
Universidade Estadual de CampinasInstituto de Química
Departamento de Físico-Química
Relaxação em Sistemas Moleculares Complexos
Milton Taidi SonodaTese de Doutoramento
Orientador: Prof. Dr. Munir S. Skaf
Departamento de Físico-Química
Universidade Estadual de Campinas
Campinas, SP
2005
i

FICHA CATALOGRÁFICA ELABORADA PELA BIBLIOTECA DOINSTITUTO DE QUÍMICA UNICAMP
Sonoda, Milton Taidi.So59r Relaxacao em sistemas moleculares complexos /
Milton Taidi Sonoda. – Campinas, SP: [s.n], 2005.
Orientador: Munir Salomao Skaf.
Tese - Universidade Estadual de Campinas, Institutode Quımica.
1. Solucoes de frutose. 2. Solucao de DNA.3. Interface agua-ar. 4. Efeito Kerr optico da agua.I. Skaf, Munir Salomao. II. Universidade Estadual deCampinas. Instituto de Quımica. III. Tıtulo.
Título em inglês: Relaxation in complex molecular systems
Palavras-chaves em inglês:Fructose solutions, DNA solution, Air-water interface,Optical Kerr effect of water
Área de concentração:Fısico-Quımica
Titulação: Doutor em Ciencias
Banca examinadora: Munir Salomao Skaf (Orientador), Mauro Carlos CostaRibeiro (IQ-USP), Leo Degreve (FFCLRP-USP), Yoshiyuki Hase (IQ-UNICAMP),Ricardo Aparıcio (IQ-UNICAMP)
Data de defesa:29/07/2005
ii

Agradecimentos
Primeiramente gostaria de agradecer ao Munir pela orientação desta Tese. Sempre
presente e pronto a ajudar, com uma visão perspicaz sobre os problemas e resul-
tados, a orientação do Munir foi vital para o sucesso deste trabalho. Muito além
de ser um excelente profissional, se desdobrando entre as obrigações com a CPG
e a orientação de seus alunos, com seu senso de humor e palavras de incentivo, o
Munir tornou-se um amigo para todas as horas.
Agradeço aos velhos amigos do grupo, Frank, Leandro, Ney, Lucimara e Sérgio,
pelo companheirismo e convivência pacífica durante todos estes anos.
Agradeço ao Prof. Daniel Laria e aos seus orientados, especialmente o Diego
Pantano, por terem me recebido gentilmente durante minha estada em Buenos
Aires. Meus sinceros agradecimentos.
Gostaria de agradecer à Unicamp, ao Instituto de Química, e a todo o seu quadro
de funcionários que, sem o qual, não seria possível realizar este trabalho.
Quero agradecer aos mais pais, Haruco e Taizo, meus irmãos Neto e Thaís, e a
minha namorada Milene, pelo apoio e carinho constante. Vocês fazem os momen-
tos alegres muito melhores, e os momentos difíceis mais suaves. Muito obrigado.
Por último gostaria de agradecer a Deus.
v

Este projeto foi financiado
pelo CNPq e pela FAPESP

Curr ıculum Vitae
• Informac oes Pessoais
Nome: Milton Taidi Sonoda
Data de nascimento: 25/07/1976
• Formac ao Universit aria
Bacharelado em Física Universidade de São Paulo, 1994-1998.
• Mestrado
Dinâmica e Genealogia de Modelos de Evolução
Orientador: Prof. Dr. José Fernando Fontanari (IFSC-USP)
Instituição: Instituto de Física, Universidade de São Paulo
Ingresso: 03/1998, Término: 02/2001.
• Trabalhos publicados em peri odicos de circulac ¸ ao internacional
1. L. Martinez, M. T. Sonoda, P. Webb, J. Baxter, M. S. Skaf, I. Polikarpov,
“Molecular Dynamics Simulations Reveal Multiple Pathways of Ligand Dis-
sociation from Thyroid Hormone Receptors”Biophysical Journal89, 2011-
2023 (2005).
2. M. S. Skaf, M. T. Sonoda “Optical Kerr Effect in Supercooled Water”Phys-
ical Review Letters94, Art. 137802 (2005).
3. D. Pantano, M. T. Sonoda, M. S. Skaf, D. Laria “Solvation of Coumarin 314
at Water/Air Interfaces Containing Anionic Surfactants. I. Low Coverage”
Journal of Physical Chemistry B109, 7365-7372 (2005).
4. M. T. Sonoda, S. M. Vechi, M. S. SKAF “A Simulation Study of the Optical
Kerr Effect in Liquid Water”Physical Chemistry Chemical Physics7, 1176-
1180 (2005).
ix

x
5. M. T. Sonoda, N. H. Moreira, L. Martínez, F. W. Fávero, S. M. Vechi, L.
R. Martins, M. S. SKAF “A Review on the dynamics of water”Brazilian
Journal of Physics34, 3-16 (2004).
6. P. R. A. Campos, M. T. Sonoda, J. F. Fontanari “On the Structure of Ge-
nealogical Trees in the Presence of Selection”Physica A (Stat. Mech. and
Applications)283, 11-16 (2000)
7. A. L. Martinez, M. T. Sonoda, R. Lebullenger, M. C. C. Custódio, A. C. Her-
nandes “Oxyfluoride glasses containing LiNbO3”Journal of Non-Crystalline
Solids247, 35-38 (1999)
• Trabalhos apresentados em congressos
1. M. T. Sonoda e M. S. Skaf “Simulação de DNA com Modelo de Solvatação
de Born Generalizado” XII Simpósio Brasileiro de Química Teórica, Cax-
ambu, MG, em Novembro de 2003.
2. Leandro Martínez, Milton T. Sonoda, Munir S. Skaf, Igor Polikarpov, “Mecan-
ismos de Dissociação do Hormônio Tireoideano de seu Receptor Nuclear.”
1o Workshop de Modelagem Molecular de Ribeirão Preto na Faculdade de
Ciências Farmacêuticas da USP-RP, Ribeirão Preto, Setembro de 2002.
3. M. T. Sonoda e M. S. Skaf “Propriedades Estruturais e Dielétricas da Solução
Aquosa de Frutose” XII Simpósio Brasileiro de Química Teórica, Caxambu,
MG, em Novembro de 2001.

Resumo
Nesta Tese são apresentados estudos por simulações de Dinâmica Molecular de al-
guns sistemas moleculares de comportamento dinâmico complexo (i.e., processos de
relaxação caracterizados por múltiplas escalas de tempo), incluindo soluções aquosas
de sacarídeos, sistemas de DNA em dupla hélice, interfaces água/vapor na presença
de surfactantes e água em estado superfrio. Tais sistemas, embora claramente distin-
tos, apresentam pontos comuns no que concerne às suas propriedades dinâmicas. Nas
soluções aquosas de sacarídeos foram identificados aglomerados de soluto estabilizados
por ligações de hidrogênio que, em função da concentração, podem estar percolados
por toda extensão do sistema criando bolsões dentro dos quais a água apresenta uma
dinâmica muito lenta devido ao confinamento. Resultados são obtidos para inúmeras
propriedades e comparados a recentes dados experimentais de RMN, espalhamento de
neutrons e relaxação dielétrica. Estudamos os processos dinâmicos de uma dupla hélice
de DNA empregando o modelo Generalizado de Solvatação de Born e comparamos a
resultados obtidos com uma descrição explícita das moléculas de solvente. Observamos
que o modelo de Born captura bem as propriedades de estrutura do DNA, bem como a
dinâmica de tempos curtos das bases nitrogenadas. No entanto, as componentes mais
lentas de relaxação da cadeia não são descritas adequadamente pelo modelo de Born.
Nos sistemas interfaciais, estudamos a dinâmica de solvatação de uma cumarina na in-
terface água/vapor na presença de surfactante aniônico (SDS) sob diferentes condições
de cobertura superficial. Encontramos que a principal componente lenta de relaxação do
solvente está associada à interconversão de duas formas predominantes de solvatação da
cumarina. As implicações deste resultado para a interpretação de recentes experimentos
de espectrocopia óptica não-linear são discutidas. Por fim, estudamos também a relax-
ação estrutural da água líquida em estados superfrios através da simulação da resposta
de efeito Kerr óptico não-linear e comparamos a recentes medidas experimentais.
xi

Abstract
In this Thesis are presented studies by Molecular Dynamics simulations on some
molecular systems presenting complex dynamics behavior (i.e., relaxation process car-
acterized by multiple time scales), including saccharides aqueous solutions, double helix
DNA systems, air/water interfaces containing surfactants, and supercooled water. These
systems, although clearly distinct, present common points concerning their dynamical
properties. In saccharides aqueous solutions, we found clusters of solute molecules sta-
bilized by hydrogen bonds which, depending on the concentration, can be percolated
through the whole extension of the system. The water molecules are located within
pools created by cavities in this cluster and present very slow dynamic due to confine-
ment. Results of various properties are computed and compared to recent experimental
NRM, neutron scattering, and dielectric relaxation data. We studied dynamical process
of a double helix DNA using the Generalized Born (GB) Solvation model and compared
with results of simulation using the explicit solvation. We observed that the GB model
captures the properties of DNA structure, as well the short time dynamics of the DNA
bases. However, the slower component of the relaxation are not well described by the
implicit solvation model. In the interfacial systems, we studied the solvation dynamics
of an adsorbed coumarin at the air/water interface in the presence of anionic surfac-
tant (SDS) at different surface coverage. We found that the principal slow component
of relaxation is associated to the interconvertion of two preferention solvation states of
the coumarin. Implications of this result to recent non-linear optical spectroscopy ex-
periment are discussed. Finally, we studied the structural relaxation of liquid water in
supercooled states through simulation of the non-linear optical Kerr effect and compared
to recent experimental findings.
xiii

Sumario
Lista de Figuras xvii
Lista de Tabelas xxi
1 Introdução Geral 1
2 Simulação por Dinâmica Molecular 3
2.1 Introdução. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
2.2 Campos de Força. . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.3 Simulação . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
3 Soluções Aquosas de Frutose: Estrutura e Dinâmica 7
3.1 Introdução. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
3.2 Detalhes Computacionais. . . . . . . . . . . . . . . . . . . . . . . 9
3.3 Propriedades Estruturais. . . . . . . . . . . . . . . . . . . . . . . . 10
3.4 Propriedades Dinâmicas. . . . . . . . . . . . . . . . . . . . . . . . 19
3.5 Conclusão. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
4 Soluções Aquosas de Frutose: Relaxação Dielétrica 29
4.1 Introdução. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4.2 Formulação Teórica. . . . . . . . . . . . . . . . . . . . . . . . . . 30
A - Vetor de onda nulo (k = 0) . . . . . . . . . . . . . . . . . . . . . . . . . 30
B - Vetor de onda não-nulo (k �= 0) . . . . . . . . . . . . . . . . . . . . . . 32
4.3 Prop. Estáticas e Relaxação independente dek . . . . . . . . . . . . 35
4.4 Propriedades Dependentes do Vetor de Ondak . . . . . . . . . . . . 44
4.5 Permissividade Dielétrica. . . . . . . . . . . . . . . . . . . . . . . 47
xv

xvi Sumario
4.6 Conclusão. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
5 Dinâmica de DNA em solvente implícito 59
5.1 Introdução. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 59
5.2 Modelo de Born Generalizado. . . . . . . . . . . . . . . . . . . . . 61
5.3 Detalhes Computacionais. . . . . . . . . . . . . . . . . . . . . . . 65
5.4 Resultados. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
5.5 Conclusão. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
6 Dinâmica de Solvatação na Interface Água-Ar 73
Solvation of Coumarin 314 at water/air interfaces. . . . . . . . . . . . . . . 77
7 Efeito Kerr da Água 85
A simulation study of the optical Kerr effect in liquid water. . . . . . . . . 89
Optical Kerr Effect in Supercooled Water. . . . . . . . . . . . . . . . . . . 95
8 Conclusão Geral 99
Referências Bibliográficas 101

Lista de Figuras
3.1 Tautômeros de frutose considerados nesse trabalho.. . . . . . . . . . . 9
3.2 Função de distribuição de pares entre os centros de massa das frutoses
nos sistemas C1, C2 e C3.. . . . . . . . . . . . . . . . . . . . . . . . 11
3.3 Função de distribuição de pares entre os oxigênios, e entre oxigênios
e hidrogênios das moléculas de água nos sistemas C1, C2 e C3.. . . . 11
3.4 Função de distribuição de pares entre o oxigênio O6 da furanose e os
sítios O e H daágua nos sistemas C1, C2 e C3. Os gráficos internos
apresentam as ampliações destes g(r) na região do primeiro pico.. . . . 12
3.5 Função distribuição de pares entre o oxigênio O4 da piranose e os
sítios O e H daágua nos sistemas C1, C2 e C3. Os gráficos internos
apresentam as ampliações destes g(r) na região do primeiro pico.. . . . 12
3.6 Distribuição do número de ligações de hidrogênio que uma molécula de água
realiza com outras moléculas de água.. . . . . . . . . . . . . . . . . . . . 15
3.7 Distribuição do número do número de ligações de hidrogênio que uma fru-
tose realiza com moléculas de água.. . . . . . . . . . . . . . . . . . . . . 15
3.8 Distribuição do número de moléculas de frutose interagindo simultanea-
mente por ligações de hidrogênio com uma frutose de referência.. . . . . . 16
3.9 Fotografias de um passo de simulação tirado aleatoriamente das tra-
jetórias dos sistemas C1 , C2 e C3.. . . . . . . . . . . . . . . . . . . . 17
3.10 Função P(N) proporcional a probabilidade de que uma molécula de frutose
pertença a um aglomerado de tamanho N.. . . . . . . . . . . . . . . . . . 18
3.11 Coeficiente de difusãoDw da água eDf das frutoses em função da
concentração, e a sua comparação com dados experimentais.. . . . . . 20
xvii

xviii Lista de Figuras
3.12 Função de correlação temporal reorientacional normalizadaCµ1 (t) do
momento de dipolo das moléculas de frutose e de água. No gráfico
interno evidencia-se o comportamento rápido.. . . . . . . . . . . . . . 21
3.13 Função de correlação temporal reorientacionalCOHw2 (t) de versor
definido pelo OH da água, e a média da funçãoCOHf2 (t) do versor
definido pelos OH’s das frutoses.. . . . . . . . . . . . . . . . . . . . 22
3.14 Probabilidade de sobrevivência de interação por ligação de hidrogênio
fHB(t) entre duas moléculas de frutose, uma molécula de frutose e
uma de água, e entre duas moléculas de água.. . . . . . . . . . . . . . 24
3.15 Ajuste por uma exponencial extirada da probabilidade de sobrevivên-
cia de interação por ligação de hidrogêniofHB(t) entre duas molécu-
las de frutose, uma molécula de frutose e uma de água, e entre duas
moléculas de água.. . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
4.1 Média cumulativa da constante dielétricaε(0) dos sistemas C1, C2 e
C3 em função do tempo de simulação.. . . . . . . . . . . . . . . . . . 36
4.2 FCT do momento de dipolo coletivo dos sistemas C1 , C2 e C3. O
gráfico interno mostra o comportamento rápido, enfatizando as os-
cilações libracionais e torcionais.. . . . . . . . . . . . . . . . . . . . 37
4.3 Função de correlação temporal do momento de dipolo total e sua
decomposição nos termos frutose-frutose, frutose-água e água-água,
para os sistemas C1, C2 e C3.. . . . . . . . . . . . . . . . . . . . . . 39
4.4 Componente frutose-frutose da FCT do momento de dipolo total nor-
malizada pelo seu valor emt = 0, ΦFF (t)/ΦFF (0), e a função de
correlação reorientacionalCµ1 (t) da frutose para as três concentrações
estudadas.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
4.5 FCT do momento de dipolo total e suas componentes frutose-frutose,
frutose-água e água-água. As componentes estão normalizadas pelos
seus valores em t=0.. . . . . . . . . . . . . . . . . . . . . . . . . . . 42
4.6 FCT normalizada da componente água-água do momento de dipolo
coletivo e FCT reorientacional C1(t) do dipolo molecular da água. . . . 43

Lista de Figuras xix
4.7 FCT ΦT (k, t) da componente transversal do momento de dipolo total do
sistema para os cinco menores vetores de onda possíveis para o sistema, e a
função de correlação totalΦ(t) parak=0. . . . . . . . . . . . . . . . . . . 45
4.8 Função de correlação temporalΦL(k, t) da componente longitudinal do mo-
mento de dipolo total do sistema para os cinco menores vetores de onda
possíveis para o sistema.. . . . . . . . . . . . . . . . . . . . . . . . . . 47
4.9 Função de correlação temporalΦL(k1, t) da componente longitudinal
do momento de dipolo total do sistema para o vetor de ondak1 para
os sistemas C1, C2 e C3.. . . . . . . . . . . . . . . . . . . . . . . . . 49
4.10 Contribuições frutose-frutose e água-água da componente longitudi-
nal da FCTΦL(k1, t) para os sistemas C1, C2, e C3.. . . . . . . . . . 50
4.11 Logaritmos da parte imaginária da permissividade dielétrica e da compo-
nente transversal da permissividade dielétrica comk = k1, em função do
logaritmo da freqüência em número de ondas.. . . . . . . . . . . . . . . . 51
4.12 Parte imaginária da permissividade dielétrica para os sistemas C1, C2 e C3,
e o resultado experimental para o sistema C3 [1].. . . . . . . . . . . . . . 52
4.13 Parte imaginária da permissividade dielétrica em função da freqüên-
cia em número de ondas para os sistemas C1, C2 e C3, e sua decom-
posição nas auto-correlações frutose-frutose, água-água e na corre-
lação cruzada frutose-água.. . . . . . . . . . . . . . . . . . . . . . . 53
4.14 Produto deω2 pela parte real do espectroΦ(ω) em função deω em
número de ondas para os sistemas C1, C2 e C3, e sua decomposição
nas auto-correlações frutose-frutose, água-água e na correlação cruzada
frutose-água.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
5.1 Dupla hélice de DNA com destaque para a sonda substituindo um par bases
nitrogenadas.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
5.2 Esquema de aproximação da Born Generalizado. . . . . . . . . . . . 62
5.3 Representação da estrutura do açúcar.. . . . . . . . . . . . . . . . . . 66
5.4 Valor médio do pseudo-ângulo de rotação da desoxirribose dos nu-
cleotídeos dos DNAs na representação explícita e implícita do solvente. 66
5.5 Estrutura das bases complementares timina e adenina.. . . . . . . . . 67

xx Lista de Figuras
5.6 Transformada de Fourier da função de correlação do vetor C2-O2 da base
timina 5. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67
5.7 Comportamento de tempos curtos e longos da FCT do vetor normal
à base citosina 1 do DNA na representação explícita e implícita do
solvente. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
6.1 Cumarina 314 adsorvida na interface água-ar na presença do surfac-
tante aniônico dodecilsulfato de sódio.. . . . . . . . . . . . . . . . . . 74
6.2 Esquema representativo da experiência de dinâmica de solvatação. . . . 75
7.1 Esquema de um experimento de OKE.. . . . . . . . . . . . . . . . . . . 86

Lista de Tabelas
3.1 Detalhes das Simulações. . . . . . . . . . . . . . . . . . . . . . . . . 9
3.2 Número de coordenação dos sítios oxigênio (Ow) e hidrogênio (Hw)
da água em torno do O3 da piranose e O6 da furanose, e o número de
coordenação de sítios oxigênio da água em torno de um oxigênio da
água. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
3.3 Parâmeros de ajuste biexponencial do comportamento pós-libracional das
FCT normalizadaCµ1 (t) para a frutose e a água (ver figura 3.12) e os tempos
de correlação reorientacional integralτx1 , x : f, w. Todos os tempos estão
em ps. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
3.4 Parâmeros de ajuste biexponencial do comportamento pós-libracional
das FCT normalizadaCOH2 (t) para a frutose e a água (ver figura 3.13)
e os tempos de correlação reorientacional integralτx2 , x : f, w.. To-
dos os tempos estão em ps.. . . . . . . . . . . . . . . . . . . . . . . . 23
3.5 Parâmetros de ajuste de biexponencial do comportamento pós-libracional
das funções probabilidade de sobrevivência de ligação de hidrogênio
fHB entre duas moléculas de frutose, entre uma molécula de frutose e
uma molécula de água, e entre duas moléculas de água (ver figura 3.14). 25
3.6 Parâmetros de ajuste da exponencial estirada nas FCT de reorientação
Cµ1 (t) eCOH
2 (t) (ver figuras 3.12 e 3.13).. . . . . . . . . . . . . . . . 27
3.7 Parâmetros de ajuste da exponencial estirada nas curvas de probabili-
dadefHB(t) de sobrevivência da ligação de H no tempot (ver figura
3.15). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
4.1 Parâmetros de ajuste biexponencial e exponencial estirada da função de cor-
relação temporalΦ(t) do momento de dipolo total do sistema (ver figura 4.2).37
xxi

xxii Lista de Tabelas
4.2 Parâmetros de ajuste das funções espectrais Cole-Cole e Debye (equação
4.31) obtidos de experimentos de relaxação dielétrica (ref. [1]).. . . . 38
4.3 Parâmetros de ajuste das componentes frutose-frutose, frutose-água e água-
água da função de correlação do momento de dipolo total do sistema. Tem-
pos em ps.. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
4.4 Magnitude dos cinco menores vetores de ondak acessíveis às simu-
lações em unidades de Å−1, de acordo com a equação 4.28.. . . . . . 46
4.5 Parâmetros de ajuste biexponencial da FCTΦ(t) e das FCTΦA(k, t)
transversal (A = T ) e longitudinal (A = L) para os cinco menores
vetores de onda acessíveis à simulação. Os valores dek utilizados são
dados na tabela 4.4.. . . . . . . . . . . . . . . . . . . . . . . . . . . . 48

Capıtulo 1
Introduc¸ ao Geral
Nesta tese são apresentados estudos teóricos a respeito da estrutura, dinâmica e es-
pectroscopia de diversos sistemas líquidos interessantes. São eles: soluções aquosas
de sacarídeos, dinâmica de DNA, sistema interfacial água-ar na presença de surfac-
tante e dinâmica da água em condições ambientes e no estado superfrio. Todos estes
sistemas foram estudados utilizando a técnica de simulação por Dinâmica Molecular
(DM). O título “Relaxação em Sistemas Moleculares Complexos” deve-se a riqueza de
propriedades estruturais e comportamentos dinâmicos apresentados por estes sistemas,
observados tanto pelos resultados experimentais como pelas simulações de DM.
As investigações de todos estes sistemas foram motivadas em parte por descober-
tas experimentais recentes cujos resultados são muito intrigantes, como ficará claro nos
próximos capítulos. Estes sistemas são bastante interessantes do ponto de vista das suas
propriedades físico-químicas básicas e têm recebido bastante atenção da comunidade
científica. Buscou-se também estudar sistemas que tivessem relevância biológica. Vale
ressaltar a variedade de técnicas experimentais que foram empregadas para estudar cada
um destes sistemas. Por exemplo, espectroscopia dielétrica no caso das soluções de
sacarídeos, deslocamento de Stokes resolvido no tempo para a dinâmica do DNA, es-
pectroscopia de geração de soma de freqüência no estudo do sistema interfacial, e es-
pectroscopia de efeito Kerr óptico para o estudo da dinâmica da água superfria. As três
últimas técnicas citadas são bastante recentes e floresceram a partir do desenvolvimento
de lases de pulsos ultracurtos que ocorreu ao longo das duas últimas décadas. A espec-
troscopia de efeito Kerr óptico em especial, é um experimento que se baseia em efeitos
ópticos não-lineares.
1

2 Capıtulo 1. Introducao Geral
Outra componente importante que resultou na variedade de sistemas estudados foi
o interesse pessoal em adquirir experiência em um espectro diversificado de temas e
alcançar assim maior versatilidade na minha formação. Os capítulos foram redigidos
de forma a conter cada qual sua própria introdução e motivação independentemente,
dispensando, acredito, descrições mais elaboradas neste estágio do texto.
O formato da tese segue a resolução CCPG 001/98 de 11/11/98, a qual permite que
o conteúdo de um ou mais capítulos da tese de doutoramento possam ser constituídos
de um breve resumo do assunto tratado, seguido dos artigos resultantes da pesquisa,
impressos na língua original da publicação.
A organização desta tese se dá da seguinte maneira. Uma apresentação concisa da
técnica de dinâmica molecular, com o principais conceitos e definições é dada no capí-
tulo 2. O capítulo 3 apresenta as propriedades estruturais e dinâmicas das soluções
aquosas de sacarídeos, enquanto que o capítulo 4 apresenta as propriedades dilétricas
destes sistemas. O capítulo 5 trata da dinâmica de DNA. O estudo do sistema interfa-
cial é apresentado no capítulo 6. O último sistema estudado, efeito Kerr óptico da água
líquida e no estado superfrio, é apresentado no capítulo 7. Por fim, uma conclusão geral
é dada no capítulo 8.

Capıtulo 2
Simulac ao por Din amica
Molecular
2.1 Introduc¸ ao
Neste capítulo, apresenta-se resumidamente a técnica de dinâmica molecular (DM) clás-
sica. O propósito aqui é apresentar uma visão superficial e simplificada visando o leitor
não especialista. Existem inúmeros excelentes textos (por exemplo, refs. [2, 3, 4]) que
abordam o tema em profundidade. Tomamos a liberdade de remeter o leitor interessado
em saber mais sobre simulações de DM a estes trabalhos.
A técnica de DM tem por objetivo o cálculo da evolução temporal de um sistema
atômico e molecular. O método, quando introduzido inicialmente por Alder e Wain-
wright para estudar interações entre esferas rígidas no final da década de 50 [5], tinha por
objetivo apoiar estudos teóricos analíticos de mecânica estatística de líquidos simples.
O próximo avanço ocorreu em 1964, quando Rahman empregou potenciais realísticos
para simular argônio líquido [6]. A partir de então as simulações têm crescido em com-
plexidade e realismo. Podem ser considerados marcos históricos da técnica, a primeira
simulação de DM realística da água realizada em 1974 por Rahman e Stillinger [7] e a
primeira simulação de proteína realizada por McCammon e colaboradores em 1977 [8].
Os desenvolvimentos dos procedimentos de simulação e de computadores cada vez mais
potentes promoveram o impulso da área. Estes são os dois principais pontos limitantes
para o tamanho e complexidade dos sistemas simulados.
Uma característica marcante da área de simulação por DM é a possibilidade da
forte sinergia com estudos experimentais. Os resultados das investigações experimentais
geralmente motivam e guiam os estudos de DM. Os resultados obtidos pela dinâmica,
por sua vez, fornecem explicações aos fenômenos observados experimentalmente no
3

4 Capıtulo 2. Simulacao por Dinamica Molecular
nível microscópico. Outra característica interessante é a diversidade de sistemas tratáveis
por esta metodologia. Podem ser estudados por DM desde sólidos cristalinos e vítreos
até lipídeos, proteínas e ácidos nucléicos e seus complexos, passando por líquidos sim-
ples e moleculares. Atualmente, encontram-se na literatura grandes simulações de pro-
teínas solvatadas, complexos proteina-DNA e sistemas lipídicos [3].
2.2 Campos de Forc¸a
Como já foi dito, o objetivo de uma simulação por DM é obter a evolução temporal de
um dado sistema molecular. Esta evolução temporal deve-se a interação entre os átomos
e moléculas que compõem o sistema. Portanto, toda simulação de DM necessita de
um conjunto de potenciais empíricos apropriados que expressem a interação entre estes
átomos e moléculas. Este conjunto de potenciais é chamadocampo de força. Existe
uma grande variedade de campos de força, desenvolvidos por grupos diferentes, que
geralmente não são muito específicos e podem ser aplicados em sistemas diferentes. A
escolha de um ou outro campo de força é parcialmente governada hoje por questões
de conveniência ou preferência do pesquisador. Existem, é claro, alguma especificidade
associada a certos sistemas moleculares e também às propriedades que se deseja estudar.
Por exemplo, alguns sistemas e/ou propriedades que exigem potenciais polarizáveis e
tratamento de graus de liberdade internos das moléculas. Os campos de força mais
populares para o estudo de sistemas líquidos e de biomoléculas são: CHARMM [9],
OPLS-UA [10], OPLS-AA [11], GROMOS96 [4], e AMBER [12].
Os campos de força mais comuns representam as interações entre átomos como uma
soma de diversos potenciais de natureza distinta. De uma forma geral os potenciais
dividem-se em dois tipos: os potenciaisnão-ligadose ospotenciais ligados. Os poten-
ciaisnão-ligadossão compostos pelas interações eletrostática e de Lennard-Jones. Por
exemplo para o conhecido campo de força OPLS [11]:
Vαβ = qαqβ/(4πε0r) + 4εαβ
[(σαβ/r)12 − (σαβ/r)6
], (2.1)
ondeα e β designam dois átomos diferentes,qα é a carga parcial sobre o átomoα, e
εαβ e σαβ são os parâmetros do potencial de Lennard-Jones. Os do segundo tipo são
chamados depotenciais ligados, pois simulam o estiramento entre dois átomos ligados
covalentemente, a distorção dos ângulos de ligação e a torsão dos ângulos diedros. Em

2.3. Simulacao 5
geral, os dois primeiros potenciais são descritos por funções harmônicas, enquanto que o
último por uma soma de funções periódicas. No campo de força OPLS esses potenciais
são dados por:
Vest. lig.= Ke (r − req)2 (2.2)
Vdob. ang.= Kθ (θ − θeq)2 (2.3)
Vtor. died.=∑n
(Vn/2) [1 + cos (nφ − γ)] , (2.4)
ondeKe e Kθ são constantes harmônicas,req e θeq são a distância de ligação e o ân-
gulo de ligação de equilíbrio,n é número de funções periódicas,Vn/2 é a amplitude
de cada uma delas eγ a sua fase. A determinação dos parâmetros para uma interação
específica é desenvolvida através de metodologias diferentes em cada campo de força.
Estas metodologias geralmente envolvem medidas espectroscópicas, cálculos quânticos
e simulações de DM e Monte Carlo. Vale notar que cada parâmetro é desenvolvido para
a interação entre um conjunto específico de átomos.
2.3 Simulac ao
Uma vez escolhido o campo de força apropriado, inicia-se a simulação propriamente
dita. O objetivo é registrar a evolução dinâmica do sistema, conseguindo a posição e o
momento de todos os átomos a intervalos de tempo regulares a partir de umaconfigu-
ração inicial, conjunto de posições de todos os átomos das moléculas que consistem o
sistema. Dada essa configuração inicial do sistema, calcula-se a configuração do sistema
num tempo∆t posterior computando-se o potencialV sobre cada átomo devido à sua
interação com todos os demais. Note que todas funções potenciais dos campos de força
devem ser diferenciáveis. Assim pode-se calcular a força resultante sobre cada átomo
através a equaçãoFi = −∇Vi, e conseqüentemente a sua aceleração através da 2a lei de
Newtonai = Fi/mi, ondemi é a massa do átomo. A partir da aceleração e da veloci-
dade sobre o átomoi computa-se a sua velocidade e posição no tempo∆t posterior, que
podem ser simplesmente representadas pelas seguintes equações de integração:
vi (t + ∆t) = vi (t) + ai (t) ∆t
xi (t + ∆t) = xi (t) + vi (t) ∆t + ai (t) ∆t2/2,(2.5)
com t = 0 e vi (0) = 0 para a configuração inicial. Estas equações se tornam cada

6 Capıtulo 2. Simulacao por Dinamica Molecular
vez mais precisas quanto menor for∆t. A partir disto o processo segue iterativamente,
recalculando o potencial, a força e a aceleração sobre todos os átomos para a nova con-
figuração e utilizando as equações 2.5 e 2.5 para computar a velocidade e posição no
intervalo de tempo seguinte. Todas configurações seqüenciais constituem atrajetória da
simulação.
A inspeção visual das trajetórias com programas gráficos constitui um tipo de análise
qualitativa do sistema. No entanto programas de análise devem ser utilizados para obter
informações quantitativas sobre a trajetória simulada. Em especial, quando se quer
comparar com alguma observável experimental, relações mecânico-estatísticas entre a
observável experimental e variáveis mecânicas microscópicas do sistema devem ser uti-
lizadas. Este é o caso para a maioria dos sistemas estudados neste trabalho, como por
exemplo, a relaxação dielétrica de soluções aquosas de frutose (capítulo 4), a função
resposta de solvatação da molécula de prova na interface (capítulo 6) e a resposta do
efeito Kerr da água em condições ambientes e no estado superfrio (capítulo 7). Os detal-
hes técnicos de cada uma das analises realizadas sobre sistemas considerados nesta tese
serão dados nos capítulos subseqüentes.

