
C
Si
Ta
b
c
d
a
ARRAA
KSSPMNTA
1
iym[flsfcoe
COGC
0
0d
Clinical Neurology and Neurosurgery 111 (2009) 708–712
Contents lists available at ScienceDirect
Clinical Neurology and Neurosurgery
journa l homepage: www.e lsev ier .com/ locate /c l ineuro
ase report
tiff person syndrome associated with lower motor neuron disease andnfiltration of cytotoxic T cells in the spinal cord
rygve Holmøy a,b,∗, Gjertrud Skorstad a, Line Sveberg Røste c, David Scheie d, Kirsti Alvik c
Department of Neurology, Oslo University Hospital Ullevål, Oslo, NorwayInstitute of Immunology, Faculty of Medicine, University of Oslo, NorwayDepartment of Neurology, Oslo University Hospital Rikshospitalet, Oslo, NorwayDepartment of Pathology, Oslo University Hospital Rikshospitalet, Oslo, Norway
r t i c l e i n f o
rticle history:eceived 15 September 2008eceived in revised form 12 June 2009ccepted 19 June 2009vailable online 17 July 2009
eywords:
a b s t r a c t
We present a 67-year-old non-diabetic male who presented with muscle cramps, paresis, atrophy andfasciculations in the left leg, followed by rapidly progressive muscle stiffness and superimposed spasmswhich subsequently also affected the right leg and the trunk. GAD65 autoantibodies were elevated inserum and CSF, compatible with systemic and intrathecal synthesis of oligoclonal and high-avidity autoan-tibodies, and GAD65 specific T cells were clonally expanded in the CSF. The patient did not respond toGABAergic and immunomodulatory treatment or plasma exchange, and died from respiratory failure after
tiff person syndrometiff limb syndromerogressive encephalomyelitis with rigidityotor neuron diseaseeuroinflammationcells
18 months. Autopsy revealed unilateral axonal swelling, chromatolysis and vacuolisation of anterior horncells of the lower spinal cord, accompanied by microglia proliferation and discrete infiltration of CD8+cytotoxic T cells. No CD4+ T helper cells, B cells or complement deposition were detected. To our knowl-edge, this is the first report of stiff person syndrome with lower motor signs restricted to a lower limb,and also the first attempt to characterize the infiltrating T cells. The finding of CD8+ cytotoxic T cells inthe absence of B cells in the inflamed area of the spinal cord suggests that the intrathecal synthesis of
kes pl
utopsy GAD65 autoantibodies ta. Introduction
Stiff person syndrome (SPS) characterized by muscular rigid-ty and superimposed spasms has been known for more than 50ears [1]. The spectrum of SPS-related disorders also comprises theore localized stiff limb subtype [2], the jerking man syndrome
3], and progressive encephalomyelitis with rigidity (PER) [4]. Therequent finding of autoantibodies against glutamic acid decarboxy-ase (GAD) 65 and the association with other autoimmune diseasesuggest an immune mediated mechanism [5,6]. Accordingly, trans-er of serum induces a SPS-like phenotype in rats [7], but it is notlear if or how these antibodies reach GAD65 within the cytoplasmaf neurons. More knowledge of the pathology related to clinicalxpressions of SPS is therefore warranted.
We have previously reported that the GAD65 autoantibodies in
SF and serum are oligoclonal and have high binding avidity [8].ne patient with substantial intrathecal production of oligoclonalAD65 autoantibodies and clonal expansion of GAD65 specificD4+ T cells [9] (patient SPS 3 in the original reports) has deceased,∗ Corresponding author at: Department of Neurology, Ullevål University Hospital,407 Oslo, Norway. Tel.: +47 23073773; fax: +47 23073510.
E-mail address: [email protected] (T. Holmøy).
