
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
A consistent multiscale bridgeconnecting atomistic and
coarse-grained models
W. G. Noid
Department of ChemistryPenn State University

AcknowledgementsProf. Gregory A. VothDr.s Yanting Wang, Pu Liu, and Vinod Krishna
Prof.s Gary S. Ayton and Jhih-Wei Chu
Prof. H.C. Andersen
Funding: NIH Ruth L. Kirschstein NRSA postdoctoral fellowship
Collaborative Research in Chemistry grant (CHE-0628257)
Dr. Buddhadev Maiti
Daryl Mains, Wayne Mullinax

“Here we tackle the [protein folding] problem differently. First, we simplify the representation of a protein by averaging over fine details. This is done both to make the calculations much more efficient and also to avoid having to distinguish between many conformations that differ only in these finer details. Second, we simulate the folding of this simple structure …”
“Our method … is based on two assumptions: (1) that much of the protein’s fine structure can be eliminated by averaging, and (2) that the overall chain folding can be obtained by considering only the most effective variables (those that vary most slowly yet cause the greatest changes in conformation).”
Inspiration

Outline0. Motivation
1. The CG Mapping
2. Consistent CG Models
3. Multiscale Coarse-graining(MS-CG) Variational Principle
4. Many-body effects
5. Numerical Illustrations
6. Transferability
Noid Group
Voth Group

CG MappingAtomistic CG
MappingOperator
The mapping operator transforms an atomistic configuration onto a CG configuration by defining the coordinates of each site as a linear combinationof the coordinates defining each site.
MRN : rn a RN =M R
N(rn)
R I =M RI (rn)= cIiri
i∑
rn RN
R I

Consistent CG ModelsAtomistic Configuration Space CG Configuration Space
For a consistent CG model that reproduces the distribution of structures generated by the atomistic model, the appropriate CG potential is a many-body PMF.
Consistency
Noid, Chu, …, Voth, AndersenJ Chem Phys (2008)
pr (rn )∝ e−u(rn ) kBT PR (R
N )∝ e−U (RN ) kBT
PR (RN )=pR (R
N )
e−U (RN ) kBT ∝ drnpr(rn)d M R
N(rn)−RN( )∫
rn RN
MRN

Mean Force Field
In a consistent model, the CG force field is the conditioned expectation value of the atomistic force field (I.e., the mean force field).
RN
rN
r1n
r2n
r3n
R1N = MR
N (r1n) = MR
N (r2n) = L
fI (r1n)
fI (r2n)
fI (r3n)
FI (R1N )
R1N
FI (RN )=
−∂U(RN )∂R I
=fI (r
n )d M RN(rn)−RN( )
d M RN(rn)−RN( )
=E fI (rn) M R
N(rn)=RN⎡⎣ ⎤⎦
fI (rn )= fi(r
n)i∈I∑
Atomistic FF:

MS-CG Variational Principle for the PMF
c 2 ′F[ ] =13N
′FI MRN (rn )( ) − fI (r
n )2
I =1
N
∑= χ 2 F[ ] + ′F − F 2
c F[ ] = F − fχ ′F[ ] = ′F − f
The MS-CG variational principle determines the many-body PMF through a geometric optimization problem in the space of CG force fields.
Noid, Chu, …, Voth, AndersenJ Chem Phys (2008)
′F
F
f
Space of force fields of r n (atomistic)
Space of force fields of R N (CG)
c F[ ]c ′F[ ]
′F − F

Linear Least Squares ProblemFMS (RN )= FDG D (R
N )D∑
FMS (RN )
FDGD (R
N )
MS-CG FF
FF parameters
FF basis fcns
c 2 F( ) =13N
FDG I ,D (MRN (rn )) − fI (r
n )D∑
2
I =1
N
∑
′F
F
Space of CG FF
FMS
GD
G ′D
Space of CG FF spanned by GD{ }
The MS-CG variational principle determines by projecting the PMF onto the space of CG force fields spanned by the given basis.
FD

