
Not Knudson’s Retinoblastoma:
One-Hit Cancer Initiated by the MYCN Oncogene?
Diane E Rushlow, Jennifer Y Kennett,* Berber M Mol,* Stephanie Yee,* Sanja Pajovic,
Brigitte L Thériault, Nadia L Prigoda-Lee, Clarellen Spencer, Helen Dimaras, Timothy W
Corson, Renee Pang,Christine Massey, Katherine Paton, Annette C Moll, Claude
Houdayer, Anthony Raizis, William Halliday, Wan L Lam, Paul C Boutros, Dietmar
Lohmann, Josephine C Dorsman, Brenda L Gallie
*These authors share second authorship
Retinoblastoma Solutions and the Toronto Western Hospital Research Institute, Campbell
Family Cancer Research Institute, Princess Margaret Cancer Centre, University Health
Network; Informatics and Biocomputing Platform, Ontario Institute for Cancer Research;
Departments of Hematology/Oncology, Ophthalmology and Visual Science and of
Pathology, Hospital for Sick Children; and Departments of Molecular Genetics,
Ophthalmology, Medical Biophysics, Pathobiology and Lab Medicine, University of Toronto,
Toronto, ON, Canada (D E Rushlow, BSc, S Yee, MSc, S Pajovic, PhD, B L Thériault, PhD, N L
Prigoda-Lee, MSc, C Spencer, BSc, R Pang, MA, C Massey, MSc,H Dimaras, PhD, P C Boutros,
PhD, W Halliday, MD, Prof B L Gallie, MD); British Columbia Cancer Research Centre and
Departments of Ophthalmology and Pathology & Laboratory Medicine, University of British
Columbia, Vancouver, BC, Canada (J Y Kennett, MSc, K Paton, MD, Prof W L Lam, PhD);
Departments of Clinical Genetics, Ophthalmology and Pediatric Oncology/Hematology, VU
University Medical Center Amsterdam, Amsterdam, The Netherlands (B M Mol, MSc, A C
Moll, MD, Prof J C Dorsman, PhD);Eugene and Marilyn Glick Eye Institute, Departments of
Ophthalmology, Biochemistry and Molecular Biology, Indiana University School of Medicine
Indianapolis, Indiana, USA (Timothy W Corson, PhD); Service de Génétique Oncologique,
Institut Curie and Université Paris Descartes Paris, France (C Houdayer, PhD); Department of

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Molecular Pathology, Canterbury Health Laboratories Christchurch, New Zealand (A Raizis,
PhD); and Institut für Humangenetik, Universitätsklinikum, Essen, Germany (Prof D
Lohmann, MD)
Correspondence to Dr Brenda L. Gallie, Campbell Family Cancer Research Institute, Princess Margaret
Cancer Centre, University Health Network, Rm 8-415, 610 University Ave, Toronto, ON, M5G 1M9, Canada,
2

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Word count 299/300
Summary
BackgroundRetinoblastoma is the childhood retinal cancer that defined tumour suppressor genes.
By analysing age of diagnosis, Knudson proposed that two “hits” initiate retinoblastoma, later
attributed to mutation of both alleles of the retinoblastoma suppressor gene, RB1, in tumours.
Persons with hereditary retinoblastoma carry a heterozygous constitutional RB1 mutation; one
additional hit initiates retinoblastoma or other cancers. Non-hereditary retinoblastoma arises when
both RB1 alleles are damaged in developing retina (RB1-/-).
MethodsOur international collaboration determined the proportion of 1054 unilateral non-familial
retinoblastomas with no evidence of RB1 mutations despite high-sensitivity assays. We analysed
clinical data, genomic copy-number changes, histology, immunohistochemistry, and gene
expression, comparing RB1+/+and RB1-/-tumours.
FindingsNo evidence of RB1 mutation (RB1+/+) was found in 2.87% (298/1054) of unilateral non-
familial retinoblastomas. Surprisingly, half of these had high-level MYCN oncogene amplification
(>10 copies), while noRB1-/-tumours showed MYCN amplification. RB1+/+MYCNAtumours had fewer
overall genomic copy-number changes and distinct, aggressive histology. Amplification of the
MYCN-encompassing region was the only change on array comparative genomic hybridization in
one RB1+/+MYCNAtumour. Median age at diagnosis of RB1+/+MYCNAtumours was 4·5 months,
compared to 24 months for non-familial unilateral RB1-/-retinoblastoma. We calculate a 1922%
chance that a child diagnosed less than six months oldwith unilateral non-familial retinoblastoma at
six months of age or less will have an RB1+/+MYCNAtumour.
InterpretationAmplification of the MYCN oncogene may initiate RB1+/+MYCNA retinoblastoma
despite normal RB1 genes. Despite their young age at diagnosis, these children and their families
are believed at population risk to develop other cancers. Since these aggressive tumours may
3

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
rapidly become extra-ocular, removal of the eye of these young children with unilateral non-familial
retinoblastoma is important.
FundingNCI-NIH; CIHR; Canadian Retinoblastoma Society; Hyland Foundation; Ontario Ministry
of Health and Long Term Care; Toronto Netralya and Doctors Lions Clubs; and Foundations
Avanti-STR and KiKa.
4

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Word count 3152/3000
Background
Retinoblastoma set the paradigm for tumour suppressor genes, with Knudson’s classic hypothesis
predicting that two rate-limiting hits initiate this childhood eye cancer.1 The two hits were later
attributed to the retinoblastoma gene (RB1).2 Approximately 40% of retinoblastoma is bilateral. A
child with bilateral retinoblastoma carries a heterozygous, constitutional RB1 mutation and is
predisposed to retinoblastoma; one additional hit, in which the second RB1 allele is damaged,
initiates retinoblastoma, and/ (~90% bilateral) or other cancers later in life. Approximately 60% of
children have unilateral retinoblastoma. Most non-familial unilateral retinoblastomas arise when
both RB1 alleles are damaged in developing retina, but 15% carry a heritable, constitutional RB1
mutation. Accepted dogma is that damage to lossboth RB1 alleles (RB1-/-) is required for
retinoblastoma development.2-4
The heterozygous mutant RB1 allele is identifiable in blood of 95% of bilaterally affected persons.
The undetected RB1 mutations in blood likely include low-level mosaicism,5 translocations, or deep
intronic mutations. RB1 mutations, or promoter methylation, are detected on both alleles (RB1-/-) in
95% of unilateral retinoblastomas.5-7 The possibility that some unilateral retinoblastomas with no
detectableRB1 mutations occur by an independent mechanism has not been previously explored.
We report the first identification of unilateral retinoblastomas with normal RB1 alleles and high-
level MYCN gene amplification (MYCNA). These unilateralRB1+/+MYCNA retinoblastomas are
characterized by distinct histology, fewer of the genomic copy-number changes characteristic of
retinoblastoma, and very early age of diagnosis. This new sub-type of retinoblastoma has immediate
diagnostic, genetic counselling, and therapeutic implications.
5

