
Practicing molecular simulations.
Master Physique Informatique2011-2012
Lucyna Firlej
Laboratoire Charles CoulombUniversité Montpellier II
Lucyna FIRLEJMPI 2011

Provide you with the background and skills needed to:Provide you with the background and skills needed to:
Appreciate and understand the use of theory and Appreciate and understand the use of theory and simulation in research simulation in research Be able to read the simulation literature and evaluate Be able to read the simulation literature and evaluate
Goals of this courseGoals of this course
Be able to read the simulation literature and evaluate Be able to read the simulation literature and evaluate it criticallyit criticallyUnderstand the difference between quantumUnderstand the difference between quantum--based based and statistical thermodynamics based calculationsand statistical thermodynamics based calculationsBe able to start an atomistic simulation researchBe able to start an atomistic simulation research
Lucyna FIRLEJMPI 2011

OutlineOutline
Introduction Introduction –– molecular simulations in research molecular simulations in research Modeling of interactionsModeling of interactionsMC program MC program –– working case: adsorption in porous media.working case: adsorption in porous media.Calculation of thermodynamic propertiesCalculation of thermodynamic propertiesCalculation of thermodynamic propertiesCalculation of thermodynamic propertiesError analysisError analysis
:�No regular exam No regular exam –– project submissionproject submission
Lucyna FIRLEJMPI 2011

Modeling Modeling –– interdisciplinary scienceinterdisciplinary science
Numerical Modeling
Numerical simulations
[atomistic]
Quantum Chemistry (electrons)
[atomistic]
(Optimization,Monte Carlo, Molecular Dynamics)
Finite elements (objects > µµµµm)
Lucyna FIRLEJMPI 2011

What is molecular simulations ?What is molecular simulations ?
Molecular simulations is a computational “experiment” conducted on a molecular (atomic) level
up to 100.000, or more atoms are simulatedand real times up to 10-50 ns
(however, with current machines it is possible to(however, with current machines it is possible tostudy N up to several million, and times up to 1 μs)
Many configurations are generated,and averages taken to yield the “measurements.”
Molecular simulation has the character of both theory and experiment
Lucyna FIRLEJMPI 2011

Based on SDSC Blue Horizon (SP3)512-1024 processors1.728 Tflops peak performanceCPU time = 1 week / processor
10-6
10-3
100
(µµµµs)
(ms)
TIME/s
Mesoscale methods
Semi-empirical
Continuum
Lattice Monte CarloBrownian dynamicsDissipative particle dyn
Methods
Atomistic Simulation Methods
Theory and Simulation ScalesTheory and Simulation Scales
10-15
10-12
10-9
10-10 10-9 10-8 10-7 10-6 10-5 10-4(nm) (µµµµm)
(fs)
(ps)
(ns)
LENGTH/meters
Semi-empiricalmethods
Ab initiomethods
Monte Carlomolecular dynamics
tight-bindingMNDO, INDO/S
From: C. Alba-Simionesco et al., Effects of confinement on freezing and meltingJ.Phys.:Cond.Matter 18 (2006) 15-68
Lucyna FIRLEJMPI 2011

1. Discrete character of variables, ( ∆x, ∆t ) – defined by the problem
2. Constant number of independent variables
Limitations due to the speed and the memory capacity of computers
N – number of independent variables
(electrons, atoms or finite elements)
nmµµµµm –nm≥µ≥µ≥µ≥µm
N finite elements
N atoms N electrons
(electrons, atoms or finite elements)
Lucyna FIRLEJMPI 2011

