
HAL Id: tel-02010574https://tel.archives-ouvertes.fr/tel-02010574
Submitted on 7 Feb 2019
HAL is a multi-disciplinary open accessarchive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come fromteaching and research institutions in France orabroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, estdestinée au dépôt et à la diffusion de documentsscientifiques de niveau recherche, publiés ou non,émanant des établissements d’enseignement et derecherche français ou étrangers, des laboratoirespublics ou privés.
Interactions et assemblages de prolamines du bléJustine Pincemaille
To cite this version:Justine Pincemaille. Interactions et assemblages de prolamines du blé. Ingénierie des aliments. Uni-versité Montpellier, 2018. Français. �NNT : 2018MONTG056�. �tel-02010574�

THÈSE POUR OBTENIR LE GRADE DE DOCTEUR
DE L’UNIVERSITÉ DE MONTPELLIER
En Biochimie et Physico-Chimie Alimentaire
École doctorale GAIA – Biodiversité, Agriculture, Alimentation, Environnement, Terre, Eau
Unité de recherche Ingénierie des Agropolymères et Technologies Emergentes, et Laboratoire Charles Coulomb, Université de Montpellier
Présentée par Justine PINCEMAILLE Le 22 novembre 2018
Sous la direction de Marie-Hélène MOREL, et Laurence RAMOS
Devant le jury composé de
Mr Antoine BOUCHOUX, Chargé de recherche, INRA Toulouse
Mr Denis RENARD, Directeur de Recherches, INRA Nantes
Mr Christophe CHASSENIEUX, Professeur des Universités, Université du Mans
Mme Marie-Hélène MOREL, Directeur de Recherches, INRA Montpellier
Mme Amélie BANC, Maître de Conférence, Université de Montpellier
Mr Paul MENUT, Maître de Conférence, SupAgro Montpellier
Rapporteur
Rapporteur
Président du jury
Directrice de thèse
Co-encadrante
Co-encadrant
Interactions et Assemblages de Prolamines du Blé


!
Interactions!et!Assemblages!de!
Prolamines!du!Blé!
Justine PINCEMAILLE
Thèse de Doctorat
Encadrée par : Marie-Hélène MOREL, Laurence RAMOS,
Amélie BANC et Paul MENUT
Novembre, 2018


Table des matières
Introduction
1
Chapitre 1 – Etat de l’art ……………………………………………….. 7
1. Description des protéines du grain de blé …………………………………. 7
1.1 Classification des protéines de blé ……………………………………………………. 8
1.2 Le gluten ……………………………………………………………………………………... 9
1.3 Les protéines de réserves du blé ………………………………………………………. 11
1.3.1 Les gliadines …………………………………………………………………………. 11
1.3.2 Les gluténines ……………………………………………………………………….. 12
1.4 Interactions des protéines du gluten ………………………………………………… 13
1.5 Extraction des protéines du gluten ………………………………………………….. 15
2. Comportement des protéines en bon solvant ……………………………. 16
2.1 Quelques notions de physique …………………………………………………………. 16
2.1.1 Chaîne idéale et chaîne réelle ………………………………………………….. 16
2.1.2 Polymère en solution ……………………………………………………………… 18
2.1.2.1 Concentration critique de recouvrement …………………………… 18
2.1.2.2 Longueur de corrélation et longueur de persistance ……………. 19
2.1.2.3 Rayon de giration et rayon hydrodynamique …………………….. 20
2.2 Modèle gli+glu ……………………………………………………………………………… 21
2.3 Modèle gli ……………………………………………………………………………………. 22
3. Transition de phases des protéines – diagrammes de phases ………. 23
3.1 Etablissement des diagrammes de phases …………………………………………. 24
3.2 Détermination de la température de points de trouble, Tcloud ………………. 26
3.3 Mécanisme de séparation de phases …………………………………………………. 28
Chapitre 2 – Matériels et méthodes …………………………………. 39
1. Matériel ………………………………………………………………………………. 39
2. Méthodes …………………………………………………………………………….. 39

2.1 Extraction des protéines du gluten ………………………………………………….. 39
2.2 SE-HPLC …………………………………………………………………………………….. 42
2.3 Electrophorèse SDS-PAGE …………………………………………………………….. 44
2.4 Développement d’un outil moyen débit de mesure des points de trouble 46
2.5 Techniques de diffusion aux petits angles …………………………………………. 46
2.5.1 Diffusion de neutrons aux petits angles (SANS) ………………………… 48
2.5.2 Diffusion de rayons X aux petits et grands angles (SAXS – WAXS) 49
2.5.3 Diffusion dynamique de la lumière (DLS) …………………………………. 51
2.6 Calorimétrie à balayage différentielle modulée (MDSC) ……………………… 53
2.7 Microscopie ………………………………………………………………………………….. 55
2.8 Rhéologie …………………………………………………………………………………….. 55
2.9 Fractionnement par flux de forces asymétrique (AsFlFFF) …………………. 58
Chapitre 3 - Mise en place d’un nouvel outil pour la
détermination des points de troubles ……………………………….. 67
1. Définition du besoin ……………………………………………………………… 68
2. Conception et calibration du montage expérimental ………………….. 73
2.1 Dispositif expérimental ………………………………………………………………….. 73
2.2 Contrôle de la température …………………………………………………………….. 74
2.2.1 Plaque lumineuse ………………………………………………………………….. 74
2.2.2 Enceinte ………………………………………………………………………………. 76
2.3 Acquisition et traitement de l’image ………………………………………………… 78
2.3.1 Caméra ………………………………………………………………………………… 78
2.3.2 Traitement de l’information ……………………………………………………. 79
2.3.3 Homogénéité de la plaque lumineuse ………………………………………… 81
2.4 Mesures de transmittance de point de trouble …………………………………… 82
2.4.1 Calibration du niveau de gris ………………………………………………….. 82
2.4.2 Prévenir l’évaporation et la condensation du solvant …………………. 84
3. Validation de l’outil pour la détermination des points de trouble .. 85
3.1 Détermination de la température de transition ………………………………….. 85
3.2 Utilisation d’un système modèle le Tx-114 ……………………………………….. 87
Conclusion ……………………………………………………………………………………. 88

Chapitre 4 – Caractérisations biochimiques des isolats de
protéines du blé ……………………………………………………………..
95
1. Solubilité des protéines du gluten en solvant éthanol/eau ………….. 95
2. Fractionnement des protéines solubles en éthanol/eau ………………. 98
2.1 Effet de la température de trempe sur la partition en masse entre les
phases dense et légère ……………………………………………………………………………………. 99
2.2 Effet de la température de trempe sur la composition protéique des
phases dense et légère ……………………………………………………………………………………. 100
2.3 Rendements du protocole de trempe ………………………………………………… 109
2.4 Préparation des solutions diluées de protéines par filtration ……………….. 110
Conclusion ……………………………………………………………………………………. 112
Chapitre 5 – Mise en évidence d’assemblages -gli+glu et
impact sur les diagrammes de phases ……………………………….. 115
1. Propriétés dynamiques des échantillons modèles ………………………. 115
2. Propriétés dynamiques et structurales des échantillons modèles
fractionnés en fonction du Rh par AsFlFFF ………………………………………. 120
3. Diagrammes de phases ………………………………………………………….. 133
Conclusion ……………………………………………………………………………………. 144
Chapitre 6 – Impact de la composition protéique sur la
rhéologie et la structure d’échantillons dilués et semi-dilués à
température ambiante ……………………………………………………..
149
1. Observations macroscopiques ………………………………………………….. 149
2. Propriétés rhéologiques …………………………………………………………. 151
3. Propriétés des microstuctures ………………………………………………… 152
3.1 En milieu dilué ……………………………………………………………………………… 152
3.2 En milieu semi-dilué……………………………………………………………………….. 155
3.2.1 Diffusion de rayons X …………………………………………………………….. 155
3.2.2 Diffusion de neutrons …………………………………………………………….. 166
Conclusion ……………………………………………………………………………………. 170

Chapitre 7 – Dynamique de séparation de phases liquide-
liquide …………………………………………………………………………..
175
1. Étude de la décomposition spinodale ………………………………. 175
1.1 Microscopie optique ………………………………………………………………………. 175
1.2 Comportement rhéologique au cours de la séparation de phases ………….. 178
2. Observation de la séparation de phases par diffusion …………………. 181
2.1 Dynamique de séparation de phases au cours d’une trempe en
température …………………………………………………………………………………………………. 181
2.2 Cinétique de croissance de la longueur caractéristique ……………………….. 183
2.2.1 Profil classique d’une décomposition spinodale ………………………….. 183
2.2.2 Cinétique de croissance de la longueur caractéristique ………………… 185
2.2.3 Séparation de phases arrêtée …………………………………………………… 188
2.2.4 Facteur de Porod ………………………………………………………………….. 188
2.3 Mise à l’échelle dynamique …………………………………………………………….. 190
2.4 Échantillons enrichis en gluténines ………………………………………………….. 192
Conclusion ……………………………………………………………………………………. 195
Conclusion générale et Perspectives …………………………………. 199
Annexes



Liste des techniques
AsFlFFF Fractionnement par flux de forces asymétrique
DLS Diffusion dynamique de la lumière
LS Diffusion statique de la lumière
MDSC Calorimétrie à balayage différentielle modulée
SANS Diffusion de neutrons à petits angles
SAXS Diffusion de rayons X aux petits angles
SDS – PAGE
Électrophorèse sur gel de polyacrylamide en présence de dodécylsulfate de
sodium
SE-HPLC Chromatographie phase liquide à haute performance par exclusion de taille
UV Spectroscopie Ultra-Violet
VSANS Diffusion de neutrons à très petits angles
WAXS Diffusion de rayons X aux grands angles
Liste des symboles
Taux de décroissance Longueur d’onde
s Viscosité Contrainte de cisaillement
Angle Densité
Taille de blob Exposant de Flory
Longueur de corrélation Paramètre de Flory-Huggins
l0 Longueur de persistance Vitesse de cisaillement
Fraction volumique

Liste des abréviations
A Absorbance N Niveau de gris
A1, A2 Amplitude 1, amplitude 2 Na Nombre d’Avogadro
C Concentration Pguinier Facteur de guinier
C* Concentration critique de
recouvrement
P(q) Facteur de forme
Cc Concentration critique q Vecteur d’onde
C1/C2 Culot 1, Culot 2 Q Débit
d Distance R Rapport
D Coefficient de diffusion Rh Rayon hydrodynamique
df Dimension fractale Rg Rayon de giration
F Coefficient de frottement S Surface
F1, F2, … Fraction 1, Fraction 2, … S1/S2 Surnageant 1, Surnageant 2
G’ Module élastique S(q) Facteur de structure
G’’ Module visqueux t Temps
Gli Gliadines T Température
Glu Gluténines Tc Température critique
HMW-SG Haut poids moléculaire Tcloud Température de points de trouble
I Intensité diffusée Téchantillon Température de l’échantillon
k Constante Tenceinte Température de l’enceinte
K Indice de consistance TP1/TP2 Sonde thermique 1 et 2
KPorod Coefficient de Porod Tr Température transition rhéologie
kB Constante de Boltzmann Tt Température de trouble
l Longueur du trajet optique U Energie de contact
LMW-SG Faible poids moléculaire V ou v Volume
m Masse V Volume exclu
ms Matière sèche Vc Flux linéaire
Mw Masse molaire Ve Volume d’exclusion
n Indice de réfraction Vt Volume total



Introduction
1
Introduction
En 2013, 720 millions d’hectares de céréales ont été cultivés dans le monde (FAO). Parmi ces
céréales, le blé, l’orge, le triticale, le seigle ou encore l’avoine contiennent naturellement du
gluten. Ce dernier a été défini pour la première fois en 1745, par le professeur Giacomo Beccari
en Italie, comme étant « une masse cohésive obtenue après lavage de la farine de blé avec de
l'eau ». Ce n’est que plusieurs années plus tard que le gluten est défini comme un ensemble de
protéines. Le gluten de blé, en particulier, est utilisé dans une large gamme de produits
alimentaires aux textures contrastées (farines, pains, pâtes, biscuits, bières, semoules, …). Ses
propriétés de transformations et sa capacité à répondre aux besoins nutritionnels de la
population font de lui une des premières ressources utilisées sur le marché européen comme
texturant et additif. Le gluten se présente donc comme un bon candidat en substitut potentiel
aux produits animaux. En effet, depuis une dizaine d’années, la consommation de viande dans
le monde (311,8 millions de tonnes en 2014) est en perpétuelle augmentation. Cette dernière
nécessite un coût à la production supérieur aux légumineuses, et l’élevage des animaux
représente la 2e source la plus importante d’émissions de CO2 dans le monde (FAO, 2014). Un
régime alimentaire plus végétal, incluant le gluten, semblerait donc être une solution plus
durable pour l’environnement.
Néanmoins, depuis les années 2000, l’industrie a dû s’adapter à l’accroissement des intolérances
et allergies, en développant des produits sans gluten. En France, 78 millions € ont été dépensés
pour le marché du sans gluten en 2015 qui continue son expansion depuis. Les produits sans
gluten représentent un secteur en forte croissance qui amènent de nombreuses interrogations.
En effet, seulement 1% de la population est réellement touchée par les allergies, et les

Introduction
2
intolérances restent à ce jour non décelables cliniquement. Cependant, depuis 2014 la
Commission Européenne a adopté une nouvelle loi, obligeant un étiquetage particulier
indiquant tout produit contenant du gluten, ce qui représente un véritable étau pour les
industriels. De plus, la suppression totale du gluten de certains produits implique des
modifications texturales et physico-chimiques non négligeables, qui demandent aux producteurs
de s’adapter continuellement pour satisfaire les clients. Une meilleure connaissance du gluten
représente donc un enjeu industriel incontestable : contrôler son comportement, et notamment
les interactions entre les protéines, pourrait permettre le développement de nouveaux produits
tout en respectant les contraintes actuelles.
Pour pallier cette difficulté, une problématique demeure : à ce jour, le gluten constitue une des
familles de protéines les plus complexes du règne végétal et sa structure n’est toujours pas
complétement connue. La difficulté à dissocier les différentes classes de protéines dont les
gliadines et les gluténines rend la caractérisation et la détermination des protéines plus
périlleuse. Cette difficulté sous-entend la présence de nombreux mécanismes au sein du système.
Cela nous amène donc à la réalisation de cette thèse qui a pour but de déterminer la structure
de ces protéines du gluten et de comprendre leurs mécanismes d’associations.
Ce projet de thèse s’inscrit dans la continuité des travaux établis précédemment par l’UMR
IATE, et le laboratoire Charles Coulomb à Montpellier. Les travaux de thèse d’Adeline Boire
avaient permis de mettre en évidence la séparation de phases liquide-liquide que subissent des
solutions riches en gliadines à basse température. Suite à ces observations, des diagrammes de
phases de gliadines ont pu être établis. Cependant, la dépendance de température similaire
observée sur un continuum entre gliadines de haute masse moléculaire ( -gliadines) et
gluténines a suggéré des mécanismes d’interactions similaires, qui n’ont pas été explorés.
D’autre part, Mohsen Dahesh par son travail de thèse axé sur les mécanismes de structuration
du gluten, a pu établir une différence rhéologique entre les modèles gliadines et gliadines +
gluténines. En effet, les systèmes modèles gliadines + gluténines présentaient des
comportements rhéologiques beaucoup plus complexes que les modèles gliadines. Ces modèles
binaires montraient également une gélification spontanée au cours du temps au-delà d’une
certaine concentration. L’étude de structure des échantillons concentrés a présenté des
caractéristiques typiques de gels de polymères en bon solvant. L’objectif de ce travail de thèse
est donc d’acquérir une connaissance et une maîtrise des processus d’assemblages des protéines
du gluten. Nous essayons de comprendre les mécanismes d’association des protéines du gluten

Introduction
3
et de caractériser les structures formées à différentes échelles. L’étude de la composition, des
propriétés rhéologiques et des conformations est rationalisé grâce aux concepts de physique de
la matière molle.
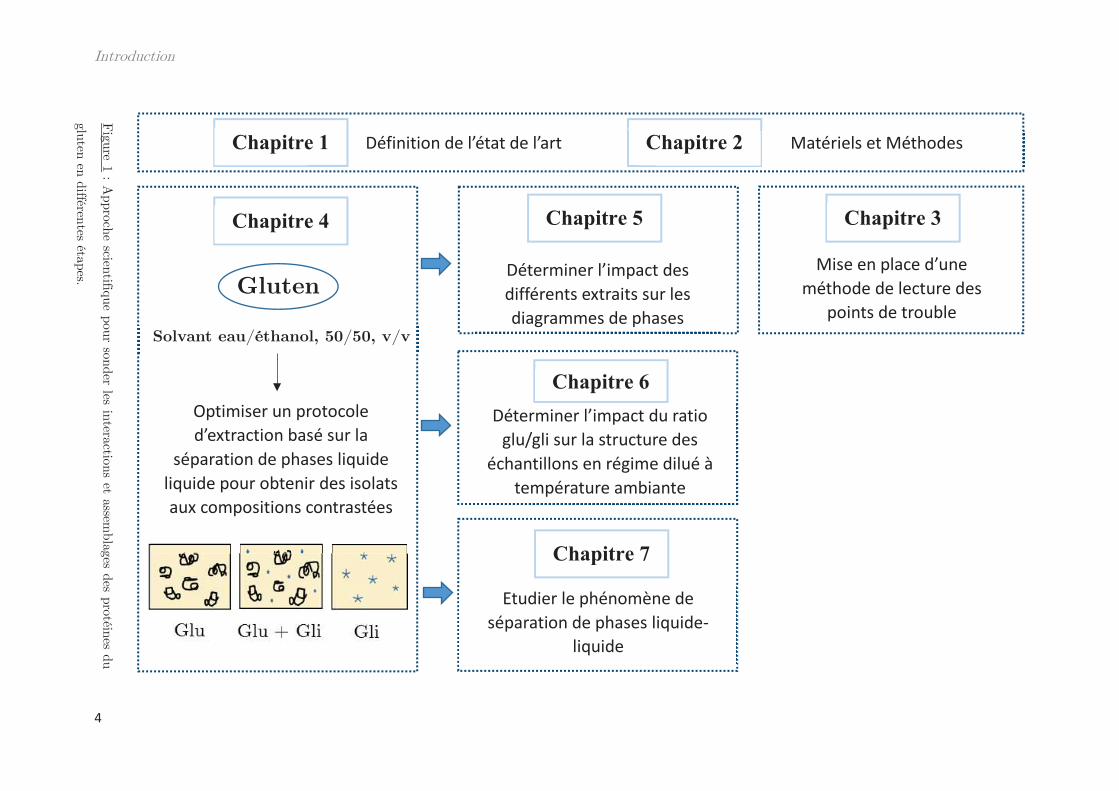
Pour atteindre cet objectif, nous avons développé une approche expérimentale découpée en 3
parties principales (Fig. 1). La première a pour but de pouvoir maîtriser certains paramètres
tels que la composition en protéines. Pour cela, nous avons optimisé le protocole d’extraction
des protéines du gluten afin d’obtenir des fractions protéiques avec un rapport massique
gluténines / gliadines contrôlé. À partir des produits obtenus, la seconde partie tend à moduler
les interactions entre les protéines notamment en jouant sur des paramètres physico-chimiques
tels que la température ou la concentration. La détermination du point de trouble des solutions
protéiques, en fonction de ces différents paramètres, permet d’établir des diagrammes de phases
des différents extraits. De plus, l’étude structurale et rhéologique du matériel permet de
caractériser les interactions entre les protéines. L’interprétation et la modélisation des résultats
se font par des outils et des modèles de la matière molle pour des polymères ou encore des gels.
Enfin, une troisième partie méthodologique, détaille la mise en place d’un montage
expérimental destiné à la lecture des points de trouble des solutions protéiques.
Introduction
4
Déterminer l’impact des
différents extraits sur les
diagrammes de phases
Mise en place d’une
méthode de lecture des
points de trouble
Optimiser un protocole
d’extraction basé sur la
séparation de phases liquide
liquide pour obtenir des isolats
aux compositions contrastées
Chapitre 7
Etudier le phénomène de
séparation de phases liquide-
liquide
Déterminer l’impact du ratio
glu/gli sur la structure des
échantillons en régime dilué à
température ambiante
Chapitre 2Définition de l’état de l’art Chapitre 2 Matériels et Méthodes
Chapitre 6
Solvant eau/éthanol, 50/50, v/v
Gluten
Chapitre 4
Chapitre 1
Chapitre 3Chapitre 5
Figure 1 : A
pproche scientifique pour sonder les interactions et assemblages des protéines du
gluten en différentes étapes.

Introduction
5
Ce manuscrit de thèse est ainsi composé de 7 chapitres :
Le Chapitre 1 est dédié à une étude bibliographique qui a pour but de rappeler les
caractéristiques et les propriétés des protéines du gluten ainsi que leurs caractéristiques physico-
chimiques. Il introduit les connaissances actuelles sur le comportement des gliadines et
gluténines en solution et rappelle les concepts fondamentaux de la physique et de la matière
molle appliqués par la suite à nos échantillons.
Le Chapitre 2 présente le matériel, techniques et méthodes expérimentales utilisés au
cours de la thèse. Il présente notamment le protocole d’extraction des protéines du
gluten qui permet d’obtenir le matériel étudié. Il est complété par le descriptif des
méthodes, avec notamment un bref rappel du fonctionnement de chaque appareil ainsi que les
conditions expérimentales adoptées pour nos expériences.
Par la suite, le Chapitre 3 met en avant le développement d’une nouvelle méthode
d’analyse destinée à établir des diagrammes de phases sur des mélanges complexes. Il décrit
notamment la mise en place de la méthode afin de l’appliquer aux isolats de protéines de blé.
L’application de la méthode sur des modèles déjà connus dans la littérature permet de valider
l’outil et de confirmer son intérêt.
Le Chapitre 4 est axé sur la caractérisation biochimique des isolats de protéines du
blé. Il tend à définir, dans un premier temps, la composition du gluten utilisé pour l’ensemble
des manipulations présenté dans le manuscrit. Dans un second temps, ce chapitre mettra en
avant la caractérisation en protéines des mélanges obtenue suite au protocole d’extraction.
Pour cela, les méthodes types telles que la chromatographie par exclusion de taille et
l’électrophorèse, couramment utilisées sur les protéines de blé, seront exploitées.
Le Chapitre 5 est axé sur la caractérisation structurale des isolats de protéines du
blé en milieu dilué et semi-dilué et l’impact sur les diagrammes de phases. L’objectif
de ce chapitre est de déterminer s’il existe des interactions ou assemblages entre les gliadines
et les gluténines. Pour cela, nous utiliserons l’association de la technique du fractionnement
par flux de force asymétrique avec celle de chromatographie par exclusion de taille.
Le Chapitre 6 présente la rhéologie et la structure des échantillons en solutions à
température ambiante à différentes concentrations. Des méthodes physiques telles que

Introduction
6
la diffusion de neutrons et de rayons X aux petits angles seront utilisées pour définir les tailles
caractéristiques des protéines et montrer s’il existe un impact de la composition protéique sur
ces dernières.
Le Chapitre 7 étudie la dynamique de séparation de phases liquide-liquide. L’objectif
de ce chapitre est de caractériser les phénomènes qui ont lieu au cours de cette séparation à
différentes échelles. Dans un premier temps, le comportement rhéologique des produits sera
étudié à l’échelle macroscopique en fonction de la température puis la décomposition spinodale
sera examinée à l’échelle microscopique via la diffusion de neutrons et de rayons X aux petits
angles.

Chapitre 1 –Etat de l’art
7
Chapitre 1 – Etat de l’art
En 2018, la production mondiale de blé est de 744 millions de tonnes (FAO). Pour cause, grâce
à leur capacité à former une masse cohésive après hydratation, les protéines de réserve que
contient le grain de blé permettent d’obtenir le gluten. Ce dernier présente des propriétés
viscoélastiques largement exploitées par les industriels. Le blé est actuellement l’une des seules
céréales dont on extrait directement le gluten pour le réincorporer comme additif ou adjuvent
dans d’autres produits alimentaires ou dans les farines. Également destiné à l’alimentation
animale et à l’amidonnerie, le blé est la 1ère céréale produite en France. Très présent dans notre
société, il fait l’objet de nombreuses études notamment pour comprendre les mécanismes
texturaux du gluten et définir ses propriétés physico-chimiques. L’objectif de ces études est de
développer de nouveaux produits issus de la culture végétale et d’améliorer ceux existants.
Ce chapitre bibliographique présente les caractéristiques biochimiques des protéines de réserve
du blé ainsi que les études récentes faites sur le comportement de ces protéines en solution. Il
se termine par la présentation des mécanismes de transitions de phases applicables aux
protéines du gluten.
1. Description des protéines du grain de blé
De nombreuses études ont tenté de décrire les protéines du grain de blé car un nombre
considérable d’informations sur ces dernières manquent encore à l’appel. Basé sur différents
paramètres, il existe aujourd’hui plusieurs moyens de les classer.
Cette partie tend à décrire les moyens utilisés pour extraire les protéines du grain, leur
classification et leurs structures.

Chapitre 1 –Etat de l’art
8
1.1 Classification des protéines du blé
En 1907, Osborne établit une description des protéines de blé selon leur fonction dans le grain
puis en fonction de leur solubilité. Il classe ainsi les protéines en 2 grandes familles.
D’une part, les protéines du métabolisme, qui sont les protéines fonctionnelles du grain de
blé, sont composées des albumines (solubles dans l’eau), et des globulines (solubles dans des
solutions salines). Cette classe de protéines représente environ 20% des protéines totales du
grain de blé déterminée sur une base de matière sèche (ms) et plus précisément de 15%
d’albumines et 5% de globulines.
D’autre part, les protéines de réserve sont composées des gliadines, (solubles en milieu
alcoolique) et des gluténines, (soluble en milieu acide ou alcalin). Ces protéines plus connues
sous le nom de prolamines représentent environ 80% des protéines du blé. Cette nomenclature
a ensuite été complétée en 1986, par les travaux de Shewry, qui proposent de les classer selon
la teneur en acides aminés soufrés des protéines insolubles. Plus précisément, ces protéines ont
été classées en sous-catégories : les gliadines, qui sont des protéines monomériques, sont
composées d’ -gliadines qui sont pauvres en soufre, des et -gliadines ainsi que des -gliadines
riches en soufre. Les gluténines forment les prolamines polymériques composées de protéines
de plus hauts poids moléculaires (HMW-SG) et de protéines plus riches en soufre (LMW-SG).
L’ensemble de ces classifications est regroupé dans la Figure 1.1. Les protéines de réserve sont
présentées plus en détails dans la suite du chapitre.

Chapitre 1 –Etat de l’art
9
Figure 1.1 : Classification des protéines de blé par Osborne (1907) et Shewry (1986)
De façon générale, les protéines ne sont pas réparties uniformément dans le grain. Cependant,
pour les plantes de blé, environ 70 à 80% des protéines totales sont retrouvées au niveau de
l’albumen amylacé. Ces teneurs en protéines restent une estimation et varient selon les
conditions climatiques ainsi que les facteurs génétiques (Campbell, 1979). La teneur en gliadines
est plus importante que celle en gluténines lors d’une augmentation du taux de protéines dans
le grain. De plus, l’apport d’azote au grain accroît les teneurs en -gliadines et en gluténines
de hauts poids moléculaires, mais diminue les teneurs en –gliadines et en gluténines de bas
poids moléculaire (Charmet et al., 2017).
1.2 Le gluten
Selon la classification précédente, les protéines de réserve correspondent aux prolamines. Les
prolamines du blé, incluant les gliadines et les gluténines forment un réseau majoritairement
constitué du gluten. Le gluten tient son origine du latin glutinum et signifie « lien », ou
« colle ». Il est essentiellement composé de constituants protéiques (75-85% ms) liés entre eux

Chapitre 1 –Etat de l’art
10
par les ponts disulfures et liaisons hydrogènes (Fig.1.4) ainsi que de quelques granules d’amidon
(10-15% ms) et de lipides (5-8% ms). Le gluten est le produit du lavage d’une pâte de blé sous
un filet d’eau. Cette étape est plus couramment appelée lixiviation. Le complexe protéique
insoluble qui se forme à la suite de cette dernière possède alors des propriétés viscoélastiques
très diverses.
D’abord extrait de la farine de blé par voie humide puis séché pour une meilleure conservation,
les industries utilisent ce produit pour diverses applications. Son premier usage est celui de la
panification où il est ajouté à la farine comme agent correcteur. En effet, la viscoélasticité du
gluten joue un rôle important sur la texture des produits tels que la pâte à pain et a fait l’objet
de nombreuses études. Les résultats ont mis en évidence que les propriétés mécaniques du
gluten dépendent essentiellement des gluténines. Les gliadines une fois hydratées possèdent une
viscosité beaucoup plus faible que celles des gluténines et l’élasticité des gliadines est
beaucoup moins importante que celles des gluténines (Cornec et al., 1994). La ténacité de la
pâte est amplifiée par l’augmentation de la quantité de gluténines par rapport à celle de
gliadines alors que la quantité de gliadines favorise l’extensibilité et le gonflement de la pâte
(Wrigley et al., 2006). Précédemment, des études plus poussées sur les propriétés mécaniques
du gluten ont démontré que la viscosité dépend essentiellement du degré de polymérisation des
gluténines (MacRitchie, 1992, Gupta et al., 1993).
De plus, de par sa capacité à se dissoudre en milieu acide ou basique, le gluten a fait aussi
l’objet de nombreuses recherches sur sa transformation en matériaux biodégradables
(Pallos, et al., 2006) ou encore en biofilms (Feillet, 2000). La conservation des propriétés
mécaniques et la barrière à la vapeur d’eau que présentent les films de gluten (Gontard, et al.,
1993) font de ces derniers un matériau intéressant pour la conservation des produits
alimentaires. Enfin, lorsque le gluten est chauffé de façon trop importante lors de l’étape de
séchage (T > 80°C), les protéines et plus particulièrement les gluténines se dénaturent. Le
gluten perd alors de sa fonctionnalité et devient un produit d’alimentation animale. Ainsi,
le gluten possède des propriétés mécaniques et physiques qui font de lui un matériau utilisé
pour de nombreuses applications mais dont la structure reste à ce jour assez mal définie.

Chapitre 1 –Etat de l’art
11
1.3 Les protéines de réserve du gluten
Les protéines de réserve représentent environ 80% des protéines totales et constituent une
réserve d’azote sous forme d’acides aminés utiles au grain lors de la germination (Wrigley et
al., 2006). D’autres céréales sont également composées majoritairement de prolamines mais
portent un nom différent tel que avénines chez l’avoine, hordéines chez l’orge, sécaline chez le
seigle ou encore zéines chez le maïs. La suite de cette partie s’intéresse à la description des
prolamines du blé qui composent le gluten : les gliadines et les gluténines.
1.3.1 Les gliadines
Les gliadines sont des protéines monomériques de faible poids moléculaire compris entre 30 et
80 kDa. Elles sont divisées en plusieurs classes selon leur mobilité électrophorétique à bas pH
(Woychik et al., 1961) : les -, -, -, et -gliadines. Les - et - gliadines sont généralement
regroupées en une seule classe ( / -gliadines) et représentent par rapport aux gliadines totales
44 à 60%, les –gliadines 30 à 46% et les -gliadines 6 à 20% (Wieser et al., 1994).
La structure primaire des différentes gliadines est très proche et peut être divisée en deux
domaines distincts : un domaine N-terminal, répétitif, riche en résidus prolines et en glutamines
et un domaine C-terminal, non répétitif. Les cystéines, présentes en nombre pair au niveau de
la séquence répétée, sont impliquées dans des liaisons intramoléculaires (Fig.1.4) (Tatham &
Shewry, 1985). Selon Shewry & Tatham, en 1990, les / -gliadines contiennent 6 résidus
cystéines contre 8 pour les -gliadines. Plus tard, une nomenclature selon laquelle les ponts
disulfures intramoléculaires s’établissent entre les cystéines 1 et 4, 5 et 7 ainsi qu’entre les
cystéines 6 et 8 a été proposée par Müller & Wieser, 1995, (Fig.1.2).
Dès 1980, Bietz et al., identifient les -gliadines comme des protéines différentes du reste des
gliadines avec un poids moléculaire spécifique plus important de l’ordre de 60 à 80 kDa (contre
28 à 35 kDa pour les / -gliadines et 31 à 38 kDa pour les -gliadines). Elles se distinguent des
/ - et -gliadines par leur absence de cystéines et de méthionines mais contiennent un nombre
plus important de glutamines, prolines et phénylalanines (Feuillet, 2000). Par leur composition,
les -gliadines sont donc incapables de former des liaisons covalentes via des ponts disulfures
intra-chaînes et forment la classe de protéines la plus hydrophobe. Par ailleurs, il existe deux
variantes principales d’ -gliadines basées sur la composition en acides aminés : les 1,2 et les

Chapitre 1 –Etat de l’art
12
5 (Kasarda et al., 1983). Les gliadines de type 5 ont un poids moyen de 44 à 55 kDa et les
gliadines de type 1,2 de l’ordre de 34 à 44 kDa (Wieser et al., 2000).
Figure 1.2 : Représentation schématique de la structure primaire des différents types de gliadines selon
Shewry & Tatham, 1997. Les bandes grises correspondent aux séquences en acides aminés répétées et les
chiffres aux cystéines établissant des ponts disulfures intramoléculaires.
Outre une structure primaire légèrement différente, la composition en acides aminés des
gliadines influence également la structure secondaire des protéines (Shewry & Tatham, 1990).
Les hélices , nombreuses dans les domaines répétés, et les coudes très présents dans les
domaines non répétés contribuent directement à la conformation de la protéine. En effet, les
hélices sont attribuées à une structure compacte et les coudes à une structure en spirale.
Via des mesures de dichroïsme circulaire, Tatham & Shewry, en 1985, estiment que les / - et
-gliadines contiennent 30-35% d’hélices et 10-20% de coudes . Les -gliadines possèdent
également quelques coudes mais principalement des tours qui ne contiennent pas d’hélices
ni de feuillets . Plus spécifiquement, l’ensemble des coudes est localisé au niveau des
domaines N-terminale et les hélices au niveau des domaines C-terminal.
1.3.2 Les gluténines
Les gluténines représentent 40 à 50% des protéines de réserve de blé. Contrairement aux
gliadines, les gluténines sont des protéines polymériques de haut poids moléculaire compris
entre 500 à 10 000 kDa, qui résultent de la polymérisation de sous-unités. Les gluténines sont
divisées en 2 groupes de sous-unités polypeptidiques : celles de hauts poids moléculaires (HMW-
SG) et celles de faibles poids moléculaires (LMW-SG).
- les LMW-SG sont les sous-unités majoritaires parmi les gluténines et représentent 60 à 80%
des gluténines. Ces dernières peuvent être divisées en trois groupes selon leur poids moléculaire :
B, C et D. Les LMW-SG de type B ont un poids moléculaire de 40 à 50 kDa, celles de types C

Chapitre 1 –Etat de l’art
13
de 30 à 40 kDa et celles de types D de 50 à 70 kDa. De même que pour les gliadines, les LMW-
SG sont composées d’un domaine N-terminal constitué de séquences répétitives et d’un
domaine C-terminal constitué de 8 cystéines capables de faire des liaisons disulfures
intramoléculaires (Fig.1.3), (Wieser, 2007).
- Les HMW-SG représentent la plus faible proportion des protéines du gluten ( 10%) mais
également les plus complexes. Leur séquence en acides aminés est composée de 80 à 100 résidus
au niveau de la partie N-terminal, et contient 3 à 5 résidus cystéines. L’autre partie de la
séquence non répétitive, au niveau C-terminal, est composée de 42 résidus dont 1 cystéine. La
séquence répétée intermédiaire peut être composée de 490 à 700 résidus (MacRitchie, 1992).
Figure 1.3 : Représentation schématique de la structure primaire des différents types de gluténines de
faibles poids moléculaires et de hauts poids moléculaires selon Shewry & Tatham, 1997. Les bandes grises
correspondent aux séquences en acides aminés répétées et les chiffres aux cystéines établissant des ponts
disulfures intramoléculaires.
Comparativement aux gliadines, Field et al., 1987 ont démontré que pour les HMW-SG, les
hélices se trouvent essentiellement dans les séquences non répétées et les coudes dans les
séquences répétées.
1.4 Interactions des protéines du gluten
Quelle que soit la protéine, la localisation des cystéines sur les chaînes polypeptidiques permet
d’expliquer la formation des ponts intra- et intermoléculaires (Keck et al., 1995), (D’Ovidio &
Masci, 2004), (Fig.1.4).

Chapitre 1 –Etat de l’art
14
Figure 1.4 : Représentation des associations des protéines via des liaisons disulfures intra- et
intermoléculaires (Feuillet, 2000).
Outre les liaisons disulfures covalentes, il existe également d’autres types de liaisons telles que
les liaisons hydrogènes non covalentes et les liaisons hydrophobes qui contribuent à la
stabilité des structures (Tatham & Shewry, en 1985).
Bien que le gluten se présente comme un réseau stable, les contraintes mécaniques et physiques
appliquées au gluten peuvent modifier le réseau. Un chauffage de l’ordre de 80°C avec la
présence de 20% d’eau minimum modifie la conformation des gluténines. Ce dernier altère les
liaisons cystéines libres, engendrant de nouvelles liaisons avec les gliadines jusqu’à la formation
de polymères, (Pallos et al., 2006). Plus particulièrement, une étude réalisée sur les protéines
en solvant eau/éthanol 70% (v/v) a démontré que le chauffage affecte la structure secondaire
des protéines (Tatham & Shewry, 1985). Il résulte une perte partielle des hélices des gliadines
qui restent néanmoins très stables grâce à la présence des liaisons hydrogènes. Ces liaisons
hydrogènes seraient également fortement présentes entre les résidus de glutamines des -
gliadines afin de stabiliser la conformation en spirale des protéines. Des interactions
hydrophobes entre les résidus aromatiques renforcent également les tours des -gliadines.
Une étude UV plus poussée des -gliadines a montré que la conformation en tours est aussi
renforcée par la présence de liaisons non-covalentes.
D’après ces différentes observations, les liaisons disulfures joueraient un rôle important : les
ponts que forment les cystéines permettent d’expliquer le comportement rhéologique des
produits alimentaires tels que celui de la pâte à pain. La rupture et la reformation de ces
liaisons engendrent une modification du réseau protéiques qui rend la ténacité et l’élasticité de
la pâte plus ou moins importante. Lors de la formation de la pâte humide, les liaisons non-
Liaisons disulfures intermoléculaires
élasticité
Liaisons covalentes intramoléculaires
viscosité

Chapitre 1 –Etat de l’art
15
covalentes et disulfures permettent de modifier le réseau de gluténines en un réseau de gluten
plus étendu (Bietz & Huebner, 1980). Ainsi, bien que les liaisons disulfures soient à l’origine
d’une grande stabilité des protéines, il n’en demeure pas moins que les liaisons hydrogènes,
hydrophobes et non-covalentes impactent également la conformation qu’adoptent les protéines
du gluten, et contribuent aux propriétés élastiques et cohésives du gluten.
1.5 Extraction des protéines du gluten
Les gliadines sont principalement solubles en milieu alcoolique alors que les gluténines le sont
dans des milieux acides ou alcalins. De nombreuses méthodes ont été mises au point pour
séparer les différentes protéines du gluten mais la plupart sont basées sur un protocole similaire
à celui établi par Osborne en 1907. L’ensemble des protocoles repose sur la mise en solution
des protéines dans différents solvants puis d’une succession de centrifugations. Le nombre
d’extraction, la force de centrifugation ou encore la température sont autant de paramètres qui
permettent d’obtenir des fractions plus ou moins enrichis en gliadines et gluténines. Il est donc
possible d’adapter la méthode d’extraction en fonction de la composition en protéines désirée.
Cependant, la plupart des études ont principalement étudié l’impact du solvant utilisé. La
Table 1.1 résume une partie non exhaustive des solvants testés dans la littérature.
Table 1.1 : Récapitulatif de différents types de solvants utilisés pour extraire les protéines du gluten
(liste non exhaustive)
Protéines solubilisées Extractant Références
Albumines / globulines puis
Gliadines
Chlorure de sodium 0,5M puis
Ethanol 70%
Bietz, et al., 1975
Gliadines issues de la farine Ethanol 55% Charbonnier, 1973
Toutes les protéines du
gluten
Tampon Tris 0,1M- HCl
pH 8,4, 2% de SDS
Bottomley et al., 1982
Toutes les protéines du
gluten
Tampon Tris 0,1M- HCl
pH 7, 2% de SDS
Danno, 1981
Gluténines Isopropanol 50% (v/v) contenant
0,08M Tris-HCl tampon, pH 6,8
Mélas et al., 1994
HMW-gluténines Propanol 60% (v/v) Marchylo et al., 1989
Prolamines Acide chlorhydrique dilué MacRitchie, 1987

Chapitre 1 –Etat de l’art
16
L’analyse des différentes fractions obtenues se fait généralement par chromatographie et
électrophorèse. Divers types d’électrophorèses ont été proposés afin d’optimiser la résolution
des pics mais l’électrophorèse en gel de polyacrylamide (que nous utiliserons par la suite) est
actuellement la plus répandue (Lookhart et al., 1982). Qu’elles soient d’exclusion (Dachkevitch
& Autran, 1989), échangeuses d’ions (Ewart, 1975) ou encore en phase inverse, les méthodes
chromatographiques nécessitent toutes d’ajouter des agents dénaturants afin de solubiliser les
prolamines, comme vu précédemment. Une alternative utilisant un traitement ultrasonore
contrôlé a été proposé pour permettre une optimisation de l’extraction tout en réduisant la
dégradation des gluténines (Morel et al., 2000).
2. Comportement des protéines en bon solvant
Les protéines du blé et plus particulièrement les gluténines sont des macromolécules dont
l’organisation dépend de la conformation qu’elles peuvent adopter dans différents solvants.
Les interactions qu’elles ont avec les gliadines pourraient être à l’origine des diverses propriétés
physico-chimiques qui dépeignent le gluten.
Par leur comportement, il est possible d’appliquer aux protéines du gluten les notions de la
matière molle. Inventée par Madeleine Veyssi dans les années 70, la matière molle désigne
tous systèmes colloïdes, surfactants, tensio-actifs et polymères pour lesquels sont mis en jeu des
énergies d’interactions faibles entre objets (Cousin et al., 2010). Rationaliser le fonctionnement
des gliadines et gluténines par les concepts de la matière molle permettra donc d’expliquer les
interactions entre ces dernières via différents modèles physiques.
2.1 Quelques notions de physiques
2.1.1 Chaîne idéale et chaîne réelle
Le modèle le plus simple pour décrire un polymère est une chaîne dite « idéale ». Les chaînes
idéales sont composées de N monomères de même longueur b qui ne sont pas en interactions
entre eux. Les différents liens entre monomères se font par des rotations libres qui sont une
suite d’événements indépendants. La distance moyenne entre les deux extrémités d’une chaine
de monomères mis bout à bout, <R²> répond à l’égalité <R2> = Nb2.
Dans le cas d’une chaîne idéale, R² est reliée au rayon de giration Rg, d’une particule. Rg qui
résulte de la distance quadratique moyenne des masses constitutives d’un polymère et de son

Chapitre 1 –Etat de l’art
17
centre de masse (Gregory Dewey, 1998) s’exprime comme le rayon de la sphère contenant une
chaîne de polymères et vaut alors <Rg²> = ²
.
Dans le cas d’une chaîne idéale où il existe des interactions, on parle alors de chaîne réelle
ou de chaîne à volume exclu. Ces interactions, qui peuvent être soit répulsives soit
attractives, impactent directement la conformation de la chaîne de polymères. Il existe 2 types
d’interactions : les interactions monomère-solvant et les interactions monomère-
monomère. Le volume exclu, V, rend compte de la distance des monomères les uns par rapport
aux autres lorsque ces derniers sont dans un solvant. En fonction du type de solvant, la chaîne
de polymères aura alors une conformation différente (Fig.1.5).
En diminuant la qualité du solvant, le volume exclu devient nul et les segments de la chaîne
s’attirent. Dans ce cas, la chaîne idéale possède des interactions nettes monomères/monomères
qui deviennent quasi nulles. Les polymères sont alors en solvant : on ne différencie plus
interactions monomères-solvants ou monomères-monomères.
Si le volume exclu est supérieur à 0 les interactions nettes monomères/monomères sont
répulsives et ce sont les interactions monomères-solvants qui sont favorisées. Les polymères
sont alors en bon solvant.
Selon la théorie de Flory, la distance bout à bout R répond à l’équation R bN
l’exposant de Flory qui mesure l’interaction effective nette entre 2 monomères. Le facteur de
prendre différentes valeurs selon la qualité du solvant avec mauvais
Figure 1.5 : Exemple de conformation tridimensionnelle d’une chaîne idéale de polymère dans différents
solvants

Chapitre 1 –Etat de l’art
18
D’autre part, un polymère est un objet fractal aléatoire qui peut prendre différentes dimensions.
dimension fractale,
df, selon laquelle df = . De façon générale, la chaîne idéale est un objet fractal aléatoire de
dimension fractale 2.
2.1.2 Polymère en solution
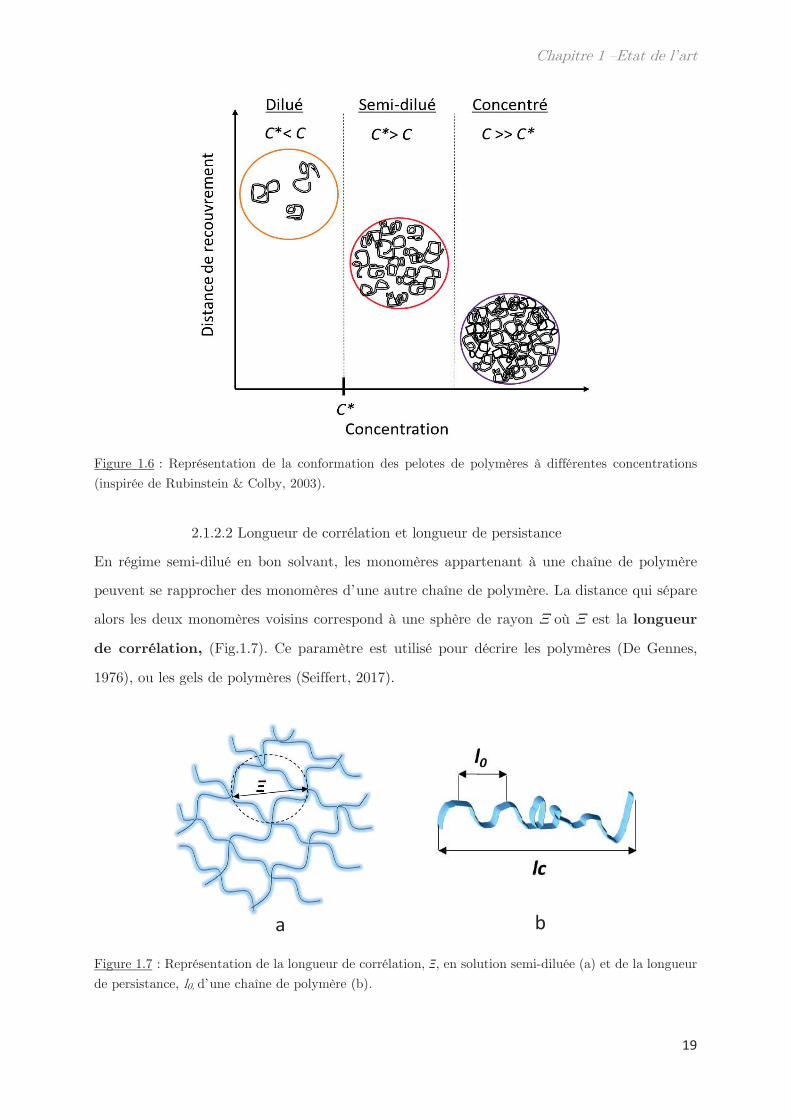
2.1.2.1 Concentration critique de recouvrement
Il existe 2 types de polymères en solutions : les fondus de polymères lorsque qu’il n’y a plus
de solvant ou les solutions de polymères. Nous nous intéressons ici uniquement aux solutions
de polymères qui peuvent être soit en régime dilué soit en régime semi-dilué.
En régime dilué, les chaînes de polymères sont éloignées les unes des autres et ne présentent
pas d’interactions entre elles. Du fait de leur isolement elles ont un comportement similaire à
ceux des polymères en chaînes idéales. Néanmoins, lors de l’augmentation de la concentration,
les interactions de type volume exclu vont devenir plus importantes et les pelotes de polymères
vont alors se rapprocher. Pour une concentration donnée, propre à chaque polymère, les pelotes
de polymères vont entrer en contact et se superposer. Cette concentration correspond à la
concentration critique de recouvrement, C*, qui marque le passage du régime dilué au
régime semi-dilué. Lorsque les interactions sont devenues trop importantes au point que les
pelotes de polymères s’enchevêtrent, la solution passe dans un régime concentré (Fig.1.6). La
concentration critique de recouvrement, C*, est définie par l’équation suivante :
C* =
(1.1)
où Mw est la masse moléculaire et Na le nombre d’Avogadro.
Chapitre 1 –Etat de l’art
19
Figure 1.6 : Représentation de la conformation des pelotes de polymères à différentes concentrations
(inspirée de Rubinstein & Colby, 2003).
2.1.2.2 Longueur de corrélation et longueur de persistance
En régime semi-dilué en bon solvant, les monomères appartenant à une chaîne de polymère
peuvent se rapprocher des monomères d’une autre chaîne de polymère. La distance qui sépare
alors les deux monomères voisins correspond à une sphère de rayon où est la longueur
de corrélation, (Fig.1.7). Ce paramètre est utilisé pour décrire les polymères (De Gennes,
1976), ou les gels de polymères (Seiffert, 2017).
Figure 1.7 : Représentation de la longueur de corrélation, , en solution semi-diluée (a) et de la longueur
de persistance, l0, d’une chaîne de polymère (b).
a b

Chapitre 1 –Etat de l’art
20
La longueur de persistance, l0, est également un paramètre utilisé pour décrire les
macromolécules ou polymères et traduit la rigidité intrinsèque de la chaîne (Fig.1.7). lo est
d’autant plus petit que la chaîne est flexible et peut être relié au rayon de giration, Rg, à partir
des équations établis par Benoit et Doty en 1953 selon lesquelles :
Rg 2 =lo 2 [ + 1 + - ² (1-exp(-a))] avec a = (1.2)
lc correspond à la longueur du contour de la chaîne (Brûlet et al., 1996).
Selon ce modèle Rg 2= lc 2/12 pour des polymères en forme de bâtons (l0>>lc) et Rg 2 =lc*lo/3
pour des pelotes statiques (lc>>l0).
2.1.2.3 Rayon de giration et rayon hydrodynamique
Les dimensions des macromolécules sont très souvent décrites en fonction de leur rayon de
giration et hydrodynamique. En règle générale, Rg est une taille associée au moment d’inertie
des objets et devient un paramètre indispensable dans la détermination du facteur de forme,
P(q), dans le régime de Guinier. En effet, en absence d’interactions, l’intensité au vecteur
d’onde, q, pour lequel q = 0 est proportionnelle au nombre total en masse ou volume d’éléments
diffusés. L’approximation de Guinier permet alors de déduire le facteur de forme selon lequel :
Pguinier(q) = 1 - ² ²
(1.3)
Le rayon hydrodynamique, Rh, ou rayon de Stockes est une grandeur hypothétique selon
laquelle la particule diffuse à la même vitesse qu’une sphère dure. Déterminé généralement par
diffusion dynamique de la lumière, il tient compte de l’hydratation de la molécule et dépend
de la viscosité du solvant dans lequel se déplace la particule. Il peut être calculé à partir de la
relation de Stokes selon laquelle F = 6 s.Rh où F représente le coefficient de frottement subit
par la particule de rayon hydrodynamique Rh, dans un solvant de viscosité s. Le rayon
hydrodynamique d’une protéine telle que le lysozyme est de l’ordre de 2 nm lorsque la protéine
est repliée (Wilkins et al., 1999) et son rayon de giration de l’ordre de 1,5 nm (Chen et al.,
1996).
Chapitre 1 –Etat de l’art
21
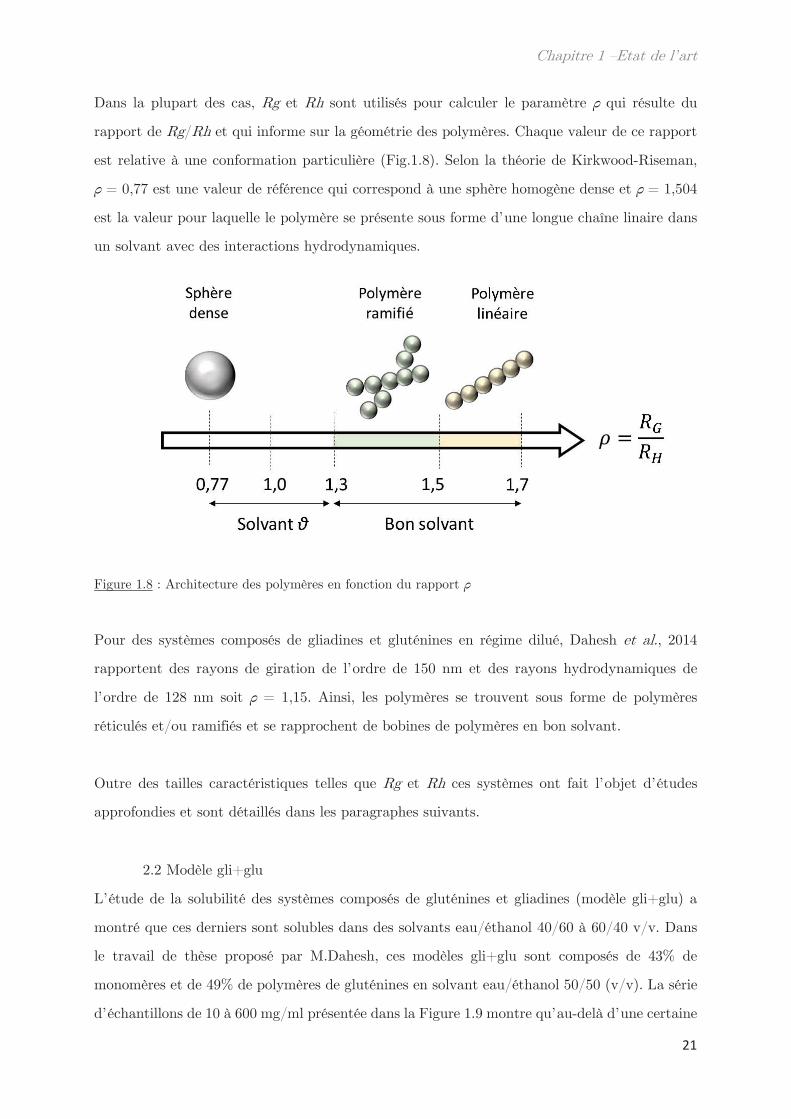
Dans la plupart des cas, Rg et Rh sont utilisés pour calculer le paramètre qui résulte du
rapport de Rg/Rh et qui informe sur la géométrie des polymères. Chaque valeur de ce rapport
est relative à une conformation particulière (Fig.1.8). Selon la théorie de Kirkwood-Riseman,
= 0,77 est une valeur de référence qui correspond à une sphère homogène dense et = 1,504
est la valeur pour laquelle le polymère se présente sous forme d’une longue chaîne linaire dans
un solvant avec des interactions hydrodynamiques.
Figure 1.8 : Architecture des polymères en fonction du rapport
Pour des systèmes composés de gliadines et gluténines en régime dilué, Dahesh et al., 2014
rapportent des rayons de giration de l’ordre de 150 nm et des rayons hydrodynamiques de
l’ordre de 128 nm soit = 1,15. Ainsi, les polymères se trouvent sous forme de polymères
réticulés et/ou ramifiés et se rapprochent de bobines de polymères en bon solvant.
Outre des tailles caractéristiques telles que Rg et Rh ces systèmes ont fait l’objet d’études
approfondies et sont détaillés dans les paragraphes suivants.
2.2 Modèle gli+glu
L’étude de la solubilité des systèmes composés de gluténines et gliadines (modèle gli+glu) a
montré que ces derniers sont solubles dans des solvants eau/éthanol 40/60 à 60/40 v/v. Dans
le travail de thèse proposé par M.Dahesh, ces modèles gli+glu sont composés de 43% de
monomères et de 49% de polymères de gluténines en solvant eau/éthanol 50/50 (v/v). La série
d’échantillons de 10 à 600 mg/ml présentée dans la Figure 1.9 montre qu’au-delà d’une certaine

Chapitre 1 –Etat de l’art
22
concentration, dans un mélange eau/éthanol 50/50 (v/v), les échantillons semblent changer de
couleur et deviennent moins turbides ce qui suppose une évolution du produit avec la
concentration. Pour C > C* (avec C* = 180 g/l) les échantillons présentent une gélification
spontanée qui dépend de la concentration et du temps. Cette gélification est due à la formation
lente de liaisons hydrogènes au sein du système.
Figure 1.9 : Photo d'échantillons de gluten à différentes concentrations en protéines, C, de 10 à 600
mg/ml, dans un mélange eau/éthanol 50/50 (v/v) (Dahesh et al., 2014).
D’autre part, la structure de ces échantillons concentrés présente des caractéristiques de gels
de polymères flexibles en bon solvant. Des mesures SANS ont permis de définir différentes
longueurs caractéristiques telles que les rayons de giration (Rg 116 nm) ou encore la longueur
de persistance (l0 0,7 nm). De plus, les complexes observés montrent une conformation de
chaîne de polymères ramifiée et non linéaire avec une dynamique interne à grands q observée
par diffusion dynamique de la lumière. Les différentes observations ont démontré la présence
d’agrégats dans le milieu sans toutefois détailler la nature de ces derniers.
2.3 Modèle gli
Contrairement aux modèles glu+gli, les modèles composés en majeure partie de gliadines
(modèle gli), (>82%) présentent des propriétés rhéologiques moins complexes. En effet, d’après
les travaux de Mohsen Dahesh ce système a la particularité rhéologique de se comporter comme
un fluide newtonien jusqu’à des concentrations très élevée de l’ordre de 560 mg/ml. D’autre
part, il ne présente aucune transition d’une solution vers un gel. Ces observations amènent
donc à conclure que la gélification des solutions de glu+gli est amenée par les gluténines ou par
leur association avec les gliadines.
Chapitre 1 –Etat de l’art
23
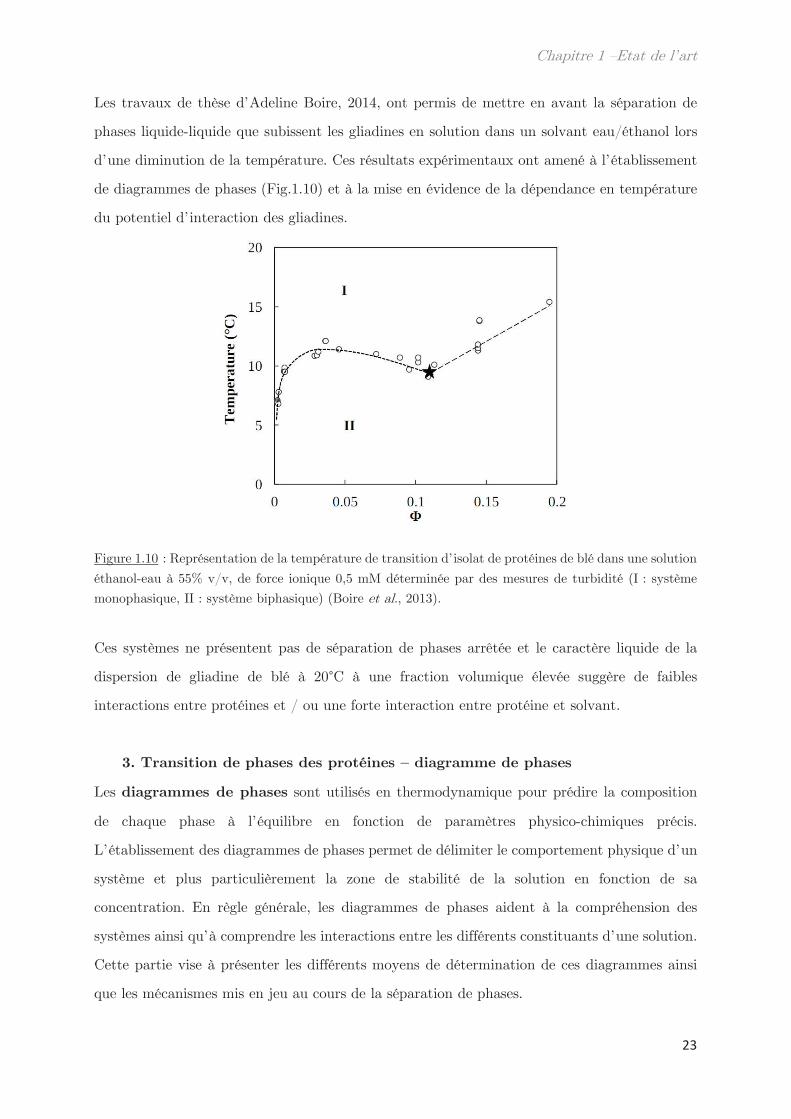
Les travaux de thèse d’Adeline Boire, 2014, ont permis de mettre en avant la séparation de
phases liquide-liquide que subissent les gliadines en solution dans un solvant eau/éthanol lors
d’une diminution de la température. Ces résultats expérimentaux ont amené à l’établissement
de diagrammes de phases (Fig.1.10) et à la mise en évidence de la dépendance en température
du potentiel d’interaction des gliadines.
Figure 1.10 : Représentation de la température de transition d’isolat de protéines de blé dans une solution
éthanol-eau à 55% v/v, de force ionique 0,5 mM déterminée par des mesures de turbidité (I : système
monophasique, II : système biphasique) (Boire et al., 2013).
Ces systèmes ne présentent pas de séparation de phases arrêtée et le caractère liquide de la
dispersion de gliadine de blé à 20°C à une fraction volumique élevée suggère de faibles
interactions entre protéines et / ou une forte interaction entre protéine et solvant.
3. Transition de phases des protéines – diagramme de phases
Les diagrammes de phases sont utilisés en thermodynamique pour prédire la composition
de chaque phase à l’équilibre en fonction de paramètres physico-chimiques précis.
L’établissement des diagrammes de phases permet de délimiter le comportement physique d’un
système et plus particulièrement la zone de stabilité de la solution en fonction de sa
concentration. En règle générale, les diagrammes de phases aident à la compréhension des
systèmes ainsi qu’à comprendre les interactions entre les différents constituants d’une solution.
Cette partie vise à présenter les différents moyens de détermination de ces diagrammes ainsi
que les mécanismes mis en jeu au cours de la séparation de phases.

Chapitre 1 –Etat de l’art
24
3.1 Établissement des diagrammes de phases
D’après la théorie de Flory-Huggins, il existe des contributions énergétiques de types
entropiques et enthalpiques spécifiques à chaque type de polymères. Pour un mélange binaire
composé d’une espèce A et d’une espèce B, l’énergie d’interaction, UA, (ou énergie de paire)
est l’énergie d’interaction d’un monomère de l’espèce A de fraction volumique A avec pour
voisins des monomères de l’espèces B de fraction volumique, B = 1 - A. Cette énergie
d’interaction est définie pour un monomère A comme UA = uAA A + uAB B et comme UB =
uBB B + uAB A pour un monomère B. uAA est l’énergie d’interaction entre deux molécules A,
uBB entre deux molécules B et uAB entre une molécule A et une molécule B. Chaque site du
réseau possède z voisins et un nombre total, n, de sites du système combiné, ce qui amène à
une énergie totale d’interaction du mélange :
U = [UA A+UB B] (1.4)
Soit U = [uAA 2 +2uAB (1- )+uBB (1- )2 ] (1.5)
Le modèle de Flory-Huggins, qui intervient dans l’expression de l’enthalpie, permet de relier le
paramètre de Flory, aux différences des énergies d’interactions. est alors défini comme un
paramètre d’interactions pour un mélange composé de 2 espèces A et B, selon lequel :
=
= E + (1.6)
Avec E représentant la dépendance à la température.
Il a été démontré que dans la plupart des cas, est inversement proportionnel à la température
et dépend de la composition en polymères.
Si F > 0, le polymère est parfaitement soluble au-dessus de la température de séparation de
phases. Le polymère a alors un comportement type UCST (Upper Critical Solution
Temperature).
Si F < 0, le polymère est soluble en-dessous de la température de séparation de phases. Le
polymère a alors un comportement type LCST (Low Critical Solution Temperature)
(Fig.1.11).

Chapitre 1 –Etat de l’art
25
Figure 1.11 : Représentation de diagrammes de phases de type UCST (a) et LCST (b) inspirée de
Teraoka, 2002.
Dans le cas des diagrammes de phases de type UCST, à faible température (et inversement
pour les diagrammes de phases de type LCST), le système se sépare en deux phases : une phase
pauvre en protéines et une phase riche en protéines qui se trouvent dans un équilibre
thermodynamique et dont les compositions sont déterminées par la courbe binodale
(Fig.1.12). En dessous de cette courbe, le système est biphasique et la courbe binodale
représente donc la limite entre un système monophasique et un système biphasique. Cette
séparation de phases peut se faire dans différentes conditions : en fonction de la température,
de la pression osmotique, de la concentration, du pH ou encore de la force ionique. Dans le
cadre de cette thèse, nous étudierons des diagrammes de phases en température en fonction de
la concentration.
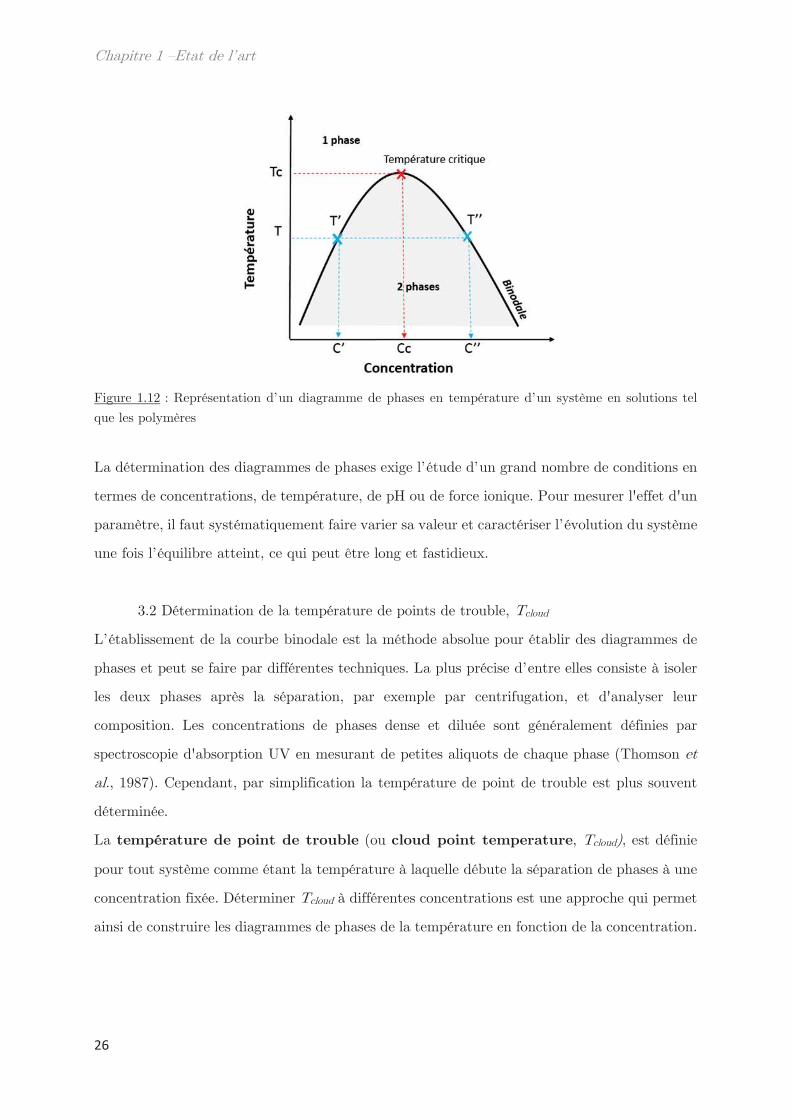
Chaque diagramme de phases est caractérisé par une température critique, Tc, qui est la
limite supérieure de la courbe binodale. À cette température critique correspond une
concentration critique, Cc, qui se lit sur l’axe des abscisses. D’autre part, lorsque le système
a passé la courbe binodale et se retrouve en 2 phases, la phase diluée a une concentration C’
correspondante à une température T’ et la phase concentrée a une concentration C’’ qui
correspond à une température T’’. Il est important de noter que T’ est alors égale à T’’.
Chapitre 1 –Etat de l’art
26
Figure 1.12 : Représentation d’un diagramme de phases en température d’un système en solutions tel
que les polymères
La détermination des diagrammes de phases exige l’étude d’un grand nombre de conditions en
termes de concentrations, de température, de pH ou de force ionique. Pour mesurer l'effet d'un
paramètre, il faut systématiquement faire varier sa valeur et caractériser l’évolution du système
une fois l’équilibre atteint, ce qui peut être long et fastidieux.
3.2 Détermination de la température de points de trouble, Tcloud
L’établissement de la courbe binodale est la méthode absolue pour établir des diagrammes de
phases et peut se faire par différentes techniques. La plus précise d’entre elles consiste à isoler
les deux phases après la séparation, par exemple par centrifugation, et d'analyser leur
composition. Les concentrations de phases dense et diluée sont généralement définies par
spectroscopie d'absorption UV en mesurant de petites aliquots de chaque phase (Thomson et
al., 1987). Cependant, par simplification la température de point de trouble est plus souvent
déterminée.
La température de point de trouble (ou cloud point temperature, Tcloud), est définie
pour tout système comme étant la température à laquelle débute la séparation de phases à une
concentration fixée. Déterminer Tcloud à différentes concentrations est une approche qui permet
ainsi de construire les diagrammes de phases de la température en fonction de la concentration.
Chapitre 1 –Etat de l’art
27
Différentes techniques ont été développées, basées sur l'évolution des propriétés optiques d'une
solution protéique lorsqu'une séparation de phases se produit dans un système initialement
homogène. Si les gouttelettes formées sont suffisamment grandes pour diffuser la lumière visible,
la solution devient trouble. Il est donc possible de déterminer le début de la séparation de
phases avec n'importe quel dispositif qui mesure soit une diffusion accrue d'un échantillon, soit
une diminution de la lumière transmise. La mesure de diminution de l’intensité transmise par
un laser est un moyen efficace de déterminer avec précision la température d'opacification de
la solution, qui correspond donc à la température de point de trouble, Tcloud. De façon générale,
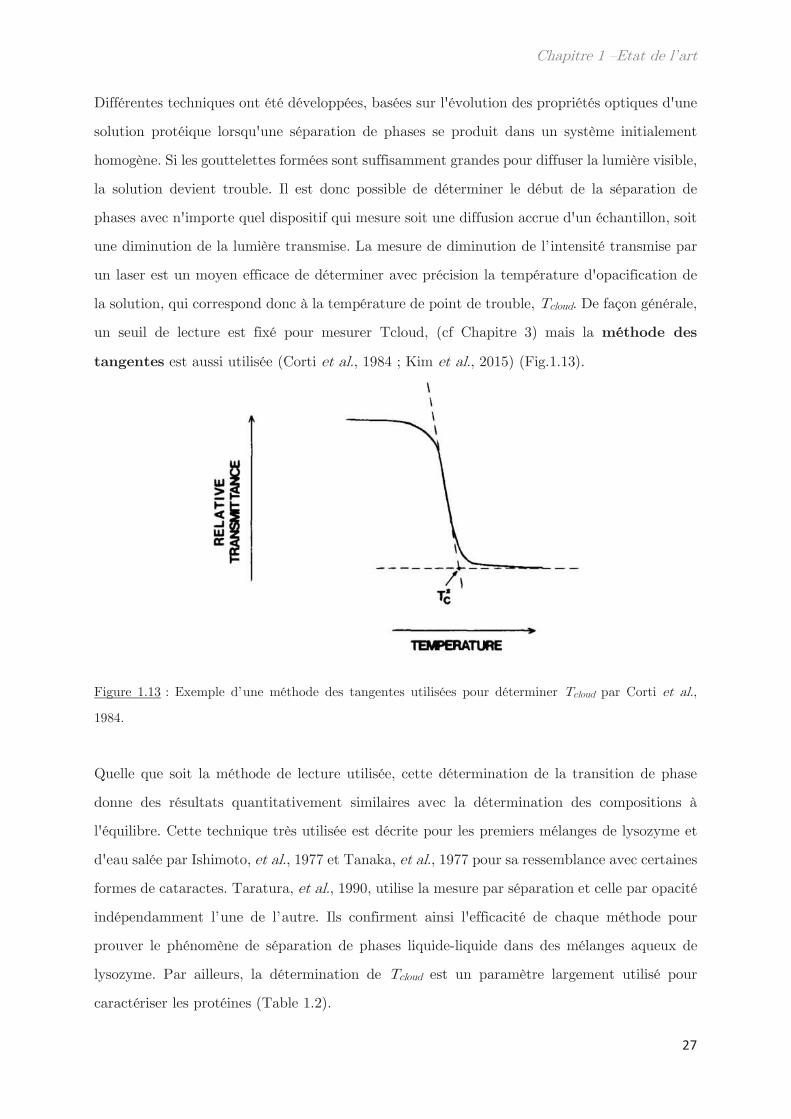
un seuil de lecture est fixé pour mesurer Tcloud, (cf Chapitre 3) mais la méthode des
tangentes est aussi utilisée (Corti et al., 1984 ; Kim et al., 2015) (Fig.1.13).
Figure 1.13 : Exemple d’une méthode des tangentes utilisées pour déterminer Tcloud par Corti et al.,
1984.
Quelle que soit la méthode de lecture utilisée, cette détermination de la transition de phase
donne des résultats quantitativement similaires avec la détermination des compositions à
l'équilibre. Cette technique très utilisée est décrite pour les premiers mélanges de lysozyme et
d'eau salée par Ishimoto, et al., 1977 et Tanaka, et al., 1977 pour sa ressemblance avec certaines
formes de cataractes. Taratura, et al., 1990, utilise la mesure par séparation et celle par opacité
indépendamment l’une de l’autre. Ils confirment ainsi l'efficacité de chaque méthode pour
prouver le phénomène de séparation de phases liquide-liquide dans des mélanges aqueux de
lysozyme. Par ailleurs, la détermination de Tcloud est un paramètre largement utilisé pour
caractériser les protéines (Table 1.2).
Chapitre 1 –Etat de l’art
28
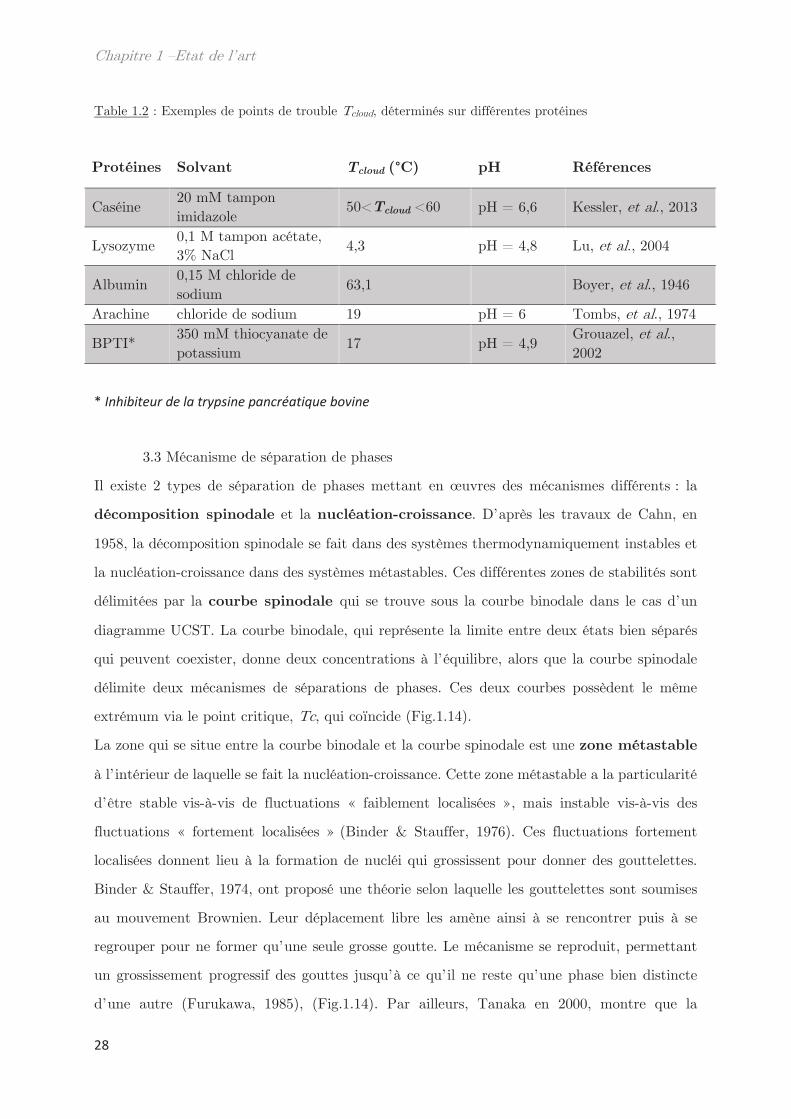
Table 1.2 : Exemples de points de trouble Tcloud, déterminés sur différentes protéines
Protéines Solvant Tcloud (°C) pH Références
Caséine 20 mM tampon imidazole
50<Tcloud <60 pH = 6,6 Kessler, et al., 2013
Lysozyme 0,1 M tampon acétate, 3% NaCl
4,3 pH = 4,8 Lu, et al., 2004
Albumin 0,15 M chloride de sodium
63,1 Boyer, et al., 1946
Arachine chloride de sodium 19 pH = 6 Tombs, et al., 1974
BPTI* 350 mM thiocyanate de potassium
17 pH = 4,9 Grouazel, et al., 2002
* Inhibiteur de la trypsine pancréatique bovine
3.3 Mécanisme de séparation de phases
Il existe 2 types de séparation de phases mettant en œuvres des mécanismes différents : la
décomposition spinodale et la nucléation-croissance. D’après les travaux de Cahn, en
1958, la décomposition spinodale se fait dans des systèmes thermodynamiquement instables et
la nucléation-croissance dans des systèmes métastables. Ces différentes zones de stabilités sont
délimitées par la courbe spinodale qui se trouve sous la courbe binodale dans le cas d’un
diagramme UCST. La courbe binodale, qui représente la limite entre deux états bien séparés
qui peuvent coexister, donne deux concentrations à l’équilibre, alors que la courbe spinodale
délimite deux mécanismes de séparations de phases. Ces deux courbes possèdent le même
extrémum via le point critique, Tc, qui coïncide (Fig.1.14).
La zone qui se situe entre la courbe binodale et la courbe spinodale est une zone métastable
à l’intérieur de laquelle se fait la nucléation-croissance. Cette zone métastable a la particularité
d’être stable vis-à-vis de fluctuations « faiblement localisées », mais instable vis-à-vis des
fluctuations « fortement localisées » (Binder & Stauffer, 1976). Ces fluctuations fortement
localisées donnent lieu à la formation de nucléi qui grossissent pour donner des gouttelettes.
Binder & Stauffer, 1974, ont proposé une théorie selon laquelle les gouttelettes sont soumises
au mouvement Brownien. Leur déplacement libre les amène ainsi à se rencontrer puis à se
regrouper pour ne former qu’une seule grosse goutte. Le mécanisme se reproduit, permettant
un grossissement progressif des gouttes jusqu’à ce qu’il ne reste qu’une phase bien distincte
d’une autre (Furukawa, 1985), (Fig.1.14). Par ailleurs, Tanaka en 2000, montre que la

Chapitre 1 –Etat de l’art
29
contrainte viscoélastique du milieu environnant peut influencer la nucléation-croissance qu’il
est important de prendre en compte lors de l’étude des séparations de phases.
En dessous de la courbe spinodale réside une zone instable où il existe de nombreuses
fluctuations de concentrations. Le transfert de matière s’opère depuis les zones de faible
concentration vers les zones de fortes concentration, ce qui qui favorise la croissance des
fluctuations de concentration. Ces fluctuations augmentent progressivement jusqu’à atteindre
un équilibre thermodynamique (Fig.1.14). Ce phénomène est la décomposition spinodale qui
est composée de 3 régimes distincts : le régime initial ou diffusif, (I), qui est le mode de
croissance des gouttelettes le plus rapide, le régime intermédiaire, (II), et le régime final,
(III).
D’après la théorie de Cahn-Hillard (Cahn, 1958 I et II; Cahn & Hillard, 1959), dans le premier
stade de croissance diffusive (I), la taille du domaine suit une loi selon laquelle la longueur
caractéristique, , augmente avec le temps selon ~ tn. Plus particulièrement, une évolution
de la longueur caractéristique qui évolue selon une loi de puissance telle que ~ t1/3 témoigne
d’une croissance diffusive des gouttelettes (Siggia, 1979). Cette évolution révèle un
grossissement contrôlé par diffusion par coalescence ou mûrissement d'Ostwald. En effet, les
mécanismes de grossissement correspondants peuvent être associés :
- soit d’un mécanisme de diffusion des petites gouttes vers les plus grosses gouttes sur des
domaines plus vastes d’une part,
- ou d’un mécanisme de coalescence des domaines d’autre part (Binder & Stauffer, 1974).
Cependant, cette pente en t1/3 ne permet pas de distinguer lequel des deux mécanismes est mis
en jeu. Dhont en 1996, souligne que l’exposant, n, varie toujours sur une gamme de 0,2 à 1,1
en fonction de l’importance des interactions hydrodynamiques.