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Methods (see webappendix for details)
Clinical samples
Tumours, blood, and clinical data were provided for identification of RB1 mutations for clinical
care of children and their families. Research Ethics Board approvals for the use of de-identified data
and tissues, after clinical analyses, are on file at each participating site. Although not required by
local REBs for de-identified use of archival tissue and data, Toronto and Essen patients provided
additional informed consent for use of de-identified samples in research.
Mutation analyses
Standard of care analyses that identify 95% of RB1 (Gen bank accession #L11910) mutant alleles5-7
were applied to tumours at each collaborating site, including DNA sequencing, quantitative
multiplex PCR (QM-PCR) or Multiplex Ligation-Dependent Probe Amplification (MLPA), RB1
promoter methylation testing, and the use of intragenic and closely-linked RB1microsatellite
markers to determine zygosity of the RB1+/+ tumours.
Genome copy-number analyses
Genomic copy-numbers of five genes were determined by QM-PCR (Toronto) (table S1, figure S1),
or MLPA and single nucleotide polymorphism (SNP) analyses (Amsterdam). Sub-megabase
resolution array comparative genomic hybridization (aCGH) or SNP array were used to assess
overall genomic copy-numbers.
6

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Protein expression studies
Paraffin-embedded sections of retinoblastomas and adjacent normal retinas were stained for full-
length pRB protein (antibodies targeting N- and C-terminal pRB) and N-Myc.8Western blots were
performed on RB1+/+MYCNA, RB1-/-and control cell lines.
RNA gene expression studies
Expression of RB1, MYCN, and genes reflecting the retinal derivation and proliferative status of the
tumours were assessed using End-Point Reverse Transcriptase PCR (RT-PCR) and/or Quantitative
Real-Time PCR (tables S2, S3).
Age of Diagnosis analysis
Ages at diagnosis vs. proportion not yet diagnosed were analysed by Weibull distributions and least
squares methodology to assess the minimal number of events for tumour initiation. Likelihood of
children having RB1+/+MYCNAtumours at different ages of diagnosis was estimated (table S4).
Role of the funding sources
The sponsors of the study had no role in study design, data collection, data analysis, data
interpretation, or writing of the manuscript. No author was paid to write this article. All authors had
full access to all data in the study; the corresponding author (BLG) had final responsibility for the
decision to submit for publication. NCI-NIH grant 5R01CA118830-05 supported the early
discovery at the Canadian site. Canadian Institutes for Health Research grants (MOP-86731, MOP-
77903, MOP-110949) supported the aCGH studies. The Canadian Retinoblastoma Society, Hyland
Foundation and Toronto Netralya and Doctors Lions Clubs provided critical funding for additional
experiments. The Ontario Ministry of Health and Long Term Care provided infrastructure.
Solutions by Sequence supported the overall project, data analysis and manuscript preparation. The
Dutch study was made possible by Avanti-STR and KiKa, while VUmc provided infrastructure.
7

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Results
The Toronto lab identifies both tumour mutations (or promoter methylation) in >95% of tumours
(as of October 10, 2012, 616 of 642, 96.0%) from unilateral probands with no known family
history. In 3-4% an RB1 mutation is identified on only one allele, and in 1.6% of tumours, no RB1
mutation is found. Low-level mosaicism is believed to account for many of the 5% "no mutation
found" blood samples from bilaterally affected patients.5 However, RB1 sequence, sensitive allele-
specific PCR screens, and microsatellite analysis make it clear that any level of mutational
mosaicism or significant normal cell contamination in retinoblastoma tumours is rare, and if noted,
is usually associated with extremely small tumours, or chemotherapy prior to enucleation. The
undetected RB1 mutations in fully tested tumours (RB1+/+ ) were believed to include translocations,
deep intronic mutations, or mutations in unknown RB1 regulatory regions.
In our clinical work in Toronto, we (DER, BLG) had found seven unilateral retinoblastoma samples
with no RB1 mutations and no loss of heterozygosity (LOH) at RB1. We investigated these tumours
further, using QM-PCR to measure copy-numbers of representative genes in 6p, 1q, 16q and 2p,
characteristically gained or lost in retinoblastoma. To our surprise, 5/7 tumours showed dramatic
MYCN oncogene amplification (MYCNA ). To validate this observation, we collaborated with RB1
clinical laboratories in Germany, France, and New Zealand, and collected DNA from RB1+/+
tumours, tested in these centres, that had no detectable RB1 mutations, no RB1 promoter
methylation, and no LOH at RB1 (table 1). The proportions of RB1+/+ and RB1+/+ MYCNA tumours
from each of the four centres was similar. After our study was complete, we discovered that an
Amsterdam lab had independently characterized three RB1+/+ MYCNA tumours. We include their
data, and data from an additional Toronto RB1+/+ MYCNA tumour (T101) found after the initial study,
in this report.
8