303-8467/$ – see front matter © 2009 Elsevier B.V. All rights reserved.oi:10.1016/j.clineuro.2009.06.005
ace in areas of the CNS not strictly related to the clinically relevant lesions.© 2009 Elsevier B.V. All rights reserved.
thereby allowing combined interpretation of clinical, immunologi-cal and histological data. The symptoms included both stiffness andspasms typical of SPS as well as lower motor signs, and autopsyrevealed unilateral infiltration of cytotoxic T cells and microgliaactivation in the anterior horn of the lower part of the spinal cord.We discuss the potential pathogenic relevance of these findings forthe clinical symptoms and signs in this patient.
2. Case history
A previously healthy 67-year-old male presented with rapidlyevolving and painful flexor cramps of the toes on his left foot, fol-lowed by fasciculations and muscle atrophy in the left leg. Thecondition progressed rapidly with marked muscular rigidity andpainful superimposed spasms. This was most prominent in the leftleg, but subsequently also involved the right limb and truncus lead-ing to frequent falls and immobilisation. Neurological examination7 months after symptom debut revealed generalized atrophy in theleft leg with paralysis of the left ankle. Muscle stiffness was promi-
nent in the left leg but was also found in the right leg and the trunk,corresponding to a stiffness score of five out of six possible points atthe SPS stiffness extent scale [10]. Moreover, auditory, somatosen-sory, emotional and visual stimuli and attempts to move the left legtriggered painful muscle cramps, corresponding to six out of seven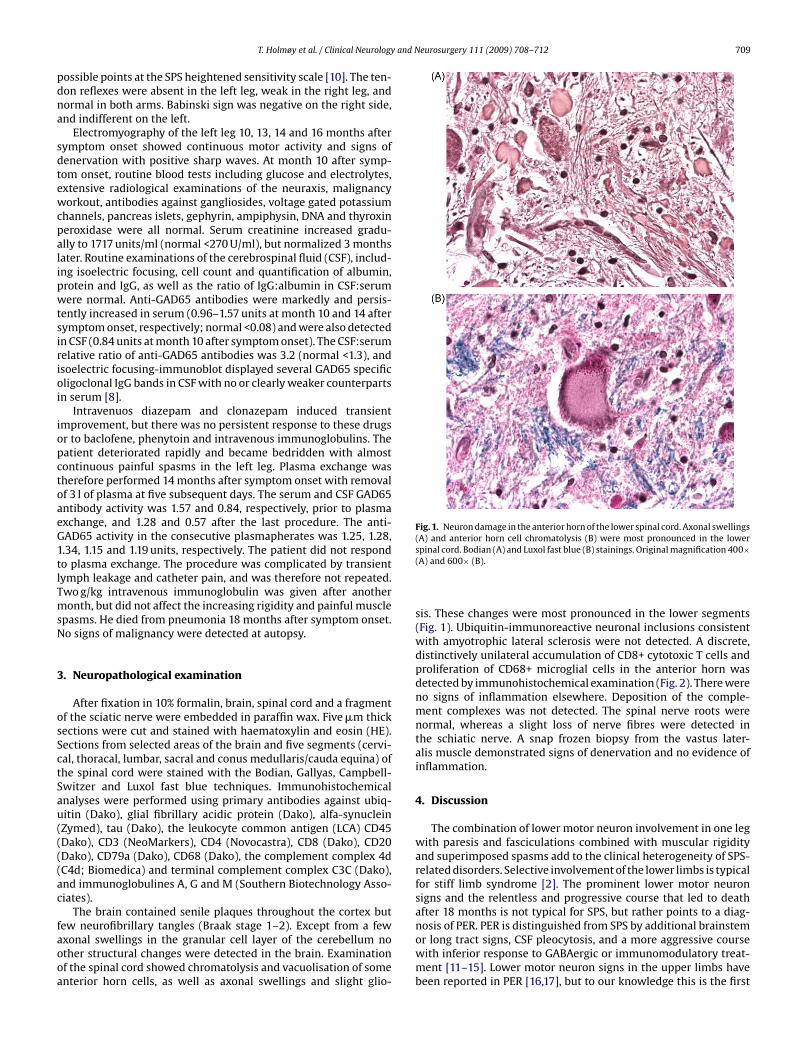
and Neurosurgery 111 (2009) 708–712 709
pdna
sdtewcpalipwtsirioi
iopctoaeG1tlTmsN
3
osSctSau((((ac
faooa
Fig. 1. Neuron damage in the anterior horn of the lower spinal cord. Axonal swellings(A) and anterior horn cell chromatolysis (B) were most pronounced in the lower
T. Holmøy et al. / Clinical Neurology
ossible points at the SPS heightened sensitivity scale [10]. The ten-on reflexes were absent in the left leg, weak in the right leg, andormal in both arms. Babinski sign was negative on the right side,nd indifferent on the left.