Ab Initio Methods
Calculate properties from first principles, solving the Schrödinger (or Dirac) equation numerically.
Pros:• Can handle processes that involve bond breaking/formation, or electronic rearrangement (e.g. chemical reactions).• Methods offer ways to systematically improve on the
Electron localization function for (a) an isolated ammonium ion and (b) an ammonium ion with its first solvation shell, from ab
initio molecular dynamics. From Y. Liu, M.E. Tuckerman, J. Phys.
Chem. B 105, 6598 (2001)
• Methods offer ways to systematically improve on the results, making it easy to assess their quality.• Can (in principle) obtain essentially exact properties without any input but the atoms conforming the system.
Cons:• Can handle only small systems, about O(102) atoms.• Can only study fast processes, usually O(10) ps.• Approximations are usually necessary to solve the eqns.
Lucyna FIRLEJMPI 2011

Semi-empirical Methods
Use simplified versions of equations from ab initio methods, e.g. only treat valence electrons explicitly; include parameters fitted to experimental data.
Pros:• Can also handle processes that involve bond breaking/formation, or electronic rearrangement.• Can handle larger and more complex systems than ab
Structure of an oligomer of polyphenylene sulfide
phenyleneamine obtained with the semiempirical method. From R. Giro, D.S. Galvão, Int. J. Quant.
Chem. 95, 252 (2003)
• Can handle larger and more complex systems than ab initio methods, often of O(103) atoms.• Can be used to study processes on longer timescales than can be studied with ab initio methods, of about O(10) ns.
Cons:• Difficult to assess the quality of the results.• Need experimental input and large parameter sets.
Lucyna FIRLEJMPI 2011

Atomistic Simulation Methods
Use empirical or ab initio derived force fields, together with semi-classical statistical mechanics (SM), to determine thermodynamic (MC, MD) and transport (MD) properties of systems. SM solved ‘exactly’.
Pros:• Can be used to determine the microscopic structure of more complex systems, O(105-6) atoms.
Structure of solid Lennard-Jones CCl4 molecules confined in a
model MCM-41 silica pore. From F.R. Hung, F.R. Siperstein, K.E.
Gubbins, in progress.
more complex systems, O(10 ) atoms.• Can study dynamical processes on longer timescales, up to O(1) ms
Cons:• Results depend on the quality of the force field used to represent the system.• Many physical processes happen on length- and timescales inaccessible by these methods, e.g. diffusion in solids, many chemical reactions, protein folding, micellization.
Lucyna FIRLEJMPI 2011

Mesoscale Methods
Introduce simplifications to atomistic methods to remove the faster degrees of freedom, and/or treat groups of atoms (‘blobs of matter’) as individual entities interacting through effective potentials.
Pros:• Can be used to study structural features of complex systems with O(108-9) atoms.• Can study dynamical processes on timescales inaccessible
Phase equilibrium between a lamellar surfactant-rich phase and a continuous surfactant-
poor phase in supercritical CO2, from a lattice MC simulation. From N. Chennamsetty, K.E.
Gubbins, in progress.
• Can study dynamical processes on timescales inaccessible to classical methods, even up to O(1) s.
Cons:• Can often describe only qualitative tendencies, the quality of quantitative results may be difficult to ascertain.• In many cases, the approximations introduced limit the ability to physically interpret the results.
Lucyna FIRLEJMPI 2011

Continuum Methods
Assume that matter is continuous and treat the properties of the system as field quantities. Numerically solve balance equations coupled with phenomenological equations to predict the properties of the systems.
Pros:• Can in principle handle systems of any (macroscopic) size and dynamic processes on longer timescales.
Temperature profile on a laser-heated surface obtained with the finite-element method. From S.M. Rajadhyaksha, P.
Michaleris, Int. J. Numer. Meth. Eng. 47, 1807 (2000)
size and dynamic processes on longer timescales.
Cons:• Require input (viscosities, diffusion coeffs., eqn of state, etc.) from experiment or from a lower-scale method that can be difficult to obtain.• Cannot explain results that depend on the electronic or molecular level of detail.