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Clinical assays in our labs establish a standard of 95% sensitivity to find an RB1 mutation in
samples expected to carry an RB1 mutation..5-7 The probability of finding no RB1 mutations in a
tumour with no LOH at the RB1 locus is equivalent to the probability of missing two independent
RB1 mutations in one sample (0·05 x 0·05) or 0·25%. By combining our data on 1054 unilateral
non-familial tumours, we identified 29 RB1+/+tumours (2·8%), about 10-fold more than expected (p
= 6 x 10-45) (table 1). This suggested that some RB1+/+ tumours might originate by a mechanism
other than two RB1 mutations.
To characterize the copy numbers of known genes commonly gained or lost in retinoblastomas,9we
used QM-PCR (Toronto) or MLPA/SNP (Amsterdam) analyses. MYCN copy-number was elevated
in 27/30 (90%) RB1+/+ vs. 60/93 (65%) RB1-/- retinoblastomas (p = 3·4 x 10-4) (two-tailed t-test with
Welch’s adjustment for heteroscedasticity). Most significantly, MYCN copy-number in the RB1+/+
tumours showed bimodal distribution, with 16/30 (53%) RB1+/+tumours showing high-level MYCN
amplification (28 to 121 copies of MYCN), called RB1+/+MYCNA retinoblastoma (tables 1, S5, figure
1). The remaining 14 RB1+/+ tumours showed between 2 and 10 MYCN copies. For ten children with
RB1+/+ MYCNA tumours, DNA from blood was available and showed the normal two MYCN copies.
In comparison to RB1-/-retinoblastoma, the 16 RB1+/+MYCNA tumours showed reduced frequency of
copy-number changes in four other genes characteristic of retinoblastoma: gain of oncogenes KIF14
(19% vs. 62%; p = 0·002), DEK and E2F3 (6% vs. 57%; p = 0·0002) and loss of tumour suppressor
gene CDH11 (13% vs. 56%; p = 0·002) (table S6, S5).
We studied DNA from 48 unilateral retinoblastomas by aCGH10 (Toronto) and 3 by SNP
(Amsterdam) analysis (14 RB1+/+MYCNA, 12 RB1+/+, and 25 RB1-/-(+) ) (tables S5,S7, figure2A).
None of the RB1+/+MYCNA retinoblastomas showed any evidence of copy-number changes or
translocations11at the RB1 locus. Except for MYCN copy-number, aCGH (figure 2A) confirmed a
reduced frequency in RB1+/+MYCNA retinoblastomas of the specific genomic copy-number changes
characteristic of RB1-/-retinoblastomas.9 The RB1+/+MYCNA retinoblastomas also showed overall
9

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
significantly fewer altered bp and aCGH clones than the RB1-/-retinoblastomas (p = 0·033; t-test
with Welch’s adjustment) (figure 2C, D, table S7).
The amplicons of the 14 RB1+/+MYCNA retinoblastomas, as well as one RB1+/-tumour (T33) with 73
copies of MYCN, and one RB1-/-primary tumour (RB381) with 9·2 copies of MYCN, were narrow,
spanning 1·1 to 6·3 Mbp encompassing MYCN (figure 2B, S2, table S7). Importantly, the sole
copy-number change detected in one RB1+/+MYCNA retinoblastoma (E4) was 48 copies of 2p24.2-
24.3 encompassing MYCN. The minimal common amplicon defined by RB1+/+(-) MYCNA
retinoblastomas T33 and P2 spanned 513 kbp containing only MYCN. RB1+/+MYCNA
retinoblastomas T5 and P2 also defined a minimal common amplicon including only MYCN. Of the
remaining 36 tumours tested by aCGH, 24 unilateral tumours showed no gain or loss at MYCN, and
12 had moderate gain spanning a broad region of at least 28 Mbp of 2p, too large to meet the
definition of amplification.12
Three (23%) RB1+/+MYCNA tumours showed 17q21.3-qter or 17q24.3-qter gain; two
RB1+/+MYCNAtumours showed 11q loss. Both regions are commonly altered in neuroblastoma,13,14
but are rare in RB1-/-retinoblastoma (present and published data15,16). Other changes in
RB1+/+MYCNA retinoblastomas not often seen in RB1-/- retinoblastoma were gains at 14q, gains at
18q, and losses at 11p (figures 2A, S3).
Retinoblastoma T33 (RB1+/-) showed high-level MYCN amplification and loss of one copy of most
of 13q, including RB1; we suspect that amplification of MYCN initiated proliferation, followed by
13q deletion. Since T33 also showed numerous characteristics of RB1+/+MYCNAtumours, we
included T33 with MYCNA retinoblastomas in many analyses (figures 2A, 2B, 4A, S2, S4).
RB1+/+MYCNA retinoblastomas expressed pRB. Primary RB1+/+MYCNA retinoblastomas and the
usual cell types in normal adjacent retina17stained for both N-terminal (figure 3A) and C-terminal
(data not shown) epitopes of the RB1 protein (pRB), while RB1-/-(+) tumours failed to stain for pRB.
Western blot on cell line A3, derived from RB1+/+ MYCNA primary retinoblastoma, showed both
10

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
phosphorylated and un-phosphorylated, full-length pRBWestern blot on three cell lines derived
from RB1+/+MYCNA primary retinoblastomas showed both phosphorylated and un-phosphorylated,
full-length pRB18 (figure 3B). Three RB1+/+MYCNA primary retinoblastomas for which mRNA was
available, expressed full-length 2·8 kbp RB1 transcripts at levels comparable to fetal retina, using
end-point and real-time RT-PCR (figure 3C, 3E, table S8). In contrast, RB1-/-retinoblastomas
expressed low levels of RB1 transcript.
RB1+/+MYCNA primary retinoblastomas (but not adjacent retina) (figure 3A) and three derived cell
lines (data not shown) stained strongly for N-Myc protein. RB1+/+MYCNA retinoblastomas showed
increased MYCN protein and transcripts compared to RB1-/-primary retinoblastomas and fetal retina
(figure3D, 3B, C, E, tableS8). MYCN and MKI67 transcripts (indicative of proliferation) were
detected in fetal retina, primary RB1+/+MYCNA retinoblastoma, and RB1-/- retinoblastomas (as
expected for embryonal neuronal tumours), but were at very low levels in adult retina (figure 3CB).
RB1+/+MYCNAtumours showed reduced expression of the oncogene KIF14,9 in contrast to normal
fetal retina, and to the high KIF14 expression in RB1-/-primary retinoblastomas and cell lines (figure
3ED, table S8).
RB1+/+MYCNAtumors expressed embryonic retinal cell markers consistent with a retinal origin. The
mRNAs of cone cell marker X-arrestin19 and CRX, a marker of retinal and pineal lineage tumours
strongly expressed in RB1-/- retinoblastoma, but not in neuroblastoma,20 were expressed in fetal
retina, human adult retina, three RB1+/+MYCNA primary retinoblastomas, and four RB1-/-
retinoblastomas (figure 3D).
Children with RB1+/+MYCNA retinoblastomas were much younger at diagnosis than children with
unilateral RB1-/-retinoblastomas. The median age at diagnosis of 17 children (16
RB1+/+MYCNAtumours, one RB1+/-MYCNA, T33) was 4·5 months, significantly younger than
children with unilateral sporadic RB1-/- (24·0 months, p < 10-4) or RB1+/+ (21·5 months, p < 10-4)
retinoblastomas (figure 4A, tableS5). Our data predict that 1922% of children diagnosed with non-
11