Capıtulo 3
Soluc oes Aquosas de
Frutose: Estrutura e
Dinamica
3.1 Introduc¸ ao
As propriedades físico-químicas de soluções de carboidratos são de considerável in-
teresse tanto por aspectos de pesquisa básica como por suas aplicações. Carboidratos
representam uma classe muito importante de compostos em sistemas biológicos e estão
envolvidos em muitos processos que sustentam a vida. Junto com os lipídeos e pro-
teínas, os carboidratos constituem uma fonte de energia para as células. Eles também
são constituintes de moléculas importantes como os ácidos nucléicos. Muitas aplicações
tecnológicas das soluções de carboidratos utilizam as suas propriedades reológicas exóti-
cas. Muitas soluções aquosas de açucares, mas mais proeminentemente a trehalose, são
usadas pela natureza para proteger esporos, e em alguns casos, organismos inteiros con-
tra a morte por desidratação [13]. Esta proteção deve-se ao aumento da viscosidade
com o aumento da concentração da solução, chegando até à imobilização estrutural do
sistema, impedindo a sua degradação química. Devido a sua biossíntese relativamente
barata, os carbohidratos tem sido aplicados pela indústria alimentícia na conservação de
alimentos, e pela indústria farmacêutica para tentar aumentar o tempo de vida de medica-
mentos [13]. As hidroxilas dos carboidratos interagem com grupos hidrofílicos das pro-
teínas, substituindo as interações com a água e atuando na estabilização da sua estrutura.
Recentemente, muita atenção tem sido dada para a rica variedade conformacional dos
carboidratos. Como parte de glicoproteínas, glicolipídeos, e outras biomoléculas, os
carboidratos oferecem um alfabeto adicional em muitos processos biológicos, como a
sinalização, reconhecimento intercelular, e comunicação molecular e celular [14]. A
hidratação dos carboidratos é uma característica chave para a compreensão das suas
7

8 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
propriedades estruturais e funcionais.
Várias propriedades exóticas também se apresentam em soluções aquosas de frutose.
Estas soluções tem sido estudadas por espectroscopia de RMN [15, 16, 17], de relaxação
dielétrica [18, 1], espalhamento de nêutrons [19] e por simulação de DM [20]. Esses
estudos mostram que estas soluções apresentam propriedades que variam de maneira
complexa em função da concentração. Dois estados para a molécula da água podem ser
definidos: as águas que estão ligadas e as que não estão ligadas às moléculas de frutose.
Como as moléculas de frutose são massivas e grandes comparativamente às da água, as
moléculas de água ligadas às de frutose apresentam uma dinâmica translacional e rota-
cional mais lenta que as moléculas de água “livres”. Com o aumento da concentração, o
número de moléculas de água ligadas ao soluto aumenta, mantendo-se o número médio
de águas ligadas por molécula de frutose constante. Deste modo, conclui-se deste mod-
elo que a taxa de relaxação da água deve diminuir linearmente com a concentração. Para
baixas concentrações, isto é de fato o que se observa experimentalmente. No entanto,
para altas concentrações interações soluto-soluto tornam-se importantes e a dinâmica
do sistema diverge do comportamento linear, apresentando propriedades extraordinari-
amente lentas. Por estudos de RMN de prótons, Padua e Schmidt [15] mostram que,
além do comportamento linear para baixas concentrações, há duas outras regiões con-
secutivas de concentração apresentando comportamento linear para a taxa de relaxação,
com as três regiões apresentando derivadas crescentes da taxa de relaxação em função
da concentração. Neste trabalho os autores sugerem que esse comportamento deva-se à
formação de agregados entre os solutos e conseqüente aumento nas barreiras rotacionais
destas moléculas. Também através de RMN de prótons, Mahawanich e Schimidt mostra
que o coeficiente de difusão da água também apresenta uma diminuição linear com a
concentração para soluções diluidas, mas para maiores concentrações, essa diminuição
torna-se cada vez mais lenta [16]. Estes resultados estão de acordo com resultados de
DM [20]. No entanto, um estudo recente de espalhamento de nêutrons em soluções
frutose-água [19] mostra que a difusão da água apresenta uma dependência linear com a
concentração em uma faixa de concentrações que se estende até próximo a concentração
de saturação deste sistema (6.19 mol/L). Em estudos de relaxação dielétrica, Fucks e
Kaatze [18] mostram que os tempos de relaxação das soluções de frutose não se ajus-
tam aos modelos comuns de interpretação, baseados em parâmetros fundamentais como
a razão entre o número de sítios hidrofílicos e hidrofóbicos, e a concentração de sítios

3.2. Detalhes Computacionais 9
receptores de ligação de hidrogênio. Duas propriedades interessantes de soluções aqu-
osas de sacarídeos, que foram estudadas tanto por RMN [17] como por DM [21], são
os desacoplamentos dos movimentos translacionais e rotacionais entre as moléculas de
água e de açúcar, e os desacoplamentos destes mesmos movimentos entre as moléculas
de água próximas e distantes das moléculas de açúcar.
Estimulados por essas observações experimentais, nesse trabalho são apresentados
resultados de propriedades estruturais (seção 3.3) e dinâmicas (seção 3.4) de soluções
aquosas de frutose em função da concentração por simulações de DM.
3.2 Detalhes Computacionais
CH2OHβ
OH
HH
H
H
OH
HO
OH
HO2
34
5
6
β-D-frutopiranose (70 %)
CH2OH
HOH2C
β
OOH
OH
H
H
H1
2HO
34
6
5
β-D-frutofuranose (30 %)
Figura 3.1. Tautômeros de frutose considerados nesse trabalho.
Tabela 3.1. Detalhes das Simulações
Sistema c (mol/L)a L (Å) b Ncpir Nd
fur Newater
C1 1.0 28.50 10 4 686
C2 3.0 31.05 37 16 647
C3 4.0 32.63 57 25 618a) Concentração. b) Comprimento da caixa cúbica
de simulação. c) Número de moléculas de piranose.
d) Número de moléculas de furanose. e) Número de
moléculas de água.
A frutose em solução se apresenta em 4 tautômeros, sendo os mais freqüentes aβ-D-
frutopiranose (67%) e aβ-D-frutofuranose (25%) [18]. Esses solutos serão designados

10 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
de agora em diante simplesmente por piranose e furanose respectivamente (ver figura
3.1), ou simplesmente de frutose, para se referir aos dois tautômeros. Nas simulações
realizadas para esse trabalho foram apenas consideradas essas espécies com freqüên-
cia de 70 % e 30 %. O campo de força OPLS desenvolvido para carboidratos [22] foi
utilizado para modelar as frutoses neste trabalho. As distâncias entre átomos ligados
quimicamente foram mantidas fixas nas suas distâncias de equilíbrio. Todos os demais
graus de liberdade foram mantidos flexíveis. Adotou-se o modelo rígido SPC para água.
[23].
As simulações foram realizadas noensembleNVE. As configurações iniciais foram
termalizadas à 298 K. As interações de Lennard-Jones foram truncadas com raio de
corte igual a metade do lado da caixa de simulação, e as interações coulômbicas foram
tratadas pelo métodoParticle Mesh Ewald. As equações de movimento foram integradas
com o algoritmoleap-frogcom passo de tempo de 2 fs. A solução foi estudada nas con-
centrações 1, 3 e 4 mol/L. Esses sistemas serão designados por C1, C2 e C3, respec-
tivamente. Outros detalhes são dados na tabela 3.1. Foram utilizadas 130 simulações
independentes de 100 ps nas análises dos sistemas C1 e C2, e 90 simulações de 200
ps para as análises do sistema C3. Esses valores de concentração foram utilizados para
fazer contato com resultados experimentais de relaxação dielétrica [18]. Todas as sim-
ulações foram realizadas utilizando o pacote GROMACS [24, 25] devido a sua rapidez
e capacidade de paralelismo. No início deste trabalho foram realizados inúmeros testes
e comparações com outros programas, incluindo o TINKER [26] e o do próprio grupo
para garantir que as simulações estão corretas.
3.3 Propriedades Estruturais
As funções radiais de paresg(r) fornecem uma maneira conveniente de se estudar a
estrutura de líquidos e soluções. A função radialgij(r) mede a correlação espacial entre
os sítiosi e j. Matematicamente ela é computada através da razão entre quantidade
Nj(r, r+∆r) de sítiosj numa casca esférica de raio internor e externor+∆r, centrado
em i, e a quantidade esperada de sítiosj nessa mesma casca esférica se estes sítios
fossem distribuídos homogeneamente no espaço:
g(r)ij =〈Nj(r, r + ∆r)〉
4πρjr2∆r, (3.1)

3.3. Propriedades Estruturais 11
0 5 10 15r (Å)
0
0.5
1
1.5
2
2.5
g(r)
C1C2C3
Figura 3.2. Função de distribuição de pares entre os centros de massa das frutoses
para os sistemas C1, C2 e C3.
0 2 4 6r (Å)
0
1
2
3
4
5
g(r)
0 2 4 6 8r (Å)
C1C2C3
Ow-O
wO
w-O
h
Figura 3.3. Função de distribuição de pares entre os oxigênios (painel esquerdo) e
oxigênios e hidrogênios (painel direito) das moléculas de água nos sistemas C1, C2 e C3.
ondeρj é a densidade de sítiosj no sistema. A figura 3.2 apresenta a função radial
de pares entre os centros de massas das frutoses. Como esperado, é evidente pelo grá-
fico que existe um maior empacotamento das moléculas do soluto com o aumento da
concentração. Og(r) dos sistemas C2 e C3 são bastante similares apresentando apenas
um máximo e um mínimo, sendo que o mínimo de C3 é mais pronunciado que o de
C2. O g(r) do sistema C1 difere bastante dos demais, apresentando o primeiro pico
mais largo, seguido de um suave mínimo e um segundo pico de baixa amplitude. Esse
perfil caracteriza uma baixa interação entre as frutoses, estando elas bem separadas na
solução.
Os g(r) entre os oxigênios das moléculas de água são apresentados no painel es-

12 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
0 2 4 6 8r (Å)
0
0.5
1
1.5
2
2.5
g(r)
2.75 31
1.5
2
0 2 4 6 8 10r (Å)
1.75 20.5
1O
f - H
wO
f - O
w
Figura 3.4. Função de distribuição de pares entre o oxigênio O6 da furanose e os
sítios O (painel esquerdo) e H (painel direito) da água. Os gráficos internos apresentam as
ampliações destes g(r) na região do primeiro pico. C1: linha contínua, C2: linha tracejada, C3:
linha pontilhada.
0 2 4 6 8r (Å)
0
0.5
1
1.5
2
2.5
3
g(r)
0 2 4 6 8 10r (Å)
2.75 31
1.5
1.7 1.8
0.75
1Op-O
w Op-H
w
Figura 3.5. Função distribuição de pares entre o oxigênio O4 da piranose e os sítios
O (painel esquerdo) e H (painel direito) da água. Os gráficos internos apresentam as ampli-
ações destes g(r) na região do primeiro pico. C1: linha contínua, C2: linha tracejada, C3: linha
pontilhada.
querdo da figura 3.3. Observa-se que a posição dos picos desta função não variam com
o aumento da concentração. Os dois primeiros picos são observados em 2.7 e 4.6 Å, car-
acterísticos da estrutura tetraédrica água. No entanto, observa-se um ligeiro alargamento
no primeiro pico dos sistemas com concentração mais alta. As mesmas observações po-
dem ser feitas sobre og(r) entre os sítios oxigênio e hidrogênio da água (painel direito
da figura 3.3). Isto nos mostra que existe uma perturbação na rede de LH da água devido

3.3. Propriedades Estruturais 13
a adição de frutose. Computa-se número de coordenaçãoNij do sítioj em torno do sítio
i através de:
Nij = 4πρj
∫ r0
0gij(r)r
2dr, (3.2)
onder0 é a posição do primeiro mínimo da funçãogij(r). O número de coordenação
NOwOw de oxigênios da água em torno de um oxigênio da água é dado na última col-
una da tabela 3.2. Também são apresentados números de coordenação de oxigênios e
hidrogênios de água ao redor de átomos específicos das frutoses (O3 da piranose e O6da furanose). Como é esperado, os números de coordenação água-açúcar e água-água
ficam cada vez menores com o aumento da concentração de frutose nesses sistemas.
Tabela 3.2. Número de coordenação dos sítios oxigênio (Ow) e hidrogênio
(Hw) da água em torno do O3 da piranose e O6 da furanose, e o número de coordenação
de sítios oxigênio da água em torno de um oxigênio da água (coluna Ow-Ow).
O3 da piranose O6 da furanoseSistema
Ow Hw Ow HwOw-Ow
C1 2.48 1.06 2.93 1.24 5.11
C2 2.53 0.74 2.48 1.14 4.40
C3 2.05 0.65 2.14 1.02 3.89
Uma característica importante dos carboidratos é a existência de diversas hidroxilas
em sua estrutura (ver figura 3.1) capazes de realizar ligações de hidrogênio com a água.
Estas interações determinam as características estruturais e dinâmicas destas soluções.
As funçõesg(r) entre os sítios O6 (oxigênio da hidroxila ligada ao carbono 6) da fu-
ranose e o O3 da piranose e os sítios da água são apresentadas nas figuras 3.4 e 3.5,
respectivamente. Várias características comuns podem ser observadas nesses gráficos.
A posição dos primeiros picos não varia com a concentração indicando que, para a faixa
de concentrações estudadas neste trabalho, a estrutura de solvatação das frutoses se man-
tém. O primeiro pico ocorre em 2.8 Å para og(r) entre os oxigênios das frutoses e da
água, e em 1.8 Å entre os oxigênios das frutoses e os hidrogênios da água. Essas distân-
cias são características de formação de ligações de H. É interessante notar que os picos
da funçãog(r) entre o O6 da furanose e os sítios da água (figura 3.4) ocorrem na mesma

14 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
posição dos picos observados nosg(r) entre os sítios da água (figura 3.3). Este sítio
pertence a uma das hidroximetilas da furanose, que está relativamente longe de outras
hidroxilas desta molécula e projetada para o interior do meio aquoso (ver figura 3.1).
Além disso, o hidrogênio e oxigênio da hidroxila podem girar com facilidade em torno
das ligações O-C6 e C6-C5, respectivamente, possibilitando que essa hidroxila se adapte,
por exemplo, à estrutura tetraédrica da água. Comparando os gráficos internos da figura
3.4 com og(r) entre os oxigênios das águas na figura 3.3, observa-se que a variação da
altura dos primeiros picos em função da concentração apresentam o mesmo padrão: a
altura do primeiro pico destas funçõesg(r) aumenta com o aumento da concentração.
Não se observa o mesmo padrão nas funçõesg(r) entre o oxigênio O4 da piranose
e os sítios da água(figura 3.5). Além disso, estruturas de longa distância (entre 4 e
8 Å) podem ser observadas em posições diferentes daquelas observadas nog(r) entre
oxigênios da água. Essas variações são causadas pela presença de moléculas de água
ligadas à hidroxilas próximas ao oxigênio O4 da piranose. O números de coordenação
entre os sítios O6 da furanose e O4 da piranose e os sítios da água são apresentados na
tabela 3.2. Observa-se que os números de coordenação são sistematicamente maiores
para a hidroximetila da furanose que para a hidroxila da piranose. Observa-se ainda que
esses números diminuem com o aumento de concentração de frutose no sistema, como
já mencionado.
As funçõesg(r) atestam que estão ocorrendo ligações de hidrogênio entre as molécu-
las do sistema, e que essa interação é determinante para as propriedades estruturais do
líquido. Passa-se então a investigar a distribuição de ligações de hidrogênio entre as
espécies presentes no sistema. É considerado uma ligação de hidrogênio (LH) quando
os sítios envolvidos satisfazem todas as seguintes condições geométricas: i) Distância
H· · ·O menor que 2.6 Å ii) Distância O· · ·O menor que 3.5 Å e iii) Ângulo O· · ·OH não
ultrapassando 30o. Esses critérios são compatíveis com critérios energéticos, porêm são
mais fáceis de se aplicar em simulação de DM. A distribuição do número de molécu-
las de água que realizam LH com uma molécula de água é apresentada na figura 3.6.
Observa-se que a introdução de frutose no sistema faz com que o número médio de LH
entre moléculas de água diminua, perturbando a rede de LH da água. Essa perturbação
é mais evidente que na análise deg(r) entre sítios da água (figura 3.3) e deve-se prin-
cipalmente às moléculas de água que estão nas primeiras camadas de solvatação das
frutoses.

3.3. Propriedades Estruturais 15
0 1 2 3 4 5 6 7número de ligações de H
0
0.1
0.2
0.3
0.4
0.5
freq
uênc
ia
C1C2C3
Figura 3.6. Distribuição do número de ligações de hidrogênio que uma molécula de
água realiza com outras moléculas de água.
0 2 4 6 8 10 12 14 16número de ligações de H
0
0.1
0.2
0.3
freq
uênc
ia
C1C2C3
Figura 3.7. Distribuição do número do número de ligações de hidrogênio que uma
frutose realiza com moléculas de água.
A distribuição do número de moléculas de água interagindo com uma molécula de
frutose via LH é apresentado na figura 3.7, enquanto que a distribuição do número de
moléculas de frutose interagindo com uma molécula de frutose é apresentado na figura
3.8. Esses gráficos mostram que, com o aumento da concentração do soluto, as molécu-
las de águas que estão interagindo com as frutoses são substituídas por outras moléculas
de frutose. Essas distribuições são relativamente largas. Por exemplo, no sistema C1
uma frutose pode estar ligada com um número entre 4 e 14moléculas de águas (figura
3.7). A presença de números tão baixos deve-se a constante quebra e reformação de LH
entre as moléculas de água e as hidroxilas das frutoses. O gráfico 3.8 mostra que no
sistema C1, as frutoses aparecem 50% do tempo isoladas umas das outras interagindo

16 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
0 1 2 3 4 5 6 7 8número de moléculas de frutose
0
0.1
0.2
0.3
0.4
0.5
freq
uêci
a
C1C2C3
Figura 3.8. Distribuição do número de moléculas de frutose interagindo simultanea-
mente por ligações de hidrogênio com uma frutose de referência.
apenas com a água. Já para os sistemas C5 e C3, as frutoses ligadas a uma ou duas outras
moléculas de frutose são mais freqüentes que as moléculas isoladas. Esse gráfico ainda
mostra que para os sistemas C2 e C3 existe uma considerável parcela de moléculas de
frutose que estão ligadas a um grande número de outras moléculas de frutose (entre 4
e 6 moléculas), apontando para a formação de aglomerados (clusters) de moléculas de
frutose interagindo via LH. Nota-se que as distribuições de LH entre frutoses e água e
entre frutoses (figuras 3.7 e 3.8) são muito mais sensíveis à variação de concentração
que a distribuição de LH entre moléculas de água (figura 3.6). Isso indica a existência
de “bolsões” ou cavidades contendo água nos aglomerados de frutose. No interior destas
cavidades a estrutura tetraédrica da água é aproximadamente mantida.
Para melhor ilustrar a estrutura das soluções, são apresentadas na figura 3.9 fo-
tografias de um passo de simulação dos sistemas C1, C2 e C3. As moléculas de frutose
são apresentadas pelas superfícies alaranjadas, enquanto que as moléculas de água são
apresentadas em azul. Podem se observar as frutoses dispersas na solução de menor con-
centração, e os aglomerados de frutoses e “bolsões” de água nas cavidades e reentrâncias
destes aglomerados nos sistemas mais concentrados.
Para estudar a estrutura destes aglomerados, considera-se que duas moléculas de fru-
tose pertencem ao mesmo aglomerado quando elas interagem diretamente por LH, ou
indiretamente, por uma cadeia de outras frutoses que interagem via LH. Considera-se
ainda que o seu tamanhoN é dado pelo número de moléculas de frutose que pertence
ao aglomerado. Computamos a distribuiçãop(N) do tamanhoN dos aglomerados. Na
figura 3.10 apresenta-se a funçãoP (N) = Np(N), que é proporcional à probabili-

3.3. Propriedades Estruturais 17
Figura 3.9. Fotografias de um passo de simulação tirado aleatoriamente das trajetórias
dos sistemas C1 (superior esquerda), C2 (superior direita) e C3 (inferior). As frutoses são repre-
sentadas pelas superfícies alaranjadas, e a água pelas moléculas em azul.
dade de que uma molécula de frutose sorteada aleatoriamente do sistema pertença a um
aglomerado de tamanhoN . Além dos sistemas C1, C2 e C3, estudamos mais dois sis-
temas intermediários, C4 e C5, com concentrações 3.5 e 3.8 M, respectivamente. As
quantidades totais de frutose nos sistemas C1, C2, C4, C5 e C3 são 14, 53, 66, 75 e 82,
respectivamente. Para o sistema C1, como já mostrado pela análise de distribuição de
LH, a maioria das moléculas de frutose estão isoladas uma das outras e solvatadas pela
água. Mas mesmo nessa concentração existe uma freqüência considerável de aglomer-
ados de tamanho 5. Para o sistema C2, novamente a maioria das frutoses permanece

18 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
0 20 40 60 80N
0
0.2
0.4
0.6
0.8
P(N
)C1C2C4C5C3
Figura 3.10. Função P(N) proporcional a probabilidade de que uma molécula de
frutose pertença a um aglomerado de tamanho N.
isolada, mas existe uma probabilidade considerável de que aglomerados de tamanhos
que variam entre 10 e 45. Já para os sistemas C3 e C4 o gráfico de P(N) mostra que
ou as moléculas de frutose estão isoladas ou pertencem a um grande aglomerado de
tamanho aproximado 65 e 72, respectivamente. A probabilidade de aglomerados de
tamanhos intermediários é pequena, mas não desprezível. As moléculas isoladas ocor-
rem quando as LH se rompem e elas se afastam do aglomerado grande. Os aglomerados
de tamanho intermediário ocorrem devido a quebra do aglomerado grande em aglomera-
dos menores. A baixa freqüência destes aglomerados de tamanho intermediários mostra
que rapidamente as LH entre eles são restabelecidas para formar um único aglomerado
grande. Entre os sistemas C4 e C5 existe uma diferença de concentração de apenas 0.3
M. No entanto, a freqüência de aglomerados de tamanho intermediário cai rapidamente
de um valor apreciável para praticamente zero, sugerindo existência de uma transição
do tipo percolativa, onde o cluster se estende de um lado a outro da amostra. Embora
a determinação da concentração crítica desta transição percolativa envolva um estudo
em função do volume do sistema, os resultados das simulações realizadas nesse trabalho
indicam que essa transição ocorre entre as concentrações 3.5 M para 4.0 M.

3.4. Propriedades Dinamicas 19
3.4 Propriedades Din amicas
O coeficiente de difusão da água possui contribuições tanto das moléculas de água nas
camadas de solvatação das frutoses como das moléculas no interior dos bolsões. Esses
dois tipos de água devem possuir dinâmica translacional muito distinta. Recentemente,
num estudo de DM de soluções aquosas diluídas de glucose, trehalose e sucrose [21],
mostrou-se que existe um retardo nas dinâmicas translacional e rotacional das moléculas
de água quando elas se aproximam das moléculas de açucar. É razoável supor que esse
fenômeno também ocorra no sistema frutose-água. O coeficiente de difusão da água de-
pende da complexa dinâmica do líquido como um todo, pois além da estrutura média dos
aglomerados, que tem influência no tamanho médio dos bolsões de água e na extensão
da interface água-aglomerado, ele também depende do tempo de vida do aglomerado,
e de como esses bolsões e interfaces evoluem no tempo. O painel esquerdo da figura
3.11 compara valores experimentais do coeficiente de auto-difusãoDw da água com os
valores obtidos pelas simulações de DM realizadas nesse trabalho. Os valores experi-
mentais foram obtidos por ressonância magnética nuclear (NMR paraNuclear Magnetic
Resonance, ref. [16]), e por espalhamento quasi-elático de neutrons (QENS paraQuasi-
Elastic Neutron Scattering, ref. [19]). Neste segundo trabalho, os autores analisaram
os dados de QENS por dois modelos distintos: modelo Gaussiano-Lorenziano (GL) e
o modelo Exponencial Estirada (SE paraStretched Exponential). Os coeficientes de di-
fusão foram computados a partir das trajetórias de DM utilizando a relação de Einstein
[27]
D = limt→∞
1
6t〈|r(t) − r(0)|2〉, (3.3)
onder(t) é a posição do centro de massa da partícula Browniana. Os comportamento de
Dw em função da concentração obtidos por NMR e QENS são discordantes na região
de altas concentrações. Os dados de NMR apresentam uma diminuição na derivada da
curva, enquanto que tanto os dados de QENS-GL como de QENS-SE apresentam um
comportamento linear por toda a faixa de concentração estudada. Tanto o comporta-
mento deDw em função da concentração obtido por QENS, como seus valores numéri-
cos, sobre tudo na região de alta concentração, estão em excelente acordo com os dados
obtidos da simulação. A discrepância para baixas concentrações deve-se ao conhecido
fato de que o coeficiente de difusão do modelo SPC,3.85 × 10−5 cm2/s, é substancial-
mente maior que o valor2.4 ± 10−5 cm2/s aceito experimentalmente para a água [19].

20 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
0 1 2 3 4 5 6 7c (mol/L)
0
1
2
3
4D
w (
10-5
cm2 / s
)NMRQENS-GLQENS-SEMD
0 1 2 3 4 5 6 7c (mol/L)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
Df (
10-5
cm
2 / s)
Figura 3.11. Painel esquerdo: Coeficiente de difusãoDw da água na solução em
função da concentração. Painel direito: Coeficiente de DifusãoDf das frutoses em função da
concentração. Dados experimentais: RMN (círculos pretos [16]), QENS-GL (círculos azuis
[19]) e QENS-SE (círculos verdes [19]). Nossos resultados: DM (quadrados vermelhos). As
linhas tracejadas servem como guia para os olhos.
É importante notar que a concordância numérica se dá na região de alta concentração,
onde ocorre a formação de aglomerados de frutose, sendo a dinâmica translacional da
água dependente da dinâmica do aglomerado.
O painel direito da figura 3.11 apresenta o coeficiente de auto-difusão Df das molécu-
las de frutoses obtidos por NMR [16] e por DM. Em baixas concentrações a difusão do
soluto é influenciada pela dinâmica translacional de água. O alto valor deDw do mod-
elo SPC puro em relação ao valor experimental novamente explica porque, para baixas
concentrações, a difusão das frutoses obtidas por DM são maiores que os valores exper-
imentais. Os coeficientes de difusão dos solutos na região de alta concentração estão em
excelente acordo com os dados experimentais, principalmente na região 3.5 M onde ex-
iste uma mudança de comportamento na curva experimental. Para altas concentrações,
a difusão das moléculas do soluto não depende tanto da difusão do solvente, mas sim da
estrutura e dinâmica do aglomerado formado.

3.4. Propriedades Dinamicas 21
0 20 40 60 80t (ps)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
C1µ (t
)0 0.1 0.2
0.8
0.9
1
F
W
Figura 3.12. Função de correlação temporal reorientacional normalizadaCµ1 (t) do
momento de dipolo das moléculas de frutose (três curvas superiores, F) e de água (três curvas
inferiores, W). No gráfico interno evidencia-se o comportamento rápido, dominado pelos movi-
mentos torcionais das hidroxilas das frutoses, e pelo movimento inercial e libracional da água.
C1: linha sólida; C2: linha tracejada; C3: linha pontilhada.
Os movimentos moleculares reorientacionais em líquidos são geralmente analisados
através das funções de correlação temporal (FCT)
C�(t) = 〈P� (cosθ(t))〉, (3.4)
ond P� refere-se ao�-ésimo polinômio de Legendre eθ(t) é o ângulo varrido por um
vetor definido na molécula em um tempot. Foram calculadosCµ1 (t) para o momento
de dipolo elétrico ( µ) da frutose e da água, eCOH2 (t) para o vetor O-H dá água e para
as hidroxilas da frutose. No caso da frutose, foi realizada a média deCOH2 (t) sobre
as diferentes hidroxilas de cada frutose, e então a média sobre os dois tautômeros con-
siderados. Os gráficos deCµ1 (t) e COH
2 (t) são apresentados nas figuras 3.12 e 3.13,
respectivamente. A funçãoCµ1 (t) está relacionada com medidas de relaxação dielétrica,
enquanto que a funçãoCOH2 (t) com medidas da relaxação por NMR. Em tempos curtos
(≈ 100 fs) essas funções apresentam um decaimento inercial gaussiano seguido por um
comportamento oscilatório rápido (ver gráfico interno da figura 3.12). Este comporta-
mento oscilatório deve-se aos movimentos torsionais das hidroxilas das frutoses, e aos
chamados movimentos libracionais, movimentos de rotação impedida, no caso da água.
O comportamento libracional emCµ1 (t) eCOH
2 (t) é seguido por um decaimento biexpo-

22 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
Tabela 3.3. Parâmeros de ajuste biexponencial do comportamento pós-libracional
das FCT normalizadaCµ1 (t) para a frutose e a água (ver figura 3.12) e os tempos de correlação
reorientacional integralτx1 , x : f, w. Todos os tempos estão em ps.
frutose
Sistema a1 t1 a2 t2 τx1
C1 0.24 6.22 0.67 130.91 89.44
C2 0.10 8.51 0.82 173.19 144.15
C3 0.11 13.90 0.81 381.28 313.71
água
C1 0.63 2.96 0.16 9.02 3.33
C2 0.58 4.95 0.18 30.86 8.65
C3 0.51 6.59 0.25 50.53 16.04
0 5 10 15 20 25 30t (ps)
0
0.2
0.4
0.6
0.8
1
C2O
Hw (
t)
0 20 40 60 80t (ps)
0
0.2
0.4
0.6
0.8
1
C2O
Hf (
t)
Figura 3.13. Função de correlação temporal reorientacionalCOHw2 (t) de versor
definido pelo OH da água (painel à esquerda), e a média da funçãoCOHf2 (t) do versor definido
pelos OH’s das frutoses (painel à direita).
nencial. Como experado, devido a sua maior massa e tamanho, e também devido a sua
camada de hidratação, os movimentos reorientacionais da frutose são muito mais lentos
que os da água. O aumento da concentração de frutose é acompanhado pelo aumento da

3.4. Propriedades Dinamicas 23
Tabela 3.4. Parâmeros de ajuste biexponencial do comportamento pós-
libracional das FCT normalizadaCOH2 (t) para a frutose e a água (ver figura 3.13) e
os tempos de correlação reorientacional integralτx2 , x : f, w.. Todos os tempos estão
em ps.
frutose água
Sistema a1 t1 a2 t2 τ f2 a1 t1 a2 t2 τw
2
C1 0.29 6.10 0.47 37.25 19.30 0.58 1.48 0.10 4.88 1.37
C2 0.19 8.26 0.59 113.71 68.95 0.50 2.40 0.15 18.74 3.93
C3 0.15 8.40 0.64 182.10 117.93 0.44 2.71 0.21 16.41 4.78
viscosidade do sistema, e por uma conseqüente reorientação mais lenta.
Para obter uma estimativa quantitativa dos tempos de correlação reorientacionais,
biexponenciais foram ajustadas às partes pós-libracionais das funçõesC�(t) (t > 0.5 ps
para a água et > 1.0 ps para a frutose):
C�(t) ≈ a1exp(−t/t1) + a2exp(−t/t2) . (3.5)
Também foram computados os tempos de correlação integral através de:
τx� =
∫ ∞
0C�(t)dt x : f, w. (3.6)
O valores dos parâmetros de ajuste da biexponencial e deτx� para as funçõesCµ
1 (t)
e COH2 (t) são apresentadas nas tabelas 3.3 e 3.4, respectivamente. De uma maneira
geral, estes resultados mostram que, tanto para a frutose como para a água, o tempo de
relaxação rápidot1 , depende muito pouco da concentração de frutose, enquanto que
o tempo de relaxação lentot2 varia aumenta bastante com a concetração. Observa-se
ainda que a importância det1, medida pelo valor dea1 diminui com o aumento da
concentração, ao passo que a importância deτ2, medida pelo valor dea2, aumenta com
a concentração.
Estudou-se também a dinâmica da rede de ligações de hidrogênio do sistema. Para
este fim computou-se a probabilidadef ijHB(t) de que, dado que uma molécula da espécie
i esteja interagindo via LH com uma molécula da espéciej no tempot0, que elas ainda
estejam interagindo no tempot0 + t, independente de possíveis quebras e reformações

24 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
0 20 40 60 80 100t (ps)
0
0.2
0.4
0.6
0.8
1
f HB(t
) FF
FW
WW
Figura 3.14. Probabilidade de sobrevivência de interação por ligação de hidrogênio
fHB(t) entre duas moléculas de frutose (3 curvas superiores, FF), uma molécula de frutose e
uma de água (3 curvas intermediárias, FW), e entre duas moléculas de água (3 curvas inferiores,
WW), para os sistemas C1 (linhas cheias), C2 (linhas tracejadas) e C3 (linhas pontilhadas).
desta ligação neste interim, ou seja,f ijHB(t) é uma medida da probabilidade de sobre-
vivência de ligações de hidrogênio entre espéciesi ej em um intervalo de tempot. Essa
função pretende capturar a dinâmica das múltiplas quebras de interações necessárias
para que uma molécula sofra difusão em um sistema líquido que forma uma rede de LH.
A figura 3.14 apresenta a probabilidadef ijHB(t) para interações entre duas moléculas
de frutose (FF), para interações entre uma molécula de frutose e uma de água (FW), e
para interações entre duas moléculas de água (WW). Parâmetros de ajuste biexponen-
cial destas curvas são apresentadas na tabela 3.5. Essas curvas e parâmetros fornecem os
tempos característicos de sobrevivência da interação entre duas moléculas por ligações
de hidrogênio e estão diretamente associados à dinâmica de rearranjo da rede de LH. O
gráfico mostra que as interações entre moléculas de frutose são extremamente estáveis.
Pode-se explicar este fato pela possibilidade de se estabelecer múltiplas LH entre um
par de frutoses simultaneamente. Além das hidroxilas, as frutoses possuem vários sítios
hidrofóbicos expostos ao solvente. Em uma eventual interação entre frutoses esses sítios
hidrofóbicos podem entrar em contato, estabilizando ainda mais a interação. As inter-
ações entre água e frutose ocorrem na camada de hidratação das frutoses e o decaimento
decorre da troca de moléculas de água com o meio aquoso. No caso do sistema mais
diluído o gráfico e os ajustes fornecem uma estimativa do tempo de vida da camada de
solvatação das moléculas de frutose. O decaimento da curva no sistema C3 ocorre pela

