 3.56 (s, 3H),
4.26-4.40 (m, 2H), 5.61 (s, 1H), 6.96 (t, J = 7.2 Hz, 2H), 7.02 (t, J = 7.2 Hz, 1H), 7.25 (s,
1H), 7.31 (d, J = 8.1 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.72 (t, J = 7.2 Hz, 1H), 7.81 (t, J
= 7.2 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.95 (d, J = 7.5 Hz, 1H), 8.59 (d, J = 7.8 Hz,
1H), 10.88 (s, 1H); 13C NMR (DMSO-d6) : 26.2, 27.3, 29.6, 51.1, 51.2, 56.1, 100.8,
109.0, 111.3, 118.1, 118.3, 120.9, 124.1, 125.3, 125.8, 127.2, 130.1, 132.3, 132.7,
134.8, 136.1, 147.3, 170.3, 172.5, 181.3, 181.5; Anal. Calcd for C27H25N3O7: C, 64.41;
H, 5.00; N, 8.35. Found: C, 64.09; H, 5.05; N, 7.98.
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-3-(1H-indol-3-yl)propanamido) -3-phenylpropanoic acid (2.28j)
Yellow crystals; yield: 81%; m. p. 121–123 ºC; 1H NMR (DMSO-d6) : 2.94-3.00
(m, 1H), 3.05-3.18 (m, 1H), 3.20-3.28 (m, 2H), 4.20-4.35 (m, 1H), 4.51-4.55 (m, 1H),

61
5.61 (s, 1H), 6.90-7.01 (m, 2H), 7.07 (t, J = 6.9 Hz, 1H), 7.12-7.25 (m, 6H), 7.33 (d, J
= 7.8 Hz, 1H), 7.64 (d, J = 7.5 Hz, 1H), 7.71 (d, J = 6.3 Hz, 1H), 7.80 (t, J = 7.5 Hz,
1H), 7.90-7.94 (m, 2H), 8.70 (d, J = 7.2 Hz, 1H), 10.88 (s, 1H); 13C NMR (DMSO-d6) :
27.9, 37.3, 54.0, 56.7, 101.4, 109.7, 111.9, 118.7, 118.8, 121.5, 124.6, 125.8, 126.4,
126.9, 127.7, 128.6, 129.6, 130.6, 132.8, 133.2, 135.4, 136.6, 137.8, 147.8, 170.7,
173.0, 181.5, 182.1; Anal. Calcd for C30H25N3O5: C, 70.99; H, 4.96; N, 8.28. Found: C,
71,26; H, 5.29; N, 7.85. HRMS calcd for C30H26N3O5: [M+H]+ 508.1867, found
508.1886.
(S)-2-((S)-2-((1,4-Dioxo-1,4-dihydronaphthalen-3-yl)amino)-4-methylpentanamido)-3-phenylpropanoic acid (2.28k)
Red crystals; yield: 78%; m. p. 161–164 ºC; 1H NMR (DMSO-d6) : 0.83 (d, J = 3.0
Hz, 3H), 0.90 (d, J = 3.0 Hz, 3H), 1.47-1.62 (m, 2H), 1.78-1.70 (m, 1H), 2.89 (dd, J =
6.3 & 13.8 Hz, 1H), 3.08 (dd, J = 4.5 & 13.8 Hz, 1H), 3.98-4.05 (m, 1H), 4.44-4.50 (m,
1H), 5.74 (s, 1H), 6.99 (d, J = 8.4 Hz, 1H), 7.11-7.06 (m, 1H), 7.20-7.15 (m, 4H), 7.75 (t,
J = 7.5 Hz, 1H), 7.85 (t, J = 7.5 Hz, 1H), 7.95-8.02 (m, 2H), 8.51 (d, J = 8.1 Hz, 1H); 13C
NMR (DMSO-d6) : 21.7, 22.4, 24.1, 36.4, 53.1, 54.1, 100.7, 125.1, 125.7, 126.0, 127.8,
128.9, 130.0, 132.2, 132.5, 134.7, 137.0, 147.2, 170.4, 172.2, 180.9, 181.5; HRMS
calcd for C25H27N2O5: [M+H]+ 435.1914; found 435.1934.
General procedure for preparation of 2-(cyclohexylthio)cyclohexa-2,5-diene-
1,4-dione (2.29). Cyclohexyl mercaptan (4.9 mL, 40 mmol) in MeOH (5 mL) was added

62
dropwise to a suspension of 1,4-benzoquinone (8.67 g, 81 mmol) in MeOH (50 mL), and
the mixture was stirred at 20 oC for 2 h. Water (100 mL) was added, and the resulting
precipitate was collected by filtration. The orange crystals were recrystallized from
CH2Cl2/MeOH to give the pure form of (2.29).
2-(Cyclohexylthio)cyclohexa-2,5-diene-1,4-dione (2.29)
Brown crystals; yield: 80%; m. p. 104–106 oC (lit. m. p. 102–106 oC);
[1991JOC5808] 1H NMR (CDCl3) : 1.28-1.59 (m, 5H), 1.61-1.72 (m, 1H), 1.75-1.90 (m,
2H), 1.97-2.14 (m, 2H), 2.98-3.20 (m, 1H), 6.42 (d, J = 1.8 Hz, 1H), 6.71 (dd, J = 6.0,
2.1 Hz, 1H), 6.81 (d, J = 10.2 Hz, 1H); 13C NMR (CDCl3) : 25.5, 25.6, 31.9, 42.5, 124.8,
136.2, 137.3, 152.0, 169.9, 184.1.
General procedure for peparation of thiol-substituted benzoquinone-amino
acid conjugates (2.31a-b). To a solution of 2-(cyclohexylsulfanyl)-p-benzoquinone
(2.29) (0.22 g, 1 mmol) in (MeCN:H2O 6: 3mL) at RT, a solution of amino acid (2.30a-b)
(0.5 mmol) and triethylamine (0.1 mL, 0.7 mmol) in water (5 mL) was added slowly. The
reaction mixture was stirred for 3 h at RT. Acetonitrile was removed under reduced
pressure, and the residue was purified by flash chromatography on silica gel eluting with
chloroform/methanol (20:1) to give the triethylamine salt of the thiol-substituted
benzoquinone-amino acid conjugates, which can be converted to free acid form via
neutralizating with 4N aqueous HCl.

63
(R)-2-((4-(Cyclohexylthio)-3,6-dioxocyclohexa-1,4-dien-1-yl)amino)propanoic
acid (2.31a)
Red microcrystals; yield: 63%; m. p. 139–141 oC; 1H NMR (CDCl3) δ: 1.21-2.12
(m, 13H), 3.0-3.28 (m, 1H), 3.93 (t, J = 6.3 Hz, 1H), 5.42 (s, 1H), 6.22 (s, 1H), 6.72 (bs,
1H); 13C NMR (CDCl3) δ: 8.1, 16.9, 25.1, 25.2, 31.5, 42.1, 44.9, 50.6, 96.8, 119.6,
143.1, 145.8, 157.6, 178.5, 180.9. Anal. Calcd for C15H19NO4S (309.39): C, 58.23; H,
6.19; N, 4.53. Found: C, 58.34; H, 6.33; 4.33.
2-((4-(Cyclohexylthio)-3,6-dioxocyclohexa-1,4-dien-1-yl)amino)propanoic acid (2.31a+2.31a')
Deep brown microcrystals; yield: 63%; m. p. 140–141 oC; 1H NMR (CDCl3) δ: 1.21-
2.22 (m, 13H), 3.01-3.15 (m, 1H), 3.99 (t, J = 7.2 Hz, 1H), 5.44 (s, 1H), 6.25 (s, 1H),
6.52 (d, J = 6.9 Hz, 1H), 8.55 (bs, 1H). 13C NMR (CDCl3) δ: 16.8, 25.1, 25.2, 31.5, 42.1,
49.9, 97.3, 119.7, 145.7, 157.4, 172.8, 178.4, 181.1. Anal. Calcd. for C15H19NO4S
(309.39): C, 58.23; H, 6.19; N, 4.53. Found: C, 58.34; H, 6.33; 4.33.
(S)-2-((4-(cyclohexylthio)-3,6-dioxocyclohexa-1,4-dien-1-yl)amino)-3-phenylpropanoic acid (2.31b)

64
Red crystals; yield: 71%; m. p. 127–129 oC; 1H NMR (DMSO-d6) : 1.00-1.58 (m,
6H), 1.58-1.70 (m, 1H), 1.70-1.80 (m, 2H), 1.80-2.15 (m, 2H), 2.95-3.10 (m, 1H), 3.15
(dd, J = 14.15 Hz, 5.22 Hz, 1H), 3.32 (dd, J = 14.15 Hz, 5.22 Hz, 1H), 4.15-4.35 (m,
1H), 5.54 (s, 1H), 6.24 (s, 1H), 6.29 (d, J = 7.69 Hz, 1H), 7.00 -7.50 (m, 5H), 8.80 (bs,
1H); 13C NMR (DMSO-d6) δ: 25.5, 25.6, 31.8, 37.1, 42.8, 55.9, 98.2, 120.1, 127.6,
128.9, 129.1, 134.7, 146.6, 158.1, 173.3, 178.2, 182.4. Anal. Calcd for C21H27NO6S
(421.52): C, 59.84; H, 6.46; N, 3.32. Found: C, 60.33; H, 5.85; 3.16.
General procedure for preparation of benzotriazole activated thiosubstituted
benzoquinone-amino acid conjugates (2.32a+2.32a'). Benzotriazol (0.17 g, 1.4
mmol) and thionyl chloride 0.05 g (0.39 mmol) were dissolved in DCM (10 mL) at 25oC
and stirred for 10 min. 2-(4-Cyclohexylsulfanyl-3,6-dioxo-cyclohexa-1,4-dienylamino)-
propionic acid (2.31a+2.31a') (0.15 g, 0.35 mmol) was added to the solution. The
reaction mixture was stirred at – 15 oC for 5 h, then at RT overnight. The reaction
mixture was filterd and dichloromethane was evaporated under reduced pressure to
give the crude residue, which was subjected to column chromatography with
dichloromethane to yield a stable, crystalline racemic acylbenzotriazole (2.32a+2.32a'),
which was recrystallized from DCM/Hexane before the elemental analysis.
2-((1-(1H-benzo[d][1,2,3]triazol-1-yl)-1-oxopropan-2-yl)amino)-5-(cyclohexylthio)cyclohexa -2,5-diene-1,4-dione (2.32a+2.32a')

65
Red crystals; yield: 65%; m. p. 172–174 oC. 1H NMR (DMSO-d6) δ: 1.20-2.30 (m,
15H), 3.00-3.20 (m, 1H), 5.46 (s, 1H), 4.40-5.70 (m, 1H), 6.31 (s, 1H), 6.50 (d, J = 7.7
Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.72 (t, J = 7.8 Hz, 1H), 8.19 (d, J = 6.9 Hz, 1H), 8.26
(d, J = 6.9 Hz); 13C NMR (DMSO-d6) δ: 18.5, 25.5, 25.6, 42.7, 51.0, 99.0, 114.2, 120.2,
120.6, 126.9, 130.9, 131.1, 145.8, 146.1, 157.8, 170.4, 178.7, 181.8; Anal. Calcd for
C21H24N4O4S.H2O (428.51): C, 58.86; H, 5.65; N, 13.07; found: C, 59.38; H, 5.18; N,
13.10.

66
CHAPTER 3 1,3,4-OXADIAZOLES FROM FUCTIONALIZED N-ACYLBENZOTRIAZOLES AND
ACYLHYDRAZIDES
3.1 Introduction
3.1.1 Oxadiazoles
Oxadiazoles are heterocyclic aromatic compounds consisting of fused 5-
membered rings containing two carbons, two nitrogen atoms and one oxygen atom. The
four possible oxadiazoles (A-D) are shown in Figure 3-1.
Figure 3-1. Four types of oxadiazoles
3.1.2 Biologically Active 1,3,4-Oxadiazoles
The 1,3,4-oxadiazole moiety is an important structural class in medicinal chemistry
due to its widespread use as a pharmacophore. [2006JOC9548, 2006T10223,
2006TL105, 2006TL4827, 2006TL6497, 2007EJMC235, 2007EJMC893,
2007EJMC934]
Oxadiazoles of type (3.1), amino-oxadiazoles of type (3.2) [2006JOC9548], and
oxadiazolinethiones of type (3.3) [1988IJC(B)542] were reported with demonstrated
bactericidal and/or fungicidal activities. The tin derivative (3.4) is a useful fungicide, and
the thione derivative (3.5) shows antimicrobial activities. Diaryloxadiazoles (3.6)
possesses certain anti-inflammatory, sedative and analgesic properties. [1984FES414]
Amino-oxadiazoles (3.7) show analgesic activity and amino-oxidazoles (3.8) exhibit both
anti-inflammatory and antiproteolytic properties. [1989JPS999] Anticonvulsant and
nervous system depressant activity was reported for amino-oxadiazoles (3.9), where R

67
is quinazolin-3-yl group. [1991PHA290] Amino-oxadiazoles (3.10) show local anesthetic
activity. [1983JIC575] Oxadiazolinone (3.11) is an orally active antiallergic agent, for
example in the treatment of asthma or allergies, and is claimed to be more potent than
sodium cromoglycate. [1984JMC121] Oxadiazolinones (3.12 and 3.13) and “oxadiazon”
(3.14) are herbicides, while oxadiazolinones (3.15 and 3.16) and oxadiazole (3.17) have
insecticidal activity (Figure 3-2).
Figure 3-2. Biologically important oxadiazoles
3.1.3 Polymeric 1,3,4-Oxadiazoles
Heat resistant polyazomethines (3.18) are used as insulators, and are obtained
from 2,5-di-(3-aminophenyl)-1,3,4-oxadiazole by reaction with aromatic dialdehydes
Ar(CHO)2. They can be converted to semiconductors by doping with iodine.
[1992JPS(A)1369] Polyazomethines having an alternative structure were prepared from

68
aromatic diamines and oxadiazole-dialdehydes. [1990JPS(A)3647] The activating effect
of the oxadiazole ring in 4-fluorophenyl- and 4-nitrophenyl-1,3,4-oxadiazoles allows
nucleophilic displacement of these subsitutents. Thus 2,5-diaryloxadiazoles react with
biphenols to give high molecular weight polyethers (3.19) (Figure 3-3). [1992MM2021]
Figure 3-3. Polymers containing 1,3,4-oxadiazoles
3.1.4 Luminescent Compounds, Dyes and Photosensitive Materials
There are various applications of 1,3,4-oxadiazoles containing three or more
conjugated rings as luminescent compounds, because oxadiazoles have strong
absorptions in the UV and strong fluorescence activity. Bis-oxadiazoles (3.20) adsorb at
267 – 299 nm, which indicates less than full conjugation, and show strong fluorescence
at 420nm in ethanol. [1990JHC1685] 2,5-Disubstituted-1,3,4-oxadiazoles often
fluorensce, which makes them potentially useful as laser dyes, optical brighteners and
scintillators. For example, oxadiazole (3.21a) [1984GPO3245202] and 1,4-bis-(5-
phenyl-1,3,4-oxadiazol-2-yl)naphthalene [1983GPO3126464] are fluorescent whiteners
on polyester fiber. Applications of oxadiazole (3.21b) (Figure 3-4) include use as a laser
dye, a blue-emitting phosphor, a wide range of applications as scintillator, and as an
electron-transport layer in thin-film electroluminescent devices. [1991CL285] 1,3,4-
Oxadiazoles were recently tested for their possible use in organic light-emitting diodes
(OLED). [2007USP085073, 2007DP641, 2007DP753].
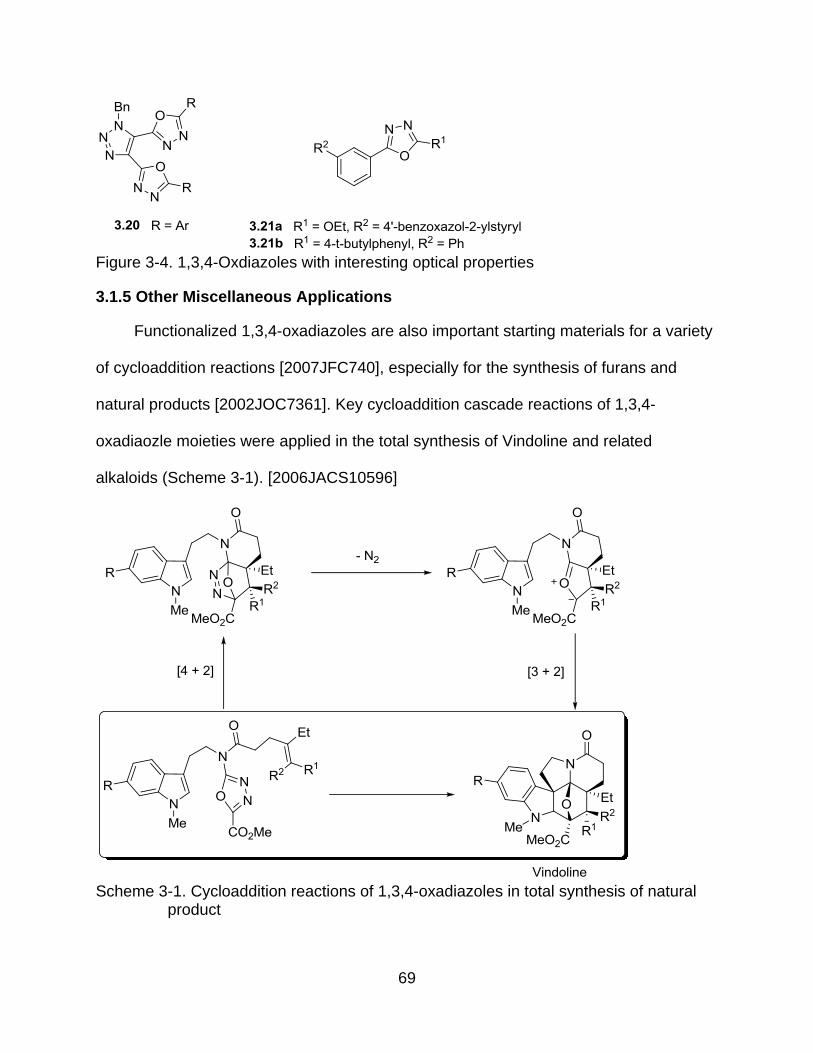
69
Figure 3-4. 1,3,4-Oxdiazoles with interesting optical properties
3.1.5 Other Miscellaneous Applications
Functionalized 1,3,4-oxadiazoles are also important starting materials for a variety
of cycloaddition reactions [2007JFC740], especially for the synthesis of furans and
natural products [2002JOC7361]. Key cycloaddition cascade reactions of 1,3,4-
oxadiaozle moieties were applied in the total synthesis of Vindoline and related
alkaloids (Scheme 3-1). [2006JACS10596]
Scheme 3-1. Cycloaddition reactions of 1,3,4-oxadiazoles in total synthesis of natural
product

70
2,5-Dipicryl-1,3,4-oxadiazole (3.22) is used as an explosive initiator
[1988USP43262] and 2,5-dimethyl-1,3,4-oxadiazole (3.23) has been used to extract
aromatic hydrocarbons from mixtures with alkanes (Figure 3-5). 4,4'-Carbonylbis-(2-
phenyl-5-oxo-1,3,4-oxadiazole) (3.24) is used as a blowing agent for foaming
thermoplastic compositions (e.g. polycarbonate). [1985USP4500653]
Figure 3-5. Other applications of 1,3,4-oxidazoles
3.1.6 Literature Preparative Methods for 1,3,4-Oxadiazoles
2,5-Disubsituted 1,3,4-oxadiazoles (3.30) are formed in the reaction of 1,2-
diacylhydrazines (3.25) with strong dehydrating agents, including chlorosulfonic acid
[1983MI406-01] or phenyl dichlorophosphite [1982RRC935] in DMF (Scheme 3-2). A
nonaqueous, nonacidic route to oxadiazoles (3.30) involves treatment of hydrazine
(3.25) with hexamethyldisilazide (HMDS) and tetrabutylammonium fluoride (TBAF), the
last step presumably being fluoride-catalyzed cyclization of intermediate bis-silyl ether
(3.26). [1986SC1665]
Scheme 3-2. Preparation of 2,5-disubstituted 1,3,4-oxadiazoles from 1,2-
diacylhydrazines

71
The cyanohydrazones (3.27), on heating in dimethyl sulfoxide, cyclized with loss of
HCN to give unsymmetrical 2,5-disubsituted oxadiazoles (3.30). [1984S146]
Benzophenone acylhydrazones (3.28) cyclized on reaction with acid chlorides RCOCl to
oxadiazoles (3.29). [1985T5187]
Scheme 3-3. Preparation of 2,5-disubstituted 1,3,4-oxadiazoles from hydrazones
Treating allyl esters (3.31) with DIPEA forms oxadiazolinones (3.33), probably via
Claisen rearrangement of an initially formed oxadiazolinone (3.32) intermediate
(Scheme 3-4). [1988JOC38]
Scheme 3-4. Preparation of 1,3,4-oxadiazolinones
Important routes to monosubstituted oxadiazoles (3.34a), amino-oxidazoles
(3.34b), oxadiazolinones (3.35a), and oxadiazolinethiones (3.35b) involve reaction of
hydrazides R1CONHNH2 with triethylorthoformate, cyanogen bromide, phosgene, or
carbon disulfide (or CSCl2) respectively. Reaction of hydrazide (3.36) with
triethylorthoformate, or with CS2/KOH, allowed the synthesis of oxadiazole (3.37)
(Scheme 3-5). [1982MC793]

72
Scheme 3-5. 1,3,4-Oxadiazole ring synthesis from acyclic precursors
Dolman et. al. reported the synthesis of 2-amino-1,3,4-oxadiazoles (3.40) via
TsCl/Py-mediated cyclization of a thiosemicarbazide (3.39), which is readily prepared by
acylation of a given hydrazide (3.38) with the appropriate isothiocyanate (Scheme 3-6).
[2006JOC9548]
Scheme 3-6. Preparation of 2-amino-1,3,4-oxadiazoles
1,3,4-Oxadiazoles are most commonly prepared by the coupling of acylhydrazides
with carboxylic acids followed by a dehydration step. [2006JOC9548, 2006SC3287,
2006TL105, 2006TL4827, 2006TL6497, 2006T10223, 2007TL1549, 2007SC1201]