Chapitre 1 –Etat de l’art
30
Quel que soit le mécanisme, la décomposition spinodale montre une évolution au cours du
temps de fluctuations de concentrations qui s’amplifient ; contrairement à la nucléation-
croissance où les objets sphériques, les nucléï, ayant une concentration à l’équilibre sous la
binodale, vont grossir. Cependant, lorsque le phénomène de séparation de phases se produit
près de la limite entre l’état métastable et l’état instable, il est difficile de distinguer nettement
le mécanisme à l’origine de la séparation.
Figure 1.14 : Schéma du mécanisme de décomposition spinodale et de nucléation-croissance inspiré des
travaux de Dhont, 1996.

Chapitre 1 –Etat de l’art
31
Bibliographie
Benoit, H., & Doty, P. M. (1953). Light Scattering from Non-Gaussian chains. The Journal of
Physical Chemistry, 57(6), 958–963.
Bietz, J. A., & Wall, J. (1975). The Effect of Various Extractant on the Subunit Composition
and Associations of wheat glutenin.
Bietz, J. A., & Huebner, F. R. (1980).
regional research center. Ann. Technol. Agric., 29(2), 249–277.
Binder, K., & Stauffer, D. (1974). Theory for the Slowing Down of the Relaxation and Spinodal
Decomposition of Binary Mixtures. Physical Review Letters, 33(17), 1006–1009.
Binder, K., Stauffer, D. (1976). Advances in Physics Statistical theory of nucleation,
condensation and coagulation Statistical theory of nucleation, condensation and
coagulation, Advances in Physics, 25(4), 343–396.
Boire, A., Menut, P., Morel, M.-H., & Sanchez, C. (2013). Phase behaviour of a wheat protein
isolate. Soft Matter, 9(47), 11417–11426.
Boire, A. (2014). Structure and dynamics of wheat gliadins dispersions: effect of the protein
concentration and solvent temperature. Thèse de doctorat, Université de Montpellier.
Bottomley, R. C., Kearns, H. F., & Schofield, J. D. (1982). Characterisation of Wheat Flour
and Gluten Proteins Using Buffers Containing Sodium Dodecyl Sulphate. J.Sci.Food
Agric., 33, 481–491.
Boyer, P.D., Lum F.G., Ballou G.A., Luck J.M., & Rice R.G. (1946). The Combination of
Fatty Acids and Related Compounds with serum Albumin. Journal of Biological
Chemistry, 162, 181–198.
Brûlet, A., Boué, F., Cotton, J., Brûlet, A., Boué, F., About, J. C. Sciences, E. D. P. (1996).
About the Experimental Determination of the Persistence Length of Wormlike Chains of
Polystyrene. Journal de Physique II, 6, 885–891.

Chapitre 1 –Etat de l’art
32
Cahn, J. W., & Hilliard, J. E. (1958). Free Energy of a Non-uniform System. I. Interfacial Free
Energy. Journal of Chemical and Engineering Data, 258(28).
Cahn, J. W. (1959). Free Energy of a Non-uniform System. II. Thermodynamic Basis. Journal
of Chemical Physics, 1121, 10–14.
Cahn, J. W., & Hilliard, J. E. (1959). Free Energy of a Non-uniform System. III. Nucleation in
a Two Component Incompressible Fluid. Journal of Chemical Physics, (31), 688–699.
Campbell, C. A., & Davidson, H. R. (1979). Effect of Temperature, Nitrogen Fertilization and
Moisture Stress on Yield, Yield Components, Protein Content and Moisture use Efficiency
of Manitou Spring Wheat. Canadian Journal of Plant Science, 4(59), 963–974.
Charbonnier, L. (1973). Etude des protéines alcoolo-solubles de la farine de blé. Biochimie,
55(10), 1217–1225.
Charmet, G., Abécassis J., Bonny S., Fardet A., Forget F., Lullien-Pellerin V. (2017).
Agriculture et Alimentation Durables – Trois enjeux dans la filière céréales. Editions Quae.
Chen, L., Hodgson, K. O., Doniach, S., & Synchrotron, S. (1996). A Lysozyme Folding
Intermediate Revealed by Solution X-ray Scattering. Journal of Molecular Biology, 658–
671.
Cornec, M., Popineau, Y., & Lefebvre J. (1994). Characterisation of Gluten Subfractions by
SE-HPLC and Dynamic Rheological Analysis in Shear. Journal of Cereal Science 19, 131-
139.
Corti, M., & Minero C. (1984). Fast measurement of the consolution curve of nonionic micellar
solutions: a turbidimetric method. Colloids and Surfaces, 12, 341–356.
Cousin, F., Grillo I., Jestin J., Oberdisse J., 2010. Une brève introduction à la matière molle.
Collection SFN 11, 1-6.

Chapitre 1 –Etat de l’art
33
Dachkevitch, T., & Autran, J. (1989). Prediction of Baking Quality of Bread Wheats in
Breeding Programs by Size-Exclusion High-Performance Liquid Chromatography. Cereal
Chem, 66(6), 448–456.
Dahesh, M., Banc, A., Duri, A., Morel, M. H., & Ramos, L. (2014). Polymeric assembly of
gluten proteins in an aqueous ethanol solvent. Journal of Physical Chemistry B, 118(38),
11065–11076.
Dahesh, M. (2014). Etude des mécanismes de structuration du gluten: Approche modèle et
multi-échelles. Thèse de doctorat, Université de Montpellier.
Danno, G. (1981). Extraction of Unreduced Glutenin from Wheat Flour with Sodium Dodecyl
Sulfate. Cereal Chemistry.
De Gennes, P. G. (1976). Dynamics of Entangled Polymer Solutions. Macromolecules, 9(4),
587–593.
Dhont, J. K. G. (1996). Spinodal decomposition of colloids in the initial and intermediate
stages. The Journal of Chemical Physics, 105(12), 5112.
Dhont, J. K. G. (1996). An introduction todynamics of colloids, Elsevier Science.
D’Ovidio, R., & Masci, S. (2004). The low-molecular-weight glutenin subunits of wheat gluten.
Journal of Cereal Science, 39(3), 321–339.
Ewart, J. A. D. (1975). Isolation of a Cappelle-Desprez Gliadin, Journal of the Sciences of Food
and Agriculture, 86, 1021–1025.
Field, J. M., Tathamt, A. S., & Shewryt, P. R. (1987). The structure of a high-Mr subunit of
durum-wheat (Triticum durum) gluten, Biochemical Journal, 247, 215–221.
Feuillet, 2000. Le grain de blé composition et utilisation. INRA, Paris.
Furukawa, H. (1985). A dynamic scaling assumption for phase separation. Advances in Physics,
34(6), 703–750.

Chapitre 1 –Etat de l’art
34
Gontard,
Mechanical and Water Vapor Barrier Properties of an Edible Wheat Gluten Film. Journal
of Food Science, 58(1), 206–211.
Gregory Dewey, T., (1998). Fractals in Molecular Biophysics. Oxford University Press, 288
pages.
Grouazel, S., Perez, J., Astier, J.-P., Bonneté, F., & Veesler, S. (2002). BPTI liquid-liquid
phase separation monitored by light and small angle X-ray scattering. Acta
Crystallographica Section D Biological Crystallography, 58(10), 1560–1563.
Gupta, R.B., Khan K., & MacRitchie, F. (1993). Biochemical Basis of Flour Properties in
Bread Wheats. I. Effects of Variation in the Quantity and Size Distribution of Polymeric
Protein. Journal of Cereal Science 18.
Ishimoto, C., & Tanaka, T. (1977). Critical behavior of a binary mixture of protein and salt
water. Physical Review Letters, 39(8), 474.
Kasarda, D. D., Autran, J.-C., Lew, E.-L., Nimmo, C. C., & Shewry, P. R. (1983). N-terminal
amino acid sequences of w- gliadins and w-secalins implications for the evolution of
prolamin genes. Biochimica and Biophysica Acta., 747, 138–150.
Keck, B., Köhler, P., & Wieser, H. (1995). Disulphide bonds in wheat gluten: cystine peptides
derived from gluten proteins following peptic and thermolytic digestion. Zeitschrift Für
Lebensmittel-Untersuchung Und -Forschung, 200(6), 432–439.
Kessler, A., Menéndez-Aguirre, O., Hinrichs, J., Stubenrauch, C., & Weiss, J. (2013). Properties
of an s-casein-rich casein fraction: Influence of dialysis on surface properties, miscibility,
and micelle formation. Journal of Dairy Science, 96(9), 5575–5590.
Kim, B., Hong, D., & Chang, W. V. (2015). LCST and UCST double-phase transitions of poly
(N-isopropylacrylamide-co-2-acrylamidoglycolicacid)/poly(dimethylaminoethyl
methacrylate) complex. Colloid and Polymer Science, 293(3), 699–708.

Chapitre 1 –Etat de l’art
35
Lookhart, G.L., Jones B.L., Hall S.B., & Finney K.F. (1982). An Improved Method for
Standardizing Polyacrylamide Gel Electrophoresis of Wheat Gliadin Proteins. Cereal
Chemistry 59(3), 178-181.
Lu, J., Carpenter, K., Li, R. J., Wang, X. J., & Ching, C. B. (2004). Cloud-point temperature
and liquid-liquid phase separation of supersaturated lysozyme solution. Biophysical
Chemistry, 109(1), 105–112.
MacRitchie, F. (1987). Evaluation of contributions from wheat protein fractions to dough
mixing and breadmaking. Journal of Cereal Science, 6(3), 259–268.
MacRitchie, F. (1992). Physicochemical Properties of Wheat Proteins In Relation To
Functionality. Asvances in Food and Nutrition Research, 36.
Melas, V., Morel, M.-H., Autran, J.-C., & Feillet, P. (1994). Simple and rapid method for
purifying low molecular weight subunits of glutenin from wheat. Cereal Chemistry.
Morel, M. H., Dehlon, P., Autran, J. C., Leygue, J. P., & Bar-L’Helgouac’H, C. (2000). Effects
of temperature, sonication time, and power settings on size distribution and extractability
of total wheat flour proteins as determined by size-exclusion high-performance liquid
chromatography. Cereal Chemistry, 77(5), 685–691.
Müller, S., & Wieser, H. (1995). The location of disulphide bonds in -type gliadins. Journal of
Cereal Science, 22(1), 21–27.
Osborne, T. B. 1907. The proteins of the wheat kernel, Washington, Carnegie Inst.
Pallos, F. M., Robertson, G. H., Pavlath, A. E., & Orts, W. J. (2006). Thermoformed wheat
gluten biopolymers. Journal of Agricultural and Food Chemistry, 54(2), 349–352.
Rubinstein, M., Colby R.H., 2003. Polymers Physics. Oxford University Press.
Seiffert, S. (2017). Scattering perspectives on nanostructural inhomogeneity in polymer network
gels. Progress in Polymer Science, 66, 1–21.

Chapitre 1 –Etat de l’art
36
Shewry, P. R., Tatham, A. S., Forde, J., Kreis, M., & Miflin, B. J. (1986). The classification
and nomenclature of wheat gluten proteins: A reassessment. Journal of Cereal Science,
4(2), 97–106.
Shewry, P. R., & Tatham, a S. (1990). The prolamin storage proteins of cereal seeds: structure
and evolution. Biochemical Journal, 267(1), 1–12.
Shewry, P. R., & Tatham, A. S. (1997). Disulphide Bonds in Wheat Gluten Proteins. Journal
of Cereal Science, 25, 207–227.
Siggia, E. D. (1979). Late stages of spinodal decomposition in binary mixtures. Physical Review
A, 20(2), 595–605.
Tanaka, T. (1977). Phase Separation of a Protein-Water Mixture in Cold Cataract in the
Young Rat Lens. Science, 197, 1010–1012.
Tanaka, H. (1996). Universality of Viscoelastic Phase Separation in Dynamically Asymmetric
Fluid Mixtures. Phys. Rev. Lett., 76(5), 787–790.
Tanaka, H. (2000). Viscoelastic phase separation. Journal of Physics Condensed Matter, 12,
207–264.
Taratura, V. G., Holschbach, A., Thurston, G. M., Blankschtein, D., & Benedek, G. B. (1990).
Liquid-liquid phase separation of aqueous lysozyme solutions: Effects of pH and salt
identity. Journal of Physical Chemistry, 94(5), 2140–2144.
Tatham, A. S., & Shewry, P. R. (1985). The conformation of wheat gluten proteins. The
secondary structures and thermal stabilities of -, -, - and -Gliadins. Journal of Cereal
Science, 3(2), 103–113.
Teraoka, I., 2002. Polymer Solutions: An Introduction to Physical Properties. John Wiley &
Sons.

Chapitre 1 –Etat de l’art
37
Thomson, J. a, Schurtenberger, P., Thurston, G. M., & Benedek, G. B. (1987). Binary liquid
phase separation and critical phenomena in a protein/water solution. Proceedings of the
National Academy of Sciences of the United States of America, 84(20), 7079–7083.
Thomson, N. H., Miles, M. J., Popineau, Y., Harries, J., Shewry, P., & Tatham, A. S. (1999).
Small angle X-ray scattering of wheat seed-storage proteins: alpha-, gamma- and omega-
gliadins and the high molecular weight (HMW) subunits of glutenin. Biochimica et
Biophysica Acta, 1430(2), 359–366.
Tombs, M. P., Newsom, B. G., & Wilding, P. (1974). Protein Solubility - Phase Separation in
Arachin-Salt-Water Systems. International Journal of Peptide and Protein Research, 6(4),
253–277.
Wieser, H., Seilmeier, W., Valdez, I., Mendez E. (2000). Characterisation of -Gliadins from
different wheat species. Wheat Gluten edited by P.R. Shewry & A.S. Tatham, 200-203.
Wieser, H. (2007). Chemistry of gluten proteins. Food Microbiology, 24(2), 115–119.
Wilkins, D. K., Grimshaw, S. B., Dobson, C. M., Jones, J. A., & Smith, L. J. (1999). Articles
Hydrodynamic Radii of Native and Denatured Proteins Measured by Pulse Field Gradient
NMR Techniques. Biochemistry, 38, 16424–16431.
Woychik, J. H., Boundy, J. A., & Dimler, R. J. (1961). Starch gel electrophoresis of wheat
gluten proteins with concentrated urea. Archives of Biochemistry and Biophysics, 94(3),
477–482.
Wrigley, C., Békés F., Bushuk W., (2006). Gliadin and Glutenin: The Unique Balance of Wheat
Quality, AACC International, Minnesot


Chapitre 2 – Matériel et méthodes
39
Chapitre 2
Matériels et méthodes
Ce chapitre présente le matériel et les méthodes utilisés au cours de la thèse. Il décrit d’une
part le matériel utilisé et les outils qui permettront sa caractérisation biochimique, et d’autre
part les techniques physiques utilisées pour répondre à la problématique.
1. Matériel
Les protéines sont extraites d’un lot de gluten fourni par Tereos Syral (Amygluten). Ce dernier
contient 73,26 % de protéines (base humide) et sa teneur en eau est de 5,89%.
Pour l’ensemble de nos mesures, le solvant éthanol/eau est préparé avec de l’éthanol absolu
99,9 % provenant du fournisseur Carbo Erba. De l’eau déionisée est utilisée à l’UMR IATE et
de l’eau MilliQ au laboratoire Charles Coulomb. Un volume d’eau est mesuré puis un volume
d’éthanol est ajouté à ce dernier dans un bécher. Le tout est agité et le volume désiré de
l’ensemble de la solution est ajouté au gluten ou aux extraits de gluten.
2. Méthodes
2.1. Extraction des protéines du gluten
Le protocole suivi est inspiré des travaux présentés par Dahesh et al., 2014, sur la séparation
liquide-liquide des protéines du gluten, en suspension dans un mélange éthanol/eau (50/50
v/v).
En routine, 6 x 20 g de gluten sont extraits par 200 mL de solvant eau/éthanol (50/50 v/v)
dans des pots à centrifuger (Nalgène, 250 mL). Les pots sont ensuite placés sur un agitateur

Chapitre 2 – Matériel et méthodes
40
dont la rotation se fait autour d’un axe vertical, à 60 rotations par minute (Reax 2, Heidolph)
pendant 19 h à 20°C.
Les pots sont ensuite centrifugés 30 min à 20°C (15 000 g ; centrifugeuse Beckman Coulter,
rotor JLA 16-250). Les différents culots (C1) issus de la centrifugation sont éliminés et les
surnageant (S1) sont transférés dans des pots de 250 ml. Ces derniers sont immergés totalement
pendant 1h dans un bain marie (V = 12 L) thermostaté à la température de trempe souhaitée
(Tt), (Tt < 12°C). Parallèlement la centrifugeuse est refroidie 1h à la même température que
celle de la trempe. Les pots sont ensuite centrifugés 33 min à 10 000 g, à Tt. Les surnageants
(S2) issus de la centrifugation sont transférés dans des pots propres de 250 ml. Les culots (C2),
quant à eux, sont recentrifugés 11 min, 3000 g à la même température que la centrifugation
précédente afin d’éliminer le reliquat du surnageant restant. Les C2 sont additionnés de 5 fois
leur masse en eau désionisée et mis à congeler à –40°C, après homogénéisation du contenu du
pot sous vortex. Les culots sont lyophilisés dans un lyophilisateur (ALPHA 2.4 LSC Christ) et
suivent une dessiccation primaire pendant 72 h (les plateaux chauffants sont à 1°C ; le piège à
-86°C et le vide de consigne est à 0,370 mbar). L’ensemble de ces opérations est représenté dans
la Figure 2.1.
Au cours de l’extraction, une aliquote frais de chaque fraction est prélevé pour analyse
quantitative par chromatographie d’exclusion stérique en phase liquide à haute performance
(SE-HPLC). Les prélèvements sont dilués dans un tampon dénaturant à base de sodium
phosphate 0,1 M à pH = 6,8 (7,02 g de dihydrogénophosphate de sodium (NaPO4, 2H2O),
(VWR) et 9,79 g de disodiumhydrogénophosphate de sodium (Na2HPO4, 2H2O), (Merck) qsp
1000 ml d’eau) contenant 1% de dodécylsufate de sodium (SDS), (VWR), auquel on ajoute
0,351 g d’urée (99,5 %), (Sigma Aldrich) (soit approximative 5,5 M d’urée final). Typiquement,
pour chacun des 6 pots, 50 µL de S1, 50 µL de S2 et 10 µL de C2 sont repris dans des tubes
Eppendorf (2,5 ml) avec respectivement 1,5 ; 2,0 et 2,0 mL de tampon dénaturant. Les tubes
sont agités 30 min, à 800 rpm sur un agitateur vibrant (Heidolph vibramax 100) et centrifugées
15 min à 14 000 g à température ambiante. 1 mL de surnageant est ensuite transféré dans un
vial HPLC pour analyse.
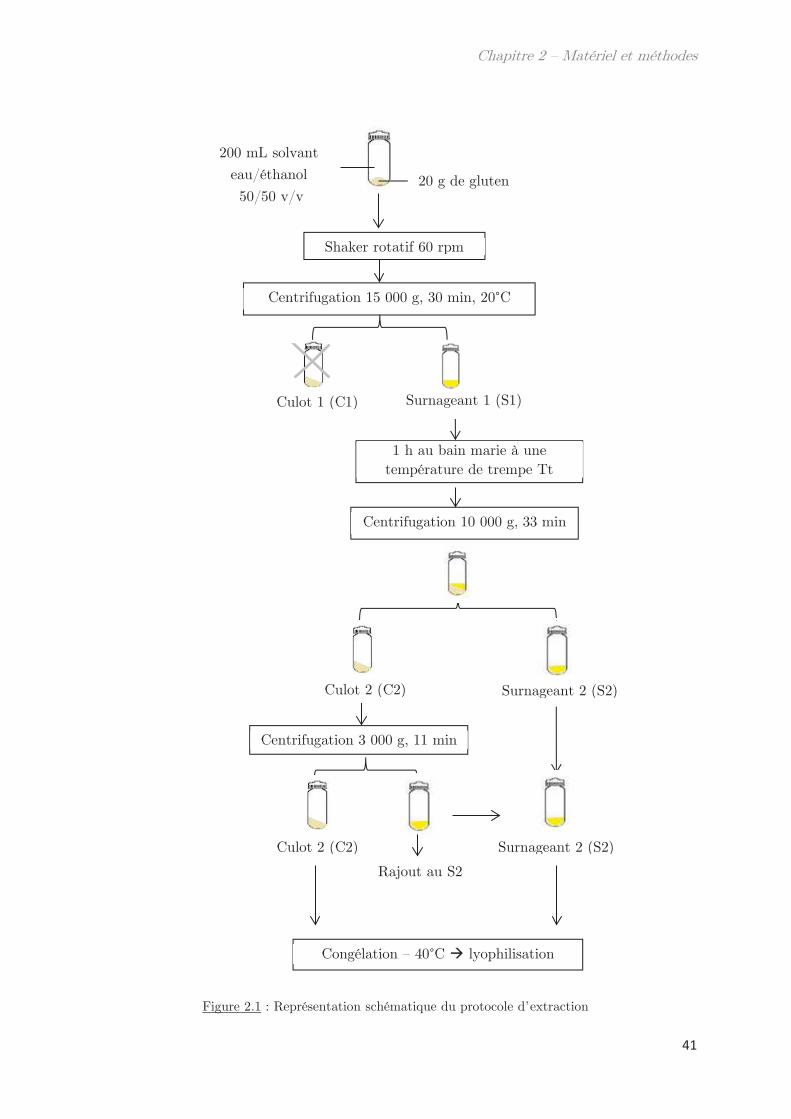
Chapitre 2 – Matériel et méthodes
41
Figure 2.1 : Représentation schématique du protocole d’extraction
Culot 2 (C2)
200 mL solvant
eau/éthanol
50/50 v/v 20 g de gluten
Shaker rotatif 60 rpm
Centrifugation 15 000 g, 30 min, 20°C
Surnageant 1 (S1)
Centrifugation 3 000 g, 11 min
Congélation – 40°C lyophilisation
1 h au bain marie à une température de trempe Tt
Centrifugation 10 000 g, 33 min
Culot 1 (C1)
Culot 2 (C2) Surnageant 2 (S2)
Rajout au S2
Surnageant 2 (S2)

Chapitre 2 – Matériel et méthodes
42
Les poudres de protéines lyophilisées issues de l’extraction sont remises en solutions à
différentes concentrations pour réaliser les mesures. Un solvant eau (milliQ) / éthanol 50 %
(v/v) est utilisé pour toutes les techniques d’analyses. La concentration, C, (g/l) tient compte
de la des protéines, de leur masse sèche, m (g), et du volume du
solvant utilisé, V, (ml) selon l’équation suivante :
C =
(2.1)
Cette concentration peut être retraduite en fraction volumique finale, , selon laquelle :
= (2.2)
2.2 SE-HPLC
La chromatographie en phase liquide à haute performance par exclusion de taille (SE-HPLC)
est une technique basée sur la séparation des protéines en fonction de leur rayon
hydrodynamique. Après injection sur la colonne, les protéines entrainées par la phase mobile
liquide (éluant) parcourent différents volumes d’élution selon la porosité de la phase
particulaire stationnaire. Avec cette technique l’ordre d’élution est inversement proportionnel
au rayon hydrodynamique des protéines. Une particularité de la méthode utilisée ici est de
mettre en jeu des protéines dénaturées par le SDS et présentant toutes, ou presque, le même
facteur de forme. De ce fait, leur rayon hydrodynamique et donc leur coefficient de partage
(kav = V(i) – Ve)/(Vt –Ve) ; Ve volume d’exclusion, Vt volume total, V(i) volume d’élution
de l’espèce (i)) est linéairement relié au logarithme de leur masse moléculaire. Les constituants
ont alors une durée de leur parcours total qui varie : plus elles sont grosses plus elles sont
rapidement éluées, et plus elles sont petites plus elles pénètrent dans les pores des billes et sont
éluées plus tard.
L’analyse est réalisée à la température de 25°C, sur une chaîne Alliance Waters équipée d’une
colonne TSK G4000 SWXL (Toso bioscience) (75 X 300 mm) et d’une pré-colonne TSK gel
PWXL (40 mm X 60 mm), éluée avec du tampon phosphate, 0,1 M, pH 6,8, SDS 0,1%. Un
volume de 20 µl (à environ 1 g.L-1 de protéine) est injecté et élué à 0,7 ml.min-1, la détection
des différentes protéines se faisant à 214 nm. La calibration en masse du dispositif est réalisée
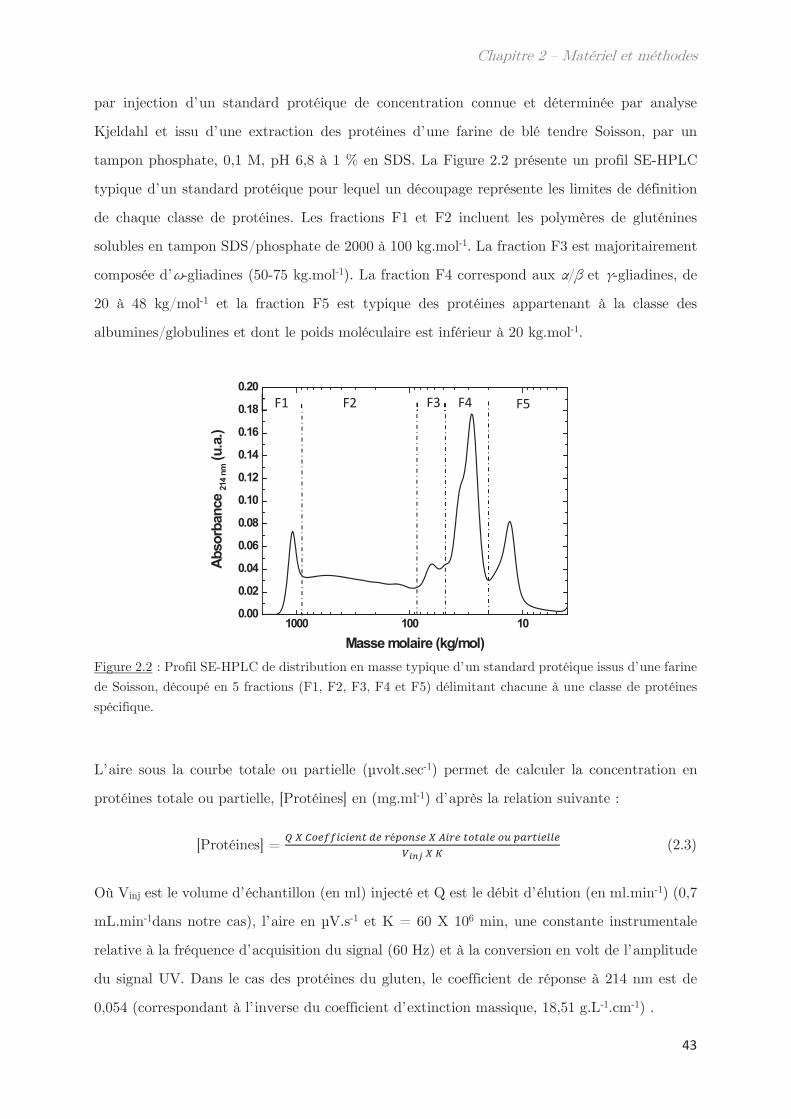
Chapitre 2 – Matériel et méthodes
43
par injection d’un standard protéique de concentration connue et déterminée par analyse
Kjeldahl et issu d’une extraction des protéines d’une farine de blé tendre Soisson, par un
tampon phosphate, 0,1 M, pH 6,8 à 1 % en SDS. La Figure 2.2 présente un profil SE-HPLC
typique d’un standard protéique pour lequel un découpage représente les limites de définition
de chaque classe de protéines. Les fractions F1 et F2 incluent les polymères de gluténines
solubles en tampon SDS/phosphate de 2000 à 100 kg.mol-1. La fraction F3 est majoritairement
composée d’ -gliadines (50-75 kg.mol-1). La fraction F4 correspond aux / et -gliadines, de
20 à 48 kg/mol-1 et la fraction F5 est typique des protéines appartenant à la classe des
albumines/globulines et dont le poids moléculaire est inférieur à 20 kg.mol-1.
Figure 2.2 : Profil SE-HPLC de distribution en masse typique d’un standard protéique issus d’une farine
de Soisson, découpé en 5 fractions (F1, F2, F3, F4 et F5) délimitant chacune à une classe de protéines
spécifique.
L’aire sous la courbe totale ou partielle (µvolt.sec-1) permet de calculer la concentration en
protéines totale ou partielle, [Protéines] en (mg.ml-1) d’après la relation suivante :
[Protéines] = é
(2.3)
Où Vinj est le volume d’échantillon (en ml) injecté et Q est le débit d’élution (en ml.min-1) (0,7
mL.min-1dans notre cas), l’aire en µV.s-1 et K = 60 X 106 min, une constante instrumentale
relative à la fréquence d’acquisition du signal (60 Hz) et à la conversion en volt de l’amplitude
du signal UV. Dans le cas des protéines du gluten, le coefficient de réponse à 214 nm est de
0,054 (correspondant à l’inverse du coefficient d’extinction massique, 18,51 g.L-1.cm-1) .
1000 100 100.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
0.20
Absorb
ance 2
14 n
m (u.a
.)
Masse molaire (kg/mol)
F2 18
F1 F3 F4 F5

Chapitre 2 – Matériel et méthodes
44
2.3 Electrophorèse SDS-PAGE
L’électrophorèse en gel de polyacrylamide en présence de dodecyl sulfate de sodium (SDS) est
une technique biochimique qui a pour but de séparer les protéines en fonction de leur taille
moléculaire sous un champ électrique (Shapiro et al., 1967). La porosité des gels est définie par
la concentration massique totale en monomères, T, et la proportion d'agent monomère
réticulant, C, déterminées par les relations suivantes :
% T = [(Acrylamide (g) + bis acrylamide (g)) X 100]/ volume de gel (2.4)
% C = [Bis acrylamide (g) x 100] /Acrylamide (g) (2.5)
Moulage des gels
2 gels de 1,5 mm d’épaisseur (16 X 18 cm) sont préparés sur un portoir réservé à cet usage. Un
premier gel de séparation (11,6 X 18 X 0,15) de 31,5 ml (T = 13,29 %, C = 0,63 %) contenant
0,380 M de tris-hydroxy-méthyl-amino-méthane (Tris-HCl), pH 8,8 et 0,1 % (w/v) de SDS est
coulé immédiatement après ajout des catalyseurs (0,038% de persulfate d’ammonium et 0,05
% de Tétraméthyléthylènediamine (TEMED)) à la solution préalablement dégazée. Quelques
gouttes de butanol saturé en eau sont introduites à la surface du gel pour éviter le contact avec
l’air et s’assurer d’un niveau égal de l’interface. Environ 30 min d’attente sont nécessaires à
la prise en masse du gel. Après élimination du butanol, et rinçage de l’interface avec quelques
mL de tampon Tris-HCl, 0,380 M, pH 8,8 et 0,1 % de SDS.
Un gel de concentration est coulé au-dessus du gel de séparation. Ce gel de concentration (T=
5,6 %, C = 10,7 %) est préparé en tampon Tris HCL, pH 6,8, 0,121 M, 0,1 % SDS. La
polymérisation du gel est déclenchée par ajout d’ammonium persulfate (0,03%) et de TEMED
(0,23 %) à 15 mL de gel dégazé. Un peigne de 10 à 15 puits est inséré rapidement après le
dépôt du gel et quelques gouttes de butanol saturées d’eau sont déposées en surface. Environ
45 min d’attente sont nécessaires pour être sûr que ce gel, qui s’opacifie en polymérisant, soit
totalement pris. Après retrait du peigne, la surface du gel est rincée plusieurs fois avec du
tampon de migration (voir plus bas) pour éliminer le butanol.
Préparation des échantillons
Les extraits protéiques (S1, S2 et C2) respectivement dilués 16 fois (S1, S2) et 85 fois (C2)
dans une solution d’extraction contenant 28,3% d’une solution stock (0,22M Tris, pH 6,8, 7,06

Chapitre 2 – Matériel et méthodes
45
% SDS, 0,035 % bleu de bromophénol, 35,5 % glycérol) contenant éventuellement -
Mercaptoéthanol, afin de réduire les liaisons disulfures des protéines. Le gluten est quant à lui
extrait dans un tube par la solution d’extraction, pendant 1H à température ambiante. Les
échantillons sont ensuite centrifugés (centrifugeuse Eppendorf, centrifuge 5427 R) à 20 800 g,
pendant 10 min, à 20°C. Les échantillons non réduits sont déposés directement sur gel et les
échantillons réduits sont chauffés 2 min 30 à 100°C. Un dépôt de 10 µL est effectué (soit environ
30 µg de protéines/par puits).
Migration
La migration s’effectue en tampon Tris, 0,025 M, glycine 0,192 M et 0,1 % SDS. Un ampérage
constant de 40 mA par plaque est appliqué sur le gel afin de faire migrer les protéines du pôle
– vers le pôle +. La migration est stoppée 10 minutes après que le colorant (bleu de Coomassie
brillant R250 à 1% dans H2O) de suivi soit sorti du gel.
Coloration et décoloration du gel
Les gels sont placés séparément dans une solution de TCA 15% dans une boîte en plastique et
agités sur une balancelle (Stuart see-saw rocker SS24). Au bout d’une heure cette solution est
remplacée par une solution colloïdale de bleu de Coomassie (brillant R250, 0,04%, TCA 15 %).
Après 24 de coloration, le gel est décoloré dans une solution de TCA (10%), puis équilibré dans
de l’eau contenant 2% de glycérol pendant 3 minutes. Le gel peut alors être séché entre deux
feuilles de cellophane sur un cadre de séchage. Une acquisition d’image est ensuite réalisée sur
un scanner de gel type Biored GS-7110.
Analyse d’image
Les images sont traitées via le logiciel ImageJ pour analyser l’intensité de chaque bande et ainsi
établir le profil électrophorétique des différentes pistes. En pratique un rectangle de 1 pixel de
hauteur et de la largeur de la piste de dépôt est déplacé de bas en haut de façon à acquérir des
tranches successives de moyenne de niveaux de gris. Les bords latéraux du rectangle sont
positionnés de sortent à ne pas mordre sur l’espace entre les pistes. Les données acquises sont
tracées en fonction de la distance de façon à obtenir un densitogramme qui est ensuite traité
sur Excel. Après ajustement de la ligne de base, une analyse de l’aire sous la courbe est
effectuée. L’intensité des différents pics (bandes) est donnée en pourcentage de l’aire totale.

Chapitre 2 – Matériel et méthodes
46
2.4 Développement d’un outil moyen débit de mesure des points de trouble
Afin de déterminer la température de point de trouble (Tcloud) des protéines en solution de
façon rapide, nous avons développé un nouveau dispositif basé sur l’observation du changement
de niveau de gris des images des différents échantillons en fonction de la température. Le
Chapitre 3 développera en détail la mise en place et le principe du dispositif expérimental.
Le montage est installé dans une chambre climatique (HPP 360 Memmert) avec un volume
interne de 0,256 m3, et une dimension interne (L x P x H : 640 x 500 x 800 mm). Un contrôle
en température via un système Peltier sur une gamme de 0 à 70°C et en humidité de 10 à 90%
HR peut être réalisé. La chambre est équipée de 2 thermocouples (1 fixe et 1 amovible) destinés
à mesurer séparément la température de l’enceinte et de l’échantillon. Une plaque lumineuse
(small Sback II-201502, 2, TPL Vision), (204 x 153 mm) éclairée par des LEDs blanches est
placée à l’intérieur de l’enceinte climatique. Cette dernière illumine une microplaque 96 puits
(Greiner Bio one, 655101) transparente, placée au-dessus de la plaque lumineuse, contenant les
échantillons. Une caméra SVS EXO174MBGEC monochrome, (1 920 x 1 200,32Hz, 1/1,2''
CMOS) avec un objectif Myutron 1.2" HF5018V-2, (5 Mega Pixel, 50 mm - f/1,8) est placée
45 cm au-dessus de la microplaque.
La caméra est connectée à un ordinateur et pilotée par un programme d’acquisition (SVCam)
fourni par le concepteur. Il permet d’acquérir des images de taille 1920 X 1200 pixels avec une
fréquence maximale de 0,1 fps. Les images de la microplaque sont acquises en temps réel au
cours d’un programme en température. L’humidité relative étant maintenue constamment à
60% HR. Le système de programmation est piloté par un logiciel fourni par le concepteur de
l’enceinte climatique (AtmoControl, Memmert).
2.5 Techniques de diffusion aux petits angles
Les techniques de diffusion aux petits angles permettent de caractériser la structure et la
dynamique de systèmes de petites tailles de l’ordre de 1 à 100 nanomètres (Rawiso, 1999).
La diffusion aux petits angles repose sur l’utilisation d’un faisceau monochromatique (lumière,
X ou neutrons) dirigé sur un échantillon, qui peut être sous toutes les formes (liquide, solide,
…), et désordonné ou partiellement ordonné. Le rayonnement du faisceau est alors réémis par
l’échantillon dans toutes les directions, sans dissipation globale d’énergie. Ce phénomène est
appelé diffusion « élastique ».

Chapitre 2 – Matériel et méthodes
47
L’intensité diffusée peut être mesurée grâce à un détecteur placé dans une direction particulière
d’observation (vecteur ) qui forme un angle par rapport à la direction incidente (vecteur
) (Fig.2.3). Le vecteur de diffusion q est défini par = – avec lkil = lkfl .
Figure 2.3 : Détermination du vecteur d’onde selon le principe de diffusion.
Le module du vecteur d’onde vaut alors :
(2.6)
, l’angle entre les directions
incidente et diffusée.
L’intensité diffusée I(q) mesurée expérimentalement, représente la section efficace de diffusion
par unité de volume :
I(q)(cm-1) = . (q) (2.7)
où d /d est le rayonnement diffusé dans un élément d’angle solide .
D’autre part, cette intensité I(q) fournit une quantification de l'interaction échantillon-
rayonnement et contient des informations sur la structure de l'échantillon. Ainsi, pour un
système composé d’objets monomériques à symétrie sphérique, il en résulte des informations
structurales définies par:
I(q) (cm-1) = ( )². . V. P(q). S(q) (2.8)
Où est le contraste de diffusion entre les phases de l'échantillon (par exemple, objets et
milieu), est la fraction volumique des objets dans le milieu, V, le volume de l'objet, P(q) le
facteur de forme des objets diffusants et S(q) est le facteur de structure. S(q) est lié à
l'arrangement spatial entre les objets et à leurs interactions.
Les caractéristiques des différentes techniques de diffusion de rayonnement permettent de
s’adapter aux différents types d’échantillons mais ont chacune des avantages et des
=4
sin( )
= –

Chapitre 2 – Matériel et méthodes
48
inconvénients. La Table 2.1 regroupe les différences entre la diffusion de neutrons et celle par
rayons X, qui ont impacté nos choix lors de nos mesures.
Table 2.1 : Différences entre les caractéristiques des techniques de diffusions de neutrons et celles de
rayons X.
Neutrons Rayons X
Dommage échantillon Non destructif Potentielle destruction
Contraste Longueur de diffusion Densité électronique
Temps d’acquisition 30 min – 1h < 5 min
2.5.1 Diffusion de neutrons aux petits angles (SANS)
Les mesures présentées dans ce manuscrit ont été réalisées sur deux centres différents :
i) au Laboratoire Léon Brillouin (LLB) du CEA de Saclay, en France, sur l’appareil
PA20. Afin de réaliser des mesures en température sur une gamme de 5°C à 35°C,
un contrôleur Peltier alimenté à un bain thermostaté était à notre disposition. Pour
confiner l’espace, une plaque de Plexis a été installée devant le porte échantillons
balayé par un flux d’air sec, afin d’éviter les phénomènes de condensation.
ii) au Jülicher Zentrum für Forschung mit Neutron (JCNS), au centre Heinz Maier-
Leibnitz Zentrum (MLZ) de Garching, en Allemagne, sur l’appareil KWS2. Un
système de contrôle en températures automatique de type Peltier a permis de faire
des mesures sur une gamme de 5°C à 35°C.
L’ensemble des échantillons ont été mesurés dans des cuves en quartz avec une épaisseur de 2
mm et les configurations utilisées sur les deux instruments sont regroupées dans la Table 2.2.
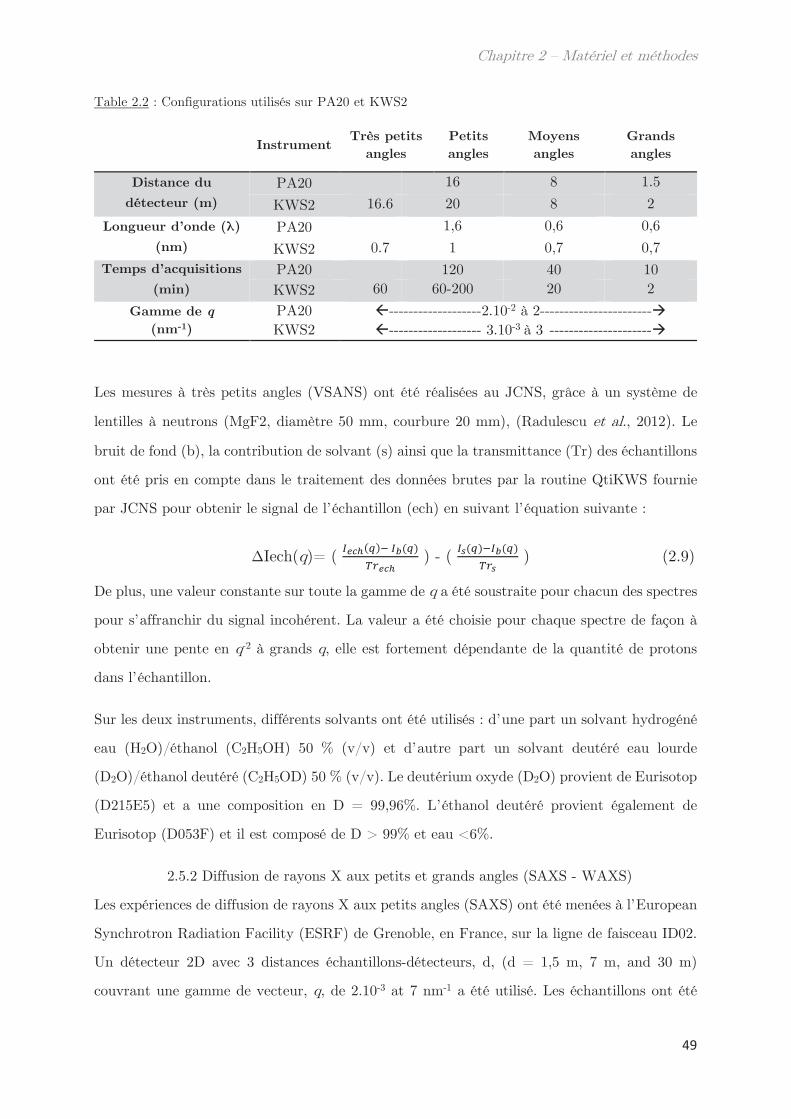
Chapitre 2 – Matériel et méthodes
49
Table 2.2 : Configurations utilisés sur PA20 et KWS2
Instrument Très petits
angles Petits angles
Moyens angles
Grands angles
Distance du
détecteur (m)
PA20 16 8 1.5
KWS2 16.6 20 8 2
Longueur d’onde (
(nm)
PA20 1,6 0,6 0,6
KWS2 0.7 1 0,7 0,7
Temps d’acquisitions
(min)
PA20 120 40 10
KWS2 60 60-200 20 2
Gamme de q (nm-1)
PA20 -------------------2.10-2 à 2----------------------- KWS2 ------------------- 3.10-3 à 3 ---------------------
Les mesures à très petits angles (VSANS) ont été réalisées au JCNS, grâce à un système de
lentilles à neutrons (MgF2, diamètre 50 mm, courbure 20 mm), (Radulescu et al., 2012). Le
bruit de fond (b), la contribution de solvant (s) ainsi que la transmittance (Tr) des échantillons
ont été pris en compte dans le traitement des données brutes par la routine QtiKWS fournie
par JCNS pour obtenir le signal de l’échantillon (ech) en suivant l’équation suivante :
Iech(q)= ( ( ) ( )
) - ( ( ) ( )
) (2.9)
De plus, une valeur constante sur toute la gamme de q a été soustraite pour chacun des spectres
pour s’affranchir du signal incohérent. La valeur a été choisie pour chaque spectre de façon à
obtenir une pente en q-2 à grands q, elle est fortement dépendante de la quantité de protons
dans l’échantillon.
Sur les deux instruments, différents solvants ont été utilisés : d’une part un solvant hydrogéné
eau (H2O)/éthanol (C2H5OH) 50 % (v/v) et d’autre part un solvant deutéré eau lourde
(D2O)/éthanol deutéré (C2H5OD) 50 % (v/v). Le deutérium oxyde (D2O) provient de Eurisotop
(D215E5) et a une composition en D = 99,96%. L’éthanol deutéré provient également de
Eurisotop (D053F) et il est composé de D > 99% et eau <6%.
2.5.2 Diffusion de rayons X aux petits et grands angles (SAXS - WAXS)
Les expériences de diffusion de rayons X aux petits angles (SAXS) ont été menées à l’European
Synchrotron Radiation Facility (ESRF) de Grenoble, en France, sur la ligne de faisceau ID02.
Un détecteur 2D avec 3 distances échantillons-détecteurs, d, (d = 1,5 m, 7 m, and 30 m)
couvrant une gamme de vecteur, q, de 2.10-3 at 7 nm-1 a été utilisé. Les échantillons ont été

Chapitre 2 – Matériel et méthodes
50
mesurés dans des capillaires de diamètre externe 1 mm. Pour éviter les phénomènes
d’évaporation, les capillaires ont été scellés avec de la colle UV (Norland Optical Adhesive 81)
réticulée sous une lampe UV afin d’assurer une étanchéité parfaite.
2 types de dispositifs mis à disposition par l’ESRF ont été utilisés :
- Un système de contrôle en température type Linkam (THMS600/TMS94) permettant
de couvrir une plage de température de -100°C à 600°C avec une vitesse de 80 K.min-1
et une stabilité de 1°C.
- Une cellule capillaire en quartz à circulation continue d’air de diamètre externe 1 mm
(flow-through cell). L’utilisation de cet appareillage permet d’effectuer une soustraction
fiable du signal du capillaire et du solvant (Fig.2.4).
Figure 2.4 : Photo du montage expérimental de mesures « flow through cell » mis à disposition par
l’ESRF sur la ligne ID02 (photo : www.ESRF.eu)
Afin de pouvoir mesurer uniquement le signal de l’échantillon, nous avons mesuré à toutes les
configurations, un capillaire vide et un capillaire contenant du solvant. Les données ont ensuite
été corrigées de façon similaire à la correction pour les neutrons via le logiciel SAXS Utilities
fourni par l’ESRF selon l’équation :
Iech(q)= ( ( ) ( )
) - ( ( ) ( )
) (2.10)
Pour atteindre une plus large gamme de vecteurs d’ondes, des mesures de diffusion de rayons
X aux grands angles (WAXS) et aux petits angles (SAXS), ont été réalisées sur un montage
expérimental disponible au laboratoire Charles Coulomb.
Un détecteur bidimensionnel et 2 distances échantillon-détecteur, d, (d = 0,19 m et 1,9 m)
couvrant une gamme de vecteur, q, de 0,07 à 20 nm-1 a été utilisé. Un four avec un contrôleur
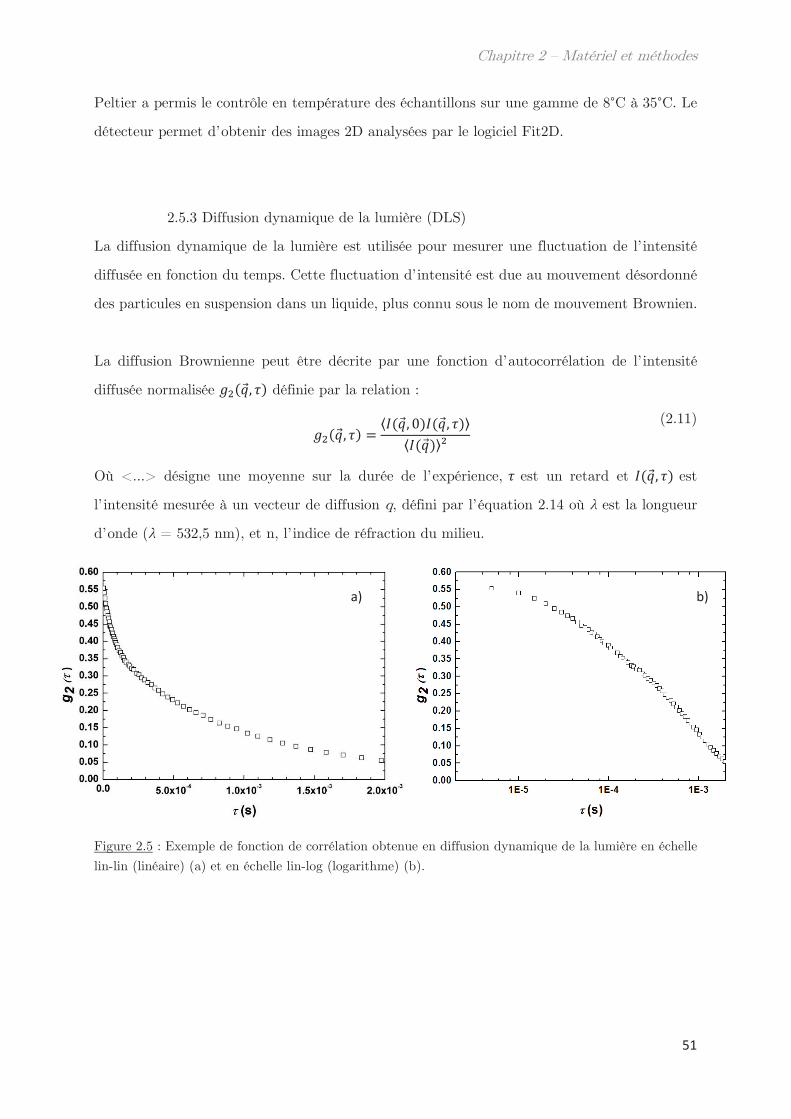
Chapitre 2 – Matériel et méthodes
51
Peltier a permis le contrôle en température des échantillons sur une gamme de 8°C à 35°C. Le
détecteur permet d’obtenir des images 2D analysées par le logiciel Fit2D.
2.5.3 Diffusion dynamique de la lumière (DLS)
La diffusion dynamique de la lumière est utilisée pour mesurer une fluctuation de l’intensité
diffusée en fonction du temps. Cette fluctuation d’intensité est due au mouvement désordonné
des particules en suspension dans un liquide, plus connu sous le nom de mouvement Brownien.
La diffusion Brownienne peut être décrite par une fonction d’autocorrélation de l’intensité
diffusée normalisée ( , ) définie par la relation :
(2.11)
Où <…> désigne une moyenne sur la durée de l’expérience, est un retard et ( , ) est
l’intensité mesurée à un vecteur de diffusion q, défini par l’équation 2.14 où est la longueur
d’onde ( = 532,5 nm), et n, l’indice de réfraction du milieu.
Figure 2.5 : Exemple de fonction de corrélation obtenue en diffusion dynamique de la lumière en échelle
lin-lin (linéaire) (a) et en échelle lin-log (logarithme) (b).
( , ) =( , 0) ( , )
( ) ²
a) b)

Chapitre 2 – Matériel et méthodes
52
Dans le cas de particules de taille monodisperse, la fonction de corrélation a la forme suivante
(Koppel, 1972) :
-2 . ) + B (2.12)
où B est la ligne de base de la fonction d’autocorrélation à un temps infini, est l’amplitude
de la fonction d’autocorrélation, et le taux de décroissance défini par :
= Dq² (2.13)
où q est défini par l’équation (2.14), qui tient compte de l’indice de réfraction, n, du milieu
dans lequel se propage le faisceau.
(2.14)
et D représente le coefficient de diffusion qui via la relation de Stockes-Einstein (Éq.2.15)
permet de déterminer le rayon hydrodynamique, Rh, d’une sphère en solution :
(2.15)
Avec la constante de Boltzmann (1.3806.10-23 m2kg s-2K-1), T la température en Kelvin, et
la viscosité du milieu.
Ces mesures ont été réalisées dans un solvant eau/éthanol 50/50 v/v avec un indice de
réfraction de 1,359 et une viscosité de 2,87.10-1 Pa.s. La viscosité du solvant a été mesurée par
un rhéomètre Anton-Paar MCR 502 dont le protocole est détaillé dans la section 2.8. L’indice
de réfraction a été mesuré par un réfractomètre d’Abbe (Carl Zeiss) à 20°C.
Le rayon hydrodynamique des protéines a été déterminé par diffusion dynamique de la lumière
sur un montage disponible au laboratoire Charles Coulomb (Fig.2.6). L’appareil utilise un
goniomètre Amtec équipé d'un corrélateur Brookhaven BI-9000AT. Les temps de retards,
varient dans l'intervalle 10-7 -3 s quel que soit l’angle de diffusion. Le laser opère à une longueur
d'onde = 514,5 nm dans la plage de puissance de 15 - 200 mW. Les mesures ont été réalisées
avec un angle de diffusion entre le faisceau incident et l’axe du détecteur compris entre 20°
et 140°. 4 mL d’échantillon, de concentration en protéines, C = 5 g.l-1, ont été filtrés par des
filtres à seringues 0,8 µm dont le détail est présenté Chapitre 4. Les échantillons filtrés sont
ensuite introduits dans des tubes en verre immergés dans une cuve remplie de toluène dont
l’indice de réfraction (n = 1,4941 à 25°C) est proche de celui du verre.
=6
=4
sin( )

Chapitre 2 – Matériel et méthodes
53
Figure 2.6 : Schéma du montage de diffusion dynamique de la lumière
2.6 Calorimétrie à balayage différentielle modulée (MDSC)
La calorimétrie à balayage différentielle repose sur la différence d’énergie thermique entre deux
capsules ; l’une contenant l’échantillon, l’autre vide, servant de référence. La différence entre
les deux capsules permet de déterminer l’absorption ou l’émission d’énergie via des phénomènes
endo- et exothermiques de l’échantillon à mesurer. La DSC est connue pour permettre d’étudier
la cristallisation (Aerts, et al., 1993), ou les températures de transition vitreuse (Micard et al.,
2000) ou encore observer un système qui sépare de phases (Arnauts, et al., 1992). Plus tard,
grâce aux travaux de Reading, en 1992, des techniques plus développées que la DSC telles que
la MDSC ont permis de déterminer plus précisément les petites variations de flux de chaleur
(Swier, et al., 2002). La MDSC permet notamment de séparer la contribution dépendant de la
température de celle dépendant de la vitesse de changement de la température dans la réponse
de l’échantillon. La réponse mesurée par cette technique permet un accroissement de la
résolution et de la sensibilité aux évènements thermiques de faible amplitude. Une seule analyse
peut ainsi révéler 2 températures de transition vitreuse ou encore dissocier les phénomènes
thermiques des phénomènes cinétiques. La MDSC a par la suite été utilisée pour mettre en
évidence des phénomènes comme la séparation de phases liquide-liquide dans des mélanges de
polymères (Dreezen et al., 2000).
Les échantillons ont été analysés sur un calorimètre à balayage différentiel modulé (DSC Q2000,
Perkin Elmer) dans des capsules en aluminium hermétiques (TzeroHermetic Lid) où étaient
Laser
Faisceau incident
la lu
Détecteur optique
Volume diffusant
Echantillon
Lentille
Photomultiplicateur
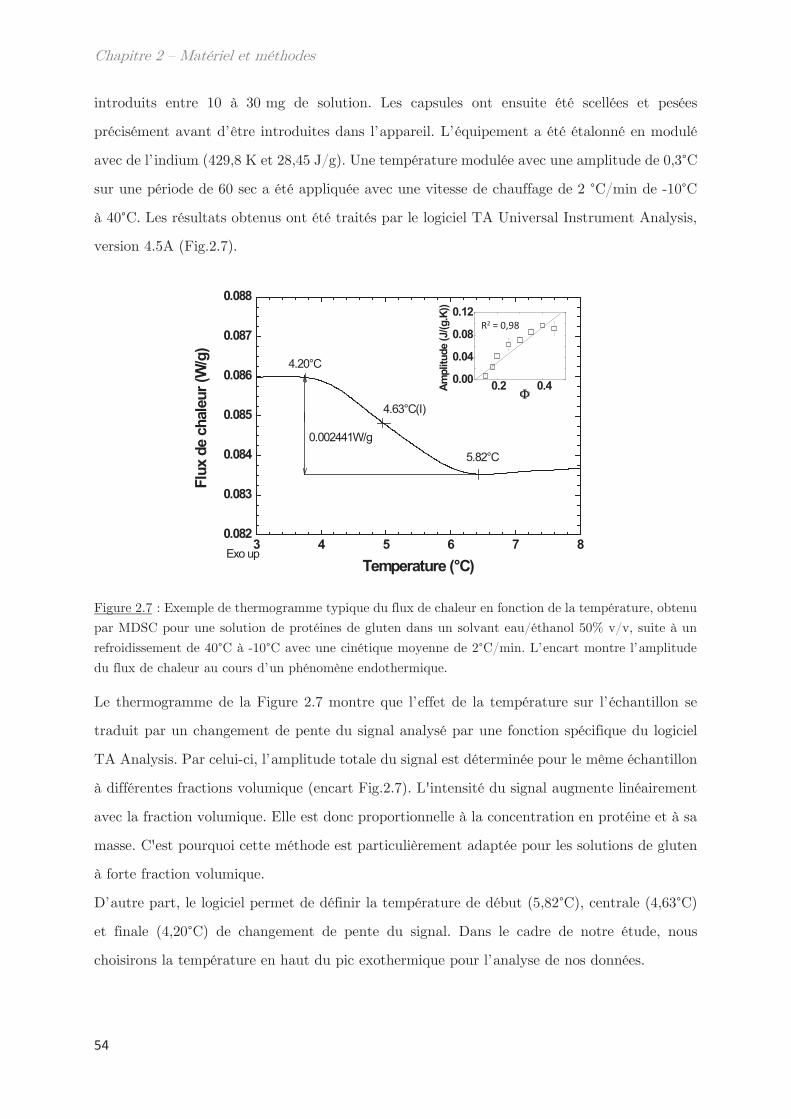
Chapitre 2 – Matériel et méthodes
54
introduits entre 10 à 30 mg de solution. Les capsules ont ensuite été scellées et pesées
précisément avant d’être introduites dans l’appareil. L’équipement a été étalonné en modulé
avec de l’indium (429,8 K et 28,45 J/g). Une température modulée avec une amplitude de 0,3°C
sur une période de 60 sec a été appliquée avec une vitesse de chauffage de 2 °C/min de -10°C
à 40°C. Les résultats obtenus ont été traités par le logiciel TA Universal Instrument Analysis,
version 4.5A (Fig.2.7).
Figure 2.7 : Exemple de thermogramme typique du flux de chaleur en fonction de la température, obtenu
par MDSC pour une solution de protéines de gluten dans un solvant eau/éthanol 50% v/v, suite à un
refroidissement de 40°C à -10°C avec une cinétique moyenne de 2°C/min. L’encart montre l’amplitude
du flux de chaleur au cours d’un phénomène endothermique.
Le thermogramme de la Figure 2.7 montre que l’effet de la température sur l’échantillon se
traduit par un changement de pente du signal analysé par une fonction spécifique du logiciel
TA Analysis. Par celui-ci, l’amplitude totale du signal est déterminée pour le même échantillon
à différentes fractions volumique (encart Fig.2.7). L'intensité du signal augmente linéairement
avec la fraction volumique. Elle est donc proportionnelle à la concentration en protéine et à sa
masse. C'est pourquoi cette méthode est particulièrement adaptée pour les solutions de gluten
à forte fraction volumique.
D’autre part, le logiciel permet de définir la température de début (5,82°C), centrale (4,63°C)
et finale (4,20°C) de changement de pente du signal. Dans le cadre de notre étude, nous
choisirons la température en haut du pic exothermique pour l’analyse de nos données.
3 4 5 6 7 80.082
0.083
0.084
0.085
0.086
0.087
0.088
4.20°C
5.82°C
4.63°C(I)
Flu
x d
e c
hale
ur
(W/g
)
Temperature (°C)Exo up
0.002441W/g
0.2 0.40.00
0.04
0.08
0.12
Am
plitu
de (J/(g.K
))
R2 = 0,98

Chapitre 2 – Matériel et méthodes
55
2.7 Microscopie
La microscopie par contraste de phase a largement été utilisée pour observer la dynamique de
la décomposition spinodale (Tanaka, 2000). C’est pourquoi nous utilisons un microscope
Olympus BX53 à contraste de phase pour examiner la séparation de phase de protéines de
gluten. Il était équipé d'une platine Linkam PE60 Peltier couvrant une plage de températures
de -20°C à 90°C. Un objectif Plan 40x avec une ouverture numérique de 0,5 a été utilisé.
L'appareil photo numérique Olympus DP26 acquiert une image toutes les 5 sec.
L'enregistrement des images a commencé à 20°C, après quoi, les températures de trempe ont
été réalisées avec une cinétique de 20K/min et filmées pendant 2 à 3h.
Afin d’obtenir des échantillons avec une épaisseur identique à chaque préparation, des feuilles
de Mylar d’une épaisseur de 50 µm sont utilisées. La lamelle est posée sur deux morceaux de
feuilles de Mylar entre lesquelles se trouve l’échantillon (Fig.2.8). Le tout est pincé quelques
secondes entre 2 pinces afin d’étaler l’échantillon de façon homogène avec l’épaisseur voulue.
Pour éviter les phénomènes d’évaporation les bords de la lamelle sont recouverts de vernis.
Figure 2.8 : Photo de préparation d’une lame de microscopie à l’aide de feuille de Mylar (50 µm) utilisé
comme espaceur entre la lame et la lamelle.
2.8 Rhéologie
Tout matériau soumis à un ensemble de forces est susceptible de se déformer, en fonction de la
répartition et de l’intensité des forces appliquées. Pour décrire et expliquer les propriétés
matériaux, il est possible de réaliser des mesures rhéologiques. La rhéologie est une science qui
traite de l’écoulement, des déformations et plus généralement de la viscosité des matériaux sous
l’action de contraintes.
Il existe différents types de grandeurs physiques :
- les contraintes de cisaillement (shear stress) : sous l’effet du mouvement relatif de deux
couches l’une par rapport à l’autre, des forces de frottement apparaissent entre les couches qui
Feuille de Mylar
Lame
Echantillon
Lamelle

Chapitre 2 – Matériel et méthodes
56
s’exercent tangentiellement à la surface de ces couches (Fig.2.9). On définit la contrainte de
cisaillement 2), par = F/S où F représente la force exercée et S la surface
soumise à la force.
- la déformation de cisaillement (shear strain) : elle s’exprime comme la variation du
déplacement dans l’espace par = où h et u ont la dimension d'une longueur avec u le
déplacement du fluide, et h la hauteur de l'entrefer.
- la vitesse de cisaillement (shear rate) : elle est définie par la dérivée par rapport au
temps de la déformation de cisaillement . Elle représente la variation de la vitesse entre les
couches limites et est définie par (s-1) = = (Fig.2.9).
Figure 2.9 : Schéma du mouvement de cisaillement entre deux surfaces planes suite à l’application d’une
force tangentielle de cisaillement.
Le comportement rhéologique des matériaux est très souvent caractérisé par la viscosité du
matériau. Il existe différents types de comportements fluides, et de viscosités différentes selon
la nature de l’échantillon : Newtonien, rhéo-fluidifiant (pseudo plastiques) et rho-épaississant.
Un fluide Newtonien est un fluide visqueux pour lequel sa courbe d’écoulement est linéaire
et sa viscosité est indépendante de la contrainte ou de la vitesse de cisaillement. Dans ce cas,
le coefficient de proportionnalité qui relie la contrainte de cisaillement, , et la vitesse de
cisaillement, avec en Pa.s.
Cette viscosité peut être influencée par différents paramètres tels que la température, la
pression, le temps, la structure physique et chimique de l’échantillon ou encore le gradient de
vitesse .
Plan fixe
Plan mobile
= h= Entrefer
Fluide
Vitesse V
DéplacementForce F

Chapitre 2 – Matériel et méthodes
57
Cependant, il existe des substances pour lesquelles dépend de . Dans ce cas la viscosité n’est
pas constante au cours d’un cisaillement. Il s’agit des fluides non Newtoniens comme la plupart
des polymères, qui sont alors qualifiés par une viscosité dite apparente.
Les fluides pseudo plastiques sont définis la relation d’Ostwald de Waele (1925) connue
aussi sous le nom de loi en puissance selon laquelle : = K( )n où K est l’indice de consistance
du liquide (N.sec2/m2) et n l’indice de comportement. Selon n, si 0 < n < 1 il s’agit d’un fluide
dit rhéo-fluidifiant ou pseudo plastique et si n > 1 il s’agit d’un fluide rhéo-épaississant
ou dilatant.
Le comportement viscoélastique des matériaux peut être mis en évidence par différents types
d’expériences. Dans notre cas, nous appliquerons des sollicitations oscillantes.
Dans le cas de module de cisaillement complexe, la contrainte et la déformation sont liés par
G* défini par l’équation (t) = G*( ). (t) avec G* = G’ + iG’’ où G’ est appelé module de
conservation (storage modulus) et G’’ le module de perte (loss modulus).
Les tests d’oscillations les plus couramment utilisés sont :
- les tests de balayage de déformation dynamique (Dynamics strain sweep test): mesure
de G’ et G’’ à une fréquence donnée, en fonction de l’amplitude de la déformation. Ces tests
permettent de déterminer le régime linéaire pour lequel G’ et G’’ sont indépendants de
l’amplitude.
- les tests de balayage en fréquence (Frequency sweep test): G’ et G’’ sont mesurés sur une
gamme de fréquences d’oscillations à une amplitude de déformation constante afin de déduire
des informations sur le comportement du matériau à différentes échelles de temps.
Ces mesures permettent notamment de déterminer si le matériau est un gel. Dans ce cas,
lorsque G’ et G’’ sont représentés en fonction de la fréquence angulaire, , G’ sera supérieur
à G’’.
D’une part, la viscosité des différents solvants utilisés pour mettre en solution les extraits de
gluten ont été mesurés. Pour cela, une géométrie cône-plan avec un entrefer de 1 mm a été
utilisée. L’échantillon est laissé reposer 10 min et recouvert d’huile pour éviter les phénomènes
d’évaporation. Un gradient de cisaillement de 1 à 100 s-1 a ensuite été appliqué à l’échantillon.

Chapitre 2 – Matériel et méthodes
58
Dans le cadre de nos mesures sur des échantillons de gluten, un rhéomètre MCR 302 équipé
d’un contrôleur en température sur une gamme de -150 à +1000 °C. Une géométrie de mesure
plan – plan est utilisée. Après avoir déposé l’échantillon, le contour de la géométrie est recouvert
d’une couche d’huile de silicone (Rhodorsil silicone oil 47V100, VWR) à faible viscosité. Cette
dernière permet d’éviter les phénomènes d’évaporations du solvant eau/éthanol et de séchage
de l’échantillon.
Le second objectif a été de déterminer la viscoélasticité des extraits de gluten plus ou moins
enrichis en gluténines et gliadines et d’observer l’évolution de cette dernière en fonction de la
température.
L’ensemble des mesures en température se fait selon le protocole expérimental suivant :
1. Dépôt de 1 ml d’échantillon au centre de la géométrie à 25°C
2. 5 min de repos pour permettre à l’échantillon de relaxer
3. Test de balayage de fréquence dynamique (déformation = 8%)
4. Rampe thermique de 25°C à -10°C (vitesse de chauffage = 3°C.min-1, fréquence = 10
rad/s, déformation = 3%)
5. Test de balayage de fréquence dynamique à -4°C (déformation = 2%)
6. Remonter en température à 25°C pour vérifier la reproductibilité – attente 10 min
7. Test de balayage de fréquence dynamique à 25°C (déformation = 8%)
8. Rampe thermique de 25°C à -10°C (vitesse de chauffage = 3°C.min-1, fréquence = 1
rad/s, déformation = 3%)
9. Vérifier la reproductibilité du balayage en fréquence à -4°C
10. Ramener la température à 25°C
L’ensemble des échantillons a été mesuré à une concentration de 237 g.l-1.
2.9 Fractionnement par flux de forces asymétrique (AsFlFFF)
Le principe fondamental de cette technique a été développé en 1966 par le professeur Calvin
Giddings à l’Université d’Utah, à Salt Lake City, aux États-Unis (Giddings, 1966). Le
fractionnement par flux de force (FFF) est une technique qui permet la séparation d’objets,
tels que les nanoparticules ou encore les protéines, en fonction de leur rayon hydrodynamique.
Cette technique a donc l’avantage de séparer les molécules de 1 nm jusqu’aux particules de
Par ailleurs, elle se présente comme un atout majeur pour analyser les

Chapitre 2 – Matériel et méthodes
59
polymères de haut poids moléculaires. En effet, les supports chromatographiques tels que la
chromatographie par exclusion de taille (SEC) ne permettent pas d’estimer de façon précise les
distributions de masses molaires les plus élevées. De plus, les forces de cisaillements appliquées
aux molécules sont moins importantes que celles provoquées en SEC qui dégradent les molécules
(Barth et Carlin, 1984).
Il existe différents types de FFF : par sédimentation, thermique, par flux ou encore électrique.
Cependant, nous développons ici uniquement le fonctionnement du fractionnement par flux de
forces asymétrique (AsFlFFF) (Fig.2.10). Le fractionnement par flux de force est une technique
basée sur l’écoulement d’un flux laminaire dans un canal, sur lequel est appliqué un champ
perpendiculaire. Ce canal est composé d’une plaque supérieure et d’une plaque inférieure jointes
et séparées entre elles par un espaceur. De façon générale, l’espaceur est une feuille de Mylar
mince ou en Téflon qui possède une épaisseur de l’ordre de 350 µm. Les plaques supérieures et
inférieures ont une forme de trapèze de telle sorte que la largeur entre l’entrée du canal et celle
de la sortie du canal diminue. Cette forme particulière permet de compenser la diminution de
la vitesse d’écoulement longitudinale qui diminue légèrement le long du canal d’écoulement.
Sur la plaque inférieure repose une membrane semi-perméable de type cellulose régénérée qui
est perméable aux molécules d’éluant mais imperméable aux plus grosses molécules tels que les
polymères.
A l’intérieur du canal, la séparation des objets se présente en 3 étapes :
1. Injection + focus : après leur entrée dans le canal via une boucle d’injection, l’effet
du flux croisé entraine les constituants le long de la paroi d’accumulation. L’échantillon
injecté se mélange alors au flux linéaire principal de la phase mobile dans le canal. Via
l’étape de focus, les échantillons sont pré-concentrés dans la tête du canal proche de la
membrane afin d’éviter leur dispersion.
2. Relaxation : lors de cette étape, un gradient de concentration se créeé, suite à
l’application du flux croisé. Le flux croisé, perpendiculaire au flux d’injection, va
permettre aux particules, en fonction de la concentration des échantillons et d’un
gradient de diffusion, de « s’organiser » dans le canal. En effet, ce gradient de diffusion
résulte d’un écoulement parabolique à l’intérieur du canal : le flux qui entraine les
molécules est un flux laminaire. Ainsi, le solvant d’élution s’écoule plus lentement sur
les bords du canal qu’en son centre.

Chapitre 2 – Matériel et méthodes
60
3. Elution : une fois le flux croisé arrêté, le flux linéaire va permettre aux molécules d’être
éluées. Cette élution va se faire en fonction du gradient de diffusion qui s’est mis en
place lors de l’étape précédente. Ainsi, les petites particules qui possèdent un coefficient
de diffusion plus élevé, vont se rapprocher de l’intérieur du canal et seront entrainées
plus rapidement par le flux linéaire : ces dernières sortiront plus vite que les grosses
particules.
Après cette dernière étape d’élution, les objets sont analysés simultanément par différentes
techniques : Ultra-Violet (UV), Diffusion dynamique de la lumière (DLS), ou encore Diffusion
statique de la lumière (LS).
Figure 2.10 : Schéma du principe de fonctionnement de l’AsFlFFF (Wahlund, et al., 1996)
Les échantillons ont été mesurés sur la plateforme FFF de l’école d’ingénieur de Purpan, à
Toulouse, en France. Les expériences ont été réalisées sur une membrane de cellulose régénérée
de 10 kDa (Superon), dans une grande cellule d’épaisseur 350 µm. Le flux linéaire Vc = 0,6
ml.min-1 et la cinétique suivie par le flux croisé est représenté dans la Figure 2.11. Le volume
d’injection est de 50 µl d’échantillon à 4 g.l-1.
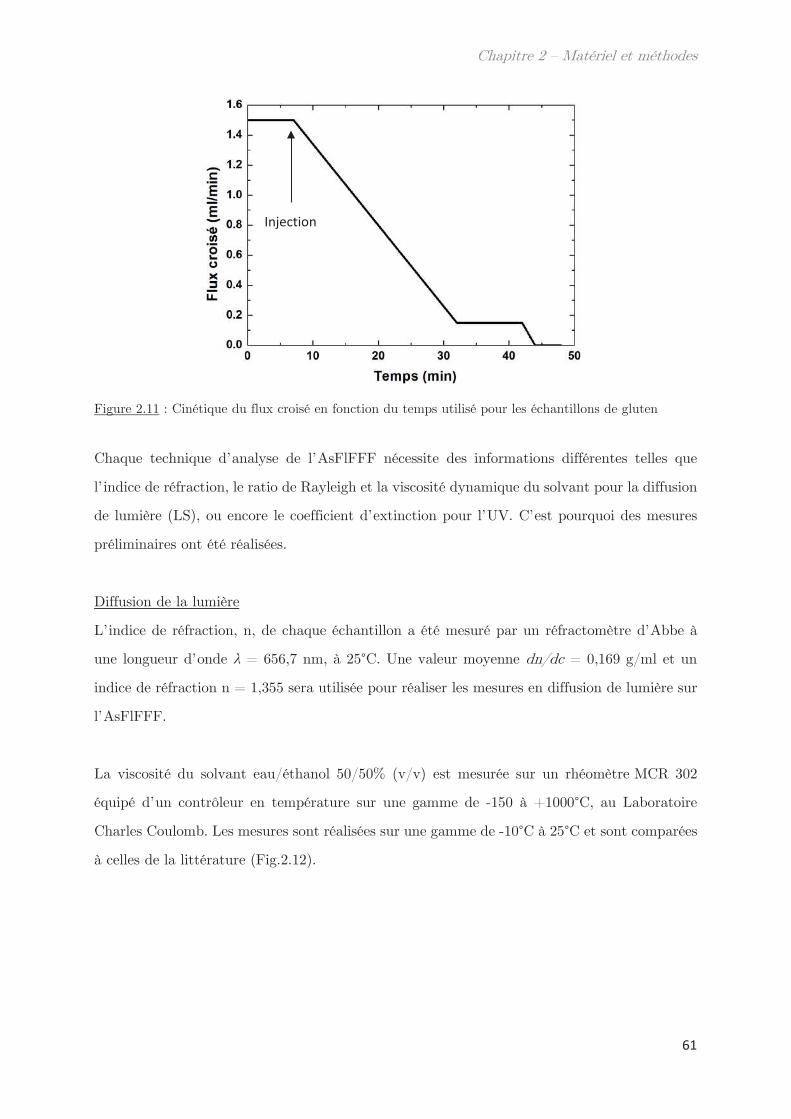
Chapitre 2 – Matériel et méthodes
61
Figure 2.11 : Cinétique du flux croisé en fonction du temps utilisé pour les échantillons de gluten
Chaque technique d’analyse de l’AsFlFFF nécessite des informations différentes telles que
l’indice de réfraction, le ratio de Rayleigh et la viscosité dynamique du solvant pour la diffusion
de lumière (LS), ou encore le coefficient d’extinction pour l’UV. C’est pourquoi des mesures
préliminaires ont été réalisées.
Diffusion de la lumière
L’indice de réfraction, n, de chaque échantillon a été mesuré par un réfractomètre d’Abbe à
une longueur d’onde = 656,7 nm, à 25°C. Une valeur moyenne dn/dc = 0,169 g/ml et un
indice de réfraction n = 1,355 sera utilisée pour réaliser les mesures en diffusion de lumière sur
l’AsFlFFF.
La viscosité du solvant eau/éthanol 50/50% (v/v) est mesurée sur un rhéomètre MCR 302
équipé d’un contrôleur en température sur une gamme de -150 à +1000°C, au Laboratoire
Charles Coulomb. Les mesures sont réalisées sur une gamme de -10°C à 25°C et sont comparées
à celles de la littérature (Fig.2.12).
Injection
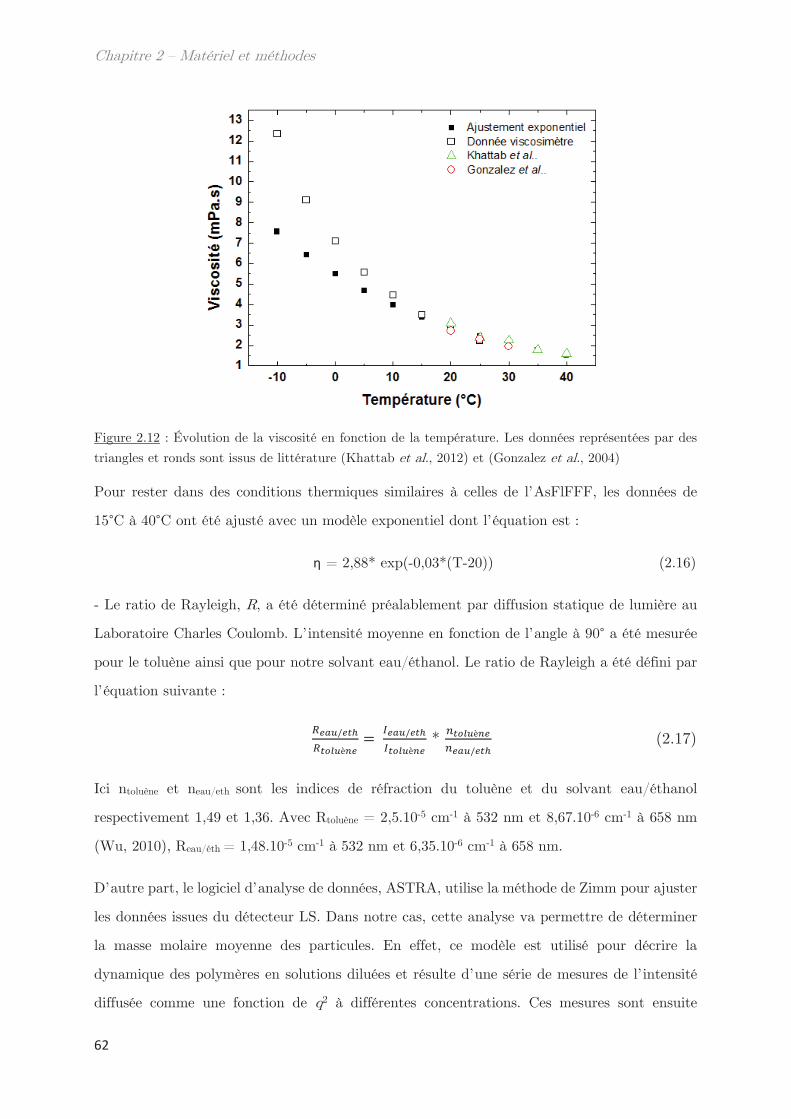
Chapitre 2 – Matériel et méthodes
62
Figure 2.12 : Évolution de la viscosité en fonction de la température. Les données représentées par des
triangles et ronds sont issus de littérature (Khattab et al., 2012) et (Gonzalez et al., 2004)
Pour rester dans des conditions thermiques similaires à celles de l’AsFlFFF, les données de
15°C à 40°C ont été ajusté avec un modèle exponentiel dont l’équation est :
= 2,88* exp(-0,03*(T-20)) (2.16)
- Le ratio de Rayleigh, R, a été déterminé préalablement par diffusion statique de lumière au
Laboratoire Charles Coulomb. L’intensité moyenne en fonction de l’angle à 90° a été mesurée
pour le toluène ainsi que pour notre solvant eau/éthanol. Le ratio de Rayleigh a été défini par
l’équation suivante :
/
è= /
è * è
/ (2.17)
Ici ntoluène et neau/eth sont les indices de réfraction du toluène et du solvant eau/éthanol
respectivement 1,49 et 1,36. Avec Rtoluène = 2,5.10-5 cm-1 à 532 nm et 8,67.10-6 cm-1 à 658 nm
(Wu, 2010), Reau/éth = 1,48.10-5 cm-1 à 532 nm et 6,35.10-6 cm-1 à 658 nm.
D’autre part, le logiciel d’analyse de données, ASTRA, utilise la méthode de Zimm pour ajuster
les données issues du détecteur LS. Dans notre cas, cette analyse va permettre de déterminer
la masse molaire moyenne des particules. En effet, ce modèle est utilisé pour décrire la
dynamique des polymères en solutions diluées et résulte d’une série de mesures de l’intensité
diffusée comme une fonction de q2 à différentes concentrations. Ces mesures sont ensuite

Chapitre 2 – Matériel et méthodes
63
extrapolées à concentration et angle nuls, (Teraoka, 2002). À la suite de ces mesures, la
représentation K*C/R( ) = f(sin²( /2)), avec K une constante optique, C, la concentration de
l’échantillon, permet notamment de définir à la fois la masse molaire, Mw, mais également le
rayon de giration, Rg, ainsi que le second coefficient du viriel, A2, selon l’équation suivante :
( )
q 0 = (1+ 2Mw A2 C) (2.18)
Spectrométrie Ultra-Violet
L’absorbance mesurée, appelée aussi densité optique, est définie par l’équation 2.19 et peut
être traduite en transmittance.
A = - log (I/I0) = - log T (2.19)
où A est l’absorbance ou densité optique, I0 est l’intensité du faisceau incident, I est l’intensité
du faisceau transmis à travers l’échantillon, et T la transmittance.
Cette absorbance est utilisée dans le cas de l’AsFlFFF pour calculer des concentrations déduites
de la loi de Beer Lambert selon laquelle :
.l.C (2.20)
-1.cm-1), l est la longueur du trajet optique
(cm) et C est la concentration (g/l).
Dans le cadre de l’AsFlFFF, l
déterminé à 214 nm pour la SE-HPLC soit 18,51 ml/(mg/cm) à 214 nm.