Electromyography of the left leg 10, 13, 14 and 16 months afterymptom onset showed continuous motor activity and signs ofenervation with positive sharp waves. At month 10 after symp-om onset, routine blood tests including glucose and electrolytes,xtensive radiological examinations of the neuraxis, malignancyorkout, antibodies against gangliosides, voltage gated potassium
hannels, pancreas islets, gephyrin, ampiphysin, DNA and thyroxineroxidase were all normal. Serum creatinine increased gradu-lly to 1717 units/ml (normal <270 U/ml), but normalized 3 monthsater. Routine examinations of the cerebrospinal fluid (CSF), includ-ng isoelectric focusing, cell count and quantification of albumin,rotein and IgG, as well as the ratio of IgG:albumin in CSF:serumere normal. Anti-GAD65 antibodies were markedly and persis-
ently increased in serum (0.96–1.57 units at month 10 and 14 afterymptom onset, respectively; normal <0.08) and were also detectedn CSF (0.84 units at month 10 after symptom onset). The CSF:serumelative ratio of anti-GAD65 antibodies was 3.2 (normal <1.3), andsoelectric focusing-immunoblot displayed several GAD65 specificligoclonal IgG bands in CSF with no or clearly weaker counterparts
n serum [8].Intravenuos diazepam and clonazepam induced transient
mprovement, but there was no persistent response to these drugsr to baclofene, phenytoin and intravenous immunoglobulins. Theatient deteriorated rapidly and became bedridden with almostontinuous painful spasms in the left leg. Plasma exchange washerefore performed 14 months after symptom onset with removalf 3 l of plasma at five subsequent days. The serum and CSF GAD65ntibody activity was 1.57 and 0.84, respectively, prior to plasmaxchange, and 1.28 and 0.57 after the last procedure. The anti-AD65 activity in the consecutive plasmapherates was 1.25, 1.28,.34, 1.15 and 1.19 units, respectively. The patient did not respondo plasma exchange. The procedure was complicated by transientymph leakage and catheter pain, and was therefore not repeated.wo g/kg intravenous immunoglobulin was given after anotheronth, but did not affect the increasing rigidity and painful muscle
pasms. He died from pneumonia 18 months after symptom onset.o signs of malignancy were detected at autopsy.
. Neuropathological examination
After fixation in 10% formalin, brain, spinal cord and a fragmentf the sciatic nerve were embedded in paraffin wax. Five �m thickections were cut and stained with haematoxylin and eosin (HE).ections from selected areas of the brain and five segments (cervi-al, thoracal, lumbar, sacral and conus medullaris/cauda equina) ofhe spinal cord were stained with the Bodian, Gallyas, Campbell-witzer and Luxol fast blue techniques. Immunohistochemicalnalyses were performed using primary antibodies against ubiq-itin (Dako), glial fibrillary acidic protein (Dako), alfa-synucleinZymed), tau (Dako), the leukocyte common antigen (LCA) CD45Dako), CD3 (NeoMarkers), CD4 (Novocastra), CD8 (Dako), CD20Dako), CD79a (Dako), CD68 (Dako), the complement complex 4dC4d; Biomedica) and terminal complement complex C3C (Dako),nd immunoglobulines A, G and M (Southern Biotechnology Asso-iates).