73
Rajapakse reported a mild and efficient one pot synthesis of 2,5-disubstituted 1,3,4-
oxadiazoles (3.41) in good yield (Scheme 3-7), from the cyclization-oxidation reaction of
acylhydrazones. Also, the synthesis was achieved by condensation of acyl hydrazides
and aromatic aldehydes in the presence of ceric ammonium nitrate in dichloromethane.
However, the conjugation of the carboxylic acid partner with -functionality such as a
styryl group gave a very low yield of 1,3,4-oxadiazoles. Moreover, incorporation of
nucleophilic functionality such as a pyridine (3.42) or phenol (3.43) moiety on the acid
partner was not feasible and the corresponding 1,3,4-oxadiazoles could not be
obtained. [2006TL4827]
Scheme 3-7. One-pot syntheses of unsymmetrical 2,5-disubstituted 1,3,4-oxadiazoles
N-Acylbenzotriazoles are easily prepared from activated derivatives of carboxylic
acids [2005SL1656] and have been applied to (i) N-acylation, (ii) O-acylation,
[2006S4135] (iii) C-acylations, [2006TL3767] [2005JOC4993] [2005JOC7792,
2005ARKIVOC329] syntheses of (iv) peptides, [2006S411, 2006MI37, 2006MI42,
2006MI326, 2007JOC407, 2007JOC4268, 2007BC994] (v) esters, [2006JOC3364] (vi)
benzodioxin-4-ones, [2007ARKIVOC6] (viii) ketones, [2006JOC9861] (xi) acyl azides,

74
[2007JOC5802] (xiii) heteroaromatics [2000JOC8069] and (xiv) heterocycles.
[2004JOC9313] Compared with acid chlorides, N-acybenzotriazoles in general showed
better functional group tolerance, ease of reaction conditions for many types of coupling
reactions, especially for constructing N-C bonds. A series of N-acybenzotriazoles
(3.45a-h) were reacted with phenylhydrazide (3.44) toward the syntheses of 2.5-
disubsituted-1,3,4-oxadiazoles (3.46a-h) (Scheme 3-6). The results are discussed in
the next section. [2008ARKIVOC62]
3.2 Results and Discussion
Reaction of (E)-1-benzotriazol-1-yl-3-phenylpropenone (3.45a) (0.5 mmol) with
benzoic acid hydrazide (3.44) (0.5 mmol) and sodium hydride (1 mmol) in
dichloromethane at RT for 12 h followed by treatment with CBr4 (1 mmol) and Ph3P (1
mmol) at RT for 12 h gave 2-phenyl-5-((E)-styryl)-1,3,4-oxadiazole (3.46a) in 84% yield
(23% yield in [2006TL4827]). The 1H NMR spectra of (3.46a) showed the
disappearance of the Bt signals in the aromatic region, indicating the loss of the
benzotriazolyl group during the reaction. The 13C NMR spectra of (3.46a) showed two
signals at 164.5 and 164.2 ppm corresponding to the two C=N functions of the product
and the disappearance of the signal at 168.8 ppm belonging to the carbonyl group at
the position of the benzotriazolyl group in the starting material. Thus, a series of
reactions of benzoic acid hydrazide with a range of N-acylbenzotriazoles (3.45a-h) were
explored to test the generality of this method. The results are shown in Table 3-1.
Reaction of heteroaryl-α,β-unsaturated acylbenzotriazoles such as (E)-1-
benzotriazol-1-yl-3-thiophen-2-ylpropenone (3.45b) and (E)-1-benzotriazol-1-yl-3-furan-
2-ylpropenone (2c) with benzoic acid hydrazide furnished novel 2-phenyl-5-((E)-2-

75
thiophen-2-yl-vinyl)-1,3,4-oxadiazole (3.46b) and 2-((E)-2-furan-2-yl-vinyl)-5-phenyl-
1,3,4-oxadiazole (3.46c) in 82% and 79% yield respectively. Similarly, reaction of 1-
benzotriazol-1-yl-3-phenylpropynone (3.45d) and benzotriazol-1-yl-naphthalen-2-yl-
methanone (3.45e) with benzoic acid hydrazide produced novel 2-phenyl-5-
phenylethynyl-1,3,4-oxadiazole (3.46d) and 2-(5-phenyl-1,3,4-oxadiazol-2-yl)-
naphthalen-1-ol (3.46e) in 73% and 76% yield respectively (Table 3-1).
Further reaction of hydroxyaryl acylbenzotriazoles including benzotriazol-1-yl-(2-
hydroxy-3-methyl-phenyl)-methanone (3.45f), 1H-benzotriazol-1-yl(1-hydroxy-2-
naphthalenyl)-methanone (3.45g) and 1H-benzotriazol-1-yl(1-hydroxy-4-bromo-2-
phenyl)methanone (3.45h) gave 2-methyl-6-(5-phenyl-1,3,4-oxadiazol-2-yl)-phenol
hydrochloride (3.46f), 2-(5-phenyl-1,3,4-oxadiazol-2-yl)-naphthalen-1-ol (3.46g) and
novel 4-bromo-2-(5-phenyl-1,3,4-oxadiazol-2-yl)-phenol (3.46h) in 86%, 66% and 89%
yields respectively (Table 3-1).
Scheme 3-8. 1,3,4-Oxadiazoles from N-acylbenzotriazoles
3.3 Conclusion
A convenient route has been developed from N-acylbenzotriazoles and acyl
hydrazides for the one pot synthesis of 1,3,4-oxadiazoles incorporating a -functionality
or a nucleophilic group in the side chain, most of which are not easily accessible by
previous methods.

76
Table 3-1. Reaction of N-acylbenzotriazoles with benzoic acid hydrazide Entry
Product
Product Structure
Yielda (%)
1 3.46a N N
OPh
Ph
84b
2 3.46b
N N
OPh
S
82
3 3.46c
N N
OPh
O
79
4 3.46d
NN
O
Ph
Ph
73
5 3.46e
NN
O
Ph
OH
76
6 3.46f
N N
OPh
OH
Me
86
7 3.46g O
NNPh
OH
66
8 3.46h O
NNPh
OH
Br
89
a Isolated yields after column purification and determined from a single experiment. b 23% [2006TL4827]
3.4 Experimental Section
Melting points were determined on a hot-stage apparatus and are uncorrected. 1H
(300 MHz, with TMS as the internal standard) and 13C NMR (75 MHz) NMR spectra

77
were recorded in CDCl3. Elemental analysis was carried out in an Eager 200 CHN
analyzer.
3.4.1 General Procedure for the Preparation of 1,3,4-Oxadiazole
To a solution of N-acylbenzotriazole (3.45a-h) (0.5 mmol) and benzoic acid
hydrazide (3.44) (68 mg, 0.5 mmol) in dichloromethane (5 mL) at RT was added sodium
hydride (60% in mineral oil, 40 mg, 1 mmol). The coupling was allowed to proceed at
RT for 12 h, then CBr4 (332 mg, 1 mmol) and Ph3P (262 mg, 1 mmol) were added in one
portion. The dehydration step was allowed to proceed at RT for 12 h and the reaction
was poured onto a silica gel column for purification (silica gel, 10-15% EtOAc/hexanes)
to afford 1,3,4-oxadiazoles (3.46a-h) in 66-89% yield.
2-Phenyl-5-((E)-styryl)-1,3,4-oxadiazole (3.46a)
N N
OPh
Ph
White microcrystals; yield: 104 mg (84%); m. p. 125–127 oC (lit. m. p. 128–130 oC
[2006TL4827]); 1H NMR (300 MHz, CDCl3) : 8.14–8.12 (m, 2H), 7.64 (d, J = 16.9 Hz,
1H), 7.58–7.54 (m, 5H), 7.44–7.42 (m, 3H), 7.12 (d, J = 16.5 Hz, 1H); 13C NMR (75
MHz, CDCl3) : 164.5, 164.2, 139.1, 135.0, 132.0, 130.2, 129.3, 129.2, 127.7, 127.2,
124.0, 110.2.
2-Phenyl-5-((E)-2-thiophen-2-yl-vinyl)-1,3,4-oxadiazole (3.46b)
N N
OPh
S
Yellow microcrystals; yield: 104 mg (82%); m. p. 110–114 oC; 1H NMR (300 MHz,
CDCl3) : 8.13 (d, J = 1.8 Hz, 1H), 8.11 (d, J = 2.7 Hz, 1H), 7.75 (d, J = 16.2 Hz, 1H),

78
7.55–7.53 (m, 3H), 7.41 (d, J = 5.1 Hz, 1H), 7.30 (d, J = 3.6 Hz, 1H), 7.10 (dd, J =5.1,
3.7 Hz , 1H), 6.91 (d, J = 16.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) : 164.2, 164.2,
140.3, 132.0, 131.8, 130.0, 129.3, 128.4, 128.2, 127.2, 124.1, 109.1. Anal. Calcd for
C14H10N2OS: C, 66.12; H, 3.96; N, 11.02. Found: C, 66.01; H, 3.85; N, 10.95.
2-((E)-2-Furan-2-yl-vinyl)-5-phenyl-1,3,4-oxadiazole (3.46c)
White microcrystals; yield: 94 mg (79%); m. p. 115–117 oC (lit. m. p. 118–119 oC
[1995CHC208]); 1H NMR (300 MHz, CDCl3) : 8.11 (d, J = 1.8Hz, 1H), 8.08 (d, J = 2.6
Hz, 1H), 7.54–7.47 (m, 4H), 7.39 (d, J = 16.2 Hz, 1H), 6.97 (d, J = 16.2 Hz, 1H), 6.62 (d,
J = 3.3 Hz, 1H), 6.50 (dd, J = 3.3, 1.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) : 164.4,
164.1, 155.2, 144.7, 131.9, 129.2, 127.1, 125.7, 124.0, 113.9, 112.5, 107.8. Anal. Calcd
for C14H10N2O2: C, 70.58; H, 4.23; N, 11.76. Found: C, 70.36; H, 4.25; N, 11.81.
2-Phenyl-5-phenylethynyl-1,3,4-oxadiazole (3.46d)
White microcrystals; yield: 94 mg (73%); m. p. 129–130 oC; 1H NMR (300 MHz,
CDCl3) : 8.13–8.10 (m, 2H), 7.68–7.65 (m, 2H), 7.60–7.40 (m, 6H); 13C NMR (75 MHz,
CDCl3) : 165.1, 151.0, 132.6, 132.4, 130.9, 129.4, 128.9, 127.4, 123.6, 120.0, 97.4,
73.3. Anal. Calcd for C16H10N2O: C, 78.03; H, 4.09; N, 11.38. Found: C, 77.75; H, 4.07;
N, 11.28.

79
2-(5-Phenyl-1,3,4-oxadiazol-2-yl)naphthalen-2-ol (3.46e)
White microcrystals; yield: 219 mg (76%); m. p. 196–198 oC; 1H NMR (300 MHz,
CDCl3) : 11.13 (bs, 1H), 8.48 (d, J = 7.7 Hz, 1H), 8.18–8.16 (m, 2H), 7.84–7.80 (m,
2H), 7.63–7.56 (m, 5H), 7.47 (d, J = 8.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) : 165.1,
163.2, 156.2, 136.2, 132.2, 129.4, 129.3, 129.1, 127.8, 127.2, 127.1, 126.4, 124.9,
123.9, 123.6, 121.8, 120.1, 101.4. Anal. Calcd for C18H12N2O2: C, 74.99; H, 4.20; N,
9.72. Found: C, 74.72; H, 4.00; N, 9.89.
2-Methyl-6-(5-phenyl-1,3,4-oxadiazol-2-yl)phenol hydrochloride (3.46f)
White microcrystals; yield: 125 mg (86%); m. p. 255–256 oC; 1H NMR (300 MHz,
CDCl3) : 10.91 (bs, 1H), 10.66 (bs, 1H), 7.97 (d, J = 7.0 Hz, 2H), 7.84 (d, J = 7.7 Hz,
1H), 7.66–7.55 (m, 4H), 7.42 (d, J = 7.1 Hz, 1H), 6.89 (t, J = 7.7 Hz, 1H), 2.22 (s, 3H);
13C NMR (75 MHz, CDCl3) : 169.8, 165.7, 159.2, 135.1, 132.1, 132.0, 128.5, 127.4,
126.1, 124.5, 118.1, 111.9, 15.4. Anal. Calcd for C15H13ClN2O2: C, 62.40; H, 4.54; N,
9.70. Found: C, 63.86; H, 5.02; N, 9.89.
2-(5-Phenyl-1,3,4-oxadiazol-2-yl)naphthalen-1-ol (3.46g)

80
Pale green microcrystals; yield: 190 mg (66%); m. p. 196–198 oC; 1H NMR (300
MHz, CDCl3) : 11.13 (bs, 1H), 8.48 (d, J = 7.7 Hz, 1H), 8.18-8.16 (m, 2H), 7.84-7.80
(m, 2H), 7.63-7.56 (m, 5H), 7.47 (d, J = 8.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) :
165.1, 163.2, 156.2, 136.2, 132.2, 129.4, 129.3, 129.1, 127.8, 127.2, 127.1, 126.4,
124.9, 123.9, 123.6, 121.8, 120.1, 101.4. Anal. Calcd for C18H12N2O2: C, 74.99; H, 4.20;
N, 9.72. Found: C, 74.72; H, 4.00; N, 9.89.
4-Bromo-2-(5-phenyl-1,3,4-oxadiazol-2-yl)phenol (3.46h)
Off-white microcrystals; yield: 282 mg (89%); m. p. 146–148 oC; 1H NMR (300
MHz, CDCl3) : 10.15 (bs, 1H), 8.08 (d, J = 6.6 Hz, 2H), 7.87 (d, J = 2.2 Hz, 1H), 7.57-
7.44 (m, 4H), 6.98 (d, J = 8.9 Hz, 1H); 13C NMR (75 MHz, CDCl3) : 163.6, 163.1, 156.7,
136.4, 132.5, 129.3, 128.7, 127.2, 123.0, 119.6, 111.7, 109.7. Anal. Calcd for
C14H9BrN2O2: C, 53.02; H, 2.86; N, 8.83. Found: C, 52.69; H, 2.79; N, 8.54.

81
CHAPTER 4 OVERVIEW OF N-HYDROXYAMIDOXIMES, N-AMINOAMIDOXIMES AND
HYDRAZIDINES
4.1 Introduction
N-Hydroxyamidoximes (4.2), N-aminoamidoximes (4.3) and hydrazidines (4.4) all
belong to the class of compounds with the general formula RC=NX(NHY) derived from
the generic structure (4.1), where X = OH or NH2, Y = OH or NH2 and R is a linear side
chain, carbocycle residue or heterocycle residue (Figure 4-1). Compound (4.2, 4.3 and
4.4) can all be considered as amidines in which one of the hydrogen atoms of the imido
group is replaced by a hydroxy or amino radical, and the amine group is replaced by a
hydroxylamine or hydrazine group. Their structures are thus similar to those of
amidoximes and amidrazones, but they possess very different synthetic utility and
pharmacological applications. Reviews published on the synthetic and biological
applications of amidrazones and amidoximes [1962CR155, 1970CR151, 1989CHC717,
2008CPD1001] do not cover N-hydroxyamidoximes, N-aminoamidoximes and
hydrazidines and their preparative methods, synthetic utility and biological applications.
The following attempts to reddress the situation in a general and comprehensive review
of the structure, synthesis and applications of these three classes of compounds.
N-Hydroxyamidoximes are derivatives of amidoximes and amidines and used as
intermediate building blocks for the construction of heterocycles; [1955HCA1560] from
the limited number of N-hydroxyamidoximes documented in the literature,
representative (4.2a-f, 4.5, 4.6) are shown in Figure 4-2 and together with two examples
of their still rarer O-substituted derivatives (4.7, 4.8)
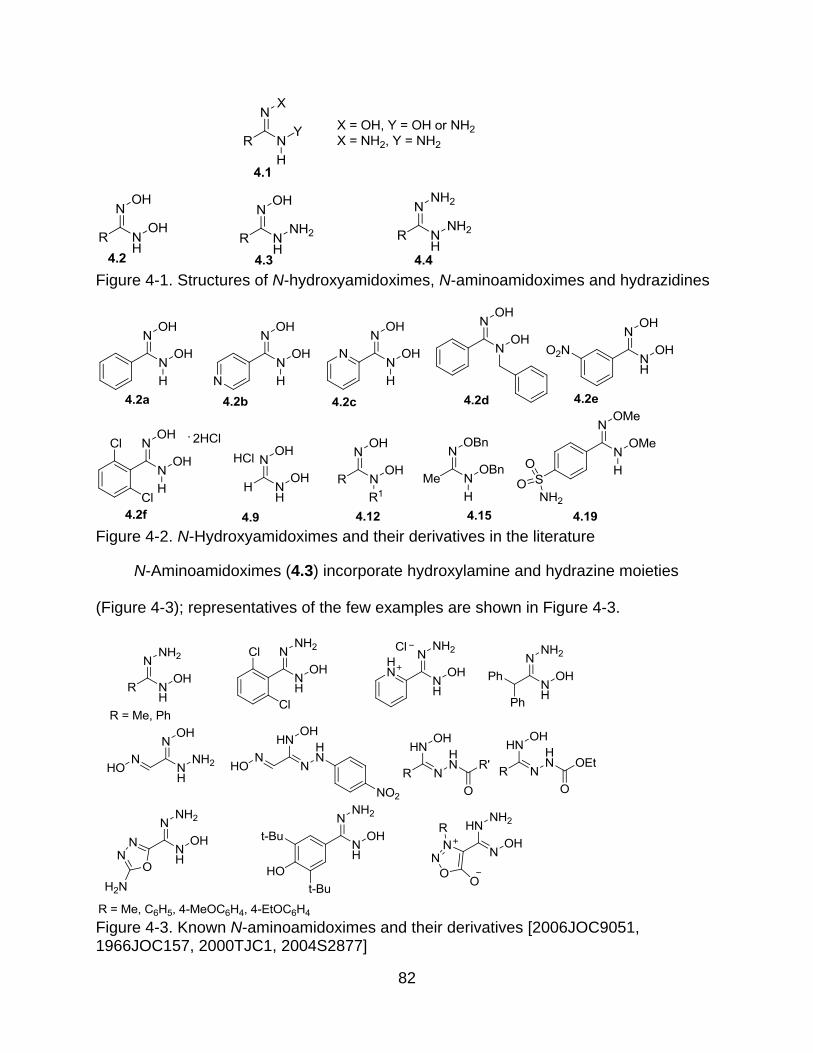
82
Figure 4-1. Structures of N-hydroxyamidoximes, N-aminoamidoximes and hydrazidines
Figure 4-2. N-Hydroxyamidoximes and their derivatives in the literature
N-Aminoamidoximes (4.3) incorporate hydroxylamine and hydrazine moieties
(Figure 4-3); representatives of the few examples are shown in Figure 4-3.
Figure 4-3. Known N-aminoamidoximes and their derivatives [2006JOC9051, 1966JOC157, 2000TJC1, 2004S2877]

83
Hydrazidines form a class of chemical compounds with the general formula
RC(NHNH2) =NNH2 (4.4) (Figure 4-4), and are derived from carboxylic acids by
replacing – OH with – NHNH2 (or N-substituted analogues) and =O with =NNH2 (or N-
substituted analogues). Hydrazidines are alternatively denoted as hydrazide-
hydrazones, dihydroxyformazans and N-aminoamidrazones. We located a total of 57
structures have been reported for diverse R groups in the acyclic (I) and (II) types
(Figure 4-4). Many more examples are known of hydrazidine moieties as part of a
heterocycle: e. g., there are 24 examples of imidazole (III), benzimidazole (IV) and
triazole (V) analogues, and 33 examples of type (I) and substituted hydrazidines (II), but
these heteocycles are outside of the scope of the present review. The preparative
methods, chemistry and applications of acyclic hydrazidines and their derivatives are
summarized in this review.
Figure 4-4. Hydrazidines and their derivatives
4.2 Structure and Configuration
4.2.1 N-Hydroxyamidoximes
N-Hydroxyamidoximes (4.2) are sometimes named as N,N'-dihydroxyimidamides
or oxyamidoximes. [1962CR155] Systematic studies reported with respect to
configurations or conformations of the many classes of N-hydroxyamidoximes are so far
limited to N-hydroxybenzamidoxime (4.2a). Clement et. al. studied and compared

84
chemical shifts and coupling constants J ( 15N, 1H) of several amidoximes with N-
hydroxyamidoxime (4.2a) via 15N NMR. As observed by the 15N NMR, benzamidoxime
(4.24) exists only in the form of an oxime with no other tautomer detected, but N-
hydroxybenzamidoxime (4.2) exists in two tautomeric forms in which a rapid equilibrium
exists between (4.2a) and (4.2b) (Figure 4-2). Two 15N signals were detected: an oxime
type nitrogen and a hydroxylamine type nitrogen. No NH coupling was observed due to
the rapid tautomerization. [1985CB3481, 2007JMC6730] Barassin et. al. studied the
configuration and conformation of N-hydroxybenzamidoxime and found that the Z-
configuration (4.2Z) is favored energetically over configuration (4.2E), and conformation
(4.2c) is the predominant form due to the stabilization by hydrogen bonding (Figure 4-
5). [1969BSCF3409] This is in agreement with the calculation results by Chem3D MM2.
The minimized total energy (-3.0061 kcal/mol) for structure (4.2Z) is much lower than
that of structure (4.2E), which is 5.6273 kcal/mol.

