Embed Size (px)
DESCRIPTION
Ejemplar de la Revista Americana de Genética Humana, Volúmen 90, Nro 4 del año 2012
Citation preview


EDITORS’ CORNER
This Month in The Journal
Sara B. Cullinan1
Genomic Privacy in GWAS?
Im et al., page 591
Recent technological advances have made it possible to
interrogate human phenotypes at a previously unimagin-
able scale. But, as with any collection of personal data, it
is important to ensure individual privacy. Indeed, previous
investigations into the ability to discern an individual’s
participation in genetic studies have led to the withdrawal
of allele frequencies from publicly available results. In this
issue, Im et al. probe deeper, questioning how much
private information can be extracted from typically re-
ported statistics, such as regression coefficients or p values.
Through a series of analyses, the authors determine that
regression coefficients can, in some cases, provide just as
much information as allele frequencies, thus creating a
situation in which even statistics that were thought to be
‘‘safe’’ can in fact identify participants and their medical
history. The possibility of membership detection is espe-
cially high in cases in which multiple phenotypes are
being reported, e.g., in multiple-omics data sets. With
exome- and whole-genome sequencing (and the large
data sets that they generate) becoming more common, it
is clear that many additional discussions between scien-
tists, clinicians, and ethicists are needed to ensure that
privacy can be maintained without sacrificing the dissem-
ination of research findings.
A Major mtDNA Shake-Up
Behar et al., page 675
In 1981, the revised Cambridge Reference Sequence was
published. It immediately became the standard against
which human mtDNA is compared and phylogenies are
derived. Indeed, its publication enabled a tremendous
amount of research aimed at better understanding human
history.However, the realization that this sequence belongs
to a recently coalescing European haplogroup creates
several concerns about inconsistencies and misinterpreta-
tion. To address these concerns, Behar et al. set out to reas-
sess and refine the human mtDNA phylogeny, and in so
doing, they constructed a new reference mtDNA sequence,
termed the Reconstructed Sapiens Reference Sequence
(RSRS). Generated through the assessment of over 18,000
human mtDNA sequences, as well as those of Homo
neanderthalensis, the RSRS performs well in molecular clock
analyses and lays the groundwork for a new way of ana-
lyzing mtDNA. Although this change will require a large
amount of rethinking, the authors put forth a coherent
plan to make this feasible, including tools to transform
previously generated data and analyses. With the amount
of deep-sequencing data that should become available in
the coming years, the RSRS presents a ‘‘next-generation’’
approach to understanding human matrilineal diversity.
First Steps toward Understanding Birth Weight
Ishida et al., page 715
Babies come in many different sizes, but being too small
is a major health concern. Indeed, intrauterine growth
restriction (IUGR) serves as a risk factor for several adult
diseases, including obesity and type 2 diabetes. Although
maternal health plays a large role in directing fetal growth,
the genetic factors that contribute to the variability in fetal
size remain poorly understood. Of interest, however, are
those genes that undergo imprinting, a process by which
the parent of origin determines monoallelic expression.
Evolutionary theory posits that expression of alleles in-
herited from the father promote in utero growth, whereas
those inherited from the mother inhibit growth. But what
happens if the maternally inherited allele exhibits an
altered expression pattern? Might the balance be tipped?
In this issue, Ishida et al. explored the possibility that
variants in PHLDA2, which is only expressed from the
maternal allele, might influence birth weight. Their studies
identified a variant in the PHLDA2 promoter region that
eliminates several consensus transcription factor binding
sites and should therefore lead to decreased expression.
Then, through a cross-sectional study of normal births,
they showed that inheritance of this variant (from the
mother), as well as maternal homozygosity, correlated
with increased birth weight. Future studies, focused specif-
ically on IUGR, should help to elucidate how variation in
PHLDA2, and potentially in other imprinted genes, con-
tributes to the regulation of birth weight and related
complications.
Evolutionary History of AD Risk Alleles
Raj et al., page 720
Alzheimer’s disease (AD) is the most common neuro-
degenerative disease, and as of yet, there are no effective
1Deputy Editor, AJHG
DOI 10.1016/j.ajhg.2012.03.008. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 575–576, April 6, 2012 575

treatments, let alone a cure. Therefore, there is great
interest in better understanding the causes of the disease
from both biochemical and genetic standpoints. The
best-characterized genetic risk factor is the ε4 haplotype
of APOE, which, interestingly, shows evidence of having
undergone positive selection, most likely because of an
effect on an unrelated phenotype. With this in mind, Raj
et al. set out to identify other possible indications of selec-
tion in loci shown to associate with AD susceptibility. They
found such evidence, all in East Asian populations, for
three loci, suggesting that the same selective pressure
might have acted on each. Given that AD is unlikely to
serve in such a role, the authors posited that pathogen
exposure might have been the driving force. Indeed,
many signatures of selection in the human genome are
attributed to interactions with pathogens. Interestingly,
the protein products generated at these loci appear to
belong to the same interaction network. This finding
suggests that additional clues about AD risk might be
found by interrogating other branches of this network.
Although much remains to be learned about the variants
that contribute to AD risk, the study of their evolution,
and possible coevolution, will no doubt yield insights
into the underlying biology of the disease.
X Marks the Spot in Breast Cancer Research
Park et al., page 734
The ubiquitous pink ribbons serve as a reminder that
many women (and some men) are affected by breast
cancer. Although well known, BRCA1 and BRCA2 muta-
tions account for a minority of hereditary cancers. There-
fore, a better understanding of the biology of breast
cancer, along with better screening tests, is sought by
many families. To help achieve these goals, Park et al.
used exome sequencing and identified rare mutations in
XRCC2 that serve as susceptibility factors for familial
breast cancer. XRCC2 is a RAD51 paralog that is required
for efficient homologous recombination (HR); its loss
leads to marked genome instability and aneuploidy.
Future studies aimed at delineating the exact role of
XRCC2 mutations, as well as mutations that lie within
the same pathway, in disease onset and/or progression
should aid in the discovery of new treatment options.
This finding adds to the list of genes whose protein prod-
ucts perform crucial roles in HR and whose mutations can
influence breast cancer risk. It also provides support for
those who seek to better understand common diseases
through sequencing studies.
576 The American Journal of Human Genetics 90, 575–576, April 6, 2012

EDITORS’ CORNER
This Month in Genetics
Kathryn B. Garber1,*
Big Gene, Big Heart
Although the cardiomyopathies have a substantial genetic
etiology, genetic testing for this class of heart disorders has
been notoriously difficult. Indeed, the causative mutation
is found in only 20%–30% of patients with dilated cardio-
myopathy. Titin is a candidate gene for cardiomyopathy
that has been examined for mutations to a limited extent
due to its massive coding sequence, which is ~100 kb
in size. Herman et al. recently published data showing
that the sequence hurdle for this gene is worth the effort.
Through next-generation sequencing, they identified
a truncating TTN mutation in ~25% of familial cases of
idiopathic dilated cardiomyopathy, moving TTN to the
forefront of genes involved in this form of the disease.
Although these mutations had very high penetrance after
age 40 in familial cases, there is also a significant amount
of TTN variation whose clinical significance is difficult to
interpret at this time. This includes missense variation,
which was not analyzed in this current paper, so its role
in cardiomyopathy is unclear. Even with truncating muta-
tions in TTN, interpretation is not always simple; these
mutations were identified, albeit at lower frequency, in
control individuals and in individuals with hypertrophic
cardiomyopathy who also had a pathogenic mutation in
a known disease gene.
Herman et al. (2012) NEJM 366, 619–628.
A Complex Balance
Perhaps it is not surprising that the more closely you look
at something, the more you see. Certainly, the advent of
whole-genome comparative genomic hybridization
(CGH) arrays taught us that many people with normal
G-banded karyotypes have cytogenetic aberrations when
we look more closely. Even high-resolution CGH arrays
don’t give us a complete picture of chromosomes, as
recently illustrated by Chiang et al. These investigators
took a set of individuals who had apparently balanced
chromosome translocations—at least based on G-banding
and whole-genome CGH arrays—and they analyzed the
breakpoints at the nucleotide level. What they found was
an unexpectedly high level of complexity to the break-
points. In almost 20% of cases, three or more breakpoints
were involved, but in some cases, a shockingly complex
interweaving of segments occurred, akin to what was
recently described in cancer cells as ‘‘chromothripsis,’’ or
chromosome shattering and reorganization. The cases
analyzed by Chiang et al. involved upward of ten break-
points with inverted segments interspersed among seg-
ments of the expected orientation. This phenomenon is
not limited to spontaneous rearrangements in humans;
analysis of transgene insertions in mice and in sheep
revealed that the sites of integration can be similarly
complex.
Chiang et al. (2012) Nat. Genet. Published online March 4,
2012. 10.1038/ng.2202.
Good News for Men
The Y chromosome is just a degenerate of its former auto-
somal self that is on its way to extinction, or so some have
proposed. If you compare the Y to the X chromosome, for
instance, the Y has lost many of the genes that the
chromosomes once shared, and without a companion
chromosome with which to fully pair itself during meiosis,
some think this sex-specific chromosome is doomed.
David Page argues otherwise. His group does species
comparisons of the Y chromosome in order to understand
its evolution and to better predict the future fate of the Y.
Page’s group previously compared the human to the chim-
panzee Y chromosome, which diverged about six million
years ago, but, in order to look at a much longer evolu-
tionary window, his group recently compared the human
and rhesus macaque Y chromosomes, which diverged
25 million years ago. This comparison yielded a surprising
level of evolutionary stability on the Y. In the majority of
the male-specific regions of the Y chromosome, rhesus
macaques and humans share the same ancestral genes,
arguing for Y chromosome stability over the long haul.
In only a very restricted segment of the Y has gene loss
occurred in humans since the split from the Old World
monkeys. Their data fit a model in which rapid degenera-
tion of segments on Y was followed by marked slowing
of this decay and chromosome stabilization. Don’t count
the Y out just yet; it looks like it may stick around a while.
Hughes et al. (2012) Nature 483, 82–86.
Enhancers Acting as Promoters
Just as we learn to group letters into words and bin words
into different parts of speech in order to extract meaning
from sentences, we try to interpret genome sequences by
picking out the nucleotide sets that comprise genes and
attempting to recognize the regulatory elements from
strings of As, Cs, Gs, and Ts. But although we might think
1Department of Human Genetics, Emory University School of Medicine, Atlanta, GA 30322, USA
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.03.009. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 577–578, April 6, 2012 577

we understand what a particular type of genetic element
does, recognition of one of its roles in gene expression
sometimes doesn’t tell the whole story. Take enhancers,
for instance. These are well-studied cis elements that
have a simple job: they bind transcription factors and
enhance expression from gene promoters, hence their
name. Kowalczyk et al. wondered whether that’s all
enhancers do, and they ended up with evidence that intra-
genic enhancers can also act as alternative tissue-specific
promoters. The resulting mRNAs are spliced and polyade-
nylated but do not appear to be translated into protein.
Because enhancers are much more common than classic
promoters and because about half of enhancers are intra-
genic, this promoter-like activity could contribute substan-
tially to the complexity of the mammalian transcriptome.
The next step is to figure out how these untranslated tran-
scripts are used.
Kowalczyk et al. (2012) Mol. Cell 45, 447–458.
A Common Turn-On
While we’re on the subject of surprising roles for noncod-
ing elements, a recent paper uncovered the coordinated
regulation of two neighboring, but nonparalogous, genes
that both tie into an identical phenotype. Joe Gleeson’s
group focuses on ciliopathies, and they recently identified
mutations in TMEM216 at the JBTS2 locus that cause Jou-
bert syndrome. Of the ten JBTS2-linked families, however,
only about half of them had a TMEM216mutation, despite
an identical phenotype to the mutation-containing
families. When they resequenced the JBTS2 locus, they
found mutations in a neighboring gene, TMEM138, that
is not related to TMEM216, although it also encodes a
transmembrane protein. Although your first thought
might be that TMEM138 simply contains a regulatory
element for TMEM216, this is not the case. Rather, both
genes are coordinately expressed via the action of an inter-
genic element, and they both encode proteins involved
in the same process, ciliogenesis. Knockdown of either
protein leads to defective ciliogenesis, which ultimately
is central to the Joubert syndrome phenotype. Thus,
despite the fact that the genes are very different, they
have evolved a system of coordinated regulation and func-
tional relatedness.
Lee et al. (2012) Science 335, 966–930.
This Month in Our Sister Journal
Yeast System for Characterization of Cystathionine-
Beta-Synthase Mutations
Although we know that individuals with deficiency of
cystathionine-beta-synthase (CBS) tend to have intellec-
tual disability, a marfanoid habitus, ectopia lentis, and
increased risk of thromboembolism, there is variable
expressivity for this disorder, and it is difficult to predict
outcome from genotype. Dietary protein and methionine
restriction is the central approach to management, and
supplementation with vitamin B6, a cofactor of CBS, can
lead to further reductions in homocystine levels in some
affected individuals, who tend to have milder disease. To
address the challenge of genotype-phenotype correlations
in CBS deficiency, Mayfield et al. used a yeast system to
characterize the function of all 84 CBS missense alleles
that had been documented as of 2010. This system, in
which the yeast ortholog of CBS is replaced by human
alleles, allows them to assess the general level of function,
as well as the responsiveness of each allele to vitamin B6
and to another cofactor, heme. The authors also propose
that glutathione deficiency should be further explored in
the context of CBS deficiency, because they noted reduced
glutathione production in their systemwhen CBS function
was disabled.
Mayfield et al. (2012) Genetics. Published online January 20,
2012. 10.1534/genetics.111.137471.
578 The American Journal of Human Genetics 90, 577–578, April 6, 2012

REVIEW
Fragile X and X-Linked Intellectual Disability:Four Decades of Discovery
Herbert A. Lubs,1 Roger E. Stevenson,1,* and Charles E. Schwartz1
X-Linked intellectual disability (XLID) accounts for 5%–10% of
intellectual disability in males. Over 150 syndromes, the most
common of which is the fragile X syndrome, have been described.
A large number of families with nonsyndromal XLID, 95 of which
have been regionally mapped, have been described as well. Muta-
tions in 102 X-linked genes have been associated with 81 of these
XLID syndromes and with 35 of the regionally mapped families
with nonsyndromal XLID. Identification of these genes has
enabled considerable reclassification and better understanding of
the biological basis of XLID. At the same time, it has improved
the clinical diagnosis of XLID and allowed for carrier detection
and prevention strategies through gamete donation, prenatal
diagnosis, and genetic counseling. Progress in delineating XLID
has far outpaced the efforts to understand the genetic basis for
autosomal intellectual disability. In large measure, this has been
because of the relative ease of identifying families with XLID
and finding the responsible mutations, as well as the determined
and interactive efforts of a small group of researchers worldwide.
Introduction
Mutations resulting in X-linked intellectual disability
(XLID) have been described in 102 genes (Table S1, avail-
able online).1 This work was accomplished over a 40 year
period during which the term X-linked mental retardation
was widely used; however, we will use intellectual
disability (ID), which is emerging as the preferred termi-
nology. Mutations in these 102 genes are responsible for
81 of the known 160 XLID syndromes and over 50 families
with nonsyndromal XLID (Table S1 and Figures 1 and 2).
An additional 30 XLID syndromes and 48 families with
nonsyndromal XLID have been regionally mapped (Table
1 and Figures 2 and 3), but the genes not yet identified.
Forty-four XLID syndromes, which remain unmapped,
have also been described (Table S2). Fewer than 400 auto-
somal genes in which mutations resulted in ID have
been identified. Of 1,640 references to ID in OMIM (as of
March 2010), 316 are entities on the X chromosome. Three
comparably sized chromosomes (6, 7, and 8) show 50, 58,
and 60 references, respectively. Several authors have
recently discussed the possibility that these striking differ-
ences might result from a relative concentration of genes
that influence intelligence on the X chromosome.2,3
Identification of the mutations in 102 genes that cause
XLID has been accomplished primarily through long-
term, planned and coordinated studies from the United
States, Europe, and Australia. These studies took advantage
of the power of pedigrees of relatively large families to
assign putative genes to the X chromosome, linkage anal-
ysis to achieve regional localizations, accumulation and
sharing of large data banks of clinical details and speci-
mens, registries of pertinent X chromosomal transloca-
tions and abnormalities, stored samples from a variety of
populations around the world with ID and effective
communication between numerous investigators. In this
setting, the continuously developing technologies were
applied and reapplied to the available clinical and spec-
imen banks effectively and rapidly. A comparable system-
atic approach to autosomal ID has not been carried out.
Publication of the first family with the marker X,4 later
renamed the fragile X (MIM 300624),5 gave an important
impetus to the field by providing a laboratory tool
which clearly identified the most prevalent XLID syn-
drome. A series of biennial international meetings on
fragile X syndrome and XLID, beginning in 1983, involved
about 100 investigators and provided a sense of unity and
progress to the field. Papers and abstracts from these meet-
ings and from other research were published (usually bien-
nially) as conference reports, special issues or updates on
XLID from 1984 to 2008.6–16
The focus of this review will be the discovery process
rather than the details of the clinical or molecular findings
in the individual XLID entities. Readers are referred to the
recently updated excellent review of the fragile X in OMIM
(MIM 300624) and OMIM entries on other XLID disorders
as detailed in Tables S1 and S2. Other reviews of different
aspects of XLID include the periodic XLID updates from
1984 to 2008, an Atlas of XLID Syndromes,1 and a number
of commentaries by individual investigators.3,17–22
XLID before Fragile X
The prelude to the current cytogenetic and molecular era
covered a century (1868–1968). It encompassed descrip-
tions of a number of clinically defined entities (Pelizaeus-
Merzbacher disease [MIM 312080], Duchenne muscular
dystrophy [MIM 310200], incontinentia pigmenti [MIM
308300], Goltz focal dermal hypoplasia [MIM 305600],
Lenz microphthalmia syndrome [MIM 309800]), inborn
errors of metabolism (Hunter syndrome [MIM 309900],
Lowe syndrome [MIM 309000], Lesch-Nyhan syndrome
[MIM 300322]), and large pedigrees in which ID segregated
with an X-linked pattern.23–28 During the same period, the
excess of males among persons with ID was observed in
1Greenwood Genetic Center, JC Self Research Institute of Human Genetics, 113 Gregor Mendel Circle, Greenwood, SC 29646, USA
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.018. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 579–590, April 6, 2012 579

census surveys and other population studies.29–31 The
magnitude of the male excess, varied from study to study
but averaged about 30 percent and was found in nearly
all studies.
These two observations—the excess of males among
persons with ID and clinical syndromes or families with
ID that segregated with an X-linked pattern—provided
compelling evidence that genes on the X chromosome
were important contributors to the overall causation of
ID and, hence, of individual, familial, and societal signifi-
cance. By virtue of having but a single X chromosome,
the male’s genome was uniquely vulnerable and that
vulnerability extended to brain development and function
as well as to other systems.
Further insights during this early period of time were
that XLID comprised syndromal entities (ID plus somatic,
metabolic, or neuromuscular manifestations) and nonsyn-
dromal entities (ID alone or with inconsistent abnormali-
ties). It also became clear that some females in XLID
pedigrees had intellectual limitations, albeit with neither
the consistency nor the severity of males. Technological
limitations (lack of tools for linkage analysis and gene
isolation) precluded a more precise genetic characteriza-
tion of XLID disorders and delayed the clinical delineation.
The Setting of the Initial Observation of the Marker X
In 1966, when a one-year-old boy and his brother were
referred to the Yale chromosome laboratory for study
because of delayed development, medical cytogenetics
was in a period of transition. The major trisomies as well
as translocations and large deletions had been defined by
nonspecific orcein or Giemsa staining. Prenatal cytoge-
netic diagnosis had begun and in order to provide more
predictive developmental information to families, there
was a need for both better, less biased clinical information
about X and Y aneuploidy and the several types of smaller
variations in the short arms of the acrocentric chromo-
somes and variant heterochromatic regions on 1, 9, 16,
and Y. The Yale laboratory had selected a minimal media
(199) for both routine diagnostic studies and for a year-
long study of 4,500 consecutive cord blood and 500
maternal samples. Special attention was given to breaks,
gaps, and chromosome variants in the year-long study.
The study also sought to identify cytogenetic markers
offin-Lowr (RPSKA3, RSK2)
Telecanthus-hypospadias (MID1)Oral-facial-digital I (OFD1)
Spermine synthase deficiency (SMS)XLID-infantile seizures, Rett like (CDKL5, STK9)
Autism (NLGN4)
MIDAS (HCCS)Turner, XLID-hydrocephaly-basal ganglia calcification
VACTERL-hydrocephalus (FANCB)
22.322.2
(AP1S2)
C y
Pyruvate dehydrogenase deficiency (PDHA1)Glycerol kinase deficiency (GKD)
Duchenne muscular dystrophy (DMD)
Ornithine transcarbamoylase deficiency (OTC)Monoamine oxidase-A deficiency (MAOA)Norrie (NDP)
Partington, West, Proud, XLAG (ARX)
Nance-Horan (NHS)
XIDE (Renin receptor; ATP6AP2
OFCD, Lenz microphthalmia (BCOR)
22.1
21.321.221.1
11 4
Ichthyosis follicularis, atrichia, photophobia (MBTPS2)
Chaissaing Lacombe chondrodysplasia (HDAC6)
XLID-nystagmus-seizures (CASK)
MEHMO (EIF2S3)
Aarskog (FGDY)
b ll d i (OPHN
( p )
XLID-choreoathetosis (HADH2)
Stocco dos Santos (SHROOM4, KIAA1202)XLID l ft li / l t (PHF8)
Epilepsy/macrocephaly (SYN1)Cornelia de Lange, X-linked (SMC1L1, SMC1A)
Renpenning, Sutherland-Haan,Cerebropalatocardiac (Hamel),
Golabi-Ito-Hall, Porteous(PQBP1)
11
11.411.3
11.1
11.2311.2211.21
Goltz (PORCN)XLID-macrocephalyJuberg-Marsidi-Brooks
(HUWE1)
-
TARP (RBM10)
-Thalassemia Intellectual DisabilityXLID-hypotonic facies, Carpenter-Waziri,Holmes-Gang, Chudley-Lowry, XLID-arch
(ATRX, XNP, XH2)
Phosphoglycerate kinase deficiency (PGK1)Menkes disease (ATP7A)
XLID-cerebellar dysgenesis -1)-cleft lip/palate
Allan-Herndon (SLC16A2, MCT8) Opitz-Kaveggia FG, Lujan (MED12, HOPA)
XLID-macrocephaly-large ears (BRWD3)
Graham coloboma (IGBP1)
Cantagrel spastic paraplegia (KIAA2022) 13
12
21.121 2
Cornelia de Lange, X-linked (HDAC8)
Pelizaeus-Merzbacher (PLP)Mohr-Tranebjaerg (TIMM8A, DDP)
Lissencephaly, X-linked (DCX)
fingerprints-hypotonia, Smith-Fineman-Myers(?)
XLID-optic atrophy (AGTR2)Arts, PRPP synthetase superactivity (PRPS1)
XLID-short stature-muscle wasting (NXF5)
Mitochondrial encephalopathy (NDUFA1) 23
21.2
21.3
22.122.222.3
XLID-hyperekplexia-seizures (ARHGEF9)
Epilepsy-intellectual disability limited to females (PCDH19)Martin-Probst (RAB40AL)
Wilson-Turner (LAS1L)
XLID-Rolandic seizures (SRPX2)
XLID-hypogonadism-tremor (CUL4B)
Lowe (OCRL1)Simpson-Golabi-Behmel (GPC3) Lesch-Nyhan (HPRT)
Fragile XA (FMR1) MASA spectrum (L1CAM)
Börjeson-Forssman-Lehmann (PHF6)
XLID-growth hormone deficiency (SOX3)
Danon cardiomyopathy (LAMP2)XLID-nail dystrophy-seizures (UBE2A)XLID-macrocephaly-Marfanoid habitus (ZDHHC9)
Christianson, Angelman-like (SLC9A6)
FG/Lujan phenotype (UPF3B)Chiyonobu XLID (GRIA3)
25
26
24
Microcephaly-pachygyria-dysmorphism (NSDHL)
Mucopolysaccharidosis IIA (IDS)Myotubular myopathy (MTM1)
Adrenoleukodystrophy (ABCD1)
Hydrocephaly-
Rett, PPM-X (MECP2)* Incontinentia pigmenti (IKBKG, NEMO)Dyskeratosis congenita (DKC1)
Periventricular nodular heterotopia, Otopalatodigital I, Otopalatodigital II, Melnick-Needles
(FLNA, FLN1)
Creatine transporter deficiency (SLC6A8) *XLID-hypotonia-recurrent infections (MECP2 dup)
Autism (RPL10)28
27
XLID-macrocephaly-seizures-autism (RAB39B)
N-Alpha acetyltransferase deficiency (NAA10)
Figure 1. Genes with Identified Mutations that Cause Syndromal XLID with Chromosomal Band Location
580 The American Journal of Human Genetics 90, 579–590, April 6, 2012
that might correlate directly with clinical conditions.32
Thus, the initial observation that the two brothers referred
to the laboratory because of ID had a consistent chromatid
break or constriction in the distal long arm of a large C
group chromosome was very pertinent to the research
goals of the laboratory. Further study revealed that their
normal mother and two maternal relatives with ID (an
uncle and great uncle of the boys) had the same marker
X chromosome.
The pedigree was, of course, consistent with X-linked ID.
Studies with H3 thymidine showed that the late repli-
cating, large C group chromosome was the same as the
chromosome with the apparent breaks and secondary
constrictions. The data led to the conclusion that ‘‘either
the secondary constriction itself or a closely linked
recessive gene may account for the pattern of X-linked
inheritance’’.4 This was, in fact, probably the first precise
localization of a gene associated with human disease. The
fragile X locus was subsequently defined as an uncoiled
region (secondary constriction) by electron microscopy.33
Studies from a number of laboratories would provide a
more precise confirmation and molecular characterization
22.322.2
22.1
CDKL5 (STK9)
( )ARX (29,32,33,
NLGN4
RPSKA3 (RSK2) (19)AP1S2 (59)CLCN4 (49)
21.321.221.1
11.411.311.23
IL1RAPL1 (21,34)( , , ,
36,38,43,54,76)
TM4SF2 (58)
PQBP1 (55)ZNF81 (45)ZNF674 (92)
(9 44)
ZNF41 (89)
13
1112
11.1
11.2211.21
OPHN1 (60)
FGDY
( )FTSJ1 (9,44)KDM5C (SMX, JARID1C)
DLG3 (8, 90)
SLC16A2 (MCT8)NLGN3
KLF8 (ZNF741)
HUWE1 (17, 31)**
IQSEC2 (1,18)
21.121.2
21.3
22.1ACSL4 (FACL4) (63 68)
ZDHHC15 (91)
SRPX2
MAGT1 (IAP)ATRX (XNP)
25
24
23
22.222.3
PAK3 (30,47)
,
ARHGEF6 ( PIX) (46)
AGTR2 (88)
UPF3B (62)NDUFA1
THOC2 (12)
28
26
27AFF2 (FMR2, FRAXE)
GDI1 (41, 48)MECP2 (16,64,79)*SLC6A8
RAB39B (72)
HCFC1 (3)
*MRX64 is due to a dupMECP2**MRX17 and MRX31 are due to dup HUWE1 and 2 adjacent genes
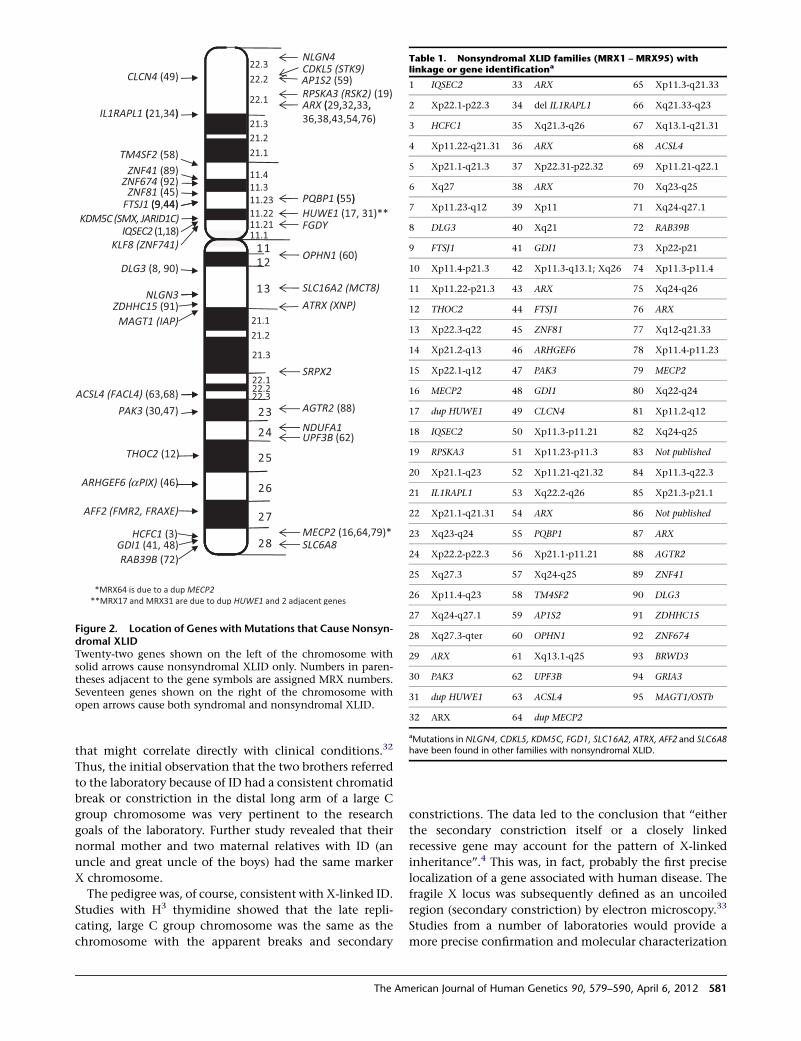
Figure 2. Location of Genes with Mutations that Cause Nonsyn-dromal XLIDTwenty-two genes shown on the left of the chromosome withsolid arrows cause nonsyndromal XLID only. Numbers in paren-theses adjacent to the gene symbols are assigned MRX numbers.Seventeen genes shown on the right of the chromosome withopen arrows cause both syndromal and nonsyndromal XLID.
Table 1. Nonsyndromal XLID families (MRX1 – MRX95) withlinkage or gene identificationa
1 IQSEC2 33 ARX 65 Xp11.3-q21.33
2 Xp22.1-p22.3 34 del IL1RAPL1 66 Xq21.33-q23
3 HCFC1 35 Xq21.3-q26 67 Xq13.1-q21.31
4 Xp11.22-q21.31 36 ARX 68 ACSL4
5 Xp21.1-q21.3 37 Xp22.31-p22.32 69 Xp11.21-q22.1
6 Xq27 38 ARX 70 Xq23-q25
7 Xp11.23-q12 39 Xp11 71 Xq24-q27.1
8 DLG3 40 Xq21 72 RAB39B
9 FTSJ1 41 GDI1 73 Xp22-p21
10 Xp11.4-p21.3 42 Xp11.3-q13.1; Xq26 74 Xp11.3-p11.4
11 Xp11.22-p21.3 43 ARX 75 Xq24-q26
12 THOC2 44 FTSJ1 76 ARX
13 Xp22.3-q22 45 ZNF81 77 Xq12-q21.33
14 Xp21.2-q13 46 ARHGEF6 78 Xp11.4-p11.23
15 Xp22.1-q12 47 PAK3 79 MECP2
16 MECP2 48 GDI1 80 Xq22-q24
17 dup HUWE1 49 CLCN4 81 Xp11.2-q12
18 IQSEC2 50 Xp11.3-p11.21 82 Xq24-q25
19 RPSKA3 51 Xp11.23-p11.3 83 Not published
20 Xp21.1-q23 52 Xp11.21-q21.32 84 Xp11.3-q22.3
21 IL1RAPL1 53 Xq22.2-q26 85 Xp21.3-p21.1
22 Xp21.1-q21.31 54 ARX 86 Not published
23 Xq23-q24 55 PQBP1 87 ARX
24 Xp22.2-p22.3 56 Xp21.1-p11.21 88 AGTR2
25 Xq27.3 57 Xq24-q25 89 ZNF41
26 Xp11.4-q23 58 TM4SF2 90 DLG3
27 Xq24-q27.1 59 AP1S2 91 ZDHHC15
28 Xq27.3-qter 60 OPHN1 92 ZNF674
29 ARX 61 Xq13.1-q25 93 BRWD3
30 PAK3 62 UPF3B 94 GRIA3
31 dup HUWE1 63 ACSL4 95 MAGT1/OSTb
32 ARX 64 dup MECP2
aMutations inNLGN4, CDKL5, KDM5C, FGD1, SLC16A2, ATRX, AFF2 and SLC6A8have been found in other families with nonsyndromal XLID.
The American Journal of Human Genetics 90, 579–590, April 6, 2012 581

of the location in the ensuing decade34–36 and identifica-
tion of the gene itself in 1991.37–40
In addition, the juxtaposition and timing of the family
study and the population survey permitted us to look for
the marker X in 5,000 individuals and over 30,000 cells
and to conclude tentatively that it was not a common
marker or variant because not even one marker X cell
was observed. Another family with a similar chromosomal
appearance at distal 16q was also ascertained in this same
interval. This was inherited in an autosomal-dominant
manner and not associated with a disease. We were, there-
fore, able to make the preliminary conclusion that such
markers did not necessarily indicate disease but that the
marker X was a significant clinical marker for a Mendelian
disease and hence a new and useful tool.
Observations in the 1970s and 1980s
More complex and folic-acid-enriched media become
popular during the 1970s and presumably made detection
of the fragile X increasingly difficult. Most early studies
gave variable results and were not published. The initial
report was confirmed by Giraud et al.34 and Harvey
et al.35 These articles and the report by Sutherland36 estab-
lished that folic acid in the culture media prevented the
expression and detection of the fragile X.
During the 1980s it became clear that a majority of XLID
families did not have fragile X, and the identification and
study of large non-fragile X XLID families with linkage
analysis began in earnest. Large scale studies began across
the globe at this time. The results summarized in Table 1,
Tables S1 and S2, and Figures 1, 2, and 3 are, therefore,
based on about 20 years of clinical and molecular studies.
Methodologies Quicken the Pace of Gene Discovery
Besides the cytogenetic methods used in the diagnosing
and confirmation of fragile X, a number of strategies
have been utilized to identify XLID genes (Table S1 and
Figures 1 and 2). Prior to 1990, these were limited to the
pursuit of genes in cases where the gene products (enzymes
in all cases: HPRT [MIM 308000], PGK1 [MIM 311800],
OTC [MIM 311250] , and PDHA1 [MIM 300582]) were
known, the molecular pathway was known (PLP [MIM
300401]) or a chromosome aberration had localized the
candidate region (DMD [MIM 300377]). Over the next
decade and a half, exploitation of chromosome rearrange-
ments and linkage coupled with candidate gene testing
dominated the field. In the past several years, X chromo-
some sequencing, microarrays (expression and genomic),
and exploration of molecular pathways have added to
the range of technologies available for XLID gene identifi-
cation. Five of the first seven gene identifications were
accomplished with a combination of known metabolic
pathways and tissue culture studies in families with inborn
errors of metabolism (Figure 4). The first identification,
Lesch-Nyhan syndrome due to mutations in HPRT, was re-
ported in 198341 and the most recent was the creatine
Aicardi
Bertini
22
Dessay
CMT, lonasescu variant
Prieto
21
XLID-blindness-seizures-spasticityWieacker-Wolff Miles-Carpenter
11
1112
Goldblatt spastic paraplegiaXLID spastic paraplegia, type 7
XLID-macrocephaly-macroorchidism
13
21
AbidiShrimpton
XLID-telecanthus-deafness
XLID-hypogammaglobulinemia23
24
22
Ahmad MRXS7
CMT, Cowchock variantXLID-panhypopituitarism
Christian
25
26
27 XLID-coarse facies
Vitale: aphasia-coarse facies
Gustavson
CraniofacioskeletalHypoparathyroidism, X-linked
ArmfieldWaisman-LaxovaHereditary bullous dystrophy
28
XLID-microcephaly-testicular failure
Figure 3. Approximate Linkage Limits for XLID Syndromes for which the Genes Have Not Been Identified
582 The American Journal of Human Genetics 90, 579–590, April 6, 2012

transporter syndrome (MIM 300352) due to mutations in
SLC6A8 [MIM 300036].42 Mutations in seven genes were
identified by this methodology.
Two workhorse approaches have been responsible for
the great majority of subsequent gene identifications.
The first of these, based on the ascertainment of a patient
with both ID and a chromosomal rearrangement involving
the X chromosome, was used successfully in identifying
the gene associated with Duchenne muscular dystrophy
in 1987. A total of 31 genes (Table S1 and Figure 4) had
been identified by the middle of 2011 with this approach.
The second and most productive ‘‘workhorse’’ approach,
linkage study of XLID families followed by molecular
analysis of appropriate candidate genes, was employed
initially by a number of investigators in detecting and
characterizing FMR1 (MIM 309550). Subsequently, its use
has resulted in the identification of 43 mutant X genes.
With increasing ease of sequencing, the pace of gene iden-
tification by this route accelerated after 2003, as shown in
Table S1 and Figure 4.
The availability of brute force sequencing capability after
completion of the Human Genome Project has brought an
additional effective method of gene identification, and 21
have been reported since 2006 (Table S1 and Figure 4).
Whether sequencing of large series of sporadic males,
male siblings, or families with clear XLID will prove to be
the most effective use of this resource remains to be deter-
mined. The selection of pedigree-based subjects for
sequencing, however, has the advantage that segregation
of gene alterations can be tested. Since this approach often
permits a relatively straight-forward path to gene identifi-
cation, continued collection of both clinical data and
blood samples remains important. Exploitation of a specific
molecular finding has accounted for four gene identifica-
tions (FANCB [MIM 300515], PORCN [MIM 300651],
SMC1A/SM1L1 [MIM 300040], NDUFA1 [MIM 300078]).
Two other new technologies, expression array and array-
comparative genomic hybridization have, surprisingly,
been applied successfully in only two and one instance,
respectively. Expression array was used in combination
with two other methods to discover the role of GRIA3
(MIM 305915) and PTCHD1 (MIM 300828) in ID. Array-
CGH was used in the isolation of the mutant gene in one
nonsyndromal family (HUWE1 [MIM 300697]).43 Many
potentially valuable combinations of array technologies
for screening followed with brute force sequencing can
Figure 4. The Year and Methodology Used to Identify Genes Associated with XLIDThe following abbreviations are used: Exp-Arr ¼ expression microarray. MCGH ¼ genomic microarray. X-seq ¼ gene sequencing.Mol-Fu ¼ follow up of a known molecular pathway. L-can ¼ candidate gene testing within a linkage interval. Chr-rea ¼ positionalcloning based on a chromosome rearrangement. Met-Fu ¼ follow up of a known metabolic pathway.
The American Journal of Human Genetics 90, 579–590, April 6, 2012 583

be envisioned. Detection of a consistent up or downregula-
tion or other abnormality in two or more XLID family
members can certainly be envisioned as a fruitful approach
to the selection of subjects for partial or complete X
sequencing. Two or more approaches were used in combi-
nation in six instances among the 102 gene identifications
shown in Table S1 and Figure 1 (FMR1, MID1 [MIM
602148], SOX3 [MIM 313430], HUWE1, CASK [MIM
300172], and GRIA3). The application of CGH and related
methods in conjunction with a variety of molecular
technologies has increasingly been used to detect du-
plications and deletions of genes associated with XLID
(Figure 5).1,43–56
In spite of the identification of mutations in 102 genes
that result in XLID, the fragile X syndrome continues to
be by far the most frequent XLID syndrome. Whether
the gradual but continuous expansion of the number of
triplet repeats in the large bank of premutation carriers,
which vary from 1/113 in Israel to 1/313–382 in the United
States) plays a role in maintaining its relatively high gene
frequency is unknown.57
Lumping, Splitting, and Reclassification Based on
Gene Discovery: A Model for Future Research
Given the variability and imprecision with which clinical
evaluations are carried out, it is inevitable that some indi-
viduals with X-linked ID will be incorrectly included in
existing diagnostic categories, whereas others will be incor-
rectly excluded. The extent to which individuals and
families can be evaluated is dependent on the setting,
access to historical information, availability and ages of
affected and nonaffected family members, and the ex-
perience and expertise of the observers. Differences in
phenotype can result frommutations in different domains
of a gene and by contributions from the balance of the
genome. The identification of mutations in many genes
associated with XLID has provided the opportunity to
compensate for some of these variables, resulting in the
lumping of entities previously considered to be separate
and the splitting of other entities previously considered
the same. In addition, the phenotypic limits of some
XLID entities were established with some degree of
objectivity.
Several XLID entities have been most instructive. Dis-
covery that mutations in ATRX (MIM 300032) (Xq21.1)
cause alpha-thalassemia ID allowed testing of large
number of males with hypotonic facies, ID, and other
features.58–60 Currently, as shown in Table S1, four other
named XLID syndromes (Carpenter-Waziri, Holmes-
Gang, XLID-Hypotonia-Arch Fingerprints, and Chudley-
Lowry syndromes [MIM 309580]) have been found to be
allelic variants of alpha-thalassemia ID as have certain
families with spastic paraplegia and nonsyndromal
XLID.1,61–65 One family clinically diagnosed as Juberg-
Marsidi syndrome was found to have an ATRX muta-
tion.66,67 This is now known to be based on misdiagnosis
of Juberg-Marsidi syndrome (MIM 300612); indeed, the
original family with this syndrome has a mutation in
HUWE1 at Xp11.22 (Friez et al., 2011, 15th International
Workshop on Fragile X and Other Early-Onset Cognitive
Disorders). One family clinically diagnosed as Smith-
Fineman-Myers syndrome was also found to harbor an
ATRX mutation, but the gene has not been analyzed in
the original family.68–70 A clinically similar condition,
Coffin-Lowry syndrome (MIM 303600), was found to be
separate from alpha-thalassemia ID and due to mutations
in RPS6KA3 (MIM 300075), which encodes a serine-threo-
nine kinase.71
Kalscheuer et al.72 found mutations in PQBP1 (MIM
300463) (Xp11.2) in two named XLID syndromes – Suther-
land-Haan syndrome (MIM 309470) and Hamel cerebropa-
latocardiac syndrome (MIM 309500)—in MRX55 and
two other families with microcephaly and other findings.
Lenski et al.,73 Stevenson et al.,74 and Lubs et al.75 added
Renpenning, Porteous, and Golabi-Ito-Hall syndromes to
the list of XLID syndromes caused by mutations in
PQBP1.73–75 The six phenotypes now attributed to muta-
tions in PQBP1 are now summarized in the allelic variants
of OMIM 300463. As with the ATRX phenotypes, a wide
variety of phenotypic expressions result from different
mutations in PQBP1 and we remain challenged to better
understand the molecular and developmental mecha-
nisms leading to these differences.
Mutations in ARX (MIM 300382) (Xp22.2) were also
found to be an important cause of XLID encompassing
Wagenstaller et al.54, Horn et al.50
Gijsbers et al.49
22.322.2
22.1
Whibley et al.55
F t l 44
21.321.221.1
11.4
Froyen et al.45
royen e a .
Bedeschi et al.481112
11.3
11.1
11.2311.2211.21
Koolen et al.46
13
21.121.2
21 3
Mimault et al.51, Woodward et al.56
Koolen et al.4623
.
22.122.222.3
Koolen et al.46
S l t
25
26
27
24
Solomon et al.53
Van Esch et al.47, Friez et al.43Rio et al.52
28
Figure 5. Location of Segmental Duplications Associated withSyndromal or Nonsyndromal XLID43–56
584 The American Journal of Human Genetics 90, 579–590, April 6, 2012

multiple phenotypes. Alterations, most commonly a 24 bp
expansion of a polyalanine tract, were found in a number
of families with nonsyndromal XLID (MRX29, 32, 33, 36,
38, 43, 54, and 76), an X-linked dystonia (Partington
syndrome [MIM 309510]), X-linked infantile spasms
(MIM 308350) (West syndrome), X-linked lissencephaly
with abnormal genitalia (MIM 300215), hydranencephaly
and abnormal genitalia (MIM 300215), and Proud
syndrome (MIM 300215).76–83
Perhaps the most prominent example of syndrome split-
ting is FG syndrome (MIM 305450). This syndrome,
initially described in 1974 by Opitz and Kaveggia,84 is
manifest by macrocephaly (or relative macrocephaly),
downslanting palpebral fissures, imperforate anus or
severe constipation, broad and flat thumbs and great
toes, hypotonia, and ID. In the ensuing years, the manifes-
tations attributed to FG syndrome have become protean,
but none was pathognomonic or required for the
diagnosis.85–88 As a result, a number of different localiza-
tions on the X chromosome were proposed for FG
syndrome.89–95
In 2007, Risheg et al.96 found a recurring mutation,
c.2881C>T (p.Arg961Trp), in MED12 (MIM 300188) in
six families with the FG phenotype, including the original
family reported by Opitz and Kaveggia.84 In addition to the
above noted manifestations, two other findings, small ears
and friendly behavior, were consistently noted.
Although most individuals who have carried the FG
diagnosis have one or more findings that overlap with
those in FG syndrome, they do not have MED12 muta-
tions.97,98 Some have been found to have mutations in
other X-linked genes (FMR1, FLNA [MIM 300017], ATRX,
CASK, and MECP2 [MIM 300005]), whereas others have
duplications or deletions of the autosomes.97 So great is
the currently existing heterogeneity within FG syndrome
that the vast majority of individuals so designated should
best be considered to have ID of undetermined cause.
In a number of instances, certain gene mutations have
been associated with nonsyndromal XLID, whereas other
mutations within the same genes have caused syndromal
XLID. Mutations in 17 genes that may cause either type
of XLID, depending on the mutation, have been identified
(Figure 2). In some cases (e.g., those with OPHN1 [MIM
300127] and ARX mutations) re-examination has found
syndromal manifestations in families previously consid-
ered to have nonsyndromal XLID.79,99,100
The frequency with which the process of lumping and
splitting in this limited field of investigation has occurred
has been extremely instructive to both clinical and molec-
ular investigators. Moreover, the process of reclassifying
and refining the XLID syndromes in light of the gene iden-
tificationsmay be one of themost important contributions
by medical genetics to clinical medicine. The underlying
mechanisms or pathways by which mutations in different
genes result in similar phenotypes and different mutations
in a single gene result in disparate phenotypes, however,
remain to be fully elucidated.
Improved Understanding of Disease Mechanisms
in XLID Disorders
Analysis of the presently known 102 genes associated with
XLID lends some insight into the numerous molecular
functions in which disruption can lead to cognitive
impairment and impaired brain development.17 Three
major functions are almost equally represented in proteins
encoded by this panel of 102 genes: 22% are involved in
regulation of transcription, 19% in signal transduction,
and 15% in metabolism. Additionally, 15% are compo-
nents of membrane-associated functions. The remainder
are equally distributed (~3%–5%) in seven other cellular
functions: cytoskeleton, RNA processing, DNA metabo-
lism, protein synthesis, ubiquitinization, cell cycle, and
cell adhesion. Regarding their localization within a cell,
the proteins encoded by genes associated with XLID are
almost equally distributed among the four major subcel-
lular fractions: 30% in the nucleus, 28% in the cytoplasm,
18% in the membranes, and 16% in cellular organelles.17
The XLID disorders offer many opportunities for under-
standing the functions of specific genes and their interac-
tions with other genes in producing disease. Studies
involving control of gene expression will necessarily be
especially complex. These have just begun, in part because
of their complexity and the rapid development of new tech-
niques. Only recently, for example, has a preliminary ex-
pressionmicroarray analysis been carried out in twoaffected
fragile X males.101 The study identified over 90 genes with
a greater than 1.5-fold change in expression. Overrepre-
sented genes were involved in signaling (both under-
and overexpression), morphogenesis (underexpression),
and neurodevelopment and function (overexpression).
Although not addressed in this study, the possibility that
a hallmark finding in the fragile X syndrome, enlargement
of the testes, might result from altered control of tubular
growth by a specific target gene is intriguing. One of the
90 genes identified, NUT (nuclear protein in testis [MIM
608963]), which is normally only expressed in the testis,
should be a candidate gene in future studies because the
BRDA-NUT fusion oncogenes are critical growth promoters
in certain aggressive carcinomas.102 Alternatively, a more
general growth-controlling gene might also explain the
prognathism, macrocephaly and large hands which occur
in some individuals with the fragile X syndrome.
Studies directed at understanding the mechanisms
underlying recurring clinical problems in XLID disorders
such as short stature, microcephaly or macrocephaly,
autistic behavior, and structural CNS abnormalities103
are also particularly appealing because they provide an
opportunity both to simultaneously understand critical
pathways, such as in dendrite development and the devel-
opment of XLID structural abnormalities, gene expression,
and phenotype. The association of autism spectrum dis-
order with mutations in at least eight of the 102 genes
listed in Table S1 is of particular current interest. This has
been reported most frequently in the fragile X syndrome
and Rett syndrome but also in disorders resulting from
The American Journal of Human Genetics 90, 579–590, April 6, 2012 585

mutations in NLGN3 (MIM 300336), NLGN4 (MIM
300427), RPL10 (MIM 312173), RAB39B (MIM 300774),
PTCHD1, and MED12. These genes, however, affect a wide
range of functions (Table S1), and the cause of the clinical
overlap is not clear. In nonsyndromal XLID, for example,
mutations have been identified in five genes involved in
the RhoGTPase cycle that affect dendritic outgrowth
(OPHN1, PAK3 [MIM 300142], ARHGEF6 [MIM 300267],
TM4SF2 [MIM 300096], and GDI1 [MIM 300104]) and are
central to the development of the nonsyndromal pheno-
type.1,17,104
The limited imaging and direct studies of macrocephaly,
microcephaly, and cerebellar hypoplasia have recently
been summarized,104 but more extensive application of
anatomical and functional brain imaging and spectros-
copy techniques that can identify variations in specific
brain regions for each disorder, in conjunction with both
clinical observations and psychometric studies, is critically
needed.
Detection of Possible Advantageous Cognitive
and Behavioral Genes
The identification of 102 X-linked genes affecting intelli-
gence has raised the probability that X chromosomal genes
(including XLID genes) might play a particularly impor-
tant role in brain structure and function as well as a specific
role in intelligence and certain cognitive abilities. Clearly,
as discussed at the beginning of this paper, the research
planned and carried out to identify XLID genes and
syndromes over the last several decades might account
for part or even all of this relative excess compared to auto-
somal loci. A number of papers, however, have addressed
the issue of active selection during evolution for X chro-
mosomal localization of important brain and cognitive
genes.2,105,106 The finding that human and mouse X chro-
mosome genes are hyperexpressed in the CNS compared to
autosomal genes provided additional important confirma-
tory data for the hypothesis of positive evolutionary selec-
tion.107 These studies showed not only that there was a
doubling of X chromosome expression (compared to auto-
somes) early in development (leading to dosage compensa-
tion), but overexpression in human CNS tissue and in
mouse CNS tissue increased by 2.83 and 2.53, respec-
tively, compared to expression in somatic tissues. These
observations also support the general idea that X genes
are particularly important for brain development and
function. Mutations significantly improving intellectual,
creative, perceptive, and leadership qualities would be
fully expressed in males and reasonably could have been
positively selected for in a relatively short period of time
in contrast to the negative selection for XLID muta-
tions.108–112 In essence, the XY males may have been the
experimental animal and the XX female, the storage
facility for both advantageous and deleterious mutations.
Medical investigations generally focus on adverse effects
and no organized searches for X-linked pedigrees with
particularly high intellectual or special cognitive talents
have been reported. Thus, the same approach that has
been effective in identifyingXLID syndrome genes, investi-
gating families with an X-linked pattern of intellectual
outliers, might also prove rewarding for studies at the other
end of the intellectual spectrum. What if we selected for
families with an X-linked pattern of high intellectual
accomplishment; special talents in art or music; unique
types of cognitive behavior involving memory, problem
solving, or, indeed, any type of special intellectual accom-
plishment such as Nobel awards in Economics or Physics?
Such families will certainly be uncommon but so are most
XLID disorders. Yet families might be identified if academi-
cians asked the pertinent family history questions during
lunch with colleagues, a dedicated, interactive home page
was available, or notices were placed in journals asking for
information about possible families. The same group of
laboratories that contributed to the data in Table S1 would
be logical sources for referral andmolecular studies because
the necessary cognitive and molecular studies are already
in place. A positive result might be even bemore important
to society than XLID disease description and provide
important insight into human evolution.
Although there is a wide array of pertinent cognitive
tests, these were not designed to detect specific familial
talents. The coapplication of a pedigree analysis with perti-
nent laboratory tests should provide sufficiently precise
initial diagnosis of the affected to carry out linkage and
array or other screening tests successfully. One family
with four to five outstanding individuals over several
generations could provide sufficient data to warrant testing
other families (or even other species) and to begin an iden-
tification process similar to that described in this paper
that has proven successful for XLID. Imagine the prospects
for investigating specific gene-environmental interactions
during learning and development!
Why, other than not having looked seriously, have
we not stumbled upon such families? Perhaps we have.
In the Inaugural Book of the new National Museum
of the American Indian, Native Universe, Voices of Indian
America,113 in which tribal leaders, writers, scholars, and
story tellers describe Indian traditions and heritages, the
following is recounted:
‘‘Story tells us that a group split from the Lenni Lenape,
perhaps a thousand years ago or more. The people then
settled on the Eastern Shore of the Chesapeake, and were
one and the same as the Nanticoke. Then, for some reason,
the first Tayac, Uttapoingassenum, led his people to the
other side of the bay. Upon their arrival, they encountered
peoples who had been living on the land for more than
8,000 years, according to various archeological estimates.
For thirteen generations prior to English settlement, as
told to Jesuit andMoravianmissionaries, the Tayac’s inher-
itance passed from brother to brother and then to the
sister’s sons. Each led the people until his death.’’
The possibility that the Nanticoke had intuitively recog-
nized and employed a quality of leadership that followed
an X-linked pattern of inheritance is intriguing to consider.
586 The American Journal of Human Genetics 90, 579–590, April 6, 2012

Although much progress has been made during the past
four decades, the clinical and molecular delineation of
XLID is far from complete. Perhaps little more than half
of the genes in which mutations will result in XLID have
been identified. The molecular pathways are incompletely
understood, the mechanisms by which brain structure and
function are deranged have not been identified, and with
few exceptions the neurobehavioral profiles and natural
history of the XLID entities have received insufficient
attention. These deficiencies notwithstanding, consider-
able benefits have been gained for individuals with XLID
and their families. Specific molecular tests, including mul-
tigene panels, are now available to more efficiently reach
a diagnosis. Carrier testing, donor eggs, prenatal diagnosis,
and preimplantation genetic testing may be used to
prevent recurrence when a specific genemutation is found.
Through these measures, reproductive confidence may be
restored for families in which XLID has occurred.114
Supplemental Data
Supplemental Data include two tables and can be found with this
article online at http://www.cell.com/AJHG/.
Web Resources
The URLs for data presented herein are as follows:
Greenwood Genetic Center, XLID Update, http://www.ggc.org/
research/molecular-studies/xlid.html
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org/
References
1. Stevenson, R.E., Schwartz, C.E., and Rogers, R.C. (2012).
Atlas of X-Linked Intellectual Disability Syndromes (New
York: Oxford University Press).
2. Skuse, D.H. (2005). X-linked genes and mental functioning.
Hum. Mol. Genet. 14 (Spec No 1), R27–R32.
3. Gecz, J., Shoubridge, C., and Corbett, M. (2009). The genetic
landscape of intellectual disability arising from chromo-
some X. Trends Genet. 25, 308–316.
4. Lubs, H.A. (1969). A marker X chromosome. Am. J. Hum.
Genet. 21, 231–244.
5. Kaiser-McCaw, B., Hecht, F., Cadien, J.D., and Moore, B.C.
(1980). Fragile X-linked mental retardation. Am. J. Med.
Genet. 7, 503–505.
6. Opitz, J.M., and Sutherland, G.R. (1984). Conference report:
International workshop on the fragile X and X-linked intel-
lectual disability. Am. J. Med. Genet. 17, 5–94.
7. Turner, G., Opitz, J.M., Brown, W.T., Davies, K.E., Jacobs,
P.A., Jenkins, E.C., Mikkelson, M., Partington, M.W., and
Sutherland, G.R. (1986). Conference report: Second interna-
tional workshop on the fragile X and on X-linked mental
retardation. Am. J. Med. Genet. 23, 11–67.
8. Neri, G., Opitz, J.M., Mikkelson, M., Jacobs, P.A., Davies, K.,
and Turner, G. (1988). Conference report: Third interna-
tional workshop on the fragile X and X-linked mental
retardation. Am. J. Med. Genet. 30, 1–29.
9. Neri, G., Gurrieri, F., Gal, A., and Lubs, H.A. (1991). XLMR
genes: Update 1990. Am. J. Med. Genet. 38, 186–189.
10. Neri,G.,Chiurazzi,P.,Arena,F.,Lubs,H.A., andGlass, I.A. (1992).
XLMR genes: Update 1992. Am. J. Med. Genet. 43, 373–382.
11. Neri, G., Chiurazzi, P., Arena, J.F., and Lubs, H.A. (1994).
XLMR genes: Update 1994. Am. J. Med. Genet. 51, 542–549.
12. Brown, W.T., Jenkins, E., Neri, G., Lubs, H., Shapiro, L.R.,
Davies, K.E., Sherman, S., Hagerman, R., and Laird, C.
(1991). Conference report: Fourth international workshop
on the fragile X and X-linked mental retardation. Am. J.
Med. Genet. 38, 158–172.
13. Lubs, H.A., Chiurazzi, P., Arena, J.F., Schwartz, C., Traneb-
jaerg, L., and Neri, G. (1996). XLMR genes: update 1996.
Am. J. Med. Genet. 64, 147–157.
14. Lubs, H., Chiurazzi, P., Arena, J., Schwartz, C., Tranebjaerg,
L., and Neri, G. (1999). XLMR genes: Update 1998. Am. J.
Med. Genet. 83, 237–247.
15. Chiurazzi, P., Hamel, B.C., and Neri, G. (2001). XLMR genes:
Update 2000. Eur. J. Hum. Genet. 9, 71–81.
16. Chiurazzi, P., Schwartz, C.E., Gecz, J., and Neri, G. (2008).
XLMR genes: Update 2007. Eur. J. Hum. Genet. 16, 422–434.
17. Ropers, H.H. (2008). Genetics of intellectual disability. Curr.
Opin. Genet. Dev. 18, 241–250.
18. Chelly, J., Khelfaoui, M., Francis, F., Cherif, B., and Bienvenu,
T. (2006). Genetics and pathophysiology of mental retarda-
tion. Eur. J. Hum. Genet. 14, 701–713.
19. Ropers, H.H., and Hamel, B.C. (2005). X-linked mental
retardation. Nat. Rev. Genet. 6, 46–57.
20. Kleefstra, T., and Hamel, B.C. (2006). X-linked mental retar-
dation: Further lumping, splitting and emerging pheno-
types. Clin. Genet. 67, 451–467.
21. Stevenson, R.E., and Schwartz, C.E. (2002). Clinical and
molecular contributions to the understanding of X-linked
mental retardation. Cytogenet. Genome Res. 99, 265–275.
22. Neri, G., and Opitz, J.M. (2000). Sixty years of X-linked
mental retardation: A historical footnote. Am. J. Med. Genet.
97, 228–233.
23. Martin, J.P., and Bell, J. (1943). A pedigree of mental defect
showing sex-linkage. J. Neurol. Psychiatry 6, 154–157.
24. Allan, W., Herndon, C.N., and Dudley, F.C. (1944). Some
examples of the inheritance ofmental deficiency: Apparently
sex-linked idiocy and microcephaly. Am. J. Ment. Defic. 48,
325–334.
25. Bickers, D.S., and Adams, R.D. (1949). Hereditary stenosis of
the aqueduct of Sylvius as a cause of congenital hydroceph-
alus. Brain 72, 246–262.
26. Losowsky,M.S. (1961). Hereditarymental defect showing the
pattern of sex influence. J. Ment. Defic. Res. 5, 60–62.
27. Renpenning, H., Gerrard, J.W., Zaleski, W.A., and Tabata, T.
(1962). Familial sex-linked mental retardation. Can. Med.
Assoc. J. 87, 954–956.
28. Dunn, H.G., Renpenning, H., Gerrard, H.W., Miller, J.R.,
Tabata, T., and Federoff, S. (1963). Mental retardation as
a sex-linked defect. Am. J. Ment. Defic. 67, 827–848.
29. Penrose, L.S. (1938). A clinical and genetic study of 1280 cases
of mental defect. Special Report Series, Medical Research
Council, No. 229 (London: His Majesty’s Stationery Office).
30. Lehrke, R.G. (1974). X-linked mental retardation and verbal
disability. Birth Defects Orig. Artic. Ser. 10, 1–100.
31. Herbst, D.S., and Miller, J.R. (1980). Nonspecific X-linked
mental retardation II: The frequency in British Columbia.
Am. J. Med. Genet. 7, 461–469.
The American Journal of Human Genetics 90, 579–590, April 6, 2012 587

32. Lubs, H.A., and Ruddle, F.H. (1970). Chromosomal abnor-
malities in the human population: estimation of rates based
on New Haven newborn study. Science 169, 495–497.
33. Harrison, C.J., Jack, E.M., Allen, T.D., and Harris, R. (1983).
The fragile X: A scanning electron microscope study. J.
Med. Genet. 20, 280–285.
34. Giraud, F., Ayme, S., Mattei, J.F., and Mattei, M.G. (1976).
Constitutional chromosomal breakage. Hum. Genet. 34,
125–136.
35. Harvey, J., Judge, C., andWiener, S. (1977). Familial X-linked
mental retardation with an X chromosome abnormality. J.
Med. Genet. 14, 46–50.
36. Sutherland, G.R. (1977). Fragile sites on human chromo-
somes: Demonstration of their dependence on the type of
tissue culture medium. Science 197, 265–266.
37. Oberle, I., Rousseau, F., Heitz, D., Kretz, C., Kevys, D., Hana-
uer, A., Boue, J., Bertheas, M.F., and Mandel, J.L. (1991).
Instability of a 550-base pair DNA segment and abnormal
methylation in fragile X syndrome. Science 252, 1097–1102.
38. Bell, M.V., Hirst, M.C., Nakahori, Y., MacKinnon, R.N.,
Roche, A., Flint, T.J., Jacobs, P.A., Tommerup, N., Tranebjaerg,
L., Froster-Iskenius, U., et al. (1991). Physical mapping across
the fragile X: hypermethylation and clinical expression of
the fragile X syndrome. Cell 64, 861–866.
39. Yu, S., Pritchard, M., Kremer, E., Lynch, M., Nancarrow, J.,
Baker, E., Holman, K., Mulley, J., Warren, S., Schlessinger,
D., et al. (1991). Fragile X genotype characterized by an
unstable region of DNA. Science 252, 1179–1181.
40. Verkerk, A.J., Pieretti, M., Sutcliffe, J.S., Fu, Y.H., Kuhl, D.P.,
Pizzuti, A., Reiner, O., Richards, S., Victoria, M.F., Zhang,
F.P., et al. (1991). Identification of a gene (FMR-1) containing
a CGG repeat coincident with a breakpoint cluster region
exhibiting length variation in fragile X syndrome. Cell 65,
905–914.
41. Jolly, D.J., Okayama, H., Berg, P., Esty, A.C., Filpula, D.,
Bohlen, P., Johnson, G.G., Shively, J.E., Hunkapillar, T., and
Friedmann, T. (1983). Isolation and characterization of
a full-length expressible cDNA for human hypoxanthine
phosphoribosyl transferase. Proc. Natl. Acad. Sci. USA 80,
477–481.
42. Salomons, G.S., van Dooren, S.J., Verhoeven, N.M., Cecil,
K.M., Ball, W.S., Degrauw, T.J., and Jakobs, C. (2001).
X-linked creatine-transporter gene (SLC6A8) defect: A new
creatine-deficiency syndrome. Am. J. Hum. Genet. 68,
1497–1500.
43. Friez, M.J., Jones, J.R., Clarkson, K., Lubs, H., Abuelo, D., Bier,
J.A., Pai, S., Simensen, R., Williams, C., Giampietro, P.F., et al.
(2006). Recurrent infections, hypotonia, and mental retarda-
tion caused by duplication of MECP2 and adjacent region in
Xq28. Pediatrics 118, e1687–e1695.
44. Froyen, G., Van Esch, H., Bauters, M., Hollanders, K., Frints,
S.G., Vermeesch, J.R., Devriendt, K., Fryns, J.P., andMarynen,
P. (2007). Detection of genomic copy number changes in
patients with idiopathic mental retardation by high-resolu-
tion X-array-CGH: Important role for increased gene dosage
of XLMR genes. Hum. Mutat. 28, 1034–1042.
45. Froyen, G., Corbett, M., Vandewalle, J., Jarvela, I., Lawrence,
O., Meldrum, C., Bauters,M., Govaerts, K., Vandeleur, L., Van
Esch, H., et al. (2008). Submicroscopic duplications of the
hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiq-
uitin ligase HUWE1 are associated with mental retardation.
Am. J. Hum. Genet. 82, 432–443.
46. Koolen, D.A., Pfundt, R., de Leeuw, N., Hehir-Kwa, J.Y., Nille-
sen, W.M., Neefs, I., Scheltinga, I., Sistermans, E., Smeets, D.,
Brunner, H.G., et al. (2009). Genomic microarrays in mental
retardation: A practical workflow for diagnostic applications.
Hum. Mutat. 30, 283–292.
47. VanEsch,H., Bauters,M., Ignatius, J., Jansen,M., Raynaud,M.,
Hollanders, K., Lugtenberg,D., Bienvenu,T., Jensen, L.R.,Gecz,
J., et al. (2005). Duplication of the MECP2 region is a frequent
causeof severemental retardation andprogressiveneurological
symptoms in males. Am. J. Hum. Genet. 77, 442–453.
48. Bedeschi, M.F., Novelli, A., Bernardini, L., Parazzini, C.,
Bianchi, V., Torres, B., Natacci, F., Giuffrida, M.G., Ficarazzi,
P., Dallapiccola, B., and Lalatta, F. (2008). Association of syn-
dromic mental retardation with an Xq12q13.1 duplication
encompassing the oligophrenin 1 gene. Am. J. Med. Genet.
A. 146A, 1718–1724.
49. Gijsbers, A.C., denHollander, N.S., Helderman-van de Enden,
A.T., Schuurs-Hoeijmakers, J.H., Vijfhuizen, L., Bijlsma, E.K.,
van Haeringen, A., Hansson, K.B., Bakker, E., Breuning,
M.H., and Ruivenkamp, C.A. (2011). X-chromosome duplica-
tions inmales withmental retardation: Pathogenic or benign
variants? Clin. Genet. 79, 71–78.
50. Horn, D., Spranger, S., Kruger, G., Wagenstaller, J., Weschke,
B., Ropers, H.H., Mundlos, S., Ullmann, R., Strom, T.M., and
Kiopocki, E. (2007). Microdeletions and microduplications
affecting the STS gene at Xp22.31 are associated with a
distinct phenotypic spectrum. Medizinische Genetik 19, 62.
51. Mimault, C., Giraud, G., Courtois, V., Cailloux, F., Boire, J.Y.,
Dastugue, B., and Boespflug-Tanguy, O.; The Clinical Euro-
pean Network on Brain Dysmyelinating Disease. (1999).
Proteolipoprotein gene analysis in 82 patients with sporadic
Pelizaeus-Merzbacher Disease: Duplications, the major cause
of the disease, originate more frequently in male germ cells,
but point mutations do not. Am. J. Hum. Genet. 65,
360–369.
52. Rio, M., Malan, V., Boissel, S., Toutain, A., Royer, G., Gobin,
S., Morichon-Delvallez, N., Turleau, C., Bonnefont, J.P.,
Munnich, A., et al. (2010). Familial interstitial Xq27.3q28
duplication encompassing the FMR1 gene but not the
MECP2 gene causes a new syndromic mental retardation
condition. Eur. J. Hum. Genet. 18, 285–290.
53. Solomon, N.M., Ross, S.A., Morgan, T., Belsky, J.L., Hol, F.A.,
Karnes, P.S., Hopwood, N.J., Myers, S.E., Tan, A.S., Warne,
G.L., et al. (2004). Array comparative genomic hybridisation
analysis of boys with X linked hypopituitarism identifies
a 3.9 Mb duplicated critical region at Xq27 containing
SOX3. J. Med. Genet. 41, 669–678.
54. Wagenstaller, J., Spranger, S., Lorenz-Depiereux, B., Kaz-
mierczak, B., Nathrath, M., Wahl, D., Heye, B., Glaser, D.,
Liebscher, V., Meitinger, T., and Strom, T.M. (2007).
Copy-number variations measured by single-nucleotide-
polymorphism oligonucleotide arrays in patients with
mental retardation. Am. J. Hum. Genet. 81, 768–779.
55. Whibley, A.C., Plagnol, V., Tarpey, P.S., Abidi, F., Fullston, T.,
Choma, M.K., Boucher, C.A., Shepherd, L., Willatt, L.,
Parkin, G., et al. (2010). Fine-scale survey of X chromosome
copy number variants and indels underlying intellectual
disability. Am. J. Hum. Genet. 87, 173–188.
56. Woodward, K., Palmer, R., Rao, K., and Malcolm, S. (1999).
Prenatal diagnosis by FISH in a family with Pelizaeus-
Merzbacher disease caused by duplication of PLP gene.
Prenat. Diagn. 19, 266–268.
588 The American Journal of Human Genetics 90, 579–590, April 6, 2012