The brain contained senile plaques throughout the cortex but
ew neurofibrillary tangles (Braak stage 1–2). Except from a fewxonal swellings in the granular cell layer of the cerebellum nother structural changes were detected in the brain. Examinationf the spinal cord showed chromatolysis and vacuolisation of somenterior horn cells, as well as axonal swellings and slight glio-spinal cord. Bodian (A) and Luxol fast blue (B) stainings. Original magnification 400×(A) and 600× (B).
sis. These changes were most pronounced in the lower segments(Fig. 1). Ubiquitin-immunoreactive neuronal inclusions consistentwith amyotrophic lateral sclerosis were not detected. A discrete,distinctively unilateral accumulation of CD8+ cytotoxic T cells andproliferation of CD68+ microglial cells in the anterior horn wasdetected by immunohistochemical examination (Fig. 2). There wereno signs of inflammation elsewhere. Deposition of the comple-ment complexes was not detected. The spinal nerve roots werenormal, whereas a slight loss of nerve fibres were detected inthe schiatic nerve. A snap frozen biopsy from the vastus later-alis muscle demonstrated signs of denervation and no evidence ofinflammation.
4. Discussion
The combination of lower motor neuron involvement in one legwith paresis and fasciculations combined with muscular rigidityand superimposed spasms add to the clinical heterogeneity of SPS-related disorders. Selective involvement of the lower limbs is typicalfor stiff limb syndrome [2]. The prominent lower motor neuronsigns and the relentless and progressive course that led to deathafter 18 months is not typical for SPS, but rather points to a diag-nosis of PER. PER is distinguished from SPS by additional brainstem
or long tract signs, CSF pleocytosis, and a more aggressive coursewith inferior response to GABAergic or immunomodulatory treat-ment [11–15]. Lower motor neuron signs in the upper limbs havebeen reported in PER [16,17], but to our knowledge this is the first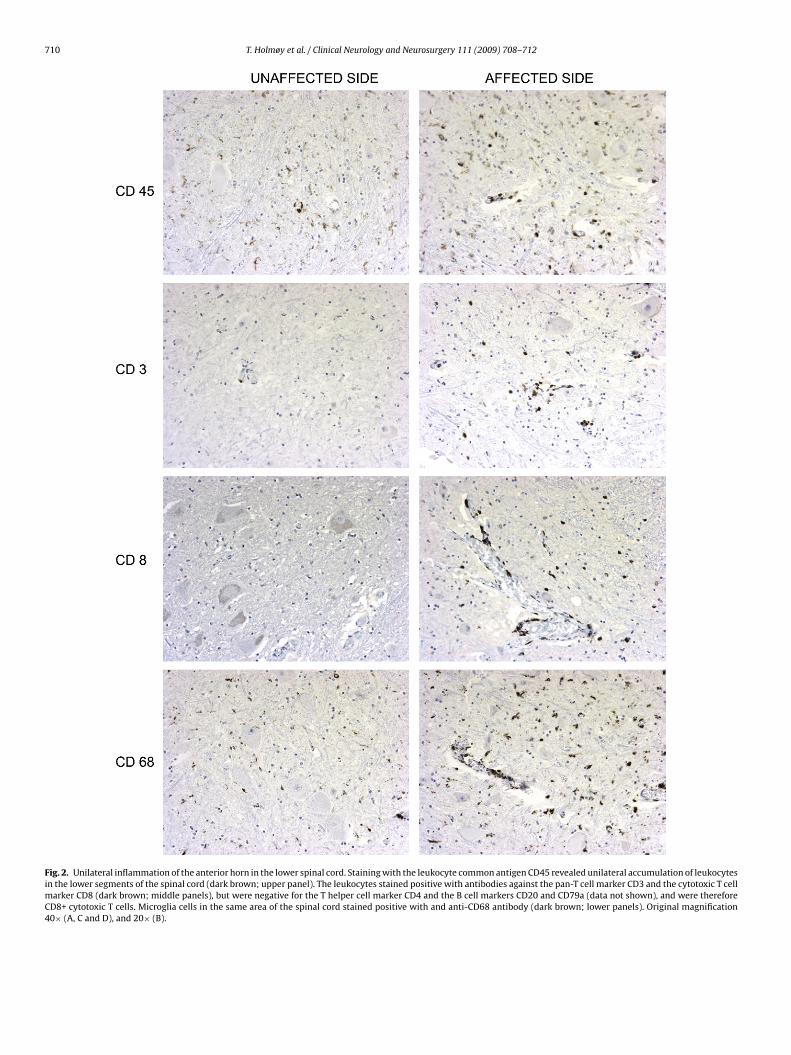
710 T. Holmøy et al. / Clinical Neurology and Neurosurgery 111 (2009) 708–712
Fig. 2. Unilateral inflammation of the anterior horn in the lower spinal cord. Staining with the leukocyte common antigen CD45 revealed unilateral accumulation of leukocytesin the lower segments of the spinal cord (dark brown; upper panel). The leukocytes stained positive with antibodies against the pan-T cell marker CD3 and the cytotoxic T cellmarker CD8 (dark brown; middle panels), but were negative for the T helper cell marker CD4 and the B cell markers CD20 and CD79a (data not shown), and were thereforeCD8+ cytotoxic T cells. Microglia cells in the same area of the spinal cord stained positive with and anti-CD68 antibody (dark brown; lower panels). Original magnification40× (A, C and D), and 20× (B).

and N
rTmo
t[cTcS[mlmwmttd[stnadPaitiHmctC
dGcTistlsd
eHtaaiamldfloua
r[t
[
[
[
[
[