85
Figure 4-5. Tautomerization, conformation and configuration of N-hydroxyamidoxime
4.2.2 N-Aminoamidoxime
In 1910, Wieland first synthesized N-aminobenzamidoxime (4.3a, 4.3b) from
benzohydroxamyl chloride and hydrazine hydrate, and named them as hydrazide
oximes. [1910Ber4199] To the best of our knowledge, there are no studies in the
literature related to the configuration or conformation of any N-aminoamidoximes, but
most papers depict them as structure (4.3a) rather than (4.3b). Again, based on the
energy minimizing calculations via Chem3D MM2, structure (4.3a) has lower total
energy (-2.7554 kcal/mol) than that of structure (4.3b) (-0.5433 kcal/mol) (Figure 4-6).
Figure 4-6. Configuration of N-aminoamidoximes
4.2.3 Hydrazidines
To the best of our knowledge, there is literature data on the structure and
configuration of hydrazidines. Chem3D MM2 energy minimizing calculations, however
found that structure (4.4b) is considerably less stable than (4.4a) (Figure 4-7).
Figure 4-7. Configuration of hydrazidines

86
4.3 Preparative Methods
4.3.1 N-Hydroxyamidoximes and Their Derivatives
4.3.1.1 From oximidoyl chlorides and hydroxyamines
N-Hydroxybenzamidoxime (4.2a) is commonly prepared from oxyimidoyl chlorides
(4.6) and hydroxylamine via the route shown in Scheme 4-1. [1980JOC3916,
1914Ber2938, 1898Ber2126] The reaction of hydroxylamine with benzaldehyde gave
benzaldoxime as the intermediate; further reaction with N-chlorosuccinimide (NCS) in
DMF gave α-chlorobenzaldoxime (4.6a), and subsequent reaction with hydroxylamine
gave N-hydroxybenzamidoxime (4.2a) (Scheme 4-1). Ley and Ulrich synthesized
compound (4.2d) and (4.2e) (Figure 4-8) in the same manner. [1914Ber2941]
Scheme 4-1. Preparation of N-hydroxybenzamidoxime
Huether et. al. prepared N-hydroxypyridylamidoxime (4.2b, 4.2c) from the
corresponding pyridyl-oxyimidoylchlorides (4.6b, 4.6c) by reaction with excess
hydroxylamine in methanol (Scheme 4-2). [1963JCED624]
Scheme 4-2. Preparation of N-hydroxypyridylamidoximes

87
Figure 4-8. N-Hydroxybenzamidoxime derivatives
Johannes et. al. synthesized alpha-hydroxylamine-2,6-dichloro-N-
hydroxybenzaldoxime hydrochloride (4.2f) from the corresponding benzaldoxime
chloride derivative (4.6f) (Scheme 4-3). [1966US3234255A]
Scheme 4-3. Preparation of 2,6-dichloro-N-hydroxybenzaldoxime hydrochloride salt
4.3.1.2 From amidoximes and hydroxyamine
Armand and Minvielle prepared formic N-hydroxyamidoxime hydrochloride (4.9)
from formic amidoxime (4.8) and hydroxylamine hydrochloride (4.7) (Scheme 4-4).
[1965CR2512]
Scheme 4-4. Preparation of formic hydroxyamidoxime hydrochloride salt
4.3.1.3 From nitrile oxides and hydroxyamines
In a different approach, Aurich et. al. reacted nitrile oxide (4.11) with N-substituted
hydroxylamine (4.10) to afford N-substituted hydroxyamidoxime (4.12). Nitrile oxides
(4.11a-c) react with hydroxylamines (4.10a-b) to give N2-hydroxyamidinyl N1-oximes
(4.12a-d), namely N-hydroxyamidoxime in 45-68% yield (Scheme 4-5). [1975CB2764]

88
Scheme 4-5. Synthesis of N-hydroxyamidoximes from nitrile oxides
4.3.1.4 Miscellaneous preparative methods for di-O-alkyl derivatives of N-hydroxyamidoximes
Benzotriazole methodology has been used to prepare N–hydroxymethylamidoxime
derivatives. Katritzky and his coworkers prepared compound (4.15), a di-O-benzyl
derivative of N-hydroxymethylamidoxime by the reaction of 1H-1,2,3-benzotriazol-1-
ylmethanone oxime (4.13) with benzyloxyhydroxylamine (4.14) under microwave
radiation (Scheme 4-6). [2006JOC9051]
Scheme 4-6. Preparation of di-O-benzyl derivative of N-hydroxymethylamidoxime
Treatment of an alcoholic solution of p-sulfamidobenzimidate hydrochloride salt
(4.16) with O-methylhydroxylamine (4.17) under pressure gave two products (4.18) and
(4.19), a di-O-methylsubstituted p-sulfamido-N-hydroxybenzamidoxime (Scheme 4-7).
[1962CR155]

89
Scheme 4-7. Synthesis of di-O-methylsubstituted p-sulfamido-N-
hydroxybenzamidoximes
4.3.2 N-Aminoamidoximes and Their Derivatives
4.3.2.1 From oxime chlorides or amidoximes
Previous preparations of aminoamidoximes include the reactants of oxime
chlorides (4.6, X=Cl) or simple amidoximes (4.6, X=NH2) with hydrazines (4.20) to give
aminoamidoximes (4.21) in 21-30% yield (Scheme 4-8). [1980CRS304, 1981PJC1253]
Scheme 4-8. General route to N-aminoamidoximes
4.3.2.2 From oximebenzotriazoles and hydrazines
N-Amino-N´-nitrophenyl benzamidoxime (4.23) was prepared by Katritzky et. al.
by the reaction of 1H-1,2,3-benzotriazol-1-ylmethanone oxime (4.7) with hydrazine
(4.22) under microwave radiation in 71% yield and isolated as a viscous oil (Scheme 4-
9). [2006JOC9051]
Scheme 4-9. Synthesis of N-amino-N´-nitrophenyl benzamidoxime

90
4.3.2.3 From N-hydroxyimidates and hydrazides
Bel Hadj and Baccar prepared N-(Ethoxycarbonyl)amide-N-
hydroxybenzamidoximes (4.26) by the reaction of hydrazide (4.25) with ethyl N-
hydroxybenzimidate (4.24) in 98% yield (Scheme 4-10). [1986JSCT9]
Scheme 4-10. Preparation of N-(ethoxycarbonyl)amide benzamidoxime
4.3.2.4 From oxyimidoylchlorides and hydrazines
The reaction of 1,2,3-oxadiazolium carbohydrazimic chloride (4.27) with hydrazine
gave N-aminoamidoxime derivative (4.28) (Scheme 4-11). [2004S2877] Hydrazino(3-
arylsydnon-4-yl)methanone oximes (4.28) are good precursors for the synthesis of
triazolyl sydnones (4.69a-f) (Scheme 4-24), some of which have important
pharmacological activities, such as antimicrobial, anti-inflammatory, analgesic and
antipyretic properties. [2004S2877]
Scheme 4-11. Preparation of 3-(3-arylsydnon-4-yl)triazole derivatives

91
4.3.2.5 From hydrazide imidate and hydroxyamine
Ikizler et. al. prepared a series of hydroxamic acid ethoxycarbonylhydrazides
(4.30) by reaction of hydrazide imidate (4.29) with hydroxylamine (Scheme 4-12).
[1992MC257]
Scheme 4-12. Preparation of hydroxamic acid ethoxycarbonylhydrazides
4.3.3 Hydrazidines
4.3.3.1 From imidate salts and hydrazines
When excess hydrazine (4.32a) was added to aliphatic imidate salt (4.31) (R =
alkyl) under anhydrous conditions at temperatures below 0 ˚C, hydrazidine (4.4) was
isolated, while at elevated temperatures (40-50 ˚C) other cyclic by-products were
produced (Scheme 4-13). [1931MC106, 1976T1031]
Scheme 4-13. Synthesis of aliphatic hydrazidines
The use of monosubstituted hydrazines (4.32) reduces the number of by-products,
and reacts smoothly with imidate salts (4.31) in alcohol at room temperature. The main
products are N-substituted amidrazones when equimolar quantities of the reactants are
used, but substituted formazans (4.33) are obtained when excess hydrazine (4.32) is
employed (Scheme 4-14). [1884Ber182, 1954JCS3319, 1955JPSJ726, 1956JCS2853,
1955CRV355]

92
Scheme 4-14. Synthesis of substituted formazans
4.3.3.2 From amidoximes and hydrazines
In the only reaction located between an amidoxime (4.34) and phenylhydrazine
(4.32b), Bamberger used excess phenylhydrazine and isolated the product as
triphenylformazan (4.35) (Scheme 4-15). [1894Ber160]
Scheme 4-15. Synthesis of triphenylformazan
4.3.3.3 From amidrazones and hydrazines
The reaction of the amidrazone hydrochlorides (4.36a-d) with anhydrous
hydrazine (4.32a) at 40 oC gives hydrazidine hydrochlorides (4.37a-d) in 40-98% yields.
(Scheme 4-16). [1972LAC16, 1975LAC1120]
Scheme 4-16. Synthesis of hydrazidine hydrochlorides
Kurzer and Douraghi-Zadeh obtained phenylaminohydrazidine (4.39) similarly via
the reaction of isothiosemicarbazide / amidrazone (4.38) with hydrazine (4.32a) at low
temperature. The triazole (4.40) was formed as a by-product in this hydrazinolysis when
the temperature was above 40 oC (Scheme 4-17). [1967JCS(C)742]

93
Scheme 4-17. Synthesis of diaminoguanidine / amino-hydrazidine
4.3.3.4 From diethoxy-N,N-dimethylethanamine and hydrazides
Glushkov et. al. prepared hydrazidine derivative (4.44) from 1,1-diethoxy-N,N-
dimethylethanamineacetyle (4.42) and isonicotinylhydrazide (INH) (4.41), known as
Isoniazid, a medication in the prevention and treatment of antituberculosis. [2004KFZ16]
The first step forms the amidine derivative (4.43), which was derivatized further to the
hydrazidine hydrochloride salt derivative (4.44) via the reaction with another equivalent
of (4.41) in refluxing acid-ethanol solution (Scheme 4-18).
Scheme 4-18. Synthesis of hydrazidine derivatives
4.3.3.5 From hydrazonyl bromides and hydrazines
Takahashi et. al. reported that the reaction of hydrazonyl bromide (4.45a-f) and
hydrazine hydrate in alcohol formed hydrazidines (4.46a-f). The Reaction of hydrazonyl
bromide (4.45a-g) with benzoylhydrazines (4.47a-d) at room temperature can yield
benzoylbenzohydrazide hydrazones (4.48a-g), which can further cyclize to N-
aminotriazoles (4.49a-g) upon heating in acetic acid (Scheme 4-19). [1977BCSJ953]

94
Scheme 4-19. Synthesis of hydrazidines from hydrazonyl bromide
4.3.3.6 From triazines
Grundmann discovered that s-triazine (4.50) reacted with dimethylhydrazine (4.51)
or hydrazine (4.53) to give hydrazidine (4.52) or amidrazone (4.54) depending on the
hydrazines used (Scheme 4-20). [1963ACIEE309]
Scheme 4-20. From triazine to hydrazidines
4.4 Chemistry and Reactions
4.4.1 N-Hydroxyamidoximes
4.4.1.1 Reduction of N-hydroxyamidoximes
Ley and Ulrich showed that N-hydroxybenzamidoxime (4.2a) may be reduced by
sulfur dioxide to benzamidoxime (4.34) (Scheme 4-21). [1914Ber2941]

95
Scheme 4-21. Conversion of N-hydroxybenamidoxime into benzamidoxime
4.4.1.2 Oxidation of N-hydroxyamidoximes
Armand and Minvielle also found that formic hydroxyamidoxime (4.55), which is
amphoteric, can be oxidized by KIO4 to potassium salt of nitrosolic acid (4.56) (Scheme
4-22). [1965CR2512]
Scheme 4-22. Conversion of formic hydroxyamidoxime to its nitrosolic acid
Armand and Minvielle reported the periodate oxidation of N-
hydroxybenzamidoxime (4.2a) to benzonitrosolate salt characterized as the potassium
salt (4.58), which is a precursor for the synthesis of 3,5-diphenyl-1,2,4-oxadiazole (4.61)
(Scheme 4-23). [1965CR2512] Quadrelli and Caramella discovered that N-
hydroxybenzamidoxime (4.2a), on treatment with alkali, gave the azo-derivative (4.57)
which disproportionated to benzamidoxime (4.34) and the deep blue potassium salt of
benzonitrosolic acid (4.58). [2007COC959]
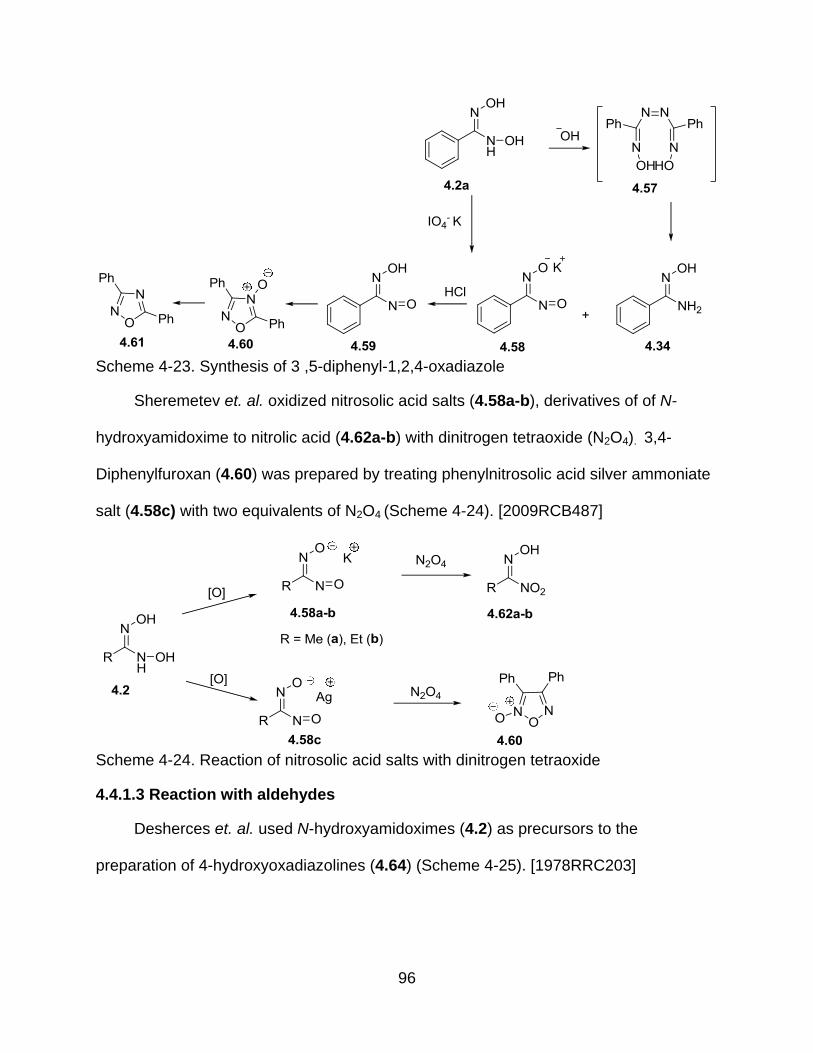
96
Scheme 4-23. Synthesis of 3 ,5-diphenyl-1,2,4-oxadiazole
Sheremetev et. al. oxidized nitrosolic acid salts (4.58a-b), derivatives of of N-
hydroxyamidoxime to nitrolic acid (4.62a-b) with dinitrogen tetraoxide (N2O4). 3,4-
Diphenylfuroxan (4.60) was prepared by treating phenylnitrosolic acid silver ammoniate
salt (4.58c) with two equivalents of N2O4 (Scheme 4-24). [2009RCB487]
Scheme 4-24. Reaction of nitrosolic acid salts with dinitrogen tetraoxide
4.4.1.3 Reaction with aldehydes
Desherces et. al. used N-hydroxyamidoximes (4.2) as precursors to the
preparation of 4-hydroxyoxadiazolines (4.64) (Scheme 4-25). [1978RRC203]

97
Scheme 4-25. Synthesis of 4-hydroxyoxadiazolines
4.4.1.4 Reaction with ketones
Desherces et. al. also found that (Z)-N-hydroxybenzamidoxime (4.2a) reacted with
benzophenone (4.65) to give hydroxamic acid (4.66) and benzophenone oxime (4.67)
(Scheme 4-26). [1978RRC203]
Scheme 4-26. Reaction of N-hydroxyamidoxime with benzophenone
4.4.2 N-Aminoamidoximes
4.4.2.1 Reaction with aldehydes
N-Aminoamidoxime (4.28), prepared as a precursor as shown in Scheme 4-11,
reacts with aromatic aldehydes (4.68) in acetonitrile in the presence of a suitable
quantity of concentrated sulfuric acid to afford the desired 3-sydnonyl triazoles (4.69a-f)
in 40-63%, as depicted in Scheme 4-27. [2004S2877] The reactions of hydrazino(3-
phenylsydnon-4-yl)methanone oxime (4.28a) with aliphatic aldehydes including hexanal
(4.68a), heptanal (4.68b) and cyclohexanecarboxaldehyde (4.68c) gave 5-alkyl-3-(3-
arylsydnon-4-yl)-1H-[1,2,4]triazoles (4.69a-c) (Scheme 4-27). [2004S2877]
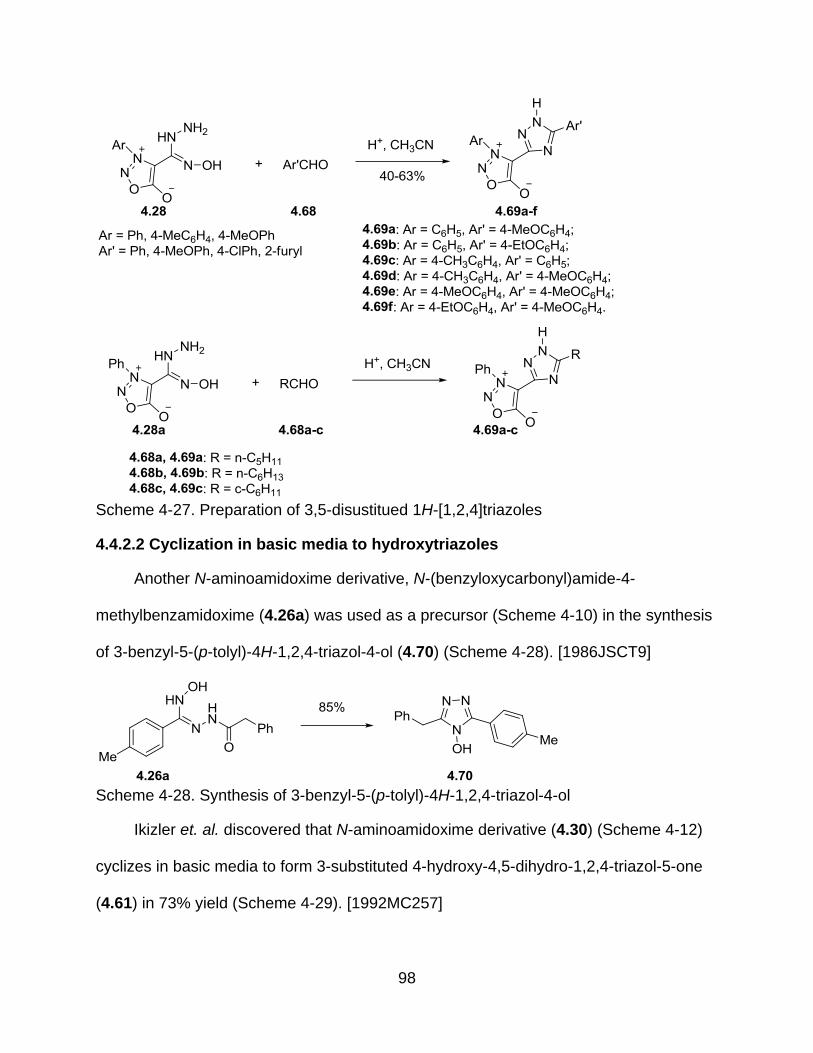
98
Scheme 4-27. Preparation of 3,5-disustitued 1H-[1,2,4]triazoles
4.4.2.2 Cyclization in basic media to hydroxytriazoles
Another N-aminoamidoxime derivative, N-(benzyloxycarbonyl)amide-4-
methylbenzamidoxime (4.26a) was used as a precursor (Scheme 4-10) in the synthesis
of 3-benzyl-5-(p-tolyl)-4H-1,2,4-triazol-4-ol (4.70) (Scheme 4-28). [1986JSCT9]
Scheme 4-28. Synthesis of 3-benzyl-5-(p-tolyl)-4H-1,2,4-triazol-4-ol
Ikizler et. al. discovered that N-aminoamidoxime derivative (4.30) (Scheme 4-12)
cyclizes in basic media to form 3-substituted 4-hydroxy-4,5-dihydro-1,2,4-triazol-5-one
(4.61) in 73% yield (Scheme 4-29). [1992MC257]

99
Scheme 4-29. Synthesis of 3-phenyl-4-hydroxy-4,5-dihydro-1,2,4-triazol-5-one
4.4.3 Hydrazidines
4.4.3.1 Reaction with aldehydes
Hydrazidines (4.4a-d) can react with benzaldehyde (4.68c) or can be used as
important synthetic auxiliaries for the synthesis of 4-amino-1,2,4-triazole hydrochlorides
(4.74a-e) by the reaction with triethoxyformate (4.72) (Scheme 4-30) [1975LAC1120]
Scheme 4-30. Synthesis of dibenzylidene hydrazidine 4-amino-1,2,4-triazole
hydrochloride
Neunhoeffer et. al. reported that the reaction of aromatic hydrazidines (4.4a) with
benzaldehyde (4.68c) gave noncyclic structure (4.77) in 79% yield as the product
(Scheme 4-31). [1992LAC115] Takahashi et. al. found that the oxidation of N-
benzylidene-N-(2-bromo-4-nitrophenyl)benzohydazidine (4.78a) formed from the
reaction of (4.75a) with aldehyde (4.76), with mercuric oxide (HgO) in refluxing ethanol
gave 4-amino-1,2,4-triazole (4.79), and 3-Alkyl and aryl-5-aryl-4-arylamino-1,2,4-