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
familial, unilateral retinoblastoma at age six months or younger will have RB1+/+MYCNA
retinoblastoma (table S4).
Analysis of age at diagnosis vs. proportion not yet diagnosed led Knudson to propose that two-hits
initiate retinoblastoma.1 Our data from79 unilaterally affected RB1-/- patients is consistent with
Knudson’s model and with unilateral RB1-/-tumours (age range 1·9 to 109 months), using birth as a
surrogate for cancer initiation, fita two hit model only when we excluded children diagnosed older
than 60 months (the oldest children included in Knudson’s analysis were 60 months) and as
expected, did not fit the one-hit model describing heritable retinoblastoma. OurRB1-/-data points fits
a two-hit curve, representative of two independent mutation events in a tumour suppressor gene, not
a one-hit curve. We similarly analysed age at diagnosis vs. proportion not yet diagnosed for
RB1+/+MYCNA tumours: whileage at diagnosis vs proportion not yet diagnosed fit both one-hit and
two-hit models poorly, perhaps reflecting that the majority of these tumours initiated before birth.
thewhile the data points of the twelve children less than 10 months old fall close to the calculated
one-hit curve, the age of diagnosis for some of the older children deviate away from it, falling on
either side of the predicted curve (figure 4B).
On clinical examination, the RB1+/+MYCNA retinoblastomas were indistinguishable from unilateral
RB1-/- retinoblastomas (figure 4C). However, on histological examination, RB1+/+MYCNAtumours
contained undifferentiated cells with large, prominent, multiple nucleoli, and necrosis, apoptosis,
and little calcification, similar to other MYCNA embryonic tumours, such as neuroblastoma21 (figures
4D, S43). They were distinctly different from prototype RB1-/-retinoblastoma, missing the typical
Flexner-Wintersteiner rosettes22 (figure 4E).
Three RB1+/+MYCNA primary retinoblastomas (RB522, T101, and A3) grew rapidly into cell lines,
unlike the usual RB1-/-retinoblastomas that grow poorly in tissue culture. One RB1+/+MYCN A
retinoblastoma had already invaded the optic nerve past the cribriform plate at age 11 months, a
feature of aggressive disease (figure 4F). The unilaterally affected patients with RB1+/+ MYCNA
12

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
tumours in this study had large tumors and early age of diagnosis, and were cured by removal of
their affected eye with no adverse outcomes; none developed retinoblastomas in the other eye.
Discussion
Knudson’s analysis of retinoblastoma established the basis to understand how normal genes
suppress cancer,1 leading to the identification of the RB1 tumour suppressor gene,2 widely assumed
to initiate all retinoblastoma. We now show a previously unrecognized type of retinoblastoma with
no detectable RB1 mutations (RB1+/+) and no LOH at RB1, but instead high-level, focal
amplification of the MYCN oncogene, aggressive behaviour, and very young age of onset.
Our international study of 1,054 unilateral retinoblastomas allowed recognition of this distinct
RB1+/+MYCNA subtype comprising 1-2% of unilateral, non-familial retinoblastomas. At least two
previously described tumours may be also examples of RB1+/+MYCNAretinoblastomas,23 although
RB1 genetic status was not defined. Notably, our discovery of a discrete retinoblastoma subtype was
validated through independent observations in different patient cohortsDespite the low incidence of
RB1+/+ MYCNA retinoblastoma, RB1+/+MYCNAtumors were independently discovered and
characterized in the Toronto and Amsterdam labs, with different patient cohorts and different
technologies, thus validating the findings.
We also identified 14 RB1+/+retinoblastomas without MYCN amplification. Although we lacked
frozen tumours to perform expression studies, aCGH of DNA showed genomic gains and losses
distinct from those seen in either RB1-/-or RB1+/+MYCNA retinoblastomas. In particular, these
RB1+/+retinoblastomas showed increased incidence of gain on 19p and q, 17 p and q, 2p, and at the
telomeric end of 9q. The RB1+/+ group is expected to be heterogeneous; mechanisms may include
translocations of oncogenes leading to tumourigenesis. This group merits further study.
Our paper highlights the use of molecular diagnoses to identify novel malignancies that previously
eluded histopathological recognition. Although pathologists have not previously recognized
13

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
RB1+/+MYCNA retinoblastoma as distinct, these tumours resemble large nucleolar neuroblastomas
with MYCN-amplification and poor outcome.21 Like neuroblastomas with MYCN-amplification,14the
RB1+/+MYCNAtumors showed less complex patterns of genomic copy-number alterations than
tumours without MYCN amplification, suggesting that MYCNA may be the critical driver of
malignancy. However, early age of diagnosis for MYCNAtumours may also factor into the reduced
copy-number alterations, since alterations have less time to accumulate. Although whole genome
sequencing has recently shown that point mutations (other than in the RB1 gene) are few inRB1-/-
retinoblastoma,24 loss of RB1 has been shown to induce mitotic changes and lagging
chromosomes,25 leading to genomic instability. The specific DNA copy-number changes associated
with RB1 loss in retinoblastoma and retinoma8,16are less frequent inRB1+/+MYCNA retinoblastomas
with intact RB1 genes.
Are RB1+/+MYCNA tumours truly retinoblastoma? Strictly speaking, the word retinoblastoma
defines ‘a blast cell tumour arising from the retina’. RB1+/+MYCNA retinoblastomas fit the basic
definition of retinoblastoma as ‘a blast cell tumour arising from the retina’, since they arise in
developing retina and express markers of embryonic retina.19,20,26 Our study demonstrated the
presence of intact RB1 genes and pRB in primary RB1+/+ MYCNA retinoblastomas, which stained
strongly for N- and C-terminal antibodies to pRb. The three cell lines derived from RB1+/+MYCNA
tumours expressed full length RB1 mRNA, and RB1+/+ MYCNA cell line A3 showed phosphorylated
and un-phosphorylated pRb, indicating normal functional pRb18.
The very early presentation of RB1+/+MYCNA retinoblastomas, the lack of RB1 mutations, and the
high MYCN-amplification in a relatively copy-number stable genome, suggests that these
retinoblastomas arise by one hit: somatic MYCN oncogene amplification in a retinal progenitor cell.
This is supported by the identification of one RB1+/+MYCNA tumour where the only genomic copy-
number change was amplification of the MYCN region. Because RB1+/+MYCNA retinoblastomas
diagnosed soon after birth are already large, they likely initiate much earlier in fetal development
14