57. Hantash, F.M.,Goos,D.G.,Tsao,D.,Quan, F., Buller-Burckle,A.,
Peng, M., Jarvis, M., Sun, W., and Strom, C.M. (2010). Qualita-
tiveassessmentof FMR1(CGG)n triplet repeat status innormal,
intermediate, premutation, full mutation, and mosaic carriers
in both sexes: Implications for fragile X syndrome carrier and
newborn screening. Genet. Med. 12, 162–173.
58. Gibbons, R.J., Brueton, L., Buckle, V.J., Burn, J., Clayton-
Smith, J., Davison, B.C., Gardner, R.J., Homfray, T., Kearney,
L., Kingston, H.M., et al. (1995a). Clinical and hematologic
aspects of the X-linked alpha-thalassemia/mental retardation
syndrome (ATR-X). Am. J. Med. Genet. 55, 288–299.
59. Gibbons, R.J., Picketts, D.J., Villard, L., and Higgs, D.R.
(1995b). Mutations in a putative global transcriptional
regulator cause X-linked mental retardation with alpha-
thalassemia (ATR-X syndrome). Cell 80, 837–845.
60. Villard,L.,Bonino,M.C.,Abidi,F.,Ragusa,A.,Belougne, J.,Lossi,
A.M., Seaver, L., Bonnefont, J.P., Romano, C., Fichera, M., et al.
(1999). Evaluationof amutation screening strategy for sporadic
cases of ATR-X syndrome. J. Med. Genet. 36, 183–186.
61. Abidi, F., Schwartz, C.E., Carpenter, N.J., Villard, L., Fontes,
M., and Curtis, M. (1999). Carpenter-Waziri syndrome results
from a mutation in XNP. Am. J. Med. Genet. 85, 249–251.
62. Lossi, A.M., Millan, J.M., Villard, L., Orellana, C., Cardoso,
C., Prieto, F., Fontes, M., and Martınez, F. (1999). Mutation
of the XNP/ATR-X gene in a family with severe mental
retardation, spastic paraplegia and skewed pattern of X inac-
tivation: Demonstration that the mutation is involved in the
inactivation bias. Am. J. Hum. Genet. 65, 558–562.
63. Abidi, F.E., Cardoso, C., Lossi, A.M., Lowry, R.B., Depetris, D.,
Mattei, M.G., Lubs, H.A., Stevenson, R.E., Fontes, M.,
Chudley, A.E., and Schwartz, C.E. (2005). Mutation in the
50 alternatively spliced region of the XNP/ATR-X gene causes
Chudley-Lowry syndrome. Eur. J. Hum. Genet. 13, 176–183.
64. Guerrini, R., Shanahan, J.L., Carrozzo, R., Bonanni, P., Higgs,
D.R., and Gibbons, R.J. (2000). A nonsense mutation of the
ATRX gene causing mild mental retardation and epilepsy.
Ann. Neurol. 47, 117–121.
65. Yntema, H.G., Poppelaars, F.A., Derksen, E., Oudakker, A.R.,
van Roosmalen, T., Jacobs, A., Obbema, H., Brunner, H.G.,
Hamel, B.C., and van Bokhoven, H. (2002). Expanding
phenotype of XNP mutations: Mild to moderate mental
retardation. Am. J. Med. Genet. 110, 243–247.
66. Mattei, J.F., Collignon, P., Ayme, S., and Giraud, F. (1983).
X-linked mental retardation, growth retardation, deafness
and microgenitalism. A second familial report. Clin. Genet.
23, 70–74.
67. Villard, L., Gecz, J., Mattei, J.F., Fontes, M., Saugier-Veber, P.,
Munnich, A., and Lyonnet, S. (1996). XNP mutation in a
large family with Juberg-Marsidi syndrome. Nat. Genet. 12,
359–360.
68. Smith, R.D., Fineman, R.M., and Myers, G.G. (1980). Short
stature, psychomotor retardation, and unusual facial appear-
ance in two brothers. Am. J. Med. Genet. 7, 5–9.
69. Ades, L.C., Kerr, B., Turner, G., and Wise, G. (1991). Smith-
Fineman-Myers syndrome in two brothers. Am. J. Med.
Genet. 40, 467–470.
70. Villard, L., Fontes, M., Ades, L.C., and Gecz, J. (2000). Identi-
fication of a mutation in the XNP/ATR-X gene in a family
reported as Smith-Fineman-Myers syndrome. Am. J. Med.
Genet. 91, 83–85.
71. Trivier, E., De Cesare, D., Jacquot, S., Pannetier, S., Zackai, E.,
Young, I., Mandel, J.L., Sassone-Corsi, P., and Hanauer, A.
(1996). Mutations in the kinase Rsk-2 associated with
Coffin-Lowry syndrome. Nature 384, 567–570.
72. Kalscheuer, V.M., Freude, K., Musante, L., Jensen, L.R.,
Yntema, H.G., Gecz, J., Sefiani, A., Hoffmann, K., Moser, B.,
Haas, S., et al. (2004). Mutations in the polyglutamine
binding protein 1 gene cause X-linked mental retardation.
Nat. Genet. 35, 313–315.
73. Lenski, C., Abidi, F., Meindl, A., Gibson, A., Platzer, M., Frank
Kooy, R., Lubs, H.A., Stevenson, R.E., Ramser, J., and
Schwartz, C.E. (2004). Novel truncating mutations in the
polyglutamine tract binding protein 1 gene (PQBP1) cause
Renpenning syndrome and X-linked mental retardation in
another family with microcephaly. Am. J. Hum. Genet. 74,
777–780.
74. Stevenson, R.E., Bennett, C.W., Abidi, F., Kleefstra, T.,
Porteous, M., Simensen, R.J., Lubs, H.A., Hamel, B.C., and
Schwartz, C.E. (2005). Renpenning syndrome comes into
focus. Am. J. Med. Genet. A. 134, 415–421.
75. Lubs, H., Abidi, F.E., Echeverri, R., Holloway, L., Meindl, A.,
Stevenson, R.E., and Schwartz, C.E. (2006). Golabi-Ito-Hall
syndrome results from a missense mutation in the WW
domain of the PQBP1 gene. J. Med. Genet. 43, e30.
76. Strømme, P., Mangelsdorf, M.E., Scheffer, I.E., and Gecz, J.
(2002). Infantile spasms, dystonia, and other X-linked
phenotypes caused by mutations in Aristaless related
homeobox gene, ARX. Brain Dev. 24, 266–268.
77. Strømme, P., Mangelsdorf, M.E., Shaw, M.A., Lower, K.M.,
Lewis, S.M., Bruyere, H., Lutcherath, V., Gedeon, A.K.,
Wallace, R.H., Scheffer, I.E., et al. (2002). Mutations in the
human ortholog of Aristaless cause X-linked mental retarda-
tion and epilepsy. Nat. Genet. 30, 441–445.
78. Bienvenu, T., Poirier, K., Friocourt, G., Bahi, N., Beaumont,
D., Fauchereau, F., Ben Jeema, L., Zemni, R., Vinet, M.C.,
Francis, F., et al. (2002). ARX, a novel Prd-class-homeobox
gene highly expressed in the telencephalon, is mutated in
X-linked mental retardation. Hum.Mol. Genet. 11, 981–991.
79. Frints, S.G., Froyen, G., Marynen, P.,Willekens, D., Legius, E.,
and Fryns, J.P. (2002). Re-evaluation of MRX36 family after
discovery of an ARX genemutation reveals mild neurological
features of Partington syndrome. Am. J. Med. Genet. 112,
427–428.
80. Kitamura, K., Yanazawa, M., Sugiyama, N., Miura, H., Iizuka-
Kogo, A., Kusaka, M., Omichi, K., Suzuki, R., Kato-Fukui, Y.,
Kamiirisa, K., et al. (2002). Mutation of ARX causes abnormal
development of forebrain and testes inmice and X-linked lis-
sencephaly with abnormal genitalia in humans. Nat. Genet.
32, 359–369.
81. Uyanik, G., Aigner, L., Martin, P., Gross, C., Neumann, D.,
Marschner-Schafer, H., Hehr, U., and Winkler, J. (2003).
ARX mutations in X-linked lissencephaly with abnormal
genitalia. Neurology 61, 232–235.
82. Kato, M., Das, S., Petras, K., Kitamura, K., Morohashi, K.,
Abuelo, D.N., Barr, M., Bonneau, D., Brady, A.F., Carpenter,
N.J., et al. (2004). Mutations of ARX are associated with
striking pleiotropy and consistent genotype-phenotype
correlation. Hum. Mutat. 23, 147–159.
83. Stepp, M.L., Cason, A.L., Finnis, M., Mangelsdorf, M., Holin-
ski-Feder, E., Macgregor, D., MacMillan, A., Holden, J.J., Gecz,
J., Stevenson, R.E., and Schwartz, C.E. (2005). XLMR in MRX
families 29, 32, 33 and 38 results from the dup24mutation in
the ARX (Aristaless related homeobox) gene. BMC Med.
Genet. 6, 16.
The American Journal of Human Genetics 90, 579–590, April 6, 2012 589

84. Opitz, J.M., and Kaveggia, E.G. (1974). Studies of malforma-
tion syndromes of man 33: the FG syndrome. An X-linked
recessive syndrome of multiple congenital anomalies and
mental retardation. Z. Kinderheilkd. 117, 1–18.
85. Opitz, J.M., Richieri-da Costa, A., Aase, J.M., and Benke, P.J.
(1988). FG syndrome update 1988: note of 5 new patients
and bibliography. Am. J. Med. Genet. 30, 309–328.
86. Romano, C., Baraitser, M., and Thompson, E. (1994). A clin-
ical follow-up of British patients with FG syndrome. Clin.
Dysmorphol. 3, 104–114.
87. Ozonoff, S., Williams, B.J., Rauch, A.M., and Opitz, J.O.
(2000). Behavior phenotype of FG syndrome: cognition,
personality, and behavior in eleven affected boys. Am. J.
Med. Genet. 97, 112–118.
88. Battaglia, A., Chines, C., and Carey, J.C. (2006). The FG
syndrome: report of a large Italian series. Am. J. Med. Genet.
A. 140, 2075–2079.
89. Briault, S., Hill, R., Shrimpton, A., Zhu, D., Till, M., Ronce, N.,
Margaritte-Jeannin, P., Baraitser, M., Middleton-Price, H.,
Malcolm, S., et al. (1997). A gene for FG syndrome maps in
the Xq12-q21.31 region. Am. J. Med. Genet. 73, 87–90.
90. Briault, S., Villard, L., Rogner, U., Coy, J., Odent, S., Lucas, J.,
Passage, E., Zhu, D., Shrimpton, A., Pembrey, M., et al.
(2000). Mapping of X chromosome inversion breakpoints
[inv(X)(q11q28)] associated with FG syndrome: A second
FG locus [FGS2]? Am. J. Med. Genet. 95, 178–181.
91. Piluso, G., Carella, M., D’Avanzo, M., Santinelli, R., Carrano,
E.M., D’Avanzo, A., D’Adamo, A.P., Gasparini, P., and Nigro,
V. (2003). Genetic heterogeneity of FG syndrome: a fourth
locus (FGS4) maps to Xp11.4-p11.3 in an Italian family.
Hum. Genet. 112, 124–130.
92. Dessay, S., Moizard, M.P., Gilardi, J.L., Opitz, J.M., Middle-
ton-Price, H., Pembrey, M., Moraine, C., and Briault, S.
(2002). FG syndrome: linkage analysis in two families sup-
porting a new gene localization at Xp22.3 [FGS3]. Am. J.
Med. Genet. 112, 6–11.
93. Jehee, F.S., Rosenberg, C., Krepischi-Santos, A.C., Kok, F.,
Knijnenburg, J., Froyen, G., Vianna-Morgante, A.M., Opitz,
J.M., and Passos-Bueno, M.R. (2005). An Xq22.3 duplication
detected by comparative genomic hybridization microarray
(Array-CGH) defines a new locus (FGS5) for FG syndrome.
Am. J. Med. Genet. A. 139, 221–226.
94. Tarpey, P.S., Raymond, F.L., Nguyen, L.S., Rodriguez, J.,
Hackett, A., Vandeleur, L., Smith, R., Shoubridge, C., Edkins,
S., Stevens, C., et al. (2007). Mutations in UPF3B, a member
of the nonsense-mediated mRNA decay complex, cause syn-
dromic and nonsyndromic mental retardation. Nat. Genet.
39, 1127–1133.
95. Unger, S., Mainberger, A., Spitz, C., Bahr, A., Zeschnigk, C.,
Zabel, B., Superti-Furga, A., and Morris-Rosendahl, D.J.
(2007). Filamin A mutation is one cause of FG syndrome.
Am. J. Med. Genet. A. 143A, 1876–1879.
96. Risheg, H., Graham, J.M., Jr., Clark, R.D., Rogers, R.C., Opitz,
J.M., Moeschler, J.B., Peiffer, A.P., May, M., Joseph, S.M.,
Jones, J.R., et al. (2007). A recurrent mutation in MED12
leading to R961W causes Opitz-Kaveggia syndrome. Nat.
Genet. 39, 451–453.
97. Lyons, M.J., Graham, J.M., Jr., Neri, G., Hunter, A.G.W.,
Clark, R.D., Rogers, R.C., Moscarda, M., Boccuto, L., Simen-
sen, R., Dodd, J., et al. (2009). Clinical experience in the
evaluation of 30 patients with a prior diagnosis of FG
syndrome. J. Med. Genet. 46, 9–13.
98. Clark, R.D., Graham, J.M., Jr., Friez, M.J., Hoo, J.J., Jones, K.L.,
McKeown, C., Moeschler, J.B., Raymond, F.L., Rogers, R.C.,
Schwartz, C.E., et al. (2009). FG syndrome, an X-linked
multiple congenital anomaly syndrome: the clinical pheno-
type and an algorithm for diagnostic testing. Genet. Med.
11, 769–775.
99. Bergmann, C., Zerres, K., Senderek, J., Rudnik-Schoneborn,
S., Eggermann, T., Hausler, M., Mull, M., and Ramaekers,
V.T. (2003). Oligophrenin 1 (OPHN1) gene mutation causes
syndromic X-linked mental retardation with epilepsy, rostral
ventricular enlargement and cerebellar hypoplasia. Brain
126, 1537–1544.
100. Philip, N., Chabrol, B., Lossi, A.M., Cardoso, C., Guerrini, R.,
Dobyns, W.B., Raybaud, C., and Villard, L. (2003). Mutations
in the oligophrenin-1 gene (OPHN1) cause X linked congen-
ital cerebellar hypoplasia. J. Med. Genet. 40, 441–446.
101. Bittel, D.C., Kibiryeva, N., and Butler, M.G. (2007). Whole
genome microarray analysis of gene expression in subjects
with fragile X syndrome. Genet. Med. 9, 464–472.
102. French, C.A., Miyoshi, I., Kubonishi, I., Grier, H.E., Perez-
Atayde, A.R., and Fletcher, J.A. (2003). BRD4-NUT fusion
oncogene: A novel mechanism in aggressive carcinoma.
Cancer Res. 63, 304–307.
103. Stevenson, R.E., and Schwartz, C.E. (2009). X-linked intellec-
tual disability: Unique vulnerability of the male genome.
Dev. Disabil. Res. Rev. 15, 361–368.
104. Renieri, A., Pescucci, C., Longo, I., Ariani, F., Mari, F., and
Meloni, I. (2005). Non-syndromic X-linked mental retarda-
tion: From a molecular to a clinical point of view. J. Cell.
Physiol. 204, 8–20.
105. Zechner, U., Wilda, M., Kehrer-Sawatzki, H., Vogel, W.,
Fundele, R., and Hameister, H. (2001). A high density of
X-linked genes for general cognitive ability: A run-away
process shapinghumanevolution?TrendsGenet.17, 697–701.
106. Graves, J.A., Gecz, J., and Hameister, H. (2002). Evolution of
the human X—a smart and sexy chromosome that controls
speciation and development. Cytogenet. Genome Res. 99,
141–145.
107. Nguyen, D.K., and Disteche, C.M. (2006). Dosage compensa-
tion of the active X chromosome in mammals. Nat. Genet.
38, 47–53.
108. Turner, G., and Partington, M.W. (1991). Genes for intelli-
gence on the X chromosome. J. Med. Genet. 28, 429.
109. Turner, G. (1996). Finding genes on the X chromosome by
which homo may have become sapiens. Am. J. Hum. Genet.
58, 1109–1110.
110. Turner, G. (1996). Intelligence and the X chromosome.
Lancet 347, 1814–1815.
111. Hedges, L.V., and Nowell, A. (1995). Sex differences in
mental test scores, variability, and numbers of high-scoring
individuals. Science 269, 41–45.
112. Lubs, H.A. (1999). The other side of the coin: a hypothesis
concerning the importance of genes for high intelligence
and evolution of the X chromosome. Am. J. Med. Genet.
85, 206–208.
113. McMaster, G., and Trafzer, C. (2004). Native Universe, Voices
of Indian America (Washington, DC: Smithsonian and
National Geographic).
114. Turner, G., Boyle, J., Partington, M.W., Kerr, B., Raymond,
F.L., and Gecz, J. (2008). Restoring reproductive confidence
in families with X-linked mental retardation by finding the
causal mutation. Clin. Genet. 73, 188–190.
590 The American Journal of Human Genetics 90, 579–590, April 6, 2012

ARTICLE
On Sharing Quantitative Trait GWAS Resultsin an Era of Multiple-omics Data and the Limitsof Genomic Privacy
Hae Kyung Im,1,* Eric R. Gamazon,2 Dan L. Nicolae,2,3,4 and Nancy J. Cox2,3,*
Recent advances in genome-scale, system-level measurements of quantitative phenotypes (transcriptome, metabolome, and proteome)
promise to yield unprecedented biological insights. In this environment, broad dissemination of results from genome-wide association
studies (GWASs) or deep-sequencing efforts is highly desirable. However, summary results from case-control studies (allele frequencies)
have been withdrawn from public access because it has been shown that they can be used for inferring participation in a study if the
individual’s genotype is available. A natural question that follows is how much private information is contained in summary results
from quantitative trait GWAS such as regression coefficients or p values. We show that regression coefficients for many SNPs can reveal
the person’s participation and for participants his or her phenotype with high accuracy. Our power calculations show that regression
coefficients contain as much information on individuals as allele frequencies do, if the person’s phenotype is rather extreme or if
multiple phenotypes are available as has been increasingly facilitated by the use of multiple-omics data sets. These findings emphasize
the need to devise a mechanism that allows data sharing that will facilitate scientific progress without sacrificing privacy protection.
Introduction
Homer et al.1 showed that it is possible to detect an individ-
ual’s presence in a complex genomic DNA mixture even
when the mixture contains only trace quantities of his or
her DNA. The study considered the implications of its find-
ings, motivated originally as an application to forensic
science, in the context of genome-wide association studies
(GWASs) fromwhich aggregate allele frequencies for a large
number of markers were being made publicly available.
Shortly after this publication, a reduction in open access
to aggregate GWAS results was implemented. Jacobs et al.2
presented an improved method using a likelihood
approach and showed that disease status could be inferred
for participants of the study. Visscher et al.3 and Sankarara-
man et al.4 calculated power estimates to understand the
limits of individual detection from sample allele frequen-
cies. They showed that the power to detect membership is
determined by the ratio between the number of markers
and the number of participants in the study.
Wepresent amethod that can infer an individual’s partic-
ipation in a study when regression coefficients from
quantitative phenotypes are available. This problem is
especially relevant now that genome-wide system-level
measurements of quantitative phenotypes (transcriptome,
proteome, and metabolome) are being widely collected
and analyzed. Undoubtedly, disseminating results from
quantitative GWAS and deep-sequencing efforts could be
of enormous benefit to research groups working on related
traits. We explore several statistics that can discriminate
study participants from nonparticipants. Notably, we find
that the use of only the direction of effects (signs of the
coefficients) enables membership inference with good
accuracy. We show the results from applying the statistics
to the Genetics of Kidneys in Diabetes (GoKinD) data
set5,6 to illustrate the level of information contained in
aggregate data. We also provide quantification of the infor-
mation content by computing the power of the method.
Furthermore, we discuss a general framework that can be
used for integrating our findings and earlier studies of
genomic privacy based on sample allele frequencies. With
the increasing use of high-throughput technologies to inte-
gratemultiple-omics data sets, these various statistics result
in a more powerful approach to the identification problem
than with the use of a single phenotype.
Material and Methods
Let us assume that we have the estimated regression coefficients
for M independent SNPs, that we use data on n individuals in a
GWAS (test sample), and that we also have the allelic dosage for
n� individuals from a reference population such as HapMap7,8 or
1000 Genomes Project.9
Membership Inference MethodWe define a statistic (a function of available data) that has a
different distribution depending on the membership status and
use this difference to infer membership. We compute this statistic
for the individual of interest, I, and for all individuals in the refer-
ence population. If the statistic falls well within the reference
distribution we will conclude that the individual is not likely to
have participated in the study, and if the statistic falls in the
extremes of the distribution, we will conclude that the individual
did participate in the study.
1Department of Health Studies, University of Chicago, Chicago, IL, 60637, USA; 2Department of Medicine, University of Chicago, Chicago, IL, 60637, USA;3Department of Human Genetics, University of Chicago, Chicago, IL, 60637, USA; 4Department of Statistics, University of Chicago, Chicago,
IL, 60637, USA
*Correspondence: [email protected] (H.K.I.), [email protected] (N.J.C.)
DOI 10.1016/j.ajhg.2012.02.008. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 591–598, April 6, 2012 591

Let bY be defined as
bYI ¼ n
M
XMj¼1
bbj
�XI;j � bXj
�; (Equation 1)
where XI;j is the allelic dosage of individual I at SNP j, bbj is the
estimated coefficient from fitting the model Yi ¼ aj þ bjXi;j þ ei,
and bXj is the estimated mean of allelic dosage (twice the allele
frequency) for SNP j computed with the reference group.
Conditional Mean and Variance of bYThe expected value and the variance of the statistic bYI conditional
on the individual’s genotype XI and demeaned phenotype YI � m
and membership status (in or out) are as follows:
E½bY jXI ;YI ; in�zðYI � mÞE½bY jXI ;YI ; out�z0
Var½bY jXI ;YI ; in�zs2 n
M
Var½bY jXI ;YI ; out�zs2 n
M
; (Equation 2)
where s2 is the variance of the phenotype, and m is the population
mean of the phenotype Y. Note that for the method to work we do
not need to make use of these expressions nor do we need to know
s2 and m because we rely on the empirical distribution from the
reference population to determine membership. These expres-
sions will serve to estimate the power of the method.
Unconditional on YI, the variance of the statistic bY is given by
Var�bY� jXI ; inzs2:
In computing these quantities we assume that the number of
markers is much larger than the number of individuals in the
test sample and the number of individuals in the reference group:
M >> n >> 1 andM >> n� >> 1. Hardy Weinberg equilibrium is
assumed. To derive these expressions, we used standard Taylor
expansions and the law of iterative expectations. We tested the
validity of these for finite samples (n between 100 and 1,000 and
M=n between 1,000 and 50,000) by fitting linear regressions
with simulated genotypes and phenotypes and computing the
sample mean and variances of the bY statistic. See Supplemental
Data, available online, to find plots of the validation.
Power of the MethodTo compute power, we define the null and alternative hypothesis.
Under the null hypothesis the individual did not participate
in the study (nor did any relatives of the individual), whereas under
the alternativehypothesis, the individual didparticipate.Using the
mean and variance under the null hypothesis and the correspond-
ingmean and variance under the alternative hypothesis computed
in Equation 2 and assuming M >> n >> 1; M >> n� >> 1,
normality of the statistic bY , and the sign of YI � m to be known,
the power will be approximately given by
powerzF
jYI � m j
s
ffiffiffiffiffiM
n
r� za
!; (Equation 3)
where a is the type I error, zx ¼ F�1ð1� xÞ is the ð1� xÞ-quantile ofthe normal distribution, and F is the normal cumulative distribu-
tion function. If the sign of bY � m is not known, a two-sided test
will be used in the derivation and the power will be given by
powerzF
jYI � m j
s
ffiffiffiffiffiM
n
r� za=2
!: (Equation 4)
See derivation in Appendix A. Because F is a strictly increasing
function the power
d increases when M, the number of SNPs, increases
d decreases when n, the study’s sample size, increases
d increases when the individual’s phenotype deviates more
from the mean (scaled by the standard deviation)
d increases when a, the type I error, increases
To facilitate comparison with Visscher et al.3 and Sankararaman
et al.,4 let us express the one-sided power Equation 3 with the
following (equivalent) implicit formula
ðza þ zbÞ2z�YI � m
s
�2M
n; (Equation 5)
where 1� b is the power (note that in Sankararaman et al.4 b is
defined as the power). Recall that in Visscher et al.3 and Sankarara-
man et al.4 power was given implicitly by
ðza þ zbÞ2zM
n: (Equation 6)
Thus, the only difference between Equations 5 and 6 is the factor
ððYI � mÞ=sÞ2. If the phenotype of the person deviates more than
one standard deviation away from the mean, i.e., jYI � mj > s
and the sign of YI � m is known, the power when regression
coefficients are used is larger than it is when allele frequencies
are used. If the person’s phenotype is close to the mean, then
the power will be much diminished. Although expectations are
computed conditional on YI � m, we do not need to know its
magnitude in order to achieve this power. However, we do need
to know the sign of YI � m in order to keep the test one-sided.
If the sign is not used, jYI � mj would need to be 1þ �ðza=2�zaÞ=
ffiffiffiffiffiffiffiffiffiffiM=n
p �times greater than the standard deviation in order
to achieve greater power than the allele frequency case. As an
example, if a ¼ 0:05 and M=n ¼ 100, jYI � mj would need to be
greater than 1.031 times s.
Individual Contribution to the Regression CoefficientIn order to get an intuitive understanding of the contribution
of each individual from the sample, we can decompose the esti-
mated regression coefficient into roughly the sum of individual
contributions:
bbj ¼�~X
0j~Xj
��1~X
0j~Y
bbj z1
ns2j
~XI;j~YI þ 1
ns2j
XisI
~Xi;j~Yi
bbj z ~bI;j þPisI
~bi;j
; (Equation 7)
defining ~bi;j ¼ ð1=ns2j Þ ~Xi;j~Yi as the individual contribution to the
regression coefficient and s2j as the variance of the allelic dosage
(under Hardy Weinberg assumption s2j ¼ 2pjð1� pjÞ where pj is
the minor allele frequency of SNP j). We use the tilde ~X for the
demeaned variable that uses themean from the sample. It is worth
comparing with the decomposition for the case whenminor allele
frequencies for the sample are available: bpjzðpI;j=nÞ þP
isI ðpi;j=nÞ,where bpj is the sample minor allele frequency and pi;j is the allelic
dosage divided by 2 of individual i for SNP j. This similarity gives
an intuitive understanding of the corresponding similarity in the
dependence of power on the ratio of the number of SNPs and
sample size of the study.
592 The American Journal of Human Genetics 90, 591–598, April 6, 2012

Combining Multiple PhenotypesIf results from multiple phenotypes such as eQTL (or other omics
data) results are available, we can combine the information
regarding the individual’s membership by using a Fisher type of
method (the sum of logarithms of p values).10
For each phenotype k, we can compute an empirical p value, pk,
defined as the proportion of reference individuals with magnitude
of the jbY j greater than the individual’s jbYI j. We can combine
p values across different phenotypes by computing
�2Xnphenok¼1
log10 pk
where npheno is the number of phenotypes to be combined. In
addition to accumulating evidence across phenotypes, this
method avoids the problem of lack of power due to one particular
phenotype being close to the population mean.
Covariate AdjustmentUsually other covariates such as age, sex, etc. are adjusted for
when performing GWASs. If the allelic dosage is independent of
the covariates (as will likely be the case for most SNPs) bY will
converge to the covariate-adjusted phenotype instead of the actual
phenotype. The standard deviation might change if the covariates
explain a substantial portion of the phenotypic variability.
However, the method will still work because under no participa-
tion bY will still be around 0, whereas if the individual participated
in the study, bY will converge to the covariate-adjusted phenotype.
Themethod does not require knowing the actual phenotype and it
will work relative to this adjusted phenotype. For the purpose of
re-identification using our method, the presence of covariates is
only a nuisance and no additional power is achieved when they
are present.
Sample Correlation StatisticEquation 7 suggests that the sample correlation between the esti-
mated beta and the individual’s genotype might be useful because
we would expect the correlation to be 0 if the individual was not in
the sample and different from 0 if the individual was part of the
study.
bC ¼
PMj¼1
�bbj � b��XI;j � bXj �XI � bX�ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiP
j
�bbj � b�2P
j
�XI;j � bXj �XI � bX�2s ;
where the long bar above an expressionmeans the samplemean of
the expression.
Sign StatisticEquation 7 also shows that the sign of the correlation coefficient
will be slightly more likely to match the sign of the demeaned
allelic dosage if the person participated in the study than other-
wise. Let bS be defined as:
bS ¼XMj¼1
sign�bb� sign�Xi;j � bXj
�We expect that strictly more than 50% of the times the product
signðbbÞ signðXi;j � bXjÞ will be positive (or negative) if the indi-
vidual participated in the study and his or her phenotype is above
(or below) average. By looking at the absolute value of the sign
statistic we expect to gain information on whether the individual
was part of the study or not.
Analysis DetailsWe used the PLINK software11 and filtered out SNP markers that
were not in Hardy Weinberg equilibrium (p < 0.001) and those
that had minor allele frequencies less than 5%. Receiver operating
characteristic (ROC) curves were generated by using the absolute
value of the statistic as the predicting variable and membership
in the sample as the labels by using the ROCR12 package for the
R statistical package.13 We used only individuals who self-reported
as white both for sample and reference.
Results
We show the performance of the statistics defined inMate-
rial and Methods ðbY ; bS; bCÞ by using data from the GoKinD
(Genetics of Kidney Disease) study.5,6 The data set was
downloaded from dbGaP14 and consisted of more than
1,800 probands with long-standing type 1 diabetes, over
300 dichotomous and quantitative phenotypes, and geno-
type from Affymetrix Genome-Wide Human SNPArray 5.0
platform.We used a subset of 1,644 individuals reported to
be Caucasian.
We show results for two of the phenotypes: cholesterol
level and body mass index (BMI). We also tested the
method on a third simulated phenotype and found at least
as good performance. The latter demonstrates that the
method does not depend on any real effect of genotype
on phenotype.
We randomly sampled 100, 500, and 1,000 individuals
from each study’s cohort and performed a GWAS including
only individuals from each random sample. The remaining
individuals were used as reference group. The statistics
ðbY ; bS; bCÞ were computed for both sample and reference
individuals.
Identifiability Statistic and Phenotype Reconstruction
Figure 1 shows bY versus the actual phenotype (rank
normalized cholesterol levels). The blue dots correspond
to individuals in the sample and the black dots correspond
to individuals in the reference group. For individuals in the
sample, bY lies close to the one-to-one line (perfect predic-
tion line), whereas the individuals in the reference popula-
tion lie close to a flat line around 0 (consistent with our
calculations of mean and variances). The sample size was
n ¼ 1; 000 and the number of SNPs was M ¼ 300;000.
The number of reference individuals was 644.
This demonstrates that for individuals who participated
in a study, their phenotype can be reconstructed with high
accuracy using the bY statistic, whereas for nonparticipants
what we get is mostly noise.
Distribution of Statistic by Membership Status
and ROC Analysis
The left panel in Figure 2 shows the distribution of the
absolute value of bY by membership status. As in Figure 1
The American Journal of Human Genetics 90, 591–598, April 6, 2012 593
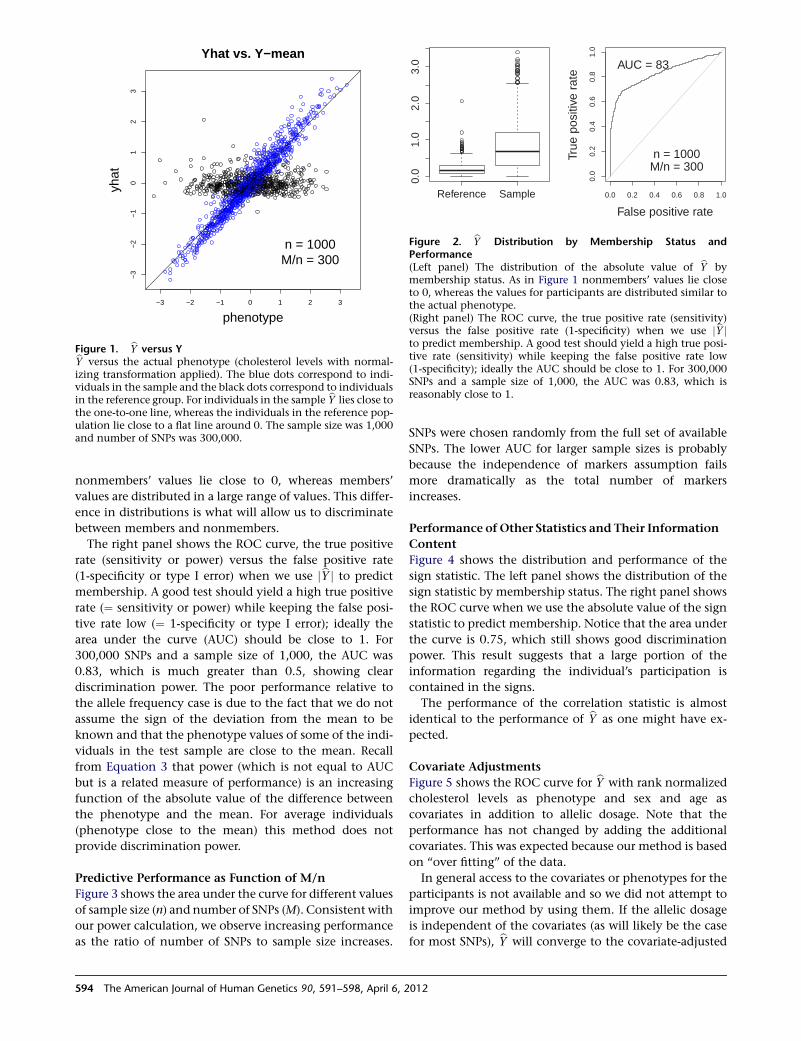
nonmembers’ values lie close to 0, whereas members’
values are distributed in a large range of values. This differ-
ence in distributions is what will allow us to discriminate
between members and nonmembers.
The right panel shows the ROC curve, the true positive
rate (sensitivity or power) versus the false positive rate
(1-specificity or type I error) when we use jbY j to predict
membership. A good test should yield a high true positive
rate (¼ sensitivity or power) while keeping the false posi-
tive rate low (¼ 1-specificity or type I error); ideally the
area under the curve (AUC) should be close to 1. For
300,000 SNPs and a sample size of 1,000, the AUC was
0.83, which is much greater than 0.5, showing clear
discrimination power. The poor performance relative to
the allele frequency case is due to the fact that we do not
assume the sign of the deviation from the mean to be
known and that the phenotype values of some of the indi-
viduals in the test sample are close to the mean. Recall
from Equation 3 that power (which is not equal to AUC
but is a related measure of performance) is an increasing
function of the absolute value of the difference between
the phenotype and the mean. For average individuals
(phenotype close to the mean) this method does not
provide discrimination power.
Predictive Performance as Function of M/n
Figure 3 shows the area under the curve for different values
of sample size (n) and number of SNPs (M). Consistent with
our power calculation, we observe increasing performance
as the ratio of number of SNPs to sample size increases.
SNPs were chosen randomly from the full set of available
SNPs. The lower AUC for larger sample sizes is probably
because the independence of markers assumption fails
more dramatically as the total number of markers
increases.
Performance of Other Statistics and Their Information
Content
Figure 4 shows the distribution and performance of the
sign statistic. The left panel shows the distribution of the
sign statistic by membership status. The right panel shows
the ROC curve when we use the absolute value of the sign
statistic to predict membership. Notice that the area under
the curve is 0.75, which still shows good discrimination
power. This result suggests that a large portion of the
information regarding the individual’s participation is
contained in the signs.
The performance of the correlation statistic is almost
identical to the performance of bY as one might have ex-
pected.
Covariate Adjustments
Figure 5 shows the ROC curve for bY with rank normalized
cholesterol levels as phenotype and sex and age as
covariates in addition to allelic dosage. Note that the
performance has not changed by adding the additional
covariates. This was expected because our method is based
on ‘‘over fitting’’ of the data.
In general access to the covariates or phenotypes for the
participants is not available and so we did not attempt to
improve our method by using them. If the allelic dosage
is independent of the covariates (as will likely be the case
for most SNPs), bY will converge to the covariate-adjusted
−3 −2 −1 0 1 2 3
−3−2
−10
12
3
phenotype
yhat
Yhat vs. Y−mean
n = 1000M/n = 300
Figure 1. bY versus YbY versus the actual phenotype (cholesterol levels with normal-izing transformation applied). The blue dots correspond to indi-viduals in the sample and the black dots correspond to individualsin the reference group. For individuals in the sample bY lies close tothe one-to-one line, whereas the individuals in the reference pop-ulation lie close to a flat line around 0. The sample size was 1,000and number of SNPs was 300,000.
Reference Sample
0.0
1.0
2.0
3.0
False positive rate
True
pos
itive
rat
e
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.2
0.4
0.6
0.8
1.0
n = 1000M/n = 300
AUC = 83
Figure 2. bY Distribution by Membership Status andPerformance(Left panel) The distribution of the absolute value of bY bymembership status. As in Figure 1 nonmembers’ values lie closeto 0, whereas the values for participants are distributed similar tothe actual phenotype.(Right panel) The ROC curve, the true positive rate (sensitivity)versus the false positive rate (1-specificity) when we use jbY jto predict membership. A good test should yield a high true posi-tive rate (sensitivity) while keeping the false positive rate low(1-specificity); ideally the AUC should be close to 1. For 300,000SNPs and a sample size of 1,000, the AUC was 0.83, which isreasonably close to 1.
594 The American Journal of Human Genetics 90, 591–598, April 6, 2012
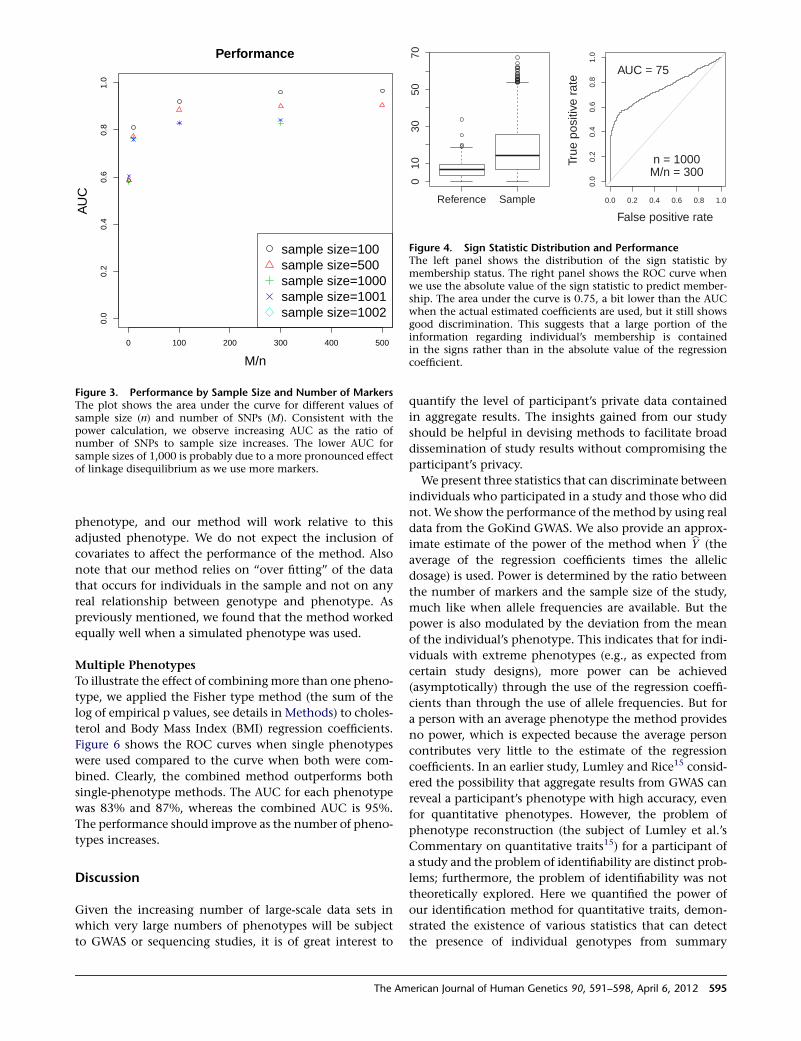
phenotype, and our method will work relative to this
adjusted phenotype. We do not expect the inclusion of
covariates to affect the performance of the method. Also
note that our method relies on ‘‘over fitting’’ of the data
that occurs for individuals in the sample and not on any
real relationship between genotype and phenotype. As
previously mentioned, we found that the method worked
equally well when a simulated phenotype was used.
Multiple Phenotypes
To illustrate the effect of combiningmore than one pheno-
type, we applied the Fisher type method (the sum of the
log of empirical p values, see details in Methods) to choles-
terol and Body Mass Index (BMI) regression coefficients.
Figure 6 shows the ROC curves when single phenotypes
were used compared to the curve when both were com-
bined. Clearly, the combined method outperforms both
single-phenotype methods. The AUC for each phenotype
was 83% and 87%, whereas the combined AUC is 95%.
The performance should improve as the number of pheno-
types increases.
Discussion
Given the increasing number of large-scale data sets in
which very large numbers of phenotypes will be subject
to GWAS or sequencing studies, it is of great interest to
quantify the level of participant’s private data contained
in aggregate results. The insights gained from our study
should be helpful in devising methods to facilitate broad
dissemination of study results without compromising the
participant’s privacy.
We present three statistics that can discriminate between
individuals who participated in a study and those who did
not. We show the performance of themethod by using real
data from the GoKind GWAS. We also provide an approx-
imate estimate of the power of the method when bY (the
average of the regression coefficients times the allelic
dosage) is used. Power is determined by the ratio between
the number of markers and the sample size of the study,
much like when allele frequencies are available. But the
power is also modulated by the deviation from the mean
of the individual’s phenotype. This indicates that for indi-
viduals with extreme phenotypes (e.g., as expected from
certain study designs), more power can be achieved
(asymptotically) through the use of the regression coeffi-
cients than through the use of allele frequencies. But for
a person with an average phenotype the method provides
no power, which is expected because the average person
contributes very little to the estimate of the regression
coefficients. In an earlier study, Lumley and Rice15 consid-
ered the possibility that aggregate results from GWAS can
reveal a participant’s phenotype with high accuracy, even
for quantitative phenotypes. However, the problem of
phenotype reconstruction (the subject of Lumley et al.’s
Commentary on quantitative traits15) for a participant of
a study and the problem of identifiability are distinct prob-
lems; furthermore, the problem of identifiability was not
theoretically explored. Here we quantified the power of
our identification method for quantitative traits, demon-
strated the existence of various statistics that can detect
the presence of individual genotypes from summary
0 100 200 300 400 500
0.0
0.2
0.4
0.6
0.8
1.0
Performance
M/n
AU
C
sample size=100sample size=500sample size=1000sample size=1001sample size=1002
Figure 3. Performance by Sample Size and Number of MarkersThe plot shows the area under the curve for different values ofsample size (n) and number of SNPs (M). Consistent with thepower calculation, we observe increasing AUC as the ratio ofnumber of SNPs to sample size increases. The lower AUC forsample sizes of 1,000 is probably due to a more pronounced effectof linkage disequilibrium as we use more markers.
Reference Sample
010
3050
70
False positive rate
True
pos
itive
rat
e
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.2
0.4
0.6
0.8
1.0
n = 1000M/n = 300
AUC = 75
Figure 4. Sign Statistic Distribution and PerformanceThe left panel shows the distribution of the sign statistic bymembership status. The right panel shows the ROC curve whenwe use the absolute value of the sign statistic to predict member-ship. The area under the curve is 0.75, a bit lower than the AUCwhen the actual estimated coefficients are used, but it still showsgood discrimination. This suggests that a large portion of theinformation regarding individual’s membership is containedin the signs rather than in the absolute value of the regressioncoefficient.
The American Journal of Human Genetics 90, 591–598, April 6, 2012 595
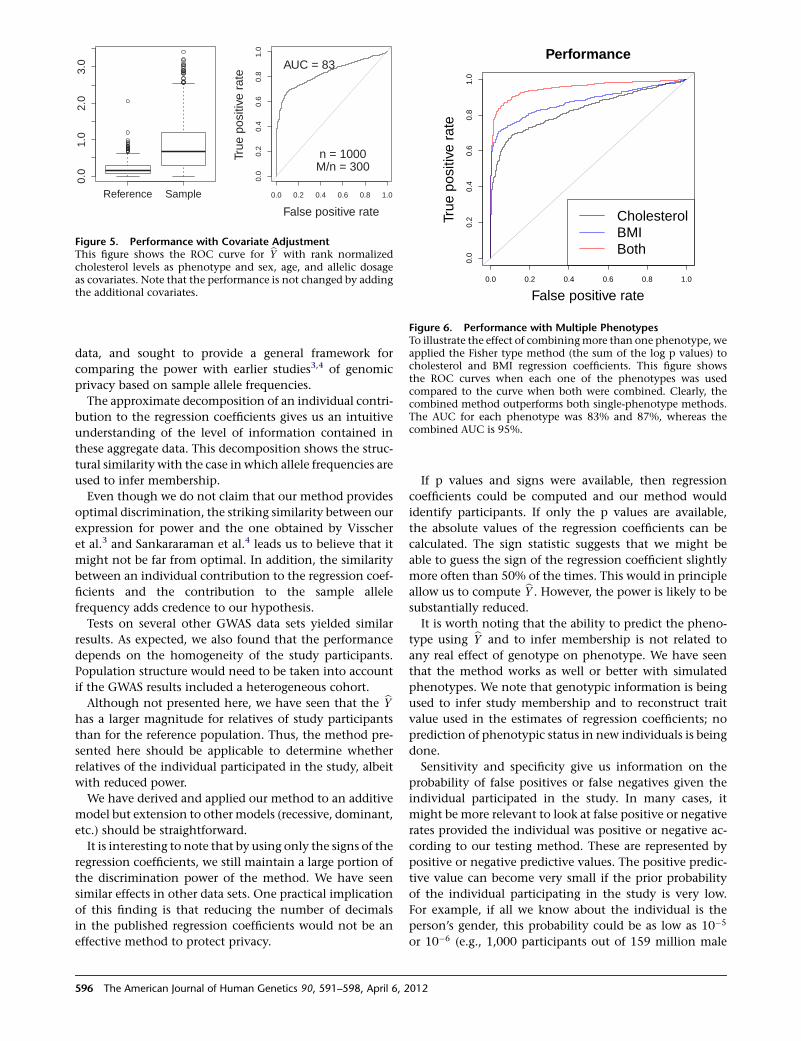
data, and sought to provide a general framework for
comparing the power with earlier studies3,4 of genomic
privacy based on sample allele frequencies.
The approximate decomposition of an individual contri-
bution to the regression coefficients gives us an intuitive
understanding of the level of information contained in
these aggregate data. This decomposition shows the struc-
tural similarity with the case in which allele frequencies are
used to infer membership.
Even though we do not claim that our method provides
optimal discrimination, the striking similarity between our
expression for power and the one obtained by Visscher
et al.3 and Sankararaman et al.4 leads us to believe that it
might not be far from optimal. In addition, the similarity
between an individual contribution to the regression coef-
ficients and the contribution to the sample allele
frequency adds credence to our hypothesis.
Tests on several other GWAS data sets yielded similar
results. As expected, we also found that the performance
depends on the homogeneity of the study participants.
Population structure would need to be taken into account
if the GWAS results included a heterogeneous cohort.
Although not presented here, we have seen that the bYhas a larger magnitude for relatives of study participants
than for the reference population. Thus, the method pre-
sented here should be applicable to determine whether
relatives of the individual participated in the study, albeit
with reduced power.
We have derived and applied our method to an additive
model but extension to other models (recessive, dominant,
etc.) should be straightforward.
It is interesting to note that by using only the signs of the
regression coefficients, we still maintain a large portion of
the discrimination power of the method. We have seen
similar effects in other data sets. One practical implication
of this finding is that reducing the number of decimals
in the published regression coefficients would not be an
effective method to protect privacy.
If p values and signs were available, then regression
coefficients could be computed and our method would
identify participants. If only the p values are available,
the absolute values of the regression coefficients can be
calculated. The sign statistic suggests that we might be
able to guess the sign of the regression coefficient slightly
more often than 50% of the times. This would in principle
allow us to compute bY . However, the power is likely to be
substantially reduced.
It is worth noting that the ability to predict the pheno-
type using bY and to infer membership is not related to
any real effect of genotype on phenotype. We have seen
that the method works as well or better with simulated
phenotypes. We note that genotypic information is being
used to infer study membership and to reconstruct trait
value used in the estimates of regression coefficients; no
prediction of phenotypic status in new individuals is being
done.
Sensitivity and specificity give us information on the
probability of false positives or false negatives given the
individual participated in the study. In many cases, it
might be more relevant to look at false positive or negative
rates provided the individual was positive or negative ac-
cording to our testing method. These are represented by
positive or negative predictive values. The positive predic-
tive value can become very small if the prior probability
of the individual participating in the study is very low.
For example, if all we know about the individual is the
person’s gender, this probability could be as low as 10�5
or 10�6 (e.g., 1,000 participants out of 159 million male
Reference Sample
0.0
1.0
2.0
3.0
False positive rate
True
pos
itive
rat
e
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.2
0.4
0.6
0.8
1.0
n = 1000M/n = 300
AUC = 83
Figure 5. Performance with Covariate AdjustmentThis figure shows the ROC curve for bY with rank normalizedcholesterol levels as phenotype and sex, age, and allelic dosageas covariates. Note that the performance is not changed by addingthe additional covariates. False positive rate
True
pos
itive
rat
e
0.0 0.2 0.4 0.6 0.8 1.0
0.0
0.2
0.4
0.6
0.8
1.0
Performance
CholesterolBMIBoth
Figure 6. Performance with Multiple PhenotypesTo illustrate the effect of combiningmore than one phenotype, weapplied the Fisher type method (the sum of the log p values) tocholesterol and BMI regression coefficients. This figure showsthe ROC curves when each one of the phenotypes was usedcompared to the curve when both were combined. Clearly, thecombined method outperforms both single-phenotype methods.The AUC for each phenotype was 83% and 87%, whereas thecombined AUC is 95%.
596 The American Journal of Human Genetics 90, 591–598, April 6, 2012

individuals from the USA). In this context, given that
the individual was positive in the test, the false negative
rate might still be very high. Naturally, because investiga-
tors have no control over how much prior information
someone can come up with, this argument cannot be
used to ignore the possible breach of confidentiality.
Results from massively parallel sequencing (in the
form of low frequency or rare genetic variations) might
enable increased power of identification. If results from
multiple phenotypes are available, as would be the case
if, for example, gene expression associations were also
conducted (and accompanying results made available),
the information from each phenotype can be combined
to achieve much greater power as suggested by the results
from combining just two phenotypes. Although the
single-phenotype method has no power for individuals
with an average phenotype, it is unlikely a person will
have an average phenotype for all the phenotypes
considered.
A recent study16 of temporal trends in the availability of
results from GWAS classified published studies according
to level of risk for potential misuse and highlights the
ongoing importance of clearer guidelines on how ‘‘data
products’’ can be appropriately shared.
With the increasing trend to collect and analyze
multiple-omics data, the need to share large amounts of
quantitative GWAS results becomes more urgent. In addi-
tion, given our finding that multiple phenotypes can be
combined to increase the power to infer membership, pro-
tecting privacy by limiting the number of significant hits
published is becoming less feasible.
Because fluid sharing of results among researchers for
legitimate scientific use would be highly desirable, our
study emphasizes the urgent need to devise protocols
and methods that facilitate this process without compro-
mising a participant’s privacy.
One mechanism to address this problem would be to
implement an annual certification process, which would
grant the certified researcher unrestricted access to study
results with the condition that the data could only be
used for research goals that do not compromise the partic-
ipants’ privacy. A researcher who does not abide by these
rules could be penalized by withdrawing further access
to data.
Appendix A
Power Calculation
To compute power, we use the same assumptions as for the
conditional mean and variance, i.e., that the number of
markers is much larger than the number of individuals
in the test sample and the number of individuals in the
reference group: M >> n >> 1 andM >> n� >> 1. Hardy
Weinberg equilibrium is assumed. Under these assump-
tions, it can be shown that bY converges to a normal variate
with mean and variance given in Equation 2.
We define the null and alternative hypothesis as follows.
Under the null hypothesis, the individual did not partici-
pate in the study (nor did any relatives of the individual),
whereas under the alternative hypothesis, the individual
did participate.
If the method uses the sign of the difference YI � m,
and we assume that the difference is greater than 0, we
will reject the null hypothesis if bYI is greater than
zasffiffiffiffiffiffiffiffiffiffin=M
p, where a is the type I error and za is the ð1� aÞ
quantile of the normal distribution. The power will be
given by the probability under the alternative thatbYI > zasffiffiffiffiffiffiffiffiffiffin=M
ppower ¼ Pin
�bYI > zas
ffiffiffiffiffin
M
r �
¼ 1� F
0BBB@zas
ffiffiffiffiffin
M
r� ðYI � mÞ
s
ffiffiffiffiffin
M
r1CCCA
(Equation 8)
¼ 1� F
za � YI � m
s
ffiffiffiffiffiM
n
r !(Equation 9)
¼ F
YI � m
s
ffiffiffiffiffiM
n
r� za
!(Equation 10)
where in Equation (8) we have used the fact that bYI is nor-
mally distributed with mean YI � m and variance s2n=M
and in Equation (10) we have used the property of the
normal CDF FðxÞ ¼ 1� Fð�xÞ.If YI � m < 0, similar arguments will give
power ¼ F
�ðYI � mÞ
s
ffiffiffiffiffiM
n
r� za
!:
Thus more generally we have
power ¼ F
jYI � m j
s
ffiffiffiffiffiM
n
r� za
!: (Equation 11)
If the sign of the difference YI � m is not used, the rejec-
tion region will be defined as jbYI j > za=2sffiffiffiffiffiffiffiffiffiffin=M
p. The
alternative distribution will be an equally weighted
mixture of normal distributions with means jYI � mj and�jYI � mj. Note that any weight other than 1/2 would
mean that we have information on whether it is more
likely that the sign is positive or negative. For example, if
we knew it was more likely to be positive, then we would
give higher weight to the normal distribution with mean
jYI � mj. The power when we do not make use of the sign
of jYI � mj is given by
power ¼ Pin
�j bYI j > za=2s
ffiffiffiffiffin
M
r �¼ Pin
�bYI > za=2s
ffiffiffiffiffin
M
r �þ Pin
�bYI < �za=2s
ffiffiffiffiffin
M
r �(Equation 12)
The American Journal of Human Genetics 90, 591–598, April 6, 2012 597

¼ 1
2
0BBB@1� F
0BBB@za=2s
ffiffiffiffiffin
M
r� jYI � m j
s
ffiffiffiffiffin
M
r1CCCA1CCCA
þ 1
2F
0BBB@�za=2s
ffiffiffiffiffin
M
rþ jYI � m j
s
ffiffiffiffiffin
M
r1CCCA
(Equation 13)
¼ 1
2F
0BBB@�za=2s
ffiffiffiffiffin
M
rþ jYI � m j
s
ffiffiffiffiffin
M
r1CCCA
þ 1
2F
0BBB@�za=2s
ffiffiffiffiffin
M
rþ jYI � m j
s
ffiffiffiffiffin
M
r1CCCA
(Equation 14)
¼ F
jYI � m j
s
ffiffiffiffiffiM
n
r� za=2
!: (Equation 15)
Supplemental Data
Supplemental Data include two figures and can be found with this
article online at http://www.cell.com/AJHG/.
Acknowledgments
This work was supported by the Genotype-Tissue Expression
project (R01 MH090937) and the University of Chicago DRTC
(Diabetes Research and Training Center; P60 DK20595). The Go-
KinD study was conducted by the GoKinD investigators and sup-
ported by the Juvenile Diabetes Research Foundation, the Centers
for Disease Control, and the Special Statutory Funding Program for
Type 1 Diabetes Research administered by the National Institute of
Diabetes and Digestive and Kidney Diseases (NIDDK). This manu-
script was not prepared in collaboration with Investigators of the
GoKinD study and does not necessarily reflect the opinions or
views of the GoKinD study or the NIDDK.
Received: November 20, 2011
Revised: January 11, 2012
Accepted: February 8, 2012
Published online: March 29, 2012
References
1. Homer, N., Szelinger, S., Redman, M., Duggan, D., Tembe, W.,
Muehling, J., Pearson, J.V., Stephan, D.A., Nelson, S.F., and
Craig, D.W. (2008). Resolving individuals contributing trace
amounts of DNA to highly complex mixtures using high-
density SNPgenotypingmicroarrays. PLoSGenet. 4, e1000167.
2. Jacobs, K.B., Yeager, M., Wacholder, S., Craig, D., Kraft, P.,
Hunter, D.J., Paschal, J., Manolio, T.A., Tucker, M., Hoover,
R.N., et al. (2009). A new statistic and its power to infer
membership in a genome-wide association study using geno-
type frequencies. Nat. Genet. 41, 1253–1257.
3. Visscher, P.M., and Hill, W.G. (2009). The limits of individual
identification from sample allele frequencies: theory and
statistical analysis. PLoS Genet. 5, e1000628.
4. Sankararaman, S., Obozinski, G., Jordan,M.I., and Halperin, E.
(2009). Genomic privacy and limits of individual detection in
a pool. Nat. Genet. 41, 965–967.
5. Pluzhnikov, A., Below, J.E., Konkashbaev, A., Tikhomirov, A.,
Kistner-Griffin, E., Roe, C.A., Nicolae, D.L., and Cox, N.J.
(2010). Spoiling the whole bunch: quality control aimed at
preserving the integrity of high-throughput genotyping. Am.
J. Hum. Genet. 87, 123–128.
6. Manolio, T.A., Rodriguez, L.L., Brooks, L., Abecasis, G., Ballin-
ger, D., Daly, M., Donnelly, P., Faraone, S.V., Frazer, K., Gabriel,
S., et al; GAIN Collaborative Research Group; Collaborative
Association Study of Psoriasis; International Multi-Center
ADHD Genetics Project; Molecular Genetics of Schizophrenia
Collaboration; Bipolar Genome Study;Major Depression Stage
1 Genomewide Association in Population-Based Samples
Study; Genetics of Kidneys in Diabetes (GoKinD) Study.
(2007). New models of collaboration in genome-wide associa-
tion studies: the Genetic Association Information Network.
Nat. Genet. 39, 1045–1051.
7. International HapMap Consortium. (2003). The international
hapmap project. Nature 426, 789–796.
8. Frazer, K.A., Ballinger, D.G., Cox, D.R., Hinds, D.A., Stuve, L.L.,
Gibbs, R.A., Belmont, J.W., Boudreau, A., Hardenbol, P., Leal,
S.M., et al; International HapMap Consortium. (2007). A
second generation human haplotype map of over 3.1 million
SNPs. Nature 449, 851–861.
9. 1000 Genomes Project Consortium. (2010). A map of human
genome variation from population-scale sequencing. Nature
467, 1061–1073.
10. Fisher, R. (1925). Statistical Methods for Research Workers,
Fifth Edition (Edinburgh: Oliver and Boyd).
11. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira,
M.A.R., Bender, D., Maller, J., Sklar, P., de Bakker, P.I.W.,
Daly, M.J., and Sham, P.C. (2007). PLINK: a tool set for
whole-genome association and population-based linkage
analyses. Am. J. Hum. Genet. 81, 559–575.
12. Sing, T., Sander, O., Beerenwinkel, N., and Lengauer, T. (2005).
ROCR: visualizing classifier performance in R. Bioinformatics
21, 3940–3941.
13. R Development Core Team. (2010). R: A Language and
Environment for Statistical Computing (Vienna: R Founda-
tion for Statistical Computing).
14. Mailman, M.D., Feolo, M., Jin, Y., Kimura, M., Tryka, K.,
Bagoutdinov, R., Hao, L., Kiang, A., Paschall, J., Phan, L.,
et al. (2007). The NCBI dbGaP database of genotypes and
phenotypes. Nat. Genet. 39, 1181–1186.
15. Lumley, T., and Rice, K. (2010). Potential for revealing indi-
vidual-level information in genome-wide association studies.
JAMA 303, 659–660.
16. Johnson, A.D., Leslie, R., and O’Donnell, C.J. (2011).
Temporal trends in results availability from genome-wide
association studies. PLoS Genet. 7, e1002269.
598 The American Journal of Human Genetics 90, 591–598, April 6, 2012