100
triazoles (4.79a-e) were prepared from N-aryl-N-arylmethylenehydrazidines (4.78a-e) in
28-75% yield in this manner. [1977BCSJ953]
Scheme 4-31. Reaction of hydrazidines with aldehydes
4.4.3.2 Reaction with anhydrides
Neunhoeffer et. al. reported that hydrazidines can react with anhydrides to
produce tetrazines (Scheme 4-32). [1979CB1981] The reaction of acetohydrazidine
(4.4a) with phthalaldehydic acid (4.87) can yield 3-methyl-1,10b-dihydro-1,2,4,5-
tetrazino[3, 2-a]isoindol-6(4H)-one) (4.88), which can be further converted to 3-methyl-
1,2,4,5-tetra-amino[3, 2-a]isoindol-6(4H)-one (4.92) upon mild oxidation. Compound
(4.92) can also be obtained by the reaction of (4.4a) with phthalic acid derivatives
(4.89), (4.90) and (4.91). The reaction of (4.4a) and nitrophthalic anhydride (4.80)
yielded two isomeric nitro-1,2,4,5-tetrazino [3, 2-a]- isoindol-6(4H)-ones (4.81a, 4.81b).
The reaction of acetohydrazidine (4.4a) with dichloromalealdehydic acid (4.82) gave
7,8-dichloro-3-methyl-1,8-dihydropyrrolo [1, 2-b]-1,2,4,5-tetrazine-6(4H)-one
hydrochloride (4.83). [1975CB3509] Other heterocycles (4.93, 4.94 and 4.95) were
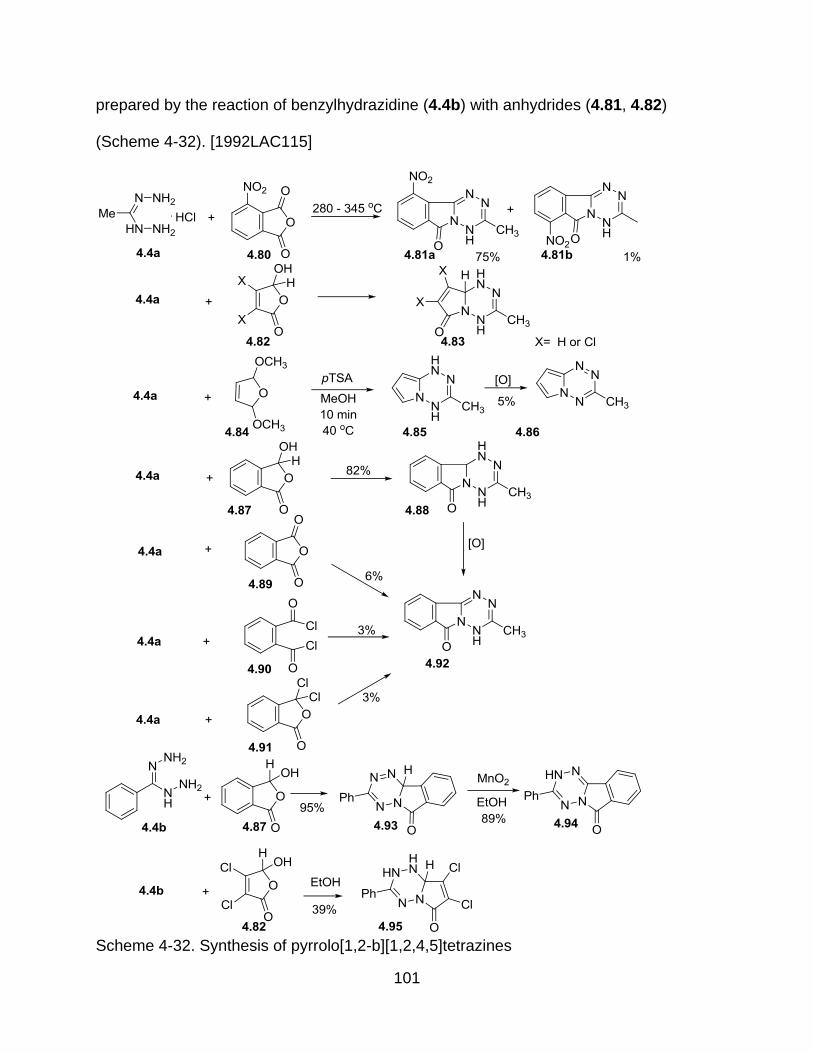
101
prepared by the reaction of benzylhydrazidine (4.4b) with anhydrides (4.81, 4.82)
(Scheme 4-32). [1992LAC115]
Scheme 4-32. Synthesis of pyrrolo[1,2-b][1,2,4,5]tetrazines

102
4.4.3.3 Reaction with diketones
Hydrazidines have been studied for the generation of different fused and nonfused
six-membered heterocyclic systems such as tetraphenylpyrazine (4.98) and 1,2,4-
triazines (4.101) (Scheme 4-33). The reaction of (4.4a) with benzoin (4.96) forms the
monocondensation product (4.97) first, then 2,3,5,6-tetraphenylpyrazine (4.98) upon
heating. The reaction of hydrazidine (4.4a) with benzil (4.99) gives preferentially 4-
amino-1,2,4-triazines (4.93). [1989LAC105] The reaction of (4.4a) with 4,4-dimethyl-1,2-
cyclopentandione (4.102) failed to produce cyclopentatriazine (4.104), but octaaza[14]-
annulen (4.103) was formed instead. Similarly, (4.4a) on reaction with diketone (4.105)
gave 14-membered structure octaazo-cyclotetradecin (4.106). (Scheme 4-33)
[1989LAC105] Neunhoeffer et. al. obtained three compounds (4.107, 4.108, 4.109) by
reaction of benzylhydrazidine (4.4a) with isophorone (4.102) (Scheme 4-33).
[1992LAC115]
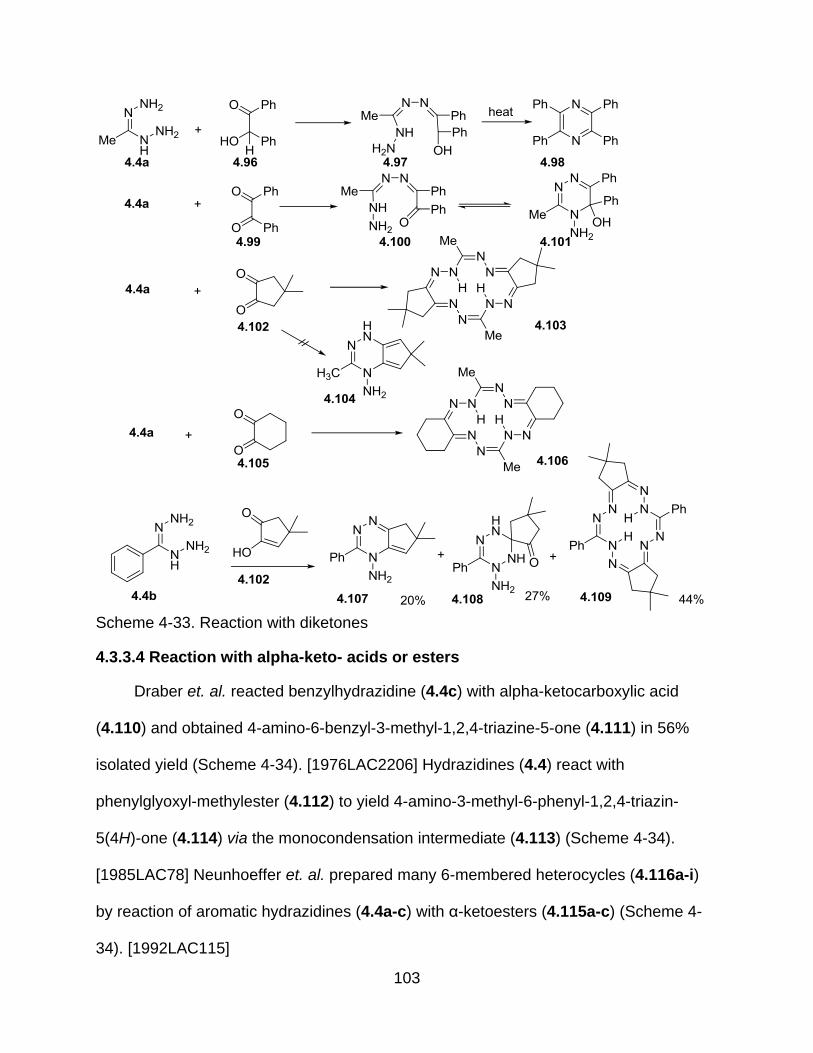
103
Scheme 4-33. Reaction with diketones
4.3.3.4 Reaction with alpha-keto- acids or esters
Draber et. al. reacted benzylhydrazidine (4.4c) with alpha-ketocarboxylic acid
(4.110) and obtained 4-amino-6-benzyl-3-methyl-1,2,4-triazine-5-one (4.111) in 56%
isolated yield (Scheme 4-34). [1976LAC2206] Hydrazidines (4.4) react with
phenylglyoxyl-methylester (4.112) to yield 4-amino-3-methyl-6-phenyl-1,2,4-triazin-
5(4H)-one (4.114) via the monocondensation intermediate (4.113) (Scheme 4-34).
[1985LAC78] Neunhoeffer et. al. prepared many 6-membered heterocycles (4.116a-i)
by reaction of aromatic hydrazidines (4.4a-c) with α-ketoesters (4.115a-c) (Scheme 4-
34). [1992LAC115]

104
Scheme 4-34. Syntheses of triazinones
4.4.3.5 Reaction with acylnitriles
The reaction of hydrazidines (4.4a-b) with benzoyl cyanide (4.117) give 4-amino-5-
imino-1,2,4-triazine (4.118), which is readily converted to triazinones (4.114) (Scheme
4-35). [1985LAC78]
Scheme 4-35. Reaction of hydrazidines with acylnitriles
4.4.3.6 Reaction with cyclopentadiene derivatives
Acetohydrazidine (4.4a) reacts with 2,3-dihydroxycyclo-pentadiene-1,4-
dicarboxylate-dimethylester (4.117a-b) to give 4-amino-4,6-dihydro-3-methyl-1H-
cyclopenta[e]1,2,4-triazin-5,7-dicarboxylester (4.118a-b). The reactions of (4.4a) with a

105
heteroaromatic systems such as 3,4-dihydroxy-2,5-furan dicarboxylate-dimethylester
(4.119a) or 3,4-dihydroxy-2,5-thiophenedicarboxylate-dimethylester (4.119b) gives
1,2,4-triazine (4.120a) and (4.120b). Likewise, 2,3- dihydroxy-5,5-dimethyl-1,3-
cyclopentadiene-1,4-dicarboxylate-dimethylester (4.119c) reacts with (4.4a) to form
(4.120c) as the major product (Scheme 4-36). [1989LAC105]
Scheme 4-36. Synthesis of 4-aminocyclopenta[e]-1,2,4-triazines
4.4.3.7 Reaction with diketoesters
The reaction of (4.4a) with dimethylester (4.121) yields diketone-triazine (4.122)
but in only 7% isolated yield. The reaction of (4.4a) with thioxamidyl methyl ester (4.123)
with triethylamine as base gives monocondensation product first, which cyclizes to
(4.124) upon heating. When (4.4a) is reacted with dimethyl acetylenedicarboxylate
(4.125) in MeOH in the presence of Et3N, crystalline pyrazolinone (4.126) was isolated
in 37% yield (Scheme 4-37). [1985LAC78]
The reaction of N-(2-bromo-4-nitrophenyl)benzohydrazidine (4.127a) with dimethyl
acetylenedicarboxylate (4.125) in tetrahydrofuran (THF) under reflux gives an orange
product, identified as 2,3,4,5-tetrahydro-1,2,4,5-tetrazine (4.128a). Other tetrahydro-
tetrazine derivatives (4.128a-e) can be prepared in a similar manner by heating the
mixture under reflux in THF (Scheme 4-37). [1977BCSJ953]

106
Scheme 4-37. Reaction of hydrazidines with diketoesters
4.4.3.8 Reaction with formic acid
3-Alkyl and arylamino-1,2,4-triazoles (4.131a and 4.131e) were first obtained upon
heating (4.127a, 4.127e) in formic acid. The reaction presumably proceeds via
formylated hydrazidine (4.130) to (4.131). However, this method produces many by-
products, and only (4.131f) and (4.131g) were reported as being isolated in pure form.
(Scheme 4-38). [1977BCSJ953]

107
Scheme 4-38. Reaction of hydrazidines with formic acid
4.3.3.9 Reaction with thioesters
S-Methylisothiocarbonohydrazide salt is used as a bis-aminoguanidine equivalent
in the synthesis of 6-aryl-3-aminotetrazines from dithio-p-benzoate esters (Scheme 4-
39). [1979JHC881] For example, dithio-p-benzoate esters (4.133) react with S-
methylisothiocarbonohydrazide hydroiodide (4.132) to form dihydrotetrazines (4.134)
which can be oxidized to (methylthio)tetrazines (4.135). The methylthio group serves to
deactivate the internal latent guanidine nitrogens for cyclization [1975JCS(PT1)1787]
and also to provide a handle for the subsequent amination to form 6-aryl-3-
aminotetrazines (4.136). [1977JHC587]
Scheme 4-39. Synthesis of unsymmetrically substituted 1,2,4,5-tetrazines

108
4.3.3.10 Reaction with hydrazine
Glushkov et. al. synthesized 3-methyl-6-pyridyl-1,2,4,5-tetrazine (4.138) by the
reaction of hydrazidine derivative (4.137) with hydrazine hydrate in methanolic media at
room temperature (Scheme 4-40). [2004KFZ16]
Scheme 4-40. Synthesis of 3-methyl-6-pyridyl-1,2,4,5-tetrazine
4.4.3.11 Reduction of hydrazidines
Bamberger et. al. discovered that ammonium sulfide in cold alcoholic solutions
reduced hydrazidines (4.139a) to amidrazones (4.140a) with amines (4.141a) as by-
products (Scheme 4-41). [1925LAC260]
Scheme 4-41. Reduction of formazans

109
Regitz and Eistert used phenylhydrazine to reduce formazan (4.139b) to
amidrazone (4.140b) at 50-100 oC (Scheme 4-41). [1963ibid3121] Hauser et. al. used
stannous chloride as a reducing agent to convert formazan (4.139) to its parent acid
(4.142a) and amines (4.141a-c), but the reaction did not give an amidrazone (Scheme
4-41). [1951CB651]
Jerchel et. al. studied the stepwise hydrogenation of tetrazolium salt (4.143) to
formazans (4.139c). [1950LAC185, 1957ibid191] Three successful methods of
reduction were reported: (i) hydrogenation using 5% palladium on barium sulfate, (ii)
Raney nickel in methanol, and (iii) sodium dithionite. The reduction process is shown in
Scheme 4-41. Hydrazidine (4.144) is only stable in solution and is oxidized back to the
formazan (4.139c) on exposure to air. Lithium aluminum hydride (LAH) has no effect on
triphenylformazan (4.139c) in ether-tetrahydrofuran (Et2O-THF) at room temperature
(RT) but cleaves it on boiling for several hours, giving the corresponding amidrazone
(4.140c). [1952CB470]
4.4.3.12 Condensation with α-halo ketones
Beyer et. al. reported that the reaction of α-bromo ketones (4.145) with N,N'-
diaminoguandine / aminohydrazidine (4.146) gave the condensation product (4.147)
(Scheme 4-42). [1968CB29]
Scheme 4-42. Reaction of α-halo ketones with hydrazidine amine

110
4.4.3.13 Miscellaneous reactions
Hydrazidine (4.148) may be readily oxidized to the blue-green free radical (4.149),
which is related to, but less stable than the cyclic verdazyl free radicals (4.150)
(Scheme 4-43). [1964ACIEE232, 1966MC517, 1968ACIEE489]
Scheme 4-43. Hydrazidine radical
Butler et. al. reported that the reaction of a hydrazidine derivative – polyhydrazine
triaminoguanidines with diketones gave hydrazidines. For example, on treatment of
triaminoguanidine nitrate (4.151) with acetylacetone (4.152), a complex reaction
occurred giving rise to products (4.153, 4.154 and 4.155), the proportions of which
varied with the conditions of the reaction. In the presence of sufficient (4.152), the
dipyrazolylmethylenehydrazono-derivative (4.155) is the main product, whereas at a
molar ratio of 1:2 for triaminoguanidine and acetylacetone, di-pyrazolylketone
hydrazone (4.154) is isolated in highest yield (Scheme 4-44). [1970JCS(C)2510]
Scheme 4-44. Reaction of hydrazine hydrazidine with acetylacetone

111
4.5 Applications
4.5.1 N-Aminoamidoximes
4.5.1.1 As a prodrug model
Clement and Reeh reported that drugs containing amidine functions could be
efficiently absorbed by the gastrointestinal tract after oral administration.
[2009USP0270440A1] N-Hydroxybenzamidoxime derivatives (4.2c) represent a new
class of prodrug to improve the oral bioavailability of medications containing amidine
functions, because they have lower basicity but higher lipophilicity than amidine
derivatives, and can be quickly absorbed, then reduced rapidly to benzamidoxime (4.24)
via N-reductases in vitro after oral administration (Scheme 4-45). [2007JMC6730] The
bioavailability of N-hydroxyamidoxime exceeds that of benzamidine after the oral
application. [2007JMC2730]
Scheme 4-45. In vitro biotransformation of N-hydroxybenzamidoxime
4.5.1.2 Applications in inorganic chemistry
The synthesis of alkali and silver nitrosolates (M[RC(NO)2], M = Metal, R = organic
substituent) was first described about a century ago. [1905Ber1445] Wieland and Hess
obtained nitrosolates from unstable N-hydroxyamidoximes by disproportion in NH3 or by
oxidation with KIO4 in basic solution. [1906Ber65, 1907LAC65, 1909Ber4175] For R =
H, these procedures lead to the formation of potassium dinitrosomethanide when KOH
is used. [1909Ber4175] Recently, salts of nitrosodicyanomethanide [(ON)C(CN)2]- and
nitrodicyanomethanide, [(O2N)C(CN) 2]- are predicted as potential propellants similar to

112
nitrite and nitrate salts respectively based on theoretical calculations. [1999IC2709] .
Brand et. al. developed a two-step synthesis of DNM salts (DNM = dinitrosomethanide)
from formamidinium nitrate. Treating a methanolic solution of (4.156) and
hydroxylammonium nitrate (4.157) (2 equiv) with a methanolic solution of KOtBu (2
equiv) resulted in the formation of the labile intermediate N,N’-dihydroxyformamidinium
nitrate (4.158) (Scheme 4-46). The reaction of (4.158 with MOtBu (2 equiv) in the
presence of oxygen yields the deep blue DNM salt (4.159). [2005JACS1360]
Scheme 4-46. Synthesis of dinitrosomethanide (DNM) salt
N-Hydroxyamidoxime derivatives are efficient ligands for transition metals in
redox systems. [1971JCPPCB601] A study of the reactions between the two redox
systems Fe(II)/Fe(III) and acetohydroximic oxime (4.160a) and ethylnitrosolic acid
(4.160b) showed a strong stabilization of Fe(II) by ethylnitrosolate (Figure 4-9). The
systems Fe(II) – (4.160a), Fe(III) – (4.160a), Fe(III) – (4.160b) are unstable and evolve
towards Fe(II) – (4.160b). [1972JCPPCB689]
Figure 4-9. Acetohydroximic oxime and ethylnitrosolic acid
4.5.2 N-Aminoamidoximes
4.5.2.1 As metal ligands for important coordination compounds
Sarikavakli et. al. prepared N-aminoamidoxime (4.162) from the hydrazimic
chloride precursor (4.161), which may be further derivatized via reaction with aldehydes

113
or ketones (4.165) to (4.163) and (4.166), both of which can complex with transition
metal ions (Ni, Cu, Co), to form novel vic-dioxime derivatives of hydrazone metal
complexes (4.164 and 4.167). (Scheme 4-47 & 4-48). [2005TJC107, 2006TJC563] vic-
Dioximes can also form stable metal complexes of transition, inner-transition or actinide
metal ions, and the ligands or their metal complexes have played a significant role in
stereochemistry, isomerism, magnetism, spectroscopy, cation exchange and ligand
exchange chromatography, analytical chemistry, catalysis, pigments and dyes.
[1974CCR1] vic-Dioximes complexes are model coordination compounds for studying
the structure of vitamin B12 and coenzyme B13, which have important roles in biology.
[2003JMS647]
Scheme 4-47. Synthesis of novel vic-dioxime derivatives of hydrazones
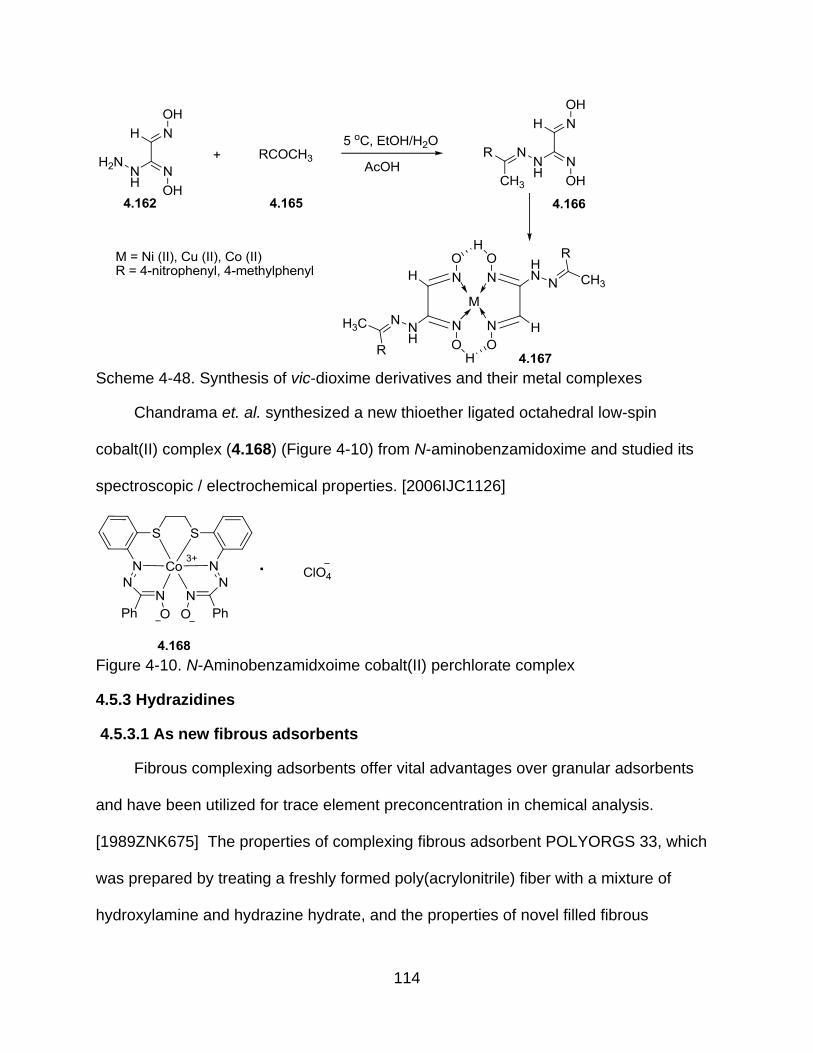
114
Scheme 4-48. Synthesis of vic-dioxime derivatives and their metal complexes
Chandrama et. al. synthesized a new thioether ligated octahedral low-spin
cobalt(II) complex (4.168) (Figure 4-10) from N-aminobenzamidoxime and studied its
spectroscopic / electrochemical properties. [2006IJC1126]
Figure 4-10. N-Aminobenzamidxoime cobalt(II) perchlorate complex
4.5.3 Hydrazidines
4.5.3.1 As new fibrous adsorbents
Fibrous complexing adsorbents offer vital advantages over granular adsorbents
and have been utilized for trace element preconcentration in chemical analysis.
[1989ZNK675] The properties of complexing fibrous adsorbent POLYORGS 33, which
was prepared by treating a freshly formed poly(acrylonitrile) fiber with a mixture of
hydroxylamine and hydrazine hydrate, and the properties of novel filled fibrous

115
adsorbents bearing hydrazidine (POLYORGS 35) groups have been studied with
respect to heavy and noble methods. It was shown that new adsorbents can be used for
the dynamic preconcentration of metals and radionuclides from aqueous solution and
these adsorbents can also be used for the preconcentration of heavy, noble, and rare
metals and radionuclides from aqueous salt solutions. [2000JAC549]
4.5.3.2 As anti-tuberculosis agents
Some hydrazidine analogues of isonicotinylhydrazine demonstrate in-vitro anti-
tuberculosis activity, with hydrazidine derivative (4.87) possessing the best in-vitro
activity against the tuberculosis pathogen. [2004KFZ16]
4.5.3.3 As environmentally friendly dyes
Dozens of patents and journals describe various hydrazidine- or formazan- derived
compounds as dye ligands that bind to metals such as Cu, Fe, Ni, Co, and they have
important applications in the textile industry. [2000EPA10, 2007DP8, 1995TCC13,
1989EPA315046A2]
Copper complexes of some hydrazidine derivatives, e.g. N, N’-bis(o-
hydroxyphenyl)-C-phenylformazan (4.169) are suitable agents for the dyeing of protein
fibers in neutral or slightly acid media, and they have fairly strong affinity to wool.
[1959ICBS532] Freeman et. al. synthesized some Fe-complexed hydrazidine
derivatives (4.170, 4.171) as environmentally friendly dyes (Figure 4-11). They can
substitute metals such as Cr and Co without adversely affecting technical and
mutagenic properties, again offering applications in the textile industry. [1995TCC13,
2007DP8]

116
Figure 4-11. Environmental friendly dye ligands
4.6 Conclusions
In summary, N-hydroxyamidoximes, N-aminoamidoximes and hydrazidines are
classes of amidine derivatives with versatile synthetic utilities and pharmacological
applications. They have been used extensively as starting materials for the preparation
of nitrogen-rich heterocycles. Typically they cyclize with various electrophiles such as
aldehydes, ketones carboxylates and acids and they have important applications in
drugs, dyes and polymers.