Chapitre 2 – Matériel et méthodes
64
Bibliographie
Aerts, L., Kunz, M., Berghmans, H., & Koningsveld, R. (1993). Relation between phase
behaviour and morphology in polyethylene/diphenyl ether systems. Die Makromolekulare
Chemie, 194(10), 2697–2712.
Arnauts, J., De Cooman, R., Vandeweerdt, P., Koningsveld, R., & Berghmans, H. (1994).
Calorimetric analysis of liquid-liquid phase separation. Thermochimica Acta, 238(C), 1–
16.
Barth H.G., Carlin Jr. F.J. (1984). A review of polymer shear degradation in size-exclusion
chromatography. Journal of Liquid Chromatography 7(9), 1717-1738.
Boire, A., Menut, P., Morel, M.-H., & Sanchez, C. (2013). Phase behaviour of a wheat protein
isolate. Soft Matter, 9(47), 11417–11426.
Chu, B. Laser Light Scattering: Basic Principles and Practice; Academic Press: Boston, 1991
Dahesh, M., Banc, A., Duri, A., Morel, M. H., & Ramos, L. (2014). Polymeric assembly of
gluten proteins in an aqueous ethanol solvent. Journal of Physical Chemistry B, 118(38),
11065–11076.
Dreezen, G., Groeninckx, G., Swier, S., & Van Mele, B. (2000). Phase separation in miscible
polymer blends as detected by modulated temperature differential scanning calorimetry.
Polymer, 42(4), 1449–1459.
Giddings, J.C., (1966). A new separation concept based on a coupling of concentration and
flow nonuniformities. Separation Science. 1(1): p. 123-125.
Gonzale Dynamic Viscosities, Densities, and Speed of
Sound and Derived Properties of the Binary Systems Acetic Acid with Water, Methanol,
Ethanol, Ethyl Acetate and Methyl Acetate at T = ( 293.15, 298.15, and 303.15 ) K at
Atmospheric Pressure. Journal of Chemical and Engineering Data, 449, 1590–1596.

Chapitre 2 – Matériel et méthodes
65
Khattab, I. S., Bandarkar, F., Fakhree, M. A. A., & Jouyban, A. (2012). Density, viscosity,
and surface tension of water+ethanol mixtures from 293 to 323K. Korean Journal of
Chemical Engineering, 29(6), 812–817.
Koppel, D. E. (1972). Analysis of macromolecular polydispersity in intensity correlation
spectroscopy: The method of cumulants. The Journal of Chemical Physics, 57(11), 4814–
4820.
Layne, E. (1957). Spectrophotometric and turbidimetric methods for measuring proteins.
Methods in Enzymology, 3(C), 447–454.
Micard, V., & Guilbert, S. (2000). Thermal behavior of native and hydrophobized wheat gluten,
gliadin and glutenin-rich fractions by modulated DSC. International Journal of Biological
Macromolecules, 27(3), 229–236.
Radulescu, A., Pipich, V., Frielinghaus, H., & Appavou, M. S. (2012). KWS-2, the high
intensity / wide Q-range small-angle neutron diffractometer for soft-matter and biology
at FRM II. Journal of Physics: Conference Series, 351(1).
Rawiso M. (1999). De l’intensité à la structure en physico-chimie des polymères. Journal
Physique IV France 9. Pr1 147-195.
Reading, M. Gill, P.S., Sauerbrunn, S.R (1993). Modulated differential scanning calorimetry,
Journal of Thermal Analysis, Vol. 40, 931-939.
Shapiro A.L., Vinuela E., & Maizel J.V. (1967). Molecular weight estimation of polypeptide
chains by electrophoresis in SDS-Polyacrylamide gels, 28(5).
Swier, S., Pieters, R., & Van Mele, B. (2002). Kinetics of demixing and remixing in
poly(ethylene oxide)/poly(ether sulphone) blends as studied by modulated temperature
differential scanning calorimetry. Polymer, 43(13), 3611–3620.
Tanaka, H. (2000). Viscoelastic phase separation. Journal of Physics: Condensed Matter,
12(15), R207.

Chapitre 2 – Matériel et méthodes
66
Teraoka I., 2002. Polymer Solutions: An Introduction to Physical Properties. John Wiley &
Sons.
Wahlund, K.-G., Gustavsson, M., MacRitchie, F., Nylander, T., & Wannerberger, L. (1996).
Size Characterisation of Wheat Proteins, Particularly Glutenin, by Asymmetrical Flow
Field-Flow Fractionation. J. Cereal Sci., 23, 113–119.
Wu, H. (2010). Correlations between the Rayleigh ratio and the wavelength for toluene and
benzene. Chemical Physics, 367(1), 44–47.

Chapitre 3 – Mise en place d’un nouvel outil
67
Chapitre 3
Mise en place d’un nouvel outil pour la
détermination des points de troubles
Ce chapitre a pour but d’expliquer les différentes étapes du développement d’un nouveau
système expérimental permettant de déterminer le point de trouble de solutions de protéines.
La conception du dispositif expérimental s’est déroulée en plusieurs étapes :
- première étape : définition du besoin pour fixer les objectifs de réalisation et de
mesures. Une étude bibliographique portant sur les méthodes actuellement utilisées pour
déterminer les points de trouble sera réalisée et mettra en évidence le besoin d’un nouvel outil.
- seconde étape : conception et calibration du montage expérimental afin d’ajuster
les différents paramètres permettant de répondre aux besoins. Des essais préliminaires ont été
réalisés pour fixer les conditions d’utilisations de chaque élément du montage. Des facteurs plus
techniques liés à la réponse des systèmes utilisés, ont également nécessité une étude. L’ensemble
de ces calibrations ont été réalisées sur des extraits de gluten.
- troisième étape : validation de l’outil pour la détermination des points de trouble.
Pour cela nous avons opté pour un surfactant, le Triton X-114 qui sépare de phases lors d’une
augmentation de température. La connaissance de ce système dans la littérature, va permettre
une comparaison avec les résultats obtenus par le montage expérimental et ainsi, de confirmer
et valider la performance et l’intérêt de la nouvelle approche expérimentale adoptée pour la
détermination de points de troubles.
Ce chapitre sera découpé selon la démarche proposée dans la Figure 3.1.

Chapitre 3 – Mise en place d’un nouvel outil
68
Figure 3.1 : Représentation schématique de la démarche utilisée pour le développement du nouvel outil
expérimental
1. Définition du besoin
Dans le cadre de ce travail de thèse, nous souhaitons établir les diagrammes de phases de
protéines de gluten en solution, et plus particulièrement, étudier les phénomènes de séparations
de phases liquides-liquides qui se manifestent par un trouble des solutions et déterminer la
température (Tcloud) à laquelle se produit ce trouble. Les points de trouble sont mesurés
lorsqu’un système passe d’un état monophasique à biphasique. Cette transition peut être
induite par un changement de concentration, de température, du pH, de la force ionique, ou
encore par l’ajout d’un composant. La détermination de plusieurs températures de points de
trouble dans différentes conditions, permet d’établir des diagrammes de phases. Cependant,
leur réalisation nécessite de nombreuses mesures qu’il n’est pas forcément aisé de réaliser. En
outre, pour de nombreuses protéines, le point de trouble est inférieur à la température
ambiante, ce qui implique de disposer d’un système permettant de travailler à basse
température.
Il existe de nombreuses méthodes permettant de mesurer Tcloud. Les plus simples sont basées
sur l’observation visuelle de la solution suite à son changement d’apparence (opacification).
C’est notamment cette méthode qu’ont utilisé Nikas, et al., 1992 ou encore Liu, et al., 1998
Définition du besoin
Conception et calibration de l’installation
Domaine de validation
ObjectifsConnaissances actuelles
Mettre en place un outil répondant à différents critères
Conception du prototype
Identification des moyens existants pour la détermination de points de trouble
Mise au point
Adaptation du matériel
Essais sur des systèmes connus (TX-114)
Validation de la
méthode
Comparaison avec la littérature
Problématique
Application
Définition du cahier des charges
Détermination des conditions opératoires, évaporation / condensation
Enceinte climatique ,caméra, plaque lumineuse
Application à d’autres
systèmes
Chapitre 5

Chapitre 3 – Mise en place d’un nouvel outil
69
sur des solutions de tensioactifs pour améliorer l’extraction de protéines membranaire par
l’ajout d’un surfactant. Cependant, ces mesures ont une précision limitée, et peuvent donner
lieu à de la variabilité entre observateurs. C’est pourquoi la mesure de la chute de transmittance
par un faisceau laser traversant la solution s’avère plus efficace est moins subjective
(Pozdnyshev et al., 1978), (Hefter et al., 1991). Pendant une rampe de température, le point
de trouble est lu lorsque la lumière transmise atteint un pourcentage arbitraire de la
transmittance initiale. Ce pourcentage arbitraire varie selon les auteurs. Chen et al., 1990 ont
défini Tcloud lorsque la lumière transmise atteint 90% de sa valeur initiale, et Broide et al., 1996,
ont fixé un seuil à 70%, mais la plupart des études optent pour une réduction du signal de 50%.
C’est par exemple ce qui a été retenu sur des polycations (Tachaboonyakiat et al., 2013), sur
des polymères, (Jain et al., 2015) ou encore sur des protéines (Asherie, 2004) et plus
particulièrement sur les gliadines (Boire et al., 2013). Une autre méthode utilisée consiste à
identifier Tcloud comme le point d’inflexion de turbidité avec la température, comme réalisé par
Miyazaki et al., en 1995.
Avoir un critère simple et quantitatif est un avantage clé par rapport à l'observation visuelle
simple, en particulier parce que la netteté de la chute de la transmittance au point de trouble
dépend des conditions expérimentales. Pour les systèmes en solutions dont le changement se
manifeste par une séparation de phases liquide-liquide, la diminution de la transmittance est
très progressive lorsque la séparation de phases est approchée à des concentrations de protéines
proches du point critique (Tc), mais nettement plus marquées des concentrations plus élevée
ou plus faibles (Liu et al., 1996). Pour les phénomènes réversibles comme une séparation de
phase liquide-liquide, certains auteurs ont pris la moyenne entre la température à laquelle la
solution devient turbide pendant le refroidissement (ou le chauffage) et la température à
laquelle elle redevient claire lors d’un retour vers la température initiale (par chauffage ou
refroidissement) (Taratura et al., 1990). Les méthodes mentionnées ci-dessus, basées sur des
mesures de transmittance ou de diffusion, permettent la détermination de la température du
point de trouble pour un échantillon à la fois. Établir le diagramme de phase complet est donc
une longue tâche.
Outres les méthodes classiques présentées plus haut, le point de trouble a été déterminé
également par viscosimètre (Nozary, et al., 2002), par résonance magnétique nucléaire à

Chapitre 3 – Mise en place d’un nouvel outil
70
transformé de Fourier (Caneba, et al., 1985), par réfractométrie (Mohsen-Nia, et al., 2006) ou
encore par diffusion de neutrons (Shibayama, et al., 2004). Bien que précises et fiables, ces
dernières techniques sont rarement exploitées. La nécessité de faire une mesure après l’autre
n’apporte en effet pas de réels avantages par rapport à d’autres méthodes plus classiques.
La calorimétrie différentielle à balayage (DSC) est une technique aussi utilisée (Boutris, et al.,
1997). Une mesure assez précise de la Tcloud est réalisée par le changement d’amplitude du
signal qui est associé à la transition de phase. Les DSC donnent des résultats similaires à des
méthodes conventionnelles telles que l’observation visuelle ou la méthode UV. (Boutris et al.,
1997 ; Yamaoka et al., 2003). La calorimétrie différentielle à balayage en mode modulée
(MDSC) permet de gagner en précision dans la détermination de Tcloud (Dreezen et al., 2001).
Cette technique permet de travailler sur des quantités relativement petites (10-30 mg) (Swier,
et al., 2002), et permet de recouvrir une large gamme de températures. Cependant, cette
technique ne permet là encore que de caractériser les échantillons un par un, et ne peut être
utilisée que pour des solutions suffisamment concentrées en protéines.
Pour réduire les temps de mesures ou pour réduire les coûts, d’autres études ont porté sur
l’amélioration de ces techniques classiques. Par exemple, l’utilisation de sources LED et de
camera disponibles sur le marché a permis la construction de systèmes de détermination des
points de trouble à bas coût (Silva, et al., 2013), (Williamson & Kiefer, 2014). Bien que ces
méthodes soient reproductibles et fiables, elles nécessitent cependant, beaucoup de matières
premières et ne permettent que d’étudier une seule condition à la fois. Or, les protéines et
autres biopolymères étudiés sont parfois disponibles en faible quantité. Récemment, un système
basé sur la millifuidique a démontré sa capacité à pouvoir sonder la séparation de phase liquide-
liquide en fonction de la température sur des mélanges de biopolymères sur de petits volumes
(Amine, et al., 2017). Cette méthode, permet de caractériser des solutions, (à une seule
température à la fois), avec de petites quantités de matière. D’autres approches pour tester
plusieurs conditions en même temps ont été proposées. Corti et ses collègues (Corti, et al.,
1984) ont développé un système basé sur la turbidimétrie pour mesurer simultanément le point
de trouble de 8 solutions. Cette technique, plus rapide que les autres, fût un premier pas pour
tester plusieurs paramètres en même temps. Plus tard, Kitabatake et al., 1994, ou encore Li et
al., 2013 ont mesuré de nombreuses conditions sur de faibles quantités grâce à un lecteur de
microplaque. Cette technique, plus conventionnelle et facilement disponible, semble la plus

Chapitre 3 – Mise en place d’un nouvel outil
71
adaptée mais n’a pourtant pas la capacité de faire des mesures à des températures inférieures
à l’ambiante. Les appareils sur le marché ne peuvent pas refroidir en dessous de l’ambiante, et
les fabricants contactés refusent de s’engager sur leur matériel si celui-ci est placé en chambre
froide ou enceinte climatique, du fait des risques de condensation.
Toutes ces méthodes ont démontré leur efficacité et chacune présente des avantages rappelés
dans la Table 3.1. Ces derniers sont représentés en fonction de critères que nous avons établi
selon un cahier des charges qui définit les fonctions auxquelles devra répondre le nouveau
montage expérimental. D’après ce cahier des charges, le nouvel outil doit :
1. Permettre de faire des mesures sur une large gamme de température (0 à 30°C)
en évitant les problèmes de condensation et/ou d’évaporation.
2. Permettre de tester simultanément plusieurs paramètres en une seule mesure
afin de réduire les temps d’expériences.
3. Utiliser des quantités d’échantillons raisonnables (inférieur au mL) afin de
pouvoir travailler sur des systèmes disponibles en faible quantité.
4. Avoir un protocole avec un temps d’expérience réduit et/ou qui ne demande
pas une surveillance continue à l’expérimentateur.
5. Pouvoir quantifier la valeur de Tcloud de façon reproductible et avec une
précision de l’ordre de +/- 0,5°C.
Comme vu dans la Table 3.1, aucune des techniques trouvées dans la littérature ne répond à
l’ensemble de tous les critères du cahier des charges. C’est pourquoi nous avons décidé de
développer un système en interne répondant aux différents critères fixés.
Par ailleurs, le montage expérimental ne doit pas dépasser le budget accordé par l’INRA et
SupAgro qui était initialement de 22k€. Enfin, le montage doit être facile à assembler et pouvoir
être utilisé dans d’autres laboratoires notamment au sein d’organismes tels que l’INRA. Ainsi,
le nouveau montage expérimental présentera l’avantage d’être accessible à un large public.
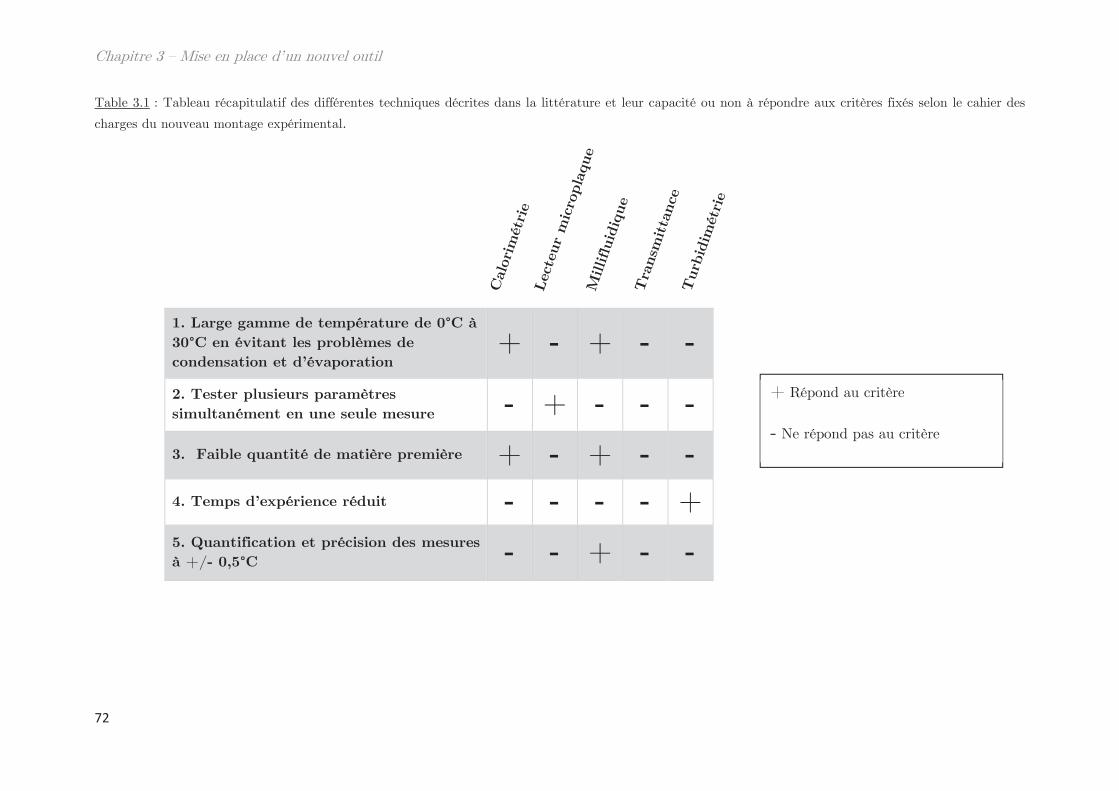
Chapitre 3 – Mise en place d’un nouvel outil
72
Table 3.1 : Tableau récapitulatif des différentes techniques décrites dans la littérature et leur capacité ou non à répondre aux critères fixés selon le cahier des
charges du nouveau montage expérimental.
1. Large gamme de température de 0°C à 30°C en évitant les problèmes de condensation et d’évaporation
+ - + - -
2. Tester plusieurs paramètres simultanément en une seule mesure - + - - -
3. Faible quantité de matière première + - + - -
4. Temps d’expérience réduit - - - - +
5. Quantification et précision des mesures à +/- 0,5°C - - + - -
+ Répond au critère
- Ne répond pas au critère

Chapitre 3 – Mise en place d’un nouvel outil
73
2. Conception et calibration du montage expérimental
Ce chapitre détaille le montage expérimental que nous avons conçu et sa calibration. Chaque
élément choisi pour le nouvel appareil a nécessité une évaluation individuelle afin d’être utilisé
dans les conditions les plus favorables pour répondre aux besoins précédemment cités.
2.1 Dispositif expérimental
Le nouveau dispositif repose sur l’acquisition d’images d’une microplaque positionnée dans une
enceinte climatique contrôlée en température. La microplaque est éclairée via une source
lumineuse placée en dessous de celle-ci, pour permettre à une caméra, reliée à un ordinateur,
de prendre des photos. Une sonde fixe (TP1) est localisée dans la paroi arrière de la chambre
et est utilisée pour contrôler et surveiller la température de l’enceinte Tenceinte. Une sonde
amovible (TP2) est placée dans un puits de la microplaque remplie avec l'échantillon (situé
hors du champ de vision de la caméra), et permet d’enregistrer en continu la température de
l'échantillon, Téchantillon. Le montage expérimental est représenté dans la Figure 3.2 par un
schéma simplifié du montage et l’ensemble du matériel est détaillé dans le Chapitre 2 section
2.4.
Figure 3.2 : Schéma du montage expérimental. Le système d'acquisition est placé dans une chambre
climatique pour réguler la température et l'humidité relative. Il est composé d'une caméra connectée à
un ordinateur (à l'extérieur de la chambre), d'une plaque lumineuse ou rétroéclairage (en jaune) et d'une
microplaque placée sur un support métallique. Un tube flexible entoure le rétroéclairage, pour extraire
les calories produites, il est relié à un bain thermostaté (à l'extérieur de la chambre). Les sondes
thermiques TP 1 (pour mesurer Tenceinte) et TP 2 (pour mesurer Téchantillon) sont indiquées par des cercles.
Circulation d’eau
Ordinateur
Microplaque
Plaque lumineuse
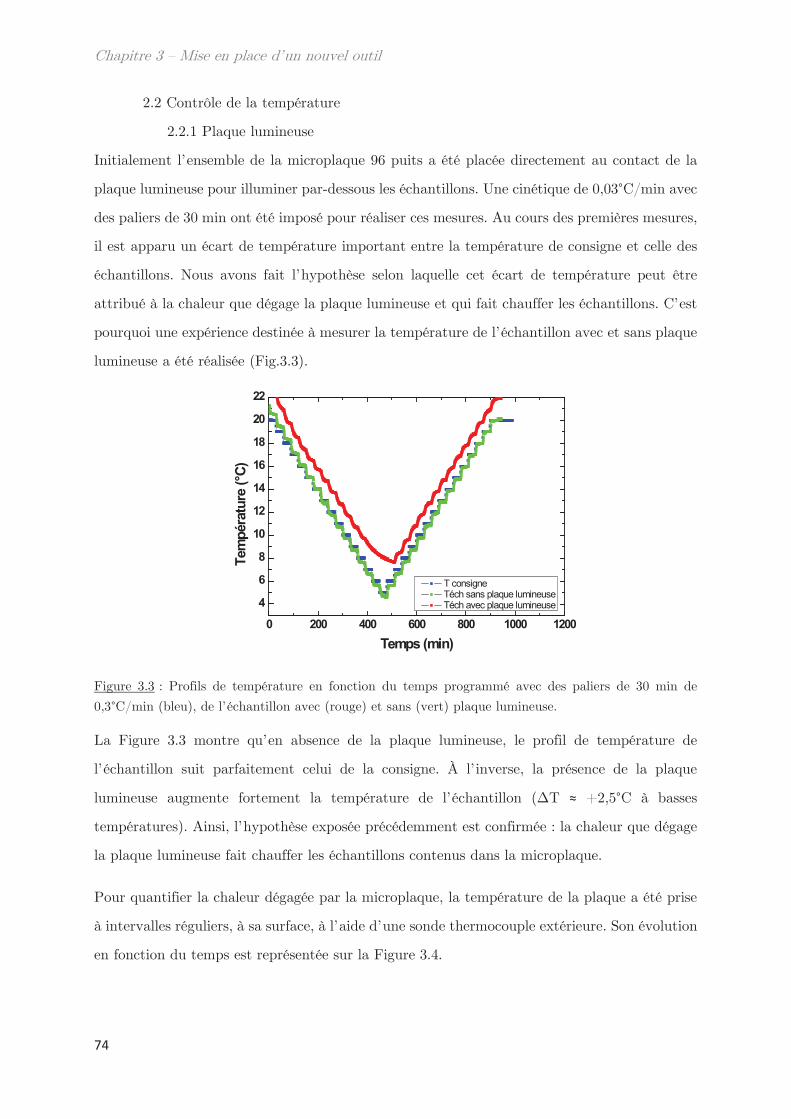
Chapitre 3 – Mise en place d’un nouvel outil
74
2.2 Contrôle de la température
2.2.1 Plaque lumineuse
Initialement l’ensemble de la microplaque 96 puits a été placée directement au contact de la
plaque lumineuse pour illuminer par-dessous les échantillons. Une cinétique de 0,03°C/min avec
des paliers de 30 min ont été imposé pour réaliser ces mesures. Au cours des premières mesures,
il est apparu un écart de température important entre la température de consigne et celle des
échantillons. Nous avons fait l’hypothèse selon laquelle cet écart de température peut être
attribué à la chaleur que dégage la plaque lumineuse et qui fait chauffer les échantillons. C’est
pourquoi une expérience destinée à mesurer la température de l’échantillon avec et sans plaque
lumineuse a été réalisée (Fig.3.3).
Figure 3.3 : Profils de température en fonction du temps programmé avec des paliers de 30 min de
0,3°C/min (bleu), de l’échantillon avec (rouge) et sans (vert) plaque lumineuse.
La Figure 3.3 montre qu’en absence de la plaque lumineuse, le profil de température de
l’échantillon suit parfaitement celui de la consigne. À l’inverse, la présence de la plaque
lumineuse augmente fortement la température de l’échantillon +2,5°C à basses
températures). Ainsi, l’hypothèse exposée précédemment est confirmée : la chaleur que dégage
la plaque lumineuse fait chauffer les échantillons contenus dans la microplaque.
Pour quantifier la chaleur dégagée par la microplaque, la température de la plaque a été prise
à intervalles réguliers, à sa surface, à l’aide d’une sonde thermocouple extérieure. Son évolution
en fonction du temps est représentée sur la Figure 3.4.
0 200 400 600 800 1000 1200
4
6
8
10
12
14
16
18
20
22
Tem
péra
ture
(°C
)
Temps (min)
T consigne Téch sans plaque lumineuse Téch avec plaque lumineuse
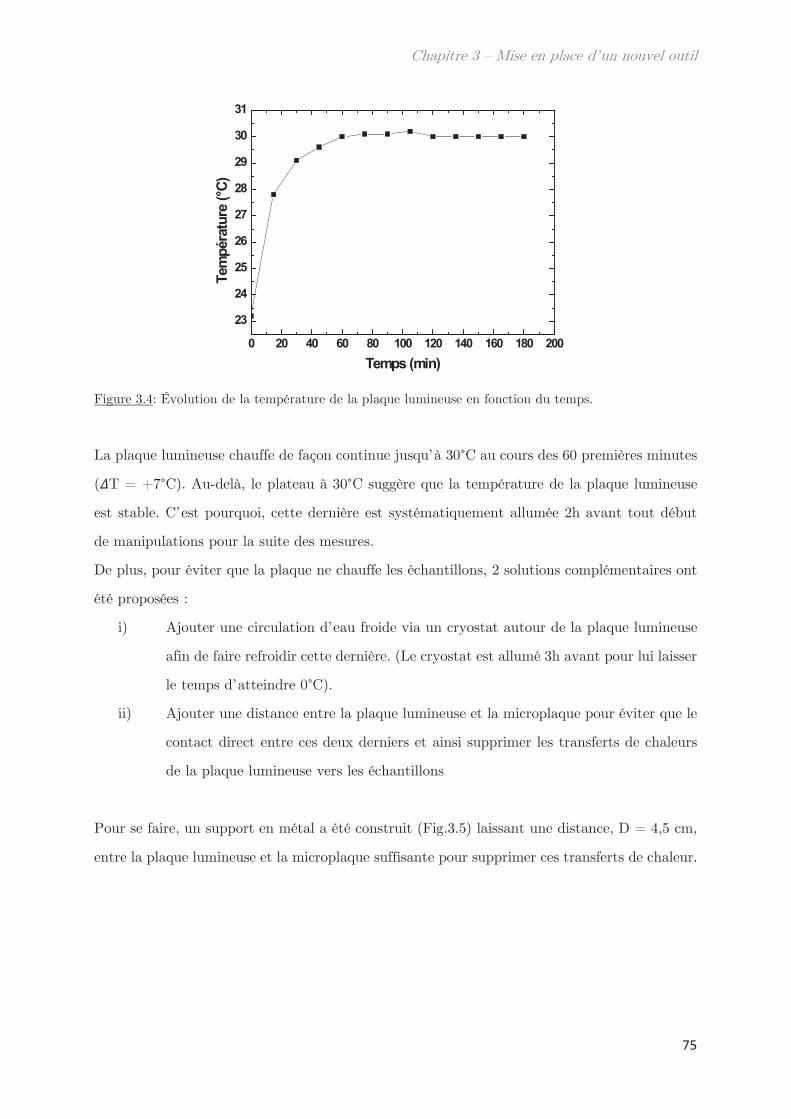
Chapitre 3 – Mise en place d’un nouvel outil
75
Figure 3.4: Évolution de la température de la plaque lumineuse en fonction du temps.
La plaque lumineuse chauffe de façon continue jusqu’à 30°C au cours des 60 premières minutes
( T = +7°C). Au-delà, le plateau à 30°C suggère que la température de la plaque lumineuse
est stable. C’est pourquoi, cette dernière est systématiquement allumée 2h avant tout début
de manipulations pour la suite des mesures.
De plus, pour éviter que la plaque ne chauffe les échantillons, 2 solutions complémentaires ont
été proposées :
i) Ajouter une circulation d’eau froide via un cryostat autour de la plaque lumineuse
afin de faire refroidir cette dernière. (Le cryostat est allumé 3h avant pour lui laisser
le temps d’atteindre 0°C).
ii) Ajouter une distance entre la plaque lumineuse et la microplaque pour éviter que le
contact direct entre ces deux derniers et ainsi supprimer les transferts de chaleurs
de la plaque lumineuse vers les échantillons
Pour se faire, un support en métal a été construit (Fig.3.5) laissant une distance, D = 4,5 cm,
entre la plaque lumineuse et la microplaque suffisante pour supprimer ces transferts de chaleur.
0 20 40 60 80 100 120 140 160 180 200
23
24
25
26
27
28
29
30
31
Tem
péra
ture
(°C
)
Temps (min)

Chapitre 3 – Mise en place d’un nouvel outil
76
Figure 3.5 : Photo du montage expérimental du support créé afin d’éviter le contact direct entre la
microplaque et la plaque lumineuse
Il est important de noter que la création du support à réduit la distance échantillon – caméra
( d = -4,5 cm). Ainsi, alors que la totalité de la microplaque (96 puits) était visible lorsque
cette dernière était placée directement sur la plaque lumineuse, seulement 77 puits restent
visibles par la caméra lorsque la microplaque est placée sur le support. Ce facteur non pris en
compte initialement réduit donc les possibilités de 96 puits à 77 puits, ce qui reste un nombre
remarquable au vu du critère fixé initialement.
Via ces différentes solutions, nous avons pallié le problème de chauffage de la plaque lumineuse
tout en conservant les différents critères fixés par le cahier des charges comme il est démontré
dans la section suivante.
2.2.2 Enceinte
Le système de programmation de l’enceinte climatique est piloté par un logiciel fourni par le
fabricant (AtmoControl, Memmert). Dans un premier temps, pour évaluer les rampes de
température utilisables avec le montage, différents profils des températures ont été testés. Les
températures relevées par la sonde TP1, Tenceinte, et celles relevées par la sonde TP2, Téchantillon,
sont représentées en fonction de la température de consigne, Tconsigne, dans la Figure 3.6 a et b.
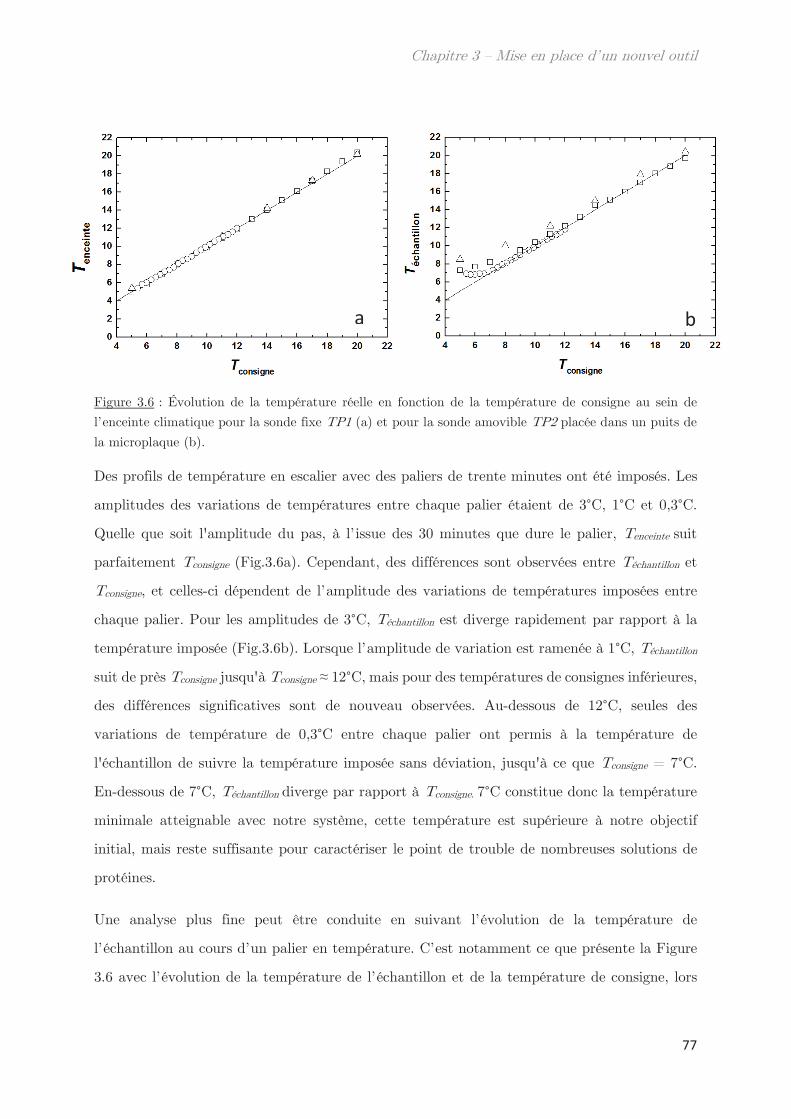
Chapitre 3 – Mise en place d’un nouvel outil
77
Figure 3.6 : Évolution de la température réelle en fonction de la température de consigne au sein de
l’enceinte climatique pour la sonde fixe TP1 (a) et pour la sonde amovible TP2 placée dans un puits de
la microplaque (b).
Des profils de température en escalier avec des paliers de trente minutes ont été imposés. Les
amplitudes des variations de températures entre chaque palier étaient de 3°C, 1°C et 0,3°C.
Quelle que soit l'amplitude du pas, à l’issue des 30 minutes que dure le palier, Tenceinte suit
parfaitement Tconsigne (Fig.3.6a). Cependant, des différences sont observées entre Téchantillon et
Tconsigne, et celles-ci dépendent de l’amplitude des variations de températures imposées entre
chaque palier. Pour les amplitudes de 3°C, Téchantillon est diverge rapidement par rapport à la
température imposée (Fig.3.6b). Lorsque l’amplitude de variation est ramenée à 1°C, Téchantillon
suit de près Tconsigne jusqu'à Tconsigne 12°C, mais pour des températures de consignes inférieures,
des différences significatives sont de nouveau observées. Au-dessous de 12°C, seules des
variations de température de 0,3°C entre chaque palier ont permis à la température de
l'échantillon de suivre la température imposée sans déviation, jusqu'à ce que Tconsigne = 7°C.
En-dessous de 7°C, Téchantillon diverge par rapport à Tconsigne. 7°C constitue donc la température
minimale atteignable avec notre système, cette température est supérieure à notre objectif
initial, mais reste suffisante pour caractériser le point de trouble de nombreuses solutions de
protéines.
Une analyse plus fine peut être conduite en suivant l’évolution de la température de
l’échantillon au cours d’un palier en température. C’est notamment ce que présente la Figure
3.6 avec l’évolution de la température de l’échantillon et de la température de consigne, lors
b a
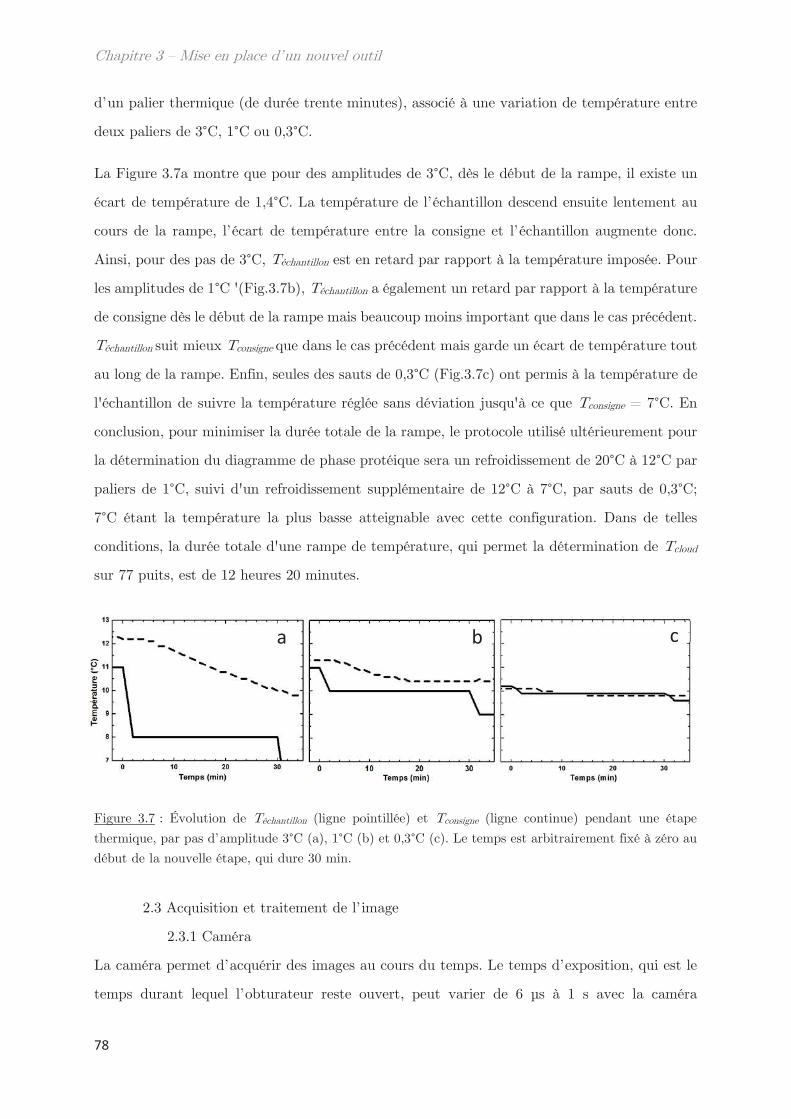
Chapitre 3 – Mise en place d’un nouvel outil
78
d’un palier thermique (de durée trente minutes), associé à une variation de température entre
deux paliers de 3°C, 1°C ou 0,3°C.
La Figure 3.7a montre que pour des amplitudes de 3°C, dès le début de la rampe, il existe un
écart de température de 1,4°C. La température de l’échantillon descend ensuite lentement au
cours de la rampe, l’écart de température entre la consigne et l’échantillon augmente donc.
Ainsi, pour des pas de 3°C, Téchantillon est en retard par rapport à la température imposée. Pour
les amplitudes de 1°C '(Fig.3.7b), Téchantillon a également un retard par rapport à la température
de consigne dès le début de la rampe mais beaucoup moins important que dans le cas précédent.
Téchantillon suit mieux Tconsigne que dans le cas précédent mais garde un écart de température tout
au long de la rampe. Enfin, seules des sauts de 0,3°C (Fig.3.7c) ont permis à la température de
l'échantillon de suivre la température réglée sans déviation jusqu'à ce que Tconsigne = 7°C. En
conclusion, pour minimiser la durée totale de la rampe, le protocole utilisé ultérieurement pour
la détermination du diagramme de phase protéique sera un refroidissement de 20°C à 12°C par
paliers de 1°C, suivi d'un refroidissement supplémentaire de 12°C à 7°C, par sauts de 0,3°C;
7°C étant la température la plus basse atteignable avec cette configuration. Dans de telles
conditions, la durée totale d'une rampe de température, qui permet la détermination de Tcloud
sur 77 puits, est de 12 heures 20 minutes.
Figure 3.7 : Évolution de Téchantillon (ligne pointillée) et Tconsigne (ligne continue) pendant une étape
thermique, par pas d’amplitude 3°C (a), 1°C (b) et 0,3°C (c). Le temps est arbitrairement fixé à zéro au
début de la nouvelle étape, qui dure 30 min.
2.3 Acquisition et traitement de l’image
2.3.1 Caméra
La caméra permet d’acquérir des images au cours du temps. Le temps d’exposition, qui est le
temps durant lequel l’obturateur reste ouvert, peut varier de 6 µs à 1 s avec la caméra
a b c
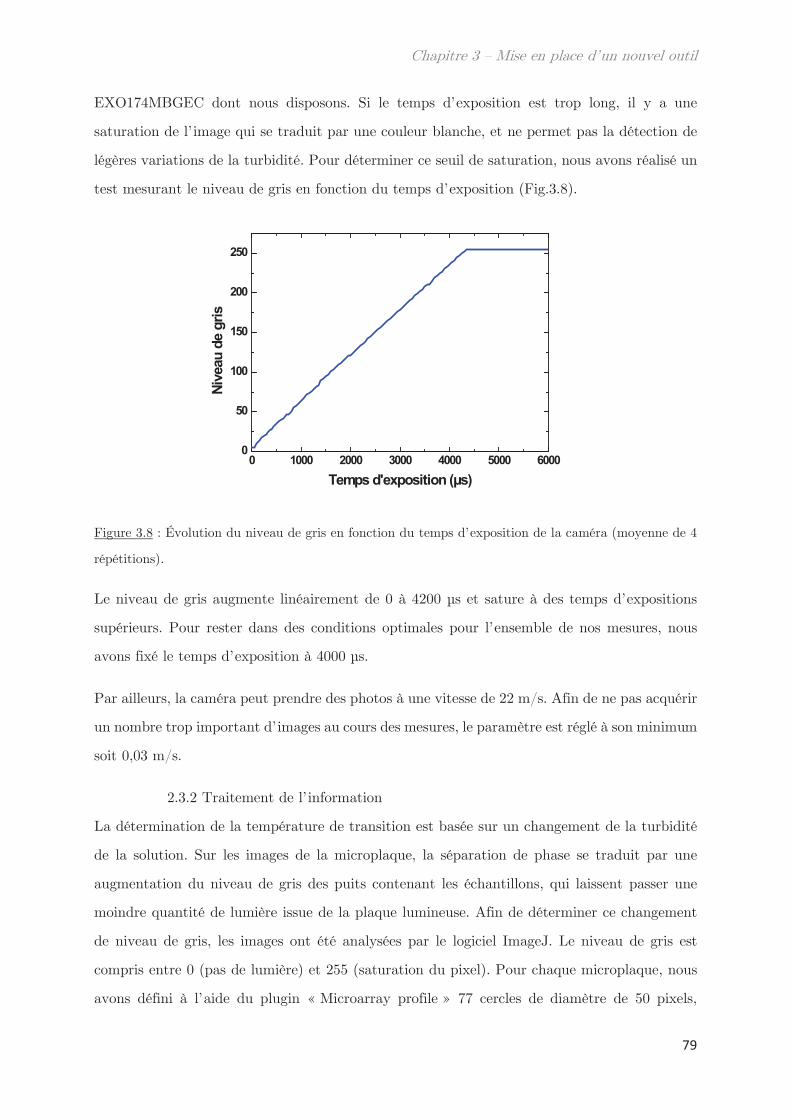
Chapitre 3 – Mise en place d’un nouvel outil
79
EXO174MBGEC dont nous disposons. Si le temps d’exposition est trop long, il y a une
saturation de l’image qui se traduit par une couleur blanche, et ne permet pas la détection de
légères variations de la turbidité. Pour déterminer ce seuil de saturation, nous avons réalisé un
test mesurant le niveau de gris en fonction du temps d’exposition (Fig.3.8).
Figure 3.8 : Évolution du niveau de gris en fonction du temps d’exposition de la caméra (moyenne de 4
répétitions).
Le niveau de gris augmente linéairement de 0 à 4200 µs et sature à des temps d’expositions
supérieurs. Pour rester dans des conditions optimales pour l’ensemble de nos mesures, nous
avons fixé le temps d’exposition à 4000 µs.
Par ailleurs, la caméra peut prendre des photos à une vitesse de 22 m/s. Afin de ne pas acquérir
un nombre trop important d’images au cours des mesures, le paramètre est réglé à son minimum
soit 0,03 m/s.
2.3.2 Traitement de l’information
La détermination de la température de transition est basée sur un changement de la turbidité
de la solution. Sur les images de la microplaque, la séparation de phase se traduit par une
augmentation du niveau de gris des puits contenant les échantillons, qui laissent passer une
moindre quantité de lumière issue de la plaque lumineuse. Afin de déterminer ce changement
de niveau de gris, les images ont été analysées par le logiciel ImageJ. Le niveau de gris est
compris entre 0 (pas de lumière) et 255 (saturation du pixel). Pour chaque microplaque, nous
avons défini à l’aide du plugin « Microarray profile » 77 cercles de diamètre de 50 pixels,
0 1000 2000 3000 4000 5000 60000
50
100
150
200
250
Niv
eau d
e g
ris
Temps d'exposition (µs)

Chapitre 3 – Mise en place d’un nouvel outil
80
localisés au centre de chaque puits. Un exemple de ces cercles nommés ‘région d’intérêt’ sont
représentés pour quelques puits dans la Figure 3.9a (à noter que les puits contenant
l’échantillon sont noirs et les puits vides sont transparents). L’intensité moyenne des pixels
contenus dans chaque cercle est exprimée en valeur de niveau de gris et exportée dans un fichier
au format Texte. Ces données sont ensuite retranscrites dans un tableur (Excel), afin de pouvoir
les analyser.
La Figure 3.9b représente un tableau dans lequel se trouve le niveau de gris associé à chaque
puits de la microplaque présentée en Figure 3.9a. Les puits apparaissant grisés sur la photo,
qui contiennent la solution de gluten, ont un niveau de gris qui varie de 94 à 109, alors que les
puits vides ont un niveau de gris qui varie de 208 à 222.
Figure 3.9 : Image d’une microplaque, à température ambiante, dont certains puits contiennent un
échantillon de gluten dans certains puits. Les cercles jaunes matérialisent l’aire utilisée pour la mesure
du niveau de gris sur ImageJ (a). Exemple de représentation Excel permettant la mesure quantitative
du niveau de gris (0 à 255) pour chaque puits (b).
Des photos de cette microplaque ont ensuite été acquise au cours d’une rampe en température,
et ce traitement a été réalisé sur toutes les images, correspondantes chacune à une température
spécifique. Les données du niveau de gris ont ensuite été tracées pour chaque puits en fonction
de la température (Fig.3.10).
Région d’intérêta
87.89 214.87 216.31 217.03 215.67 96.97 216.97 216.91 213.83 211 90.83
214.9 217.89 218.26 97.21 217.92 218.99 218.73 95.6 213.84 211.74 210.74
89.11 219.79 220.82 220.16 99.95 221.09 220.52 219.39 216.51 214.27 89.25
214.44 218.66 221.76 221.38 220.07 101.04 221.57 220.89 218.41 215.73 212.96
214.54 216.31 96.15 220.61 218.84 219.54 102.33 220.61 218.48 215.02 87.54
87.22 214.08 222.34 217.96 216.12 98.08 217.88 218.7 215.6 212.73 208.13
213.13 87.48 215.03 216.32 93.21 217.55 219.44 93.69 217.62 211.19 89.38
b
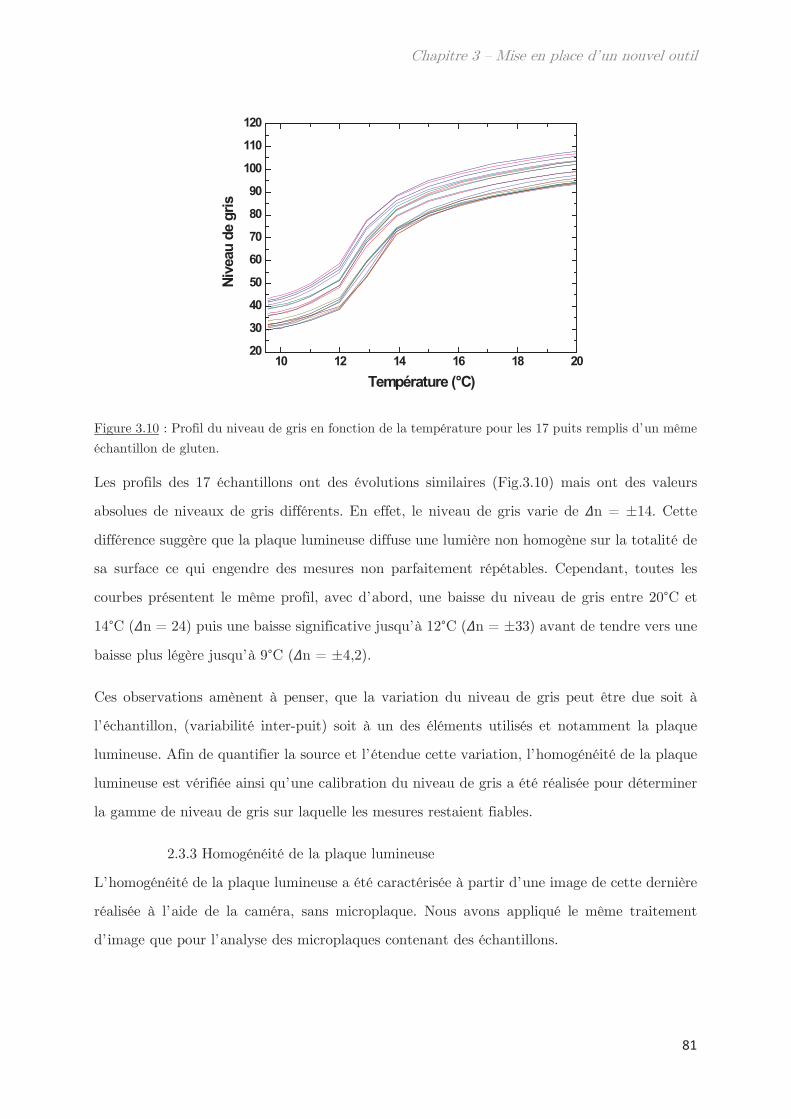
Chapitre 3 – Mise en place d’un nouvel outil
81
Figure 3.10 : Profil du niveau de gris en fonction de la température pour les 17 puits remplis d’un même
échantillon de gluten.
Les profils des 17 échantillons ont des évolutions similaires (Fig.3.10) mais ont des valeurs
absolues de niveaux de gris différents. En effet, le niveau de gris varie de n = ±14. Cette
différence suggère que la plaque lumineuse diffuse une lumière non homogène sur la totalité de
sa surface ce qui engendre des mesures non parfaitement répétables. Cependant, toutes les
courbes présentent le même profil, avec d’abord, une baisse du niveau de gris entre 20°C et
14°C ( n = 24) puis une baisse significative jusqu’à 12°C ( n = ±33) avant de tendre vers une
baisse plus légère jusqu’à 9°C ( n = ±4,2).
Ces observations amènent à penser, que la variation du niveau de gris peut être due soit à
l’échantillon, (variabilité inter-puit) soit à un des éléments utilisés et notamment la plaque
lumineuse. Afin de quantifier la source et l’étendue cette variation, l’homogénéité de la plaque
lumineuse est vérifiée ainsi qu’une calibration du niveau de gris a été réalisée pour déterminer
la gamme de niveau de gris sur laquelle les mesures restaient fiables.
2.3.3 Homogénéité de la plaque lumineuse
L’homogénéité de la plaque lumineuse a été caractérisée à partir d’une image de cette dernière
réalisée à l’aide de la caméra, sans microplaque. Nous avons appliqué le même traitement
d’image que pour l’analyse des microplaques contenant des échantillons.
10 12 14 16 18 2020
30
40
50
60
70
80
90
100
110
120
Niv
eau d
e g
ris
Température (°C)
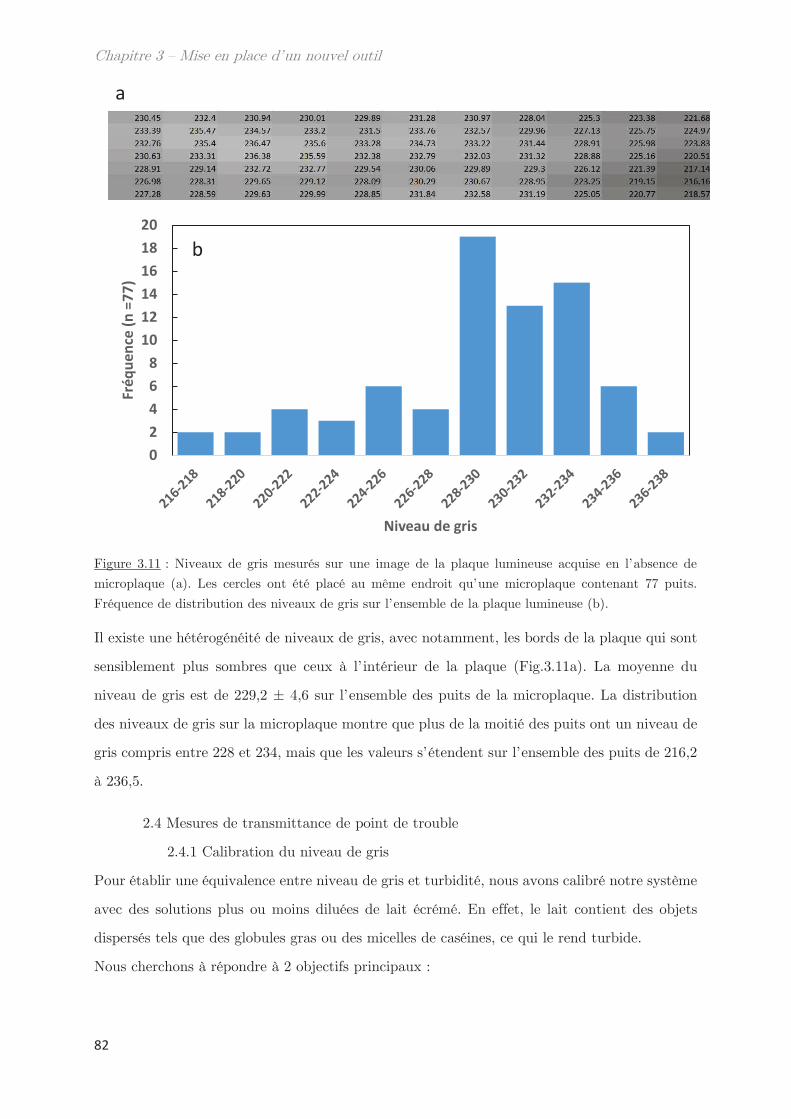
Chapitre 3 – Mise en place d’un nouvel outil
82
Figure 3.11 : Niveaux de gris mesurés sur une image de la plaque lumineuse acquise en l’absence de
microplaque (a). Les cercles ont été placé au même endroit qu’une microplaque contenant 77 puits.
Fréquence de distribution des niveaux de gris sur l’ensemble de la plaque lumineuse (b).
Il existe une hétérogénéité de niveaux de gris, avec notamment, les bords de la plaque qui sont
sensiblement plus sombres que ceux à l’intérieur de la plaque (Fig.3.11a). La moyenne du
niveau de gris est de 229,2 ± 4,6 sur l’ensemble des puits de la microplaque. La distribution
des niveaux de gris sur la microplaque montre que plus de la moitié des puits ont un niveau de
gris compris entre 228 et 234, mais que les valeurs s’étendent sur l’ensemble des puits de 216,2
à 236,5.
2.4 Mesures de transmittance de point de trouble
2.4.1 Calibration du niveau de gris
Pour établir une équivalence entre niveau de gris et turbidité, nous avons calibré notre système
avec des solutions plus ou moins diluées de lait écrémé. En effet, le lait contient des objets
dispersés tels que des globules gras ou des micelles de caséines, ce qui le rend turbide.
Nous cherchons à répondre à 2 objectifs principaux :
a
0
2
4
6
8
10
12
14
16
18
20
Fré
qu
en
ce (
n =
77
)
Niveau de gris
b
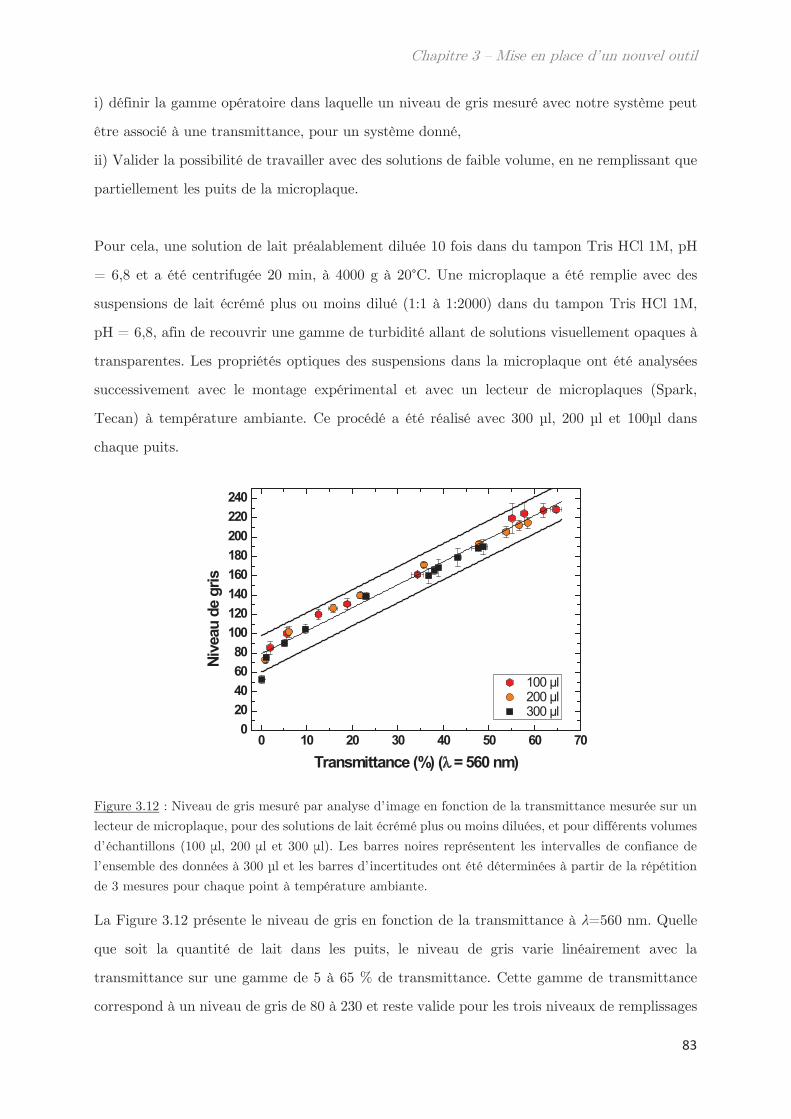
Chapitre 3 – Mise en place d’un nouvel outil
83
i) définir la gamme opératoire dans laquelle un niveau de gris mesuré avec notre système peut
être associé à une transmittance, pour un système donné,
ii) Valider la possibilité de travailler avec des solutions de faible volume, en ne remplissant que
partiellement les puits de la microplaque.
Pour cela, une solution de lait préalablement diluée 10 fois dans du tampon Tris HCl 1M, pH
= 6,8 et a été centrifugée 20 min, à 4000 g à 20°C. Une microplaque a été remplie avec des
suspensions de lait écrémé plus ou moins dilué (1:1 à 1:2000) dans du tampon Tris HCl 1M,
pH = 6,8, afin de recouvrir une gamme de turbidité allant de solutions visuellement opaques à
transparentes. Les propriétés optiques des suspensions dans la microplaque ont été analysées
successivement avec le montage expérimental et avec un lecteur de microplaques (Spark,
Tecan) à température ambiante. Ce procédé a été réalisé avec 300 µl, 200 µl et 100µl dans
chaque puits.
Figure 3.12 : Niveau de gris mesuré par analyse d’image en fonction de la transmittance mesurée sur un
lecteur de microplaque, pour des solutions de lait écrémé plus ou moins diluées, et pour différents volumes
d’échantillons Les barres noires représentent les intervalles de confiance de
l’ensemble des données à 300 µl et les barres d’incertitudes ont été déterminées à partir de la répétition
de 3 mesures pour chaque point à température ambiante.
La Figure 3.12 présente le niveau de gris en fonction de la transmittance à =560 nm. Quelle
que soit la quantité de lait dans les puits, le niveau de gris varie linéairement avec la
transmittance sur une gamme de 5 à 65 % de transmittance. Cette gamme de transmittance
correspond à un niveau de gris de 80 à 230 et reste valide pour les trois niveaux de remplissages
0 10 20 30 40 50 60 700
20
40
60
80
100
120
140
160
180
200
220
240
100 µl 200 µl 300 µl
Niv
eau d
e g
ris
Transmittance (%) ( = 560 nm)

Chapitre 3 – Mise en place d’un nouvel outil
84
(100, 200 et 300 µl) des puits. Ainsi, ces mesures montrent qu’il est possible d’associer une
transmittance à un niveau de gris. De plus, les résultats similaires pour les trois niveaux de
remplissages démontrent qu’il est possible de réaliser des mesures avec 100 µl de solution.
2.4.2 Prévenir l’évaporation et la condensation du solvant
La principale difficulté rencontrée pour mesurer Tcloud en dessous de la température ambiante
est la condensation de la vapeur de solvant qui affecte les mesures optiques. Cette difficulté est
renforcée par le fait que les échantillons de gluten sur lesquels nous travaillons sont dispersés
dans un solvant eau/éthanol 50% (v/v), l’éthanol étant un composé très volatil.
La condensation se produit lorsque localement, la pression de vapeur partielle dépasse la
pression de vapeur saturante, qui dépend de la température. Pour éviter la condensation de
l'eau dans l’enceinte, nous réglons l'humidité relative de la chambre à 40%. Éviter l'évaporation
du solvant contenu dans le puits est cependant beaucoup plus difficile, notamment si des
solvants volatils sont utilisés, comme c'est le cas pour les prolamines du blé, qui ne sont solubles
que dans les mélanges eau / éthanol (Osborn, 1907). Pour quantifier cette évaporation, tous
les puits de la microplaque ont été rempli avec une solution de protéines en solvant eau/éthanol
(50% v/v). Cette dernière a été introduite dans la chambre climatique et le programme en
température défini dans la section 1.2.1 a été imposé. La perte de poids a été estimée en
mesurant la microplaque avant et après le cycle en température. À la fin de la mesure, les puits
apparaissent inhomogènes (Fig.3.13a) et la perte de masse totale de l’échantillon est de 43,68%.
Pour éviter l'évaporation du solvant, deux configurations ont été évaluées :
i) un film collant transparent a été appliqué sur la microplaque. Cette solution a permis de
réduire la perte de masse en fin de mesures à 2,30%. Cependant, le faible volume d’air
emprisonné entre le film et la solution n’a pas permis d’empêcher l'évaporation du solvant et
sa recondensation à la surface interne du film, rendant impossible toutes mesures sur l’image
(Fig.3.13b).
ii) une couche d’huile de paraffine ( = 0,87) a été déposée directement à la surface de chaque
échantillon. Les puits sont alors apparus homogènes en fin de mesures et la perte de masse
était limitée à 0,12% (Fig.3.13c).

Chapitre 3 – Mise en place d’un nouvel outil
85
Figure 3.13 : Image de puits de la microplaque remplis de 300 µl de solvant eau / éthanol (50/50 v/v).
La surface de la microplaque reste ouverte (a), la microplaque est recouverte d’un film plastique
transparent autocollant (b), chaque puit est recouvert d’une fine couche d’huile (75µl d’huile de
paraffine) (c). Pour plus de clarté les images sont limitées à 4 puits (le diamètre d’un puits est de 6,96
mm). La perte de masse est déterminée après 15 heures.
En conséquence, pour l’utilisation de notre système, 75 µl d’huile de paraffine (quantité jugée
suffisante pour recouvrir l’intégralité de la surface de la solution), seront systématiquement
déposés à la surface de chaque puits contenant la solution pour la suite des expériences.
Cette méthode nous permettra de travailler avec des échantillons en solution dans des solvants
plus ou moins volatiles sans avoir d’effets d’évaporation et/ou de condensation
3. Validation de l’outil pour la détermination des points de trouble
3.1 Détermination de la température de transition
La détermination du point de trouble est basée sur l’évolution typique du niveau de gris des
puits au cours d’une trempe en température. Les courbes présentées en Figure 3.14a montrent
une variation discontinue de l’intensité du niveau de gris au cours d’une baisse de température,
pour un échantillon de gluten. L’observation d’un puit montre que l’échantillon subit une
transition de phase qui se manifeste par une diminution du niveau de gris ( ). À l’inverse, si
le puits ne contient que du solvant, le niveau de gris reste constant au cours du changement
thermique ( ). Cette observation confirme que la variation de gris de l’échantillon résulte
uniquement de l’évolution de structure du produit et non de potentiels facteurs extérieurs
comme de la fine couche d’huile.
Sans protection
Perte de masse :
43,68%
n
:
Film protection
Perte de masse :
2,30%
n
:
Huile
Perte de masse :
0,12%
a b c
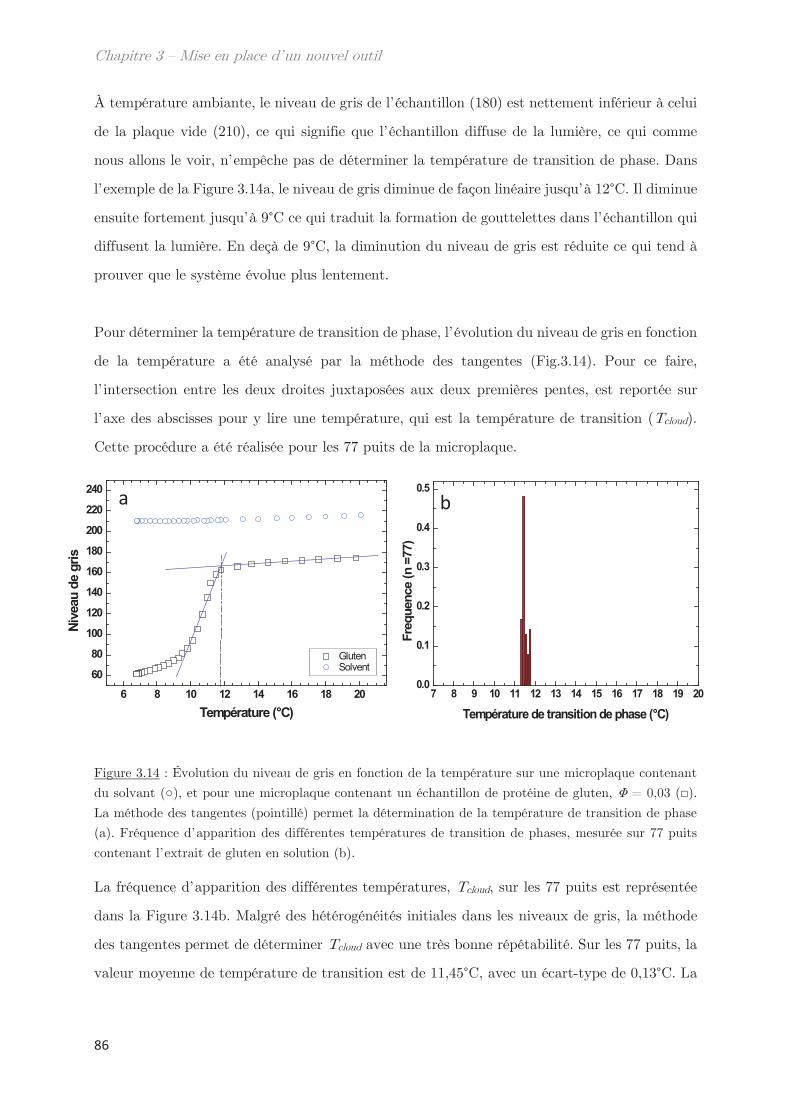
Chapitre 3 – Mise en place d’un nouvel outil
86
À température ambiante, le niveau de gris de l’échantillon (180) est nettement inférieur à celui
de la plaque vide (210), ce qui signifie que l’échantillon diffuse de la lumière, ce qui comme
nous allons le voir, n’empêche pas de déterminer la température de transition de phase. Dans
l’exemple de la Figure 3.14a, le niveau de gris diminue de façon linéaire jusqu’à 12°C. Il diminue
ensuite fortement jusqu’à 9°C ce qui traduit la formation de gouttelettes dans l’échantillon qui
diffusent la lumière. En deçà de 9°C, la diminution du niveau de gris est réduite ce qui tend à
prouver que le système évolue plus lentement.
Pour déterminer la température de transition de phase, l’évolution du niveau de gris en fonction
de la température a été analysé par la méthode des tangentes (Fig.3.14). Pour ce faire,
l’intersection entre les deux droites juxtaposées aux deux premières pentes, est reportée sur
l’axe des abscisses pour y lire une température, qui est la température de transition (Tcloud).
Cette procédure a été réalisée pour les 77 puits de la microplaque.
Figure 3.14 : Évolution du niveau de gris en fonction de la température sur une microplaque contenant
du solvant ( ), et pour une microplaque contenant un échantillon de protéine de gluten, = 0,03 ( ).
La méthode des tangentes (pointillé) permet la détermination de la température de transition de phase
(a). Fréquence d’apparition des différentes températures de transition de phases, mesurée sur 77 puits
contenant l’extrait de gluten en solution (b).
La fréquence d’apparition des différentes températures, Tcloud, sur les 77 puits est représentée
dans la Figure 3.14b. Malgré des hétérogénéités initiales dans les niveaux de gris, la méthode
des tangentes permet de déterminer Tcloud avec une très bonne répétabilité. Sur les 77 puits, la
valeur moyenne de température de transition est de 11,45°C, avec un écart-type de 0,13°C. La
a b
6 8 10 12 14 16 18 20
60
80
100
120
140
160
180
200
220
240
Gluten Solvent
Niv
eau d
e g
ris
Température (°C)
7 8 9 10 11 12 13 14 15 16 17 18 19 200.0
0.1
0.2
0.3
0.4
0.5
Fre
quence (n =
77)
Température de transition de phase (°C)

Chapitre 3 – Mise en place d’un nouvel outil
87
valeur de Tcloud minimale étant de 11,30°C et la maximale de 11,70°C, la méthode des tangentes
semble robuste et parfaitement appropriée à notre application.
3.2 Utilisation d’un système modèle le TX-114
Pour valider la fiabilité de notre système expérimental, nous avons choisis de travailler sur un
modèle connu : le Triton X-114 (TX-114).
Le TX-114 est un détergent non ionique possédant des têtes d’oxyde d’éthylène (n = 7 ou 8),
utilisé pour l’extraction des protéines membranaires (Pryde, 1986 ; Bordier, 1981 ; Hinze et al.,
2012). Suite au chauffage des protéines membranaires dans une solution de tensioactifs, il y a
séparation de phases qui permet d’obtenir une phase pauvre et une phase riche, dans laquelle
se retrouvent les protéines membranaires.
Afin d’observer la séparation de phase du TX-114 (Sigma Aldrich, X114) sur notre montage
expérimental, nous avons mélangé ce dernier à du poly(éthylène glycol) (PEG 4000, Flucka
Analytical), avec une masse molaire moyenne de 4000 g/l. La préparation des échantillons est
basée sur le travail décrit par Qiao et al., 1998. Dans un premier temps, une solution mère de
TX-114 à 200 g/l et une solution mère de PEG 4000 à 150 g/l ont été préparées. Ces solutions
ont ensuite été agitées une nuit, à 20°C sur un agitateur rotatif (Reax 2, Heidolph) à 60
rotations par minutes. La solution mère de PEG est ensuite diluée afin d’obtenir des solutions
filles sur une gamme de 13 à 89 g/l. 1 ml de chacune des solutions filles est ensuite ajouté à 1
ml de solution mère de TX-114 pour obtenir des solutions finales contenant du TX-114 à 100
g/l et du PEG 4000 sur une gamme de 6,5 à 44,5 g/l. 300 µl de chaque solution ont ensuite été
déposés dans 3 puits de la microplaque. Pour rester dans les mêmes conditions que celle de
Qiao et al., la fine couche d’huile n’est pas ajoutée comme dans les expériences précédentes et
la cinétique programmée est de 0,5°C.min-1 en 30 min. Le point de trouble est ensuite déterminé
par la méthode des tangentes exposées précédemment et l’ensemble des résultats est représenté
dans la Figure 3.15.
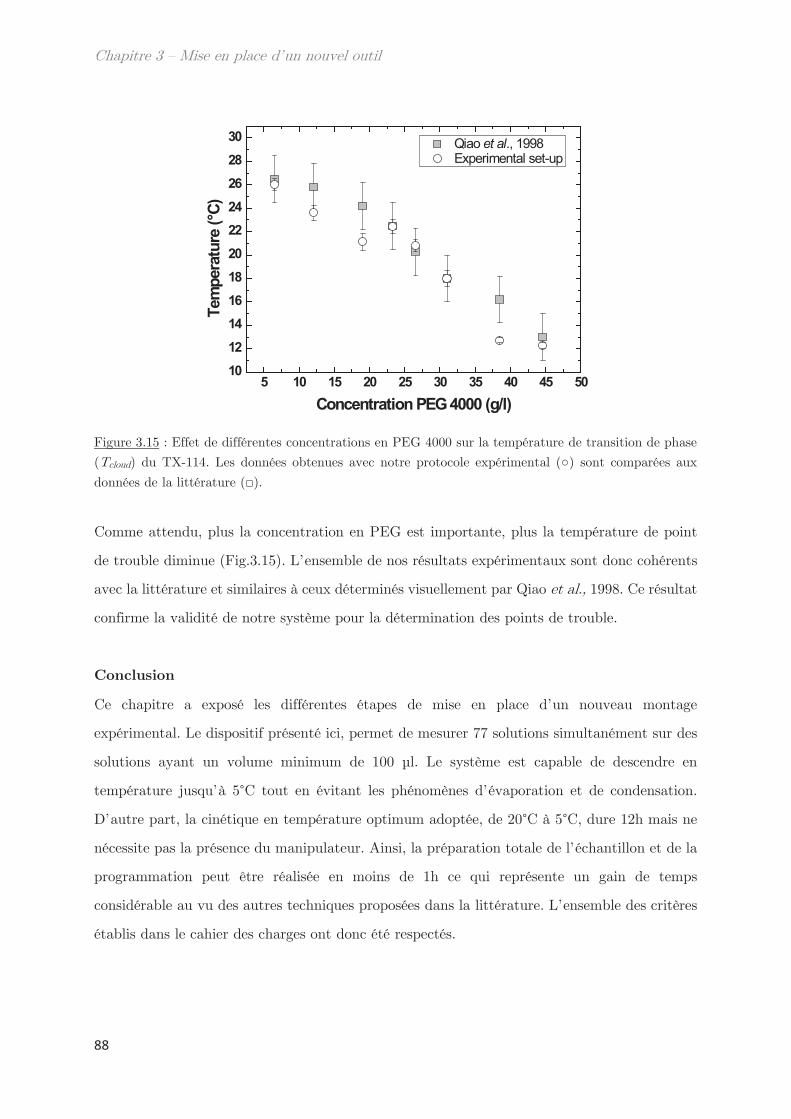
Chapitre 3 – Mise en place d’un nouvel outil
88
Figure 3.15 : Effet de différentes concentrations en PEG 4000 sur la température de transition de phase
(Tcloud) du TX-114. Les données obtenues avec notre protocole expérimental ( ) sont comparées aux
données de la littérature ( ).
Comme attendu, plus la concentration en PEG est importante, plus la température de point
de trouble diminue (Fig.3.15). L’ensemble de nos résultats expérimentaux sont donc cohérents
avec la littérature et similaires à ceux déterminés visuellement par Qiao et al., 1998. Ce résultat
confirme la validité de notre système pour la détermination des points de trouble.
Conclusion
Ce chapitre a exposé les différentes étapes de mise en place d’un nouveau montage
expérimental. Le dispositif présenté ici, permet de mesurer 77 solutions simultanément sur des
solutions ayant un volume minimum de 100 µl. Le système est capable de descendre en
température jusqu’à 5°C tout en évitant les phénomènes d’évaporation et de condensation.
D’autre part, la cinétique en température optimum adoptée, de 20°C à 5°C, dure 12h mais ne
nécessite pas la présence du manipulateur. Ainsi, la préparation totale de l’échantillon et de la
programmation peut être réalisée en moins de 1h ce qui représente un gain de temps
considérable au vu des autres techniques proposées dans la littérature. L’ensemble des critères
établis dans le cahier des charges ont donc été respectés.
5 10 15 20 25 30 35 40 45 5010
12
14
16
18
20
22
24
26
28
30 Qiao et al., 1998 Experimental set-up
Tem
pera
ture
(°C
)
Concentration PEG 4000 (g/l)

Chapitre 3 – Mise en place d’un nouvel outil
89
Ces critères (balayage d’une large gamme en température, nombre important de paramètres à
tester simultanément, utilisation d’une faible quantité de matière première, temps d’expérience
réduit et protocole expérimental accessible à tous) sont néanmoins des critères qui peuvent
éventuellement être améliorés.
En effet, dans notre cas ce nouvel outil fourni des résultats fiables au regard de la littérature,
mais pour étendre un peu plus son application nous proposons les solutions suivantes :
i) Pour élargir encore plus le nombre d’analyses simultanées (77 possibilités
actuellement, ce qui est non négligeable), il suffirait pour un futur montage de
pouvoir remonter la caméra. En effet, la création du support métallique qui a permis
de réduire les transferts de chaleur a amené une distance échantillon-caméra de 4,5
cm, ce qui entraîné ‘la perte’ de 19 puits de la microplaque.
ii) La plaque lumineuse a nécessité plusieurs vérifications qui ont démontré que
l’homogénéité de cette dernière n’était pas parfaite. En effet, pourtant vendu
comme telle, cette homogénéité pourrait poser des problèmes dans le cas d’une
mesure autre que la lecture du point de trouble. C’est pourquoi il serait peut-être
intéressant de changer de fournisseur de plaque lumineuse.
iii) Pour limiter l’utilisation de matériel tel que le bain thermostaté qui permet de
refroidir la plaque lumineuse, il serait intéressant d’utiliser un système déclencheur,
allumant la plaque au moment de la prise de la photo puis éteignant la plaque une
fois la prise terminée. Pour répondre à cette exigence il est nécessaire de posséder
un mode « strobe » sur la plaque lumineuse et de créer un code type Matlab ou
Python avec nos paramètres de mesures.
iv) Enfin, pour élargir la gamme de température, l’utilisation d’une enceinte climatique
qui ne fonctionne pas à effet Peltier pourrait être une solution.
Ces points d’améliorations restent cependant, une ouverture pour l’application à d’autres
domaines et ne sont pas déterminants à la mise en pratique du montage.
La nouvelle méthode expérimentale fournit donc une approche reproductible pour la mise en
place de la détermination de diagrammes de phases. La similitude des résultats avec ceux de
la littérature permettent de valider le modèle et de l’appliquer à tous systèmes susceptibles de
faire une séparation de phase liquide-liquide. Cet outil permet de limiter les phénomènes de

Chapitre 3 – Mise en place d’un nouvel outil
90
condensation et d’évaporation ainsi que celui de tester rapidement différents critères en une
seule mesure. La technique peut en effet, permettre de dépister diverses conditions physico-
chimiques simultanément telles que le pH, la force ionique, la concentration en protéines et
d’identifier les conditions pour lesquelles le système devient trouble. Ainsi, ce dispositif de
lecture de point de trouble apparaît comme un outil qui répond à la fois aux demandes sur des
matrices alimentaires mais qui peut être aussi une application aux domaines cosmétiques ou
biomédicales.
L’objectif pratique final de cet outil semble donc bien adapté à l’élaboration de diagrammes
de phases sur des protéines et plus particulièrement sur le gluten que nous souhaitons étudier.
En effet, la connaissance des diagrammes de phases de différents extraits de gluten va permettre
la compréhension et la caractérisation des interactions et des assemblages de prolamines du
blé.
L’ensemble de ces résultats a conduit à une publication scientifique « Methods for screening
cloud point temperature, Justine Pincemaille, Amélie Banc, Edouard Chauveau, Jean-Marc
Fromental, Laurence Ramos, Marie-Hélène Morel, Paul Menut, Food Biophysics, 2018 ».