ARTICLE
Resolving the Breakpointsof the 17q21.31 Microdeletion Syndromewith Next-Generation Sequencing
Andy Itsara,1 Lisenka E.L.M. Vissers,2,3 Karyn Meltz Steinberg,1 Kevin J. Meyer,4 Michael C. Zody,5
David A. Koolen,2,3 Joep de Ligt,2,3 Edwin Cuppen,6,7 Carl Baker,1 Choli Lee,1 Tina A. Graves,8
Richard K. Wilson,8 Robert B. Jenkins,4 Joris A. Veltman,2,3 and Evan E. Eichler1,9,*
Recurrent deletions have been associatedwith numerous diseases and genomic disorders. Few, however, have been resolved at themolec-
ular level because their breakpoints often occur in highly copy-number-polymorphic duplicated sequences.We present an approach that
uses a combination of somatic cell hybrids, array comparative genomic hybridization, and the specificity of next-generation sequencing
todeterminebreakpoints that occurwithin segmental duplications. Applyingour technique to the17q21.31microdeletion syndrome,we
used genome sequencing to determine copy-number-variant breakpoints in three deletion-bearing individualswithmolecular resolution.
For two cases, we observed breakpoints consistent with nonallelic homologous recombination involving only H2 chromosomal haplo-
types, as expected. Molecular resolution revealed that the breakpoints occurred at different locations within a 145 kbp segment
of >99% identity and disrupt KANSL1 (previously known as KANSL1). In the remaining case, we found that unequal crossover occurred
interchromosomally between theH1 andH2haplotypes and that this eventwasmediated by ahomologous sequence thatwas once again
missing from the human reference. Interestingly, the breakpoints mapped preferentially to gaps in the current reference genome
assembly, which we resolved in this study. Our method provides a strategy for the identification of breakpoints within complex regions
of the genomeharboringhigh-identity and copy-number-polymorphic segmental duplication. The approach should become particularly
useful ashigh-quality alternate reference sequences becomeavailable andgenome sequencingof individuals’DNAbecomesmore routine.
Introduction
Structural variation, including copy-number variation,
accounts for a significant proportion of human genetic
diversity.1–4 A notable feature of copy-number variation is
the potential for recurrent events to occur at ‘‘hotspots’’
within the human genome as a resultof nonallelic homolo-
gous recombination (NAHR) between repetitive sequences.
Most notable in this regard are segmental duplications
(SDs)—contiguous regions (>1 kbp) with high sequence
identity (>90%).5,6 Recurrent, de novo copy-number vari-
ants (CNVs) have been associated with a variety of pheno-
types, including schizophrenia (MIM 181500),7 autism
(MIM 209850),8 epilepsy (MIM 604827),9 intellectual
disability,10 congenital anomalies (MIM 612474 and
187500),11,12 severe obesity (MIM 613444),13 and renal
disease (MIM 137920).14
Although there have been significant advances in CNV
discovery and genotyping, precise breakpoint delineation
within SDs remains challenging. This information is,
however, essential if we are to further our fundamental
understanding of genome plasticity and processes under-
lying genomic rearrangements. Traditionally, breakpoint
resolution of genomic rearrangements required a combina-
tion of pulse-field gel electrophoresis and Southern blot
analysis to reveal an atypical hybridizing band that
harbored the breakpoint of interest.15,16 Sequence-level
breakpoint identification of the genome has advanced
considerably with more modern molecular methods that
leverage the high quality of the human reference
genome.17 For unique regions, the procedure is relatively
straightforward and typically includes array comparative
genomic hybridization (arrayCGH) followed by long-range
PCR,18 subcloning, and direct Sanger sequencing.19,20
More recently, next-generation methods have allowed
researchers to rapidly capture breakpoints by using split-
read21 and paired-end-read mapping approaches.19,20,22
In contrast, few breakpoints mapping to repetitive
regions, particularly those with large and highly identical
duplications (>10 kbp and >95%), have been cloned
and sequenced.16,23 Unlike unique regions, breakpoints
that map to repeated sequences are much more problem-
atic. Array CGH is unable to localize CNV breakpoints
within blocks of near-perfect sequence identity, which
may span hundreds of kilobases, because of probe cross-
hybridization. Long-range PCR is relatively ineffective
over such large distances of high sequence identity. Simi-
larly, paired-end-read or split-read approaches generally
fail to identify the breakpoints because of short library
inserts and short read lengths that cannot successfully
1Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA; 2Department of Human Genetics, Nijmegen Centre for Molecular
Life Sciences, Radboud University NijmegenMedical Centre, Nijmegen, The Netherlands; 3Institute for Genetic andMetabolic Disease, Radboud University
NijmegenMedical Centre, Nijmegen, The Netherlands; 4Division of Laboratory Genetics, Department of Laboratory Medicine and Pathology, Mayo Clinic,
Rochester, MN 55905, USA; 5Broad Institute, Cambridge, MA 02142, USA; 6Hubrecht Institute, University Medical Center Utrecht, 3584 CT, The
Netherlands; 7Royal Netherlands Academy of Arts and Science, NL-1000 GC Amsterdam, The Netherlands; 8The Genome Institute at Washington Univer-
sity, Washington University School of Medicine, St. Louis, MO 63108, USA; 9Howard Hughes Medical Institute
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.013. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 599–613, April 6, 2012 599

traverse the distances needed to anchor PCR primers to
unique identifiers on either side of the breakpoint. Break-
point resolution is further complicated by both structural
polymorphisms and gaps in the human genome reference
sequence, which often occur precisely at the breakpoints of
interest. Such differences make determination of the true
breakpoint particularly difficult because both variation
and sequences exist at these sites, which are not present
in the human reference sequence.
Here, we present an approach for determining sequence-
level breakpoints occurring within SDs by using a
combination of somatic cell hybrids, array CGH, and
high-throughput sequencing. We take advantage of the
specificity of next-generation sequencing data and the
fact that large duplicated sequences with near-perfect
sequence identity will still carry hundreds of sequence
variants that distinguish the copies. A singly unique nucle-
otide (SUN) identifier is defined as a paralogous sequence
variant (PSV) that tags a specific sequence paralog by
uniquely distinguishing it from all other paralogs in the
human genome. Such variants allow for interrogation of
individual paralogs that are otherwise difficult to distin-
guish. In practice, SUNs are identified from next-genera-
tion sequencing data with SUN k-mers (SUNKs), sequences
that have length k and map to exactly one genomic
location containing one or more SUNs. Previously, we
developed a catalog of these variants, and here we apply
them to define breakpoints24 in individuals. We examine
recurrent microdeletions on 17q21.31, one of the most
structurally complex regions of the genome, as a model
locus. Structural variation at this locus has been exten-
sively characterized, most notably in haplotype-specific
sequence assemblies of the H1 and H2 haplotypes, making
the locus ideal for further study.25,26
Material and Methods
H2 Reference AssemblyAnalysis of the H1 and H2 haplotypes was based on previously
reported haplotype-specific sequence assemblies.26
Generation of Somatic Cell HybridsSomatic cell hybrids were generated at MayoMedical Laboratories.
After electrofusion of Epstein-Barr Virus (EBV) cells with E2 cells,
mouse-human hybrid colonies were observed at 18 days. Subse-
quently, 88 clones were selected for initial expansion and genotyp-
ing. Six A and six B chromosome 17 homologs were selected for
additional subculture. At pass three, all 12 hybrid clones were
tested for chromosome 17 by FISH. On the basis of the FISH
results, two A and two B hybrid clones were selected for confirma-
tory genotyping, and all cases confirmed retention of the appro-
priate A or B genotype. This study was approved by the institu-
tional review board of the University of Washington and
Radboud University, and all subjects provided informed consent.
Sample GenotypingAs previously described,27 H1/H2 genotyping was determined via
gel electrophoresis on the basis of a deletion in intron 9 of MAPT.
After generation of somatic cell hybrids, initial confirmatory
genotyping was performed at Mayo Medical Laboratories
(AFMa061za9, AFM192yh2, AFMa154za9, and AFM044xg3). Addi-
tional markers, AFM298wg5, AFMb364yh9, AFM155xd12, and
AFMa110wb5, were identified as being close to the 17q21.31 dele-
tion on the basis of the Marshfield genetic map,28 and these were
subsequently genotyped at the University of Washington with the
primers specified in the UniSTS marker database. To examine
microsatellites within SDs, we chose a subset of the reported
markers and primers used in a previously reported BAC assembly
of the H2 haplotype.25 After amplification, all microsatellite geno-
types were determined with an ABI 3730 DNA analyzer. All
primers used are listed in Table S1.
Haplotype-Specific Array CGHBy using hybrid cell line DNA, we performed array CGH to
compare the H1, H2, and 17q21.31 deletion-bearing chromo-
somes to one another. Because the hybrid cell lines are haploid
for human chromosome 17, unique regions of the human genome
removed by deletion have an extremely low signal, corresponding
to copy number 0. In contrast, deletions within SDs, regions of the
genome for which there exist additional paralogs, display interme-
diate levels of signal loss proportional to the number of paralogous
copies elsewhere in the genome. Although a mouse genome is
present in hybrid cells, we expected minimal cross-hybridization
because even single mismatches are known to affect probe hybrid-
ization,29–31 and at exons within 17q21.31, the average human-
mouse identity is ~85%, corresponding to nine mismatches on
a 60 bp probe.32 Finally, we visualized array CGH data on the H2
haplotype by remapping probes.26
Array Design and AnalysisWe designed a custom 244K Agilent array specifically to interro-
gate 17q21.31 contained within hybrid cell lines (Table S2; GEO
accession code GSE34867). At the deletion locus and flanking
sequence (NCBI build 36, chr17:40.25M–42.75M), probes were
placed at high density at 1 probe per 100 bp. Sample labeling
was achieved with Roche NimbleGen Dual-Color DNA Labeling
kits according to the manufacturer’s protocol, but half (500 ng)
the input DNA was used, and the protocol was scaled appropri-
ately. For array hybridization, 25 ng each of labeled test and
reference DNA was then brought to a 158 ml volume. Subse-
quently, the labeled DNA was hybridized to a custom Agilent
array according to the Agilent hybridization protocol. In brief,
the recommended hybridization master mix for a 13 microarray
was prepared and added to the labeled DNA, and hybridization
at 65�C on a rotator rack (20 rpm) followed for 72 hr. Array
wash and scanning proceeded according to the manufacturer’s
protocol. However, feature extraction was carried out with a
normalization set consisting of probes on human chromosome
17 but outside of 17q21.31.
Array CGH oligonucleotide probes were remapped to the H2
assembly with BLAST (blastn parameters �e 1e�10 �m 8 �W
7).33 Partial BLAST hits were extended without gaps to encompass
the entire probe sequence, and probes with no BLAST hits were
aligned with JAligner (see Web Resources), an implementation of
the Smith-Waterman algorithm (NUC.4.4 matrix; gap open and
extension penalties were equal to 10). Finally, probes weremapped
to a given location on the H2 assembly if and only if the global
alignment mapped with a %1 bp mismatch and a %1 bp gap.
Using these criteria, we mapped 11,967 distinct probes to 18,914
positions in the H2 assembly. To calculate the haploid copy
600 The American Journal of Human Genetics 90, 599–613, April 6, 2012

number of probes mapping to the H2 assembly, we aligned each
probe to the human genome (build 36), mouse genome (mm8),
and the H2 assembly by using BLAST (with the same parameters
as those used in probe mapping). To avoid double-counting
between the human genome and the H2 assembly, we excluded
human genome BLAST hits to the 17q21 deletion region (chr17:
40799295–42204344). To provide a ceiling on the copy number
of a given probe, we defined a probe’s copy number as the number
of BLAST hits covering R90% of the probe with %3 mismatches
and%1 bp gap. Consistent with a tendency to overestimate probe
copy number, for the 3,231 probes that were within the H2
assembly between 700,000–1,000,000 bp, a region predicted to
be almost entirely unique sequence in a haploid human genome,
99% (3,186/3,231) of probes were predicted to have a copy
number of 1, and the remaining probes were predicted to have
a copy number >1.
We determined copy-number loss at each probe given NAHR
between a particular pair of paralogous sequences. The expected
relative copy number for a given probe was defined as the copy
number of a probe after the deletion divided by the estimated
probe copy number in the H2 assembly. We compared expected
changes in relative copy number to observed log2 ratios to deter-
mine the most likely pair of paralogous sequences mediating
each deletion (Figure S1B).
Gap ClosureTo close gap 2, we used the previously identified BAC RP11-84A7
(AC243906). To close gap 1, we screened for clones mapping to
gap regions by using a method similar to that previously reported
for placing fosmids in the genome.20 We locally aligned fosmid
end sequences to the H1 assembly and H2 pseudo-assembly by
using MegaBLAST.34 Clones under consideration were subse-
quently limited to those with an alignment either within the
spacer sequence (represented in AC217768) or at the proximal
end of AC139677. Local alignments were then extended into
global alignments with needle, a Needleman-Wunsch algorithm
implementation from the EMBOSS software suite.35 We scored
global alignments for mismatches and gaps by only using bases
with Q30 or higher quality. Paired end-sequence placements
were then screened on the basis of concordant clone-end orienta-
tion and estimated insert size. Subsequently, clone-end orienta-
tion and size-concordant placements were assigned to the H1
haplotype, other paralogous sequence in the H2 haplotype, or
sequence that mapped adjacent to or within the proximal gap;
sequence identity was used as a tie-breaker. Importantly, for all
clones chosen, end sequences were best assigned to sequence adja-
cent to the gap or inferred sequence within the gap and not at
paralogous sequence elsewhere in the H1 or H2 assemblies. We
selected three clones for sequencing: two clones extending proxi-
mally and distally from the spacer sequence on AC217768
(1134622_I19 and 50932900_K17; AC244164 and AC244161,
respectively) and one clone (1013914_P2; AC244163) extending
proximally from the proximal end of AC139677 (Figure S2). The
three fosmids and the BAC clone used for closing gaps in the H2
haplotype were sequenced and assembled at The Genome Insti-
tute at Washington University. Consistent with our hypothesized
structure for RP11-374-N3, distal portions of 50932900_K17
(AC244161) and proximal portions of 1013914_P2 (AC244163),
which mapped to gap 1, were paralogous and in direct orientation
to SDs on the H1 and H2 haplotypes proximal to unique deleted
sequence (Figure S2, Figure S3, and Figure S4). Similarly,
1134622_I19 (AC244164) mapped entirely to finished sequence
(all from AC217768; Figure S5) in the H2 assembly and contained
sequence that was paralogous, but of inverted orientation (based
on end-sequence placement), to SDs on the H1 and H2 haplotypes
proximal to unique deleted sequence.
Next-Generation Sequencing, Complete Genome
Sequencing, and Breakpoint Mapping with SUNsMassively parallel sequence data were generated from three
probands with both SOLiD and Illumina sequencing platforms.
Formembers of family 2, longmate-paired libraries were generated
from 100 mg of genomic DNA, which was isolated from peripheral
blood samples via QIAampmini columns (QIAGEN). Library prep-
aration was essentially as described in the SOLiDv3.5 library prep-
arationmanual (Applied Biosystems). Of note, we performed DNA
size selections directly after CAP adaptor ligation to select genomic
fragments between 2 and 3 kbp and, moreover, to reduce the pres-
ence of concatamers. Additionally, we performed a size selection
after library amplification. To assess the presence of adaptors and
determine the average insert sizes, we cloned libraries and chose
384 clones per library for capillary sequencing. Initially, we
sequenced two 50 bp mates for each library (F3 and R3 tags) on
a SOLiD 3PLUS instrument and thereby used a single quadrant
for the father and mother of the sequencing slide, but two quad-
rants for the proband. To obtain additional read depth for the
mother and proband, we subsequently performed a 50-bp-frag-
ment run on the same libraries by using a full sequencing slide
for each on a SOLiD4 instrument.
For the family 1 proband (31928) and family 3 proband (31873),
3 mg of genomic DNA was sheared, end-repaired, an A-tail added,
and adaptors were ligated to the fragments as described in Igartua
et al.36 After ligation, the samples were run on a 6% pre-cast
polyacrylamide gel (Invitrogen, catalog number EC6265BOX).
The band at 400 bp was excised, diced, and incubated. Size-
selected fragments were amplified with 0.5 ml of primers, 25 ml of
23 iProof, 0.25 ml of SYBR Green, and 8.25 ml of dH2O under the
following conditions: 98�C for 30 s, 30 cycles of 98�C for 10 s,
60�C for 30 s, 72�C for 30 s, 72�C for 15 s, and 72�C for 2 min.
Fluorescence was assessed between the 30 and 15 s 72�C step.
Amplified, size-selected libraries were quantified with an Agilent
2100 Bioanalyzer and paired-end sequenced (101 bp reads) on
an Illumina HiSeq 2000.
Using a pipeline similar to that previously described,24 we identi-
fied 36-mer SUNKs that uniquely distinguish paralogs potentially
mediating 17q21.31 deletions in the H2 assembly. We identified
PSVs by one of two methods: First, for sequence present in the
current assembly, we used whole-genome assembly comparison
(WGAC)-defined global alignments to identify single-base-pair
differences between paralogs (Figure S6). Second, for sequence in
the proximal gap, we identified and sequenced fosmids
(AC244161 and AC244163) extending into either side of the gap.
We subsequently identified PSVs from alignment of fosmid draft
sequences against inferred regions of paralogy on the H1 and H2
haplotypes (H1:219,599–261,693 and H2:452,165–261,693,
respectively) by using stretcher, a Needleman-Wunsch algorithm
implementation from the EMBOSS software suite (Figure S6).35
For each identified PSV, we generated all possible 36-mers incor-
porating the variant. Subsequently, we passed the 36-mers
through a series of filters. First, those containing repeat sequence
as identified by RepeatMasker and TandemRepeatFinder37 or those
within 36 bp of such sequence were excluded. Second, we used
mrFAST38 to identify all possible mappings, including that to the
H2 haplotype (GRCh37), of each 36-mer to the mouse (mm8)
The American Journal of Human Genetics 90, 599–613, April 6, 2012 601

and human reference assembly, allowing for up to two mis-
matches, insertions, or deletions (edit distance %2). For PSVs
outside gap 1, we identified SUNKs as those reads with one exact
match in the human reference assembly or the H2 haplotype,
no exact matches to the mouse genome, %10 mrFAST hits with
edit distance %2 in the human genome, and %10 mrFAST hits
with edit distance %2 in the mouse genome. SUNKs within gap
1 were defined similarly, but no matches to the current reference
assembly or H2 haplotype were allowed.
Because of high sequence identity within AC217768 in the
current H2 assembly, relatively few SUNs were identified in
gap 1. However, because all sequence in AC217768 is lost in
NAHR-mediated 17q21.31 deletions, gap 1 PSVs that are only
present elsewhere in the genome within AC217768 are still break-
point-informative for H2/H2 NAHR. Similarly, gap 1 PSVs that are
only present on the H2 haplotype proximal to or within
AC217768 are breakpoint-informative for H1/H2 NAHR. Using
these criteria, we identified additional H1/H2 or H2/H2 break-
point-informative PSVs.
Finally, we empirically validated the presence or absence of SUNs
by using data from the 1000 Genomes Project.39 As a positive
control, we identified candidate SUNKs in the combined sequence
data from nine H1/H2 CEU (Utah residents with ancestry from
northern and western Europe from the CEPH collection) individ-
uals (mean coverage 33), and H2-specific candidate SUNs without
observed mapped reads were excluded. As a negative control, we
identified candidate SUNKs in combined sequence data from
a CEU trio (mean coverage 27.63; NA12878, NA12891, and
NA12892) and from an YRI trio (mean coverage 21x; NA19238,
NA19239, and NA19240), all with H1/H1 genotypes. H2-specific
candidate SUNs were discarded if observed at a read depth above
theminimumH1-specific SUN read depth in two ormore samples.
A similar validation procedurewas carriedout forH1-specific SUNs.
We used next-generation sequencing data from probands to refine
the breakpoints of the rearrangement on the basis of the absence
or presence of reads mapping to these unique identifiers.
Results
We briefly review the structural features of the 17q21.31
microdeletion locus. Within the current reference
assembly (GRCh37), the locus is defined approximately
by chr17:43.4–44.8 Mbp. The locus encompasses ~600 kbp
of unique sequence. This sequence contains several genes,
including MAPT, CRHR1, and KANSL1 (previously known
as KIAA1267), and is flanked by extensive SDs. The
17q21.31 locus has two major structural haplotypes span-
ning ~1.5Mbp: the H1 haplotype, which is most common,
and the H2 haplotype, which is present at a frequency of
20% in Europeans.25,27,40 BAC-based, haplotype-specific
sequence assemblies of the H1 and H2 haplotypes have
previously been created from the BAC library RP11, which
was derived from an H1/H2 individual.26 The reference
assembly at 17q21.31 represents the H1 haplotype, and
the H2 is presented as an alternate haplotype
(chr17_ctg5_hap1). These two haplotypes are distin-
guished by the presence of an approximately 970 kbp
inversion in addition to more than 300 kbp of differences
in the copy number and content of SDs (Figure S7).25,26
Importantly, the H2 haplotype contains 95 kbp of SD in
direct orientation flanking the unique region, whereas no
such sequence is observed in the H1 haplotype. Recurrent
deletions at this locus cause the 17q21.31 microdeletion
syndrome (MIM 610433), in which deletions only arise
in parents with one or more H2-bearing chromo-
somes.41–43 NAHR involving only this H2-specific duplica-
tion is hypothesized to underlie the H2 predisposition to
microdeletion.26
Our goal was to localize the breakpoints of recurrent
17q21.31 deletions in six individuals of European descent.
This set included three families wherein de novo microde-
letions had been previously identified41 and for which
transformed cell lines had been constructed from the
proband and both parents, as well as three unrelated
probands with the 17q21.31 deletion, for further anal-
ysis.9 To assess the accuracy of our experiments, we pro-
ceeded in a series of steps whereby we developed genomic
resources to simplify and validate our findings as needed.
To remove the potential confounding effects of large-scale
differences on different structural haplotypes on chromo-
some 17, we initially isolated deletion-bearing chromo-
somes by using somatic cell hybrids (reviewed in Trask
et al.44) from both the transmitting parent and the
proband (Figure 1). This allowed us to design the ideal
array CGH experiment, where duplicated sequences flank-
ing the critical region could be compared in the isolated
donor and deleted chromosomes (Figure 1B, Figure 2).
Once we refined the location of the paralogous segments
where breakpoints were likely to occur, we focused on
obtaining sequence-level breakpoint resolution in the
three probands with parental information. It then became
necessary to discover and characterize sequence that map-
ped to gaps within the H2 haplotype; the additional
sequence allowed us to attain sequence-level breakpoint
delineation by using a combination of next-generation
sequencing and SUN identifiers.24 This breakpoint delinea-
tion was consistent with results obtained by array CGH of
somatic cell hybrids. These results give us confidence that
genome sequencing of individuals in conjunction with
SUN mapping will provide a robust method for routine
breakpoint characterization in the future.
Somatic Cell Hybrid Characterization
We constructed 36 somatic cell hybrids derived from three
parent-child trios in which the child harbored a de novo
17q21.31 deletion and from three unrelated 17q21.31-
deletion-bearing probands for whom no parental DNA
samples were available (Figure 1; Table S3). H1/H2 haplo-
type status was determined with a previously described
238 bp deletion marker within intron 9 of MAPT.27 In all
three cases for which parental DNA samples were available,
one parent was either homozygous or heterozygous for the
H2 haplotype, and the other was homozygous for the H1
haplotype. For each of the six probands and the parents
containing an H2 haplotype, we constructed at least two
human-mouse somatic cell hybrid cell lines such that
602 The American Journal of Human Genetics 90, 599–613, April 6, 2012

each of the chromosome 17 homologs (referred to as A
and B; see Material and Methods) was isolated. The crea-
tion of somatic cell hybrids isolates the 17q21.31 dele-
tion-bearing chromosome and the progenitor parental
chromosome prior to deletion and thereby facilitates
breakpoint detection (Figure 1).
We initially genotyped the somatic cell hybrids by using
eight microsatellite markers (Figure S8 and Table S3) to
assess the integrity of each chromosome 17 homolog and
confirm that deletions originated from the parent carrying
the H2 haplotype. In family 1, markers immediately flank-
ing the deletion locus in the proband (31928) indicate that
it probably arose as a result of interchromatidal NAHR
(between sister chromatids), as expected. In family 2, the
deletion occurred in the gamete of the mother (31918),
who is homozygous for the H2 chromosomes and is also
suggestive of interchromatidal NAHR. Finally, in family
3, crossover between the H1 and H2 haplotypes and the
17q21.31 deletion co-occur within a genetic distance of
less than 0.54–1.32 cM, as determined by the Marshfield
map and HapMap, respectively.28,45 Because of the short
genetic distance separating the events, these preliminary
results suggested the possibility that unequal crossover
between the H1 and H2 haplotypes generated the deletion
within this family. We tested an additional seven microsa-
tellite markers flanking the deletion locus (Table S3).
The results remained consistent with interchromosomal
but not intrachromatidal NAHR for family 3 (Figures S8
and S9).
Haplotype-Specific Array CGH
We next performed haplotype-specific array CGH by using
matched chromosome 17 hybrid cell lines (Figure 1B;
Material and Methods; GEO accession code GSE34867).
For each family, we hybridized DNA from a line containing
the 17q21.31-deletion-bearing chromosome of the child
against the corresponding H2-haplotype-bearing hybrid
cell line from the parent. As expected, deletions within
the unique portion of 17q21.31 were readily apparent
(relative copy number 0; Figure 2). Deletions within the
SDs were detectable but displayed intermediate levels of
signal loss proportional to the number of paralogous
copies elsewhere on chromosome 17. We observed similar
patterns and log2 ratio signal intensity for both families 1
A unaffectedchr17
17q21-delchr17
electrofusion
human/mousehybrid cells loss of
unaffectedchr17
loss of17q21-delchr17
mousegenome
isolated 17q21 delchr17
isolated unaffectedchr17
phased markergenotyping
haplotype-specificarray CGH
haplotype-specific high-throughput sequencing
B
locus-specific PSVs
C
inferred region of crossover
maximum extent of deletion
isolated H2chromosomefrom parent
isolated 17q21 delchr17 from proband
haplotype-specific array CGH
genomic position
log2
rat
io
relativecopy number = 0
relativecopy number = 0.5
17q21 del(test)
unaffected H2 chromosome(reference)
inferred region of crossover(high coverage)
or
Figure 1. Schematic of SD-Breakpoint Detection Approach(A) After the creation of human/mouse hybrid cells, clonal populations that carried only one of two chromosome 17 homologs wereselected. The 17q21.31 deletion-bearing chromosome could then be studied in isolation from the unaffected chromosome 17.(B) Hybrid cell lines permit haplotype-specific array CGH. NAHR-mediated deletions (bottom schematic, gray box) remove both uniquesequence and SD (block arrows). Deletions in unique sequence are seen as extremely low signal representing relative copy number 0 (log2ratio plot schematic). Copy-number loss in SD displays intermediate signal loss proportional to the number of remaining paralogouscopies elsewhere in the genome (in the schematic, relative copy number ¼ 0.5).(C) For NAHR-mediated deletions, unequal crossover within SDs (rectangles) removes PSVs specific to the proximal and distal duplicons(vertical hashes in upper and lower rectangle halves, respectively), which can be used to infer themaximal extent of the deletion and theregion of crossover. At low coverage, the absence of reads mapping to a PSV might reflect lack of sequence coverage. At sufficiently highcoverage, however, the absence of reads mapping to a PSV (gray vertical hashes) implies the absence of the PSV in the sample and canfurther refine the crossover region.
The American Journal of Human Genetics 90, 599–613, April 6, 2012 603
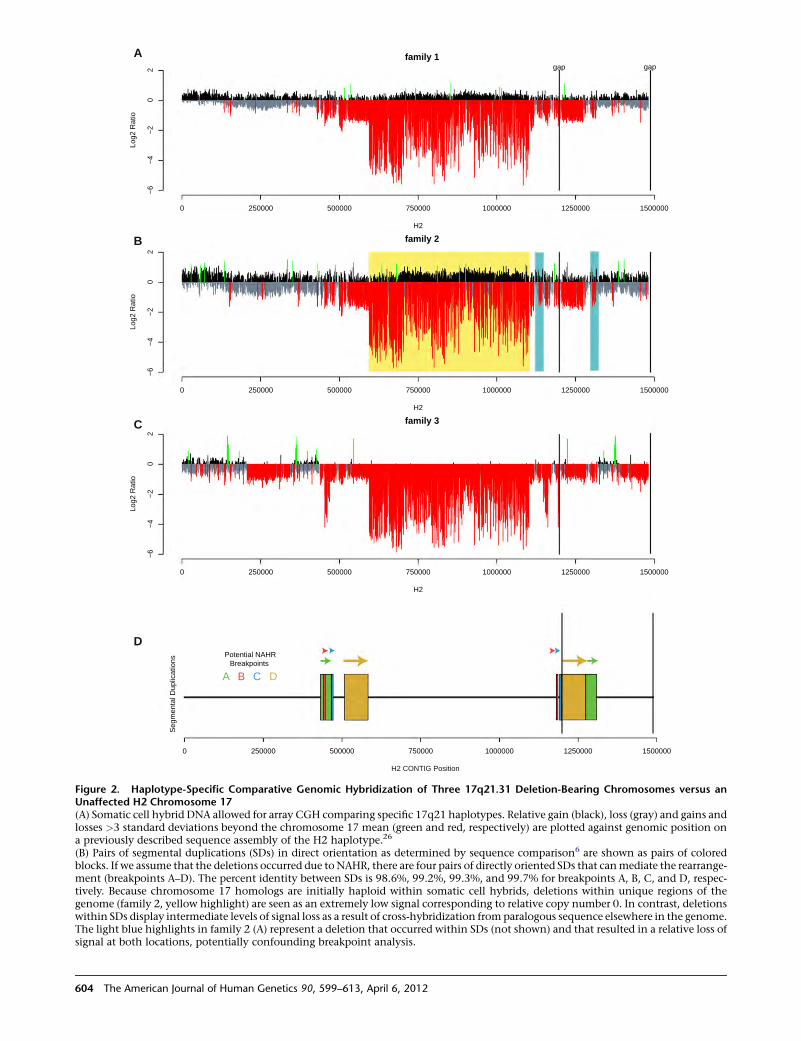
6 − 4 −
2 − 0
2
family 1
H2
o i t a R
2 g o L
0 250000 500000 750000 1000000 1250000 1500000
gap
6 − 4 −
2 − 0
2
family 2
H2
o i t a R
2 g o L
0 250000 500000 750000 1000000 1250000 1500000
6 − 4 −
2 − 0
2
family 3
0 250000 500000 750000 1000000 1250000 1500000
gap
s n o i t a c i l p u D
l a t n e
m
g e S
0 250000 500000 750000 1000000 1250000 1500000
H2
o i t a R
2 g o L
D C B A
Potential NAHRBreakpoints
H2 CONTIG Position
A
B
C
D
Figure 2. Haplotype-Specific Comparative Genomic Hybridization of Three 17q21.31 Deletion-Bearing Chromosomes versus anUnaffected H2 Chromosome 17(A) Somatic cell hybrid DNA allowed for array CGH comparing specific 17q21 haplotypes. Relative gain (black), loss (gray) and gains andlosses >3 standard deviations beyond the chromosome 17 mean (green and red, respectively) are plotted against genomic position ona previously described sequence assembly of the H2 haplotype.26
(B) Pairs of segmental duplications (SDs) in direct orientation as determined by sequence comparison6 are shown as pairs of coloredblocks. If we assume that the deletions occurred due to NAHR, there are four pairs of directly oriented SDs that canmediate the rearrange-ment (breakpoints A–D). The percent identity between SDs is 98.6%, 99.2%, 99.3%, and 99.7% for breakpoints A, B, C, and D, respec-tively. Because chromosome 17 homologs are initially haploid within somatic cell hybrids, deletions within unique regions of thegenome (family 2, yellow highlight) are seen as an extremely low signal corresponding to relative copy number 0. In contrast, deletionswithin SDs display intermediate levels of signal loss as a result of cross-hybridization from paralogous sequence elsewhere in the genome.The light blue highlights in family 2 (A) represent a deletion that occurred within SDs (not shown) and that resulted in a relative loss ofsignal at both locations, potentially confounding breakpoint analysis.
604 The American Journal of Human Genetics 90, 599–613, April 6, 2012

and 2, whereas the deletion in family 3 showed a different
pattern by array CGH. We noted, for example, that some
signal loss proximal to 340 kbp and distal to 1.38 Mbp
was not observed in the other individuals (Figure 2;
Figure S10).
We hypothesized that the array CGH signature observed
in family 3 was a consequence of interchromosomal NAHR
and sought to assess its relative frequency in 17q21.31-
deletion-bearing probands. Further examination of the
three additional unrelated 17q21.31-deletion-bearing
probands by array CGH showed log2 ratios similar to those
in families 1 and 2 (Figure S11). The breakpoints for these
three additional individuals had been previously analyzed
by array CGH of diploid DNA42 and provided a benchmark
for comparison. We also surveyed 12 additional 17q21.31
spontaneous deletions by using a combination of a
lower-resolution array CGH platform and marker segrega-
tion and noted only one further case, which was consistent
with the H1/H2 recombination pattern identified in family
3. Thus, on the basis of our analysis with somatic cell
hybrids (1/6) and examination of other data (1/12), H1/H2
deletions account for ~10% of cases.
Under the assumption that the 17q21.31 deletions arose
as a result of NAHR between high-identity SDs, we devel-
oped a breakpoint analysis method that compares the
array CGH signal intensity to the expected changes in
relative copy number of high-identity SDs bracketing the
critical region (see Material and Methods). Analysis of the
H2 assembly predicted four possible pairs of paralogous
sequences (breakpoint regions A–D; Figure 2; Figures S7
and S10) under a model of H2 interchromatidal NAHR.
Examining SDs at the proximal deletion breakpoint, we
observed a predicted region of copy number 0 (yellow
highlight, Figures S10A and S10C) for breakpoints A–C.
Although array CGH data from family 3 demonstrated
a log2 signal consistent with a copy number of 0 in this
region, the same degree of signal loss was not observed
in either family 1 or family 2. This suggests that deletions
for both family 1 and family 2 are mediated by sequences
at breakpoint D. Similarly, the distal breakpoint, a region
of predicted copy number 0 (yellow highlight, Figures
S10B and S10D), for breakpoints A–C is inconsistent with
the log2 ratios observed in families 1 and 2. Thus, the
most likely sequences mediating NAHR for families 1 and
2 are those of breakpoint D, corresponding to a pair of
directly oriented SDs with >99% identity and a length of
~75 kbp in the current H2 assembly.
In contrast to that in families 1 and 2, relative copy-
number loss proximal to 340 kbp and distal to 1.38 Mbp
in family 3 (orange highlight, Figure S10) was not consis-
tent with intrachromosomal NAHR involving any of the
breakpoints A–D but was consistent with the previous
microsatellite data suggesting that the family 3 deletion
might be mediated by interchromosomal NAHR between
the H1 and H2 haplotypes. This was paradoxical; it
would require sequence proximal to the unique deleted
sequence on the H1 haplotype to directly orient with
paralogous sequence distal to the unique deleted sequence
on the H2 haplotype. However, such sequences are
not currently observed in the current H2 assembly
(Figure S7 and Table S4).26 This suggested several possible
hypotheses. If the H1/H2 crossover and the deletion
were separate events, then the family 3 deletion could
have occurred on an H2 haplotype with altered copy
number within SDs or might not have been the result
of NAHR. Alternatively, interchromosomal crossover
between the H1 and H2 haplotypes might have occurred
as a result of sequences not currently represented in the
H2 assembly.
We performed array CGH between hybrid cell lines con-
taining the H2 chromosome from the mother in family 3
and the mother in family 2 and observed no copy-number
differences across the region (Figure S12). This suggested
that the unusual log2 ratio observed for the deletion in
family 3 was not the result of structural variation or poly-
morphism on the H2 haplotype.
Closing the Sequence Gaps in the H2 Assembly
We explored the possibility that crossover between H1 and
H2 haplotypes is mediated by previously unrepresented
sequence in the current haplotype assembly. There are
two gaps within the current H2 assembly in GRCh37
(gap 1 and gap 2; Figure 3), both of which lie distal
to the unique deleted sequence (Figure 3; Figure S7).
Previously reported marker data suggested that gap 2
(spanned by RP11-84A7) does not contain sequence that
can mediate 17q21.31 deletions by H1/H2 NAHR.25 In
contrast, a draft sequence of RP11-374N3 (AC048388) con-
tained sequence paralogous to SDs proximal to the unique
deleted sequence on both the H1 and H2 haplotypes,
in agreement with our hypothesis that H1/H2 NAHR
might occur. This was additionally supported by the pres-
ence and orientation of microsatellites DG17S133 and
DG17S435 in RP11-374N3 (Figure S9).25
We noted that, to close gap 2 (~130 kbp), Steffansson
et al.25 had placed RP11-84A7, which was not used in
the H2 sequence assembly,26 in a BAC assembly to
connect the distal end of the H2 haplotype to the refer-
ence assembly. To reconfirm placement of RP11-84A7
(AC243906) on the H2 haplotype, we first end-sequenced
the clone and noted that the T7 end maps to the distal
portion of either the H1 or H2 assembly from Zody
et al.26 and that the SP6 end maps to AC019319 in build
36. In order to distinguish placement of RP11-84A7 on
the H2 haplotype versus the H1 haplotype, we compared
microsatellites on RP11-84A7 with those on RP11-619A10
(AC217775), the last BAC in the H2 assembly, by using
RP11-113E17, a clone assigned by Stefansson et al.25 to
the H1 haplotype, as a negative control (Figure S13 and
Table S5).Marker genotyping confirmed the predicted over-
lap between RP11-619A10 andRP11-84A7 and also demon-
strated RP11-84A7 and RP11-113E17 to be on opposite
haplotypes. Finally, the size of gap 2 was estimated as the
average size of a BAC from RP11 minus its overlap, based
The American Journal of Human Genetics 90, 599–613, April 6, 2012 605

on end-sequence placement, with sequence on either side
of the gap (130 kbp ¼ 180 kbp – 25 kbp – 25 kbp).
RP11-374N3 (AC048388) was previously determined to
span gap 1 (~70 kbp) in the H2 assembly but could not
be assembled by shotgun sequencing alone.26 We hypoth-
esized that this was due to the presence of two arms of
oppositely oriented, highly identical sequence separated
by a spacer sequence unique within the clone (Figure S2).
Importantly, the hypothesized structure suggested that
gap 1 contains sequence paralogous to SDs on the H1 and
H2 haplotypes and that this sequence might mediate
NAHR. If sequence in gap 1 largely corresponds to one of
two highly identical arms of sequence in RP11-374N3
(Figure S2), then the other duplicated arm of sequence,
entirely contained within the neighboring finished clone
AC217768, provides a good approximation of the sequence
in gap 1. On the basis of this hypothesized structure, we
estimated that gap 1 contains 40 kbp and 70 kbp of
sequence with ~99% identity to the H1 and H2 haplotypes
proximal to the unique deleted sequence, respectively.
We sequenced RP11-84A7 and additional clone-
based resources to aid in the assembly of RP11-374N3
(Figure 3). A draft assembly of RP11-84A7 (spanning gap
2; AC243906) did not contain sequence that couldmediate
17q21.31 deletions. Because RP11-374N3 (spanning gap 1)
previously could not be assembled by shotgun sequencing
alone,26 we identified three additional smaller clones of
a fosmid clone library (ABC14) from an H1/H2 individual
to effectively provide subassembly and resolve near-perfect
local duplications of the larger BAC (Material and Methods
and additional references19,46,47). As predicted, draft
sequences from these clones (AC244161, AC244163, and
AC244164) identified the presence of an additional ~70
kbp of SD in direct orientation (~99% estimated identity)
between gap 1 and the H2 haplotype proximal to unique
deleted sequence and an additional ~40 kbp of SD in direct
orientation (>99% estimated identity) between gap 1 and
the H1 haplotype. This confirmed our hypothesized struc-
ture of RP11-374N3 and therefore that previously unchar-
acterized sequence in the H2 assembly could mediate
NAHR between the H1 and H2 haplotypes in family 3.
Additionally, it suggested that the length of breakpoint
D, which probably mediated deletions in the remaining
five probands, is nearly twice as large (~145 kbp versus
75 kbp) as what is annotated in the human genome
reference.
Identification of Breakpoint-Informative Paralogous
Sequence Variants
To achieve sequence-level resolution, we identified SUNs,
PSVs unique to specific loci in the genome (Figure 1C), as
well as other breakpoint-informative PSVs within the SDs
mediating the observed 17q21.31 deletions.24 We used
two different techniques to identify breakpoint-informa-
tive PSVs (Figure S6). For sequences present in the current
H2 assembly, we identified PSVs by using WGAC as
described previously6 to generate alignments of paralogous
sequence. To create SUNs, we then filtered PSVs by deter-
mining which PSVs could generate unique 36 bp reads
with respect to the human and mouse genomes (Material
and Methods). For sequences mapping to gaps in the
current H2 assembly, PSVs were identified from the align-
ment of the fosmid draft sequences mapping to gap 1
with the expected regions of paralogous sequence on the
H1 and H2 haplotypes (Material and Methods). This
technique could be useful with other regions that have
alternate structural haplotypes and where a haplotype-
specific sequence assembly might not exist, yet where
the haplotype of a given clone is known. Subsequent
filtering of these PSVs revealed relatively few SUNs in gap
1 (Table 1). This was due to the near identity of sequence
within gap 1 to sequence immediately proximal on
AC217768 in the H2 assembly (Figure S2). This sequence,
however, would be lost in the event of H1/H2 or H2/H2
NAHR. Therefore, gap 1 PSVs present elsewhere only
within AC217768 would still be breakpoint informative
H2 haplotype
AC217778
AC217769
AC138688
AC127032
AC217772
AC217779
BX544879
AC217770
AC225613
AC217768
AC139677
AC217775
AC019319(build 36)
build 36, chr17: 40799295
RP11-84A7
100kb
RP11-374N3RP11 BACs sequenced
ABC14 fosmids sequenced
Figure 3. Completion of the H2 Contig with Clone-Based ResourcesTwo gaps exist in the H2 contig (dotted vertical lines). The distal gap (gap 2, ~130 kbp) is spanned by the previously placed BAC RP11-84A7.25 So that the proximal gap (gap 1, ~70 kbp) can be closed, assembly of RP11-374N3 will be completed with the assistance of addi-tional clones from the fosmid library of an H1/H2 individual (ABC14, NA12156).
606 The American Journal of Human Genetics 90, 599–613, April 6, 2012

in the event of H2/H2NAHR andwould thus effectively act
as SUNs (Material and Methods). Similarly, gap 1 PSVs
present elsewhere in the genome but exclusively on the
H2 haplotype within or proximal to AC217768 would
effectively act as SUNs in the event of H1/H2 NAHR.
After quality control (Material and Methods), we identi-
fied 4,680 36-mers corresponding to 187 distinct PSVs that
can be used to distinguish deletions due to H2 interchro-
matidal NAHR and 3,912 36-mers corresponding to 142
distinct PSVs that can be used to distinguish H1/H2 inter-
chromosomal NAHR (Table S6).
Resolution of CNV Breakpoints within Paralogous
Sequence
We leveraged the specificity of next-generation sequence
data to achieve sequence-level breakpoint resolution in
the three parent-child trios by mapping genome sequence
data to this set of SUN identifiers. We initially compared
sequence patterns between the proband and mother for
family 2 by generating whole-genome sequence from
both individuals. We generated ~26 Gbp of sequence (~9-
fold coverage) for the family 2 mother, who was an H2-
homozygote, by using the SOLIDv4 sequencing platform.
As expected, reads aligned to breakpoint-informative
PSVs across both the proximal and distal paralogs of the
breakpoint D region: an ~145 kbp region of near-perfect
sequence identity including previously uncharacterized
sequence mapping to the gap in the H2 assembly. This
finding is consistent with the finding, from array CGH
results from somatic cell hybrids, that themother is diploid
across the 17q21.31 microdeletion region (Figure 4). In
stark contrast, when genome sequence (44 Gbp, ~15-fold
coverage) was generated from the proband in family 2 and
mapped to these variants, we observed no aligned reads to
PSVs on the proximal paralog of breakpoint D past the H2
position at 508,415 bp and no aligned reads to PSVs before
the H2 position at 1,209,274 bp on the distal paralog. This
localizes the crossover between the paralogs and refines
the deletion breakpoints from a 145 kbp region based on
array CGH to a ~22 kbp window (H2:508,415–529,961 on
the proximal paralog and Gap 1: 56,251 to H2:1,209,274
on the distal paralog; chr17_ctg5_hap1:567,056–588,595
on the proximal paralog and gap 1 to chr17_
ctg5_hap1:1,317,189 in the GRCh37 genomic sequence).
This breakpoint includes the 50 UTR of KANSL1.
We repeated this mapping strategy by focusing on the
remaining two probands. We generated ~42 Gbp of
whole-genome sequence (~14-fold coverage) for the
proband from family 1 (31928) and ~46 Gbp of sequence
(~15-fold coverage) for the proband from family 3 (31873)
by using Illumina Hi-Seq2000 platform. In family 1, we
narrowed the deletion breakpoints to a ~4 kbp window
(H2:554,425–558,503 and H2:1,233,725–1,237,776 on
the proximal and distal paralogs, respectively; chr17_ctg5_
hap1:613,066–617,144 and chr17_ctg5_hap1:1,341,640–
1,345,691 in the GRCh37 genomic sequence) that includes
the first coding exonofKANSL1. Althoughweobserve a few
sequence read alignments to PSVs outside of these break-
point intervals, the hits are not collinear, and we attribute
these to either polymorphisms between the H1 and H2
haplotypes or spurious PCR-induced mutations that arose
during library prep. Finally, we observed no reads aligning
to PSVs from the proximal segment of breakpoint D in
family 3, but we did observe sequence alignments after
the gap 1 position at 45,302 bp on the distal paralog, which
aligns to thepositionat 248,866bpon theH1assembly.The
first PSVobserved on the H1 assembly proximal to this is at
theH1position at 224,601 bp. This places the breakpoint in
a ~24 kbp window (chr17:43,668,073–43,692,338 in the
GRCh37 genomic sequence) upstream of CRHR1 on the
H1 chromosome and completelywithin the gap 1 sequence
of the H2 chromosome. This pattern is consistent with our
previous hypothesis of H1/H2-mediated NAHR because
such a crossover occurs within the expected region of
directly oriented H1/H2 SDs and would remove the prox-
imal paralog of breakpoint D in its entirety.
Discussion
We employed a combination of technologies and analyses
that allow for breakpoint delineation within genomic
regions previously refractory to analysis. We note three
key components of our analysis. First, generation of
Table 1. Summary of Identified Breakpoint-Informative PSVs
NameH2 Proximaland Distal
H1/H2 InferredProximal
H1/H2 InferredDistal
H2/H2 InferredProximal
H2/H2 InferredDistal
H2/H2Informative
H1/H2Informative
Region(s) H2:519,560–593,627 bp, H2:1,198,880–1,273,881 bp
H1:219,599–261,693 bp
H2, gap 1 H2:452,165–519,559 bp
H2, gap 1 NA NA
Description breakpoint D,proximal anddistal paralogs
inferred H1paralog to gap 1
PSVs inferredfrom alignmentto H1
inferred H1paralog to gap 1
PSVs inferredfrom alignmentto H2
H2 proximal,H2 distal, andH2/H2 inferredproximaland distal
H2 proximal,H2 distal, andH1/H2 inferredproximaland distal
k-mers 2,627 845 440 858 1,195 4,680 3,912
PSVs (SUNs) 86 (86) 37 (37) 19 (1) 40 (40) 61 (2) 187 142
The American Journal of Human Genetics 90, 599–613, April 6, 2012 607
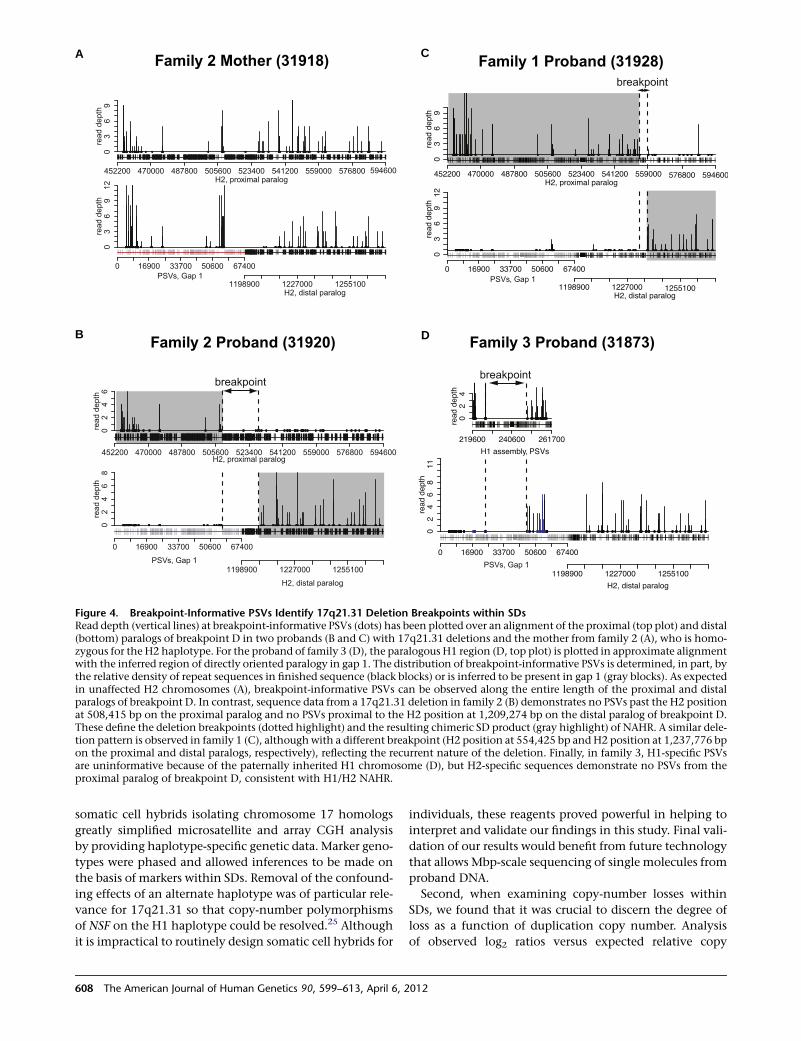
somatic cell hybrids isolating chromosome 17 homologs
greatly simplified microsatellite and array CGH analysis
by providing haplotype-specific genetic data. Marker geno-
types were phased and allowed inferences to be made on
the basis of markers within SDs. Removal of the confound-
ing effects of an alternate haplotype was of particular rele-
vance for 17q21.31 so that copy-number polymorphisms
of NSF on the H1 haplotype could be resolved.25 Although
it is impractical to routinely design somatic cell hybrids for
individuals, these reagents proved powerful in helping to
interpret and validate our findings in this study. Final vali-
dation of our results would benefit from future technology
that allows Mbp-scale sequencing of single molecules from
proband DNA.
Second, when examining copy-number losses within
SDs, we found that it was crucial to discern the degree of
loss as a function of duplication copy number. Analysis
of observed log2 ratios versus expected relative copy
A
B D
C
Figure 4. Breakpoint-Informative PSVs Identify 17q21.31 Deletion Breakpoints within SDsRead depth (vertical lines) at breakpoint-informative PSVs (dots) has been plotted over an alignment of the proximal (top plot) and distal(bottom) paralogs of breakpoint D in two probands (B and C) with 17q21.31 deletions and the mother from family 2 (A), who is homo-zygous for the H2 haplotype. For the proband of family 3 (D), the paralogous H1 region (D, top plot) is plotted in approximate alignmentwith the inferred region of directly oriented paralogy in gap 1. The distribution of breakpoint-informative PSVs is determined, in part, bythe relative density of repeat sequences in finished sequence (black blocks) or is inferred to be present in gap 1 (gray blocks). As expectedin unaffected H2 chromosomes (A), breakpoint-informative PSVs can be observed along the entire length of the proximal and distalparalogs of breakpoint D. In contrast, sequence data from a 17q21.31 deletion in family 2 (B) demonstrates no PSVs past the H2 positionat 508,415 bp on the proximal paralog and no PSVs proximal to the H2 position at 1,209,274 bp on the distal paralog of breakpoint D.These define the deletion breakpoints (dotted highlight) and the resulting chimeric SD product (gray highlight) of NAHR. A similar dele-tion pattern is observed in family 1 (C), althoughwith a different breakpoint (H2 position at 554,425 bp andH2 position at 1,237,776 bpon the proximal and distal paralogs, respectively), reflecting the recurrent nature of the deletion. Finally, in family 3, H1-specific PSVsare uninformative because of the paternally inherited H1 chromosome (D), but H2-specific sequences demonstrate no PSVs from theproximal paralog of breakpoint D, consistent with H1/H2 NAHR.
608 The American Journal of Human Genetics 90, 599–613, April 6, 2012
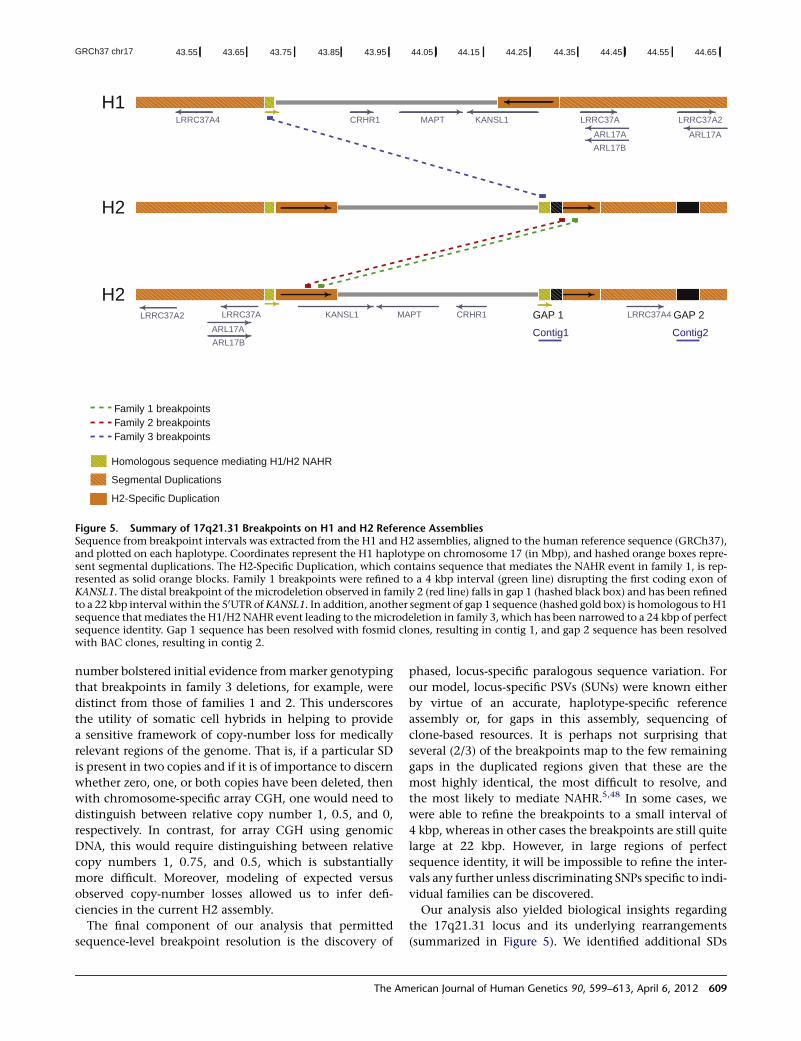
number bolstered initial evidence frommarker genotyping
that breakpoints in family 3 deletions, for example, were
distinct from those of families 1 and 2. This underscores
the utility of somatic cell hybrids in helping to provide
a sensitive framework of copy-number loss for medically
relevant regions of the genome. That is, if a particular SD
is present in two copies and if it is of importance to discern
whether zero, one, or both copies have been deleted, then
with chromosome-specific array CGH, one would need to
distinguish between relative copy number 1, 0.5, and 0,
respectively. In contrast, for array CGH using genomic
DNA, this would require distinguishing between relative
copy numbers 1, 0.75, and 0.5, which is substantially
more difficult. Moreover, modeling of expected versus
observed copy-number losses allowed us to infer defi-
ciencies in the current H2 assembly.
The final component of our analysis that permitted
sequence-level breakpoint resolution is the discovery of
phased, locus-specific paralogous sequence variation. For
our model, locus-specific PSVs (SUNs) were known either
by virtue of an accurate, haplotype-specific reference
assembly or, for gaps in this assembly, sequencing of
clone-based resources. It is perhaps not surprising that
several (2/3) of the breakpoints map to the few remaining
gaps in the duplicated regions given that these are the
most highly identical, the most difficult to resolve, and
the most likely to mediate NAHR.5,48 In some cases, we
were able to refine the breakpoints to a small interval of
4 kbp, whereas in other cases the breakpoints are still quite
large at 22 kbp. However, in large regions of perfect
sequence identity, it will be impossible to refine the inter-
vals any further unless discriminating SNPs specific to indi-
vidual families can be discovered.
Our analysis also yielded biological insights regarding
the 17q21.31 locus and its underlying rearrangements
(summarized in Figure 5). We identified additional SDs
Family 1 breakpointsFamily 2 breakpointsFamily 3 breakpoints
H1
H2
KANSL1MAPTCRHR1 LRRC37A
ARL17B
LRRC37A2
ARL17A
LRRC37A4
ARL17A
43.8543.55 43.95 44.05 44.1543.7543.65 44.25 44.35 44.45 44.55 44.65GRCh37 chr17
Segmental Duplications
H2-Specific Duplication
Homologous sequence mediating H1/H2 NAHR
H2GAP 1LRRC37A KANSL1 MAPT CRHR1
ARL17B
LRRC37A2 LRRC37A4
ARL17A
GAP 2
Contig1 Contig2
Figure 5. Summary of 17q21.31 Breakpoints on H1 and H2 Reference AssembliesSequence from breakpoint intervals was extracted from the H1 and H2 assemblies, aligned to the human reference sequence (GRCh37),and plotted on each haplotype. Coordinates represent the H1 haplotype on chromosome 17 (in Mbp), and hashed orange boxes repre-sent segmental duplications. The H2-Specific Duplication, which contains sequence that mediates the NAHR event in family 1, is rep-resented as solid orange blocks. Family 1 breakpoints were refined to a 4 kbp interval (green line) disrupting the first coding exon ofKANSL1. The distal breakpoint of the microdeletion observed in family 2 (red line) falls in gap 1 (hashed black box) and has been refinedto a 22 kbp interval within the 50UTR of KANSL1. In addition, another segment of gap 1 sequence (hashed gold box) is homologous to H1sequence thatmediates the H1/H2 NAHR event leading to themicrodeletion in family 3, which has been narrowed to a 24 kbp of perfectsequence identity. Gap 1 sequence has been resolved with fosmid clones, resulting in contig 1, and gap 2 sequence has been resolvedwith BAC clones, resulting in contig 2.
The American Journal of Human Genetics 90, 599–613, April 6, 2012 609

critical to understanding the genetic basis for the unequal
crossing over that mapped to the gap of the H2 assembly.
First of all, we find that ~90% of 17q21.31 rearrangement
events (16/18 based on specific screening for the H1/H2
events) occurring as a result of interchromatidal NAHR
are driven by European-specific SDs on the H2 haplotype.
Second, all interchromatidal events were mediated by
a single pair of SDs that were ~145 kbp and had ~99% iden-
tity, which accounts for 84% of the directly oriented SDs
flanking the unique deleted sequence. In the two cases
where we refined these breakpoints by using genome
sequence data, the exact breakpoints differed but both
localized to the same 99% identity segment. In both cases
the rearrangements are predicted to disrupt KANSL1—for
example, the family 2 breakpoints occur precisely in the
first exon of this gene. It is noteworthy that the same dupli-
cations are highly stratified and have risen to high
frequency in individuals of European descent.25
We also show that 17q21.31 deletions can occur as
a result of interchromosomal NAHR between the H1 and
H2 haplotypes. Our limited survey of 17q21.31 break-
points indicates that interchromosomal NAHR is relatively
uncommon. One case was previously identified,43 and
we observed it independently twice in 18 probands, sug-
gesting that such events account for ~10% of 17q21.31
microdeletions. This is also compatible with previous
population genetic data and theoretical predictions that
crossovers between the H1 and H2 haplotypes are effec-
tively suppressed. Interchromatidal deletions are probably
more common than interchromosomal deletions for
several reasons. First, sperm typing has shown NAHR due
to interchromatidal deletions to be the predominant class
of NAHR.49 Second, the interchromosomal paralogous seg-
ments mediating unequal crossover are smaller (40 kbp
versus 145 kbp) and less numerous than those that can
mediate interchromatidal NAHR. Finally, most crossover
events between H1 and H2 in this region would be
between allelic sequences in inverted orientation, creating
the classic acentric and dicentric chromosomal products of
a paracentric inversion, and are therefore inviable.
17q21.31 represents one of the most studied human
genomic loci for which a complex alternate structural
haplotype has been generated. Additional loci have either
been implicated in pathogenic deletions or have been
shown to have structural haplotypes predisposing an
individual to such deletions.50,51 Unlike the 17q21.31
locus, none of these regions, to our knowledge, yet have
haplotype-specific sequence assemblies. Although this
presents a challenge, the methods we have developed
provide a clear path forward to fine-mappingof breakpoints
within segmental regions both in basic research and, ulti-
mately, in a clinical setting. We propose the following
strategy. In lieu of somatic cell hybrids, recently developed
methods involving next-generation sequencing of flow-
sorted chromosomes52 or pooled fosmids53 could be em-
ployed for the rapid generation of haplotype-specific
sequence data, recovery of sequence information within
the gaps, and discovery of large structural polymorphisms.
Phased, locus-specific paralogous sequence variation could
be generated through targeted sequencing of clone-based
resources that now exist for more than 30 human
genomes19,51,54 or through conventional46 or massively
parallel sequencing53 methods. This would allow the estab-
lishment of high-quality alternate reference haplotypes of
the human reference genome as is being pursued by
the Genome Reference Consortium (Online Resources).
These data could be used in the creation of a catalog of
SUN identifiers so that breakpoints in deletion probands
could be refined. Once such a catalog was established,
itwouldbe relatively trivial to routinely delineate thebreak-
points of duplication anddeletionprobandswith extraordi-
nary precision by mapping complete genome sequencing
to this catalog of sequence variants. This is important
clinically for distinguishing breakpoints that are superfi-
cially similar (by array CGH) but that have different func-
tional consequences with respect to breakpoints within
duplicated genes or portions of genes (e.g., CHRNA7,55
SIRPB119 orKANSL1 [present study]). It is possible that these
differences in breakpoints contribute to the variability of
expressivity for genomic disorders and, as such, that it will
be important to distinguish between them in the future.
Supplemental Data
The Supplemental Data include 13 figures and six tables and can
be found with this article online at http://www.cell.com/AJHG.
Acknowledgments
We thank B. Coe, S. Ng and J. Hehir-Kwa for thoughtful
discussion, T. Brown for assistance with manuscript preparation,
A. Mackenzie, C. Igartua, C. Fields, S. Casadei, L. Vives, members
of the Mayo Medical Laboratories, members of The Genome Insti-
tute at Washington University, and members of the Hubrecht
Institute for assistance with data generation, and B. de Vries for
clinical collection and evaluation of individuals with 17qmicrode-
letions and their parents. K.M.S. was supported by a Ruth L.
Kirschstein National Research Service Award (NRSA) Fellowship
(F32GM097807). This work was supported by National Institutes
of Health grants HG002385 and HG004120 to E.E.E, and the
Netherlands Organization for Health Research and Development
(ZonMW 916.86.016 to L.E.L.M.V., and 917.66.363 to JAV).
E.E.E. is an investigator of the Howard Hughes Medical Institute.
E.E.E. is on the scientific advisory boards for Pacific Biosciences,
Inc. and SynapDx Corp.
Received: November 30, 2011
Revised: January 23, 2012
Accepted: February 16, 2012
Published online: March 29, 2012
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
The EMBOSS software suite, http://emboss.sourceforge.net/
610 The American Journal of Human Genetics 90, 599–613, April 6, 2012

Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/
Genome Reference Consortium, http://www.ncbi.nlm.nih.gov/
projects/genome/assembly/grc/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
JAligner Java implementation of the Smith-Waterman algorithm,
http://jaligner.sourceforge.net/
Marshfield Genetic Maps, http://research.marshfieldclinic.org/
genetics/
mrFAST, http://mrfast.sourceforge.net/
NCBI nucleotide database, http://www.ncbi.nlm.nih.gov/unists
NCBI BLAST and megaBLAST, http://blast.ncbi.nlm.nih.gov/
NCBI UniSTS database, http://www.ncbi.nlm.nih.gov/unists
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org/
RepeatMasker, http://www.repeatmasker.org/
Tandem Repeats Finder, http://tandem.bu.edu/trf/trf.html
UCSC Human Genome Browser (human reference genomes),
http://genome.ucsc.edu
Accession Numbers
The NCBI nucleotide accession numbers for the four clone
sequences reported in this paper are AC244161, AC244163,
AC244164, and AC243906.
The GEO accession numbers for the nine microarray experi-
ments in this paper are GSE34867.
References
1. Stankiewicz, P., and Lupski, J.R. (2010). Structural variation in
the human genome and its role in disease. Annu. Rev. Med.
61, 437–455.
2. Conrad, D.F., Pinto, D., Redon, R., Feuk, L., Gokcumen, O.,
Zhang, Y., Aerts, J., Andrews, T.D., Barnes, C., Campbell, P.,
et al; Wellcome Trust Case Control Consortium. (2010).
Origins and functional impact of copy number variation in
the human genome. Nature 464, 704–712.
3. Vissers, L.E., de Vries, B.B., and Veltman, J.A. (2010). Genomic
microarrays in mental retardation: From copy number varia-
tion to gene, from research to diagnosis. J. Med. Genet. 47,
289–297.
4. Girirajan, S., and Eichler, E.E. (2010). Phenotypic variability
and genetic susceptibility to genomic disorders. Hum. Mol.
Genet. 19 (R2), R176–R187.
5. Lupski, J.R. (1998). Genomic disorders: structural features of
the genome can lead to DNA rearrangements and human
disease traits. Trends Genet. 14, 417–422.
6. Bailey, J.A., Yavor, A.M., Massa, H.F., Trask, B.J., and Eichler,
E.E. (2001). Segmental duplications: organization and impact
within the current human genome project assembly. Genome
Res. 11, 1005–1017.
7. Xu, B., Roos, J.L., Levy, S., van Rensburg, E.J., Gogos, J.A., and
Karayiorgou, M. (2008). Strong association of de novo copy
number mutations with sporadic schizophrenia. Nat. Genet.
40, 880–885.
8. Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C.,
Walsh, T., Yamrom, B., Yoon, S., Krasnitz, A., Kendall, J., et al.
(2007). Strong association of de novo copy number mutations
with autism. Science 316, 445–449.
9. Sharp, A.J., Mefford, H.C., Li, K., Baker, C., Skinner, C., Steven-
son, R.E., Schroer, R.J., Novara, F., De Gregori, M., Ciccone, R.,
et al. (2008). A recurrent 15q13.3 microdeletion syndrome
associated with mental retardation and seizures. Nat. Genet.
40, 322–328.
10. de Vries, B.B., Pfundt, R., Leisink, M., Koolen, D.A., Vissers,
L.E., Janssen, I.M., Reijmersdal, S., Nillesen, W.M., Huys,
E.H., Leeuw, N., et al. (2005). Diagnostic genome profiling in
mental retardation. Am. J. Hum. Genet. 77, 606–616.
11. Mefford, H.C., Sharp, A.J., Baker, C., Itsara, A., Jiang, Z.,
Buysse, K., Huang, S., Maloney, V.K., Crolla, J.A., Baralle, D.,
et al. (2008). Recurrent rearrangements of chromosome
1q21.1 and variable pediatric phenotypes. N. Engl. J. Med.
359, 1685–1699.
12. Greenway, S.C., Pereira, A.C., Lin, J.C., DePalma, S.R., Israel,
S.J., Mesquita, S.M., Ergul, E., Conta, J.H., Korn, J.M., McCar-
roll, S.A., et al. (2009). De novo copy number variants identify
new genes and loci in isolated sporadic tetralogy of Fallot. Nat.
Genet. 41, 931–935.
13. Bochukova, E.G., Huang, N., Keogh, J., Henning, E., Purmann,
C., Blaszczyk, K., Saeed, S., Hamilton-Shield, J., Clayton-
Smith, J., O’Rahilly, S., et al. (2010). Large, rare chromosomal
deletions associated with severe early-onset obesity. Nature
463, 666–670.
14. Mefford, H.C., Clauin, S., Sharp, A.J., Moller, R.S., Ullmann,
R., Kapur, R., Pinkel, D., Cooper, G.M., Ventura, M., Ropers,
H.H., et al. (2007). Recurrent reciprocal genomic rearrange-
ments of 17q12 are associated with renal disease, diabetes,
and epilepsy. Am. J. Hum. Genet. 81, 1057–1069.
15. Lupski, J.R., de Oca-Luna, R.M., Slaugenhaupt, S., Pentao, L.,
Guzzetta, V., Trask, B.J., Saucedo-Cardenas, O., Barker, D.F.,
Killian, J.M., Garcia, C.A., et al. (1991). DNA duplication asso-
ciated with Charcot-Marie-Tooth disease type 1A. Cell 66,
219–232.
16. Chen, K.S., Manian, P., Koeuth, T., Potocki, L., Zhao, Q., Chi-
nault, A.C., Lee, C.C., and Lupski, J.R. (1997). Homologous
recombination of a flanking repeat gene cluster is a mecha-
nism for a common contiguous gene deletion syndrome.
Nat. Genet. 17, 154–163.
17. Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody,
M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh,
W., et al; International Human Genome Sequencing Consor-
tium. (2001). Initial sequencing and analysis of the human
genome. Nature 409, 860–921.
18. Lee, J.A., Carvalho, C.M., and Lupski, J.R. (2007). A DNA
replication mechanism for generating nonrecurrent rear-
rangements associated with genomic disorders. Cell 131,
1235–1247.
19. Kidd, J.M., Cooper, G.M., Donahue, W.F., Hayden, H.S.,
Sampas, N., Graves, T., Hansen, N., Teague, B., Alkan, C.,
Antonacci, F., et al. (2008). Mapping and sequencing of struc-
tural variation fromeight humangenomes.Nature453, 56–64.
20. Tuzun, E., Sharp, A.J., Bailey, J.A., Kaul, R., Morrison, V.A.,
Pertz, L.M., Haugen, E., Hayden, H., Albertson, D., Pinkel,
D., et al. (2005). Fine-scale structural variation of the human
genome. Nat. Genet. 37, 727–732.
21. Ye, K., Schulz, M.H., Long, Q., Apweiler, R., and Ning, Z.
(2009). Pindel: a pattern growth approach to detect break
points of large deletions and medium sized insertions from
paired-end short reads. Bioinformatics 25, 2865–2871.
22. Korbel, J.O., Urban, A.E., Affourtit, J.P., Godwin, B., Grubert,
F., Simons, J.F., Kim, P.M., Palejev, D., Carriero, N.J., Du, L.,
et al. (2007). Paired-end mapping reveals extensive structural
variation in the human genome. Science 318, 420–426.
The American Journal of Human Genetics 90, 599–613, April 6, 2012 611