Simple Liquid AnalogFIJ =R̂ IJF(RIJ)
= FD R̂ IJd(RIJ −RD )D∑
FI = FDG I ,D (RN )
D∑
G I ,D = R̂ IJd(RIJ −RD )J∑
R [nm]
RD
FD
F(R)Pair force
RD
R ¢D
R̂IJ
GI , ¢D
GI ,D
R̂IK
I
J ¢J
K
¢K
For a simple liquid with central pair potentials, the basis function describes the distribution of sites at a given distance, , around a given site. RD
GI ,D

Role of Three-Body Correlations
GD ⋅f=G D ⋅F= G D ⋅G ′D( )F ′D′D
∑Projecting the atomistic FF onto the CG basis
− ′w (R) = F(R) + d ′R F( ′R )ρ [3]( ′R ;R)∫pair MF
CG pair force
direct indirect
conditioned 3-particle density
Projecting the atomistic force field onto a central pair potential results in the Yvon-Born-Green equation.
Yvon-Born-Green Equation
R ¢R
J K
I
Noid, Chu, Ayton, Voth. J Phys Chem B (2007)

Role of 3-Body Correlations II− ′w (R) = F(R) + d ′R F( ′R ) cosθ ( ′R ;R)∫
cosθ( ′R ;R)
F− ′w
R ¢R
J K
I
θ( ′R ;R)
The optimal approximation to the many-body PMF is obtained by treating the non-orthogonality (3-particle correlations) among the basis functions.
Noid, Chu, Ayton, Voth. J Phys Chem B (2007)

Results - MethanolCG pair potential CG pair structure
Calculations by Pu LiuNoid, Liu, Wang, …, Andersen, Voth. J Chem Phys (2008)
The approximate decomposition of the many-body PMF into central pair potentials quantitatively reproduces the pair distribution function.

Consistent Momenta Distribution
Restrictions upon the model:
1. CG Mapping: No atom can be involved in more than one site.
2. Site Masses:
In order to generate the correct momenta distribution, the mass of the CG site is equal to the net mass of the associated atoms only for the center-of-mass mapping.
Calculation by Pu Liu.Noid, Liu, Wang, …, Andersen, Voth. J Chem Phys (2008)
M I =cIi
2
m ii∑⎛⎝⎜
⎞⎠⎟
−1