1990
2000
Evolution of Computational MethodsEvolution of Computational Methods
Combinationwith DFT
Molecular
Gradient corrections
Car-Parinello
Multiscale methodslink to macroscopic models
Quantum Monte Carlo
2010 Mesoscale modeling
Ab initio molecular dynamics
1960
1980
1970
Solid State Physics Quantum ChemistryStatistical Mechanics
Molecular dynamics andMonte Carlo
Molecular Mechanics
Force fieldsSemi-empirical
parameters
Total energies
Analytical forcesgeometry optimization
Analytical frequencies
SCF
Hartree-Fock
Total energies
ForcesCar-Parinello
Density functional
Tight-binding
Energy band structures

Simulationsnumériques
Expérience
Real systems(nature)
Theories(analytical solution)
Model (parameters, interaction, ...)
Numerical experiments
experiment
Why simulations ?Why simulations ?
1. Fundamental studies, e.g. to determine range of validity of :Fick’s law of diffusion, Newton’s law of viscosity,
Exact solution(trajectories of all particles)
Tests of model Tests of theories
data (spectra)experimental
Approximate solutions
viscosity, etc.
2. Test theories by comparing theory and simulation
3. Test model by comparing simulated and experimental properties. Then use model in further simulations to carry out “experiments” not possible in the laboratory, e.g. critical points for molecules that decompose below Tc, properties of molten salts, long-chain hydrocarbon properties at very high pressures, properties of confined nano-phases, etc.
Lucyna FIRLEJMPI 2011

Construction of model
Method
Model of interaction
Numerical simulations: how ?Numerical simulations: how ?
Real systems
Pores structure and adsorbed atoms (neutral system)
Van der Waals and repulsive, no
Numerical Simulationsin statistical ensemble
Generation of states
Trajectory analysis
interaction
Interaction with
environment
Equation of motion
T = const.N variable
and repulsive, no electrostatics
Stochastic (Monte Carlo)
Grand CanonicalMonte Carlo algorithm
Mean values and fluctuations of:
EnergiesNumber of atoms
Lucyna FIRLEJMPI 2011
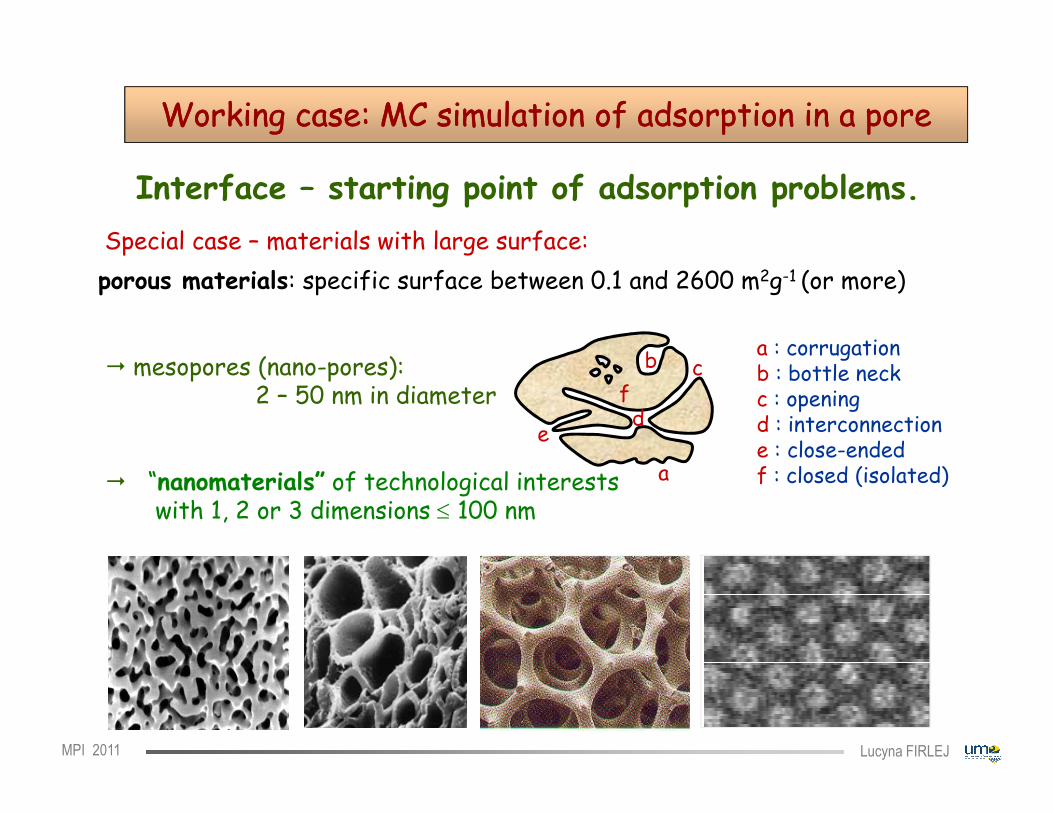
Working case: MC simulation of adsorption in a poreWorking case: MC simulation of adsorption in a pore
Interface – starting point of adsorption problems.Special case – materials with large surface:
cfd
b a : corrugationb : bottle neckc : opening
porous materials: specific surface between 0.1 and 2600 m2g-1 (or more)
� mesopores (nano-pores):2 – 50 nm in diameter
a
dc : openingd : interconnectione : close-endedf : closed (isolated)
2 – 50 nm in diameter
� “nanomaterials” of technological interestswith 1, 2 or 3 dimensions ≤ 100 nm
Lucyna FIRLEJMPI 2011