23. Pentao, L., Wise, C.A., Chinault, A.C., Patel, P.I., and Lupski,
J.R. (1992). Charcot-Marie-Tooth type 1A duplication appears
to arise from recombination at repeat sequences flanking the
1.5 Mb monomer unit. Nat. Genet. 2, 292–300.
24. Sudmant, P.H., Kitzman, J.O., Antonacci, F., Alkan, C., Malig,
M., Tsalenko, A., Sampas, N., Bruhn, L., Shendure, J., and
Eichler, E.E.; 1000 Genomes Project. (2010). Diversity of
human copy number variation and multicopy genes. Science
330, 641–646.
25. Stefansson, H., Helgason, A., Thorleifsson, G., Steinthorsdot-
tir, V., Masson, G., Barnard, J., Baker, A., Jonasdottir, A.,
Ingason, A., Gudnadottir, V.G., et al. (2005). A common
inversion under selection in Europeans. Nat. Genet. 37,
129–137.
26. Zody, M.C., Jiang, Z., Fung, H.C., Antonacci, F., Hillier, L.W.,
Cardone, M.F., Graves, T.A., Kidd, J.M., Cheng, Z., Abouelleil,
A., et al. (2008). Evolutionary toggling of the MAPT 17q21.31
inversion region. Nat. Genet. 40, 1076–1083.
27. Baker, M., Litvan, I., Houlden, H., Adamson, J., Dickson, D.,
Perez-Tur, J., Hardy, J., Lynch, T., Bigio, E., and Hutton, M.
(1999). Association of an extended haplotype in the tau
gene with progressive supranuclear palsy. Hum. Mol. Genet.
8, 711–715.
28. Broman, K.W., Murray, J.C., Sheffield, V.C., White, R.L., and
Weber, J.L. (1998). Comprehensive human genetic maps:
individual and sex-specific variation in recombination. Am.
J. Hum. Genet. 63, 861–869.
29. Sharp, A.J., Itsara, A., Cheng, Z., Alkan, C., Schwartz, S., and
Eichler, E.E. (2007). Optimal design of oligonucleotide micro-
arrays for measurement of DNA copy-number. Hum. Mol.
Genet. 16, 2770–2779.
30. Benovoy, D., Kwan, T., and Majewski, J. (2008). Effect of
polymorphisms within probe-target sequences on olignonu-
cleotide microarray experiments. Nucleic Acids Res. 36,
4417–4423.
31. Lee, I., Dombkowski, A.A., and Athey, B.D. (2004). Guidelines
for incorporating non-perfectly matched oligonucleotides
into target-specific hybridization probes for a DNAmicroarray.
Nucleic Acids Res. 32, 681–690.
32. Frazer, K.A., Pachter, L., Poliakov, A., Rubin, E.M., and
Dubchak, I. (2004). VISTA: Computational tools for compara-
tive genomics. Nucleic Acids Res. 32, W273–W279.
33. Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman,
D.J. (1990). Basic local alignment search tool. J. Mol. Biol.
215, 403–410.
34. Zhang, Z., Schwartz, S., Wagner, L., and Miller, W. (2000). A
greedy algorithm for aligning DNA sequences. J. Comput.
Biol. 7, 203–214.
35. Rice, P., Longden, I., and Bleasby, A. (2000). EMBOSS: The
European Molecular Biology Open Software Suite. Trends
Genet. 16, 276–277.
36. Igartua, C., Turner, E.H., Ng, S.B., Hodges, E., Hannon, G.J.,
Bhattacharjee, A., Rieder, M.J., Nickerson, D.A., and Shendure,
J. (2010). Targeted enrichment of specific regions in the
human genome by array hybridization. Curr. Prot. Hum.
Genet., Chapter 18, Unit 18.3.
37. Benson, G. (1999). Tandem repeats finder: a program to
analyze DNA sequences. Nucleic Acids Res. 27, 573–580.
38. Alkan, C., Kidd, J.M., Marques-Bonet, T., Aksay, G., Antonacci,
F., Hormozdiari, F., Kitzman, J.O., Baker, C., Malig, M., Mutlu,
O., et al. (2009). Personalized copy number and segmental
duplication maps using next-generation sequencing. Nat.
Genet. 41, 1061–1067.
39. Durbin, R.M., Abecasis, G.R., Altshuler, D.L., Auton, A.,
Brooks, L.D., Gibbs, R.A., Hurles, M.E., and McVean, G.A.;
1000 Genomes Project Consortium. (2010). A map of human
genome variation from population-scale sequencing. Nature
467, 1061–1073.
40. Conrad, C., Andreadis, A., Trojanowski, J.Q., Dickson, D.W.,
Kang, D., Chen, X., Wiederholt, W., Hansen, L., Masliah, E.,
Thal, L.J., et al. (1997). Genetic evidence for the involvement
of tau in progressive supranuclear palsy. Ann. Neurol. 41,
277–281.
41. Koolen, D.A., Vissers, L.E., Pfundt, R., de Leeuw, N., Knight,
S.J., Regan, R., Kooy, R.F., Reyniers, E., Romano, C., Fichera,
M., et al. (2006). A new chromosome 17q21.31 microdeletion
syndrome associated with a common inversion polymor-
phism. Nat. Genet. 38, 999–1001.
42. Sharp, A.J., Hansen, S., Selzer, R.R., Cheng, Z., Regan, R.,
Hurst, J.A., Stewart, H., Price, S.M., Blair, E., Hennekam,
R.C., et al. (2006). Discovery of previously unidentified
genomic disorders from the duplication architecture of the
human genome. Nat. Genet. 38, 1038–1042.
43. Shaw-Smith, C., Pittman, A.M., Willatt, L., Martin, H., Rick-
man, L., Gribble, S., Curley, R., Cumming, S., Dunn, C., Kalait-
zopoulos, D., et al. (2006). Microdeletion encompassing
MAPT at chromosome 17q21.3 is associated with develop-
mental delay and learning disability. Nat. Genet. 38, 1032–
1037.
44. Trask, B.J. (2002). Human cytogenetics: 46 chromosomes, 46
years and counting. Nat. Rev. Genet. 3, 769–778.
45. Frazer, K.A., Ballinger, D.G., Cox, D.R., Hinds, D.A., Stuve,
L.L., Gibbs, R.A., Belmont, J.W., Boudreau, A., Hardenbol, P.,
Leal, S.M., et al; International HapMap Consortium. (2007).
A second generation human haplotype map of over
3.1 million SNPs. Nature 449, 851–861.
46. Kidd, J.M., Cheng, Z., Graves, T., Fulton, B., Wilson, R.K., and
Eichler, E.E. (2008). Haplotype sorting using human fosmid
clone end-sequence pairs. Genome Res. 18, 2016–2023.
47. Kidd, J.M., Sampas, N., Antonacci, F., Graves, T., Fulton, R.,
Hayden, H.S., Alkan, C., Malig, M., Ventura, M., Giannuzzi,
G., et al. (2010). Characterization of missing human genome
sequences and copy-number polymorphic insertions. Nat.
Methods 7, 365–371.
48. Cooper, G.M., Nickerson, D.A., and Eichler, E.E. (2007). Muta-
tional and selective effects on copy-number variants in the
human genome. Nat. Genet. 39(7, Suppl), S22–S29.
49. Turner, D.J., Miretti, M., Rajan, D., Fiegler, H., Carter, N.P.,
Blayney, M.L., Beck, S., and Hurles, M.E. (2008). Germline
rates of de novo meiotic deletions and duplications causing
several genomic disorders. Nat. Genet. 40, 90–95.
50. Sharp, A.J., Cheng, Z., and Eichler, E.E. (2006). Structural vari-
ation of the human genome. Annu. Rev. Genomics Hum.
Genet. 7, 407–442.
51. Antonacci, F., Kidd, J.M., Marques-Bonet, T., Teague, B.,
Ventura, M., Girirajan, S., Alkan, C., Campbell, C.D., Vives,
L., Malig, M., et al. (2010). A large and complex structural
polymorphism at 16p12.1 underlies microdeletion disease
risk. Nat. Genet. 42, 745–750.
52. Fan, H.C., Wang, J., Potanina, A., and Quake, S.R. (2011).
Whole-genome molecular haplotyping of single cells. Nat.
Biotechnol. 29, 51–57.
612 The American Journal of Human Genetics 90, 599–613, April 6, 2012

53. Kitzman, J.O.,Mackenzie,A.P.,Adey,A.,Hiatt, J.B., Patwardhan,
R.P., Sudmant, P.H.,Ng, S.B., Alkan,C.,Qiu, R., Eichler, E.E., and
Shendure, J. (2011). Haplotype-resolved genome sequencing
of a Gujarati Indian individual. Nat. Biotechnol. 29, 59–63.
54. Kidd, J.M., Graves, T., Newman, T.L., Fulton, R., Hayden, H.S.,
Malig, M., Kallicki, J., Kaul, R., Wilson, R.K., and Eichler, E.E.
(2010). A human genome structural variation sequencing
resource reveals insights into mutational mechanisms. Cell
143, 837–847.
55. Shinawi,M., Schaaf, C.P., Bhatt, S.S., Xia, Z., Patel, A., Cheung,
S.W., Lanpher, B., Nagl, S., Herding, H.S., Nevinny-Stickel, C.,
et al. (2009). A small recurrent deletion within 15q13.3 is
associated with a range of neurodevelopmental phenotypes.
Nat. Genet. 41, 1269–1271.
The American Journal of Human Genetics 90, 599–613, April 6, 2012 613

ARTICLE
Primate Genome Gain and Loss: A Bone Dysplasia,Muscular Dystrophy, and Bone Cancer Syndrome Resultingfrom Mutated Retroviral-Derived MTAP Transcripts
Olga Camacho-Vanegas,1 Sandra Catalina Camacho,1 Jacob Till,1 Irene Miranda-Lorenzo,1
Esteban Terzo,1 Maria Celeste Ramirez,1 Vern Schramm,2 Grace Cordovano,2 Giles Watts,3 Sarju Mehta,3
Virginia Kimonis,3 Benjamin Hoch,4 Keith D. Philibert,5 Carsten A. Raabe,6 David F. Bishop,1
Marc J. Glucksman,5 and John A. Martignetti1,7,8,*
Diaphyseal medullary stenosis with malignant fibrous histiocytoma (DMS-MFH) is an autosomal-dominant syndrome characterized by
bone dysplasia, myopathy, and bone cancer. We previously mapped the DMS-MFH tumor-suppressing-gene locus to chromosomal
region 9p21–22 but failed to identify mutations in known genes in this region. We now demonstrate that DMS-MFH results frommuta-
tions in the most proximal of three previously uncharacterized terminal exons of the gene encoding methylthioadenosine phosphor-
ylase, MTAP. Intriguingly, two of these MTAP exons arose from early and independent retroviral-integration events in primate genomes
at least 40 million years ago, and since then, their genomic integration has gained a functional role. MTAP is a ubiquitously expressed
homotrimeric-subunit enzyme critical to polyamine metabolism and adenine and methionine salvage pathways and was believed to be
encoded as a single transcript from the eight previously described exons. Six distinct retroviral-sequence-containing MTAP isoforms,
each of which can physically interact with archetype MTAP, have been identified. The disease-causing mutations occur within one of
these retroviral-derived exons and result in exon skipping and dysregulated alternative splicing of all MTAP isoforms. Our results identify
a gene involved in the development of bone sarcoma, provide evidence of the primate-specific evolution of certain parts of an existing
gene, and demonstrate that mutations in parts of this gene can result in human disease despite its relatively recent origin.
Introduction
Diaphyseal medullary stenosis with malignant fibrous
histiocytoma (DMS-MFH [MIM 112250]) is a rare, auto-
somal-dominant bone dysplasia and cancer syndrome of
unknown etiology.1–3 The disorder has a unique bone-
dysplasia phenotype characterized by cortical growth
abnormalities, including diffuse diaphyseal medullary
stenosis with overlying endosteal cortical thickening,
metaphyseal striations, and scattered infarctions within
the bone marrow. Affected individuals endure pathologic
fractures that subsequently heal poorly, progressive
wasting, bowing of the lower extremities, painful debilita-
tion, and the development of presenile cataracts. We
recently expanded the known clinical features of the
syndrome by characterizing two new unrelated families
affected by a progressive form of muscular disease consis-
tent with facioscapulohumeral muscular dystrophy
(FSHD [MIM 158900]) (see below). Among DMS-MFH-
affected individuals, approximately 35% develop a form
of bone sarcoma consistent with the diagnosis of malig-
nant fibrous histiocytoma (MFH).1–4
Using a positional-cloning approach, we originally local-
ized the disease-associated allele locus to chromosomal
region 9p21–22 and established a 3.5 cM critical locus
between markers D9S1778 and D9S171.4 Given the cancer
component of the syndrome, the 9p21–22 region is of
particular interest in that it is one of the most frequently
deleted and/or translocated chromosomal regions in
human cancer.5 A diverse group of human cancers
demonstrate loss of this region and include gliomas,6,7
melanomas,8 non-small-cell lung cancers,9 acute leuke-
mias,10,11 and, of direct significance to this study, osteosar-
comas.12,13 In an attempt to further narrow the region as
well as establish a link between hereditary and sporadic
tumor forms, we performed loss of heterozygosity (LOH)
analysis of sporadic MFH samples. This analysis supported
a shared genetic etiology between hereditary and sporadic
MFH cases and mapped the smallest region of overlap to
the 2.9 Mb region between markers D9S736 and
D9S171.14
A number of DMS-MFH candidate genes were originally
screened by DNA sequencing and were excluded because
they lacked mutations. These genes included the cyclin-
dependent kinase inhibitor 2A (CDKN2A [p16] [MIM
600160]) and its alternatively spliced product p14-ARF,
CDKN2B (p15), members of the interferon (IFN) super-
family, and the methylthioadenosine phosphorylase
gene (MTAP [MIM 156540]).4 MTAP has been thought
to consist of eight exons and seven introns15 and encode
1Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY 10029, USA; 2Department of Biochemistry, Albert Einstein
College of Medicine, Bronx, NY 10461, USA; 3University of California Irvine, Irvine, CA 92868, USA; 4Department of Pathology, Mount Sinai School of
Medicine, New York, NY 10029, USA; 5Midwest Proteome Center and Department of Biochemistry and Molecular Biology, Rosalind Franklin University
of Medicine and Science, ChicagoMedical School, Chicago, IL, 60064, USA; 6Institute of Experimental Pathology, University of Muenster, 48149Muenster,
Germany; 7Department of Pediatrics, Mount Sinai School of Medicine, New York, NY 10029, USA; 8Department of Oncological Sciences, Mount Sinai
School of Medicine, New York, NY 10029, USA
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.024. �2012 by The American Society of Human Genetics. All rights reserved.
614 The American Journal of Human Genetics 90, 614–627, April 6, 2012

a ubiquitously expressed enzyme that plays a crucial role
in the salvage pathway for adenine and methionine in
all tissues.16 In the salvage pathways, methylthioadeno-
sine (MTA), a by-product of the polyamine pathway, is
recovered through its phosphorolysis into adenine and
methylthioribose-1-phosphate by MTAP.17 Through a
series of reactions, methylthioribose-1-phosphate is then
converted into methionine.17,18 It is suggested that loss
of MTAP activity plays a role in human cancer because its
loss has been reported in a number of cancers, including
osteosarcoma,12,13 leukemia,19 non-small-cell lung
cancer,20 malignant melanoma,21 biliary-tract cancer,22
breast cancer,23 pancreatic cancer,24 and gastrointestinal
stromal tumors.25 Reintroduction of MTAP expression
into the MCF7 breast adenocarcinoma cell line, which
lacks endogenous MTAP gene expression and enzymatic
activity, inhibits the cells’ ability to grow both in vitro
and in vivo;23 the fact that MTAP inhibits cell growth is
consistent with its presumed role as a tumor suppressor.
We have now identified and characterized the genetic
defect underlying DMS-MFH. All affected members of
five unrelated DMS-MFH-affected families possess synony-
mous mutations in the most proximal of three terminal
MTAP exons identified and characterized in these studies.
Interestingly, DNA-sequence analysis revealed that at
least two of the exons are remnants of retroviral insertions
into the primate genome. Both disease-causing mutations
in exon 9, the most proximal of these exons, result in
exon skipping and subsequent loss of this exon in alterna-
tively spliced, biologically active isoforms. Biochemical
studies provide evidence that isoforms containing exon 7
have MTAP activity. Altogether, these findings identify
a gene associated with both hereditary bone dysplasia
and osteosarcoma and also highlight the importance of
evolutionarily co-opted gene parts for both health and
disease.
Material and Methods
Linkage and Haplotype MappingAfter participants gave informed consent for these studies, which
were approved by the Human Research Protection Program at
the Mount Sinai School of Medicine, blood samples were obtained
from affected and unaffected family members. Genomic DNA was
extracted with the Puregene kit according to the manufacturer’s
(Minneapolis, MN) protocol, and individuals were genotyped
with a panel of markers spanning the length of chromosome 9;
included were markers from the Single Chromosome Scan Human
Screening Set (Research Genetics, Carlsbad, CA) and a number of
polymorphic markers that were custom generated. Family 1
from the original study was not included in this reanalysis. Allelic
determination was performed with the ABI 3130xl Genetic
Analyzer and GeneMapper 4.0 software (Applied Biosystems,
Foster City, CA). We handled the generated data by using Mega
2,26 and linkage analysis was computed by SIMWALK2 v.2.8327
with the following parameters: all families are genetically homog-
enous, inheritance pattern is autosomal dominant and has 80%
penetrance, there are no phenocopies, and the disease allele
frequency is 0.0001. Marker positions were obtained from the
Marshfield database and the UCSC Genome Browser March 2006
assembly. We further refined areas of positive location scores by
using custom-generated microsatellite markers within the defined
region. In brief, we designed the microsatellite markers by identi-
fying simple tandem repeats (STRs) in DNA sequences of clone
fragments from the Human Genome Assembly (UCSC Genome
Browser) by using the Tandem Repeats Finder Program.28 Fluores-
cently labeled primers were then designed to amplify these repeat
regions, and allele separation and analyses were performed as
previously described29 (Table S1, available online).
DNA Sequence AnalysisWe used PCR to amplify all MTAP exons by using Amplitaq-Gold
(Applied Biosystems, Foster City, CA), and we purified them by
using the QIAquick Spin PCR Purification Kit (QIAGEN, Valencia,
CA); the exons were amplified and purified according to themanu-
facturers’ protocol, and they were directly sequenced on an ABI
Prism 3700 automated DNA Analyzer (Applied Biosystems, Foster
City, CA). Data were analyzed with the program Sequencher v3.0
(Gene Codes Corporation, Ann Arbor, MI). Sets of intronic primers
that we used to amplify the coding region and intron and exon
boundaries are listed in Table S1. The PCR cycling conditions
were the following: 94�C (10 min) for 1 cycle, 94�C (30 s), 55�C(30 s), and 72�C (60 s) for 35 cycles each, and a final extension
of 72�C (10 min).
Computational DNA AnalysisWe performed computational analysis to detect aberrant splice
sites by using the NetGene2 Server30,31 and the splicing-enhancer
motif-prediction program ESEfinder Release 2.0,32 for which we
used the default parameters for the following SR proteins: SF2/
ASF, SC35, SRp40, and SRp55.
RNA Isolation, Semiquantitative Reverse
Transcription PCR, and Quantitative Real Time PCRWe extracted cultured-cell-line and tumor RNA by using the
RNeasy Mini kits, and we treated it with DNase according to the
manufacturer’s (QIAGEN, Valencia, CA) protocol. For semiquanti-
tative reverse transcription PCR (RT-PCR), we reverse transcribed
a total of 1 mg of RNA per reaction by using first-strand cDNA
synthesis with random primers (Promega, Madison, WI). To eval-
uate the transcription level of the MTAP splice variants, we per-
formed RT-PCR by using combinations of isoform-specific primers
listed in Table S1. We electrophoretically separated and visualized
RT-PCR products on a 1.5% agarose gel by using ethidium
bromide. We excised the bands and cloned and inserted them
into the pCR4-TOPO vector (Invitrogen, Carlsbad, CA), and we
sequenced 20 independent clones from each of the isolated bands
to establish their identity. For quantitative RT-PCR (qRT-PCR), we
reverse transcribed a total of 1 mg of RNA per reaction by
using iSCRIPT cDNA Synthesis according to the manufacturer’s
(Bio-Rad, Hercules, CA) protocol. We performed qRT-PCR by using
iQ SYBR Green Supermix (Bio-Rad, Hercules, CA), according to the
manufacturer’s protocol, on an ABI PRISM 7900HT Sequence
Detection System (Applied Biosystems, Foster City, CA) and by
using the primers listed in Table S1. All values were normalized
to either GAPDH or HPRT levels. All experiments were done in
triplicate, and all cell-culture experiments were independently
validated at least three times.
The American Journal of Human Genetics 90, 614–627, April 6, 2012 615

RACE: Rapid Amplification of cDNA 30 EndsTotal RNA was isolated from five primary human fibroblast cell
lines. We used a total of 1 mg of RNA per cell line, the PowerScript
Reverse Transcriptase, and the BD SMART IIA primer from the BD
SMART RACE cDNA amplification kit (according to the manufac-
turer’s [Franklin Lakes, NJ] protocol) to prepare corresponding
first-strand cDNAs. In brief, each of the 30 RACE-ready cDNAs
was used in PCR-amplification reactions with the SMART RACE
kit universal primers and sense gene-specific primers (GSP). First-
round PCR and second-round PCR were performed with the
specific primers listed in Table S1, Outer Primer (SMART RACE
kit), and Inner Primer (SMART RACE kit).
Cell Culture and TransfectionsPatient-derived osteosarcoma, fibroblast, and lymphoblast cell
lines and other commercially available cell lines (obtained from
ATCC) were maintained in Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10% fetal bovine serum, 100 U/ml
penicillin, and 100 mg/ml streptomycin and were grown at 37�Cin 5% CO2. For the expression studies, we transfected the cells
24 hr after plating by using lipofectamine 2000 according to
the manufacturer’s (Invitrogen, Carlsbad, CA) recommended
protocol.
Expression ConstructsGST and V5 Expression Vectors
To generate the vectors expressing each of the MTAP splice
variants (archetype MTAP and MTAP_v1, _v2, _v3, _v4, _v5,
and _v6), we used RT-PCR to amplify the cDNA of each splice
variant from normal fibroblasts by using the relevant primers
listed in Table S1. In brief, the exon 1 forward primer was used
for the amplification of all variants, and the reverse primers were
designed to delete the stop codon so that a fusion protein would
be generated with the V5 epitope. The amplified products were
cloned and inserted into the pcDNA3.1/V5-His TOPO TA expres-
sion vector (Invitrogen, Carlsbad, CA). The resulting clones were
completely sequenced in both orientations prior to their use.
Partial Minigene Expression Vectors
We used genomic DNAs obtained from a normal fibroblast cell line
and two patient-derived fibroblast cell lines to amplify the
following fragments: an 8 kb fragment containing exons 6–8
(flanked by introduced SalI and NheI restriction sites; subcloned
into the pcDNA3.1/V5-His TOPO TA vector), 2.1 kb fragments
containing exon 9 and carrying the wild-type (WT) DNA or either
the c.813-2A>G or c.885A>G mutation (flanked by introduced
SpeI and XhoI restriction sites; cloned into the pCR4-TOPO
vector), and a 3 kb fragment containing exons 10 and 11 (flanked
by introduced XhoI and SacII restriction sites; cloned into the
pCR4-TOPO vector). Each 2.1 kb fragment was digested with
SpeI and XhoI, and we cloned the fragments and inserted them
into a SpeI/XhoI-digested pcDNA3.1 (exons 6–8) vector to
generate the following three pcDNA3.1 (exons 6–8 and 9)
constructs:WT, c.813-2A>G, and c.885A>G. Finally, the 3 kb frag-
ment was digested with XhoI and SacII and cloned and inserted
into each one of the three pcDNA3.1 (exons 6–8 and 9) vectors di-
gested with XhoI and SacII. The following three pcDNA3.1 (exons
6–8 and 9–11) minigene expression vectors were created: WT,
c.813-2A>G, and c.885A>G. The primers that we used are listed
in Table S1. We performed PCR amplifications by using the
EXPAND Long Template PCR system according to the manufac-
turer’s (Roche, Indianapolis, IN) protocol. We sequenced all exons
and approximately 500 base pairs of the intron-exon flanking
boundaries of the generated plasmids.
Coimmunoprecipitation AssayRelevant combinations of GST- and V5-tagged constructs for
archetype MTAP and MTAP_v1, _v2, _v3, _v4, _v5, and _v6 were
cotransfected into PC3M cells. All of the following procedures
were performed at 4�C. Cell extracts for immunoblotting were har-
vested in NP40 lysis buffer (Santa Cruz Biotechnology, standard
protocol), and insoluble material was removed by centrifugation
(10 min at 13,000 rpm). One tenth of the cell extracts were
reserved for subsequent immunoblot analysis. Cell extracts were
incubated with V5 antibody (1 mg/ml) or GST antibody (1 mg/ml)
for 4 hr and Protein A-Sepharose (Invitrogen, Carlsbad, CA). Beads
were washed three times with 1% lysis buffer, and coimmunopre-
cipitates were released by being boiled in 100 ml 23 SDS Reducing
Sample Buffer (Invitrogen, Carlsbad, CA) for 5 min. Coimmuno-
precipitates were analyzed by immunoblot analysis (see below).
Immunoblot and Densitometric AnalysisCell extracts for immunoblotting were harvested in radioimmuno-
precipitation assay (RIPA) buffer (Santa Cruz Biotechnology,
standard protocol). Equal amounts of protein (50 mg) as deter-
mined by the BioRad DC Protein quantification assay were loaded
and separated by polyacrylamide gel electrophoresis and trans-
ferred to nitrocellulose membranes. We performed immunoblot-
ting by using a goat polyclonal antibody to actin (SC-1615), a
monoclonal (0.2 mg/ml) antibody to GST (BD PharMingen), and
a monoclonal antibody to the V5 tag (Santa Cruz Biotechnology).
We analyzed enhanced chemiluminescent images of immuno-
blots by using a scanning densitometer and quantifying the bands
(BIOQUANT NOVA imaging system). All values were normalized
to actin and expressed as fold changes relative to the control.
MTA QuantificationBlood-serum and cell-extract lysates were mixed with 63 pmol
[50-2H3] MTA, neutralized with KOH, and centrifuged. MTA frac-
tions were purified by HPLC (SymmetryShield RP18 column),
concentrated, dissolved in 10% methanol with 0.1% TFA, and
subjected to LCQ ESI-MS analysis. MTA was quantitated with an
internal mass standard of [50-2H3] (301 amu) relative to the peak
area for authentic MTA (298 amu). Samples were analyzed in
triplicate.
MTAP Enzymatic Activity AssayCells were trypsinized and washed twice with PBS, and the cell
pellets were frozen at�80�C. On the day of the analysis, the pellets
were thawed on ice and resuspended in MTAP lysis buffer (20 mM
potassium phosphate, pH 7.4, 1 mM dithiothreitol, and Roche
complete, EDTA-free protease cocktail in a dilution equivalent to
1 tablet for 125 ml of lysate buffer), sonicated three times for
15 s each and cooled on ice between sonications, and centrifuged
at 13,000 3 g for 15 min at 4�C. Protein concentrations were
measured with the Bio-Rad DC Protein Assay. The MTAP-activity
assay was as described33 and had the following modifications:
Cell lysates or an enzyme blank consisting of an equal volume
of lysis buffer was preincubated at 37�C for 5 min in 86 ml of a
solution containing 116 mM potassium phosphate, 58 mM KCl,
and 0.23 mM dithiothreitol in quartz microcuvettes. The enzyme
reactions were started with the addition of 4 ml xanthine oxidase
(0.15 units) and 10 ml of 5 mM MTA. Data was collected for
616 The American Journal of Human Genetics 90, 614–627, April 6, 2012

1,300 s, and the rates were calculated by a linear fit to the data
between 800 and 1,100 s. The blank rate was subtracted from all
the enzyme rates. MTAP-assay rates were linear; the lysate protein
concentration ranged from 10 to 500 ng protein per assay. One
unit of MTAP activity is the amount of enzyme that catalyzed
the formation of 1 mmole of adenine per minute under the condi-
tions of the assay.
Results
Positional Cloning of the DMS-MFH-Associated Gene
Previously, we performed genome-wide linkage analysis
and haplotype reconstruction on three unrelated families
to establish the critical region of the DMS-MFH-associated
gene as a 3.5 cM locus in chromosomal region 9p21–22.
The centromeric boundary was defined by D9S1778, and
the telomeric end was defined by D9S171.4 Since the
original mapping, we identified two additional families
(families 4 and 5) (Figure 1). Interestingly, both of the
new families had evidence of a progressive myopathic
disease (Martignetti et al., unpublished observations34)
not previously noted in the three original families. The
disease-associated gene for one of these later multigenera-
tional families (this family was originally described by
Henry et al.35 as having a history of bone disease, patho-
logic fractures, fibrosarcoma, cataracts, and myopathy
(MIM 609940]) had been independently mapped to a
15 Mb region at 9p21–22.34 This region broadly overlap-
ped the DMS-MFH critical region. To determine whether
these two additional families affected by myopathic
disease were allelic and supported the previously identified
DMS-MFH critical region, we performed multipoint
parametric linkage analysis by using markers spanning
chromosome 9.
We analyzed all five families by using a combination of
previously described and laboratory-developed microsatel-
lite markers. A maximal combined location score of 4.27
was obtained for marker D9SB3 (Figure 2A). We then per-
formed haplotype analysis of additional markers within
the shared region and reconstruction of haplotypes to
tested/unaffected
bone dysplasia
bone tumor
Suspected by history
I
II
III
V
2 3
1
Family 4
IV 1
1
I
II
III
IV
V
6 7 8
Family 3
3
I
II
III
IV
V
VI
Family 2
7 8
1 2 3 4 5
1 4 5 6 9 13
I
II
III
IV
V
VI
Family 5
1 3 5
VII
I
II
III
IV
V
VI
Family 1
5 6
7 8 9
Figure 1. Pedigrees of the Five DMS-MFH-Affected FamiliesFamilies 1, 2, and 3 were originally described as the ‘‘American,’’ ‘‘Australian,’’ and ‘‘New York’’ families, respectively.1–3 Family 4 isa previously undescribed DMS-MFH-affected family from New York. Family 5 has been described as having autosomal-dominantbone fragility and limb-girdle myopathy.34 Family members classified as ‘‘suspected by history’’ were unavailable for radiological diag-nosis but had a history of multiple (>2) pathological fractures and/or had children whowere clinically diagnosed with DMS-MFH on thebasis of plain-film X-rays and family history.
The American Journal of Human Genetics 90, 614–627, April 6, 2012 617
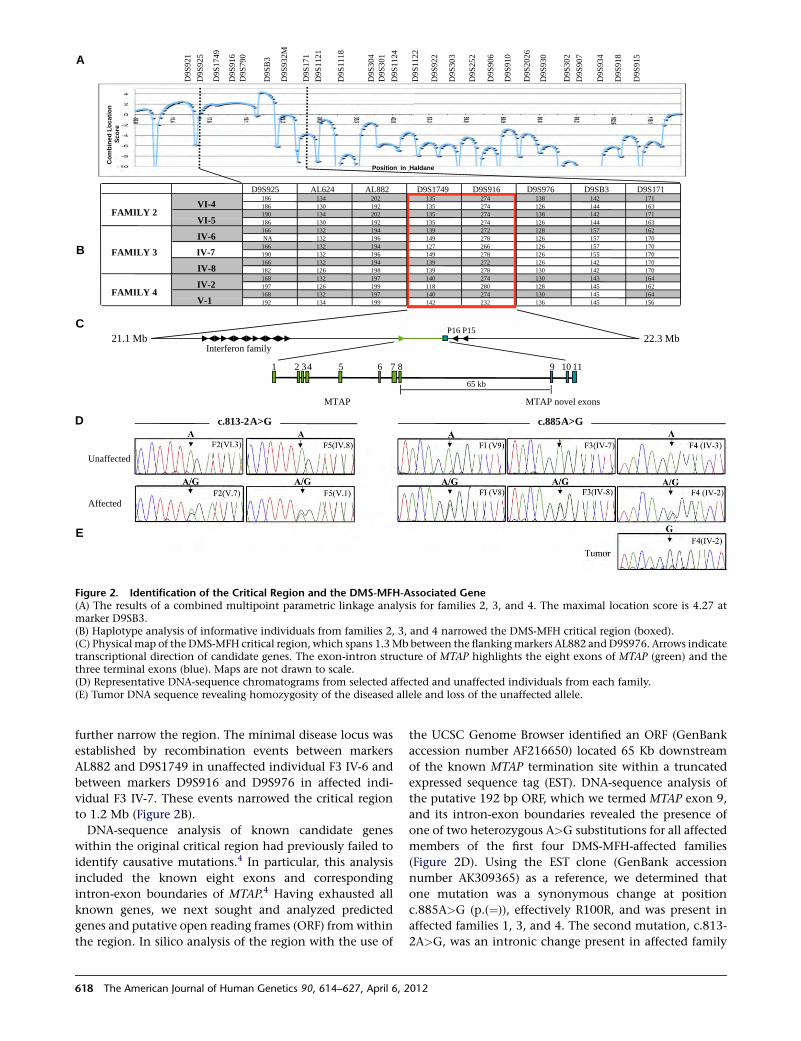
further narrow the region. The minimal disease locus was
established by recombination events between markers
AL882 and D9S1749 in unaffected individual F3 IV-6 and
between markers D9S916 and D9S976 in affected indi-
vidual F3 IV-7. These events narrowed the critical region
to 1.2 Mb (Figure 2B).
DNA-sequence analysis of known candidate genes
within the original critical region had previously failed to
identify causative mutations.4 In particular, this analysis
included the known eight exons and corresponding
intron-exon boundaries of MTAP.4 Having exhausted all
known genes, we next sought and analyzed predicted
genes and putative open reading frames (ORF) from within
the region. In silico analysis of the region with the use of
the UCSC Genome Browser identified an ORF (GenBank
accession number AF216650) located 65 Kb downstream
of the known MTAP termination site within a truncated
expressed sequence tag (EST). DNA-sequence analysis of
the putative 192 bp ORF, which we termed MTAP exon 9,
and its intron-exon boundaries revealed the presence of
one of two heterozygous A>G substitutions for all affected
members of the first four DMS-MFH-affected families
(Figure 2D). Using the EST clone (GenBank accession
number AK309365) as a reference, we determined that
one mutation was a synonymous change at position
c.885A>G (p.(¼)), effectively R100R, and was present in
affected families 1, 3, and 4. The second mutation, c.813-
2A>G, was an intronic change present in affected family
A
B
21.1 Mb 22.3 MbInterferon family
P16 P15
snoxe levon PATMPATM
65 kb
1 3 11019876542
D9S925 AL624 AL882 D9S1749 D9S916 D9S976 D9SB3 D9S171186 134 202 135 274 138 142 171186 130 192 135 274 126 144 163190 134 202 135 274 138 142 171186 130 192 135 274 126 144 163166 132 194 139 272 128 157 162
132 196 149 278 126 157 170166 132 194 127 266 126 157 170190 132 196 149 278 126 155 170166 132 194 139 272 126 142 170182 126 198 139 278 130 142 170168 132 197 140 274 130 143 164197 126 199 118 280 128 145 162168 132 197 140 274 130 145 164192 134 199 142 232 136 145 156
FAMILY 4
IV-6
IV-7
IV-2
V-1
FAMILY 2
FAMILY 3
IV-8
VI-4
VI-5
NA
Unaffected
Affected
Co
mb
in
ed
Lo
ca
tio
n
Sc
ore
D
Position in Haldane
D9S
925
D9S
1749
D9S
916
D9S
790
D9S
B3
D9S
932M
D9S
171
D9S
1121
D9S
1118
D9S
304
D9S
301
D9S
1124
D9S
1122
D9S
922
D9S
303
D9S
252
D9S
906
D9S
910
D9S
2026
D9S
915
D9S
930
D9S
302
D9S
907
D9S
918
D9S
934
D9S
921
A>G A>G
C
E
c.813-2 c.885
Figure 2. Identification of the Critical Region and the DMS-MFH-Associated Gene(A) The results of a combined multipoint parametric linkage analysis for families 2, 3, and 4. The maximal location score is 4.27 atmarker D9SB3.(B) Haplotype analysis of informative individuals from families 2, 3, and 4 narrowed the DMS-MFH critical region (boxed).(C) Physical map of the DMS-MFH critical region, which spans 1.3Mb between the flankingmarkers AL882 and D9S976. Arrows indicatetranscriptional direction of candidate genes. The exon-intron structure of MTAP highlights the eight exons of MTAP (green) and thethree terminal exons (blue). Maps are not drawn to scale.(D) Representative DNA-sequence chromatograms from selected affected and unaffected individuals from each family.(E) Tumor DNA sequence revealing homozygosity of the diseased allele and loss of the unaffected allele.
618 The American Journal of Human Genetics 90, 614–627, April 6, 2012
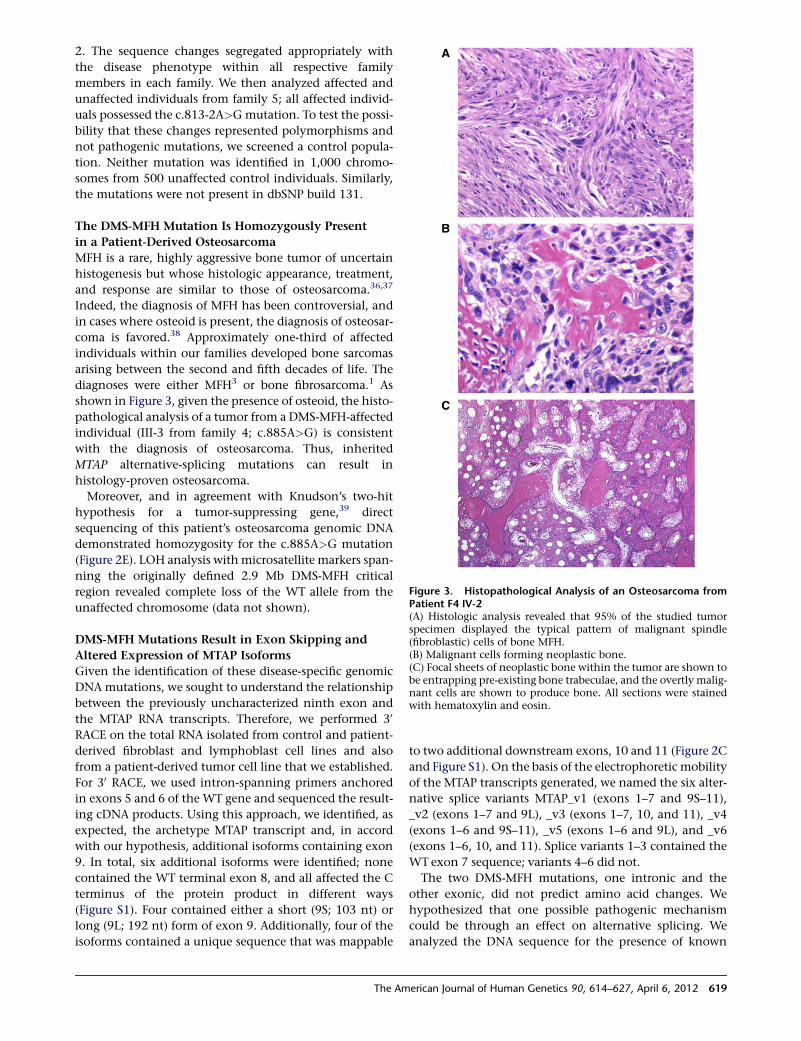
2. The sequence changes segregated appropriately with
the disease phenotype within all respective family
members in each family. We then analyzed affected and
unaffected individuals from family 5; all affected individ-
uals possessed the c.813-2A>Gmutation. To test the possi-
bility that these changes represented polymorphisms and
not pathogenic mutations, we screened a control popula-
tion. Neither mutation was identified in 1,000 chromo-
somes from 500 unaffected control individuals. Similarly,
the mutations were not present in dbSNP build 131.
The DMS-MFH Mutation Is Homozygously Present
in a Patient-Derived Osteosarcoma
MFH is a rare, highly aggressive bone tumor of uncertain
histogenesis but whose histologic appearance, treatment,
and response are similar to those of osteosarcoma.36,37
Indeed, the diagnosis of MFH has been controversial, and
in cases where osteoid is present, the diagnosis of osteosar-
coma is favored.38 Approximately one-third of affected
individuals within our families developed bone sarcomas
arising between the second and fifth decades of life. The
diagnoses were either MFH3 or bone fibrosarcoma.1 As
shown in Figure 3, given the presence of osteoid, the histo-
pathological analysis of a tumor from a DMS-MFH-affected
individual (III-3 from family 4; c.885A>G) is consistent
with the diagnosis of osteosarcoma. Thus, inherited
MTAP alternative-splicing mutations can result in
histology-proven osteosarcoma.
Moreover, and in agreement with Knudson’s two-hit
hypothesis for a tumor-suppressing gene,39 direct
sequencing of this patient’s osteosarcoma genomic DNA
demonstrated homozygosity for the c.885A>G mutation
(Figure 2E). LOH analysis withmicrosatellite markers span-
ning the originally defined 2.9 Mb DMS-MFH critical
region revealed complete loss of the WT allele from the
unaffected chromosome (data not shown).
DMS-MFH Mutations Result in Exon Skipping and
Altered Expression of MTAP Isoforms
Given the identification of these disease-specific genomic
DNA mutations, we sought to understand the relationship
between the previously uncharacterized ninth exon and
the MTAP RNA transcripts. Therefore, we performed 30
RACE on the total RNA isolated from control and patient-
derived fibroblast and lymphoblast cell lines and also
from a patient-derived tumor cell line that we established.
For 30 RACE, we used intron-spanning primers anchored
in exons 5 and 6 of the WT gene and sequenced the result-
ing cDNA products. Using this approach, we identified, as
expected, the archetype MTAP transcript and, in accord
with our hypothesis, additional isoforms containing exon
9. In total, six additional isoforms were identified; none
contained the WT terminal exon 8, and all affected the C
terminus of the protein product in different ways
(Figure S1). Four contained either a short (9S; 103 nt) or
long (9L; 192 nt) form of exon 9. Additionally, four of the
isoforms contained a unique sequence that was mappable
to two additional downstream exons, 10 and 11 (Figure 2C
and Figure S1). On the basis of the electrophoretic mobility
of the MTAP transcripts generated, we named the six alter-
native splice variants MTAP_v1 (exons 1–7 and 9S–11),
_v2 (exons 1–7 and 9L), _v3 (exons 1–7, 10, and 11), _v4
(exons 1–6 and 9S–11), _v5 (exons 1–6 and 9L), and _v6
(exons 1–6, 10, and 11). Splice variants 1–3 contained the
WT exon 7 sequence; variants 4–6 did not.
The two DMS-MFH mutations, one intronic and the
other exonic, did not predict amino acid changes. We
hypothesized that one possible pathogenic mechanism
could be through an effect on alternative splicing. We
analyzed the DNA sequence for the presence of known
Figure 3. Histopathological Analysis of an Osteosarcoma fromPatient F4 IV-2(A) Histologic analysis revealed that 95% of the studied tumorspecimen displayed the typical pattern of malignant spindle(fibroblastic) cells of bone MFH.(B) Malignant cells forming neoplastic bone.(C) Focal sheets of neoplastic bone within the tumor are shown tobe entrapping pre-existing bone trabeculae, and the overtly malig-nant cells are shown to produce bone. All sections were stainedwith hematoxylin and eosin.
The American Journal of Human Genetics 90, 614–627, April 6, 2012 619
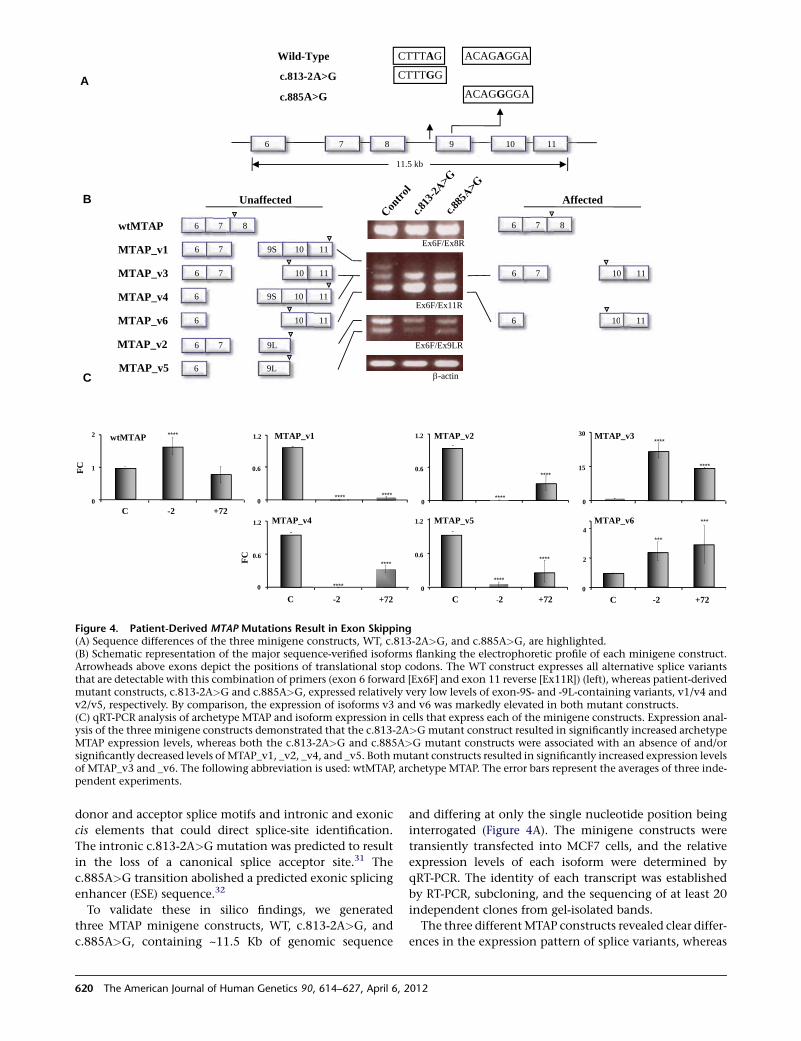
donor and acceptor splice motifs and intronic and exonic
cis elements that could direct splice-site identification.
The intronic c.813-2A>G mutation was predicted to result
in the loss of a canonical splice acceptor site.31 The
c.885A>G transition abolished a predicted exonic splicing
enhancer (ESE) sequence.32
To validate these in silico findings, we generated
three MTAP minigene constructs, WT, c.813-2A>G, and
c.885A>G, containing ~11.5 Kb of genomic sequence
and differing at only the single nucleotide position being
interrogated (Figure 4A). The minigene constructs were
transiently transfected into MCF7 cells, and the relative
expression levels of each isoform were determined by
qRT-PCR. The identity of each transcript was established
by RT-PCR, subcloning, and the sequencing of at least 20
independent clones from gel-isolated bands.
The three differentMTAP constructs revealed clear differ-
ences in the expression pattern of splice variants, whereas
A
B
C
A>G
A>G
6 7 8 9 10 11
11.5 kb
Wild-Type
AffectedUnaffected
wtMTAP
MTAP_v4
MTAP_v1
MTAP_v6
MTAP_v3
6 7 8
6 7 10 11
6 9S 10 11
6 10 11
6 7 8
6 7 10 11
6 10 11
Ex6F/Ex8R
Ex6F/Ex11R
-actin
6 7 9S 10 11
MTAP_v1
0
2
4
C -2 +72
***
***
MTAP_v6
MTAP_v2
0.6
1.2
C -2 +720
****
MTAP_v5
****
0
0.6
1.2
********
0.6
1.2
0
****
****
FC
0
0.6
1.2
C -2 +72
****
MTAP_v4
****
0
15
30****
****
MTAP_v3wtMTAP
FC
0
1
2
Ex6F/Ex9LRMTAP_v2
MTAP_v5
6 7 9L
6 9L
C -2 +72
****
β
c.813-2
c.885
c.813
-2c.8
85
CTTTAG
CTTTGG
ACAGAGGA
ACAGGGGA
Figure 4. Patient-Derived MTAP Mutations Result in Exon Skipping(A) Sequence differences of the three minigene constructs, WT, c.813-2A>G, and c.885A>G, are highlighted.(B) Schematic representation of the major sequence-verified isoforms flanking the electrophoretic profile of each minigene construct.Arrowheads above exons depict the positions of translational stop codons. The WT construct expresses all alternative splice variantsthat are detectable with this combination of primers (exon 6 forward [Ex6F] and exon 11 reverse [Ex11R]) (left), whereas patient-derivedmutant constructs, c.813-2A>G and c.885A>G, expressed relatively very low levels of exon-9S- and -9L-containing variants, v1/v4 andv2/v5, respectively. By comparison, the expression of isoforms v3 and v6 was markedly elevated in both mutant constructs.(C) qRT-PCR analysis of archetype MTAP and isoform expression in cells that express each of the minigene constructs. Expression anal-ysis of the three minigene constructs demonstrated that the c.813-2A>Gmutant construct resulted in significantly increased archetypeMTAP expression levels, whereas both the c.813-2A>G and c.885A>G mutant constructs were associated with an absence of and/orsignificantly decreased levels of MTAP_v1, _v2, _v4, and _v5. Bothmutant constructs resulted in significantly increased expression levelsof MTAP_v3 and _v6. The following abbreviation is used: wtMTAP, archetype MTAP. The error bars represent the averages of three inde-pendent experiments.
620 The American Journal of Human Genetics 90, 614–627, April 6, 2012
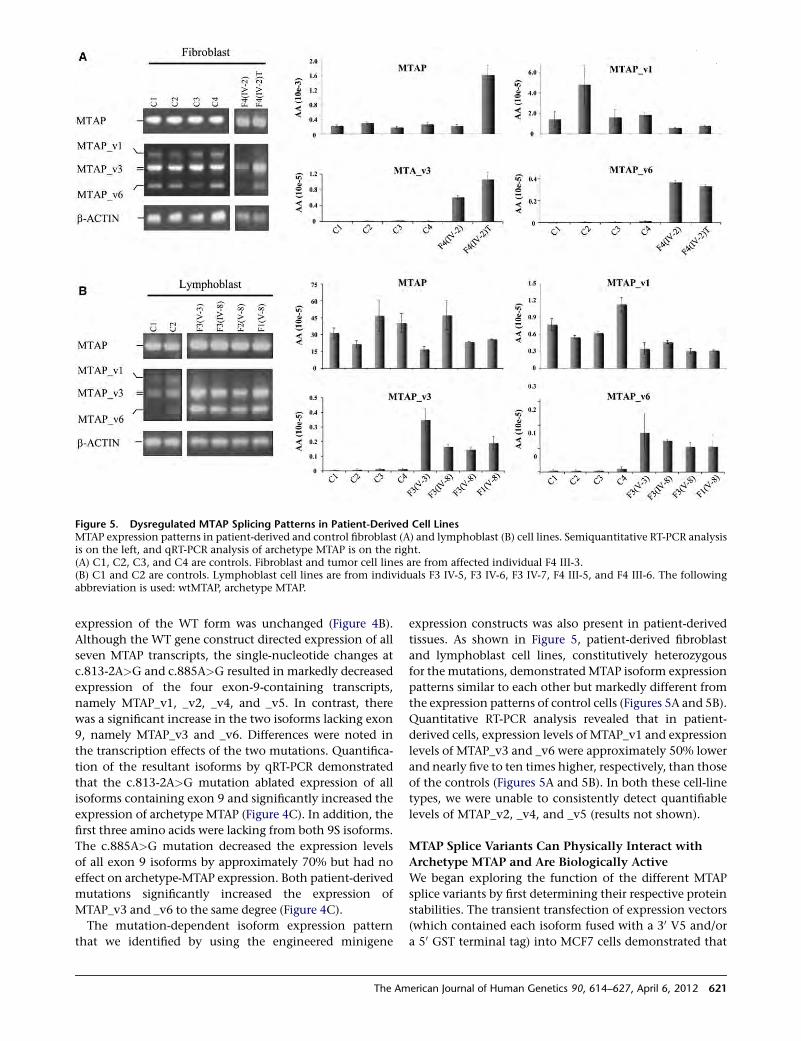
expression of the WT form was unchanged (Figure 4B).
Although the WT gene construct directed expression of all
seven MTAP transcripts, the single-nucleotide changes at
c.813-2A>G and c.885A>G resulted in markedly decreased
expression of the four exon-9-containing transcripts,
namely MTAP_v1, _v2, _v4, and _v5. In contrast, there
was a significant increase in the two isoforms lacking exon
9, namely MTAP_v3 and _v6. Differences were noted in
the transcription effects of the two mutations. Quantifica-
tion of the resultant isoforms by qRT-PCR demonstrated
that the c.813-2A>G mutation ablated expression of all
isoforms containing exon 9 and significantly increased the
expression of archetype MTAP (Figure 4C). In addition, the
first three amino acids were lacking from both 9S isoforms.
The c.885A>G mutation decreased the expression levels
of all exon 9 isoforms by approximately 70% but had no
effect on archetype-MTAP expression. Both patient-derived
mutations significantly increased the expression of
MTAP_v3 and _v6 to the same degree (Figure 4C).
The mutation-dependent isoform expression pattern
that we identified by using the engineered minigene
expression constructs was also present in patient-derived
tissues. As shown in Figure 5, patient-derived fibroblast
and lymphoblast cell lines, constitutively heterozygous
for the mutations, demonstrated MTAP isoform expression
patterns similar to each other but markedly different from
the expression patterns of control cells (Figures 5A and 5B).
Quantitative RT-PCR analysis revealed that in patient-
derived cells, expression levels of MTAP_v1 and expression
levels of MTAP_v3 and _v6 were approximately 50% lower
and nearly five to ten times higher, respectively, than those
of the controls (Figures 5A and 5B). In both these cell-line
types, we were unable to consistently detect quantifiable
levels of MTAP_v2, _v4, and _v5 (results not shown).
MTAP Splice Variants Can Physically Interact with
Archetype MTAP and Are Biologically Active
We began exploring the function of the different MTAP
splice variants by first determining their respective protein
stabilities. The transient transfection of expression vectors
(which contained each isoform fused with a 30 V5 and/or
a 50 GST terminal tag) into MCF7 cells demonstrated that
Figure 5. Dysregulated MTAP Splicing Patterns in Patient-Derived Cell LinesMTAP expression patterns in patient-derived and control fibroblast (A) and lymphoblast (B) cell lines. Semiquantitative RT-PCR analysisis on the left, and qRT-PCR analysis of archetype MTAP is on the right.(A) C1, C2, C3, and C4 are controls. Fibroblast and tumor cell lines are from affected individual F4 III-3.(B) C1 and C2 are controls. Lymphoblast cell lines are from individuals F3 IV-5, F3 IV-6, F3 IV-7, F4 III-5, and F4 III-6. The followingabbreviation is used: wtMTAP, archetype MTAP.
The American Journal of Human Genetics 90, 614–627, April 6, 2012 621

all six MTAP splice variants were translated. Although RNA
levels were essentially equivalent (data not shown), there
was, however, a large variability in protein expression
levels (Figure S2). Archetype-MTAP and MTAP_v1, _v2,
and _v3 proteins (i.e., those containing exon 7) were
expressed at comparable levels, whereas MTAP_v4, _v5,
and _v6 were expressed at markedly lower levels.
These findings suggest a possible difference in protein
stability that might be regulated through the proteosome
pathway. To test this hypothesis, we treated the transfected
cells with the proteosome inhibitor MG132. The v4, v5,
and v6 isoforms accumulated, whereas archetype MTAP
and variants v1, v2, and v3 were relatively unaffected
(Figure S2A). The half-life of each splice variant was esti-
mated by cyclohexamide treatment. The half-life of arche-
type MTAP and MTAP_v1, _v2, and _v3 was R12 hr. It was
significantly less than 6 hr for MTAP_v4, _v5, and _v6
(Figure S2B).
Given these findings and the knowledge that MTAP
exists as a trimeric40–42 protein complex, we tested the
ability of the MTAP splice variants to physically interact
with archetype MTAP. We performed coimmuprecipita-
tions on cell extracts from cotransfected PC3M cells by
using combinations of V5- andGST-tagged fusion proteins.
All MTAP splice variants were able to physically interact
with archetype MTAP (Figure S3).
We next sought to determine whether MTAP splice vari-
ants have MTAP activity. We directly measured the ability
of each isoform to convert MTA to adenine by using a
biochemical assay after we stably transfected them into
two MTAP-null cell lines, MCF7 and MNNG-HOS (a
human-osteosarcoma-derived cell line). Cellular lysates
from mock-transfected cells demonstrated negligible
MTAP activity. Against this null-activity background,
only MTAP isoforms v1, v2, and v3 demonstrated MTAP
activity (Figure S4). Given that variants v4, v5, and v6
had appreciably shorter half-lives, we also performed these
studies in the presence of MG132. Despite the presence of
these isoforms, as determined by immunoblot analysis, we
were unable to detect MTAP activity within the limits of
our assay system (data not shown).
MTA Serum Levels Are Increased in DMS-MFH
MTA is not normally present in human serum. Cells lack-
ing MTAP activity are unable to metabolize MTA,43 and
functional inhibition44 or dysregulation of MTAP activity
would therefore be expected to result in intracellular MTA
accumulation and secretion. When tumor cells are MTAP
deficient, excess MTA would be expected to be cleared by
surrounding MTAP-normal stromal cells. If MTAP expres-
sion or activity is globally affected in all tissues, for
example, in an individual with an inherited germline
MTAP deficiency, then serum levels of MTA should be
increased. To establish whether decreased expression of
exon-9-containing isoforms affects MTAP activity, we
measured MTA serum levels in DMS-MFH-affected family
members and controls. Serum samples from two affected
adults (F4 III-1 and F4 IV-1), one unaffected familymember
(F4 IV-3), and two unrelated controls were analyzed in a
blinded fashion. All three serum samples from unaffected
individuals had no detectable MTA levels. In marked
contrast, both affected individuals had accumulations of
MTA detectable in their serum (Table 1).
Molecular Modeling of MTAP Isoforms
The high-resolution structure of human archetype MTAP
has been previously determined by X-ray crystallography
with several ligands. Most relevant is the trimeric enzyme
complex including 50-deoxy-50-methylthioadenosine sul-
fate (PDB ID 1CG6) and the MTAP apoprotein (PDB ID
1CB0).36 These structures of human 50-deoxy-50-methyl-
thioadenosine phosphorylase at 1.7 A resolution provide
insights into substrate binding (Figure 6A) and catalysis
and serve as a template for modeling potential interactions
among the subunits. On the basis of this model, the amino
acid sequences providing the substrate binding site are en-
coded primarily by exons 6 and 7. Only the L279 residue is
provided by exon 8 (Figure S5). The structure reveals that
each of the three identical subunits is comprised of an
alpha-beta domain containing an eight-stranded and a
five-stranded mixed beta sheet with six dispersed alpha
helices similar to the family of purine nucleoside phos-
phorylases (Figure 6B). On the basis of the results of our
coimmunoprecipitation experiments, the MTAP trimer
could exist as a heterologous assembly of different subunits
comprised of archetype and splice variants. In the MTAP
splice variants, the 75 amino acids of v1 are inserted after
K271, and those of v4 are inserted after A230. In the
MTAP monomer, v1–v3 and v4–v6 oppose each other,
but most importantly, at the interfaces of different sub-
units of the trimer, all of the splice variants are relatively
close to each other spatially, regardless of in which exon
amino acids are inserted within the symmetrically equiva-
lent subunits. As seen in Figure 6C, exon 6 is in juxtaposi-
tion with exon 7 of the adjacent subunit. The trimeric
subunit interface of MTAP does appear to be affected by
the alternate splicing events or, possibly, the MTA active
site (Figure 6C). Secondary-structure and disulfide-bond
prediction analyses predict the generation of a disulfide
bond between cysteine residues in exons 9 and 10; these
prediction analyses require future biochemical analysis
for validation.
Table 1. MTA Serum Levels
Serum Donor MTA Level (pmol/100 ul)
F4 III-1 11.5
F4 IV-2 4.3
F4 IV-3 not detected
Control 1 not detected
Control 2 not detected
622 The American Journal of Human Genetics 90, 614–627, April 6, 2012