Chapitre 3 – Mise en place d’un nouvel outil
91
Bibliographie
Asherie, N. (2004). Protein crystallization and phase diagrams. Methods, 34(3), 266–272.
Amine, C., Boire, A., Davy, J., Marquis, M., & Renard, D. (2017). Droplets-based millifluidic
for the rapid determination of biopolymers phase diagrams. Food Hydrocolloids, 70, 134–
142.
Boire, A., Menut, P., Morel, M.-H., & Sanchez, C. (2013). Phase behaviour of a wheat protein
isolate. Soft Matter, 9(47), 11417–11426.
Boutris, C., Chatzi, E. G., & Kiparissides, C. (1997). Characterization of the LCST behaviour
of aqueous poly(N-isopropylacrylamide) solutions by thermal and cloud point techniques.
Polymer, 38(10), 2567–2570.
Bordier, C. (1981). Phase separation of integral membrane proteins in Triton X-114 solution.
Journal of Biological Chemistry.
Broide, M. L., Tominc, T. M., & Saxowsky, M. D. (1996). Using phase transitions to investigate
the effect of salts on protein interactions. Phys Rev E Stat Phys Plasmas Fluids Relat
Interdiscip Topics, 53(6), 6325–6335.
Caneba, G.T., and Soong, D.S., (1985). Polymer membrane formation through the thermal-
inversion process. Experimental study of membrane structure formation, Macromolecules,
18, p.2538-2545.
Chen, J. P., Yang, H. J., & Hoffman, A. S. (1990). Polymer-protein conjugates. Biomaterials
Science, 11, 625–630.
Corti M., & Minero C. (1984). Fast measurement of the consolution curve of nonionic micellar
solutions: a turbidimetric method. Colloids and Surfaces, 12, 341–356.
Dreezen, G., Groeninckx, G., Swier, S., & Van Mele, B. (2001). Phase separation in miscible
polymer blends as detected by modulated temperature differential scanning calorimetry.
Polymer, 42(4), 1449–1459.

Chapitre 3 – Mise en place d’un nouvel outil
92
Hefter, G. T., Barton, A. F. M., & Chand, A. L. B.-S.-R. (1991). Semi-automated Apparatus
for the Determination of Liyuid Solubilities: Mutual Solubilities of Water and Butan-2-ol.
J. Chem. Soc. Faraday Trans., 87(4), 591–596.
Hinze, W. L., Pramauro, E., & Poole, C. F. (1993). A Critical Review of Surfactant-Mediated
Phase Separations (Cloud-Point Extractions): Theory and Applications. Critical Reviews
in Analytical Chemistry.
Jain, K., Vedarajan, R., Watanabe, M., Ishikiriyama, M., & Matsumi, N. (2015). Tunable
LCST behavior of poly(N-isopropylacrylamide/ionic liquid) copolymers. Polym. Chem.,
6(38), 6819–6825.
Kitabatake, N., Doi, E., & Kinekawa, Y. (1994). Simple and Rapid Method for Measuring
Turbidity in Gels and Sols from Milk Whey Protein. Journal of Food Science, 59(4), 769–
772.
Li, L., Kantor, A., & Warne, N. (2013). Application of a PEG precipitation method for
solubility screening: A tool for developing high protein concentration formulations. Protein
Science, 22(8), 1118–1123.
Liu, C. L., Kamei, D. T., King, J. A., Wang, D. I., & Blankschtein, D. (1998). Separation of
proteins and viruses using two-phase aqueous micellar systems. Journal of
Chromatography. B, Biomedical Sciences and Applications, 711(1–2), 127–138.
Liu, C., Asherie, N., Lomakin, a, Pande, J., Ogun, O., & Benedek, G. B. (1996). Phase
separation in aqueous solutions of lens gamma-crystallins: special role of gamma s.
Proceedings of the National Academy of Sciences of the United States of America, 93(1),
377–382.
Mohsen-Nia, M., Rasa, H., & Modarress, H. (2006). Cloud-point measurements for (water +
poly(ethylene glycol) + salt) ternary mixtures by refractometry method. Journal of
Chemical and Engineering Data, 51(4), 1316–1320.

Chapitre 3 – Mise en place d’un nouvel outil
93
Nikas, Y. J., Liu, C. L., Srivastava, T., Abbott, N. L., & Blankschtein, D. (1992). Protein
Partitioning in Two-Phase Aqueous Nonionic Micellar Solutions. Macromolecules, 25(18),
4797–4806.
Nozary, S., Modarress, H., & Eliassi, A. (2002). Cloud-Point Measurements for Salt + Poly
(ethylene glycol ) + Water Systems by Viscometry and Laser Beam Scattering Methods,
2–9.
Pozdnyshev, G. N., Emkov, A. A., Shagibekova, M. M., & Voronchikhina, D. P. (1978). Method
for determination of cloud point of solutions of substances in water and oil media, (7), 59–
61.
Pryde J.G. (1986). Triton X-114: a detergent that has come in from the cold. Biochem. J, 233,
525–533.
Qiao, L., & Easteal, A. J. (1998). The interaction between triton X series surfactants and poly
(ethylene glycol) in aqueous solutions. Colloid and Polymer Science.
Shibayama, M., Isono, K., Okabe, S., Karino, T., & Nagao, M. (2004). SANS Study on
Pressure-Induced Phase Separation of Poly(N-isopropylacrylamide) Aqueous Solutions
and Gels. Macromolecules, 37(8), 2909–2918.
Silva, A. D. P. M. Da, De Oliveira, P. B., Bandini, T. B., Barreto Junior, A. G., De Sena, R.
C., & Silva, J. F. C. Da. (2013). Low-cost system based on image analysis to determine
solubility curves. Sensors and Actuators, B: Chemical, 177, 1071–1074.
Swier, S., Pieters, R., & Van Mele, B. (2002). Kinetics of demixing and remixing in
poly(ethylene oxide)/poly(ether sulphone) blends as studied by modulated temperature
differential scanning calorimetry. Polymer, 43(13), 3611–3620.
Tachaboonyakiat, W., Ajiro, H., & Akashi, M. (2013). Synthesis of a thermosensitive polycation
by random copolymerization of N-vinylformamide and N-vinylbutyramide. Polymer
Journal, 45(9), 971–978.

Chapitre 3 – Mise en place d’un nouvel outil
94
Taratura, V. G., Holschbach, A., Thurston, G. M., Blankschtein, D., & Benedek, G. B. (1990).
Liquid-liquid phase separation of aqueous lysozyme solutions: Effects of pH and salt
identity. Journal of Physical Chemistry, 94(5), 2140–2144.
Voorn, M. (1959). Phase separation in polymer solutions. Fortschritte Der Hochpolymeren-Forschung, 3, 192–233.
Williamson, A. P., & Kiefer, J. (2014). Automatic low-cost method to determine the solubility
of liquid-liquid mixtures by continuous-flow cloud point titration. Chemical Engineering
and Technology, 37(10), 1736–1740.
Yamaoka, T., Tamura, T., Seto, Y., Tada, T., Kunugi, S., & Tirrell, D. A. (2003). Mechanism
for the phase transition of a genetically engineered elastin model peptide (VPGIG)40 in
aqueous solution. Biomacromolecules, 4(6), 1680–1685.

Chapitre 4 – Caractérisations biochimiques
95
Chapitre 4
Caractérisations biochimiques des isolats de
protéines du blé
Ce chapitre est consacré à la description du lot gluten utilisé dans ce travail et des extraits
protéiques qui en découlent après séparation de phases liquide/liquide. Il se concentre plus
particulièrement sur la composition en protéines, analysée par chromatographie d’exclusion
stérique et par électrophorèse.
1. Solubilité des protéines du gluten en solvant éthanol/eau (50/50, v/v)
La composition en protéines du gluten est déterminée par analyse chromatographique des 2
extraits séquentiels réalisée en tampon dénaturant (SDS/phosphate), sans, puis avec sonication
(respectivement extraits « soluble » et « insoluble », (cf. Chapitre 2)). La Figure 4.1 présente
les profils chromatographiques de ces deux extraits, en fonction de leur masse moléculaire
apparente, obtenus par calibration de la colonne (cf. Chapitre 2). Les fractions F1 et F2 incluent
les polymères de gluténines solubles en tampon SDS/phosphate de 2000 à 100 kg.mol-1. La
fraction F3 est majoritairement composée d’ -gliadines (50-75 kg.mol-1) tandis que la fraction
F4 inclut les autres types de gliadines ( / et -gliadines, de 20 à 48 kg/mol-1). Enfin dans la
fraction F5 se trouvent des protéines appartenant à la classe des albumines/globulines et dont
le poids moléculaire est inférieur à 20 kg.mol-1.
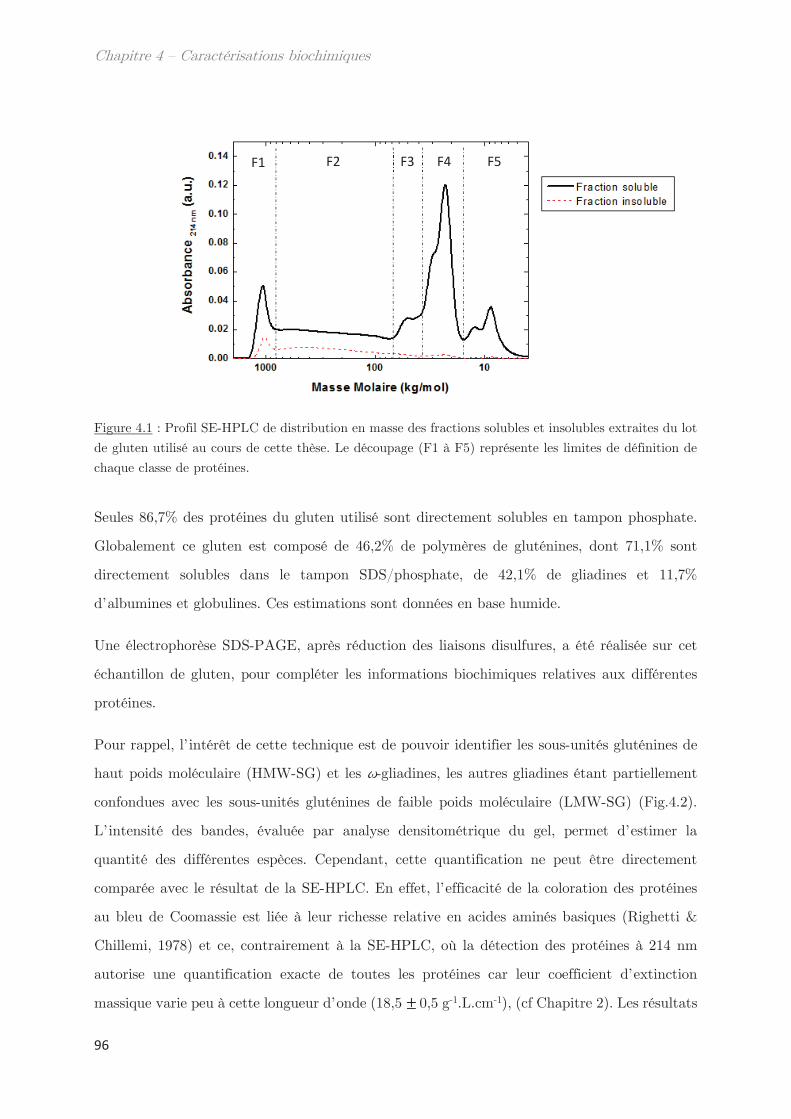
Chapitre 4 – Caractérisations biochimiques
96
Figure 4.1 : Profil SE-HPLC de distribution en masse des fractions solubles et insolubles extraites du lot
de gluten utilisé au cours de cette thèse. Le découpage (F1 à F5) représente les limites de définition de
chaque classe de protéines.
Seules 86,7% des protéines du gluten utilisé sont directement solubles en tampon phosphate.
Globalement ce gluten est composé de 46,2% de polymères de gluténines, dont 71,1% sont
directement solubles dans le tampon SDS/phosphate, de 42,1% de gliadines et 11,7%
d’albumines et globulines. Ces estimations sont données en base humide.
Une électrophorèse SDS-PAGE, après réduction des liaisons disulfures, a été réalisée sur cet
échantillon de gluten, pour compléter les informations biochimiques relatives aux différentes
protéines.
Pour rappel, l’intérêt de cette technique est de pouvoir identifier les sous-unités gluténines de
haut poids moléculaire (HMW-SG) et les -gliadines, les autres gliadines étant partiellement
confondues avec les sous-unités gluténines de faible poids moléculaire (LMW-SG) (Fig.4.2).
L’intensité des bandes, évaluée par analyse densitométrique du gel, permet d’estimer la
quantité des différentes espèces. Cependant, cette quantification ne peut être directement
comparée avec le résultat de la SE-HPLC. En effet, l’efficacité de la coloration des protéines
au bleu de Coomassie est liée à leur richesse relative en acides aminés basiques (Righetti &
Chillemi, 1978) et ce, contrairement à la SE-HPLC, où la détection des protéines à 214 nm
autorise une quantification exacte de toutes les protéines car leur coefficient d’extinction
massique varie peu à cette longueur d’onde (18,5 ± 0,5 g-1.L.cm-1), (cf Chapitre 2). Les résultats
F5 F4 F2 F1 F3
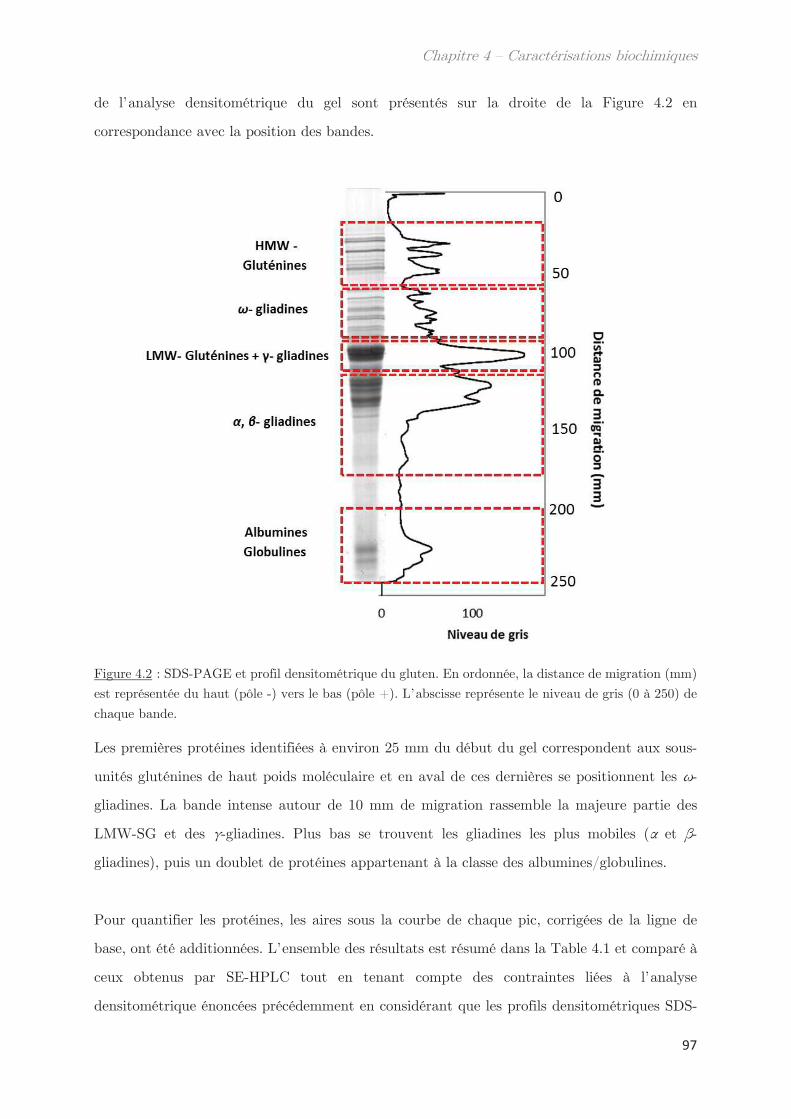
Chapitre 4 – Caractérisations biochimiques
97
de l’analyse densitométrique du gel sont présentés sur la droite de la Figure 4.2 en
correspondance avec la position des bandes.
Figure 4.2 : SDS-PAGE et profil densitométrique du gluten. En ordonnée, la distance de migration (mm)
est représentée du haut (pôle -) vers le bas (pôle +). L’abscisse représente le niveau de gris (0 à 250) de
chaque bande.
Les premières protéines identifiées à environ 25 mm du début du gel correspondent aux sous-
unités gluténines de haut poids moléculaire et en aval de ces dernières se positionnent les -
gliadines. La bande intense autour de 10 mm de migration rassemble la majeure partie des
LMW-SG et des -gliadines. Plus bas se trouvent les gliadines les plus mobiles ( et -
gliadines), puis un doublet de protéines appartenant à la classe des albumines/globulines.
Pour quantifier les protéines, les aires sous la courbe de chaque pic, corrigées de la ligne de
base, ont été additionnées. L’ensemble des résultats est résumé dans la Table 4.1 et comparé à
ceux obtenus par SE-HPLC tout en tenant compte des contraintes liées à l’analyse
densitométrique énoncées précédemment en considérant que les profils densitométriques SDS-

Chapitre 4 – Caractérisations biochimiques
98
PAGE donnent des compositions apparentes et qui sont des estimations. En confrontant les
analyses SE-HPLC et SDS-PAGE et en faisant l’hypothèse que l’intensité relative des bandes
HMW-SG reflète leur pourcentage massique, l’estimation du rapport HMW-SG/LMW-SG du
lot de gluten est de 0,57. Cette valeur est relativement faible mais correspond à la gamme (0,31
- 0,93) des valeurs rapportées pour les farines de qualité boulangère (Dhaka & Khatar, 2014).
Table 4.1 : Composition (%) en protéines par rapport à la composition en protéines totale du gluten de
blé déduite du profil densitométrique du gel obtenu par SDS-PAGE et SE-HPLC.
Suite à la première étape du protocole d’extraction (cf Chapitre 2), le surnageant, S1, a été
analysé dans des conditions SE-HPLC et SDS-PAGE similaires à celles du gluten. En
considérant comme précédemment que l’intensité relative des bandes HMW-SG reflète leur
pourcentage massique, l’estimation du rapport HMW-SG/LMWS-SG de S1 est de 0,68.
Le rapport HMW-SG/LMWS-SG de S1 est donc légèrement supérieur à celui du lot de gluten
initial mais reste néanmoins très proche (0,57).
En connaissance de cette information, nous avons étudié les différentes fractions issues de S1
dans la suite du protocole d’extraction.
2. Fractionnement des protéines solubles en éthanol/eau (50/50 v/v)
En 1925, Dill & Alsberg, rapportent que les extraits éthanol/eau de farine de blé deviennent
turbides en dessous d’une température critique et ce de façon réversible. Ce phénomène, qui
signale la démixtion liquide-liquide de l’extrait protéique, a récemment été étudié en détail par
Adeline Boire (2014) sur des gliadines, quasi exemptes de gluténines et d’albumines/globulines,
dispersées en solvant eau/éthanol 55%, v/v, et 0,5 mM NaCl. Par analyse SE-HPLC, des phases
dense et légère ont été obtenues à différentes concentrations et températures. De l’analyse de
leur composition en protéines, elle a conclu que le coefficient de partition des différentes classes
de gliadines dépendait de leur masse moléculaire, tandis que la plus haute température à
HMW-SG -gliadine LMW-SG + -gli / -gliadine Alb/glo
SDS-PAGE 16,75 16,26 21,73 38,19 7,06 Polymère de gluténines -gliadine , -gliadine Alb/glo
SE-HPLC 46,18 6,78 35,32 11,73

Chapitre 4 – Caractérisation biochimique
99
laquelle peut être observée la démixtion (Tc) variait peu (en moyenne 11,7 ±0,1°C, pour
l’ensemble des et -gliadines et les polymères de gluténines de moins de 200 kg.mol-1
également présents à l’état de trace). Sur la base de ces résultats, nous avons envisagé la
partition liquide-liquide comme moyen permettant d’obtenir des fractions protéiques de
compositions variables en gliadines et gluténines.
2.1 Effet de la température de trempe sur la partition en masse entre les phases dense
et légère
La Figure 4.3a présente la partition des protéines contenues dans l’extrait éthanol/eau du
gluten (S1) dans les phases dense (C2) et légère (S2) selon la température de trempe. À 2°C,
la plupart des protéines se retrouvent dans la phase dense. À l’inverse, à 12°C, la quasi-totalité
des protéines se retrouvent dans la phase légère. La gamme de températures étudiée (de -2°C
à 12°C) permet de balayer l’ensemble des possibilités en matière de partition des protéines dans
les phases dense et légère.
Pour l’ensemble des expériences réalisées, la masse de protéines contenue dans C2 augmente
avec la profondeur de la trempe (Fig.4.3a) de même que le volume de C2 normalisé par celui
de S1 (Fig.4.3b). La concentration de la phase dense augmente lorsque la trempe en
température est plus profonde; elle est multipliée par 3 entre 12°C et 0,2°C passant de 120 g/l
à 367 g/l (Fig.4.3c). D’autre part, la concentration en C2 est environ 100 fois supérieur à la
concentration en S2 à des trempes très profondes (-0,8°C) et 2,5 fois plus importante à des
trempes moins profondes (12°C). L’ensemble des données de la Figure 4.3 est regroupé dans un
l’Annexe 1 pour plus de détail.
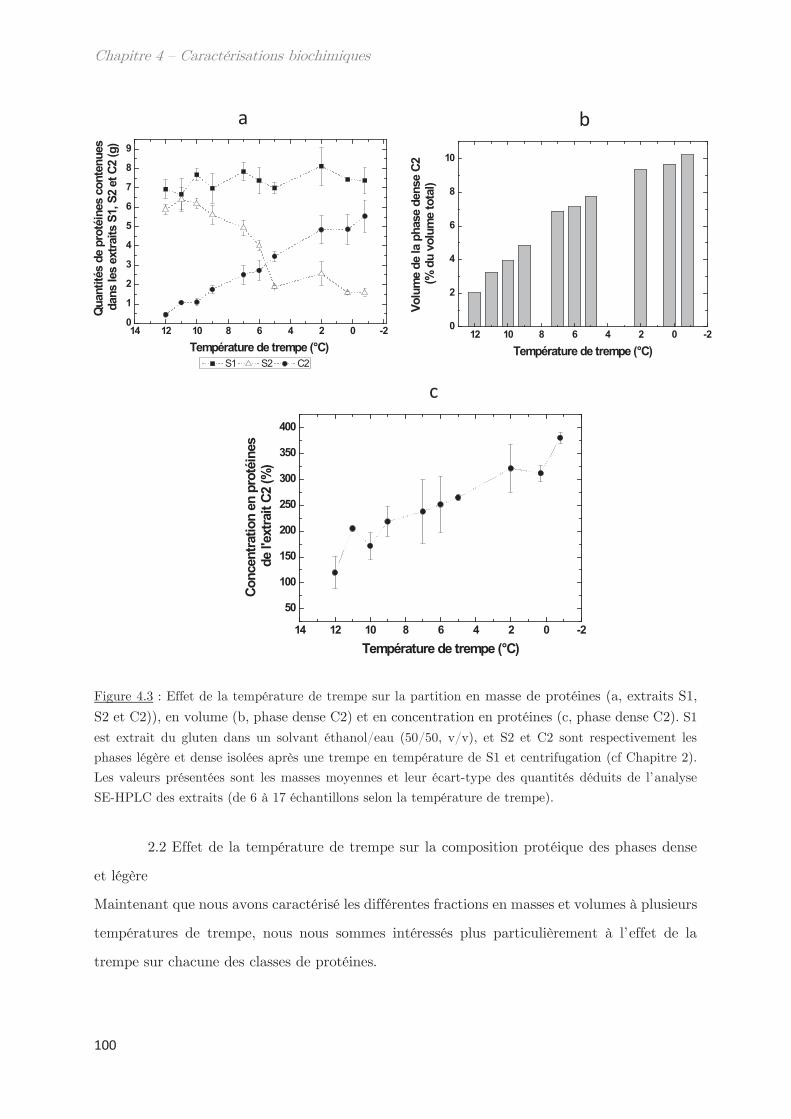
Chapitre 4 – Caractérisations biochimiques
100
Figure 4.3 : Effet de la température de trempe sur la partition en masse de protéines (a, extraits S1,
S2 et C2)), en volume (b, phase dense C2) et en concentration en protéines (c, phase dense C2). S1
est extrait du gluten dans un solvant éthanol/eau (50/50, v/v), et S2 et C2 sont respectivement les
phases légère et dense isolées après une trempe en température de S1 et centrifugation (cf Chapitre 2).
Les valeurs présentées sont les masses moyennes et leur écart-type des quantités déduits de l’analyse
SE-HPLC des extraits (de 6 à 17 échantillons selon la température de trempe).
2.2 Effet de la température de trempe sur la composition protéique des phases dense
et légère
Maintenant que nous avons caractérisé les différentes fractions en masses et volumes à plusieurs
températures de trempe, nous nous sommes intéressés plus particulièrement à l’effet de la
trempe sur chacune des classes de protéines.
a b
c
12 10 8 6 4 2 0 -20
2
4
6
8
10
Volu
me d
e la p
hase d
ense C
2
(% d
u v
olu
me tota
l)
Température de trempe (°C)
14 12 10 8 6 4 2 0 -2
50
100
150
200
250
300
350
400
Concentr
ation e
n p
roté
ines
de l'e
xtr
ait C
2 (%
)
Température de trempe (°C)
14 12 10 8 6 4 2 0 -20
1
2
3
4
5
6
7
8
9
Quantité
s d
e p
roté
ines c
onte
nues
dans les e
xtr
aits S
1, S
2 e
t C
2 (g)
Température de trempe (°C)
S1 S2 C2

Chapitre 4 – Caractérisation biochimique
101
La Figure 4.4 présente le profil SE-HPLC des phases obtenues après une trempe à 6°C. Par
rapport à l’extrait initial de protéines du gluten (S1), qui inclut 30,8% de polymères de
gluténines (F1+F2) et 58% de gliadines (F3+F4), la phase dense est sur-concentrée en
polymères de gluténines (51,7% ± 2,1%), alors que ces derniers sont quasi-inexistants dans la
phase légère (11,3% ± 0,8%). À l’inverse, les gliadines sont surreprésentées dans cette dernière
(74,1% ± 0,8%). Le comportement des gliadines (F3+F4) n’est pas homogène et les -gliadines
présentes dans le pic F3 s’accumulent préférentiellement dans la phase dense. Le comportement
thermodynamique des -gliadines apparait ainsi plus proche de celui des gluténines, que de
celui des autres gliadines. Ce résultat pourrait être mis sur le compte du différentiel de taille
des différentes protéines. En effet, plus la masse moléculaire des polymères est élevée, plus ils
se retrouvent dans la phase dense. Cette observation est en accord avec les arguments
théoriques développés par P.J.Flory, en 1944, qui rendent compte des phénomènes de démixtion
de solutions de polymères de tailles hétérogènes, lorsque l’interaction avec le solvant décroît.
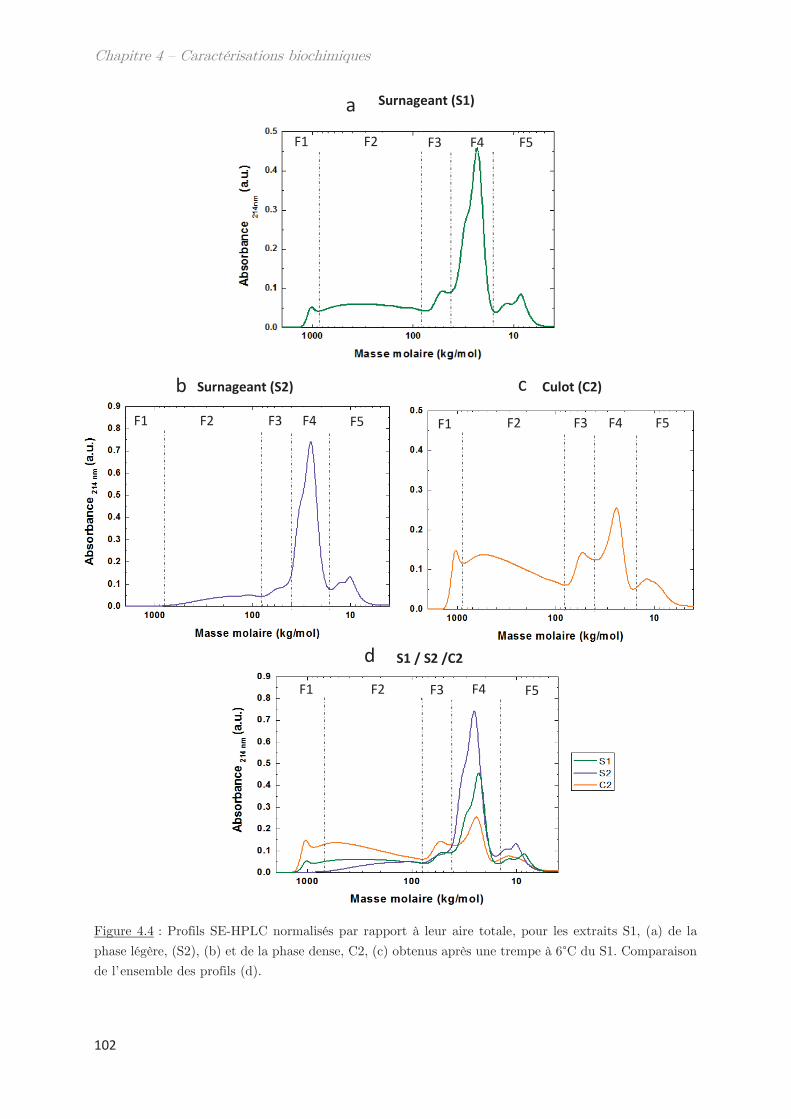
Chapitre 4 – Caractérisations biochimiques
102
Figure 4.4 : Profils SE-HPLC normalisés par rapport à leur aire totale, pour les extraits S1, (a) de la
phase légère, (S2), (b) et de la phase dense, C2, (c) obtenus après une trempe à 6°C du S1. Comparaison
de l’ensemble des profils (d).
S1 / S2 /C2
Culot (C2) Surnageant (S2)
a
Sub
Surnageant (S1)
c
d
F1 F2 F3 F4 F5
F1 F2 F3 F4 F5
F1 F2 F3 F4 F5
F1 F2 F3 F4 F5
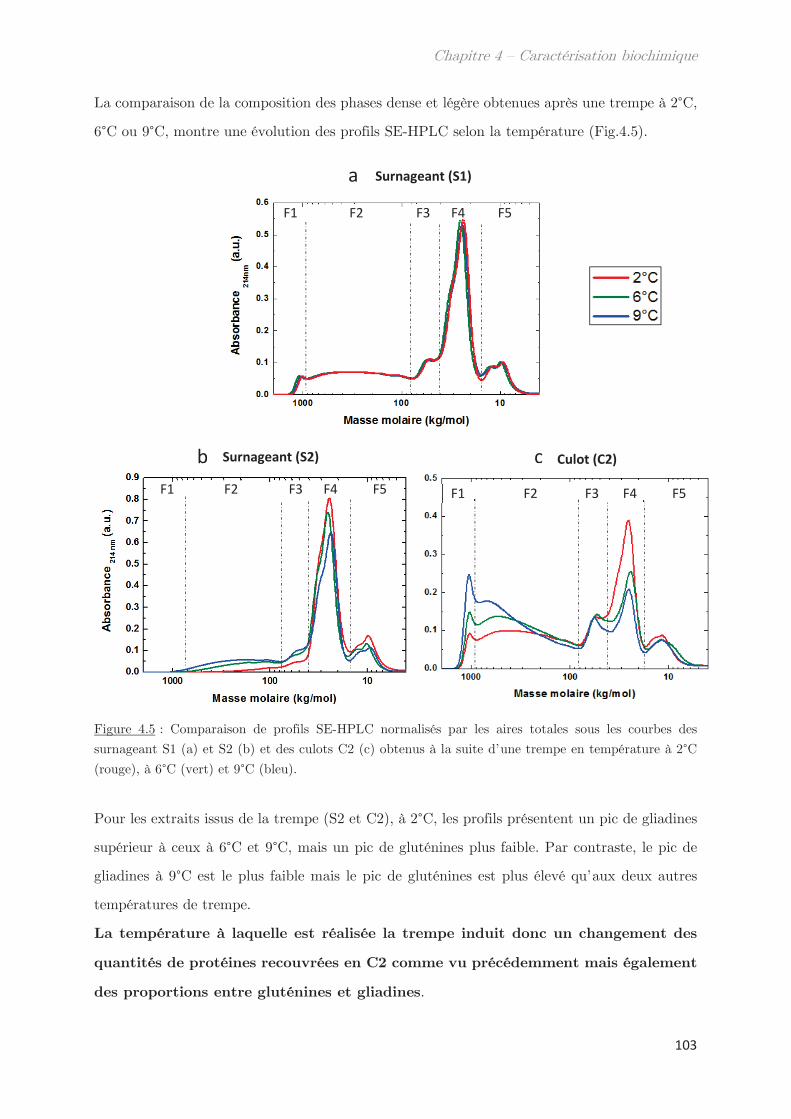
Chapitre 4 – Caractérisation biochimique
103
La comparaison de la composition des phases dense et légère obtenues après une trempe à 2°C,
6°C ou 9°C, montre une évolution des profils SE-HPLC selon la température (Fig.4.5).
Figure 4.5 : Comparaison de profils SE-HPLC normalisés par les aires totales sous les courbes des
surnageant S1 (a) et S2 (b) et des culots C2 (c) obtenus à la suite d’une trempe en température à 2°C
(rouge), à 6°C (vert) et 9°C (bleu).
Pour les extraits issus de la trempe (S2 et C2), à 2°C, les profils présentent un pic de gliadines
supérieur à ceux à 6°C et 9°C, mais un pic de gluténines plus faible. Par contraste, le pic de
gliadines à 9°C est le plus faible mais le pic de gluténines est plus élevé qu’aux deux autres
températures de trempe.
La température à laquelle est réalisée la trempe induit donc un changement des
quantités de protéines recouvrées en C2 comme vu précédemment mais également
des proportions entre gluténines et gliadines.
a
c
Surnageant (S1)
F1 F2 F3 F4 F5
b
F1 F2 F3 F4 F5 F1 F2 F3 F4 F5
b Surnageant (S2) c Culot (C2)
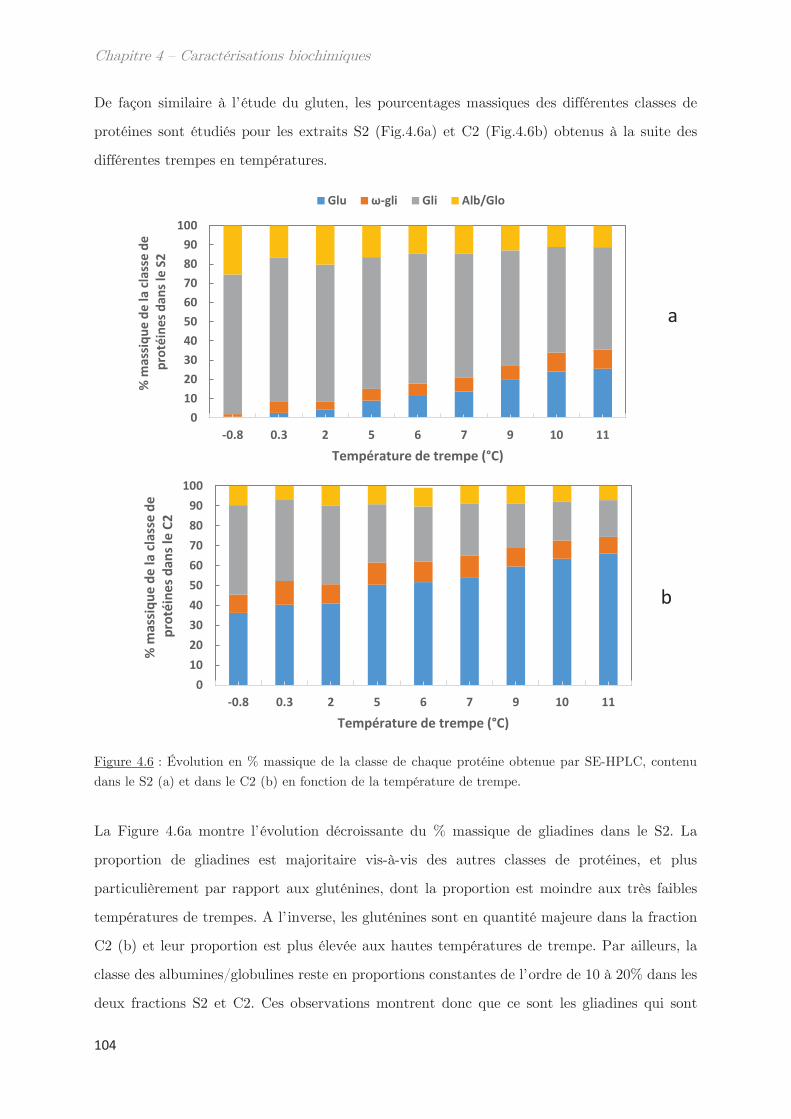
Chapitre 4 – Caractérisations biochimiques
104
De façon similaire à l’étude du gluten, les pourcentages massiques des différentes classes de
protéines sont étudiés pour les extraits S2 (Fig.4.6a) et C2 (Fig.4.6b) obtenus à la suite des
différentes trempes en températures.
Figure 4.6 : Évolution en % massique de la classe de chaque protéine obtenue par SE-HPLC, contenu
dans le S2 (a) et dans le C2 (b) en fonction de la température de trempe.
La Figure 4.6a montre l’évolution décroissante du % massique de gliadines dans le S2. La
proportion de gliadines est majoritaire vis-à-vis des autres classes de protéines, et plus
particulièrement par rapport aux gluténines, dont la proportion est moindre aux très faibles
températures de trempes. A l’inverse, les gluténines sont en quantité majeure dans la fraction
C2 (b) et leur proportion est plus élevée aux hautes températures de trempe. Par ailleurs, la
classe des albumines/globulines reste en proportions constantes de l’ordre de 10 à 20% dans les
deux fractions S2 et C2. Ces observations montrent donc que ce sont les gliadines qui sont
0
10
20
30
40
50
60
70
80
90
100
-0.8 0.3 2 5 6 7 9 10 11
% m
ass
iqu
e d
e l
a c
lass
e d
e
pro
téin
es
da
ns
le C
2
Température de trempe (°C)
Glu -gli Gli Alb/Glo
0
10
20
30
40
50
60
70
80
90
100
-0.8 0.3 2 5 6 7 9 10 11
% m
ass
iqu
e d
e l
a c
lass
e d
e
pro
téin
es
da
ns
le S
2
Température de trempe (°C)
Glu -gli Gli Alb/Glo
b
a
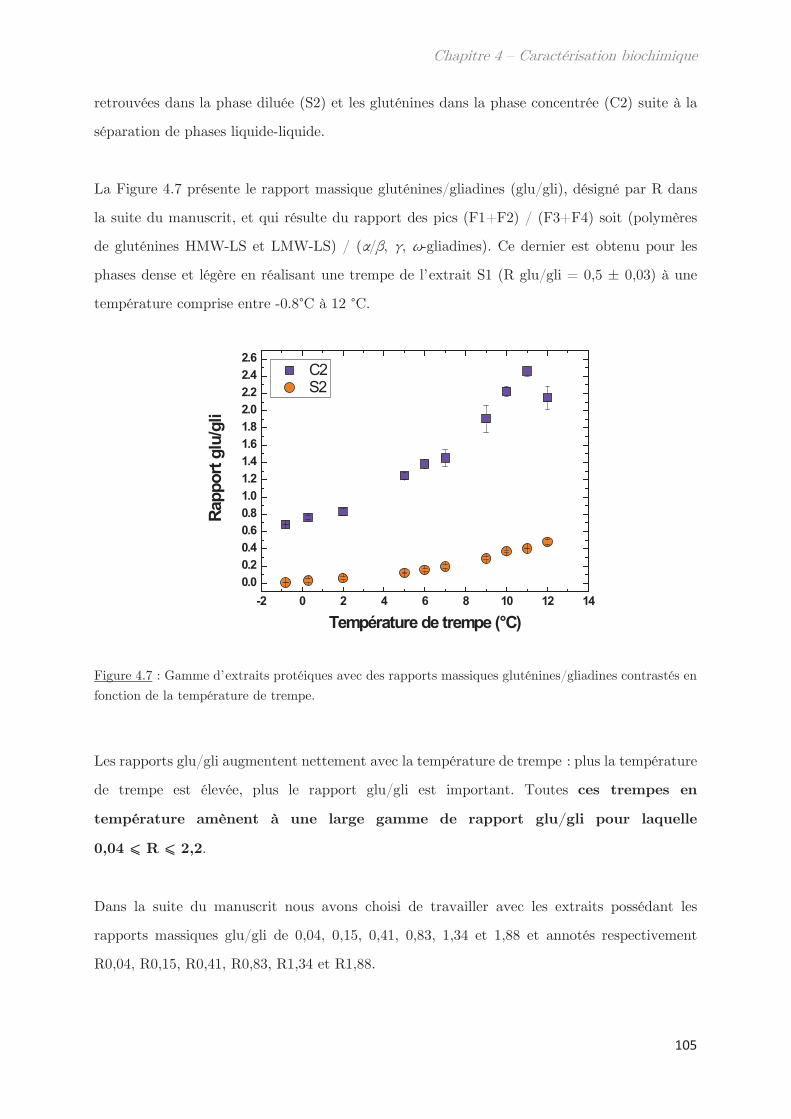
Chapitre 4 – Caractérisation biochimique
105
retrouvées dans la phase diluée (S2) et les gluténines dans la phase concentrée (C2) suite à la
séparation de phases liquide-liquide.
La Figure 4.7 présente le rapport massique gluténines/gliadines (glu/gli), désigné par R dans
la suite du manuscrit, et qui résulte du rapport des pics (F1+F2) / (F3+F4) soit (polymères
de gluténines HMW-LS et LMW-LS) / ( / , , -gliadines). Ce dernier est obtenu pour les
phases dense et légère en réalisant une trempe de l’extrait S1 (R glu/gli = 0,5 ± 0,03) à une
température comprise entre -0.8°C à 12 °C.
Figure 4.7 : Gamme d’extraits protéiques avec des rapports massiques gluténines/gliadines contrastés en
fonction de la température de trempe.
Les rapports glu/gli augmentent nettement avec la température de trempe : plus la température
de trempe est élevée, plus le rapport glu/gli est important. Toutes ces trempes en
température amènent à une large gamme de rapport glu/gli pour laquelle
0,04 R 2,2.
Dans la suite du manuscrit nous avons choisi de travailler avec les extraits possédant les
rapports massiques glu/gli de 0,04, 0,15, 0,41, 0,83, 1,34 et 1,88 et annotés respectivement
R0,04, R0,15, R0,41, R0,83, R1,34 et R1,88.
-2 0 2 4 6 8 10 12 14
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
2.2
2.4
2.6
Rapport
glu
/gli
C2 S2
Température de trempe (°C)
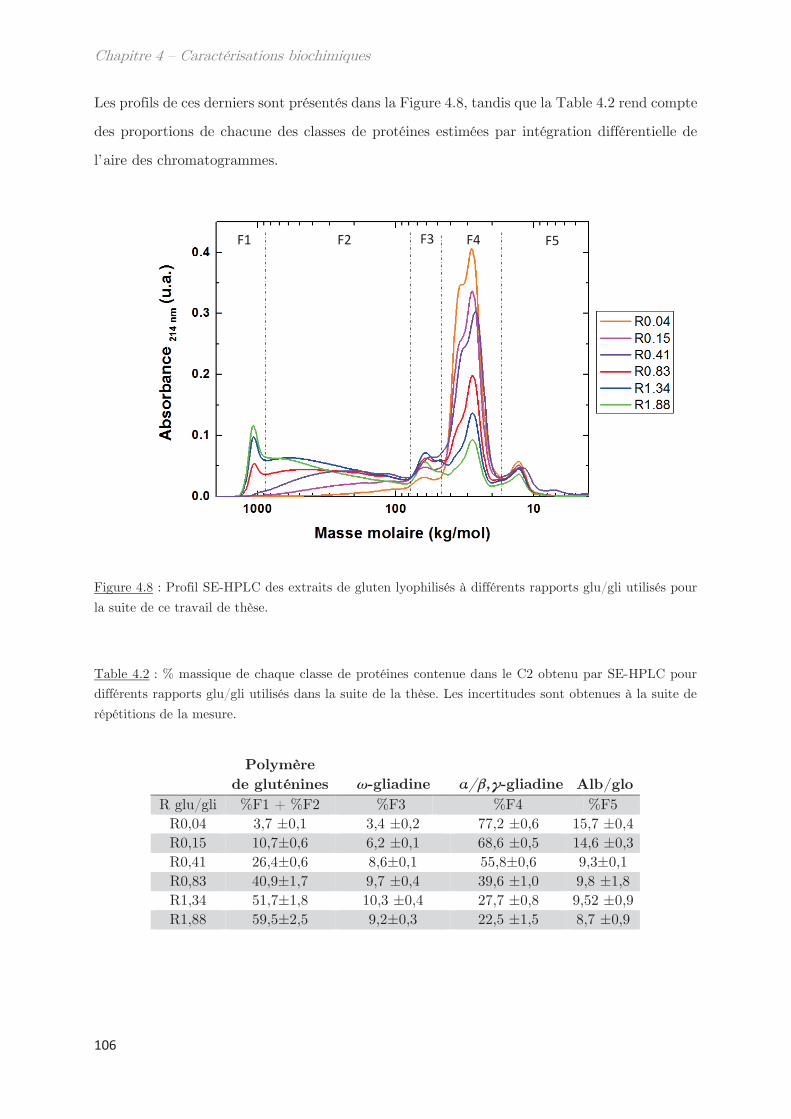
Chapitre 4 – Caractérisations biochimiques
106
Les profils de ces derniers sont présentés dans la Figure 4.8, tandis que la Table 4.2 rend compte
des proportions de chacune des classes de protéines estimées par intégration différentielle de
l’aire des chromatogrammes.
Figure 4.8 : Profil SE-HPLC des extraits de gluten lyophilisés à différents rapports glu/gli utilisés pour
la suite de ce travail de thèse.
Table 4.2 : % massique de chaque classe de protéines contenue dans le C2 obtenu par SE-HPLC pour
différents rapports glu/gli utilisés dans la suite de la thèse. Les incertitudes sont obtenues à la suite de
répétitions de la mesure.
Polymère de gluténines -gliadine / -gliadine Alb/glo
R glu/gli %F1 + %F2 %F3 %F4 %F5 R0,04 3,7 ±0,1 3,4 ±0,2 77,2 ±0,6 15,7 ±0,4 R0,15 10,7±0,6 6,2 ±0,1 68,6 ±0,5 14,6 ±0,3 R0,41 26,4±0,6 8,6±0,1 55,8±0,6 9,3±0,1 R0,83 40,9±1,7 9,7 ±0,4 39,6 ±1,0 9,8 ±1,8 R1,34 51,7±1,8 10,3 ±0,4 27,7 ±0,8 9,52 ±0,9 R1,88 59,5±2,5 9,2±0,3 22,5 ±1,5 8,7 ±0,9
F1 F3 F2 F4 F5
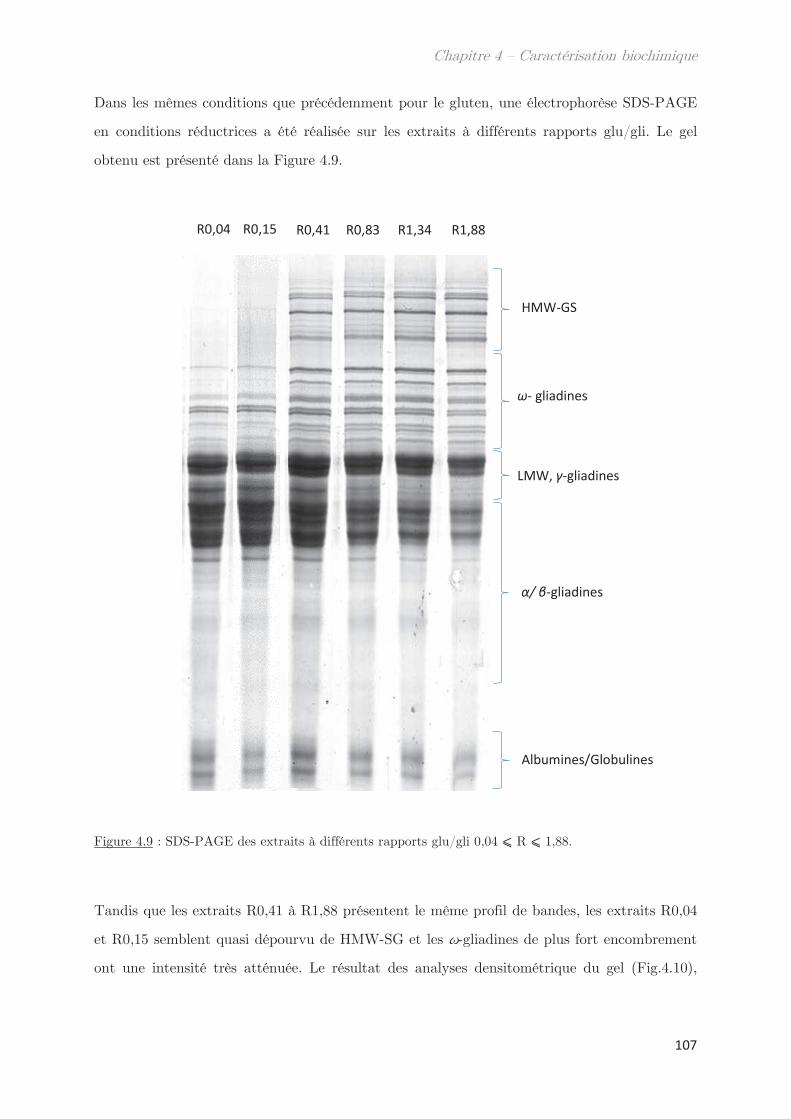
Chapitre 4 – Caractérisation biochimique
107
Dans les mêmes conditions que précédemment pour le gluten, une électrophorèse SDS-PAGE
en conditions réductrices a été réalisée sur les extraits à différents rapports glu/gli. Le gel
obtenu est présenté dans la Figure 4.9.
Figure 4.9 : SDS-PAGE des extraits à différents rapports glu/gli 0,04 R 1,88.
Tandis que les extraits R0,41 à R1,88 présentent le même profil de bandes, les extraits R0,04
et R0,15 semblent quasi dépourvu de HMW-SG et les -gliadines de plus fort encombrement
ont une intensité très atténuée. Le résultat des analyses densitométrique du gel (Fig.4.10),
R0,15 R0,04 R0,41 R0,83 R1,34 R1,88
HMW-GS
- gliadines
LMW, -gliadines
Albumines/Globulines
-gliadines
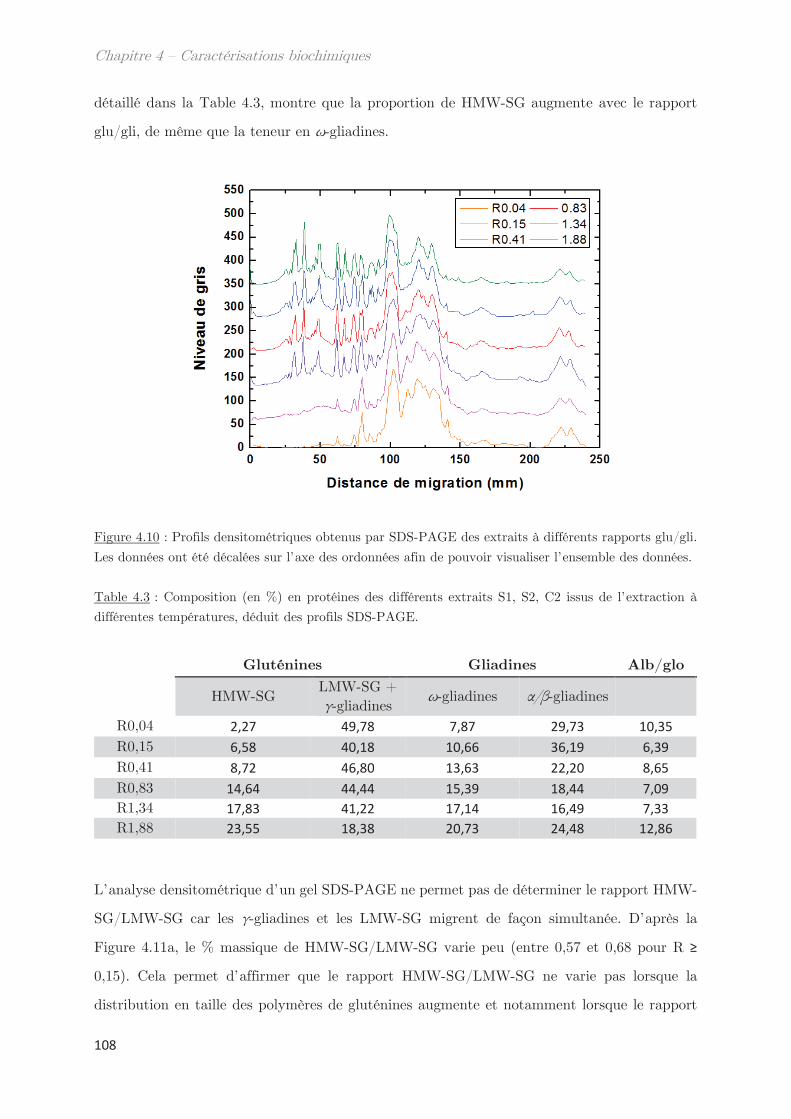
Chapitre 4 – Caractérisations biochimiques
108
détaillé dans la Table 4.3, montre que la proportion de HMW-SG augmente avec le rapport
glu/gli, de même que la teneur en -gliadines.
Figure 4.10 : Profils densitométriques obtenus par SDS-PAGE des extraits à différents rapports glu/gli.
Les données ont été décalées sur l’axe des ordonnées afin de pouvoir visualiser l’ensemble des données.
Table 4.3 : Composition (en %) en protéines des différents extraits S1, S2, C2 issus de l’extraction à
différentes températures, déduit des profils SDS-PAGE.
Gluténines Gliadines Alb/glo
HMW-SG LMW-SG +
-gliadines -gliadines -gliadines
R0,04 2,27 49,78 7,87 29,73 10,35
R0,15 6,58 40,18 10,66 36,19 6,39
R0,41 8,72 46,80 13,63 22,20 8,65
R0,83 14,64 44,44 15,39 18,44 7,09
R1,34 17,83 41,22 17,14 16,49 7,33
R1,88 23,55 18,38 20,73 24,48 12,86
L’analyse densitométrique d’un gel SDS-PAGE ne permet pas de déterminer le rapport HMW-
SG/LMW-SG car les -gliadines et les LMW-SG migrent de façon simultanée. D’après la
Figure 4.11a, le % massique de HMW-SG/LMW-SG varie peu (entre 0,57 et 0,68 pour R
0,15). Cela permet d’affirmer que le rapport HMW-SG/LMW-SG ne varie pas lorsque la
distribution en taille des polymères de gluténines augmente et notamment lorsque le rapport
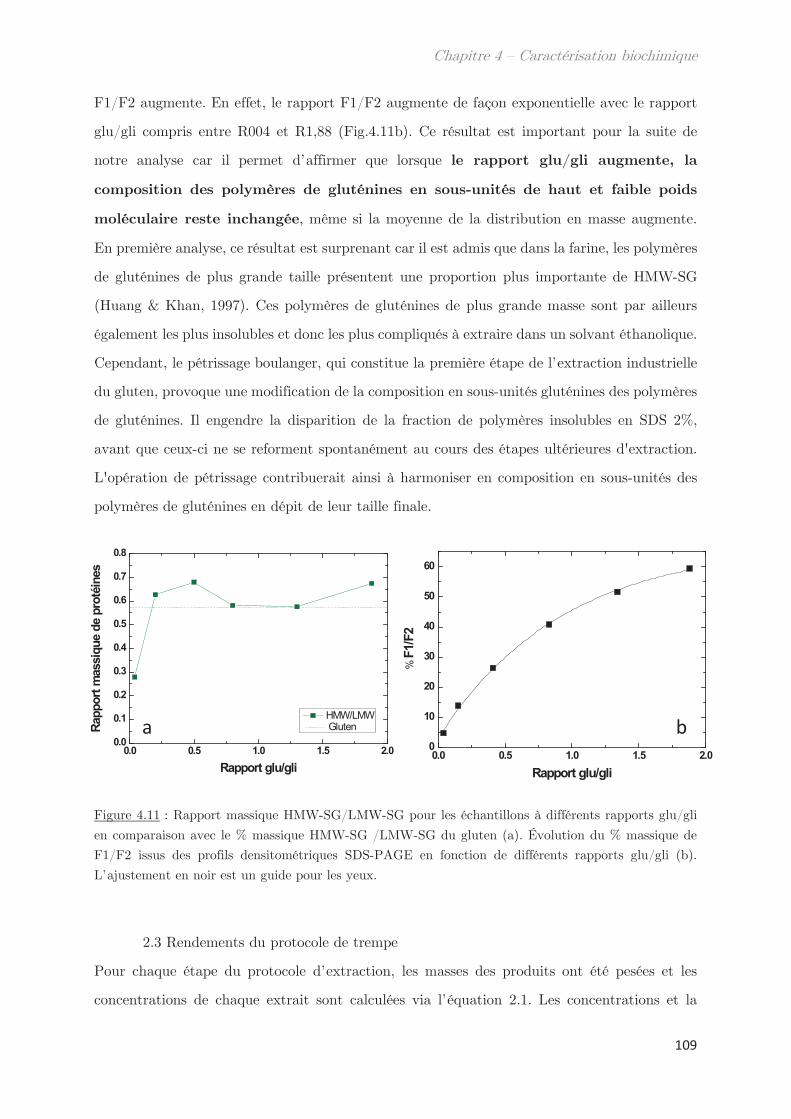
Chapitre 4 – Caractérisation biochimique
109
F1/F2 augmente. En effet, le rapport F1/F2 augmente de façon exponentielle avec le rapport
glu/gli compris entre R004 et R1,88 (Fig.4.11b). Ce résultat est important pour la suite de
notre analyse car il permet d’affirmer que lorsque le rapport glu/gli augmente, la
composition des polymères de gluténines en sous-unités de haut et faible poids
moléculaire reste inchangée, même si la moyenne de la distribution en masse augmente.
En première analyse, ce résultat est surprenant car il est admis que dans la farine, les polymères
de gluténines de plus grande taille présentent une proportion plus importante de HMW-SG
(Huang & Khan, 1997). Ces polymères de gluténines de plus grande masse sont par ailleurs
également les plus insolubles et donc les plus compliqués à extraire dans un solvant éthanolique.
Cependant, le pétrissage boulanger, qui constitue la première étape de l’extraction industrielle
du gluten, provoque une modification de la composition en sous-unités gluténines des polymères
de gluténines. Il engendre la disparition de la fraction de polymères insolubles en SDS 2%,
avant que ceux-ci ne se reforment spontanément au cours des étapes ultérieures d'extraction.
L'opération de pétrissage contribuerait ainsi à harmoniser en composition en sous-unités des
polymères de gluténines en dépit de leur taille finale.
Figure 4.11 : Rapport massique HMW-SG/LMW-SG pour les échantillons à différents rapports glu/gli
en comparaison avec le % massique HMW-SG /LMW-SG du gluten (a). Évolution du % massique de
F1/F2 issus des profils densitométriques SDS-PAGE en fonction de différents rapports glu/gli (b).
L’ajustement en noir est un guide pour les yeux.
2.3 Rendements du protocole de trempe
Pour chaque étape du protocole d’extraction, les masses des produits ont été pesées et les
concentrations de chaque extrait sont calculées via l’équation 2.1. Les concentrations et la
b 0.0 0.5 1.0 1.5 2.00
10
20
30
40
50
60
%F1/F
2
Rapport glu/gli
a 0.0 0.5 1.0 1.5 2.0
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
Rapport
massiq
ue d
e p
roté
ines
Rapport glu/gli
HMW/LMW Gluten

Chapitre 4 – Caractérisations biochimiques
110
quantité de protéines totales ont été déterminées et récapitulées dans la Table 4.4 pour chaque
température de trempe.
De façon générale, la concentration et la masse en protéines totales issues de S1 ne varient pas.
Cette observation rejoint celle précédemment faite sur les profils SE-HPLC. Les températures
de trempe plus profondes semblent faire diminuer les concentrations et masses en protéines de
l’extrait S2 mais augmenter ceux de l’extrait C2.
Les rendements d’extractions des protéines sont déterminés pour la seconde étape d’extraction
à la suite de laquelle le rendement répond à l’égalité : Surnageant S1 = Surnageant S2 + Culot
C2.
Table 4.4 : Rendements en protéines totales, des gluténines et des gliadines des phases S2 et C2 issus de
la phase S1 suite au protocole d’extraction.
L’ensemble des résultats montrent des rendements en moyenne de 93,8% et supérieur à 80%
ce qui représente une estimation acceptable. Le fait que les rendements n’atteignent pas 100%
peut s’expliquer par des erreurs de pesées lors de la récupération des phases ou d’erreurs de
dilution en préalable au dosage de la concentration en protéine par SE-HPLC.
2.4 Préparation des solutions diluées de protéines par filtration
Pour éliminer les particules de poussière et résidus indésirables, les échantillons ont été filtrés
avec des filtres à seringue de cellulose hydrophile (Rotilabo, n.sté
mm) avec une taille de pores de 0,8 µm. Pour cela, les échantillons ont d’abord été préparés à
20 g/l ( = 0,015) puis dilués à 5 g/l ( = 0,003) et ensuite filtrés. Cette manipulation a été
T trempe (°C) Nombre d’essais Rprot.tot. (%) RGlu (%) RGli (%) -0,8 2 96,4 ± 0,4 88,6 ± 5,7 98,3 ± 2,72 0,3 6 86,7 ± 11,5 102,7 ± 10,2 82,3 ± 13,5 2 18 92,1 ± 10,9 86,9 ± 13,9 91,9 ± 11,5 5 6 104,1 ± 5,1 90,7 ± 5,1 125,1 ± 9,4 6 18 92,1 ± 9,3 85,2 ± 12,5 94,3 ± 8,5 7 6 95,6 ± 11,4 80,7 ± 17,4 98,6 ± 9,1 9 24 101,5 ± 11,3 97,8 ± 12,8 102,2 ± 11,1 10 6 95,1 ± 5,7 95,2 ± 10,9 96,4 ± 5,5 11 6 113,5 ± 16,5 117,2 ± 19,9 112,6 ± 16,6 12 12 91,7 ± 5,8 92,2 ± 5,7 90,5 ± 5,4

Chapitre 4 – Caractérisation biochimique
111
réalisée dans le cas des échantillons dilués étudiés en DLS, SANS, SAXS et AsFlFFF qui sont
exposés dans la suite du manuscrit.
Afin de déterminer s’il y a eu des protéines retenues sur le filtre, nous avons analysé par SE-
HPLC, les échantillons avant la filtration puis après la filtration. La Figure 4.12 présente la
soustraction des profils avant et après filtration suite à 2 expériences réalisées dans les mêmes
conditions, ainsi que le pourcentage de perte en protéines totales pour les différents échantillons,
calculé à partir des spectres SE-HPLC. Les échantillons sont exprimés en terme de rapport
massique glu/gli comme exposé précédemment dans ce chapitre.
La Figure 4.12a montre que plus l’échantillon est enrichi en gluténines (R1,88) plus la différence
entre le profil de l’échantillon non filtré et celui de l’échantillon filtré est importante entre 8 et
14 min d’élution. D’après le découpage établie dans la Figure 4.1, cela suggère qu’il y a d’autant
plus de perte en polymères de gluténines dans les échantillons qui en contiennent beaucoup
initialement. La Figure 4.12b montre des profils légèrement différents mais vont dans le même
sens que la Figure 4.12a. La différence de profil suggère que cette étape de filtration n’est pas
totalement contrôlée et que la quantité de protéines et principalement des plus grosses protéines
sont retenues sur le filtre de façon aléatoire. Néanmoins, il apparait dans les 2 cas des pertes
de protéines totales inférieures à 13 % pour tous les échantillons (Fig.4.12c). Ainsi, il est
important de noter que pour les mesures en diluées, les rapports glu/gli réels sont donc en
réalité légèrement inférieurs aux rapports glu/gli nominaux. Cependant, afin de pouvoir
comparer plus simplement l’ensemble des échantillons en milieu dilué et en milieu concentré
nous exprimerons l’ensemble des résultats en fonction des rapports nominaux dans la suite du
manuscrit.
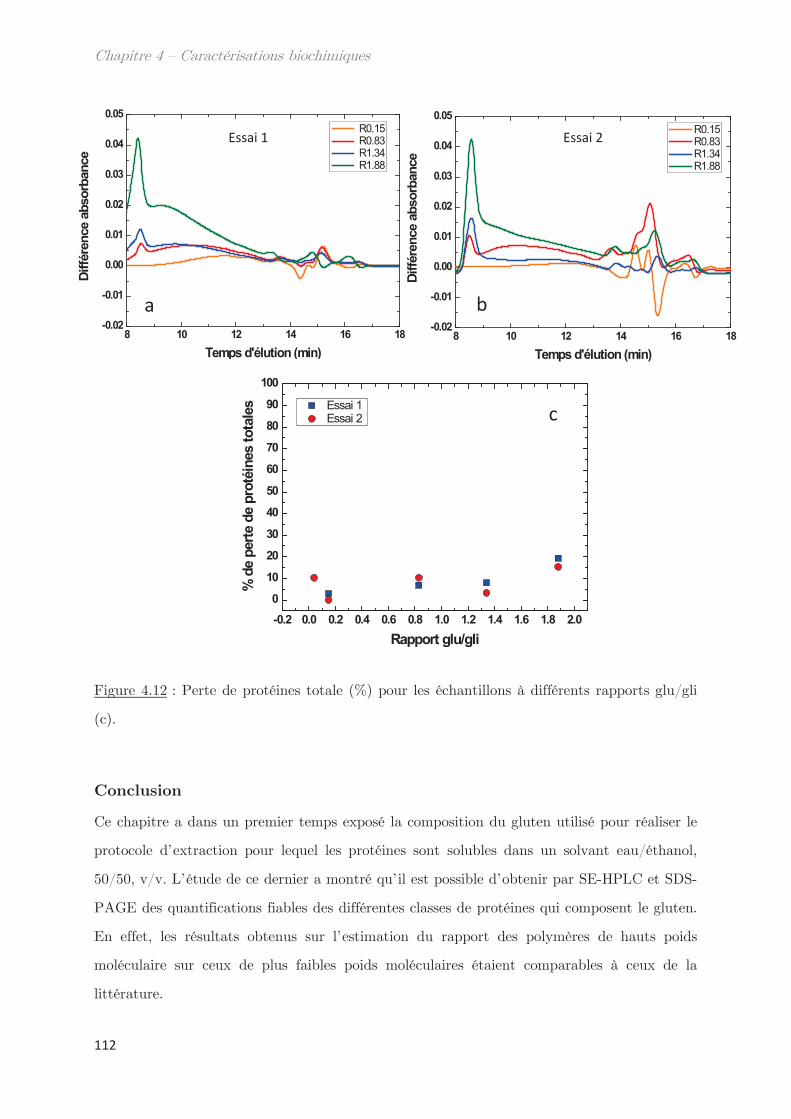
Chapitre 4 – Caractérisations biochimiques
112
Figure 4.12 : Perte de protéines totale (%) pour les échantillons à différents rapports glu/gli
(c).
Conclusion
Ce chapitre a dans un premier temps exposé la composition du gluten utilisé pour réaliser le
protocole d’extraction pour lequel les protéines sont solubles dans un solvant eau/éthanol,
50/50, v/v. L’étude de ce dernier a montré qu’il est possible d’obtenir par SE-HPLC et SDS-
PAGE des quantifications fiables des différentes classes de protéines qui composent le gluten.
En effet, les résultats obtenus sur l’estimation du rapport des polymères de hauts poids
moléculaire sur ceux de plus faibles poids moléculaires étaient comparables à ceux de la
littérature.
8 10 12 14 16 18-0.02
-0.01
0.00
0.01
0.02
0.03
0.04
0.05
Diffé
rence a
bsorb
ance
Temps d'élution (min)
R0.15 R0.83 R1.34 R1.88
8 10 12 14 16 18-0.02
-0.01
0.00
0.01
0.02
0.03
0.04
0.05
Diffé
rence a
bsorb
ance
Temps d'élution (min)
R0.15 R0.83 R1.34 R1.88
Essai 1 Essai 2
-0.2 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0
0
10
20
30
40
50
60
70
80
90
100
Essai 1 Essai 2
% d
e p
ert
e d
e p
roté
ines tota
les
Rapport glu/gli
a b
c

Chapitre 4 – Caractérisation biochimique
113
Par la suite, l’étude de l’effet de la température de trempe sur la partition des différents extraits
issus de la séparation de phases, a permis d’obtenir des échantillons contrastés en composition
protéique. En effet, d’après nos observations, en abaissant la température, la séparation de
phases qui se produit donne deux phases : une enrichie en gliadines (S2) et une enrichie en
polymères de gluténines (C2). Il est important de noter qu’au cours de ce phénomène, il est
apparu que la classe des albumines/globulines restait en proportions constantes et que seules
les proportions de gluténines et gliadines variaient. C’est pourquoi la suite de notre étude
portera uniquement sur ces 2 classes de protéines.
Par les mêmes techniques analytiques que celles utilisées sur le gluten, l’étude de ces extraits
a montré que plus la trempe en température est profonde (basse température), plus les phases
diluées et concentrées seront enrichies en gliadines alors qu’une température de trempe peu
profonde donnera des extraits enrichis en gluténines. Ce résultat semble donc indiquer que la
température de trempe a un impact direct sur la séparation de phases.
Ainsi, la variation de la température lors de la procédure d’extraction a permis d’obtenir une
large gamme d’extraits de rapport glu/gli différents, (0,04 R 2,2). Les extraits
obtenus les plus enrichis en gliadines (R0,04) possèdent ainsi 88% de gliadines et ceux enrichis
en gluténines (R1,88), 60% de gluténines.
Maintenant que nous avons à disposition des extraits qui se distinguent nettement par leur
composition en gliadines et gluténines, il va être possible de caractériser structuralement ces
extraits (Chapitre 6) mais aussi d’établir des diagrammes de phases pour comprendre les
interactions entre ces 2 classes de protéines (Chapitre 5). Enfin, nous porterons notre attention
sur la séparation de phases qui se produit suite à la trempe en température et qui est le
phénomène majeur dont découle notre étude (Chapitre 7).

Chapitre 4 – Caractérisations biochimiques
114
Bibliographie
Boire, A., Menut, P., Morel, M.-H., & Sanchez, C. (2013). Phase behavior of a wheat protein
isolate, Soft Matter, 9(47), 11417–11426.
Boire A. (2014). Structure and dynamics of wheat gliadins dispersions: effect of the protein
concentration and solvent temperature. Thèse de doctorat, Université de Montpellier.
Dhaka, V., & Khatkar, B. S. (2014). Effects of Gliadin / Glutenin and HMW-GS / LMW-GS
Ratio on Dough Rheological Properties and Bread-Making Potential of Wheat Varieties,
71–82.
Dill, D.B., & Alsberg, C.L. (1925). Preparation, Solubility, and Specific Rotation of Wheat
Gliadin, The Journal of Biological Chemistry, 65(2), 279–304.
Flory, P.J. (1944). Thermodynamics of heterogeneous polymers and their solutions. The
Journal of Chemical Physics, 12(11), 425–438.
Huang, D. Y., & Khan, K. (1997). Quantitative Determination of High Molecular Weight
Glutenin Subunits of Hard Red Spring Wheat by SDS-PAGE. I. Quantitative Effects of
Total Amounts on Breadmaking Quality Characteristics. Cereal Chemistry Journal, 74(6),
781–785.
Righetti, P.G., & Chillemi, F. (1978). Isoelectric Focusing of Peptides. Journal of
Chromatography A, 157, 243–251.