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
than RB1-/- retinoblastomas, possibly even earlier than hereditary retinoblastomas, so Knudson’s
analysis using birth as a surrogate for tumour initiation may not be applicable. In addition, the
aggressive growth ofthese retinoblastomas may confound determination of how many hits initiate
RB1+/+MYCNAtumours, using Knudson's analysis.How the MYCN-amplification is initiated, and
whether MYCN-amplification alone suffices to initiate these retinoblastoma remains to be formally
demonstrated.
It has been shown that RB1-/- retinoblastoma cell lines are sensitive to MYCN knockdown, an effect
that may be more significant in RB1+/+ MYCNA retinoblastomas.29 The relative genomic stability of
RB1+/+ MYCNA retinoblastomas suggests that anti-Myc therapeutic agents28 may not evoke the
resistance to therapy acquired by genomic rearrangements in RB1-/- retinoblastoma. We have
performed preliminary experiments to knock down MYCN expression and observed rapid death of
RB1+/+ MYCNA cell lines. Globally, many children less than one year of age with extra-ocular
retinoblastoma30 might have RB1+/+MYCNA retinoblastomas and benefit from anti-MYCN therapy.
For intraocular disease, enucleation is presently the only way to diagnose RB1+/+MYCNAtumors, so
anti-MYCN therapy for intraocular tumours is unlikely.
Young age at diagnosis of unilateral retinoblastoma frequently suggests heritable retinoblastoma,
and is often considered a reason to try to cure retinoblastoma without enucleation. However,
attempts to salvage an eye with RB1+/+MYCNA retinoblastoma could be dangerous. One
RB1+/+ MYCNA retinoblastoma showed early significant optic nerve invasion, a predictor of high
mortality through tumour invasion into brain.27 The patients with unilateral RB1+/+ MYCNA tumours
in this study were cured by removal of their affected eye with no adverse outcomes.
Standard care for unilateral non-familial retinoblastoma is identification of the RB1 mutant alleles
in tumour, and examination of blood to determine whether either mutant allele is germline. Our
study suggests that when no RB1 mutation is detected in a retinoblastoma tumor, especially for
children less than 12 months old, determining MYCN copy-number assists in on-going care. The
15

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
diagnosis of RB1+/+MYCNA retinoblastoma strongly suggests non-hereditary disease with normal
population risks for retinoblastomas in the other eye and other cancers later in life.
Our findings challenge the long-standing dogma that all retinoblastomas are initiated by RB1 gene
mutations (Panel). RB1+/+MYCNA retinoblastoma provides an intriguing contrast to classical
retinoblastoma, of immediate importance to patients.
Panel: Research in Context
Systematic Review
We systematically searched the published, peer-reviewed literature on PubMed
(http://www.ncbi.nlm.nih.gov/pubmed/) using the search terms “retinoblastoma”, “initiation” and
“genetics”; “retinoblastoma tumour genetics”; “retinoblastoma development”; and “retinoblastoma
initiation”. We reviewed publications with a main focus on genetic initiation and development of
human retinoblastoma. We found no data that challenged Knudson’s 1971 conclusion that two rate-
limiting events1, later shown to be loss of both RB1 gene alleles,2-5,7 are essential but not necessarily
sufficient for development of retinoblastoma. We found no suggestion of another form of
retinoblastoma.
Interpretation
Our collaborative studies identify a previously unrecognized disease: retinoblastoma apparently
driven by MYCN oncogene amplification. This newly recognised form of retinoblastoma has
immediate clinical implications for patients. RB1+/+MYCNA retinoblastoma can only be diagnosed
by molecular study of the tumour after removal of the eye of very young children with unilateral
non-familial disease. The children with RB1+/+MYCNA retinoblastoma and their relatives are
predicted to be at normal population risk for other cancers. Attempts to salvage the eye on the
assumption of heritable disease in such young children, could incur high treatment morbidity and
failure to cure these aggressive oncogene-driven retinoblastomas.
16

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Tables
Table 1: Frequency of RB1+/+ unilateral retinoblastoma at five international sites.
Test Site Total RB1+/+ Proportion RB1+/+ P-value*Number
RB1+/+MYCNA
Proportion
RB1+/+MYCNA
Canada 441 7 1.6% 0.06 5 1.1%
Germany 400 12 3.0% 0.31 4 1.0%
France 150 5 3.3% 0.22 2 1.3%
New Zealand 30 2 6.7% 0.06 1 3.3%
Netherlands 33 3 9.1% 0.03** 3 9.1%
Total 1054 29 2.8% 15# 1.4%
*Pair-wise proportion test: frequency of RB1+/+tumours in each site compared to all other sites.
**A larger percentage of Netherlands patients were RB1+/+ compared to Canadian patients (p = 0.03).
# One additional RB1+/+MYCNA and one RB1+/-MYCNA (T33) tumours were subsequently included for a total of 17 included in some analyses
.
17