Ancestral Retroposition Events Gave Rise to Specific
MTAP Exons in Humans and Possibly Other
Anthropoid Primates
Sequence analysis of the three terminal exons revealed that
exons 9 and 10 shared high homology with different
primate-specific retroviral sequences, which are known to
have integratedmultiple times into different chromosomes
throughout the genome. In general, human endogenous
retrovirus (HERV) sequences are the result of ancestral infec-
tion events that can become incorporated into the genome
and transmitted vertically through speciation events.45 As
shown in Figure 7A, an analysis (with the use of programs
RepeatMasker andRetrosearch) of exons 9 and10 suggested
that these exons arose from integrated retroviruses from
distinct subfamilies. Exon 9 arose from part of a MER50I
element, and exon 10 arose from part of a THE1A element,
one of several families of primate-specific long terminal
repeat (LTR) retrotransposons.46 Both the c.813-2A>G
and c.885A>Gmutations in exon 9 represent G>A nucleo-
tide transitions from the consensus MER50I sequence.
To more precisely establish the evolutionary age of exon
9, which contains the DMS-MFH mutations, we amplified
and sequenced this exon and its intron-exon boundaries
from a panel of primate genomic DNA. PCR amplicons
were obtained and sequenced for confirmation in great
apes and Old and New World monkeys. No product was
amplified from the ring-tailed lemur, a member of an
ancestral extant primate lineage (Figure 7). From this
analysis, we could determine that the MER50I remnant,
which now encodes MTAP exon 9, was integrated over
40 million years ago into the lineage leading to anthropoid
primates.
Discussion
Hereditary cancer syndromes represent a powerful and
tractable biologic system for identifying cancer-causing
mutations.47 Although the syndromes themselves are
rare, their study can provide insight into the basis of the
sporadic forms of the cancers. DMS-MFH represents the
only known hereditary form of MFH, which has been
thought to exist along a spectrum of bone sarcomas with
osteosarcoma. Osteosarcoma itself has been linked to
several hereditary disorders, including Li-Fraumeni
syndrome (MIM 151623), Rothmund-Thomson syndrome
(MIM 268400), Bloom syndrome (MIM 210900), Werner
syndrome (MIM 277700), Paget disease (MIM 602080),
and retinoblastoma, albeit the incidence of cancer devel-
opment associated with these syndromes is lower when
compared to the >30% associated with DMS-MFH.
Recently, one of our affected individuals developed histo-
logically proven osteosarcoma (Figure 3), thus further sup-
porting a genetic link between these tumor types.
The 9p21 region containing theMTAP locus is one of the
most frequently deleted and/or translocated chromosomal
regions in human cancer.5 The facts that MTAP is more
complex than previously recognized and that its terminal
coding exon lies within 25 kb of the p15/p16 locus has
immediate significance to LOHmapping and copy number
variation (CNV) studies in human cancer. Deletions
including the p15/p16 locus will more than likely also
include the 30 region of MTAP and therefore might
affect MTAP biochemical activity. Thus, the interpretation
of many of these studies with regard to the genes
being affected should be reevaluated. Ultimately, the
Figure 6. Molecular Modeling of MTAPModeling is based on the coordinates of the trimeric enzyme complex including 50-deoxy-50-methylthioadenosine sulfate (PDB ID1CG6) and the MTAP apoprotein (PDB ID 1CB0).42 Rendering was done with the program O v.13 (courtesy of Dr. Alwyn Jones, Univer-sity of Uppsala, Uppsala, Sweden), and visualization was done with the PyMOL molecular graphics system v.1.3 (Delano Scientific).(A) Overview of the top of the MTAP trimer. The stick representation (arrow) indicates the MTA substrate.(B) View of an MTAP monomer. The insertion positions of exon 7 (salmon) and exon 6 (tan) are at opposite ends.(C) View of the bottom of the MTAP trimer. Subunits and the insertion positions of exon 7 (salmon) and exon 6 (tan; discontinuouselectron density) are indicated. Displayed is the close proximity of one of these junctions: exon 7 in subunit 2 with exon 8 in subunit3. Because of the three-fold symmetry, there is the possibility of three splice-variant insertion points in the trimer.
The American Journal of Human Genetics 90, 614–627, April 6, 2012 623
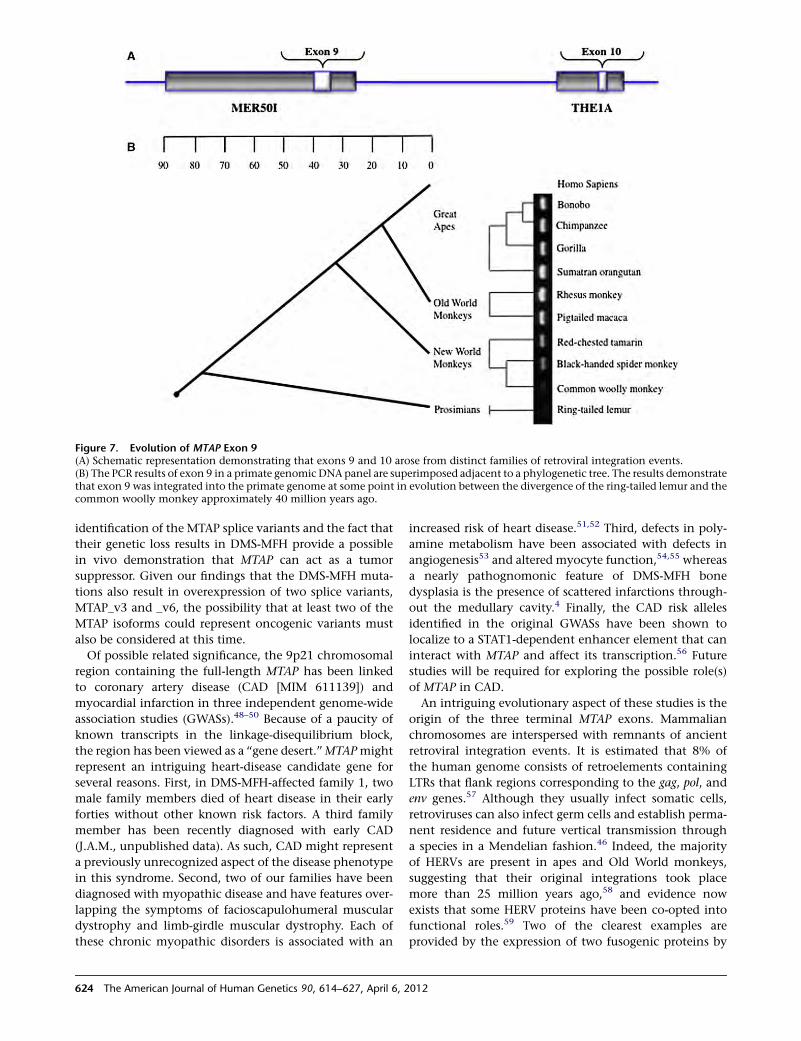
identification of the MTAP splice variants and the fact that
their genetic loss results in DMS-MFH provide a possible
in vivo demonstration that MTAP can act as a tumor
suppressor. Given our findings that the DMS-MFH muta-
tions also result in overexpression of two splice variants,
MTAP_v3 and _v6, the possibility that at least two of the
MTAP isoforms could represent oncogenic variants must
also be considered at this time.
Of possible related significance, the 9p21 chromosomal
region containing the full-length MTAP has been linked
to coronary artery disease (CAD [MIM 611139]) and
myocardial infarction in three independent genome-wide
association studies (GWASs).48–50 Because of a paucity of
known transcripts in the linkage-disequilibrium block,
the region has been viewed as a ‘‘gene desert.’’MTAPmight
represent an intriguing heart-disease candidate gene for
several reasons. First, in DMS-MFH-affected family 1, two
male family members died of heart disease in their early
forties without other known risk factors. A third family
member has been recently diagnosed with early CAD
(J.A.M., unpublished data). As such, CAD might represent
a previously unrecognized aspect of the disease phenotype
in this syndrome. Second, two of our families have been
diagnosed with myopathic disease and have features over-
lapping the symptoms of facioscapulohumeral muscular
dystrophy and limb-girdle muscular dystrophy. Each of
these chronic myopathic disorders is associated with an
increased risk of heart disease.51,52 Third, defects in poly-
amine metabolism have been associated with defects in
angiogenesis53 and altered myocyte function,54,55 whereas
a nearly pathognomonic feature of DMS-MFH bone
dysplasia is the presence of scattered infarctions through-
out the medullary cavity.4 Finally, the CAD risk alleles
identified in the original GWASs have been shown to
localize to a STAT1-dependent enhancer element that can
interact with MTAP and affect its transcription.56 Future
studies will be required for exploring the possible role(s)
of MTAP in CAD.
An intriguing evolutionary aspect of these studies is the
origin of the three terminal MTAP exons. Mammalian
chromosomes are interspersed with remnants of ancient
retroviral integration events. It is estimated that 8% of
the human genome consists of retroelements containing
LTRs that flank regions corresponding to the gag, pol, and
env genes.57 Although they usually infect somatic cells,
retroviruses can also infect germ cells and establish perma-
nent residence and future vertical transmission through
a species in a Mendelian fashion.46 Indeed, the majority
of HERVs are present in apes and Old World monkeys,
suggesting that their original integrations took place
more than 25 million years ago,58 and evidence now
exists that some HERV proteins have been co-opted into
functional roles.59 Two of the clearest examples are
provided by the expression of two fusogenic proteins by
Figure 7. Evolution of MTAP Exon 9(A) Schematic representation demonstrating that exons 9 and 10 arose from distinct families of retroviral integration events.(B) The PCR results of exon 9 in a primate genomic DNA panel are superimposed adjacent to a phylogenetic tree. The results demonstratethat exon 9 was integrated into the primate genome at some point in evolution between the divergence of the ring-tailed lemur and thecommon woolly monkey approximately 40 million years ago.
624 The American Journal of Human Genetics 90, 614–627, April 6, 2012

trophoblasts. Syncytin 1 had been derived from a HERV-W
envelope glycoprotein, is expressed in all trophoblastic
cells, and mediates trophoblast cell fusion into the multi-
nucleated syncytiotrophoblast layer. Syncytin 2, derived
from a HERV-FRD envelope glycoprotein, is expressed in
human placenta.60–62 Our studies reveal that two of the
three terminal exons of MTAP are derived from indepen-
dent retroviral integration events during primate evolu-
tion. Although the integration of exon-9-associated
material most likely occurred after prosimian and New
World monkey divergence more than 40 million years
ago (Figure 7), the timing of its being functionally co-opted
is presently unknown.
The retroviral origins of the terminal exons of MTAP
suggest a number of interesting a priori conclusions. First,
given the evolutionary species restriction of these HERVs,
the existence of MTAP variants and possible biochemical
regulation resulting from their expression must be unique
to primates. Second, the exon 9 mutations that result in
DMS-MFH highlight not only the existence of MTAP
isoforms but also that their heterozygous loss (v1, v2, v4,
and v5) and/or overexpression (v3 and v6) results in
disease. This functionally demonstrates the importance
of the acquired domains in MTAP function and their fixa-
tion into normal human physiology. This phenomenon,
the recruitment and fixation over evolutionary time of a
nucleic-acid sequence as a functional gene, or part thereof,
is termed exaptation, and a number of examples are
known.63–65 However, we are unaware of any other
example wherein the loss of a co-opted gene and/or
protein domain results in a disease phenotype.
Finally, our results continue to demonstrate the dynamic
nature of the genome and the dual reality of disease asso-
ciations and beneficial implications for both alternative
splicing and retroelements. Future studies will now be
required for defining the exact biochemical function(s)
and regulation of these isoforms, their association with
bone dysplasia, cancer initiation, and CAD, and their inter-
action with archetype MTAP.
Supplemental Data
Supplemental Data include five figures and one table and can be
found with this article online at http://www.cell.com/AJHG.
Acknowledgments
The authors gratefully acknowledge the families that participated
in this study. We also thank D. Springfield and J. Brosius for their
thoughtful comments about the patients and manuscript, respec-
tively, and X. Xu, J. Solis, J. Moorjani, C. Meret, S. Tam, and
N. Kham for technical assistance.
Received: October 14, 2011
Revised: January 19, 2012
Accepted: February 16, 2012
Published online: March 29, 2012
Web Resources
The URLs for data presented herein are as follows:
ESEfinder Release 3.0, http://rulai.cshl.edu/cgi-bin/tools/ESE3/
esefinder.cgi
Marshfield database, http://research.marshfieldclinic.org/genetics/
GeneticResearch/compMaps.asp
NetGene2 Server, http://www.cbs.dtu.dk/services/NetGene2/
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
RepeatMasker, http://www.repeatmasker.org/
Retrosearch, http://www.daimi.au.dk/~biopv/herv/
UCSC Genome Browser, March 2006 assembly, http://genome.
ucsc.edu/
Accession Numbers
The EMBL-Bank accession numbers for the translated protein
MTAP variants reported in this paper are: HE654772 (MTAP_v1),
HE654773 (MTAP_v2), HE654774 (MTAP_v3), HE654775
(MTAP_v4), HE654776 (MTAP_v5), and HE654777 (MTAP_v6).
References
1. Arnold, W.H. (1973). Hereditary bone dysplasia with sarcoma-
tous degeneration. Study of a family. Ann. Intern. Med. 78,
902–906.
2. Hardcastle, P., Nade, S., and Arnold, W. (1986). Hereditary
bone dysplasia with malignant change. Report of three fami-
lies. J. Bone Joint Surg. Am. 68, 1079–1089.
3. Norton, K.I., Wagreich, J.M., Granowetter, L., andMartignetti,
J.A. (1996). Diaphyseal medullary stenosis (sclerosis) with
bone malignancy (malignant fibrous histiocytoma): Hardcas-
tle syndrome. Pediatr. Radiol. 26, 675–677.
4. Martignetti, J.A., Desnick, R.J., Aliprandis, E., Norton, K.I.,
Hardcastle, P., Nade, S., and Gelb, B.D. (1999). Diaphyseal
medullary stenosis with malignant fibrous histiocytoma: A
hereditary bone dysplasia/cancer syndrome maps to 9p21-
22. Am. J. Hum. Genet. 64, 801–807.
5. Mitelman, F. (1994). Catalog of Chromosome Aberrations in
Cancer (New York: Wiley/Liss).
6. Miyakoshi, J., Dobler, K.D., Allalunis-Turner, J., McKean, J.D.,
Petruk, K., Allen, P.B., Aronyk, K.N., Weir, B., Huyser-Wier-
enga, D., Fulton, D., et al. (1990). Absence of IFNA and IFNB
genes from human malignant glioma cell lines and lack of
correlation with cellular sensitivity to interferons. Cancer
Res. 50, 278–283.
7. Olopade, O.I., Jenkins, R.B., Ransom, D.T., Malik, K., Pomy-
kala, H., Nobori, T., Cowan, J.M., Rowley, J.D., and Diaz,
M.O. (1992). Molecular analysis of deletions of the short
arm of chromosome 9 in human gliomas. Cancer Res. 52,
2523–2529.
8. Fountain, J.W., Karayiorgou, M., Ernstoff, M.S., Kirkwood,
J.M., Vlock, D.R., Titus-Ernstoff, L., Bouchard, B., Vijayasar-
adhi, S., Houghton, A.N., Lahti, J., et al. (1992). Homozygous
deletions within human chromosome band 9p21 in mela-
noma. Proc. Natl. Acad. Sci. USA 89, 10557–10561.
9. Lukeis, R., Irving, L., Garson, M., and Hasthorpe, S. (1990).
Cytogenetics of non-small cell lung cancer: Analysis of consis-
tent non-random abnormalities. Genes Chromosomes Cancer
2, 116–124.
The American Journal of Human Genetics 90, 614–627, April 6, 2012 625

10. Diaz, M.O., Ziemin, S., Le Beau, M.M., Pitha, P., Smith, S.D.,
Chilcote, R.R., and Rowley, J.D. (1988). Homozygous deletion
of the alpha- and beta 1-interferon genes in human leukemia
and derived cell lines. Proc. Natl. Acad. Sci. USA 85, 5259–
5263.
11. Diaz, M.O., Rubin, C.M., Harden, A., Ziemin, S., Larson, R.A.,
Le Beau, M.M., and Rowley, J.D. (1990). Deletions of inter-
feron genes in acute lymphoblastic leukemia. N. Engl. J.
Med. 322, 77–82.
12. Garcıa-Castellano, J.M., Villanueva, A., Healey, J.H., Sowers,
R., Cordon-Cardo, C., Huvos, A., Bertino, J.R., Meyers, P.,
and Gorlick, R. (2002). Methylthioadenosine phosphorylase
gene deletions are common in osteosarcoma. Clin. Cancer
Res. 8, 782–787.
13. Miyazaki, S., Nishioka, J., Shiraishi, T., Matsumine, A., Uchida,
A., and Nobori, T. (2007). Methylthioadenosine phosphory-
lase deficiency in Japanese osteosarcoma patients. Int. J. On-
col. 31, 1069–1076.
14. Martignetti, J.A., Gelb, B.D., Pierce, H., Picci, P., and Desnick,
R.J. (2000). Malignant fibrous histiocytoma: Inherited and
sporadic forms have loss of heterozygosity at chromosome
bands 9p21-22-evidence for a common genetic defect. Genes
Chromosomes Cancer 27, 191–195.
15. Nobori, T., Takabayashi, K., Tran, P., Orvis, L., Batova, A., Yu,
A.L., and Carson, D.A. (1996). Genomic cloning of methyl-
thioadenosine phosphorylase: A purine metabolic enzyme
deficient in multiple different cancers. Proc. Natl. Acad. Sci.
USA 93, 6203–6208.
16. Kamatani, N., Nelson-Rees, W.A., and Carson, D.A. (1981).
Selective killing of human malignant cell lines deficient in
methylthioadenosine phosphorylase, a purine metabolic
enzyme. Proc. Natl. Acad. Sci. USA 78, 1219–1223.
17. Trackman, P.C., and Abeles, R.H. (1981). The metabolism of
1-phospho-5-methylthioribose. Biochem. Biophys. Res. Com-
mun. 103, 1238–1244.
18. Trackman, P.C., and Abeles, R.H. (1983). Methionine
synthesis from 50-S-Methylthioadenosine. Resolution of
enzyme activities and identification of 1-phospho-5-S methyl-
thioribulose. J. Biol. Chem. 258, 6717–6720.
19. Kamatani, N., Yu, A.L., and Carson, D.A. (1982). Deficiency of
methylthioadenosine phosphorylase in human leukemic cells
in vivo. Blood 60, 1387–1391.
20. Schmid, M., Malicki, D., Nobori, T., Rosenbach, M.D., Camp-
bell, K., Carson, D.A., and Carrera, C.J. (1998). Homozygous
deletions of methylthioadenosine phosphorylase (MTAP) are
more frequent than p16INK4A (CDKN2) homozygous dele-
tions in primary non-small cell lung cancers (NSCLC). Onco-
gene 17, 2669–2675.
21. Stevens, A.P., Spangler, B., Wallner, S., Kreutz, M., Dettmer, K.,
Oefner, P.J., and Bosserhoff, A.K. (2009). Direct and tumor
microenvironment mediated influences of 50-deoxy-50-(meth-
ylthio)adenosine on tumor progression of malignant mela-
noma. J. Cell. Biochem. 106, 210–219.
22. Karikari, C.A., Mullendore, M., Eshleman, J.R., Argani, P.,
Leoni, L.M., Chattopadhyay, S., Hidalgo, M., and Maitra, A.
(2005). Homozygous deletions of methylthioadenosine phos-
phorylase in human biliary tract cancers. Mol. Cancer Ther. 4,
1860–1866.
23. Christopher, S.A., Diegelman, P., Porter, C.W., and Kruger, W.D.
(2002). Methylthioadenosine phosphorylase, a gene frequently
codeleted with p16(cdkN2a/ARF), acts as a tumor suppressor in
a breast cancer cell line. Cancer Res. 62, 6639–6644.
24. Subhi, A.L., Tang, B., Balsara, B.R., Altomare, D.A., Testa, J.R.,
Cooper, H.S., Hoffman, J.P., Meropol, N.J., and Kruger, W.D.
(2004). Loss of methylthioadenosine phosphorylase and
elevated ornithine decarboxylase is common in pancreatic
cancer. Clin. Cancer Res. 10, 7290–7296.
25. Huang, H.Y., Li, S.H., Yu, S.C., Chou, F.F., Tzeng, C.C., Hu,
T.H., Uen, Y.H., Tian, Y.F., Wang, Y.H., Fang, F.M., et al.
(2009). Homozygous deletion of MTAP gene as a poor prog-
nosticator in gastrointestinal stromal tumors. Clin. Cancer
Res. 15, 6963–6972.
26. Mukhopadhyay, N., Almasy, L., Schroeder, M., Mulvihill,W.P.,
and Weeks, D.E. (2005). Mega2: Data-handling for facilitating
genetic linkage and association analyses. Bioinformatics 21,
2556–2557.
27. Sobel, E., and Lange, K. (1996). Descent graphs in pedigree
analysis: Applications to haplotyping, location scores, and
marker-sharing statistics. Am. J. Hum. Genet. 58, 1323–1337.
28. Benson, G. (1999). Tandem repeats finder: A program to
analyze DNA sequences. Nucleic Acids Res. 27, 573–580.
29. Dowling, O., Difeo, A., Ramirez, M.C., Tukel, T., Narla, G.,
Bonafe, L., Kayserili, H., Yuksel-Apak, M., Paller, A.S., Norton,
K., et al. (2003). Mutations in capillary morphogenesis gene-2
result in the allelic disorders juvenile hyaline fibromatosis
and infantile systemic hyalinosis. Am. J. Hum. Genet. 73,
957–966.
30. Hebsgaard, S.M., Korning, P.G., Tolstrup, N., Engelbrecht, J.,
Rouze, P., and Brunak, S. (1996). Splice site prediction in Ara-
bidopsis thaliana pre-mRNA by combining local and global
sequence information. Nucleic Acids Res. 24, 3439–3452.
31. Brunak, S., Engelbrecht, J., and Knudsen, S. (1991). Prediction
of human mRNA donor and acceptor sites from the DNA
sequence. J. Mol. Biol. 220, 49–65.
32. Cartegni, L., Wang, J., Zhu, Z., Zhang, M.Q., and Krainer, A.R.
(2003). ESEfinder: A web resource to identify exonic splicing
enhancers. Nucleic Acids Res. 31, 3568–3571.
33. Savarese, T.M., Crabtree, G.W., and Parks, R.E., Jr. (1981).
50-Methylthioadenosine phosphorylase-L. Substrate activity
of 50-deoxyadenosine with the enzyme from Sarcoma 180
cells. Biochem. Pharmacol. 30, 189–199.
34. Watts, G.D., Mehta, S.G., Zhao, C., Ramdeen, S., Hamilton,
S.J., Novack, D.V., Mumm, S., Whyte, M.P., Mc Gillivray, B.,
and Kimonis, V.E. (2005). Mapping autosomal dominant
progressive limb-girdle myopathy with bone fragility to chro-
mosome 9p21-p22: A novel locus for a musculoskeletal
syndrome. Hum. Genet. 118, 508–514.
35. Henry, E.W., Auckland, N.L., McINTOSH, H.W., and Starr, D.E.
(1958). Abnormality of the long bones and progressive
muscular dystrophy in a family. Can. Med. Assoc. J. 78,
331–336.
36. Picci, P., Bacci, G., Ferrari, S., and Mercuri, M. (1997). Neoad-
juvant chemotherapy in malignant fibrous histiocytoma of
bone and in osteosarcoma located in the extremities: Analo-
gies and differences between the two tumors. Ann. Oncol. 8,
1107–1115.
37. Jeon, D.G., Song, W.S., Kong, C.B., Kim, J.R., and Lee, S.Y.
(2010). MFH of Bone and Osteosarcoma Show Similar
Survival and Chemosensitivity. Clin. Orthop. Relat. Res. 469,
584–590.
38. Ghandur-Mnaymneh, L., Zych, G., and Mnaymneh, W.
(1982). Primary malignant fibrous histiocytoma of bone:
Report of six cases with ultrastructural study and analysis of
the literature. Cancer 49, 698–707.
626 The American Journal of Human Genetics 90, 614–627, April 6, 2012

39. Knudson, A.G. (2001). Two genetic hits (more or less) to
cancer. Nat. Rev. Cancer 1, 157–162.
40. Della Ragione, F., Cartenı-Farina, M., Gragnaniello, V., Schet-
tino,M.I., and Zappia, V. (1986). Purification and characteriza-
tion of 50-deoxy-50-methylthioadenosine phosphorylase from
human placenta. J. Biol. Chem. 261, 12324–12329.
41. Della Ragione, F., Oliva, A., Gragnaniello, V., Russo, G.L.,
Palumbo, R., and Zappia, V. (1990). Physicochemical and
immunological studies on mammalian 50-deoxy-50-methyl-
thioadenosine phosphorylase. J. Biol. Chem. 265, 6241–6246.
42. Appleby, T.C., Erion, M.D., and Ealick, S.E. (1999). The struc-
ture of human 50-deoxy-50-methylthioadenosine phosphory-
lase at 1.7 A resolution provides insights into substrate
binding and catalysis. Structure 7, 629–641.
43. Williams-Ashman, H.G., Seidenfeld, J., and Galletti, P. (1982).
Trends in the biochemical pharmacology of 50-deoxy-50-meth-
ylthioadenosine. Biochem. Pharmacol. 31, 277–288.
44. Schramm, V.L. (2007). Enzymatic transition state theory and
transition state analogue design. J. Biol. Chem. 282, 28297–
28300.
45. Bannert, N., and Kurth, R. (2006). The evolutionary dynamics
of human endogenous retroviral families. Annu. Rev. Geno-
mics Hum. Genet. 7, 149–173.
46. Smit, A.F. (1993). Identification of a new, abundant super-
family of mammalian LTR-transposons. Nucleic Acids Res.
21, 1863–1872.
47. Fearon, E.R. (1997). Human cancer syndromes: Clues to the
origin and nature of cancer. Science 278, 1043–1050.
48. McPherson, R., Pertsemlidis, A., Kavaslar, N., Stewart, A., Rob-
erts, R., Cox, D.R., Hinds, D.A., Pennacchio, L.A., Tybjaerg-
Hansen, A., Folsom, A.R., et al. (2007). A common allele on
chromosome 9 associated with coronary heart disease. Science
316, 1488–1491.
49. Helgadottir, A., Thorleifsson, G., Manolescu, A., Gretarsdottir,
S., Blondal, T., Jonasdottir, A., Jonasdottir, A., Sigurdsson, A.,
Baker, A., Palsson, A., et al. (2007). A common variant on chro-
mosome 9p21 affects the risk of myocardial infarction.
Science 316, 1491–1493.
50. Willer, C.J., Sanna, S., Jackson, A.U., Scuteri, A., Bonnycastle,
L.L., Clarke, R., Heath, S.C., Timpson, N.J., Najjar, S.S., String-
ham, H.M., et al. (2008). Newly identified loci that influence
lipid concentrations and risk of coronary artery disease. Nat.
Genet. 40, 161–169.
51. van der Kooi, A.J., Ledderhof, T.M., de Voogt, W.G., Res, C.J.,
Bouwsma, G., Troost, D., Busch, H.F., Becker, A.E., and de
Visser, M. (1996). A newly recognized autosomal dominant
limb girdle muscular dystrophy with cardiac involvement.
Ann. Neurol. 39, 636–642.
52. Kimonis, V.E., Kovach, M.J., Waggoner, B., Leal, S., Salam, A.,
Rimer, L., Davis, K., Khardori, R., and Gelber, D. (2000). Clin-
ical and molecular studies in a unique family with autosomal
dominant limb-girdle muscular dystrophy and Paget disease
of bone. Genet. Med. 2, 232–241.
53. Takigawa, M., Nishida, Y., Suzuki, F., Kishi, J., Yamashita, K.,
and Hayakawa, T. (1990). Induction of angiogenesis in chick
yolk-sac membrane by polyamines and its inhibition by tissue
inhibitors of metalloproteinases (TIMP and TIMP-2). Bio-
chem. Biophys. Res. Commun. 171, 1264–1271.
54. Harris, S.P., Patel, J.R., Marton, L.J., and Moss, R.L. (2000).
Polyamines decrease Ca(2þ) sensitivity of tension and
increase rates of activation in skinned cardiac myocytes. Am.
J. Physiol. Heart Circ. Physiol. 279, H1383–H1391.
55. Tantini, B., Fiumana, E., Cetrullo, S., Pignatti, C., Bonavita, F.,
Shantz, L.M., Giordano, E., Muscari, C., Flamigni, F., Guar-
nieri, C., et al. (2006). Involvement of polyamines in
apoptosis of cardiac myoblasts in a model of simulated
ischemia. J. Mol. Cell. Cardiol. 40, 775–782.
56. Harismendy, O., Notani, D., Song, X., Rahim, N.G., Tanasa, B.,
Heintzman, N., Ren, B., Fu, X.D., Topol, E.J., Rosenfeld, M.G.,
and Frazer, K.A. (2011). 9p21 DNA variants associated with
coronary artery disease impair interferon-g signalling
response. Nature 470, 264–268.
57. Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody,
M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh,
W., et al; International Human Genome Sequencing Consor-
tium. (2001). Initial sequencing and analysis of the human
genome. Nature 409, 860–921.
58. Mayer, J., and Meese, E. (2005). Human endogenous retrovi-
ruses in the primate lineage and their influence on host
genomes. Cytogenet. Genome Res. 110, 448–456.
59. Volff, J.N., and Brosius, J. (2007). Modern genomes with retro-
look: Retrotransposed elements, retroposition and the origin
of new genes. In Gene and Protein Evolution. Genome
Dynamics, J.-N. Volff, ed. (Basel: Karger), pp. 175–190.
60. Blaise, S., de Parseval, N., Benit, L., and Heidmann, T. (2003).
Genomewide screening for fusogenic human endogenous
retrovirus envelopes identifies syncytin 2, a gene conserved
on primate evolution. Proc. Natl. Acad. Sci. USA 100,
13013–13018.
61. Blond, J.L., Lavillette, D., Cheynet, V., Bouton, O., Oriol, G.,
Chapel-Fernandes, S., Mandrand, B., Mallet, F., and Cosset,
F.L. (2000). An envelope glycoprotein of the human endoge-
nous retrovirus HERV-W is expressed in the human placenta
and fuses cells expressing the type D mammalian retrovirus
receptor. J. Virol. 74, 3321–3329.
62. Mi, S., Lee, X., Li, X., Veldman, G.M., Finnerty, H., Racie,
L., LaVallie, E., Tang, X.Y., Edouard, P., Howes, S., et al.
(2000). Syncytin is a captive retroviral envelope protein
involved in human placental morphogenesis. Nature 403,
785–789.
63. Brosius, J., and Gould, S.J. (1992). On ‘‘genomenclature’’: A
comprehensive (and respectful) taxonomy for pseudogenes
and other ‘‘junk DNA’’. Proc. Natl. Acad. Sci. USA 89, 10706–
10710.
64. Krull, M., Brosius, J., and Schmitz, J. (2005). Alu-SINE exoniza-
tion: En route to protein-coding function. Mol. Biol. Evol. 22,
1702–1711.
65. Baertsch, R., Diekhans, M., Kent, W.J., Haussler, D., and Bro-
sius, J. (2008). Retrocopy contributions to the evolution of
the human genome. BMC Genomics 9, 466.
The American Journal of Human Genetics 90, 614–627, April 6, 2012 627

ARTICLE
Large-Scale Population Analysis Challengesthe Current Criteria for the Molecular Diagnosisof Fascioscapulohumeral Muscular Dystrophy
Isabella Scionti,1 Francesca Greco,1 Giulia Ricci,2 Monica Govi,1 Patricia Arashiro,3 Liliana Vercelli,4
Angela Berardinelli,5 Corrado Angelini,6 Giovanni Antonini,7 Michelangelo Cao,6 Antonio Di Muzio,8
Maurizio Moggio,9 Lucia Morandi,10 Enzo Ricci,11 Carmelo Rodolico,12 Lucia Ruggiero,13
Lucio Santoro,13 Gabriele Siciliano,2 Giuliano Tomelleri,14 Carlo Pietro Trevisan,15 Giuliana Galluzzi,16
Woodring Wright,17 Mayana Zatz,18 and Rossella Tupler1,19,*
Facioscapulohumeral muscular dystrophy (FSHD) is a common hereditary myopathy causally linked to reduced numbers (%8) of 3.3
kilobase D4Z4 tandem repeats at 4q35. However, because individuals carrying D4Z4-reduced alleles and no FSHD and patients with
FSHD and no short allele have been observed, additionalmarkers have been proposed to support an FSHDmolecular diagnosis. In partic-
ular a reduction in the number of D4Z4 elements combined with the 4A(159/161/168)PAS haplotype (which provides the possibility of
expressing DUX4) is currently used as the genetic signature uniquely associated with FSHD. Here, we analyzed these DNA elements in
more than 800 Italian and Brazilian samples of normal individuals unrelated to any FSHD patients. We find that 3% of healthy subjects
carry alleles with a reduced number (4–8) of D4Z4 repeats on chromosome 4q and that one-third of these alleles, 1.3%, occur in combi-
nation with the 4A161PAS haplotype. We also systematically characterized the 4q35 haplotype in 253 unrelated FSHD patients. We find
that only 127 of them (50.1%) carry alleles with 1–8 D4Z4 repeats associated with 4A161PAS, whereas the remaining FSHD probands
carry different haplotypes or alleles with a greater number of D4Z4 repeats. The present study shows that the current genetic signature
of FSHD is a common polymorphism and that only half of FSHD probands carry this molecular signature. Our results suggest that
the genetic basis of FSHD, which is remarkably heterogeneous, should be revisited, because this has important implications for genetic
counseling and prenatal diagnosis of at-risk families.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD [MIM
158900]), a common myopathy, has a prevalence of 1 in
20,000.1,2 The disease is characterized byweakness of selec-
tive muscle groups and wide variability of clinical expres-
sion.1,3,4 The onset of the disease is in the second or third
decade of life and usually involves the weakening of facial
and limb-girdle muscles. The mode of inheritance of
classical FSHD is considered to be autosomal dominant,
with complete penetrance by age 20.4,5 No biochemical,
histological, or instrumental markers are available to inde-
pendently confirm a specific FSHD diagnosis that remains
mainly clinical.
The FSHD genetic defect does not reside in any protein-
coding gene.6 Instead, FSHD has been genetically linked
to the reduction of an integral number of tandem 3.3-kb
D4Z4 repeats located on chromosome 4q35.7,8 Although
nearly identical D4Z4 sequences reside on chromosome
10q26,9 only subjects with a reduced number of D4Z4
repeats on chromosome 4, but not chromosome 10,
develop FSHD.10–12 Based on these results, p13E-11 EcoRI
alleles larger than 50 kb (R11 D4Z4 repeats) originating
from chromosome 4 have been considered normal,
whereas alleles of 35 kb or less (%8 D4Z4 repeats) have
been considered diagnostic for the disease.8,13
Because there are individuals with reduced D4Z4 alleles
that do not have clinical signs of FSHD,14,15 it has been pro-
posed that additional DNA sequences flanking the D4Z4
repeat array are necessary for disease development.16–18
These studies concluded that D4Z4 reduction is pathogenic
only in a few genetic backgrounds, which include a specific
simple sequence length polymorphism (SSLP) proximal
to the D4Z4 repeat and the 4qA polymorphism distal to
1Department of Biomedical Sciences, University of Modena and Reggio Emilia, Modena 41125, Italy; 2Department of Neuroscience, Neurological Clinic,
University of Pisa, Pisa 56126, Italy; 3Program in Genomics, Division of Genetics, Informatics Program, Children’s Hospital, The Howard Hughes Medical
Institute, Harvard Medical School, Boston, MA 02115, USA; 4Department of Neuroscience, Center for Neuromuscular Diseases, University of Turin, Turin
10126, Italy; 5Unit of Child Neurology and Psychiatry, IRCCS ‘‘C. Modino’’ Foundation, University of Pavia, Pavia 27100, Italy; 6Department of Neurosci-
ences, University of Padua, Padua 35129, Italy; 7Department of Neuroscience, Salute Mentale e Organi di Senso, S. Andrea Hospital, University of Rome
‘‘Sapienza,’’ Rome 00189, Italy; 8Center for Neuromuscular Disease, University ‘‘G. d’Annunzio,’’ Chieti 66013, Italy; 9Neuromuscular Unit, IRCCS
Foundation Ca Granda Ospedale Maggiore Policlinico, Dino Ferrari Center, University of Milan, Milan 20122, Italy; 10Unit of Muscular Pathology and
Immunology, Neurological Institute Foundation ‘‘Carlo Besta,’’ Milano 20133, Italy; 11Department of Neurosciences, Universita Cattolica Policlinico
A. Gemelli, Rome 00168, Italy; 12Department of Neurosciences, Psychiatry and Anaesthesiology, University of Messina, Messina 98125, Italy; 13Department
of Neurological Sciences, University ‘‘Federico II,’’ Naples 80131, Italy; 14Department of Neurological Sciences and Vision, University of Verona, Verona
37134, Italy; 15Department of Neurological and Psychiatric Sciences, University of Padua, Padua 35100, Italy; 16Molecular Genetics Laboratory of UILDM,
Lazio Section, IRCCS Santa Lucia Foundation, Rome 00179, Italy; 17Department of Cell Biology, University of Texas Southwestern Medical Center, Dallas,
TX 75390, USA; 18Human Genome Research Center, Department of Genetics and Evolutionary Biology, Institute of Biosciences, University of Sao Paulo,
Sao Paulo 05508-090, Brazil; 19Program in Gene Function and Expression, University of Massachusetts Medical School, Worcester, MA 01605, USA
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.019. �2012 by The American Society of Human Genetics. All rights reserved.
628 The American Journal of Human Genetics 90, 628–635, April 6, 2012

the repeat (Figure 1). These haplotypes, named 4A159,
4A161, and 4A168, have been proposed to be uniquely
associated with FSHD. Recently it has been shown that
a single-nucleotide polymorphism (SNP) in the pLAM
sequence of the 4qA alleles provides a polyadenylation
signal (PAS; ATTAAA) for the DUX4 transcript from the
most distal D4Z4 unit on 4qA chromosomes. Thus, the
molecular signature, named 4A(159,161,168)PAS, has
been proposed to define alleles causally related to FSHD.
This signature results from the combination of (1) a reduc-
tion in the number of D4Z4 elements, (2) the presence of
the 4qA allele, and (3) the PAS in the pLAM sequence. In
this scenario, FSHD arises from a specific genetic setting
enabling the normally silent double homeobox protein 4
gene (DUX4 [MIM606009]) tobe expressed.18On this basis,
healthy subjects carrying reduced D4Z4 alleles would be
explained by the absence of the 4A(159,161,168)PAS.
This model does not apply to all FSHD cases. For
example, nonpenetrant carriers have been reported in
FSHD families,14,15 and there are FSHD patients carrying
full-length D4Z4 alleles (R11 repeats) that are clinically
indistinguishable from patients carrying D4Z4 alleles of
reduced size (%8 repeats).19 Rare exceptions could be ex-
plained by a variety of mechanisms that do not challenge
the basic hypothesis. However, recently we found that
2.7% of cases in the Italian National Registry for FSHD
(which contains over 1,100 unrelated FSHD patients)
were compound heterozygotes carrying two D4Z4-reduced
alleles (0.5% were homozygotes for the 4A161 haplotype).
Based on this finding, we estimated that the population
frequency of the 4A161PAS haplotype associated with a
D4Z4-reduced allele could be higher than 1%.20
The correlation between genotype and phenotype in
FSHD thus appears to be more complex than just the
Figure 1. Schematic Representation of Polymorphisms at the 4q and 10q Subtelomeres(A) Schematic representation of the method used to calculate D4Z4 repeat numbers from EcoRI fragment sizes. The D4Z4 repeat array isindicated with triangles. Seven and eight D4Z4 repeats (31–36 kb EcoRI fragment size) were defined to be the upper diagnostic range forFSHD. D4Z4 repeat units on chromosomes 4 and 10 can be distinguished because all repeats on 10q contain BlnI restriction sites(B within the black triangles), whereas all D4Z4 repeats on 4q contain XapI restriction sites (X within the white triangles).(B) Schematic representation of the current view of pathogenic haplotypes.(C) Elements examined in the present study. In addition to the number of D4Z4 repeats, elements that distinguish subjects include: (1)the chromosomal localization of the D4Z4 repeat, chromosome 4q35 or 10q26; (2) the SSLP, which is a combination of five variablenumber tandem repeats, an 8 bp insertion/deletion, and two SNPs localized 3.5 kb proximal to D4Z4; it varies in length between 157and 182 bp; (3) the AT(T/C)AAA SNP in the pLAM region; (4) a large sequence variation (termed 4qA or B) that is distal to D4Z4. Inthe 4qB variant, the terminal 3.3 kb repeat contains only 570 bp of a complete repeat, whereas in the 4qA variant the terminal repeatis a divergent 3.3 kb repeat named pLAM. 4q chromosomes that do not hybridize to probes for (A) and (B) are termed ‘‘null,’’ and theirsequences vary from case to case.
The American Journal of Human Genetics 90, 628–635, April 6, 2012 629

presence of the 4APAS signature. In order to confirm the
high frequency of this signature in the normal population
and reevaluate the allele distribution in FSHD patients, we
performed a systematic unbiased clinical and molecular
study of 801 normal control subjects from Italy and Brazil
and 253 FSHD probands from the Italian Registry for
FSHD. Our results establish that the 4APAS structure is
a frequent genetic polymorphism that is neither sufficient
nor necessary for the development of FSHD. This result is
not incompatible with evidence implicating DUX4 or
other factors as important mediators of the disease.
However, it does demonstrate that the pathogenesis is
more complex than currently thought and that the current
genetic signature is insufficient for diagnosis.
Subjects and Methods
Control PopulationThe control group consisted of 801 unrelated healthy subjects
with no family history of muscular dystrophy. Subjects were
recruited from the Italian and Brazilian populations through
advertisements. Italian controls subjects were equally distributed
among Northern, Central, and Southern regions. The local ethics
committee approved the study. All subjects enrolled in the
study were clinically and molecularly characterized after giving
informed consent to participate (see Table S1 available online).
FSHD PatientsTwo-hundred-fifty-three unrelated FSHD patients were accrued
through the Italian National Registry for FSHD. All subjects were
clinically and molecularly characterized. In particular, we consid-
ered patients to have typical FSHD if: (1) disease onset occurred
in facial or shoulder girdle muscles; (2) there was facial and/or
scapular fixator weakness; and (3) there was absence of atypical
signs suggesting an alternative diagnosis (including extraocular,
masticatory, pharyngeal, or lingual muscle weakness and cardio-
myopathy).21,22 Clinical data were collected with the FSHD
clinical form. The clinical severity of the disease was measured
according to the FSHD score, as previously described.23 Briefly,
the FSHD score quantifies the degree of weakness and defines
the level of disability affecting six separate muscle groups: facial
(score 0–2), shoulder girdle (score 0–3), upper limbs (score 0–2),
pelvic girdle (score 0–5), leg muscles (score 0–3), and Beevor’s
sign (score 0–1).23 The final clinical evaluation score, calculated
by summing the single scores, ranged from 0, when no signs of
muscle weakness are present, to 15, when all muscle groups tested
are severely impaired.23 All selected subjects were evaluated using
a standard protocol, and each subject received the standardized
FSHD score previously described.23
Molecular Genetic AnalysisDNA was prepared from isolated lymphocytes according to stan-
dard procedures. In brief, restriction endonuclease digestion of
DNA was performed in agarose plugs with the appropriate restric-
tion enzyme: EcoRI, EcoRI/BlnI, XapI (p13E-11 probe), Hind III
(4qA/4qB probes), and NotI (B31 probe). Digested DNA was
separated by pulsed-field gel electrophoresis (PFGE) in 1% agarose
gels. Allele sizes were estimated by southern hybridization with
probe p13E-11 of 7 mg of EcoRI-, EcoRI/BlnI-, and XapI-digested
genomic DNA extracted from peripheral blood lymphocytes,
electrophoresed in a 0.4% agarose gel for 45–48 hr at 35 V, along-
side an 8–48 kb marker (Bio-Rad). To assess the chromosomal
origin of the two D4Z4-reduced alleles, DNA from each proband
was analyzed by NotI digestion and hybridization with the
B31 probe (Figure S1). Restriction fragments were detected
by autoradiography or by using a Typhoon Trio system (GE
Healthcare).
4qA/4qB allelic variants were defined using 7 mg of HindIII-
digested DNA, PFGE electrophoresis, and Southern blot hybridiza-
tion with radiolabeled 4qB and 4qA probes according to standard
procedures. The 4qA/4qB variants were attributed to each chromo-
some based on the size of EcoRI restriction fragments (Figure S1).
To define the SSLP and the pLAM SNP (AT(T/C)AAA) sequences
flanking the D4Z4 repeat units, linear gel electrophoresis of
EcoRI-digested DNA was used to isolate each D4Z4-reduced allele.
The SSLP sequence was determined after PCR amplification using
specific oligonucleotides (forward primer 50-GGTGGAGTTCTGGT
TTCAGC-30 labeled with hexachlorofluorescein [HEX], reverse
primer 50-CCTGTGCTTCAGAGGCATTTG-30) as previously re-
ported.17,18 Analysis of the pLAM SNP was performed on PCR-
amplified DNA using specific oligonucleotides (forward primer
50-ACGCTGTCTAGGCAAACCTG-30, reverse primer 50-TGCAC
TCATCACACAAAAGATG-30). SSLP size differences and pLAM
sequences were analyzed using an ABI Prism 3130 Genetic
Analyzer.17,18
Results
Presence of FSHD-Sized Alleles in the Healthy
Population
Previous smaller investigations of the Dutch population
suggested that 3% of healthy subjects had D4Z4 alleles of
35–38 kb in size, and one-third of these might present
a potentially pathogenic 4A allele.11,24 An additional
recent genetic criteria is that, in order to have FSHD, the
reduction of D4Z4 repeats must be associated with
a specific chromosomal background, 4A(159/161/168)
PAS, allowing the expression of the DUX4 gene.18
However, the frequency of compound heterozygotes in
patients with FSHD suggested that the frequency of
D4Z4-reduced 24–35 kb alleles associated with the
4A161PAS in the Italian population would be >1%.20
Because this prediction has crucial implications for clinical
practice, we searched for D4Z4-reduced alleles associated
with the 4A161PAS haplotype in 801 healthy individuals,
560 from Italy and 241 from Brazil. Figure 2 shows that
25 of these 801 subjects carry D4Z4 alleles ranging from
21 to 35 kb (4 to 8 D4Z4 units); 17 of these 25 alleles are
associated with 4qA (Figure 2, groups 1 and 2), and 11 carry
the 4A161PAS haplotype (Figure 2, group 1; Figures S2–S7).
Therefore, 3% (25 of 801) of normal controls carry D4Z4
alleles of reduced size, and ~1.3% (11) have the supposedly
pathogenic 4A161PAS haplotype. The age of all these
healthy carriers ranged between 40 and 78 years, an age
in which FSHD is considered to be fully penetrant. On
this basis, we conclude that the haplotype 4A161PAS has
the frequency of a common polymorphism and that it
630 The American Journal of Human Genetics 90, 628–635, April 6, 2012
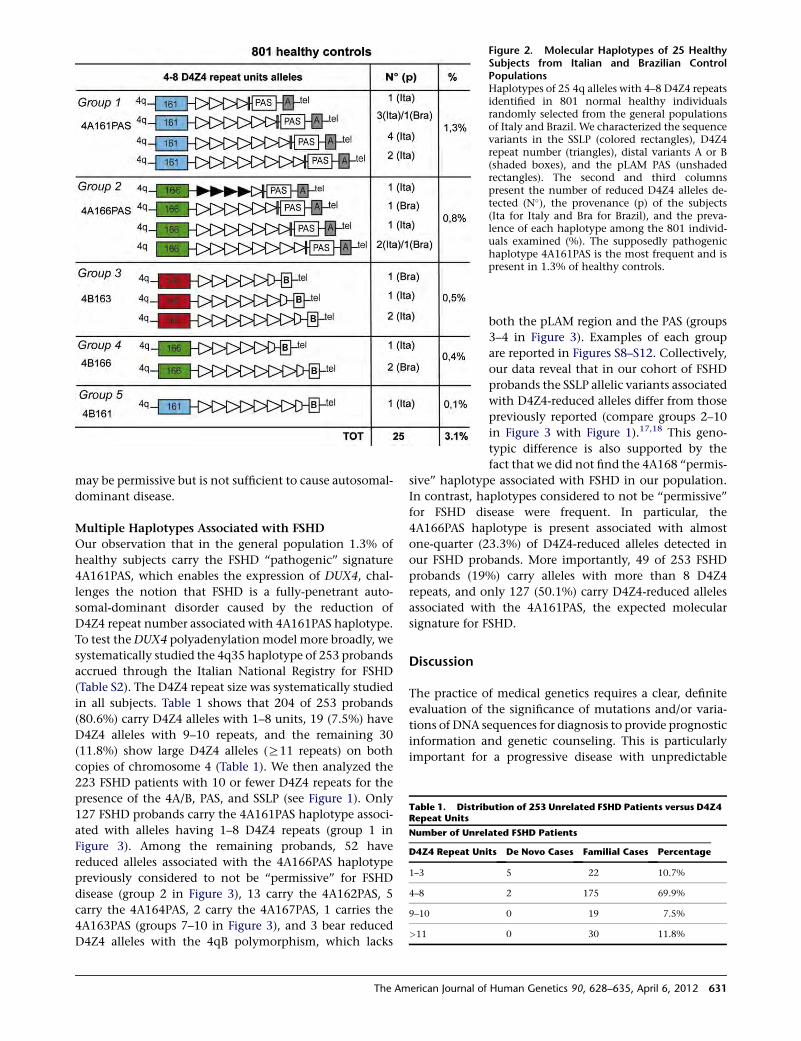
may be permissive but is not sufficient to cause autosomal-
dominant disease.
Multiple Haplotypes Associated with FSHD
Our observation that in the general population 1.3% of
healthy subjects carry the FSHD ‘‘pathogenic’’ signature
4A161PAS, which enables the expression of DUX4, chal-
lenges the notion that FSHD is a fully-penetrant auto-
somal-dominant disorder caused by the reduction of
D4Z4 repeat number associated with 4A161PAS haplotype.
To test theDUX4 polyadenylation model more broadly, we
systematically studied the 4q35 haplotype of 253 probands
accrued through the Italian National Registry for FSHD
(Table S2). The D4Z4 repeat size was systematically studied
in all subjects. Table 1 shows that 204 of 253 probands
(80.6%) carry D4Z4 alleles with 1–8 units, 19 (7.5%) have
D4Z4 alleles with 9–10 repeats, and the remaining 30
(11.8%) show large D4Z4 alleles (R11 repeats) on both
copies of chromosome 4 (Table 1). We then analyzed the
223 FSHD patients with 10 or fewer D4Z4 repeats for the
presence of the 4A/B, PAS, and SSLP (see Figure 1). Only
127 FSHD probands carry the 4A161PAS haplotype associ-
ated with alleles having 1–8 D4Z4 repeats (group 1 in
Figure 3). Among the remaining probands, 52 have
reduced alleles associated with the 4A166PAS haplotype
previously considered to not be ‘‘permissive’’ for FSHD
disease (group 2 in Figure 3), 13 carry the 4A162PAS, 5
carry the 4A164PAS, 2 carry the 4A167PAS, 1 carries the
4A163PAS (groups 7–10 in Figure 3), and 3 bear reduced
D4Z4 alleles with the 4qB polymorphism, which lacks
Figure 2. Molecular Haplotypes of 25 HealthySubjects from Italian and Brazilian ControlPopulationsHaplotypes of 25 4q alleles with 4–8 D4Z4 repeatsidentified in 801 normal healthy individualsrandomly selected from the general populationsof Italy and Brazil. We characterized the sequencevariants in the SSLP (colored rectangles), D4Z4repeat number (triangles), distal variants A or B(shaded boxes), and the pLAM PAS (unshadedrectangles). The second and third columnspresent the number of reduced D4Z4 alleles de-tected (N�), the provenance (p) of the subjects(Ita for Italy and Bra for Brazil), and the preva-lence of each haplotype among the 801 individ-uals examined (%). The supposedly pathogenichaplotype 4A161PAS is the most frequent and ispresent in 1.3% of healthy controls.
Table 1. Distribution of 253 Unrelated FSHD Patients versus D4Z4Repeat Units
Number of Unrelated FSHD Patients
D4Z4 Repeat Units De Novo Cases Familial Cases Percentage
1–3 5 22 10.7%
4–8 2 175 69.9%
9–10 0 19 7.5%
>11 0 30 11.8%
both the pLAM region and the PAS (groups
3–4 in Figure 3). Examples of each group
are reported in Figures S8–S12. Collectively,
our data reveal that in our cohort of FSHD
probands the SSLP allelic variants associated
with D4Z4-reduced alleles differ from those
previously reported (compare groups 2–10
in Figure 3 with Figure 1).17,18 This geno-
typic difference is also supported by the
fact that we did not find the 4A168 ‘‘permis-
sive’’ haplotype associated with FSHD in our population.
In contrast, haplotypes considered to not be ‘‘permissive’’
for FSHD disease were frequent. In particular, the
4A166PAS haplotype is present associated with almost
one-quarter (23.3%) of D4Z4-reduced alleles detected in
our FSHD probands. More importantly, 49 of 253 FSHD
probands (19%) carry alleles with more than 8 D4Z4
repeats, and only 127 (50.1%) carry D4Z4-reduced alleles
associated with the 4A161PAS, the expected molecular
signature for FSHD.
Discussion
The practice of medical genetics requires a clear, definite
evaluation of the significance of mutations and/or varia-
tions of DNA sequences for diagnosis to provide prognostic
information and genetic counseling. This is particularly
important for a progressive disease with unpredictable
The American Journal of Human Genetics 90, 628–635, April 6, 2012 631

onset and a high variability of clinical expression, such as
FSHD.
The extensive use over the past 20 years of DNA anal-
ysis for studying Mendelian disorders has revealed many
complex mechanisms in addition to single mutant genes
that cause disease. Identical phenotypes may be produced
by mutations in different genes,25 the same mutation can
cause different phenotypes,26 and distinct mutations in
the same gene may result in different disorders that
segregate with diverse Mendelian or even multifactorial
patterns.27 In addition, the incomplete penetrance of
certain mutations argues for the importance of modifying
loci or epigenetic mechanisms influencing the clinical
expression in many Mendelian disorders.28 Thus, estab-
lishing the value of mutational events underlying genetic
diseases may be complex even when there are simple
patterns of inheritance in diseases with a well-character-
ized pathologic course.
FSHD seems to fall in this complex pattern, even
though it is currently considered to be a fully penetrant
disease with a wide variability in clinical spectrum,
ranging from subjects with very mild muscle weakness
to wheelchair-bound patients.5,13 The molecular test
initially used for FSHD diagnosis was based on the obser-
vation that 95% of FSHD patients carry a reduction of
integral numbers of D4Z4 repeats at 4q35 with full pene-
trance.10 However, the wide use of this test revealed
several exceptions to the original model. Through the
years, the threshold size of D4Z4 alleles has been
increased from the original 28 kb7 (6 repeats) to 35 kb10
(8 repeats), with FSHD cases carrying D4Z4 alleles of 38–
41 kb (9–11 repeats), considered borderline alleles.22,29
Figure 3. Molecular Haplotypes of 223 Unrelated FSHD PatientsOverview of haplotypes of the D4Z4-reduced alleles (%10 repeats) on chromosome 4 found in 223 unrelated FSHD patients. Alleles areseparated into 1–3 (column 1), 4–8 (column 3), and 9–10 (column 5) D4Z4 repeat units. Within these columns, we group the observedhaplotypes based on the type of SSLP and PAS. N� indicates the number of alleles found in each haplotype, and TOT(%) represents thetotal number of alleles found and the prevalence of each haplotype. The 4A haplotypes not previously observed in FSHDpatients include4A166 (group 2), 4A162 (group 7), 4A164 (group 8), 4A167 (group 9), and 4A163 (group 10), and the 4B haplotypes include 4B163 (group3) and 4B166 (group 4). Frequencies of 4A161 (group 1, 63.7%) and 4A166 (group 2, 25.1%) are different than previously reported inother normal populations. Chromosomes with 4qB haplotypes (groups 3–4) lack the pLAM and PAS.
632 The American Journal of Human Genetics 90, 628–635, April 6, 2012

Additional genotype-phenotype studies led to the identifi-
cation of subjects carrying D4Z4-reduced alleles with no
sign of muscle weakness in FSHD families,14,15 as well as
of healthy unrelated subjects without family history of
FSHD.11,30 The present results from our systematic clinical
and molecular analysis of FSHD patients from the Italian
National Registry for FSHD, as well as a large number of
healthy controls, challenge the current model for FSHD
diagnosis.
Remarkably, our data establish as a general rule rather
than an exception that detection of a D4Z4-reduced
allele is not sufficient to diagnose FSHD. Although
the majority of FSHD patients (70%) carry D4Z4
alleles with 4–8 units, this size range is carried by 3% of
healthy subjects from the general population. Addition-
ally, there is little predictive value of the 4qA161PAS
haplotype in the absence of family history, because
1.3% of healthy subjects carry this haplotype, which
therefore has the frequency of a common polymorphism
(Figure 1) rather than a rare mutation. Finally, 49 of 253
probands (19%) do not carry D4Z4 alleles with 1–8
repeats, and only 50% of the probands carry the 4A161
permissive haplotype.
In summary, our study indicates that a profound
rethinking of the genetic disease mechanism and modes
of inheritance of FSHD are now required and that entirely
newmodels and approaches are needed. Our results do not
exclude an important pathogenic role for DUX4 or other
candidate factors but do establish a complex mechanism
beyond current understanding. Indeed, our data point
at the possibility that in the heterozygous state a D4Z4
reductionmight produce a subclinical sensitized condition
that requires other epigenetic mechanisms or a contrib-
uting factor to cause overt myopathy. In some rare cases,
that could be by becoming homozygous20 and doubling
the dose of a dominant factor such as DUX4. In others, it
might be by the simultaneous heterozygosity for a different
and recessive myopathy, as suggested by many reports
in which the FSHD contractions are found in association
with a second molecular defect.31–44 This possibility is
also consistent with previous reports of expression
changes of candidate proteins such as CRYM that were
associated with FSHD in some families but that were
unchanged when other families were examined. Finally,
it is also plausible that drugs or toxic agents might
contribute to the disease onset and clinical variability.
This would explain the observation of discordant mono-
zygotic twins carrying the FSHD reduction.45,46 It is hoped
that broadening the scope of investigations, including
next-generation deep sequencing in particular in families
with asymptomatic and clinically affected members
carrying the same FSHD allele, may finally lead to an
understanding of the molecular pathogenesis of this
complex disease. These findings have important clinical
implications for genetic counseling of patients and fami-
lies with FSHD, with particular regard to the interpretation
of data in prenatal diagnosis.
Supplemental Data
Supplemental Data include twelve figures and two tables and can
be found with this article online at http://www.cell.com/AJHG/.
Acknowledgments
We are indebted to all FSHD patients and their families for partici-
pating in this study. The Associazione Amici del Centro Dino
Ferrari-University of Milan is gratefully acknowledged. We thank
Paul D. Kaufman and Michael R. Green for their in-depth critique
of the manuscript. DNA from Brazilian controls was kindly
provided by Naila Lourenco and Antonia Cerqueira (Department
of Genetics and Evolutionary Biology, Institute of Biosciences,
University of Sao Paulo, Brazil). This work was supported by Tele-
thon GUP08004 and GUP11009, by Association Francaise Contre
les Myopathies 14339, by National Institute of Health-National
Institutes of Neurological Disorders and Stroke grant RO1
NS047584, by Centros de Pesquisa, Inovacao e Difusao/Fundacao
de Amparo a Pesquisa do Estado de Sao Paulo, by Institutos
Nacionais de Ciencia e Tecnologia, and by Conselho Nacional de
Desenvolvimento Cientıfico e Tecnologico.
Received: November 12, 2011
Revised: January 27, 2012
Accepted: February 16, 2012
Published online: April 4, 2012
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.
nlm.nih.gov/Omim/
References
1. Padberg, G.W. (1982). Facioscapulohumeral disease. PhD
thesis, Leiden University, Leiden, Holland.
2. Mostacciuolo, M.L., Pastorello, E., Vazza, G., Miorin, M.,
Angelini, C., Tomelleri, G., Galluzzi, G., and Trevisan, C.P.
(2009). Facioscapulohumeral muscular dystrophy: epidemio-
logical and molecular study in a north-east Italian population
sample. Clin. Genet. 75, 550–555.
3. Flanigan, K.M. (2004). Facioscapulohumeral muscular dys-
trophy and scapuloperoneal disorders. In Myology, A. Engel
and C. Franzini-Armstrong, eds. (New York: McGrow Hill
Professional), pp. 1123–1133.
4. Lunt, P.W., and Harper, P.S. (1991). Genetic counselling in
facioscapulohumeral muscular dystrophy. J. Med. Genet. 28,
655–664.
5. Tawil, R., van der Maarel, S., Padberg, G.W., and van Engelen,
B.G. (2010). 171st ENMC international workshop: Standards
of care and management of facioscapulohumeral muscular
dystrophy. Neuromuscul. Disord. 20, 471–475.
6. Hewitt, J.E., Lyle, R., Clark, L.N., Valleley, E.M., Wright, T.J.,
Wijmenga, C., van Deutekom, J.C., Francis, F., Sharpe, P.T.,
Hofker, M., et al. (1994). Analysis of the tandem repeat locus
D4Z4 associated with facioscapulohumeral muscular
dystrophy. Hum. Mol. Genet. 3, 1287–1295.
7. Wijmenga, C., Hewitt, J.E., Sandkuijl, L.A., Clark, L.N.,Wright,
T.J., Dauwerse, H.G., Gruter, A.M., Hofker, M.H., Moerer, P.,
The American Journal of Human Genetics 90, 628–635, April 6, 2012 633

Williamson, R., et al. (1992). Chromosome 4q DNA rearrange-
ments associated with facioscapulohumeral muscular
dystrophy. Nat. Genet. 2, 26–30.
8. van Deutekom, J.C.T., Wijmenga, C., van Tienhoven, E.A.,
Gruter, A.M., Hewitt, J.E., Padberg, G.W., van Ommen, G.J.,
Hofker, M.H., and Frants, R.R. (1993). FSHD associated DNA
rearrangements are due to deletions of integral copies of
a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 2, 2037–
2042.
9. Deidda, G., Cacurri, S., Grisanti, P., Vigneti, E., Piazzo, N., and
Felicetti, L. (1995). Physicalmapping evidence for a duplicated
region on chromosome 10qter showing high homology with
the facioscapulohumeral muscular dystrophy locus on chro-
mosome 4qter. Eur. J. Hum. Genet. 3, 155–167.
10. van Deutekom, J.C.T., Bakker, E., Lemmers, R.J., van der Wie-
len, M.J., Bik, E., Hofker, M.H., Padberg, G.W., and Frants, R.R.
(1996). Evidence for subtelomeric exchange of 3.3 kb
tandemly repeated units between chromosomes 4q35 and
10q26: implications for genetic counselling and etiology of
FSHD1. Hum. Mol. Genet. 5, 1997–2003.
11. van Overveld, P.G.M., Lemmers, R.J., Deidda, G., Sandkuijl, L.,
Padberg, G.W., Frants, R.R., and van der Maarel, S.M. (2000).
Interchromosomal repeat array interactions between chromo-
somes 4 and 10: a model for subtelomeric plasticity. Hum.
Mol. Genet. 9, 2879–2884.
12. Matsumura, T., Goto, K., Yamanaka, G., Lee, J.H., Zhang, C.,
Hayashi, Y.K., and Arahata, K. (2002). Chromosome 4q;10q
translocations; comparison with different ethnic populations
and FSHD patients. BMC Neurol. 2, 7.
13. Lunt, P.W., Jardine, P.E., Koch, M.C., Maynard, J., Osborn, M.,
Williams,M., Harper, P.S., and Upadhyaya, M. (1995). Correla-
tion between fragment size at D4F104S1 and age at onset or at
wheelchair use, with a possible generational effect, accounts
for much phenotypic variation in 4q35-facioscapulohumeral
muscular dystrophy (FSHD). Hum. Mol. Genet. 4, 951–958.
14. Ricci, E., Galluzzi, G., Deidda, G., Cacurri, S., Colantoni, L.,
Merico, B., Piazzo, N., Servidei, S., Vigneti, E., Pasceri, V.,
et al. (1999). Progress in themolecular diagnosis of facioscapu-
lohumeral muscular dystrophy and correlation between the
number of KpnI repeats at the 4q35 locus and clinical pheno-
type. Ann. Neurol. 45, 751–757.
15. Tonini, M.M., Passos-Bueno, M.R., Cerqueira, A., Matioli, S.R.,
Pavanello, R., and Zatz, M. (2004). Asymptomatic carriers and
gender differences in facioscapulohumeral muscular dys-
trophy (FSHD). Neuromuscul. Disord. 14, 33–38.
16. Lemmers, R.J., Wohlgemuth, M., Frants, R.R., Padberg, G.W.,
Morava, E., and van der Maarel, S.M. (2004). Contractions of
D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral
muscular dystrophy. Am. J. Hum. Genet. 75, 1124–1130.
17. Lemmers, R.J., Wohlgemuth, M., van der Gaag, K.J., van der
Vliet, P.J., van Teijlingen, C.M., de Knijff, P., Padberg, G.W.,
Frants, R.R., and van der Maarel, S.M. (2007). Specific
sequence variations within the 4q35 region are associated
with facioscapulohumeral muscular dystrophy. Am. J. Hum.
Genet. 81, 884–894.
18. Lemmers, R.J., van der Vliet, P.J., Klooster, R., Sacconi, S., Ca-
mano, P., Dauwerse, J.G., Snider, L., Straasheijm, K.R., van
Ommen, G.J., Padberg, G.W., et al. (2010). A unifying genetic
model for facioscapulohumeral muscular dystrophy. Science
329, 1650–1653.
19. de Greef, J.C., Lemmers, R.J., Camano, P., Day, J.W., Sacconi,
S., Dunand, M., van Engelen, B.G., Kiuru-Enari, S., Padberg,
G.W., Rosa, A.L., et al. (2010). Clinical features of facioscapu-
lohumeral muscular dystrophy 2. Neurology 75, 1548–1554.
20. Scionti, I., Fabbri, G., Fiorillo, C., Ricci, G., Greco, F., D’Amico,
R., Termanini, A., Vercelli, L., Tomelleri, G., Cao, M., et al.
(2012). Facioscapulohumeral muscular dystrophy: new
insights from compound heterozygotes and implication for
prenatal genetic counselling. J. Med. Genet. 49, 171–178.
21. Padberg, G.W., Lunt, P.W., Koch, M., and Fardeau, M. (1991).
Diagnostic criteria for facioscapulohumeral muscular
dystrophy. Neuromuscul. Disord. 1, 231–234.
22. Butz, M., Koch, M.C., Muller-Felber, W., Lemmers, R.J., van
der Maarel, S.M., and Schreiber, H. (2003). Facioscapulohum-
eral muscular dystrophy. Phenotype-genotype correlation in
patients with borderline D4Z4 repeat numbers. J. Neurol.
250, 932–937.
23. Lamperti, C., Fabbri, G., Vercelli, L., D’Amico, R., Frusciante,
R., Bonifazi, E., Fiorillo, C., Borsato, C., Cao, M., Servida, M.,
et al. (2010). A standardized clinical evaluation of patients
affected by facioscapulohumeral muscular dystrophy: The
FSHD clinical score. Muscle Nerve 42, 213–217.
24. Wohlgemuth, M., Lemmers, R.J., van der Kooi, E.L., van der
Wielen, M.J., van Overveld, P.G., Dauwerse, H., Bakker, E.,
Frants, R.R., Padberg, G.W., and van der Maarel, S.M. (2003).
Possible phenotypic dosage effect in patients compound het-
erozygous for FSHD-sized 4q35 alleles. Neurology 61, 909–913.
25. Casasnovas, C., Cano, L.M., Albertı, A., Cespedes, M., and
Rigo, G. (2008). Charcot-Marie-tooth disease. Foot Ankle
Spec 1 (6, Spec.), 350–354.
26. Takahashi, M., Asai, N., Iwashita, T., Murakami, H., and Ito, S.
(1998). Mechanisms of development of multiple endocrine
neoplasia type 2 and Hirschsprung’s disease by ret mutations.
Recent Results Cancer Res. 154, 229–236.
27. Kanagawa, M., and Toda, T. (2006). The genetic andmolecular
basis of muscular dystrophy: roles of cell-matrix linkage in the
pathogenesis. J. Hum. Genet. 51, 915–926.
28. Chahwan, R., Wontakal, S.N., and Roa, S. (2011). The multidi-
mensional nature of epigenetic information and its role in
disease. Discov. Med. 11, 233–243.
29. Vitelli, F., Villanova, M., Malandrini, A., Bruttini, M., Piccini,
M., Merlini, L., Guazzi, G., and Renieri, A. (1999). Inheritance
of a 38-kb fragment in apparently sporadic facioscapulohum-
eral muscular dystrophy. Muscle Nerve 22, 1437–1441.
30. Weiffenbach, B., Bagley, R., Falls, K., Hyser, C., Storvick, D., Ja-
cobsen, S.J., Schultz, P., Mendell, J., Willems van Dijk, K., Mil-
ner, E.C., et al. (1992). Linkage analyses of five chromosome 4
markers localizes the facioscapulohumeral muscular
dystrophy (FSHD) gene to distal 4q35. Am. J. Hum. Genet.
51, 416–423.
31. Lecky, B.R., MacKenzie, J.M., Read, A.P., and Wilcox, D.E.
(1991). X-linked and FSH dystrophies in one family. Neuro-
muscul. Disord. 1, 275–278.
32. Felice, K.J., North, W.A., Moore, S.A., and Mathews, K.D.
(2000). FSH dystrophy 4q35 deletion in patients presenting
with facial-sparing scapular myopathy. Neurology 54, 1927–
1931.
33. van der Kooi, A.J., Visser, M.C., Rosenberg, N., van den Berg-
Vos, R., Wokke, J.H., Bakker, E., and de Visser, M. (2000).
Extension of the clinical range of facioscapulohumeral
dystrophy: report of six cases. J. Neurol. Neurosurg. Psychiatry
69, 114–116.
34. Krasnianski, M., Eger, K., Neudecker, S., Jakubiczka, S., and
Zierz, S. (2003). Atypical phenotypes in patients with
634 The American Journal of Human Genetics 90, 628–635, April 6, 2012

facioscapulohumeral muscular dystrophy 4q35 deletion.
Arch. Neurol. 60, 1421–1425.
35. Chuenkongkaew, W.L., Lertrit, P., Limwongse, C., Nilanont,
Y., Boonyapisit, K., Sangruchi, T., Chirapapaisan, N., and Su-
phavilai, R. (2005). An unusual family with Leber’s hereditary
optic neuropathy and facioscapulohumeral muscular
dystrophy. Eur. J. Neurol. 12, 388–391.
36. Filosto, M., Tonin, P., Scarpelli, M., Savio, C., Greco, F., Man-
cuso, M., Vattemi, G., Govoni, V., Rizzuto, N., Tupler, R., and
Tomelleri, G. (2008). Novel mitochondrial tRNA Leu(CUN)
transitionandD4Z4partialdeletion inapatientwitha faciosca-
pulohumeral phenotype. Neuromuscul. Disord. 18, 204–209.
37. Rudnik-Schoneborn, S., Weis, J., Kress, W., Hausler, M., and
Zerres, K. (2008). Becker’s muscular dystrophy aggravating
facioscapulohumeral muscular dystrophy—double trouble as
an explanation for an atypical phenotype. Neuromuscul.
Disord. 18, 881–885.
38. Korngut, L., Siu, V.M., Venance, S.L., Levin, S., Ray, P., Lem-
mers, R.J., Keith, J., and Campbell, C. (2008). Phenotype of
combined Duchenne and facioscapulohumeral muscular
dystrophy. Neuromuscul. Disord. 18, 579–582.
39. Zouvelou, V., Manta, P., Kalfakis, N., Evdokimidis, I., and Vas-
silopoulos, D. (2009). Asymptomatic elevation of serum crea-
tine kinase leading to the diagnosis of 4q35 facioscapulohum-
eral muscular dystrophy. J. Clin. Neurosci. 16, 1218–1219.
40. Tsuji, M., Kinoshita, M., Imai, Y., Kawamoto, M., and Kohara,
N. (2009). Facioscapulohumeral muscular dystrophy present-
ing with hypertrophic cardiomyopathy: a case study. Neuro-
muscul. Disord. 19, 140–142.
41. Reilich, P., Schramm,N., Schoser, B., Schneiderat, P., Strigl-Pill,
N., Muller-Hocker, J., Kress, W., Ferbert, A., Rudnik-Schone-
born, S., Noth, J., et al. (2010). Facioscapulohumeral muscular
dystrophy presenting with unusual phenotypes and atypical
morphological features of vacuolar myopathy. J. Neurol.
257, 1108–1118.
42. Jordan, B., Eger, K., Koesling, S., and Zierz, S. (2011). Campto-
cormia phenotype of FSHD: a clinical and MRI study on six
patients. J. Neurol. 258, 866–873.
43. Tonini, M.M., Passos-Bueno, M.R., Cerqueira, A., Pavanello,
R., Vainzof, M., Dubowitz, V., and Zatz, M. (2002). Facioscapu-
lohumeral (FSHD1) and other forms of muscular dystrophy in
the same family: is there more in muscular dystrophy than
meets the eye? Neuromuscul. Disord. 12, 554–557.
44. Ricci, G., Scionti, I., Alı, G., Volpi, L., Zampa, V., Fanin, M.,
Angelini, C., Politano, L., Tupler, R., and Siciliano, G. (2012).
Rippling muscle disease and facioscapulohumeral dystrophy-
like phenotype in a patient carrying a heterozygous CAV3
T78Mmutation and a D4Z4 partial deletion: Further evidence
for ‘‘double trouble’’ overlapping syndromes. Neuromuscul.
Disord., in press. Published online January 13, 2012. 10.
1016/j.nmd.2011.12.001.
45. Griggs, R.C., Tawil, R., McDermott, M., Forrester, J., Figlewicz,
D., andWeiffenbach, B.; FSH-DY Group. (1995). Monozygotic
twins with facioscapulohumeral dystrophy (FSHD): implica-
tions for genotype/phenotype correlation. Muscle Nerve 2,
S50–S55.
46. Tupler, R., Barbierato, L., Memmi, M., Sewry, C.A., De Grandis,
D., Maraschio, P., Tiepolo, L., and Ferlini, A. (1998). Identical
de novo mutation at the D4F104S1 locus in monozygotic
male twins affected by facioscapulohumeral muscular
dystrophy (FSHD) with different clinical expression. J. Med.
Genet. 35, 778–783.
The American Journal of Human Genetics 90, 628–635, April 6, 2012 635