Working case: MC simulation of adsorption in a poreWorking case: MC simulation of adsorption in a pore
(Examples of porous systems)
Lucyna FIRLEJMPI 2011

Some (evident) applications of porous materials:Some (evident) applications of porous materials:(only those related to adsorption phenomenon)(only those related to adsorption phenomenon)(only those related to adsorption phenomenon)(only those related to adsorption phenomenon)
Purification of gases (closed atmosphere)or liquids (edible fluids)or solids (cleaning of the sol)
Storage of gas (natural, responsible for greenhouse effect, hydrogen)
Catalysis (carried out in confined geometry)
Working case: MC simulation of adsorption in a poreWorking case: MC simulation of adsorption in a pore
Catalysis (carried out in confined geometry)
Separation of molecules (molecular sieves)
Storage of energy (adsorption is exothermic)Potable water production (elimination of humid acids, detergents,
insecticides… by adsorption on carbons) Purification of edible liquids and of effluents from industrial plantsRetention by soil (especially by clays) of fertilizers, water, insecticides,
radioactive waste...Detergency and cleaning in liquid medium (adsorption of surfactants, softening of water by adsorption, dissolution of stains and re-adsorption on porous powders…)
Lucyna FIRLEJMPI 2011

Adsorption – a phenomenon general which involves the preferentialpartitioning of substances from the gaseous or liquid phase onto thesurface of a solid substrate
Gaz adsorbable«adsorbable»
Working case: MC simulation of adsorption in a poreWorking case: MC simulation of adsorption in a pore
Adsorbant
«adsorbable»
Phase adsorbée, «adsorbat»
Physical adsorption is caused mainly by van der Waals forces and electrostatic forces between adsorbate molecules and the atoms which compose the adsorbent surface. Thus adsorbents are characterized first by surface properties such as surface area, structure (roughness) and polarity.
Lucyna FIRLEJMPI 2011

Fundamental questions :1. Mechanism of adsorption – influence of
the geometry and pore structure2. Influence of confined geometry
(interaction with walls) on structural transformations
3. Capillary condensation4. Hysteresis and metastability
nσ/ms
B
B
I II III
IV V VI
nσ/ms
B
B
I II III
IV V VI
Working case: MC simulation of adsorption in a poreWorking case: MC simulation of adsorption in a pore
Numerical challenge:1. Simulations of equilibrium between gas and adsorbed phase
2. Modeling of interaction between pore walls and adsorbed particles
4. Hysteresis and metastability
p/p0p/p0
p/p°
nσ/ms
Lucyna FIRLEJMPI 2011