EMIM+ cation
NO3- anion
Results - Ionic Liquids1-ethyl-3-methylimidazolium nitrate (EMIM+/NO3
-) ionic liquid (IL) pair
CG Potential
The many-body PMF is approximated with a CG potential including both bonded and pair-additive non-bonded terms.
Calculations by Yanting WangNoid, Liu, Wang, …, Andersen, Voth. J Chem Phys (2008)
U(RN )≈ U IJnb(RIJ)+
QIQJ
4pe0RIJ
⎡⎣⎢
⎤⎦⎥I >J
pairs
∑
+ U ib(di)+
i
bonds
∑ U iθ (θi)+
i
angles
∑ U iψ (ψ i)
i
dihedrals
∑
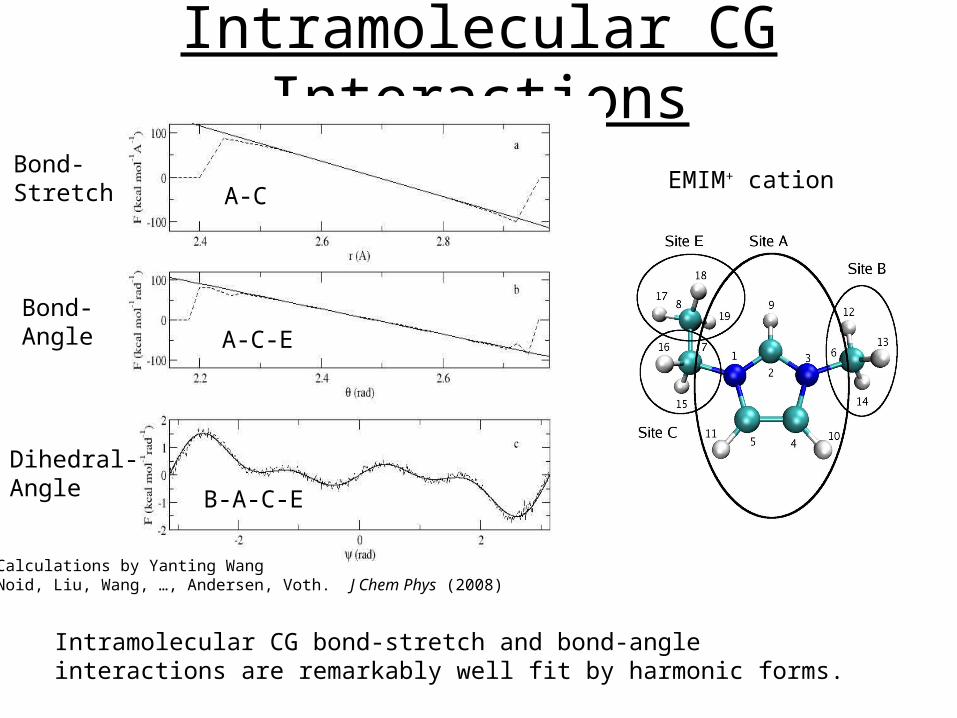
Intramolecular CG Interactions
EMIM+ cationBond-Stretch
Bond-Angle
A-C
A-C-E
B-A-C-E
Dihedral-Angle
Calculations by Yanting WangNoid, Liu, Wang, …, Andersen, Voth. J Chem Phys (2008)
Intramolecular CG bond-stretch and bond-angle interactions are remarkably well fit by harmonic forms.

Intermolecular CG InteractionsShort-ranged Nonbonded
A-A A-D
D-D E-E
EMIM+ cation
NO3- anion
Calculations by Yanting WangNoid, Liu, Wang, …, Andersen, Voth. J Chem Phys (2008)
CG pair potentials are primarily repulsive but not Lennard-Jones in form.

Bond-Stretch
Bond-Angle
Dihedral-Angle
A-C
A-C-E
B-A-C-E
EMIM+ cation
Intramolecular CG Distributions
CG
Atomistic
exp[−U iψ (ψ i) / kBT]
Calculations by Yanting WangNoid, Liu, Wang, …, Andersen, Voth. J Chem Phys (2008)
While the bond-stretch and bond-angle distributions are nearly quantitatively reproduced, the bond dihedral angle distribution is not well reproduced.

EMIM+ cation
NO3- anion
Intermolecular CG Distributions
CGAtomistic
Calculations by Yanting WangNoid, Liu, Wang, …, Andersen, Voth. J Chem Phys (2008)
A-A A-D
D-D E-E
Nonbonded pair distributions in the IL system are reproduced with reasonable accuracy.

Temperature Transferability
%cT2 F(RN ;T )⎡⎣ ⎤⎦=
13N
FI (RN ;T ) − λ (rn;T , ′T )fI (r
n )2
I =1
N
∑′T
Krishna, Noid, Voth (In preparation)
F(R)
R
dF(R)

Summary• CG models “liberate” us from the shackles of atomistic MD, but may be misleading
unless they are consistent with the underlying physics.
• The MS-CG variational principle, in principle, determines the many-body PMF, which
is the appropriate potential for a consistent CG model.
• When implemented with a finite basis set, the MS-CG variational principle
determines an optimal approximation to the many-body PMF.
• Many-body correlations must be considered when determining an optimal pair
decomposition of the many-body PMF.
• The MS-CG method systematically incorporates such correlations in determining a
molecular CG force field.
• A generalized MS-CG variational principle incorporates temperature transferrability.
Recommended

