ARTICLE
Combined Analysis of Genome-wide AssociationStudies for Crohn Disease and PsoriasisIdentifies Seven Shared Susceptibility Loci
David Ellinghaus,1 Eva Ellinghaus,1 Rajan P. Nair,2 Philip E. Stuart,2 Tonu Esko,3,4 Andres Metspalu,3,4
Sophie Debrus,5 John V. Raelson,6 Trilokraj Tejasvi,2 Majid Belouchi,7 Sarah L. West,8 Jonathan N. Barker,8
Sulev Koks,9 Kulli Kingo,10 Tobias Balschun,1 Orazio Palmieri,11 Vito Annese,11,12 Christian Gieger,13
H. Erich Wichmann,14,15,16 Michael Kabesch,17 Richard C. Trembath,8 Christopher G. Mathew,8
Goncalo R. Abecasis,18 Stephan Weidinger,19 Susanna Nikolaus,20,21 Stefan Schreiber,1,21 James T. Elder,2,22
Michael Weichenthal,19Michael Nothnagel,23,24 and Andre Franke1,24,*
Psoriasis (PS) and Crohn disease (CD) have been shown to be epidemiologically, pathologically, and therapeutically connected, but little
is known about their shared genetic causes. We performedmeta-analyses of five published genome-wide association studies on PS (2,529
cases and 4,955 controls) and CD (2,142 cases and 5,505 controls), followed up 20 loci that showed strongest evidence for shared disease
association and, furthermore, tested cross-disease associations for previously reported PS and CD risk alleles in additional 6,115 PS cases,
4,073 CD cases, and 10,100 controls. We identified seven susceptibility loci outside the human leukocyte antigen region (9p24 near
JAK2, 10q22 at ZMIZ1, 11q13 near PRDX5, 16p13 near SOCS1, 17q21 at STAT3, 19p13 near FUT2, and 22q11 at YDJC) shared between
PS and CD with genome-wide significance (p< 53 10�8) and confirmed four already established PS and CD risk loci (IL23R, IL12B, REL,
and TYK2). Three of the shared loci are also genome-wide significantly associated with PS alone (10q22 at ZMIZ1, prs1250544 ¼3.53 3 10�8, 11q13 near PRDX5, prs694739 ¼ 3.71 3 10�09, 22q11 at YDJC, prs181359 ¼ 8.02 3 10�10). In addition, we identified one
susceptibility locus for CD (16p13 near SOCS1, prs4780355 ¼ 4.99 3 10�8). Refinement of association signals identified shared genome-
wide significant associations for exonic SNPs at 10q22 (ZMIZ1) and in silico expression quantitative trait locus analyses revealed that
the associations at ZMIZ1 and near SOCS1 have a potential functional effect on gene expression. Our results show the usefulness of joint
analyses of clinically distinct immune-mediated diseases and enlarge the map of shared genetic risk loci.
Introduction
Psoriasis (PS [MIM 177900]) and Crohn disease (CD [MIM
266600]) are both chronic inflammatory epithelial disorders
that are triggered by an activated cellular immune system
and have an estimated sibling relative risk (ls) of 4–111,2
and 25–42,3 respectively, and a prevalence of 2%–3% and
about 0.1%, respectively, in populations of European
ancestry.4,5 PS is a common hyperproliferative disorder of
the skin, characterized by red scaly plaques, typically occur-
ringon the elbows, knees, scalp, and lowerback.6 Incontrast,
CD is primarily a gut disorder affecting any aspect of the
gastrointestinal tract butwith extraintestinalmanifestations
which might also affect the skin (e.g., erythema nodosum
and pyoderma gangraenosum).7 It results from the interac-
tion of environmental factors, including the commensal
microflora, with host immune mechanisms in a genetically
susceptible host.8 Although PS and CD are clinically
distinct diseases, they are observed togethermore frequently
than expected by chance, which could indicate shared
genetic factors acting in the etiology of both diseases.9–11
Recently, several genome-wide association studies (GWASs)
have successfully been carried out separately for CD
and PS12–20 and identified shared susceptibility genes,
such as IL23R (MIM 607562), IL12B (MIM 161561),
REL (MIM 164910), and TYK2 (MIM 176941), thereby
providing further evidence for a genetic overlap of
both diseases.13,16,21–23 One of the best characterized
risk loci for bothCDandPS is IL23R, located in a drug-target-
able pathway.24 IL-23, a pro-inflammatory cytokine, is
1Institute of Clinical Molecular Biology, Christian-Albrechts-University, 24105 Kiel, Germany; 2Department of Dermatology, University of Michigan, Ann
Arbor, MI 48109, USA; 3Estonian Genome Center, University of Tartu, 50409 Tartu, Estonia; 4Institute of Molecular and Cell Biology, University of Tartu,
50409 Tartu, Estonia; 5Gatineau, QC J9J 2X6, Canada; 6PGX-Services, Montreal, QC H2T 1S1, Canada; 7Genizon BioSciences, Inc., St. Laurent, QC H4T
2C7, Canada; 8Division of Genetics and Molecular Medicine, King’s College London, London SE1 9RT, UK; 9Department of Physiology, Centre of Trans-
lational Medicine and Centre of Translational Genomics, University of Tartu, 50409 Tartu, Estonia; 10Department of Dermatology and Venerology, Univer-
sity of Tartu, 50409 Tartu, Estonia; 11Division of Gastroenterology, Istituto di Ricovero e Cura a Carattere Scientifico-Casa Sollievo della Sofferenza Hospital,
San Giovanni Rotondo 71013, Italy; 12Unit of Gastroenterology SOD2, Azienda Ospedaliero Universitaria Careggi, Florence 50134, Italy; 13Institute of
Genetic Epidemiology, Helmholtz Centre Munich, German Research Center for Environmental Health, 85764 Neuherberg, Germany; 14Institute of Epide-
miology I, Helmholtz Centre Munich, German Research Center for Environmental Health, 85764 Neuherberg, Germany; 15Institute of Medical Infor-
matics, Biometry and Epidemiology, Ludwig-Maximilians-University, 81377 Munich, Germany; 16Klinikum Grosshadern, 81377 Munich, Germany;17Department of Paediatric Pneumology, Allergy and Neonatology, Hannover Medical School, 30625 Hannover, Germany; 18Department of Biostatistics,
Center for Statistical Genetics, University of Michigan, Ann Arbor, MI 48109, USA; 19Department of Dermatology, Allergology, and Venerology, University
Hospital Schleswig-Holstein, Christian-Albrechts-University, 24105 Kiel, Germany; 20PopGen Biobank, Christian-Albrechts-University Kiel, 24105 Kiel,
Germany; 21Department of General Internal Medicine, University Hospital Schleswig-Holstein, 24105 Kiel, Germany; 22Ann Arbor Veterans Affairs Hopital,
Ann Arbor, MI 48105, USA; 23Institute of Medical Informatics and Statistics, Christian-Albrechts University, 24105 Kiel, Germany24These authors contributed equally to this work
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.020. �2012 by The American Society of Human Genetics. All rights reserved.
636 The American Journal of Human Genetics 90, 636–647, April 6, 2012

thought to be a key player driving autoimmunity in
human disease.25 A recent functional characterization of
the amino acid substitution R381Q in IL23R suggests that
IL-23-induced Th17 cell effector function is reduced in
protective allele carriers and leads to protection against
several autoimmune diseases, including PS, CD, and
ankylosing spondylitis.26
So far, shared susceptibility loci for CD and PS have been
identified by single-disease GWAS for CD or PS separately,
and established risk SNPs for one disease are usually tested
for association in another disease,27–31 rather than in a
combined systematic approach. Combined GWASs were
only conducted across clinically related phenotypes, such
as CD and ulcerative colitis (UC),32 CD and sarcoidosis
(SA)27 or CD and celiac disease (CelD).33 Recently, Zherna-
kova et al.34 performed a meta-analysis with a similar
systematic approach that combined genome-wide geno-
type data from two autoimmune diseases affecting dif-
ferent organs, namely CelD and rheumatoid arthritis
(RA). They identified eight shared risk loci outside the
human leukocyte antigen (HLA) region for CelD and RA,
four of them previously not known to be associated with
either CelD or RA. Based on genome-wide SNP data for
CelD and RA, the authors identified an increased proba-
bility for CelD risk SNPs to confer also an increase in risk
for RA and vice versa. Zhernakova et al.34 postulated
criteria for declaring a SNP as being a shared risk factor
for two clinically distinct diseases, namely that shared
SNPs (1) have to reach genome-wide significance in the
combined analysis of the initial GWAS screening stage
and the replication stage of the two distinct diseases
(pGWASþRepl < 5 3 10�8) and (2) have to achieve, for each
disease separately, nominal significance in the replication
stage (pRepl < 0.05) as well as pGWASþRepl < 10�3 in the
combined analysis of screening and replication stage.
We used the criteria proposed by Zhernakova et al.34 in
a genome-wide association analysis combining CD and
PS to systematically identify shared risk loci associated
with both diseases. A two-fold strategy was employed: in
a first approach (OVERLAP), we tested established non-
HLA CD risk SNPs for association with PS and vice versa,
thereby seeking confirmation of whether known risk loci
for one disease also play a role in the etiology of the other
disease, disregarding the direction of effect. In a second
approach (COMBINED), we performed a meta-analysis for
the combined phenotype based on genome-wide data
sets of both CD and PS in order to increase power for the
detection of new shared risk alleles because of an increased
sample size. The latter approach allows consideration of
same-direction as well as opposing-direction allelic effects
of putative shared markers between CD and PS through
the use of suitable allele coding. We followed up 20 loci
that showed the strongest association in the COMBINED
approach and that had not been previously reported
as being risk factors for either CD or PS. Follow-up
was performed in independent replication panels from
Germany, Estonia, Italy, United Kingdom, and the United
States (see Table S1, available online).
Subjects and Methods
Study SubjectsWe analyzed a collection of different data sets. Figure 1 details the
different panels and their use in this study. For discovery, we
combined genome-wide case-control data of single-nucleotide
polymorphisms (SNP) for psoriasis (PS; panel A) and Crohn disease
(CD; panel B) (see Table S1) respectively, conducted genome-wide
meta-analyses on PS and CD, respectively, and employed two
strategies (COMBINED and OVERLAP, see below) to systematically
identify risk loci associated with both diseases. Replication was
Figure 1. Study Design for the CombinedAnalysis of CD and PSFor discovery, we conducted a PS and a CDGWASmeta-analysis (panel A and B in Table S1), respec-tively, and employed two strategies (OVERLAPand COMBINED) to systematically search forshared risk loci. In a first approach (OVERLAP),we tested established non-HLA PS risk SNPs forpotential association (p < 0.01) with CD andvice versa. In a second approach (COMBINED),we selected SNPs from 20 loci for being nomi-nally associated in each of the single-diseasemeta-analyses (ppanel A < 0.05, ppanel B < 0.05)and for being significantly associated in thecombined-phenotype association analysis at the10�4 level (ppanel A&B < 10�4). For replication,follow-up SNP genotyping was performed forCD and PS in independent replication panels(panels C–E in Table S1). The following abbrevia-tions are used: PS-GER, German PS GWAS; PS-US,United States PS GWAS; PS-Canada, CanadianPS GWAS; CD-GER, German CD GWAS; andCD-UK, United Kingdom CD GWAS. For eachpanel, numbers of cases/controls are displayedin parentheses.
The American Journal of Human Genetics 90, 636–647, April 6, 2012 637

performed in independent replication panels from Germany (see
panels C–E), Estonia (panels C and D), Italy (panel E), United
Kingdom (panels C and E), and the United States (panel C) (see
Table S1). Written, informed consent was obtained from all study
participants and all protocols were approved by the institutional
ethical review committees of the participating centers.
Initial GWAS and German Replication Data
All German CD patients in discovery and replication panels (B and
C–E, respectively) were recruited either at the Department of
General Internal Medicine, Christian-Albrechts-University, Kiel,
and the Charite University Hospital, Berlin; through local outpa-
tient services; or nationwide with the support of the German
Crohn and Colitis Foundation. German PS cases in discovery
and replication panels (A and C andD, respectively) were recruited
either at the Department of Dermatology, Christian-Albrechts-
University, Kiel, or the Department of Dermatology and Allergy,
Technical University, Munich, or through local outpatient
services. Individuals were considered to be affected by PS if chronic
plaque or guttate psoriasis lesions covered more than 1% of the
total body surface area or if at least two skin, scalp, nail, or joint
lesions were clinically diagnosed as psoriasis. The 4,680 German
healthy control individuals in discovery and replication panels
(A–E) were obtained from the Popgen biobank.35 The additional
3,391 German healthy controls (after quality control measures)
in the discovery panels (A and B) were selected from the KORA
S3þS4 survey, an independent population-based sample from
the general population living in the region of Augsburg, southern
Germany.36 Another 674 German healthy controls in the
discovery panels (A and B) were selected from ISAAC Phase II
study.37 German GWAS controls of the discovery phase were
randomly assigned to panels A and B at equal proportions, while
ensuring that controls in panel A did not overlap with German
GWAS controls used in the independent genome-wide meta-anal-
ysis on CD.13 The Collaborative Association Study of Psoriasis
(CASP) samples14 consisted of 1,303 PS cases and 1,322 controls
after quality control measures and are part of panel A in our study.
The data sets used for the analyses described in this manuscript
were obtained from the database of Genotype and Phenotype
(dbGaP). The genotyping of samples was provided through the
Genetic Association Information Network (GAIN).38 The Cana-
dian samples (from Genizon BioSciences andM. Belouchi, unpub-
lished data) consisted of 757 PS cases and 987 controls sampled
from the Quebec founder population (QFP) after quality control
measures. They are part of panel A. Membership in the QFP was
defined as having four grandparents with French-Canadian family
names who were born in the Province of Quebec, Canada, or in
adjacent areas in the provinces of New Brunswick and Ontario
or in New England or New York state. This criterion assured that
all subjects were descendants of French-Canadians living before
the 1960s, after which time admixture with non-French-Cana-
dians became more common. CD cases and controls from the
United Kingdom were recruited from the 1958 birth cohort and
UK National Blood Service for the Welcome Trust Case Controls
Consortium (WTCCC) (described in details in WTCCC28). The
WTCCC1 CD samples consisted of 1,662 CD cases and 2,860
healthy controls after quality control procedures and entered the
analysis as part of panel B.
Additional Replication Data
Anumber of collaborative data sets were used as replication panels.
The Estonian samples used in the OVERLAP and COMBINED
approaches (part of panels C and D) were collected at the Depart-
ment of Dermatology and Venerology and at the Department of
Physiology and Centre of Translational Medicine at the University
of Tartu.
Additional Estonian samples (part of panel C) used for replica-
tion in the OVERLAP approach consisted of samples provided by
the population-based biobank of the Estonian Genome Center,
University of Tartu. Subjects were recruited by general practi-
tioners (GP) and physicians in the hospitals. Participants in the
hospitals were randomly selected from individuals visiting GP
offices or hospitals. Diagnosis of PS on the basis of clinical symp-
toms was posed by a general practitioner and confirmed by
a dermatologist. At the moment of recruitment, the controls did
not report diagnosis of osteoarthritis, psoriasis, or autoimmune
diseases. The United States samples (part of panel C) used for repli-
cation in the OVERLAP approach consisted of 2,137 PS cases and
1,903 controls of white European ancestry from the United States.
The Italian samples (panel E) used in the COMBINED approach
consisted of 688 CD cases and 879 healthy controls that were
used in the independent genome-wide meta-analysis on CD.13
The psoriasis data set from the United Kingdom consisted of
2,178 PS cases collected through the Genetic Analysis of Psoriasis
Consortium (GAPC) and 2,657 controls from the WTCCC2
common control set, used as the GWAS discovery set described
in Strange et al. in 2010.16 Only controls that did not overlap
with WTCCC1 controls were used. UK cases and controls entered
the analysis as part of panel C and E.
Quality Control and Genome-wide Genotype
ImputationQuality control (QC) was performed for each sample set separately.
In each sample set samples with more than 5% missing data
were excluded before genotype imputation. We also excluded
individuals from each pair of unexpected duplicates or relatives,
as well as outlier individuals with average marker heterozygosities
of55 standard deviation away from the samplemean. The remain-
ing samples were tested for population stratification with the
principal components stratification method as implemented in
EIGENSTRAT,39 and population outliers were subsequently
excluded. SNPs that hadmore than 5%missing data, aminor allele
frequency less than 1% or deviated from Hardy-Weinberg equilib-
rium (exact p < 10�4 in controls) per sample set were excluded
with thePLINKsoftwareversion1.07.40 SNP imputationwascarried
out with the BEAGLE v.3.1.141 software package and 690HapMap3
referencehaplotypes fromtheCEU,TSI,MEX, andGIHcohorts42 to
predict missing autosomal genotypes in silico. We subsequently
analyzed only those SNPs that could be imputed with moderate
confidence (INFO score r2 > 0.3) and had a minor allele frequency
more than 1% in cases or in controls. To take imputation uncer-
tainty into account, phenotypic association was tested for allele
dosage data separately for each of the five GWAS data sets in panels
AandB through theuseof PLINK’s logistic regression framework for
dosage data. To control potentially confounding effects due to pop-
ulation stratification, we adjusted for the top ten eigenvectors from
EIGENSTRAT in the regression analysis. The genomic inflation
factor l is defined as the ratio of the medians of the sample c2 test
statistics and the 1 degree of freedom c2 distribution (0.455).43
Because the estimated genomic inflation factor l scaleswith sample
size, it is informative to report the inflation factor for an equivalent
study of 1,000 cases and 1,000 controls (l1000) by rescaling l.44
Meta-AnalysesMeta-analyses were performed with PLINK’s meta-analysis func-
tion and with its standard error of odds ratio weighting option
638 The American Journal of Human Genetics 90, 636–647, April 6, 2012

(inverse variance weighting), which implicitly deals with imputa-
tion uncertainty. For the combined-phenotype analysis, we per-
formed two sorts of meta-analysis in order to detect associations
of SNPs with either the same or opposite allelic effects in the two
diseases. For the same-effect analysis, themeta-analysis was carried
out as usual. For the opposite-effect analysis, first we flippedminor
andmajor alleles of each biallelic SNP in the CD data sets tomimic
an opposite-direction effect of the allele in CD and performed
a meta-analysis afterward. For both effects models, we considered
only those SNPs whose genotypes were available from at least
four out of the five GWAS data sets in panels A and B.
Follow-Up GenotypingGenotyping was carried out with our Sequenom iPlex plat-
form from Sequenom and TaqMan technology from Applied
Biosystems. Individuals with more than 3% missing data were
removed. SNPs that hadmore than 3%missing data, a minor allele
frequency less than 1% or deviated from Hardy-Weinberg
equilibrium (exact p < 10�4 in controls) per sample set were
excluded. p values for allele-based tests of phenotypic association
for each single-replication sample sets (panels C–E) were calcu-
lated with PLINK. PLINK’s meta-analysis function was used to
obtain p values for the replication data set (pRepl) and for the
combined discovery-replication data set (pGWASþRepl).
Regional Imputation Based on the 1000 Genomes
Project ReferenceTo enable imputation based on the 1000 Genomes project
data, SNPpositions referring toNCBIbuild36weremapped tobuild
37. SNP imputation was carried out with the BEAGLE software
package v.3.1.141 and 566 EUR (European) haplotypes generated
by the 1000 Genomes Project.45 We analyzed only imputed SNPs
with moderate imputation confidence (INFO score r2 > 0.3) and
a minor allele frequency more than1% in cases or in controls.
Gene Relationships across Implicated Loci Pathway
AnalysisThe Gene Relationships Across Implicated Loci (GRAIL) software46
quantifies functional similarity between genes by applying
established statistical text mining methods to the PubMed
database of published scientific abstracts. As input we used
the following list of SNPs: rs2201841, rs2082412, rs702873,
rs12720356, rs10758669, rs694739, rs281379, rs181359,
rs4780355, rs744166, and rs1250544. GRAIL was run with the
following settings: HapMap release ¼ HapMap release 22/hg18;
HapMap population ¼ CEU (Utah residents with ancestry from
northern and western Europe from the Centre d0Etude du Poly-
morphisme Humain collection); functional data source¼ PubMed
Text (April 2011); and gene size correction ¼ on. GRAIL output
results were visualized with VIZ-GRAIL.
Results
Preparation of Single-Disease Meta-Analyses for
Discovery Phase by Means of HapMap3 Imputation
The overall study workflow for the combined analysis of
CD and PS is displayed in Figure 1. For discovery, we con-
ducted a meta-analysis on PS comprising 2,529 PS cases
and 4,955 controls from three previously published
GWASs,14,15 all of European descent (panel A in Table
S1). SNP data were combined with genotype imputation
based on the HapMap3 reference. We subsequently used
standard meta-analysis methodology (see Subjects and
Methods). In total, 1,121,166 quality-controlled auto-
somal-imputed SNPmarkers were available for the analysis
on PS. To control for potential population stratification, we
adjusted association test statistics by means of principal
component analysis (PCA) (see Subjects and Methods). A
quantile-quantile (Q-Q) plot of the meta-analysis revealed
a marked excess of significant associations in the tail of
the distribution (Figure S1A), which is primarily due to
thousands of highly significant association signals from
the HLA region. Genetic heterogeneity was low; there
was an estimated genomic inflation factor of l1000 ¼1.02743,44 (see Subjects and Methods). Results of the
meta-analysis on PS are summarized in Figure S2A.
In the same way, we performed ameta-analysis on CD by
using 1,034,639 quality-controlled autosomal-imputed
markers fromaGermanGWAS13 andapreviouslypublished
UK GWAS,28 consisting of 2,142 CD cases and 5,505
controls in total (panel B in Table S1). Again, we observed
low genomic inflation (l1000 ¼ 1.032, Figure S1B). Results
of the meta-analysis on CD are summarized in Figure S2B.
OVERLAP Approach: Cross-Disease Analysis of
Established Risk SNPs
So far, four established GWAS risk loci that are located
outside the HLA region and shared between CD and PS
have been reported in the literature, namely IL23R, IL12B,
REL, andTYK2 (Table 1). Although themarkers that showed
the strongest association differed between the two diseases
at each of the first three loci, we found the same SNP
rs12720356 at TYK2 to be associated with both CD and
PS. To seek confirmation of whether known risk loci for
CD also play a role in the etiology of PS and vice versa, we
checked whether markers that were significant (p < 0.01)
in our PS meta-analysis were among the 71 established
risk SNPs previously implicated inCD13 andwhether signif-
icant markers (p < 0.01) from our CD meta-analysis were
among the25 establishedPS risk SNPs.14–20 The four already
established shared risk SNPs (Table 1) were excluded. Given
the well-established and heterogeneous allelic associations
of the HLA region on chromosome 6 with both CD and
PS, we also excluded all markers from the extended HLA
region (chr6:25-34 Mb). Although none of the known PS
SNPs were significantly associated with CD in our analysis,
five out of the 71 known CD risk SNPs met our criterion
of significance, namely rs10758669 (JAK2 [MIM
147796]), rs694739 (PRDX5 [MIM 606583]), rs281379
(FUT2 [MIM 182100]), rs744166 (STAT3 [MIM 102582]),
and rs181359 (YDJC;HGNC 27158). We genotyped these
five SNPs by using TaqMan technology in a large indepen-
dent replication panel comprising 3,937 PS cases and
4,847 controls but also used summary statistics data of
the five SNPs from an independent GWAS on PS16
comprising 2,178 PS cases and 2,657 controls. The overall
replication panel consisted of 6,115 PS cases and 7,504
The American Journal of Human Genetics 90, 636–647, April 6, 2012 639
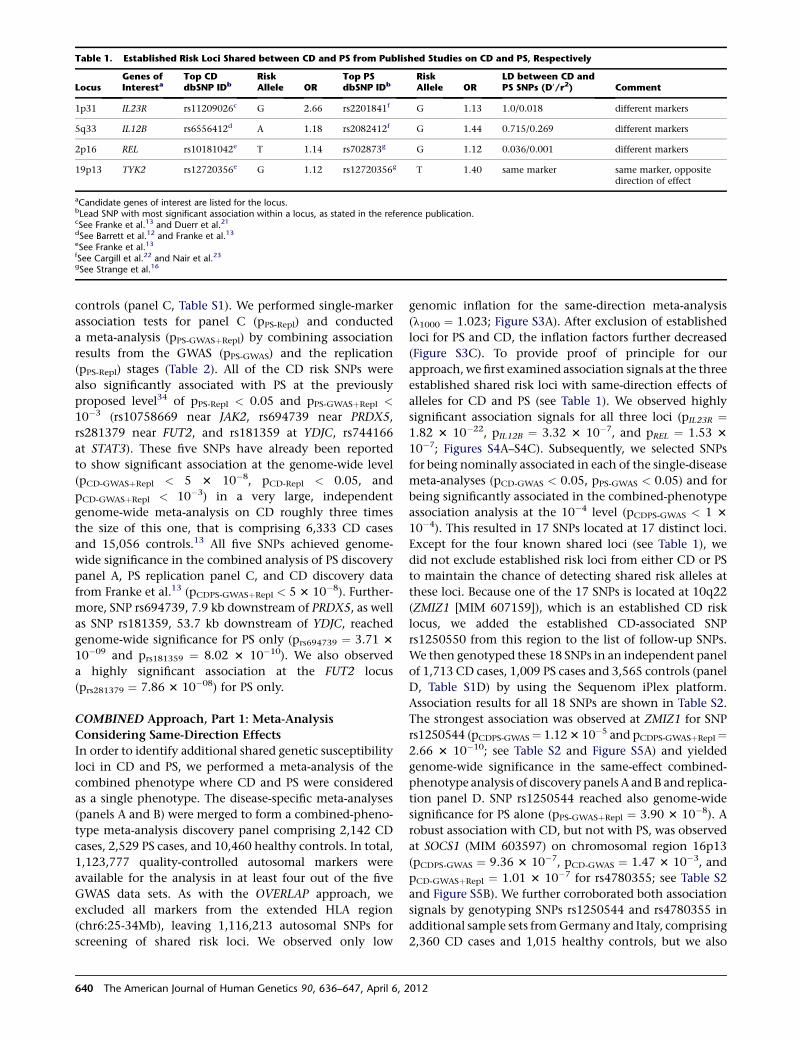
controls (panel C, Table S1). We performed single-marker
association tests for panel C (pPS-Repl) and conducted
a meta-analysis (pPS-GWASþRepl) by combining association
results from the GWAS (pPS-GWAS) and the replication
(pPS-Repl) stages (Table 2). All of the CD risk SNPs were
also significantly associated with PS at the previously
proposed level34 of pPS-Repl < 0.05 and pPS-GWASþRepl <
10�3 (rs10758669 near JAK2, rs694739 near PRDX5,
rs281379 near FUT2, and rs181359 at YDJC, rs744166
at STAT3). These five SNPs have already been reported
to show significant association at the genome-wide level
(pCD-GWASþRepl < 5 3 10�8, pCD-Repl < 0.05, and
pCD-GWASþRepl < 10�3) in a very large, independent
genome-wide meta-analysis on CD roughly three times
the size of this one, that is comprising 6,333 CD cases
and 15,056 controls.13 All five SNPs achieved genome-
wide significance in the combined analysis of PS discovery
panel A, PS replication panel C, and CD discovery data
from Franke et al.13 (pCDPS-GWASþRepl < 5 3 10�8). Further-
more, SNP rs694739, 7.9 kb downstream of PRDX5, as well
as SNP rs181359, 53.7 kb downstream of YDJC, reached
genome-wide significance for PS only (prs694739 ¼ 3.71 3
10�09 and prs181359 ¼ 8.02 3 10�10). We also observed
a highly significant association at the FUT2 locus
(prs281379 ¼ 7.86 3 10�08) for PS only.
COMBINED Approach, Part 1: Meta-Analysis
Considering Same-Direction Effects
In order to identify additional shared genetic susceptibility
loci in CD and PS, we performed a meta-analysis of the
combined phenotype where CD and PS were considered
as a single phenotype. The disease-specific meta-analyses
(panels A and B) were merged to form a combined-pheno-
type meta-analysis discovery panel comprising 2,142 CD
cases, 2,529 PS cases, and 10,460 healthy controls. In total,
1,123,777 quality-controlled autosomal markers were
available for the analysis in at least four out of the five
GWAS data sets. As with the OVERLAP approach, we
excluded all markers from the extended HLA region
(chr6:25-34Mb), leaving 1,116,213 autosomal SNPs for
screening of shared risk loci. We observed only low
genomic inflation for the same-direction meta-analysis
(l1000 ¼ 1.023; Figure S3A). After exclusion of established
loci for PS and CD, the inflation factors further decreased
(Figure S3C). To provide proof of principle for our
approach, we first examined association signals at the three
established shared risk loci with same-direction effects of
alleles for CD and PS (see Table 1). We observed highly
significant association signals for all three loci (pIL23R ¼1.82 3 10�22, pIL12B ¼ 3.32 3 10�7, and pREL ¼ 1.53 3
10�7; Figures S4A–S4C). Subsequently, we selected SNPs
for being nominally associated in each of the single-disease
meta-analyses (pCD-GWAS < 0.05, pPS-GWAS < 0.05) and for
being significantly associated in the combined-phenotype
association analysis at the 10�4 level (pCDPS-GWAS < 1 3
10�4). This resulted in 17 SNPs located at 17 distinct loci.
Except for the four known shared loci (see Table 1), we
did not exclude established risk loci from either CD or PS
to maintain the chance of detecting shared risk alleles at
these loci. Because one of the 17 SNPs is located at 10q22
(ZMIZ1 [MIM 607159]), which is an established CD risk
locus, we added the established CD-associated SNP
rs1250550 from this region to the list of follow-up SNPs.
We then genotyped these 18 SNPs in an independent panel
of 1,713 CD cases, 1,009 PS cases and 3,565 controls (panel
D, Table S1D) by using the Sequenom iPlex platform.
Association results for all 18 SNPs are shown in Table S2.
The strongest association was observed at ZMIZ1 for SNP
rs1250544 (pCDPS-GWAS¼ 1.123 10�5 and pCDPS-GWASþRepl¼2.66 3 10�10; see Table S2 and Figure S5A) and yielded
genome-wide significance in the same-effect combined-
phenotype analysis of discovery panels A andB and replica-
tion panel D. SNP rs1250544 reached also genome-wide
significance for PS alone (pPS-GWASþRepl ¼ 3.90 3 10�8). A
robust association with CD, but not with PS, was observed
at SOCS1 (MIM 603597) on chromosomal region 16p13
(pCDPS-GWAS ¼ 9.36 3 10�7, pCD-GWAS ¼ 1.47 3 10�3, and
pCD-GWASþRepl ¼ 1.01 3 10�7 for rs4780355; see Table S2
and Figure S5B). We further corroborated both association
signals by genotyping SNPs rs1250544 and rs4780355 in
additional sample sets fromGermany and Italy, comprising
2,360 CD cases and 1,015 healthy controls, but we also
Table 1. Established Risk Loci Shared between CD and PS from Published Studies on CD and PS, Respectively
LocusGenes ofInteresta
Top CDdbSNP IDb
RiskAllele OR
Top PSdbSNP IDb
RiskAllele OR
LD between CD andPS SNPs (D0/r2) Comment
1p31 IL23R rs11209026c G 2.66 rs2201841f G 1.13 1.0/0.018 different markers
5q33 IL12B rs6556412d A 1.18 rs2082412f G 1.44 0.715/0.269 different markers
2p16 REL rs10181042e T 1.14 rs702873g G 1.12 0.036/0.001 different markers
19p13 TYK2 rs12720356e G 1.12 rs12720356g T 1.40 same marker same marker, oppositedirection of effect
aCandidate genes of interest are listed for the locus.bLead SNP with most significant association within a locus, as stated in the reference publication.cSee Franke et al.13 and Duerr et al.21dSee Barrett et al.12 and Franke et al.13eSee Franke et al.13fSee Cargill et al.22 and Nair et al.23gSee Strange et al.16
640 The American Journal of Human Genetics 90, 636–647, April 6, 2012

used summary statistics data of the two SNPs from the inde-
pendent GWAS on PS16 comprising 2,178 PS cases and
2,657 controls (panel E in Table S1, the same cases and
controls from the United Kingdom, as described in panel
C). In the combined analysis of discovery panels A
and B and replication panels D and E (Tables 3 and 4),
SNP rs4780355 achieved genome-wide significance
(pCDPS-GWASþRepl ¼ 1.37 3 10�13) but also attained
genome-wide significance for CD alone (pCD-GWASþRepl ¼4.99 3 10�8).
COMBINED Approach, Part 2: Meta-Analysis
Assuming Opposite-Direction Effects
An allele might confer a risk for CD while protecting
against PS and vice versa, as is the case for TYK2. Therefore,
we also screened our combined-phenotype meta-analysis
data (panels A and B) while coding alleles in such a way
as to consider the opposite effects of them in the two
diseases (see Subjects and Methods). We observed low
genomic inflation for the opposite-direction meta-analysis
(l1000 ¼ 1.009, Figure S3B). After excluding established
shared loci for PS and CD, the inflation factors further
decreased (Figure S3D). In a first step, we checked the
known risk SNP rs12720356 (TYK2; see Table 1) for oppo-
site direction of effects. SNP rs12720356 had a p value of
4.09 3 10�5 in the combined analysis of panels A and B
(Figure S4D); there was an odds ratio (OR) of 1.29 (95%
confidence interval [CI] [1.10,1.51]) for allele A in panel A
(pPS-GWAS ¼ 1.39 3 10�3) and of 0.78 (95% CI [0.65,0.94])
in panel B (pCD-GWAS¼ 1.013 10�2).We then selected three
SNPs for subsequent genotyping (with Sequenom) and
testing in replication panel D (see Table S1D). The selection
criteria were the same as for the same-direction effect meta-
analysis. However, none of the three SNPs replicated in
both diseases at p value < 0.05 (see Table S3).
In Silico Fine-Mapping: Refinement of Association
Signals of COMBINED Approach
For refinement of the association signals at ZMIZ1 and
SOCS1, we imputed a region of about 51 Mb around the
strongest signals from the discovery panels A and B (see
Table S1) by using the EUR reference from the 1000
Genomes Project45 (see Subjects and Methods). In silico
fine-mapping of the region around SOCS1 via standard
meta-analysis methodology (see Subjects and Methods)
confirmed rs4780355 to be highly significant in this region
(pGWAS ¼ 4.04 3 10�7). Additionally, another SNP,
rs2021511, which is located in the same intron as
rs4780355 (2.9 kb downstream of rs4780355) showed
the same magnitude of association (pGWAS ¼ 1.58 3 10�7;
Figure 2) but was not selected for further replication
because of the high linkage disequilibrium (LD) between
SNPs rs4780355 and rs2021511 (r2 ¼ 0.934) according to
the 1000 Genomes Project EUR reference. Screening of
the imputed region of SOCS1 for coding SNPs revealed
one missense SNP with p < 10�4 within the TNP2 gene,
namely rs11640138. We genotyped this SNP in replicationTable
2.
AssociationResu
ltsofOVERLA
PAppro
achfrom
Cro
ss-D
isease
Compariso
nofEstablish
edRiskMark
ers
Chr
SNP
A1
Locus
CD
GW
ASMeta
-Analysisa
(6,333/15,056)
PSGW
AS
(2,529/4,955)
PSReplication
(6,115/7,504)
PSGW
ASand
Repl(8
,644/12,459)
CD
GW
ASMeta
-Analysisa
þPSGW
AS
andRepl(1
4,977/27,515)
Sta
tusNow
bp
OR
pOR
pOR
pOR
pOR
9rs10758669
CJAK2
1.0
310�13
1.18
2.433
10�03
1.13
2.473
10�03
1.08
2.693
10�05
1.10
1.303
10�16
1.14
CD-PS,
CD
11
rs694739
GPRDX5
3.4
310�07
0.89
1.133
10�04
0.86
6.123
10�06
0.89
3.713
10�09
0.88
2.413
10�14
0.89
PS,
CD-PS,
CD
19
rs281379
AFUT2
8.6
310�10
1.13
3.223
10�03
1.13
7.123
10�06
1.13
7.863
10�08
1.12
1.323
10�17
1.13
CD-PS,
CD
22
rs181359
GYDJC
6.3
310�13
0.83
4.833
10�03
0.88
3.543
10�08
0.84
8.023
10�10
0.85
1.333
10�21
0.84
PS,
CD-PS,
CD
17
rs744166c
AST
AT3
1.1
310�07
1.13
2.443
10�04
0.87
1.493
10�02
0.94
5.303
10�05
0.92
5.483
10�11
0.90
CD-PS,
CD
Thefollo
wingabbreviationsare
used:Chr,ch
romosomeofmarker;SNP,rsID;A1,minorallele;Lo
cus,onecandidate
genein
theregion;p/O
R,pvalueandco
rrespondingoddsratiowithrespect
tominoralleleforthelarge
GWASmeta-analysisofCD,13GWASmeta-analysisofPS(panelA),PSreplicationanalysis(panelC),co
mbinedanalysisofPSGWASmeta-analysis(panelA)andPSreplication(panelC
),andco
mbinedanalysisofCDGWAS
meta-analysis1
3andpanelsAandC.Fo
reach
panel,numbers
ofcases/co
ntrolsare
displayedin
parentheses.
aSeeFranke
etal.13
bStatusnow:new
statusofassociationwithCDand/orPS.AllSNPsare
establishedCDrisk
SNPswithp<
53
10�813thatwere
significant(p
<0.01)in
ourPSmeta-analysis.AllSNPs,exceptforrs744166,showedthesame
directionofeffect
forCD
andPS.NoneoftheSNPsshowedanexact
Hardy-W
einberg
pvalue<
0.01in
thePSreplication(panelC).
cMinorandmajorallelesofrs744166were
flippedin
theCD
GWASmeta-analysisin
orderto
calculate
theco
mbined-phenotypepvalueandoddsratio.
The American Journal of Human Genetics 90, 636–647, April 6, 2012 641
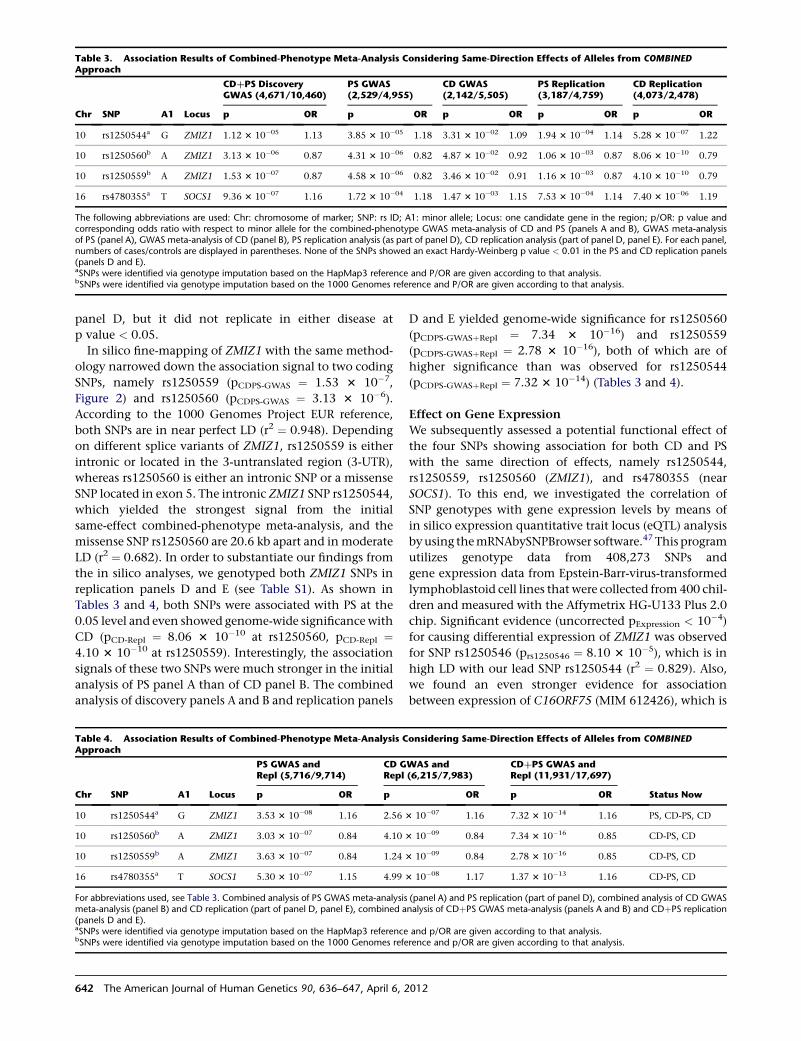
panel D, but it did not replicate in either disease at
p value < 0.05.
In silico fine-mapping of ZMIZ1 with the same method-
ology narrowed down the association signal to two coding
SNPs, namely rs1250559 (pCDPS-GWAS ¼ 1.53 3 10�7,
Figure 2) and rs1250560 (pCDPS-GWAS ¼ 3.13 3 10�6).
According to the 1000 Genomes Project EUR reference,
both SNPs are in near perfect LD (r2 ¼ 0.948). Depending
on different splice variants of ZMIZ1, rs1250559 is either
intronic or located in the 3-untranslated region (3-UTR),
whereas rs1250560 is either an intronic SNP or a missense
SNP located in exon 5. The intronic ZMIZ1 SNP rs1250544,
which yielded the strongest signal from the initial
same-effect combined-phenotype meta-analysis, and the
missense SNP rs1250560 are 20.6 kb apart and in moderate
LD (r2 ¼ 0.682). In order to substantiate our findings from
the in silico analyses, we genotyped both ZMIZ1 SNPs in
replication panels D and E (see Table S1). As shown in
Tables 3 and 4, both SNPs were associated with PS at the
0.05 level and even showed genome-wide significance with
CD (pCD-Repl ¼ 8.06 3 10�10 at rs1250560, pCD-Repl ¼4.10 3 10�10 at rs1250559). Interestingly, the association
signals of these two SNPs were much stronger in the initial
analysis of PS panel A than of CD panel B. The combined
analysis of discovery panels A and B and replication panels
D and E yielded genome-wide significance for rs1250560
(pCDPS-GWASþRepl ¼ 7.34 3 10�16) and rs1250559
(pCDPS-GWASþRepl ¼ 2.78 3 10�16), both of which are of
higher significance than was observed for rs1250544
(pCDPS-GWASþRepl ¼ 7.32 3 10�14) (Tables 3 and 4).
Effect on Gene Expression
We subsequently assessed a potential functional effect of
the four SNPs showing association for both CD and PS
with the same direction of effects, namely rs1250544,
rs1250559, rs1250560 (ZMIZ1), and rs4780355 (near
SOCS1). To this end, we investigated the correlation of
SNP genotypes with gene expression levels by means of
in silico expression quantitative trait locus (eQTL) analysis
byusing themRNAbySNPBrowser software.47 This program
utilizes genotype data from 408,273 SNPs and
gene expression data from Epstein-Barr-virus-transformed
lymphoblastoid cell lines that were collected from 400 chil-
dren and measured with the Affymetrix HG-U133 Plus 2.0
chip. Significant evidence (uncorrected pExpression < 10�4)
for causing differential expression of ZMIZ1 was observed
for SNP rs1250546 (prs1250546 ¼ 8.10 3 10�5), which is in
high LD with our lead SNP rs1250544 (r2 ¼ 0.829). Also,
we found an even stronger evidence for association
between expression of C16ORF75 (MIM 612426), which is
Table 4. Association Results of Combined-Phenotype Meta-Analysis Considering Same-Direction Effects of Alleles from COMBINEDApproach
Chr SNP A1 Locus
PS GWAS andRepl (5,716/9,714)
CD GWAS andRepl (6,215/7,983)
CDþPS GWAS andRepl (11,931/17,697)
Status Nowp OR p OR p OR
10 rs1250544a G ZMIZ1 3.53 3 10�08 1.16 2.56 3 10�07 1.16 7.32 3 10�14 1.16 PS, CD-PS, CD
10 rs1250560b A ZMIZ1 3.03 3 10�07 0.84 4.10 3 10�09 0.84 7.34 3 10�16 0.85 CD-PS, CD
10 rs1250559b A ZMIZ1 3.63 3 10�07 0.84 1.24 3 10�09 0.84 2.78 3 10�16 0.85 CD-PS, CD
16 rs4780355a T SOCS1 5.30 3 10�07 1.15 4.99 3 10�08 1.17 1.37 3 10�13 1.16 CD-PS, CD
For abbreviations used, see Table 3. Combined analysis of PS GWAS meta-analysis (panel A) and PS replication (part of panel D), combined analysis of CD GWASmeta-analysis (panel B) and CD replication (part of panel D, panel E), combined analysis of CDþPS GWAS meta-analysis (panels A and B) and CDþPS replication(panels D and E).aSNPs were identified via genotype imputation based on the HapMap3 reference and p/OR are given according to that analysis.bSNPs were identified via genotype imputation based on the 1000 Genomes reference and p/OR are given according to that analysis.
Table 3. Association Results of Combined-Phenotype Meta-Analysis Considering Same-Direction Effects of Alleles from COMBINEDApproach
Chr SNP A1 Locus
CDþPS DiscoveryGWAS (4,671/10,460)
PS GWAS(2,529/4,955)
CD GWAS(2,142/5,505)
PS Replication(3,187/4,759)
CD Replication(4,073/2,478)
p OR p OR p OR p OR p OR
10 rs1250544a G ZMIZ1 1.12 3 10�05 1.13 3.85 3 10�05 1.18 3.31 3 10�02 1.09 1.94 3 10�04 1.14 5.28 3 10�07 1.22
10 rs1250560b A ZMIZ1 3.13 3 10�06 0.87 4.31 3 10�06 0.82 4.87 3 10�02 0.92 1.06 3 10�03 0.87 8.06 3 10�10 0.79
10 rs1250559b A ZMIZ1 1.53 3 10�07 0.87 4.58 3 10�06 0.82 3.46 3 10�02 0.91 1.16 3 10�03 0.87 4.10 3 10�10 0.79
16 rs4780355a T SOCS1 9.36 3 10�07 1.16 1.72 3 10�04 1.18 1.47 3 10�03 1.15 7.53 3 10�04 1.14 7.40 3 10�06 1.19
The following abbreviations are used: Chr: chromosome of marker; SNP: rs ID; A1: minor allele; Locus: one candidate gene in the region; p/OR: p value andcorresponding odds ratio with respect to minor allele for the combined-phenotype GWAS meta-analysis of CD and PS (panels A and B), GWAS meta-analysisof PS (panel A), GWAS meta-analysis of CD (panel B), PS replication analysis (as part of panel D), CD replication analysis (part of panel D, panel E). For each panel,numbers of cases/controls are displayed in parentheses. None of the SNPs showed an exact Hardy-Weinberg p value < 0.01 in the PS and CD replication panels(panels D and E).aSNPs were identified via genotype imputation based on the HapMap3 reference and P/OR are given according to that analysis.bSNPs were identified via genotype imputation based on the 1000 Genomes reference and P/OR are given according to that analysis.
642 The American Journal of Human Genetics 90, 636–647, April 6, 2012
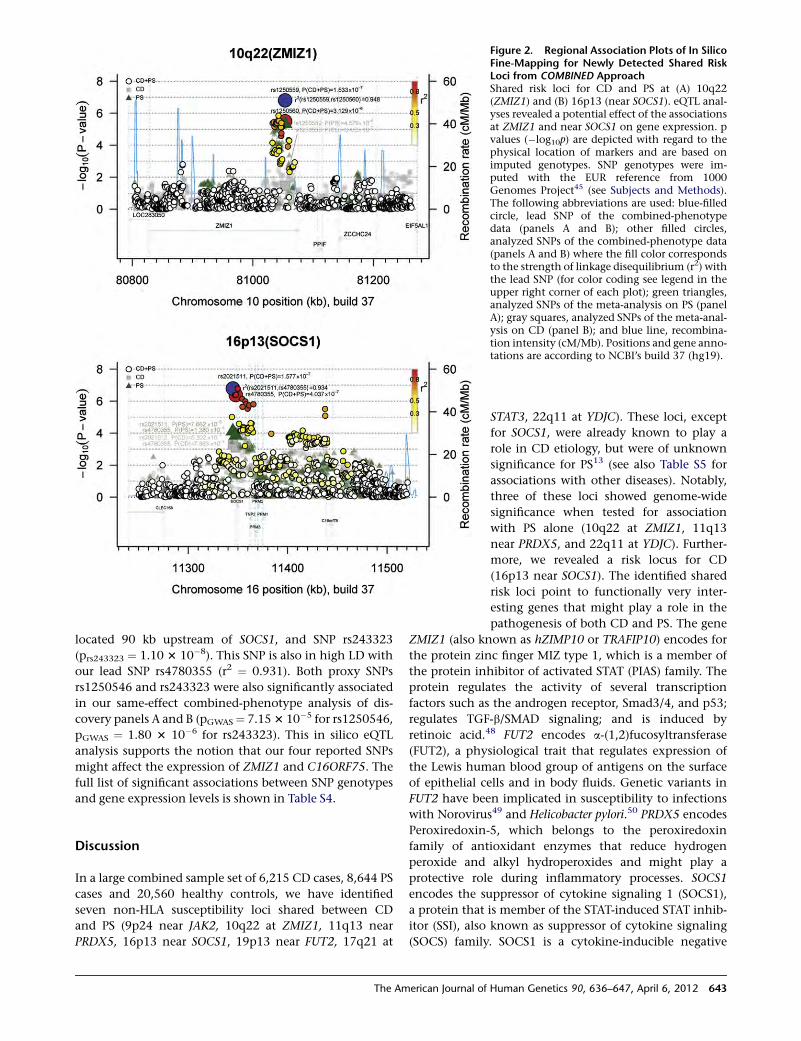
located 90 kb upstream of SOCS1, and SNP rs243323
(prs243323 ¼ 1.10 3 10�8). This SNP is also in high LD with
our lead SNP rs4780355 (r2 ¼ 0.931). Both proxy SNPs
rs1250546 and rs243323 were also significantly associated
in our same-effect combined-phenotype analysis of dis-
covery panels A and B (pGWAS¼ 7.153 10�5 for rs1250546,
pGWAS ¼ 1.80 3 10�6 for rs243323). This in silico eQTL
analysis supports the notion that our four reported SNPs
might affect the expression of ZMIZ1 and C16ORF75. The
full list of significant associations between SNP genotypes
and gene expression levels is shown in Table S4.
Discussion
In a large combined sample set of 6,215 CD cases, 8,644 PS
cases and 20,560 healthy controls, we have identified
seven non-HLA susceptibility loci shared between CD
and PS (9p24 near JAK2, 10q22 at ZMIZ1, 11q13 near
PRDX5, 16p13 near SOCS1, 19p13 near FUT2, 17q21 at
Figure 2. Regional Association Plots of In SilicoFine-Mapping for Newly Detected Shared RiskLoci from COMBINED ApproachShared risk loci for CD and PS at (A) 10q22(ZMIZ1) and (B) 16p13 (near SOCS1). eQTL anal-yses revealed a potential effect of the associationsat ZMIZ1 and near SOCS1 on gene expression. pvalues (�log10p) are depicted with regard to thephysical location of markers and are based onimputed genotypes. SNP genotypes were im-puted with the EUR reference from 1000Genomes Project45 (see Subjects and Methods).The following abbreviations are used: blue-filledcircle, lead SNP of the combined-phenotypedata (panels A and B); other filled circles,analyzed SNPs of the combined-phenotype data(panels A and B) where the fill color correspondsto the strength of linkage disequilibrium (r2) withthe lead SNP (for color coding see legend in theupper right corner of each plot); green triangles,analyzed SNPs of the meta-analysis on PS (panelA); gray squares, analyzed SNPs of the meta-anal-ysis on CD (panel B); and blue line, recombina-tion intensity (cM/Mb). Positions and gene anno-tations are according to NCBI’s build 37 (hg19).
STAT3, 22q11 at YDJC). These loci, except
for SOCS1, were already known to play a
role in CD etiology, but were of unknown
significance for PS13 (see also Table S5 for
associations with other diseases). Notably,
three of these loci showed genome-wide
significance when tested for association
with PS alone (10q22 at ZMIZ1, 11q13
near PRDX5, and 22q11 at YDJC). Further-
more, we revealed a risk locus for CD
(16p13 near SOCS1). The identified shared
risk loci point to functionally very inter-
esting genes that might play a role in the
pathogenesis of both CD and PS. The gene
ZMIZ1 (also known as hZIMP10 or TRAFIP10) encodes for
the protein zinc finger MIZ type 1, which is a member of
the protein inhibitor of activated STAT (PIAS) family. The
protein regulates the activity of several transcription
factors such as the androgen receptor, Smad3/4, and p53;
regulates TGF-b/SMAD signaling; and is induced by
retinoic acid.48 FUT2 encodes a-(1,2)fucosyltransferase
(FUT2), a physiological trait that regulates expression of
the Lewis human blood group of antigens on the surface
of epithelial cells and in body fluids. Genetic variants in
FUT2 have been implicated in susceptibility to infections
with Norovirus49 and Helicobacter pylori.50 PRDX5 encodes
Peroxiredoxin-5, which belongs to the peroxiredoxin
family of antioxidant enzymes that reduce hydrogen
peroxide and alkyl hydroperoxides and might play a
protective role during inflammatory processes. SOCS1
encodes the suppressor of cytokine signaling 1 (SOCS1),
a protein that is member of the STAT-induced STAT inhib-
itor (SSI), also known as suppressor of cytokine signaling
(SOCS) family. SOCS1 is a cytokine-inducible negative
The American Journal of Human Genetics 90, 636–647, April 6, 2012 643
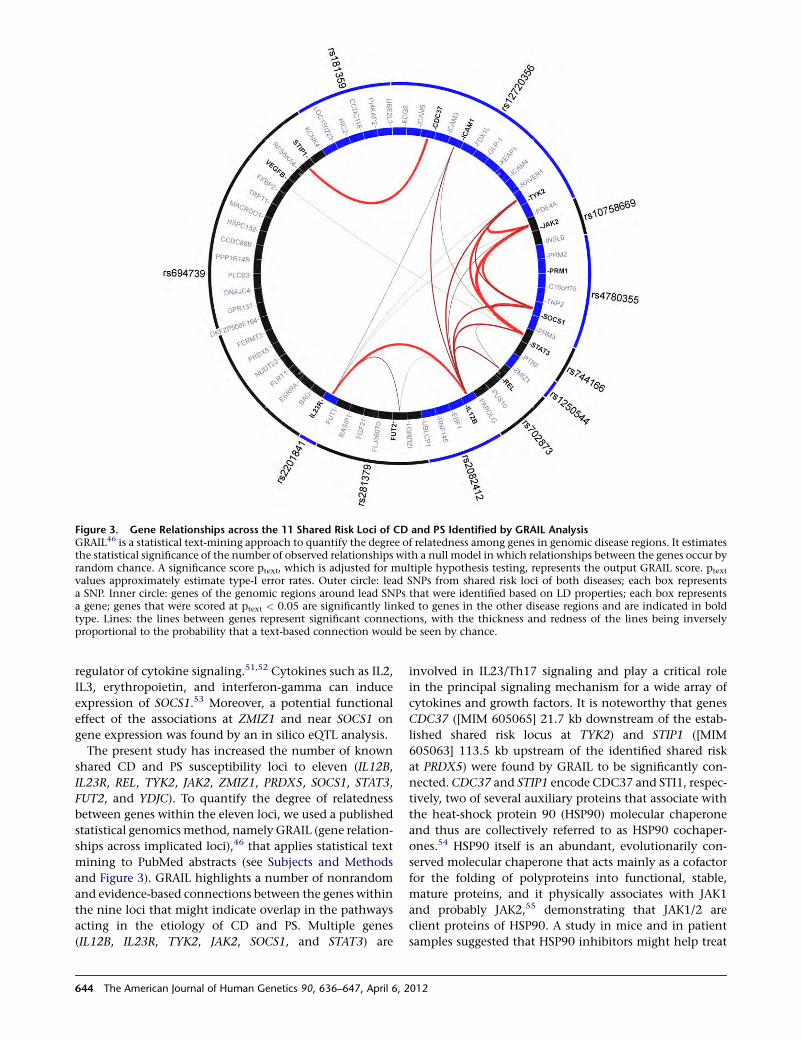
regulator of cytokine signaling.51,52 Cytokines such as IL2,
IL3, erythropoietin, and interferon-gamma can induce
expression of SOCS1.53 Moreover, a potential functional
effect of the associations at ZMIZ1 and near SOCS1 on
gene expression was found by an in silico eQTL analysis.
The present study has increased the number of known
shared CD and PS susceptibility loci to eleven (IL12B,
IL23R, REL, TYK2, JAK2, ZMIZ1, PRDX5, SOCS1, STAT3,
FUT2, and YDJC). To quantify the degree of relatedness
between genes within the eleven loci, we used a published
statistical genomicsmethod, namely GRAIL (gene relation-
ships across implicated loci),46 that applies statistical text
mining to PubMed abstracts (see Subjects and Methods
and Figure 3). GRAIL highlights a number of nonrandom
and evidence-based connections between the genes within
the nine loci that might indicate overlap in the pathways
acting in the etiology of CD and PS. Multiple genes
(IL12B, IL23R, TYK2, JAK2, SOCS1, and STAT3) are
involved in IL23/Th17 signaling and play a critical role
in the principal signaling mechanism for a wide array of
cytokines and growth factors. It is noteworthy that genes
CDC37 ([MIM 605065] 21.7 kb downstream of the estab-
lished shared risk locus at TYK2) and STIP1 ([MIM
605063] 113.5 kb upstream of the identified shared risk
at PRDX5) were found by GRAIL to be significantly con-
nected. CDC37 and STIP1 encode CDC37 and STI1, respec-
tively, two of several auxiliary proteins that associate with
the heat-shock protein 90 (HSP90) molecular chaperone
and thus are collectively referred to as HSP90 cochaper-
ones.54 HSP90 itself is an abundant, evolutionarily con-
served molecular chaperone that acts mainly as a cofactor
for the folding of polyproteins into functional, stable,
mature proteins, and it physically associates with JAK1
and probably JAK2,55 demonstrating that JAK1/2 are
client proteins of HSP90. A study in mice and in patient
samples suggested that HSP90 inhibitors might help treat
Figure 3. Gene Relationships across the 11 Shared Risk Loci of CD and PS Identified by GRAIL AnalysisGRAIL46 is a statistical text-mining approach to quantify the degree of relatedness among genes in genomic disease regions. It estimatesthe statistical significance of the number of observed relationships with a null model in which relationships between the genes occur byrandom chance. A significance score ptext, which is adjusted for multiple hypothesis testing, represents the output GRAIL score. ptext
values approximately estimate type-I error rates. Outer circle: lead SNPs from shared risk loci of both diseases; each box representsa SNP. Inner circle: genes of the genomic regions around lead SNPs that were identified based on LD properties; each box representsa gene; genes that were scored at ptext < 0.05 are significantly linked to genes in the other disease regions and are indicated in boldtype. Lines: the lines between genes represent significant connections, with the thickness and redness of the lines being inverselyproportional to the probability that a text-based connection would be seen by chance.
644 The American Journal of Human Genetics 90, 636–647, April 6, 2012