3.4. Propriedades Dinamicas 25
Tabela 3.5. Parâmetros de ajuste de biexponencial do comportamento pós-
libracional das funções probabilidade de sobrevivência de ligação de hidrogênio fHB
entre duas moléculas de frutose, entre uma molécula de frutose e uma molécula de água,
e entre duas moléculas de água (ver figura 3.14).
frutose-frutose
Sistema a1 t1 a2 t2C1 0.21 23.84 0.58 421.18
C2 0.11 23.38 0.73 436.41
C3 0.07 13.71 0.76 587.33
frutose-água
C1 0.44 10.80 0.31 53.60
C2 0.30 13.36 0.46 95.04
C3 0.28 14.33 0.51 122.87
água-água
C1 0.57 3.15 0.06 22.07
C2 0.44 4.73 0.09 36.96
C3 0.46 6.43 0.15 47.00
troca de moléculas de água que estão na interface do aglomerado com aquelas que es-
tão no interior dos “bolsões” aquosos. Também observa-se do gráfico que, mesmo para
o sistema mais diluído, as interações entre a frutose e a água são mais estáveis que as
interações entre moléculas de água, sustentando a hipótese de que a dinâmica da água
ligada à frutose é mais lenta que a dinâmica da água pura. A formação dos aglomerados
e o conseqüente retardo da dinâmica do sistema com o aumento da concentração leva ao
aumento no tempo de interação entre as moléculas. O decaimento nas curvas das inter-
ações entre frutoses para os sistemas C3 é uma medida indireta da relaxação estrutural
dos aglomerados.
As FCT de reorientaçãoCµ1 (t) e COH
2 (t) da frutose e da água, e a probabilidade
fHB(t) de sobrevivência de LH entre as diversas espécies do sistema tiveram um exce-
lente ajuste ajuste com a exponencial estirada
f(t) ≈ a exp
[−
(t
τ
)β]. (3.7)

26 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
0
0.2
0.4
0.6
0.8
1
f HB(t
)
0 20 40 60 80 100t (ps)
0.001
0.01
0.1
1
f HB(t
)
FF
FW
WW
FF
FW
WW
Figura 3.15. Painel superior: Ajuste por uma exponencial estirada da probabilidade
de sobrevivência de interação por ligação de hidrogêniofHB(t) entre duas moléculas de frutose
(3 curvas superiores, FF), uma molécula de frutose e uma de água (3 curvas intermediárias,
FW), e entre duas moléculas de água (3 curvas inferiores, WW), para os sistemas C1 (circulos
cheios), C2 (quadrados vazios) e C3 (triângulos cheios). Painel inferior: Mesmo gráfico do
painel superior com o eixo das ordenadas em escala logarítmica.
Para mostrar a precisão do ajuste da exponencial estirada nestas funções, apresenta-se no
painel superior da figura 3.15, os dados da DM e as curvas ajustadas para afHB(t). No
painel inferior da mesma figura é apresentado o mesmo gráfico com o eixo da ordenada
em escala logarítmica. Neste painel observa-se que a diferença entre a curva e o ajuste
ocorre na funções de ligações entre moléculas de águas de decaimento mais rápido,
e numa região onde o valor da função é muito próximo de zero. O ajuste emCµ1 (t) e
COH2 (t) tiveram precisão semelhante às das curvas do gráfico apresentado. Sistemas que

3.5. Conclusao 27
Tabela 3.6. Parâmetros de ajuste da exponencial estirada nas FCT de reorien-
taçãoCµ1 (t) eCOH
2 (t) (ver figuras 3.12 e 3.13).
frutose água
Sistema a τ β a τ β
C1 0.98 86.81 0.45 0.92 2.98 0.75
Cµ1(t) C2 0.93 181.01 0.68 1.04 4.40 0.52
C3 0.95 559.12 0.51 1.04 7.07 0.46
C1 0.85 17.11 0.62 0.91 1.15 0.69
COH2 (t) C2 0.86 74.43 0.51 1.08 1.37 0.43
C3 0.87 166.24 0.45 0.99 2.25 0.47
Tabela 3.7. Parâmetros de ajuste da exponencial estirada nas curvas de proba-
bilidadefHB(t) de sobrevivência da ligação de H no tempot (ver figura 3.15).
frutose-frutose frutose-água água-água
a τ β a τ β a τ β
C1 0.89 249.18 0.43 0.95 14.75 0.57 1.12 1.45 0.51
C2 0.88 504.91 0.55 0.89 38.56 0.55 1.12 1.45 0.39
C3 0.86 1331.98 0.48 0.91 53.94 0.52 1.10 3.15 0.41
exibem relaxação descrita por exponenciais estiradas, cujo decaimento se estentem por
uma vasta faixa de tempos, é uma indicação de que dinâmica complexa está ocorrendo
no seu interior, com processos caracterizados por uma variedade de escalas de tempo.
Observa-se que, para todos os ajustes, os valores do parâmetro de estiramentoβ diferem
muito de 1, caso especial onde a exponencial estirada equivaleria a um decaimento ex-
ponencial simples.
3.5 Conclus ao
Foram estudadas as propriedades básicas de estrutura, estatística de LH e dinâmica de
soluções aquosas de frutose por DM. Este estudo mostrou que este sistema apresenta
propriedades estruturais muito interessantes em função da concentração, sendo o ponto

28 Capıtulo 3. Solucoes Aquosas de Frutose: Estrutura e Dinamica
mais interessante a formação de aglomerados de frutoses e uma possível transição per-
colativa na faixa de concentração entre 3.5 e 4.0 mol/L. As moléculas de água deste
sistema ficam agrupadas em cavidades no interior destes aglomerados. O coeficiente de
difusão da água e da frutose em função de concentração estão em excelente acordo com
dados experimentais disponíveis, sobretudo na faixa próxima à concentração de tran-
sição percolativa. Esta concordância é surpreendente, pois a dinâmica translacional das
moléculas torna-se dependente da estrutura média e da relaxação estrutural dos aglomer-
ados, onde estão envolvidos movimentos correlacionados de várias moléculas do soluto.
A análise do tempo de sobrevivência da interação entre moléculas de frutose por ligações
de hidrogênio mostra que estas simulações capturam pelo menos parte desta relaxação.

Capıtulo 4
Soluc oes Aquosas de
Frutose: Relaxac¸ ao
Diel etrica
4.1 Introduc¸ ao
Neste capítulo continuamos com os estudos das soluções aquosas de sacarídeos através
da análise de suas propriedades dielétricas. O principal interesse no fenômeno dielétrico
é o estudo da dinâmica de rotações moleculares. O estudo destas propriedades funda-
mentais de líquidos moleculares são uma área de pesquisaper se, mas outros campos
da físico-química de líquidos têm bastante interesse na compreensão das propriedades
dielétricas de um determinado sistema. Por exemplo, existe grande interesse na com-
preensão do efeito de um ambiente dielétrico flutuante sobre o curso de reações quími-
cas. A energia livre de solvatação caracterizando a transferência de elétrons ou de out-
ras partículas carregadas dependem da polarização instantânea do meio no local onde
a reação ocorre. A dinâmica de solvatação pode ser modelada aproximadamente uti-
lizando como dado de entrada as propriedades dielétricas do solvente. Como a resposta
de solvatação depende da distância do solvente ao soluto, esses modelos necessitam do
tensor permissividadeε (k, ω), dependente do vetor de ondak. Muito embora aborda-
gens analíticas tenham sido empregadas com sucesso, propriedades dielétricas de líqui-
dos são geralmente estudadas através de simulações de dinâmica molecular. Essa abor-
dagem permite acessar os rápidos mecanismos de relaxação não-difusivos que são de es-
pecial importância para a dinâmica de solvatação. Além disto, esta abordagem permite
o estudo de sistemas que se estruturam através de uma rede de ligações de hidrogênio.
Com as simulações de DM, pode-se estudar o efeito da dinâmica da rede de ligações de
hidrogênio nas propriedades dielétricas do sistema.
Neste trabalho foi computada a permissividade dielétricaε(ω) e a permissividade
29

30 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
dielétrica dependente do vetor de ondaε(k, ω) das soluções aquosas de frutose. Além
da sua utilização em modelos teóricos de dinâmica de solvatação, existem dois outros
motivos práticos para o cálculo das propriedades dielétricas dependentes do vetor de
onda. Primeiramente, com uma mesma quantidade de trajetórias,ε(k, ω) pode ser obtido
de forma mais precisa queε(ω), pois podem ser tomadas médias sobre vetores de onda
ortogonais de mesma magnitude. Segundo, com o estudo deε(k, ω) pode-se saber se o
sistema simulado é suficientemente grande para computar suas propriedades dielétricas.
Na próxima seção é apresentada a formulação teórica sobre qual a abordagem deste
trabalho é baseada.
4.2 Formulac ao Teorica
A - Vetor de onda nulo ( k = 0)
A quantidade de interesse é a permissividade dielétrica dependente da freqüênciaε(ω),
acessível por experimentos de espectroscopia dielétrica. Ela está ligada à suscetibili-
dade dielétricaχ(ω), que relaciona a polarização de um material com o campo elétrico
aplicado, por [28]:
χ(ω) = ε(ω) − 1. (4.1)
A conexão entre a propriedade macroscópicaχ(ω) e propriedades microscópicas acessíveis
por simulações de DM, se dá através da função de correlação do dipolo coletivo do sis-
tema
Φ(t) =〈M(t) · M(0)〉〈|M(0)|2〉 (4.2)
e pela equação, obtida pela teoria de resposta linear [29]
χ(ω) =〈|M(0)|2〉3KBTV ε0
L[−Φ(t)
], (4.3)
ondeKB é a constante de Boltzmann, T é a temperatura do sistema, V é volume do
sistema,ε0 é a permissividade de vácuo, eM(t) é o momento de dipolo coletivo do
sistema, dado por
M(t) =∑α
Nα∑j=1
µjα(t), (4.4)

4.2. Formulacao Teorica 31
ondeNα é o numero de moléculas da espécieα, e µjα(t) é o momento de dipolo daj-
ésima molécula da espécieα no tempot. A funçãoΦ(t) retrata a relaxação de flutuações
no momento de dipolo coletivo do sistema. O símboloL[f ] significa a transformada de
Fourier-Laplace da funçãof(t), ou seja
L[f(t)] =∫ ∞
0dteiωtf(t) = f(ω). (4.5)
Utilizando a propriedadeL[−Φ(t)] = Φ(0) + iωΦ(ω) em 4.3 tem-se:
χ(ω) =〈|M(0)|2〉3KBTV ε0
[1 − ωΦ(ω)
](4.6)
Esta equação mostra que, no regime de resposta linear, a suscetibilidade do sistema a
um campo de freqüênciaω está relacionado a flutuações no momento de dipolo total
do sistema com a mesma freqüência. Usualmente, a parte real deχ(ω) é chamada de
suscetibilidade dependente da freqüência e a parte imaginária deχ(ω) é chamada de ab-
sorção dielétrica. Sumarizando, para obterε(ω), basta computar a função de correlação
do momento de dipolo total do sistemaΦ(t), tomar sua transformada de Fourier-Laplace,
substituir na equação 4.6, e por fim substituir esta última em 4.1.
Para o caso especial queω = 0, a equação 4.3 se reduz a:
χ(0) =Φ(0)
3KBTV ε0
=〈|M(0)|2〉3KBTV ε0
, (4.7)
o que define a suscetibilidade dielétrica estáticaχ(0). A constante dielétrica é com-
putada através da equação 4.1
ε(0) = χ(0) + 1. (4.8)
Portanto a constante dielétrica de um sistema depende basicamente da flutuação média
do seu momento de dipolo total.
O momento de dipolo total do sistema para misturas binárias pode ser decomposto
nas contribuições de cada uma das espécies. Por exemplo, seguindo notação da equação
4.4, numa mistura de frutose (F ) e água (W ), temos
M(t) = MF (t) + MW (t) =NF∑i=1
µiF (t) +
NW∑i=1
µiW (t), (4.9)

32 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
Desta forma podemos decompor a função de correlação temporalΦ(t) em:
Φ(t) = ΦFF (t) + ΦFW (t) + ΦWW (t), (4.10)
onde
ΦFF (t) = 〈MF (t) · MF (0)〉/〈|M(0)|2〉 (4.11)
ΦWW (t) = 〈MW (t) · MW (0)〉/〈|M(0)|2〉 (4.12)
ΦFW (t) =(〈MF (t) · MW (0)〉 + 〈MW (t) · MF (0)〉
)/〈|M(0)|2〉. (4.13)
As funçõesΦFF (t) e ΦWW (t) são chamadas funções de autocorrelação das espéciesF
e W , enquanto queΦWF (t) é chamada função de correlação cruzada entre as espécies.
Portanto, a relaxação da funçãoΦ(t) depende da relaxação destas funções separadas
e a constante dielétrica do sistema depende da flutuação média do dipolo coletivo das
águas, do dipolo coletivo das frutoses e da correlação destes dipolos coletivos. Tomando
a transformada de Fourier-Laplace das três componentes deΦ(t) na equação 4.10, e
utilizando as equações 4.6 e 4.1, pode-se obter a decomposição deε(ω) nos termos de
autocorrelação e de correlação cruzada
ε(ω) = εFF (ω) + εFW (ω) + εWW (ω). (4.14)
A autocorrelação da especieα é dada por
〈Mα(t) · Mα(t)〉 =Nα∑i=1
µiα(t) · µi
α(0) +Nα∑i�=j
µiα(t) · µj
α(0). (4.15)
O primeiro termo do lado direito da igualdade é a FCT reorientacionalCµ1 (t) das molécu-
las da espécieα. As correlações cruzadas entre as espéciesα eβ são dadas por
〈Mα(t) · Mβ(t)〉 =Nα∑i=1
Nβ∑j=1
µiα(t) · µj
β(0). (4.16)
B - Vetor de onda n ao-nulo ( k �= 0)
A quantidade de interesse neste caso é a permissividade dielétricaε(k, ω) dependente
do vetor de ondak e da freqüênciaω, e pode ser decomposto nos termos transversal e
longitudinal, respectivamente perpendicular e paralelo ao vetor de ondak:
ε(k, ω) = εT (k, ω)(1 − kk) + εL(k, ω)kk, (4.17)

4.2. Formulacao Teorica 33
ondek = k/k, 1 é a diade unitária, e os subscritosT e L referem-se respectivamente
às componentes transversal e longitudinal deε(k, ω). As funçõesεT (k, ω) e εL(k, ω)
não são em geral acessíveis experimentalmente, mas são importantes do ponto de vista
teórico conforme mencionamos no início deste capítulo.
Novamente faz-se uso da suscetibilidadeχ(k, ω), desta vez dependente do vetor
de ondak e da freqüênciaω. As componentes deχ(k, ω) são relacionadas com as
componentes do tensor permissividade dielétrica por
χT (k, ω) = εT (k, ω) − 1. (4.18)
e
χL(k, ω) = 1 − 1
εL(k, ω)(4.19)
Essas componentes podem ser expressas em termos de correlações moleculares. As
propriedades microscópicas acessíveis por DM são as componentes transversal e lon-
gitudinal da transformada de Fourier espacial do momento de dipolo total do sistema
M(k, t):
M(k, t) =∑α
Nα∑j=1
µjα(t)exp
[ik · rj
α(t)], (4.20)
ondeNα é número de moléculas da espécieα, µjα é o momento de dipolo daj-ésima
molécula da espécieα, e rjα(t) é a posição do centro de massa desta molécula com
relação ao sistema de referência fixo ao laboratório. A relação entre as componentes
transversal (A = T ) e longitudinal (A = L) deχ(k, ω) e as componentes deM(k, t)
são dadas por:
χA(k, ω) =〈|MA(k, 0))|2〉νAKBTV ε0
[1 + iωΦA(k, ω)
], (4.21)
ondeνT = 2, νL = 1, e as demais constantes possuem seus significados usuais. Esta
equação mostra que a suscetibilidade de uma amostra a um campo com vetor de onda
k e freqüênciaω está relacionado com flutuações no momento de dipolo coletivo em
regiões da amostra de tamanho característico2π/k e que possuem freqüênciaω. A
funçãoΦA(k, ω) é a transformada de Fourier-Laplace
ΦA(k, ω) =∫ ∞
0dtΦA(k, t)exp(iωt) (4.22)

34 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
da função de correlação temporal normalizadaΦA(k, t) das componentes transversal e
longitudinal do momento de dipolo totalMA(k, t)
ΦA(k, t) =〈MA(k, t) · MA(−k, 0)〉
〈|MA(k, 0)|2〉 , A : T, L (4.23)
Assim, como no casok = 0, o momento de dipolo total dependente do vetor de onda
MA(k, t) também possui contribuições das moléculas de frutose (F ) e da água (W ):
MA(k, t) = MFA(k, t) + MW
A (k, t). (4.24)
Substituindo a equação 4.24 em 4.23 facilmente concluímos queΦA(k, t) possui con-
tribuições das autocorrelações de cada espécie, e das correlações cruzadas entre espé-
cies. Assim a equação 4.23 pode ser reescrita como:
ΦA(k, t) = ΦFFA (k, t) + ΦFW
A (k, t) + ΦWWA (k, t), A : T, L (4.25)
onde
ΦααA (k, t) =
〈MαA(k, t) · Mα
A(−k, 0)〉〈|MA(k, 0)|2〉 (4.26)
e
ΦαβA (k, t) =
〈MαA(k, t) · Mβ
A(−k, 0)〉 + 〈MβA(k, t) · Mα
A(−k, 0)〉〈|MA(k, 0)|2〉 , α �= β(4.27)
Neste trabalho foram computadas funções de correlações para várias magnitudes de k
próximas de zero. Como foram utilizadas condições de contorno periódicas em nossas
simulações, os vetores de onda são dadas por
k = (l,m, n)2π
L(4.28)
ondel,m e n são inteiros não-negativos eL é o lado da caixa cúbica de simulação. É
importante lembrar que o valor deL é dependente da concentração (ver tabela 3.1).
De forma geral, dado um determinado valor dek que satisfaça a equação 4.28, para
se obterε(k, ω), devemos inicialmente computar as componentes transversal e longitudi-
nal do momento de dipolo total dependente do vetor de ondaM(k, t) pela equação 4.20
a partir das trajetórias de DM. Destas componentes deM(k, t) computamos as funções

4.3. Prop. Estaticas e Relaxacao independente de k 35
de correlaçãoΦA:T,L(k, t) pela equação 4.23. Computa-se a transformada de Fourier-
Laplace destas funções de correlação e substitui-se em 4.21 para obterχA:T,L(k, ω).
Por fim computamos as componentes transversal e longitudinal deε(k, ω) substituindo
χA:T,L(k, ω) nas equações 4.18 e 4.19, respectivamente.
Para um sistema infinito e isotrópico, a permissividade dielétricaε(ω) pode ser obtida
do limitek → 0 deεT (k, ω) ou εL(k, ω)
ε(ω) = limk→0
εT (k, ω) = limk→0
εL(k, ω) (4.29)
No entanto, como em simulações de DM o sistema não exatamente infinito, mas sim in-
finitamente periódico, a menor magnitude de vetor de onda acessível nestas simulações
é dado pork1 = 2π/L. Segundo a equação 4.28, existem três vetores de onda inde-
pendentes que possuem essa magnitude. Assim, se os tamanhos dos sistemas utiliza-
dos neste trabalho forem macroscópicos o suficientes para poder computar propriedades
dielétricas tem-se que
ε(ω) ∼= εT (k1, ω) ∼= εL(k1, ω). (4.30)
Desta forma, tem-se uma maneira prática para testar se o sistema possui tamanho sufi-
cientemente grande para computar suas propriedades dielétricas ou se existem inomo-
geneidades espaciais no sistema. Maiores detalhes sobre as simulações encontram-se no
capítulo anterior.
4.3 Propriedades Est aticas e Relaxac¸ ao Independente doVetor de Onda
Por tratar-se de uma propriedade coletiva, a constante dielétricaε(0) converge lenta-
mente. A figura 4.1 apresenta a convergência da constante dielétricaε(0) em função do
tempo de simulação, computados pelas equações 4.7 e 4.8. Os valores deε(0) para os
sistemas C1, C2 e C2 são 64±3, 57±3 e 50±3, respectivamente, enquanto que a con-
stante dielétrica para o modelos SPC puro é 65.0 [23]. Os valores experimentais para
esses sistemas são 74.4, 66.2, e 62.4 [18], enquanto que a constante dielétrica da água
pura é 78.4 [23]. É sabido que o valor da constante dielétrica do modelo SPC é bem
menor que o valor aceito experimentalmente. Como resultado, todos os valores teóricos
computados para as soluções são menores que os valores obtidos experimentalmente.

36 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
0 2 4 6 8 10 12t (ns)
0
20
40
60
80
ε 0C1C2C3
Figura 4.1. Média cumulativa da constante dielétricaε(0) dos sistemas C1, C2 e C3
em função do tempo de simulação. Oε(0) é computado pela equação 4.8.
Apesar disto, as constantes dielétricas experimentais e teóricas possuem comportamen-
tos semelhantes em função da concentração. Note também que foram necessárias longas
simulações (12 ns) para garantir convergência.
A figura 4.2 apresenta a FCT do momento de dipolo coletivo dos sistemas C1, C2 e
C3. Como esperado, com o aumento da concentração de frutose a relaxação do momento
de dipolo coletivo torna-se mais lenta. O gráfico interno da figura 4.2 enfatiza o com-
portamento de tempos curtos da FCT, mostrando as rápidas oscilações iniciais devido ao
movimento libracional da água e aos movimentos torcionais das hidroxilas das frutoses.
O comportamento pós-libracional foi ajustado por uma biexponencialf(t) ≈ A1exp(-
t/t1) + A2 exp(-t/t2) e por uma exponencial estiradaf(t) ≈ A exp[-(t/τ )β] . Os valores
dos parâmetros de ajuste para os três sistemas são apresentados na tabela 4.1. Os valores
do tempo de relaxação rápidot1 para os três sistemas se assemelha ao valor do tempo
de relaxação dielétrica do modelo SPC puro de 7.6 ps [23], principalmente o tempo do
sistema C1. A diminuição da importância deste processo de relaxação, medido por A1,
correlaciona bem com a diminuição da quantidade de água nesses sistemas. Estes tem-
pos revelam a existência de moléculas de água no sistema cuja dinâmica se assemelha à
dinâmica da água pura. No sistema mais diluído, esta dinâmica deve-se às águas que não
pertencem às primeiras camadas de solvatação do sacarídeo. Para o sistema mais con-
centrado, esse tempo de relaxação reflete a dinâmica das águas no interior dos “bolsões”
formados na estrutura dos aglomerados de açúcar. Höchtl, Boresche e Steinhauser [30]
estudaram as propriedades dielétricas de soluções diluídas de glucose por DM. Naquele
trabalho, eles utilizaram modelo de água TIP3P [31] e campos de força AMBER [12]

4.3. Prop. Estaticas e Relaxacao independente de k 37
0 0.2 0.40.96
0.98
1
0 20 40 60 80t (ps)
0
0.2
0.4
0.6
0.8
1
1.2
Φ(t
)
C1
C2
C3
Figura 4.2. FCT do momento de dipolo coletivo dos sistemas C1 (linha cheia), C2
(linha tracejada) e C3 (linha pontilhada). O gráfico interno mostra o comportamento rápido,
enfatizando as oscilações libracionais e torcionais.
Tabela 4.1. Parâmetros de ajuste biexponencial e exponencial estirada da função de
correlação temporalΦ(t) do momento de dipolo total do sistema (ver figura 4.2).
biexponencial exponencial estirada
Sistema A1 t1 A2 t2 A τ β
C1 0.78 7.16 0.18 58.72 1.20 6.95 0.54
C2 0.44 10.06 0.50 142.54 1.17 31.38 0.39
C3 0.28 8.93 0.67 183.96 1.07 100.38 0.40
para os solutos, desenvolvidos com metodologias diferentes dos campos de força empre-
gados aqui. Assim como a frutopiranose, a glucose possui um anel de seis membros com
uma hidroximetila e quatro hidroxilas ligadas a esse anel. A frutopiranose e a glucose
diferem apenas na posição relativa das hidroxilas, e na posição da hidroximetila, que no
caso da glucose, está ligado ao carbono 5. Naquele artigo, os autores estudaram soluções
bem mais diluídas que as do presente deste trabalho, 0.11 e 0.41 mol/L, e encontraram
tempos de relaxação rápido iguais á 6.5 e 6.2 ps, respectivamente, estando próximos dos
valores encontrados da simulações com a frutose. Os autores atribuíram esses tempos á
relaxação das moléculas de água que não pertencem a primeira camada de solvatação da
glucose.

38 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
Tabela 4.2. Parâmetros de ajuste das funções espectrais Cole-Cole e Debye
(equação 4.31) obtidos de experimentos de relaxação dielétrica (ref. [1]).
Sistema ε(∞) ∆εD τD (ps) ∆εC τC (ps) h ε(0)
C1 3.5 - - 70.9 10.6 0.081 74.4
C2 6.0 16.0 11.6 44.0 37 0.147 66.1
C3 5.9 11.0 17.0 45.0 72 0.205 62.3
O tempo de relaxação lentoτ2 claramente depende da concentração de frutose no
sistema. Este tempo depende da relaxação estrutural do aglomerado de frutose, que
são formados com o aumento da concentração. A importância deste processo de relax-
ação, medido porA2 correlaciona-se bem com o aumento da concentração de frutose na
solução.
Em um trabalho experimental de espectroscopia dielétrica [1], Fuchs e Kaatze estu-
daram soluções aquosas em diversas concentrações de alguns dissacarídeos e monosacarídeos,
dentre eles a frutose. Neste trabalho, os autores ajustaram suas curvas experimentais dos
sistemas com concentração igual ou maior a 2 mol/L por uma soma de funções espectrais
ε(ω) = ε(∞) +∆εC
1 + (iωτC)1−h +∆εD
1 + iωτD
. (4.31)
O segundo termo à direita da igualdade na equação 4.31 é conhecido como função es-
pectral Cole-Cole e baseia-se em uma distribuição contínua de tempos de relaxação ao
redor de um tempo principalτC , e cuja largura depende do parâmetroh. O terceiro
termo à direita da igualdade é conhecido como função espectral de Debye e reflete um
processo de relaxação exponencial caracterizado por um único tempo característicoτD.
A relaxação dielétrica da água pura à 25oC é descrita apenas por uma função espectral
de Debye com tempo característicoτD = τW = 8.27 ps. A necessidade de se repre-
sentar os dados experimentais por uma soma de dois espectros mostra que ao menos
dois processos distintos estão envolvidos na relaxação dos sistemas mais concentrados.
Para sistemas menos concentrados (c < 2 mol/L) Fucks e Kaatze utilizaram apenas
a função Cole-Cole. Os parâmetros de ajuste para as soluções de frutose publicados
neste artigo são reproduzidos na tabela 4.2. No sistema C1, tanto o valor reduzido deh,
quanto a semelhança deτC com osτD dos outros sistemas levam a crer que, apesar de

4.3. Prop. Estaticas e Relaxacao independente de k 39
0
0.2
0.4
0.6
0.8
1
FFFWWWtotal
0
0.2
0.4
0.6
0.8
1Φ
(t)
0 20 40 60 80t (ps)
0
0.2
0.4
0.6
0.8
1
C1
C2
C3
Figura 4.3. Função de correlação temporal do momento de dipolo total e sua decom-
posição nos termos frutose-frutose (FF), frutose-água (FW) e água-água (WW), para os sistemas
C1 (painel superior), C2 (painel intermediário) e C3 (painel inferior).
ter sido analisada com uma função espectral Cole-Cole, a relaxação desse sistema deve
ser muito próxima do tipo Debye. Esses dados mostram uma dependência fraca deτD
e uma dependência forte deτC com a concentração, similar ao que é observado pelas
simulações deste trabalho. Os valores deτD são maiores que, mas da ordem de,τW e de
t1. Os valores deτC diferem bastante det2, o que pode ser parcialmente explicado pelo
aumento do parâmetroh.
A decomposição da função de correlação temporal totalΦ(t) nos termos de autocor-
relaçãoΦFF (t) e ΦWW (t) e no termo de correlação cruzadaΦFW (t), descrita na seção

40 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
Tabela 4.3. Parâmetros de ajuste das componentes frutose-frutose, frutose-água e
água-água da função de correlação do momento de dipolo total do sistema. Tempos em ps.
frutose-frutose frutose-água
Sistema A1 t1 A2 t2 A1 t1C1 0.004 7.10 0.02 105.33 0.07 50.21
C2 0.01 17.38 0.09 288.59 0.22 140.03
C3 0.01 9.26 0.14 395.94 0.13 220.67
água-água
Sistema A1 t1 A2 t2C1 0.77 7.25 0.09 56.63
C2 0.40 9.23 0.21 90.56
C3 0.25 8.26 0.27 103.42
4.2, é apresentada na figura 4.3. Para uma análise quantitativa, os termos de autocor-
relação foram ajustados por biexponenciais e o termo de correlação cruzada por uma
exponencial. Os valores dos parâmetros são apresentados na tabela 4.3. No sistema C1,
tanto a constante dielétrica quanto a relaxação deΦ(t) dependem basicamente da estru-
turação e relaxação das moléculas de água. Apesar das frutoses possuírem momento de
dipolo e, portanto, poderem contribuir para a relaxação dielétrica, nesta concentração
elas estão praticamente descorrelacionadas por permanecem basicamente isoladas uma
das outras interagindo apenas com as moléculas de água. Nesta situação, a relaxação do
termoΦFF (t) é praticamente igual a função de correlação reorientacionalCµF1 (t) da fru-
tose. Isto está ilustrado na figura 4.4 que comparaΦFF (t)/ΦFF (0) eCµF1 (t) da frutose.
Diferentemente do termoΦFF (0), a estruturação da água em torno das frutoses devido
ao estabelecimento de ligações de hidrogênio entre estas espécies faz com que o termo
ΦFW (0) contribua de uma quantidade apreciável para a constante dielétrica mesmo na
concentração mais diluída. A relaxação deΦFW (t) nessa concentração decorre basica-
mente pela relaxação da estrutura de solvatação das frutoses e pela troca entre moléculas
de água que estão ligadas às frutoses com as que estão no meio aquoso.
Com o aumento da concentração, observa-se pelos gráficos da figura 4.3 que a con-

4.3. Prop. Estaticas e Relaxacao independente de k 41
0 20 40 60 80t (ps)
0.5
0.75
1
1.25
1.5
ΦF
F (t)/
ΦF
F (0);
C 1µ F
(t)
C1
C2
C3
Figura 4.4. Componente frutose-frutose da FCT do momento de dipolo total normal-
izada pelo seu valor emt = 0, ΦFF (t)/ΦFF (0) (linha cheia), e a função de correlação reori-
entacionalCµ1 (t) da frutose (linha tracejada) para as três concentrações estudadas. Para melhor
visualização, as curvas dos sistemas C2 e C3 foram deslocadas de 0.2 e 0.4, respectivamente.
tribuição para a constante dielétrica do termoΦWW (0) torna-se cada vez menor, en-
quanto que os termosΦFW (0) e ΦFF (0) tornam-se cada vez mais importantes. A for-
mação dos aglomerados de açucar permite flutuações estruturais que envolvem várias
frutoses, fazendo com que o termoΦFF (0) contribua substancialmente para a constante
dielétrica. A maior disponibilidade de hidroxilas com o aumento da concentração de
frutose faz com que haja um aumento no número total de LH entre moléculas de fru-
toses e água, aumentando também a contribuição do termoΦFW (0) para a constante
dielétrica. Os gráficos mostram que a relaxação inical deΦ(t) depende basicamente da
relaxação deΦWW (t), enquanto que o comportamento de tempo longos deΦ(t) deve-se
a uma sobreposição dos tempos de relaxação longos dos termosΦWW (t) e ΦFF (t), e
do termo cruzadoΦFW (t). Esse comportamento de tempos longos não é perceptível
para o sistemas C1 pois a contribuição destes termos para a função total é bastante re-
duzida. É interessante notar que nos sistemas C1 e C2, cujas concentrações estão abaixo
da concentração estimada para a transição percolativa, o termoΦFW (t) é maior que o
termoΦFF (t), enquanto que no sistema C3, o inverso é observado. Outro ponto interes-
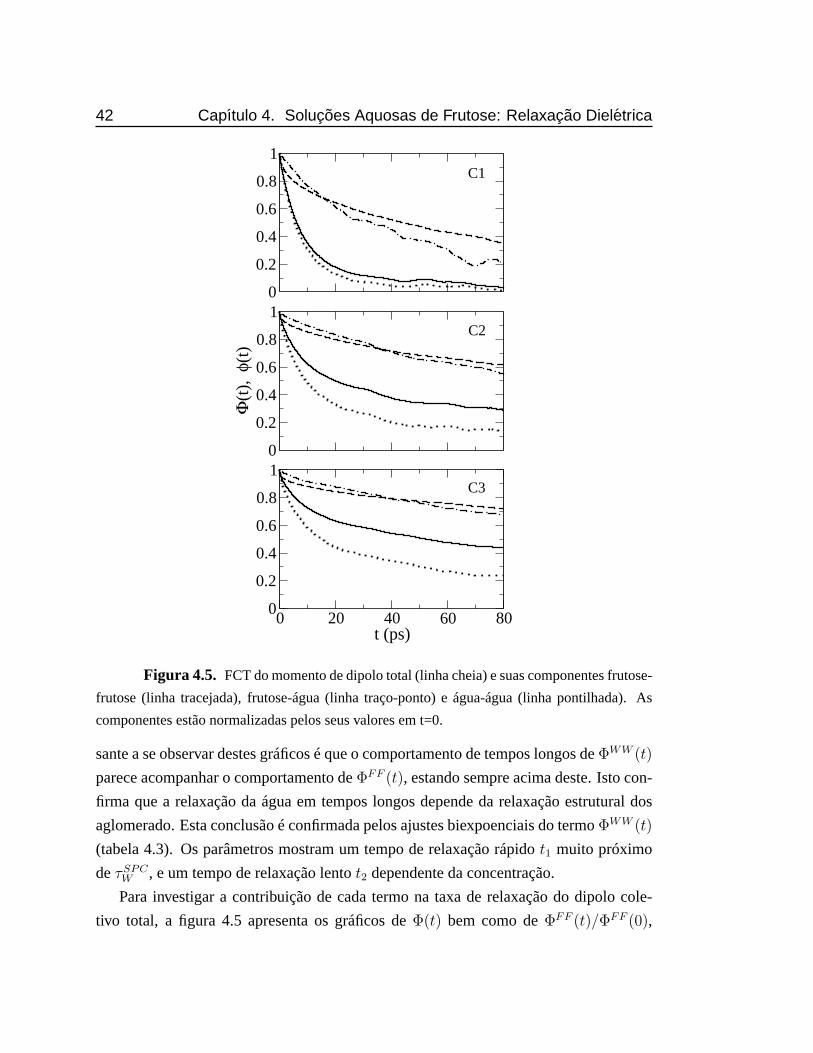
42 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0.8
1
Φ(t
),φ(
t)
0 20 40 60 80t (ps)
0
0.2
0.4
0.6
0.8
1
C1
C2
C3
Figura 4.5. FCT do momento de dipolo total (linha cheia) e suas componentes frutose-
frutose (linha tracejada), frutose-água (linha traço-ponto) e água-água (linha pontilhada). As
componentes estão normalizadas pelos seus valores em t=0.
sante a se observar destes gráficos é que o comportamento de tempos longos deΦWW (t)
parece acompanhar o comportamento deΦFF (t), estando sempre acima deste. Isto con-
firma que a relaxação da água em tempos longos depende da relaxação estrutural dos
aglomerado. Esta conclusão é confirmada pelos ajustes biexpoenciais do termoΦWW (t)
(tabela 4.3). Os parâmetros mostram um tempo de relaxação rápidot1 muito próximo
deτSPCW , e um tempo de relaxação lentot2 dependente da concentração.
Para investigar a contribuição de cada termo na taxa de relaxação do dipolo cole-
tivo total, a figura 4.5 apresenta os gráficos deΦ(t) bem como deΦFF (t)/ΦFF (0),