117
CHAPTER 5 SUMMARY OF ACHIEVEMENTS
Heterocyclic chemistry is an intriguing aspect of organic chemistry. It is a subject
with major studies focused on cyclic organic compounds made up of carbon, oxygen,
nitrogen and sulphur atoms. Many natural products, pharmaceutical intermediates,
drugs, textile dyes and polymers are heterocyclic molecules. Heterocyclic chemistry is
important to mankind and society for its immense applications that touch all of our daily
lives. 1H-Benzotriazole and its derivatives are known and used as important synthetic
auxiliaries in heterocyclic synthesis. Many important aspects of 1H-benzotriazole
chemistry have been explored over the last 30 years. My graduate studies aimed to
further apply the benzotriazole methodology to synthesize different heterocyclic
compounds with important applications. To summarize, Chapter 1 provides an overview
of 1H-benzotriazole methodology and some recent applications of 1H-benzotriazole and
its derivatives in heterocyclic synthesis. In Chapter 2, an efficient N-acylbenzotriazole
mediated synthesis of naphthoquinone-dipeptide conjugates is reported. Chapter 3
presents a straightforward approach to the synthesis of 1,3,4-oxadiazoles from
functionalized N-acylbenzotriazoles and acylhydrazides, which is an extension of the N-
acylbenzotriazole methodology. Chapter 4 provides a systematic review of the structure,
synthesis, reactivity and utility of N-hydroxy and N-amino- amidoximes and
hydrazidines, which are important classes of nitrogen-rich building blocks.
Apart from the above mentioned work, I participated in and completed the
synthesis and studies of the mechanical property of highly-filled crosslinked
polytriazoles, which is described in Appendix A.

118
APPENDIX A HIGHLY FILLED CROSSLINKED 1,2,3-TRIAZOLE POLYMERS AS NOVEL ROCKET
PROPELLANT BINDERS
A-1 Introduction
A-1-1 Rocket Propellant Binders
Rockets propellants are materials that give spacecraft a forward push via
producing large volumes of hot gas upon burning. Commonly, rocket propellants consist
of fuels and the oxidizers. [1997JP36, 1999JEM1, 2007PEP213] The fuel is generally
aluminum; the oxidizer is often finely ground ammonium perchlorate powder, which
constitutes 60% - 90% of the mass of the propellant. The other important ingredient for
rocket propellant is a polymeric binder which binds fuel, oxidizer and other additives
together. The most commonly used binders are polyurethane (PU), polybutadiene
acrylic acid acrylonitrile (PBAN), and hydroxy-terminator polybutadiene (HTPB) (Figure
A-1). Recently, 1,2,3-triazole-polymers prepared via “Click Chemistry” [2001ACIE2004,
2007MRC15, 2007ACIE1018] were reported as novel binders for high-energy explosive
and propellant materials, with advantages in terms of lower tensile stress and modulus
comparable to the polyurethanes used extensively as rocket propellant binders.
[2008JPS(A)PC238, 2001HPP313, 1992EPA481838]
Figure A-1. Common rocket propellant binders
Reproduced in part with permission from Jounal of Applied Polymer Sciences, 2010, 117, 121-127. Copyright © 2010 John Wiley and Sons.

119
The mechanical properties of solid rocket propellants and binders are important for
the functioning of rocket motors. [2003JTAC921] Polyurethanes were found to have
good physical properties, and aluminum powder could be incorporated for higher
specific impulse. However, polyurethanes are so viscous that the amount of oxidizers
and other solid additives that could be incorporated is limited. Polyurethane can
undergo side reactions during and after polymerization that degrade the mechanical
properties of the resulting propellant, e.g., loss of elasticity. Polyurethane propellants
tend to possess low tensile stress and modulus. [2000USP6103029] Polybutadiene-
based propellants such as PBAN and (HTPB) have physical properties superior to those
of polyurethanes. However, PBAN propellant is difficult to process and requires an
elevated curing temperature, and HTPB uses isocyanates for curing, which are very
toxic for the environment.
A-1-2 Triazole Polymers as Novel Rocket Propellant Binders
1,2,3-Triazole polymers (Figure A-1) are novel macromolecules that have received
growing interest in the area of polymer chemistry and material science. [2007ACIE1018]
Typically, they are synthesized by Huisgen 1,3-dipolar cycloaddition of azides with
terminal alkynes, which has been utilized for the synthesis of functionalized monomers
[2008JPS(A)PC2897], polymers [2008JPS(A)PC2316], chromophores [2005CC2029],
conjugated polymers [2005CC4333], glyco-polymers [2005EJOC3182] and macrocyclic
polymers [2006JACS4238]. Reed et. al. synthesized crosslinked triazoles as energetic
binders with improved mechanical properties and stability. [1992EPA481838]
[2001HPP313] Huang et. al. synthesized and characterized several series of novel low-
temperature curing and heat-resistant poly-triazole resins as advanced composites.
[2007PAT556, 2007JAPS1038, 2007JMS(PA)PAC175] Our group has developed

120
strategies for low-temperature synthesis of oligo-triazoles as binder ingredients.
[2006ARKIVOC43] Triazole-cured polymers were prepared with various alkynes and
azides without any solvent or copper catalysts under mild conditions near room
temperature. [1996JAPS2347] However, the mechanical properties of those triazole
polymers were not quantified. To meet the requirements of the specification of rocket
propellant binders, the monomers are required to polymerize at low temperatures (room
temperature to 60 o
C) with no or little side reactions. The polymerization process should
proceed in the absence of any solvent or heavy metal catalysts. In addition, the
polymerization should be capable of being scaled up easily. Polymerization through
triazole linkages proceeds readily and the components of the triazole cure (ethynyl
groups and azido groups) react preferentially with each other [2005EJOC3182], which
largely avoids the possibility of side reactions.
Since crosslinkers provide less mobility and increase the stiffness of the polymer,
the addition of crosslinkers can modify polymer mechanical properties such as tensile
strength, modulus and elasticity by limiting the mobility of individual polymer chains. It is
known that the mechanical properties of triazole polymers are significantly influenced by
their molecular structures such as the chain length between the triazole groups.
[1996JAPS2347] Crosslinker effects on mechanical properties of conventional rubbers
have been studied for many years and are well understood. [2001JAPS710] However,
such studies have not been systematically performed on triazole polymers. Hence, we
are interested in investigating the crosslinker effect on the mechanical properties of
formed triazole polymers. Triazole polymer formation is a good model to understand the
relationship between crosslinking and polymer mechanical properties, because

121
acetylene and azide groups should react with each other at 1:1 molar ratio, no small
molecules are produced, the reaction should not be influenced by residual moisture,
and side reactions should not occur. Thus, syntheses were carried out to investigate the
relationship between crosslinker concentration and mechanical properties of unfilled
and filled triazole polymers in terms of elongation strain (% elongation at break) and
elastic modulus (Young’s modulus).
Another aim of the project was to investigate the filler effects (types, sizes) on the
mechanical properties of the crosslinker triazole polymers in order to maximize the
amount of fillers in the polymeric binders based on military requirements. The binder for
propellants should possess a low reactivity to the filler ingredients and to the oxygen in
the air over long period storage at ambient temperature. In addition, the binder must be
able to tolerate a high loading of particulate solid ingredients. All else being equal, the
more solids one can blend into a given binder, the higher the performance of the
energetic formulation. The inclusion of particulate fillers in polymeric materials is an
established industrial practice which is performed in order to enhance polymer
properties such as modulus, fracture resistance and toughness while reducing the
overall component cost. [1977JMS1605] The effects of using different weight
percentages of fillers such as carbon black, silica, aluminum oxide, zirconium oxide
[1998JAPS1057], metal or metal clad fillers [2003USP113531], carbon nanotubes
[2006MCP132], glass-ceramic [2004B949] and sodium sulfate [1991RCT181] on the
thermal and mechanical properties of elastomers have been reported in detail by
several groups. Aluminum powder is a commonly used filler that improves mechanical,
electrical and thermal properties of polythiourethane-modified epoxy adhesives

122
[2008POC133], low density polyethylene (LDPE) [2007JAPS2436] and high density
polyethylene (HDPE) composites [2006JAPS2161], natural rubber (NR) composites
[2007PPTE667], polymethylmethacrylate (PMMA) [2001JP5267] and ethylene-
propylene-diene terpolymer (EPDM) composites [2007PPTE1201].
The mechanical properties of a composite material depend strongly on particle-
matrix interface adhesion, particle size and particle loading. [2008CBE933] Landon et al
studied the importance of adhesion between filler and the matrix phase in explaining the
mechanical behavior of the composite. [1977JMS1605] The dependence of filler particle
size on mechanical properties has also been studied with Chalk-filled PP models
[1993CM509]. Bhattarcharya et. al. examined the effect of particle size ratios of the
polymer to metal particles on the mechanical properties of PVC-Cu composites.
[1978JMS2109] Significant improvements in the mechanical properties were achieved
by incorporating a few weight percent of inorganic exfoliated clay minerals consisting of
layered silicate into polymer matrices. [2004JAPS2144, 1999JAPS1133, 2004P7579]
Ozkar et. al. systematically studied the effect of the use of additional fillers apart from
the main filler, in improving the thermal, rheological and tensile properties of
polyurethane elastomers. [1998JAPS1057] However, no such studies on the use of
mixed fillers on the mechanical properties of triazole polymers have been conducted to
date.
Therefore, experiments were designed to study the effect of filler loading on the
mechanical properties of crosslinked triazole polymers obtained by the selected model
polymerization reaction of E300 dipropiolate (A-1), diazide (A-2) and tetraacetylene-
functionalized crosslinker (A-3). Aluminum (10-14 micron) was used as the main filler

123
during the formulations; the effect of using secondary fillers such as aluminum (<75
micron), NaCl (45-50 and 83-105 micron) was monitored with the increase in the total
filler loading. The modulus of the aluminum-filled crosslinked triazole polymers
increases with increase of the filler content while using either of the two particle sizes of
aluminum powder. The use of Al (particle size < 75 micron) and NaCl (particle size 45-
50 micron and 83-105 micron) as secondary or additional fillers while using aluminum
(10-14 micron) as the main filler, has a diminishing effect on the modulus and strain of
the crosslinked triazole polymers. Triazole polymers described here have the ability to
wet and adhere large quantities of inorganic salts and thus the mechanical properties of
the composite remain comparable to typical polyurethane elastomeric matrices,
regardless of the chemistry of the oxidizer, which imparts them with important and
necessary binder characteristics for energetic composites. My research carried out
extensive studies on the use of two different particle sized aluminum fillers and mixtures
of different particle sized aluminum and sodium chloride fillers, on the mechanical
properties of crosslinked triazole polymers. These experiments are intended to evaluate
the degree to which the triazole-cured binder candidates can tolerate increases in the
loadings of various solids (especially that of sodium chloride, the model for inorganic
oxidizing salts in general) and still maintain good stress and a reasonable strain
capability.
In summary, my research efforts were a continuation of the work of developing a
novel robust binder cure system, with improved mechanical property comparable to that
of the urethane cure and with minimum possible incompatibility with new high-energy
ingredients. The optimum parameters were investigated for the triazole polymer with

124
desired mechanical properties by 1,3-dipolar cycloadditions between bis- or
polyacetylenes and polyazides in terms of crosslinker concentration, filler types, sizes
and concentrations.
A-2 Results and Discussion
A-2-1 Selection of Model Polymer System
Based on the criteria required by standard rocket propellants such as the
appreciable modulus and elasticity, nature and availability of the starting monomers,
time of reaction, temperature conditions and ease of scaling up, different diazides,
diacetylenes and crosslinkers were screened and thirteen different polymers were
synthesized and compared by our group members (Dr. Yuming Song, Ms. Reena
Gyanda and Dr. Rajeev Sakhuja). [2009JPS(A)PC3748] Accordingly, the reaction of
E300 dipropiolate (A-1), tetraethylene glycol-derived diazide (A-2), and tetrapropiolate
crosslinker (A-3) was selected as a model binder system to study the relationship
between the effects of crosslinker and filler on the mechanical properties of triazole
polymers.
Scheme A-1. Triazole polymer model system
A-2-2 Preparation of Monomers
For each series of studies, I prepared three monomers E300 dipropiolate (A-1),
tetraethylene glycol diazide (A-2) and tetrapropiolate crosslinker (A-3) in large quantities

125
(>200g each) following literature methods (Scheme A-2). [2006ARKIVOC43,
2008JPS(A)PC238, 2009JPS(A)PC3748, 2010JAPS121]
Scheme A-2. Preparation of monomers
A-2-3 Preparation of Dogbone Samples
Each dogbone sample was prepared by thoroughly mixing the three reactants (A-
1, A-2 and A-3) manually in an aluminum pan (~ 1 h per sample), then transferring the
uniform mixture to the dogbone molds before the curing process. The mechanical
properties - strain (percentage elongation at break) and elastic modulus (Young’s
modulus) for dogbone samples (Figure A-2) of filled and unfilled triazole polymers were
measured at a strain rate of 50 mm/min by Instron universal tensile testing machine
located at Department of Material Science, University of Florida. (Figure A-3)

126
Figure A-2. Dogbone mold containing filled and unfilled triazole polymers
Figure A-3. Instron universal tensile testing machine
A-2-4 Filler Loading Effect
The earlier studies conducted by our group members (Dr. Yongming Song, Ms.
Reena Gyanda, Dr. Rajeev Sakhuja) [2010JAPS2612] found that with the use of
43wt% aluminum filler in a crosslinked triazole polymerization process, the polymer had
good processability and a better modulus compared to unfilled triazole polymers. In
continuation of the development a robust polymeric triazole system with improved
mechanical properties, I studied the effect of increase in the filler type and content on
the mechanical properties of the crosslinked triazole polymers obtained by mixing E300
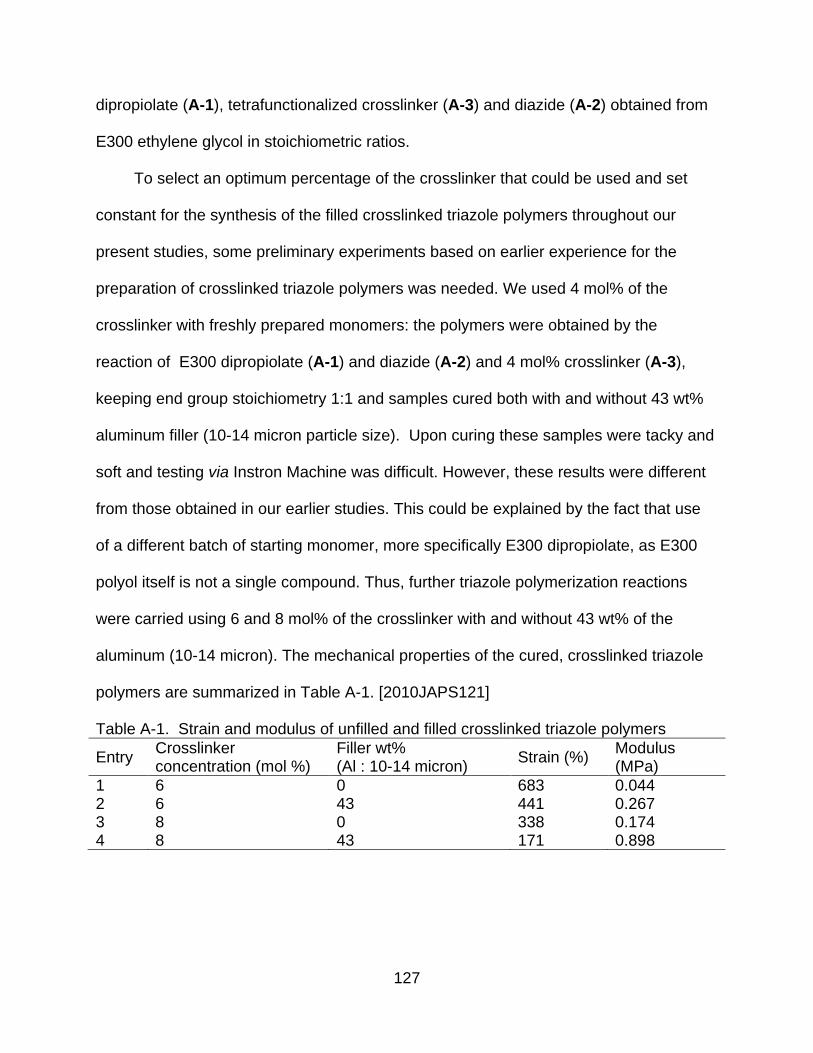
127
dipropiolate (A-1), tetrafunctionalized crosslinker (A-3) and diazide (A-2) obtained from
E300 ethylene glycol in stoichiometric ratios.
To select an optimum percentage of the crosslinker that could be used and set
constant for the synthesis of the filled crosslinked triazole polymers throughout our
present studies, some preliminary experiments based on earlier experience for the
preparation of crosslinked triazole polymers was needed. We used 4 mol% of the
crosslinker with freshly prepared monomers: the polymers were obtained by the
reaction of E300 dipropiolate (A-1) and diazide (A-2) and 4 mol% crosslinker (A-3),
keeping end group stoichiometry 1:1 and samples cured both with and without 43 wt%
aluminum filler (10-14 micron particle size). Upon curing these samples were tacky and
soft and testing via Instron Machine was difficult. However, these results were different
from those obtained in our earlier studies. This could be explained by the fact that use
of a different batch of starting monomer, more specifically E300 dipropiolate, as E300
polyol itself is not a single compound. Thus, further triazole polymerization reactions
were carried using 6 and 8 mol% of the crosslinker with and without 43 wt% of the
aluminum (10-14 micron). The mechanical properties of the cured, crosslinked triazole
polymers are summarized in Table A-1. [2010JAPS121]
Table A-1. Strain and modulus of unfilled and filled crosslinked triazole polymers
Entry Crosslinker concentration (mol %)
Filler wt% (Al : 10-14 micron)
Strain (%) Modulus (MPa)
1 6 0 683 0.044 2 6 43 441 0.267 3 8 0 338 0.174 4 8 43 171 0.898

128
The physical nature of the polymers and the modulus and strain values suggested
that the use of 8 mol% crosslinker would be better in a study of the effect of different
filler loadings on the mechanical properties of the resulting triazole polymers.
The content of the aluminum filler with particle size 10-14 micron and <75 micron
was systematically increased (Scheme A-1) from 34.18 to 74.14 weight percent (Table
A-2 and Table A-3) resulting in two separate sets of gumstock samples, which were
cured in standard dogbone molds at 55 oC for 72 h. The cured polymers were tested
using Instron tensile testing machine.
Figures A-4 and Figure A-5 show the variation of the modulus and strain values of
these two sets of filled gumstock samples with the increase in filler loading.
Table A-2. Effect of filler loading (Al: 10-14 micron) on strain and modulus of crosslinked triazole polymers
Entry Crosslinker concentration (mol %)
Filler Wt % (Al : 10-14 micron)
Strain (%) Modulus (MPa)
1 8 34.2 205.4 0.64 2 8 43.0 171.0 0.90 3 8 58.1 97.5 2.33 4 8 67.5 48.3 5.06 5 8 71.7 29.6 11.31 6 8 74.2 18.7 13.68
Table A-3. Effect of filler loading (Al: < 75 micron) on strain and modulus of crosslinked
triazole polymers
Entry Crosslinker concentration (mol %)
Filler Wt % (Al : < 75 micron)
Strain (%) Modulus (MPa)
1 8 34.2 108.1 0.88 2 8 43.0 93.7 1.68 3 8 58.1 41.0 4.70 4 8 67.5 33.1 9.72 5 8 71.7 10.7 25.39 6 8 74.2 9.2 32.07