T. Holmøy et al. / Clinical Neurology
eport of predominant lower motor involvement in the lower limb.he patient also suffered typical symptoms of SPS with tonicallyaintained rigidity and muscle cramps, supporting the relevance
f the concept “SPS plus” syndromes [16].The immunopathological findings comprised intrathecal syn-
hesis of high-avidity oligoclonal IgG antibodies against GAD658], evidence of clonally expanded GAD65125–136 specific CD4+ Tells restricted by DRB1*0801 [9], and infiltration of cytotoxic CD8+
cells and microglia activation in the spinal cord. Lymphocyteuffing of small CNS vessels is a typical finding in PER [12–14].imilar findings have been reported in some cases with typical SPS12,14,18–21], whereas others have not found evidence of inflam-
ation [22,23]. Vacuolar degeneration of anterior horn cells at theumbar segments of the spinal cord associated with prominent
icroglia proliferation has previously been described in a patientith atypical SPS [24]. T cell infiltration and microglia activationay play a primary pathogenic role, but could also be secondary
o neuronal damage caused by other factors. Thus, T cell infil-ration and microglia activation is observed also in degenerativeiseases like amyotrophic lateral sclerosis and its animal model25]. This distinction has therapeutic implications, because aggres-ive anti-inflammatory treatment would hardly be beneficial unlesshe inflammation is pathogenic. CD8+ cytotoxic T cells may recog-ize antigen presented on HLA class I molecules by neurons [26],nd are believed to be important effector cells in inflammatory CNSisorders [27]. HLA class I expression has not been studied in SPS orER, but has been demonstrated on neurons in both inflammatorynd degenerative CNS diseases [28]. It is therefore conceivable thatnfiltrating T cells could contribute to the irreversible and destruc-ive changes observed in some patients with SPS-related disease,ncluding the focal neurodegeneration observed in our patient.owever, although the co-localization of inflammation and lowerotor neuron involvement makes it tempting to speculate on a
ausal relationship, it should be emphasized that the T cell infil-ration was more discrete than usually observed in inflammatoryNS disorders.
The intrathecal synthesis of oligoclonal antibodies and the evi-ence of clonally expanded HLA class II restricted T cells specific forAD65 previously demonstrated in this patient implies intrathe-al persistence of B cell clones producing these antibodies [8,9].he lack of B cells, plasma cells and T helper cells within the
nflamed section of the spinal cord suggests that the intrathecalynthesis of GAD65 antibodies takes place elsewhere, either inhe CNS or in the meninges. This needs to be investigated in aarger sample set, but may indicate that the intrathecal synthe-is of GAD65 autoantibodies is not directly related to the tissueestruction.