4.3. Prop. Estaticas e Relaxacao independente de k 43
0 20 40 60 80t (ps)
0
0.5
1
1.5
2
2.5
3
ΦW
W(t
)/Φ
WW
(0);
C 1µ W
(t)
C1
C2
C3
Figura 4.6. FCT normalizada da componente água-água do momento de dipolo co-
letivo (linha cheia) e FCT reorientacional C1(t) do dipolo molecular da água (linha tracejada).
Para melhor visualização os gráficos dos sistemas C2 e c3 foram deslocados de 0.5 e 1.0, respec-
tivamente.
ΦWW (t)/ΦWW (0) eΦFW (t)/ΦFW (0). Estes gráficos apresentam o mesmo padrão para
as três concentrações estudadas, mostrando mais claramente que o tempo de relaxação
rápidot1 deΦ(t) deve-se principalmente à relaxação da água, enquanto que o tempo de
relaxação lentat2 deve-se tanto ao termo de autocorrelação da frutose, com da corre-
lação cruzada. Dentro da janela temporal analisada, esses dois termos possuem tempos
de relaxação da mesma ordem. Para o sistema mais concentrado, o tempot2 do termo
de autocorrelação da frutose é o tempo característico da relaxação estrutural dos aglom-
erados. Inicialmente, a taxa de relaxação do termo de autocorrelação da frutose é maior
que a do termo cruzado, mas para tempos longos essa situação se inverte. Esse com-
portamento deve-se provavelmente às trocas entre as moléculas de água da camada de
solvatação e do seio aquoso. Inicialmente as moléculas de água pertencentes à camada
de solvatação das moléculas de frutose participam do seu processo de difusão rotacional,
fazendo com que a taxa de relaxação do termo cruzado seja bastante pequeno. À medida

44 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
que essas moléculas são trocadas pelas moléculas da seio aquoso, a taxa de relaxação do
termo cruzado começa a crescer. A análise de sobrevivência de ligações de hidrogênio
mostra que o tempo característico para que essa troca ocorra é da ordem de 10 ps (ver
tabela 3.5).
Na seção 3.3, comparando a dependência da distribuições de LH com a concentração
de frutose nas figuras 3.7, 3.8 e 3.6, conclui-se que, além de aglomerados de frutose, ex-
iste a formação de “bolsões” de água no interior destes aglomerados. Em decorrência
da rede de ligações de hidrogênio, dentro desses bolsões as correlações dinâmicas entre
moléculas de água é muito grande, equivalente à correlação encontrada na água pura.
Segundo a equação 4.16, caso essas correlações não existissem, o termo de autocorre-
lação da águaΦww(t) deveria ser igual à função de correlação de reorientação molec-
ular Cµw1 (t) da água. A figura 4.6 comparaΦww(t)/Φww(0) e Cµw
1 (t). Estes gráficos
mostram que, na verdade, as correlações dinâmicas entre moléculas de água tornam-se
cada vez mais importantes com o aumento da concentração, mostrando que mesmo na
solução mais concentrada as moléculas de água ficam agrupadas, ao contrário do que
se poderia imaginar caso as moléculas de frutose se dispersassem homogeneamente na
solução.
4.4 Propriedades Dependentes do Vetor de Onda k
Apresenta-se agora as propriedades dielétrica dependentes do vetor de ondak. Na figura
4.7 são apresentadas a função de correlação totalΦ(t) e sua componente transversal
ΦT (k, t) para os cinco menores vetores de onda acessíveis para os tamanhos de sistema
simulados nas três concentrações. As magnitudes dos vetores de onda são apresentados
na tabela 4.4. Observa-se que, para cada mistura,ΦT (k, t) relaxa mais lentamente com
a diminuição dek. Isto é esperado, pois uma região maior do espaço é provado com a
diminuição da magnitude do vetor de onda, de forma que a relaxação deΦT (k, t) tende
a Φ(t) quandok → 0. Relaxação mais lenta com a diminuição dek também foram
encontradas em simulações de DM de metanol e água puros e na misturas destas duas
substâncias [32]. As FCTΦA(k, t) (A = T, L) foram ajustadas por biexponenciais,
cujos valores dos parâmetros são apresentados na tabela 4.5.
Na figura 4.8 são apresentadas a componente longitudinalΦL(k, t) deΦ(k, t) para
os cinco menoresk possíveis para os sistemas estudados. Como se sabe da literatura

4.4. Propriedades Dependentes do Vetor de Onda k 45
0
0.2
0.4
0.6
0.8
1k = 0k = 1k = 2k = 3k = 4k = 5
0
0.2
0.4
0.6
0.8
1Φ
(t),
ΦT (
κ,t)
0 20 40 60 80t (ps)
0
0.2
0.4
0.6
0.8
1
C1
C2
C3
Figura 4.7. FCT ΦT (k, t) da componente transversal do momento de dipolo total do
sistema para os cinco menores vetores de onda possíveis para o sistema, e a função de correlação
totalΦ(t) parak=0.
[33, 34], a difusão rotacional contribui muito pouco para a componente longitudinal,
ΦL(k, t), tornando-a bastante sensível às rápidas oscilações do momento de dipolo cole-
tivo. Diferente do que se observa na componente transversal, a componente longitudinal
não apresenta uma dependência clara comk. A taxa de relaxação deΦL(k, t) para tem-
pos longos (t > 12 ps) parece ser quase independente dek. Já para o sistema C2 e a
parte de tempos curtos do sistema C1 (t < 12 ps),ΦL(k, t) possui um máximo no tempo
de relaxação parak = k2, enquanto que para o sistema C3,ΦL(k, t) relaxa cada vez

46 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
Tabela 4.4. Magnitude dos cinco menores vetores de ondak acessíveis às sim-
ulações em unidades de Å−1, de acordo com a equação 4.28.
k1 k2 k3 k4 k5
C1 0.239 0.338 0.414 0.585 0.717
C2 0.202 0.286 0.350 0.495 0.607
C3 0.192 0.272 0.333 0.471 0.577
mais lentamente com o aumento dek, exatamente o oposto do que se observa para a
componente transversal. Não existe uma razão evidente para essas
observações, mas pode-se especular que a microheterogeneidade do sistema possa
causar uma relaxação mais lenta em determinados comprimentos de onda. Na figura
4.9 são apresentados os comportamentos de tempos curtos das funções de correlação da
componente logitudinal do momento de dipolo coletivoΦL(k1, t) para o menor vetor
de ondak = k1 possível para cada sistema. Este gráfico mostra que esta componente
relaxa mais lentamente para a concentração intermediária C2. Ainda, as amplitudes das
oscilações são menores para esta concentração. A decomposição nos termos de autocor-
relação e correlação cruzada mostra que as rápidas oscilações observadas devem-se aos
movimentos libracionais da água e torcionais das hidroxilas das frutoses, sendo que o
primeiro possui uma contribuição maior e com oscilações mais pronunciadas que o se-
gundo para esta característica doΦL(k, t) (figura 4.10). A posição dos máximos das os-
cilações das componentes frutose-frutose e água-água deΦL(k1, t) permanecem inalter-
adas com o aumento da concentração. A amplitude das oscilações do termoΦFFL (k1, t)
aumenta continuamente com o aumento da fração de frutose no sistema. Para a compo-
nenteΦWWL (k1, t), observa-se um mímimo da amplitude das oscilações para a concen-
tração C2. Estudando soluções de monossacarídeos por simulação de DM [20], Roberts
e Debenedetti mostraram que existe uma concentração intermediária de açucar para o
qual a estrutura da água é a mais próxima da rede de LH tetraédrica ideal. Muito em-
bora nenhuma das análises realizadas no presente trabalho aponte para um máximo na
estruturação da água, esta hipótese é consistente com o mínimo da amplitude das os-
cilações deΦWWL (k1, t) observado em uma concentração intermediária. Com uma maior
estruturação na rede de ligações de hidrogênio, um número grande de moléculas de água
realizaria 4 ligações de LH com outras moléculas de água simultaneamente. Estas lig-

4.5. Permissividade Dieletrica 47
0 10 20 30 400
0.2
0.4
0.6
0.8
1
k = 1k = 2k = 3k = 4k = 5
0 10 20 30 400
0.2
0.4
0.6
0.8
1Φ
L(κ
,t)
0 10 20 30 40t (ps)
0
0.2
0.4
0.6
0.8
1
C1
C2
C3
Figura 4.8. Função de correlação temporalΦL(k, t) da componente longitudinal do
momento de dipolo total do sistema para os cinco menores vetores de onda possíveis para o
sistema.
ações aumentariam as restrições para as oscilações libracionais da água, diminuindo a
amplitude do decaimento inercial.
4.5 Permissividade Diel etrica
A relaxação do dipolo coletivo é acessível experimentalmente no domínio de freqüência
através de medidas de perda dielétrica
e absorção de infravermelho distante. Na seção teórica está descrito como obter o
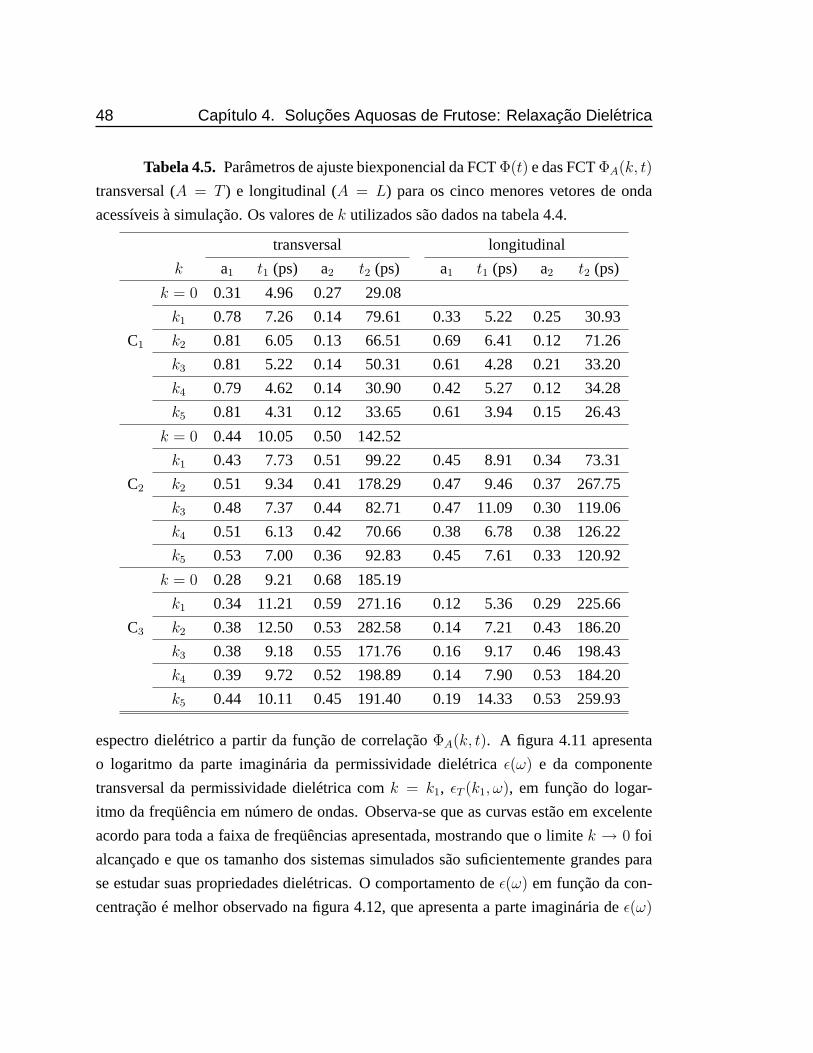
48 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
Tabela 4.5. Parâmetros de ajuste biexponencial da FCTΦ(t) e das FCTΦA(k, t)
transversal (A = T ) e longitudinal (A = L) para os cinco menores vetores de onda
acessíveis à simulação. Os valores dek utilizados são dados na tabela 4.4.
transversal longitudinal
k a1 t1 (ps) a2 t2 (ps) a1 t1 (ps) a2 t2 (ps)
k = 0 0.31 4.96 0.27 29.08
k1 0.78 7.26 0.14 79.61 0.33 5.22 0.25 30.93
C1 k2 0.81 6.05 0.13 66.51 0.69 6.41 0.12 71.26
k3 0.81 5.22 0.14 50.31 0.61 4.28 0.21 33.20
k4 0.79 4.62 0.14 30.90 0.42 5.27 0.12 34.28
k5 0.81 4.31 0.12 33.65 0.61 3.94 0.15 26.43
k = 0 0.44 10.05 0.50 142.52
k1 0.43 7.73 0.51 99.22 0.45 8.91 0.34 73.31
C2 k2 0.51 9.34 0.41 178.29 0.47 9.46 0.37 267.75
k3 0.48 7.37 0.44 82.71 0.47 11.09 0.30 119.06
k4 0.51 6.13 0.42 70.66 0.38 6.78 0.38 126.22
k5 0.53 7.00 0.36 92.83 0.45 7.61 0.33 120.92
k = 0 0.28 9.21 0.68 185.19
k1 0.34 11.21 0.59 271.16 0.12 5.36 0.29 225.66
C3 k2 0.38 12.50 0.53 282.58 0.14 7.21 0.43 186.20
k3 0.38 9.18 0.55 171.76 0.16 9.17 0.46 198.43
k4 0.39 9.72 0.52 198.89 0.14 7.90 0.53 184.20
k5 0.44 10.11 0.45 191.40 0.19 14.33 0.53 259.93
espectro dielétrico a partir da função de correlaçãoΦA(k, t). A figura 4.11 apresenta
o logaritmo da parte imaginária da permissividade dielétricaε(ω) e da componente
transversal da permissividade dielétrica comk = k1, εT (k1, ω), em função do logar-
itmo da freqüência em número de ondas. Observa-se que as curvas estão em excelente
acordo para toda a faixa de freqüências apresentada, mostrando que o limitek → 0 foi
alcançado e que os tamanho dos sistemas simulados são suficientemente grandes para
se estudar suas propriedades dielétricas. O comportamento deε(ω) em função da con-
centração é melhor observado na figura 4.12, que apresenta a parte imaginária deε(ω)

4.5. Permissividade Dieletrica 49
0 0.2 0.4 0.6t (ps)
0.5
0.6
0.7
0.8
0.9
1
ΦL(κ
1,t)
0 20 40 600
0.5
1
C1
C2
C3
Figura 4.9. Função de correlação temporalΦL(k1, t) da componente longitudinal do
momento de dipolo total do sistema para o vetor de ondak1 para os sistemas C1, C2 e C3.
Importante notar que o módulo do vetor de ondak1 depende do lado da caixa de simulação, que
varia com a concentração do sistema.
em função deω. Para o sistema C1, observa-se um perfil bimodal emε(ω), com um
pico 0.712 cm−1 e um leve ombro de baixa freqüência em 0.083 cm−1. Com o aumento
da concentração, o perfil bimoldal torna-se mais evidente, com a diminuição do pico de
alta freqüência acompanhado pelo aumento do pico de baixa freqüência. No sistema C2,
estes picos são observados em 0.567 cm−1 e 0.053 cm−1, enquanto que no sistema C3
eles são observados em 0.549 cm−1 e 0.029 cm−1. O deslocamento destas bandas para
freqüências mais baixas com o aumento da concentração correlaciona-se bem com o au-
mento de viscosidade do sistema. A figura 4.12 também apresenta o gráfico de Im[ε(ω)]
experimental obtido por Fucks e Kaatze utilizando a soma das funções espectrais Cole-
Cole com uma Debye [1] para a concentração C3. Apesar de possuir dois tempos car-

50 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
0.4
0.6
0.8
1
ΦL
FF (t
)
0 0.1 0.2 0.3 0.4t (ps)
1
1.2
1.4
1.6
ΦL
WW
(t)
C1C2C3
Figura 4.10. Contribuições frutose-frutose (painel superior) e água-água (painel infe-
rior) da componente longitudinal da FCTΦL(k1, t) para os sistemas C1, C2, e C3.
acterísticos na sua descrição, a curva experimental não apresenta característica bimodal
encontrada neste trabalho. Uma possível explicação seria a de que dinâmica do açúcar
na simulação estaria muito mais lenta que a do açúcar no sistema real. No entanto, é
importante ressaltar que no trabalho de Höchtl, Boresch e Steinhauser, simulação de
DM de soluções diluidas de sacarídeos, onde foram utilizandos outros açúcares e outros
campos de força, um ombro de baixa freqüência também foi observado [30]. Portanto
permanece aberta a questão sobre a existência desta banda de baixa freqüência.
Pode-se obter informações interessantes decompondoε(ω) nos termos de autocor-
relação frutose-frutose e água-água, e no termo de correlação cruzada frutose-água (ver
equação 4.14). Esta decomposição para os três sistemas é apresentado na figura 4.13.
No sistema C1, o perfil da banda deve-se basicamente ao termo água-água. O ombro de
baixa freqüência deve-se em sua maior parte ao termo de correlação cruzada e em menor

4.5. Permissividade Dieletrica 51
-4 -3 -2 -1 0 1 2 3log(ω / cm
-1)
-1
0
1
2
3
4
5
log(
Im[ε
(ω)])
, log
(Im
[εT(k
1,ω)])
C1
C2
C3
Figura 4.11. Logaritmos da parte imaginária da permissividade dielétrica e da compo-
nente transversal da permissividade dielétrica comk = k1, em função do logaritmo da freqüência
em número de ondas.
extensão ao termo frutose-frutose. Estas são exatamente as mesmas observações feitas
em estudos de DM de propriedades dielétricas de soluções aquosas diluídas de glucose
e maltose [30]. A diminuição do pico de alta freqüência acompanha a diminuição da
fração de água no sistema. Mais interessante, com o aumento da concentração a de-
composição deixa evidente que além dos termos FF e FW, o termo de autocorrelação
da água também contribui para a composição do pico de baixa freqüência, evidenciando
a existência de dois tipos de dinâmica da água no sistema: uma rápida, com dinâmica
semelhante a da água pura, e uma lenta, associada às moléculas de água que estão na
camada de solvatação das frutoses. Esta contribuição está em acordo com resultados de
simulação de DM soluções aquosas de diversos sacarídeos que mostram que as molécu-

52 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
0.0001 0.001 0.01 0.1 1 10 100 1000
ω (cm-1
)
0
5
10
15
20
25
30Im
[ε(ω
)]C1 MDC2 MDC3 MDC3 Kaatze et. al
Figura 4.12. Parte imaginária da permissividade dielétrica para os sistemas C1, C2 e
C3, e o resultado experimental para o sistema C3 [1].
las de água apresentam modos dinâmicos mais lentos que os observados em água pura
quando estão próximos de moléculas de açúcar [21]. No sistema C3, a dinâmica ráp-
ida deve-se às moléculas de água que se encontram nos bolsões de água no interior dos
aglomerados de frutose. Percebe-se ainda que o deslocamento do espectro total para
menores freqüências com o aumento da concentração ocorre devido ao deslocamento de
todas as componentes. Entretanto, este deslocamento não é igual para todas as compo-
nentes. Em decorrência da crescente interação entre moléculas de açucar com o aumento
da concentração, culminando na formação do aglomerado no sistema C3, a rotação das
frutoses torna-se extremamente lenta, resultando num deslocamento de grande extensão
do termo FF emε(ω). Do ponto de vista da água, a formação das cavidades aquosas
no sistema C3 permite a troca da maior parte das moléculas de água que estão na inter-
façe com o aglomerado. Nos sistemas de menor concentração, essas trocas se tornam
mais facilitadas, como se observou pela análise de sobrevivência de interaçãofHB(t)
entre moléculas frutose e água do capítulo anterior. A possibilidade destas trocas en-
tre moléculas de água livres e solvatação dos solutos reflete-se em um deslocamento
levemente perceptível do termo WW deε(ω).
Outro ponto interessante do efeito da concentração sobreε(ω) que pode ser obser-
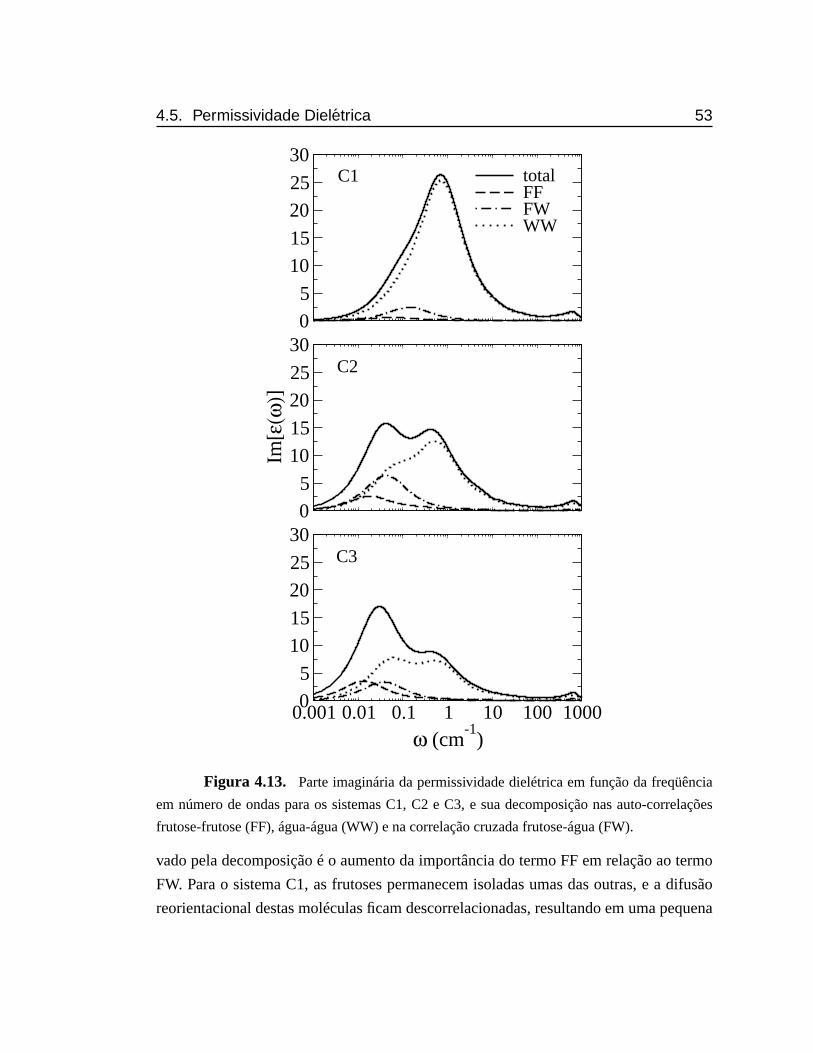
4.5. Permissividade Dieletrica 53
0
5
10
15
20
25
30totalFFFWWW
0
5
10
15
20
25
30
Im[ε
(ω)]
0.001 0.01 0.1 1 10 100 1000ω (cm
-1)
0
5
10
15
20
25
30
C1
C2
C3
Figura 4.13. Parte imaginária da permissividade dielétrica em função da freqüência
em número de ondas para os sistemas C1, C2 e C3, e sua decomposição nas auto-correlações
frutose-frutose (FF), água-água (WW) e na correlação cruzada frutose-água (FW).
vado pela decomposição é o aumento da importância do termo FF em relação ao termo
FW. Para o sistema C1, as frutoses permanecem isoladas umas das outras, e a difusão
reorientacional destas moléculas ficam descorrelacionadas, resultando em uma pequena

54 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
contribuição do termo FF para o espectro total. Por outro lado, as ligações de hidrogênio
entre as águas e frutoses resulta em uma grande contribuição do termo cruzado. No sis-
tema C3, a amplitude do termo FF chega e ser levemente maior que o termo FW. As in-
terações entre frutoses tornam-se importantes no sistema mais concentrado e a relaxação
estrutural depende de um número grande de frutoses. Observando atentamente a decom-
posição de figura 4.13, nota-se que o pico do termo FF aumenta continuamente com o
aumento da concentração enquanto que o termo FW apresenta um pico máximo para o
sistema C2, voltando a diminuir em C3. Isto também pode ser reflexo da formação dos
aglomerados. Com o aumento inicial da concentração as frutoses tendem a permanecer
dispersas no sistema. O aumento da concentração levaria a um aumento monotônico no
número de interações entre moléculas de açúcar e água, caso as moléculas de frutose se
distribuíssem homogeneamente no meio aquoso, resultando num aumento na importân-
cia do termo cruzado. Isto é o que se observa quando a concentração é aumentada do
sistema C1 para C2. A formação do aglomerado no sistema C3 leva a uma diminuição
na interface frutose-água, refletindo na diminuição da contribuição do termo cruzado
paraε(ω).
Analisa-se agora as características de alta freqüência do espectro. Essa faixa de fre-
qüências está relacionada a rápidos movimentos intra e intermoleculares. Na figura ante-
rior (4.13) essas características estão presentes no pico a extrema direita destes gráficos,
entre 100 e 1000 cm−1. Para que as estruturas de alta freqüência fiquem mais evidentes,
a figura 4.14 apresenta o gráfico deω2Φ(ω) em função deω, e a sua decomposição nos
termo de autocorrelação e correlação cruzada. A expressãoω2Φ(ω) é proporcional ao
produtoα(ω)n(ω) do coeficiente de absorção do infravermelho distante com o índice
de refração dependente da freqüência [33]. Analisando inicialmente o espectro total nas
três concentrações, observa-se duas bandas: uma banda larga de baixa freqüência, cen-
trada em 650 cm−1, e outra de alta freqüência entre 1150 a 1500 cm−1. A posição das
duas bandas independe da concentração. Entretanto, o aumento da fração de frutose pro-
move a diminuição da largura da primeira banda e o aumento da intensidade da segunda
banda. Quando se decompõemΦ(ω) do sistema C1 nos termos de autocorrelação FF e
WW, e no termo de correlação cruzada FW, observa-se que o pico de baixa freqüência
deve-se quase que na sua totalidade a modos dinâmicos da água. Uma banda de baixa
freqüência semelhante é observada em vários experimentos com água pura, bem como
em simulações de DM com diversos campos de força [35]. Trata-se da amplamente

4.5. Permissividade Dieletrica 55
0
1
2
3totalFFWFWW
0
1
2
3
ω2 R
e[Φ
(ω)]
0 500 1000 1500ω (cm
-1)
0
1
2
3
C1
C2
C3
~
Figura 4.14. Produto deω2 pela parte real do espectroΦ(ω) em função deω em
número de ondas para os sistemas C1, C2 e C3, e sua decomposição nas auto-correlações frutose-
frutose (FF), água-água (WW) e na correlação cruzada frutose-água (FW).
reconhecida banda libracional da água. Interessante notar que não há praticamente alter-
ação na posição da banda libracional com a composição, refletindo o fato das moléculas
de água interagirem com o açucar via LH similares às da água pura. Seguindo com a

56 Capıtulo 4. Solucoes Aquosas de Frutose: Relaxacao Dieletrica
decomposição deΦ(ω) nos sistemas C2 e C3 fica claro que a aparente independência
da altura da banda libracional com a concentração deve a troca de modos dinâmicos da
água por modos dinâmicos da frutose. Esses modos muito provavelmente são os movi-
mentos torcionais das hidroxilas das frutoses. Também se observa pela decomposição
que a banda de alta freqüência deve-se exclusivamente a modos dinâmicos da frutose.
Simulações auxiliares mostram que esta banda deve-se à distorção dos ângulos C-O-H
das hidroxilas das frutoses. Nestas simulações os ângulos C-O-H das hidroxilas foram
mantidos fixos nos seus valores de equilíbrio. A banda de alta freqüência é completa-
mente inexistente no espectro destas trajetórias adicionais. É importante lembrar que
neste trabalho foi adotado um modelo de água rígida. Caso um modelo de água flexível
fosse utilizado este também contribuiria para a banda em 1400 cm−1.
4.6 Conclus ao
Neste capítulo demos continuidade aos estudos de soluções aquosas de frutose, en-
focando o comportamento dielétrico estático e de relaxação. A constante dielétrica
destes sistemas obtido das simulações comparam-se bem com os dados experimentais
disponíveis. É importante ressaltar que os campos de força utilizados nestas simulações
não foram desenvolvidos para reproduzir as propriedades dielétridas dos sistemas reais.
O estudo da permissividade dielétricaε(k, ω) dependente do vetor de onda mostrou que
os sistemas simulados são suficientemente grandes para se computar suas propriedades
dielétricas. As análises das FCT do momento de dipolo coletivo destes sistemas, e sua
decomposições nas componentes de autocorrelação e correlação cruzada, são interpre-
tados em termos da solvatação dos sacarídeos no sistema C1 e C2, e na formação dos
aglomerados de açúcar e cavidades aquosas, no sistema C3. Dois tempos característicos
para a relaxação da água pode ser identificado: um tempo rápido e independente da con-
centração e aproximadamente igual ao tempo de relaxação da água pura, e um tempo
lento e fortemente dependente da concentração. Este tempo lento decorre da dinâmica
das moléculas de água que solvatam a frutose, como da relaxação da estrutura do aglom-
erado, e conseqüêntemente das suas cavidades. A presença do tempo de relaxação rápido
no sistema C3 é um reflexo das cavidades aquosas no interior dos aglomerados.
O aumento da concentração leva ao aparecimento de um pico de baixa freqüência
em torno de 0.053 cm−1 no gráfico da absorção dielétrica, acompanhando a diminuição

4.6. Conclusao 57
do pico de alta freqüência devido a difusão rotacional da água. A decomposição deste
espectro mostra que o pico de baixa freqüência possui contribuição de todos os termos,
inclusive do termo de autocorrelação da água. Esta observação está de acordo com
dois resultados obtidos por DM. Um deles mostra o retardo da dinâmica das molécu-
las de água próximas às moléculas de açucar [21], e o outro com estudos de relaxação
dielétrica de soluções diluídas de glucose e maltose [30]. No entanto, os espectros ex-
perimentais de relaxação dielétrica são unimodais. Duas hipóteses são levantadas: ou as
simulações superestimam as interações frutose-frutose ou a relaxação dielétrica experi-
mental disponível não foi capaz de capturar a componente dos sacarídeos.