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Figures
Figure 1:The 5-genecopynumber signature of retinoblastoma
(A) Box-plot of genomic copy-numbers of indicated genes in unilateral retinoblastoma, categorized
by RB1 mutation status: 11 of the original 25 RB1+/+tumours showed high-level amplification of
MYCN (RB1+/+MYCNA) (median MYCN copy-number: 54), and few copy-number changes in the
other genes typically gained or lost in retinoblastoma. Median MYCN copy of the other 14 (non
MYCNA) RB1+/+ tumors was 3.09. T33 is an outlier of the RB1+/-group, showing an RB1+/+MYCNA-
like profile; gray line, 2-copies; on each boxplot, vertical line marks the maximum and minimum
copy-numbers observed while the box bounds second and third quartiles, and horizontal line within
the box represents the median.(B) Copy-number heat-map for the profile genes (red, increased and
blue, decreased copy-number; gray, 2 copies; white, not tested; n, number in each group, including
five more recently discovered RB1+/+MYCNAtumours (black triangles) not included in (A). (C)
MYCN copy-number assessment in 30 RB1-/-and 3 RB1+/+ tumours studied in Amsterdam, showing
high MYCN copy-number in the RB1+/+tumours.
18

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
19

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
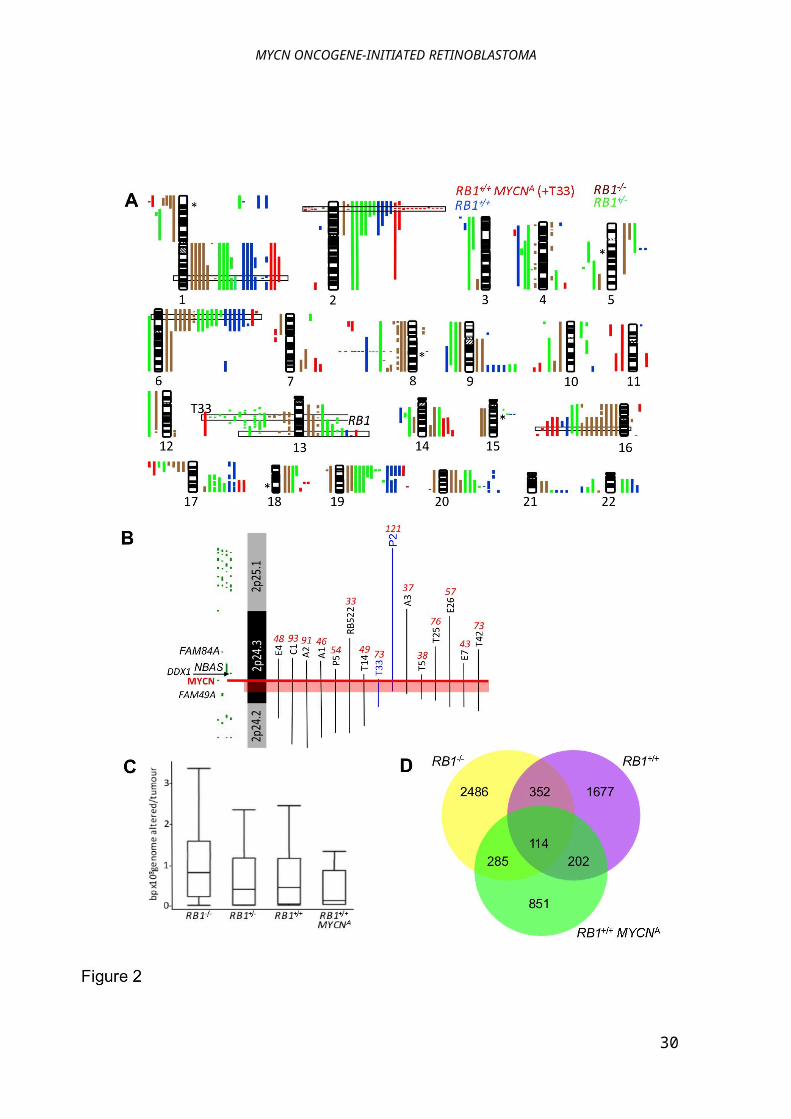
Figure 2:Fewer genomic copy-number alterations in RB1+/+MYCNA than RB1-/-tumours
(A) aCGH on 12 RB1+/+MYCNA (including T33), 12 RB1+/+, 13 RB1+/-, and 11 RB1-/-tumours; gains,
right; losses, left; minimal commonly gained/lost regions in RB1-/- tumours boxed; *normally
occurring copy-number variations. The RB1+/- MYCNA tumour T33 shows loss of most of 13q; this
may not be an initiating event. (B) The minimal amplicon of 513 kbp is defined by two MYCNA
tumours (pink band); MYCNcopy-number by QM-PCR, red italics; aCGH individual probes, green
bars. (C) Boxplot of bp altered shows fewer changes in RB1+/+MYCNA than RB1-/-tumours (p =
0.033; t-test with Welch’s adjustment); vertical line marks the maximum and minimum copy-
numbers observed, box bounds second and third quartiles, and horizontal line within the box
represents the median. (D) fewer aCGH clones are altered in RB1+/+ and RB1+/+MYCNA, than RB1-/-
tumours; each class has more unique clones altered than in common.
20

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
21

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Figure 3: RB1+/+MYCNA tumours express pRB and MYCN
(A) RB1+/+MYCNA retinoblastomas stained positive with pRB N-terminus antibody and for MYCN
protein; controls, normal retina and adjacent normal retina (*); RB-/-tumour is negative for pRB. (B)
Western blot shows the characteristic two bands of unphoshorylated and phosphorylated pRB in
fetal retina (17 weeks), RB1+/+ MYCNA cell line A3, and neuroblastoma cell lines SH-SY5Y and
BE(2)-M17, while the two RB1-/- cell lines (WERI-Rb1 and Y79) show no full length pRb. Low
expression of MYCN protein is detected in fetal retina (week 17) and WERI-Rb-1, while high
expression of MYCN protein is detected in RB1+/+ MYCNA cell lines A3, Y79 and BE(2)-M17 (with
MYCN amplification), but not in SH-SY5Y (which has no MYCN amplification). (C) Primary
RB1+/+MYCNA retinoblastomas express full-length RB1, MYCN and Ki67 transcripts (end point RT-
PCR); Ki67 mRNA indicated proliferation; TBP endogenous control. (D) Expression of retinal
progenitor cell marker CRX and cone cell maker X-arrestin in human fetal retina, human adult
retina, primary RB1+/+MYCNA tumours, and primary RB1-/-tumours with between two and ten
MYCN copies; end point RT-PCR; TBP endogenous control. (E) RB1, MYCN and KIF14 mRNA
expression in human fetal (FR) and (HR) adult retina, RB1+/+ MYCNA, RB1-/-, or RB+/-primary
tumours and RB1-/-cell lines; real-time RT-PCR, triplicate measurements normalized against
GAPDH, relative to FR; MYCN DNA copy-numbers in italics; #, not done; *Y79 has a homozygous
RB1 del exons 2 to 6 that results in increased expression of shortened RB1 mRNA. Cell lines Y79
and RB381 have MYCN amplification.
22