Chapitre 5 – Assemblages et diagrammes de phases
115
Chapitre 5
Mise en évidence d’assemblages -gli+glu et
impact sur les diagrammes de phases
Cette partie décrit tout d’abord la structure et la dynamique des échantillons à différents
rapports gluténines/gliadines dans un solvant eau/éthanol, 50/50, v/v, dans un régime dilué.
Des protéines sous forme monomérique et des assemblages sont mis en évidence par DLS et
AsFlFFF. Un fractionnement sous flux croisé combiné à une analyse SE-HPLC permet
d’identifier la composition protéique de ces assemblages. Enfin la mise en évidence de ces
assemblages permet de rationnaliser la séparation de phases liquide-liquide utilisée pour
fractionner le gluten, décrite dans le Chapitre 4 et le comportement de phases de différentes
fractions protéiques homogénéisées.
1. Propriétés dynamiques des échantillons modèles
Une première estimation de la taille des objets diffusants de fractions modèles de gluten en
solutions, à = 0,0152, a été réalisée par diffusion dynamique de la lumière (DLS). Ces
solutions ont été diluées à = 0,0038 en eau/éthanol 50/50, v/v, et filtrées par des filtres à
seringue de cellulose hydrophile avec une taille de pores de 0,8 µm. Les données recueillies par
ce type de technique sont des fonctions d’autocorrélation de l’intensité diffusée g2 -1 en
fonction du temps caractéristique, . Un exemple de données obtenues pour un échantillon de
gluten R0,83 sur une gamme de détection angulaire de 20° à 140° est présenté dans la Figure
5.1.
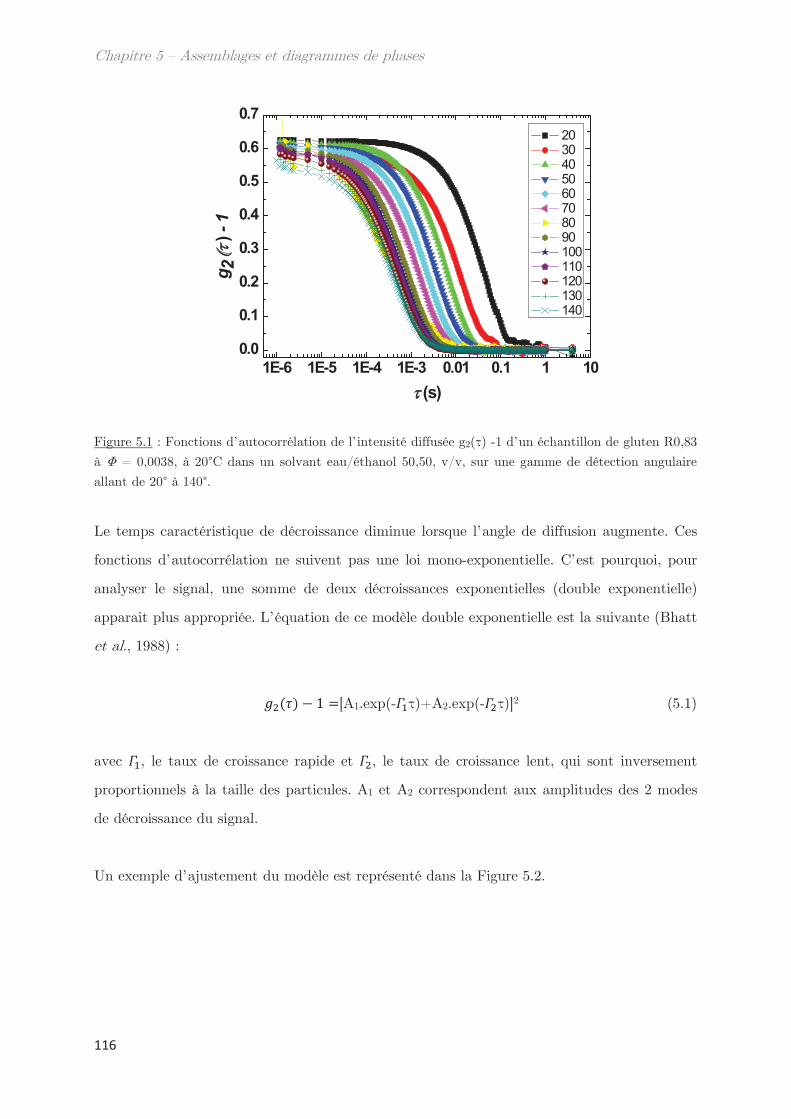
Chapitre 5 – Assemblages et diagrammes de phases
116
Figure 5.1 : Fonctions d’autocorrélation de l’intensité diffusée g2 -1 d’un échantillon de gluten R0,83
à = 0,0038, à 20°C dans un solvant eau/éthanol 50,50, v/v, sur une gamme de détection angulaire
allant de 20° à 140°.
Le temps caractéristique de décroissance diminue lorsque l’angle de diffusion augmente. Ces
fonctions d’autocorrélation ne suivent pas une loi mono-exponentielle. C’est pourquoi, pour
analyser le signal, une somme de deux décroissances exponentielles (double exponentielle)
apparait plus appropriée. L’équation de ce modèle double exponentielle est la suivante (Bhatt
et al., 1988) :
( ) 1 =[A1.exp(- 2.exp(- 2 (5.1)
avec , le taux de croissance rapide et , le taux de croissance lent, qui sont inversement
proportionnels à la taille des particules. A1 et A2 correspondent aux amplitudes des 2 modes
de décroissance du signal.
Un exemple d’ajustement du modèle est représenté dans la Figure 5.2.
1E-6 1E-5 1E-4 1E-3 0.01 0.1 1 10
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
g2
)- 1
(s)
2030405060708090100110120130140
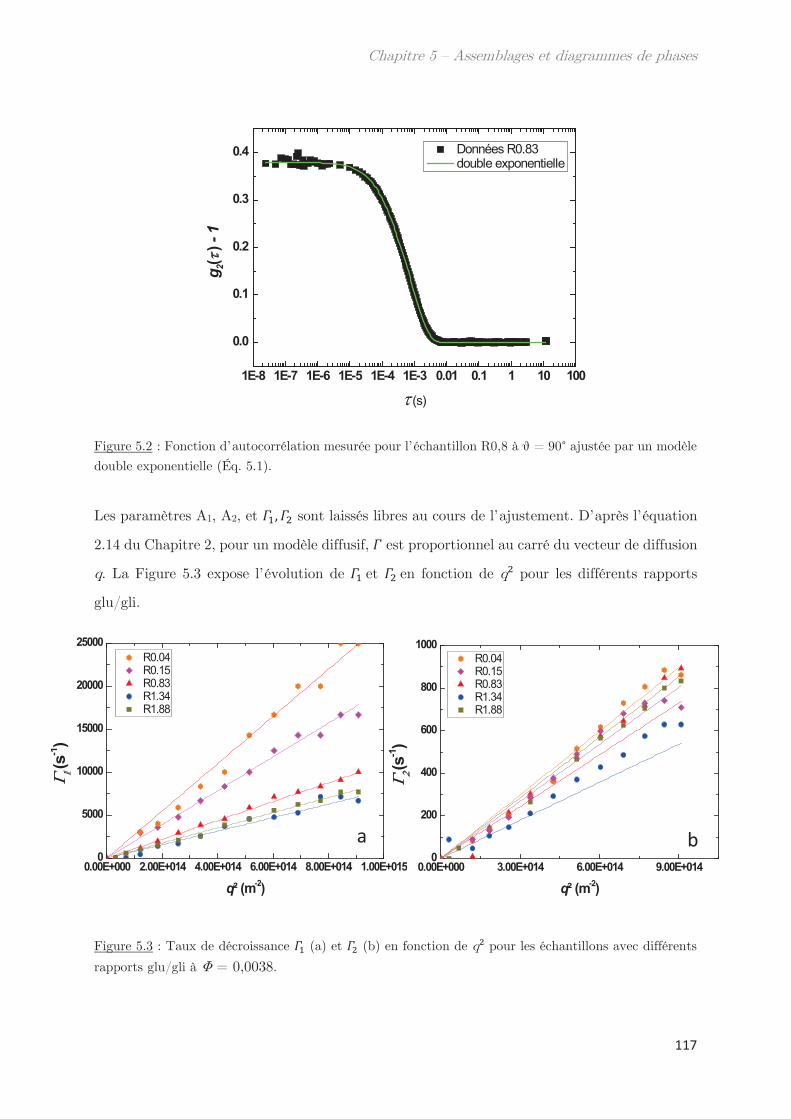
Chapitre 5 – Assemblages et diagrammes de phases
117
Figure 5.2 : F ajustée par un modèle
double exponentielle (Éq. 5.1).
Les paramètres A1, A2, et , sont laissés libres au cours de l’ajustement. D’après l’équation
2.14 du Chapitre 2, pour un modèle diffusif, est proportionnel au carré du vecteur de diffusion
q. La Figure 5.3 expose l’évolution de et en fonction de q² pour les différents rapports
glu/gli.
Figure 5.3 : Taux de décroissance (a) et (b) en fonction de q² pour les échantillons avec différents
rapports glu/gli à = 0,0038.
1E-8 1E-7 1E-6 1E-5 1E-4 1E-3 0.01 0.1 1 10 100
0.0
0.1
0.2
0.3
0.4 Données R0.83 double exponentielle
g2(
)- 1
(s)
0.00E+000 2.00E+014 4.00E+014 6.00E+014 8.00E+014 1.00E+0150
5000
10000
15000
20000
25000
R0.04 R0.15 R0.83 R1.34 R1.88
(s-1)
q² (m-2)
0.00E+000 3.00E+014 6.00E+014 9.00E+0140
200
400
600
800
1000
R0.04 R0.15 R0.83 R1.34 R1.88
(s-1)
q² (m-2)
a b
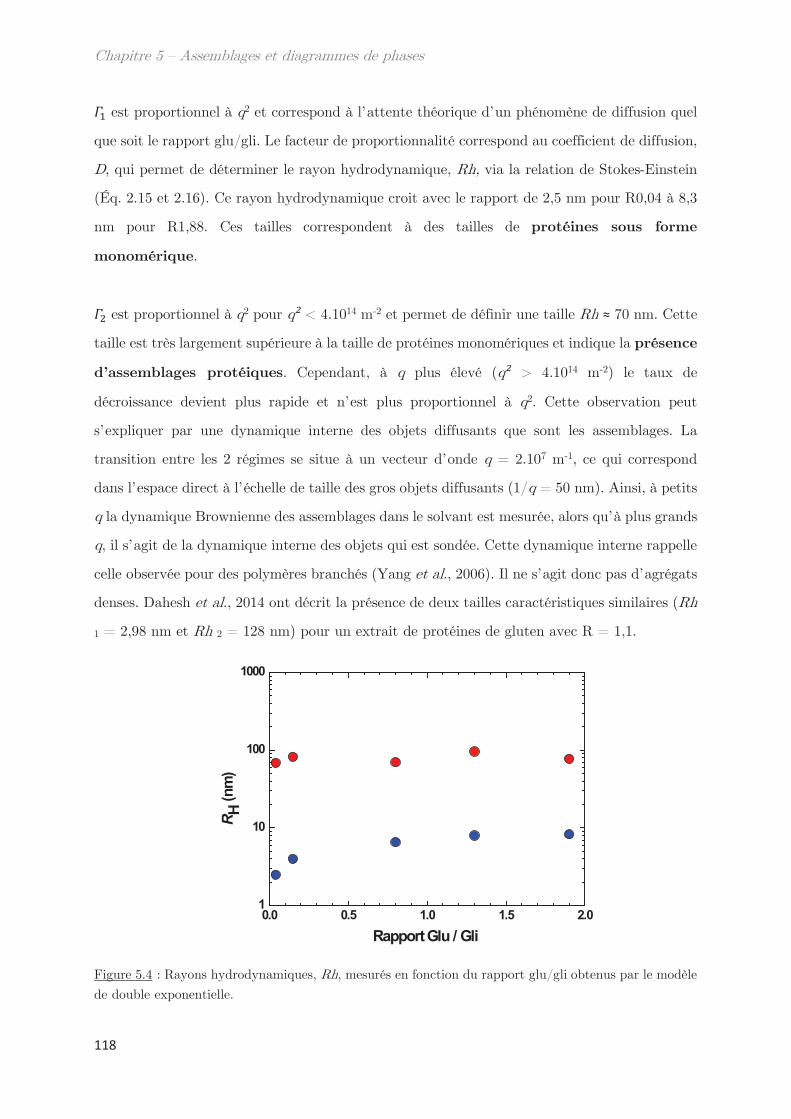
Chapitre 5 – Assemblages et diagrammes de phases
118
est proportionnel à q2 et correspond à l’attente théorique d’un phénomène de diffusion quel
que soit le rapport glu/gli. Le facteur de proportionnalité correspond au coefficient de diffusion,
D, qui permet de déterminer le rayon hydrodynamique, Rh, via la relation de Stokes-Einstein
(Éq. 2.15 et 2.16). Ce rayon hydrodynamique croit avec le rapport de 2,5 nm pour R0,04 à 8,3
nm pour R1,88. Ces tailles correspondent à des tailles de protéines sous forme
monomérique.
est proportionnel à q2 pour q² < 4.1014 m-2 et permet de définir une taille Rh 70 nm. Cette
taille est très largement supérieure à la taille de protéines monomériques et indique la présence
d’assemblages protéiques. Cependant, à q plus élevé (q² > 4.1014 m-2) le taux de
décroissance devient plus rapide et n’est plus proportionnel à q2. Cette observation peut
s’expliquer par une dynamique interne des objets diffusants que sont les assemblages. La
transition entre les 2 régimes se situe à un vecteur d’onde q = 2.107 m-1, ce qui correspond
dans l’espace direct à l’échelle de taille des gros objets diffusants (1/q = 50 nm). Ainsi, à petits
q la dynamique Brownienne des assemblages dans le solvant est mesurée, alors qu’à plus grands
q, il s’agit de la dynamique interne des objets qui est sondée. Cette dynamique interne rappelle
celle observée pour des polymères branchés (Yang et al., 2006). Il ne s’agit donc pas d’agrégats
denses. Dahesh et al., 2014 ont décrit la présence de deux tailles caractéristiques similaires (Rh
1 = 2,98 nm et Rh 2 = 128 nm) pour un extrait de protéines de gluten avec R = 1,1.
Figure 5.4 : Rayons hydrodynamiques, Rh, mesurés en fonction du rapport glu/gli obtenus par le modèle
de double exponentielle.
0.0 0.5 1.0 1.5 2.01
10
100
1000
RH
(nm
)
RapportGlu / Gli

Chapitre 5 – Assemblages et diagrammes de phases
119
La Figure 5.4 montre l’évolution des 2 tailles en fonction du rapport R. Pour tous les
échantillons, les solutions sont composées de deux types de particules possédant des rayons
hydrodynamiques de l’ordre de 5 nm et de 80 nm. Les échantillons enrichis en gliadines (R0,04
et R0,18) semblent avoir des Rh de petites tailles légèrement inférieurs à ceux des autres
échantillons aux rapports glu/gli plus élevés. R0,04 et R0,18 contiennent essentiellement des
/ , -gliadines qui ont des masses molaires de l’ordre de 35 000 g/mol (Rg = 3,55 à 3,80 nm
selon Thomson et al., 1999) alors que les R0,83, R1,35 et R1,88 contiennent également des
gluténines dont les sous-unités ont des masses molaires plus élevées (80 000 < M < 100 0000
g/mol). L’évolution de la petite taille suggère que la population monomérique comprend à la
fois des gliadines et des gluténines. Cependant, la taille des structures associatives apparait
constante avec le rapport glu/gli (<Rh> = 78,9 ± 11,2 nm). Il est impossible à ce stade
d’analyse de déterminer la composition de ces assemblages.
D’autre part, en supposant que la densité est la même pour les petits et les gros objets, il est
possible d’évaluer le rapport du nombre de petits objets sur celui des gros objets dans les
différents mélanges. Ainsi, l’amplitude relative du signal des deux populations (A1/A2) a été
analysée et représentée dans la Figure 5.5. Pour les échantillons R0,83, R1,34 et R1,88, le
rapport d’amplitude est identique, plutôt faible (0,1 < A1/A2 < 0,2) et évolue peu avec q.
Ainsi, ces échantillons ont un signal dominé par les gros objets de façon indifférenciée. Le
rapport A1/A2 des échantillons aux rapports glu/gli plus faibles (R0,04 et R0,18) est plus élevé
et augmente avec q. À petits q, le signal est dominé par les gros objets alors qu’à grands q le
signal est dominé par les petits objets. L’augmentation du signal à grands q indique qu’il y a
plus de petits objets dans les échantillons à faible rapport glu/gli. Ainsi, la proportion d’objets
monomériques est plus importante dans ces échantillons et suggère que l’existence
d’assemblages est associée à la présence de gluténines dans la composition protéique, bien qu’on
s’attendrait à un rapport A1/A2 plus élevé pour R0,04 que R0,15.
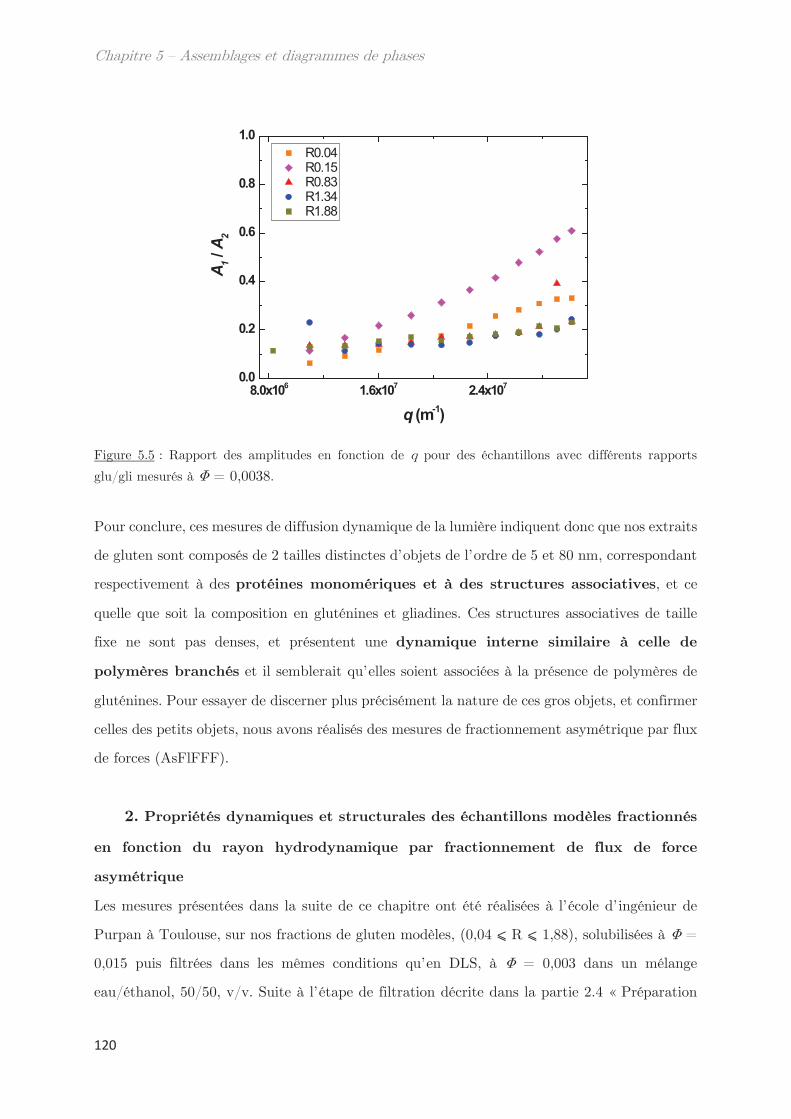
Chapitre 5 – Assemblages et diagrammes de phases
120
Figure 5.5 : Rapport des amplitudes en fonction de q pour des échantillons avec différents rapports
glu/gli mesurés à = 0,0038.
Pour conclure, ces mesures de diffusion dynamique de la lumière indiquent donc que nos extraits
de gluten sont composés de 2 tailles distinctes d’objets de l’ordre de 5 et 80 nm, correspondant
respectivement à des protéines monomériques et à des structures associatives, et ce
quelle que soit la composition en gluténines et gliadines. Ces structures associatives de taille
fixe ne sont pas denses, et présentent une dynamique interne similaire à celle de
polymères branchés et il semblerait qu’elles soient associées à la présence de polymères de
gluténines. Pour essayer de discerner plus précisément la nature de ces gros objets, et confirmer
celles des petits objets, nous avons réalisés des mesures de fractionnement asymétrique par flux
de forces (AsFlFFF).
2. Propriétés dynamiques et structurales des échantillons modèles fractionnés
en fonction du rayon hydrodynamique par fractionnement de flux de force
asymétrique
Les mesures présentées dans la suite de ce chapitre ont été réalisées à l’école d’ingénieur de
Purpan à Toulouse, sur nos fractions de gluten modèles, (0,04 R 1,88), solubilisées à =
0,015 puis filtrées dans les mêmes conditions qu’en DLS, à = 0,003 dans un mélange
eau/éthanol, 50/50, v/v. Suite à l’étape de filtration décrite dans la partie 2.4 « Préparation
8.0x106
1.6x107
2.4x107
0.0
0.2
0.4
0.6
0.8
1.0
R0.04 R0.15 R0.83 R1.34 R1.88
A1/A
2
q (m-1)
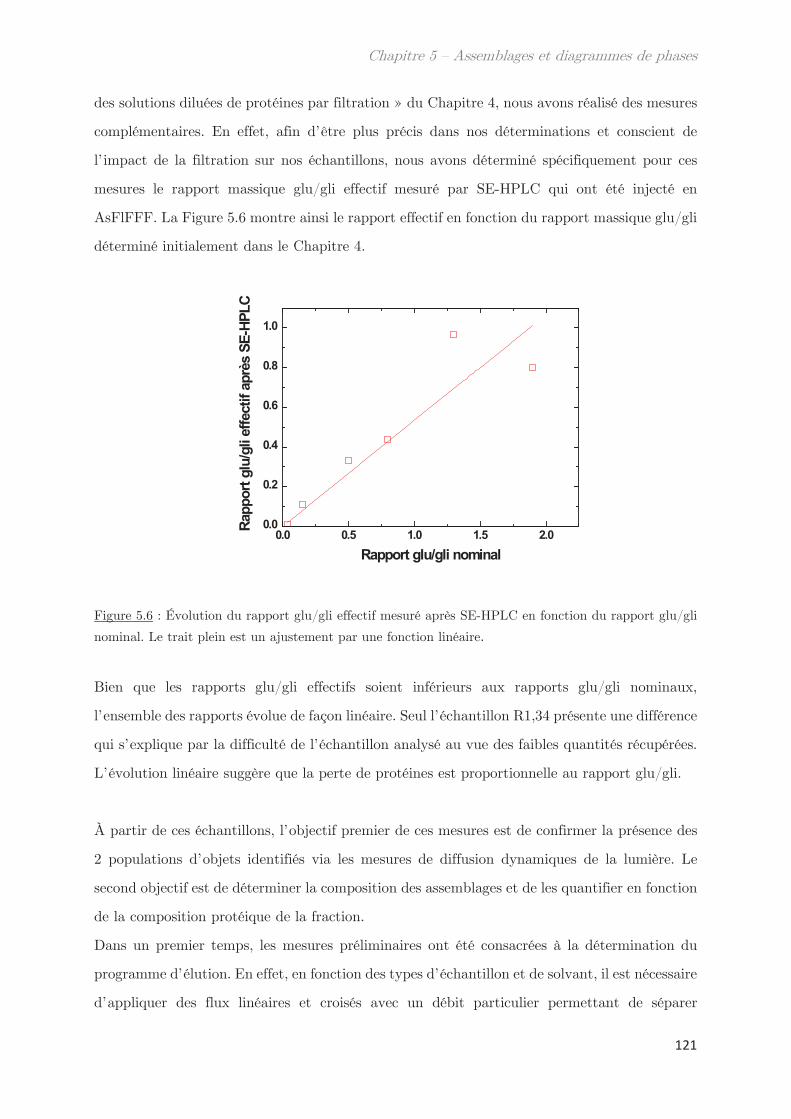
Chapitre 5 – Assemblages et diagrammes de phases
121
des solutions diluées de protéines par filtration » du Chapitre 4, nous avons réalisé des mesures
complémentaires. En effet, afin d’être plus précis dans nos déterminations et conscient de
l’impact de la filtration sur nos échantillons, nous avons déterminé spécifiquement pour ces
mesures le rapport massique glu/gli effectif mesuré par SE-HPLC qui ont été injecté en
AsFlFFF. La Figure 5.6 montre ainsi le rapport effectif en fonction du rapport massique glu/gli
déterminé initialement dans le Chapitre 4.
Figure 5.6 : Évolution du rapport glu/gli effectif mesuré après SE-HPLC en fonction du rapport glu/gli
nominal. Le trait plein est un ajustement par une fonction linéaire.
Bien que les rapports glu/gli effectifs soient inférieurs aux rapports glu/gli nominaux,
l’ensemble des rapports évolue de façon linéaire. Seul l’échantillon R1,34 présente une différence
qui s’explique par la difficulté de l’échantillon analysé au vue des faibles quantités récupérées.
L’évolution linéaire suggère que la perte de protéines est proportionnelle au rapport glu/gli.
À partir de ces échantillons, l’objectif premier de ces mesures est de confirmer la présence des
2 populations d’objets identifiés via les mesures de diffusion dynamiques de la lumière. Le
second objectif est de déterminer la composition des assemblages et de les quantifier en fonction
de la composition protéique de la fraction.
Dans un premier temps, les mesures préliminaires ont été consacrées à la détermination du
programme d’élution. En effet, en fonction des types d’échantillon et de solvant, il est nécessaire
d’appliquer des flux linéaires et croisés avec un débit particulier permettant de séparer
0.0 0.5 1.0 1.5 2.00.0
0.2
0.4
0.6
0.8
1.0
Rapport
glu
/gli e
ffectif aprè
s S
E-H
PLC
Rapport glu/gli nominal
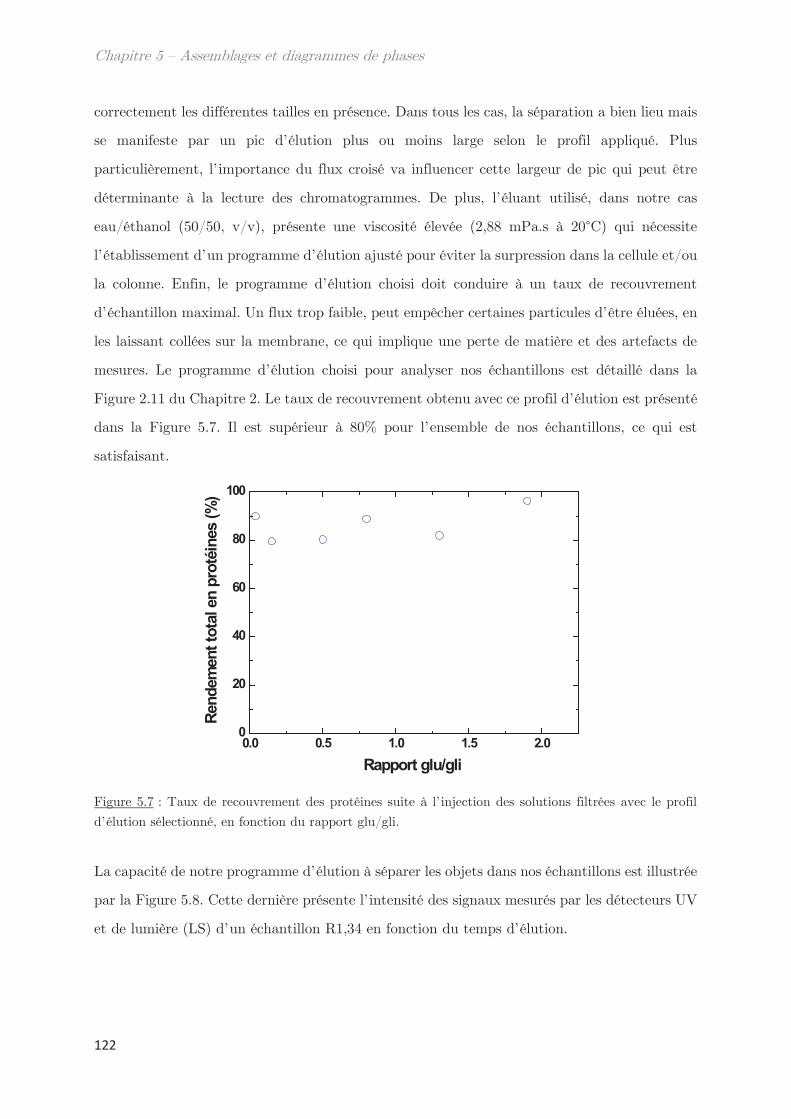
Chapitre 5 – Assemblages et diagrammes de phases
122
correctement les différentes tailles en présence. Dans tous les cas, la séparation a bien lieu mais
se manifeste par un pic d’élution plus ou moins large selon le profil appliqué. Plus
particulièrement, l’importance du flux croisé va influencer cette largeur de pic qui peut être
déterminante à la lecture des chromatogrammes. De plus, l’éluant utilisé, dans notre cas
eau/éthanol (50/50, v/v), présente une viscosité élevée (2,88 mPa.s à 20°C) qui nécessite
l’établissement d’un programme d’élution ajusté pour éviter la surpression dans la cellule et/ou
la colonne. Enfin, le programme d’élution choisi doit conduire à un taux de recouvrement
d’échantillon maximal. Un flux trop faible, peut empêcher certaines particules d’être éluées, en
les laissant collées sur la membrane, ce qui implique une perte de matière et des artefacts de
mesures. Le programme d’élution choisi pour analyser nos échantillons est détaillé dans la
Figure 2.11 du Chapitre 2. Le taux de recouvrement obtenu avec ce profil d’élution est présenté
dans la Figure 5.7. Il est supérieur à 80% pour l’ensemble de nos échantillons, ce qui est
satisfaisant.
Figure 5.7 : Taux de recouvrement des protéines suite à l’injection des solutions filtrées avec le profil
d’élution sélectionné, en fonction du rapport glu/gli.
La capacité de notre programme d’élution à séparer les objets dans nos échantillons est illustrée
par la Figure 5.8. Cette dernière présente l’intensité des signaux mesurés par les détecteurs UV
et de lumière (LS) d’un échantillon R1,34 en fonction du temps d’élution.
0.0 0.5 1.0 1.5 2.00
20
40
60
80
100
Rendem
ent to
tal en p
roté
ines (%
)
Rapport glu/gli
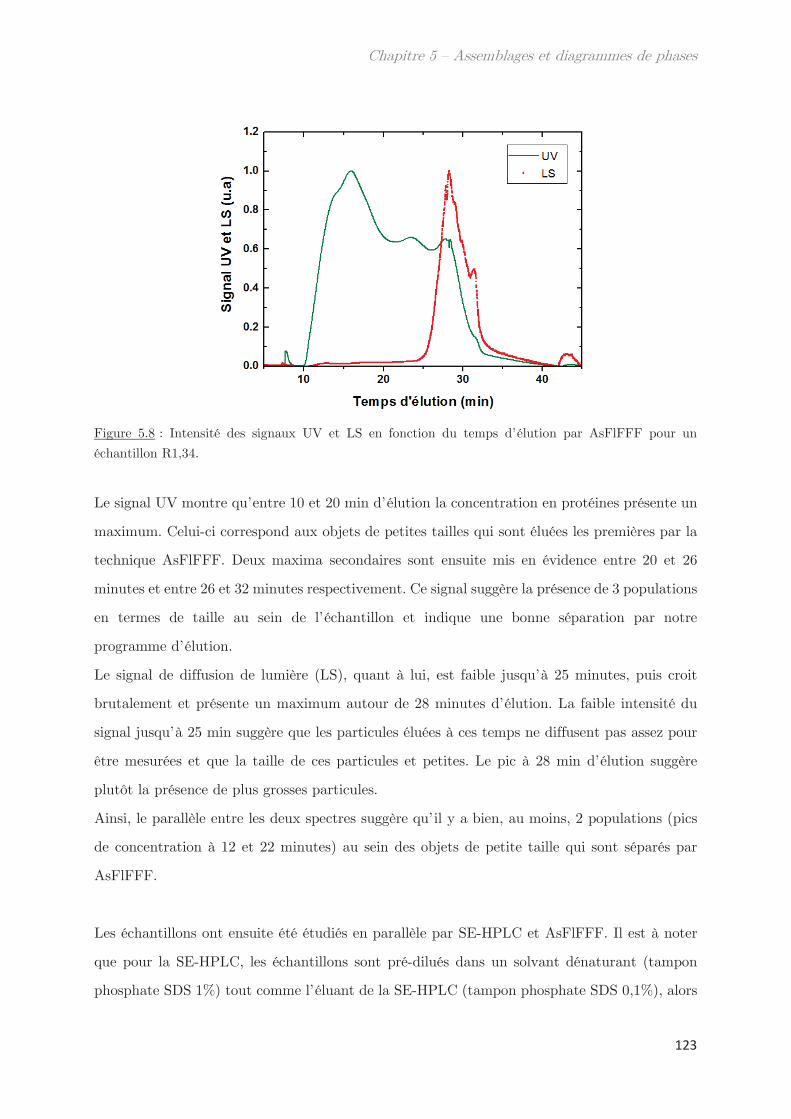
Chapitre 5 – Assemblages et diagrammes de phases
123
Figure 5.8 : Intensité des signaux UV et LS en fonction du temps d’élution par AsFlFFF pour un
échantillon R1,34.
Le signal UV montre qu’entre 10 et 20 min d’élution la concentration en protéines présente un
maximum. Celui-ci correspond aux objets de petites tailles qui sont éluées les premières par la
technique AsFlFFF. Deux maxima secondaires sont ensuite mis en évidence entre 20 et 26
minutes et entre 26 et 32 minutes respectivement. Ce signal suggère la présence de 3 populations
en termes de taille au sein de l’échantillon et indique une bonne séparation par notre
programme d’élution.
Le signal de diffusion de lumière (LS), quant à lui, est faible jusqu’à 25 minutes, puis croit
brutalement et présente un maximum autour de 28 minutes d’élution. La faible intensité du
signal jusqu’à 25 min suggère que les particules éluées à ces temps ne diffusent pas assez pour
être mesurées et que la taille de ces particules et petites. Le pic à 28 min d’élution suggère
plutôt la présence de plus grosses particules.
Ainsi, le parallèle entre les deux spectres suggère qu’il y a bien, au moins, 2 populations (pics
de concentration à 12 et 22 minutes) au sein des objets de petite taille qui sont séparés par
AsFlFFF.
Les échantillons ont ensuite été étudiés en parallèle par SE-HPLC et AsFlFFF. Il est à noter
que pour la SE-HPLC, les échantillons sont pré-dilués dans un solvant dénaturant (tampon
phosphate SDS 1%) tout comme l’éluant de la SE-HPLC (tampon phosphate SDS 0,1%), alors
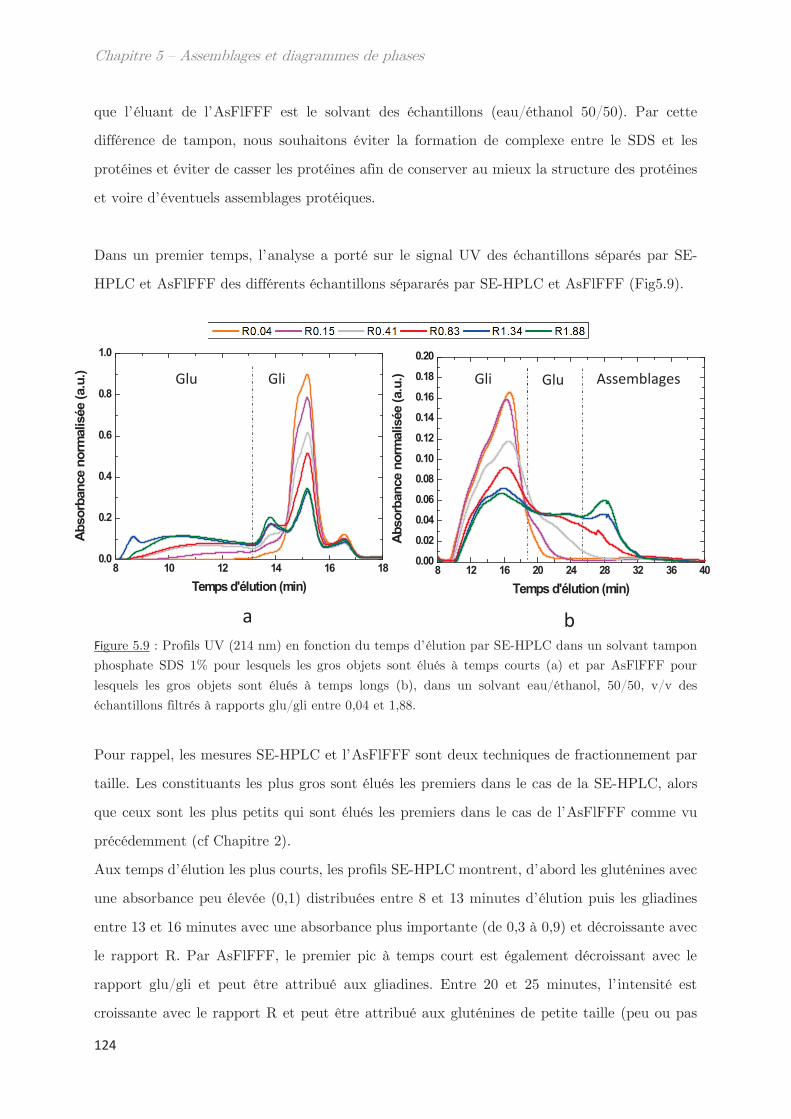
Chapitre 5 – Assemblages et diagrammes de phases
124
que l’éluant de l’AsFlFFF est le solvant des échantillons (eau/éthanol 50/50). Par cette
différence de tampon, nous souhaitons éviter la formation de complexe entre le SDS et les
protéines et éviter de casser les protéines afin de conserver au mieux la structure des protéines
et voire d’éventuels assemblages protéiques.
Dans un premier temps, l’analyse a porté sur le signal UV des échantillons séparés par SE-
HPLC et AsFlFFF des différents échantillons sépararés par SE-HPLC et AsFlFFF (Fig5.9).
Figure 5.9 : Profils UV (214 nm) en fonction du temps d’élution par SE-HPLC dans un solvant tampon
phosphate SDS 1% pour lesquels les gros objets sont élués à temps courts (a) et par AsFlFFF pour
lesquels les gros objets sont élués à temps longs (b), dans un solvant eau/éthanol, 50/50, v/v des
échantillons filtrés à rapports glu/gli entre 0,04 et 1,88.
Pour rappel, les mesures SE-HPLC et l’AsFlFFF sont deux techniques de fractionnement par
taille. Les constituants les plus gros sont élués les premiers dans le cas de la SE-HPLC, alors
que ceux sont les plus petits qui sont élués les premiers dans le cas de l’AsFlFFF comme vu
précédemment (cf Chapitre 2).
Aux temps d’élution les plus courts, les profils SE-HPLC montrent, d’abord les gluténines avec
une absorbance peu élevée (0,1) distribuées entre 8 et 13 minutes d’élution puis les gliadines
entre 13 et 16 minutes avec une absorbance plus importante (de 0,3 à 0,9) et décroissante avec
le rapport R. Par AsFlFFF, le premier pic à temps court est également décroissant avec le
rapport glu/gli et peut être attribué aux gliadines. Entre 20 et 25 minutes, l’intensité est
croissante avec le rapport R et peut être attribué aux gluténines de petite taille (peu ou pas
Gli Glu Gli
8 10 12 14 16 180.0
0.2
0.4
0.6
0.8
1.0
Absorb
ance n
orm
alisée (a.u
.)
Temps d'élution (min)
a b
Glu Assemblages
8 12 16 20 24 28 32 36 400.00
0.02
0.04
0.06
0.08
0.10
0.12
0.14
0.16
0.18
0.20
Absorb
ance n
orm
alisée (a.u
.)
Temps d'élution (min)
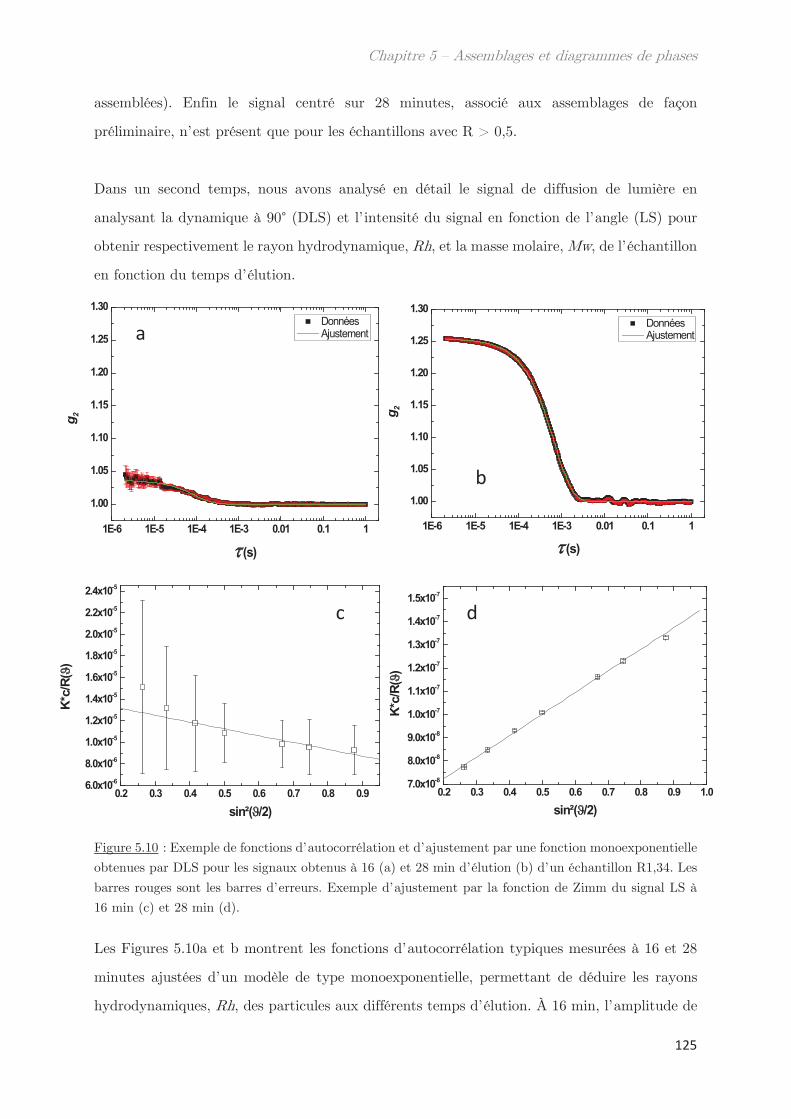
Chapitre 5 – Assemblages et diagrammes de phases
125
assemblées). Enfin le signal centré sur 28 minutes, associé aux assemblages de façon
préliminaire, n’est présent que pour les échantillons avec R > 0,5.
Dans un second temps, nous avons analysé en détail le signal de diffusion de lumière en
analysant la dynamique à 90° (DLS) et l’intensité du signal en fonction de l’angle (LS) pour
obtenir respectivement le rayon hydrodynamique, Rh, et la masse molaire, Mw, de l’échantillon
en fonction du temps d’élution.
Figure 5.10 : Exemple de fonctions d’autocorrélation et d’ajustement par une fonction monoexponentielle
obtenues par DLS pour les signaux obtenus à 16 (a) et 28 min d’élution (b) d’un échantillon R1,34. Les
barres rouges sont les barres d’erreurs. Exemple d’ajustement par la fonction de Zimm du signal LS à
16 min (c) et 28 min (d).
Les Figures 5.10a et b montrent les fonctions d’autocorrélation typiques mesurées à 16 et 28
minutes ajustées d’un modèle de type monoexponentielle, permettant de déduire les rayons
hydrodynamiques, Rh, des particules aux différents temps d’élution. À 16 min, l’amplitude de
b
c d
a
0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.96.0x10
-6
8.0x10-6
1.0x10-5
1.2x10-5
1.4x10-5
1.6x10-5
1.8x10-5
2.0x10-5
2.2x10-5
2.4x10-5
K*c
/R(
)
sin²( /2)
0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.07.0x10
-8
8.0x10-8
9.0x10-8
1.0x10-7
1.1x10-7
1.2x10-7
1.3x10-7
1.4x10-7
1.5x10-7
K*c
/R(
)
sin²( /2)
1E-6 1E-5 1E-4 1E-3 0.01 0.1 1
1.00
1.05
1.10
1.15
1.20
1.25
1.30
g2
(s)
Données Ajustement
1E-6 1E-5 1E-4 1E-3 0.01 0.1 1
1.00
1.05
1.10
1.15
1.20
1.25
1.30
Données Ajustement
g2
(s)

Chapitre 5 – Assemblages et diagrammes de phases
126
la fonction d’autocorrélation est très faible (1,04) et les barres d’erreurs sur la mesure de g2
sont importantes notamment aux temps très courts. Ces dernières suggèrent qu’ils existent de
plus gros objets qui diffusent, comptabilisés en nombre supérieurs vis-à-vis des petits objets, ce
qui ne permet pas de déterminer précisément les tailles d’objets à 16 min. Cependant, à 28
min, l’intensité diffusée est plus importante au temps courts (1,25) et les barres d’erreurs sont
négligeables ce qui suggèrent qu’il y a moins de gros objets qui diffusent ce qui laisse la
possibilité de calculer une taille pour les plus petits objets.
Les Figures 5.10c et d montrent des données LS ajustées par un modèle de Zimm (cf Chapitre
2). À temps courts (16 min), l’intensité diffusée est faible (donc K*C/R( ) élevé) et les barres
d’erreurs sont importantes. L’évolution de K*C/R( ) en fonction de sin2( /2) n’est pas
parfaitement linéaire et l’incertitude sur la pente (associée à Rg) est grande. Cependant, le
niveau permet tout de même d’évaluer un ordre de grandeur de la masse molaire par l’analyse
de Zimm (c). En revanche, aux temps plus longs (26 min), l’intensité diffusée est beaucoup
plus élevée et K*C/R( ) = f(sin2( /2)) est parfaitement linéaire. La qualité des données permet
ainsi une bonne évalution de Mw par l’analyse de Zimm (d).
L’ensemble des échantillons à différents rapports glu/gli a été analyss en suivant ce traitement
du signal aux différents temps d’élution. La Figure 5.11 présente les résultats obtenus en terme
de rayon hydrodynamique, Rh, et de masse molaire, Mw.
Deux classes de rayons hydrodynamiques, Rh, se distinguent sur la Figure 5.11a : une première
classe de taille Rh 10 nm qui croit légèrement avec le temps d’élution (de 5 à 20 nm entre 10
et 25 minutes). La seconde classe d’objets autour de 100 nm croit de 70 à 200 nm entre 25 et
40 minutes d’élution. Cependant, d’après le signal UV, il est à noter que la quantité de
protéines éluées au-delà de 32 minutes est négligeable. La taille maximale des objets peut donc
être évaluée à 120 nm.
De même pour la masse molaire, Mw, il existe deux populations différentes : la première aux
temps d’élution les plus faibles d’environ 105 g/mol, et une seconde aux temps d’élution les
plus longs à environ 2.107 g/mol. On remarque qu’il y a davantage d’incertitude sur les valeurs
obtenues pour des objets de petite taille (temps d’élution courts) et aux temps d’élution où la
concentration protéique est faible (au-délà de 22 min pour R < 0,41 et 32 min pour R > 0,41),
du fait de la faible intensité du signal diffusé.
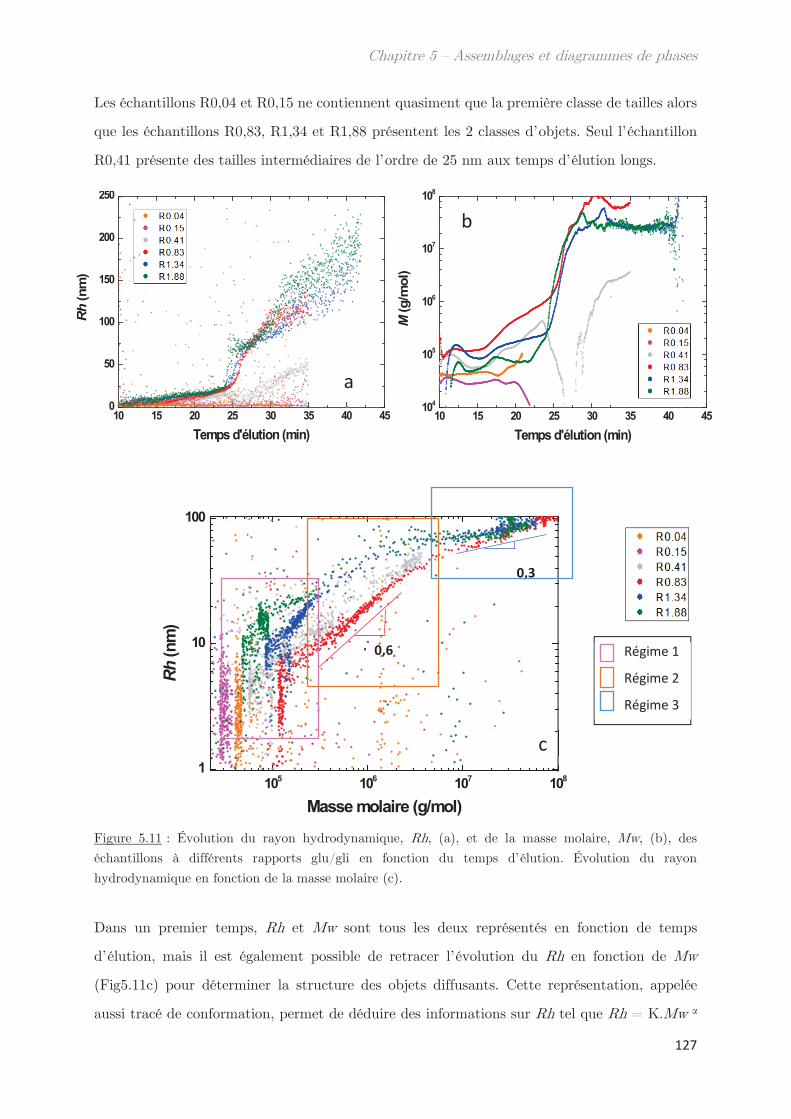
Chapitre 5 – Assemblages et diagrammes de phases
127
Les échantillons R0,04 et R0,15 ne contiennent quasiment que la première classe de tailles alors
que les échantillons R0,83, R1,34 et R1,88 présentent les 2 classes d’objets. Seul l’échantillon
R0,41 présente des tailles intermédiaires de l’ordre de 25 nm aux temps d’élution longs.
Figure 5.11 : Évolution du rayon hydrodynamique, Rh, (a), et de la masse molaire, Mw, (b), des
échantillons à différents rapports glu/gli en fonction du temps d’élution. Évolution du rayon
hydrodynamique en fonction de la masse molaire (c).
Dans un premier temps, Rh et Mw sont tous les deux représentés en fonction de temps
d’élution, mais il est également possible de retracer l’évolution du Rh en fonction de Mw
(Fig5.11c) pour déterminer la structure des objets diffusants. Cette représentation, appelée
aussi tracé de conformation, permet de déduire des informations sur Rh tel que Rh = K.Mw
0,3
10 15 20 25 30 35 40 4510
4
105
106
107
108
M (g/m
ol)
Temps d'élution (min)
105
106
107
108
1
10
100
Rh (nm
)
Masse molaire (g/mol)
Régime 1
Régime 2
Régime 3
a
b
c
0,6
10 15 20 25 30 35 40 450
50
100
150
200
250
Rh (nm
)
Temps d'élution (min)

Chapitre 5 – Assemblages et diagrammes de phases
128
paramètre caractéristique de la conformation des protéines. Dans notre cas, 3 régimes
peuvent être distingués :
- un premier aux masses molaires inférieures à 2.105 g/mol pour lequel les Rh sont de l’ordre
de 2 à 20 nm. Dans ce régime, les échantillons présentent peu d’évolution de masse molaire et
les mesures de rayons hydrodynamiques sont bruités.
- un régime intermédiaire de 2.105 à 5.106 g/mol au cours duquel la taille des Rh augmente
entre 10 et 70 nm. Ces tailles sont observées en majeure partie pour les échantillons R0,41 et
R0,83 qui présentent une évolution selon Rh Mw 0,6. D’après les valeurs théoriques (Smidsrod,
1969), 0,5 est attendu pour une conformation de type pelote. Ainsi, les objets de ce régime
semblent bien correspondre à des assemblages polymériques peu denses.
- un troisième régime de plus grandes masses molaires (5.106 à 1.108 g/mol) qui correspond à
des objets de l’ordre de 70 à 100 nm. Dans ce régime, l’évolution de Rh avec la masse molaire
est plus faible et indique des assemblages plus denses. Plus précisément, la pente évolue selon
Rh Mw 0,3. Or d’après la littérature, est la valeur théorique attendue pour des
été précédemment décrites
dans la littérature pour des polymères de gluténines en solution dans un tampon citrique pH
4,0 extraits de farine de blé de printemps Cheyenne et Chinois (Mendichi et al., 2008). Des
solubilisé dans un tampon sodium de phosphate 0,1 M (pH 6,9) et 2% de SDS (Carceller &
Aussenac, 2001).
L’analyse du Rh en fonction de M informe donc qu’il existe dans nos échantillons de gluten, 3
populations : la première est composée essentiellement de monomères, une seconde
d’assemblages de structures coils et une troisième composée d’assemblages plus denses.
Par ailleurs, via l’analyse des données AsFlFFF, il est possible de calculer précisémment la
quantité de protéines impliquées dans les assemblages polymériques pour les échantillons à
différents rapport glu/gli en considérant que les assemblages sont élués entre 25 et 35 minutes.
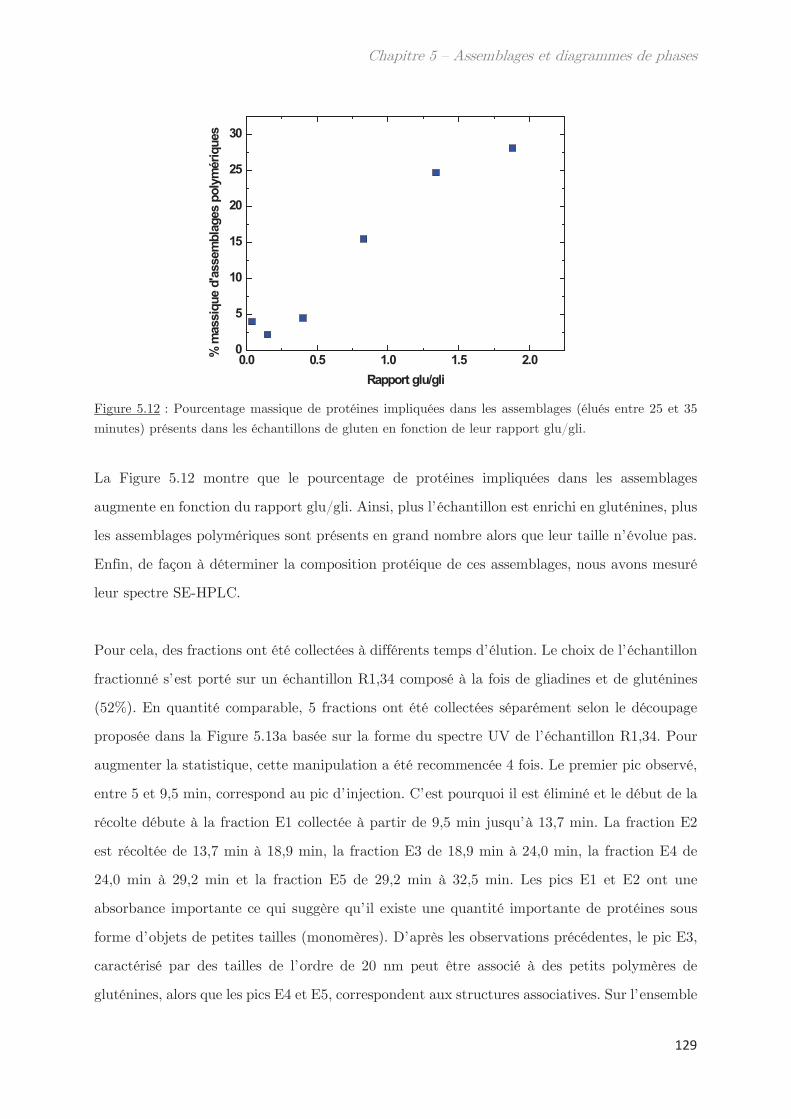
Chapitre 5 – Assemblages et diagrammes de phases
129
Figure 5.12 : Pourcentage massique de protéines impliquées dans les assemblages (élués entre 25 et 35
minutes) présents dans les échantillons de gluten en fonction de leur rapport glu/gli.
La Figure 5.12 montre que le pourcentage de protéines impliquées dans les assemblages
augmente en fonction du rapport glu/gli. Ainsi, plus l’échantillon est enrichi en gluténines, plus
les assemblages polymériques sont présents en grand nombre alors que leur taille n’évolue pas.
Enfin, de façon à déterminer la composition protéique de ces assemblages, nous avons mesuré
leur spectre SE-HPLC.
Pour cela, des fractions ont été collectées à différents temps d’élution. Le choix de l’échantillon
fractionné s’est porté sur un échantillon R1,34 composé à la fois de gliadines et de gluténines
(52%). En quantité comparable, 5 fractions ont été collectées séparément selon le découpage
proposée dans la Figure 5.13a basée sur la forme du spectre UV de l’échantillon R1,34. Pour
augmenter la statistique, cette manipulation a été recommencée 4 fois. Le premier pic observé,
entre 5 et 9,5 min, correspond au pic d’injection. C’est pourquoi il est éliminé et le début de la
récolte débute à la fraction E1 collectée à partir de 9,5 min jusqu’à 13,7 min. La fraction E2
est récoltée de 13,7 min à 18,9 min, la fraction E3 de 18,9 min à 24,0 min, la fraction E4 de
24,0 min à 29,2 min et la fraction E5 de 29,2 min à 32,5 min. Les pics E1 et E2 ont une
absorbance importante ce qui suggère qu’il existe une quantité importante de protéines sous
forme d’objets de petites tailles (monomères). D’après les observations précédentes, le pic E3,
caractérisé par des tailles de l’ordre de 20 nm peut être associé à des petits polymères de
gluténines, alors que les pics E4 et E5, correspondent aux structures associatives. Sur l’ensemble
0.0 0.5 1.0 1.5 2.00
5
10
15
20
25
30
% m
assiq
ue d
'assem
bla
ges p
oly
méri
ques
Rapport glu/gli

Chapitre 5 – Assemblages et diagrammes de phases
130
des répétitions, les spectres présentent des profils similaires sauf pour le pic E3 qui semble
légérement différent en fonction des répétitions. L’ensemble des résultats présenté dans la suite
résulte donc de la moyenne de ces 4 spectres issus des 4 répétitions (chacune des 5 fractions est
composée des 4 répétitions).
À la suite de la récolte les 5 fractions totales ont été séchées par un système SpeedVac. Les
échantillons ont ensuite été repris en eau dans 120 µl, 150 µl, 100 µl, 80 µl et 40 µl
respectivement pour les fractions E1, E2, E3, E4 et E5. La quantité d’eau ajoutée a été
déterminée en fonction de la masse de produit sec de chaque fraction. Une fois solubilisés, ils
ont été 2M d’urée afin de pouvoir être analysés
par SE-HPLC dans les conditions standards (cf Chapitre 2 et 4).
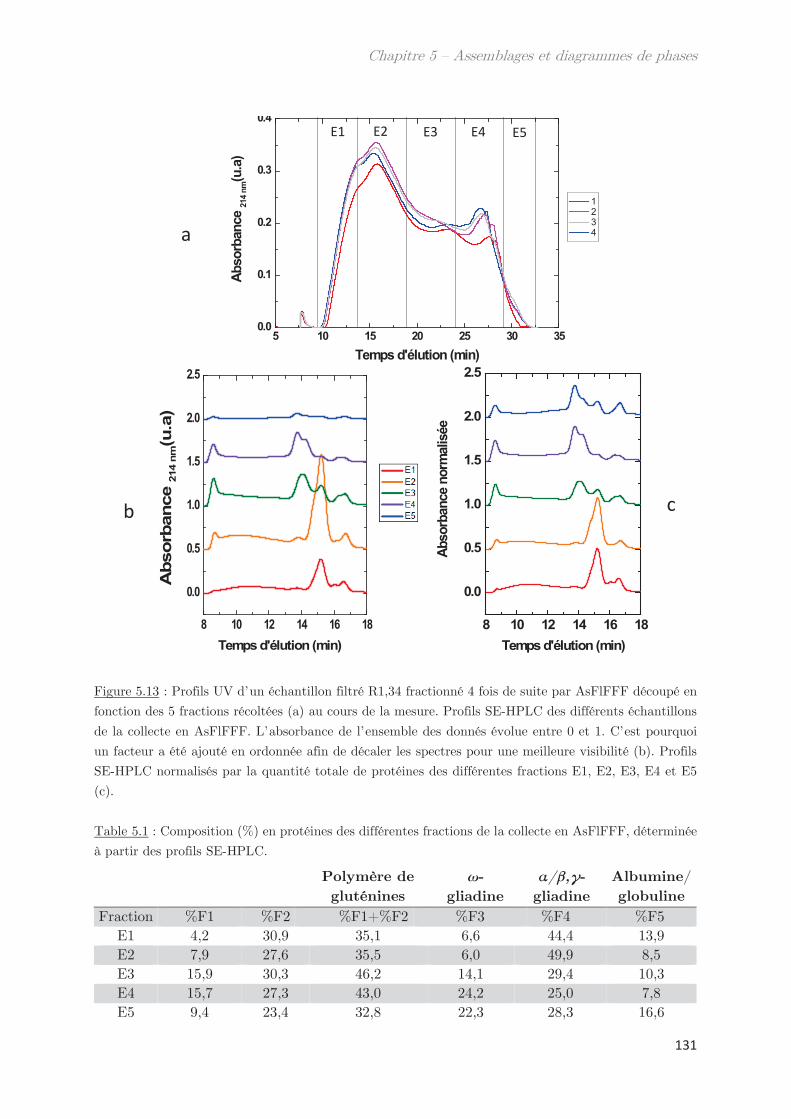
Chapitre 5 – Assemblages et diagrammes de phases
131
Figure 5.13 : Profils UV d’un échantillon filtré R1,34 fractionné 4 fois de suite par AsFlFFF découpé en
fonction des 5 fractions récoltées (a) au cours de la mesure. Profils SE-HPLC des différents échantillons
de la collecte en AsFlFFF. L’absorbance de l’ensemble des donnés évolue entre 0 et 1. C’est pourquoi
un facteur a été ajouté en ordonnée afin de décaler les spectres pour une meilleure visibilité (b). Profils
SE-HPLC normalisés par la quantité totale de protéines des différentes fractions E1, E2, E3, E4 et E5
(c).
Table 5.1 : Composition (%) en protéines des différentes fractions de la collecte en AsFlFFF, déterminée
à partir des profils SE-HPLC.
Polymère de gluténines
-gliadine
/ -gliadine
Albumine/ globuline
Fraction %F1 %F2 %F3 %F4 %F5 E1 4,2 30,9 35,1 6,6 44,4 13,9 E2 7,9 27,6 35,5 6,0 49,9 8,5 E3 15,9 30,3 46,2 14,1 29,4 10,3 E4 15,7 27,3 43,0 24,2 25,0 7,8 E5 9,4 23,4 32,8 22,3 28,3 16,6
a
E1 E2 E3 E4 E5
b c
8 10 12 14 16 18
0.0
0.5
1.0
1.5
2.0
2.5
Absorb
ance n
orm
alisée
Temps d'élution (min)
5 10 15 20 25 30 350.0
0.1
0.2
0.3
0.4
1 2 3 4
Absorb
ance 2
14 n
m(u
.a)
Temps d'élution (min)
Temps d'élution (min)
8 10 12 14 16 18
0.0
0.5
1.0
1.5
2.0
2.5
Absorb
ance 2
14 n
m(u
.a)

Chapitre 5 – Assemblages et diagrammes de phases
132
D’après la Figure 5.13b, plus la fraction est prélevée tard (de E1 vers E5) plus la proportion
du pic de gluténines, observable entre 8 et 12 min d’élution en SE-HPLC, devient importante,
contrairement au pic de de gliadines, observable entre 13 et 16 min d’élution, qui devient de
plus en plus faible.
En étudiant plus précisément les spectres et les quantités protéiques (Table 5.1), il apparait de
façon significative, que les fractions E1 et E2, éluées à 14-16 min d’après les profils SE-HPLC,
sont très enrichies en / et -gliadines, (44,4% et 49,9% respectivement) et appauvries en -
gliadines, éluées à 13-14 min, (6,6% et 6,0% respectivement) mais qui sont bien présentes. Elles
contiennent cependant, une quantité non négligeable de polymères de gluténines, (35,1% et
35,5% respectivement) ce qui suggère d’après l’ensemble des profils SE-HPLC et AsFlFFF,
qu’il y a bien 3 types de population composés de monomères de gluténines et de gliadines. Il
apparait ensuite que le pic d’ -gliadines (de 13 à 14,3 min en SE-HPLC) augmente lorsque la
fraction est récoltée tardivement. Ce dernier passe de 6,0% à 24,2% entre E2 et E4. Cette
observation montre donc que les assemblages, élués tardivement en AsFlFFF, sont
majoritairement composées de gluténines mais aussi d’ -gliadines. En effet, la fraction E3 est
celle la plus riche en gluténines (46,2%) et contient une quantité importante d’ -gliadines
(14,1%). Enfin, on remarque que E5 qui est élué en dernier est majoritairement composé de
gros objet que sont les polymères de gluténines (32,8%).
Les différentes mesures réalisées par AsFlFFF ont ainsi confirmé la présence des 2 populations
de tailles dans les échantillons. Plus précisément, nous retrouvons bien une taille d’objet de
l’ordre de 5-20 nm comme conclu précédemment avec la DLS. Ces objets peuvent être associés
à des monomères. La seconde population correspond à des objets de plus grosses tailles de
l’ordre de 100 nm qui ont une structure pelote plus ou moins dense. De plus, via l’analyse plus
spécifique des fractions récoltées en fonction du temps d’élution sur un échantillon R1,34, nous
avons appris que les petits objets sont principalement des / et -gliadines mais aussi des
gluténines de faible masse molaire. Quant aux gros objets de l’ordre de 100 nm, ils résultent
de l’association de polymères de gluténines avec des -gliadines. À la lumière de ces
résultats, la séparation de phases liquide-liquide utilisée pour fractionner le gluten et les
diagrammes de phases des différentes fractions modèles sont étudiés dans la suite de ce chapitre.
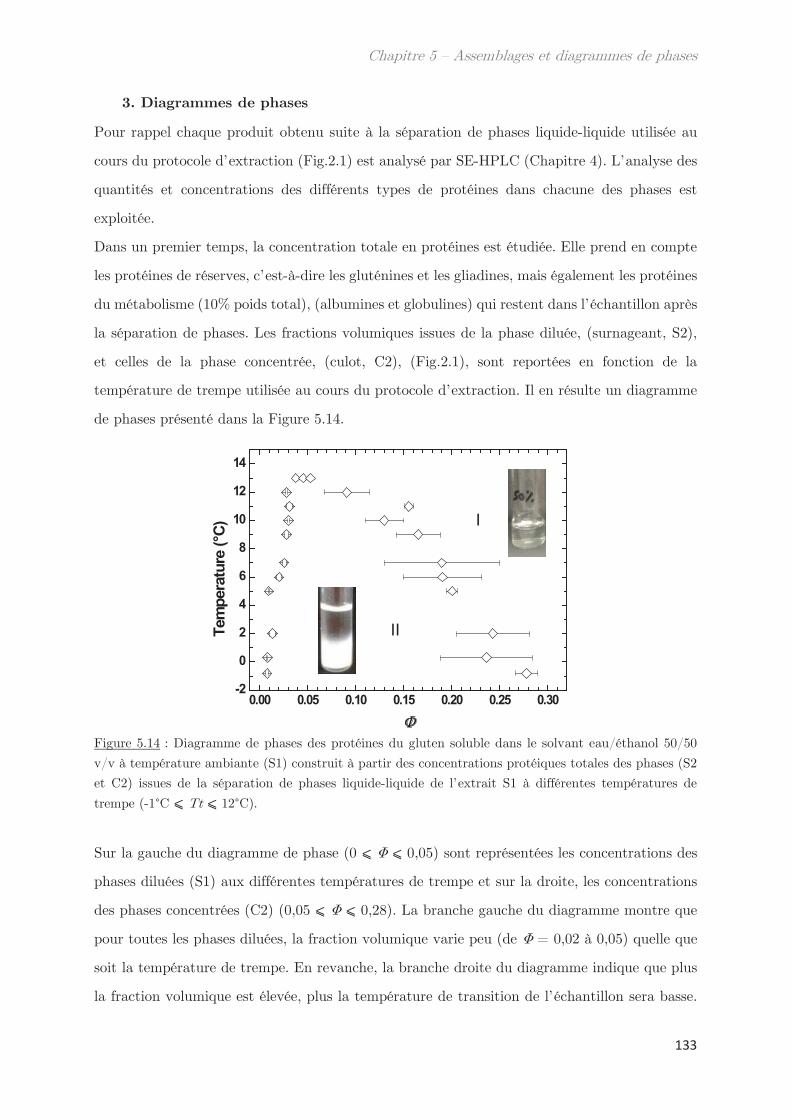
Chapitre 5 – Assemblages et diagrammes de phases
133
3. Diagrammes de phases
Pour rappel chaque produit obtenu suite à la séparation de phases liquide-liquide utilisée au
cours du protocole d’extraction (Fig.2.1) est analysé par SE-HPLC (Chapitre 4). L’analyse des
quantités et concentrations des différents types de protéines dans chacune des phases est
exploitée.
Dans un premier temps, la concentration totale en protéines est étudiée. Elle prend en compte
les protéines de réserves, c’est-à-dire les gluténines et les gliadines, mais également les protéines
du métabolisme (10% poids total), (albumines et globulines) qui restent dans l’échantillon après
la séparation de phases. Les fractions volumiques issues de la phase diluée, (surnageant, S2),
et celles de la phase concentrée, (culot, C2), (Fig.2.1), sont reportées en fonction de la
température de trempe utilisée au cours du protocole d’extraction. Il en résulte un diagramme
de phases présenté dans la Figure 5.14.
Figure 5.14 : Diagramme de phases des protéines du gluten soluble dans le solvant eau/éthanol 50/50
v/v à température ambiante (S1) construit à partir des concentrations protéiques totales des phases (S2
et C2) issues de la séparation de phases liquide-liquide de l’extrait S1 à différentes températures de
trempe (-1°C Tt 12°C).
Sur la gauche du diagramme de phase (0 0,05) sont représentées les concentrations des
phases diluées (S1) aux différentes températures de trempe et sur la droite, les concentrations
des phases concentrées (C2) (0,05 0,28). La branche gauche du diagramme montre que
pour toutes les phases diluées, la fraction volumique varie peu (de = 0,02 à 0,05) quelle que
soit la température de trempe. En revanche, la branche droite du diagramme indique que plus
la fraction volumique est élevée, plus la température de transition de l’échantillon sera basse.
0.00 0.05 0.10 0.15 0.20 0.25 0.30-2
0
2
4
6
8
10
12
14
Tem
pera
ture
(°C
) I
II

Chapitre 5 – Assemblages et diagrammes de phases
134
La forme et la pente de cette diminution dépend donc de . L’ensemble de cette courbe délimite
2 régions : une première homogène comprenant une seule phase (I), une seconde comprenant
deux phases (II).
Dans un second temps, la composition des phases S2 et C2 a été étudiée à partir des profils
SE-HPLC des S2 et des C2 issus des différentes températures de trempe. Ces profils sont
représentés sous forme normalisée par l’aire totale sous la courbe dans la Figure 5.15. Pour
rappel, la quantification des sous classes de protéines sont détaillés dans le Chapitre 4.
Les profils normalisés des surnageants, S2, montrent que plus la température de trempe est
basse, plus le surnageant est enrichi en / , -gliadines et déplété en gluténines et -gliadines.
De plus, la distribution de masse des gluténines évolue avec la température de trempe : la
masse molaire médiane de la distribution se décale vers les faibles masses lorsque la température
de trempe diminue.
Par ailleurs, les profils normalisés des culots, C2, montrent que plus la température de trempe
est élevée, plus le culot est enrichi en gluténines de hautes masses molaires, et déplété en / ,
-gliadines. La proportion de -gliadines quant à elle ne présente pas d’évolution significative.
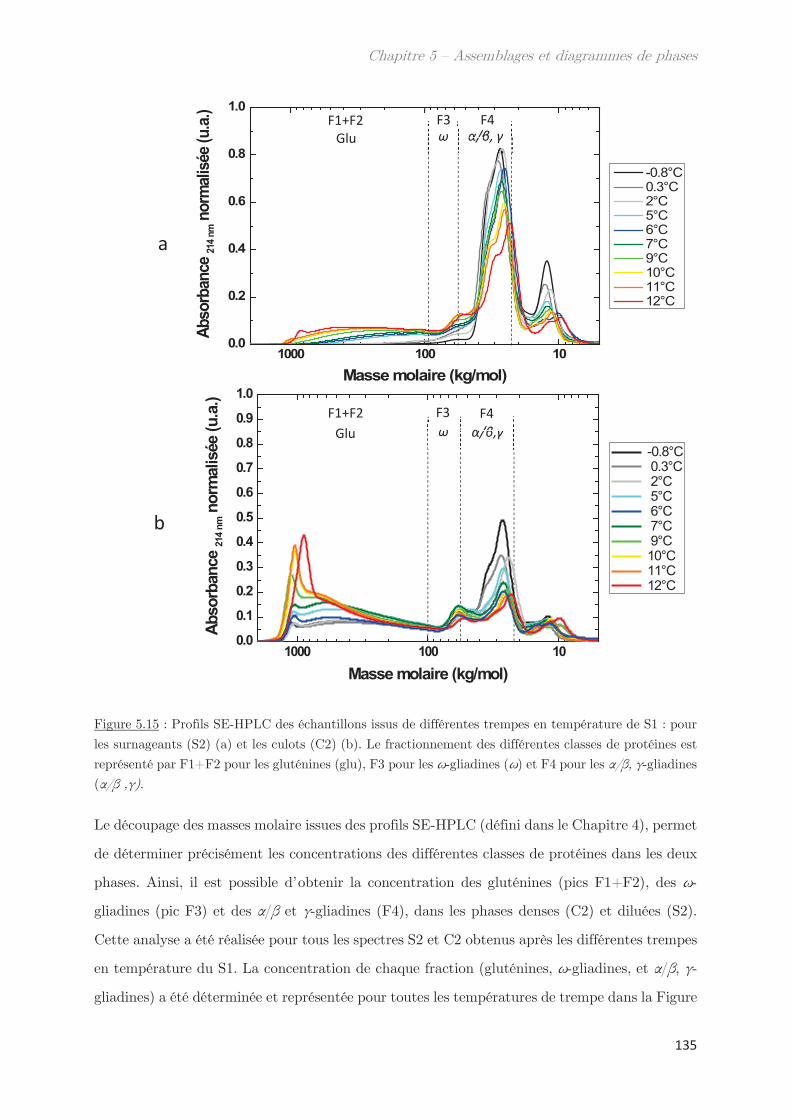
Chapitre 5 – Assemblages et diagrammes de phases
135
Figure 5.15 : Profils SE-HPLC des échantillons issus de différentes trempes en température de S1 : pour
les surnageants (S2) (a) et les culots (C2) (b). Le fractionnement des différentes classes de protéines est
u), F3 pour les -gliadines ( ) et F4 pour les -gliadines
(
Le découpage des masses molaire issues des profils SE-HPLC (défini dans le Chapitre 4), permet
de déterminer précisément les concentrations des différentes classes de protéines dans les deux
phases. Ainsi, il est possible d’obtenir la concentration des glu -
gliadines (pic F3) et des / et -gliadines (F4), dans les phases denses (C2) et diluées (S2).
Cette analyse a été réalisée pour tous les spectres S2 et C2 obtenus après les différentes trempes
en température du S1. La concentration de chaque fraction (gluténines, -gliadines, et / , -
gliadines) a été déterminée et représentée pour toutes les températures de trempe dans la Figure
F1+F2
1000 100 100.0
0.2
0.4
0.6
0.8
1.0
Abso
rban
ce 21
4 nm n
orm
alis
ée (u.a
.)
Masse molaire (kg/mol)
-0.8°C 0.3°C 2°C 5°C 6°C 7°C 9°C 10°C 11°C 12°C
a
b
Glu
Glu
F3 F4
Glu
F1+F2 F3 F4
1000 100 100.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
Absorb
ance 2
14 n
m n
orm
alisée (u.a
.)
Masse molaire (kg/mol)
-0.8°C 0.3°C 2°C 5°C 6°C 7°C 9°C 10°C 11°C 12°C
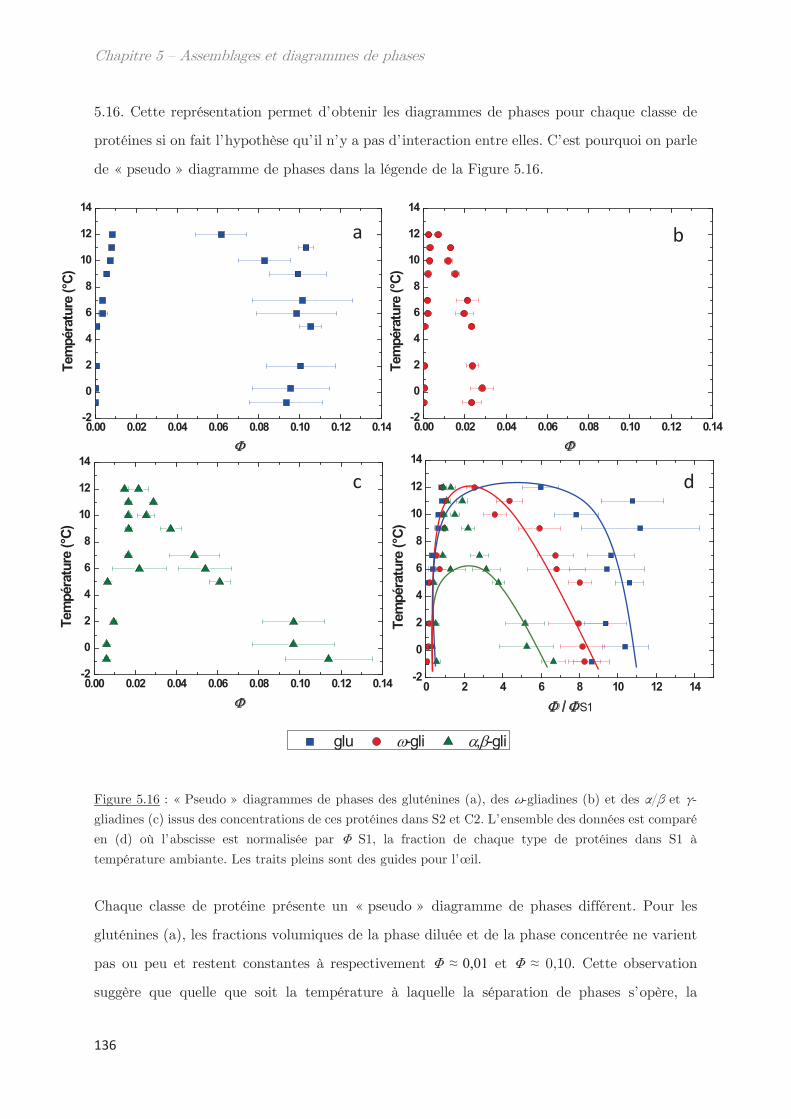
Chapitre 5 – Assemblages et diagrammes de phases
136
5.16. Cette représentation permet d’obtenir les diagrammes de phases pour chaque classe de
protéines si on fait l’hypothèse qu’il n’y a pas d’interaction entre elles. C’est pourquoi on parle
de « pseudo » diagramme de phases dans la légende de la Figure 5.16.
Figure 5.16 : « Pseudo » diagrammes de phases des gluténines (a), des -gliadines (b) et des / et -
gliadines (c) issus des concentrations de ces protéines dans S2 et C2. L’ensemble des données est comparé
en (d) où l’abscisse est normalisée par S1, la fraction de chaque type de protéines dans S1 à
température ambiante. Les traits pleins sont des guides pour l’œil.
Chaque classe de protéine présente un « pseudo » diagramme de phases différent. Pour les
gluténines (a), les fractions volumiques de la phase diluée et de la phase concentrée ne varient
pas ou peu et restent constantes à respectivement 0,01 et 0,10. Cette observation
suggère que quelle que soit la température à laquelle la séparation de phases s’opère, la
0.00 0.02 0.04 0.06 0.08 0.10 0.12 0.14-2
0
2
4
6
8
10
12
14
Tem
péra
ture
(°C
)
a b
c d
0.00 0.02 0.04 0.06 0.08 0.10 0.12 0.14-2
0
2
4
6
8
10
12
14
Tem
péra
ture
(°C
)
0.00 0.02 0.04 0.06 0.08 0.10 0.12 0.14-2
0
2
4
6
8
10
12
14
Tem
péra
ture
(°C
)
0 2 4 6 8 10 12 14-2
0
2
4
6
8
10
12
14
Tem
péra
ture
(°C
)
S1
glu -gli , -gli