Capıtulo 5
Estrutura e Din amica do
DNA com modelos de
solvatac ao expl ıcita e
impl ıcita
5.1 Introduc¸ ao
O desenvolvimento de campos de força e protocolos de simulação otimizados, bem
como o desenvolvimento de processadores cada vez mais rápidos, possibilitou a obtenção
de trajetórias de DM de solução de ácidos nucléicos por tempos da ordem de algumas
dezenas de nanosegundos nos últimos anos. Ao contrário do que as experiências de
difração de raio-X poderiam levar a crer, simulações de DM revelam flutuações rela-
tivamente grandes na estrutura do DNA em torno de sua estrutura média, com tempo
de vida que variam de dezenas de ps a vários ns. A existência destas flutuações tem
profundas implicações biológicas. Conjectura-se que a dependência de estrutura média
e flexibilidade da molécula de DNA com sua seqüência específica tem um papel no re-
conhecimento de genes na interação DNA/proteinas. Flutuações na estrutura do DNA
poderiam afetar drasticamente as taxas de transferência de elétrons, tanto pela modu-
lação do acoplamento interbase como sendo uma coordenada de reorganização capaz de
localizar carga [36]. A dinâmica de flutuações estruturais do DNA são importantes para
as taxas de reação ocorrendo dentro do DNA, pois ela determina o quão rápido o DNA
pode se adaptar à nova geometria do produto [36].
Recentemente, um interessante estudo experimental de Deslocamento de Stokes Re-
solvido no Tempo (DSRT) foi conduzido no grupo do Prof. Berg [36]. O foco do es-
tudo foram as flutuações estruturais no interior do DNA. Para realizar este experimento,
diferentes seqüências de DNA foram especialmente sintetizadas. Nestas seqüências, um
par de bases nitrogenadas no centro da seqüência é substituida por uma sonda fluores-
cente (cumarina 102, C102). Diferentemente de outros estudos de espectroscopia de
59

60 Capıtulo 5. Dinamica de DNA em solvente implıcito
fluorescência, onde a sonda complexa-se ao DNA, neste experimento a sonda liga-se
covalentemente ao açúcar do nucleotídeo (ver figura 5.1). O experimento DSRT con-
siste em monitorar a evolução temporal do espectro da emissão fluorescente da sonda
após excitação por pulso laser ultracurto (∼ 40 fs). O DSRT é sensível a flutuações nas
coordenadas de sítios providos de carga parcial localizados relativamente próximos à
molécula de prova, como as bases adjacentes à sonda, hidroxilas do açucar e oxigênios
dos fosfatos, pertencentes à espinha dorsal, bem como movimentos do solvente e contra-
íons.
Figura 5.1. Dupla hélice de DNA com destaque para a sonda substituindo um par
bases nitrogenadas.
Naquele estudo, Berg e seus colaboradores observaram o deslocamento da banda
fluorescente na janela temporal de 40 ps a 100 ns. O resultados mais inusitados deste
experimento são: (i) a presença de um decaimento logarítmico na função resposta de
solvatação, indicando a existência de uma dinâmica complexa com vários processos
de relaxação com tempos característicos distintos, e (ii) a taxa de decaimento independe
da seqüência de nucleotídeos específicos do DNA mostrando que esses processos devem
depender apenas das características gerais da dupla hélice. Geralmente, em líquidos sim-
ples, a relaxação pode ser bem descrita por uma exponencial ou uma soma de exponen-
ciais, indicando múltiplos processos de relaxação simples. Alguns sistemas complexos,
onde os processos dinâmicos se extendem continuamente por uma larga distribuição
de tempos característicos, apresentam relaxação descrita por uma exponencial estirada.
Tanto a soma de exponenciais como a exponencial estirada foram observadas nesta tese

5.2. Modelo de Born Generalizado 61
nos capítulos 3 e 4 que trata das soluções aquosas de frutose. Decaimentos logarítmicos
representam uma relaxação ainda mais severa em termos de complexidade, com uma
distribuição de tempos característicos bastante larga. A independência da taxa de relax-
ação com a seqüência específica de DNA entre 40 ps e 100 ns também tem implicações
bastante intrigantes para a biologia. Conjectura-se que as variações de estrutura média
e de flexibilidade do DNA em função da seqüência de nucleotídeos têm um papel im-
portante no reconhecimento destas seqüências por proteínas [36]. Assim, a dinâmica de
reconhecimento de uma seqüência de DNA por uma proteína deve acontecer em tempos
menores que 40 ps. A complexação de determinadas proteínas em posições específicas
do DNA são responsáveis por vários processos biológicos fundamentais que sustentam
a vida [37].
Estas descobertas experimentais e suas importantes implicações biológicas moti-
varam o estudo deste sistema. Em princípio, é possível estudar o sistema DNA-sonda
via simulação de DM, inclusive computando a resposta de solvatação através de uma
expressão bem estabelecida, derivada da teoria da resposta linear [38]. Entretanto, para
o devido tratamento do sistema, um número extremamente grande de moléculas de água
deve ser empregado, resultando em um custo computacional excessivamente alto para os
recursos disponíveis na época em que o estudo se iniciou. Para contornar este problema
estudou-se a dinâmica do DNA utilizando um modelo de solvatação implícita: o modelo
de Born Generalizado (BG). Para familiarização com simulações de DNA e sua análise,
bem como do modelo de solvatação BG, iniciou-se o estudo com uma seqüência menor
que as utilizadas experimentalmente, desprovido da sonda espectroscópica. Este estudo
preliminar produziu resultados interessantes que são apresentados neste capítulo.
5.2 Modelo de Born Generalizado
A importância do efeito de solvatação sobre aspectos conformacionais, energéticos e
funcionais de biomoléculas polares e iônicas é conhecido há muito tempo. A presença
de cargas parciais em biomoléculas, como uma proteína ou um ácido nucléico, pertur-
bam a estrutura e a dinâmica de moléculas de água que estão posicionadas a vários
angstroms de distância da sua superfície. Com efeito, para a simulação apropriada de
uma biomolécula um número extremamente grande de moléculas de solvente deve ser
empregado explicitamente, exigindo um alto esforço computacional. Para contornar essa

62 Capıtulo 5. Dinamica de DNA em solvente implıcito
Figura 5.2. Esquema da aproximação de Born Generalizado, onde o soluto é rep-
resentado por cargas em cavidades com constante dielétricaεin em um contínuo de constante
dielétricaεex.
dificuldade diversos modelos simplificados do efeito do solvente sobre o soluto têm sido
propostos. Um dos mais populares, especialmente para a água, trata o solvente como
um contínuo de alta constante dielétrica e provido de cavidades de constante dielétrica
menor, onde os átomos do soluto estão dispostos (ver figura 5.2).
Um tipo dessa classe de modelos que tem recebido bastante atenção em DM é o
Modelo de Born Generalizado(BG) [39]. Para se obter o potencial eletrostáticoφ do
modelo de dois dielétricos, com uma carga posicionada no interior da cavidade de con-
stante dielétrica menor, deve-se resolver a equação de Poisson
∇ [ε (r)∇φ (r)] = −4πρ (r) , (5.1)
ondeρ é a distribuição de carga. A constante dielétricaε valeεin no interior da cavidade
onde se encontram as moléculas do soluto eεex no exterior da cavidade. Para o soluto no
gás tem-seεex = 1, enquanto que para a solução aquosaεex = εw, a constante dielétrica
da água. Resolvendo a equação 5.1 para essas duas situações encontram-se os potenciais
no vácuoφvac e em soluçãoφsol. A diferença entre esses dois potenciais é ocampo de
reação, φreac = φsol−φvac, relacionado com a componente eletrostática da energia livre
de solvatação por
∆Gpol = 1/2∫
φreac (r) ρ (r) dV
= 1/2∑
i
qiφreac (ri) , (5.2)
onde a segunda igualdade é válida para um conjunto discreto de cargas parciais. Para o
caso de um único íon de raioa e cargaq, os potenciais podem ser calculados analitica-
mente e a componente eletrostática da energia livre de solvatação é dada pela conhecida

5.2. Modelo de Born Generalizado 63
fórmula de Born [39]
∆GBorn = −q2/2a (1 − 1/εw) . (5.3)
Para o caso de um conjunto de N cargasq1, . . .,qN com raiosa1, . . . ,aN , e as distâncias
rij entre as cargasi e j são suficientemente maiores que os seus raios, a energia livre de
solvatação é expressa por uma soma de termos de Born e de Coulomb
∆Gpol =N∑i
q2i
2ai
(1
εw
− 1)
+1
2
N∑i
N∑j �=i
qiqj
rij
(1
εw
− 1)
, (5.4)
onde o fator(1/εw − 1) deve-se ao efeito de blindagem da água sobre as interações
eletrostáticas entre os pares de carga.
O objetivo da teoria de BG pode ser pensado como um esforço de encontrar uma fór-
mula analítica simples, como a equação 5.4, que retenha o máximo da física da equação
de Poisson para estruturas moleculares realísticas. Pela abordagem BG, a contribuição
eletrostática da energia livre de solvatação é dada por:
∆Gpol = −1
2
(1 − 1
εw
) ∑i,j
qiqj
fBG
, (5.5)
ondefBG é uma função que interpola entre oraio de Born efetivoRi do átomoi, quando
a distância rij é pequena, e a própria distância rij para grande separação entre os átomos
i e j. A escolha de uma forma funcional apropriada defBG não é uma tarefa trivial. A
partir de soluções numéricas da equação de Poisson 5.1 sabe-se que o efeito do solvente
não está restrito sobre a forma de um prefator1/εw − 1, nem é um resultado geral que
a constante dielétrica da cavidadeεin não tenha nenhum efeito na energia de solvatação.
A forma mais comum parafBG é:
fBG (rij) =[r2ij + RiRjexp
(−r2
ij/4RiRj
)]1/2. (5.6)
A água influencia a energia livre de um soluto de duas maneiras. Ela solvata cada carga
individual da molécula e blinda a interação eletrostática entre pares de carga. Esses dois
efeitos são relacionados, pois quanto melhor uma carga é solvatada, mais blindada ela
está da interação com as outras cargas. O raio efetivo de BornRi pode ser pensado como

64 Capıtulo 5. Dinamica de DNA em solvente implıcito
uma medida da distância média da carga à interface com o dielétrico externo. Quanto
mais no interior do soluto e protegido da interação com o solvente está uma carga, maior
será o seu raio de Born e menor será sua blindagem da interação com as outras cargas
do soluto que estão no interior deste raio. Portanto,Ri não depende apenas do raio
intrínsecoai da carga, mas também da posição relativa e dos raios de todas as outras
cargas da molécula.
Diversos modelos têm sido desenvolvidos para calcularRi de forma eficiente. Um
dos mais famosos é o modelo proposto por Hawkins, Cramer e Truhlar (HCT). Por esse
modelo, o raio de Born possui a forma:
R−1i = a−1
i − ∑j
H (rij, Sijaj) , (5.7)
onde H é uma expressão relativamente complexa, dada pela equação 13 da referência
[40], e está aqui apenas para enfatizar (i) a dependência do parâmetro de escalaSij, que
tem como objetivo levar em conta a blindagem devido a sobreposição parcial de átomos
ligados covalentemente, e (ii) queRi depende da posição e dos raios de todas as demais
cargas do sistema.
Apesar de introduzir um número grande de novos parâmetros ao campo de força, o
modelo BG é capaz de reproduzir relativamente bem a estrutura e a energia de solvatação
de diversas classes de moléculas. Esses parâmetros são desenvolvidos comparando-se
a estrutura do soluto observada utilizando modelos de solvatação explícita, e sua ener-
gia livre de solvatação obtida através da soluções numéricas da equação de Poisson 5.1,
com a estrutura e energia livre obtida adotando-se o modelo de solvatação de BG. A ex-
pressão analítica da energia livre de solvatação∆Gpol fornecido pelo modelo HCT pode
ser derivada explicitamente, podendo ser utilizada diretamente como potencial efetivo
em cálculos de mecânica e dinâmica molecular. Do ponto de vista computacional, a apli-
cação do modelo BG em simulações de DM é extremamente atraente, já que, enquanto
o tempo de CPU de um sistema com solvatação explícita é muitas ordens de grandeza
maior que uma simulação do soluto em fase gasosa, o tempo de CPU com a solvatação
BG é apenas um pouco maior do que em vácuo.

5.3. Detalhes Computacionais 65
5.3 Detalhes Computacionais
Comparou-se a dinâmica torsional e vibracional das bases da seqüência de DNA 3’-
CTGATCAG-5’ utilizando modelos de solvatação explícita e implícita. Nestas simu-
lações foi empregado o campo de força AMBER [12]. Esse campo é extensamente
utilizado em simulações de moléculas orgânicas. Além disso a escolha desse campo
de força deveu-se principalmente à disponibilidade de parâmetros para simulações com
tratamento implícito do solvente desenvolvido especialmente para ácidos nucléicos [41,
42], apresentando boa concordância estrutural e energética.
Nas simulações com solvente explícito utilizou-se o modelo TIP3P [31] para a água,
na presença de contra-íons Na+. Uma caixa inicial de 34 x 35 x 45 Å3 contendo o DNA,
14 contra-íons e 1544 moléculas de água foi gerada [43]. Durante as simulações, a
estrutura interna das moléculas de água foi mantida rígida. Usou-se condições periódicas
de contorno, com raio de corte de 10 Å para as interações dispersivas. O método de
Particle Mesh Ewald(PME) foi utilizado para computar as interações eletrostáticas entre
as cargas da caixa principal e as cargas das imagens periódicas. Foram simuladas 5
trajetórias de 750 ps no ensemble NVE a partir de configurações iniciais termalizadas
em 298 K, com passos de 2 fs e salvando coordenadas a cada 6 fs. Nos simulações com
solvente implícito, 4 trajetórias de 1 ns foram simuladas para análise.
5.4 Resultados
Existe uma série de parâmetros que caracterizam estruturalmente o DNA [44]. Um
desses parâmetros é opseudo-ângulo de rotaçãoP , que depende da conformação do
anel da desoxirribose do nucleotídeo. O nitrogênio (N) faz parte da base, enquanto que
os átomosC ′5, C ′
4, C ′3 e O′
3 fazem parte da espinha dorsal do DNA. Uma representação
esquemática do açucar é apresentada na figura 5.3. O pseudo-ângulo de rotaçãoP é
definido em função dos valores dos ângulos diedros do anel do açúcar:
ν0 = C ′4 − O − C ′
1 − C ′2
ν1 = O − C ′1 − C ′
2 − C ′3
ν2 = C ′1 − C ′
2 − C ′3 − C ′
4
ν3 = C ′2 − C ′
3 − C ′4 − O
ν4 = C ′3 − C ′
4 − C ′5 − C ′
1.

66 Capıtulo 5. Dinamica de DNA em solvente implıcito
Figura 5.3. Representação da estrutura do açúcar, com a nomenclatura usual de seus
átomos utilizada na literatura.
C1 T1 G1 A1 T1 C1 A1 G1 C2 T2 G2 A2 T2 C2 A2 G2base nitrogenada
0
25
50
75
100
125
150
175
200
grau
s
solvente explícitosolvente implícitoforma B (C2’ endo)forma A (C3’ endo)
Figura 5.4. Valor médio do pseudo-ângulo de rotação da desoxirribose dos nu-
cleotídeos dos DNAs na representação explícita e implícita do solvente. São apresentados tam-
bém os valores ideais para as formas A (linha traço-ponto) e B (linha pontilhada) do DNA.
O valor deP é definido pela seguinte expressão
tan(P ) =(ν4 + ν1) − (ν3 + ν0)
2 · ν2 · (sen(36o) + sen(72o)). (5.8)
Devido às interações estéricas e ao posicionamento das bases em relação ao eixo do
DNA e sua espinha dorsal, a conformação do anel é sensível à conformação do DNA
como um todo. A figura 5.4 apresenta o valor médio do pseudo-ângulo de rotação e
o seu desvio padrão sobre 200 conformações da desoxirribose de cada nucleotídeo do
DNA com solvatação explícita e implícita. O gráfico também apresenta os valores ideais
para as duas conformações de DNA mais encontradas em experimentos de raio-X, as

5.4. Resultados 67
Figura 5.5. Estrutura das bases complementares timina e adenina, com as ligações
de hidrogênio (pontilhado vermelho) realizadas entre elas. São mostrados os átomos da timina
utilizados para definir os vetores no interior desta base.
0 2 4 6 8 10
ω (x 102 cm
-1)
0
25
50
75
100
125
ω2 F(
ω)
x 10
2
solv. explícitosolv. implícito
Figura 5.6. Transformada de Fourier da função de correlação do vetor C2-O2 da base
timina 5.
chamadas formas A e B. Observa-se que os valores de pseudo-rotação para as duas rep-
resentações de solvente estão dentro de uma mesma faixa de valores, mostrando que a
utilização do tratamento implícito de solvatação reproduz satisfatoriamente bem as es-
truturas observadas em tratamento explícito. O gráfico mostra ainda que a conformação
da molécula de DNA se aproxima da forma B em ambas as simulações, em acordo com
estruturas resolvidas por difração de raio-X para essa seqüência [44].
Tendo obtido boa comparação de propriedades estruturais do DNA com solvatação

68 Capıtulo 5. Dinamica de DNA em solvente implıcito
implícita e explícita, compara-se agora a dinâmica das flutuações conformacionais das
bases. Para isso comparou-se as FCT de vetores definidos por pares sítios no interior de
cada base. Existe uma variedade de formas de se escolher esses sítios. Escolheu-se pares
de sítios ligados covalentemente para definir o vetor. Além disso, os sítios escolhidos ou
participam de LH com sua base complementar, ou estão expostos para realizar LH com
o solvente ou interagir com os contra-íons. Esta segunda situação é o caso do sítio O2
da timina (figura 5.5). Uma comparação mais clara pode ser feita entre as transformadas
de FourierF (ω) das FCT do que com as FCT diretamente. O produtoω2F (ω) da FCT
do vetor definido pela ligação C2-O2 da base timina 5 são apresentados na figura 5.6. A
primeira característica que se observa quando se compara as TF é que a largura dos picos
na representação explícita é muito maior e a altura dos picos muito menor do que aquela
observada nas representação implícita. A largura do pico aumenta com a diversidade de
ambientes moleculares aos quais a base é submetida. No caso do solvente implícito, as
bases interagem diretamente apenas com as bases adjacentes e a sua base complemen-
tar. No caso do tratamento explícito, além destas interações, as bases interagem com as
moléculas do solvente e os contra-íons presentes nas cavidades maior e menor do DNA.
A posição das moléculas de água em torno das bases varia constantemente, alterando as
freqüências dos seus modos normais. A figura 5.6 mostra que alguns picos são comuns
às representações implícita e explícita, mas muitos picos apresentados em uma represen-
tação não encontram correspondente na outra. Esse efeito pode ser explicado por dois
ou mais estados preferenciais de solvatação, gerando um desdobramento de linhas do
tratamento implícito quando a base passa a interagir explicitamente com as moléculas
de água, ou uma dissipação de energia de alguns modos normais da base para modos vi-
bracionais e translacionais do solvente. Por fim, daqueles picos observados em ambos os
tratamentos, observam-se leves deslocamentos para freqüência mais alta no caso de sol-
vatação explícita. Este fato também é um reflexo da interação da base com as moléculas
de água. A presença da água em torno da base reduz suas flutuações conformacionais. O
sítio O2 da timina não participa das ligações de hidrogênio com seu par complementar, a
adenina, porém interage fortemente com as moléculas do solvente ou com os contra-íons
que eventualmente entrem na cavidade menor do DNA [44]. No entanto as conclusões
apresentadas aqui são as mesmas no caso de outro vetor específico ser escolhido, como
por exemplo o vetor definido pela ligação C4-O4 da timina (ver figura 5.5). Apesar da
diferença observada entre os espectros obtidos das simulações com solvatação explícita

5.4. Resultados 69
e implícita, pode-se dizer que, apesar das drásticas aproximações do modelo BG, a rep-
resentação implícita captura boa parte da dinâmica rápida de flutuações estruturais das
bases.
0 0.5 1 1.5 2t (ps)
0
0.5
1
φ(t)
0 10 20 30 40 50t (ps)
0
0.5
1
solv. explícitosolv. implícito
Figura 5.7. Comportamento de tempos curtos (painel à esquerda) e longos (painel à
direita) da FCT do vetor normal à base citosina 1 do DNA na representação explícita e implícita
do solvente.
Compara-se agora a dinâmica de rotações das bases dos nucleotídieos do DNA, que
está intimamente ligada ao espectro obtido em experimentos de decaimento fluores-
cente. Pretende-se obter informação a respeito das flutuações estruturais globais do
DNA. Para isso comparou-se a função de correlação de vetores normais às bases nitro-
genadas. Como os diedros e os ângulos não são rígidos, a estrutura das bases distorce
durante as simulações. Para obter o vetor normal posicionou-se a mesma base com sua
estrutura padrão, plana e não distorcida, sobre a base distorcida obtida da DM de forma
que as duas bases tivessem centros de massa coincidentes. Depois se utilizou um pro-
cedimento que obtém a matriz de rotação que minimiza o desvio quadrático médio das
posições atômicas das duas bases [45]. Um vez feito isso, toma-se a normal à base não
distorcida. As funções de correlação para a base citosina 1, posicionada no início da
seqüência, são apresentadas na figura 5.7. Observa-se para tempos curtos (< 0.5 ps) um
decaimento inicial rápido para ambas as funções (painel à esquerda), mas para tempos
maiores ( > 0.5 ps ) as duas funções diferem destacadamente em comportamento (painel

70 Capıtulo 5. Dinamica de DNA em solvente implıcito
à direita). Enquanto o decaimento do sistema com solvente explícito é exponencial típico
de sistemas difusivos, a função de correlação obtida utilizando tratamento implícito do
solvente mostra oscilações com tempo característico de aproximadamente 17 ps que se
mantém por tempos muito maiores de 1 ns (não mostrado no gráfico). Este comporta-
mento se deve à ausência de efeitos dissipativos no sistema com solvatação implícita, o
que é esperado.
5.5 Conclus ao
O modelo BG de solvatação implícita fornece um potencial efetivo analítico e diferen-
ciável para a interação soluto-solvente produzindo resultados que comparam quantitati-
vamente com cálculos mais sofisticados e computacionalmente caros, como a resolução
numérica da equação de Poisson [41]. Muito se tem trabalhado na parametrização do
modelo BG para que ele possa ser utilizado como um modelo de solvatação efetivo
para simulações de DM. O modelo de solvatação implícito procura computar o mais
próximo possível as interações do soluto com o solvente e a influência da presença do
solvente sobre as interações entre os átomos do soluto. Mostra-se que o comportamento
dinâmico das bases para tempos rápidos é relativamente bem descrito pelo modelo im-
plícito. No entanto, ele não leva em conta de forma apropriada a fricção mecânica ou
dielétrica do soluto com o solvente, pertinentes na escala de tempos longos, permitindo
oscilações periódicas na estrutura do DNA ao invés de um decaimento gradual típico
de decorrelação em regimes de tempos longos. Desta forma, efeitos dissipativos, sejam
hidrodinâmicos ou dielétricos, não estão presentes nas simulações com solvatação im-
plícita. Além disso, nesse tratamento a resposta dielétrica do solvente a uma flutuação
conformacional do soluto é instantânea. No entanto, sabe-se que em condições de alta
viscosidade a dinâmica do DNA é limitada pela dinâmica do solvente [46]. Portanto, a
abordagem implícita da solvatação não é adequada para o estudo da dinâmica do DNA.
A utilização deste modelo em conjunto com uma simulação estocástica, resolvendo a
equação de Langevin, parece promissora [47]. No entanto, essa abordagem necessita
de uma parametrização dos coeficientes de fricção, o que foge completamente ao es-
copo deste estudo, constituindo por si só em um problema que levaria meses para ser
satisfatoriamente abordado.
A disponibilidade de novos computadores permitiu que o sistema DNA-sonda sol-

5.5. Conclusao 71
vatado explicitamente pudesse ser tratado. Estas simulações estão atualmente em curso.
No entanto, o computo da resposta de solvatação possui convergência extremamente
lenta, e a quantidade de trajetórias obtidas até o momento não foi suficiente para um
resultado estatisticamente relevante. O estudo preliminar, com a seqüência menor e sol-
vatação implícita, foi extremamente valioso para a familiarização com simulações de
DNA e seus métodos de análise, possibilitando o desenvolvimento até o estágio atual do
estudo do sistema DNA-sonda solvatado explicitamente.

Capıtulo 6
Dinamica de Solvatac¸ ao
na Interface Agua-Ar na
presenc a de Surfactante
Ani onico
Neste capítulo são apresentados resultados de estudos estruturais e dinâmicos da sol-
vatação de uma molécula de prova espectroscópica, cumarina 314 (C314), adsorvida
na interface água-ar na presença do surfactante aniônico dodecilsulfato de sódio (SDS,
[CH3(CH2)11OSO3]− e Na+). Um esquema representativo do sistema é apresentado na
figura 6.1. O surfactante possui uma cabeça hidrofílica carregada e uma longa cauda
hidrofóbica, tornando-a uma molécula tensoativa. Este trabalho foi motivado por ex-
perimentos recentes de geração de segundo harmônico (SHG, paraSecond Harmonic
Generation) conduzidos pelo grupo de K. Eisenthal para este sistema [48]. São in-
vestigadas propriedades estruturais e dinâmicas da C314 e da interface em função da
concentração superficial de SDS. Neste trabalho são utilizadas densidades superficiais
ρs =0.002, 0.005 e 0.01 moléculas de SDS/Å−2, abaixo da densidade de recobrimento
total da interface estimada emρs ≈ 1.25 x 10−2 Å−2. Estes valores são utilizados para
comparar com os valores utilizados experimentalmente. Nesta situação, onde a densi-
dade é insuficiente para o recobrimento total da superfície, as moléculas de surfactante
agrupam-se em domínios espaciais na interface. Estes domínios são energeticamente
favoráveis e se formam a um custo entrópico bastante reduzido. A evolução dinâmica
destes domínios resulta em grande flutuação local de surfactante.
Estudos da evolução temporal da orientação da C314 em relação à interface, e a in-
teração de Lennard-Jones entre a C314 e as caudas dos surfactantes mostram a existência
de dois estados de solvatação da molécula de prova. Em um deles, denominado estado
A, a C314 fica posicionada adjacente ou exterior aos aglomerados bidimensionais de
surfactantes que ocorrem na interface. Neste estado o plano da sonda permanece aprox-
73

74 Capıtulo 6. Dinamica de Solvatacao na Interface Agua-Ar
Figura 6.1. Cumarina 314 (C314) adsorvida na interface água-ar na presença do
surfactante aniônico dodecilsulfato de sódio (SDS). É apresentada a orientação do momento de
dipolo do C314 em relação ao versor normal à interface.
imadamente paralelo ao plano da interface. No outro estado, denominado estadoB, a
C314 fica no interior dos aglomerados de surfactante envolto nas suas caudas hidrofóbi-
cas. Neste estado o plano da sonda tende a permanecer perpendicular ao plano da inter-
face. Além da orientação média, a dinâmica de reorientação da sonda difere muito nos
dois estados de solvatação. O aumento da concentração de surfactante faz com que o
comportamento da sonda na interface se assemelhe ao comportamento do estadoB, que
correspondente a uma ambiente molecular similar ao do recobrimento total da interface.
Valores de orientação média da sonda em relação ao plano da interface são acessíveis
por experimentos de SHG [48]. A orientação média experimental é intermediária a ori-
entação média dos estadosA eB para todas as concentrações estudadas, o que fornece
bastante suporte para os resultados da simulação.
No experimento de deslocamento de Stokes resolvido no tempo, sondas moleculares,
como a C314, são dissolvidas no sistema e excitadas eletronicamente. Um esquema de
experimento é apresentado no painel superior da figura 6.2. As curvas nesta figura rep-
resentam a energia livre do sistema para os estados fundamental e excitado da sonda em
função da estrutura de solvatação do sistema, representado por uma coordenada gener-
alizada. Em geral, o momento de dipolo destas sondas no estado fundamental e excitado
diferem muito. A estrutura do solvente em torno da sonda, inicialmente em equilíbrio
com a distribuição de carga do estado fundamental, deve se reorganizar para alcançar
o equilíbrio para nova distribuição de carga do estado excitado. À medida que a sonda

75
Figura 6.2. Painel superior: dinâmica de solvatação - As curvas representam a en-
ergia livre do sistema em função da coordenada de reação, representado pelo eixox para os
estados eletrônicos fundamental (S0) e excitado (S1) da sonda espectroscópica. Painel inferior:
Deslocamento da banda fluorescente em função do tempo.
decai para estado o eletrônico fundamental durante o processo de reorganização, a banda
fluorescente desloca-se. Ou seja, como o processo de reorganização do solvente se dá
continuamente até o equilíbrio do estado excitado, a banda de emissão desloca-se para a
direção de freqüências menores à medida que a reorganização ocorre (figura 6.2, painel
inferior). Portanto, o deslocamento da banda de emissão em função do tempo contem

76 Capıtulo 6. Dinamica de Solvatacao na Interface Agua-Ar
informações sobre o processo de reorganização do meio reacional. Este processo de
reorganização pode ser estudado computando-se a resposta de solvataçãoCs(t)
Cs =ν(t) − ν(0)
ν(∞) − ν(0), (6.1)
ondeν(t) é a freqüência média ou máxima da banda de emissão no tempot. Esta técnica
experimental é utilizada para se estudar a dinâmica de líquidos puros, pois é provável
que o processo reorganização ocorra, pelo menos em parte, por processos dinâmicos
presentes nestes líquidos.
Pode-se computar a função resposta de solvataçãoCs(t) utilizando as trajetórias de
dinâmica molecular no equilíbrio a partir de uma expressão derivada da teoria da re-
sposta linear. Esta expressão é apresentada na equação 7 do artigo deste capítulo. Esta
expressão baseia-se na diferença da flutuação da interação eletrostática entre o solvente
e a sonda nos estados fundamental e excitado. Foi computada a resposta de solvatação
Cs(t) da interface frente a uma mudança no estado eletrônico da sonda para os estados
de solvataçãoA. eB. Mostra-se que no estadoA, como a molécula fica adjacente ao
aglomerado de surfactante, a resposta de solvataçãoCs(t) independe da concentração
de surfactante. Isto é, mesmo para o sistema mais concentrado, a respostaCs(t) de-
pende apenas da dinâmica de solvatação da água. Para o estado de solvataçãoB, como a
sonda fica envolta nas caudas de surfactante, a resposta torna-se dependente da concen-
tração, ficando cada vez mais lenta com o aumento da fração de surfactante no sistema.
Grandes barras de erros são encontradas nas respostas no estado de solvataçãoB. Es-
sas barras de erro devem-se a uma rica variedade de mecanismos dinâmicos, como por
exemplo o movimento das caudas dos SDS e da própria sonda, as flutuações estruturais
dos aglomerados, ou as flutuações na densidade superficial de SDS.
Este trabalho foi iniciado durante um estágio de 3 semanas no grupo do Prof. Daniel
Laria, da Universidade de Buenos Aires (Argentina), durante o qual trabalhei juntamente
com seu então aluno de Doutorado Diego Pantano. Segue o artigo publicado resultante
da pesquisa neste sistema.

Solvation of Coumarin 314 at Water/Air Interfaces Containing Anionic Surfactants. I. LowCoverage
Diego A. Pantano,† Milton T. Sonoda,‡ Munir S. Skaf,‡ and Daniel Laria* ,†,§
Departamento de Quı´mica Inorganica, Analıtica y Quımica-Fısica e INQUIMAE, Facultad de Ciencias Exactasy Naturales, UniVersidad de Buenos Aires, Ciudad UniVersitaria, Pabellon II, 1428 Buenos Aires, Argentina,Instituto de Quı´mica, UniVersidade Estadual de Campinas, Cx. P. 6154, Campinas, SP 13083-970, Brazil, andUnidad ActiVidad Quımica, Comisio´n Nacional de Energı´a Atomica, AVenida Libertador 8250,1429 Buenos Aires, Argentina
ReceiVed: September 22, 2004; In Final Form: February 3, 2005
Through the use of molecular dynamics techniques, we analyze equilibrium and dynamical aspects of thesolvation of Coumarin 314 adsorbed at water/air interfaces in the presence of sodium dodecyl sulfate surfactantmolecules. Three different coverages in the submonolayer regime were considered, 500, 250, and 100 Å2/SDS molecule. The surfactant promotes two well-differentiated solvation environments, which can be clearlydistinguished in terms of their structures for the largest surfactant coverage considered. The first one ischaracterized by the probe lying adjacent or exterior to two-dimensional spatial domains formed by clusteredsurfactant molecules. A second type of solvation environment is found in which the coumarin appears embeddedwithin compact surfactant domains. Equilibrium and dynamical aspects of the interfacial orientation of theprobe are investigated. Our results show a gradual transition from parallel to perpendicular dipolar alignmentof the probe with respect to the interface as the concentration of surfactantFs increases. The presence of thesurfactant leads to an increase in the roughness and in the characteristic width of the water/air interface.These modifications are also manifested by the decorrelation times for the probe reorientational dynamics,which become progressively slower withFs in both solvation states, although much more pronounced for theembedded ones. The dynamical characteristics of the solvation responses of the charged interfaces are alsoanalyzed, and the implications of our findings to the interpretation of available experimental measurementsare discussed.
I. Introduction
Liquid-vapor interfaces constitute nonuniform environmentswhere most variables controlling chemical reactivity presentabrupt changes over length scales comparable to typicalmolecular sizes. These sharp gradients open new interestingscenarios, where reaction mechanisms are well-differentiatedfrom what is normally encountered in bulk liquid or gaseousisotropic phases.1-3 Clear manifestations of these phenomenacan be found in displacements of chemical equilibria,4 inmodifications of rates and mechanisms5 of surface reactions,and in local enhancements of pH,6 to list just a few importantexamples. In all of these cases, interfacial solvation plays afundamental role.
Most of the direct experimental information about solvationdynamics in condensed phases is obtained through signals frommolecular probes; among which, coumarins stand out as a groupof versatile solvatochromic dyes commonly used in steady-stateand time-resolved fluorescence spectroscopy. Without beingexhaustive, the list of different environments that have beeninvestigated using these probes includes polar7 and nonpolar8
homogeneous liquid phases and inhomogeneous environments,such as nanoclusters,9,10 self-assembled structures,11 and
interfaces.12-22 Microscopic descriptions related to equilibriumsolvation structures and solvent dynamics are essential for thecorrect interpretation of the chemical reactivity characteristicsprevailing in each specific environment.
The study of surfactant-coated, charged interfaces has re-ceived considerable attention in recent times.18,23-27 The interestin these systems resides mainly in the fact that charged interfacesmay incorporate the basic functional and structural groups thatallow for the stabilization of more complex supramolecularassemblies of biological interest, such as micelles, lipidicbilayers, and cell membranes. The presence of adsorbed ionicsurfactant introduces new characteristics to the solvationstructures found at bare interfaces. Most notable are thoseassociated with the strong local electric fields created by thesurfactant headgroups and counterions. In many cases, theresulting electrostatic coupling between the chemical speciesat the interface may be significant in comparison to that involvedin a typical aqueous hydrogen bond. Therefore, it is reasonableto expect alterations in thesalready modified with respect tobulk phasessintermolecular connectivity and orientational cor-relations of interfacial water. The presence of charged speciesalso affects the interfacial dynamics in important ways. The mainphysical effect promoted by the surfactant electric fields is theoverall slowing down of the dynamical modes of the watermolecules in close contact with the surfactant.28
In this work, we investigate equilibrium and dynamicalaspects of the solvation of dyes adsorbed at charged liquid/airinterfaces. More specifically, we report results from molecular
* Author to whom correspondence should be addressed. E-mail: [email protected].
† Universidad de Buenos Aires.‡ Universidade Estadual de Campinas.§ Comision Nacional de Energı´a Atomica.
7365J. Phys. Chem. B2005,109,7365-7372
77