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
23

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Figure 4: RB1+/+MYCNA tumours in very young children are clinically distinct. (A) Children
with RB1+/+MYCNA retinoblastoma are diagnosed significantly younger than children with RB1-/-
tumours (p<0.0001, Wilcoxon rank sum test). (B) Using our data, tThe Knudson plot of proportion
not yet diagnosed vs. age at diagnosis, using birth as a surrogate for initiation, fits the two-hit curve
(blue) but not the one-hit curve (red) for RB1-/- disease, as expected. F; for RB1+/+MYCNA
retinoblastoma, while the data points of the twelve children less than 10 months of age fall close to
the calculated one-hit curve (red), the age of diagnosis for some of the older children deviate away
from it, falling on either side of the predicted curve (data points for identical ages overlap). (C)
Fundus image of an RB1+/+MYCNA unilateral tumour in a 4 month-old child shows characteristic
calcification on ultrasound. (D) Round nuclei with large prominent multiple nucleoli shown on
primary pathology, in comparison to (E) RB1-/-tumour showing classic retinoblastoma pathology:
Flexner-Wintersteiner rosettes and nuclear molding; hematoxylin-eosin staining. (F)
RB1+/+MYCNAretinoblastoma in an 11 month-old child shows extra-ocular extension into the optic
nerve (white arrows) (2.5x, hematoxylin-eosin staining).
24

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
25

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
Contributors
Diane E Rushlow recognized the initial connection between MYCNA amplification and RB1
mutation status, performed literature search and QM-PCR analysis and supervised RB1 mutation
analysis, coordinated collaborations with the other sites and was the major contributor to manuscript
preparation. Jennifer Y Kennett performed aCGH and analysed aCGH data. Berber M Mol
determined MYCN status by MLPA experiments and performed SNP array data analysis,
immunohistochemistry imaging and Western blots. Stephanie Yee performed analysis of aCGH
data, the MYCNA alignment, immunohistochemistry and reverse transcriptase PCR. Sanja Pajovic
performed literature search, reverse transcriptase PCR and immunohistochemistry. Brigitte L
Thériault performed literature search and RNA expression studies. Nadia L Prigoda-Lee performed
literature search, statistical analysis and contributed to figure and manuscript preparation. Clarellen
Spencer performed immunohistochemistry. Helen Dimaras and Timothy W Corson performed
literature searches, assisted in data analysis and conceptualization of discussion, and contributed to
figure preparation. Renee Pang performed statistical and bioinformatic analyses on the aCGH data.
Christine Massey performed statistical analysis on age of diagnosis data. Katherine Paton and
Annette C Moll provided clinical features and material, and conceptual discussion. Claude
Houdayer and Anthony Raizis provided RB1 mutation analysis, and clinical features. William
Halliday recognized and characterized the unique histological features of the
RB1+/+MYCNAretinoblastomas and prepared digital images for publication. Wan L Lam supervised
aCGH experiments. Paul C Boutros performed detailed and novel analysis of the aCGH data, and
statistical analyses throughout the project. Dietmar Lohmann performed literature search, provided
RB1 mutation analysis, and contributed to figure construction and development of concepts.
Josephine C Dorsman coordinated the Amsterdam study, recognized the RB1 and MYCN mutation
status of the Amsterdam samples, and supervised Berber Mol. Brenda L Gallie supervised overall,
performed literature search, provided critical guidance on all components of the project, and
26

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
contributed extensively to figure and manuscript preparation. All authors contributed to manuscript
preparation.
Conflicts of interest
BLG is part-owner of Solutions by Sequence. All other authors declare that they have no conflicts
of interest.
Acknowledgments
This study was conducted with the support of the Ontario Institute for Cancer Research to PCB
through funding provided by the Government of Ontario. SY was funded by the Vision Science
Research Program of the University Health Network and the University of Toronto. RP was funded
in part by a Great West Life Studentship from Queen’s University School of Medicine. BMM was
funded by a grant from CCA/V-ICI/ Avanti-STR (to JCD, J. Cloos and ACM), the Dutch research
was also funded in part by KIKA (JCD, H. te Riele, J. Cloos, ACM). We thank Leslie MacKeen for
the montage of RetCam image in figure 3B. We thank Dr. Valerie White of U. British Columbia for
providing clinical and pathological details and images. We thank members of the VU University
Medical Center/The Netherlands Cancer Institute, Institut Curie, Toronto retinoblastoma teams and
other wise colleagues for useful discussions. We thank the children and families who donated
tissues for these studies for the benefit of future families.
27