Chapitre 5 – Assemblages et diagrammes de phases
137
profondeur de la trempe en température a toujours le même impact sur la répartition des
protéines dans les phases diluées et denses.
Le diagramme des -gliadines (b) montre une évolution similaire à celle des gluténines alors
que le diagramme des / , -gliadines (c) montre une forte évolution de la fraction volumique
de la phase dense avec la température de trempe. À T > 8°C, les concentrations en gliadines
dans les phases denses et diluées sont peu différentes alors qu’elles sont beaucoup plus marquées
à plus basses températures de trempes. Les différents « pseudo » diagrammes sont comparés
dans la Figure 5.16d en normalisant la fraction volumique par la fraction volumique de chaque
type de protéines dans la solution initiale à température ambiante de S1. Cette figure montre
que suite à une trempe à 0°C la phase dense est enrichie en gluténines, -gliadines et / , -
gliadines d’un facteur 10, 8 et 5 respectivement, par rapport à la solution initiale S1. De plus,
de par cette représentation, la proximité de forme des diagrammes de gluténines et -gliadines
est mise en avant et suggère que ces deux types de protéines sont en interactions. Les -
gliadines quant à elles se séparent de façon significative seulement à partir de 6°C et montrent
en première approximation un comportement indépendant des -gliadines et gluténines. Cette
observation peut être mise en parallèle avec la conclusion faite précédemment en AsFlFFF
selon laquelle les gros objets sont composés principalement d’associations entre les -gliadines
et les gluténines. Les assemblages auraient donc une température critique de 12°C alors que les
gliadines sépareraient de phases à plus basse température. La séparation de phases serait
donc pilotée par la masse molaire des objets en solution.
Pour confirmer cette dernière conclusion nous avons établi les diagrammes de phase des
différentes fractions modèles : R0,04, R0,15, R0,41, R0,83, R1,3 et R1,9.
Établir des diagrammes de phases à partir des concentrations obtenues via des mesures de SE-
HPLC, est une technique précise mais coûteuse qui demande beaucoup de matière première.
L’objectif de la méthode expérimentale développée dans le Chapitre 3 permet de pallier ces
problèmes. Nous avons donc analysé les différents extraits de protéines plus ou moins enrichis
en gluténines à partir du nouvel outil. Cependant, la gamme de température du système mis
en place ne permet pas de descendre en dessous de 5°C. Or, d’après nos essais préliminaires,
les températures de transitions Tcloud des échantillons enrichis en gliadines (R0,04 et R0,15)
sont inférieures à 5°C. L’étude des diagrammes de phases par cette technique s’applique donc
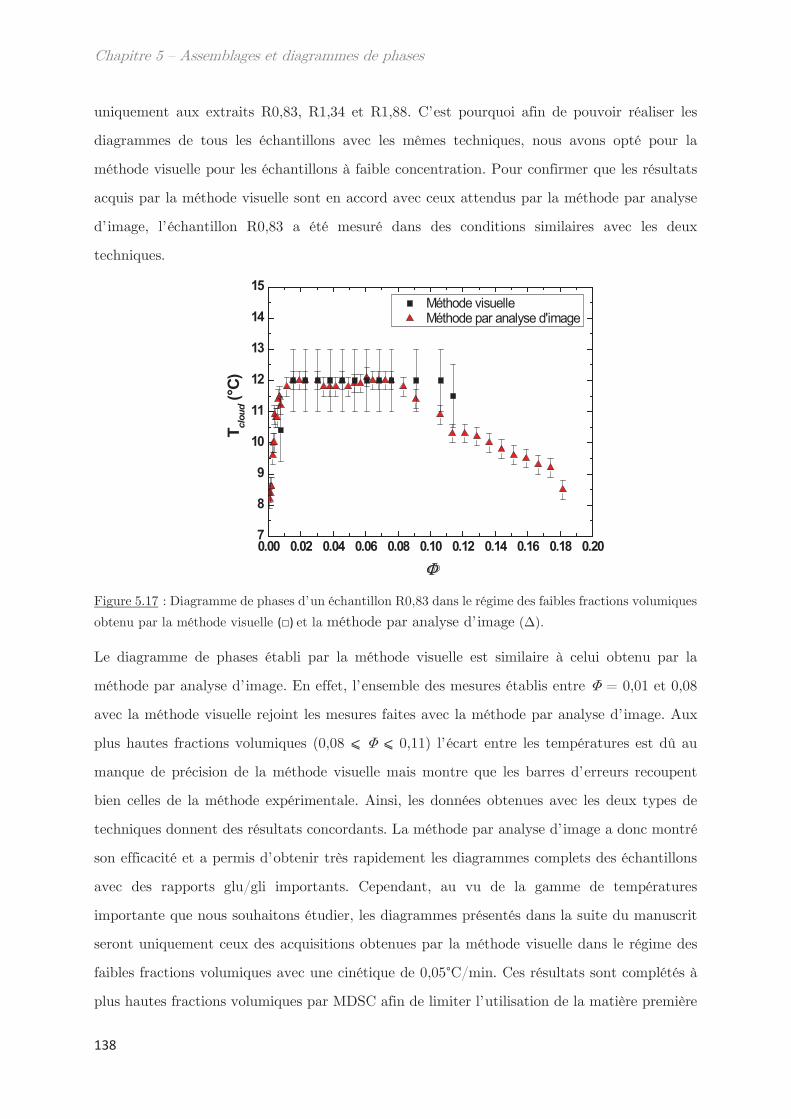
Chapitre 5 – Assemblages et diagrammes de phases
138
uniquement aux extraits R0,83, R1,34 et R1,88. C’est pourquoi afin de pouvoir réaliser les
diagrammes de tous les échantillons avec les mêmes techniques, nous avons opté pour la
méthode visuelle pour les échantillons à faible concentration. Pour confirmer que les résultats
acquis par la méthode visuelle sont en accord avec ceux attendus par la méthode par analyse
d’image, l’échantillon R0,83 a été mesuré dans des conditions similaires avec les deux
techniques.
Figure 5.17 : Diagramme de phases d’un échantillon R0,83 dans le régime des faibles fractions volumiques
obtenu par la méthode visuelle ( ) et la méthode par analyse d’image
Le diagramme de phases établi par la méthode visuelle est similaire à celui obtenu par la
méthode par analyse d’image. En effet, l’ensemble des mesures établis entre = 0,01 et 0,08
avec la méthode visuelle rejoint les mesures faites avec la méthode par analyse d’image. Aux
plus hautes fractions volumiques (0,08 0,11) l’écart entre les températures est dû au
manque de précision de la méthode visuelle mais montre que les barres d’erreurs recoupent
bien celles de la méthode expérimentale. Ainsi, les données obtenues avec les deux types de
techniques donnent des résultats concordants. La méthode par analyse d’image a donc montré
son efficacité et a permis d’obtenir très rapidement les diagrammes complets des échantillons
avec des rapports glu/gli importants. Cependant, au vu de la gamme de températures
importante que nous souhaitons étudier, les diagrammes présentés dans la suite du manuscrit
seront uniquement ceux des acquisitions obtenues par la méthode visuelle dans le régime des
faibles fractions volumiques avec une cinétique de 0,05°C/min. Ces résultats sont complétés à
plus hautes fractions volumiques par MDSC afin de limiter l’utilisation de la matière première
0.00 0.02 0.04 0.06 0.08 0.10 0.12 0.14 0.16 0.18 0.207
8
9
10
11
12
13
14
15
Méthode visuelle Méthode par analyse d'image
Tclo
ud (°C
)
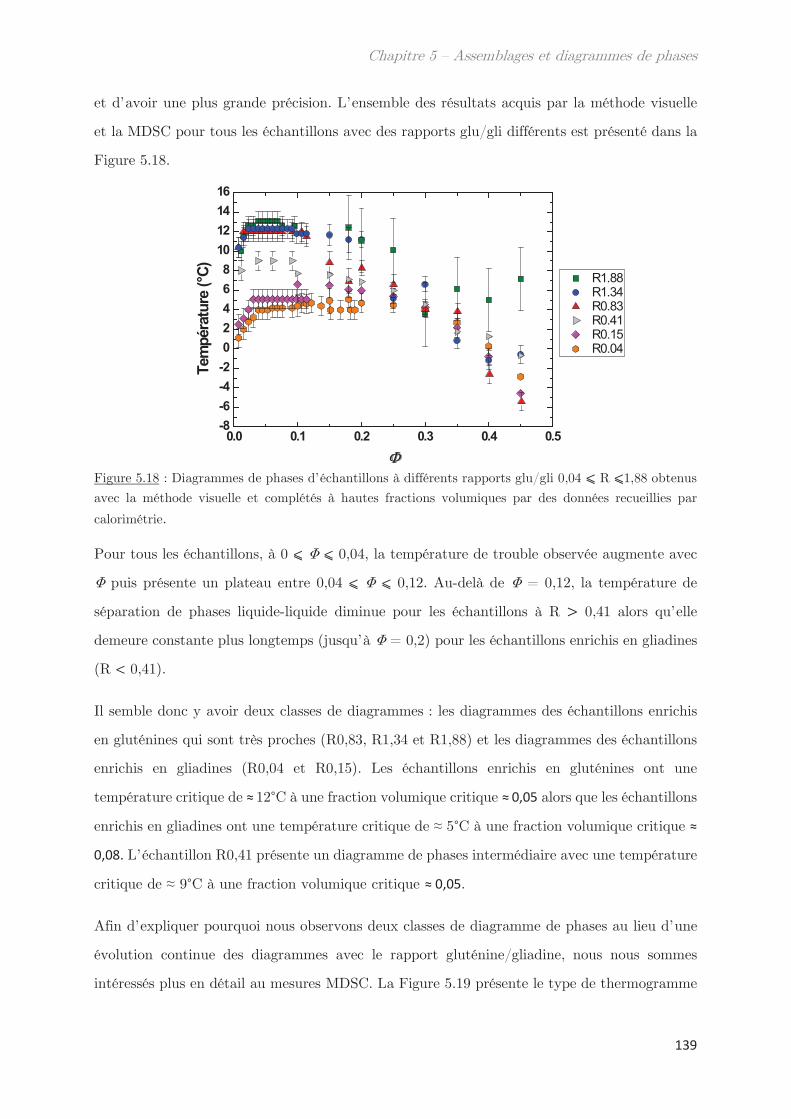
Chapitre 5 – Assemblages et diagrammes de phases
139
et d’avoir une plus grande précision. L’ensemble des résultats acquis par la méthode visuelle
et la MDSC pour tous les échantillons avec des rapports glu/gli différents est présenté dans la
Figure 5.18.
Figure 5.18 : Diagrammes de phases d’échantillons à différents rapports glu/gli 0,04 R 1,88 obtenus
avec la méthode visuelle et complétés à hautes fractions volumiques par des données recueillies par
calorimétrie.
Pour tous les échantillons, à 0 0,04, la température de trouble observée augmente avec
puis présente un plateau entre 0,04 0,12. Au-delà de = 0,12, la température de
séparation de phases liquide-liquide diminue pour les échantillons à R > 0,41 alors qu’elle
demeure constante plus longtemps (jusqu’à = 0,2) pour les échantillons enrichis en gliadines
(R < 0,41).
Il semble donc y avoir deux classes de diagrammes : les diagrammes des échantillons enrichis
en gluténines qui sont très proches (R0,83, R1,34 et R1,88) et les diagrammes des échantillons
enrichis en gliadines (R0,04 et R0,15). Les échantillons enrichis en gluténines ont une
température critique de 12°C à une fraction volumique critique alors que les échantillons
enrichis en gliadines ont une température critique de 5°C à une fraction volumique critique
L’échantillon R0,41 présente un diagramme de phases intermédiaire avec une température
critique de 9°C à une fraction volumique critique .
Afin d’expliquer pourquoi nous observons deux classes de diagramme de phases au lieu d’une
évolution continue des diagrammes avec le rapport gluténine/gliadine, nous nous sommes
intéressés plus en détail au mesures MDSC. La Figure 5.19 présente le type de thermogramme
0.0 0.1 0.2 0.3 0.4 0.5-8
-6
-4
-2
0
2
4
6
8
10
12
14
16
R1.88 R1.34 R0.83 R0.41 R0.15 R0.04
Tem
péra
ture
(°C
)
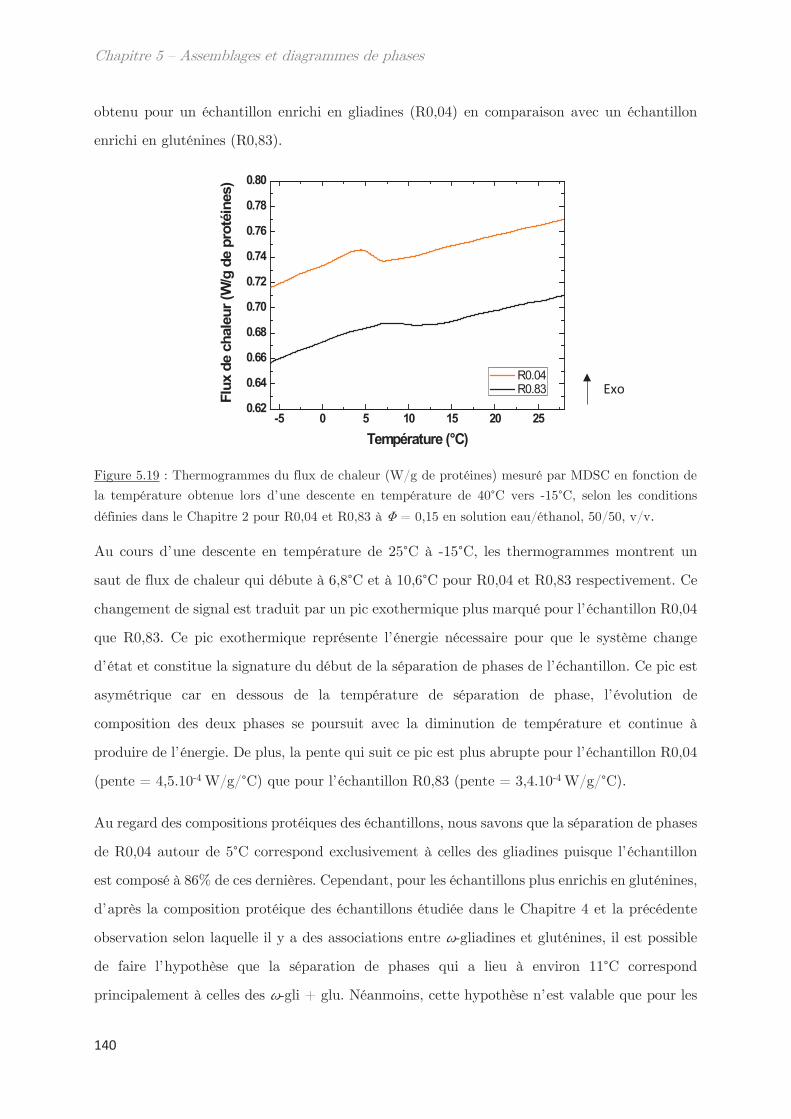
Chapitre 5 – Assemblages et diagrammes de phases
140
obtenu pour un échantillon enrichi en gliadines (R0,04) en comparaison avec un échantillon
enrichi en gluténines (R0,83).
Figure 5.19 : Thermogrammes du flux de chaleur (W/g de protéines) mesuré par MDSC en fonction de
la température obtenue lors d’une descente en température de 40°C vers -15°C, selon les conditions
définies dans le Chapitre 2 pour R0,04 et R0,83 à = 0,15 en solution eau/éthanol, 50/50, v/v.
Au cours d’une descente en température de 25°C à -15°C, les thermogrammes montrent un
saut de flux de chaleur qui débute à 6,8°C et à 10,6°C pour R0,04 et R0,83 respectivement. Ce
changement de signal est traduit par un pic exothermique plus marqué pour l’échantillon R0,04
que R0,83. Ce pic exothermique représente l’énergie nécessaire pour que le système change
d’état et constitue la signature du début de la séparation de phases de l’échantillon. Ce pic est
asymétrique car en dessous de la température de séparation de phase, l’évolution de
composition des deux phases se poursuit avec la diminution de température et continue à
produire de l’énergie. De plus, la pente qui suit ce pic est plus abrupte pour l’échantillon R0,04
(pente = 4,5.10-4 W/g/°C) que pour l’échantillon R0,83 (pente = 3,4.10-4 W/g/°C).
Au regard des compositions protéiques des échantillons, nous savons que la séparation de phases
de R0,04 autour de 5°C correspond exclusivement à celles des gliadines puisque l’échantillon
est composé à 86% de ces dernières. Cependant, pour les échantillons plus enrichis en gluténines,
d’après la composition protéique des échantillons étudiée dans le Chapitre 4 et la précédente
observation selon laquelle il y a des associations entre -gliadines et gluténines, il est possible
de faire l’hypothèse que la séparation de phases qui a lieu à environ 11°C correspond
principalement à celles des - Néanmoins, cette hypothèse n’est valable que pour les
Exo
-5 0 5 10 15 20 250.62
0.64
0.66
0.68
0.70
0.72
0.74
0.76
0.78
0.80
Flu
x d
e c
hale
ur
(W/g
de p
roté
ines)
Température (°C)
R0.04 R0.83
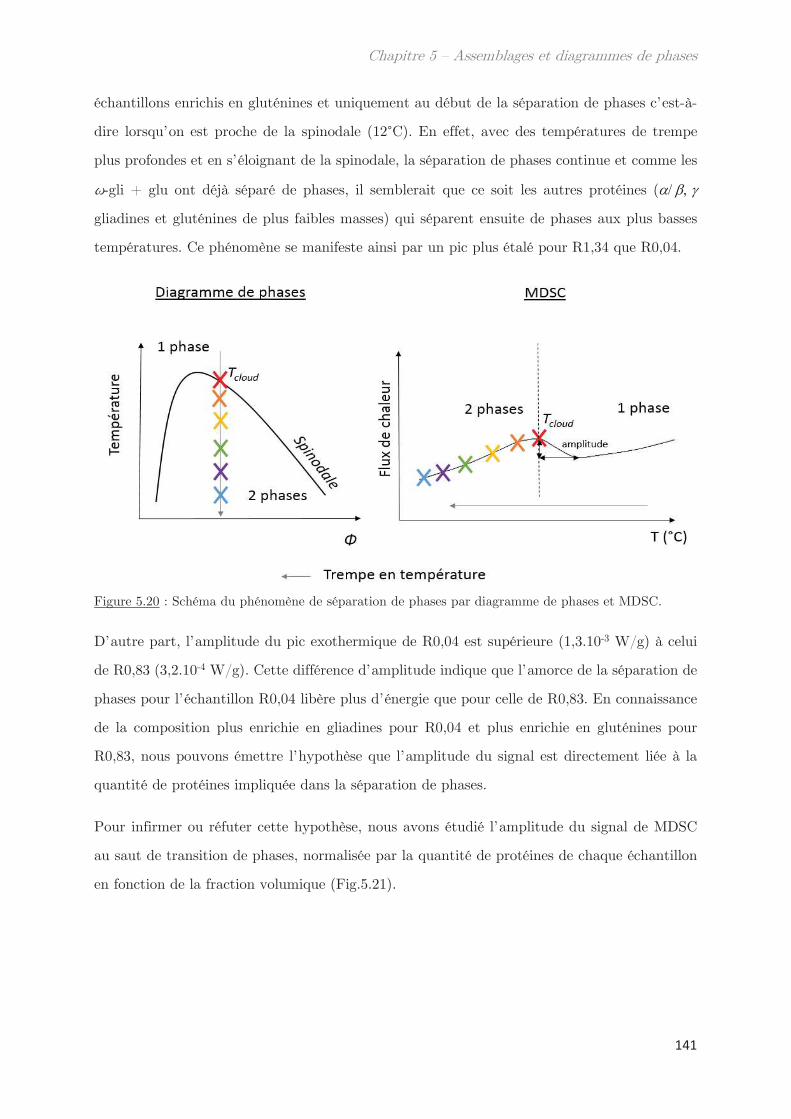
Chapitre 5 – Assemblages et diagrammes de phases
141
échantillons enrichis en gluténines et uniquement au début de la séparation de phases c’est-à-
dire lorsqu’on est proche de la spinodale (12°C). En effet, avec des températures de trempe
plus profondes et en s’éloignant de la spinodale, la séparation de phases continue et comme les
- ont déjà séparé de phases, il semblerait que ce soit les autres protéines (
gliadines et gluténines de plus faibles masses) qui séparent ensuite de phases aux plus basses
températures. Ce phénomène se manifeste ainsi par un pic plus étalé pour R1,34 que R0,04.
Figure 5.20 : Schéma du phénomène de séparation de phases par diagramme de phases et MDSC.
D’autre part, l’amplitude du pic exothermique de R0,04 est supérieure (1,3.10-3 W/g) à celui
de R0,83 (3,2.10-4 W/g). Cette différence d’amplitude indique que l’amorce de la séparation de
phases pour l’échantillon R0,04 libère plus d’énergie que pour celle de R0,83. En connaissance
de la composition plus enrichie en gliadines pour R0,04 et plus enrichie en gluténines pour
R0,83, nous pouvons émettre l’hypothèse que l’amplitude du signal est directement liée à la
quantité de protéines impliquée dans la séparation de phases.
Pour infirmer ou réfuter cette hypothèse, nous avons étudié l’amplitude du signal de MDSC
au saut de transition de phases, normalisée par la quantité de protéines de chaque échantillon
en fonction de la fraction volumique (Fig.5.21).
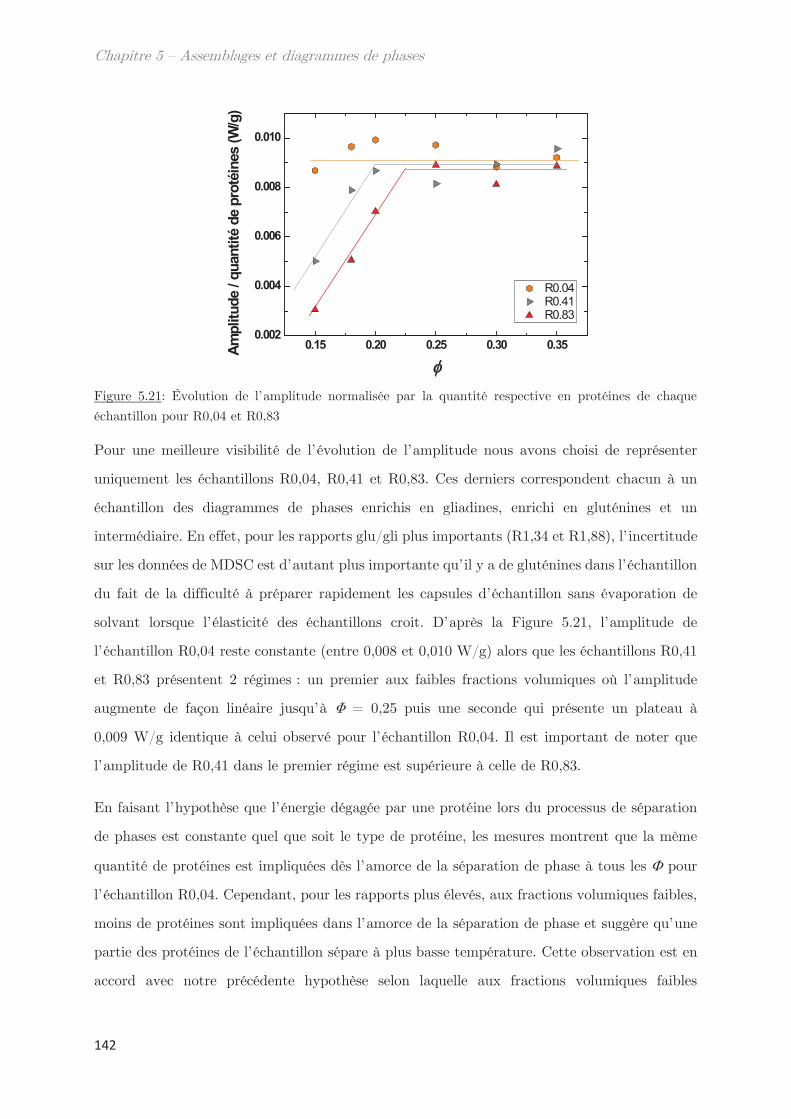
Chapitre 5 – Assemblages et diagrammes de phases
142
Figure 5.21: Évolution de l’amplitude normalisée par la quantité respective en protéines de chaque
échantillon pour R0,04 et R0,83
Pour une meilleure visibilité de l’évolution de l’amplitude nous avons choisi de représenter
uniquement les échantillons R0,04, R0,41 et R0,83. Ces derniers correspondent chacun à un
échantillon des diagrammes de phases enrichis en gliadines, enrichi en gluténines et un
intermédiaire. En effet, pour les rapports glu/gli plus importants (R1,34 et R1,88), l’incertitude
sur les données de MDSC est d’autant plus importante qu’il y a de gluténines dans l’échantillon
du fait de la difficulté à préparer rapidement les capsules d’échantillon sans évaporation de
solvant lorsque l’élasticité des échantillons croit. D’après la Figure 5.21, l’amplitude de
l’échantillon R0,04 reste constante (entre 0,008 et 0,010 W/g) alors que les échantillons R0,41
et R0,83 présentent 2 régimes : un premier aux faibles fractions volumiques où l’amplitude
augmente de façon linéaire jusqu’à = 0,25 puis une seconde qui présente un plateau à
0,009 W/g identique à celui observé pour l’échantillon R0,04. Il est important de noter que
l’amplitude de R0,41 dans le premier régime est supérieure à celle de R0,83.
En faisant l’hypothèse que l’énergie dégagée par une protéine lors du processus de séparation
de phases est constante quel que soit le type de protéine, les mesures montrent que la même
quantité de protéines est impliquées dès l’amorce de la séparation de phase à tous les pour
l’échantillon R0,04. Cependant, pour les rapports plus élevés, aux fractions volumiques faibles,
moins de protéines sont impliquées dans l’amorce de la séparation de phase et suggère qu’une
partie des protéines de l’échantillon sépare à plus basse température. Cette observation est en
accord avec notre précédente hypothèse selon laquelle aux fractions volumiques faibles
0.15 0.20 0.25 0.30 0.350.002
0.004
0.006
0.008
0.010
R0.04 R0.41 R0.83
Am
plitu
de / q
uantité
de p
roté
ines (W
/g)
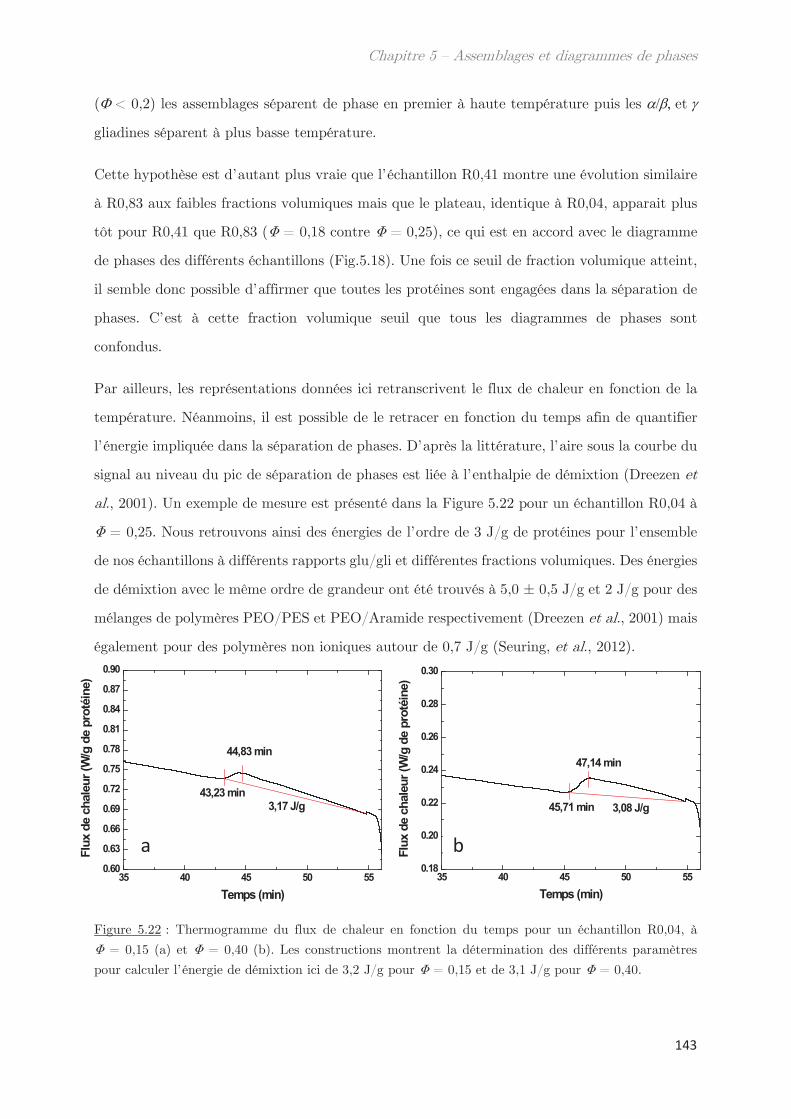
Chapitre 5 – Assemblages et diagrammes de phases
143
( < 0,2) les assemblages séparent de phase en premier à haute température puis les et
gliadines séparent à plus basse température.
Cette hypothèse est d’autant plus vraie que l’échantillon R0,41 montre une évolution similaire
à R0,83 aux faibles fractions volumiques mais que le plateau, identique à R0,04, apparait plus
tôt pour R0,41 que R0,83 ( = 0,18 contre = 0,25), ce qui est en accord avec le diagramme
de phases des différents échantillons (Fig.5.18). Une fois ce seuil de fraction volumique atteint,
il semble donc possible d’affirmer que toutes les protéines sont engagées dans la séparation de
phases. C’est à cette fraction volumique seuil que tous les diagrammes de phases sont
confondus.
Par ailleurs, les représentations données ici retranscrivent le flux de chaleur en fonction de la
température. Néanmoins, il est possible de le retracer en fonction du temps afin de quantifier
l’énergie impliquée dans la séparation de phases. D’après la littérature, l’aire sous la courbe du
signal au niveau du pic de séparation de phases est liée à l’enthalpie de démixtion (Dreezen et
al., 2001). Un exemple de mesure est présenté dans la Figure 5.22 pour un échantillon R0,04 à
= 0,25. Nous retrouvons ainsi des énergies de l’ordre de 3 J/g de protéines pour l’ensemble
de nos échantillons à différents rapports glu/gli et différentes fractions volumiques. Des énergies
de démixtion avec le même ordre de grandeur ont été trouvés à 5,0 ± 0,5 J/g et 2 J/g pour des
mélanges de polymères PEO/PES et PEO/Aramide respectivement (Dreezen et al., 2001) mais
également pour des polymères non ioniques autour de 0,7 J/g (Seuring, et al., 2012).
Figure 5.22 : Thermogramme du flux de chaleur en fonction du temps pour un échantillon R0,04, à
= 0,15 (a) et = 0,40 (b). Les constructions montrent la détermination des différents paramètres
pour calculer l’énergie de démixtion ici de 3,2 J/g pour = 0,15 et de 3,1 J/g pour = 0,40.
35 40 45 50 550.60
0.63
0.66
0.69
0.72
0.75
0.78
0.81
0.84
0.87
0.90
3,17 J/g43,23 min
44,83 min
Flu
x d
e c
hale
ur
(W/g
de p
roté
ine)
Temps (min)
35 40 45 50 550.18
0.20
0.22
0.24
0.26
0.28
0.30
3,08 J/g
47,14 min
Flu
x d
e c
hale
ur
(W/g
de p
roté
ine)
Temps (min)
45,71 min
b a
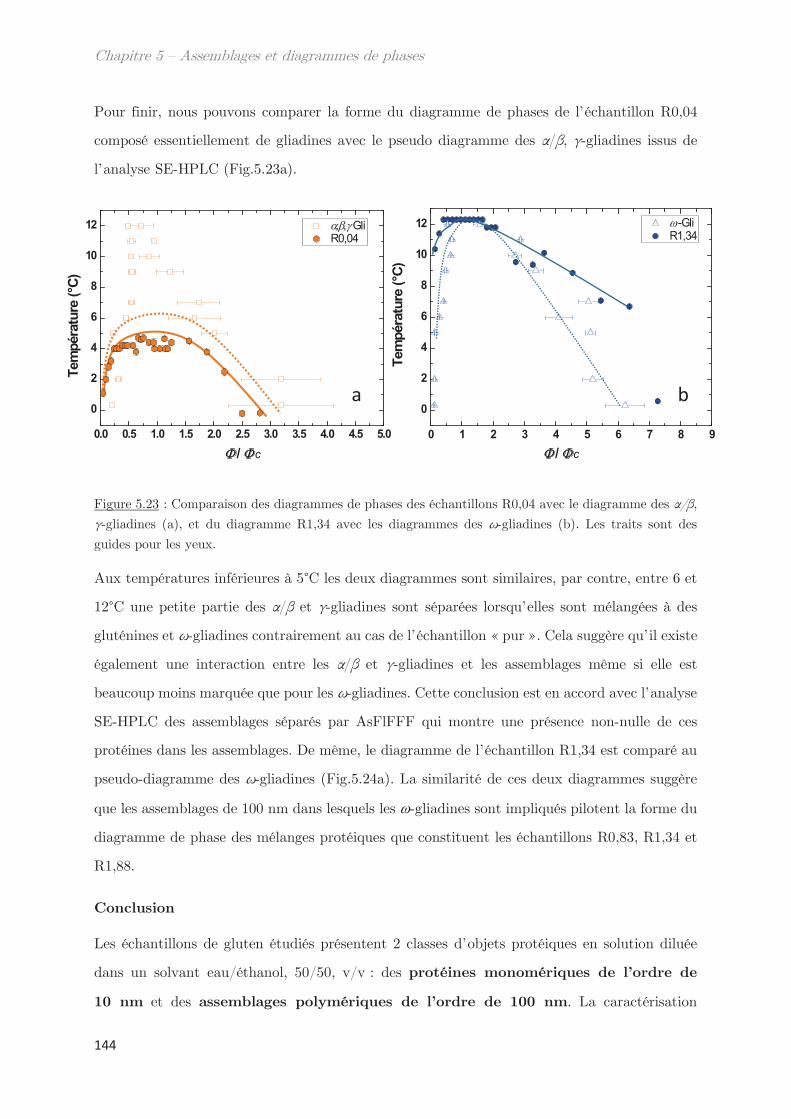
Chapitre 5 – Assemblages et diagrammes de phases
144
Pour finir, nous pouvons comparer la forme du diagramme de phases de l’échantillon R0,04
composé essentiellement de gliadines avec le pseudo diagramme des / , -gliadines issus de
l’analyse SE-HPLC (Fig.5.23a).
Figure 5.23 : Comparaison des diagrammes de phases des échantillons R0,04 avec le diagramme des / ,
-gliadines (a), et du diagramme R1,34 avec les diagrammes des -gliadines (b). Les traits sont des
guides pour les yeux.
Aux températures inférieures à 5°C les deux diagrammes sont similaires, par contre, entre 6 et
12°C une petite partie des / et -gliadines sont séparées lorsqu’elles sont mélangées à des
gluténines et -gliadines contrairement au cas de l’échantillon « pur ». Cela suggère qu’il existe
également une interaction entre les / et -gliadines et les assemblages même si elle est
beaucoup moins marquée que pour les -gliadines. Cette conclusion est en accord avec l’analyse
SE-HPLC des assemblages séparés par AsFlFFF qui montre une présence non-nulle de ces
protéines dans les assemblages. De même, le diagramme de l’échantillon R1,34 est comparé au
pseudo-diagramme des -gliadines (Fig.5.24a). La similarité de ces deux diagrammes suggère
que les assemblages de 100 nm dans lesquels les -gliadines sont impliqués pilotent la forme du
diagramme de phase des mélanges protéiques que constituent les échantillons R0,83, R1,34 et
R1,88.
Conclusion
Les échantillons de gluten étudiés présentent 2 classes d’objets protéiques en solution diluée
dans un solvant eau/éthanol, 50/50, v/v : des protéines monomériques de l’ordre de
10 nm et des assemblages polymériques de l’ordre de 100 nm. La caractérisation
0 1 2 3 4 5 6 7 8 9
0
2
4
6
8
10
12 -Gli R1,34
Tem
péra
ture
(°C
)
/ c
b
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
0
2
4
6
8
10
12 Gli R0,04
Tem
péra
ture
(°C
)
/ c
a

Chapitre 5 – Assemblages et diagrammes de phases
145
biochimique par SE-HPLC des différentes fractions collectées en AsFlFFF, a permis de mettre
en évidence que ces assemblages résultent d’interactions fortes entre -gliadines et
gluténines qui conduisent à la formation des assemblages d’une centaine de nanomètres de
rayon alors que les protéines monomériques sont majoritairement des , -gliadines. D’après
la littérature, des rayons hydrodynamiques de 8 nm et 35 nm, associées respectivement à des
gliadines et à des gluténines de faibles poids moléculaires, ont été également observés en
AsFlFFF sur du gluten en tampon SDS (Wahlund et al., 1995). Plus tard, des protéines de
farines de blé en tampon sodium de phosphate 0,1 M, (pH 6,9) contenant 2% de SDS (Lemelin
et al., 2005) ont également montré la présence de protéines monomériques (Mw < 105 g/mol)
et polymériques (105 Mw < 108 g/mol). Ainsi, les résultats de la littérature sont en accords
avec nos observations. Cependant, dans la plupart des études déjà réalisées sur le gluten, le
solvant utilisé contient du SDS alors que dans notre cas le solvant des échantillons (eau/éthanol
50/50) est moins dénaturant. L’utilisation d’un tampon SDS a prouvé, par les études
antérieures, son efficacité pour caractériser les tailles des gliadines et des gluténines mais cela
au prix d’un changement de conformation des protéines, alors que, d’après nos mesures, le
solvant eau/éthanol conserve mieux la structure des protéines. En effet, ce dernier permet non
seulement de mettre en évidence les tailles caractéristiques des sous classes de protéines mais
également de montrer les assemblages entre -gliadines et gluténines.
D’autre part, ces assemblages ont également été confirmés par l’étude des diagrammes de
phases qui montrent des résultats très proches pour les -gliadines et les gluténines. Néanmoins,
la présence de ces assemblages n’exclue pas totalement les interactions avec les autres gliadines.
L’existence de ces assemblages de forte masse molaire conduit à un comportement de séparation
de phase des assemblages bien distinct des protéines monomériques, mis à profit dans notre
protocole de préparation des différents extraits au rapport glu/gli contrasté. En effet, au cours
de la séparation de phases, chaque classe de protéines est séparée différemment
(Fig.5.24). De façon similaire aux observations faites sur des solutions de polystyrène (Daoud
et al., 1976), il est possible d’établir des diagrammes de phases différents en fonction de la
masse molaire des polymères. D’après le schéma de Daoud et al., lorsque N (le nombre de
monomères par chaîne) augmente, la température critique de séparation de phase augmente
alors que la concentration critique diminue. Il en est de même pour les diagrammes des gliadines
et des assemblages qui nous permettent d’expliquer la répartition des différentes protéines au
cours de notre protocole d’extraction et ainsi l’obtention des extraits à différents rapports
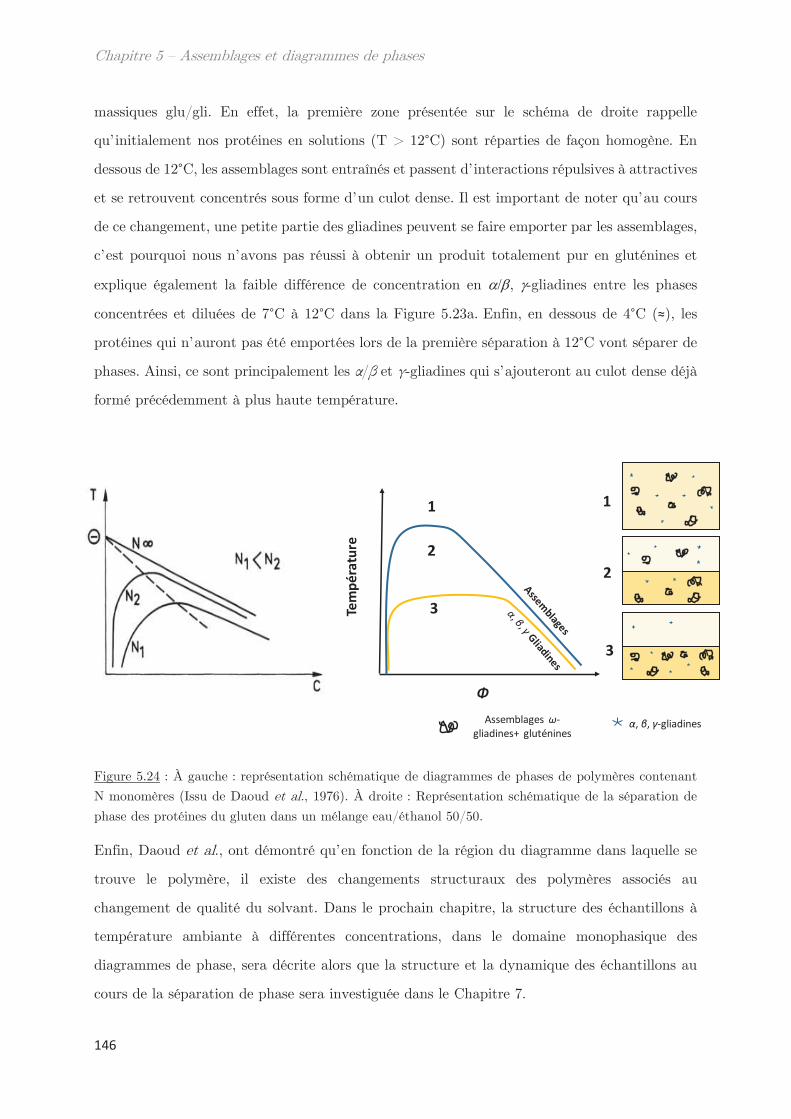
Chapitre 5 – Assemblages et diagrammes de phases
146
massiques glu/gli. En effet, la première zone présentée sur le schéma de droite rappelle
qu’initialement nos protéines en solutions (T > 12°C) sont réparties de façon homogène. En
dessous de 12°C, les assemblages sont entraînés et passent d’interactions répulsives à attractives
et se retrouvent concentrés sous forme d’un culot dense. Il est important de noter qu’au cours
de ce changement, une petite partie des gliadines peuvent se faire emporter par les assemblages,
c’est pourquoi nous n’avons pas réussi à obtenir un produit totalement pur en gluténines et
explique également la faible différence de concentration en , -gliadines entre les phases
concentrées et diluées de 7°C à 12°C dans la Figure 5.23a. Enfin, en dessous de 4°C ( ), les
protéines qui n’auront pas été emportées lors de la première séparation à 12°C vont séparer de
phases. Ainsi, ce sont principalement les / et -gliadines qui s’ajouteront au culot dense déjà
formé précédemment à plus haute température.
Figure 5.24 : À gauche : représentation schématique de diagrammes de phases de polymères contenant
N monomères (Issu de Daoud et al., 1976). À droite : Représentation schématique de la séparation de
phase des protéines du gluten dans un mélange eau/éthanol 50/50.
Enfin, Daoud et al., ont démontré qu’en fonction de la région du diagramme dans laquelle se
trouve le polymère, il existe des changements structuraux des polymères associés au
changement de qualité du solvant. Dans le prochain chapitre, la structure des échantillons à
température ambiante à différentes concentrations, dans le domaine monophasique des
diagrammes de phase, sera décrite alors que la structure et la dynamique des échantillons au
cours de la séparation de phase sera investiguée dans le Chapitre 7.
3
1
v v
2 vv
v
v
vAssemblages -
gliadines+ gluténines-gliadines
Tem
pé
ratu
re
1
2
3

Chapitre 5 – Assemblages et diagrammes de phases
147
Bibliographie
Carceller, J. L., & Aussenac, T. (2001). Size characterisation of glutenin polymers by HPSEC-
MALLS. Journal of Cereal Science, 33(2), 131–142.
Dahesh, M., Banc, A., Duri, A., Morel, M. H., & Ramos, L. (2014). Polymeric assembly of
gluten proteins in an aqueous ethanol solvent. Journal of Physical Chemistry B, 118(38),
11065–11076.
Daoud, M., & Jannink G. (1976). Temperature-Concentration Diagram of polymer solutions.
Le Journal de Physique, 37, 973–979.
Dreezen, G., Groeninckx, G., Swier, S., & Van Mele, B. (2001). Phase separation in miscible
polymer blends as detected by modulated temperature differential scanning calorimetry.
Polymer, 42(4), 1449–1459.
Lemelin, E., Aussenac, T., Violleau, F., Salvo, L., & Lein, V. (2005). Impact of cultivar and
environment on size characteristics of wheat proteins using asymmetrical flow field-flow
fractionation and multi-angle laser light scattering. Cereal Chemistry, 82(1), 28–33.
Mendichi, R., Fisichella, S., & Savarino, A. (2008). Molecular weight, size distribution and
conformation of Glutenin from different wheat cultivars by SEC e MALLS, 48, 486–493.
Seuring, J., Bayer, F. M., Huber, K., & Agarwal, S. (2012). Upper Critical Solution
Temperature of Poly( N -acryloyl glycinamide) in Water: A Concealed Property.
Macromolecules, 45(1), 374–384.
Smidsrod O. (1970). Solution Properties of Alginate. Carbohydrate Research, 13, 359–372.
Thomson, N. H., Miles, M. J., Popineau, Y., Harries, J., Shewry, P., & Tatham, A. S. (1999).
Small angle X-ray scattering of wheat seed-storage proteins: alpha-, gamma- and omega-
gliadins and the high molecular weight (HMW) subunits of glutenin. Biochimica et
Biophysica Acta, 1430(2), 359–366.

Chapitre 5 – Assemblages et diagrammes de phases
148
Yang, C., Meng, B., Chen, M., Liu, X., Hua, Y., & Ni, Z. (2006). Laser-light-scattering study
of structure and dynamics of waxy corn amylopectin in dilute aqueous solution, 64, 190–
196.
Wahlund, K.-G., Gustavsson, M., MacRitchie, F., Nylander, T., & Wannerberger, L. (1996).
Size Characterisation of Wheat Proteins, Particularly Glutenin, by Asymmetrical Flow
Field-Flow Fractionation. J. Cereal Sci., 23, 113–119.

Chapitre 6 – Rhéologie et structure
149
Chapitre 6
Impact de la composition protéique sur la
rhéologie et la structure d’échantillons dilués et
semi-dilués à température ambiante
1. Observations macroscopiques
Pour l’ensemble des expérimentations présentées dans le Chapitre 5, les extraits de gluten à
différents rapports glu/gli (0,15 R 1,88) ont été dissous en solutions diluées ( = 0,0038)
dans un solvant eau/éthanol 50/50% v/v puis filtrés sur des filtres à seringues, de cellulose
régénérée, 0,8 µm. Comme le témoigne la photo de la Figure 6.1, dès leur mise en solution, les
extraits présentent une turbidité différente.
Figure 6.1 : Photos représentant des solutions de gluten à différents rapports glu/gli à = 0,0038 après
filtration.
R = 0,15 R = 0,83 R = 1,34 R = 1,88
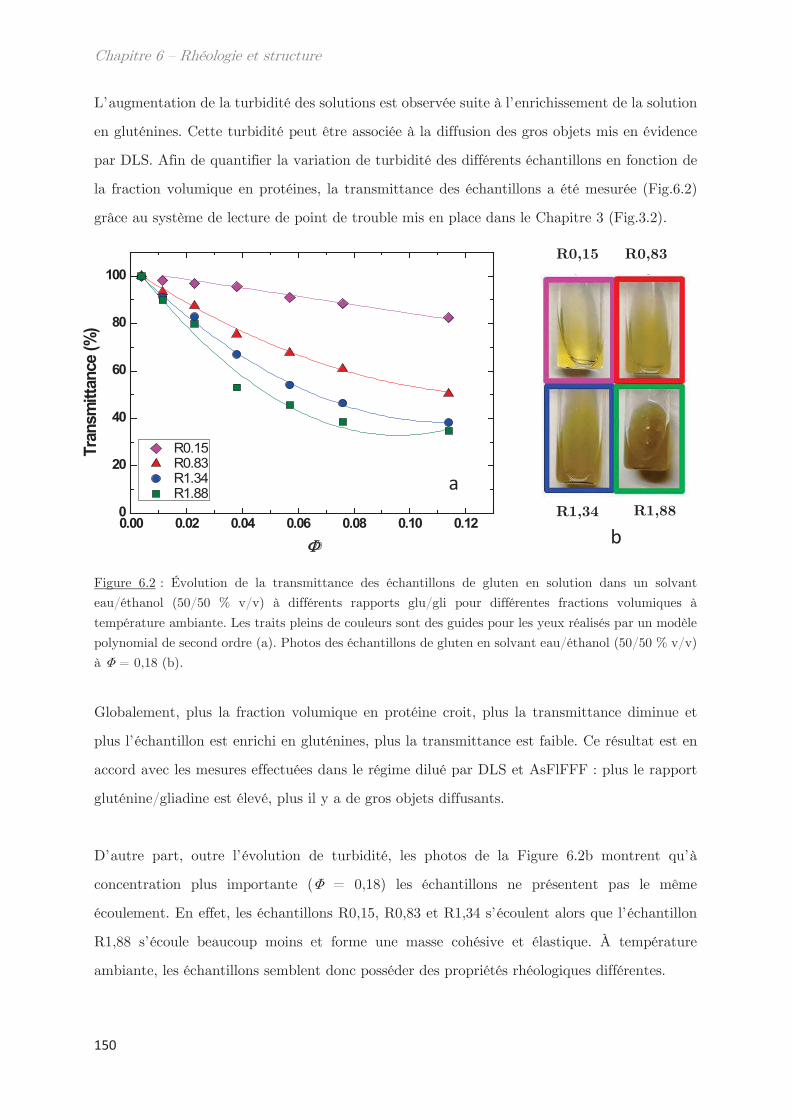
Chapitre 6 – Rhéologie et structure
150
L’augmentation de la turbidité des solutions est observée suite à l’enrichissement de la solution
en gluténines. Cette turbidité peut être associée à la diffusion des gros objets mis en évidence
par DLS. Afin de quantifier la variation de turbidité des différents échantillons en fonction de
la fraction volumique en protéines, la transmittance des échantillons a été mesurée (Fig.6.2)
grâce au système de lecture de point de trouble mis en place dans le Chapitre 3 (Fig.3.2).
Figure 6.2 : Évolution de la transmittance des échantillons de gluten en solution dans un solvant
eau/éthanol (50/50 % v/v) à différents rapports glu/gli pour différentes fractions volumiques à
température ambiante. Les traits pleins de couleurs sont des guides pour les yeux réalisés par un modèle
polynomial de second ordre (a). Photos des échantillons de gluten en solvant eau/éthanol (50/50 % v/v)
à = 0,18 (b).
Globalement, plus la fraction volumique en protéine croit, plus la transmittance diminue et
plus l’échantillon est enrichi en gluténines, plus la transmittance est faible. Ce résultat est en
accord avec les mesures effectuées dans le régime dilué par DLS et AsFlFFF : plus le rapport
gluténine/gliadine est élevé, plus il y a de gros objets diffusants.
D’autre part, outre l’évolution de turbidité, les photos de la Figure 6.2b montrent qu’à
concentration plus importante ( = 0,18) les échantillons ne présentent pas le même
écoulement. En effet, les échantillons R0,15, R0,83 et R1,34 s’écoulent alors que l’échantillon
R1,88 s’écoule beaucoup moins et forme une masse cohésive et élastique. À température
ambiante, les échantillons semblent donc posséder des propriétés rhéologiques différentes.
b
a
R0,15 R0,83
R1,34 R1,88 0.00 0.02 0.04 0.06 0.08 0.10 0.120
20
40
60
80
100
Tra
nsm
itta
nce (%
)
R0.15 R0.83 R1.34 R1.88
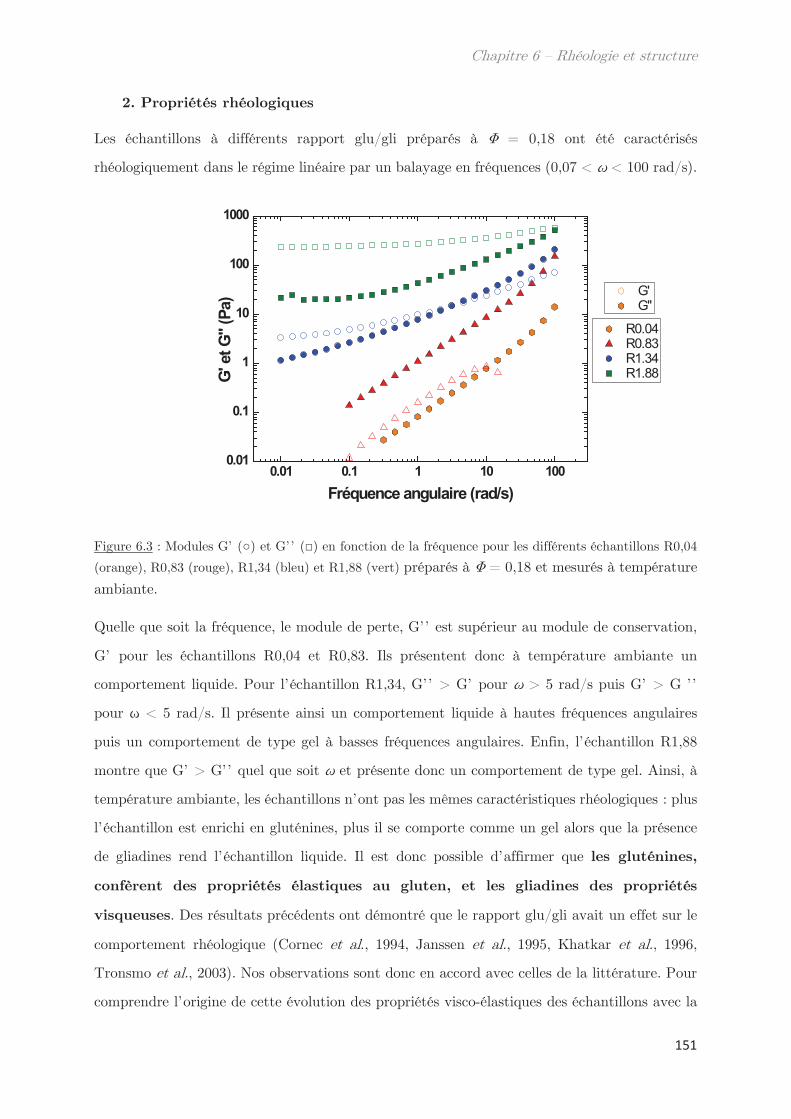
Chapitre 6 – Rhéologie et structure
151
2. Propriétés rhéologiques
Les échantillons à différents rapport glu/gli préparés à = 0,18 ont été caractérisés
rhéologiquement dans le régime linéaire par un balayage en fréquences (0,07 < < 100 rad/s).
Figure 6.3 : Modules G’ ( ) et G’’ ( ) en fonction de la fréquence pour les différents échantillons R0,04
(orange), R0,83 (rouge), R1,34 (bleu) et R1,88 (vert) préparés à = 0,18 et mesurés à température
ambiante.
Quelle que soit la fréquence, le module de perte, G’’ est supérieur au module de conservation,
G’ pour les échantillons R0,04 et R0,83. Ils présentent donc à température ambiante un
comportement liquide. Pour l’échantillon R1,34, G’’ > G’ pour > 5 rad/s puis G’ > G ’’
< 5 rad/s. Il présente ainsi un comportement liquide à hautes fréquences angulaires
puis un comportement de type gel à basses fréquences angulaires. Enfin, l’échantillon R1,88
montre que G’ > G’’ quel que soit et présente donc un comportement de type gel. Ainsi, à
température ambiante, les échantillons n’ont pas les mêmes caractéristiques rhéologiques : plus
l’échantillon est enrichi en gluténines, plus il se comporte comme un gel alors que la présence
de gliadines rend l’échantillon liquide. Il est donc possible d’affirmer que les gluténines,
confèrent des propriétés élastiques au gluten, et les gliadines des propriétés
visqueuses. Des résultats précédents ont démontré que le rapport glu/gli avait un effet sur le
comportement rhéologique (Cornec et al., 1994, Janssen et al., 1995, Khatkar et al., 1996,
Tronsmo et al., 2003). Nos observations sont donc en accord avec celles de la littérature. Pour
comprendre l’origine de cette évolution des propriétés visco-élastiques des échantillons avec la
0.01 0.1 1 10 1000.01
0.1
1
10
100
1000
G' G"
R0.04 R0.83 R1.34 R1.88G
' et G
" (P
a)
Fréquence angulaire (rad/s)
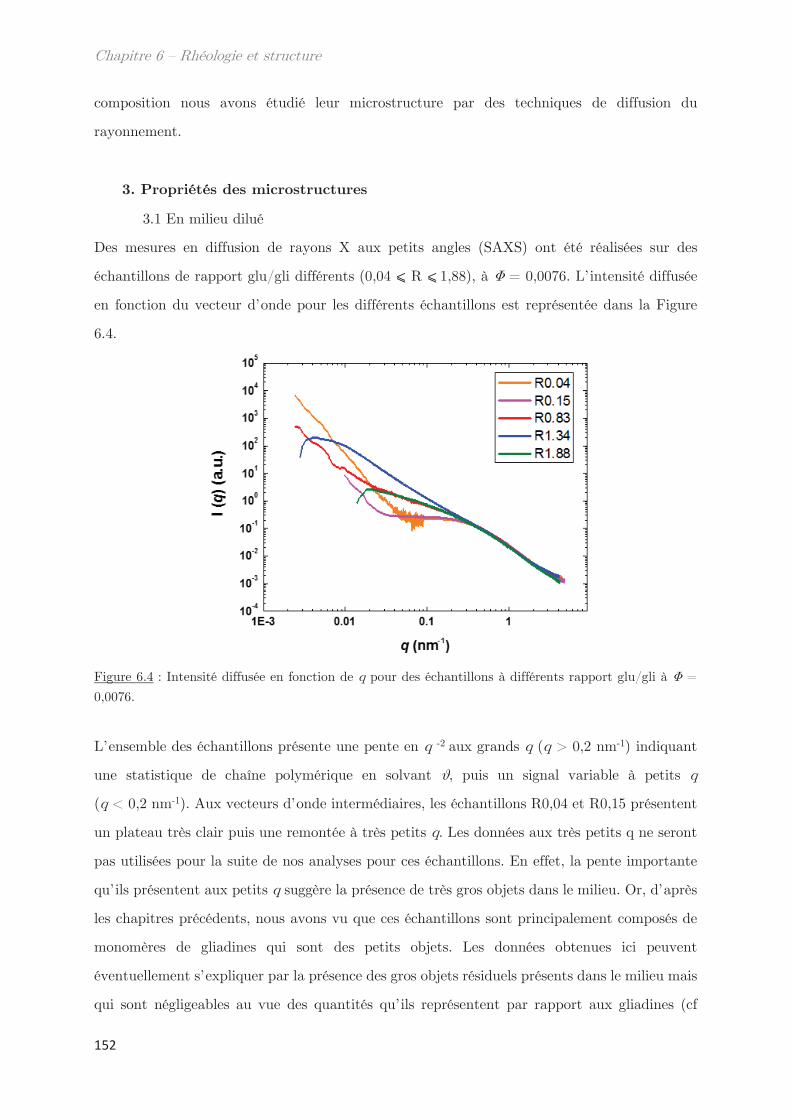
Chapitre 6 – Rhéologie et structure
152
composition nous avons étudié leur microstructure par des techniques de diffusion du
rayonnement.
3. Propriétés des microstructures
3.1 En milieu dilué
Des mesures en diffusion de rayons X aux petits angles (SAXS) ont été réalisées sur des
échantillons de rapport glu/gli différents (0,04 R 1,88), à = 0,0076. L’intensité diffusée
en fonction du vecteur d’onde pour les différents échantillons est représentée dans la Figure
6.4.
Figure 6.4 : Intensité diffusée en fonction de q pour des échantillons à différents rapport glu/gli à =
0,0076.
L’ensemble des échantillons présente une pente en q -2 aux grands q (q > 0,2 nm-1) indiquant
une statistique de chaîne polymérique en solvant , puis un signal variable à petits q
(q < 0,2 nm-1). Aux vecteurs d’onde intermédiaires, les échantillons R0,04 et R0,15 présentent
un plateau très clair puis une remontée à très petits q. Les données aux très petits q ne seront
pas utilisées pour la suite de nos analyses pour ces échantillons. En effet, la pente importante
qu’ils présentent aux petits q suggère la présence de très gros objets dans le milieu. Or, d’après
les chapitres précédents, nous avons vu que ces échantillons sont principalement composés de
monomères de gliadines qui sont des petits objets. Les données obtenues ici peuvent
éventuellement s’expliquer par la présence des gros objets résiduels présents dans le milieu mais
qui sont négligeables au vue des quantités qu’ils représentent par rapport aux gliadines (cf

Chapitre 6 – Rhéologie et structure
153
Chapitre 4 et 5). C’est pourquoi nous limiterons l’analyse des données de ces échantillons à
q > 0,1 nm-1.
Les échantillons enrichis en gluténines (R0,83, R1,34 et R1,88) ne présentent pas de plateau
dans la gamme de q mesurée. Cela peut s’expliquer par la présence de gros objets que sont les
assemblages de gluténines + -gliadines précédemment identifiés en AsFlFFF (Chapitre 5). En
effet, pour des objets de 100 nm de rayon, le plateau serait attendu à q < 10-3 nm-1, gamme
non couverte par la technique. Cependant, une évolution croissante de l’intensité diffusée à
petits q avec le rapport gluténine/gliadine serait attendue avec l’augmentation du nombre
d’assemblages protéiques, mais n’est pas observée avec l’échantillon R1,88. Cela peut être dû
à la filtration des échantillons dilués qui n’est pas vraiment maitrisée, comme vu au Chapitre
2.
Seuls les échantillons enrichis en gliadines (R0,04 et R0,15) sont ajustés sur une gamme
0,1 < q < 1 nm-1 par un modèle de Debye, qui est le facteur de forme des chaînes gaussiennes
(Pedersen et al., 2004) dont l’équation est définie par :
( ) = 2( )
² (5.1)
Avec x = (q.Rg), Rg étant le rayon de giration et I0 l’intensité diffusée à très petits q.
Un exemple d’ajustement de ce modèle est présenté dans la Figure 6.5.
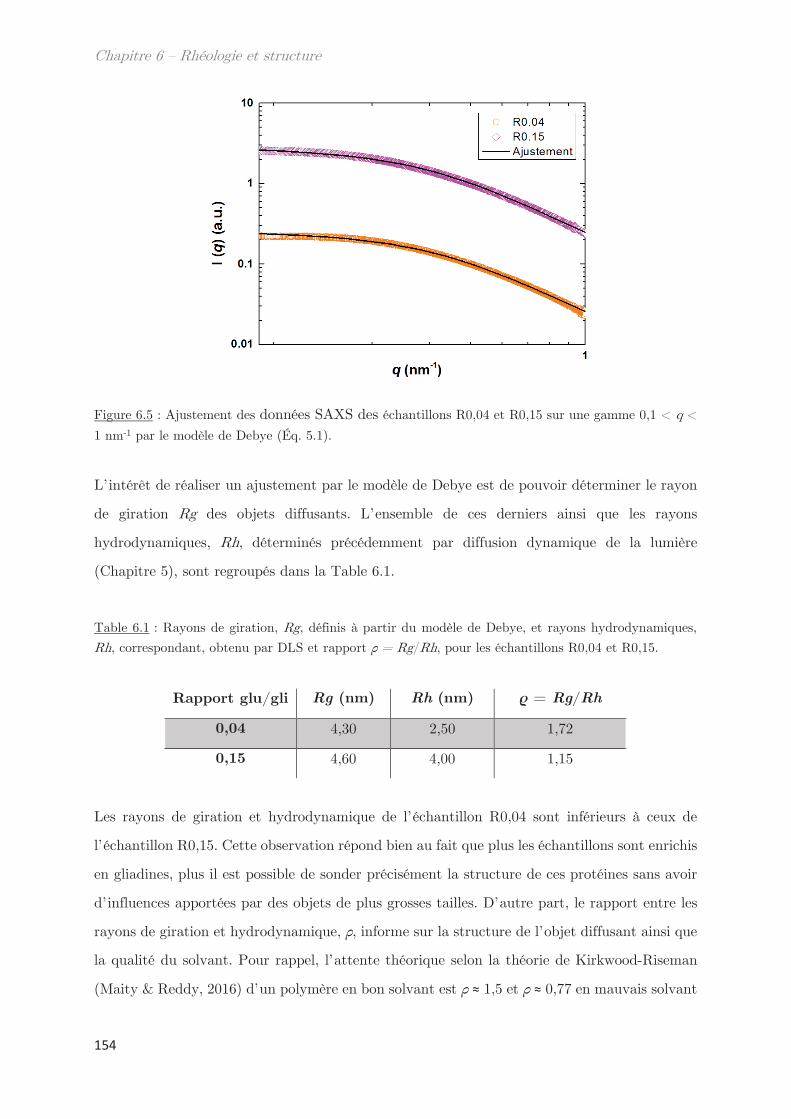
Chapitre 6 – Rhéologie et structure
154
Figure 6.5 : Ajustement des données SAXS des échantillons R0,04 et R0,15 sur une gamme 0,1 < q <
1 nm-1 par le modèle de Debye (Éq. 5.1).
L’intérêt de réaliser un ajustement par le modèle de Debye est de pouvoir déterminer le rayon
de giration Rg des objets diffusants. L’ensemble de ces derniers ainsi que les rayons
hydrodynamiques, Rh, déterminés précédemment par diffusion dynamique de la lumière
(Chapitre 5), sont regroupés dans la Table 6.1.
Table 6.1 : Rayons de giration, Rg, définis à partir du modèle de Debye, et rayons hydrodynamiques,
Rh, correspondant, obtenu par DLS et rapport = Rg/Rh, pour les échantillons R0,04 et R0,15.
Rapport glu/gli Rg (nm) Rh (nm) = Rg/Rh
0,04 4,30 2,50 1,72
0,15 4,60 4,00 1,15
Les rayons de giration et hydrodynamique de l’échantillon R0,04 sont inférieurs à ceux de
l’échantillon R0,15. Cette observation répond bien au fait que plus les échantillons sont enrichis
en gliadines, plus il est possible de sonder précisément la structure de ces protéines sans avoir
d’influences apportées par des objets de plus grosses tailles. D’autre part, le rapport entre les
rayons de giration et hydrodynamique, , informe sur la structure de l’objet diffusant ainsi que
la qualité du solvant. Pour rappel, l’attente théorique selon la théorie de Kirkwood-Riseman
(Maity & Reddy, 2016) d’un polymère en bon solvant est 1,5 et 0,77 en mauvais solvant

Chapitre 6 – Rhéologie et structure
155
(Chapitre 1). Dans notre cas, deux valeurs de différentes sont obtenues pour les échantillons
R0,04 et R0,15. Cependant ces valeurs ne sont pas significativement différentes étant donné
l’incertitude qu’il y a sur la détermination du rayon hydrodynamique par DLS (notamment en
utilisant un modèle de double exponentielle). Nous pouvons simplement conclure que ces
valeurs de sont compatibles avec celles attendues pour un polymère en solvant , trouvées
également sur du poly(methyl methacrylate), ( = 1,16), selon Schmidt et al., 1981 et = 1,78
pour un polymère linéaire en solvant selon Burchard W., 1999).
D’autre part, la connaissance du rayon de giration, Rg, permet de remonter à C*, la
concentration critique de recouvrement, qui représente le passage du régime dilué au régime
semi-dilué (Ying et al., 1987). Au-delà de la concentration critique, les chaînes de polymères
commencent à s’enchevêtrer et à se recouvrir pour former un réseau.
Cette concentration, C*, est définie par :
C* =
(5.4)
où Mw est la masse molaire et Na le nombre d’Avogadro. Dans notre cas, Mw 40 000 g/mol
ce qui permet de déduire que la concentration limite de recouvrement est égale à 199,4 g/l
soit * = 0,15 avec Rg = 4,3 nm et 162,9 g/l soit * = 0,12 avec Rg = 4,6 nm. Cette
approximation confirme que les solutions étudiées ici appartiennent au régime dilué.
3.2 En milieu semi-dilué
3.2.1 Diffusion de rayons X
Les solutions de gluten R0,04 à R1,88 ont été mesurées en SAXS, à différentes fractions
volumiques sur une gamme 0,038 < < 0,304 en solvant eau/éthanol 50/50 (v/v). L’ensemble
des résultats pour chaque rapport est présenté dans la Figure 6.6.
Pour tous les échantillons, il existe deux échelles bien distinctes : à petits q (q < 0,9 nm-1)
l’intensité normalisée par la fraction volumique diminue avec l’augmentation de la fraction
volumique. Cette évolution peut être attribuée au facteur de structure, S(q). Pour rappel,
l’intensité mesurée répond à l’équation I(q) (cm-1) = )². . V. P(q). S(q) avec le
contraste de diffusion, , la fraction volumique des objets dans le milieu, V, le volume de