It is expected that five plasma exchanges removing 3 l of plasmaach would deplete at least 90% of the serum autoantibodies.owever, plasma exchange did only induce a discrete drop in
he GAD65 antibody activity in serum, plasmapherates and CSF,nd did not induce clinical improvement in our patient. This mayppear as a paradox, as 15 l of plasma with high GAD65 activ-ty was removed from the patient. The persistence of GAD65ntibody activity in serum and CSF may reflect that the radioim-unoassay is relatively insensitive to changes in GAD65 antibody
evels at this order of magnitude, or that GAD65 antibodiesepleted from serum is replaced from tissues and extracellularuid. We can therefore not exclude that more aggressive removalf autoantibodies could have been helpful, although this seemsnlikely given the signs of neurodegeneration detected shortly
fterwards.The etiology of SPS is enigmatic and most likely multifacto-ial. SPS has been reported after infection with West Nile virus29], and T cells may cross-recognize GAD65 and microbial pro-eins [30]. Our patient had no previous history of serious infections
[
[
eurosurgery 111 (2009) 708–712 711
or exposure to toxins. There was no family history of SPS-relateddisorders, but he did carry the HLA-DRB1*0301 allele reportedto be associated with autoimmune diseases including SPS [10].This allele is, however, carried by almost one third of Norwe-gians. The GAD65 specific T cells cloned from the CSF of thispatient was not restricted by HLA-DR�1*0301 [9], and the signifi-cance of HLA-DRB1*0301 in this patient should be interpreted withcare.
Conflicts of interest
We have no conflicts of interest.
References
[1] Moersch FP, Woltman HW. Progressive fluctuating muscular rigidity and spasm(“stiff-man” syndrome); report of a case and some observations in 13 othercases. Mayo Clin Proc 1956;31:421–7.
[2] Brown P, Rothwell JC, Marsden CD. The stiff leg syndrome. J Neurol NeurosurgPsychiatry 1997;62:31–7.
[3] Leigh PN, Rothwell JC, Traub M, Marsden CD. A patient with reflex myoclonusand muscle rigidity: “jerking stiff-man syndrome”. J Neurol Neurosurg Psychi-atry 1980;43:1125–31.
[4] Whiteley AM, Swash M, Urich H. Progressive encephalomyelitis with rigidity.Brain 1976;99:27–42.
[5] Folli F, Solimena M, Cofiell R, Austoni M, Tallini G, Fassetta G, et al. Autoanti-bodies to glutamic acid decarboxylase in a patient with stiff-man syndrome,epilepsy, and type I diabetes mellitus. N Engl J Med 1988;318:1012–20.
[6] Raju R, Rakocevic G, Chen Z, Hoehn G, Semino-Mora C, Shi W, et al. Autoim-munity to GABAA-receptor-associated protein in stiff-person syndrome. Brain2006;129:3270–326.
[7] Manto MU, Laute MA, Aguera M, Rogemond V, Pandolfo M, Honnorat J. Effectsof anti-glutamic acid decarboxylase antibodies associated with neurologicaldiseases. Ann Neurol 2007;61:544–51.
[8] Skorstad G, Hestvik AL, Torjesen P, Alvik K, Vartdal F, Vandvik B, et al. GAD65IgG autoantibodies in stiff person syndrome: clonality, avidity and persistence.Eur J Neurol 2008;15:973–80.
[9] Skorstad G, Hestvik AL, Vartdal F, Holmøy T. Cerebrospinal fluid T cell responsesagainst glutamic acid decarboxylase 65 in stiff person syndrome. J Autoimmun2009;32:24–32.
[10] Dalakas MC, Fujii M, Li M, McElroy B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology2000;55:1531–5.
[11] Campbell AM, Garland H. Subacute myoclonic spinal neuronitis. J Neurol Neu-rosurg Psychiatry 1956;19:268–74.
12] Kasperek S, Zebrowski S. Stiff-man syndrome and encephalomyelitis. Report ofa case. Arch Neurol 1971;24:22–30.