129
In general, it may be concluded that the modulus of the aluminum-filled
crosslinked triazole polymers increases with increase of the filler content using either of
the two particle sized aluminum powders. The value of the modulus increased from 0.64
MPa to 13.68 MPa as the aluminum with particle size 10-14 micron is loaded from 34.2
to 74.2 wt. percent of the binder. The 74.2 value is the maximum percentage of the filler
resulting in polymers which are processable. Beyond this point the binder does not
completely wet all the filler particles resulting in a highly viscous, non uniform material
difficult to cast into molds before curing. The same result was inferred from the
mechanical data generated by using <75 micron aluminum powder. The addition of rigid
particles to a polymeric matrix improves the modulus since the rigidity of the inorganic
fillers is generally higher than that of organic polymers. [2008CBE933] Anuar and
coworkers observed similar trends with the use of aluminum on natural rubber (NR) and
ethylene-propylene-diene-terpolymer (EPDM) composites. [2007PPTE1201]
However, there is an increase in the value of the modulus while shifting from 10-14
micron to <75 micron particle size for the same filler content (for example 0.64 to 0.88
MPa; 0.90 to 1.68 MPa). For lower weight percentages of filler, the modulus is almost
independent of particle size, but the difference in modulus values increases with
increase of the filler loading. [2008CBE933]
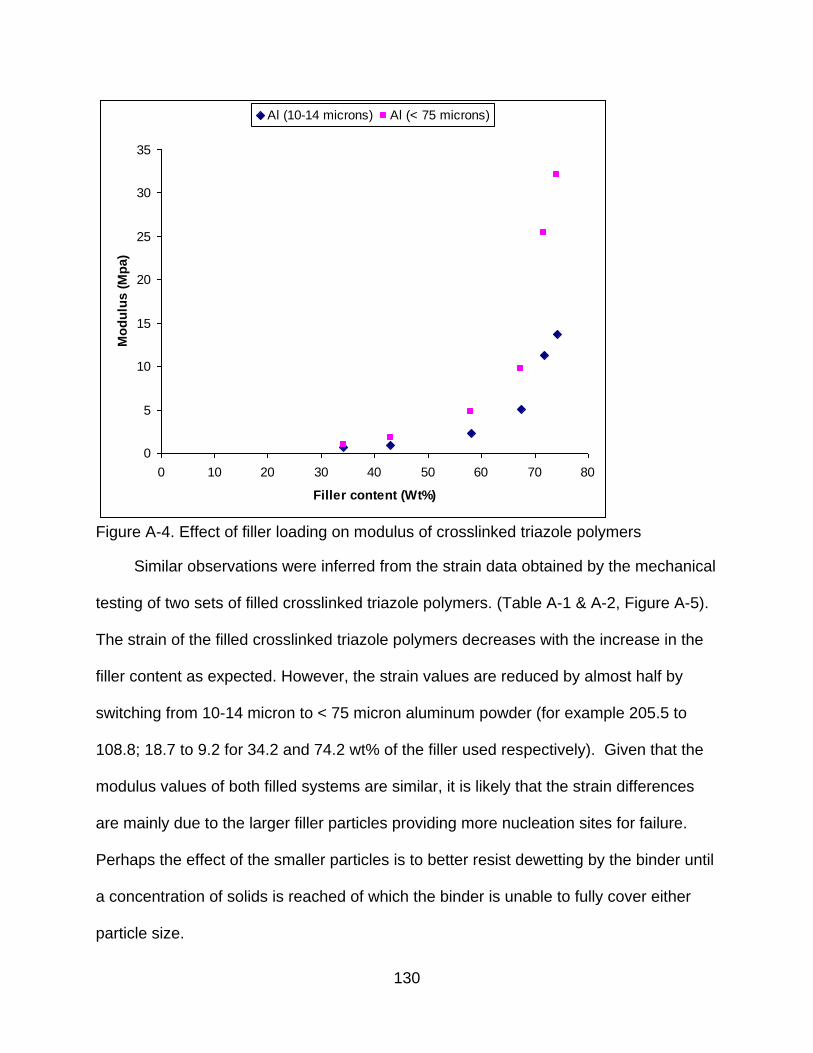
130
0
5
10
15
20
25
30
35
0 10 20 30 40 50 60 70 80
Filler content (Wt%)
Mo
du
lus (
Mp
a)
Al (10-14 microns) Al (< 75 microns)
Figure A-4. Effect of filler loading on modulus of crosslinked triazole polymers
Similar observations were inferred from the strain data obtained by the mechanical
testing of two sets of filled crosslinked triazole polymers. (Table A-1 & A-2, Figure A-5).
The strain of the filled crosslinked triazole polymers decreases with the increase in the
filler content as expected. However, the strain values are reduced by almost half by
switching from 10-14 micron to < 75 micron aluminum powder (for example 205.5 to
108.8; 18.7 to 9.2 for 34.2 and 74.2 wt% of the filler used respectively). Given that the
modulus values of both filled systems are similar, it is likely that the strain differences
are mainly due to the larger filler particles providing more nucleation sites for failure.
Perhaps the effect of the smaller particles is to better resist dewetting by the binder until
a concentration of solids is reached of which the binder is unable to fully cover either
particle size.

131
0
50
100
150
200
250
0 10 20 30 40 50 60 70 80
Filler content (Wt%)
Str
ain
(%
)Al (10-14 microns) Al (< 75 microns)
Figure A-5. Effect of filler loading on strain of crosslinked triazole polymers
The studies were extended by using two different particle sizes of NaCl powder as
fillers. Tables A-4 and A-5 show the variation of the modulus and strain of the filled
crosslinked triazole polymers with NaCl as the main filler with the increase in the
content and particle size of NaCl (Scheme A-3). The samples prepared were somewhat
sticky and non-uniform, and the mechanical testing was therefore difficult. However, the
trend for the modulus and the strain with increase in the filler content was similar to that
obtained with aluminum, but the values of the modulus were quite low. Thus it made
sense to study the effect of NaCl as secondary/additional filler while retaining aluminum
as the main filler.
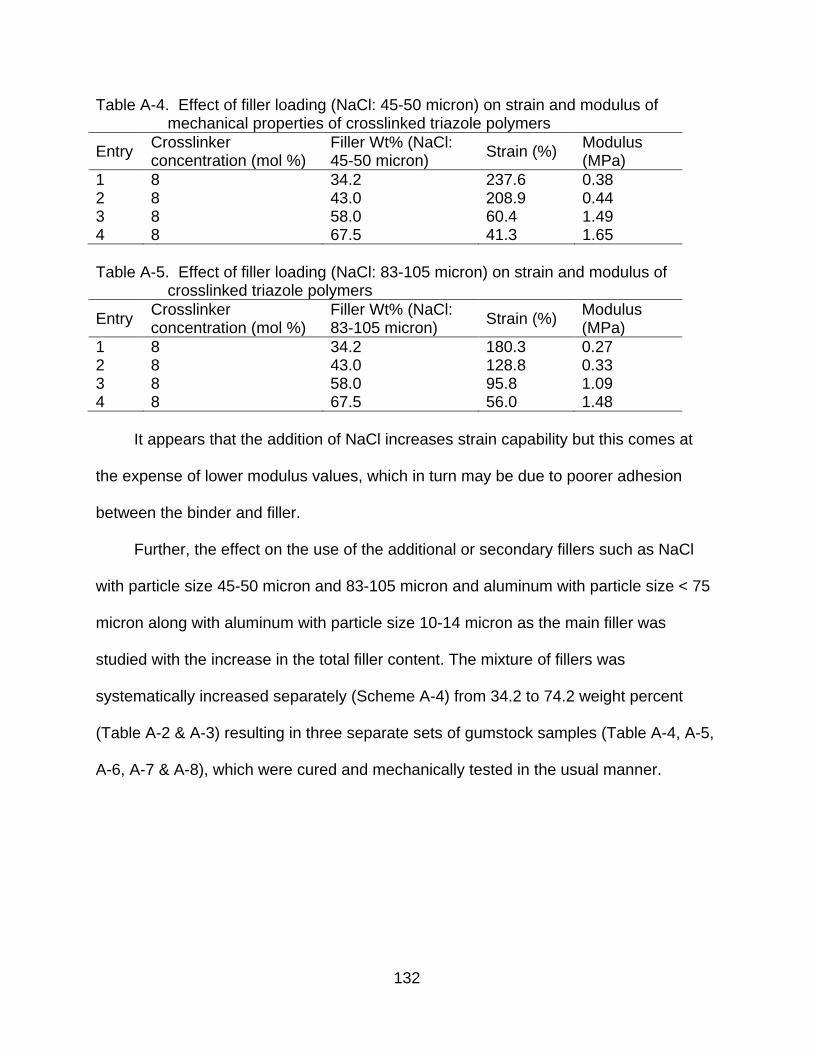
132
Table A-4. Effect of filler loading (NaCl: 45-50 micron) on strain and modulus of mechanical properties of crosslinked triazole polymers
Entry Crosslinker concentration (mol %)
Filler Wt% (NaCl: 45-50 micron)
Strain (%) Modulus (MPa)
1 8 34.2 237.6 0.38 2 8 43.0 208.9 0.44 3 8 58.0 60.4 1.49 4 8 67.5 41.3 1.65
Table A-5. Effect of filler loading (NaCl: 83-105 micron) on strain and modulus of
crosslinked triazole polymers
Entry Crosslinker concentration (mol %)
Filler Wt% (NaCl: 83-105 micron)
Strain (%) Modulus (MPa)
1 8 34.2 180.3 0.27 2 8 43.0 128.8 0.33 3 8 58.0 95.8 1.09 4 8 67.5 56.0 1.48
It appears that the addition of NaCl increases strain capability but this comes at
the expense of lower modulus values, which in turn may be due to poorer adhesion
between the binder and filler.
Further, the effect on the use of the additional or secondary fillers such as NaCl
with particle size 45-50 micron and 83-105 micron and aluminum with particle size < 75
micron along with aluminum with particle size 10-14 micron as the main filler was
studied with the increase in the total filler content. The mixture of fillers was
systematically increased separately (Scheme A-4) from 34.2 to 74.2 weight percent
(Table A-2 & A-3) resulting in three separate sets of gumstock samples (Table A-4, A-5,
A-6, A-7 & A-8), which were cured and mechanically tested in the usual manner.

133
Table A-6. Effect of mixed filler loading (mixture of two different particle sized Aluminum) on strain and modulus of crosslinked triazole polymers
Entry Crosslinker concentration (mol %)
Filler Wt % (Al : < 75 micron + Al: 10-14 micron)(1:1)
Strain (%) Modulus (MPa)
1 8 34.2 100.8 0.78 2 8 43.0 91.2 1.47 3 8 58.0 63.6 3.51 4 8 67.5 36.2 4.70 5 8 71.7 21.3 7.21 6 8 74.2 19.4 9.37
Table A-7. Effect of mixed filler loading (mixture of Aluminum and NaCl) on strain and modulus of crosslinked triazole polymers
Entry Crosslinker concentration (mol %)
Filler Wt % (Al : 10-14 micron + NaCl: 45-50 micron)(1:1)
Strain (%) Modulus (MPa)
1 8 34.2 79.8 0.73 2 8 43.0 57.7 0.91 3 8 58.0 40.9 1.90 4 8 67.5 34.8 3.52 5 8 71.7 26.7 4.68
Table A-8. Effect of mixed filler loading (mixture of Aluminum and NaCl) on strain and modulus of crosslinked triazole polymers
Entry Crosslinker concentration (mol %)
Filler Wt % (Al : 10-14 micron + NaCl: 83-105 micron)(1:1)
Strain (%) Modulus (MPa)
1 8 34.2 68.6 0.71 2 8 43.0 55.4 0.95 3 8 58.0 44.9 1.32 4 8 67.5 35.7 3.59 5 8 71.7 24.8 4.45
Figures A-6 and A-7 compare the results on the use of additional fillers with the
main filler in the ratio 1:1 on the mechanical properties of crosslinked triazole polymers.
These data include equal weights of mixed fillers of two different particle sized
aluminum powders and aluminum with two different particle sized NaCl powder. In

134
general, the use of Al (particle size < 75 micron) and NaCl (particle size 45-50 micron
and 83-105 micron) as secondary or additional fillers while using aluminum (10-14
micron) as the main filler, has a diminishing effect on the modulus and strain of the
crosslinked triazole polymers. Perhaps the effect of the larger aluminum particles in
reducing strain capability is overriding the effect of the smaller aluminum.
0
1
2
3
4
5
6
7
8
9
10
0 10 20 30 40 50 60 70 80
Filler content (Wt%)
Mo
du
lus (
Mp
a)
(Al : 10-14 microns + Al: <75 microns)(1:1)
(Al : 10-14 microns + NaCl: 45-50 microns)(1:1)
(Al : 10-14 microns + NaCl: 83-105 microns)(1:1)
Figure A-6. Effect of mixed filler loading on modulus of crosslinked triazole polymers
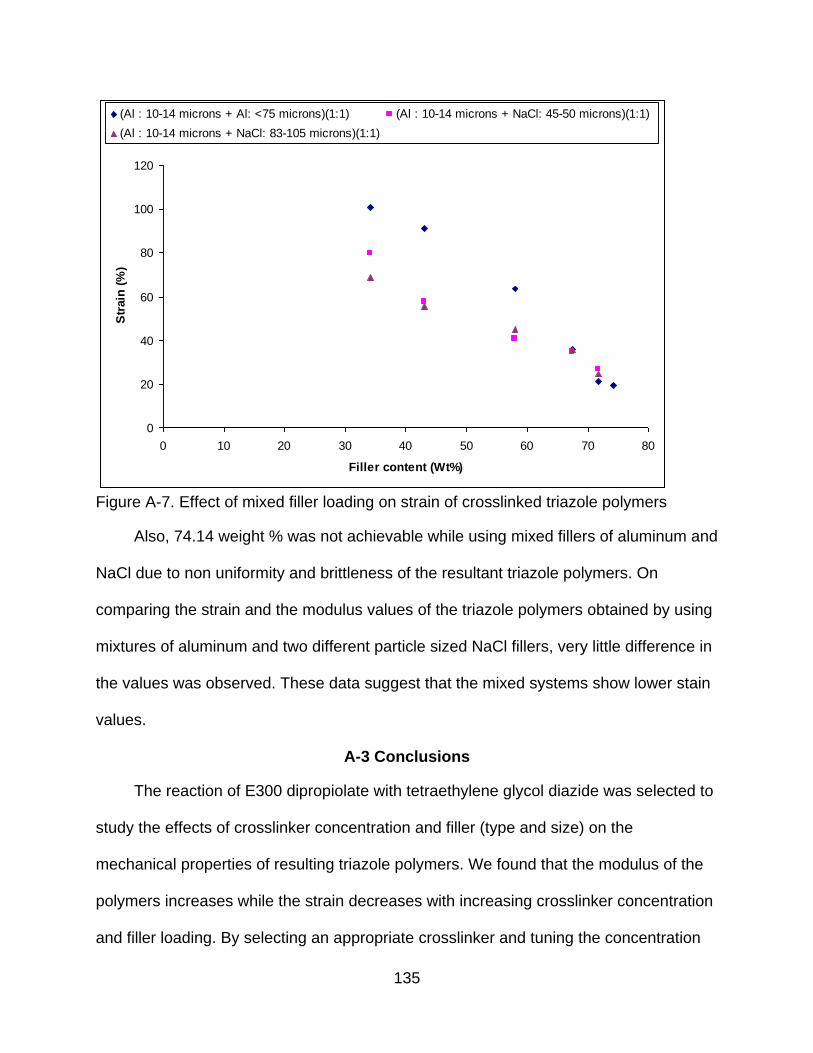
135
0
20
40
60
80
100
120
0 10 20 30 40 50 60 70 80
Filler content (Wt%)
Str
ain
(%
)(Al : 10-14 microns + Al: <75 microns)(1:1) (Al : 10-14 microns + NaCl: 45-50 microns)(1:1)
(Al : 10-14 microns + NaCl: 83-105 microns)(1:1)
Figure A-7. Effect of mixed filler loading on strain of crosslinked triazole polymers
Also, 74.14 weight % was not achievable while using mixed fillers of aluminum and
NaCl due to non uniformity and brittleness of the resultant triazole polymers. On
comparing the strain and the modulus values of the triazole polymers obtained by using
mixtures of aluminum and two different particle sized NaCl fillers, very little difference in
the values was observed. These data suggest that the mixed systems show lower stain
values.
A-3 Conclusions
The reaction of E300 dipropiolate with tetraethylene glycol diazide was selected to
study the effects of crosslinker concentration and filler (type and size) on the
mechanical properties of resulting triazole polymers. We found that the modulus of the
polymers increases while the strain decreases with increasing crosslinker concentration
and filler loading. By selecting an appropriate crosslinker and tuning the concentration

136
of the crosslinker and filler, the triazole polymers with desired mechanical properties
could be obtained. The mechanical properties of these triazole polymers are superior to
the typical polyurethane elastomeric matrices for rocket propellant binders, and some
highly filled crosslinked triazole polymers possess properties of potential rocket
propellant binders. Overall, the study suggests, as expected, that the aluminum fillers
give rise to better mechanical properties than inorganic materials (sodium chloride).
The data suggest that the smaller metal particles act to produce enhanced mechanical
properties whereas the mixed metal/inorganic filler simply produce samples of
intermediate mechanical properties over the compositions tested. Triazole polymers
described have the ability to wet and adhere large quantities of inorganic salts and thus
maintain the tensile strength of the composite, regardless of the chemistry of the
oxidizer, thus imparting important binder characteristic for energetic composites.
A-4 Experimental Section
General methods. NMR spectra were recorded in CDCl3 or DMSO-d6 with TMS
for 1H (300 MHz) and 13C (75 MHz) as internal reference. Elemental analyses were
performed on a Carlo Erba-1106 instrument. Commercially obtained reagents were
used without further purification. E300 dipropiolate (A-1), diazide (A-2) derived from
ethylene glycol and the tetrafunctional crosslinker 3-(propioloyloxy)-2,2-
bis[(propioloylxy)methyl]propylpropiolate (A-3) were prepared following reported
procedures. [2008JPS(A)PC238] In view of the stringent stoichiometry requirements for
step polymerization, the monomers were systematically dried by azeotropic distillation
and lyophilization. The uni-axial test specimen was a standard micro-tensile dogbone
with dimensions of 0.88’’× 0.19’’× 0.13’’ inches (Figure A-8). [1992JPCE] The dogbone
mold containing filled and unfilled triazole polymers is shown in Figure A-2. Strain

137
(percentage elongation at break) and elastic modulus (Young’s modulus) were
measured by an Instron universal tensile testing machine (model number 4301) with a
strain rate of 50 mm/min. Aluminum (10-14 micron and < 75 micron) was purchased
from Aldrich. Anhydrous sodium chloride was ground and passed through a series of
sieves of different pore sizes to obtain NaCl in 45-50 micron and 83-105 micron mono-
disperse particle sizes. Each data entry in the Tables (Table A-1 – Table A-8) is an
average of at least three measurements.
Figure A-8. Dimensions of dogbone mold and dogbone sample
Preparation of tetraethyleneglycoldipropiolate (A-1). A solution of E300
polyethylene glycol (10 g, 51.5 mmol), propiolic acid (7.9 g, 113.0 mmol) and p-
toluenesulfonic acid (0.5 g, 2.63 mmol) in toluene (100 mL) was heated under reflux
using a Dean Stark apparatus for 48 h. The reaction mixture was cooled to room
temperature and the solvent was removed under reduced pressure. The residue was
dissolved in CHCl3 (150 mL) washed with saturated NaHCO3 (70 mL), water (50 mL)
and brine (50 mL). The chloroform layer was dried over anhydrous MgSO4, filtered and
the solvent was evaporated to give tetraethyleneglycoldipropiolate (13.94 g, 91%) as
yellow oil.

138
Tetraethyleneglycoldipropiolate (A-1). Yellow oil; yield: 13.94 g (91%); 1H NMR
(CDCl3) : 2.93 (s, 2H), 3.67 (s, 8H), 3.75 (t, J = 4.8 Hz, 4H), 4.35 (t, J = 4.8 Hz, 4H);
13C NMR (CDCl3) : 65.2, 68.5, 70.6, 70.6, 74.5, 75.2, 152.6; Anal. Calcd for C14H18O7:
C, 56.37; H, 6.08; Found: C, 56.07; H, 6.22.
Preparation of 1-azido-2-{2-[2-(2-azidoethoxy)ethoxy]ethoxy}ethane (A-2). A
mixture of tetraethylene glycol dimesylate (14.4 g, 41.43 mmol) and NaN3 (10.77 g,
165.72 mmol) in 100 mL CH3CN /H2O (9:1) was refluxed at 80 oC for 24 h. The mixture
was filtered, the filtrate diluted with water and extracted with ethyl acetate. The organic
layer was dried over anhydrous magnesium sulfate and solvent was removed under
vacuum to obtain pure 1-azido-2-{2-[2-(2-azidoethoxy)ethoxy]ethoxy}ethane (7.88 g,
78%) as a light yellow oil.
1-Azido-2-{2-[2-(2-azidoethoxy)ethoxy]ethoxy}ethane(A-2). Colorless oil; yield:
7.88 g (78%); 1H NMR (CDCl3) : 3.70-3.67 (m, 12H), 3.40 (t, J = 4.8Hz, 4H); 13C NMR
(CDCl3) : 70.5, 69.8, 50.5.
Preparation of 3-(propioloyloxy)-2,2-bis[(propioloyloxy)methyl]propyl
propiolate (tetrapropiolate) (A-3). A solution of pentaerythritol (5 g, 36.72 mmol),
propiolic acid (14.91 g, 213 mmol) and conc. H2SO4 (0.5 ml) in benzene (75 ml) was
heated under reflux using a Dean Stark apparatus for 12.5 h. The reaction mixture was
cooled in ice and neutralized with solid Na2CO3, filtered, washed with ether and the
filtrate was evaporated to obtain a solid. The solid was dissolved in CH2Cl2 (100 ml)
washed with saturated NaHCO3 (50 ml), water (50 ml) and brine (25 ml). The
dichloromethane layer was dried over anhydrous MgSO4, filtered and the solvent was
evaporated to give pentaerythritol tetrapropiolate as a white powder.

139
3-(Propioloyloxy)-2,2-bis[(propioloyloxy)methyl]propyl propiolate (A-3).
White microcrystals; yield 6.5 g (51 %); 1H NMR (CDCl3) : 2.98 (s, 4H), 4.31 (s, 8H);
13C NMR (CDCl3) : 41.7, 63.3, 73.6, 76.4, 151.8; Anal. Calcd for C17H12O8: C, 59.31; H,
3.51; Found: C, 59.05; H, 3.57.
Procedure for the preparation of dogbone samples for mechanical studies.
In an aluminum pan, E300 diacetylene (A-1) was weighed and different concentrations
of crosslinker (A-3) were added and stirred until the crosslinker dissolved. The time
required to dissolve crosslinker varied from 5-20 min with the increase in the
concentration of the crosslinker. This was followed by the addition of diazide (A-2),
which on stirring gave a homogeneous mixture (Scheme A-3). The reactions were
carried out a scale of 2 g (including the three reactants for each dogbone sample) in
aluminium pans by taking 100mol% of diazide (A-2) and calculating the concentrations
of diacetylene (A-1) and the crosslinker (A-3) as shown in Scheme A-3, keeping the
overall end group stoichiometric ratios 1:1. The mixture was cast into dogbone molds
(Figure A-14), and the dogbone molds were degassed under vacuum at room
temperature for 15 minutes and left at room temperature for 3-4 h. The curing was then
carried out in a vacuum oven at 55 oC for 72 h. The dogbone samples were carefully
removed from the mold. After the cooling, they were tested using a Universal Tensile
Test Machine with a 200 lb load cell and 50 mm/min test speed. For the filled systems,
aluminum powder was added to the homogeneous mixture of diacetylene (A-1), diazide
(A-2) and crosslinker (A-3), then mixed uniformly and degassed followed by curing in a
vacuum oven at 55 oC for 1h. The mixture was then stirred again and cured at 55 oC for
an additional 71 h.