Chapitre 6 – Rhéologie et structure
156
l’objet, et P(q), le facteur de forme. De plus, S(q) = 1 en milieu dilué et S(q) 1 dans des
milieux concentrés. À grands q (0,9 < q < 5 nm-1), l’évolution est la même quelle que soit la
fraction volumique. À petite échelle, l’intensité du signal est donc simplement proportionnelle
à la fraction volumique des échantillons, ce qui permet d’affirmer que la structure locale des
échantillons est identique.
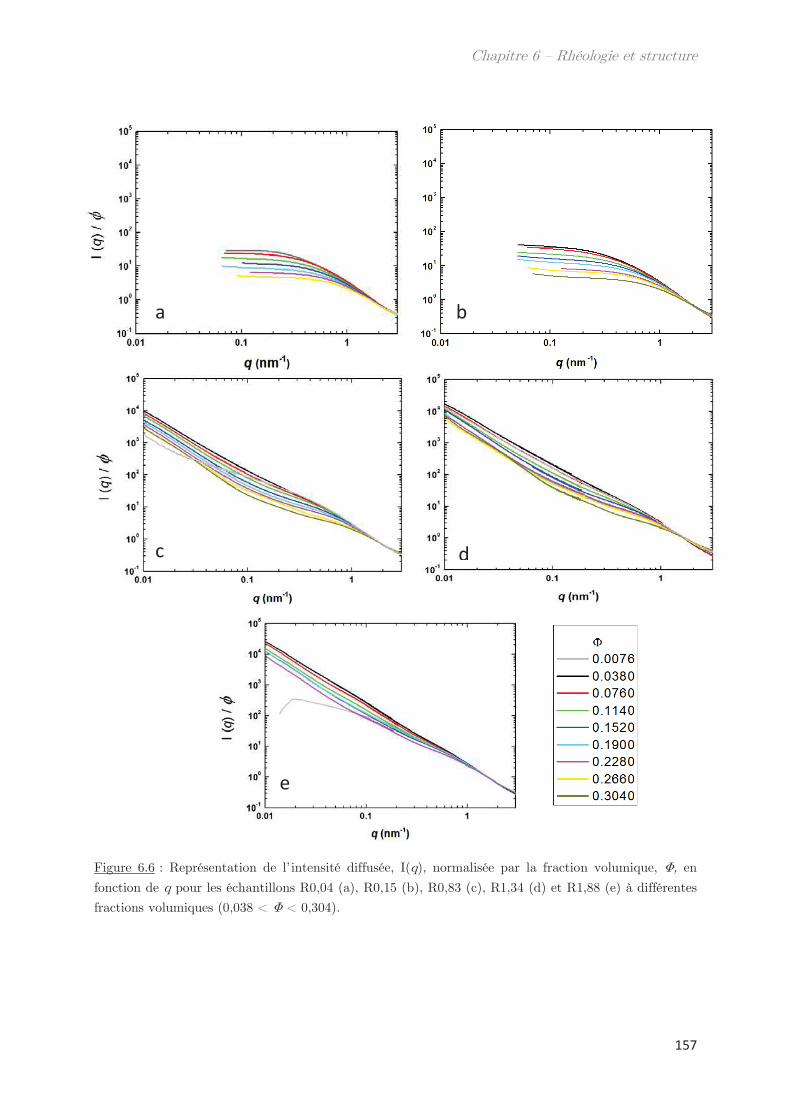
Chapitre 6 – Rhéologie et structure
157
Figure 6.6 : Représentation de l’intensité diffusée, I(q), normalisée par la fraction volumique, , en
fonction de q pour les échantillons R0,04 (a), R0,15 (b), R0,83 (c), R1,34 (d) et R1,88 (e) à différentes
fractions volumiques (0,038 < < 0,304).
a b
c d
e
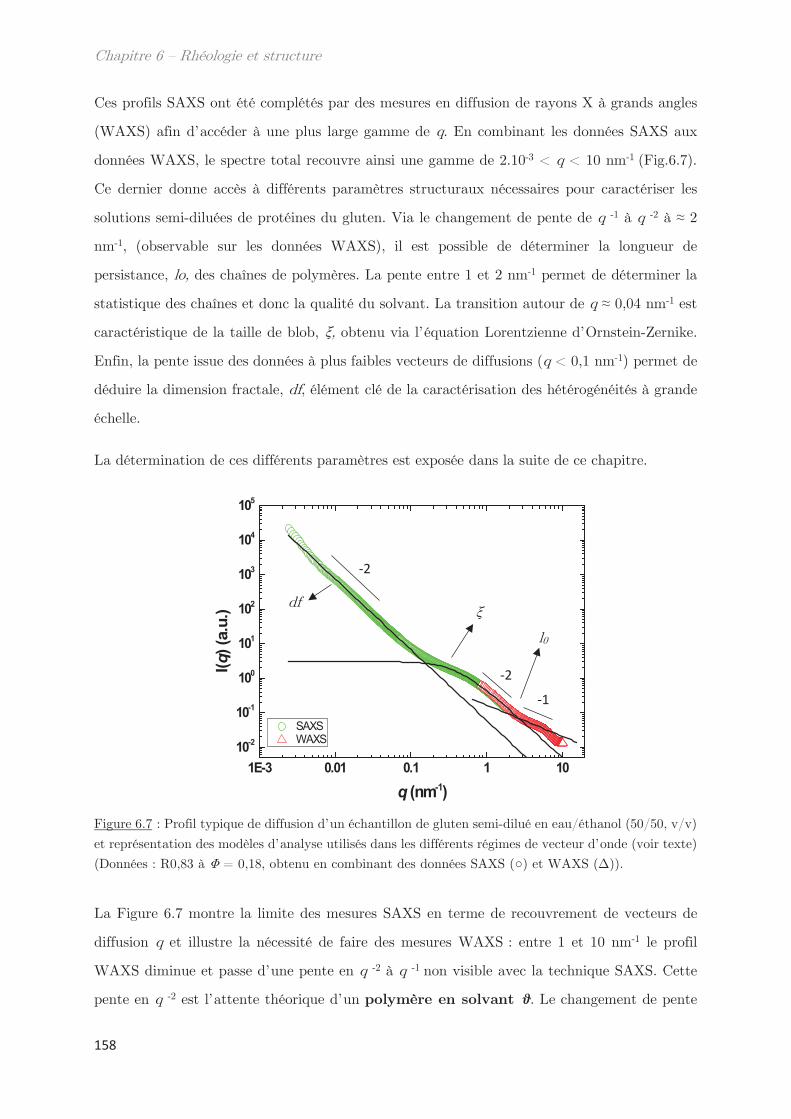
Chapitre 6 – Rhéologie et structure
158
Ces profils SAXS ont été complétés par des mesures en diffusion de rayons X à grands angles
(WAXS) afin d’accéder à une plus large gamme de q. En combinant les données SAXS aux
données WAXS, le spectre total recouvre ainsi une gamme de 2.10-3 < q < 10 nm-1 (Fig.6.7).
Ce dernier donne accès à différents paramètres structuraux nécessaires pour caractériser les
solutions semi-diluées de protéines du gluten. Via le changement de pente de q -1 à q -2 à 2
nm-1, (observable sur les données WAXS), il est possible de déterminer la longueur de
persistance, lo, des chaînes de polymères. La pente entre 1 et 2 nm-1 permet de déterminer la
statistique des chaînes et donc la qualité du solvant. La transition autour de q 0,04 nm-1 est
caractéristique de la taille de blob, , obtenu via l’équation Lorentzienne d’Ornstein-Zernike.
Enfin, la pente issue des données à plus faibles vecteurs de diffusions (q < 0,1 nm-1) permet de
déduire la dimension fractale, df, élément clé de la caractérisation des hétérogénéités à grande
échelle.
La détermination de ces différents paramètres est exposée dans la suite de ce chapitre.
Figure 6.7 : Profil typique de diffusion d’un échantillon de gluten semi-dilué en eau/éthanol (50/50, v/v)
et représentation des modèles d’analyse utilisés dans les différents régimes de vecteur d’onde (voir texte)
(Données : R0,83 à = 0,18, obtenu en combinant des données SAXS (
La Figure 6.7 montre la limite des mesures SAXS en terme de recouvrement de vecteurs de
diffusion q et illustre la nécessité de faire des mesures WAXS : entre 1 et 10 nm-1 le profil
WAXS diminue et passe d’une pente en q -2 à q -1 non visible avec la technique SAXS. Cette
pente en q -2 est l’attente théorique d’un polymère en solvant . Le changement de pente
1E-3 0.01 0.1 1 10
10-2
10-1
100
101
102
103
104
105
SAXS WAXS
I(q) (a
.u.)
q (nm-1)
-2
-1
df
l0
-2
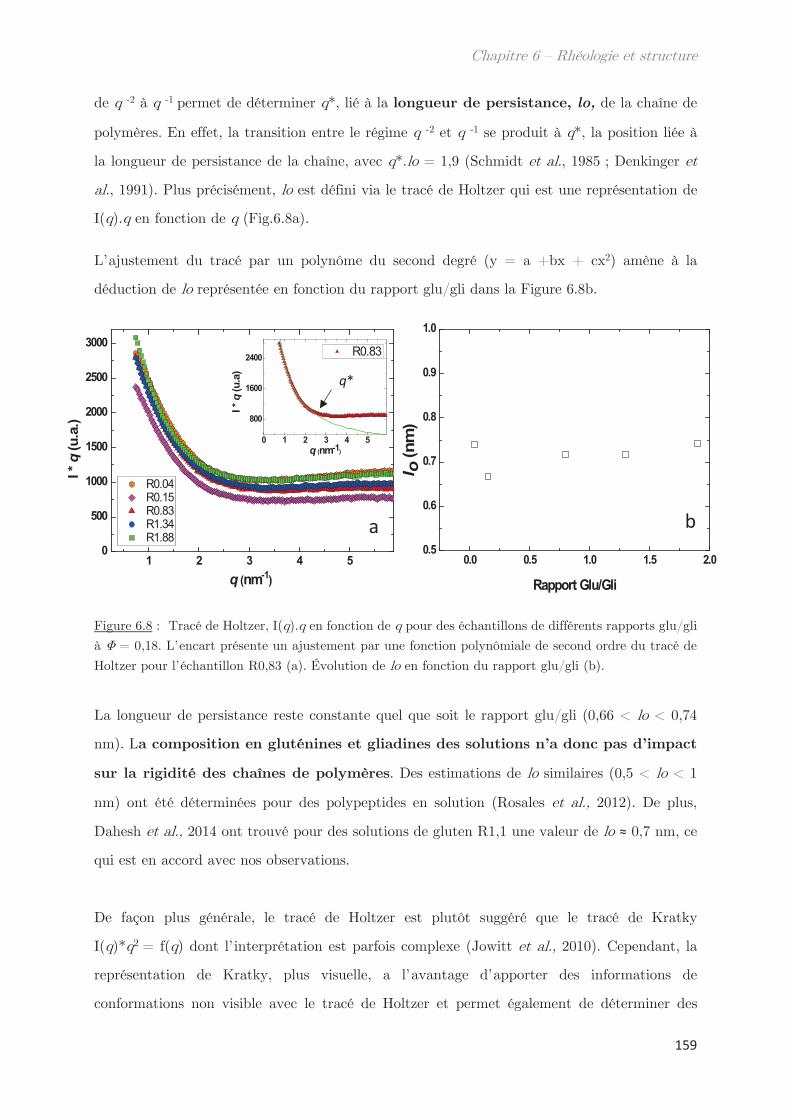
Chapitre 6 – Rhéologie et structure
159
de q -2 à q -1 permet de déterminer q*, lié à la longueur de persistance, lo, de la chaîne de
polymères. En effet, la transition entre le régime q -2 et q -1 se produit à q*, la position liée à
la longueur de persistance de la chaîne, avec q*.lo = 1,9 (Schmidt et al., 1985 ; Denkinger et
al., 1991). Plus précisément, lo est défini via le tracé de Holtzer qui est une représentation de
I(q).q en fonction de q (Fig.6.8a).
L’ajustement du tracé par un polynôme du second degré (y = a +bx + cx2) amène à la
déduction de lo représentée en fonction du rapport glu/gli dans la Figure 6.8b.
Figure 6.8 : Tracé de Holtzer, I(q).q en fonction de q pour des échantillons de différents rapports glu/gli
à = 0,18. L’encart présente un ajustement par une fonction polynômiale de second ordre du tracé de
Holtzer pour l’échantillon R0,83 (a). Évolution de lo en fonction du rapport glu/gli (b).
La longueur de persistance reste constante quel que soit le rapport glu/gli (0,66 < lo < 0,74
nm). La composition en gluténines et gliadines des solutions n’a donc pas d’impact
sur la rigidité des chaînes de polymères. Des estimations de lo similaires (0,5 < lo < 1
nm) ont été déterminées pour des polypeptides en solution (Rosales et al., 2012). De plus,
Dahesh et al., 2014 ont trouvé pour des solutions de gluten R1,1 une valeur de lo 0,7 nm, ce
qui est en accord avec nos observations.
De façon plus générale, le tracé de Holtzer est plutôt suggéré que le tracé de Kratky
I(q)*q2 = f(q) dont l’interprétation est parfois complexe (Jowitt et al., 2010). Cependant, la
représentation de Kratky, plus visuelle, a l’avantage d’apporter des informations de
conformations non visible avec le tracé de Holtzer et permet également de déterminer des
a b
q*
1 2 3 4 50
500
1000
1500
2000
2500
3000
R0.04 R0.15 R0.83 R1.34 R1.88
I *
q (u.a
.)
q (nm-1)
0 1 2 3 4 5
800
1600
2400
I *
q (u.a
)
R0.83
q (nm-1)
0.0 0.5 1.0 1.5 2.00.5
0.6
0.7
0.8
0.9
1.0
I o (nm
)
Rapport Glu/Gli
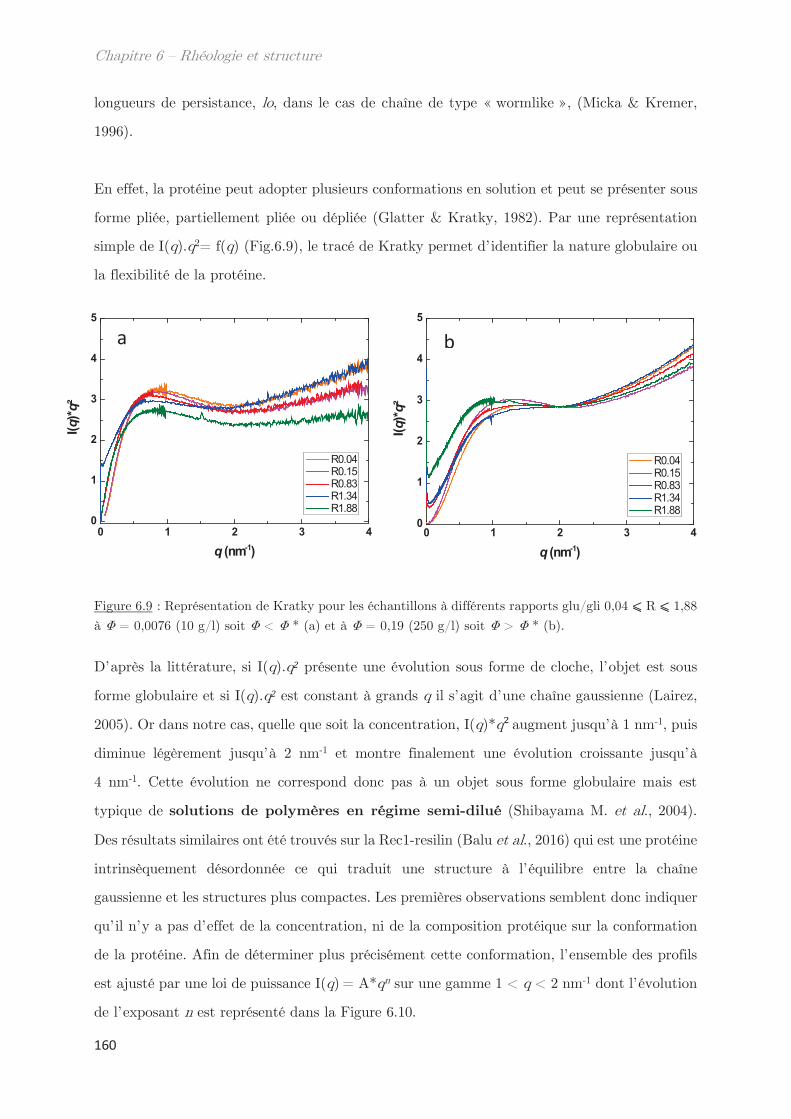
Chapitre 6 – Rhéologie et structure
160
longueurs de persistance, lo, dans le cas de chaîne de type « wormlike », (Micka & Kremer,
1996).
En effet, la protéine peut adopter plusieurs conformations en solution et peut se présenter sous
forme pliée, partiellement pliée ou dépliée (Glatter & Kratky, 1982). Par une représentation
simple de I(q).q2= f(q) (Fig.6.9), le tracé de Kratky permet d’identifier la nature globulaire ou
la flexibilité de la protéine.
Figure 6.9 : Représentation de Kratky pour les échantillons à différents rapports glu/gli 0,04 R 1,88
à = 0,0076 (10 g/l) soit < * (a) et à = 0,19 (250 g/l) soit > * (b).
D’après la littérature, si I(q).q2 présente une évolution sous forme de cloche, l’objet est sous
forme globulaire et si I(q).q2 est constant à grands q il s’agit d’une chaîne gaussienne (Lairez,
2005). Or dans notre cas, quelle que soit la concentration, I(q)*q² augment jusqu’à 1 nm-1, puis
diminue légèrement jusqu’à 2 nm-1 et montre finalement une évolution croissante jusqu’à
4 nm-1. Cette évolution ne correspond donc pas à un objet sous forme globulaire mais est
typique de solutions de polymères en régime semi-dilué (Shibayama M. et al., 2004).
Des résultats similaires ont été trouvés sur la Rec1-resilin (Balu et al., 2016) qui est une protéine
intrinsèquement désordonnée ce qui traduit une structure à l’équilibre entre la chaîne
gaussienne et les structures plus compactes. Les premières observations semblent donc indiquer
qu’il n’y a pas d’effet de la concentration, ni de la composition protéique sur la conformation
de la protéine. Afin de déterminer plus précisément cette conformation, l’ensemble des profils
est ajusté par une loi de puissance I(q) = A*qn sur une gamme 1 < q < 2 nm-1 dont l’évolution
de l’exposant n est représenté dans la Figure 6.10.
a b
0 1 2 3 40
1
2
3
4
5
I(q)*
q²
q (nm-1)
R0.04 R0.15 R0.83 R1.34 R1.88
0 1 2 3 40
1
2
3
4
5
I(q)*
q²
q (nm-1)
R0.04 R0.15 R0.83 R1.34 R1.88
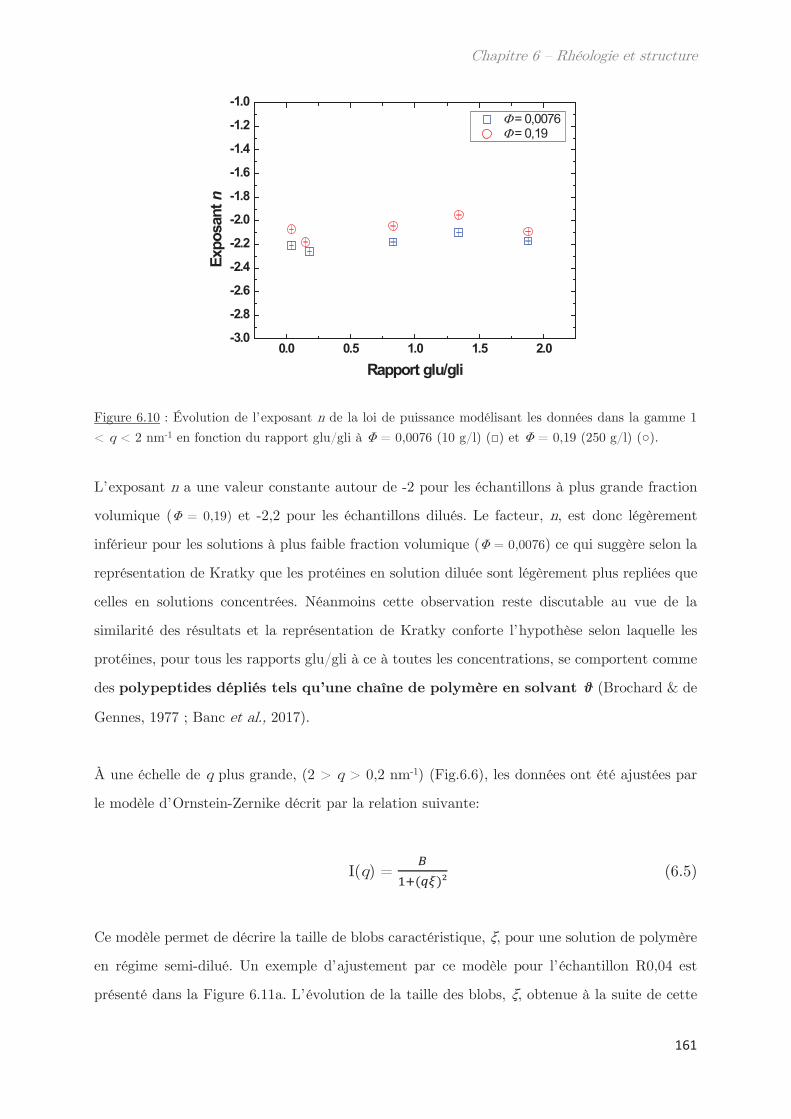
Chapitre 6 – Rhéologie et structure
161
Figure 6.10 : Évolution de l’exposant n de la loi de puissance modélisant les données dans la gamme 1
< q < 2 nm-1 en fonction du rapport glu/gli à = 0,0076 (10 g/l) ( ) et = 0,19 (250 g/l) ( ).
L’exposant n a une valeur constante autour de -2 pour les échantillons à plus grande fraction
volumique ( = 0,19) et -2,2 pour les échantillons dilués. Le facteur, n, est donc légèrement
inférieur pour les solutions à plus faible fraction volumique ( = 0,0076) ce qui suggère selon la
représentation de Kratky que les protéines en solution diluée sont légèrement plus repliées que
celles en solutions concentrées. Néanmoins cette observation reste discutable au vue de la
similarité des résultats et la représentation de Kratky conforte l’hypothèse selon laquelle les
protéines, pour tous les rapports glu/gli à ce à toutes les concentrations, se comportent comme
des polypeptides dépliés tels qu’une chaîne de polymère en solvant (Brochard & de
Gennes, 1977 ; Banc et al., 2017).
À une échelle de q plus grande, (2 > q > 0,2 nm-1) (Fig.6.6), les données ont été ajustées par
le modèle d’Ornstein-Zernike décrit par la relation suivante:
I(q) = ( )²
(6.5)
Ce modèle permet de décrire la taille de blobs caractéristique, , pour une solution de polymère
en régime semi-dilué. Un exemple d’ajustement par ce modèle pour l’échantillon R0,04 est
présenté dans la Figure 6.11a. L’évolution de la taille des blobs, , obtenue à la suite de cette
0.0 0.5 1.0 1.5 2.0-3.0
-2.8
-2.6
-2.4
-2.2
-2.0
-1.8
-1.6
-1.4
-1.2
-1.0
= 0,0076 = 0,19
Exposant n
Rapport glu/gli
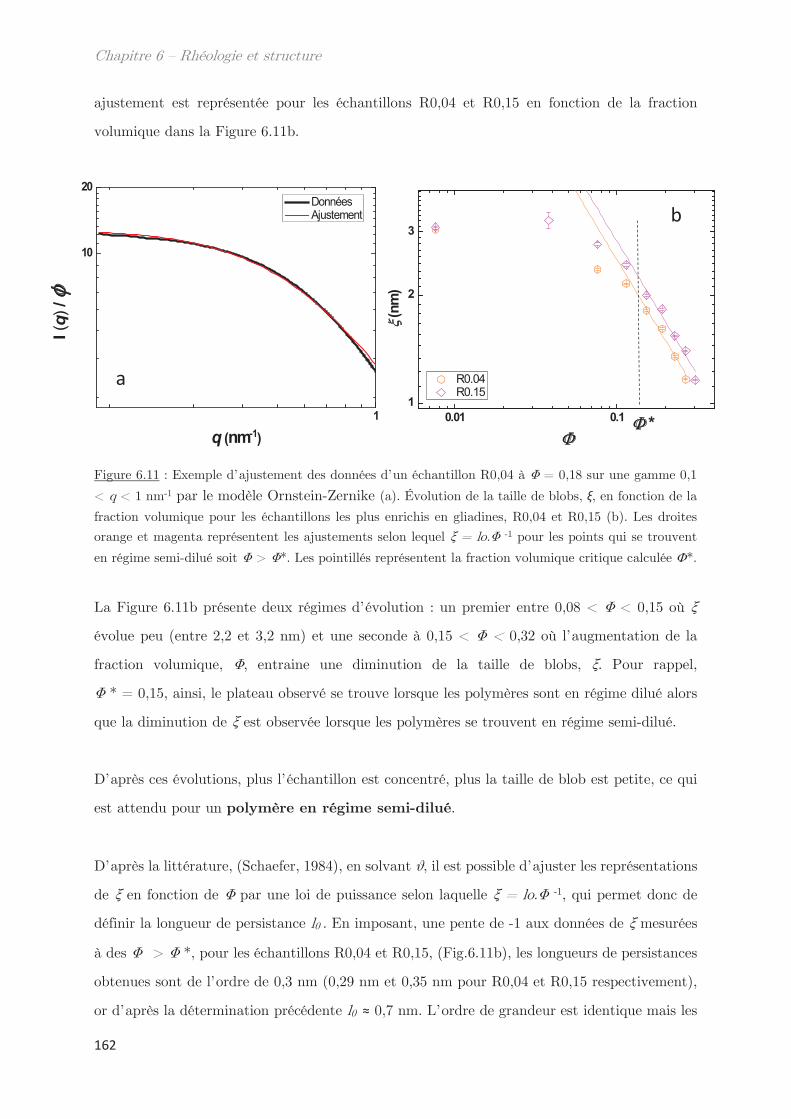
Chapitre 6 – Rhéologie et structure
162
ajustement est représentée pour les échantillons R0,04 et R0,15 en fonction de la fraction
volumique dans la Figure 6.11b.
Figure 6.11 : Exemple d’ajustement des données d’un échantillon R0,04 à = 0,18 sur une gamme 0,1
< q < 1 nm-1 par le modèle Ornstein-Zernike (a). Évolution de la taille de blobs, , en fonction de la
fraction volumique pour les échantillons les plus enrichis en gliadines, R0,04 et R0,15 (b). Les droites
orange et magenta représentent les ajustements selon lequel -1 pour les points qui se trouvent
en régime semi-dilué soit > *. Les pointillés représentent la fraction volumique critique calculée *.
La Figure 6.11b présente deux régimes d’évolution : un premier entre 0,08 < < 0,15 où
évolue peu (entre 2,2 et 3,2 nm) et une seconde à 0,15 < < 0,32 où l’augmentation de la
fraction volumique, , entraine une diminution de la taille de blobs, . Pour rappel,
* = 0,15, ainsi, le plateau observé se trouve lorsque les polymères sont en régime dilué alors
que la diminution de est observée lorsque les polymères se trouvent en régime semi-dilué.
D’après ces évolutions, plus l’échantillon est concentré, plus la taille de blob est petite, ce qui
est attendu pour un polymère en régime semi-dilué.
D’après la littérature, (Schaefer, 1984), en solvant , il est possible d’ajuster les représentations
de en fonction de par une loi de puissance selon laquelle = lo. -1, qui permet donc de
définir la longueur de persistance l0 . En imposant, une pente de -1 aux données de mesurées
à des > *, pour les échantillons R0,04 et R0,15, (Fig.6.11b), les longueurs de persistances
obtenues sont de l’ordre de 0,3 nm (0,29 nm et 0,35 nm pour R0,04 et R0,15 respectivement),
or d’après la détermination précédente l0 0,7 nm. L’ordre de grandeur est identique mais les
0.01 0.1
1
2
3
*
R0.04 R0.15
(nm
)a
b
1
10
20
I(q
) /
q (nm-1)
Données Ajustement
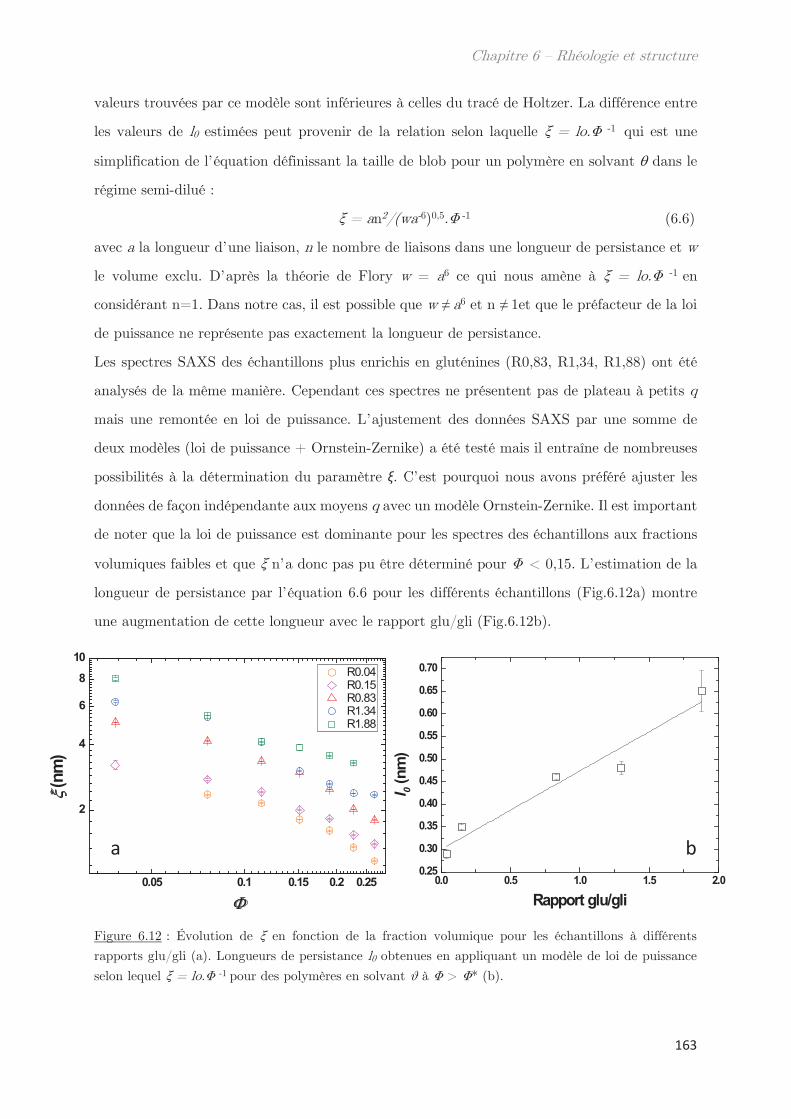
Chapitre 6 – Rhéologie et structure
163
valeurs trouvées par ce modèle sont inférieures à celles du tracé de Holtzer. La différence entre
les valeurs de l0 estimées peut provenir de la relation selon laquelle -1 qui est une
simplification de l’équation définissant la taille de blob pour un polymère en solvant dans le
régime semi-dilué :
= an2/(wa-6)0,5 -1 (6.6)
avec a la longueur d’une liaison, n le nombre de liaisons dans une longueur de persistance et w
le volume exclu. D’après la théorie de Flory w = a6 ce qui nous amène à -1 en
considérant n=1. Dans notre cas, il est possible que w a6 et n 1et que le préfacteur de la loi
de puissance ne représente pas exactement la longueur de persistance.
Les spectres SAXS des échantillons plus enrichis en gluténines (R0,83, R1,34, R1,88) ont été
analysés de la même manière. Cependant ces spectres ne présentent pas de plateau à petits q
mais une remontée en loi de puissance. L’ajustement des données SAXS par une somme de
deux modèles (loi de puissance + Ornstein-Zernike) a été testé mais il entraîne de nombreuses
possibilités à la détermination du paramètre . C’est pourquoi nous avons préféré ajuster les
données de façon indépendante aux moyens q avec un modèle Ornstein-Zernike. Il est important
de noter que la loi de puissance est dominante pour les spectres des échantillons aux fractions
volumiques faibles et que n’a donc pas pu être déterminé pour < 0,15. L’estimation de la
longueur de persistance par l’équation 6.6 pour les différents échantillons (Fig.6.12a) montre
une augmentation de cette longueur avec le rapport glu/gli (Fig.6.12b).
Figure 6.12 : Évolution de en fonction de la fraction volumique pour les échantillons à différents
rapports glu/gli (a). Longueurs de persistance l0 obtenues en appliquant un modèle de loi de puissance
selon lequel -1 pour des polymères en solvant à > * (b).
0.0 0.5 1.0 1.5 2.00.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
0.70
l 0(n
m)
Rapport glu/gli
a b
0.05 0.1 0.15 0.2 0.25
2
4
6
8
10
R0.04 R0.15 R0.83 R1.34 R1.88
(nm
)

Chapitre 6 – Rhéologie et structure
164
Ces résultats sont en désaccord avec ceux observés dans la Figure 6.10 selon laquelle lo est
constante ( 0,7 nm). Différentes hypothèses sont proposées pour expliquer ces
résultats contradictoires:
i) La détermination de la taille de est erronée par la remontée à petits q dont
il est difficile de s’affranchir. Ainsi l0 croit avec R de façon artificielle.
ii) La détermination de la taille de l0 est erronée par une mauvaise estimation de la
valeur de la fraction volumique pour les échantillons au rapport glu/gli les plus
élevés. En effet, les remontées aux petits q croissantes avec R indiquent une
croissance des hétérogénéités de concentrations au sein des solutions qui se
manifestent localement par des zones plus enrichies en protéines et d’autres zones
appauvries en protéines. En considérant que la détermination du paramètre est
dominée par les zones appauvries des échantillons, la fraction volumique associée à
la longueur de persistance mesurée se retrouve inférieure à la fraction volumique
nominale. Cela conduirait à une surestimation des longueurs de persistance en
présence d’hétérogénéités à grande échelle.
Avec l’analyse du tracé de Holtzer qui est indépendante de la diffusion à petits q, et de la
fraction volumique de l’échantillon, l’évolution de l0 n’est pas mesurée. Nous donnons ainsi
plus de crédit à l’analyse faite aux grands q et concluons que la longueur de persistance des
échantillons est constante avec le rapport glu/gli.
Enfin, à petits q (q > 0,1 nm-1) (Fig.6.6), les échantillons enrichis en gluténines, ont été ajustés
par un modèle de loi de puissance selon l’équation 6.7 :
I(q) = Aq –df (6.7)
où A est une constante et df une dimension fractale.
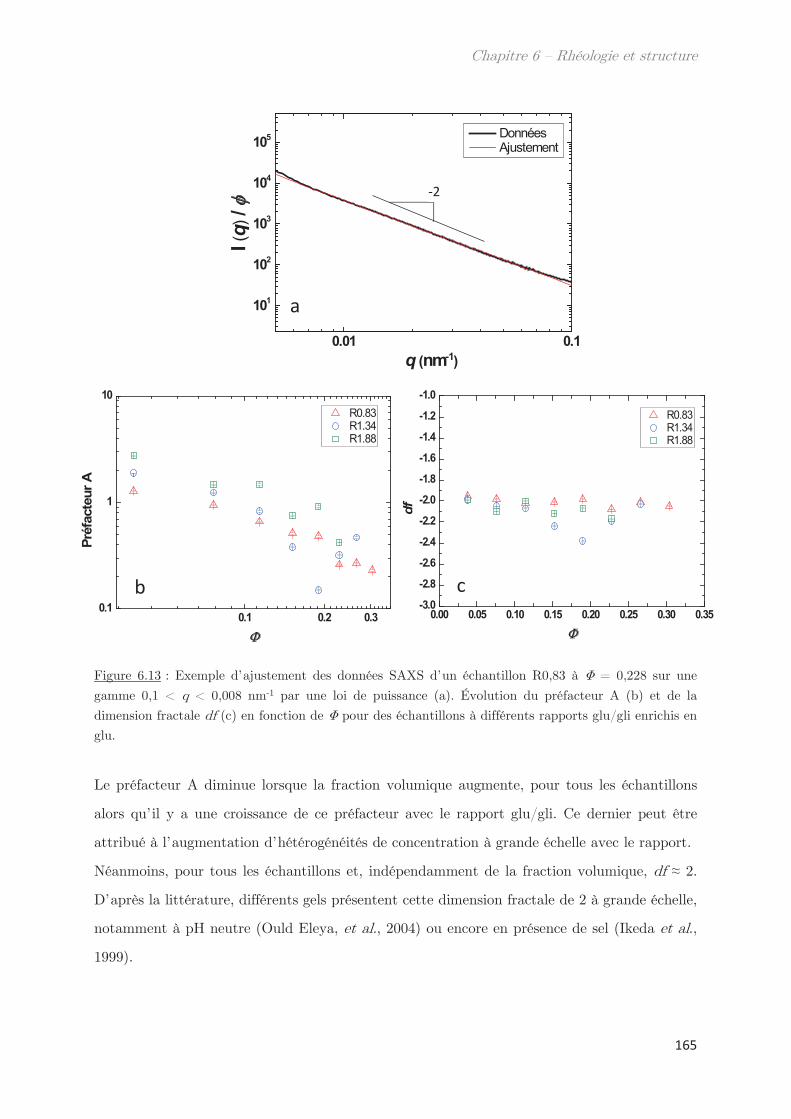
Chapitre 6 – Rhéologie et structure
165
Figure 6.13 : Exemple d’ajustement des données SAXS d’un échantillon R0,83 à = 0,228 sur une
gamme 0,1 < q < 0,008 nm-1 par une loi de puissance (a). Évolution du préfacteur A (b) et de la
dimension fractale df (c) en fonction de pour des échantillons à différents rapports glu/gli enrichis en
glu.
Le préfacteur A diminue lorsque la fraction volumique augmente, pour tous les échantillons
alors qu’il y a une croissance de ce préfacteur avec le rapport glu/gli. Ce dernier peut être
attribué à l’augmentation d’hétérogénéités de concentration à grande échelle avec le rapport.
Néanmoins, pour tous les échantillons et, indépendamment de la fraction volumique, df 2.
D’après la littérature, différents gels présentent cette dimension fractale de 2 à grande échelle,
notamment à pH neutre (Ould Eleya, et al., 2004) ou encore en présence de sel (Ikeda et al.,
1999).
0.1 0.2 0.30.1
1
10
R0.83 R1.34 R1.88
Pré
facte
ur
A
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35-3.0
-2.8
-2.6
-2.4
-2.2
-2.0
-1.8
-1.6
-1.4
-1.2
-1.0
R0.83 R1.34 R1.88
df
a
c
-2
b
0.01 0.1
101
102
103
104
105 Données
Ajustement
I(q
) /
q (nm-1)

Chapitre 6 – Rhéologie et structure
166
3.2.2 Diffusion de neutrons
Les mesures de diffusion de neutrons aux petits angles (SANS) ont été réalisées sur les
échantillons de gluten à différents rapports glu/gli à température ambiante. De façon générale,
les mesures de SANS se font en solvant deutéré à la place d’un solvant hydrogéné, ce qui
permet d’obtenir un contraste entre les objets diffusants hydrogénés et le solvant deutéré. Afin
de comparer l’effet du solvant hydrogéné, H, et du solvant deutéré, D, deux séries d’échantillons
à = 0,18 ont été préparées. L’une a été dispersée dans un solvant eau/éthanol deutéré 50%
(v/v) et l’autre dans un solvant eau/éthanol hydrogéné 50% (v/v). Les résultats SANS
présentés ici ont été réalisés sur KWS2 au MLZ de Garching en Allemagne sur une gamme
3.10-5 < q < 2.10-3 nm-1 (Chapitre 2). La Figure 6.14 met en évidence la comparaison des
extraits solubilisés à = 0,18 en solvant hydrogéné (a) et en solvant deutéré (b) avec la
technique SANS. La forme des spectres diffère fortement entre les deux solvants. En effet, à
faibles q (q < 0,1 nm-1) les échantillons deutérés présentent une pente en q -4 alors qu’une pente
en q -2 est visible pour les échantillons hydrogénés.
Par ailleurs, les Figures 6.14c et d présentent les mêmes échantillons respectivement en solvant
H et D mesurés en diffusion de rayon X. De même que l’échantillon hydrogéné en SANS (a),
les profils SAXS sont identiques quel que soit le solvant et présentent une pente en q -2 à q <
0,1 nm-1. Il n’existe donc pas de différence entre les échantillons deutérés et hydrogénés en
SAXS. Il est important de rappeler ici que le contraste observé en SAXS résulte des variations
de densités électroniques alors qu’en SANS, le contraste est associé à la variation de densité de
longueur de diffusion qui est très grande entre les espèces hydrogénées et deutérées.
Ainsi la similarité des spectres SAXS des échantillons hydrogénés et deutérés montre que la
structure des protéines au sein des échantillons n’est pas affectée par la deutération du solvant.
Cependant, l’évolution forte du signal avec le solvant deutéré en SANS suggère l’existence
d’autres phénomènes mis en jeu qui seront l’objet d’étude de la suite de ce chapitre.
D’après la Figure 6.14, les données SANS des échantillons préparés en solvant deutéré montrent
une allure identique pour les échantillons R0,83, R1,34 et R1,88 avec une pente en q -4 entre
2.10-2 nm-1 et 0,2 nm-1, ainsi qu’un niveau croissant d’intensité avec le rapport R et un quasi
plateau à plus petits q.
.
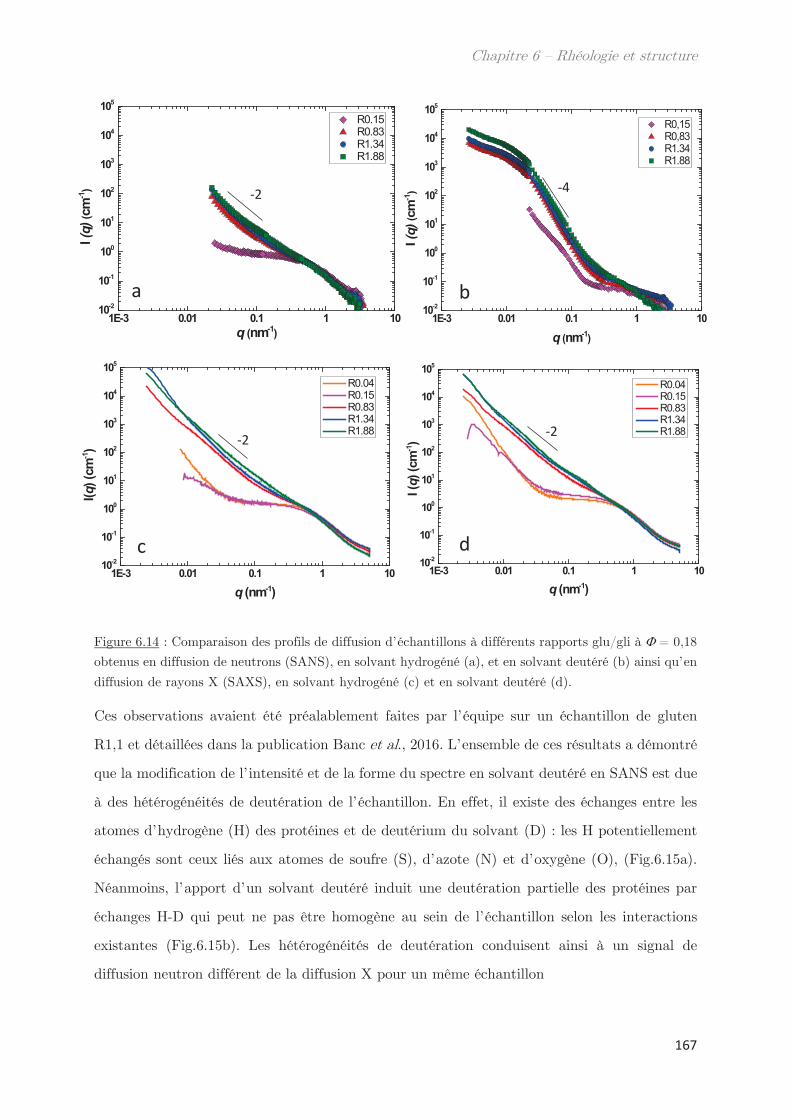
Chapitre 6 – Rhéologie et structure
167
Figure 6.14 : Comparaison des profils de diffusion d’échantillons à différents rapports glu/gli à = 0,18
obtenus en diffusion de neutrons (SANS), en solvant hydrogéné (a), et en solvant deutéré (b) ainsi qu’en
diffusion de rayons X (SAXS), en solvant hydrogéné (c) et en solvant deutéré (d).
Ces observations avaient été préalablement faites par l’équipe sur un échantillon de gluten
R1,1 et détaillées dans la publication Banc et al., 2016. L’ensemble de ces résultats a démontré
que la modification de l’intensité et de la forme du spectre en solvant deutéré en SANS est due
à des hétérogénéités de deutération de l’échantillon. En effet, il existe des échanges entre les
atomes d’hydrogène (H) des protéines et de deutérium du solvant (D) : les H potentiellement
échangés sont ceux liés aux atomes de soufre (S), d’azote (N) et d’oxygène (O), (Fig.6.15a).
Néanmoins, l’apport d’un solvant deutéré induit une deutération partielle des protéines par
échanges H-D qui peut ne pas être homogène au sein de l’échantillon selon les interactions
existantes (Fig.6.15b). Les hétérogénéités de deutération conduisent ainsi à un signal de
diffusion neutron différent de la diffusion X pour un même échantillon
1E-3 0.01 0.1 1 1010
-2
10-1
100
101
102
103
104
105
R0.04 R0.15 R0.83 R1.34 R1.88
I(q)(c
m-1)
q (nm-1)
ba
c d
-4
-2-2
-2
1E-3 0.01 0.1 1 1010
-2
10-1
100
101
102
103
104
105
R0,15 R0,83 R1.34 R1.88
I (q
)(c
m-1
)
q (nm-1)
1E-3 0.01 0.1 1 1010
-2
10-1
100
101
102
103
104
105
R0.15 R0.83 R1.34 R1.88
I(q
)(c
m-1
)
q (nm-1)
1E-3 0.01 0.1 1 1010
-2
10-1
100
101
102
103
104
105
I(q
)(c
m-1)
q (nm-1)
R0.04 R0.15 R0.83 R1.34 R1.88
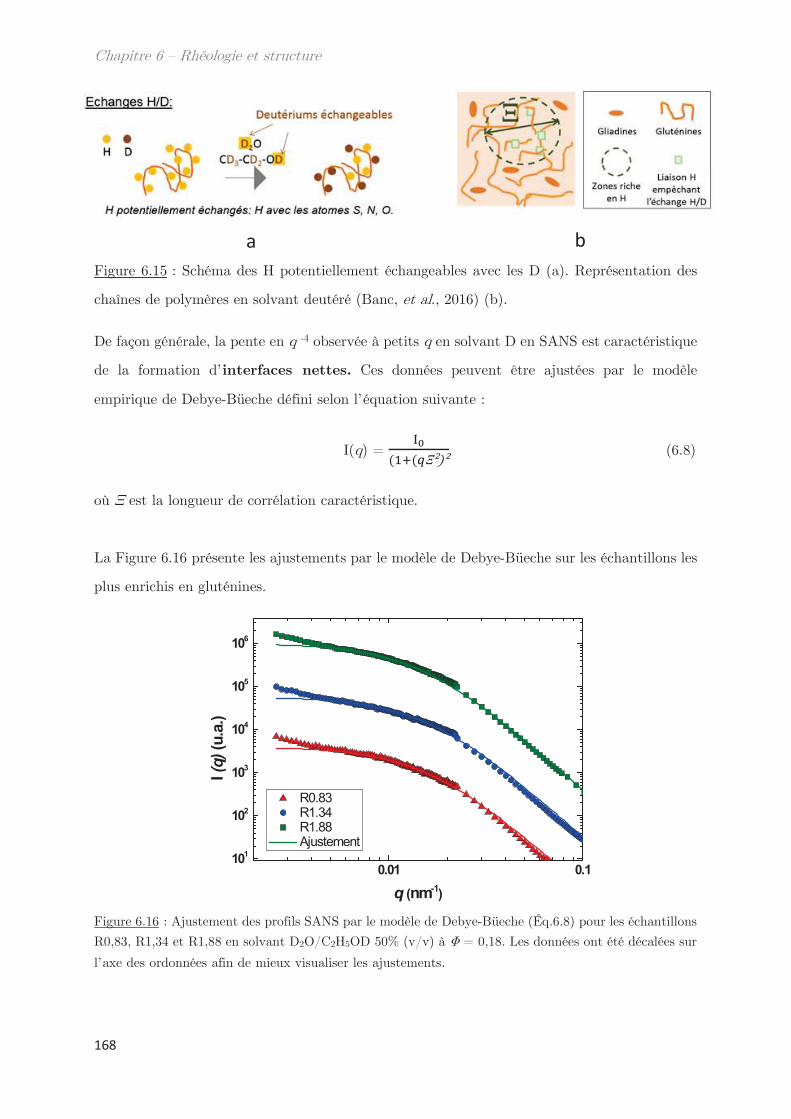
Chapitre 6 – Rhéologie et structure
168
Figure 6.15 : Schéma des H potentiellement échangeables avec les D (a). Représentation des
chaînes de polymères en solvant deutéré (Banc, et al., 2016) (b).
De façon générale, la pente en q -4 observée à petits q en solvant D en SANS est caractéristique
de la formation d’interfaces nettes. Ces données peuvent être ajustées par le modèle
empirique de Debye-Büeche défini selon l’équation suivante :
I(q) = ( ( ²)²
(6.8)
où est la longueur de corrélation caractéristique.
La Figure 6.16 présente les ajustements par le modèle de Debye-Büeche sur les échantillons les
plus enrichis en gluténines.
Figure 6.16 : Ajustement des profils SANS par le modèle de Debye-Büeche (Éq.6.8) pour les échantillons
R0,83, R1,34 et R1,88 en solvant D2O/C2H5OD 50% (v/v) à = 0,18. Les données ont été décalées sur
l’axe des ordonnées afin de mieux visualiser les ajustements.
0.01 0.110
1
102
103
104
105
106
R0.83 R1.34 R1.88 Ajustement
I (q
) (u
.a.)
q (nm-1)
a b
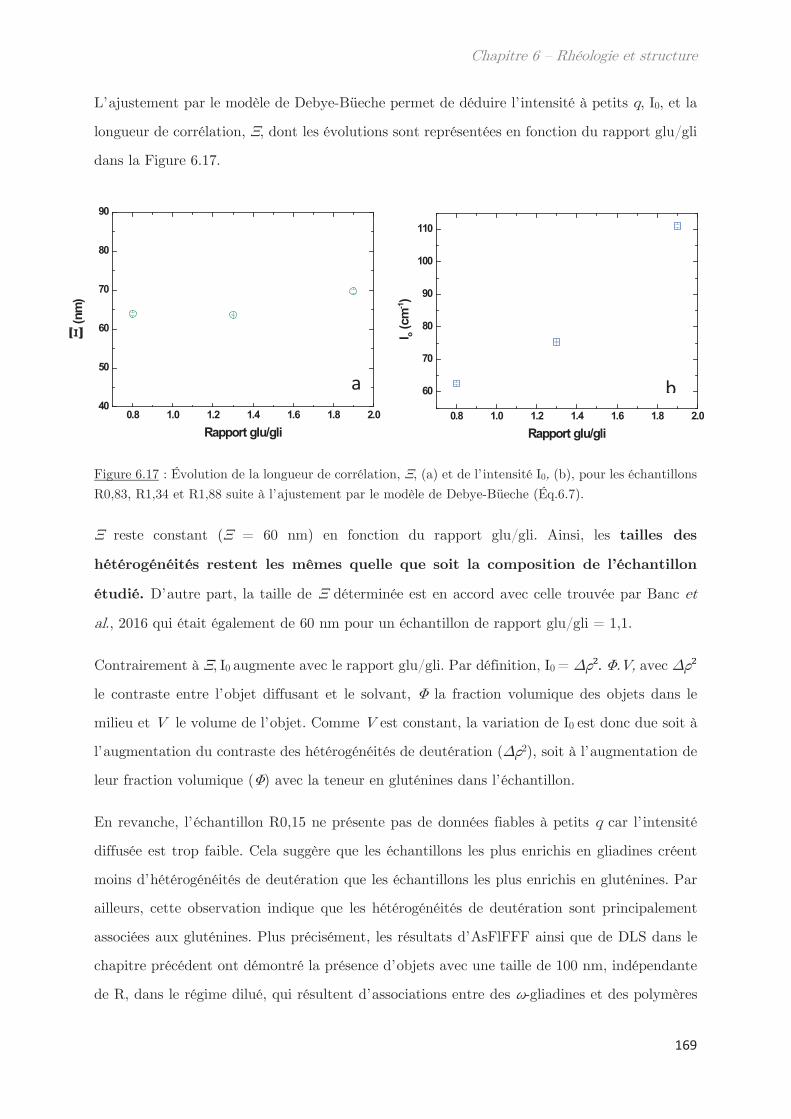
Chapitre 6 – Rhéologie et structure
169
L’ajustement par le modèle de Debye-Büeche permet de déduire l’intensité à petits q, I0, et la
longueur de corrélation, , dont les évolutions sont représentées en fonction du rapport glu/gli
dans la Figure 6.17.
Figure 6.17 : Évolution de la longueur de corrélation, , (a) et de l’intensité I0, (b), pour les échantillons
R0,83, R1,34 et R1,88 suite à l’ajustement par le modèle de Debye-Büeche (Éq.6.7).
reste constant ( = 60 nm) en fonction du rapport glu/gli. Ainsi, les tailles des
hétérogénéités restent les mêmes quelle que soit la composition de l’échantillon
étudié. D’autre part, la taille de déterminée est en accord avec celle trouvée par Banc et
al., 2016 qui était également de 60 nm pour un échantillon de rapport glu/gli = 1,1.
Contrairement à , I0 augmente avec le rapport glu/gli. Par définition, I0 = ². .V, avec ²
le contraste entre l’objet diffusant et le solvant, la fraction volumique des objets dans le
milieu et V le volume de l’objet. Comme V est constant, la variation de I0 est donc due soit à
l’augmentation du contraste des hétérogénéités de deutération ( 2), soit à l’augmentation de
leur fraction volumique ( ) avec la teneur en gluténines dans l’échantillon.
En revanche, l’échantillon R0,15 ne présente pas de données fiables à petits q car l’intensité
diffusée est trop faible. Cela suggère que les échantillons les plus enrichis en gliadines créent
moins d’hétérogénéités de deutération que les échantillons les plus enrichis en gluténines. Par
ailleurs, cette observation indique que les hétérogénéités de deutération sont principalement
associées aux gluténines. Plus précisément, les résultats d’AsFlFFF ainsi que de DLS dans le
chapitre précédent ont démontré la présence d’objets avec une taille de 100 nm, indépendante
de R, dans le régime dilué, qui résultent d’associations entre des -gliadines et des polymères
a b 0.8 1.0 1.2 1.4 1.6 1.8 2.0
60
70
80
90
100
110
I o(c
m-1)
Rapport glu/gli
0.8 1.0 1.2 1.4 1.6 1.8 2.040
50
60
70
80
90
(nm
)
Rapport glu/gli

Chapitre 6 – Rhéologie et structure
170
de gluténines. Ces assemblages pourraient être à l’origine des hétérogénéités de deutération qui
sont de taille du même ordre de grandeur et également indépendante de R. Les assemblages
seraient moins susceptibles d’échanger leurs protons avec les deutériums du solvant que les
protéines monomériques et conduiraient au signal SANS observé, différent du signal SAXS.
L’intensité du signal croissant avec le rapport glu/gli peut être associé au nombre croissant des
hétérogénéités et/ou à un contraste plus élevé de celles-ci dû à une densité plus importante des
assemblages. Ces 2 hypothèses sont en accord avec les observations faites par AsFlFFF.
Conclusion
Dans ce présent travail, il apparait que les gluténines modifient les propriétés rhéologiques des
échantillons. L’augmentation de gluténines amène les extraits de gluten en solutions à gélifier
alors que la présence de gliadines rend les échantillons plus visqueux. Cette observation répond
bien à la capacité des pâtes à pain à former un réseau élastique et semble donc en accord avec
la littérature (Cornec et al., 1994, Janssen et al., 1995, Khatkar et al., 1996, Tronsmo et al.,
2003). À plus petites échelles, qu’elles soient en régime dilué ou semi-dilué, les protéines se
comportent comme des chaînes de polymères en solvant avec une longueur de
persistance constante ( 0,7 nm) quel que soit le rapport glu/gli et la fraction volumique.
D’autre part, l’intensité diffusée mesurée en diffusion de rayons X aux petits angles montre
une évolution croissante avec l’augmentation du rapport glu/gli qui indique une augmentation
des hétérogénéités de concentration de dimension fractale 2. De plus, en jouant sur le
contraste du solvant utilisé pour les échantillons mesurés en SANS, le solvant deutéré a permis
de mettre en avant des hétérogénéités de deutérations dans le régime semi-dilué qui ont
toujours la même taille quel que soit le rapport glu/gli. Ces hétérogénéités de deutérations se
manifestent par des zones de protéines partiellement deutérées, de taille qui résultent de
fortes liaisons H qui empêchent l'échange H/D entre le solvant et certains polypeptides de
gluten, en particulier les gluténines. Ces hétérogénéités de concentrations et de deutérations
dans le régime semi-dilué peuvent être attribuées aux assemblages précédemment identifiés
dans le régime dilué.
Enfin, les mesures en SANS, à très petits q, sur les échantillons les plus enrichis en gluténines,
ont permis de déterminer des tailles, , de 60 nm qui sont légèrement plus petites que celles

Chapitre 6 – Rhéologie et structure
171
trouvées dans le chapitre précédent ( 100 nm). Ces tailles peuvent être dû à la répartition de
densité des protéines au sein des assemblages observés dans le chapitre précédent par AsFlFFF.

Chapitre 6 – Rhéologie et structure
172
Bibliographie
Balu, R., Mata, J. P., Knott, R., Elvin, C. M., Hill, A. J., Choudhury, N. R., & Dutta, N. K.
(2016). Effects of Crowding and Environment on the Evolution of Conformational
Ensembles of the Multi-Stimuli-Responsive Intrinsically Disordered Protein, Rec1-
A Small-Angle Scattering Investigation. The Journal of Physical Chemistry, 120, 6490–
6503.
Banc, A., Charbonneau, C., Dahesh, M., Appavou, M.-S., Fu, Z., Morel, M.-H., & Ramos, L.
(2016). Small angle neutron scattering contrast variation reveals heterogeneities of
interactions in protein gels. Soft Matter, 1–28.
Banc, A., Dahesh, M., Wolf, M., Morel, M., & Ramos, L. (2017). Model gluten gels. Journal of
Cereal Science, 75, 175–178.
Brochard, F., & Gennes, P. G. d. (1977). Dynamical Scaling for Polymers in Theta Solvents.
Macromolecules, 10(5), 1157–1161.
Burchard, W. (1999). Solution Properties of Branched Macromolecules. Branched Polymers II,
143, 113–194.
Cornec, M., Popineau, Y., & Lefebvre J. (1994). Characterisation of Gluten Subfractions by
SE-HPLC and Dynamic Rheological Analysis in Shear.
Dahesh, M., Banc, A., Duri, A., Morel, M. H., & Ramos, L. (2014). Polymeric assembly of
gluten proteins in an aqueous ethanol solvent. Journal of Physical Chemistry B, 118(38),
11065–11076.
Denkinger, P., & Burchard, W. (1991). Determination of chain stiffness and polydispersity from
static light scattering. Journal of Polymer Science Part B: Polymer Physics, 29(5), 589–
600.
Glatter O., & Kratky, O. (1982). Small Angle X-ray Scattering. Academic Press.
Ikeda, S., Foegeding, E. A., & Hagiwara, T. (1999). Rheological Study on the Fractal Nature
of the Protein Gel. Langmuir, (15), 8584–8589.

Chapitre 6 – Rhéologie et structure
173
Janssen, A. M., van Vliet, T., & Vereijken, J. M. (1996). Rheological Behavior of Wheat
Glutens at Small and Large Deformations. Effect of Gluten Composition. Journal of Cereal
Science, 23(1), 33–42.
Jowitt, T. A., Murdoch, A. D., Baldock, C., Berry, R., Day, J. M., & Hardingham, T. E.
(2010). Order within disorder: Aggrecan chondroitin sulphate-attachment region provides
new structural insights into protein sequences classified as disordered. Proteins: Structure,
Function and Bioinformatics, 78(16), 3317–3327.
Khatkar, B. S., Bell, A. E., & Schofield, J. D. (1995). The dynamic rheological properties of
glutens and gluten sub-fractions from wheats of good and poor bread making quality.
Journal of Cereal Science, 22(1), 29–44.
Lairez
des macromolécules biologiques en solution. Journal de Physique IV (Proceedings), 130,
39–62.
Maity, H., & Reddy, G. (2016). Folding of Protein L with Implications for Collapse in the
Denatured State Ensemble. Journal of the American Chemical Society, 138(8), 2609–2616.
Micka, U., & Kremer, K. (1996). The persistence length of polyelectrolyte chains. Journal of
Physics Condensed Matter, 8(47), 9463–9470.
Ould Eleya, M. M., Ko, S., & Gunasekaran, S. (2004). Scaling and fractal analysis of viscoelastic
properties of heat-induced protein gels. Food Hydrocolloids, 18(2), 315–323.
Rosales, A. M., Murnen, H. K., Kline, S. R., Zuckermann, R. N., & Segalman, R. A. (2012).
Determination of the persistence length of helical and non-helical polypeptoids in solution.
Soft Matter, 8, 3673–3680.
Schaefer, D. W. (1984). A unified model for the structure of polymers in semidilute solution.
Polymer, 25, 386–394.
Schmidt, M., & Burchard, W. (1981). Translational Diffusion and Hydrodynamic Radius of
Unperturbed Flexible Chains. Macromolecules, 479(4), 210–211.

Chapitre 6 – Rhéologie et structure
174
Schmidt, M., Paradossi, G., & Burchard, W. (1985). Remarks on the determination of chain
stiffness from static scattering experiments. Die Makromolekulare Chemie, Rapid
Communications, 6(11), 767–772.
Shibayama, M., Isono, K., Okabe, S., Karino, T., & Nagao, M. (2004). SANS Study on
Pressure-Induced Phase Separation of Poly(N-isopropylacrylamide) Aqueous Solutions
and Gels. Macromolecules, 37(8), 2909–2918.
Tronsmo, K. M., Magnus, E. M., Baardseth, P., Schofield, J. D., Aamodt, a., & Faergestad, E.
M. (2003). Comparison of small and large deformation rheological properties of wheat
dough and gluten, 80(5), 1–4.
Ying, Q., & Chu, B. (1987). Overlap Concentration of Macromolecules in Solution.
Macromolecules, 20(2), 362–366.

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
175
Chapitre 7
Dynamique de séparation de phases liquide-
liquide
Les chapitres précédents ont mis en avant les interactions et assemblages des gliadines et des
gluténines, ainsi que leurs comportements différents suite à la séparation de phases. L’objectif
de ce chapitre est d’analyser en détail ce phénomène de séparation de phases à différentes
échelles. Dans un premier temps, des études microscopiques par microscopie de contraste de
phases permettront de caractériser le phénomène de décomposition spinodale en fonction des
différents rapports glu/gli. En parallèle, le comportement rhéologique des solutions au cours de
la transition de phase sera étudié. Enfin, les mesures par diffusion de neutrons et rayons X
permettront d’expliquer les mécanismes structuraux à l’échelle des chaînes de polymères.
1. Étude de la décomposition spinodale
1.1 Microscopie optique
La microscopie de contraste de phase contrôlée en température permet d’illustrer l’évolution
des microstructures au cours du temps et plus particulièrement au cours des phénomènes de
séparation de phases des extraits de gluten. L’objectif est de sonder la dynamique interne des
échantillons mais également d’observer si le phénomène se manifeste de la même façon pour
tous les extraits à différents rapports glu/gli. Les échantillons R0,83, R1,34 et R1,88 ont été
analysés en milieu concentré ( = 0,18), après une trempe en température. Il est indispensable
de choisir une température de trempe (Tt) pour laquelle Tt < Tc afin d’être sûr que le système
soit dans le domaine biphasique du diagramme de phases. Pour rappel, Tc = 12°C pour R0,83,

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
176
12,5°C pour R1,34 et 13°C pour R1,88 d’après les diagrammes de phases établis dans le Chapitre
5. La Figure 7.1 présente des images au cours du temps pour chaque rapport glu/gli.
Figure 7.1 : Observation au cours du temps des solutions protéiques ( = 0,18) à 20°C, puis suite à une
séparation de phases après des trempes en températures à 8°C pour les échantillons R0,83, à 10°C pour
l’échantillon R1,34 et 10,5°C pour l’échantillon R1,88 puis après remontée en température à 20°C.
20
20°C Avant Tt
R0,83 R1,88R1,34
Tt = 8°C Tt = 10°C Tt = 10.5°C
t > 2000 sec
1535 sec
1325 sec
20°C Après Tt
20µm

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
177
Des premières photos ont été prises à 20°C pour tous les échantillons avant de commencer la
trempe en température. Quel que soit l’échantillon, les photos montrent un milieu parfaitement
homogène pour lesquels il n’y a pas d’évolution particulière du système.
Une fois la température de trempe Tt atteinte, dans le cas des échantillons les plus enrichis en
gliadines, R0,83 et R1,34, il apparait des microstructures dès les premières secondes (1325 sec)
qui semblent peu évoluer jusqu’à 1535 sec. Il y a donc une structure bi-continue qui se forme
à la suite de la trempe en température. Celle de l’échantillon R0,83 montre même un début de
gouttelettes qui se forment.
Après 1535 sec, un grand nombre de gouttelettes est nettement observable pour les 2
échantillons. Ces dernières grossissent au cours du temps et leur nombre diminue comme exposé
par les photos prises à t> 2000 sec. Cependant, cette étape nécessite plus de temps pour les
échantillons les plus enrichis en gluténines. En effet, pour l’échantillon R0,83 de petites
gouttelettes de l’ordre de 5 à 10 µm sont visibles après 1500 sec alors que ces dernières ne sont
visibles qu’après 2000 sec pour l’échantillon R1,34. Dans la limite des mesures réalisées, nous
avons observé que les gouttelettes de l’échantillons R0,83 grossissent jusqu’à 20 µm après 2000
sec et dépassent largement cette taille ( 40 µm) à 7200 sec (non montré ici). En effet, les
gouttes suivent un phénomène de coalescence, se rapprochent et fusionnent au cours du temps.
Ce phénomène de grossissement est également observé pour l’échantillon R1,34 mais à des
temps plus longs que pour R0,83.
Ainsi, les microstructures semblent évoluer de la même façon pour des échantillons plus ou
moins enrichis en gliadines et gluténines mais avec des temps d’évolutions différents. Cette
observation peut s’expliquer par la température de trempe, Tt : plus celle-ci est proche de la
ligne spinodale, moins le contraste entre la phase concentrée et la phase diluée est important.
Ces différentes observations suggèrent que pour les échantillons R0,83 et R1,34 dans ces
conditions, le début de la séparation de phases à des temps très courts se produit par
décomposition spinodale.
L’échantillon le plus enrichi en gluténines, R1,88, se comporte différemment des autres
échantillons : la dynamique est très lente et le début de la séparation de phase difficile à
détecter. En effet, il y a présence d’un motif continu dès 535 sec qui reste constant au cours
du temps. La matrice qui est formée dès les premières secondes semble bloquée. Cette
observation nous permet de faire l’hypothèse que soit la phase concentrée est nettement
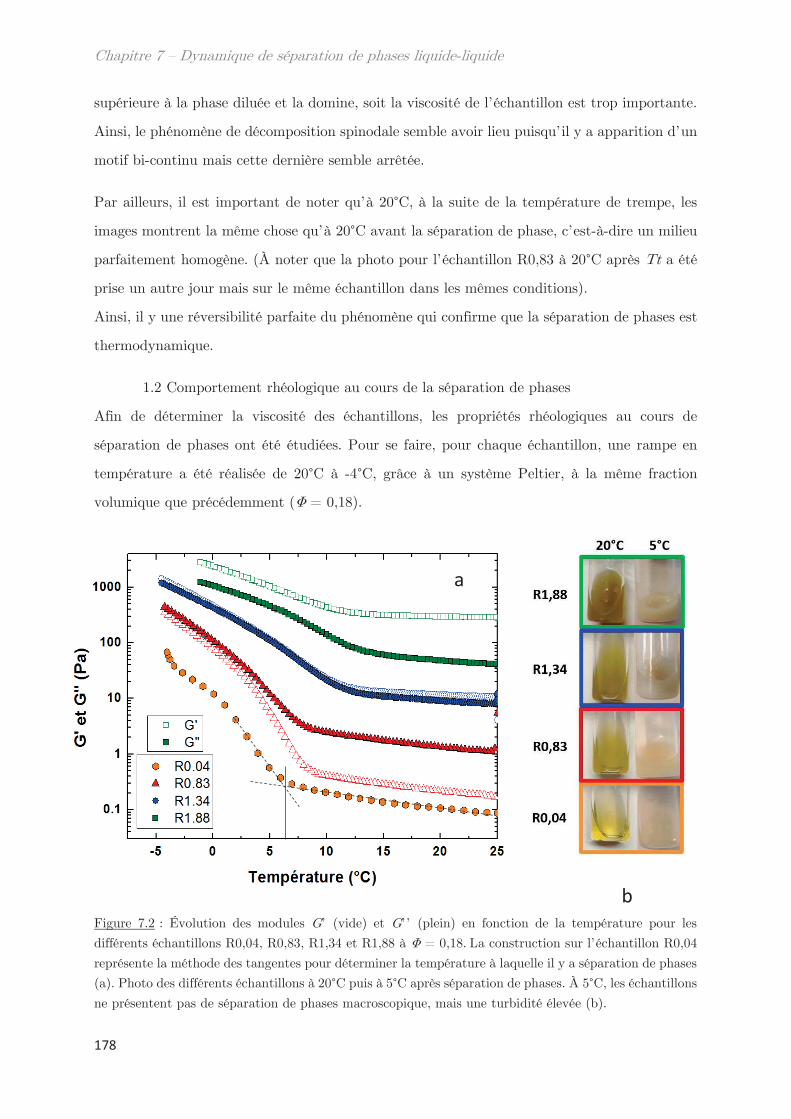
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
178
supérieure à la phase diluée et la domine, soit la viscosité de l’échantillon est trop importante.
Ainsi, le phénomène de décomposition spinodale semble avoir lieu puisqu’il y a apparition d’un
motif bi-continu mais cette dernière semble arrêtée.
Par ailleurs, il est important de noter qu’à 20°C, à la suite de la température de trempe, les
images montrent la même chose qu’à 20°C avant la séparation de phase, c’est-à-dire un milieu
parfaitement homogène. (À noter que la photo pour l’échantillon R0,83 à 20°C après Tt a été
prise un autre jour mais sur le même échantillon dans les mêmes conditions).
Ainsi, il y une réversibilité parfaite du phénomène qui confirme que la séparation de phases est
thermodynamique.
1.2 Comportement rhéologique au cours de la séparation de phases
Afin de déterminer la viscosité des échantillons, les propriétés rhéologiques au cours de
séparation de phases ont été étudiées. Pour se faire, pour chaque échantillon, une rampe en
température a été réalisée de 20°C à -4°C, grâce à un système Peltier, à la même fraction
volumique que précédemment ( = 0,18).
Figure 7.2 : Évolution des modules G’ (vide) et G’’ (plein) en fonction de la température pour les
différents échantillons R0,04, R0,83, R1,34 et R1,88 à = 0,18. La construction sur l’échantillon R0,04
représente la méthode des tangentes pour déterminer la température à laquelle il y a séparation de phases
(a). Photo des différents échantillons à 20°C puis à 5°C après séparation de phases. À 5°C, les échantillons
ne présentent pas de séparation de phases macroscopique, mais une turbidité élevée (b).
a
b

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
179
Suite à la diminution de température, le module de stockage (G’) tend à se rapprocher du
module de perte (G’’), pour tous les échantillons (Fig.7.2a). La rampe en température induit
une gélification des solutions de protéines. De plus, les profils montrent une augmentation
brutale de (G’) et de (G’’) à différentes températures au-delà de laquelle il y a un changement
de pente des modules. Ces changements peuvent être attribués à la température à laquelle le
système sépare de phases.
Ces températures de transitions, nommées Tr, sont définies par la méthode des tangentes,
représentée sur l’échantillon R0,04 (Fig.7.2), comme utilisée dans les Chapitres 3 et sont
récapitulée dans la Table 7.1.
Table 7.1 : Détermination de la température, Tr, à laquelle les pentes des modules G’ ’ changent pour
les différents échantillons de gluten, comparée aux températures de transitions, Tt, à = 0,18 déduites
des diagrammes de phases établis par MDSC (cf Chapitre 5, Fig.5.18).
Échantillons Tr rhéologie (°C) Tt MDSC (°C)
R0,04 7 ± 1 5 ± 1
R0,83 8 ± 0,5 8 ± 1
R1,34 11 ± 0,5 11 ± 1
R1,88 12 ± 0,5 11 ± 1
Les températures de transitions déterminées par MDSC sont très proches, voire identique des
températures de transitions définies en rhéologie ce qui confirme que le changement de pente
des modules correspond au phénomène de séparation de phases.
D’après les images de la Figure 7.2b, à 5°C, les échantillons semblent former une masse plus
élastique qu’à 20°C. En accord avec les mesures rhéologiques, les échantillons gélifient (sans
toutefois former totalement des gels pour les échantillons enrichis en gliadines, R0,04 et R0,83).
En effet, l’échantillon R0,83 présente une plus forte viscosité à 5°C et met du temps à s’écouler
alors que l’échantillon R0,04 reste liquide. De plus, les photos montrent un changement de
couleur des échantillons qui passent d’une teinte jaunâtre transparente (20°C) à blanc
translucide, caractéristique d’une diffusion (5°C). Ces observations suggèrent qu’il n’y a pas
de séparation de phases macroscopique mais les changements de couleurs et de propriétés
rhéologiques suggèrent plutôt une séparation de phases microscopique (Fig.7.3).
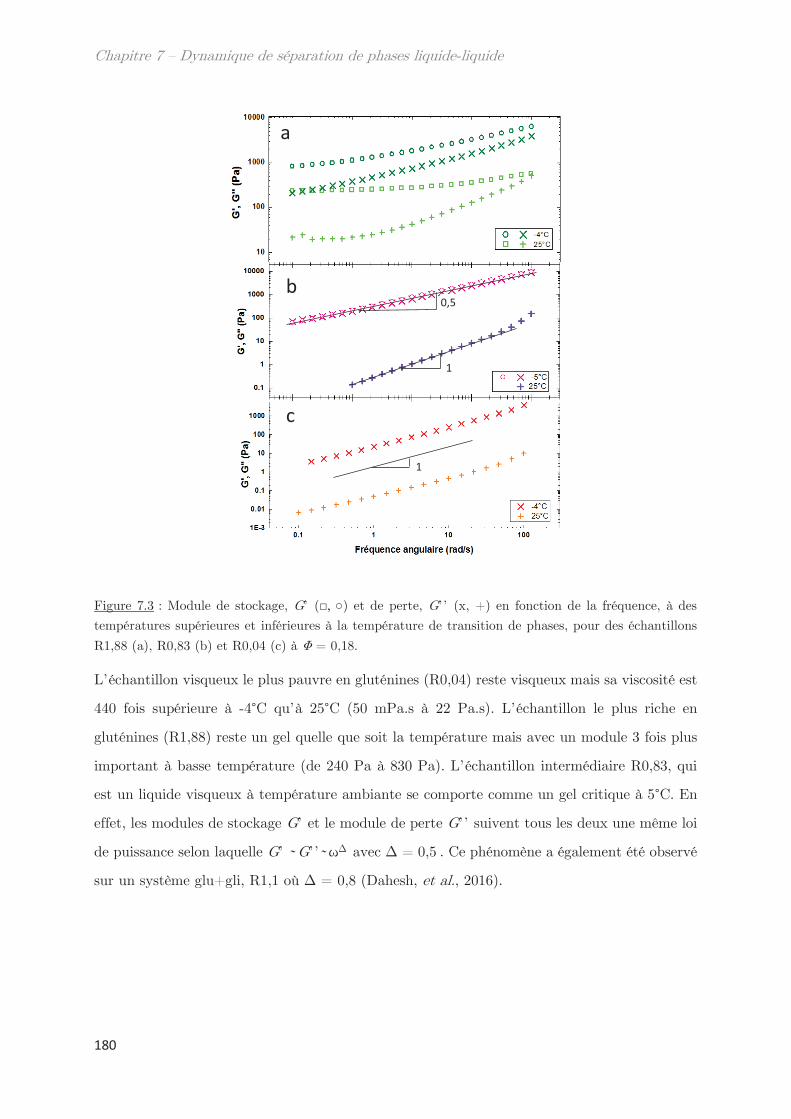
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
180
Figure 7.3 : Module de stockage, G’ ( ) et de perte, G’ ’ (x, +) en fonction de la fréquence, à des
températures supérieures et inférieures à la température de transition de phases, pour des échantillons
R1,88 (a), R0,83 (b) et R0,04 (c) à = 0,18.
L’échantillon visqueux le plus pauvre en gluténines (R0,04) reste visqueux mais sa viscosité est
440 fois supérieure à -4°C qu’à 25°C (50 mPa.s à 22 Pa.s). L’échantillon le plus riche en
gluténines (R1,88) reste un gel quelle que soit la température mais avec un module 3 fois plus
important à basse température (de 240 Pa à 830 Pa). L’échantillon intermédiaire R0,83, qui
est un liquide visqueux à température ambiante se comporte comme un gel critique à 5°C. En
effet, les modules de stockage G’ et le module de perte G’’ suivent tous les deux une même loi
de puissance selon laquelle G’ G’’ avec . Ce phénomène a également été observé
sur un système glu+gli, R1,1 où (Dahesh, et al., 2016).
a
b
c
0,5
1
1

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
181
2. Observation de la séparation de phases par diffusion
2.1 Dynamique de séparation de phases au cours d’une trempe en température
Les mesures à température ambiante ont démontré une forte évolution du signal en solvant
deutéré par rapport au solvant hydrogéné (Chapitre 6). C’est pourquoi, des mesures ont été
réalisées en température au CEA de Saclay, sur un échantillon R1,88 à = 0,18 en solvant
deutéré. Les résultats préliminaires ont montré qu’il n’existe aucune évolution du signal quelle
que soit la température (Fig.7.4a). Or, d’après nos précédentes observations, l’échantillon
R1,88 sépare de phases à 11°C ce qui suggère un réarrangement des polymères que nous
supposions possible d’observer avec la technique SANS. C’est pourquoi pour avoir un contraste
protéines/solvant différent, les mesures ont été réalisées dans les mêmes conditions en solvant
hydrogéné. Les premiers essais ont montré une évolution du signal en fonction de la
température. Dans un premier temps, nous avons vérifié la réversibilité du phénomène pour
confirmer qu’il s’agissait bien d’une séparation de phases ainsi que pour voir si l’échantillon
n’était pas dénaturé suite à la mesure en température. Pour cela, l’échantillon R1,88, = 0,18,
en solvant hydrogéné, a été mesuré à grands q (2.10-3 < q < 0,2 nm-1) à 20°C. Il a ensuite subi
une trempe en température à la suite de laquelle il a été remonté à 20°C et remesuré dans les
mêmes conditions que précédemment (Fig.7.4b). Comme la réversibilité du phénomène a été
confirmé, nous avons voulu compléter ces résultats sur une plus large gamme de q et de
températures. C’est pourquoi nous avons réalisé ces mêmes mesures au JCNS de Garching en
Allemagne. La Figure 7.4c expose l’ensemble des profils obtenus à différentes températures
pour l’échantillon R1,88 à = 0,18.
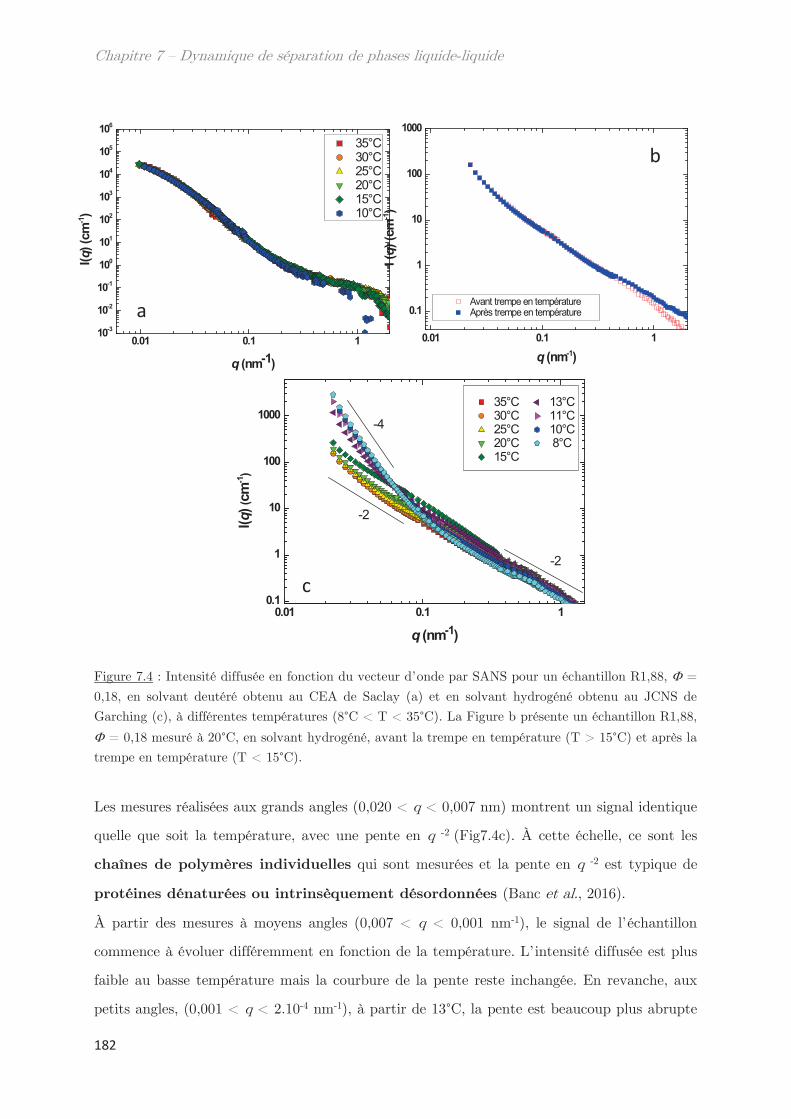
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
182
Figure 7.4 : Intensité diffusée en fonction du vecteur d’onde par SANS pour un échantillon R1,88, =
0,18, en solvant deutéré obtenu au CEA de Saclay (a) et en solvant hydrogéné obtenu au JCNS de
Garching (c), à différentes températures (8°C < T < 35°C). La Figure b présente un échantillon R1,88,
= 0,18 mesuré à 20°C, en solvant hydrogéné, avant la trempe en température (T > 15°C) et après la
trempe en température (T < 15°C).
Les mesures réalisées aux grands angles (0,020 < q < 0,007 nm) montrent un signal identique
quelle que soit la température, avec une pente en q -2 (Fig7.4c). À cette échelle, ce sont les
chaînes de polymères individuelles qui sont mesurées et la pente en q -2 est typique de
protéines dénaturées ou intrinsèquement désordonnées (Banc et al., 2016).
À partir des mesures à moyens angles (0,007 < q < 0,001 nm-1), le signal de l’échantillon
commence à évoluer différemment en fonction de la température. L’intensité diffusée est plus
faible au basse température mais la courbure de la pente reste inchangée. En revanche, aux
petits angles, (0,001 < q < 2.10-4 nm-1), à partir de 13°C, la pente est beaucoup plus abrupte
0.01 0.1 1
0.1
1
10
100
1000
Avant trempe en température Après trempe en température
I(q
)(c
m-1)
q (nm-1)
0.01 0.1 110
-3
10-2
10-1
100
101
102
103
104
105
106
35°C 30°C 25°C 20°C 15°C 10°C
I(q)(c
m-1)
q (nm-1)
a
b
c 0.01 0.1 1
0.1
1
10
100
100035°C 13°C30°C 11°C25°C 10°C20°C 8°C15°C
I(q)
(cm
-1)
q (nm-1)
-4
-2
-2