dynamics (MD) computer experiments performed on modelsystems consisting of a Coumarin 314 (C314) molecule (Figure1) lying at water/air interfaces in the presence of differentconcentrations of the anionic surfactant sodium dodecyl sulfate(SDS). This study is largely motivated by recent time-resolvedsecond harmonic generation29 (TRSHG) spectroscopy measure-ments performed on these systems by Benderskii and Eisenthal.23
It has been found that the diffusive component of the TRSHGsolvation response is described by a biexponential decay inwhich the corresponding characteristic times exhibit a complexdependence on the degree of surfactant coverage (i.e., the surfacecharge density). At very low (500 Å2 /SDS molecule) andintermediate (250 Å2/SDS molecule) surface coverages, the fastcomponents of the measured responses remain similar to thosefound for the bare interface (τ1 250 fs) and bulk water( 130-250 fs) but slow to 600 fs at higher coverages (100Å2/SDS molecule). The slower component of the solvationresponse, however, changes fromτ2 ) 2.0 ps for bare interfacesto nearly 4.4 ps at the lowest sulfate surface coverage considered(500 Å2/SDS molecule) and to 5.4 ps at 100 Å2/SDS molecule.
The complex behavior displayed by the solvation dynamicsat such charged surfaces is far from being understood. Inparticular, the solvent dynamics underlying the relaxationcomponents are unknown. Benderskii and Eisenthal haveproposed a correspondence between the experimental TRSHGtime scales and two microscopic features involving the inter-facial water molecules, the enhancement in the hydrogenbonding network connectivity and modifications of the dynami-cal modes. In an effort to provide additional information fromamolecular perspective, we present simulation results showingevidence that, in addition to the above-mentioned effects, theinterfacial solvation dynamics of C314 are also affected in asensible fashion by the different solvation environments gener-ated by the surfactant spatial domains. The systems we considerhere correspond to surfaces below the full-monolayer surfactantcoverage (g100 Å2/SDS molecule) and represent a naturalextension of our recent work on the solvation of similar probesat clean interfaces.30 Yet, the present simulations reveal interest-ing new features of the dynamics at the interface, which mayhave implications for the interpretation of the experimental data.In particular, we find that the interfacial solvation of the C314molecule exhibits two predominant types of structures, thosein which the C314 molecule sits at the edge of a two-dimensional surfactant spatial domain and those where the soluteprobe sits in the interior of a surfactant patch. The intercon-version between these two types of solvation environments andthe overall dynamics of the probe itself constitute importantaspects of the simulated solvation dynamics.
This paper is organized as follows. Details of the simulation
results and discussion are presented in section III. Our conclud-ing remarks appear in section IV.
II. Model and Simulation Procedure
The MD experiments described in this paper were performedon systems composed of a C314 molecule lying at one of theliquid/air interfaces of an aqueous slab comprisingNw ) 1000water molecules. The slab was generated from a fully periodic,cubic system of density 1 g cm-3, in which periodic boundaryconditions were suppressed along one of the main axes of thesimulation box (hereafter referred to as thez-axis). After aninitial equilibration period of 100 ps, equal numbersNs ofdodecyl sulfate [CH3(CH2)11OSO3]- and Na+ counterions weredistributed across the interface containing the C314. The initialintramolecular arrangement of the surfactant molecules cor-responded to that of a fully trans conformer, with their head-to-tail vectors oriented perpendicularly to the surface. Threedifferent surface coverages were considered,Ns ) 2, 5, and 10,which expressed in terms of the overall interfacial density,Fs,correspond roughly toFs ) 0.002, 0.005, and 0.01 Å-2,respectively. This density regime is below the estimated valuefor the saturated monolayer,31 Fs 1.25 10-2 Å-2 and waschosen so as to coincide with available experimental informationunder similar conditions.23
Water molecules and the chromophore were modeled as rigidbodies containing a collection of interaction sites. For the watermolecules, we adopted the simple point charge (SPC) model.32
Charge distributions for the C314 ground and first excitedelectronic states and molecular geometry were identical to theones we considered previously,30 which were obtained from theAM1 semiempirical parametrization using the AMPAC pack-age.33-36 Within this level of description, the C314 dipolemoment in the ground state isµ ) 8.4 D, in reasonableagreement to the experimental value (8.20( 0.02 D).37 Acomplete description of the partial charges and the moleculargeometry of the dye is provided in ref 30. For the surfactant,we used the fully flexible model proposed by Dominguezetal.25 Each surfactant comprises a total of 17 interaction sites,using the united atom description for CH2 and CH3 groups.Intramolecular interactions include stretching, bending, anddihedral contributions. Additional details of the surfactantmolecules and the atomic parameters for the counterions canbe obtained from ref 25.
We implemented a MD scheme that included a reversiblemultiple time step algorithm to integrate Newton’s equation ofmotion. The dynamical trajectories corresponded to microca-nonical runs at temperatures close toT 298 K. In thistemperature regime, all slabs presented stable structures, withnegligible evaporation. After initial equilibration runs of typi-cally 500 ps, statistics were collected along equilibriumtrajectories of 3-5 ns. Two different time steps were used,∆t ) 1 fs for the intermolecular motions andδt ) ∆t/3 to evolvethe rest of the fast dynamical variables associated to thesurfactant internal degrees of freedom. Intramolecular constraintsin water and C314 were handled using the RATTLE algorithm.38
All interactions were truncated atRc ) 15.5 Å and brought tozero in an interval of 1 Å by a fourth-degree spline.39
III. Results and Discussion
A. Slab Structures. The starting point of our analysis willbe the consideration of a set of density fields associated withthe relevant chemical species. For purposes of clarity, watermolecules will be referred to as “solvent”, whereas the term“solute” will be used to indistinctly denote the rest of the
Figure 1. Coumarin 314 molecular structure and labeling.
7366 J. Phys. Chem. B, Vol. 109, No. 15, 2005 Pantano et al.
78

kinds of spatial correlations. (i) For the solvent, we havecomputedFw(z) dz, the number of water molecules per unit ofarea in thexyplane,A, with their centers of mass lying betweenz andz + dz
In this equation,⟨‚‚‚⟩ denotes an equilibrium ensemble average;Zi andZCM represent thez-coordinates of the centers of massof the ith water molecule and the water slab, respectively. (ii)For the solute species, we consider spatial correlations of thetype
whereziR represents thez-coordinate of theR site in the ith
molecule (i ) 1, 2, ...,NR). Densities of four selected solutespecies were examined, the Na+ counterions, the center of massof the C314 molecule, and two distal surfactant sites, namely,the S atom in the headgroup and the terminal CH3 group in thehydrophobic aliphatic tail.
Results for spatial correlation functions in different chargedinterfaces are shown in Figure 2, which also includes resultsfor C314 adsorbed at bare interfaces as a reference benchmark.Results forFw(z) show that the four aqueous slabs present similarwidths, ∆L 31 Å, measured from the positions of the twoGibbs dividing surfaces. Charge localization on the right-handside interfaces promotes a barely discernible enhancement ofthe water local density, as one gradually moves from thenegative to the positive portion of thez-axis. However, allaverage water densities at the middle points of the slabs fallwithin the intervalFw(z) 0) ) 0.034( 0.001 Å-3, which is inclose agreement to the bulk water density at ambient conditions,
The degree of penetration of the C314 center of mass intothe water slabs remains practically unchanged at low surfactantcoverages, and only forFs ) 0.01 Å-2, one observes a mildshift in the position of the probe toward the interior of theaqueous phase (fromz 17 Å down to 15 Å). At this surfacecoverage, one can also notice somewhat larger fluctuations inthe probability distribution functions. These changes go hand-in-hand with an increase in the characteristic length scale thatdescribes the width of the interface,L. The direct comparisonbetweenFw(z) for clean and charged interfaces shown in Figure3 is instructive. All profiles are practically identical on thesurfactant-free side (left panel), whereas for charged interfaces(right panel),Fw(z) presents a gradual widening as the densityof surface charge increases. To acquire a quantitative idea ofthis effect, we took thez-coordinates at 10% and 90% of themaximum density value to compute the width for the largestsurfactant coverage,L(Fs ) 0.01 Å -2) 8.2 Å. This width isapproximately twice that of the bare interface,L 3.6 Å.
In the concentration range considered, the penetration of thepolar headgroups into the aqueous phase is found to besomewhat deeper in comparison to the coumarin (z 11-12Å), while the average positions of the distal CH3 groups werefound to be approximately at∆z 7-8 Å outward. Thesestructures are accordant to that reported for the case of infinitelydiluted SDS.25 From the average positions of the distal groupsof the surfactant molecules (S and CH3), it is possible to get acrude estimate of the overall orientation of the amphiphiles atthe surface. Considering a typical head-to-tail length ofLs
15 Å, one can estimate the overall orientationθs of the surfactanttails with respect to thez-axis asθs cos-1(∆z/Ls) 60°. Thisshows that in this regime of surface coverage, the hydrophobictails still lie close to the aqueous interface. In all cases, thedistributions of couterions present two peaks. The inner ones,located near the center of the aqueous phase,z -5 Å,correspond to “bulk” solvated ions, while the most prominentones are located at typical valuesz 9-10 Å. This particularstructure is the combined result of the attraction exerted by theadsorbed polar headssgenerating both, contact and solventseparated, Na+-SO3
- ion pairssand the cation avoidance ofthe bare liquid/vapor (surfactant free) interface.43
B. Coumarin Interfacial States.Having established the mainstructural features of the different slabs, we will now proceedto the microscopic characteristics of the interfacial solvationenvironments. We will first examine orientational correlations.
Figure 2. Water local density (thick solid lines, left axis) andprobability densities (right axis) for different solute species alongdirections perpendicular to the interface: C314 center of mass (dot-dashed lines); counterions (thin solid lines); S (dotted lines); CH3
(dashed lines). Top-left panel, bare interfaces; top-right panel,Fs )0.002 Å-2; bottom-left panel,Fs ) 0.005 Å-2; bottom-right panel,Fs
) 0.01 Å-2.
Figure 3. Local water density in the vicinity of the Gibbs dividingsurfaces for bare (left panel) and charged (right panel) interfaces.Circles,Fs ) 0.01 Å-2; squares,Fs ) 0.005 Å-2; diamonds,Fs ) 0.002Å-2; triangles, bare interface. The origins of thez-axis coincide withthe positions of the corresponding Gibbs dividing surfaces.
Fw(z) )1
A⟨∑
i)1
Nw
δ(Zi - ZCM - z)⟩ (1)
PR(z) )1
NR
⟨∑i)1
NR
δ(ziR - ZCM - z)⟩ (2)
Solvation of Coumarin 314 at Water/Air Interfaces J. Phys. Chem. B, Vol. 109, No. 15, 20057367
79

system of coordinates (x′, y′, z′), as shown in Figure 1. Notethat, in this frame, the atomic coordinates of the probe lie mostlyon thez′ ) 0 plane, while the molecular dipole moment makesan angle of 18° with thex′ direction. Previous simulation studieshave shown that, in the absence of surfactant, the plane of thecoumarin remains mostly parallel to the interface.30
Figure 4 includes details of the time evolution of the angularvariable related to the out-of-plane dynamics of the probe,namely
for charged and uncharged interfaces. The results for the latter(top-left panel) are practically identical to those presented inour previous study30 and are accordant to the value reported inref 15 obtained from null-angle, SHG experiments,44 cosθ0.17. The prevalence of positive values for cosθ in most ofthe trajectories indicates a moderate preferential solvation ofthe negatively charged end of the probe.
Orientational correlations at charged interfaces contrastsharply with the previous picture. The three trajectories presenttwo well-differentiated temporal domains. During the firstsegments (hereafter denoted as statesA), and specially at lowsurfactant coverage, the time history of cosθ(t) closelyresembles the one already described for bare interfaces, namely,moderate orientation fluctuations with cosθ typically betweenapproximately-0.2 and 0.5. Sudden transitions take place att) 0 when the values of cosθ increase abruptly, suggesting astronger alignment of the coumarin dipole along directionsperpendicular to the interface. These configurations will behereafter denoted as statesB. Differences between statesAand B, as reflected from the orientation of the probe, becomeeven more evident from the inspection of the distributionsdepicted in Figure 5 for the quantity
where⟨‚‚‚⟩O represents a restricted time average for episodesof type O ) A, B. Note that, asFs increases, there is a gradualtendency toward perpendicular alignment of the C314 molecularplane with respect to the interface normal and a considerable
A similar description, but based on energetic grounds, canalso be readily obtained by monitoring the coupling betweenthe probe and the surfactant molecules. The most clear exampleis provided byVLJ, the Lennard-Jones contribution to thatcoupling, whose time evolution in shown in Figure 6. Throughthe use of this energetic order parameter, one observes thatsmaller C314-surfactant LJ couplings are involved in statesof typeA. The overall analysis of these observations suggeststhat a distinctive signature to differentiate these two statesislikely to be found in modifications of the solvation structureprovided by the SDS hosting of the probe.
To shed light on this point, it will be instructive to pause fora moment to establish a clearer distinction between the typesof surface environments associated with statesand . Typical
Figure 4. Temporal evolution of the out-of-plane orientation ofCoumarin 314 adsorbed at different charged water/air interfaces. Top-left panel, bare interface; top-right panel,Fs ) 0.002 Å-2; bottom-leftpanel, Fs ) 0.005 Å-2; bottom-right panel,Fs ) 0.01 Å-2. Thetrajectories were time-aligned so that transitions between solvation statesA andB take place at approximatelyt ) 0
cosθ(t) ) µ(t)‚z (3)
PO(cosθ0) ) ⟨δ(cosθ - cosθ0)⟩O (4)
Figure 5. Probability distributions for the out-of-plane orientation foradsorbed Coumarin 314 for solvation statesA (solid lines) andB(dashed lines) at different surfactant concentrations. (a) Bare interface;(b) Fs ) 0.005 Å-2; (c) Fs ) 0.002 Å-2; (d) Fs ) 0.01 Å-2.
Figure 6. Time evolution of the Lennard-Jones contributions to thesurfactant-coumarin coupling for different surfactant concentrations.Top panel,Fs ) 0.005 Å-2; middle panel,Fs ) 0.002 Å-2; lower panel,Fs ) 0.01 Å-2.
7368 J. Phys. Chem. B, Vol. 109, No. 15, 2005 Pantano et al.
80

surfactant concentration analyzed are shown in Figure 7. Themain differences are self-evident. In statesA, the coumarinappears external or adjacent to a two-dimensional spatial domainformed by a clustered group of SDS molecules. For statesB,however, the coumarin appears completely surrounded (“sol-vated”) by the SDS molecules within one of these two-dimensional domains. Such a molecular view allows a sensiblephysical interpretation for the C314 average orientation relativeto the surface that we have discussed above. Namely, in thecompact surfactant structureB, the C314 aligns perpendicularto the surface, and there is a reduction of the contact area withthe aqueous substrate. Conversely, a direct inspection of a largeseries of configurations for solvation states of typeA at highsurfactant concentrations showed that, despite the propensitytoward perpendicular alignment, the probe maintains closecontact with the aqueous phase. This is made possible via alocal enhancement of the roughness of the water surface, whichwe have already referred to in connection to the widthsL shownin Figure 3. Incidentally, the degree of the surface roughnesscan be clearly visualized from the snapshots in the lower portionof Figure 7, where the surfactants have been omitted forpurposes of clarity.
Several important conclusions can be drawn from the analysescarried out so far. First, the characteristics of the two solvationenvironments and the mechanisms that drive the interconversionsbetween them will be intimately connected to those governinglocal interfacial concentration fluctuations of the surfactant.Second, packing effects within the surfactant domains lead tolarger values of cosθ and to a lesser extent of the correspondingorientational fluctuations in states of typeB. Third, in termsof the dynamics, one observes that spontaneous transitionsbetween the two solvation states for the adsorbed C314 take
Given these scenarios, it is clear that a rigorous analysis ofthe resulting distribution of surfactant cluster sizes and theinterconversion dynamics between these structures for differentvalues ofFs are well beyond the scope of the present study.Limitations in the largest length scale accessible in the presentsimulations as well as in the maximum number of particlespreclude an adequate treatment of all relevant surfactant densityfluctuations. Besides, although in the course of a typical 5 nstrajectory we did observe severalA-B transitions in bothdirections, the magnitudes of the time scales governing con-centration fluctuations for surfactant species were comparableto the total length of our simulation runs. In view of theserestrictions, we have not proceeded any further in thesedirections. Nevertheless, we still believe that, based on theprevious observations, that the following physical picture holds.At low SDS concentrations, the degree of surface coverage issufficiently low to allow for large fluctuations in the local, two-dimensional surfactant density fields. This would promote adistribution of energetically favorable clustering of SDS, at arelatively minor entropic cost. This dynamical pattern of surface“patches”, including zones with low and high local concentra-tions of surfactant molecules, permits the existence of inner andouter solvation environments for the coumarin. Of course, asthe SDS coverage approaches the full-monolayer limit, spatialsurfactant distributions should become more homogeneous, localfluctuations in the density fields should drop considerably, andthe dual solvation status should converge into a single one,characterized by the full embedding of the probe within thesurfactant structure.
Finally, to provide an estimate for the quality of ourpredictions, we present results for the average values of cosθobtained from our simulations and those reported by Benderskii
Figure 7. Top, snapshots of solvation statesA and B. Surfactants, Coumarin 314 and water are rendered in black, dark gray, and light gray,respectively. No atomistic details are depicted in the aqueous phase. Bottom, same as top figures with the surfactant omitted.
Solvation of Coumarin 314 at Water/Air Interfaces J. Phys. Chem. B, Vol. 109, No. 15, 20057369
81

Note that, in all cases, the experimental values from SHGexperiments are intermediate between the simulated values fortheA andB environments, which adds support to the solvationdynamics picture presented below.
C. Dynamical Results.The occurrence of two interfacialsolvation states also has consequences in the solvation dynamics.Our analysis is based on an equilibrium time-correlation functionapproach and includes dynamical aspects of both the motionsof the adsorbed probe and the solvation response of the interfaceto a change in the electronic state of the dye. For the adsorbedprobe, we computed time correlations for the out-of-planemotion. The quantity of interest is
whereδ[cos θ(t)] ) cos θ(t) - ⟨cos θ⟩O. Results forCcosθ(t)are shown in Figure 8. All plots present similar characteristics.During the first picosecond, the decays present Gaussian-likeprofiles and account for 10-20% of the total relaxation. Thisregime is followed by a much slower one, which, for allFs
considered, can be reasonably well-approximated by singleexponentials with characteristic timesτr. Results for thecharacteristic rotational time scales are presented in the fifthand sixth columns of Table 1. These were obtained assumingthat Ccosθ(t) can be written as
Note that the presence of the surfactant does not affect the freerotational timesωr
-1/2 which, in turn, are between 1 and 2 ordersof magnitude smaller than the molecular reorientation timesτr.In addition, the computedτr values are considerably longer forstatesB compared to those ofA, suggesting that the dynamicalhindrance imposed by the surfactant is stronger in the former.Of course, the difference between these characteristic times,expressed for example in terms of the ratioτr
B/τrA, decreases
from 3.5 down to 1.5, as one moves to larger surfactantconcentrations, where the distinction between solvation statesA andB should progressively disappear.
To analyze the solvation dynamics at the interface, we focuson time-correlation functions for the energy gap45 derived fromthe interactions of the dye with the solvent, counterions, andsurfactant molecules, namely
The energy gap∆E(t) is defined as
where∆qR denotes the change in the partial charge of siteR inthe C314 as the dye is electronically excited andVR representsthe Coulomb potential exerted by the solvent, the surfactantheads, and the counterions on the tagged site. An analysis basedon the linear response theory45 is justified by the fact that, forclean interfaces, our previous studies found good agreementbetweenCs(t) and the solvation dynamics response functionobtained from full nonequilibrium simulations.30
Results forCs(t) for solvation states of typeA are shown inthe bottom panel of Figure 9. One observes that all interfacespresent similar dynamical characteristics, regardless of thesurfactant content. The corresponding overall solvation timesobtained from the time integrals ofCs(t) are in the range ofτs
0.8-0.9 ps (see also entries in the seventh column of Table1). This behavior can be interpreted by considering that (i) thesolvation responses are essentially governed by molecules in
TABLE 1: Solvation Parameters for Interfacial Solvation ofCoumarin 314
Fs(Å-2) state ⟨cosθ⟩ ωr-1/2 (ps) τr (ps) τs (ps)
0.0 0.15 0.17a 0.35 11.9 0.80A 0.19 0.37 8.4 0.75
0.002 B 0.54 0.42a 0.52 29.7 bA 0.37 0.38 25.6 0.83
0.005 B 0.54 0.50a 0.44 45.3 bA 0.52 0.42 30.3 0.84
0.01 B 0.72 0.71a 0.34 46.6 b
a From ref 23.b Error bars are exceedingly large to provide physicallymeaningful values.
Figure 8. Time correlation function for the out-of-plane dynamics ofCoumarin 314 adsorbed at charged interfaces. Top-left panel, bareinterface; top-right panel,Fs) 0.002 Å-2; bottom-left panel,Fs) 0.005Å-2; bottom-right panel,Fs ) 0.01 Å-2. Results for solvation statesA(B) are shown in solid (dashed) lines.
Ccosθ(t) )⟨δ[cosθ(t)]δ[cosθ(0)]⟩O
⟨(δ[cosθ])2⟩O
(5)
Figure 9. Equilibrium time correlation functions for the solvent energygap for solvation statesA (bottom panel) andB (top panel) at differentsurfactant concentrations.
Cs(t) )⟨δ[∆E(t)]δ[∆E(0)]⟩O
⟨(δ[∆E])2⟩O
(7)
∆E(t) ) ∑R
∆qRVR(t) (8)
7370 J. Phys. Chem. B, Vol. 109, No. 15, 2005 Pantano et al.
82

mainly correspond to interfacial water with practically no contactwith surfactant molecules and (ii) the persistence of large contactareas between the probe and the aqueous interface, despite themodifications in the overall orientation of the probe with respectto thez-axis.
The analysis of the dynamical responses of the interfaces forsolvation states of typeB is much more complex. Thecorrelation functions for these cases are shown in the top panelof Figure 9. Given the magnitude of the error bars, it is evidentthat our attempts to extract characteristic time scales from theseplots were hampered by poor statistics. Although the reasonsfor this failure still remain somewhat obscure to us, we tend tobelieve that they can be ascribed to the wide variety of relevanttime scales involved in the interfacial dynamical response. Notethat, in these solvation states, in addition to the dynamical modesof the water substrate and surfactant (normally characterizedby time scales of the order of a few picoseconds), the overallsolvation response is also indirectly modulated by the dynamicalmodes of the probe itself, which are typically 1 or 2 orders ofmagnitude slower. Moreover, we would like to point out thatthe distinctions between states of typeA and B, clearlyestablished in terms of the behaviors of the order parameterscos θ(t) (Figure 4) orVLJ(t) (Figure 6), become much moreambiguous if one wishes to discriminate solvation states basedon the time evolution of∆E(t) (not shown). In fact, the lattercan be pictured as a sequence of episodes taking place at avariety of time intervals, ranging from 2-5 ps up to even afew hundreds of picoseconds, during which∆E(t) fluctuatesaround well-differentiated mean values. Although we failed tocorrelate these episodes with changes in the order parameterscos θ(t) or VLJ(t) in a clear manner, we could establish acorrespondence with global changes in shape and size. There-fore, to explore the manifold of relevant time scales that dictatethe dynamical evolution of the solvationscovering practicallyfrom the picosecond up to several nanosecondssmuch longersimulations and elaborate analysis would be required, somethingthat we will postpone to a future study.
IV. Conclusions
The computer simulations presented in this paper demonstratethat the addition of ionic surfactants, even at low coverages,affect the solvation of adsorbed dyes at water/air interfaces ina sensible fashion. The presence of two, well-differentiated,interfacial environments is one key feature of our results. Thesestates can be reasonably well-discriminated by monitoringappropriate structural and energetic order parameters. For thehighest surfactant concentration considered, it is possible todescribeA and B in terms of the position of the coumarinwith respect to the surfactant spatial domains. This dual solvationstatus is the result of large surfactant concentration fluctuationsthat prevail at low coverages, a regime that is normally referredto as the “two-dimensional gas-liquid coexistence” region.46
Not surprisingly, we came across similar difficulties to thoseusually encountered whenever one intends to characterizesviacomputer simulation experimentssequilibrium and dynamicalaspects of density fluctuations in the gas-liquid coexistenceregion of ordinary fluids. Under these circumstances, theappropriate sampling of the wide variety of relevant length andtime scales normally requires collecting statistics of prohibitivelylarge systems during long time spans. In the present context,we do not discard that due to finite size effects, our quantitativeestimates may be affected by a nonnegligible degree ofuncertainty. These effects should be more pronounced at the
two molecules. As such, the agreement between the simulatedand the experimental data for the probe orientation shown inTable 1 forFs ) 0.002 Å-2 may be fortuitous.
We have verified that the presence of the surfactant modifiesthe reorientational correlations of the probe compared to thosefound at clean interfaces. As a result, the distribution for thedipolar orientation of the C314 becomes narrower and graduallyshifts toward more parallel alignments of the dipole with respectto the surface normal. Two reasons can be invoked to accountfor the latter feature: (i) for states of typeA, the enhancementof the roughness of the interface and the persistence of largecontact areas between the dye and the aqueous phase; (ii) forsolvation states of typeB, the competition for contact with theaqueous substrate between the dye and the surfactant polarheadgroups. In passing, we also note that a similar analysis,focused on the modifications of the localization of the probe atthe interface, shows less marked surfactant effects.
Our simulations also provide dynamical information relatedto the solvation of the dye. The presence of surfactant leads toa gradual slowing down of the probe’s reorientational motion,most likely due to the interactions with the surroundingsurfactants. The solvation dynamics for statesA are littleaffected by the degree of surfactant surface coverage and presenta characteristic time scale close to 0.8 ps. The solvationdynamics for states of typeB, however, turned out to be muchmore complex and apparently governed by a rich variety ofdynamical mechanisms, which would require further work tobe fully understood. Nevertheless, our results suggest that thedynamics of density fluctuations of the surfactant domains andthe motions of the probe itself are key elements in the correctinterpretation of the solvation responses of these chargedinterfacial systems.
Given the present status of our investigations, we can thinkof several important unanswered questions whose elucidationwill lead to a more comprehensive picture of solvation atcharged interfaces. First, one should still concentrate on thenature of the two-dimensional surfactant spatial domains inabsence of the dye. More specifically, a detailed analysis ofthe size and shape distributions of the domains will surely benecessary, along with an evaluation of the time scales governingthe interconversions between them. Equally important will bethe analysis of the relative stabilization of dyes in each solvationenvironment. Finally, we want to mention the extension of thepresent study into the full-monolayer regime, which is currentlybeing carried out in our laboratory. We are confident that theseinvestigations will open new and interesting perspectives of thesolvation of probes localized at more complex biointerfaces.
Acknowledgment. D.L. and M.S. acknowledge financialsupport from the CAPES/SPU Brazil-Argentina BinationalProgram (CAPG/BA-03/02). D.L. is a staff member of CONICET(Argentina). M.S. also thanks the Brazilian agencies FAPESP(03/09361-4) and CNPq.
References and Notes
(1) References 2 and 3 provide recent review articles on chemicalreactivity at interfaces and nonuniform environments.
(2) Benjamin, I.Acc. Chem. Res.1995, 28, 233. Benjamin, I.Chem.ReV. 1996, 96, 1449. Benjamin, I.Annu. ReV. Phys. Chem.1997, 48, 407.
(3) Nandi, N.; Bhattacharyya, K.; Bagchi, B.Chem. ReV. 2000, 100,2013.
(4) Smart, J. L.; McCammon, J. A.J. Am. Chem. Soc.1996, 118, 2283.Wang, H.; Zhao, X.; Eisenthal, K. B.J. Phys. Chem. B2000, 104, 8855.
(5) Benjamin, I.; Pohorile, A.J. Chem. Phys.1993, 98, 236.
Solvation of Coumarin 314 at Water/Air Interfaces J. Phys. Chem. B, Vol. 109, No. 15, 20057371
83

(7) Walker, G. C.; Harzeb, W.; Kang, T. J.; Johnson, A. W.; Barbara,P. F.J. Opt. Soc. Am. B1990, 369, 1521. Jimenez, R.; Fleming, G.; Kumar,P.; Maroncelli, M.Nature1994, 369, 471.
(8) Reynolds, L.; Gardecki, J. A.; Frankland, S. J. V.; Horng, M. L.;Maroncelli, M.J. Phys. Chem.1996, 100, 10337.
(9) Pryor, B. A.; Palmer, P. M.; Andrews, P. M.; Berger, M. B.; Topp,M. R. J. Phys. Chem. A1998, 102, 3284. Palmer, P. M.; Chen, Y.; Topp,M. R. Chem. Phys. Lett.2000, 318, 440. Palmer, P. M.; Chen, Y.; Topp,M. R. Chem. Phys. Lett.2000, 321, 62.
(10) Tamashiro, A.; Rodriguez, J.; Laria, D.J. Phys. Chem. A2002,106, 215.
(11) Sakar, N.; Datta, A.; Das, S.; Bhattacharyya, K.J. Phys. Chem.1996, 100, 15483. Datta, A.; Pal, S. K.; Mandal, D.; Bhattacharyya, K.J.Phys. Chem. B1998, 102, 6114. Datta, A.; Pal, S. K.; Mandal, D.;Bhattacharyya, K.J. Phys. Chem.1996, 100, 10523. Rau, B. B.; Costa, S.M. B. J. Phys. Chem. B1999, 103, 4309. Shirota, H. J. Horie, K.J. Phys.Chem. B1999, 103, 1437.
(12) Eisenthal, K.Annu. ReV. Phys. Chem.1992, 43, 627.(13) Goh, M. C.; Hicks, J. M.; Kemnitz, K.; Pinto, G. R.; Heinz, T. F.;
Eisenthal, K. B.; Bhattacharyya, K.J. Phys. Chem.1988, 92, 5074.(14) Zimdars, D.; Dadap, J. I.; Eisenthal, K. B.; Heinz, T. F.Chem.
Phys. Lett.1999, 301, 112.(15) Zimdars, D.; Dadap, J. I.; Eisenthal, K. B.; Heinz, T. F.J. Phys.
Chem. B.1999, 103, 3425.(16) Zimdars, D.; Eisenthal, K. B.J. Phys. Chem. A1999, 103, 10567.(17) Zimdars, D.; Eisenthal, K. B.J. Phys. Chem. B.2001, 105, 3393.(18) Benderskii, A. V.; Eisenthal, K. B.J. Phys. Chem. B. 2000, 103,
11723. Benderskii, A. V.; Eisenthal, K. B.J. Phys. Chem. B. 2001, 105,6698.
(19) Bessho, K.; Uchida, T.; Yamaguchi, A.; Shioya, T.; Teramae, N.Chem. Phys. Lett.1997, 264, 381.
(20) Yanagimachi, M.; Tamai, N.; Masuhara, H.Chem. Phys. Lett.1992,200, 469.
(21) Pant, D.; Levinger, N. E.Chem. Phys. Lett.1998, 292, 200. Pant,D.; Levinger, N. E.J. Phys. Chem. B1999, 103, 7846.
(22) Martins, L. R.; Skaf, M. S.; Ladanyi, B. M.J. Phys. Chem. B2004,108, 19687.
(23) Benderskii, A. V.; Eisenthal, K. B.J. Phys. Chem. A2002, 106,7482.
(24) Schweighofer, K. J.; Essman, U.; Berkowitz, M.J. Phys. Chem. B1997, 101, 3793.
(25) Dominguez, H.; Berkowitz, M. L.J. Phys. Chem. B2000, 104,5302.
(26) Pohorile, A.; Benjamin, I.J. Phys. Chem.1993, 97, 2664. Tarek,M.; Tobias, D. J.; Klein, M. L.J. Phys. Chem.1995, 99, 1393. Bocker, J.;Schlenkrich, M.; Bopp, P.; Brickmann, J.J. Phys. Chem.1992, 96, 9915.
(27) Conboy, J. C.; Messmer, M. C.; Richmond, G.J. Phys. Chem.1996,100, 7617.
(28) Carlstro¨m, G.; Halle, B. Langmuir 1988, 4, 1346. Hauser, H.;Haering, G.; Pande, A.; Luisi, P. L.J. Phys. Chem.1989, 93, 7869. Zinsli,
P. E.J. Phys. Chem.1979, 83, 3223. Wong, M.; Thomas, J. K.; Nowak, T.J. Am. Chem. Soc.1997 , 99, 4730.
(29) Shen, Y. R.The Principles of Nonlinear Optics; Wiley: New York,1984. Shen, Y. R.Annu. ReV. Phys. Chem.1989, 40, 327. T. F. Heinz InNonlinear Surface Electromagnetic Phenomena; Ponath, H., Stegeman, G.,Eds.; Elsevier: Amsterdam, 1991. Bervet, P. F., Girault, H. H.SecondHarmonic Generation at Liquid/Liquid Interfaces; CRC Press: Boca Raton,FL, 1996. Eisenthal, K. B.Acc. Chem. Res.1993, 26, 636.
(30) Pantano, D. A.; Laria, D.J. Phys. Chem. B2003, 107, 297.(31) In absence of surface active dyes, the density of the saturated SDS
monolayer rises up to 2 10-2 Å-2.(32) Berendsen, H. J. C.; Postma, J. P. M.; Von Gunsteren, W. F.;
Hermans, J.Intermolecular Forces; Reidel: Dordrecht, 1981.(33) Liotard, D. A.; Healey, E. F.; Ruiz, J. M.; Dewar, M. J. S.QPCE
Bull. 1989, 9, 123. AMPAC, version 2.1i; Quantum Chemistry ProgramExchange; Program No. 506.
(34) McCarthy, P. K.; Blanchard, G. J.J. Phys. Chem.1993, 97, 12205.(35) Rechthaler, K.; Ko¨hler, G.Chem. Phys.1994, 189, 99.(36) A similar parametrization to that used in the present study was
employed in ref 30.(37) Moylan, C. R.J. Phys. Chem.1994, 98, 13513.(38) Allen M. P.; Tildesley, D. J.Computer Simulation of Liquids;
Clarendon: Oxford, 1987.(39) Although the implementation of Ewald summation techniques and
variants thereof (see refs 40 and 41) is known to provide more accuratedescriptions for the electrostatics of periodically replicated systems in twodimensions, solvation dynamics results seem to be only mildly affected bythe truncation of long-range forces, an approximation that introducesaconsiderable savings in computational effort. See ref 42.
(40) Essmann, U.; Parera, L.; Berkowitz, M. L.; Darden, T.; Lee, H.;Pedersen, J. G.J. Chem. Phys.1995, 103, 8577. Yeh, I.-C.; Berkowitz, M.L. J. Chem. Phys.1999, 111, 3155.
(41) Feller, S. E.; Pastor, R. W.; Rojnucharin, A.; Bogusz, S.; Brooks,B. R. J. Phys. Chem.1996, 100, 17011.
(42) Vieceli J.; Benjamin, I.J. Phys. Chem. B2003, 107, 4801. VieceliJ.; Benjamin, I.J. Phys. Chem. B2002, 106, 7898.
(43) Jungwirth, P.; Tobias, D.J. Phys. Chem. B2000, 104, 7702.Jungwirth, P.; Tobias, D.J. Phys. Chem. B2001, 105, 10468.
(44) The local coordinate system reported in ref 15 is slightly differentfrom that adopted here. In particular, cosθ is measured with respect to the� direction, taken along the molecular transition dipole moment. Still, webelieve that this difference should not preclude a direct comparison betweensimulation and SHG results.
(45) Carter, E. A.; Hynes, J. T.J. Chem. Phys.1991, 94, 5961. Seealso Chandler, D.An Introduction to Modern Statistical Mechanics; OxfordUniversity Press: New York, 1987. Chapter 8.
(46) Gaines, F. L., Jr.Insoluble Monolayers at Liquid-Gas Interfaces;J. Wiley & Sons: New York, 1966.
7372 J. Phys. Chem. B, Vol. 109, No. 15, 2005 Pantano et al.
84