140
Procedure for the preparation of linear triazole polymer P-1. The monomers
diacetylene (A-1) and diazides (A-2) were mixed manually in 1:1 equivalent in an
aluminum pan until a homogenous mixture was obtained. The pan was cured in a
vacuum oven for 72 h.
Unfilled Triazole Polymer (P-1). Light yellow rubbery polymer; 1H NMR (CDCl3)
: 3.56-3.67 (m, (-O-CH2-CH2-O-)), 3.81-3.95 (m, (-COO-CH2-CH2-O) & (-triazole-CH2-
CH2-O-)), 4.48-4.51 (m, (-CH2-triazole-)), 4.60-4.63 (m, -(triazole-COO-CH2-)), 8.34 (s,
(-triazole-H)); 13C NMR (CDCl3) : 50.4, 64.1, 68.9, 70.3, 70.4, 70.5, 77.2, 130.0, 139.7,
160.8. Anal. Calcd for C26H42N6O12: C, 49.52; H, 6.71; N, 13.33 Found: C, 49.38; H,
6.72; N, 13.00.
General procedure for preparation of crosslinked triazole polymers. E300
Dipropiolate (A-1) and crosslinker (A-3) were weighed into an aluminum pan, and
stirred until homogeneous. The time for dissolving the crosslinker varied from 15 to 30
minutes. Diazide (A-2) was added with stirring to give a homogeneous mixture. The
reactions were carried on a total scale of 2 g (comprising the three reactants for each
dogbone sample) in aluminum pans by taking 100mol% of (A-2) and calculating the
concentrations of (A-1) and the crosslinker (A-3) as shown in Scheme A-3, keeping the
overall end group stoichiometric ratios 1:1. (Scheme A-3) The filler (or mixture of fillers)
was then added to the homogeneous mixture and mixed uniformly by hand for about 45
minutes. The mixtures were cast into a dogbone molds, degassed under vacuum at
room temperature for 15 minutes and then cured in an oven at 55 oC for 72 h. The
dogbone samples were carefully removed from the mold. After the cooling, they were

141
tested at ambient temperature using a Universal Tensile Test Machine with a 22 lb load
cell and 50 mm/min test speed.
Scheme A-3. General route to crosslinked 1,2,3-triazole polymers with fillers

142
LIST OF REFERENCES
[1884Ber182] A. Pinner, Ber., 17, 182 (1884).
[1894Ber160] E. Bamberger, Ber., 27, 160 (1894).
[1898Ber2126] H. Ley, Ber., 31, 2126 (1898).
[1905Ber1445] H. Wieland, Ber., 38, 1445 (1905).
[1906Ber65] H. Wieland, Ber., 39, 1480 (1906).
[1907LAC65] H. Wieland, Liebigs Ann. Chem., 353, 65 (1907).
[1909Ber4175] H. Wieland and H. Hess, Ber., 42, 4175 (1909).
[1910Ber4199] H. Wieland, Ber., 42, 4199 (1910).
[1914Ber2941] H. Ley and M. Ulrich, Ber., 47, 2941 (1914).
[1914Ber2938] H. Ley and M. Ulrich, Ber., 47, 2938 (1914).
[1925LAC260] E. Bamberger, R. Padova, and E. Ormerod, Liebigs Ann.
Chem., 446, 260 (1925).
[1931MC106] W. Oberhummer, Monatsh. Chem., 57, 106 (1931).
[1948JCS2240] A. Albert, R. Goldacre and J. L. Philips, J. Chem. Soc., 2240
(1948).
[1950LAC185] D. Jerchel and R. Kuhn, Liebigs Ann. Chem., 568, 185
(1950).
[1951CB651] I. Hauser, D. Jerchel and R. Kuhn, Chem. Ber., 84,
651(1951).
[1952CB470] W. Reid and F. Muller, Chem. Ber., 85, 470 (1952).
[1954JCS3319] M. R. Atkinson and J. B. Polya, J. Chem. Soc., 3319 (1954).
[1955CRV355] A. W. Nineham, Chem. Rev., 55, 355 (1955).

143
[1955HCA1473] A. Marxer, Helv. Chim. Acta., 38, 1473 (1955).
[1955HCA1560] P. Schmidt and J. Druey, Helv. Chim. Acta., 188, 1560
(1955).
[1955JPSJ726] N. Kunimine and K. Itano, J. Pharm. Soc. Jap., 74, 726
(1954); Chem. Abstr., 49, 11, 627 (1955).
[1956JCS2853] F. P. Doyle, W. Ferrier, D., Holland, M. D. Mehta, and J. H. C.
Nayler, J. Chem. Soc., 2853 (1956).
[1957ibid191] D. Jerchel and W. Wotichy, ibid., 605, 191 (1957).
[1959ICBS532] H. Ziegler, Ind. Chim. Belge. Suppl., 2, 532 (1959).
[1962CR155] F. Eloy and R. Lenaers, Chem. Rev., 62, 155 (1962).
[1963ACIEE309] C. Grundmann, Angew. Chem. Intern. Ed. Engl., 2, 309
(1963).
[1963ibid3121] M. Regitz and B. Eistert, ibid., 96, 3121 (1963).
[1963JCED624] C. Huether, J.M. Cretsos, J.M. Shackelford, J. Chem. Engin.
Data, 8, 624 (1963).
[1964ACIEE232] R. Kuhn, F. A. Neugebauer, and H. Trischmann, Angew.
Chem. Intern. Ed. Ennl., 3, 232 (1964).
[1965CR2512] J. Armand and R. M. Minvielle, Compt. Rend., 260, 2512
(1965).
[1965JOC931] F. H. Case, J. Org. Chem., 931 (1965).
[1966JOC157] C. Grundmann and H. –D. Frommeld, J. Org. Chem., 31, 157
(1966).
[1966MC517] R. Kuhn and G. Fischer-Schwarz, Monatsh. Chem., 97, 517

144
(1966).
[1966US3234255A] T. H. Johannes, A. H. Paulus and P. R. Herbert, U. S. Pat.,
3,234,255 A (1966).
[1967JCS(C)742] F. Kurzer and K. Douraghi-Zadeh, J. Chem. Soc., C, 742
(1967).
[1968ACIEE489] O. Westphal, Angew. Chem. Intern. Ed. Engl., 7, 489 (1968).
[1968CB29] T. Pyl, L. Seidl, and H. Beyer, Chem. Ber., 101, 29 (1968).
[1969BSCF3409] J, Barassin, J. Armand and H. Lumbroso, Bull. Soc. Chem.
Fr., 10, 3409 (1969).
[1970CR151] D. G. Neilson, R. Roger, J. W. M. Heatlie and L. R.
Newlands, Chem. Rev., 70, 151 (1970).
[1970JCS(C)2510]
R. N. Butler, F. L. Scott and R. D. Scott, J. Chem. Soc. C,
2510 (1970).
[1971JCPPCB601]
F. Valentini, P. Gouzerh and P. Souchay, J. Chim. Physique
Physico-Chim. Biologique, 68, 601 (1971).
[1972JCPPCB689] F. Valentini, P. Gouzerh and P. Souchay, J. Chim. Physique
Physico-Chim. Biologique, 69, 689 (1972).
[1972LAC16] H. Neunhoeffer and F. Weischedel, Leibigs Ann. Chem., 749,
16 (1972).
[1973JOC514] H. O. House, R. A. Auerbach, M. Gall and N. P. Peet, J. Org.
Chem., 514 (1973).
[1974CCR1] A. Chakravorty, Coord. Chem. Rev., 13, 1 (1974).
[1975CB3509] H. Neunhoeffer and H. –J. Degen, Chem. Ber., 108, 3509

145
(1975).
[1975CB2764] H. G. Aurich and K. Stork, Chem. Ber., 108, 2764 (1975).
[1975LAC1120] H. Neunhoeffer, H. –J. Degen and J. J. Kohler, Leibigs Ann.
Chem., 1120 (1975).
[1975JCS(PT1)1787]
Esmail, R.; Kurzer, F. J. Chem. Soc., Perkin Trans. 1, 1787
(1975).
[1975JCS(PT1)1181] J. R. L. Smith and J. S. Sadd, J. Chem. Soc., Perkin Trans. 1,
1181 (1975).
[1976LAC2206] W. Draber, H. Timmler, K. Dickore and W. Donner, Liebigs
Ann. Chem., 2206 (1976).
[1976T1031] K.M. Doyle and F. Kurzer, Tetrahedron, 32, 1031 (1976).
[1977BBRC387] J. R. White, Biochem. Biophys. Res. Comm., 77, 387 (1977).
[1977BCSJ953]
M. Takahashi, H. Tan, K. Fukushima and H. Yamazaki, Bull.
Chem. Soc. Jpn., 50, 953 (1977).
[1977JHC587] A. Mangia, F. Bortesi and U. Amendola, J. Heterocycl.
Chem., 14, 587 (1977).
[1977JMS1605] G. Landon, G. Lewis and G. F. Boden, J. Mater. Sci., 12,
1605 (1977).
[1978JMS2109] S. K. Bhattacharyya, S. Basu and S. K. De, J. Mater. Sci., 13,
2109 (1978).
[1978RRC203] S. Desherces, J. Barrans and J. L. Roubaty, Revue
Roumaine Chim., 23, 203 (1978).
[1979CB1981] H. -J. Degen, S. Haller, K. Heeg and H. Neunhoeffer, Chem.

146
Ber., 112, 1981 (1979).
[1979JHC881] L. Werbel, J. MaNamara, N. Colbry and J. Johnson, J.
Heterocycl. Chem., 16, 881 (1979).
[1979JMC1024] K. C. Murdock, R. G. Child, P. F. Fabio, R. D. Angier, R. E.
Wallace, F. E. Durr and R. V. Citarella, J. Med. Chem., 22,
1024 (1979).
[1980CRS304] J. Armand and P. Bassinet, J. Chem. Res. Synop., 304
(1980).
[1980JOC3916] K.-C. Liu, B. R. Shelton and R. K. Howe, J. Org. Chem., 45,
3916 (1980).
[1981PJC1253] E. Zygmunt, R. Dorota, R. Bany and P. Jelenski, Polish J.
Chem., 55, 1253 (1981).
[1981USP4299965] M. S. Chan and W. E. Hunter, U. S. Pat., 4,299,965 (1981).
[1982MC793] B. Kuebel, Monatsh. Chem., 113, 793 (1982).
[1983GPO3126464] E. Schinzel, T. Martini, W. Spatzier and H.Probst, Ger. Pat.
Offen. 3,126,464 (1983); Chem. Abstr., 98, 199850 (1983).
[1983P683] T. W. Moody, V. Bertness and D. N. Carney, Peptides, 4, 683
(1983).
[1984FES414] G. Mazzone, F. Bonina, G. Puglisi, A. M. Panico and R. R.
Arrigo, Farmaco Ed. Sci., 39, 414 (1984).
[1984JMC121] J. H. Musser, J. Med. Chem., 21, 1807 (1984).
[1984S146] F. Pochat, Synthesis, 146 (1984).
[1984GPO3245202] U. Eckstein, Ger. Pat. Offen., 3,245,202 (1984); Chem.

147
Abstr., 101, 173038 (1984).
[1985CB3481] B. Clement and T. Kampchen, Chem. Ber., 118, 3481 (1985).
[1985LAC78] H. Neunhoeffer, G. Köhler and H. –J. Degen, Liebigs Ann.
Chem., 78 (1985).
[1985T5187] L. Somogyi, Tetrahedron, 41, 5187 (1985).
[1985USP4500653] M. Schmidt, M. W. Witman, G. E. Reinert and I. C. Lim, U. S.
Pat., 4,500,653 (1985); Chem. Abstr., 102, 166761 (1985).
[1986JSCT9]
A. A. Bel Hadj and B. Baccar, J. Soc. Chim. Tunisie, 2, 9
(1986).
[1986SC1665] B. Rigo, P. Cauliez, D. Fasseur and D. Couturier, Syn.
Commun., 16, 1665 (1986).
[1987CL223] M. N., Pollak, J. F. Perdue, R. G. Margolese, K. Baer and M.
Richard, Cancer Lett., 38, 223 (1987).
[1987JCS(PT1)2673] A. R. Katritzky, K. Yannakopoulou, W. Kuzmierkiewicz, J. M.
Aurrecoechea, G. J. Palenik, A. E. Koziol, M. Szczesniak and
R. Skarjune, J. Chem. Soc., Perkin Trans. 1, 2673 (1987).
[1988IJC(B)542] V. A. Adhikari and V. V. Badiger, Indian J. Chem., Sect. B,
27, 542 (1988).
[1988JOC38] K. H. Pilgram, R. D. Skiles and D. A. Kleier, J. Org. Chem.,
53, 38 (1988).
[1988JOC5854] A. R. Katritzky, M. Drewniak and P. Lue, J. Org. Chem., 54,
5854 (1989).
[1988USP43262] M. F. Sitzmann, U. S. Pat., 43,262 (1988).

148
[1989CHC717] K. N. Zelenin, O. V. Solod and V. A. Khrustalev, Chem.
Heterocycl. Comp., 25, 717 (1989).
[1989EPA315046A2]
K. Pandl and M. Patsch, Eur. Pat. Appl., EP 315,046 A2, 7
(1989).
[1989H1121] A. R. Katritzky and K. Yannakopoulou, Heterocycl., 28, 1121
(1989).
[1989JOC6022] A. R. Katritzky, S. Rachwal and B. Rachwal, J. Org. Chem.,
54, 6022 (1989).
[1989JPS999] K. Raman, S. S. Parmar and S. K. Salzman, J. Pharm. Sci.,
78, 999 (1989).
[1989LAC105] H. Neunhoeffer, W. Diehl and U. Karafiat, Liebigs Ann.
Chem., 105 (1985).
[1989PNASU6318] S. Bajusz, T. Janaky, V. J. Csernus, L. Bokser, M. Fekete, G.
Srkalovic, T. W. Redding and A. V. Schally, Proc. Natl. Acad.
Sci. USA, 86, 6318 (1989).
[1989ZNK675] I.Y. Andreeva, E.K. Bocharova, E.A. Danilova, Zh. Anal.
Khim, 44, 675 (1989).
[1990B2538] C. A. Frederick, L. D. Williams, G. Ughetto, G. A. van der
Marel, B. J. H. Van, A. Rich and A. H. Wang, Biochemistry,
29, 2538 (1990).
[1990HC21] A. R. Katritzky and J. N. Lam, Heteroat. Chem., 1, 21 (1990).
[1990JHC1685] A. B. Theocharis and N. E. Alexandrou, J. Heterocycl. Chem.,
27, 1685 (1990).

149
[1990JOC5683] A. R. Katritzky, K. Yannakopoulou, E. Anders, J. Stevens and
M. Szafran, J. Org. Chem., 55, 5683 (1990).
[1990JPS(A)3647] Y. Saegusa, K. Sekiba and S. Nakamura, J. Polym. Sci.,
Polym. Chem., Part A, 28, 3647 (1990).
[1990P1205] M. Lee, R. T. Jensen, G. Bepler, L. Y. Korman and T. W.
Moody, Peptides, 11, 1205 (1990).
[1990JSBMB1083] A. Manni, A. E. Boucher, L. M. Demers, H. A. Harvey, A.
Lipton, M. A. Simmonds and M. Bartholomew, J. Steroid
Biochem. Mol. Biol., 37, 1083 (1990).
[1990JCS(PT2)2059] A. R. Katritzky, S. Perumal and W.-Q. Fan, J. Chem. Soc.
Perkin Trans. 2, 2059 (1990).
[1991CB1819] A. R. Katritzky, X. Lan and J. N. Lam, Chem. Ber., 123, 1819
(1991).
[1991CL285] Y. Saitoh, M. Matsuoka, Y. Nakao and T. Kitao, Chem. Lett.,
285 (1991).
[1991HCA1936] A. R. Katritzky, W. Kuzmierkiewicz and S. Perumal, Helv.
Chim. Acta, 74, 1936 (1991).
[1991IJI393] M. L. Bourguet-Kondraki, A. Longeon, E. Morel and M.
Guyot, Int. J. Immunopharmacol., 13, 393 (1991).
[1991JOC5808] H. Grennberg, A. Gogoll and J.-E, Backvall, J. Org. Chem.,
56, 5808 (1991).
[1991PHA290] N. Ergenc, S. Buyuktimkin, G. Capan, G. Baktir and S.
Rollas, Pharmazi, 46, 290 (1991).

150
[1991RCT181] R. D., Vargo and F. N. Kelley, Rubber Chem. Technol., 64,
181 (1991).
[1991S69] A. R. Katritzky, S. Rachwal and B. Rachwal, Synthesis, 69
(1991).
[1991T2683] A. R. Katritzky, S. Rachwal and G. J. Hitchings, Tetrahedron,
47, 2683 (1991).
[1992AJC513] J. L. Flippen-Anderson, R. D. Gilardi, A. M. Pitt and W. S.
Wilson, Australian J. Chem., 45, 513 (1992).
[1992EPA481838] M. Andre, J. P. Mazer and B. Nouguez, Eur. Pat. Appl.,
481,838 (1992).
[1992JPCE]
D. Riser, J. Hunter and R. Rast, AIAA/SAE/ASME/ASEE 28th
Joint Propulsion Conference and Exhibit, Nashville, TN, July
6 (1992).
[1992JCS(PT1)1111] A. R. Katritzky, M. F. Gordeev, J. V. Greenhill and P. J. Steel,
J. Chem. Soc. Perkin Trans. 1, 1111 (1992).
[1992JOC4932] A. R. Katritzky, S. Rachwal, B. Rachwal and P. J. Steel, J.
Org. Chem., 57, 4932 (1992).
[1992JPS(A)1369] Y. Saegusa, T. Koshikawa and S. Nakamura, J. Polym. Sci.,
Polym. Chem., Part A, 30, 1369 (1992).
[1992LAC843] A. R. Katritzky, J. Li and N. Malhotra, Liebigs Ann. Chem.,
843 (1992).
[1992LAC115] H. Neunhoeffer, U. Karafiat, G. Köhler and B. Sowa, Liebigs
Ann. Chem., 115 (1992).

151
[1992MC257] A. A. Ikizler and K. Sancak, Monatsh. Chem., 123, 257
(1992).
[1992MM2021] J. L. Hendrick and R. Twieg, Marcromolecules, 25, 2021
(1992).
[1992PNAS972] T. Janaky, A. Juhasz, S. Bajusz, V. Csernus, G. Srkalovic, L.
Bokser, S. Milovanovic, T. W. Redding, Z. Rekasi, A. Nagy
and A. V. Schally, Proc. Natl. Acad. Sci., 89, 972 (1992).
[1992T7817] A. R. Katritzky, N. Shobana, J. Pernak, A. S. Afridi and W.-Q.
Fan, Tetrahedron, 48, 7817 (1992).
[1993CM509] J. M. Adams, Clay Mineral, 28, 509 (1993).
[1994CSR363] A. R. Katritzky and X. Lan, Chem. Soc. Rev., 363 (1994).
[1994JHC917] A. R. Katritzky, B. Galuszka, S. Rachwal and M. Black, J.
Heterocycl. Chem., 31, 917 (1994).
[1994LAC1] A. R. Katritzky, J. Wu, W. Kuzmierkiewicz and S. Rachwal,
Liebigs Ann. Chem., 1 (1994).
[1995CHC208] R. A. Karakhanov, V. I. Kelarev, V. N. Koshelev, G. V.
Morozova, A. Dibi, Chem. Heterocycl. Compounds, 31, 208
(1995).
[1995JANYAS402] D. LeRoith, H. Werner, S. Neuenschwander, T. Kalebic and
L. Helman, J. Ann. New York Acad. Sci., 766, 402 (1995).
[1995JOC7612] A. R. Katritzky and H. Lang, J. Org. Chem., 60, 7612 (1995).
[1995JOC7619] A. R. Katritzky, H. Lang, Z. Wang, Z. Zhang and H. Song, J.
Org. Chem., 60, 7619 (1995).

152
[1995JOC7625] A. R. Katritzky, G. Zhang and J. Jiang, J. Org. Chem., 60,
7625 (1995).
[1995S1315] A. R. Katritzky, H. Wu, L. Xie, S. Rachwal, B. Rachwal, J.
Jiang, G. Zhang and H. Lang, Synthesis, 1315 (1995).
[1995TCC13] H. S. Freeman, J. Sokolowska-Gajda and A. Reife, Text.
Chem. Color, 27, 13 (1995).
[1996JAPS2347] S. B. Haska, E. Bayramli, F. Pekel and S. Ozkar, J. Appl.
Polym. Sci., 64, 2347 (1996).
[1996JOC1624] A. R. Katritzky and J. Li, J. Org. Chem., 61, 1624 (1996).
[1996LAC745] A. R. Katritzky, A. Jesorka, J. Wang, B. Yang, J. Wu and P.
J. Steel, Liebigs Ann. Chem., 745 (1996).
[1996LPS263] S. Rahimipour, L. Weiner, M. Fridkin, P. B. Shrestha-Dawadi,
S. Bittner, Lett. Pept. Sci., 3, 263 (1996).
[1996S1468] P. B. Shreshta-Dawadi, S. Bittner, M. Fridkin and S.
Rahimipour, Synthesis, 12, 1468 (1996).
[1997JOC706] A. R. Katritzky, O. Feng and H. Lang, J. Org. Chem., 62, 706
(1997).
[1997JOC4148] A. R. Katritzky, C. N. Fali and J. Li, J. Org. Chem., 62, 4148
(1997).
[1997JP36] J. Louwers, J. Pyrotechnics, 6, 36 (1997).
[1997TL6771] S. Fustero, B. Pina and A. Simón-Fuentes, Tetrahedron Lett.,
38, 6771 (1997).
[1998AA35] A. R. Katritzky and S. A. Belyakov, Aldrichimica Acta, 31, 35

153
(1998).
[1998CR409] A. R. Katritzky, X. Lan, J. Z. Yang and O. V. Denisco, Chem.
Rev., 98, 409 (1998).
[1998L139] E. J. Lee, S. L. George, P. C. Amrein, P. A. Paciucci, S. L.
Allen and C. A. Schiffer, Leukemia, 12, 139 (1998).
[1998LPS421] S. Rahimipour, L. Weiner, P. B. Shrestha-Dawadi, S. Bittner,
Y. Koch and M. Fridkin, Lett. Pept. Sci., 5, 421 (1998).
[1998JAPS1057] S. Benli, U. Yilmazer, F. Pekel and S. Ozkar, J. Appl. Polym.
Sci., 68, 1057 (1998).
[1999IC2709] N. Arulsamy, D. S. Bohle and B. Doletski, Inorg. Chem., 38,
2709 (1999).
[1999JAPS1133] L. Liu, Z. Qi and X. Zhu, J. Appl. Polym. Sci., 71, 1133
(1999).
[1999JEM1] D. M. Hanson-Parr and T. P. Parr, J. of Energ. Mater., 17, 1
(1999).
[1999JHC777] A. R. Katritzky, A. Pastor and M. V. Voronkov, J. Heterocycl.
Chem., 36, 777 (1999).
[1999OL977] M. Garcia de la Torre, A. Navarro, C. Ramírez de Arellano
and A. Simón, Org. Lett., 1, 977 (1999).
[1999T8263] A. R. Katritzky, J. Li and L. Xie, Tetrahedron, 55, 8263
(1999).
[2000AA439] S. Bittner, E. Gorohovsky, E. Lozinsky and A. I. Shames,
Amino Acids, 19, 439 (2000).