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
183
et passe de q -2 à q -4. Ce changement de pente témoigne de la formation d’interfaces
nettes entre les deux phases formées (Dhont, 1996).
Cependant, ces mesures ne permettent pas de déterminer précisément la taille caractéristique
des régions avec des interfaces nettes. C’est pourquoi, nous avons réalisé des mesures
complémentaires en diffusion de rayons X aux très petits angles (0,002 < q < 10 nm-1) (SAXS)
pour en apprendre davantage sur les mécanismes de séparation.
2.2 Cinétique de croissance de la longueur caractéristique
2.2.1 Profil classique d’une décomposition spinodale
Les mesures SAXS, ont également été réalisées sur des gels de gluten, = 0,18, sur une gamme
de température de 20°C à -12°C afin d’avoir une température de trempe T < Tc pour tous les
échantillons. Pour recouvrir l’ensemble de la gamme de rapport glu/gli, les échantillons les plus
enrichis en gliadines (R0,04), les plus enrichis en gluténines (R1,88) et un rapport intermédiaire,
(R0,83) ont été étudiés. Par ailleurs, la structure de l’échantillon est sondée après une trempe
en température de 20°C vers une température plus basse, en suivant une cinétique de -
80°C/min. La dynamique est suivie sur une durée d'environ 270 s avec un espacement
logarithmique de l'acquisition des points de données. Cela permet d'atteindre une large plage
dynamique tout en ne mesurant que 50 points de données et en limitant la dégradation de
l’échantillon par le faisceau X. Chaque mesure permet d’obtenir un spectre de l’intensité
diffusée en fonction du vecteur d’onde q. L’intensité de l’ensemble de ces spectres est corrigée
par l’intensité du spectre mesuré à 20°C (Fig.7.5).
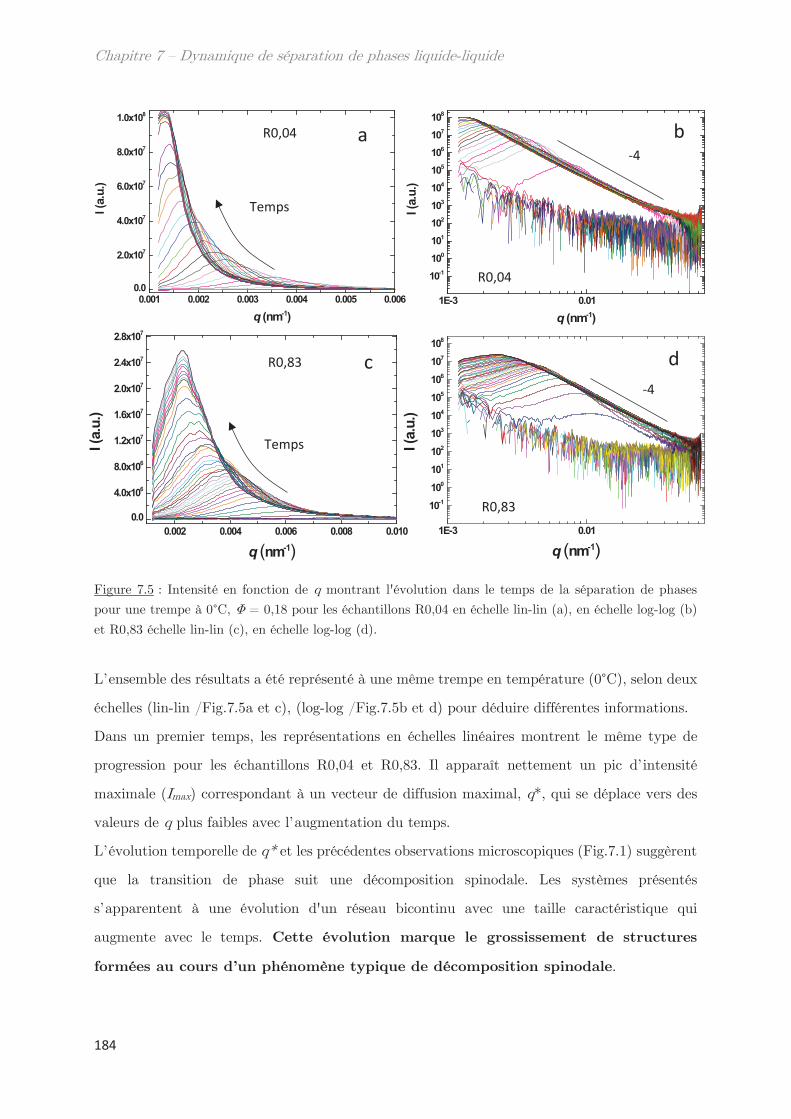
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
184
Figure 7.5 : Intensité en fonction de q montrant l'évolution dans le temps de la séparation de phases
pour une trempe à 0°C, = 0,18 pour les échantillons R0,04 en échelle lin-lin (a), en échelle log-log (b)
et R0,83 échelle lin-lin (c), en échelle log-log (d).
L’ensemble des résultats a été représenté à une même trempe en température (0°C), selon deux
échelles (lin-lin /Fig.7.5a et c), (log-log /Fig.7.5b et d) pour déduire différentes informations.
Dans un premier temps, les représentations en échelles linéaires montrent le même type de
progression pour les échantillons R0,04 et R0,83. Il apparaît nettement un pic d’intensité
maximale (Imax) correspondant à un vecteur de diffusion maximal, q*, qui se déplace vers des
valeurs de q plus faibles avec l’augmentation du temps.
L’évolution temporelle de q* et les précédentes observations microscopiques (Fig.7.1) suggèrent
que la transition de phase suit une décomposition spinodale. Les systèmes présentés
s’apparentent à une évolution d'un réseau bicontinu avec une taille caractéristique qui
augmente avec le temps. Cette évolution marque le grossissement de structures
formées au cours d’un phénomène typique de décomposition spinodale.
-4
a b
Temps
Temps
R0,83 c
R0,83
-4
d
R0,04
R0,04
0.001 0.002 0.003 0.004 0.005 0.0060.0
2.0x107
4.0x107
6.0x107
8.0x107
1.0x108
I (a
.u.)
q (nm-1)
1E-3 0.01
10-1
100
101
102
103
104
105
106
107
108
I (a
.u.)
q (nm-1)
0.002 0.004 0.006 0.008 0.010
0.0
4.0x106
8.0x106
1.2x107
1.6x107
2.0x107
2.4x107
2.8x107
I (a
.u.)
q (nm-1)
1E-3 0.01
10-1
100
101
102
103
104
105
106
107
108
I (a
.u.)
q (nm-1)
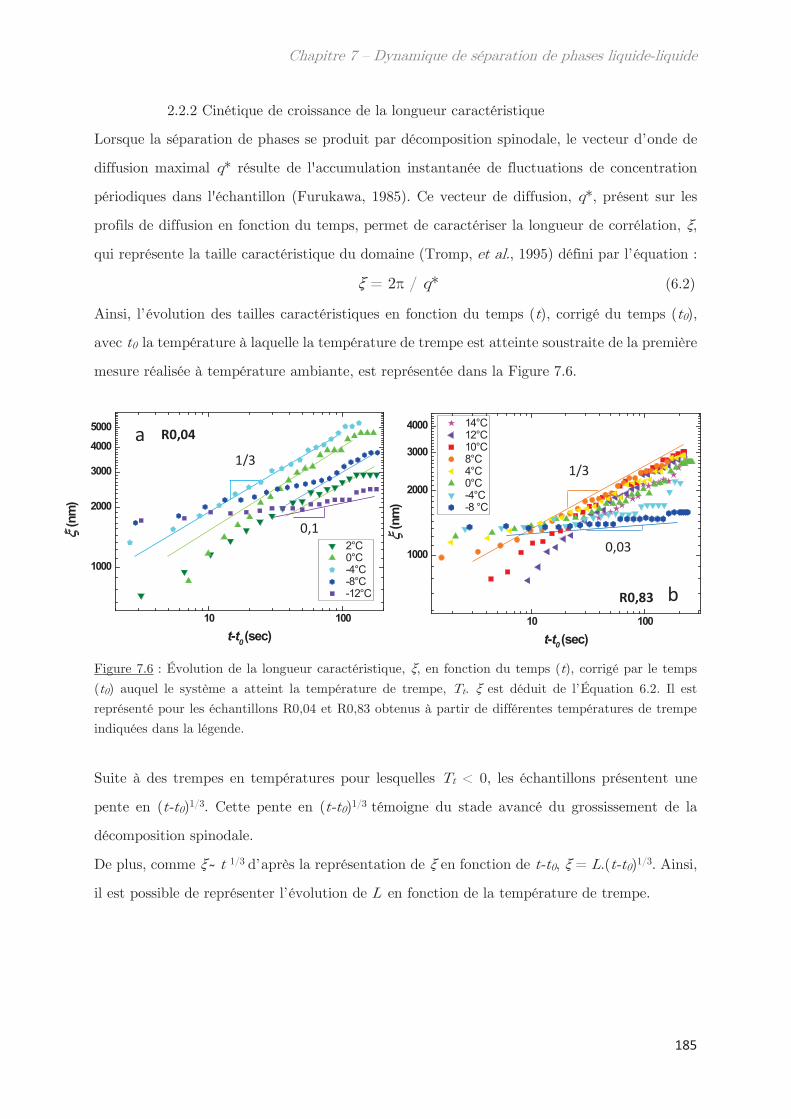
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
185
2.2.2 Cinétique de croissance de la longueur caractéristique
Lorsque la séparation de phases se produit par décomposition spinodale, le vecteur d’onde de
diffusion maximal q* résulte de l'accumulation instantanée de fluctuations de concentration
périodiques dans l'échantillon (Furukawa, 1985). Ce vecteur de diffusion, q*, présent sur les
profils de diffusion en fonction du temps, permet de caractériser la longueur de corrélation, ,
qui représente la taille caractéristique du domaine (Tromp, et al., 1995) défini par l’équation :
q* (6.2)
Ainsi, l’évolution des tailles caractéristiques en fonction du temps (t), corrigé du temps (t0),
avec t0 la température à laquelle la température de trempe est atteinte soustraite de la première
mesure réalisée à température ambiante, est représentée dans la Figure 7.6.
Figure 7.6 : Évolution de la longueur caractéristique, , en fonction du temps (t), corrigé par le temps
(t0) auquel le système a atteint la température de trempe, Tt. est déduit de l’Équation 6.2. Il est
représenté pour les échantillons R0,04 et R0,83 obtenus à partir de différentes températures de trempe
indiquées dans la légende.
Suite à des trempes en températures pour lesquelles Tt < 0, les échantillons présentent une
pente en (t-t0)1/3. Cette pente en (t-t0)1/3 témoigne du stade avancé du grossissement de la
décomposition spinodale.
De plus, comme t 1/3 d’après la représentation de en fonction de t-t0, = L.(t-t0)1/3. Ainsi,
il est possible de représenter l’évolution de L en fonction de la température de trempe.
a
b
R0,04
R0,83
1/31/3
0,03
0,1
10 100
1000
2000
3000
4000 14°C 12°C 10°C 8°C4°C0°C-4°C-8 °C
(nm
)
t-t0(sec)
10 100
1000
2000
3000
4000
5000
2°C0°C-4°C-8°C-12°C
(nm
)
t-t0(sec)
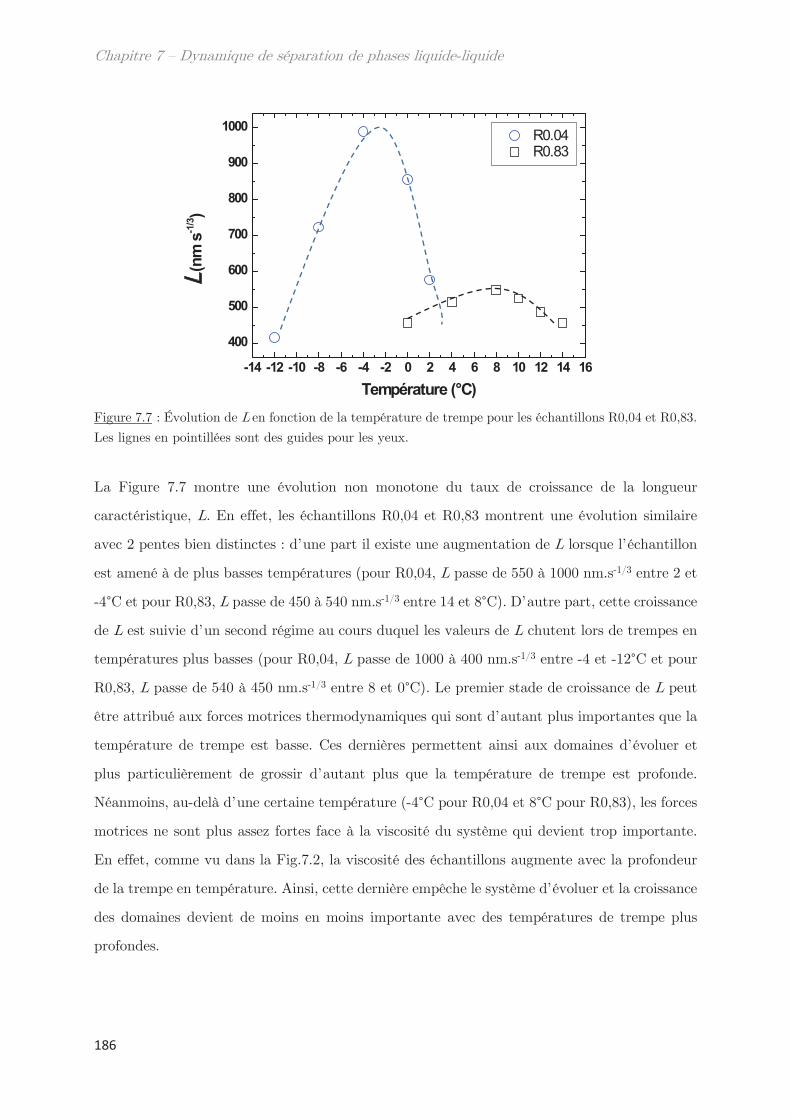
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
186
Figure 7.7 : Évolution de L en fonction de la température de trempe pour les échantillons R0,04 et R0,83.
Les lignes en pointillées sont des guides pour les yeux.
La Figure 7.7 montre une évolution non monotone du taux de croissance de la longueur
caractéristique, L. En effet, les échantillons R0,04 et R0,83 montrent une évolution similaire
avec 2 pentes bien distinctes : d’une part il existe une augmentation de L lorsque l’échantillon
est amené à de plus basses températures (pour R0,04, L passe de 550 à 1000 nm.s-1/3 entre 2 et
-4°C et pour R0,83, L passe de 450 à 540 nm.s-1/3 entre 14 et 8°C). D’autre part, cette croissance
de L est suivie d’un second régime au cours duquel les valeurs de L chutent lors de trempes en
températures plus basses (pour R0,04, L passe de 1000 à 400 nm.s-1/3 entre -4 et -12°C et pour
R0,83, L passe de 540 à 450 nm.s-1/3 entre 8 et 0°C). Le premier stade de croissance de L peut
être attribué aux forces motrices thermodynamiques qui sont d’autant plus importantes que la
température de trempe est basse. Ces dernières permettent ainsi aux domaines d’évoluer et
plus particulièrement de grossir d’autant plus que la température de trempe est profonde.
Néanmoins, au-delà d’une certaine température (-4°C pour R0,04 et 8°C pour R0,83), les forces
motrices ne sont plus assez fortes face à la viscosité du système qui devient trop importante.
En effet, comme vu dans la Fig.7.2, la viscosité des échantillons augmente avec la profondeur
de la trempe en température. Ainsi, cette dernière empêche le système d’évoluer et la croissance
des domaines devient de moins en moins importante avec des températures de trempe plus
profondes.
-14 -12 -10 -8 -6 -4 -2 0 2 4 6 8 10 12 14 16
400
500
600
700
800
900
1000 R0.04 R0.83
(nm
s-1
/3)
Température (°C)

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
187
De plus, cette représentation montre également que l’amplitude de L est beaucoup plus
importante pour l’échantillon R0,04 que R0,83. Cette observation correspond aux attentes
théoriques selon lesquelles, comme vu dans la Figure 6.3, l’échantillon R0,04 est beaucoup
moins visqueux que l’échantillon R0,83. Ainsi, basé sur les mêmes observations que
précédemment, l’échantillon le moins visqueux permettra au taux de croissance de la longueur
caractéristique d’être plus important.
Par ailleurs, le décalage entre les deux échantillons est principalement dû à la dépendance de
la température du diagramme de phases pour chaque échantillon. Comme observé
précédemment avec un rapport glu/gli plus faible, la séparation de phases se déroule à plus
basse température (cf Chapitre 5). D’autre part, cette représentation permet de déterminer la
température à laquelle le système commence à séparer de phases. Les valeurs sont reportées
dans la Table 7.2 qui reprend également pour comparaison les températures de transitions à
= 0,18 de la Table 7.1, obtenues par rhéologie et par MDSC. (À noter que la valeur de la
température de séparation de phases pour R1,88 n’est pas présentée sur la Figure 7.7 mais sera
détaillé un peu plus loin dans ce chapitre).
Table 7.2 : Comparaison de la température de séparation de phases, Tr, obtenue par rhéologie, à la
température Tt, obtenue par MDSC et de la première température, Ts, à laquelle le système commence
à séparer de phases en SAXS, = 0,18.
Échantillons Tr rhéologie (°C) Tt MDSC (°C) Ts SAXS (°C)
R0,04 7 ± 1 5 ± 1 4 ± 2
R0,83 8 ± 0,5 8 ± 1 14 ± 2
R1,34 11 ± 0,5 11 ± 1 /
R1,88 12 ± 0,5 11 ± 1 12 ± 2
La Table 7.2 montre que les températures de séparation de phases à = 0,18 en SAXS sont
identiques à celles trouvées par les autres techniques pour les échantillons R0,04 ( 5°C) et
R1,88 ( 12°C). En revanche, la température trouvée en SAXS pour l’échantillon R0,83 est
nettement supérieure (14°C) à celles trouvées par rhéologie et MDSC (8°C). Cette différence
peut s’expliquer par une évaporation d’une partie du solvant qui compose la solution. En effet,
cette évaporation a pu avoir lieu au moment de la préparation de l’échantillon qu’il est difficile
de mettre dans des capillaires (de par son caractère visqueux) ou d’un mauvais scellage du
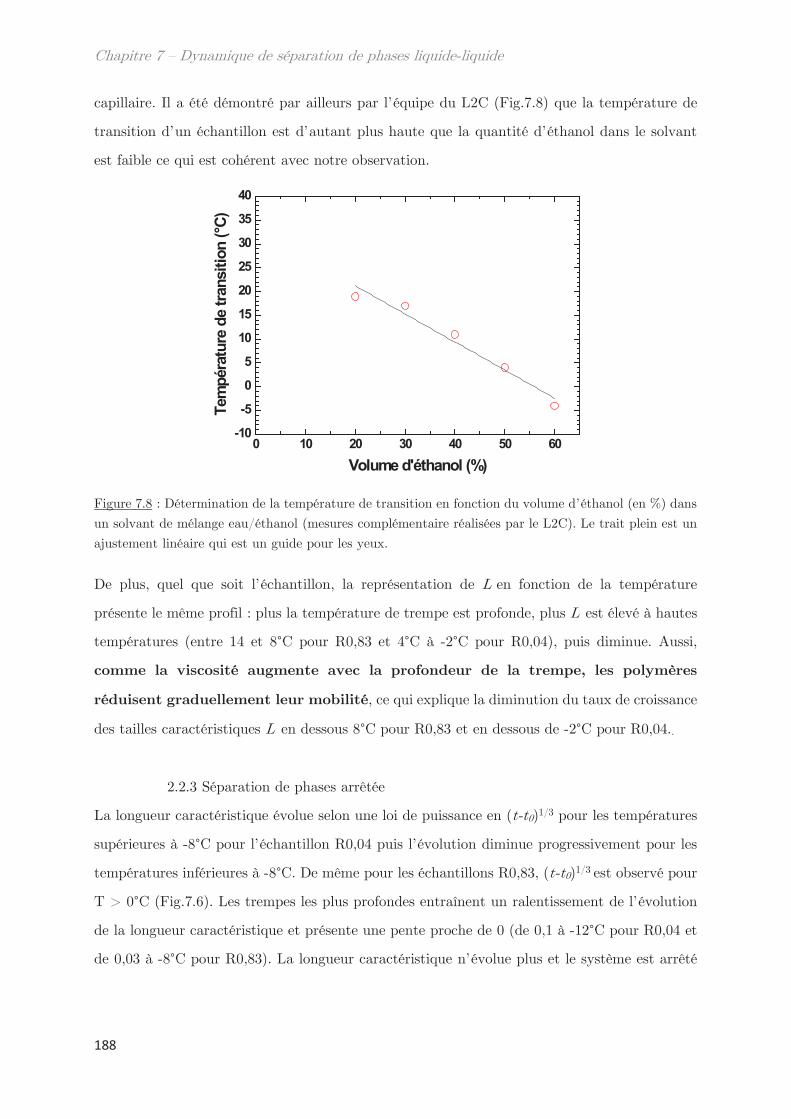
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
188
capillaire. Il a été démontré par ailleurs par l’équipe du L2C (Fig.7.8) que la température de
transition d’un échantillon est d’autant plus haute que la quantité d’éthanol dans le solvant
est faible ce qui est cohérent avec notre observation.
Figure 7.8 : Détermination de la température de transition en fonction du volume d’éthanol (en %) dans
un solvant de mélange eau/éthanol (mesures complémentaire réalisées par le L2C). Le trait plein est un
ajustement linéaire qui est un guide pour les yeux.
De plus, quel que soit l’échantillon, la représentation de L en fonction de la température
présente le même profil : plus la température de trempe est profonde, plus L est élevé à hautes
températures (entre 14 et 8°C pour R0,83 et 4°C à -2°C pour R0,04), puis diminue. Aussi,
comme la viscosité augmente avec la profondeur de la trempe, les polymères
réduisent graduellement leur mobilité, ce qui explique la diminution du taux de croissance
des tailles caractéristiques L en dessous 8°C pour R0,83 et en dessous de -2°C pour R0,04..
2.2.3 Séparation de phases arrêtée
La longueur caractéristique évolue selon une loi de puissance en (t-t0)1/3 pour les températures
supérieures à -8°C pour l’échantillon R0,04 puis l’évolution diminue progressivement pour les
températures inférieures à -8°C. De même pour les échantillons R0,83, (t-t0)1/3 est observé pour
T > 0°C (Fig.7.6). Les trempes les plus profondes entraînent un ralentissement de l’évolution
de la longueur caractéristique et présente une pente proche de 0 (de 0,1 à -12°C pour R0,04 et
de 0,03 à -8°C pour R0,83). La longueur caractéristique n’évolue plus et le système est arrêté
0 10 20 30 40 50 60-10
-5
0
5
10
15
20
25
30
35
40
Tem
péra
ture
de tra
nsitio
n (°C
)
Volume d'éthanol (%)

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
189
cinétiquement. Il s’agit d’un phénomène de séparation de phases arrêtée qui se produit
au cours de la séparation spinodale.
Il est important de noter que la séparation de phases arrêtée est plus couramment observée
lorsque la vitesse de trempe est importante. En effet, dans notre cas la cinétique de descente
en température est très rapide (-80 K/min) ce qui explique ce phénomène.
2.2.4 Facteur de Porod
D’autre part, les représentations en échelles log-log (Fig.7.5b et 7.5d) montrent une pente en
q -4 aux grands vecteurs de diffusions, 0,003 < q < 0,06 nm, pour les deux échantillons. Cette
loi d’échelle, précédemment observée en SANS, confirme à nouveau le passage à des interfaces
nettes. L’intensité diffusée par ces interfaces peut être quantifiée par la loi de Porod
(Koberstein, et al., 1980) selon laquelle:
IPorod(q) = (6.3)
avec KPorod = ²
(6.4)
où ² est la différence de densité de diffusion entre la phase riche et la phase pauvre en
protéines, S, la surface totale d’interface pour un volume, V, d’échantillon. La Figure 7.9
montre l’évolution du préfacteur ² * de Porod suite à la séparation phase à différentes
températures de trempe.
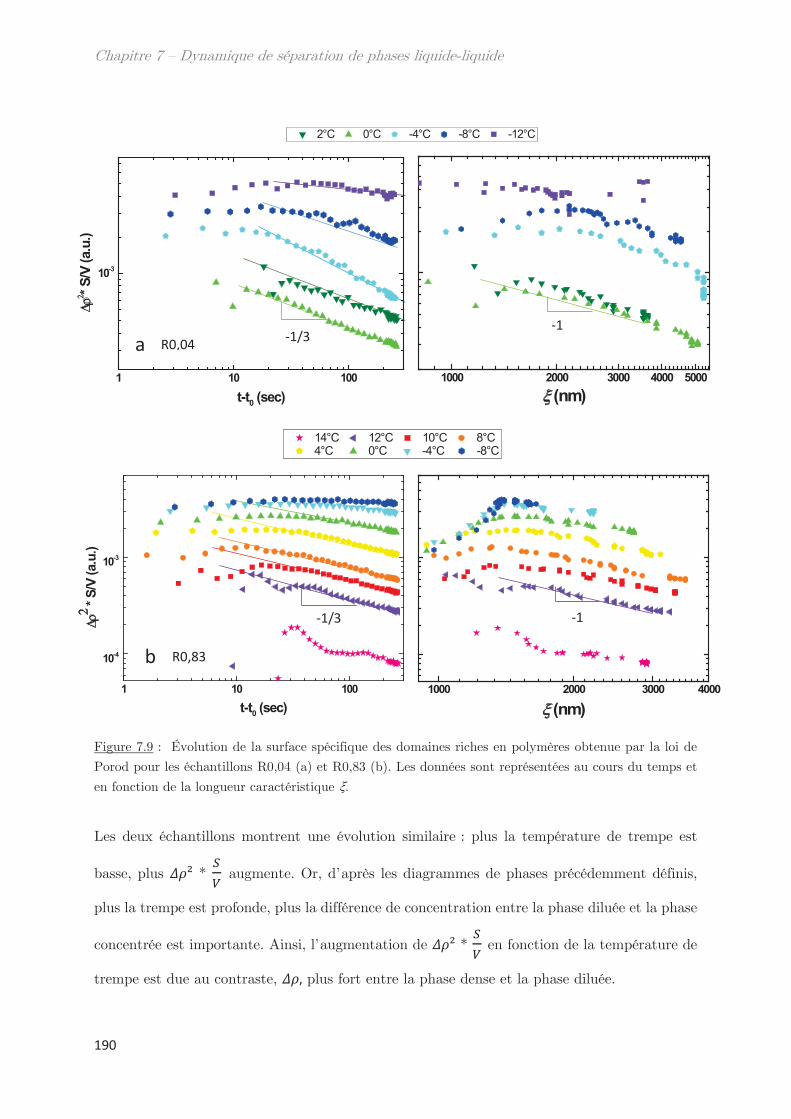
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
190
Figure 7.9 : Évolution de la surface spécifique des domaines riches en polymères obtenue par la loi de
Porod pour les échantillons R0,04 (a) et R0,83 (b). Les données sont représentées au cours du temps et
en fonction de la longueur caractéristique .
Les deux échantillons montrent une évolution similaire : plus la température de trempe est
basse, plus ² * augmente. Or, d’après les diagrammes de phases précédemment définis,
plus la trempe est profonde, plus la différence de concentration entre la phase diluée et la phase
concentrée est importante. Ainsi, l’augmentation de ² * en fonction de la température de
trempe est due au contraste, , plus fort entre la phase dense et la phase diluée.
a R0,04
R0,83 b
-1/3
-1/3 -1
-1
1 10 100
10-3
* S
/V (a.u
.)
t-t0 (sec)
2°C 0°C -4°C -8°C -12°C
14°C 12°C 10°C 8°C 4°C 0°C -4°C -8°C
1 10 100
10-4
10-3
* S
/V (a.u
.)
t-t0 (sec)
1000 2000 3000 4000
(nm)
1000 2000 3000 4000 5000
(nm)
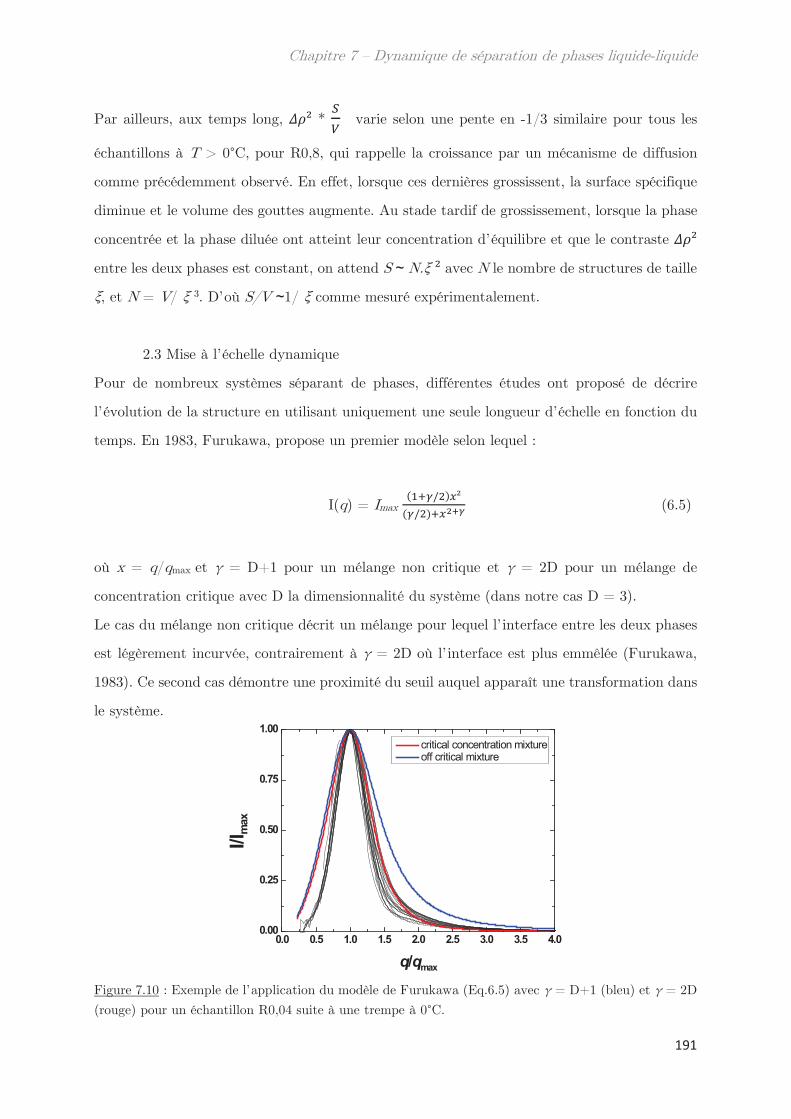
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
191
Par ailleurs, aux temps long, ² * varie selon une pente en -1/3 similaire pour tous les
échantillons à T > 0°C, pour R0,8, qui rappelle la croissance par un mécanisme de diffusion
comme précédemment observé. En effet, lorsque ces dernières grossissent, la surface spécifique
diminue et le volume des gouttes augmente. Au stade tardif de grossissement, lorsque la phase
concentrée et la phase diluée ont atteint leur concentration d’équilibre et que le contraste ²
entre les deux phases est constant, on attend S ~ N. ² avec N le nombre de structures de taille
, et N = V/ 3. D’où S/V ~1/ comme mesuré expérimentalement.
2.3 Mise à l’échelle dynamique
Pour de nombreux systèmes séparant de phases, différentes études ont proposé de décrire
l’évolution de la structure en utilisant uniquement une seule longueur d’échelle en fonction du
temps. En 1983, Furukawa, propose un premier modèle selon lequel :
I(q) = Imax ( / ) ²
( / ) (6.5)
où x = q/qmax et = D+1 pour un mélange non critique et = 2D pour un mélange de
concentration critique avec D la dimensionnalité du système (dans notre cas D = 3).
Le cas du mélange non critique décrit un mélange pour lequel l’interface entre les deux phases
est légèrement incurvée, contrairement à = 2D où l’interface est plus emmêlée (Furukawa,
1983). Ce second cas démontre une proximité du seuil auquel apparaît une transformation dans
le système.
Figure 7.10 : Exemple de l’application du modèle de Furukawa (Eq.6.5) avec = D+1 (bleu) et = 2D
(rouge) pour un échantillon R0,04 suite à une trempe à 0°C.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.00.00
0.25
0.50
0.75
1.00
I/I m
ax
q/qmax
critical concentration mixture off critical mixture
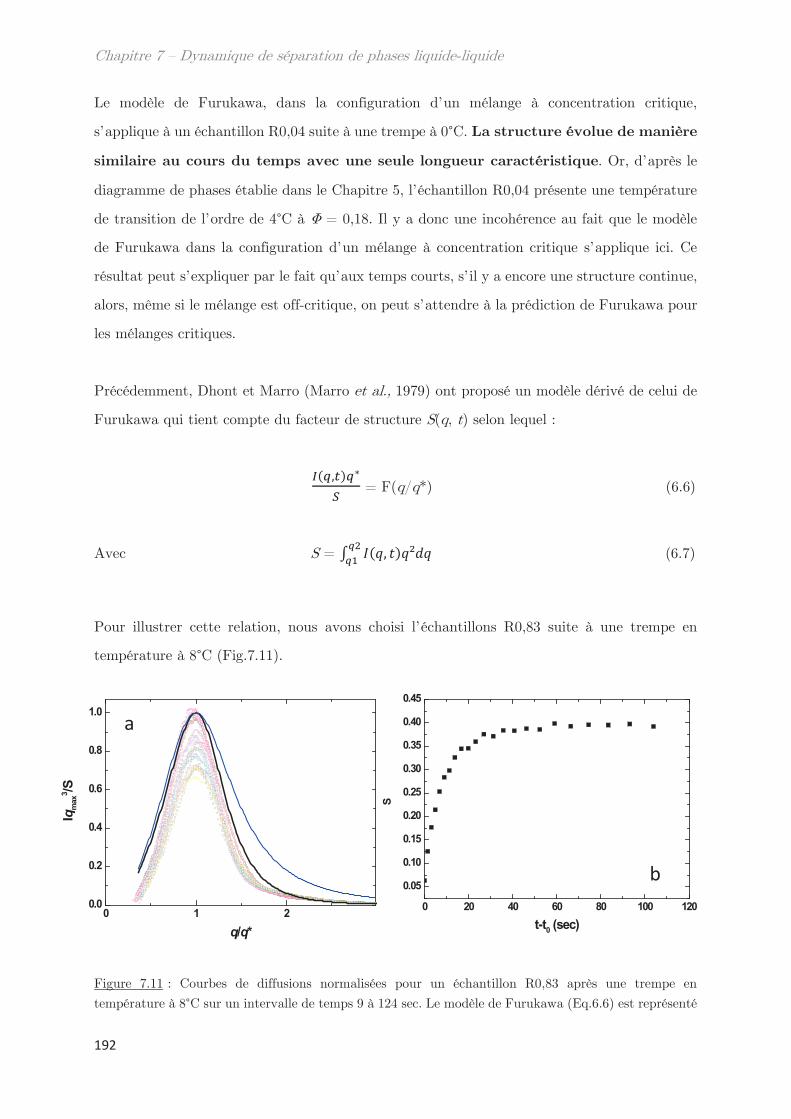
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
192
Le modèle de Furukawa, dans la configuration d’un mélange à concentration critique,
s’applique à un échantillon R0,04 suite à une trempe à 0°C. La structure évolue de manière
similaire au cours du temps avec une seule longueur caractéristique. Or, d’après le
diagramme de phases établie dans le Chapitre 5, l’échantillon R0,04 présente une température
de transition de l’ordre de 4°C à = 0,18. Il y a donc une incohérence au fait que le modèle
de Furukawa dans la configuration d’un mélange à concentration critique s’applique ici. Ce
résultat peut s’expliquer par le fait qu’aux temps courts, s’il y a encore une structure continue,
alors, même si le mélange est off-critique, on peut s’attendre à la prédiction de Furukawa pour
les mélanges critiques.
Précédemment, Dhont et Marro (Marro et al., 1979) ont proposé un modèle dérivé de celui de
Furukawa qui tient compte du facteur de structure S(q, t) selon lequel :
( , )
= F(q/q*) (6.6)
Avec S = ( , ) ² (6.7)
Pour illustrer cette relation, nous avons choisi l’échantillons R0,83 suite à une trempe en
température à 8°C (Fig.7.11).
Figure 7.11 : Courbes de diffusions normalisées pour un échantillon R0,83 après une trempe en
température à 8°C sur un intervalle de temps 9 à 124 sec. Le modèle de Furukawa (Eq.6.6) est représenté
0 20 40 60 80 100 120
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
S
t-t0 (sec)
0 1 20.0
0.2
0.4
0.6
0.8
1.0
Iqm
ax
3/S
q/q*
a
b
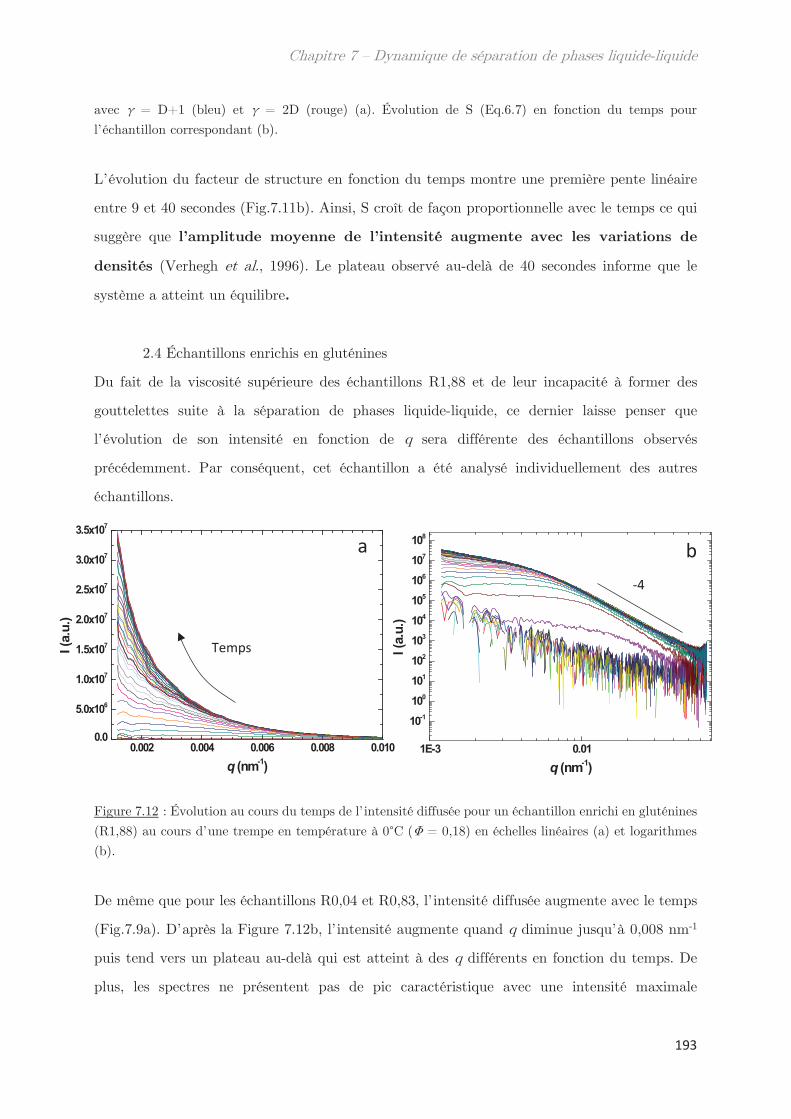
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
193
avec = D+1 (bleu) et = 2D (rouge) (a). Évolution de S (Eq.6.7) en fonction du temps pour
l’échantillon correspondant (b).
L’évolution du facteur de structure en fonction du temps montre une première pente linéaire
entre 9 et 40 secondes (Fig.7.11b). Ainsi, S croît de façon proportionnelle avec le temps ce qui
suggère que l’amplitude moyenne de l’intensité augmente avec les variations de
densités (Verhegh et al., 1996). Le plateau observé au-delà de 40 secondes informe que le
système a atteint un équilibre.
2.4 Échantillons enrichis en gluténines
Du fait de la viscosité supérieure des échantillons R1,88 et de leur incapacité à former des
gouttelettes suite à la séparation de phases liquide-liquide, ce dernier laisse penser que
l’évolution de son intensité en fonction de q sera différente des échantillons observés
précédemment. Par conséquent, cet échantillon a été analysé individuellement des autres
échantillons.
Figure 7.12 : Évolution au cours du temps de l’intensité diffusée pour un échantillon enrichi en gluténines
(R1,88) au cours d’une trempe en température à 0°C ( = 0,18) en échelles linéaires (a) et logarithmes
(b).
De même que pour les échantillons R0,04 et R0,83, l’intensité diffusée augmente avec le temps
(Fig.7.9a). D’après la Figure 7.12b, l’intensité augmente quand q diminue jusqu’à 0,008 nm-1
puis tend vers un plateau au-delà qui est atteint à des q différents en fonction du temps. De
plus, les spectres ne présentent pas de pic caractéristique avec une intensité maximale
b a
-4
Temps
1E-3 0.01
10-1
100
101
102
103
104
105
106
107
108
I (a
.u.)
q (nm-1)
0.002 0.004 0.006 0.008 0.0100.0
5.0x106
1.0x107
1.5x107
2.0x107
2.5x107
3.0x107
3.5x107
I (a
.u.)
q (nm-1)
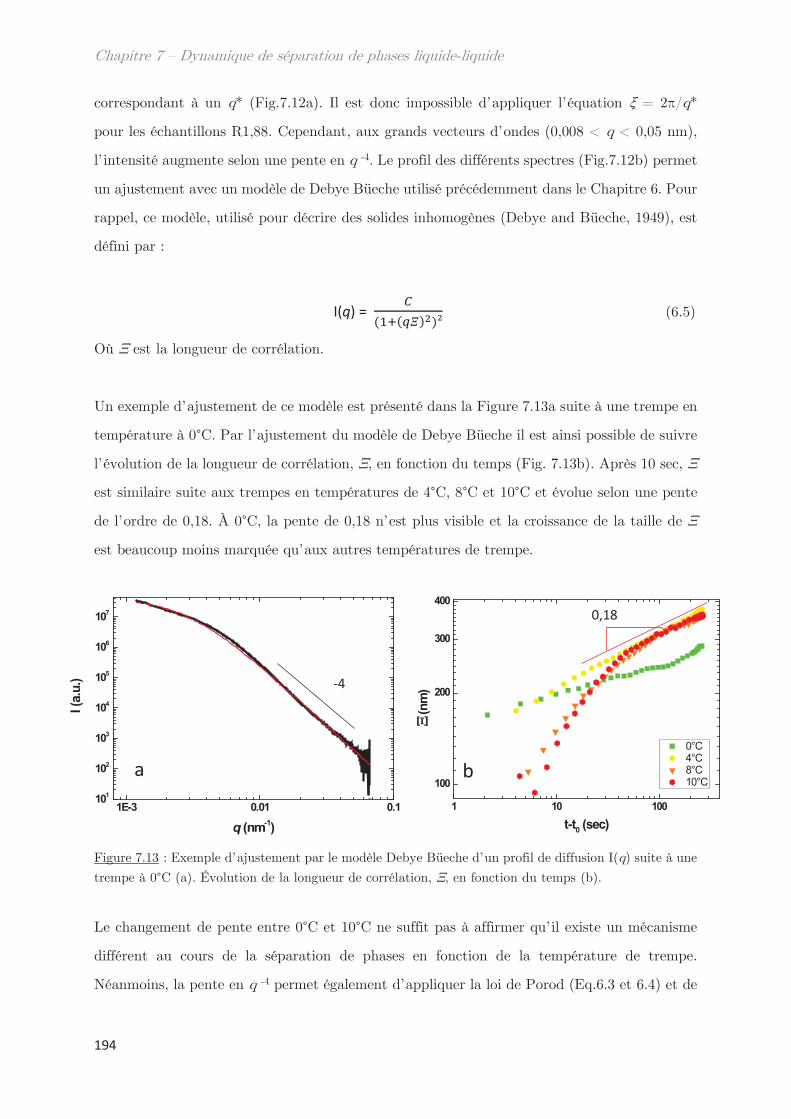
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
194
correspondant à un q* (Fig.7.12a). Il est donc impossible d’appliquer l’équation = q*
pour les échantillons R1,88. Cependant, aux grands vecteurs d’ondes (0,008 < q < 0,05 nm),
l’intensité augmente selon une pente en q -4. Le profil des différents spectres (Fig.7.12b) permet
un ajustement avec un modèle de Debye Büeche utilisé précédemment dans le Chapitre 6. Pour
rappel, ce modèle, utilisé pour décrire des solides inhomogènes (Debye and Büeche, 1949), est
défini par :
I(q) = ( ( ) )²
(6.5)
Où est la longueur de corrélation.
Un exemple d’ajustement de ce modèle est présenté dans la Figure 7.13a suite à une trempe en
température à 0°C. Par l’ajustement du modèle de Debye Büeche il est ainsi possible de suivre
l’évolution de la longueur de corrélation, , en fonction du temps (Fig. 7.13b). Après 10 sec,
est similaire suite aux trempes en températures de 4°C, 8°C et 10°C et évolue selon une pente
de l’ordre de 0,18. À 0°C, la pente de 0,18 n’est plus visible et la croissance de la taille de
est beaucoup moins marquée qu’aux autres températures de trempe.
Figure 7.13 : Exemple d’ajustement par le modèle Debye Büeche d’un profil de diffusion I(q) suite à une
trempe à 0°C (a). Évolution de la longueur de corrélation, , en fonction du temps (b).
Le changement de pente entre 0°C et 10°C ne suffit pas à affirmer qu’il existe un mécanisme
différent au cours de la séparation de phases en fonction de la température de trempe.
Néanmoins, la pente en q -4 permet également d’appliquer la loi de Porod (Eq.6.3 et 6.4) et de
a
-4
0,18
b
1 10 100
100
200
300
400
0°C 4°C 8°C 10°C
(nm
)
t-t0 (sec)
1E-3 0.01 0.110
1
102
103
104
105
106
107
I (a
.u.)
q (nm-1)
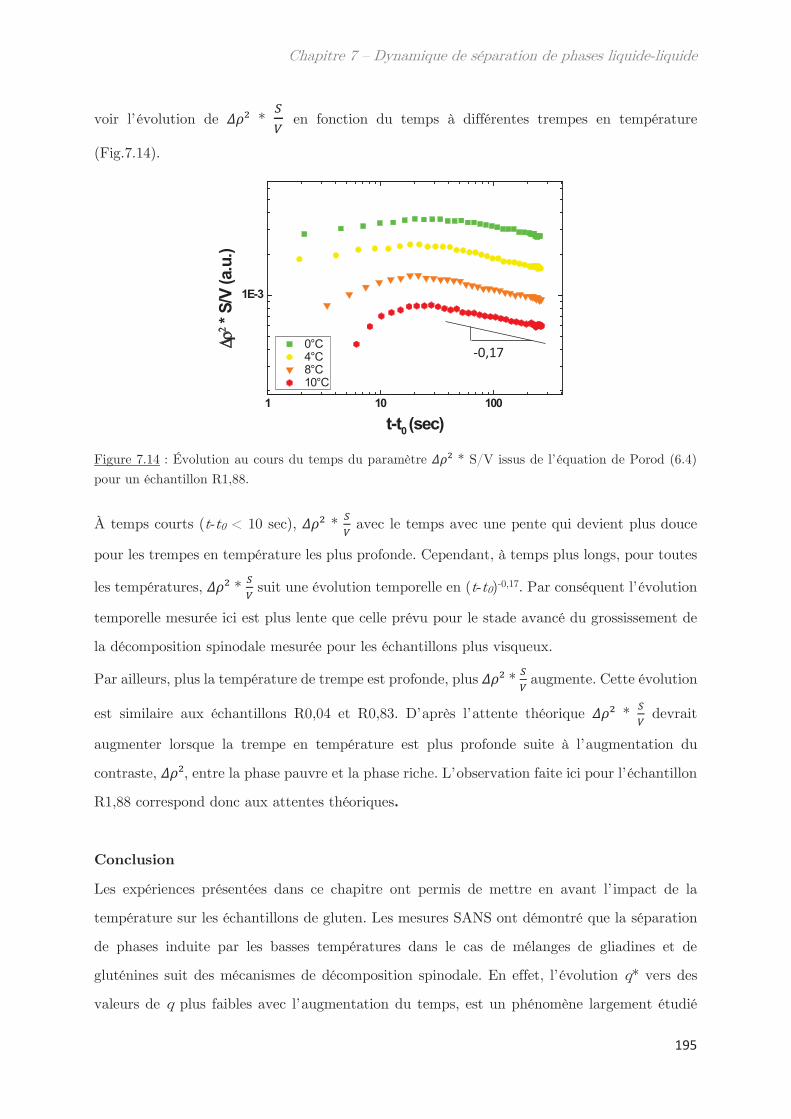
Chapitre 7 – Dynamique de séparation de phases liquide-liquide
195
voir l’évolution de ² * en fonction du temps à différentes trempes en température
(Fig.7.14).
Figure 7.14 : Évolution au cours du temps du paramètre ² * S/V issus de l’équation de Porod (6.4)
pour un échantillon R1,88.
À temps courts (t-t0 < 10 sec), ² * avec le temps avec une pente qui devient plus douce
pour les trempes en température les plus profonde. Cependant, à temps plus longs, pour toutes
les températures, ² * suit une évolution temporelle en (t-t0)-0,17. Par conséquent l’évolution
temporelle mesurée ici est plus lente que celle prévu pour le stade avancé du grossissement de
la décomposition spinodale mesurée pour les échantillons plus visqueux.
Par ailleurs, plus la température de trempe est profonde, plus ² * augmente. Cette évolution
est similaire aux échantillons R0,04 et R0,83. D’après l’attente théorique ² * devrait
augmenter lorsque la trempe en température est plus profonde suite à l’augmentation du
contraste, ², entre la phase pauvre et la phase riche. L’observation faite ici pour l’échantillon
R1,88 correspond donc aux attentes théoriques.
Conclusion
Les expériences présentées dans ce chapitre ont permis de mettre en avant l’impact de la
température sur les échantillons de gluten. Les mesures SANS ont démontré que la séparation
de phases induite par les basses températures dans le cas de mélanges de gliadines et de
gluténines suit des mécanismes de décomposition spinodale. En effet, l’évolution q* vers des
valeurs de q plus faibles avec l’augmentation du temps, est un phénomène largement étudié
-0,17
1 10 100
1E-3
0°C 4°C 8°C 10°C
* S
/V (a.u
.)
t-t0(sec)

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
196
sur d’autres systèmes. D’après la littérature, un comportement similaire a été observé pour
d’autres protéines telles que l’albumine de sérum bovin (Da Vela, et al., 2016) et le lysozyme
(Gibaud, et al., 2009). D’autres systèmes tels que des solutions colloïdales (Verhaegh, et al.,
1996) ont également montré ce type de résultats. Les échantillons présentent une évolution
temporelle de la taille caractéristique, , qui suit une loi de puissance en t 1/3 distinctif
d’une décomposition spinodale. Ce phénomène est notamment confirmé et illustré par les
images de microscopie qui montrent une évolution bi-continue caractéristique de la
décomposition spinodale dès les premières secondes après que la température de trempe soit
atteinte.
Néanmoins, au-delà d’une certaine température de trempe (Tt < -8°C pour R0,04 et Tt < 0°C
pour R0,83), c’est-à-dire pour des trempes plus profondes, l’évolution de ne suit plus cette
même loi. D’après les mesures de rhéologie, suite à une trempe en température les modules
élastiques augmentent pour tous les échantillons. Quelle que soit la composition en gluténines
et gliadines, les faibles températures font augmenter la viscosité jusqu’à obtenir un gel critique
pour les échantillons R0,83. Ainsi, la viscosité du système devient si importante que la
séparation de phases est stoppée suite à des trempes en température profondes. Ce phénomène,
connu sous le nom « séparation de phases arrêtée », a également été observé sur le
lysozyme (Gibaud, et al., 2009) ou encore sur des solutions de polypeptides (Glassman, et al.,
2015). Il suggère que la mobilité des protéines est réduite, empêchant le mécanisme de
décomposition spinodale de se faire. Néanmoins, cette séparation de phases arrêtée permet de
créer des gels en contrôlant la longueur de corrélation résultante du réseau bi-continu. En effet,
les échantillons viscoélastiques comme R1,88 montrent que l’effet de la séparation de phases
arrêtée est encore plus fort et se manifeste dès les premières trempes en température (Tt =
10°C).
La diversité de composition protéique des extraits étudiés montre que la séparation de phases
est différente en fonction de la teneur en gluténines et gliadines présente dans l’échantillon. La
séparation de phases observée sur nos systèmes multi-composants s’apparente à celle réalisée
par des mélanges binaires de micelles de caséines et de biopolymères (Bhat et al., 2006) ou
encore de mélanges de polymères (Hashimoto et al., 1986). Ces systèmes complexes valident la
similarité de la mise à l’échelle dynamique de la même façon que nos mélanges de protéines
du gluten.

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
197
Bibliographie
Bhat, S., Tuinier, R., & Schurtenberger, P. (2006). Spinodal decomposition in a food colloid-
biopolymer mixture: evidence for a linear regime. Journal of Physics: Condensed Matter,
18(26), L339–L346.
Banc, A., Charbonneau, C., Dahesh, M., Appavou, M.-S., Fu, Z., Morel, M.-H., & Ramos, L.
(2016). Small angle neutron scattering contrast variation reveals heterogeneities of
interactions in protein gels. Soft Matter, 1–28.
Cahn, J. W., & Hilliard, J. E. (1958). Free Energy of a Non-uniform System. I. Interfacial Free
Energy. Journal of Chemical and Engineering Data, 258(28).
Cahn, J. W. (1959). Free Energy of a Non-uniform System. II. Thermodynamic Basis. Journal
of Chemical Physics, 1121, 10–14.
Cahn, J. W., & Hilliard, J. E. (1959). Free Energy of a Non-uniform System. III. Nucleation in
a Two Component Incompressible Fluid. Journal of Chemical Physics, (31), 688–699.
Dahesh, M., Banc, A., Duri, A., Morel, M. H., & Ramos, L. (2016). Spontaneous gelation of
wheat gluten proteins in a food grade solvent. Food Hydrocolloids, 52, 1–10.
Da Vela, S., Braun, M. K., Dörr, A., Greco, A., Möller, J., Fu, Z. Schreiber, F. (2016). Kinetics
of liquid–liquid phase separation in protein solutions exhibiting LCST phase behavior
studied by time-resolved USAXS and VSANS. Soft Matter, 12(46), 9334–9341.
Debye, P., & Bueche, A. M. (1949). Scattering by an Inhomogeneous Solid. Journal of Applied
Physics, 20, 518–525.
Dhont, J. K. G. (1996). Spinodal decomposition of colloids in the initial and intermediate
stages. The Journal of Chemical Physics, 105(12), 5112.
Furukawa, H. (1985). A dynamic scaling assumption for phase separation. Advances in Physics,
34(6), 703–750.

Chapitre 7 – Dynamique de séparation de phases liquide-liquide
198
Gibaud, T., & Schurtenberger, P. (2009). A closer look at arrested spinodal decomposition in
protein solutions. Journal of Physics. Condens ,
21(32), 322201.
Glassman, M. J., & Olsen, B. D. (2015). Arrested Phase Separation of Elastin-like Polypeptide
Solutions Yields Stiff, Thermoresponsive Gels. Biomacromolecules, 16(12), 3762–3773.
Hashimoto, T., Itakura, M., & Hasegawa, H. (1986). Late stage spinodal decomposition of a
binary polymer mixture. I. Critical test of dynamical scaling on scattering function. The
Journal of Chemical Physics, 85(10), 6118.
Koberstein, J. T., Morra, B., & Stein, R. S. (1980). The determination of diffuse boundary
thickness of polymers by small angle x-ray scattering. Journal of Applied Crystallography,
13, 34–45.
Marro, J., Lebowitz, J. L., & Kalos, M. H. (1979). Computer simulation of the time evolution
of a quenched model alloy in the nucleation region. Physical Review Letters, 43(4), 282–
285.
Tromp, R. H., Rennie, A. R., & Jones, R. A. L. (1995). Kinetics of the Simultaneous Phase
Separation and Gelation in Solutions of Dextran and Gelatin. Macromolecules, 28(12),
4129–4138.
Verhegh N., Dhont, J. K. G., & Lekkerkerker, H. N. W. (1996). Fluid-fluid phase separation
in colloid-polymer mixtures studied with small angle light scattering and light microscopy, 230.

Conclusion et Perspectives
199
Conclusion générale et Perspectives
L’objectif principal de ce travail de thèse était d’étudier les protéines du gluten en solution
pour obtenir des informations sur leur structure, leur mécanisme d’assemblage et également
leurs interactions. Plus précisément, la thèse s’est axée autour de 3 questions :
1. Quel est le rôle de chaque classe de protéine sur les propriétés rhéologiques et de
structure des échantillons en solutions ?
2. Quel(s) mécanisme(s) sont mis en jeu lors d’une variation de la température du
système ?
3. Y a-t’il des interactions entre les gliadines et les gluténines ?
Pour répondre à ces questions nous avons choisis d’optimiser un protocole d’extraction basé
sur la séparation de phases liquide-liquide d’une solution de protéines du gluten soluble
en solvant eau/éthanol 50/50, v/v. L’application d’une trempe en température sur le système
a démontré que ce dernier passe d’un état monophasique à un état biphasique, composé d’une
phase diluée et d’une phase dense. En jouant sur la température de trempe, la caractérisation,
par chromatographie d’exclusion de tailles (SE-HPLC) des extraits obtenus, a permis de
montrer que la composition protéique des 2 phases variait : à des températures de trempes
proches de 12°C, la phase dense est enrichie en polymères de gluténines et déplétée en gliadines
alors qu’à des températures de trempes plus profondes, proche de 0°C, la phase diluée est
composée principalement de gliadines. Ainsi, dans le cas des protéines du gluten, la température
met en œuvre des mécanismes thermodynamiques qui influencent la partition des protéines au
sein des 2 phases. À ce stade de nos recherches, nous avons alors exprimer les résultats en
fonction du rapport massique de gluténines/gliadines, R, allant de 0,04 à 2,2, déduit de l’analyse

Conclusion et Perspectives
200
SE-HPLC. Ces différents extraits ont ensuite été solubilisés en solvant eau/éthanol, 50/50, v/v,
pour réaliser différentes mesures :
i) en milieu dilué (avec une fraction volumique, = 0,0038) pour estimer une taille
de rayon hydrodynamique des particules en fonction du rapport glu/gli, par
diffusion dynamique de la lumière (DLS),
ii) en milieu concentré ( = 0,18) pour étudier, d’une part, l’effet du rapport glu/gli
sur la prise en gel des solutions par rhéologie. D’autre part, pour estimer des tailles
caractéristiques par diffusion de neutrons et de rayons X aux petits angles ainsi que
sonder la dynamique de séparation de phases liquide-liquide.
D’après les mesures par diffusion dynamique de la lumière, deux tailles de particules de l’ordre
de 10 nm et 100 nm ont été mises en avant. Ces dernières ont été associées respectivement à
des monomères de gliadines et des polymères de gluténines + -gliadines via le couplage de la
technique SE-HPLC avec celle du fractionnement par flux de force asymétrique (AsFlFFF).
Plus particulièrement, l’analyse par SE-HPLC de chaque fraction récoltée, à l’issue du
fractionnement des protéines du gluten par AsFlFFF, a démontré la présence d’assemblages
entre -gliadines et gluténines. Ces derniers ont été confirmés par l’établissement de
diagrammes de phases de type UCST réalisé par lecture du point de trouble. La courbe
binodale établie a suggéré un comportement différent pour chaque classe de protéines : les -
gliadines présentent un diagramme de phases proche de celui des polymères de gluténines alors
que celui des / , - gliadines est différent et présente une température de transition critique
inférieure (Tc 5°C) à celle des assemblages (Tc 12°C).
D’autre part, la caractérisation rhéologique de ces différents extraits a démontré que plus le
rapport glu/gli est important, plus l’échantillon tend à gélifier. Cette observation permet
d’expliquer notamment, le comportement des échantillons suite à la séparation de phases
liquide-liquide : lors d’une trempe profonde en température, la séparation de phases s’opère
selon un mécanisme de décomposition spinodale, démontré par les mesures réalisées par
diffusion de rayons X aux petits angles (SAXS). Les échantillons R1,88, enrichis en gluténines,
présentent cependant un comportement différent de ceux les plus enrichis en gliadines (R0,04
et R0,83). Dès les premières secondes suivant la trempe en température, le système se trouve
sous forme d’un motif dense qui semble bloqué : ces échantillons ne présentent pas de taille
caractéristique qui évolue comme pour R0,04 et R0,83 mais une distribution de tailles figée,

Conclusion et Perspectives
201
plus large, composée, quelle que soit la température de trempe, de la juxtaposition de domaines
plus ou moins concentrés. Les échantillons R1,88 révèlent ainsi un phénomène de séparation
de phases arrêtées, tout comme c’est le cas pour les échantillons R0,04 et R0,83 soumis à
des trempes en température profondes. Le phénomène est donc directement influencé par une
viscosité importante dû à la présence des assemblages. Néanmoins, il est apparu que la présence
de ces assemblages n’impactait pas les grandeurs caractéristiques telles que la longueur de
persistance qui reste constante (lo 0,7 nm) ou encore les tailles d’hétérogénéités de deutération
(60 nm), identiques à tous les R. Ainsi, la caractérisation structurale réalisée par diffusion de
neutrons et rayons X aux petits angles (SANS et SAXS) montre que toutes les protéines se
comportent comme des chaînes de polymères en solvant et que les tailles
caractéristiques sont indépendantes de la fraction volumique en gluténines.
Sur la base de l’ensemble de ces résultats et conclusions, il apparait que l’expression de la
composition des extraits par le rapport massique polymères de gluténines/ ( / , + -
gliadines) ne semble pas être la plus approprié pour décrire les fractions modèles étudiées. Il
serait intéressant pour la suite d’exprimer les résultats en fonction des polymères de (gluténines
+ - gliadines) / ( / , - gliadines), ou plus précisément en terme d’assemblages/protéines
monomériques.
Perspectives
Ce travail a donc démontré des interactions et des assemblages entre différentes classes de
protéines du gluten et ce, à partir d’extraits plus ou moins pures en gliadines et gluténines +
-gliadines ce qui amènent de nouvelles interrogations :
1. Comment obtenir des extraits encore plus purs ?
2. Comment préciser la nature des interactions ?
Concernant la pureté protéique des extraits, une trempe en température proche de 12°C permet
d’obtenir un extrait enrichis en assemblages. Il reste cependant dans cet extrait une partie de
gliadines résiduelles. Or nous savons que les gliadines rejoignent la phase dense à plus basse
température. Ainsi, il est possible de se demander qu’adviendrait-il d’un échantillon enrichis
en assemblages, qui subirait une nouvelle trempe en température? À partir de ces réflexions, il
est possible de proposer une nouvelle expérience (Fig.2) qui consiste à obtenir des extraits
enrichis en assemblages via notre protocole d’extraction déjà mis en place, puis de renouveler
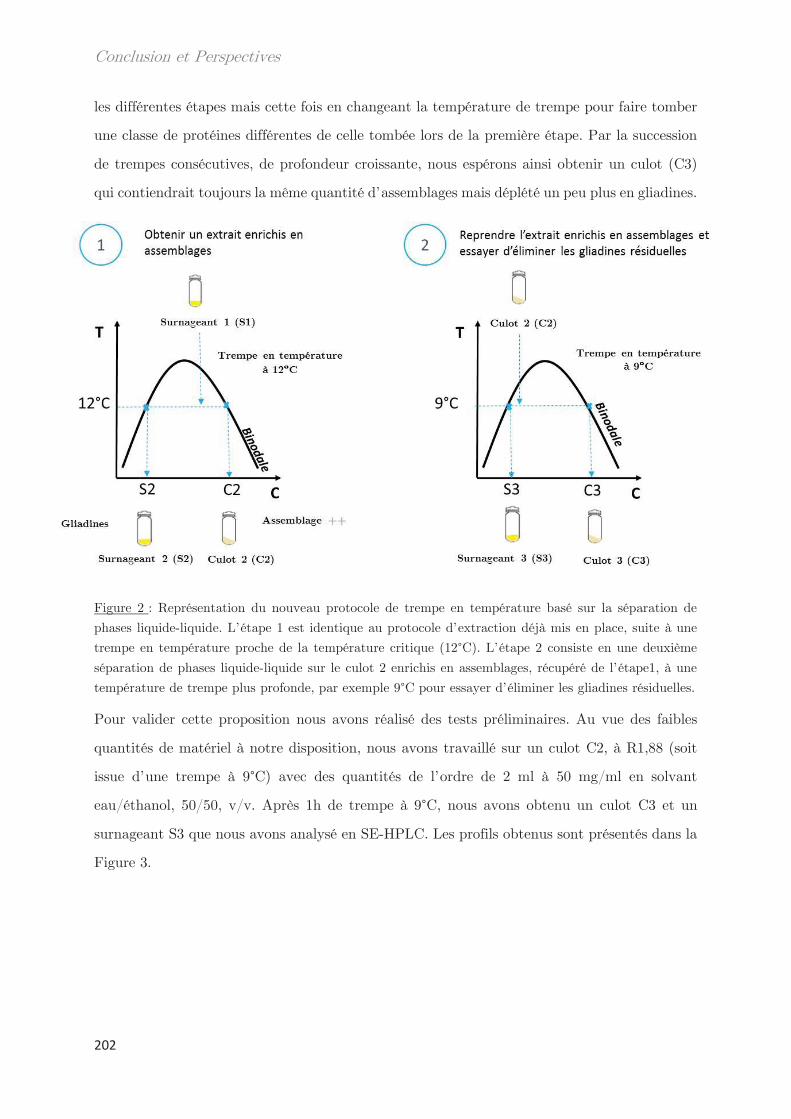
Conclusion et Perspectives
202
les différentes étapes mais cette fois en changeant la température de trempe pour faire tomber
une classe de protéines différentes de celle tombée lors de la première étape. Par la succession
de trempes consécutives, de profondeur croissante, nous espérons ainsi obtenir un culot (C3)
qui contiendrait toujours la même quantité d’assemblages mais déplété un peu plus en gliadines.
Figure 2 : Représentation du nouveau protocole de trempe en température basé sur la séparation de
phases liquide-liquide. L’étape 1 est identique au protocole d’extraction déjà mis en place, suite à une
trempe en température proche de la température critique (12°C). L’étape 2 consiste en une deuxième
séparation de phases liquide-liquide sur le culot 2 enrichis en assemblages, récupéré de l’étape1, à une
température de trempe plus profonde, par exemple 9°C pour essayer d’éliminer les gliadines résiduelles.
Pour valider cette proposition nous avons réalisé des tests préliminaires. Au vue des faibles
quantités de matériel à notre disposition, nous avons travaillé sur un culot C2, à R1,88 (soit
issue d’une trempe à 9°C) avec des quantités de l’ordre de 2 ml à 50 mg/ml en solvant
eau/éthanol, 50/50, v/v. Après 1h de trempe à 9°C, nous avons obtenu un culot C3 et un
surnageant S3 que nous avons analysé en SE-HPLC. Les profils obtenus sont présentés dans la
Figure 3.
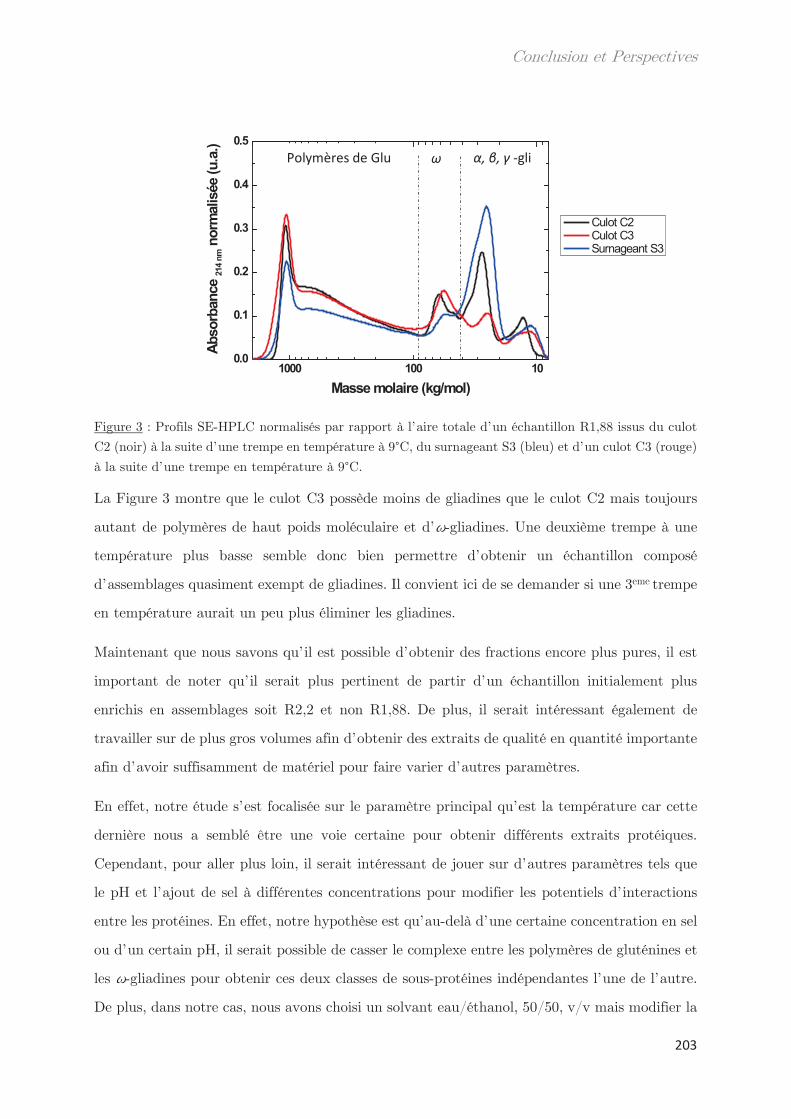
Conclusion et Perspectives
203
Figure 3 : Profils SE-HPLC normalisés par rapport à l’aire totale d’un échantillon R1,88 issus du culot
C2 (noir) à la suite d’une trempe en température à 9°C, du surnageant S3 (bleu) et d’un culot C3 (rouge)
à la suite d’une trempe en température à 9°C.
La Figure 3 montre que le culot C3 possède moins de gliadines que le culot C2 mais toujours
autant de polymères de haut poids moléculaire et d’ -gliadines. Une deuxième trempe à une
température plus basse semble donc bien permettre d’obtenir un échantillon composé
d’assemblages quasiment exempt de gliadines. Il convient ici de se demander si une 3eme trempe
en température aurait un peu plus éliminer les gliadines.
Maintenant que nous savons qu’il est possible d’obtenir des fractions encore plus pures, il est
important de noter qu’il serait plus pertinent de partir d’un échantillon initialement plus
enrichis en assemblages soit R2,2 et non R1,88. De plus, il serait intéressant également de
travailler sur de plus gros volumes afin d’obtenir des extraits de qualité en quantité importante
afin d’avoir suffisamment de matériel pour faire varier d’autres paramètres.
En effet, notre étude s’est focalisée sur le paramètre principal qu’est la température car cette
dernière nous a semblé être une voie certaine pour obtenir différents extraits protéiques.
Cependant, pour aller plus loin, il serait intéressant de jouer sur d’autres paramètres tels que
le pH et l’ajout de sel à différentes concentrations pour modifier les potentiels d’interactions
entre les protéines. En effet, notre hypothèse est qu’au-delà d’une certaine concentration en sel
ou d’un certain pH, il serait possible de casser le complexe entre les polymères de gluténines et
les -gliadines pour obtenir ces deux classes de sous-protéines indépendantes l’une de l’autre.
De plus, dans notre cas, nous avons choisi un solvant eau/éthanol, 50/50, v/v mais modifier la
1000 100 100.0
0.1
0.2
0.3
0.4
0.5
Absorb
ance 2
14 n
m n
orm
alisée (u.a
.)
Masse molaire (kg/mol)
Culot C2 Culot C3 Surnageant S3
Polymères de Glu -gli

Conclusion et Perspectives
204
qualité du solvant eau/éthanol tout en restant dans une gamme suffisante pour obtenir une
bonne solubilité des protéines permettrait également de modifier les liaisons entre les protéines
: nous savons que la quantité d’éthanol présente dans le solvant impacte la température de
transition (moins il y a d’éthanol, plus la température de transition est haute). La
complémentarité de ces études devrait permettre d’affiner la séparation des sous-classes de
protéines mais également de préciser la nature des interactions entre ces dernières.
Enfin, maintenant que nous avons établi l’existence d’assemblages entre les gluténines et les
- gliadines, et validé un protocole permettant de les isoler, il serait intéressant d’étudier la
texture et la rhéologie de produits dont la composition en sous classes de protéines est contrôlée.
Le travail présenté ici n’est donc qu’une première étape dont la finalité présente un challenge
à plus long terme.

Annexe
205
Index des annexes
Annexe 1 – Tableau récapitulatif des concentration (g/l), et masse totale de protéines (g),
et en volume (ml) pour chaque extrait S1, S2 et C2 aux différentes
températures de trempes relatif au Chapitre 4.
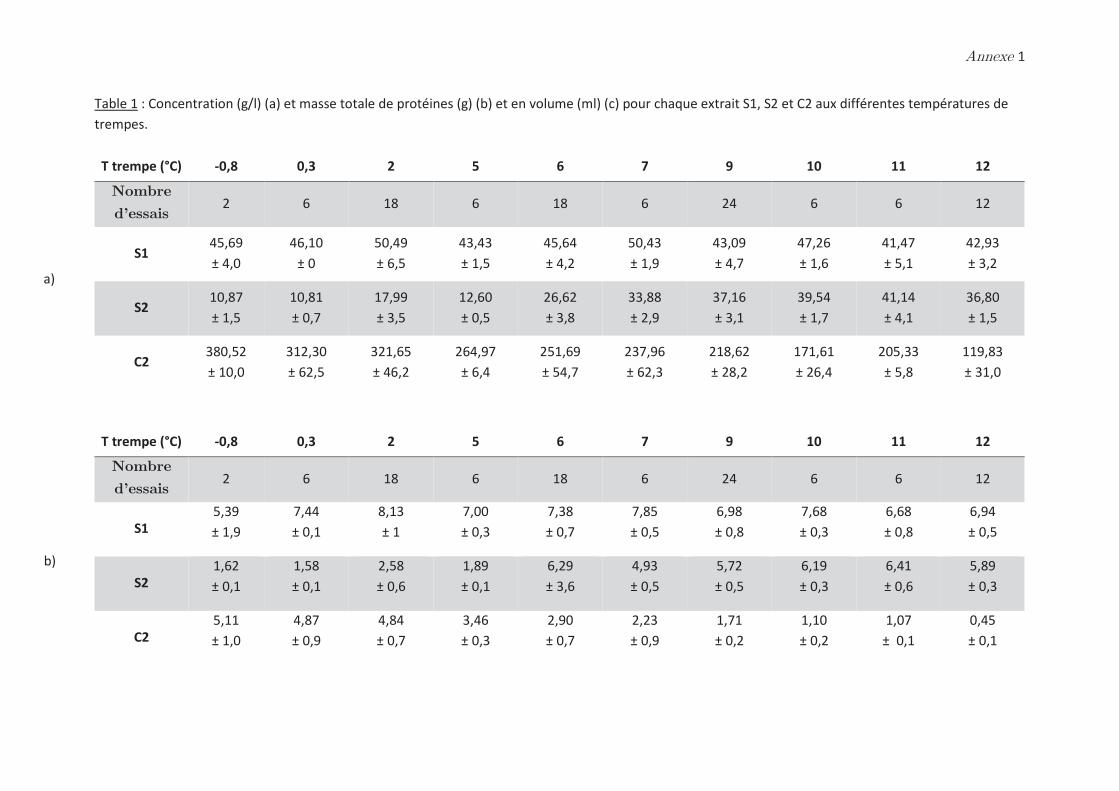
Annexe 1
Table 1 : Concentration (g/l) (a) et masse totale de protéines (g) (b) et en volume (ml) (c) pour chaque extrait S1, S2 et C2 aux différentes températures de
trempes.
T trempe (°C) -0,8 0,3 2 5 6 7 9 10 11 12
Nombre
d’essais 2 6 18 6 18 6 24 6 6 12
S1 45,69
± 4,0
46,10
± 0
50,49
± 6,5
43,43
± 1,5
45,64
± 4,2
50,43
± 1,9
43,09
± 4,7
47,26
± 1,6
41,47
± 5,1
42,93
± 3,2
S2 10,87
± 1,5
10,81
± 0,7
17,99
± 3,5
12,60
± 0,5
26,62
± 3,8
33,88
± 2,9
37,16
± 3,1
39,54
± 1,7
41,14
± 4,1
36,80
± 1,5
C2 380,52
± 10,0
312,30
± 62,5
321,65
± 46,2
264,97
± 6,4
251,69
± 54,7
237,96
± 62,3
218,62
± 28,2
171,61
± 26,4
205,33
± 5,8
119,83
± 31,0
T trempe (°C) -0,8 0,3 2 5 6 7 9 10 11 12
Nombre
d’essais 2 6 18 6 18 6 24 6 6 12
S1
5,39
± 1,9
7,44
± 0,1
8,13
± 1
7,00
± 0,3
7,38
± 0,7
7,85
± 0,5
6,98
± 0,8
7,68
± 0,3
6,68
± 0,8
6,94
± 0,5
S2
1,62
± 0,1
1,58
± 0,1
2,58
± 0,6
1,89
± 0,1
6,29
± 3,6
4,93
± 0,5
5,72
± 0,5
6,19
± 0,3
6,41
± 0,6
5,89
± 0,3
C2
5,11
± 1,0
4,87
± 0,9
4,84
± 0,7
3,46
± 0,3
2,90
± 0,7
2,23
± 0,9
1,71
± 0,2
1,10
± 0,2
1,07
± 0,1
0,45
± 0,1
a)
b)
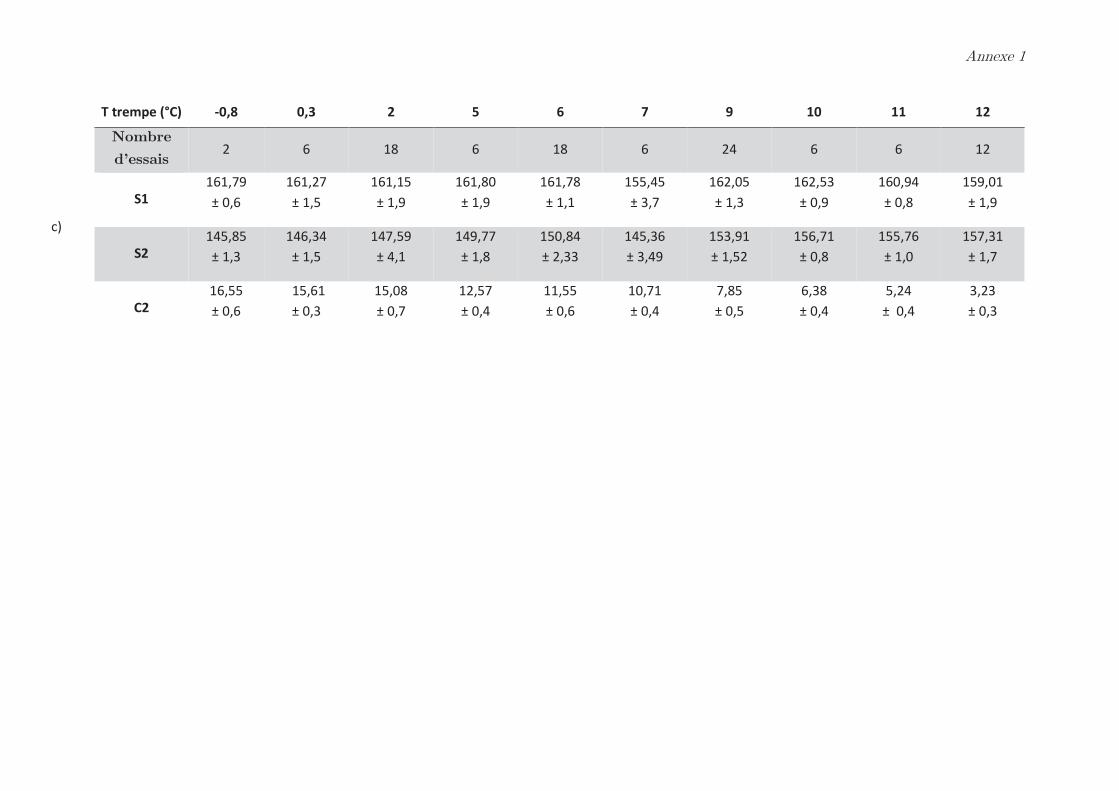
Annexe 1
T trempe (°C) -0,8 0,3 2 5 6 7 9 10 11 12
Nombre
d’essais 2 6 18 6 18 6 24 6 6 12
S1
161,79
± 0,6
161,27
± 1,5
161,15
± 1,9
161,80
± 1,9
161,78
± 1,1
155,45
± 3,7
162,05
± 1,3
162,53
± 0,9
160,94
± 0,8
159,01
± 1,9
S2
145,85
± 1,3
146,34
± 1,5
147,59
± 4,1
149,77
± 1,8
150,84
± 2,33
145,36
± 3,49
153,91
± 1,52
156,71
± 0,8
155,76
± 1,0
157,31
± 1,7
C2
16,55
± 0,6
15,61
± 0,3
15,08
± 0,7
12,57
± 0,4
11,55
± 0,6
10,71
± 0,4
7,85
± 0,5
6,38
± 0,4
5,24
± 0,4
3,23
± 0,3
c)



Résumé
Ce travail de thèse vise à apporter des connaissances structurales et fonctionnelles sur les protéines du
gluten. Pour cela, nous utilisons les concepts et méthodes de la physique des polymères et de la matière
molle. Plus précisément, nous optimisons un protocole d’extraction basé sur la séparation de phases
liquide-liquide. Ce dernier permet d’obtenir des isolats de protéines à différents rapports massiques
gluténines/gliadines que nous étudions ensuite dans un solvant eau/éthanol 50/50 (v/v). Les résultats,
montrent que les protéines se comportent comme des chaînes de polymères en solvant , en régime dilué
et semi-dilué avec des tailles caractéristiques définis par diffusion de rayons X et de neutrons aux petits
angles. De plus, 2 tailles d’objets sont distinguées en régime dilué par diffusion dynamique de la lumière:
d’une part des protéines monomériques de l’ordre d’une dizaine de nanomètres associées aux et -
gliadines et à des polymères de gluténines de faibles masses molaires et d’autre part des assemblages
polymériques de l’ordre de 100 nm, principalement composés de -gliadines et polymères de gluténines
de haute masse molaire. Ces assemblages sont mis en avant par une combinaison de mesures réalisées
par chromatographie d’exclusion de taille et par fractionnement par flux de forces asymétrique et
permettent de rationaliser les diagrammes de phases de ces mélanges protéiques, en fonction de la
température. L’étude de la dynamique de séparation de phases de ces mélanges protéiques, par diffusion
de rayons X aux petits angles, montre que celle-ci est pilotée par un phénomène de décomposition
spinodale. Cette décomposition peut être arrêtée lors de trempes en température profondes mais également
observée à toutes les températures de trempe, pour les échantillons les plus riches en gluténines, formant
un gel dès le régime monophasique, d’après leur étude par rhéologie.
Mots clés : protéines, gluten, assemblages, diffusion de rayonnement, séparation de phases
liquide-liquide
Abstract
The aim of this thesis is to provide structural and functional knowledge on wheat gluten proteins. For
that, we use the physical methods and the concept of soft matter. We optimize an extraction protocol
based on a liquid-liquid phase separation. With this protocol, we obtain protein batches with different
glutenin/gliadin mass ratios, which we then study in a 50/50 water/ethanol solvent (v/v). We show that
proteins behave like polymer chains in solvent in dilute and semi-dilute regime, whose characteristic
size are extracted by small angle X-ray and neutron scattering. Moreover, two sizes of objects are
evidenced in dilute regime by dynamic light scattering: monomeric proteins with a size around 10 nm
which can be associated to and -gliadins and polymeric glutenins with low molecular weight and
polymeric assemblies with a size around 100 nm composed of -gliadins and glutenins polymers with
high molecular weight. These assemblies are revealed by a combination of size exclusion chromatography
and asymmetric flow field flow fractionation and allow one to rationalize the phase diagrams of the
protein mixtures with temperature. The study of the dynamics of the phase separation of these protein
mixtures by small angle X-ray scattering shows that the phase separation proceeds through a spinodal
decomposition phenomenon. An arrested phase separation is observed for deep quenches but also at all
temperature quenches for the most glutenin rich samples, which are gels in the monophasic regime, as
confirmed by rheology.
Key word: proteins, gluten, assemblies, scattering techniques, liquid-liquid phase separation
Recommended

