Pathways and difficulties for H2 use in transportationPathways and difficulties for H2 use in transportation
• Hydrogen production– Hopefully from renewable sources (wind, solar), or non-CO2 producing
(nuclear)• Fuel cells
– Serious cost problem, alternatives to Pt necessary• Storage (in particular in a vehicle)
– Weight & volume– Temperature– Vibrations– Safety, etc
• “Chicken and the egg”– No vehicles ⇔ No infrastructure
Lucyna FIRLEJMPI 2011

Gas storage (past & present)Gas storage (past & present)
First NG vehicle 1910 (USA) ~1930 (France)
Current NG vehicle with high-pressure tank in trunk
Problem: Trunk space is gone!
~200 bar (~3000 psi)
Lucyna FIRLEJMPI 2011

”Magic numbers” for: 2010 2015 UltimateEnergetic capacity
(kWh kg−1) (specific energy) 1.5 1.8 2.5
Gravimetric capacity(g H2 / kg) 45 (4.5 wt%) 55 (5.5 wt%) 75 (7.5 wt%)
DOE targets for hydrogen storage systemDOE targets for hydrogen storage system
Volumetric capacity (energy density) (kWh l−1) 0.9 1.3 2.3
Volumetric capacity(g H2 / l) 28 40 70
Lucyna FIRLEJMPI 2011

23
31
71
� �����
��������� ���������
in g/L
pressure
Gas
den
sity
Basic definitionsBasic definitions
23
���������������������
���������������������
VHmvsc )( 2= [kgH2/m3]
)( )()(
2
2CmHm
Hmgsc += [%wt]
� ����������������� �
� ���������������
� ��������������������� �
)( )( )()( )(
22
22
CmHmHmHmHmExcess o
o
+−−= [%wt]
Lucyna FIRLEJMPI 2011

Activated carbons.Activated carbons.
R.E.Franklin, Proc.Roy.Soc.London 209. 1951
more or less locally parallel graphite-like structures
N.A.Seaton. Carbon 27, 1989
J.Romanos,APS 2010 F.Rodriguez Reinoso, 2007
Lucyna FIRLEJMPI 2011

Adsorbent modelAdsorbent model
���������������ø < 2 nm – nanopores
2nm< ø < 50 nm - mezoporesø > 50 nm – macropores
�������������������������������������
5 10 15 20 25 30 35 40 45 500.00
0.05
0.10
0.15
0.20
Differential Pore Volume (cm3 /(g·Å))
Pore Width (Å)
Batch 5.1 S-33/k
Nanptechnology 20, 2009
H2H2H2 H2
~ 100 Ǻ~ 90 Ǻ
szerokość6-20 Ǻ
Lucyna FIRLEJMPI 2011

2
4
6
8
10
T = 298 KE = 4.5 kJ/molw
eigh
t %
298 K
DOE 2010
DOE 2015
Result : Adsorption limits vs. DOE requirements.
6 8 10 12 140
2
DOE 2010
DOE 2015
77 K
6 8 10 12 1402468
10121416
wei
ght %
pore width (Å)
T = 77 KE = 4.5 kJ/mol
Lucyna FIRLEJMPI 2011

Problem: Fluid adsorption in cylindrical pores.Problem: Fluid adsorption in cylindrical pores.Problem: Fluid adsorption in cylindrical pores.Problem: Fluid adsorption in cylindrical pores.
µVT- constant
Grand Canonical Monte Carlo
Working case: MC simulation of adsorption in a poreWorking case: MC simulation of adsorption in a pore
µ(gas) = µ(adsorbate)
µ(gas, ideal) = µ0(gas) + kBT ln(P)
µVT PVT - constant
External ideal gas pressure P
Lucyna FIRLEJMPI 2011

Recommended

