JAK2-dependent myeloproliferative neoplasms (MPNs).56
Moreover, inhibition of HSP90 was found to block Nod2-
mediated activation of the transcription factor NF-kB and
reduce NALP3-mediated gout-like inflammation in
mice,57 and mutations in the gene encoding NALP3, a
member of the Nod-like receptor (NLR) protein family,
are associated with several autoinflammatory disor-
ders.58,59 Our hypothesis that CDC37 and STIP1 are poten-
tial joint risk factors for CD and PS is substantiated by an
association peak within CDC37 in our same-effect com-
bined-phenotype analysis of discovery panels A and B
(pGWAS ¼ 1.61 3 10�3 for rs11879191, Figure S6).
It is worth noting that we used a two tier strategy to iden-
tify shared disease risk loci: Both approaches turned out to
be effective and complementary tools for gaining insights
into the postulated shared pathogenesis of CD and PS.
Application of only a single strategy would have decreased
the number of identified loci. Although the OVERLAP
approach represents a simple and cost-effective strategy
(cross-disease comparison of known risk SNPs), the
COMBINED approach provides the power to identify
shared susceptibility loci even if association signals are
heterogeneous between diseases, that is the particular
SNP showing the smallest p value at the considered locus,
as was the case, for example, for the identified risk locus at
ZMIZ1. This heterogeneity of most strongly associated
SNPs could be due to interactions with other genetic
variants or environmental factors, to differences in the
distribution or effect size of causal alleles, or to the fact
the identified SNPs show an association signal only
because they are in LD with the actual causal variant. In
particular, the increase of power due to increased sample
sizes makes the COMBINED approach a potentially power-
ful tool to detect shared risk loci that might be missed in
disease-specific GWASs that are often underpowered
because of their comparatively smaller sample sizes.
Because we did not search for loci harboring association
signals with different and independent SNPs in terms of
LD associated with CD and PS, there is room for improve-
ment. For instance, in a simple rank approach with regard
to single-marker associationpvalues, different disease-asso-
ciated markers for the same locus could be determined
when they rank high with regard to their p value in associ-
ation scans of CD and PS, respectively. This would allow
detecting shared susceptibility loci even if association
signals are heterogeneous between diseases. An approach
tomeet the challenge of theheterogeneity of genetic effects
of the same markers between different diseases was
proposed by Morris et al.60 The authors developed a test of
association within a multinomial regression framework
and demonstrated the improved power of their multino-
mial regression-based analysis over existing methods.
It is likely that future studies will identify additional
shared disease loci for CD and PS by further increasing
the sample size of analyzed case-control panels or, for
example, by applying the suggested rank approach.
Evidence for a shared etiological basis among several auto-
immune and inflammatory diseases is growing. For Crohn
disease, for example, Lees and colleagues currently re-
ported that 51 of the known 71 loci overlap with more
than 23 distinct diseases, comprising also several nonau-
toimmune conditions.61 Given the success of this study,
we expect the same for the investigation of further combi-
nations of such diseases for shared risk factors.
Supplemental Data
Supplemental Data include six figures and five tables and can be
found with this article online at http://www.cell.com/AJHG/.
Acknowledgments
We thank all individuals with psoriasis or CD, their families,
control individuals and clinicians for their participation in this
project. We thank the WTCCC consortium for the access to the
CDcase/control data.Weacknowledge the cooperationofGenizon
Biosciences.Wewish to thank TanjaWesse, TanjaHenke and Susan
Ehlers for expert technical help. We acknowledge EGCUT and
Estonian Biocentre personnel, especially Ms. M. Hass and Mr. V.
Soo. A list of funding sources is included in the Supplemental Data.
Received: October 28, 2011
Revised: January 30, 2012
Accepted: February 16, 2012
Published online: April 5, 2012
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
BEAGLE, http://faculty.washington.edu/browning/beagle/beagle.html
dbGaP, http://www.ncbi.nlm.nih.gov/gap
EIGENSTRAT, http://genepath.med.harvard.edu/~reich/Software.htm
GRAIL, http://www.broadinstitute.org/mpg/grail/
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
PLINK, http://pngu.mgh.harvard.edu/~purcell/plink/
PopGen Biobank, http://www.popgen.de
VIZ-GRAIL, http://www.broadinstitute.org/mpg/grail/vizgrail.html
References
1. Bhalerao, J., and Bowcock, A.M. (1998). The genetics of psori-
asis: A complex disorder of the skin and immune system.
Hum. Mol. Genet. 7, 1537–1545.
2. Elder, J.T., Nair, R.P., Guo, S.W., Henseler, T., Christophers, E.,
and Voorhees, J.J. (1994). The genetics of psoriasis. Arch. Der-
matol. 130, 216–224.
3. Russell, R.K., and Satsangi, J. (2004). IBD: A family affair. Best
Pract. Res. Clin. Gastroenterol. 18, 525–539.
4. Griffiths, C.E., and Barker, J.N. (2007). Pathogenesis and clin-
ical features of psoriasis. Lancet 370, 263–271.
5. Logan, I., andBowlus,C.L. (2010). Thegeoepidemiologyof auto-
immune intestinal diseases. Autoimmun. Rev. 9, A372–A378.
6. Sagoo, G.S., Cork, M.J., Patel, R., and Tazi-Ahnini, R. (2004).
Genome-wide studies of psoriasis susceptibility loci: A review.
J. Dermatol. Sci. 35, 171–179.
The American Journal of Human Genetics 90, 636–647, April 6, 2012 645

7. Najarian, D.J., and Gottlieb, A.B. (2003). Connections
between psoriasis and Crohn’s disease. J. Am. Acad. Dermatol.
48, 805–821, quiz 822–804.
8. Khor, B., Gardet, A., andXavier, R.J. (2011). Genetics and path-
ogenesis of inflammatory bowel disease. Nature 474, 307–317.
9. Yates, V.M., Watkinson, G., and Kelman, A. (1982). Further
evidence for an association between psoriasis, Crohn’s disease
and ulcerative colitis. Br. J. Dermatol. 106, 323–330.
10. Bernstein, C.N., Wajda, A., and Blanchard, J.F. (2005). The
clustering of other chronic inflammatory diseases in inflam-
matory bowel disease: A population-based study. Gastroenter-
ology 129, 827–836.
11. Weng, X., Liu, L., Barcellos, L.F., Allison, J.E., and Herrinton,
L.J. (2007). Clustering of inflammatory bowel disease with
immunemediated diseases amongmembers of a northern cal-
ifornia-managed care organization. Am. J. Gastroenterol. 102,
1429–1435.
12. Barrett, J.C., Hansoul, S., Nicolae, D.L., Cho, J.H., Duerr, R.H.,
Rioux, J.D., Brant, S.R., Silverberg, M.S., Taylor, K.D., Barmada,
M.M., et al; NIDDK IBD Genetics Consortium; Belgian-French
IBD Consortium; Wellcome Trust Case Control Consortium.
(2008). Genome-wide association defines more than 30 distinct
susceptibility loci for Crohn’s disease. Nat. Genet. 40, 955–962.
13. Franke, A., McGovern, D.P., Barrett, J.C., Wang, K., Radford-
Smith, G.L., Ahmad, T., Lees, C.W., Balschun, T., Lee, J., Rob-
erts, R., et al. (2010). Genome-wide meta-analysis increases to
71 the number of confirmed Crohn’s disease susceptibility
loci. Nat. Genet. 42, 1118–1125.
14. Nair, R.P., Duffin, K.C., Helms, C., Ding, J., Stuart, P.E., Gold-
gar, D., Gudjonsson, J.E., Li, Y., Tejasvi, T., Feng, B.J., et al;
Collaborative Association Study of Psoriasis. (2009).
Genome-wide scan reveals association of psoriasis with IL-23
and NF-kappaB pathways. Nat. Genet. 41, 199–204.
15. Ellinghaus, E., Ellinghaus, D., Stuart, P.E., Nair, R.P., Debrus, S.,
Raelson, J.V., Belouchi, M., Fournier, H., Reinhard, C., Ding, J.,
et al. (2010). Genome-wide association study identifies a psori-
asis susceptibility locus at TRAF3IP2. Nat. Genet. 42, 991–995.
16. Strange, A., Capon, F., Spencer, C.C., Knight, J., Weale, M.E.,
Allen, M.H., Barton, A., Band, G., Bellenguez, C., Bergboer,
J.G., et al; Genetic Analysis of Psoriasis Consortium & the
Wellcome Trust Case Control Consortium 2. (2010). A
genome-wide association study identifies new psoriasis
susceptibility loci and an interaction between HLA-C and
ERAP1. Nat. Genet. 42, 985–990.
17. Sun, L.D., Cheng, H., Wang, Z.X., Zhang, A.P., Wang, P.G., Xu,
J.H., Zhu, Q.X., Zhou, H.S., Ellinghaus, E., Zhang, F.R., et al.
(2010). Association analyses identify six new psoriasis suscepti-
bility loci in theChinesepopulation.Nat.Genet.42, 1005–1009.
18. Stuart, P.E., Nair, R.P., Ellinghaus, E., Ding, J., Tejasvi, T.,
Gudjonsson, J.E., Li, Y., Weidinger, S., Eberlein, B., Gieger,
C., et al. (2010). Genome-wide association analysis identifies
three psoriasis susceptibility loci. Nat. Genet. 42, 1000–1004.
19. Huffmeier, U., Uebe, S., Ekici, A.B., Bowes, J., Giardina, E., Ko-
rendowych, E., Juneblad, K., Apel, M., McManus, R., Ho, P.,
et al. (2010). Common variants at TRAF3IP2 are associated
with susceptibility to psoriatic arthritis and psoriasis. Nat.
Genet. 42, 996–999.
20. Zhang, X.J., Huang, W., Yang, S., Sun, L.D., Zhang, F.Y., Zhu,
Q.X., Zhang, F.R., Zhang, C., Du, W.H., Pu, X.M., et al.
(2009). Psoriasis genome-wide association study identifies
susceptibility variants within LCE gene cluster at 1q21. Nat.
Genet. 41, 205–210.
21. Duerr, R.H., Taylor, K.D., Brant, S.R., Rioux, J.D., Silverberg,
M.S., Daly, M.J., Steinhart, A.H., Abraham, C., Regueiro, M.,
Griffiths, A., et al. (2006). A genome-wide association study
identifies IL23R as an inflammatory bowel disease gene.
Science 314, 1461–1463.
22. Cargill, M., Schrodi, S.J., Chang, M., Garcia, V.E., Brandon, R.,
Callis, K.P., Matsunami, N., Ardlie, K.G., Civello, D., Catanese,
J.J., et al. (2007). A large-scale genetic association study
confirms IL12B and leads to the identification of IL23R as
psoriasis-risk genes. Am. J. Hum. Genet. 80, 273–290.
23. Nair, R.P.,Ruether,A., Stuart, P.E., Jenisch, S., Tejasvi, T.,Hirema-
galore, R., Schreiber, S., Kabelitz, D., Lim, H.W., Voorhees, J.J.,
et al. (2008). Polymorphisms of the IL12B and IL23R genes are
associated with psoriasis. J. Invest. Dermatol. 128, 1653–1661.
24. Mannon, P.J., Fuss, I.J., Mayer, L., Elson, C.O., Sandborn, W.J.,
Present, D., Dolin, B., Goodman, N., Groden, C., Hornung,
R.L., et al; Anti-IL-12 Crohn’s Disease Study Group. (2004).
Anti-interleukin-12 antibody for active Crohn’s disease. N.
Engl. J. Med. 351, 2069–2079.
25. Abraham, C., and Cho, J.H. (2009). IL-23 and autoimmunity:
New insights into the pathogenesis of inflammatory bowel
disease. Annu. Rev. Med. 60, 97–110.
26. Di Meglio, P., Di Cesare, A., Laggner, U., Chu, C.C., Napolitano,
L., Villanova, F., Tosi, I., Capon, F., Trembath, R.C., Peris, K., and
Nestle, F.O. (2011). The IL23R R381Q gene variant protects
against immune-mediated diseases by impairing IL-23-induced
Th17 effector response in humans. PLoS ONE 6, e17160.
27. Franke, A., Fischer, A., Nothnagel, M., Becker, C., Grabe, N.,
Till, A., Lu, T., Muller-Quernheim, J., Wittig, M., Hermann,
A., et al. (2008). Genome-wide association analysis in sarcoid-
osis and Crohn’s disease unravels a common susceptibility
locus on 10p12.2. Gastroenterology 135, 1207–1215.
28. Wellcome Trust Case Control Consortium. (2007). Genome-
wide association study of 14,000 cases of seven common
diseases and 3,000 shared controls. Nature 447, 661–678.
29. Franke, A., Balschun, T., Karlsen, T.H., Hedderich, J., May, S.,
Lu, T., Schuldt, D., Nikolaus, S., Rosenstiel, P., Krawczak, M.,
and Schreiber, S. (2008). Replication of signals from recent
studies of Crohn’s disease identifies previously unknown
disease loci for ulcerative colitis. Nat. Genet. 40, 713–715.
30. Wang, K., Baldassano, R., Zhang, H., Qu, H.Q., Imielinski, M.,
Kugathasan, S., Annese, V., Dubinsky, M., Rotter, J.I., Russell,
R.K., et al. (2010). Comparative genetic analysis of inflamma-
tory bowel disease and type 1 diabetes implicates multiple loci
with opposite effects. Hum. Mol. Genet. 19, 2059–2067.
31. Cotsapas, C., Voight, B.F., Rossin, E., Lage, K., Neale, B.M., Wal-
lace, C., Abecasis, G.R., Barrett, J.C., Behrens, T., Cho, J., et al;
FOCiS Network of Consortia. (2011). Pervasive sharing of
genetic effects inautoimmunedisease.PLoSGenet.7, e1002254.
32. Imielinski, M., Baldassano, R.N., Griffiths, A., Russell, R.K.,
Annese, V., Dubinsky, M., Kugathasan, S., Bradfield, J.P.,
Walters, T.D., Sleiman, P., et al; Western Regional Alliance for
Pediatric IBD; International IBDGenetics Consortium;NIDDK
IBD Genetics Consortium; Belgian-French IBD Consortium;
Wellcome Trust Case Control Consortium. (2009). Common
variants at five new loci associated with early-onset inflamma-
tory bowel disease. Nat. Genet. 41, 1335–1340.
33. Festen, E.A., Goyette, P., Green, T., Boucher, G., Beauchamp,
C., Trynka, G., Dubois, P.C., Lagace, C., Stokkers, P.C.,
Hommes, D.W., et al. (2011). A meta-analysis of genome-
wide association scans identifies IL18RAP, PTPN2, TAGAP,
646 The American Journal of Human Genetics 90, 636–647, April 6, 2012

and PUS10 as shared risk loci for Crohn’s disease and celiac
disease. PLoS Genet. 7, e1001283.
34. Zhernakova, A., Stahl, E.A., Trynka, G., Raychaudhuri, S.,
Festen, E.A., Franke, L., Westra, H.J., Fehrmann, R.S., Kurree-
man, F.A., Thomson, B., et al. (2011). Meta-analysis of
genome-wide association studies in celiac disease and rheu-
matoid arthritis identifies fourteen non-HLA shared loci.
PLoS Genet. 7, e1002004.
35. Krawczak, M., Nikolaus, S., von Eberstein, H., Croucher, P.J., El
Mokhtari, N.E., and Schreiber, S. (2006). PopGen: Population-
based recruitment of patients and controls for the analysis of
complex genotype-phenotype relationships. Community
Genet. 9, 55–61.
36. Wichmann, H.E., Gieger, C., and Illig, T.; MONICA/KORA
Study Group. (2005). KORA-gen—resource for population
genetics, controls and a broad spectrum of disease pheno-
types. Gesundheitswesen 67 (Suppl 1 ), S26–S30.
37. Weiland, S.K., Bjorksten, B., Brunekreef, B., Cookson, W.O.,
von Mutius, E., and Strachan, D.P.; International Study of
Asthma and Allergies in Childhood Phase II Study Group.
(2004). Phase II of the International Study of Asthma and
Allergies in Childhood (ISAAC II): Rationale and methods.
Eur. Respir. J. 24, 406–412.
38. Manolio, T.A., Rodriguez, L.L., Brooks, L., Abecasis, G., Ballin-
ger, D., Daly, M., Donnelly, P., Faraone, S.V., Frazer, K., Gabriel,
S., et al; GAIN Collaborative Research Group; Collaborative
Association Study of Psoriasis; International Multi-Center
ADHD Genetics Project; Molecular Genetics of Schizophrenia
Collaboration; Bipolar Genome Study; Major Depression
Stage 1 Genomewide Association in Population-Based Sam-
ples Study; Genetics of Kidneys in Diabetes (GoKinD) Study.
(2007). New models of collaboration in genome-wide associa-
tion studies: The Genetic Association Information Network.
Nat. Genet. 39, 1045–1051.
39. Price, A.L., Patterson, N.J., Plenge, R.M., Weinblatt, M.E.,
Shadick, N.A., and Reich, D. (2006). Principal components
analysis corrects for stratification in genome-wide association
studies. Nat. Genet. 38, 904–909.
40. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira,
M.A., Bender, D., Maller, J., Sklar, P., de Bakker, P.I., Daly,
M.J., and Sham, P.C. (2007). PLINK: A tool set for whole-
genome association and population-based linkage analyses.
Am. J. Hum. Genet. 81, 559–575.
41. Browning, B.L., and Browning, S.R. (2009). A unified approach
to genotype imputation and haplotype-phase inference for
large data sets of trios and unrelated individuals. Am. J.
Hum. Genet. 84, 210–223.
42. Altshuler, D.M., Gibbs, R.A., Peltonen, L., Altshuler, D.M.,
Gibbs, R.A., Peltonen, L., Dermitzakis, E., Schaffner, S.F., Yu,
F., Peltonen, L., et al; International HapMap 3 Consortium.
(2010). Integrating common and rare genetic variation in
diverse human populations. Nature 467, 52–58.
43. Devlin, B., and Roeder, K. (1999). Genomic control for associ-
ation studies. Biometrics 55, 997–1004.
44. de Bakker, P.I., Ferreira, M.A., Jia, X., Neale, B.M., Raychaud-
huri, S., and Voight, B.F. (2008). Practical aspects of imputa-
tion-drivenmeta-analysis of genome-wide association studies.
Hum. Mol. Genet. 17 (R2), R122–R128.
45. 1000 Genomes Project Consortium. (2010). A map of human
genome variation from population-scale sequencing. Nature
467, 1061–1073.
46. Raychaudhuri, S., Plenge, R.M., Rossin, E.J., Ng, A.C., Purcell,
S.M., Sklar, P., Scolnick, E.M., Xavier, R.J., Altshuler, D., and
Daly, M.J.; International Schizophrenia Consortium. (2009).
Identifying relationships among genomic disease regions:
Predicting genes at pathogenic SNP associations and rare dele-
tions. PLoS Genet. 5, e1000534.
47. Dixon, A.L., Liang, L., Moffatt, M.F., Chen, W., Heath, S.,
Wong, K.C., Taylor, J., Burnett, E., Gut, I., Farrall, M., et al.
(2007). A genome-wide association study of global gene
expression. Nat. Genet. 39, 1202–1207.
48. Li, X., Thyssen, G., Beliakoff, J., and Sun, Z. (2006). The novel
PIAS-like protein hZimp10 enhances Smad transcriptional
activity. J. Biol. Chem. 281, 23748–23756.
49. Carlsson, B., Kindberg, E., Buesa, J., Rydell, G.E., Lidon, M.F.,
Montava, R., Abu Mallouh, R., Grahn, A., Rodrıguez-Dıaz, J.,
Bellido, J., et al. (2009). The G428A nonsense mutation
in FUT2 provides strong but not absolute protection against
symptomatic GII.4 Norovirus infection. PLoS ONE 4, e5593.
50. Ikehara, Y., Nishihara, S., Yasutomi,H., Kitamura, T.,Matsuo, K.,
Shimizu, N., Inada, K., Kodera, Y., Yamamura, Y., Narimatsu, H.,
et al. (2001). Polymorphisms of two fucosyltransferase genes
(Lewis and Secretor genes) involving type I Lewis antigens are
associated with the presence of anti-Helicobacter pylori IgG
antibody. Cancer Epidemiol. Biomarkers Prev. 10, 971–977.
51. Starr, R., Willson, T.A., Viney, E.M., Murray, L.J., Rayner, J.R.,
Jenkins, B.J., Gonda, T.J., Alexander, W.S., Metcalf, D., Nicola,
N.A., and Hilton, D.J. (1997). A family of cytokine-inducible
inhibitors of signalling. Nature 387, 917–921.
52. Yasukawa, H., Sasaki, A., and Yoshimura, A. (2000). Negative
regulation of cytokine signaling pathways. Annu. Rev. Immu-
nol. 18, 143–164.
53. Krebs, D.L., and Hilton, D.J. (2000). SOCS: Physiological
suppressors of cytokine signaling. J. Cell Sci. 113, 2813–2819.
54. Abbas-Terki, T., Briand, P.A., Donze, O., and Picard, D. (2002).
The Hsp90 co-chaperones Cdc37 and Sti1 interact physically
and genetically. Biol. Chem. 383, 1335–1342.
55. Shang, L., and Tomasi, T.B. (2006). The heat shock protein 90-
CDC37 chaperone complex is required for signaling by types I
and II interferons. J. Biol. Chem. 281, 1876–1884.
56. Marubayashi, S., Koppikar, P., Taldone, T., Abdel-Wahab, O.,
West, N., Bhagwat, N., Caldas-Lopes, E., Ross, K.N., Gonen,
M., Gozman, A., et al. (2010). HSP90 is a therapeutic target
in JAK2-dependent myeloproliferative neoplasms in mice
and humans. J. Clin. Invest. 120, 3578–3593.
57. Mayor, A., Martinon, F., De Smedt, T., Petrilli, V., and Tschopp,
J. (2007). A crucial function of SGT1 and HSP90 in inflamma-
some activity links mammalian and plant innate immune
responses. Nat. Immunol. 8, 497–503.
58. Hoffman, H.M., Mueller, J.L., Broide, D.H., Wanderer, A.A., and
Kolodner, R.D. (2001). Mutation of a new gene encoding a puta-
tive pyrin-like protein causes familial cold autoinflammatory
syndromeandMuckle-Wells syndrome.Nat.Genet.29, 301–305.
59. Hawkins,P.N.,Lachmann,H.J.,Aganna,E., andMcDermott,M.F.
(2004). Spectrum of clinical features in Muckle-Wells syndrome
and response to anakinra. Arthritis Rheum. 50, 607–612.
60. Morris, A.P., Lindgren,C.M., Zeggini, E., Timpson,N.J., Frayling,
T.M., Hattersley, A.T., and McCarthy, M.I. (2010). A powerful
approach to sub-phenotype analysis in population-based
genetic association studies. Genet. Epidemiol. 34, 335–343.
61. Lees, C.W., Barrett, J.C., Parkes, M., and Satsangi, J. (2011).
New IBD genetics: Common pathways with other diseases.
Gut 60, 1739–1753.
The American Journal of Human Genetics 90, 636–647, April 6, 2012 647

ARTICLE
Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 asSusceptibility Loci for Systemic Lupus Erythematosusin a Large-Scale Multiracial Replication Study
Christopher J. Lessard,1,2 Indra Adrianto,1 John A. Ice,1 Graham B. Wiley,1 Jennifer A. Kelly,1
Stuart B. Glenn,1 Adam J. Adler,1 He Li,1,2 Astrid Rasmussen,1 Adrienne H. Williams,3 Julie Ziegler,3
Mary E. Comeau,3 Miranda Marion,3 Benjamin E. Wakeland,4 Chaoying Liang,4 Paula S. Ramos,5
Kiely M. Grundahl,1 Caroline J. Gallant,6 Marta E. Alarcon-Riquelme for the BIOLUPUS andGENLES Networks,1,7 Graciela S. Alarcon,8 Juan-Manuel Anaya,9 Sang-Cheol Bae,10 Susan A. Boackle,11
Elizabeth E. Brown,8 Deh-Ming Chang,12 Soo-Kyung Cho,10 Lindsey A. Criswell,13 Jeffrey C. Edberg,8
Barry I. Freedman,14 Gary S. Gilkeson,5 Chaim O. Jacob,15 Judith A. James,1,2,16 Diane L. Kamen,5
Robert P. Kimberly,8 Jae-Hoon Kim,10 Javier Martin,17 Joan T. Merrill,18 Timothy B. Niewold,19
So-Yeon Park,10 Michelle A. Petri,20 Bernardo A. Pons-Estel,21 Rosalind Ramsey-Goldman,22
John D. Reveille,23 R. Hal Scofield,1,2,16,24 Yeong Wook Song,25 Anne M. Stevens,26,27 Betty P. Tsao,28
Luis M. Vila,29 Timothy J. Vyse,30 Chack-Yung Yu,31,32 Joel M. Guthridge,1 Kenneth M. Kaufman,1,33,34
John B. Harley,33,34 Edward K. Wakeland,4 Carl D. Langefeld,3 Patrick M. Gaffney,1,2
Courtney G. Montgomery,1 and Kathy L. Moser1,2,*
Systemic lupus erythematosus (SLE) is a chronic heterogeneous autoimmune disorder characterized by the loss of tolerance to self-anti-
gens and dysregulated interferon responses. The etiology of SLE is complex, involving both heritable and environmental factors.
Candidate-gene studies and genome-wide association (GWA) scans have been successful in identifying new loci that contribute to
disease susceptibility; however, much of the heritable risk has yet to be identified. In this study, we sought to replicate 1,580 variants
showing suggestive association with SLE in a previously published GWA scan of European Americans; we tested a multiethnic
population consisting of 7,998 SLE cases and 7,492 controls of European, African American, Asian, Hispanic, Gullah, and Amerindian
ancestry to find association with the disease. Several genes relevant to immunological pathways showed association with SLE. Three
loci exceeded the genome-wide significance threshold: interferon regulatory factor 8 (IRF8; rs11644034; pmeta-Euro ¼ 2.08 3 10�10),
transmembrane protein 39A (TMEM39A; rs1132200; pmeta-all ¼ 8.62 3 10�9), and 17q21 (rs1453560; pmeta-all ¼ 3.48 3 10�10)
between IKAROS family of zinc finger 3 (AIOLOS; IKZF3) and zona pellucida binding protein 2 (ZPBP2). Fine mapping, resequencing,
imputation, and haplotype analysis of IRF8 indicated that three independent effects tagged by rs8046526, rs450443, and rs4843869,
respectively, were required for risk in individuals of European ancestry. Eleven additional replicated effects (5 3 10�8 < pmeta-Euro <
9.99 3 10�5) were observed with CFHR1, CADM2, LOC730109/IL12A, LPP, LOC63920, SLU7, ADAMTSL1, C10orf64, OR8D4,
FAM19A2, and STXBP6. The results of this study increase the number of confirmed SLE risk loci and identify others warranting further
investigation.
1Arthritis and Clinical Immunology Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK 73104, USA; 2Department of
Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA; 3Department of Biostatistical Sciences, Wake Forest University
Health Sciences, Winston-Salem, NC 27157, USA; 4Department of Immunology, University of Texas Southwestern Medical Center at Dallas, Dallas, TX
75390, USA; 5Division of Rheumatology and Immunology, Department of Medicine, Medical University of South Carolina, Charleston, SC 29425, USA;6Department of Genetics and Pathology, Rudbeck Laboratory, Uppsala University, Uppsala 75105, Sweden; 7Centro de Genomica e Investigaciones
Oncologicas, Pfizer-Universidad de Granada-Junta de Andalucıa, Granada 18100, Spain; 8Division of Clinical Immunology and Rheumatology, Department
of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA; 9Center for Autoimmune Diseases Research, Universidad del Rosario,
Bogota, Colombia; 10Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 133-792, Korea; 11Division of
Rheumatology, University of Colorado Denver, Aurora, CO 80045, USA; 12National Defense Medical Center, Taipei 114, Taiwan; 13Rosalind Russell Medical
Research Center for Arthritis, University of California, San Francisco, San Francisco, CA 94143, USA; 14Section on Nephrology, Department of Internal
Medicine, Wake Forest School of Medicine, Winston-Salem, NC 27157, USA; 15Department of Medicine, University of Southern California, Los Angeles,
CA 90089, USA; 16Department of Medicine, University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA; 17Instituto de Parasitologıa
y Biomedicina Lopez-Neyra, Consejo Superior de Investigaciones Cientificas, Granada 18100, Spain; 18Clinical Pharmacology, OklahomaMedical Research
Foundation, Oklahoma City, OK 73104, USA; 19Section of Rheumatology and Gwen Knapp Center for Lupus and Immunology Research, University of Chi-
cago, Chicago, IL 60637, USA; 20Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21287, USA; 21Sanatorio Parque,
Rosario 2000, Argentina; 22Division of Rheumatology, Northwestern University Feinberg School ofMedicine, Chicago, IL 60611, USA; 23Rheumatology and
Clinical Immunogenetics, University of Texas Health Science Center at Houston, Houston, TX 77030, USA; 24US Department of Veterans Affairs Medical
Center, Oklahoma City, OK 73104, USA; 25Division of Rheumatology, Seoul National University, Seoul 110-799, Korea; 26Division of Rheumatology,
Department of Pediatrics, University of Washington, Seattle, WA 98105, USA; 27Center for Immunity and Immunotherapies, Seattle Children’s Research
Institute, Seattle, WA 98105, USA; 28Division of Rheumatology, Department of Medicine, University of California, Los Angeles, Los Angeles, CA 90095,
USA; 29Division of Rheumatology, Department of Medicine, University of Puerto Rico Medical Sciences Campus, San Juan 00936-5067, Puerto Rico;30Division of Genetics and Molecular Medicine and Division of Immunology, Infection, and Inflammatory Disease, King’s College London, London SE1
9RT, UK; 31Center for Molecular and Human Genetics, The Research Institute, Nationwide Children’s Hospital, Columbus, OH 43205, USA; 32Department
of Pediatrics, Ohio State University, Columbus, OH 43205, USA; 33Division of Rheumatology, Cincinnati Children’s Hospital Medical Center, Cincinnati,
OH 45229, USA; 34US Department of Veterans Affairs Medical Center, Cincinnati, OH 45220, USA
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.023. �2012 by The American Society of Human Genetics. All rights reserved.
648 The American Journal of Human Genetics 90, 648–660, April 6, 2012

Introduction
Systemic lupus erythematosus (SLE [MIM 152700]) is a
chronic autoimmune disease that is classically character-
ized by inflammation, dysregulated type 1 interferon
responses, and autoantibodies directed to the nuclear
compartment. Women of childbearing age are preferen-
tially affected at a rate nine times that of men, and those
of African American and Asian ancestries are affected
more frequently and manifest more severe disease than
those of European ancestry.1 Although the etiology of SLE
is largely unknown, its pathogenesis most likely involves
a complex interplay between environmental (e.g., UV light,
Epstein-Barr virus infection, etc.) and genetic (e.g., MHC,
IRF5 [MIM 607218], etc.) components.2 A sibling risk ratio
(ls) of approximately 30 in SLE illustrates a strong genetic
component,3 and the fact that observational studies have
identified many families with multiple cases of SLE and
other autoimmune conditions suggests the potential for
shared genetic predisposition.4–6
Candidate-gene studies and, more recently, genome-
wide association (GWA) scans have been highly successful
in identifyingmultiple susceptibility loci.2,7 The histocom-
patibility leukocyte antigen (HLA) region has been known
to contribute to the risk of SLE and other related autoim-
mune diseases since the 1970s.8–11 In the early 2000s,
gene expression studies determined that, compared to
healthy controls, individuals with SLE overexpress genes
in the interferon pathway.12–14 Association between SLE
and variants in the region of IRF5 was first reported in
2005 and has since been replicated in most GWA scans
of SLE.15–19 In 2008, four GWA scans of SLE cases of
European descent were published, and the first GWA
scan of Asian descent was published in 2009.16,17,19–21
Collectively, these studies have identified and confirmed
~35 loci that contribute to the pathogenesis of SLE. These
data highlight the importance of several pathways,
including those involving lymphocyte activation and
function, immune-complex clearance, innate immune
response, and adaptive immune responses.2 However,
a substantial portion of the heritable risk has yet to be iden-
tified.17,22 The lack of causal variants, rare variants, and/or
other loci yet to be discovered might account for the
missing heritability.
In an effort to identify regions contributing to SLE risk,
we sought to replicate suggestive association signals in
our previously published European American SLE GWA
scan.19 We evaluated 1,580 single-nucleotide polymor-
phisms (SNPs) in an independent population of 7,998
SLE cases and 7,492 controls of European, African Amer-
ican, Asian, Hispanic, Gullah, and Amerindian ancestry
(Tables S1–S3, available online). Three loci, interferon
regulatory factor 8 (IRF8 [MIM 601565]), transmembrane
protein 39A (TMEM39A), and the region between IKAROS
family of zinc finger 3 (AIOLOS; IKZF3 [MIM 606221]) and
zona pellucida binding protein 2 (ZPBP2 [MIM 608449])
exceeded the genome-wide significance threshold (p <
5 3 10�8). Through fine mapping, resequencing, and
imputation of the IRF8 region, we identified three inde-
pendent effects required for risk. Moreover, we replicated
11 other loci, several of which had been previously re-
ported in related conditions.
Subjects and Methods
GWA ScanGenotyping, quality control, procedures for data analysis, and
summary statistics for the GWA scan were described previously
in Graham et al., 2008.19
Study DesignThe genotype data used in this study were generated as a part of
a joint effort of more than 40 investigators from around the world.
These investigators contributed samples, funding, and hypotheses
on a combined array containing ~35,000 SNPs (Figure S1). The
Oklahoma Medical Research Foundation (OMRF) served as the
coordinating center, ran the arrays, and sent the data to a central
facility for quality control at Wake Forest Medical Center. These
data were then distributed back to the investigators, who re-
quested the SNPs for final analysis and publication.23–28
SubjectsThemultiracial replication study consisted of 17,003 total samples
(8,922 SLE cases and 8,077 controls) and included individuals
of self-reported African American, Asian, European, Gullah,
Hispanic, and Amerindian ancestry (Table S1). A total of 374
samples were common between the GWA scan and the replication
study so that genotypes generated by the two platforms could
be confirmed and so that genotypes of SNPs not present on the
Affymetrix 5.0 array could be obtained. These data were only
used as observed data for the imputation analysis of specific
genomic regions, as described below; to maintain independence
between the GWA scan and replication samples, we did not
include the data generated on these shared samples in the replica-
tion or fine-mapping analyses. The OMRF gathered the samples
from consenting subjects (according to the guidelines of the ethics
committees at the respective institutions where the samples were
collected) and prepared them for genotyping. All cases used in this
study fulfilled at least 4 of the 11 American College of Rheuma-
tology criteria for SLE, whereas the healthy, population-based
controls did not have any family history of SLE or any other auto-
immune disease.29
Genotyping and Sample Quality ControlA total of 1,580 SNPs that attained p < 0.05 in the previously pub-
lished GWA scan were selected for replication. In addition, 287
SNPs (chosen to capture all variation with aminimum r2 threshold
of 0.8 via the TAGGER algorithm in HAPLOVIEW30) within the
IRF8 region and 347 ancestral-informative markers (AIMs) span-
ning the genome were genotyped. Genotyping of SNPs was per-
formed at OMRF with Infinium chemistry on an Illumina iSelect
custom array according to the manufacturer’s protocol. The
following quality-control procedures were implemented prior to
the analysis (Table S2): there were well-defined clusters within the
scatter plots, the SNP call rate was >90% across all samples geno-
typed, the minor allele frequency was >1%, the sample call rate
was >90%, p > 0.05 for differential missingness between cases
and controls, the total proportion missing was <5%, and the
The American Journal of Human Genetics 90, 648–660, April 6, 2012 649

Hardy-Weinberg proportions were p > 0.01 in controls and p >
0.0001 in cases.
Samples exhibiting excess heterozygosity (>5 standard devia-
tions [SDs] from the mean) or a <90% call rate were excluded
from the analysis. The remaining individuals were examined for
excessive allele sharing as estimated by identity-by-descent
(IBD). In sample pairs with excess relatedness (IBD > 0.4), one
individual was removed from the analysis on the basis of the
following criteria: (1) remove the sample with the lower call rate,
(2) remove the control and retain the case, (3) remove the male
sample before the female sample, (4) remove the younger control
before the older control, and (5) in a situation with two cases, re-
move the case with fewer phenotype data available. Discrepancies
between self-reported and genetically determined gender were
evaluated. Males were required to be heterozygous at rs2557523
(given that the G allele for this SNP is only observed on the
Y chromosome and the A allele appears only on the X chromo-
some) and to have%10% chromosome X heterozygosity. Females
were required to be homozygous for the A allele at rs2557523 and
to have >10% chromosome X heterozygosity.
Ascertainment of Population StratificationGenetic outliers from each ethnic and/or racial group were
removed from further analysis as determined by principal-compo-
nent analysis and admixture estimates (Figure S6).31,32 Using the
163 AIMs that passed quality control in both EIGENSTRAT31 and
ADMIXMAP33,34 to distinguish the four continental ancestral pop-
ulations—Africans, Europeans, Amerindians, and East Asians—
allowed identification of the substructure within the sample
set (Figure S6A).35,36 We utilized principal components from
EIGENSTRAT outputs to identify outliers >4 SDs from the mean
of each of the first three principal components (PC) for the indi-
vidual population clusters. After quality control, a total of 1,139
samples were excluded (Figure S6B and Table S3). Overall, 2,586
subjects were included in the GWA scan, and 15,490 subjects
were included in the replication study, resulting in a total of
18,076 subjects.
Statistical AnalysisTesting for SNP-SLE association in the replication studywas carried
out through the computation of logistic regression as imple-
mented in PLINK v.1.07.37 The calculation of the additive genetic
model included adjustments for the first three PCs and gender.
Models were also adjusted for ancestry with estimates provided
by ADMIXMAP and resulted in no observable difference in associ-
ation as comparedwith PC adjustment.We conducted conditional
likelihood-ratio tests by using the extended WHAP functionality
in PLINK v.1.07. The genome-wide p value threshold for all data
replicating GWA-scan results was p< 53 10�8 after meta-analysis.
For the finemapping and imputation of IRF8, we utilized a Bonfer-
roni-corrected p value threshold of p< 1.093 10�4 on the basis of
the maximum number of tests across all populations (460 inde-
pendent variants with r2 < 0.8). Using METAL,38 we performed
meta-analyses of the SNPs observed in both the GWA scan and
the multiracial replication study with a weighted Z score. Each
racial group was weighted by the square root of its sample size
so that sample-size differences could be controlled for between
studies. We combined all data generated unless the variant failed
quality control in a given racial group.
To test for meta-analysis heterogeneity, we utilized both Co-
chran’s Q test and the I2 index. Cochran’s Q test is a classical
method that calculates the weighted sum of the squared devia-
tions between individual study effects and the overall effect across
studies.39 It follows a chi-square distribution with k-1 degrees of
freedom (k is the number of studies). A value of p < 0.05 was
considered significant evidence of heterogeneity. The I2 index
measures the degree or percentage of inconsistency—due to
heterogeneity rather than random chance—across studies.40 The
I2 index ranges from 0% to 100% and can equal 0%–25% (low
heterogeneity), 26% –50% (moderate heterogeneity), 51%–75%
(high heterogeneity), or 76%–100% (very high heterogeneity).
Linkage disequilibrium (LD) and probable haplotypes were
determined with HAPLOVIEW v.4.2.30 We calculated haplotype
blocks for those haplotypes present at >3% frequency by using
the solid-spine of LD algorithms with minimum r2 values of
0.8.30
ResequencingWe resequenced the IRF8 region (chromosome 16: 84,488,150–
84,539,352 bp) in 206 (92 SLE cases and 114 healthy controls)
European and 46 (25 SLE cases and 21 healthy controls) African
American subjects. For each sample, 3–5 mg of whole genomic
DNA were sheared and prepared for sequencing with an Illumina
Paired-End Genomic DNA Sample Prep Kit. Targeted regions of
interest from each sample were then enriched with a SureSelect
Target Enrichment System utilizing a custom-designed bait pool
(Agilent Technologies). Resequencing was undertaken with an
Illumina GAIIx platform according to standard procedures. Post-
sequence data were processed with Illumina’s Pipeline software
v.1.7. All samples were sequenced to minimum average fold
coverage of 253.
Variant Detection and Quality ControlUnique sequences corresponding to an individual nucleotide
molecule were aligned to the human genome reference (hg18)
with the Burrows-Wheeler Aligner (BWA)41 alignment tool. Reads
were locally realigned around known and suspected insertion and
deletion sites with the Genome Analysis Toolkit (GATK) analysis
suite so that the best possible read alignment could be gener-
ated.42 Recalculation of the correct quality score for each base
within the alignment was then performed empirically with the
GATK suite. This process served to correct overestimated high-
quality scores initially reported by the sequencer itself.
After local realignment around deletion-insertion polymor-
phism (DIP) sites and base quality-score recalibration, SNP and
DIP genotypes were generated for each sample individually as
well as for the samples as a whole. Finally, SNP and DIP genotypes
were hard-filtered against a set of criteria designed to remove any
remaining low-quality calls. For a variant to be included in the call
list, we required a Phred quality score>30, a quality-by-depth ratio
of >5.0, a strand bias score of <�0.10, and a homopolymer
run of <5 bases. The variant phase was determined with the
program BEAGLE.43 Variants meeting call parameters were output
to files compatible with PLINK and other genotyping tools via the
VCFtools analysis suite.
To assess the accuracy of sequence-based SNP calling, we cross-
referenced the sequenced and genotyped allele calls. We observed
~99% concordance between genotypes and sequence-based var-
iant detection, suggesting high-quality sequence data. We manu-
ally inspected the samples withR5% of variants differing between
sequencing and genotyping to determine where sequence quality
was poor. As an additional quality-control measure, we confirmed
650 The American Journal of Human Genetics 90, 648–660, April 6, 2012

each variant identified by our automated workflow by manual
inspection of the assembled contig by using the Integrative
Genomics Viewer (IGV) program.44
ImputationTo increase the informativeness of the IRF8 region, we conducted
imputation in subjects of European, African American, and Asian
ancestry over a 100 kb interval spanning the IRF8 locus. Imputa-
tion of the replication data across chromosome 16 (84.45–84.46
Mb) was performed with IMPUTE2 and the reference panels
provided in Table S7.45–47 Imputed genotypes were required to
meet or exceed a probability threshold of 0.8, an information
measure of >0.4, and the same quality-control-criteria thresholds
described above for inclusion in the analyses.
Results
Two SNPs, rs11648084 and rs11644034, telomeric to IRF8
at 16q24.1 were suggestive of association with SLE in
our published GWA scan (p ¼ 5.99 3 10�4 and 2.29 3
10�3, respectively; odds ratio [OR] ¼ 0.76 and 0.66,
respectively; Table 1). Both rs11648084 and rs11644034
were replicated in the current, independent population
of SLE cases and controls of European ancestry and
exceeded the genome-wide threshold (pmeta-Euro ¼ 2.34 3
10�9 and pmeta-Euro ¼ 2.08 3 10�10, respectively; Table 1).
However, neither SNP was significantly associated with
SLE in any other population studied, perhaps as a result
of the reduced sample size, clinical and/or genetic
heterogeneity, decreased minor allele frequency, and/or
reduced correlation with the causal variants (Table 1 and
Table S4).
To better refine the association signal, 287 additional
SNPs covering ~100 kb encompassing the IRF8 coding
region were genotyped (see Subjects and Methods; Figures
1A and 1D, Table 2, and Table S4). The most significant
association in the European population was with
rs9936079 (p ¼ 3.96 3 10�9, OR ¼ 0.77; Figures 1A and
1D and Table 2), located ~11 kb telomeric to IRF8 and
found to be in strong LD with rs11644034 (r2 ¼ 0.92;
Figures 2B and 2C). The Asian population also exhibited
association with rs9936079 (p ¼ 2.95 3 10�3, OR ¼0.73); however, rs9936079 failed to pass quality-control
measures (see Subjects and Methods; differential missing-
ness p ¼ 10�7) in individuals of African ancestry and
was not associated with disease in patients of Hispanic
or Amerindian ancestry. Meta-analysis yielded pmeta-all ¼9.28 3 10�11 (Table 2 and Table S4).
We observed a modest association in the African Ameri-
cans at rs2934498 (p¼ 3.923 10�4, OR¼ 0.83), which was
also significant in those individuals of European ancestry
(p ¼ 5.96 3 10�6, OR ¼ 1.19) but not in those of Asian
ancestry (Figure 1B,D, Table 2, and Table S4). The strongest
Asian association was observed in a region (rs11117427,
p ¼ 1.99 3 10�5, OR ¼ 0.64) ~34 kb telomeric to IRF8
(Figures 1C and 1D, Table 2, and Table S4). Association
was also observed with rs11117427 (p ¼ 3.46 3 10�4,
OR ¼ 0.84) in the Europeans (Figures 1A and 1D and Table
2). Interestingly, this SNP is only ~2 kb away from
rs12444486, which has been reported by Gateva et al.22
as being suggestive of association with SLE in Europeans
(Figure 1D and Table S4).
We resequenced the IRF8 region in 206 subjects of
European ancestry and in 46 subjects of African American
ancestry to identify variants not previously evaluated
within the IRF8 region and to assess their association
with SLE (see Subjects and Methods). Thirty-eight and
85 variants not present in dbSNP 130 were identified in
European and African American individuals, respectively.
After imputing these data into our larger European and
African American datasets (see Subjects and Methods),
the most significantly associated region within the
European population was ~19 kb telomeric to IRF8
(rs4843869, p ¼ 7.61 3 10�10, OR ¼ 0.76; Table 2 and
Figures 1A and 1D). Ultimately, three strongly correlated
(r2 > 0.90) SNPs emerged as the most significantly associ-
ated with SLE in the Europeans: rs11644034 (identified
via GWA scan), rs9936079 (identified by fine mapping),
and rs4843869 (imputed on the basis of resequencing).
Interestingly, our targeted resequencing revealed a DIP
(rs11347703, p ¼ 1.113 10�8, OR ¼ 0.78) that was located
less than 100 bp from a genotyped SNP, rs8052690
(p ¼ 5.69 3 10�8, OR ¼ 0.79, Table 2). This DIP,
which has high biological plausibility, was in strong LD
with the peak European SNP (rs4843869) and rs8052690
(r2/D’ > 0.9; Figures 2B and 2C). Of note, some of
the African Americans resequenced in this study did
harbor the DIP (rs11347703) identified in the Europeans.
However, neither rs11347703 nor any SNP correlated
with it was found to be significantly associated with
SLE in African Americans, which could be due to the
decrease in power and/or a decrease in the minor allele
frequency.
The peak association in African Americans after imputa-
tion was at rs450443 (p ¼ 1.41 3 10�4, OR ¼ 0.82; Table 2
and Figures 1B and 1D) and was in strong LD (r2 ¼ 0.88)
with rs2934498. Patients of European but not Asian
ancestry showed association with rs450443 (p ¼ 9.73 3
10�6, OR ¼ 1.18; Table 2 and Figures 1A, 1C, and 1D).
We conducted imputation in the Asian population by
using 1,000 Genomes phased haplotypes;48 however,
rs11117427 remained the peak signal (Table 2 and Figures
1C and 1D).
To assess the independence of variants in the European
population, we used logistic regression models that
adjusted for the best tagging SNP at each signal. When
we adjusted for rs4843869 in Europeans, the association
persisted at rs450443, and variants correlated to it.
However, adjusting for rs4843869 negated the association
with rs11117427 and its correlated variants (Figures 2B,
2C, and 3, Table S5, and Figure S2A). Adjusting for either
rs450443 or rs11117427 was only able to negate the asso-
ciations of the polymorphisms that were correlated with
each of these SNPs (Figures 2B, 2C, and 3, Table S5, and
The American Journal of Human Genetics 90, 648–660, April 6, 2012 651
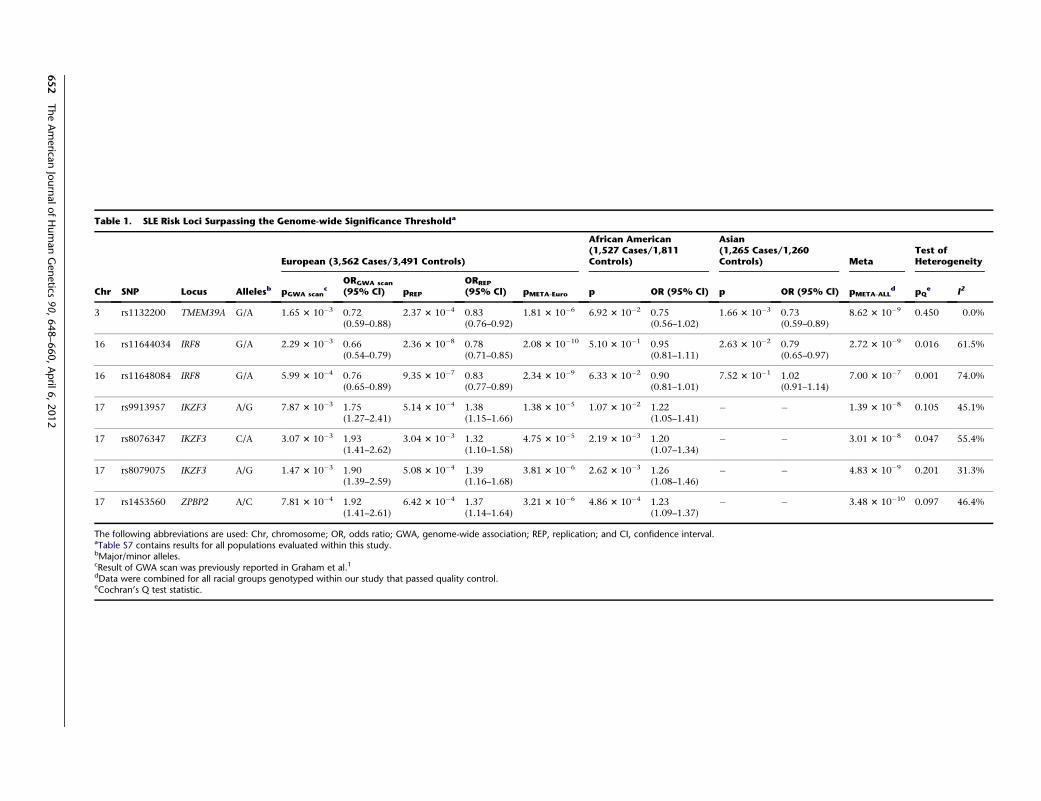
Table 1. SLE Risk Loci Surpassing the Genome-wide Significance Thresholda
Chr SNP Locus Allelesb
European (3,562 Cases/3,491 Controls)
African American(1,527 Cases/1,811Controls)
Asian(1,265 Cases/1,260Controls) Meta
Test ofHeterogeneity
pGWA scanc
ORGWA scan
(95% Cl) pREP
ORREP
(95% Cl) pMETA-Euro p OR (95% Cl) p OR (95% Cl) pMETA-ALLd pQ
e I2
3 rs1132200 TMEM39A G/A 1.65 3 10�3 0.72(0.59–0.88)
2.37 3 10�4 0.83(0.76–0.92)
1.81 3 10�6 6.92 3 10�2 0.75(0.56–1.02)
1.66 3 10�3 0.73(0.59–0.89)
8.62 3 10�9 0.450 0.0%
16 rs11644034 IRF8 G/A 2.29 3 10�3 0.66(0.54–0.79)
2.36 3 10�8 0.78(0.71–0.85)
2.08 3 10�10 5.10 3 10�1 0.95(0.81–1.11)
2.63 3 10�2 0.79(0.65–0.97)
2.72 3 10�9 0.016 61.5%
16 rs11648084 IRF8 G/A 5.99 3 10�4 0.76(0.65–0.89)
9.35 3 10�7 0.83(0.77–0.89)
2.34 3 10�9 6.33 3 10�2 0.90(0.81–1.01)
7.52 3 10�1 1.02(0.91–1.14)
7.00 3 10�7 0.001 74.0%
17 rs9913957 IKZF3 A/G 7.87 3 10�3 1.75(1.27–2.41)
5.14 3 10�4 1.38(1.15–1.66)
1.38 3 10�5 1.07 3 10�2 1.22(1.05–1.41)
� � 1.39 3 10�8 0.105 45.1%
17 rs8076347 IKZF3 C/A 3.07 3 10�3 1.93(1.41–2.62)
3.04 3 10�3 1.32(1.10–1.58)
4.75 3 10�5 2.19 3 10�3 1.20(1.07–1.34)
� � 3.01 3 10�8 0.047 55.4%
17 rs8079075 IKZF3 A/G 1.47 3 10�3 1.90(1.39–2.59)
5.08 3 10�4 1.39(1.16–1.68)
3.81 3 10�6 2.62 3 10�3 1.26(1.08–1.46)
� � 4.83 3 10�9 0.201 31.3%
17 rs1453560 ZPBP2 A/C 7.81 3 10�4 1.92(1.41–2.61)
6.42 3 10�4 1.37(1.14–1.64)
3.21 3 10�6 4.86 3 10�4 1.23(1.09–1.37)
� � 3.48 3 10�10 0.097 46.4%
The following abbreviations are used: Chr, chromosome; OR, odds ratio; GWA, genome-wide association; REP, replication; and CI, confidence interval.aTable S7 contains results for all populations evaluated within this study.bMajor/minor alleles.cResult of GWA scan was previously reported in Graham et al.1dData were combined for all racial groups genotyped within our study that passed quality control.eCochran’s Q test statistic.
652
TheAmerica
nJournalofHumanGenetics
90,648–660,April
6,2012
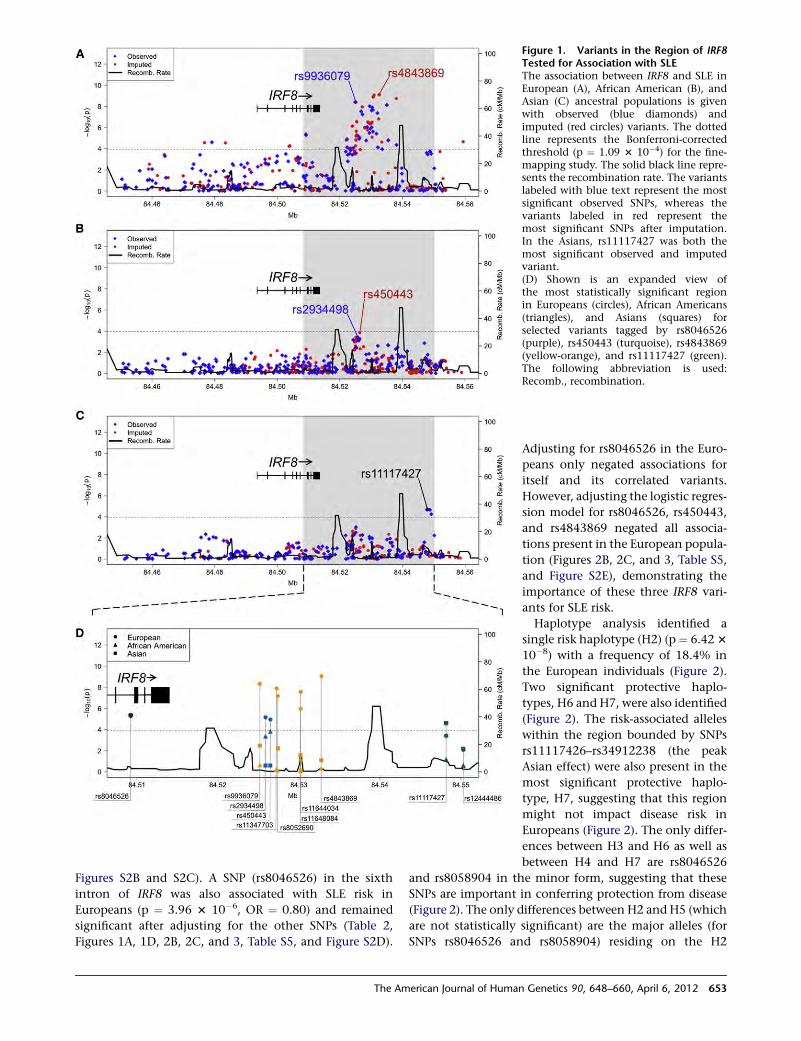
Figures S2B and S2C). A SNP (rs8046526) in the sixth
intron of IRF8 was also associated with SLE risk in
Europeans (p ¼ 3.96 3 10�6, OR ¼ 0.80) and remained
significant after adjusting for the other SNPs (Table 2,
Figures 1A, 1D, 2B, 2C, and 3, Table S5, and Figure S2D).
Figure 1. Variants in the Region of IRF8Tested for Association with SLEThe association between IRF8 and SLE inEuropean (A), African American (B), andAsian (C) ancestral populations is givenwith observed (blue diamonds) andimputed (red circles) variants. The dottedline represents the Bonferroni-correctedthreshold (p ¼ 1.09 3 10�4) for the fine-mapping study. The solid black line repre-sents the recombination rate. The variantslabeled with blue text represent the mostsignificant observed SNPs, whereas thevariants labeled in red represent themost significant SNPs after imputation.In the Asians, rs11117427 was both themost significant observed and imputedvariant.(D) Shown is an expanded view ofthe most statistically significant regionin Europeans (circles), African Americans(triangles), and Asians (squares) forselected variants tagged by rs8046526(purple), rs450443 (turquoise), rs4843869(yellow-orange), and rs11117427 (green).The following abbreviation is used:Recomb., recombination.
Adjusting for rs8046526 in the Euro-
peans only negated associations for
itself and its correlated variants.
However, adjusting the logistic regres-
sion model for rs8046526, rs450443,
and rs4843869 negated all associa-
tions present in the European popula-
tion (Figures 2B, 2C, and 3, Table S5,
and Figure S2E), demonstrating the
importance of these three IRF8 vari-
ants for SLE risk.
Haplotype analysis identified a
single risk haplotype (H2) (p¼ 6.423
10�8) with a frequency of 18.4% in
the European individuals (Figure 2).
Two significant protective haplo-
types, H6 and H7, were also identified
(Figure 2). The risk-associated alleles
within the region bounded by SNPs
rs11117426–rs34912238 (the peak
Asian effect) were also present in the
most significant protective haplo-
type, H7, suggesting that this region
might not impact disease risk in
Europeans (Figure 2). The only differ-
ences between H3 and H6 as well as
between H4 and H7 are rs8046526
and rs8058904 in the minor form, suggesting that these
SNPs are important in conferring protection from disease
(Figure 2). The only differences between H2 and H5 (which
are not statistically significant) are the major alleles (for
SNPs rs8046526 and rs8058904) residing on the H2
The American Journal of Human Genetics 90, 648–660, April 6, 2012 653
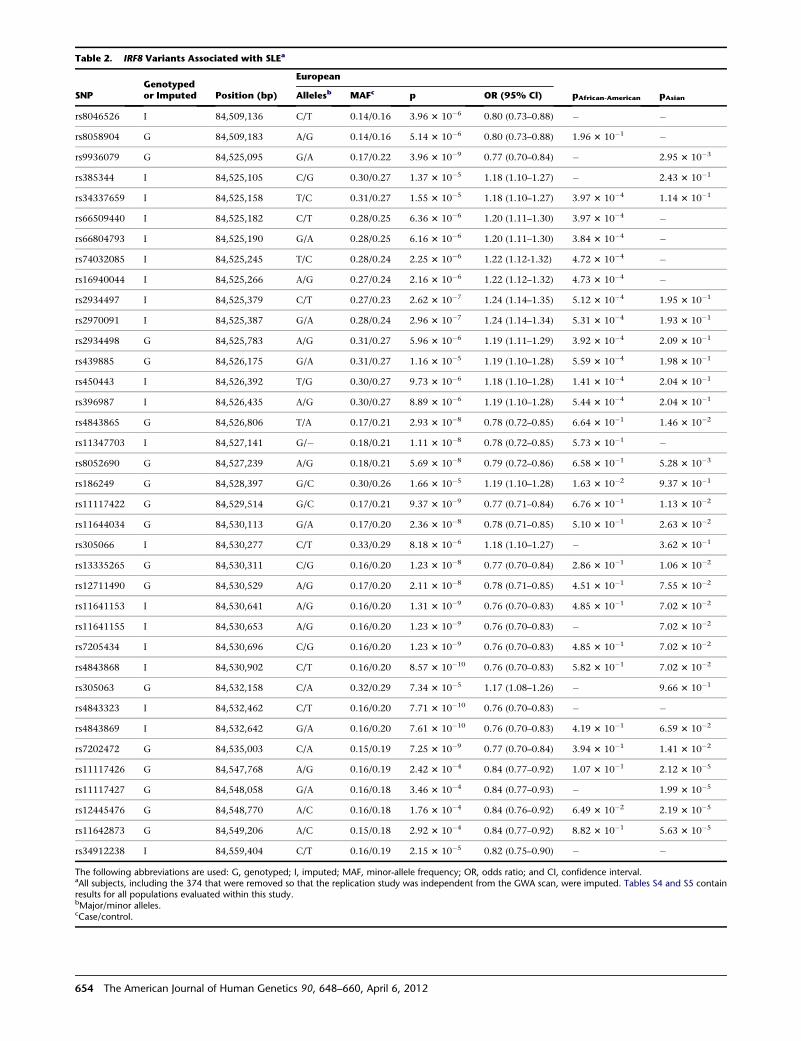
Table 2. IRF8 Variants Associated with SLEa
SNPGenotypedor Imputed Position (bp)
European
pAfrican-American pAsianAllelesb MAFc p OR (95% Cl)
rs8046526 I 84,509,136 C/T 0.14/0.16 3.96 3 10�6 0.80 (0.73–0.88) � �
rs8058904 G 84,509,183 A/G 0.14/0.16 5.14 3 10�6 0.80 (0.73–0.88) 1.96 3 10�1 �
rs9936079 G 84,525,095 G/A 0.17/0.22 3.96 3 10�9 0.77 (0.70–0.84) � 2.95 3 10�3
rs385344 I 84,525,105 C/G 0.30/0.27 1.37 3 10�5 1.18 (1.10–1.27) � 2.43 3 10�1
rs34337659 I 84,525,158 T/C 0.31/0.27 1.55 3 10�5 1.18 (1.10–1.27) 3.97 3 10�4 1.14 3 10�1
rs66509440 I 84,525,182 C/T 0.28/0.25 6.36 3 10�6 1.20 (1.11–1.30) 3.97 3 10�4 �
rs66804793 I 84,525,190 G/A 0.28/0.25 6.16 3 10�6 1.20 (1.11–1.30) 3.84 3 10�4 �
rs74032085 I 84,525,245 T/C 0.28/0.24 2.25 3 10�6 1.22 (1.12-1.32) 4.72 3 10�4 �
rs16940044 I 84,525,266 A/G 0.27/0.24 2.16 3 10�6 1.22 (1.12–1.32) 4.73 3 10�4 �
rs2934497 I 84,525,379 C/T 0.27/0.23 2.62 3 10�7 1.24 (1.14–1.35) 5.12 3 10�4 1.95 3 10�1
rs2970091 I 84,525,387 G/A 0.28/0.24 2.96 3 10�7 1.24 (1.14–1.34) 5.31 3 10�4 1.93 3 10�1
rs2934498 G 84,525,783 A/G 0.31/0.27 5.96 3 10�6 1.19 (1.11–1.29) 3.92 3 10�4 2.09 3 10�1
rs439885 G 84,526,175 G/A 0.31/0.27 1.16 3 10�5 1.19 (1.10–1.28) 5.59 3 10�4 1.98 3 10�1
rs450443 I 84,526,392 T/G 0.30/0.27 9.73 3 10�6 1.18 (1.10–1.28) 1.41 3 10�4 2.04 3 10�1
rs396987 I 84,526,435 A/G 0.30/0.27 8.89 3 10�6 1.19 (1.10–1.28) 5.44 3 10�4 2.04 3 10�1
rs4843865 G 84,526,806 T/A 0.17/0.21 2.93 3 10�8 0.78 (0.72–0.85) 6.64 3 10�1 1.46 3 10�2
rs11347703 I 84,527,141 G/� 0.18/0.21 1.11 3 10�8 0.78 (0.72–0.85) 5.73 3 10�1 �
rs8052690 G 84,527,239 A/G 0.18/0.21 5.69 3 10�8 0.79 (0.72–0.86) 6.58 3 10�1 5.28 3 10�3
rs186249 G 84,528,397 G/C 0.30/0.26 1.66 3 10�5 1.19 (1.10–1.28) 1.63 3 10�2 9.37 3 10�1
rs11117422 G 84,529,514 G/C 0.17/0.21 9.37 3 10�9 0.77 (0.71–0.84) 6.76 3 10�1 1.13 3 10�2
rs11644034 G 84,530,113 G/A 0.17/0.20 2.36 3 10�8 0.78 (0.71–0.85) 5.10 3 10�1 2.63 3 10�2
rs305066 I 84,530,277 C/T 0.33/0.29 8.18 3 10�6 1.18 (1.10–1.27) � 3.62 3 10�1
rs13335265 G 84,530,311 C/G 0.16/0.20 1.23 3 10�8 0.77 (0.70–0.84) 2.86 3 10�1 1.06 3 10�2
rs12711490 G 84,530,529 A/G 0.17/0.20 2.11 3 10�8 0.78 (0.71–0.85) 4.51 3 10�1 7.55 3 10�2
rs11641153 I 84,530,641 A/G 0.16/0.20 1.31 3 10�9 0.76 (0.70–0.83) 4.85 3 10�1 7.02 3 10�2
rs11641155 I 84,530,653 A/G 0.16/0.20 1.23 3 10�9 0.76 (0.70–0.83) � 7.02 3 10�2
rs7205434 I 84,530,696 C/G 0.16/0.20 1.23 3 10�9 0.76 (0.70–0.83) 4.85 3 10�1 7.02 3 10�2
rs4843868 I 84,530,902 C/T 0.16/0.20 8.57 3 10�10 0.76 (0.70–0.83) 5.82 3 10�1 7.02 3 10�2
rs305063 G 84,532,158 C/A 0.32/0.29 7.34 3 10�5 1.17 (1.08–1.26) � 9.66 3 10�1
rs4843323 I 84,532,462 C/T 0.16/0.20 7.71 3 10�10 0.76 (0.70–0.83) � �
rs4843869 I 84,532,642 G/A 0.16/0.20 7.61 3 10�10 0.76 (0.70–0.83) 4.19 3 10�1 6.59 3 10�2
rs7202472 G 84,535,003 C/A 0.15/0.19 7.25 3 10�9 0.77 (0.70–0.84) 3.94 3 10�1 1.41 3 10�2
rs11117426 G 84,547,768 A/G 0.16/0.19 2.42 3 10�4 0.84 (0.77–0.92) 1.07 3 10�1 2.12 3 10�5
rs11117427 G 84,548,058 G/A 0.16/0.18 3.46 3 10�4 0.84 (0.77–0.93) � 1.99 3 10�5
rs12445476 G 84,548,770 A/C 0.16/0.18 1.76 3 10�4 0.84 (0.76–0.92) 6.49 3 10�2 2.19 3 10�5
rs11642873 G 84,549,206 A/C 0.15/0.18 2.92 3 10�4 0.84 (0.77–0.92) 8.82 3 10�1 5.63 3 10�5
rs34912238 I 84,559,404 C/T 0.16/0.19 2.15 3 10�5 0.82 (0.75–0.90) � �
The following abbreviations are used: G, genotyped; I, imputed; MAF, minor-allele frequency; OR, odds ratio; and CI, confidence interval.aAll subjects, including the 374 that were removed so that the replication study was independent from the GWA scan, were imputed. Tables S4 and S5 containresults for all populations evaluated within this study.bMajor/minor alleles.cCase/control.
654 The American Journal of Human Genetics 90, 648–660, April 6, 2012
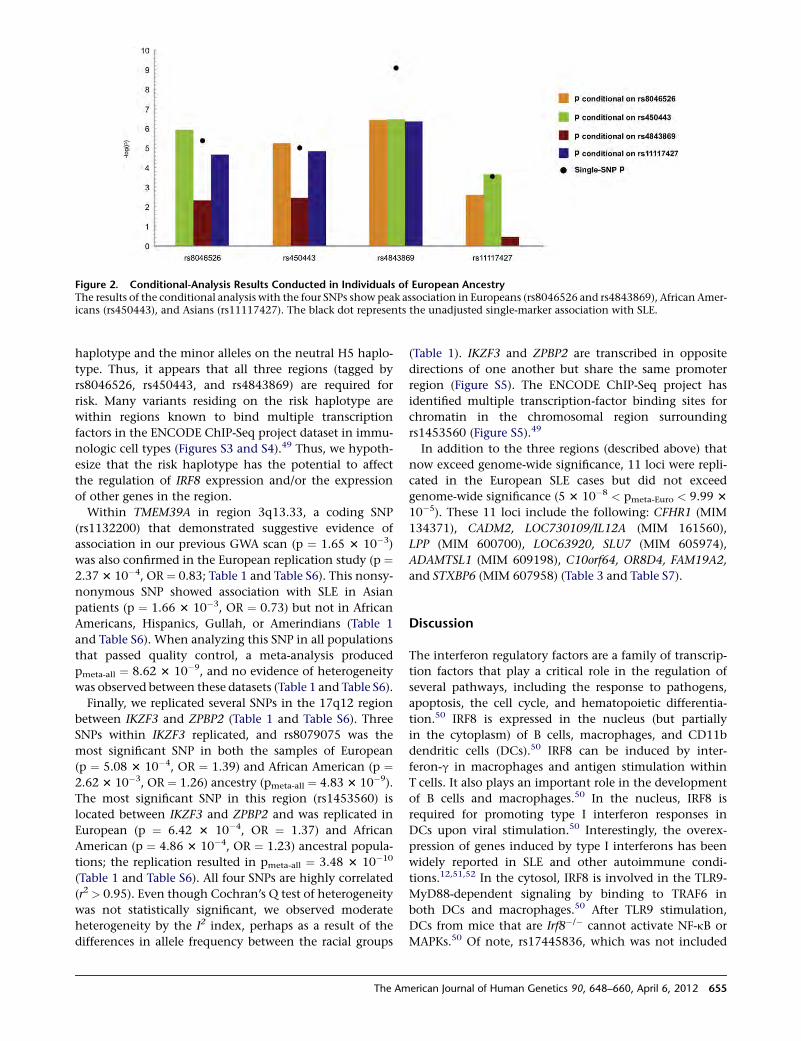
haplotype and the minor alleles on the neutral H5 haplo-
type. Thus, it appears that all three regions (tagged by
rs8046526, rs450443, and rs4843869) are required for
risk. Many variants residing on the risk haplotype are
within regions known to bind multiple transcription
factors in the ENCODE ChIP-Seq project dataset in immu-
nologic cell types (Figures S3 and S4).49 Thus, we hypoth-
esize that the risk haplotype has the potential to affect
the regulation of IRF8 expression and/or the expression
of other genes in the region.
Within TMEM39A in region 3q13.33, a coding SNP
(rs1132200) that demonstrated suggestive evidence of
association in our previous GWA scan (p ¼ 1.65 3 10�3)
was also confirmed in the European replication study (p ¼2.37 3 10�4, OR ¼ 0.83; Table 1 and Table S6). This nonsy-
nonymous SNP showed association with SLE in Asian
patients (p ¼ 1.66 3 10�3, OR ¼ 0.73) but not in African
Americans, Hispanics, Gullah, or Amerindians (Table 1
and Table S6). When analyzing this SNP in all populations
that passed quality control, a meta-analysis produced
pmeta-all ¼ 8.62 3 10�9, and no evidence of heterogeneity
was observed between these datasets (Table 1 and Table S6).
Finally, we replicated several SNPs in the 17q12 region
between IKZF3 and ZPBP2 (Table 1 and Table S6). Three
SNPs within IKZF3 replicated, and rs8079075 was the
most significant SNP in both the samples of European
(p ¼ 5.08 3 10�4, OR ¼ 1.39) and African American (p ¼2.62 3 10�3, OR ¼ 1.26) ancestry (pmeta-all ¼ 4.83 3 10�9).
The most significant SNP in this region (rs1453560) is
located between IKZF3 and ZPBP2 and was replicated in
European (p ¼ 6.42 3 10�4, OR ¼ 1.37) and African
American (p ¼ 4.86 3 10�4, OR ¼ 1.23) ancestral popula-
tions; the replication resulted in pmeta-all ¼ 3.48 3 10�10
(Table 1 and Table S6). All four SNPs are highly correlated
(r2 > 0.95). Even though Cochran’s Q test of heterogeneity
was not statistically significant, we observed moderate
heterogeneity by the I2 index, perhaps as a result of the
differences in allele frequency between the racial groups
(Table 1). IKZF3 and ZPBP2 are transcribed in opposite
directions of one another but share the same promoter
region (Figure S5). The ENCODE ChIP-Seq project has
identified multiple transcription-factor binding sites for
chromatin in the chromosomal region surrounding
rs1453560 (Figure S5).49
In addition to the three regions (described above) that
now exceed genome-wide significance, 11 loci were repli-
cated in the European SLE cases but did not exceed
genome-wide significance (5 3 10�8 < pmeta-Euro < 9.99 3
10�5). These 11 loci include the following: CFHR1 (MIM
134371), CADM2, LOC730109/IL12A (MIM 161560),
LPP (MIM 600700), LOC63920, SLU7 (MIM 605974),
ADAMTSL1 (MIM 609198), C10orf64, OR8D4, FAM19A2,
and STXBP6 (MIM 607958) (Table 3 and Table S7).
Discussion
The interferon regulatory factors are a family of transcrip-
tion factors that play a critical role in the regulation of
several pathways, including the response to pathogens,
apoptosis, the cell cycle, and hematopoietic differentia-
tion.50 IRF8 is expressed in the nucleus (but partially
in the cytoplasm) of B cells, macrophages, and CD11b
dendritic cells (DCs).50 IRF8 can be induced by inter-
feron-g in macrophages and antigen stimulation within
T cells. It also plays an important role in the development
of B cells and macrophages.50 In the nucleus, IRF8 is
required for promoting type I interferon responses in
DCs upon viral stimulation.50 Interestingly, the overex-
pression of genes induced by type I interferons has been
widely reported in SLE and other autoimmune condi-
tions.12,51,52 In the cytosol, IRF8 is involved in the TLR9-
MyD88-dependent signaling by binding to TRAF6 in
both DCs and macrophages.50 After TLR9 stimulation,
DCs from mice that are Irf8�/� cannot activate NF-kB or
MAPKs.50 Of note, rs17445836, which was not included
Figure 2. Conditional-Analysis Results Conducted in Individuals of European AncestryThe results of the conditional analysis with the four SNPs show peak association in Europeans (rs8046526 and rs4843869), African Amer-icans (rs450443), and Asians (rs11117427). The black dot represents the unadjusted single-marker association with SLE.
The American Journal of Human Genetics 90, 648–660, April 6, 2012 655
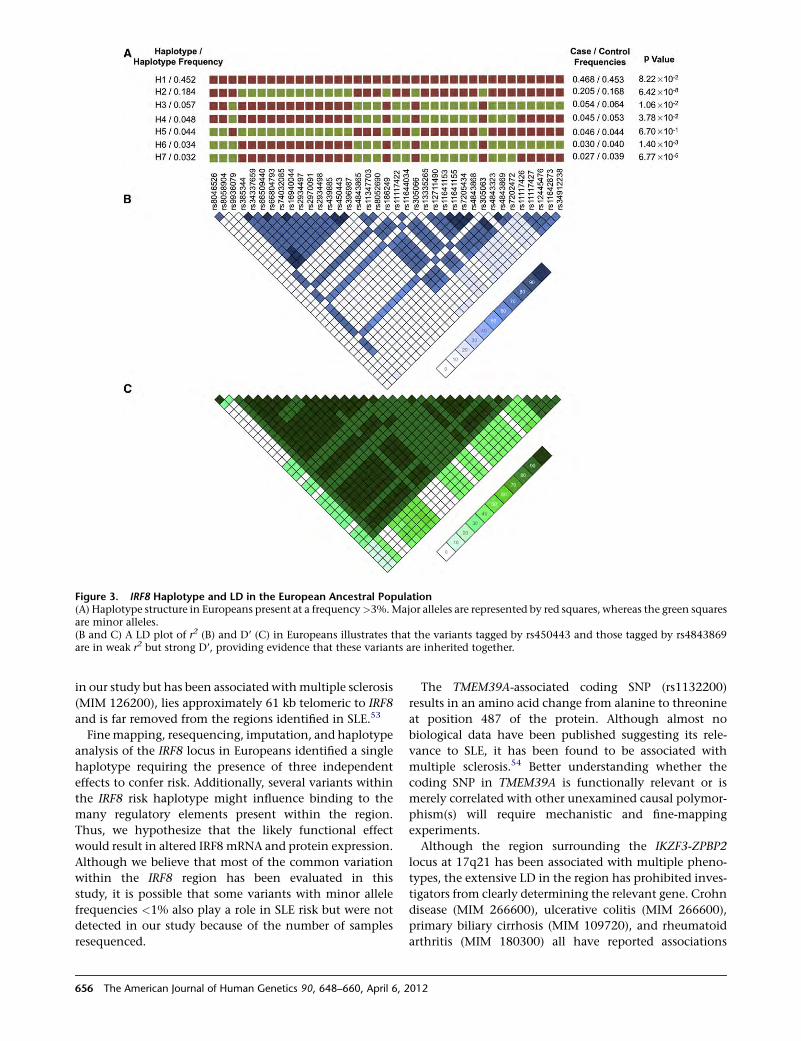
in our study but has been associated withmultiple sclerosis
(MIM 126200), lies approximately 61 kb telomeric to IRF8
and is far removed from the regions identified in SLE.53
Finemapping, resequencing, imputation, and haplotype
analysis of the IRF8 locus in Europeans identified a single
haplotype requiring the presence of three independent
effects to confer risk. Additionally, several variants within
the IRF8 risk haplotype might influence binding to the
many regulatory elements present within the region.
Thus, we hypothesize that the likely functional effect
would result in altered IRF8 mRNA and protein expression.
Although we believe that most of the common variation
within the IRF8 region has been evaluated in this
study, it is possible that some variants with minor allele
frequencies <1% also play a role in SLE risk but were not
detected in our study because of the number of samples
resequenced.
The TMEM39A-associated coding SNP (rs1132200)
results in an amino acid change from alanine to threonine
at position 487 of the protein. Although almost no
biological data have been published suggesting its rele-
vance to SLE, it has been found to be associated with
multiple sclerosis.54 Better understanding whether the
coding SNP in TMEM39A is functionally relevant or is
merely correlated with other unexamined causal polymor-
phism(s) will require mechanistic and fine-mapping
experiments.
Although the region surrounding the IKZF3-ZPBP2
locus at 17q21 has been associated with multiple pheno-
types, the extensive LD in the region has prohibited inves-
tigators from clearly determining the relevant gene. Crohn
disease (MIM 266600), ulcerative colitis (MIM 266600),
primary biliary cirrhosis (MIM 109720), and rheumatoid
arthritis (MIM 180300) all have reported associations
Figure 3. IRF8 Haplotype and LD in the European Ancestral Population(A) Haplotype structure in Europeans present at a frequency>3%.Major alleles are represented by red squares, whereas the green squaresare minor alleles.(B and C) A LD plot of r2 (B) and D’ (C) in Europeans illustrates that the variants tagged by rs450443 and those tagged by rs4843869are in weak r2 but strong D’, providing evidence that these variants are inherited together.
656 The American Journal of Human Genetics 90, 648–660, April 6, 2012
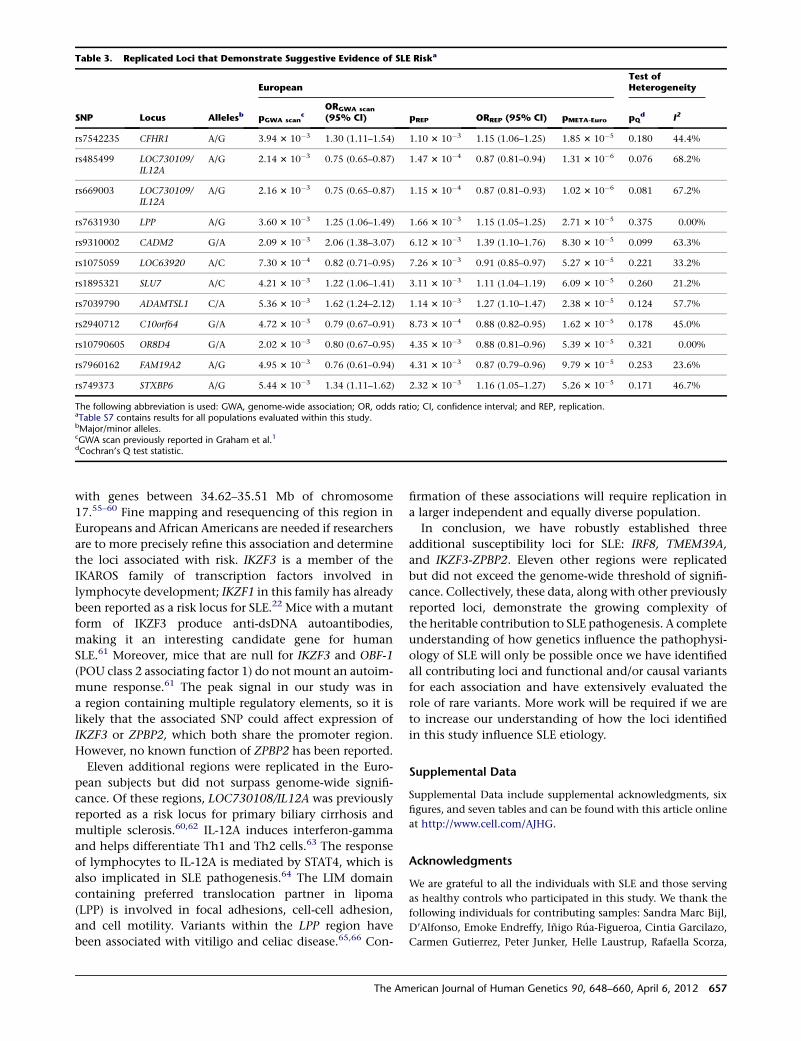
with genes between 34.62–35.51 Mb of chromosome
17.55–60 Fine mapping and resequencing of this region in
Europeans and African Americans are needed if researchers
are to more precisely refine this association and determine
the loci associated with risk. IKZF3 is a member of the
IKAROS family of transcription factors involved in
lymphocyte development; IKZF1 in this family has already
been reported as a risk locus for SLE.22 Mice with a mutant
form of IKZF3 produce anti-dsDNA autoantibodies,
making it an interesting candidate gene for human
SLE.61 Moreover, mice that are null for IKZF3 and OBF-1
(POU class 2 associating factor 1) do not mount an autoim-
mune response.61 The peak signal in our study was in
a region containing multiple regulatory elements, so it is
likely that the associated SNP could affect expression of
IKZF3 or ZPBP2, which both share the promoter region.
However, no known function of ZPBP2 has been reported.
Eleven additional regions were replicated in the Euro-
pean subjects but did not surpass genome-wide signifi-
cance. Of these regions, LOC730108/IL12A was previously
reported as a risk locus for primary biliary cirrhosis and
multiple sclerosis.60,62 IL-12A induces interferon-gamma
and helps differentiate Th1 and Th2 cells.63 The response
of lymphocytes to IL-12A is mediated by STAT4, which is
also implicated in SLE pathogenesis.64 The LIM domain
containing preferred translocation partner in lipoma
(LPP) is involved in focal adhesions, cell-cell adhesion,
and cell motility. Variants within the LPP region have
been associated with vitiligo and celiac disease.65,66 Con-
firmation of these associations will require replication in
a larger independent and equally diverse population.
In conclusion, we have robustly established three
additional susceptibility loci for SLE: IRF8, TMEM39A,
and IKZF3-ZPBP2. Eleven other regions were replicated
but did not exceed the genome-wide threshold of signifi-
cance. Collectively, these data, along with other previously
reported loci, demonstrate the growing complexity of
the heritable contribution to SLE pathogenesis. A complete
understanding of how genetics influence the pathophysi-
ology of SLE will only be possible once we have identified
all contributing loci and functional and/or causal variants
for each association and have extensively evaluated the
role of rare variants. More work will be required if we are
to increase our understanding of how the loci identified
in this study influence SLE etiology.
Supplemental Data
Supplemental Data include supplemental acknowledgments, six
figures, and seven tables and can be found with this article online
at http://www.cell.com/AJHG.
Acknowledgments
We are grateful to all the individuals with SLE and those serving
as healthy controls who participated in this study. We thank the
following individuals for contributing samples: Sandra Marc Bijl,
D’Alfonso, Emoke Endreffy, Inigo Rua-Figueroa, Cintia Garcilazo,
Carmen Gutierrez, Peter Junker, Helle Laustrup, Rafaella Scorza,
Table 3. Replicated Loci that Demonstrate Suggestive Evidence of SLE Riska
SNP Locus Allelesb
EuropeanTest ofHeterogeneity
pGWA scanc
ORGWA scan
(95% Cl) pREP ORREP (95% Cl) pMETA-Euro pQd I2
rs7542235 CFHR1 A/G 3.94 3 10�3 1.30 (1.11–1.54) 1.10 3 10�3 1.15 (1.06–1.25) 1.85 3 10�5 0.180 44.4%
rs485499 LOC730109/IL12A
A/G 2.14 3 10�3 0.75 (0.65–0.87) 1.47 3 10�4 0.87 (0.81–0.94) 1.31 3 10�6 0.076 68.2%
rs669003 LOC730109/IL12A
A/G 2.16 3 10�3 0.75 (0.65–0.87) 1.15 3 10�4 0.87 (0.81–0.93) 1.02 3 10�6 0.081 67.2%
rs7631930 LPP A/G 3.60 3 10�3 1.25 (1.06–1.49) 1.66 3 10�3 1.15 (1.05–1.25) 2.71 3 10�5 0.375 0.00%
rs9310002 CADM2 G/A 2.09 3 10�3 2.06 (1.38–3.07) 6.12 3 10�3 1.39 (1.10–1.76) 8.30 3 10�5 0.099 63.3%
rs1075059 LOC63920 A/C 7.30 3 10�4 0.82 (0.71–0.95) 7.26 3 10�3 0.91 (0.85–0.97) 5.27 3 10�5 0.221 33.2%
rs1895321 SLU7 A/C 4.21 3 10�3 1.22 (1.06–1.41) 3.11 3 10�3 1.11 (1.04–1.19) 6.09 3 10�5 0.260 21.2%
rs7039790 ADAMTSL1 C/A 5.36 3 10�3 1.62 (1.24–2.12) 1.14 3 10�3 1.27 (1.10–1.47) 2.38 3 10�5 0.124 57.7%
rs2940712 C10orf64 G/A 4.72 3 10�3 0.79 (0.67–0.91) 8.73 3 10�4 0.88 (0.82–0.95) 1.62 3 10�5 0.178 45.0%
rs10790605 OR8D4 G/A 2.02 3 10�3 0.80 (0.67–0.95) 4.35 3 10�3 0.88 (0.81–0.96) 5.39 3 10�5 0.321 0.00%
rs7960162 FAM19A2 A/G 4.95 3 10�3 0.76 (0.61–0.94) 4.31 3 10�3 0.87 (0.79–0.96) 9.79 3 10�5 0.253 23.6%
rs749373 STXBP6 A/G 5.44 3 10�3 1.34 (1.11–1.62) 2.32 3 10�3 1.16 (1.05–1.27) 5.26 3 10�5 0.171 46.7%
The following abbreviation is used: GWA, genome-wide association; OR, odds ratio; CI, confidence interval; and REP, replication.aTable S7 contains results for all populations evaluated within this study.bMajor/minor alleles.cGWA scan previously reported in Graham et al.1dCochran’s Q test statistic.
The American Journal of Human Genetics 90, 648–660, April 6, 2012 657