[13] Armon C, Swanson JW, McLean JM, Westbrook PR, Okazaki H, KurtinPJ, et al. Subacute encephalomyelitis presenting as stiff-person syndrome:clinical, polygraphic, and pathologic correlations. Mov Disord 1996;11:701–9.
[14] Warren JD, Scott G, Blumbergs PC, Thompson PD. Pathological evidence ofencephalomyelitis in the stiff man syndrome with anti-GAD antibodies. J ClinNeurosci 2002;9:328–9.
[15] Meinck HM, Thompson PD. Stiff man syndrome and related conditions. MovDisord 2002;17:853–66.
[16] Brown P, Marsden CD. The stiff man and stiff man plus syndromes. J Neurol1999;246:648–52.
[17] Thompson PD. The stiff-man syndrome and related disorders. ParkinsonismRelat Disord 2001;8:147–53.
[18] Nakamura N, Fujiya S, Yahara O, Fujioka Y, Kawakami Y. Stiff-man syndromewith spinal cord lesion. Clin Neuropathol 1986;5:40–6.
[19] Mitsumoto H, Schwartzman MJ, Estes ML, Chou SM, La Franchise EF, De CamilliP, et al. Sudden death and paroxysmal autonomic dysfunction in stiff-man syn-drome. J Neurol 1991;238:91–6.
20] Meinck HM, Ricker K, Hulser PJ, Schmid E, Peiffer J, Solimena M. Stiffman syndrome: clinical and laboratory findings in eight patients. J Neurol1994;241:157–66.
21] Ishizawa K, Komori T, Okayama K, Qin X, Kaneko K, Sasaki S, et al. Large motorneuron involvement in stiff-man syndrome: a qualitative and quantitativestudy. Acta Neuropathol 1999;97:63–70.
22] Martinelli P, Pazzaglia P, Montagna P, Coccagna G, Rizzuto N, Simonati S, etal. Stiff-man syndrome associated with nocturnal myoclonus and epilepsy. JNeurol Neurosurg Psychiatry 1978;41:458–62.
23] Warich-Kirches M, Von Bossanyi P, Treuheit T, Kirches E, Dietzmann K, Feistner
H, et al. Stiff-man syndrome: possible autoimmune etiology targeted againstGABA-ergic cells. Clin Neuropathol 1997;16:214–9.24] Saiz A, Minguez A, Graus F, Marin C, Tolosa E, Cruz-Sanchez F. Stiff-man syn-drome with vacuolar degeneration of anterior horn motor neurons. J Neurol1999;246:858–60.
25] Holmoy T. T cells in amyotrophic lateral sclerosis. Eur J Neurol 2008;15:360–6.

7 and N
[
[
[
[JJ, et al. Cytomegalovirus in autoimmunity: T cell crossreactivity to viral anti-gen and autoantigen glutamic acid decarboxylase. Proc Natl Acad Sci USA
12 T. Holmøy et al. / Clinical Neurology
26] Neumann H, Medana IM, Bauer J, Lassmann H, Cytotoxic. T lymphocytes inautoimmune and degenerative CNS diseases. Trends Neurosci 2002;25:313–9.
27] Junker A, Ivanidze J, Malotka J, Eiglmeier I, Lassmann H, Wekerle H, et al.
Multiple sclerosis: T-cell receptor expression in distinct brain regions. Brain2007;130:2789–99.28] Höftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmid-bauer M, et al. Expression of major histocompatibility complex class Imolecules on the different cell types in multiple sclerosis lesions. Brain Pathol2004;14:43–50.
[
eurosurgery 111 (2009) 708–712
29] Hiemstra HS, Schloot NC, van Veelen PA, Willemen SJ, Franken KL, van Rood
2001;98:3988–91.30] Hassin-Baer S, Kirson ED, Shulman L, Buchman AS, Bin H, Hindiyeh M, et al.
Stiff-person syndrome following West Nile fever. Arch Neurol 2004;61:938–41.
Recommended

