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
REFERENCES
1. Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Science, USA. 1971;68(4):820-3. 2. Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986 Oct 16-22;323(6089):643-6. 3. Cavenee WK, Hansen MF, Nordenskjold M, Kock E, Maumenee I, Squire JA, Phillips RA, Gallie BL. Genetic origin of mutations predisposing to retinoblastoma. Science (New York, NY. 1985;228(4698):501-3. 4. Lohmann DR, Gallie BL. Retinoblastoma: Revisiting the model prototype of inherited cancer. Am J Med Genet. 2004 Aug 15;129C(1):23-8. 5. Rushlow D, Piovesan B, Zhang K, Prigoda-Lee NL, Marchong MN, Clark RD, Gallie BL. Detection of mosaic RB1 mutations in families with retinoblastoma. Human mutation. 2009 May;30(5):842-51. 6. Schuler A, Weber S, Neuhauser M, Jurklies C, Lehnert T, Heimann H, Rudolph G, Jockel KH, Bornfeld N, Lohmann DR. Age at diagnosis of isolated unilateral retinoblastoma does not distinguish patients with and without a constitutional RB1 gene mutation but is influenced by a parent-of-origin effect. Eur J Cancer. 2005 Mar;41(5):735-40. 7. Houdayer C, Gauthier-Villars M, Lauge A, Pages-Berhouet S, Dehainault C, Caux-Moncoutier V, Karczynski P, Tosi M, Doz F, Desjardins L, Couturier J, Stoppa-Lyonnet D. Comprehensive screening for constitutional RB1 mutations by DHPLC and QMPSF. Human mutation. 2004 Feb;23(2):193-202. 8. Dimaras H, Khetan V, Halliday W, Orlic M, Prigoda NL, Piovesan B, Marrano P, Corson TW, Eagle RC, Jr., Squire JA, Gallie BL. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008 May 15;17(10):1363-72. 9. Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007 Apr 16;46(7):617-34. 10. Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP, Snijders A, Albertson DG, Pinkel D, Marra MA, Ling V, MacAulay C, Lam WL. A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet. 2004 Mar;36(3):299-303. 11. Watson SK, deLeeuw RJ, Horsman DE, Squire JA, Lam WL. Cytogenetically balanced translocations are associated with focal copy number alterations. Human genetics. 2007 Feb;120(6):795-805. 12. Myllykangas S, Bohling T, Knuutila S. Specificity, selection and significance of gene amplifications in cancer. Seminars in cancer biology. 2007 Feb;17(1):42-55. 13. O'Neill S, Ekstrom L, Lastowska M, Roberts P, Brodeur GM, Kees UR, Schwab M, Bown N. MYCN amplification and 17q in neuroblastoma: evidence for structural association. Genes Chromosomes Cancer. [Case Reports]. 2001 Jan;30(1):87-90. 14. Mosse YP, Diskin SJ, Wasserman N, Rinaldi K, Attiyeh EF, Cole K, Jagannathan J, Bhambhani K, Winter C, Maris JM. Neuroblastomas have distinct genomic DNA profiles that predict clinical phenotype and regional gene expression. Genes Chromosomes Cancer. 2007 Oct;46(10):936-49. 15. Chen D, Gallie BL, Squire JA. Minimal regions of chromosomal imbalance in retinoblastoma detected by comparative genomic hybridization. Cancer Genet Cytogenet. 2001;129(1):57-63.
28

MYCN ONCOGENE-INITIATED RETINOBLASTOMA
16. Sampieri K, Amenduni M, Papa FT, Katzaki E, Mencarelli MA, Marozza A, Epistolato MC, Toti P, Lazzi S, Bruttini M, De Filippis R, De Francesco S, Longo I, Meloni I, Mari F, Acquaviva A, Hadjistilianou T, Renieri A, Ariani F. Array comparative genomic hybridization in retinoma and retinoblastoma tissues. Cancer Sci. 2009 Mar;100(3):465-71. 17. Spencer C, Pajovic S, Devlin H, Dinh QD, Corson TW, Gallie BL. Distinct patterns of expression of the RB gene family in mouse and human retina. Gene Expr Patterns. 2005 Jun;5(5):687-94. 18. Buchkovich K, Duffy LA, Harlow E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell. [Research Support, U.S. Gov't, P.H.S.]. 1989 Sep 22;58(6):1097-105. 19. Murakami A, Yajima T, Sakuma H, McLaren MJ, Inana G. X-arrestin: a new retinal arrestin mapping to the X chromosome. FEBS letters. [Comparative Study Research Support, Non-U.S. Gov't]. 1993 Nov 15;334(2):203-9. 20. Terry J, Calicchio ML, Rodriguez-Galindo C, Perez-Atayde AR. Immunohistochemical Expression of CRX in Extracranial Malignant Small Round Cell Tumors. Am J Surg Pathol. 2012 Aug;36(8):1165-9. 21. Tornoczky T, Semjen D, Shimada H, Ambros IM. Pathology of peripheral neuroblastic tumors: significance of prominent nucleoli in undifferentiated/poorly differentiated neuroblastoma. Pathol Oncol Res. 2007;13(4):269-75. 22. Flexner S. A peculiar glioma (neuroepithelioma?) of the retina. Johns Hopkins Hosp Bull. 1891;2:115. 23. Lillington DM, Goff LK, Kingston JE, Onadim Z, Price E, Domizio P, Young BD. High level amplification of N-MYC is not associated with adverse histology or outcome in primary retinoblastoma tumours. British journal of cancer. 2002 Sep 23;87(7):779-82. 24. Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J, Brennan R, Rusch M, Manning AL, Ma J, Easton J, Shurtleff S, Mullighan C, Pounds S, Mukatira S, Gupta P, Neale G, Zhao D, Lu C, Fulton RS, Fulton LL, Hong X, Dooling DJ, Ochoa K, Naeve C, Dyson NJ, Mardis ER, Bahrami A, Ellison D, Wilson RK, Downing JR, Dyer MA. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. 2012 Jan 19;481(7381):329-34. 25. Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes & development. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. 2010 Jul 1;24(13):1364-76. 26. Kobayashi M, Takezawa S, Hara K, Yu RT, Umesono Y, Agata K, Taniwaki M, Yasuda K, Umesono K. Identification of a photoreceptor cell-specific nuclear receptor. Proc Natl Acad Sci U S A. [Research Support, Non-U.S. Gov't]. 1999 Apr 27;96(9):4814-9. 27. Chantada GL, Casco F, Fandino AC, Galli S, Manzitti J, Scopinaro M, Schvartzman E, de Davila MT. Outcome of patients with retinoblastoma and postlaminar optic nerve invasion. Ophthalmology. 2007 Nov;114(11):2083-9. 28. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, Sims RJ, 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011 Oct 4;108(40):16669-74. 29. Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, Liu A, Jhanwar SC, Abramson DH, Cobrinik D. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009 Jun 12;137(6):1018-31. 30. Dimaras H, Kimani K, Dimba EA, Gronsdahl P, White A, Chan HS, Gallie BL. Retinoblastoma. Lancet. [Research Support, Non-U.S. Gov't]. 2012 Apr 14;379(9824):1436-46.
29
MYCN ONCOGENE-INITIATED RETINOBLASTOMA
30
Recommended

