154
[2000EPA10] R. Pedemonte and W. Russ, Eur. Pat. Appl., 10 (2000).
[2000JAC549] G. V. Myasoedova, V. A. Nikashina, N. P. Molochnikova and
L. V. Lileeva, J. Anal. Chem., 55, 6, 549 (2000).
[2000JOC3679] A. R. Katritzky and A. Pastor, J. Org. Chem., 65, 3679
(2000).
[2000JOC8210] A. R. Katritzky, H. Y. He and K. Suzuki, J. Org. Chem., 65,
8210 (2000).
[2000USP6103029] R. Reed, U.S. Pat., 6,103,029 (2000).
[2000S2029] A. R. Katritzky, Y. Fang, A. Donkor and J. Xu, Synthesis,
2029 (2000).
[2001AA381] M.; Alnabari and S. Bittner, Amino Acids, 20, 381 (2001).
[2001AA135] S. Gorohovsky and S. Bittner, Amino Acids, 20, 135 (2001).
[2001ACIE2004] H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem.
Int. Ed., 40, 2004 (2001).
[2001BCSJ2133] S. H. Mashraqui, S. Kumar and C. D. Mudaliar, Bull. Chem.
Soc. Jpn., 74, 2133 (2001).
[2001HPP313] C. M. Thompson and P. M. Hergenrother, High Perform
Polym., 13, 313 (2001).
[2001HYDX350] B. Du and H. Han, Henan Yike Daxue Xuebao, 36, 350
(2001).
[2001JAPS710] R. Fan, Y. Zhang, C. Huang, Y. Zhang, Y. Fan and K. Sun, J.
Appl. Polym. Sci., 81, 710 (2001).
[2001JN158S] J. M. Jamison, J. Gilloteaux, H. S. Taper and J. L. Summers,

155
J. Nutr., 131, 158S (2001).
[2001JP5267] S. N. Goyanes, J. D. Marconi, P. G. Konig, G. H. Rubiolo, C.
L. Matteo and A. Marzocca, J. Polymer, 42, 5267 (2001).
[2001RCT 915] A. R. Katritzky, H. H. Odens, M. V. Voronkov, C. J. Rostek
and O. W. Maender, Rubber Chem. Technol. 74, 915 (2001).
[2001T407] G. R.; Sridhar, V. S. Murty, S. H. Lee, I. A. Blair and T. M.
Penning, Tetrahedron, 57, 407 (2001).
[2002AA71] S. Bittner, S. Gorohovsky, O. Paz-Tal and J. Y. Becker,
Amino Acids, 22, 71 (2002).
[2002CSR324] G. Mehta and V. Singh, Chem. Soc. Rev., 31, 324 (2002).
[2002JOC4667] S. Fustero, B. Pina, E. Salavert, A. Navarro, M. C. Ramírez
de Arellano and A. Simón Fuentes, J. Org. Chem., 67, 4667
(2002).
[2002JOC7361] S. E. Wolkenberg and D. L. Boger, J. Org. Chem., 67, 7361
(2002).
[2003AAPP34] G. C. Barrett and J. S. Davies, Amino Acids, Peptides and
Proteins, 34 (2003).
[2003ACIE3996] L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem.
Int. Ed., 42, 3996 (2003).
[2003CEJ4586] A. R. Katritzky and B. V. Rogovoy, Chem. Eur. J., 9, 4586
(2003).
[2003JOC4932] A. R. Katritzky, S. Rachwal, B. Rachwal and P. J. Steel, J.
Org. Chem., 57, 4932 (2003).

156
[2003JOC5720] A. R. Katritzky, K. Suzuki, S. K. Singh and H. Y. He, J. Org.
Chem., 68, 5720 (2003).
[2003JMS647] T. Ozpozan, D. Kucukusta and Z. Buyukumumcu, J. Mol.
Struct., 661-662, 647 (2003).
[2003JTAC921] G. Herder, F. P. Weterings and W. P. C. de Klerk, J. Therm.
Analy. Calorim., 72, 921 (2003).
[2003S2795] A. R. Katritzky, Y. Zhang and S. K. Singh, Synthesis, 2795
(2003).
[2003USP113531] K. Hajmrle, B. W. Callen and J. K. Mah, U. S. Pat., 113,531
(2003).
[2004B949] J. A. Juhasz, S. M. Best, R. Brooks, M. Kawashita, N. Miyata,
T. Kokubo, T. Nakamura and W. Bonfield, Biomaterials, 25,
949 (2004).
[2004CC2356] C. Galli, P. Gentili, O. Lanzalunga, M. Lucarini and G. F.,
Pedulli, Chem. Commun., 2356 (2004).
[2004COC1720] M. Aguilar-Martinez, N. A. Macias-Ruvalcaba, J. A. Bautista-
Martinez, M. Gomez, F. J. Gonzalez and I. Gonzalez, Curr.
Org. Chem., 8, 1721 (2004).
[2004JAPS2144] M. H. Kim, C. I. Park, W. M. Choi, J. W. Lee, J. G. Lim, O. O.
Park and J. M. Kim, J. Appl. Polym. Sci., 92, 2144 (2004).
[2004KFZ16] R. G. Glushkov, G. A. Modnikova, A. I. L'vov, L. Yu. Krylova,
T. V. Pushkina, T. A. Gus'kova and N. P. Solov'eva, Khim.-
Farm. Zh., 38, 16 (2004).

157
[2004P7579] Y. H. Zhang, J. T. Wu, S. Y. Fu, S. Y. Yang, Y. Li, L. Fan, R.
Li, L. F. Li and Q. Yan, Polymer, 45, 7579 (2004).
[2004S2877] M.-H. Shih, M.-Y. Yeh, M.-J. Lee and Y.-S. Su, Synthesis, 17,
2877 (2004).
[2005ARKIVOC329] A. R. Katritzky, A. A. Abdel-Fattah and R. G. Akhmedova,
ARKIVOC, vi, 329 (2005).
[2005BMCL5324] V. K. Tandon, D. B. Yadav, R. V. Singh, A. K. Chaturvedi and
P. K. Shukla, Bioorg. Med. Chem. Lett., 15, 5324 (2005).
[2005CC2029] M. Parent, O. Mongin, K. Kamada, C. Katan and M.
Blanchard-Desce, Chem. Commun., 2029 (2005).
[2005CC4333] D. J. V. C. Van Steenis, O. R. P. David, G. P. F. Van
Strijdonck, J. H. Van Maarseveen and J. N. H. Reek, Chem.
Commun., 4333 (2005).
[2005EJOC3182] J. A. F. Joosten, N. T. H. Tholen, F. A. Ei Maate, A. J.
Brouwer, G. W. Van Esse, D. T. S. Rijkers, R. M. J. Liskamp
and R. J. Pieters, Eur. J. Org. Chem., 3182 (2005).
[2005JACS1360] H. Brand, P. Mayer, K. Polborn, A. Schulz and J. J. Weigand,
J. Am. Chem. Soc., 127, 1360 (2005).
[2005JOC4993] A. R. Katritzky, R. Jiang and K. Suzuki, J. Org. Chem., 70,
4993 (2005).
[2005JOC7792] A. R. Katritzky, N. K. Meher and S. K. Singh, J. Org. Chem.,
70, 7792 (2005).
[2005JOC9521] P. Brandi, C. Galli and P. Gentili, J. Org. Chem., 70, 9521

158
(2005).
[2005MRMC449] C. Asche, Mini-Rev. Med. Chem., 5, 449 (2005).
[2005SL1656] A. R. Katritzky, K. Suzuki and Z. Wang, Synlett, 1656 (2005).
[2005S397] A. R. Katritzky, P. Angrish, D. Hür and K. Suzuki, Synthesis,
397 (2005).
[2005TJC107] N. Sarikavakli, I. Nursabah and G. Irez, Turk. J. Chem., 29,
107 (2005).
[2006AA173] J. A. McCourt and R. G. Duggleby, Amino Acids, 31, 173
(2006).
[2006ARKIVOC43] A. R. Katritzky, S. K. Singh, N. K. Meher, J. Doskocz, K.
Suzuki, R. Jiang, G. L. Sommen, D. A. Ciaramitaro and P. J.
Steel, ARKIVOC, 5, 43 (2006).
[2006IJC1126] B. Chandrama, C. Santanu and M. Soma, Indian Journal of
Chemistry, Section A: Inorganic, Bio-inorganic, Physical,
Theoretical & Analytical Chemistry, 45A, 1126 (2006).
[2006JACS4238] B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 128,
4238 (2006).
[2006JACS10596] H. Ishikawa, G. I. Elliott, J. Velcicky,Y. Choi and D. L. Boger,
J. Am. Chem. Soc., 128, 10596 (2006).
[2006JAPS2161] I. H. Tavman, J. Appl. Polym. Sci., 62, 2161 (1996).
[2006JOC3364] A. R. Katritzky, S. K. Singh, C. Cai and S. Bobrov, J. Org.
Chem., 71, 3364 (2006).
[2006JOC9861] A. R. Katritzky, K. N. B. Le, L. Khelashvili, and P. P.

159
Mohapatra, J. Org. Chem., 71, 9861 (2006).
[2006JOC9051] A. R. Katritzky, N. M. Khashab, N. Kirchenko and A. Singh, J.
Org. Chem., 71, 9051 (2006).
[2006JOC9548] Dolman, S. J.; Gosselin, F.; O’Shea, P. D.; Davies, I. W. J.
Org. Chem., 71, 9548 (2006).
[2006MCP132] G. D. Liang and S. C. Tjong, Mater. Chem. Phys., 100, 132
(2006).
[2006S411] A. R. Katritzky, P. Angrish and K. Suzuki, Synthesis, 3, 411
(2006).
[2006S3231] A. R. Katritzky, S. R. Tala and S. K. Singh, Synthesis, 3231
(2006).
[2006S4135] A. R. Katritzky and P. Angrish, Synthesis, 4135 (2006).
[2006SC3287] Z. Li, Y. Xing, X. Ma, S. Xiao and Z. Lu, Synth. Commun., 36,
3287 (2006).
[2006Steroids660] A. R. Katritzky and P. Angrish, Steroids, 71, 660 (2006).
[2006T10223] S. Cesarini, N. Colombo, M. Pulici, E. R. Felder and W. K.-D.
Brill, Tetrahedron, 62, 10223 (2006).
[2006TJC563] I. Babahan, H. Anil and N. Sarikavakli, Turk. J. Chem., 30(5),
563 (2006).
[2006TL105] Y. Wang, D. R. Sauer and S.W. Djuric, Tetrahedron Lett., 47,
105 (2006).
[2006TL3767] X. Zou, X. Wang, C. Cheng, L. Kong and H. Mao,
Tetrahedron Lett., 47, 3767 (2006).

160
[2006TL4827] H. A. Rajapakse, H. Zhu, M. B. Young and B. T. Mott,
Tetrahedron Lett., 47, 4827 (2006).
[2006TL6497] C. O. Kangani, D. E. Kelley and B. W. Day, Tetrahedron Lett.,
47, 6497 (2006).
[2007ACIE1018] J.-F. Lutz, Angew. Chem. Int. Ed., 46, 1018 (2007).
[2007BC994] A. R. Katritzky, P. Angrish and T. Narindoshvili, Bioconjugate
Chem., 18, 994 (2007).
[2007BMCL2340] C. Valente, R. Moreira, R. C. Guedes, J. Iley, M. Jaffarc and
K. Douglasc, Bioorg. Med. Chem. Lett., 15, 2340 (2007).
[2007USP085073] H. Inoue, M. Egawa and S. Seo, U. S. Pat., 085073; Chem.
Abstr., 146, 402092 (2007).
[2007COC959] P. Quadrelli and P. Caramella, Curr. Org. Chem., 11, 959
(2007).
[2007DP8] M. Szymczyk, A. El-Shafei and H. S. Freeman, Dyes and
Pigments, 72, 8 (2007).
[2007DP753] E. Wolarz, E. Chrzumnicka, T. Fischer and J. Stumpe, Dyes
Pigments, 75, 753 (2007).
[2007DP641] Y. Qian, Z. Lu, C. Lu, Z. Chen and Y. Cui, Dyes Pigments,
75, 641 (2007).
[2007EJOC1244] S. Kotha, K. Mandal, S. Banerjee and S. M. Mobin, Eur. J.
Org. Chem., 1244 (2007).
[2007EOTP649] F. J. Alcain, and J. M. Villalba, Expert Opin. Ther. Pat. 17,
649 (2007).

161
[2007EJMC235] A. A. Kadi, N. R. El-Brollosy, O. A. Al-Deeb, E. E. Habib, T.
M. Ibrahim and A. A. El-Emam, Eur. J. Med. Chem., 42, 235
(2007).
[2007EJMC893] S. G. Kuecuekguezel, I. Kuecuekguezel, E. Tatar, S. Rollas,
F. Sahin, M. Guelluece, E. De Clercq and L. Kabasakal, Eur.
J. Med. Chem., 42, 893 (2007).
[2007EJMC934] A. I. Hashem, A. S. A. Youssef, K. A. Kandeel and W. S.
Abou-Elmagd, Eur. J. Med. Chem., 42, 934 (2007).
[2007JAPS1038] L. Wan, Y. Luo, L. Xue, J. Tian, Y. Hu, H. Qi, X. Shen, F.
Huang, L. Du and X. Chen, J. Appl. Polym. Sci., 104, 1038
(2007).
[2007JAPS2436] M. D. Dasture and D. S. Kelkar, J. Appl. Polym. Sci., 106,
2436 (2007).
[2007JOC407] A. R. Katritzky, H. Tao, R. Jiang, K. Suzuki and K.
Kirichenko, J. Org. Chem , 72, 407 (2007).
[2007JOC4268] A. R. Katritzky, P. P. Mohapatra, D. Fedoseyenko, M.
Duncton and P. J. Steel, J. Org. Chem., 72, 4268 (2007).
[2007JOC5802] A. R. Katritzky, K. Widyan and K. Kirichenko, J. Org. Chem.,
72, 5802 (2007).
[2007JFC740] N. V. Vasil'ev, D. V. Romanov, A. A. Bazhenov, K. A.
Lyssenko and G. V. Zatonsky, J. Fluorine Chem., 128, 740
(2007).
[2007JMC6730] C. Reeh, J. Wundt and B. Clement, J. Med. Chem., 50, 6730

162
(2007).
[2007JMS(PA)PAC175] L. Wan, J. Tian, J. Huang, Y. Hu, F. Huang and L. Du, J. M.
Sci., Part A: Pure and Appl. Chem., 44, 175 (2007).
[2007MRC15] W. H. Binder and R. Sachsenhofer, Macromol. Rapid.
Comm., 28, 15 (2007).
[2007MRMC481] L. Garuti, M. Roberti and D. Pizzirani, Mini-Rev. Med. Chem.,
7, 481(2007).
[2007PAT556] J. Tian, L. Wan, J. Huang, Y. Hu, F. Huang and L. Du, Polym.
Adv. Tech., 18, 556 (2007).
[2007PEP213] V. Weiser, N. Eisenreich, A. Koleczko and E. Roth,
Propellants, Explosives, Pyrotechnics, 32(2), 213 (2007).
[2007PPTE667] J. Anuar, M. Mariatti and H. Ismail, Polym.-Plast. Tech. Eng.,
46, 667 (2007).
[2007PPTE1201]
J. Anuar, M. Mariatti and H. Ismail, Polym.-Plast. Tech. Eng.,
46, 1201 (2007).
[2007S3141] A. R. Katritzky, K. N. B. Le and P. P. Mohapatra, Synthesis,
3141 (2007).
[2007SC1201] M. Dabiri, P. Salehi, M. Baghbanzadeh, M. A. Zolfigol and M.
Bahramnejad, Synth. Comm., 37, 1201 (2007).
[2007TL1549] A. Souldozi and A. Ramazani, Tetrahedron Lett., 48, 1549
(2007).
[2008ARKIVOC62] A. R. Katritzky, P. P. Mohapatra and L. Huang, ARKIVOC, ix,
62 (2008).

163
[2008CPD1001] K. C. Fylaktakidou, D. J. Hadjipavlou-Litina, K. E. Litinas, E.
A. Varella and D. Nicolaides, Curr. Pharm. Design, 14,
1001(2008).
[2008CBE933] S. Y. Fu, X. Q. Feng, B. Lauke and Y. Mai, Compos. B: Eng.,
39, 933 (2008).
[2008JPS(A)PC238] A. R. Katritzky, N. K. Meher, S. Hanci, R. Gyanda, S. R. Tala,
S. Mathai, R. S. Duran, S. Bernard, F. Sabri, S. K. Singh, J.
Doskocz and D. A. Ciaramitaro, J. Polym. Sci. Part A: Polym.
Chem., 46, 238 (2008).
[2008JPS(A)PC2316]
A. Nagal, Y. Kamel, X. Wang, M. Omura, A. Sudo, H.
Nishida, E. Kawamoto and T. Endo, J. Polym. Sci. Part A:
Polym. Chem., 46, 2316 (2008).
[2008JPS(A)PC2897] K. Takizawa, H. Nulwala, R. J. Thibault, P. Lowenhielm, K.
Yoshinaga, K. Wooley and C. J. Hawker, J. Polym. Sci. Part
A: Polym. Chem., 46, 2897 (2008).
[2008OBC637] M. A. Colucci, G. D. Couch and C. Moody, J. Org. Biomol.
Chem., 6, 637 (2008).
[2008OBC2400] A. R. Katritzky, Q.-Y. Chen and S. R. Tala, Org. Biomol.
Chem., 6, 2400 (2008).
[2008POC133] K. Strzelec and P. Pospiech, Prog. Org. Coat., 63, 133
(2008).
[2009JAE269] D. Gopi, K. M. Govindaraju, V. Collins Arun Prakash, V.
Manivannan and L. Kavitha, J. Appl. Electrochem., 39, 269

164
(2009).
[2009JMPT1729] H. Lee, B. Park and H. Jeong, J. Mater. Process Tech., 209,
1729 (2009).
[2009JOC7165] A. R. Katritzky, S. Tala, N. Abo-Dya, K. Gyanda, B. El-Gendy,
Z. Abdel-Samii and P. Steel, J. Org. Chem, 74, 7165 (2009).
[2009JPS(A)PC3748] A. R. Katritzky, Y. Song, R. Sakhuja, R. Gyanda, N. K.
Meher, L. Wang, R. S. Duran, D. A. Ciaramitaro and C. D.
Bedford, J. Poly. Sci., Part A: Poly. Chem., 47, 3748 (2009).
[2009ME367] S. Pandija, D. Roy and S. V. Babu, Microelec. Eng., 86, 367
(2009).
[2009RCB487] A. B. Sheremetev, N. S. Aleksandrova, T. R. Tukhbatshin, S.
V. Penzin and P. A. Belyakov, Russ.Chem. Bull. Int. Ed., 58,
487 (2009).
[2009USP0270440A1] B. Clement and C. Reeh, U. S. Pat., 0,270,440 A1 (2009).
[2010CR1564] A. R. Katritzky and S. Rachwal, Chem. Rev., 110, 1564
(2010).
[2010JAPS121] A. R. Katritzky, R. Sakhuja, L. Huang, R. Gyanda, L. Wang,
D. C. Jackson, D. A. Ciaramitaro, C. D. Bedford and R. S.
Duran, J. Appl. Poly. Sci., 117, 121 (2010).
[2010JAPS2612] L. Wang, R. Gyanda, R. Sakhuja, N. K. Meher, S. Hanci, K.
Gyanda, S. Mathai, F. Sabri, D. A. Ciaramitaro, C. D.
Bedford, A. R. Katritzky and R. S. Duran, J. Appl. Poly. Sci.,
117, 2612 (2010).

165
[2010JOC3938] A. R. Katritzky, K. Bajaj, M. Charpentier and E. G. Zadeh, J.
Org. Chem., 75, 3938 (2010)
[2010OBC2316] A. R. Katritzky, S. R. Tala and N. Abo-Dya, Org. Biomed.
Chem., 8, 2316 (2010).
[2010S2011] A. R. Katritzky, L. Huang, R. Sakhuja, Synthesis, 12, 2011
(2010).
[2010SL1337] A. R. Katritzky, N. Abo-Dya, S. R. Tala, E. G. Zadeh, K. Bajaj
and S. A. El-Feky, Synlett, 9, 1337 (2010).

166
BIOGRAPHICAL SKETCH
Longchuan Huang was born in October 1981, in Yuantan, Anhui province, China.
She was the first of daughter of Fayuan Huang and Miaorong Zhu. From 1986 to 1997,
she attended Yuantan Primary school and later Yuantan High School. Afterward, she
did her undergraduate studies at Beijing Institute of Petrochemical Technology (BIPT)
where she received a Bachelor of Science in Polymer Chemistry. Upon graduation in
July 2001, she attended Hongkong Polytechnic University, Hangzhou campus majoring
in Hotel and Tourism Management, and graduated with a Master of Science degree in
July 2003. She worked briefly in Crown Plaza (Beijing) shortly where she realized her
interest is not in the hospitality industry. She started applying for graduate schools in the
USA, while meanwhile working as a part-time English teacher at Xicheng Foreign
Language School of Beijing. In 2004, she received an admission offer from Florida
Institute of Technology (FIT) (Melbourne, FL) with full teaching assistantship for a
Master’s degree in Chemistry. After some preparation, she traveled to the USA in
January 2005, and started her studies and research specializing in bioorganic chemistry
in Dr. Nasri Nesnas’s laborotary, where she synthesized a type of goldfish pheromone.
She completed her MS in Chemistry from FIT in December 2006, then she joined
Professor Alan R. Katritzky’s research group at the Florida Center for Heterocyclic
Chemistry, University of Florida, pursuring her Doctoral research studies in the
synthesis of heterocyclic compounds. In January 2011, she looks forward to joining the
group of Professor Amos B. Smith III at University of Pennsylvania as a postdoctoral
research associate.
Recommended

