Capıtulo 7
Efeito Kerr Optico da
Agua em Condic¸ oes
Ambientes e Superfrias
A dinâmica intermolecular da água tem sido estudada por diversas técnicas experimen-
tais. Nas últimas duas décadas, baseados na tecnologia de laser de pulsos ultracurtos,
foram desenvolvidas novas técnicas experimentais que permitem investigar em grande
detalhe fenômenos ultra-rápidos, que ocorrem na escala temporal de algumas dezenas
de fentosegundos. Entre elas estão a espectroscopia terahertz [49] e técnicas de óptica
não-linear, como a espectroscopia Raman Echo [50] e o efeito Kerr óptico (EKO) [51].
Seguindo esse desenvolvimento, estudos de simulação de DM tem enriquecido a
visão sobre a dinâmica da água, tanto em condições ambientes como em condições não
ambientes de temperatura e pressão. Várias simulações foram realizadas focando aspec-
tos distintos da dinâmica da água. Espectroscopias de geração de segundo harmônico
e soma de freqüência vibracional de sistemas interfaciais também foram estudadas por
DM. O efeito Kerr foi estudado por Saito e Ohmine [52], utilizando o modelo TIPS2
da água à temperatura ambiente, e Bursulaya e Kim [53], utilizando uma abordagem
semi-clássica para a polarizabilidade eletrônica.
O experimento de efeito Kerr Óptico está esquematizado na figura 7.1. Quando uma
amostra líquida é submetida a um forte campo elétricoE, uma anisotropia óptica é in-
duzida no meio. A amostra fica caracterizada por dois índices de refração diferentesn‖en⊥, um para luz com polarização paralela àE e outra para polarização perpendicular à
E. Considerando-se agora um feixe de luz que atravessa um par de polarizadores cruza-
dos colocados antes e depois de uma amostra líquida, tem-se que, na ausência de bir-
refringência da amostra, nenhuma luz deve ser detectada depois do segundo polarizador.
Se um campo elétrico for aplicado na amostra, deverá ocorrer uma despolarização par-
85

86 Capıtulo 7. Efeito Kerr da Agua
Figura 7.1. Esquema de um experimento de OKE.
cial e alguma intensidade luminosa será observada depois do segundo polarizador. No
efeito Kerr óptico (OKE paraOptical Kerr Effect) os campos que induzem e provam a
birrefringência da amostra são pulsos laser ultra-rápidos. Pode-se acompanhar a relax-
ação da birrefringência variando-se o tempo entre os dois pulsos, possibilitando extrair
informações a respeito da dinâmica do sistema no nível microscópico. Este experimento
pertence a uma vasta classe de técnicas conhecidas como experimentos “excita e prova”
(pump-probe). A resposta do sistema a excitações longe de qualquer freqüência óptica
possui basicamente duas contribuições: (i) uma contribuição puramente eletrônica, cuja
duração é da ordem da duração do pulso de excitação, causada pela distorção da densi-
dade de elétrons em torno dos átomos e moléculas do sistema, e (ii) uma contribuição
nuclear, causada pela reorientação parcial das moléculas ao campo de excitação. É esta
segunda contribuição, onde a reorientação das moléculas é não isotrópica, que é respon-
sável pela birrefringência transiente. Para tempos longos (t > 1 ps), a contribuiçãoii é
dominada pelo movimento rotacional espontâneo das moléculas, que trás o sistema de
volta ao estado isotrópico de equilíbrio. Para tempos curtos (t < 1 ps) outros modos

87
dinâmicos importantes dominam o sinal observado. Um destes modos são as oscilações
libracionais das moléculas na gaiola formada por seus primeiros vizinhos, portanto, é
resultado de interações intermoleculares. Outro modo dinâmico importante resultante
das interações intermoleculares são flutuações na densidade local do líquido e, portanto,
o sinal observado deve-se à anisotropia da polarizabilidade molecular induzida por col-
isão. Um último processo importante que pode afetar o sinal observado está relacionado
com as vibrações intramoleculares. Todos esses modos dinâmicos influenciam na bir-
refringência do líquido.
Apresenta-se neste capítulo os resultados de estudos por simulação de DM da relax-
ação da anisotropia da polarizabilidade total da água, que é provada diretamente pelos
experimentos de espalhamento Raman despolarizado e espectroscopia de efeito Kerr
óptico. São realizados estudos da água em condições ambientes e no estado superfrio.
Os fundamentos teóricos, métodos de simulação e de análise estão bem descritos nos
artigos que se seguem.
As características de tempo curto do efeito Kerr da água SPC/E em condições ambi-
entes compara bem com resultados experimentais e de simulação com modelos de água
mais sofisticados. O acordo é menos satisfatório para a componente difusiva.
A dinâmica complexa da água superfria observada em medidas experimentais de
efeito Kerr óptico realizadas por R. Torre e colaboradores [54] motivaram o estudo deste
sistema. Neste trabalho, os autores mostram que a dinâmica da relaxação estrutural água
em diferentes temperaturas do estado superfrio obedece a uma exponencial estirada,
f(t) ≈ a exp[−(t/τ)β], com o parâmetro de estiramentoβ praticamente independente
da temperatura. Ainda, os autores observaram que a dependência do tempo de relaxação
τ com a temperaturaT obedece a uma lei de potências do tipoτ(T ) = a (T − TS)−γ,
onde a temperatura singularTS está relacionada com a imobilização estrutural do sis-
tema em baixas temperaturas. Estas propriedades conseguem ser capturadas nas simu-
lações de DM realizadas neste trabalho, e seus resultados estão bem descritos no segundo
artigo que acompanha este capítulo.

A simulation study of the optical Kerr effect in liquid water
Milton T. Sonoda, Sergio M. Vechi and Munir S. Skaf*
Instituto de Quımica, Universidade Estadual de Campinas, Cx. P. 6154, Campinas, SP,13084-971, Brazil. E-mail: [email protected]
Received 10th November 2004, Accepted 20th January 2005First published as an Advance Article on the web 1st February 2005
A molecular dynamics simulation study is presented for the dynamics of the polarizability anisotropy of liquidwater using the SPC/E model and a dipolar induction scheme that involves the intrinsic polarizability and firsthyperpolarizability tensors obtained from ab initio quantum chemical calculations at the MP2/6-311þþG(d,p)level. The time-correlation functions for the collective polarizability anisotropy, the optical Kerr effect response,and the frequency spectra are analyzed in terms of the intrinsic and induced polarizability contributions. At shorttimes, the simulated Kerr nuclear response exhibits maxima near 15, 50 and 180 fs, followed by a diffusive tailwhich has been fitted by a bi-exponential with time constants ca. 0.4 and 2.5 ps. The short time features are ingood agreement with available simulation and experimental results. The agreement with experiments is lesssatisfactory for the diffusive components. The main features of the frequency spectrum include a rotational-diffusion peak centered around 3 cm�1, a collision-induced (hindered translations) band near 200 cm�1, and abroad librational band at 450 cm�1. The simulation results are in good agreement with experimental frequencyspectra obtained from Kerr effect and related spectroscopies, but fail to reproduce the experimental band near60 cm�1.
1. Introduction
The intermolecular dynamics of liquid water has long been asubject of intense experimental and theoretical research.1–5
Numerous experimental studies have been reported on thedynamics of liquid water in the last two or three decades usinga variety of different techniques, including NMR,6 microwavedielectric relaxation,7 far-infrared,8 Raman9 and light scatter-ing spectroscopies,10 and small angle neutron scattering.11
More recently, new experimental developments based on ultra-fast laser techniques have made it possible to investigateintermolecular dynamical processes of water at much shortertimescales with substantially higher level of detail. Amongthese, terahertz spectroscopy12 and nonlinear optical methodssuch as Raman echo13 and optical Kerr effect14,15 spectro-scopies stand out for providing remarkably valuable informa-tion directly in the time domain.
Parallel to these developments, advances in molecular dy-namics (MD) computer simulations have markedly enrichedour views on the dynamics of water, both at ambient andnonambient conditions of temperature and pressure.1 A varietyof MD simulation studies have been performed on liquid waterfocusing on different aspects of its dynamics. In particular,detailed simulation analyses have been reported for dielectricrelaxation, depolarized Raman or light scattering, time re-solved fluorescence spectroscopy,5 and nonlinear optical Kerrresponse for bulk phases,16,17 as well as second harmonicgeneration and vibrational sum frequency18 signals forstudying interfacial aqueous systems more specifically. MDsimulations have been carried out for the Kerr effect bySaito and Ohmine16 on the TIPS2 potential at room tempera-ture and by Bursulaya and Kim17 on a mixed quantum-classical model for the electronic polarizability. The depolar-ized Raman spectrum of TIP4P water was studied at severaltemperatures as well.19 Very recently, we have reported MDsimulations of the Kerr response of binary dimethylsulfoxide–water mixtures, in which we investigate the effects of theH-bonding between water and DMSO on the mixtures’Kerr response.20
In this paper, we report MD simulations results for thepolarizability anisotropy relaxation of liquid water at ambientconditions. Our goal is to examine, from a microscopic per-spective, the characteristics of the intermolecular dynamicsthat are directly probed by depolarized Raman scattering oroptical Kerr effect (OKE) spectroscopy.14 Here we use Torii’sextended dipole–induced-dipole model21 for the collective po-larizability and keep separate the contributions from theintrinsic and induced polarizabilities in order to identify themolecular origin of the different features that characterizewater’s OKE response.In Section 2 we summarize the main theoretical relationships
involved in the calculation of the OKE signals from thesimulations and describe the computational details. Resultsand discussions appear in Section 3. Our concluding remarksare presented in Section 4.
2. Theory and computational details
2.1. Theoretical background
The relevant dynamical variable for the spectroscopic proper-ties of interest is the collective electronic polarizability tensor ofthe system, P:22,23
P ¼XNi
ai ¼ PM þPI; ð1Þ
where the total polarizability has been separated into intrinsic(molecular), PM, and induced, PI contributions. The intrinsicpart is the sum of unperturbed, gas phase molecular polariz-abilities,
PM ¼XNi
aMi ; ð2Þ
while the induced part comprises polarizability enhancementsdue to the intermolecular interactions. In this work, thedynamical modulation of the polarizabilities arises from a
PCC
Pw
ww
.rsc.org/pccp
P h y s . C h e m . C h e m . P h y s . , 2 0 0 5 , 7 , 1 1 7 6 – 1 1 8 0 T h i s j o u r n a l i s & T h e O w n e r S o c i e t i e s 2 0 0 5
89

center-to-center dipolar coupling between molecules:
PI ¼XNi
Xj 6¼i
aMi T ij aj þ bMi T ij lj
� �: ð3Þ
The enhanced polarizability tensor and dipole vector of themolecules are obtained by iteratively solving the equations:
ai ¼ aMi þXj 6¼i
aMi T ij aj ð4Þ
li ¼ lMi þXj 6¼i
aMi T ij lj ; ð5Þ
where liM and bi
M are the gas phase dipole and hyperpolariz-ability of molecule i, respectively, and Tij ¼ rirj | ri � rj |
�1 isthe dipole tensor between molecules i and j. All vectors andtensors are expressed in the lab frame. This enhancementmechanism, named extended dipole–induced-dipole (XDID),21
includes contributions from the first hyperpolarizability of themolecule and has been used in recent simulations of depolar-ized Raman spectrum and OKE responses of formamide andN-methylformamide,21 and DMSO/water mixtures.20 Byneglecting the ‘‘bTm’’ term, one recovers the usual dipole–induced-dipole (DID) mechanism.24
The dynamics of interest here is given by time-correlationfunction (TCF) of the off-diagonal elements of the collectivepolarizability tensor:
C(t) ¼ hPxz(t) Pxz (0)i/ (Ng2)/15) (6)
where g2 ¼ 12[(a1 � a2)
2 þ (a1 � a3)2 þ (a2 � a3)
2] is the square ofthe ideal polarizability anisotropy, with aj as the principalpolarizability components. To improve statistics, C(t) is com-puted from all the independent TCFs of off-diagonal elements(xy, xz and yz).
2.2. Models and simulations
The intrinsic molecular polarizability and first hyperpolariz-ability tensors were obtained from ab initio quantum chemicalcalculations with electronic correlation taken into account atthe MP2 level and basis set 6-311þþG(d,p) using the Gaus-sian98 program package.25 The nonzero elements of the aM
and bM tensors for water are collected in Table 1. Simulationswere also performed using aM and bM tensors computed at theMP3/6-31þG(2df,p) level, yielding practically identical resultsfor the spectroscopic quantities of interest. The molecularcoordinate axes are defined with the y-axis normal to themolecular plane and the z-axis along the dipole. The polariz-ability and first hyperpolarizability tensors obtained from ourcalculations are consistent with the quantum calculations ofGubskaya and Kusalik.26 There are, however, appreciabledifferences between our bM tensor (including the sign of some
components) and the results obtained from higher levelcoupled-cluster calculations.27,28 The effects of different bM
tensors on the OKE response was not fully investigated, butare expected to be small since the DID contribution is mark-edly predominant.The MD trajectories were generated using the well-known
SPC/E potential for water,29 which treats the molecules asnonpolarizable, rigid bodies interacting via a pairwise sum ofsite-site potentials involving standard (12–6) Lennard-Jonesplus Coulombic terms. The simulations were performed in theNVE ensemble with a total of N ¼ 500 molecules placed inperiodically replicated cubic boxes dimensioned to reproducethe experimental density at ambient conditions (0.997 g cm�3).Lennard-Jones forces were cut-off at half the box length and
Coulomb forces were treated via Ewald sums with conductingboundaries.30 The equations of motion were integrated withSHAKE31 and leap-frog algorithms32 with a timestep of 3 fs.Total energy is conserved within 0.2% during unperturbed 12ps runs. Approximately 200 of such runs, separated by occa-sional intervals for velocity rescaling, were used to compute thequantities of interest.
3. Results and discussion
According to eqn. (1)–(6), C(t) has contributions from mole-cular and induced autocorrelations plus the cross-correlationbetween molecular and induced polarizabilities:
C(t) ¼ CMM(t) þ CII(t) þ CMI(t), (7)
with
CMM(t) ¼ hPxzM(t)Pxz
M(0)i/(Ng2/15);
CII(t) ¼ hPxzI(t)Pxz
I(0)i/(Ng2/15) (8)
CMI(t) ¼ (hPxzM(t)Pxz
I(0)i þ hPxzI(t)Pxz
M(0)i)/(Ng2/15) (9)
Being an intrinsically collective quantity, C(t) may be aslowly converging property. To assure that the simulations arelong enough to yield converged anisotropy correlations, we havecomputed the cumulative averages for CMM(0), CII(0), andCMI(0) as functions of the simulation time. The results areshown in Fig. 1, which clearly indicate that convergence hasbeen reached. The estimated relative errors are less than 5%. The
Table 1 Nonzero components of the molecular polarizability and first
hyperpolarizability tensor obtained from electronic structure computa-
tions at the MP2/6–311þþG(d,p) level. The components of aM and bM
are given in units of A3 and A5/e, respectively
axxM axy
M ayyM azz
M
Watera 1.04 0 1.00 1.17
bxxxM bxxz
M bxyyM byyz
M bzzzM
Watera 0.80 �0.19 0.06 0.05 0.86
a The z- and y-directions for water are along the C2v symmetry axis and
normal to the molecular plane, respectively. The byzz components is
smaller than 0.05 A5/e and the remainders are zero.
Fig. 1 Cumulative average of the static fluctuations of the collectivepolarizability anisotropy for SPC/E water versus simulation time. Theintrinsic molecular (MM) fluctuation is shown by a solid line, theinduced (II) and cross-correlation (MI) contributions are shown bydashed and dashed-dotted lines, respectively.
P h y s . C h e m . C h e m . P h y s . , 2 0 0 5 , 7 , 1 1 7 6 – 1 1 8 0T h i s j o u r n a l i s & T h e O w n e r S o c i e t i e s 2 0 0 5
90

total static correlation, C(0) ¼ 2.33, is substantially higher thanthe uncorrelated value of Cideal ¼ 1.0 because of the intrinsicstructure of liquid water. The molecular and induced autocorre-lations at t ¼ 0 contribute nearly 46 and 26%, respectively, whilethe cross-correlation represents the remaining 28%.
Fig. 2, upper panel, shows the relaxation of the totalanisotropy, C(t) (solid line) and its separate contributions,CMM, CII and CMI. The insert shows details of the initialstages of the relaxation in which librational oscillations areevident. After the librational transient, the relaxation rates ofall three components are similar, which suggests that theinduced polarizabilities tend to follow the reorientationaldynamics of the molecules. The post-librational decay is well-described by a bi-exponential relaxation, a1exp(�t/t1) þ a2exp(�t/t2), in the time interval 0.2–6.0 ps. Our best fittingparameters are given in Table 2. The characteristic decay timesfor the total C(t), 0.37 and 2.46 ps, are roughly comparable tothe experimental values reported by Castner and co-workers(0.50 and 1.19 ps).33 In this time interval, C(t) can be equallywell fitted by a stretched exponential function, a0 exp[�(t/t0)
b],with t0 ¼ 1.01 ps and b ¼ 0.59. Very recent experimental OKEdata yield t0 ¼ 0.35 ps and b ¼ 0.58 for water at 294 K.34 Therotational-diffusion relaxation of the simulated OKE signal istherefore slower than the experimentally observed dynamics.How much of the total TCF is due to reorientational processescan be determined by means of the projection scheme used toanalyze the dielectric relaxation of polar-polarizable fluids.35
Namely, the collective polarizability tensor components areseparated into ‘‘local-field’’ and ‘‘collision-induced’’ portionsaccording to:35,36
PxzI(t) ¼ GPxz
M(t) þ DPxz(t), (10)
where
G ¼ hPxzM(0)Pxz
I(0)i/h |PxzM(0) |2i (11)
is the static projection of the induced polarizability over themolecular polarizability. From the simulations we obtainG ¼ 0.30. The quantity
DPxz(t) ¼ PxzI(t) � GPxz
M(t) (12)
represents the time-dependent fluctuations of the inducedpolarizability around the intrinsic counterpart renormalizedby the induced effects. Thus, the projection allows the separa-tion of the translational contributions (collision-induced) fromthe reorientational components of the polarizability anisotropyrelaxation.In terms of projected variables, the total TCF can be
separated into a purely ‘‘reorientational’’ (R), a ‘‘collision-induced’’ contribution (D), and their cross correlation (X):
C(t) ¼ CR(t) þ CX(t) þ CD(t), (13)
with
CR(t) ¼ (1 þ G)2CMM(t) (14)
and
CX(t) ¼ 2(1 þ G)hDPxz(t)PxzM(0)i/(Ng2/15) (15)
CD(t) ¼ hDPxz(t)DPxz(0)i/(Ng2/15). (16)
The lower panel of Fig. 2 shows the total TCF (solid line) andthe projected contributions. The data clearly shows that therenormalized intrinsic contribution CR(t) (dashed line) predomi-nates over the other components. Although the magnitude of thecollision-induced contribution CD at early times is less than athird of the reorientational counterpart, there are some impor-tant features due to the translational dynamics that will appear inthe OKE signal itself, as discussed below. The results depicted inFig. 2, lower panel, also show that the simulated anisotropyrelaxation in the rotational-diffusion regime (t > 2 ps), isessentially given by the reorientational component CR(t).The optically heterodyne-detected OKE signal in the time
domain provides valuable information about the intermolecu-lar dynamics through the nuclear response function, Rnuc(t),which is experimentally obtained from the raw OKE transientsafter removal of the instantaneous electronic response anddeconvolution from the instrument function.14,24,37 Simplyput, Rnuc(t) is related to the time derivative of the anisotropypolarizability TCF through:
RnucðtÞ ¼ � 1
kBT
@CðtÞ@t
: ð17Þ
Fig. 3 shows the total nuclear response function (thick solidline) and the separate contributions from qCMM(t)/qt, qCII(t)/qt, and qCMI(t)/qt. The cross-correlation between molecularand induced polarizabilities makes a negligible contribution toRnuc(t) because CMI is a slowly varying function of time (cf.Fig. 2). The nuclear response function and its RMM componentpresent a rapid initial rise to their maxima within the first 15 fs,followed by a brief period of oscillatory decay, which subse-quently merges into a smooth relaxation. Like the case ofacetonitrile38 and polar protic solvents,17,36,39 the simulated
Fig. 2 Total collective polarizability anisotropy relaxation (solid line),intrinsic molecular contributions (MM; dashed line), induced polariz-ability auto-correlation (II; dotted line), and cross-correlation (MI;dashed-dotted line). The insert shows features of the short-timedynamics. The lower panel shows the total TCF and the separatecontributions in terms of projected variables. Solid line: C(t); dashedline: CR(t); Dotted line: CD(t); dashed-dotted line: CX(t).
Table 2 Bi-exponential, a1 exp(�t/t1) þ a2 exp(�t/t2), fits to the
polarizability anisotropy TCF in the time intervals 0.2–6.0 ps
a1 t1/ps a2 t2/ps
C (t) 0.76 0.37 1.14 2.46
CMM (t) 0.30 0.45 0.56 2.90
CII (t) 0.24 0.16 0.25 1.16
CMI (t) 0.23 0.33 0.42 2.36
P h y s . C h e m . C h e m . P h y s . , 2 0 0 5 , 7 , 1 1 7 6 – 1 1 8 0 T h i s j o u r n a l i s & T h e O w n e r S o c i e t i e s 2 0 0 5
91

Rnuc(t) function shows prominent oscillatory features asso-ciated to fast hindered reorientational motions because ofwater’s small moments of inertia and strong intermolecularassociation. The behavior of the simulated OKE nuclearresponse presented here lies somewhere in-between the nuclearresponses reported by Bursulaya and Kim17 obtained fromsimulations with the TAB/10D polarizable water potential anda fixed, nonfluctuating (NF) molecular polarizability model.Specifically, our Rnuc(t) shows less pronounced oscillations inthe time interval of Fig. 3 than does TAB/10D water, but isnoticeably more oscillatory than the response from the NFmodel.17 The features near 12–20 and 150 fs from both TAB/10D and NF simulations, are also present here. The structurenear 50 fs is due to librational motions40 and has been observedwith the TAB/10D potential but not with the NF model.17
These results imply that important dynamical fluctuations ofwater polarizability, particularly the effects of the solventnonlinear electronic response, are only partially accountedfor by our simulations through the ‘‘bTm’’ term, with quanti-tative consequences to the OKE transients and also to thefrequency spectra, discussed below. Overall, there is reasonableagreement with the experimental nuclear response reported byCastner et al.,15 which shows peaks at ca. 20, 57 and 197 fs.Interestingly, the simulation results shown in Fig. 3 indicatesthat the peak between 150–200 fs is mainly due to collision-induced dynamics, which corroborates the experimental inter-pretation of this feature in terms of hindered translationalmodes within the cage of neighbors.
Further comparison with available experimental data can bemade in terms of the frequency spectra. The spectrum ofinterest here is provided by the imaginary part of the Fouriertransform of the nuclear response according to:17,37
~RðoÞ ¼ 21� e�b�ho
1þ e�b�ho
Z 1
0
sinðotÞRnucðtÞdt ; ð18Þ
where 1/b ¼ kBT. The detailed balance desymmetrizationfactor 2/(1 þ e�b‘o), which accounts for quantum correctionsto the classical spectrum, has been explicitly indicated.17,41 Thisrelationship indicates that depolarized Raman or light scatter-ing and OKE spectra are closely related quantities.37 The long-time portion of the Rnuc(t) response, is ca. �q[a1exp(�t/t1) þa2exp(�t/t2)]/qt was used in order to improve the accuracy ofthe numerical Fourier transforms. Results for R(o) are shownin Fig. 4. The spectrum of the total nuclear response (solid line)presents peaks at different frequencies corresponding to theintricate motions of water.
The slow rotational-diffusion dynamics is mainly character-ized by a central band near 3 cm�1 and a shoulder between 20and 30 cm�1 (Fig. 4, insert). The band shape in this frequencyregion is given essentially by the molecular (MM) polarizabilityauto-correlations. The rotational-diffusion relaxation obtainedfrom our SPC/E-based simulations is slower than that reportedfor the TAB/10D model, which has a maximum in the reor-ientational band near 8 cm�1.17 For higher frequencies, twomain peaks are identified in the total spectrum at ca. 230 and500 cm�1, which are due to induced (II) and molecular (MM)polarizability auto-correlations, respectively. The simulatedspectrum is qualitatively compared with the experimental15
analyses from different groups, which ascribe the measuredbroad band in the range 400–800 cm�1 to hindered reorienta-tional motions (librations) about the different molecular axes,and the peaks at ca. 60 and 170 cm�1 to collision-inducedeffects from hindered translational motions associated with thestretching and bending of hydrogen bonds.15,16 The experi-mentally observed feature near 60 cm�1 is not resolved in oursimulations because the intrisinc polarizability (MM) contri-bution picks up intensity as o falls below ca. 60 cm�1. Thissuggests that the intrinsic polarizability tensor used in thesimulations may overestimate the anisotropy of water.
4. Concluding remarks
In this work we report an MD simulation study of thepolarizability anisotropy relaxation of SPC/E water, focusingon the Kerr nuclear responses in the time- and frequency-domains. The simulations require a model for the collectivepolarizability which has been described by a sum of individualmolecular polarizabilities obtained from ab initio electronicstructure calculations with a dynamical modulation given by adipolar coupling. Some degree of nonlinear electronic responseis incorporated through fixed (intrinsic) values of molecularhyperpolarizability tensors. We have analyzed the relaxation ofthe polarizability anisotropy in terms of the dynamics ofintrinsic and induced polarizabilities and interpreted theOKE response and its spectrum in the light of the differentcontributions. Our results are compared with available experi-mental data and previous simulation results.
Acknowledgements
We gratefully acknowledge financial support from the Brazi-lian agencies FAPESP (Grants 01/09374-3 and 03/09163-4)and CNPq, and CAPES for a Graduate Fellowship to M.T.S.
Fig. 3 OKE nuclear response for SPC/E water within the XDIDscheme. Total, molecular, induced and cross-correlation contributionsare as indicated.
Fig. 4 OKE frequency spectra for SPC/E water within the XDIDscheme. Solid, dashed-dotted, and dashed lines correspond to total,induced, and cross-correlation spectra. The insert depicts the spectraversus o on a logarithmic scale, which shows the low-frequency band ingreater detail.
P h y s . C h e m . C h e m . P h y s . , 2 0 0 5 , 7 , 1 1 7 6 – 1 1 8 0T h i s j o u r n a l i s & T h e O w n e r S o c i e t i e s 2 0 0 5
92

References
1 Over the years, there have appeared numerous excellent reviewson water and aqueous systems. Refs. 2–5 serve as a starting pointto the literature. The reader may also see the recent Special Issues:Chem. Phys., 258 (2000) and Braz. J. Phys., 34 (2004).
2 (a) F. H. Stillinger, Adv. Chem. Phys., 1975, 31, 1; (b) F. H.Stillinger, Science, 1980, 209, 451.
3 P. J. Rossky, Annu. Rev. Phys. Chem., 1985, 36, 321.4 I. Ohmine and H. Tanaka, Chem. Rev., 1993, 7, 2545.5 B. M. Ladanyi and M. S. Skaf, Annu. Rev. Phys. Chem., 1993, 44,
335.6 J. Jonas, T. DeFries and D. J. Wilbur, J. Chem. Phys., 1981, 65,
581.7 U. Kaatze and V. Uhlendorf, Z. Phys. Chem., Neue Folge, 1981,
126, 151.8 (a) M. N. Afsar and J. B. Hasted, J. Opt. Soc. Am., 1977, 67, 902;
(b) J. B. Hasted, S. K. Husain, F. A. M. Frescura and J. R. Birch,Chem. Phys. Lett., 1985, 118, 622; (c) A. N. Rusk, D. Williams andM. R. Querry, J. Opt. Soc. Am., 1971, 61, 895.
9 (a) A. de Santis, M. Sampoli, V. Mazzacurati and M. A. Ricci,Chem. Phys. Lett., 1987, 133, 381; (b) A. de Santis, R. Frattini, M.Sampoli, V. Mazzacurati, M. Nardone, M. A. Ricci and G.Ruocco, Mol. Phys., 1987, 61, 1199; (c) K. Mizoguchi, Y. Horiand Y. Tominaga, J. Chem. Phys., 1992, 97, 1961.
10 C. J. Montrose, J. A. Bucaro, J. Marshall-Coakley and T. A.Litowitz, J. Chem. Phys., 1974, 60, 5025.
11 (a) A. K. Soper and A. Luzar, J. Phys. Chem., 1996, 100, 1357; (b)A. K. Soper and A. Luzar, J. Chem. Phys., 1992, 97, 1320.
12 (a) J. E. Bertie and Z. Lan, Appl. Spectrosc., 1996, 50, 1047; (b)J. T. Kindt and C. A. Schmuttenmaer, J. Phys. Chem., 1996, 100,10373.
13 M. Berg and D. A. Vanden Bout, Acc. Chem. Res., 1997, 30, 65.14 (a) D. McMorrow, W. T. Lotshaw and G. Kenney-Wallace, IEEE
J. Quantum Electron., 1988,QE–24, 443; (b) S. J. Rosenthal, N. F.Scherer, M. Cho, X. Xie, M. E. Schmidt and G. R. Fleming, inUltrafast Phenomena, ed. J.-L. Martin, A. Migus, G. A. Mourouand A. H. Zewail, Springer, Berlin, 1993; (c) R. Righini, Science,1993, 262, 1386.
15 (a) S. Palese, L. Schilling, R. J. D. Miller, P. R. Staver and W. T.Lotshaw, J. Phys. Chem., 1994, 98, 6308; (b) E. W. Castner, Jr, Y.J. Chang, Y. C. Chu and G. E. Walrafen, J. Chem. Phys., 1995,102, 653; (c) K. Winkler, J. Lindner, H. Bursing and P. Vohringer,J. Chem. Phys., 2000, 113, 4674; (d) C. J. Fecko, J. D. Eaves andA. Tokmakoff, J. Chem. Phys., 2002, 117, 1139.
16 (a) S. Saito and I. Ohmine, J. Chem.Phys., 1997, 106, 4889; (b)I. Ohmine and S. Saito, Acc. Chem. Res., 1999, 32, 741.
17 B. D. Bursulaya and H. J. Kim, J. Chem. Phys., 1998, 109, 4911.18 A. Perry, H. Ahlborn, B. Space and P. B. Moore, J. Chem. Phys.,
2003, 118, 8411.19 M. A. Ricci, G. Ruocco and M. Sampoli,Mol. Phys., 1989, 67, 19.20 M. S. Skaf and S. M. Vechi, J. Chem. Phys., 2003, 119, 2181.21 H. Torii, Chem. Phys. Lett., 2003, 353, 431.22 B. J. Berne and R. Pecora, Dynamic Light Scattering, Dover,
Mineola, 2000.23 S. Mukamel, Principles of Nonlinear Optical Spectroscopy, Oxford
University Press, New York, 1999.
24 (a) L. C. Geiger and B. M. Ladanyi, J. Chem. Phys., 1987, 87, 191;(b) L. C. Geiger and B. M. Ladanyi, Chem. Phys. Lett., 1989, 159,413; (c) B. M. Ladanyi, Chem. Phys. Lett., 1985, 121, 351; (d) P. A.Madden, in Ultrafast Phenomena, ed. D. H. Auston and K. B.Eisenthal, Springer, Berlin, 1985, vol. IV, p. 244; (e) T. L. C.Jansen, J. G. Snijders and K. Duppen, J. Chem. Phys., 2001, 114,10910; (f) K. Kiyohara, K. Kamada and K. Ohta, J. Chem. Phys.,2000, 112, 6338; (g) H. Stassen, Th. Dorfmuller and B. M.Ladanyi, J. Chem. Phys., 1994, 100, 6318; (h) O. FaurskovNielsen, Annu. Rep. Prog. Chem., Sect. C, 1993, 90, 3.
25 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A.Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr.,R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D.Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V.Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C.Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala,Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Ragha-vachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul,B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R.Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M.W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M.Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98(Revision A.7), Gaussian, Inc., Pittsburgh, PA, 1998.
26 A. V. Gubskaya and P. G. Kusalik, Mol. Phys., 2001, 99, 1107.27 (a) G. Maroulis, Chem. Phys. Lett., 1998, 289, 403; (b) G.
Maroulis, J. Chem. Phys., 2000, 113, 1813.28 (a) J. Kongsted, A. Osted, K. V. Mikkelsen and O. Christiansen, J.
Chem. Phys., 2003, 119, 10519; (b) J. Kongsted, A. Osted, K. V.Mikkelsen and O. Christiansen, J. Chem. Phys., 2004, 120, 3787.
29 H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys.Chem., 1987, 91, 6269.
30 (a) S. W. de Leeuw, J. M. Perram and E. R. Smith, Annu. Rev.Phys. Chem., 1986, 37, 245; (b) S. W. de Leeuw, J. M. Perram andE. R. Smith, Proc. R. Soc. London, Ser. A, 1980, 373, 27; (c) S. W.de Leeuw, J. M. Perram and E. R. Smith, Proc. R. Soc. London,Ser. A, 1980, 373, 57.
31 J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput.Phys., 1977, 23, 327.
32 (a) M. P. Allen and D. J. Tildesley, Computer Simulation ofLiquids, Clarendon, Oxford, 1989; (b) R. W. Hockney, MethodsComput. Phys., 1970, 9, 136.
33 P. P. Wiewior, H. Shirota and E. W. Castner Jr, J. Chem. Phys.,2002, 116, 4643.
34 R. Torre, P. Bartolini and R. Righini, Nature, 2004, 428, 296.35 (a) D. M. F. Edwards and P. A. Madden, Mol. Phys., 1984, 51,
1163; (b) M. S. Skaf, T. Fonseca and B. M. Ladanyi, J. Chem.Phys., 1993, 98, 8929.
36 B. M. Ladanyi and Y. Q. Liang, J. Chem. Phys., 1995, 103, 6325.37 M. Cho, M. Du, N. F. Scherer, G. R. Fleming and S. Mukamel,
J. Chem. Phys., 1993, 99, 2410.38 B. M. Ladanyi and S. Klein, J. Chem. Phys., 1996, 105, 1552.39 Y. J. Chang and E. W. Castner Jr, J. Chem. Phys., 1993, 99, 7289.40 B. D. Bursulaya and H. J. Kim, J. Phys. Chem. B, 1997, 101,
10994.41 J. Borysow, M. Moraldi and L. Frommhold,Mol. Phys., 1985, 56,
913.
P h y s . C h e m . C h e m . P h y s . , 2 0 0 5 , 7 , 1 1 7 6 – 1 1 8 0 T h i s j o u r n a l i s & T h e O w n e r S o c i e t i e s 2 0 0 5
93

Optical Kerr Effect in Supercooled Water
Munir S. Skaf* and Milton T. SonodaInstitute of Chemistry, State University of Campinas, Cedex P. 6154, Campinas-SP, 13084-971, Brazil
(Received 7 July 2004; published 7 April 2005)
We present molecular dynamics simulations of the optical Kerr effect in liquid and supercooled waterand compare with recent time-resolved Kerr spectroscopy measurements [R. Torre et al., Nature (London)428, 296 (2004)]. The short time features of the Kerr response, characterized by peaks near 15, 60, and160 fs, are weakly temperature dependent. The long-time decay is well described by a stretched exponen-tial with a nearly constant stretch parameter and relaxation times that follow a power law ��T�TS���,with TS � 198:3 K and � � 2:35. Our findings are discussed in the light of the spectroscopy data andprevious simulation analyzes of the structural relaxation in supercooled water.
DOI: 10.1103/PhysRevLett.94.137802 PACS numbers: 61.20.Lc, 61.20.Ja, 82.53.-k
There is a large body of experimental measurementsshowing that supercooled water exhibits critical behaviortypical of fragile glass forming liquids [1]. The tempera-ture dependence of several properties diverges as a powerlaw ��T � TS��x for T not too close to TS. A variety ofexperimental estimates places the singularity temperaturein the range TS � 220–230 K, slightly below the homoge-neous nucleation temperature (235 K), and exponents thatdepend on whether the measurement refers to a thermo-dynamic (x� 0:02–0:35) or a transport property (x�1:5–2:5) [2–4]. The nature of this critical behavior, theaqueous glass transition, and the putative relationship be-tween structural, thermodynamic, and dynamic super-cooled anomalies are subjects of intense research activitycurrently [5–11].
Molecular dynamics (MD) simulations and experimen-tal works have been reported in recent years which suc-cessfully interpret the dynamics of weakly supercooledwater (T * 250 K) within the framework of the modecoupling theory (MCT) of ordinary glass forming liquids[12–14]. The underlying physical picture of the MCT isthat the slowing down of the dynamics of supercooledstates arises from density fluctuation modes over lengthscales comparable to the distance between nearest neigh-bors (transient caging effects). In short, the main pre-dictions of the MCT are: (1) The long-time decay (�relaxation) of dynamical correlations follows a stretchedexponential form,
F�t� � A exp���t=����; (1)
with a temperature independent stretch exponent �;(2) The characteristic relaxation time exhibits a powerlaw temperature dependence of the type,
��T� � �0
�TTS
� 1���
; (2)
divergent at some temperature related to the liquid’s struc-tural arrest.
Through extensive MD simulations on the extendedsimple point charge (SPC/E) potential, Sciortino, Stanley,and co-workers investigated the time-dependent structuralcorrelations of supercooled water over a wide range oftemperatures [15], the diffusivity along different isobars[16], and the spatially heterogeneous dynamics [17] inconnection to the Adam-Gibbs theory of supercooled dy-namics [3]. The structural relaxation in supercooled statesshows two time separated processes [15], the slowest ofwhich is identified with the � relaxation. In this regime, therelaxation is well described by Eq. (1) in which the wavevector dependence of the parameters A, �, and � are nicelycorrelated with the oscillations in the static structure factor,in agreement with the MCT predictions. The first experi-mental confirmation of these MD predictions were ob-tained by Bellissent-Funel and co-workers through time-domain neutron spin-echo spectroscopy, although only twosupercooled temperatures were studied [18]. Other experi-mental measurements based on time-resolved ultrafastlaser techniques, such as time-domain terahertz (THz)and Raman induced optical Kerr effect (OKE) spectros-copies, have been reported in recent years for varioustemperatures in the range 270–370 K. The analysis andinterpretation of the observed dynamics differ: The THzdynamics [19] has been characterized as an intermolecularstructural relaxation with a singular point estimated at228 K, whereas the OKE relaxation was interpreted assingle particle diffusive reorientational motions [20].
In contrast, Torre et al. [21] have very recently reportednew state-of-the-art OKE measurements, which fully sup-port the scenario predicted by the MCT. The observedOKE relaxation rates are notably consistent with theMCT structural relaxation behavior: Stretched exponentialdecay with � � 0:6, independently of temperature, and apower law dependence for the relaxation time with TS �221 5 K and � � 2:2 0:3.
In this Letter, we report MD simulations for the OKEresponse of liquid and weakly supercooled water andcompare the results directly to the most recent experimen-tal Kerr study [21]. We find that the short time features of
PRL 94, 137802 (2005) P H Y S I C A L R E V I E W L E T T E R S week ending8 APRIL 2005
0031-9007=05=94(13)=137802(4)$23.00 137802-1 2005 The American Physical Society
95

the Kerr response have a limited temperature dependence,whereas the behavior of the long-time relaxation is welldescribed by Eqs. (1) and (2), with a nearly constant stretchparameter �� 0:6, TS � 198:3 K, and � � 2:35, in verygood agreement with experiments. Our simulations wereperformed in the NVE ensemble on systems containingN � 500 SPC=E [22] water molecules at a fixed density of1 g=cm3 and several (average) temperatures in the range215–323 K. We use a cutoff at half the box length forLennard-Jones forces and Ewald sums for Coulombinteractions.
The dynamics of interest here is the relaxation of thepolarizability anisotropy expressed by the time correlationfunction (tcf) of the off-diagonal elements of the collectivepolarizability tensor [23]:
��t� � h�xz�t��xz�0�i=�N�2=15�; (3)
with �2 � 1=2���1 � �2�2 ��1 � �3�2 ��2 � �3�2�being the square of the ideal polarizability anisotropyand �j the principal polarizability components. The col-lective polarizability tensor, � � �M �I, is comprisedby the sum of gas phase molecular polarizabilities andinduced polarizability contributions computed accordingto [24]:
�I � XNi
Xj�i
��Mi � Tij � �j �M
i � Tij � �j�: (4)
The enhanced polarizability tensor and dipole vectors areobtained by iteratively solving the equations:
� i � �Mi X
j�i
�Mi � Tij � �j; (5)
� i � �Mi X
j�i
�Mi � Tij � �j; (6)
where �Mi , �M
i , and �Mi are the gas phase dipole, polar-
izability, and hyperpolarizability of molecule i, respec-tively. Tij � rirjjri � rjj�1 is the dipole tensor betweenmolecules i and j. The molecular �M and �M tensorsneeded in this scheme were computed from ab initio quan-tum chemical calculations [25] and are shown in Table I.The hyperpolarizability contributions are very small in
liquid water, but some degree of nonlinear electronic re-sponse is incorporated through the ‘‘�T�’’ term. Theoptically heterodyne-detected OKE signal obtained fromthe time-resolved spectroscopy provides the intermolecu-lar dynamics through the nuclear response function, givenby the time derivative of the polarizability anisotropy tcf[26,27]:
Rnuc�t� � ���t�kBT
@��t�@t
; (7)
where ��t� is the Heaviside step function.We start by discussing the short time behavior of the
OKE relaxation, shown in Fig. 1(a) for several tempera-tures. The response functions show a fast rise to a maxi-mum at �15 fs, followed by a satellite peak near 60 fs anda third, broader, local maximum at 160 fs. The remainingdecay is comprised by slowly relaxing components. Theshort time features are in agreement with previous simu-lation results for near ambient conditions [28,29] andexperimental OKE signals [30,31], which report peaksnear 20, 60, and 200 fs. The first peak is mainly due toinertial and librational motions of the Hydrogen atoms,while the peaks at 60 and 200 fs have been ascribed to a
TABLE I. Molecular polarizability and first hyperpolarizabil-ity tensors obtained from restricted Hartree-Fock electronicstructure calculations at the MP2=6� 311G�d; p� level.The z and y directions are along the main symmetry axis andnormal to the molecular plane, respectively. �y33 is smaller than0:05 �A=e and the remainders are zero.
�M11 �M
12 �M22 �M
33
� �A3� 1.04 � � � 1.00 1.17
�Mx11 �M
x13 �Mx22 �M
y23 �Mz33
� �A5=e� 0.80 -0.19 0.06 0.05 0.86
0 0.1 0.2 0.3 0.4
t / ps
-1
0
1
2
3
4
5
TotalInducedCrossIntrinsic
0 0.1 0.2 0.3 0.4
0
1
2
3
4
5
100
xk B
Rnu
c (t)
/ K-1
ps-1
323 K270 K250 K215 K
(a)
(b)
FIG. 1. (a) Simulated Kerr nuclear responses for differenttemperatures. The effect of T on the short time features isrestricted to a sharpening of the peaks without affecting theirpositions. (b) Total and separate contributions to the Kerr re-sponse at ambient conditions. For comparison, the intrinsic andinduced contributions at 250 K are also shown (dotted lines).
PRL 94, 137802 (2005) P H Y S I C A L R E V I E W L E T T E R S week ending8 APRIL 2005
137802-2
96

superposition of librational and hindered translationalmodes within the cage of neighbors [31]. This picture issupported by the behavior of the separate contributions toRnuc depicted in Fig. 1(b). The intrinsic part is mainlyresponsible for the rapid rise of the response, whereas thecollision induced contribution accounts for most of thelibrational and hindered translation peaks. In frequencydomain, such translational motions appear at �60 and�180 cm�1 [31,32]. The temperature dependence of theshort time oscillations in Rnuc�t� [Fig. 1(a)] is limited to anenhancement in the definition of the translational peaks,which are sharper at lower temperatures. Essentially nochanges in the peak positions are observed, indicating thatthe underlying ‘‘intracage’’ motions are not affected by T.This trend is very consistent with the experimental Kerrsignals obtained by Vohringer and co-workers [20] and theearlier light scattering spectra by Sokolov et al. [33].
The slow relaxation of the response is best seen from thepolarizability anisotropy tcf itself. The normalized tcfswere computed for times up to 12 ps at several differenttemperatures and are shown in Fig. 2 (symbols). The ��t�tcfs are very well described by stretched exponentials(lines) and our best fit parameters are listed for eachtemperature, accordingly. The stretch parameter � � 0:6,independent of T within the range of temperatures consid-ered, is in very good agreement with the OKE experiments[21]. Other studies, theoretical and experimental, of thestructural relaxation of supercooled water based on theframework of MCT report very similar values of �[15,18]. The time scale of the simulated OKE relaxationcorresponds to the early part of the structural � relaxationand is roughly 2 orders of magnitude smaller than the timescales involved in previous studies of the intermediatescattering functions and diffusion constants. This is an
important feature, not only because of the shorter MDruns that are required here, but mainly because it hasenabled experimental observation [21] of the structuralrelaxation through a technique that works best for timesshorter than a few tens of picoseconds due to the nearlyisotropic polarizability of the water molecule, whichrenders OKE signals of very low intensity.
The relaxation time � is strongly temperature dependentand spans a range substantially larger than the correspond-ing experimental range, which goes from 0.22 ps at 314 Kto 2.20 ps at 254 K [21]. Nevertheless, the temperaturedependence of � is very well reproduced by the power lawof Eq. (2), with �0 � 0:242 ps, TS � 198:3 K, and � �2:35, as shown in Fig. 3. These results are in good agree-ment with the time-resolved Kerr experiments (Texp:
S �221 5 K and �exp: � 2:2 0:3). The experimental char-acterisitc time is �exp:0 � 0:03 ps. The TS obtained from thesimulations is �22 K lower than the experimental esti-mate. This shift is comparable to the maximum densitytemperature difference ( � 35 K) between experimentsand MD simulations using the SPC/E model under similarconditions (P � 0 MPa). Our results for TS and � agreeremarkably well with the MD results (199 K, � � 2:73)reported by Sciortino et al. [15] from the temperaturedependence of the incoherent scattering function andmean square displacement. Our findings therefore provideunambiguous support to the interpretation of the Kerrexperiments in terms of an underlying dynamics that isclosely tied to the liquid’s structural relaxation.
In summary, we have reported for the first time an MDsimulation analysis of the optical Kerr effect relaxation forweakly supercooled water and compared directly to veryrecent time-resolved Kerr spectroscopy measurements.The short time dynamics is quite insensitive to cooling,whereas the long-time relaxation follows the behaviorpredicted by the MCT in the temperature range considered,
0 2 4 6 8t / ps
0
0.5
1
Ψ(t
) / Ψ
(0)
215 81.55 0.62
220 40.34 0.56
225 28.05 0.64
230 17.12 0.62
240 10.71 0.64
245 7.19 0.62
250 5.36 0.65
260 4.67 0.60
270 2.64 0.61
298 1.00 0.59
323 0.65 0.68
T / K τ / ps β
FIG. 2. Collective polarizability anisotropy relaxation forSPC/E water at � � 1:0 g=cm3 and different temperatures (sym-bols). The lines are exp���t=���� fits to the data (t > 0:2 ps) andthe best-fitting parameters are tabulated.
200 220 240 260 280 300 320 340
T / K
0
20
40
60
80
100
τ / p
s
200 220 240 260 280 300 320
T / K
0
0.2
0.4
0.6
0.8
(τ /
τ 0)−1/γ
τ(T) = τ0 (T/T
S - 1)
-γ
TS = 198.3 K γ = 2.35
τ0 = 0.24 ps
FIG. 3. Temperature dependence of the Kerr relaxation time(symbols). The lines correspond to the power law fit to ��T� withTS � 198:3 K and � � 2:35.
PRL 94, 137802 (2005) P H Y S I C A L R E V I E W L E T T E R S week ending8 APRIL 2005
137802-3
97

in a remarkable agreement with the experimental observa-tion. The characteristic parameters �, TS, and � we obtainagree well with the parameters obtained from other de-scriptors of the structural relaxation in supercooled water,thus demonstrating that MCT is a useful framework toanalyze the temperature dependence of the post-librationalrelaxation as probed by time-domain ultrafast spectros-copies. The close relationship between the liquid’s re-sponses obtained from the optically heterodyne-detectedOKE signal and the solvation responses extracted from thewell-known time-resolved fluorescence spectroscopy tech-nique, opens the interesting possibility of investigating thestructural relaxation of supercooled water and other liquidsby means of fluorescence up-conversion experiments usingsoluble dyes as spectroscopic probes.
This work is supported by the Brazilian agenciesFAPESP (01/09374-3 and 03/09361-4) and CNPq. Wethank Professor Renato Torre for sending us Ref. [21].
*Corresponding author.E-mail: [email protected]
[1] See references [2–4] for comprehensive reviews on theproperties of supercooled and glassy states of water.
[2] A. Angell, Annu. Rev. Phys. Chem. 55, 559 (2004); 34,593 (1983); Chem. Rev. 102, 2627 (2002).
[3] P. G. Debenedetti, Metastable Liquids. Concepts andPrinciples (Princeton University Press, Princeton, 1996).
[4] P. G. Debenedetti, J. Phys. Condens. Matter 15, R1669(2003); See also the special issue edited by A. K. Soperand P. J. Rossky [Chem. Phys. 258, (2000)].
[5] P. G. Debenedetti and F. H. Stillinger, Nature (London)410, 259 (2001).
[6] P. G. Debenedetti and H. E. Stanley, Phys. Today 56, 6, 40(2003).
[7] M. S. Shell, P. G. Debenedetti, and A. Z. Panagiotopoulos,Phys. Rev. Lett. 92, 035506 (2004); Phys. Rev. Lett. 92,169902 (2004).
[8] S. Whitelam, L. Berthier, and J. P. Garrahan, Phys. Rev.Lett. 92, 185705 (2004).
[9] J. P. Garrahan and D. Chandler, Phys. Rev. Lett. 89,035704 (2002); Proc. Natl. Acad. Sci. U.S.A. 100, 9710(2003).
[10] X. Xia and P. G. Wolynes, Proc. Natl. Acad. Sci. U.S.A.97, 2990 (2000).
[11] F. W. Starr et al., Biophys. Chem. 105, 573 (2003).[12] W. Gotze and L. Sjogren, Rep. Prog. Phys. 55, 241
(1992).[13] L. Fabbian et al., Phys. Rev. E 60, 5768 (1999).[14] As T ! TS, the dynamics depart from the MCT behavior.
The reasons why are not fully understood.[15] P. Gallo et al., Phys. Rev. Lett. 76, 2730 (1996);
F. Sciortino et al., Phys. Rev. E 54, 6331 (1996); 56,5397 (1997).
[16] F. W. Starr et al., Phys. Rev. Lett. 82, 3629 (1999).[17] N. Giovambattista et al., Phys. Rev. Lett. 90, 085506
(2003); A. Scala et al. Nature (London) 406, 166 (2000).[18] M. C. Bellissent-Funel et al., Phys. Rev. Lett. 85, 3644
(2000).[19] C. Ronne, P.-O. Astrand, and S. R. Keiding, Phys. Rev.
Lett. 82, 2888 (1999).[20] K. Winkler, J. Lindner, H. Bursing, and P. Vohringer,
J. Chem. Phys. 113, 4674 (2000).[21] R. Torre, P. Bartolini, and R. Righini, Nature (London)
428, 296 (2004).[22] H. J. C. Berendsen, J. R. Grigera, and T. P. Straatsma,
J. Phys. Chem. 91, 6269 (1987).[23] S. Mukamel, Principles of Nonlinear Optical
Spectroscopy (Oxford University Press, New York, 1999).[24] H. Torii, Chem. Phys. Lett. 353, 431 (2002).[25] M. J. Frisch et al., GAUSSIAN 98 (Gaussian, Inc., Pittsburgh,
PA, 1998), Revision A.7.[26] D. McMorrow, W. T. Lotshaw, and G. Kenney-Wallace,
IEEE J. Quantum Electron. 24, 443 (1988); S. J. Rosenthalet al., in Ultrafast Phenomena, edited by J.-L. Martin,A. Migus, G. A. Mourou, and A. H. Zewail (Springer,Berlin, 1993); R. Righini, Science 262, 1386 (1993).
[27] M. Cho et al., J. Chem. Phys. 99, 2410 (1993).[28] S. Saito and I. Ohmine, J. Chem. Phys. 106, 4889 (1997);
I. Ohmine and S. Saito, Acc. Chem. Res. 32, 741 (1999).[29] B. D. Bursulaya and H. J. Kim, J. Chem. Phys. 109, 4911
(1998); J. Phys. Chem. B 101, 10994 (1997).[30] S. Palese et al., J. Phys. Chem. 98, 6308 (1994).[31] E. W. Castner, Jr. et al., J. Chem. Phys. 102, 653 (1995).[32] K. Mizoguchi, Y. Hori, and Y. Tominaga, J. Chem. Phys.
97, 1961 (1992).[33] A. P. Sokolov, J. Hurst, and D. Quitmann, Phys. Rev. B 51,
12865 (1995).
PRL 94, 137802 (2005) P H Y S I C A L R E V I E W L E T T E R S week ending8 APRIL 2005
137802-4
98

Capıtulo 8
Conclus ao Geral
Neste trabalho foram estudados vários sistemas distintos, focando diversas propriedades.
Estudamos as propriedades estruturais, dinâmicas e dielétricas em função da concen-
tração de soluções aquosas de frutose, onde foi encontrada a formação de aglomerados
de soluto nos sistemas mais concentrados. Foi observada a existência de cavidades no
interior destes aglomerados onde as moléculas de água ficam confinadas.
Comparamos os efeitos da solvatação implícita e explícita sobre as propriedades es-
truturais e dinâmicas de uma dupla hélice de DNA. Neste trabalho conclui-se que a rep-
resentação implícita do solvente descreve bem a estrutura e a dinâmica rápida do DNA.
No entanto, apesar de ser muito menos custoso computacionalmente que a simulação
com representação explícita, devido à ausência dos efeitos de dissipação, a dinâmica de
tempos longos não é bem representada na solvatação implícita.
Estudamos a estrutura, dinâmica e a solvatação de uma cumarina adsorvida na inter-
face água-ar na presença de surfactante aniônico. Neste trabalho observou-se a aglomer-
ação dos surfactantes, formando regiões de alta densidade interfacial. Observou-se ainda
a existência de dois estados de solvatação preferencial de sonda na interface com pro-
priedades estruturais e dinâmicas bastante distintas: um estado onde a sonda permanece
fora ou adjacente ao aglomerado de surfactante, e outro onde a sonda permanece no
interior do aglomerado.
Simulamos o efeito Kerr óptico da água líquida e no estado superfrio, comparando
com diversos resultados experimentais e de simulação. No caso da simulação da água
no estado superfrio em várias temperaturas, observa-se que a sua relaxação estrutural
é descrita por uma exponencial estiradaf(t) ≈ a exp[−(t/τ)β], com parâmetro de es-
99

100 Capıtulo 8. Conclusao Geral
tiramentoβ praticamente independente da temperaturaT e com o tempo de relaxação
τ dependendo da temperatura por uma lei de potências do tipoτ(T ) = a (T − TS)−γ.
Estas observações estão em excelente acordo com resultados experimentais recentes e
com o cenário descrito pela Teoria de Acoplamentos de Modos, utilizada para descrever
líquidos simples.
A quantidade e diversidade de sistemas e de propriedades estudadas foi um ponto
extremamente positivo deste trabalho, me dando muita experiência em simulação de
DM e na sua análise, me dando uma formação bastante diversificada. Em todos eles
procurou-se, com bastante êxito, diria, comparar os resultados deste trabalho com resul-
tados experimentais. A escolha por alguns sistemas de interesse biológico faz parte de
uma trilha pessoal que venho seguindo desde o começo do meu mestrado e alguns dos
trabalhos apresentados nesta Tese representam um passo decisivo nesta direção.

Refer encias Bibliogr aficas
[1] K. Fucks e U. Kaatze. Dielectric spectra of mono- and disaccharide aqueous solu-
tions. Journal of Chemical Physics, 116:7137–7144, 2002.
[2] M. P. Allen e D. Tildesley. Computer Simulation of Liquids. Clarendon Press,
1987.
[3] A. R. Leach.Molecular Modelling: Principles and Applications. Pearson Educa-
tion Limited, segunda edicao edition, 2001.
[4] W. F. van Gunsteren e H. J. C. Berendsen. Computer simulation of molecular
dynamics - methodology, applications, and perspectives in chemistry.Angewandte
Chemie - International Edition in English, 29:992–1023, 1990.
[5] B. J. Alder e T. E. Wainwright. Phase transitions for a hard sphere system.Journal
of Chemical Physics, 27:1208–1209, 1957.
[6] A. Rahman. Correlations in motion of atoms in liquid argon.Physics Review A -
General Physics, 136:A405, 1964.
[7] F. H. Stillinger e A. Rahman. Inproved simulation of liquid water by molecular
dynamics.Journal of Chemical Physics, 60:1545–1557, 1974.
[8] J. A. McCammon and B. R. Gelin e M. Karplus. Dynamics of folded proteins.
Nature, 267:585–590, 1977.
[9] B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, and S. Swaminathan
e M. Karplus. Charmm: A program for macromolecular energy, minimization,
and dynamics calculations.Journal of Computational Chemistry, 4:187–217,
http://yuri.harvard.edu/ 1983.
101

102 Referencias Bibliograficas
[10] W. L. Jorgensen e J. Tirado-Rives. The opls potential functions for proteins. energy
minimizations for crystals of cyclic peptides and crambin.Journal of the American
Chemical Society, 110:1657–1666, 1988.
[11] W. L. Jorgensen and D. S. Maxwell e Julian Tirado-Rives. Development and test-
ing of the opls all-atom force field on conformational energetics and properties
of organic liquids.Journal of the American Chemical Society, 118:11225–11236,
1996.
[12] D. A. Pearlman, D. A. Case, J. W. Caldwell, W. R. Ross, T. E. Cheatham III,
S. DeBolt, D. Ferguson, and G. Seibel e P. Kollman. Amber, a computer program
for applying molecular mechanics, normal mode analysis, molecular dynamics and
free energy calculations to elucidate the structures and energies of molecules.Com-
puter Physics Communications, 91:1–41, 1995.
[13] C. A. Angell. Liquid fragility and the glass transition in water and aqueous solu-
tions. Chemical Reviews, 102:2627–2650, 2002.
[14] P. Sears e C. Wong. Carbohydrate mimetics: A new strategy for tackling the prob-
lem of carbohydrate-mediated biological recognition.Angewandte Chemie Inter-
national Edition, 38:2300–2324, 1999.
[15] G. W. Padua e S. J. Schmidt. Proton nuclear magnetic resonance measurements
on various sugar solutions.Journal of Agricuture Food Chemistry, 40:1524–1527,
1992.
[16] T. Mahawanich e S. J. Schmidt. Molecular mobility and the perceived sweetness
of sucrose, fructose, and glucose soluctions.Food Chemistry, 84:169–179, 2004.
[17] M. Rampp and C. Buttersack e H. Lüdemann. c,t-dependence of the viscosity and
the self-diffusion coefficients in some aqueous carbohydrate solutions.Carbohy-
drate Research, 328:561–572, 2000.
[18] K. Fucks e U. Kaatze. Molecular dynamics of carbohydrate aqueous solutions.
dielectric relaxation as a function of glucose and fructose concentration.Journal
of Physical Chemistry B, 105:2036–2042, 2001.

Referencias Bibliograficas 103
[19] M. Feeney, C. Brown, A. Tsai, and D. Neumann e P. G. Debenedetti. Incoherent
quasi-elastic neutron scattering from frutose-water solutions.Journal of Physical
Chemistry B, 105:7799–7804, 2001.
[20] C. J. Roberts e P. G. Debenedetti. Structure and dynamics in concentrated, amor-
phous carbohydrate-water system by molecular dynamics simulation.Journal of
Physical Chemistry B, 103:7308–7318, 1999.
[21] S. L. Lee and P. G. Debenedetti e J. R. Errington. A computational study of hy-
dration, solution structure, dynamics in dilute carbohydrate solutions.Journal of
Chemical Physics, 122:204511, 2005.
[22] W. Damm, A. Frontera, and J. Tirado-Rives e W. L. Jorgensen. Opls all-atom force
field for carbohydrates.Jounal of Computational Chemistry, 18:1955–1970, 1997.
[23] D. van der Spoel and P. J. van Maaren e H. J. C. Berendsen. A systematic study of
water models for molecular simulation: Derivation of water models optimized for
use with a reaction field.Journal of Chemical Physics, 108:10220–10230, 1998.
[24] H. J. C. Berendsen and D. van der Spoel e R. van Drunen. Gromacs: A message-
passing parallel molecular dynamics implementation.Computer Physics Commu-
nications, 91:43–56, www.gromacs.org 1995.
[25] E. Lindahl and B. Hess e D. van der Spoel. Gromacs 3.0: a package for molecular
simulation and trajectory analysis.Journal of Molecular Moldeling, 7:306–317,
2001.
[26] J. W. Ponder.http://dasher.wustl.edu/tinker/.
[27] D. A. McQuarrie.Statistical Mechanics. University Science Books, 2000.
[28] G. Löffler and H. Schreiber e O. Steinhauser. Calculation of the dielectric prop-
erties of a protein and its solvent: Theory and a case study.Journal of Molecular
Biology, 270:520–534, 1997.
[29] P. Madden e D. Kivelson. A consistent molecular treatment of dielectric phenom-
ena.Advances in Chemical Physics, 56:467–566, 1984.

104 Referencias Bibliograficas
[30] P. Höchtl and S. Boresh e O. Steinhauser. Dielectric properties of glucose and
maltose solutions.Journal of Chemical Physics, 112:9810–9821, 2000.
[31] W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, and R. W. Impey e M. L. Klein.
Comparison of simple potential functions for simulating liquid water.Journal of
Chemical Physics, 79:926–935, 1983.
[32] B. M. Ladanyi e M. S. Skaf. Computer simulation of hydrogen-bonding liquids.
Annual Review of Physical Chemistry, 44:335–368, 1993.
[33] B. M. Ladanyi e M. S. Skaf. Wave vector-dependent dielectric relaxation of
methanol-water mixtures.Journal of Physical Chemistry, 100:1368–1380, 1996.
[34] D. Kivelson e H. L. Friedman.Journal of Physical Chemistry, 93:7026, 1989.
[35] B. Guillot. A molecular dynamics study of the far infrared spectrum of liquid
water.Journal of Chemical Physics, 95:1543–1551, 1991.
[36] E. B. Brauns, M. L. Madaras, R. S. Coleman, and C. J. Murphy e M. A. Berg.
Complex local dynamics in dna on the picosecond and nanosecond time scales.
Physical Review Letters, 88:158101–4, 2002.
[37] H. Gronemeyer and J. Gustafsson e V. Laudet. Principles for modulation of the
nuclear receptor superfamily.Nature, 3:950–964, 2004.
[38] E. A. Carter e J. T. Hynes. Solvation dynamics for an ion pair in a polar solvent:
Time-dependent fluorescence and photochemical charge transfer.Jounal of Chem-
ical Physics, 94:5961–5979, 1991.
[39] D. Bashford e D. A. Case. Generalized born models of macromolecular solvation
effects.Annual Review of Physical Chemistry, 51:129–152, 2000.
[40] G. D. Hawkins and C. J. Cramer e D. G. Thrular. Pairwise solute descreening of
solute charges from a dielectric medium.Chemical Physics Letters, 246:122–129,
1995.
[41] V. Tsui e D. A. Case. Molecular dynamics simulation of nucleic acids with a gener-
alized born solvation model.Journal of the American Chemical Society, 122:2489–
2498, 2000.

Referencias Bibliograficas 105
[42] D. A. Case and V. Tsui. Theory and applications of the generalized born solvation
model in macromolecular simulations.Biopolymers, 56:275–291, 2000.
[43] J. M. Martínez e L. Martínez. Packing optimization for automated generation of
complex system’s initial configurations for molecular dynamics and docking.Jour-
nal of Computational Chemistry, 24:819–825, http://www.ime.unicamp.br/ mar-
tinez/packmol/ 2003.
[44] W. Saenger.Principles of Nucleic Acid Structure. Spring-Verlag, 1988.
[45] A. D. McLachlan. Gene duplications in the structural evolution of chymotrypsin.
Journal of Molecular Biology, 128:49–79, 1979.
[46] S. Georghiou, T. D. Bradrick, and A. Philippetis e J. M. Beechem. Large-amplitude
picosecond anisotropy decay of the intrinsic fluorescence of double-stranded dna.
Biophysical Journal, 70:1909–1922, 1996.
[47] D. J. Williams e K. B. Hall. Unrestrained stochastic dynamics simulations of the
uucg tetraloop using an implicit solvation model.Biophysical Journal, 76:3192–
3205, 1999.
[48] A. V. Benderskii e K. B. Eisenthal. Dynamical time scales of aqueous solva-
tion at negatively charged lipid/water interfaces.Journal of Physical Chemistry
A, 106:7482–7490, 2002.
[49] J. T. Kindt e C. A. Schmuttenmaer. Far-infrared dielectric properties of polar liq-
uids probed by femtosecond terahertz pulse spectroscopy.Journal of Physical
Chemistry, 100:10373–10379, 1996.
[50] M. Berg e D. A. Vanden Bout. Ultrafast raman echo measurements of vibrational
dephasing and the nature of solvent-solute interactions.Accounts of Chemical
Research, 30:65–71, 1997.
[51] Roberto Righini. Ultrafast optical kerr effect in liquids and solids.Science,
262:1386–1390, 1993.
[52] S. Saito e I. Ohmine. Third order nonlinear response of liquid water.Journal of
Chemical Physics, 106:4889–4893, 1997.

106 Referencias Bibliograficas
[53] B. D. Bursulaya e H. J. Kim. Spectroscopic and dielectric properties of liquid
water: A molecular dynamics simulation study.Journal of Chemical Physics,
109:4911–4919, 1998.
[54] R. Torre and P. Bartolini e R. Righini. Structural relaxation in supercooled water
by time-resolved spectroscopy.Nature, 428:296–299, 2004.
Recommended

