BertaMartins da Silva, Ana Suarez, and Carlos Vasconcelos. For the
GENLES collaboration, we thank Eduardo Acevedo, Mario Cardiel,
Ignacio Garcıa de la Torre, Mabel Busajm, Cecilia Castel, Marco
Maradiaga, Jose F. Moctezuma, and Jorge Musuruana. For the
Asociacion Andaluza de Enfermedades Autoimmunes collabora-
tion, we thank Juan Jimenez-Alonso, Norberto Ortego-Centeno,
Enrique de Ramon, and Julio Sanchez-Roman. We would like to
thank Summer Frank and Mei Li Zhu for their assistance in geno-
typing, quality-control analyses, and clinical data management.
We would also like to thank Emily Cole for her assistance in
preparing figures. Grant support information is provided in the
Supplemental Acknowledgments available online.
Received: October 26, 2011
Revised: February 22, 2012
Accepted: February 22, 2012
Published online: March 29, 2012
Web Resources
The URLs for data presented herein are as follows:
dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/index.html
Encyclopedia of DNA Elements (ENCODE), http://genome.ucsc.
edu/ENCODE/
HUGO Gene Nomenclature Committee, http://www.genenames.
org/
LocusZoom, http://csg.sph.umich.edu/locuszoom/
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
VCFtools, http://www.vcftools.sourceforge.net
References
1. Petri, M. (2002). Epidemiology of systemic lupus erythemato-
sus. Best Pract. Res. Clin. Rheumatol. 16, 847–858.
2. Moser, K.L., Kelly, J.A., Lessard, C.J., and Harley, J.B. (2009).
Recent insights into the genetic basis of systemic lupus eryth-
ematosus. Genes Immun. 10, 373–379.
3. Alarcon-Segovia, D., Alarcon-Riquelme, M.E., Cardiel, M.H.,
Caeiro, F., Massardo, L., Villa, A.R., and Pons-Estel, B.A.; Grupo
Latinoamericano de Estudio del Lupus Eritematoso (GLADEL).
(2005). Familial aggregation of systemic lupus erythematosus,
rheumatoid arthritis, and other autoimmune diseases in 1,177
lupus patients from the GLADEL cohort. Arthritis Rheum. 52,
1138–1147.
4. Anaya, J.M., Tobon, G.J., Vega, P., and Castiblanco, J. (2006).
Autoimmune disease aggregation in families with primary
Sjogren’s syndrome. J. Rheumatol. 33, 2227–2234.
5. Arora-Singh, R.K., Assassi, S., del Junco, D.J., Arnett, F.C., Perry,
M., Irfan, U., Sharif, R., Mattar, T., and Mayes, M.D. (2010).
Autoimmune diseases and autoantibodies in the first degree
relatives of patients with systemic sclerosis. J. Autoimmun.
35, 52–57.
6. Sestak, A.L., Shaver, T.S., Moser, K.L., Neas, B.R., and Harley,
J.B. (1999). Familial aggregation of lupus and autoimmunity
in an unusual multiplex pedigree. J. Rheumatol. 26, 1495–
1499.
7. Deng, Y., and Tsao, B.P. (2010). Genetic susceptibility to
systemic lupus erythematosus in the genomic era. Nat. Rev.
Rheumatol. 6, 683–692.
8. Reinertsen, J.L., Klippel, J.H., Johnson, A.H., Steinberg, A.D.,
Decker, J.L., and Mann, D.L. (1978). B-lymphocyte alloanti-
gens associated with systemic lupus erythematosus. N. Engl.
J. Med. 299, 515–518.
9. Nies, K.M., Brown, J.C., Dubois, E.L., Quismorio, F.P., Friou,
G.J., and Terasaki, P.I. (1974). Histocompatibility (HL-A) anti-
gens and lymphocytotoxic antibodies in systemic lupus
erythematosus (SLE). Arthritis Rheum. 17, 397–402.
10. Graham, R.R., Ortmann, W.A., Langefeld, C.D., Jawaheer, D.,
Selby, S.A., Rodine, P.R., Baechler, E.C., Rohlf, K.E., Shark,
K.B., Espe, K.J., et al. (2002). Visualizing human leukocyte
antigen class II risk haplotypes in human systemic lupus
erythematosus. Am. J. Hum. Genet. 71, 543–553.
11. McCulloch, D.K., Klaff, L.J., Kahn, S.E., Schoenfeld, S.L.,
Greenbaum, C.J., Mauseth, R.S., Benson, E.A., Nepom, G.T.,
Shewey, L., and Palmer, J.P. (1990). Nonprogression of subclin-
ical beta-cell dysfunction among first-degree relatives of
IDDM patients. 5-yr follow-up of the Seattle Family Study.
Diabetes 39, 549–556.
12. Baechler, E.C., Batliwalla, F.M., Karypis, G., Gaffney, P.M., Ort-
mann, W.A., Espe, K.J., Shark, K.B., Grande, W.J., Hughes,
K.M., Kapur, V., et al. (2003). Interferon-inducible gene
expression signature in peripheral blood cells of patients
with severe lupus. Proc. Natl. Acad. Sci. USA 100, 2610–2615.
13. Bennett, L., Palucka, A.K., Arce, E., Cantrell, V., Borvak, J., Ban-
chereau, J., and Pascual, V. (2003). Interferon and granulopoi-
esis signatures in systemic lupus erythematosus blood. J. Exp.
Med. 197, 711–723.
14. Kirou, K.A., Lee, C., George, S., Louca, K., Papagiannis, I.G.,
Peterson, M.G., Ly, N., Woodward, R.N., Fry, K.E., Lau, A.Y.,
et al. (2004). Coordinate overexpression of interferon-alpha-
induced genes in systemic lupus erythematosus. Arthritis
Rheum. 50, 3958–3967.
15. Sigurdsson, S., Nordmark, G., Goring, H.H., Lindroos, K.,
Wiman, A.C., Sturfelt, G., Jonsen, A., Rantapaa-Dahlqvist, S.,
Moller, B., Kere, J., et al. (2005). Polymorphisms in the tyro-
sine kinase 2 and interferon regulatory factor 5 genes are
associated with systemic lupus erythematosus. Am. J. Hum.
Genet. 76, 528–537.
16. Hom, G., Graham, R.R., Modrek, B., Taylor, K.E., Ortmann,
W., Garnier, S., Lee, A.T., Chung, S.A., Ferreira, R.C., Pant,
P.V., et al. (2008). Association of systemic lupus erythematosus
with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 358,
900–909.
17. Harley, J.B., Alarcon-Riquelme, M.E., Criswell, L.A., Jacob,
C.O., Kimberly, R.P., Moser, K.L., Tsao, B.P., Vyse, T.J., Lange-
feld, C.D., Nath, S.K., et al; International Consortium for
Systemic Lupus Erythematosus Genetics (SLEGEN). (2008).
Genome-wide association scan in womenwith systemic lupus
erythematosus identifies susceptibility variants in ITGAM,
PXK, KIAA1542 and other loci. Nat. Genet. 40, 204–210.
18. Yang, W., Shen, N., Ye, D.Q., Liu, Q., Zhang, Y., Qian, X.X.,
Hirankarn, N., Ying, D., Pan, H.F., Mok, C.C., et al; Asian
Lupus Genetics Consortium. (2010). Genome-wide associa-
tion study in Asian populations identifies variants in ETS1
and WDFY4 associated with systemic lupus erythematosus.
PLoS Genet. 6, e1000841.
19. Graham, R.R., Cotsapas, C., Davies, L., Hackett, R., Lessard,
C.J., Leon, J.M., Burtt, N.P., Guiducci, C., Parkin, M., Gates,
C., et al. (2008). Genetic variants near TNFAIP3 on 6q23 are
associated with systemic lupus erythematosus. Nat. Genet.
40, 1059–1061.
658 The American Journal of Human Genetics 90, 648–660, April 6, 2012

20. Kozyrev, S.V., Abelson, A.K., Wojcik, J., Zaghlool, A., Linga
Reddy, M.V., Sanchez, E., Gunnarsson, I., Svenungsson, E.,
Sturfelt, G., Jonsen, A., et al. (2008). Functional variants in
the B-cell gene BANK1 are associated with systemic lupus
erythematosus. Nat. Genet. 40, 211–216.
21. Han, J.W., Zheng, H.F., Cui, Y., Sun, L.D., Ye, D.Q., Hu, Z., Xu,
J.H., Cai, Z.M., Huang, W., Zhao, G.P., et al. (2009). Genome-
wide association study in a Chinese Han population identifies
nine new susceptibility loci for systemic lupus erythematosus.
Nat. Genet. 41, 1234–1237.
22. Gateva, V., Sandling, J.K., Hom, G., Taylor, K.E., Chung, S.A.,
Sun, X., Ortmann, W., Kosoy, R., Ferreira, R.C., Nordmark,
G., et al. (2009). A large-scale replication study identifies
TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for
systemic lupus erythematosus. Nat. Genet. 41, 1228–1233.
23. Lessard, C.J., Adrianto, I., Kelly, J.A., Kaufman, K.M., Grun-
dahl, K.M., Adler, A., Williams, A.H., Gallant, C.J., Anaya,
J.M., Bae, S.C., et al; Marta E. Alarcon-Riquelme on behalf of
the BIOLUPUS and GENLES Networks. (2011). Identification
of a systemic lupus erythematosus susceptibility locus at
11p13 between PDHX and CD44 in a multiethnic study.
Am. J. Hum. Genet. 88, 83–91.
24. Namjou, B., Kothari, P.H., Kelly, J.A., Glenn, S.B., Ojwang,
J.O., Adler, A., Alarcon-Riquelme, M.E., Gallant, C.J., Boackle,
S.A., Criswell, L.A., et al. (2011). Evaluation of the TREX1 gene
in a large multi-ancestral lupus cohort. Genes Immun. 12,
270–279.
25. Adrianto, I., Wen, F., Templeton, A., Wiley, G., King, J.B.,
Lessard, C.J., Bates, J.S., Hu, Y., Kelly, J.A., Kaufman, K.M.,
et al; BIOLUPUS and GENLES Networks. (2011). Association
of a functional variant downstream of TNFAIP3 with systemic
lupus erythematosus. Nat. Genet. 43, 253–258.
26. Tan,W., Sunahori, K., Zhao, J., Deng, Y., Kaufman, K.M., Kelly,
J.A., Langefeld, C.D., Williams, A.H., Comeau, M.E., Ziegler,
J.T., et al; BIOLUPUS Network; GENLES Network. (2011).
Association of PPP2CA polymorphisms with systemic lupus
erythematosus susceptibility in multiple ethnic groups.
Arthritis Rheum. 63, 2755–2763.
27. Zhao, J., Wu, H., Khosravi, M., Cui, H., Qian, X., Kelly, J.A.,
Kaufman, K.M., Langefeld, C.D., Williams, A.H., Comeau,
M.E., et al; BIOLUPUS Network; GENLES Network. (2011).
Association of genetic variants in complement factor H and
factor H-related genes with systemic lupus erythematosus
susceptibility. PLoS Genet. 7, e1002079.
28. Sanchez, E., Nadig, A., Richardson, B.C., Freedman, B.I., Kauf-
man, K.M., Kelly, J.A., Niewold, T.B., Kamen, D.L., Gilkeson,
G.S., Ziegler, J.T., et al; BIOLUPUS andGENLES. (2011). Pheno-
typic associations of genetic susceptibility loci in systemic
lupus erythematosus. Ann. Rheum. Dis. 70, 1752–1757.
29. Hochberg, M.C. (1997). Updating the American College of
Rheumatology revised criteria for the classification of systemic
lupus erythematosus. Arthritis Rheum. 40, 1725.
30. Barrett, J.C., Fry, B., Maller, J., and Daly, M.J. (2005). Haplo-
view: Analysis and visualization of LD and haplotype maps.
Bioinformatics 21, 263–265.
31. Price, A.L., Patterson, N.J., Plenge, R.M., Weinblatt, M.E.,
Shadick, N.A., and Reich, D. (2006). Principal components
analysis corrects for stratification in genome-wide association
studies. Nat. Genet. 38, 904–909.
32. McKeigue, P.M., Carpenter, J.R., Parra, E.J., and Shriver, M.D.
(2000). Estimation of admixture and detection of linkage in
admixed populations by a Bayesian approach: Application
to African-American populations. Ann. Hum. Genet. 64,
171–186.
33. Hoggart, C.J., Parra, E.J., Shriver, M.D., Bonilla, C., Kittles,
R.A., Clayton, D.G., and McKeigue, P.M. (2003). Control of
confounding of genetic associations in stratified populations.
Am. J. Hum. Genet. 72, 1492–1504.
34. Hoggart, C.J., Shriver, M.D., Kittles, R.A., Clayton, D.G., and
McKeigue, P.M. (2004). Design and analysis of admixture
mapping studies. Am. J. Hum. Genet. 74, 965–978.
35. Smith, M.W., Patterson, N., Lautenberger, J.A., Truelove, A.L.,
McDonald, G.J., Waliszewska, A., Kessing, B.D., Malasky, M.J.,
Scafe, C., Le, E., et al. (2004). A high-density admixture map
for disease gene discovery in african americans. Am. J. Hum.
Genet. 74, 1001–1013.
36. Halder, I., Shriver, M., Thomas, M., Fernandez, J.R., and
Frudakis, T. (2008). A panel of ancestry informative markers
for estimating individual biogeographical ancestry and
admixture from four continents: Utility and applications.
Hum. Mutat. 29, 648–658.
37. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira,
M.A., Bender, D., Maller, J., Sklar, P., de Bakker, P.I., Daly,
M.J., and Sham, P.C. (2007). PLINK: A tool set for whole-
genome association and population-based linkage analyses.
Am. J. Hum. Genet. 81, 559–575.
38. Willer, C.J., Li, Y., and Abecasis, G.R. (2010). METAL: Fast
and efficient meta-analysis of genomewide association scans.
Bioinformatics 26, 2190–2191.
39. Cochran, W.G. (1954). The Combination of Estimates from
Different Experiments. Biometrics 10, 101–129.
40. Higgins, J.P., Thompson, S.G., Deeks, J.J., and Altman, D.G.
(2003). Measuring inconsistency in meta-analyses. BMJ 327,
557–560.
41. Li, H., and Durbin, R. (2009). Fast and accurate short read
alignment with Burrows-Wheeler transform. Bioinformatics
25, 1754–1760.
42. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis,
K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly,
M., and DePristo, M.A. (2010). The Genome Analysis Toolkit:
A MapReduce framework for analyzing next-generation DNA
sequencing data. Genome Res. 20, 1297–1303.
43. Browning, S.R., and Browning, B.L. (2007). Rapid and accurate
haplotype phasing and missing-data inference for whole-
genome association studies by use of localized haplotype clus-
tering. Am. J. Hum. Genet. 81, 1084–1097.
44. Robinson, J.T., Thorvaldsdottir, H., Winckler, W., Guttman,
M., Lander, E.S., Getz, G., and Mesirov, J.P. (2011). Integrative
genomics viewer. Nat. Biotechnol. 29, 24–26.
45. Frazer, K.A., Ballinger, D.G., Cox, D.R., Hinds, D.A., Stuve,
L.L., Gibbs, R.A., Belmont, J.W., Boudreau, A., Hardenbol, P.,
Leal, S.M., et al; International HapMap Consortium. (2007).
A second generation human haplotype map of over 3.1
million SNPs. Nature 449, 851–861.
46. Howie, B.N., Donnelly, P., and Marchini, J. (2009). A flexible
and accurate genotype imputationmethod for the next gener-
ation of genome-wide association studies. PLoS Genet. 5,
e1000529.
47. Via, M., Gignoux, C., and Burchard, E.G. (2010). The 1000
Genomes Project: New opportunities for research and social
challenges. Genome Med. 2, 3.
48. 1000 Genomes Project Consortium. (2010). A map of human
genome variation from population-scale sequencing. Nature
467, 1061–1073.
The American Journal of Human Genetics 90, 648–660, April 6, 2012 659

49. Birney, E., Stamatoyannopoulos, J.A., Dutta, A., Guigo, R.,
Gingeras, T.R., Margulies, E.H., Weng, Z., Snyder, M., Dermit-
zakis, E.T., Thurman, R.E., et al; ENCODE Project Consortium;
NISC Comparative Sequencing Program; Baylor College of
Medicine Human Genome Sequencing Center; Washington
University Genome Sequencing Center; Broad Institute;
Children’s Hospital Oakland Research Institute. (2007).
Identification and analysis of functional elements in 1% of
the human genome by the ENCODE pilot project. Nature
447, 799–816.
50. Tamura, T., Yanai, H., Savitsky, D., and Taniguchi, T. (2008).
The IRF family transcription factors in immunity and onco-
genesis. Annu. Rev. Immunol. 26, 535–584.
51. Baechler, E.C., Gregersen, P.K., and Behrens, T.W. (2004).
The emerging role of interferon in human systemic lupus
erythematosus. Curr. Opin. Immunol. 16, 801–807.
52. Baechler, E.C., Batliwalla, F.M., Reed, A.M., Peterson, E.J.,
Gaffney, P.M., Moser, K.L., Gregersen, P.K., and Behrens,
T.W. (2006). Gene expression profiling in human autoimmu-
nity. Immunol. Rev. 210, 120–137.
53. De Jager, P.L., Jia, X., Wang, J., de Bakker, P.I., Ottoboni, L., Ag-
garwal, N.T., Piccio, L., Raychaudhuri, S., Tran, D., Aubin, C.,
et al; International MS Genetics Consortium. (2009). Meta-
analysis of genome scans and replication identify CD6, IRF8
and TNFRSF1A as new multiple sclerosis susceptibility loci.
Nat. Genet. 41, 776–782.
54. International Multiple Sclerosis Genetics Consortium
(IMSGC). (2010). Comprehensive follow-up of the first
genome-wide association study of multiple sclerosis identifies
KIF21B and TMEM39A as susceptibility loci. Hum. Mol.
Genet. 19, 953–962.
55. Franke, A., McGovern, D.P., Barrett, J.C., Wang, K., Radford-
Smith, G.L., Ahmad, T., Lees, C.W., Balschun, T., Lee, J., Rob-
erts, R., et al. (2010). Genome-wide meta-analysis increases to
71 the number of confirmed Crohn’s disease susceptibility
loci. Nat. Genet. 42, 1118–1125.
56. Liu, X., Invernizzi, P., Lu, Y., Kosoy, R., Lu, Y., Bianchi, I.,
Podda, M., Xu, C., Xie, G., Macciardi, F., et al. (2010).
Genome-wide meta-analyses identify three loci associated
with primary biliary cirrhosis. Nat. Genet. 42, 658–660.
57. Hirschfield, G.M., Liu, X., Han, Y., Gorlov, I.P., Lu, Y., Xu, C.,
Lu, Y., Chen, W., Juran, B.D., Coltescu, C., et al. (2010).
Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated
with primary biliary cirrhosis. Nat. Genet. 42, 655–657.
58. Stahl, E.A., Raychaudhuri, S., Remmers, E.F., Xie, G., Eyre, S.,
Thomson, B.P., Li, Y., Kurreeman, F.A., Zhernakova, A., Hinks,
A., et al; BIRAC Consortium; YEAR Consortium. (2010).
Genome-wide association studymeta-analysis identifies seven
new rheumatoid arthritis risk loci. Nat. Genet. 42, 508–514.
59. Anderson, C.A., Boucher, G., Lees, C.W., Franke, A., D’Amato,
M., Taylor, K.D., Lee, J.C., Goyette, P., Imielinski, M., Latiano,
A., et al. (2011). Meta-analysis identifies 29 additional ulcera-
tive colitis risk loci, increasing the number of confirmed
associations to 47. Nat. Genet. 43, 246–252.
60. Hirschfield, G.M., Liu, X., Xu, C., Lu, Y., Xie, G., Lu, Y., Gu, X.,
Walker, E.J., Jing, K., Juran, B.D., et al. (2009). Primary biliary
cirrhosis associated with HLA, IL12A, and IL12RB2 variants.
N. Engl. J. Med. 360, 2544–2555.
61. Sun, J., Matthias, G., Mihatsch, M.J., Georgopoulos, K., and
Matthias, P. (2003). Lack of the transcriptional coactivator
OBF-1 prevents the development of systemic lupus erythema-
tosus-like phenotypes in Aiolos mutant mice. J. Immunol.
170, 1699–1706.
62. International Multiple Sclerosis Genetics Conssortium
(IMSGC). (2010). IL12A, MPHOSPH9/CDK2AP1 and RGS1
are novel multiple sclerosis susceptibility loci. Genes Immun.
11, 397–405.
63. Peluso, I., Pallone, F., and Monteleone, G. (2006). Inter-
leukin-12 and Th1 immune response in Crohn’s disease:
Pathogenetic relevance and therapeutic implication. World
J. Gastroenterol. 12, 5606–5610.
64. Remmers, E.F., Plenge, R.M., Lee, A.T., Graham, R.R., Hom, G.,
Behrens, T.W., de Bakker, P.I., Le, J.M., Lee, H.S., Batliwalla, F.,
et al. (2007). STAT4 and the risk of rheumatoid arthritis and
systemic lupus erythematosus. N. Engl. J. Med. 357, 977–986.
65. Jin, Y., Birlea, S.A., Fain, P.R., Gowan, K., Riccardi, S.L.,
Holland, P.J., Mailloux, C.M., Sufit, A.J., Hutton, S.M.,
Amadi-Myers, A., et al. (2010). Variant of TYR and autoimmu-
nity susceptibility loci in generalized vitiligo. N. Engl. J. Med.
362, 1686–1697.
66. Hunt, K.A., Zhernakova, A., Turner, G., Heap, G.A., Franke, L.,
Bruinenberg, M., Romanos, J., Dinesen, L.C., Ryan, A.W.,
Panesar, D., et al. (2008). Newly identified genetic risk variants
for celiac disease related to the immune response. Nat. Genet.
40, 395–402.
660 The American Journal of Human Genetics 90, 648–660, April 6, 2012

ARTICLE
Attenuated BMP1 Function Compromises Osteogenesis,Leading to Bone Fragility in Humans and Zebrafish
P.V. Asharani,1,10 Katharina Keupp,2,3,4,10 Oliver Semler,5 Wenshen Wang,1 Yun Li,2,3,4 Holger Thiele,6
Gokhan Yigit,2,3,4 Esther Pohl,2,3,4 Jutta Becker,3 Peter Frommolt,4,6 Carmen Sonntag,7,12
Janine Altmuller,6 Katharina Zimmermann,3 Daniel S. Greenspan,8 Nurten A. Akarsu,9
Christian Netzer,3 Eckhard Schonau,5 Radu Wirth,3 Matthias Hammerschmidt,2,4,7
Peter Nurnberg,2,4,6 Bernd Wollnik,2,3,4,11,* and Thomas J. Carney1,11,*
Bone morphogenetic protein 1 (BMP1) is an astacin metalloprotease with important cellular functions and diverse substrates, including
extracellular-matrix proteins and antagonists of some TGFb superfamily members. Combining whole-exome sequencing and filtering
for homozygous stretches of identified variants, we found a homozygous causative BMP1 mutation, c.34G>C, in a consanguineous
family affected by increased bone mineral density and multiple recurrent fractures. The mutation is located within the BMP1 signal
peptide and leads to impaired secretion and an alteration in posttranslationalmodification.We also characterize a zebrafish bonemutant
harboring lesions in bmp1a, demonstrating conservation of BMP1 function in osteogenesis across species. Genetic, biochemical, and
histological analyses of this mutant and a comparison to a second, similar locus reveal that Bmp1a is critically required for mature-
collagen generation, downstream of osteoblast maturation, in bone.We thus define themolecular and cellular bases of BMP1-dependent
osteogenesis and show the importance of this protein for bone formation and stability.
Introduction
Osteogenesis imperfecta (OI), also known as ‘‘brittle-bone
disease’’ is a rare genetic collagenopathy primarily charac-
terized by dramatically increased bone fragility causing
susceptibility to numerous fractures.1,2 Individuals often
show distinctive features including reduced bone mass,
short stature, blue sclerae, and/or dentinogenesis imper-
fecta. The severity of this disorder varies from profound
forms with intrauterine fractures and perinatal lethality to
milder phenotypic expression such as rare fractures or
even no fractures.3,4 Most OI cases are inherited in an auto-
somal-dominant manner and are caused by mutations in
COL1A1 (MIM 120150) and COL1A2 (MIM 120160);5,6
these two genes encode for the two a chains of collagen
type I, the predominant protein component of the bone
matrix. For a minority of OI individuals, an autosomal-
recessive inheritance is described as including underlying
mutations in CRTAP7 (MIM 605497), LEPRE18 (MIM
610339), SERPINH19 (MIM 600943), PPIB10 (MIM
123841), SP711 (MIM 606633), SERPINF112 (MIM 172860),
and FKBP1013 (MIM 607063). Mutations in COL1A1 or
COL1A2 result in primary structural or quantitative defects
of collagen I molecules, whereas genetic mutations causa-
tive for recessive forms mainly lead to defects in collagen I
biosynthesis. Defects in collagen I have been associated
with reduced bone mineralization and bone fragility not
only in individuals suffering from congenital OI but also
in elderly individuals who have developed osteoporosis.14
Type I collagen belongs to the fibril-forming collagens
and is composed of a triple helix consisting of two a1(I)
chains and one a2(I) chain. The a chains are synthesized
at the rough endoplasmic reticulum (ER). They are highly
post-translationally modified in the ER lumen and are
subsequently assembled to a triple helix. This premature
collagen helix, which contains globular appendages at
the amino (N-) and carboxyl (C-) ends, is then transported
through the trans-golgi network to the extracellular matrix
(ECM), where N- and C-proteinases catalyze proteolytic
cleavage of these propeptides. After this final processing
step, the released mature collagen I triple-helical monomer
can be assembled into highly ordered collagen fibrils.15,16
Bonemorphogenetic protein 1 (BMP1) is an astacin met-
alloprotease,17,18 the physiological function of which has
been of considerable interest for some time. This protein
has been suggested to play essential roles in osteogenesis
and ECM formation; it has also been described as exerting
influence over dorsal-ventral patterning through the indi-
rect activation of some TGFb-like proteins.19–21 Of partic-
ular interest has been the role of BMP1 in proteolytic
removal of the C-propeptides from procollagen precursors
of the major fibrillar collagen types I–III. This processing is
1Institute of Molecular and Cell Biology, Proteos, Singapore 138673, Singapore; 2Center for Molecular Medicine Cologne, University of Cologne, Cologne
D-50931, Germany; 3Institute of Human Genetics, University Hospital Cologne, University of Cologne, Cologne D-50931, Germany; 4Cologne Excellence
Cluster on Cellular Stress Responses in Aging-Associated Diseases, University of Cologne, Cologne D-50674, Germany; 5Children’s Hospital, University of
Cologne, Cologne D-50937, Germany; 6Cologne Center for Genomics, University of Cologne, Cologne D-50931, Germany; 7Institute of Developmental
Biology, University of Cologne, Cologne D-50674, Germany; 8Department of Cell and Regenerative Biology, School of Medicine and Public Health,
University of Wisconsin, Madison, Wisconsin 53706, USA; 9Department of Medical Genetics, Hacettepe University Medical Faculty, 06100 Ankara, Turkey10These authors contributed equally to this work11These authors contributed equally to this work12Present address: Australian Regenerative Medicine Institute, Monash University, Victoria 3800, Australia
*Correspondence: [email protected] (B.W.), [email protected] (T.J.C.)
DOI 10.1016/j.ajhg.2012.02.026. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 661–674, April 6, 2012 661

essential for the self-assembly of mature collagen mono-
mers into fibrils.22 The precise functional requirement of
BMP1 in vivo is unclear. To date, Bmp1 loss of function
has only been analyzed in a knock-out mouse, in which
it was found to be lethal around birth and for which no
detailed analysis of osteogenesis was presented.23 Thus,
the role of BMP1 in bone formation and organogenesis
remains obscure.
Here, we describe two siblings with a high-bone-density
form of OI, identify a causative mutation in BMP1, and
provide detailed analysis of the effect of the mutation on
BMP1modification and secretion. We show that the hypo-
functional nature of the mutation is demonstrated in vivo
by using two assays that test substrate cleavage in zebra-
fish. We further describe a zebrafish Bmp1a mutant with
skeletal defects comparable to those seen in individuals
with OI, demonstrating conservation of important BMP1
function in osteogenesis across species. Our analysis of
these mutants has demonstrated that loss of Bmp1a affects
neither osteoblast formation nor activity but rather the
ability to generate mature collagen fibrils. This finding
therefore indicates that BMP1 is very much required in
the process of bone formation.
Material and Methods
Whole-Exome SequencingGenomic DNA was enriched from exonic and adjacent splice-site
sequences by the use of the Agilent SureSelect Human Exome
Kit and run on the Illumina Genome Analyzer IIX Sequencer.
Further data analysis was performed with an in-house bioinfor-
matics pipeline in combination with SAMTOOLS v.0.1.7 for SNP
and indel detection. In-house-developed scripts were applied for
the detection of protein changes, splice-site affections, and over-
laps with known variations (Ensembl build 61 and 1,000 Genomes
Project release 2010_3).
Mutation ScreeningThe identified mutation was resequenced in an independent
experiment, tested for cosegregation with the phenotype within
the family, and then screened in 300 healthy control individuals
from Turkey by PCR and restriction digestion (AvaI; Fermentas,
St. Leon-Rot, Germany). All subjects or their legal representatives
gave written informed consent for the study. The study was per-
formed in accordance with the Declaration of Helsinki protocols
and was approved by the local institutional review boards.
Generation of BMP1 ConstructsTheBMP1-FLAGpcDNA3.1 construct contained full-lengthhuman
cDNA of BMP1 (RefSeq accession number NM_001199.3) and was
fused to a C-terminal FLAG tag; it was provided by the group of
Karl E. Kadler (University of Manchester, UK) and was used for
BMP1 expression studies. The identified substitution, p.Gly12Arg,
was introduced by site-directed PCR mutagenesis with the use of
a primer containing the specific nucleotide substitution.
Cell Culture and Transient TransfectionHuman embryonic kidney (HEK) 293T cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) containing 10%
fetal bovine serum (FBS, GIBCO) and antibiotics. Cells were
transiently transfected with Lipofectamine 2000 (Invitrogen,
Karlsruhe, Germany) and vectors containing wild-type (WT) and
mutant variants of BMP1 cDNA. Transfections were performed
according to the manufacturer’s instructions.
Secretion Assay and Immunoblot Analysis30 hr after transient transfection, cells and untransfected control
cells were maintained in serum-free medium for 18 hr at 37�C.After starvation, supernatant-containing secreted proteins were
precipitated by trichloroacetic acid, and cells were lysed with ice-
cold lysis buffer. The total protein concentration of the extracts
was determined by the BCA (bicinchoninic acid) Protein Assay
Kit (Pierce Protein Research Products, Thermo Fischer Scientific,
Rockford, IL, USA), and proteins were separated by gradient
(4%–12%) SDS-PAGE (Invitrogen) under reducing conditions
and transferred to a nitrocellulose membrane by immunoblotting.
Immunoblots were blocked in 5% milk powder in TBS containing
0.1% Tween20, and they were probed with Flag antibody (Agilent
Technologies, Waldbronn, Germany). Equal protein amounts
were confirmed by b-actin detection in whole-cell lysates or by
Coomassie staining of a ~65 kDa protein in supernatant of
serum-free medium. A peroxidase-conjugated secondary antibody
(goat anti-mouse) was purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA), and blots were developed with an
enhanced chemiluminescence system, ECL Plus (Amersham,
UK); exposure on autoradiographic film (GE Healthcare, Mun-
chen, Germany) followed.
Zebrafish StudiesRadioimmunoprecipitation (RIPA) buffer (50 mM Tris-HCl, pH
7.6, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and
1% NP-40) was used for the extraction of total protein from zebra-
fish larvae (6 days old) or fins (4 months old), and the protein was
measured with a standard Bradford assay. Proteins (20 mg) were
separated on a 6% denaturing polyacrylamide gel, transferred to
a nitrocellulose membrane, and probed with a rabbit polyclonal
antibody raised against a peptide of zebrafish Collagen1a1a.
Goat anti-rabbit antibody conjugated with horseradish peroxidase
(HRP) was employed as a secondary antibody, and the bands were
visualized with chemiluminescence detection (Millipore, Billerica,
MA, USA). For the loading control, blots were stripped and then
reprobed with a rabbit anti-b-actin antibody (Cell Signaling
Technology, Danvers, MA, USA).
N-glycosidase AssayFor the N-glycosylation studies, 20 mg of whole-cell lysates of
transiently transfected and untransfected HEK 293T cells were
either treated with N-glycosidase F (PNGaseF) (New England
BioLabs, Frankfurt, Germany) or left untreated. The enzyme reac-
tion was performed according to the manufacturer’s instructions.
Proteins were subjected to SDS-PAGE and analyzed by anti-Flag
immunoblotting. Equal protein amounts were confirmed by
b-actin detection.
Fish Lines and MappingEmbryos were obtained by natural crosses and were staged as per
Parichy et al.24 Themicrowaved (med tt281), dino (chd tt250), and frilly
fins (frf tm317a, frf tf5, frf tp34, and frf ty68) alleles that we used have
been described previously25 and were isolated in an N-ethyl-N-
nitrosourea (ENU) screen in Freiburg (frf fr24). frf tm317 was used
662 The American Journal of Human Genetics 90, 661–674, April 6, 2012

for all analyses. The Tg(sp7:mCherry) transgenic line has been re-
ported previously.26 We performed genetic mapping by crossing
frfþ/� and medþ/� fish to the WIK strain and by subjecting the
F2 progeny to simple-sequence-length polymorphism (SSLP)
meiotic mapping as outlined by Geisler.27 Heat shocks were per-
formed at 37�C for 1 hr at 30 and 56 hr postfertilization (hpf).
MicroscopyFluorescent images were taken on an Olympus Fluoview confocal
microscope, whereas brightfield and Nomarski micrographs were
taken on a Zeiss Axioimager or a Leica MZ16FA. For live imaging,
larvae were anesthetized in Tricaine and mounted in 3% methyl
cellulose or 1% low-melting-point agarose. All whole-mount stain-
ings using alizarin red, in situ hybridization, or immunodetection
were cleared in glycerol prior to mounting. Stained ultrathin
sections of the fins were employed for transmission electron
microscopy (TEM) on a Jeol JEM-1010 electron microscope.
RNA and DNA Isolation, cDNA Synthesis,
and SequencingTrizol (Invitrogen, CA, USA) was used for the isolation of RNA
from WT or mutant larvae, and SuperscriptIII Reverse Transcrip-
tase (Invitrogen, CA, USA) was used for the generation of cDNA
by reverse transcription. RT-PCR was used for the amplification
of bmp1a cDNA from all frilly fins mutated alleles and correspond-
ing siblings. Similarly, col1a1a cDNA was amplified from the
microwaved mutant allele and siblings. Resulting PCR fragments
were purified and sequenced directly. To identify mutations at
the genomic level, we extracted larval genomic DNA and directly
sequenced the region of interest from amplified PCR products.
DNA Construct GenerationGateway cloning technology and entry constructs from the
Tol2Kit28 were used for the generation of zebrafish and Myc-
tagged human Bmp1 heat-shock constructs. Bmp1 coding regions
were cloned into a middle entry vector via standard cloning
methods or through a BP Gateway reaction.
DNA and RNA InjectionsDNA and RNAs were diluted in Danieau buffer and Phenol
red before being injected into 1-cell embryos by a Pico-Injector
(Harvard Apparatus, MA, USA). Sense RNAs for overexpression
or rescue were transcribed from cDNAs and cloned into pCS2þ.Plasmids were linearized with NotI, and capped mRNA was
synthesized with mMessage mMachine SP6 kit (Ambion, Applied
Biosystems, Austin, TX, USA). Full-length cDNA for BMP1 was
amplified from a HeLa-cell cDNA library and cloned into pCS2þ.A primer harboring the p.Gly12Arg signal-peptide substitution
was used for the generation of the mutant BMP1 cDNA version
by PCR. chordin RNA was transcribed as previously reported.29
chordin and BMP1 RNA were injected at a concentration of 60
and 450 pg, respectively. To generate adult dino mutants, we
rescued early patterning defects by injecting 30 pg of chordin
RNA. Tol2 RNA was generated as published.30
In Situ HybridizationSingle- and double-stranded-RNA in situ hybridizations were
performed and developed with either chromogenic substrate31
or fluorescent tyramide signal amplification.32 Probes for sp7,
osteopontin, and collagen10a1 were synthesized as described.33
The bmp1a probe was generated with EcoRI by linearizing clone
IMAGp998H0417161 from ImaGenes (Berlin, Germany) and was
transcribed with T7 Polymerase. In situ hybridization of 8 dpf
(days postfertilization) larvae required extended 40min proteinase
K (15 mg/ml) digestions at room temperature, 48 hr hybridization
in the antisense probe, and 2 days of signal development at room
temperature.
Skeletal and Matrix StainingBones of larvae and adult fish were stained with alizarin red alone
or in combinationwith the cartilage stain, alcian blue, as described
by Walker et al.34 and Spoorendonk et al.26 For microscopic anal-
ysis of fibrillar collagen organization, fins and larvae were fixed in
4% paraformaldehyde and embedded in 1% agarose for cryosec-
tioning. Sections were stained with picrosirius red as previously
described35 and were visualized by birefringence under polariza-
tion filters.
Antibody StainingMyc-tagged BMP1 proteins were detected by whole-mount
immunofluorescent staining with the 9E10 monoclonal antibody
(Santa Cruz, CA, USA). 3 dpf embryos were fixed in 4% paraformal-
dehyde overnight at 4�C and were then washed with 0.1% PBS
Triton X-100. Embryos were permeabilized by incubation in
100% acetone for 7 min at �20�C, were rewashed, and were
blocked overnight in PBS/Triton with 0.5% goat serum and 0.1%
dimethyl sulfoxide. After extensive washing, the embryos
were incubated with Alexa488-conjugated secondary antibodies
(Invitrogen) diluted in blocking solution. Finally, embryos were
rewashed before they were cleared in glycerol. Osteoblasts of
the adult fins were stained with the zns5 monoclonal antibody
(obtained from the Zebrafish International Resource Center
[Eugene, OR, USA]) either in the wholemount or after cryosection-
ing. Theywere then counterstainedwith either alizarin red (for the
whole mounts) or DAPI (for the cryosections) as per previous
methods.36
Retinoic-Acid TreatmentWe purchased all-trans retinoic acid (RA) from Sigma (MI, USA),
and we made a 1 mM stock solution by dissolving it in ethanol.
Larvae were treated with 1 mM RA diluted in egg water at 4 dpf.
The controls were exposed to an equivalent amount of just the
carrier (ethanol). Fresh solution was added every other day until
the fish were fixed at 11 dpf for alizarin-red processing.
MicroCT Scanning of Adult FishAdult fish were euthanized in Tricaine and scanned immediately
at 40 kV, 130 mAwith a Siemens Inveon PET-CT (positron emission
tomography-computed tomography) scanner. The images were
reconstructed and analyzed with Inveon Acquisition Workplace
1.4 and Inveon Research Workplace 3.0, respectively. Bone
mineral density was calculated with the density-phantoms stan-
dards supplied by the company.
Results
A Homozygous BMP1 Mutation Causes High Bone
Mineral Density and Multiple Fractures
Weusedwhole-exome sequencing to identify the causative
mutation underlying an autosomal-recessive form of bone
fragility in a consanguineous family from Turkey. Both
The American Journal of Human Genetics 90, 661–674, April 6, 2012 663
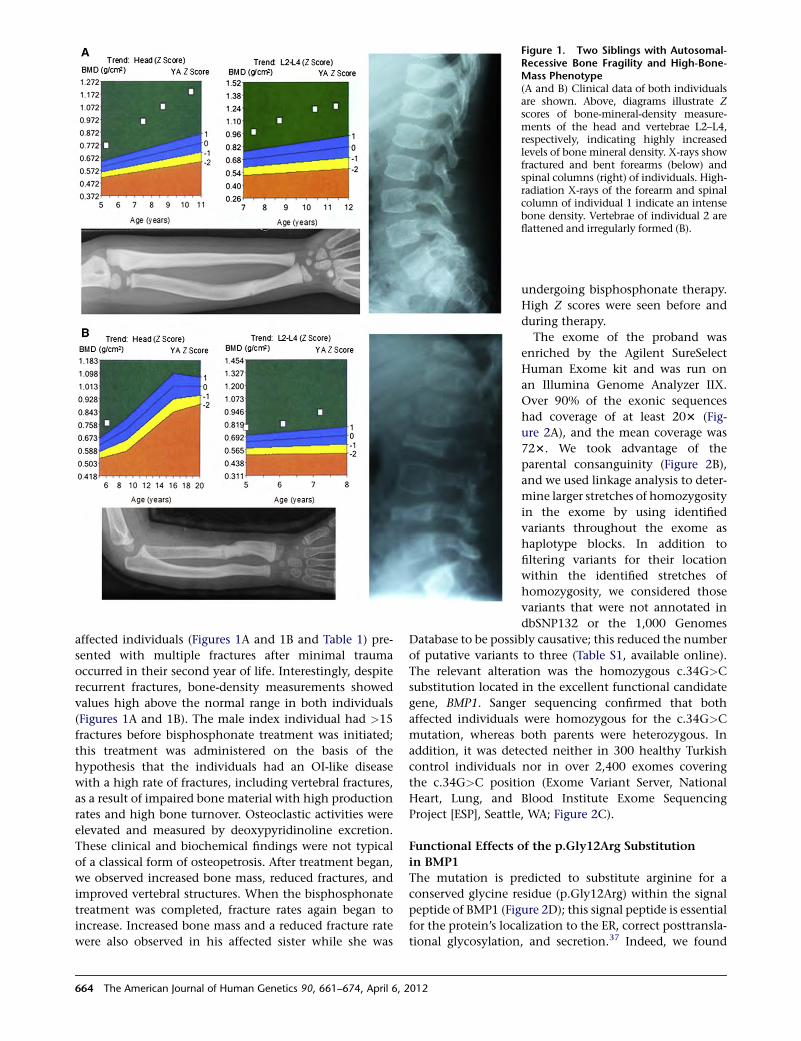
affected individuals (Figures 1A and 1B and Table 1) pre-
sented with multiple fractures after minimal trauma
occurred in their second year of life. Interestingly, despite
recurrent fractures, bone-density measurements showed
values high above the normal range in both individuals
(Figures 1A and 1B). The male index individual had >15
fractures before bisphosphonate treatment was initiated;
this treatment was administered on the basis of the
hypothesis that the individuals had an OI-like disease
with a high rate of fractures, including vertebral fractures,
as a result of impaired bone material with high production
rates and high bone turnover. Osteoclastic activities were
elevated and measured by deoxypyridinoline excretion.
These clinical and biochemical findings were not typical
of a classical form of osteopetrosis. After treatment began,
we observed increased bone mass, reduced fractures, and
improved vertebral structures. When the bisphosphonate
treatment was completed, fracture rates again began to
increase. Increased bone mass and a reduced fracture rate
were also observed in his affected sister while she was
Figure 1. Two Siblings with Autosomal-Recessive Bone Fragility and High-Bone-Mass Phenotype(A and B) Clinical data of both individualsare shown. Above, diagrams illustrate Zscores of bone-mineral-density measure-ments of the head and vertebrae L2–L4,respectively, indicating highly increasedlevels of bone mineral density. X-rays showfractured and bent forearms (below) andspinal columns (right) of individuals. High-radiation X-rays of the forearm and spinalcolumn of individual 1 indicate an intensebone density. Vertebrae of individual 2 areflattened and irregularly formed (B).
undergoing bisphosphonate therapy.
High Z scores were seen before and
during therapy.
The exome of the proband was
enriched by the Agilent SureSelect
Human Exome kit and was run on
an Illumina Genome Analyzer IIX.
Over 90% of the exonic sequences
had coverage of at least 203 (Fig-
ure 2A), and the mean coverage was
723. We took advantage of the
parental consanguinity (Figure 2B),
and we used linkage analysis to deter-
mine larger stretches of homozygosity
in the exome by using identified
variants throughout the exome as
haplotype blocks. In addition to
filtering variants for their location
within the identified stretches of
homozygosity, we considered those
variants that were not annotated in
dbSNP132 or the 1,000 Genomes
Database to be possibly causative; this reduced the number
of putative variants to three (Table S1, available online).
The relevant alteration was the homozygous c.34G>C
substitution located in the excellent functional candidate
gene, BMP1. Sanger sequencing confirmed that both
affected individuals were homozygous for the c.34G>C
mutation, whereas both parents were heterozygous. In
addition, it was detected neither in 300 healthy Turkish
control individuals nor in over 2,400 exomes covering
the c.34G>C position (Exome Variant Server, National
Heart, Lung, and Blood Institute Exome Sequencing
Project [ESP], Seattle, WA; Figure 2C).
Functional Effects of the p.Gly12Arg Substitution
in BMP1
The mutation is predicted to substitute arginine for a
conserved glycine residue (p.Gly12Arg) within the signal
peptide of BMP1 (Figure 2D); this signal peptide is essential
for the protein’s localization to the ER, correct posttransla-
tional glycosylation, and secretion.37 Indeed, we found
664 The American Journal of Human Genetics 90, 661–674, April 6, 2012
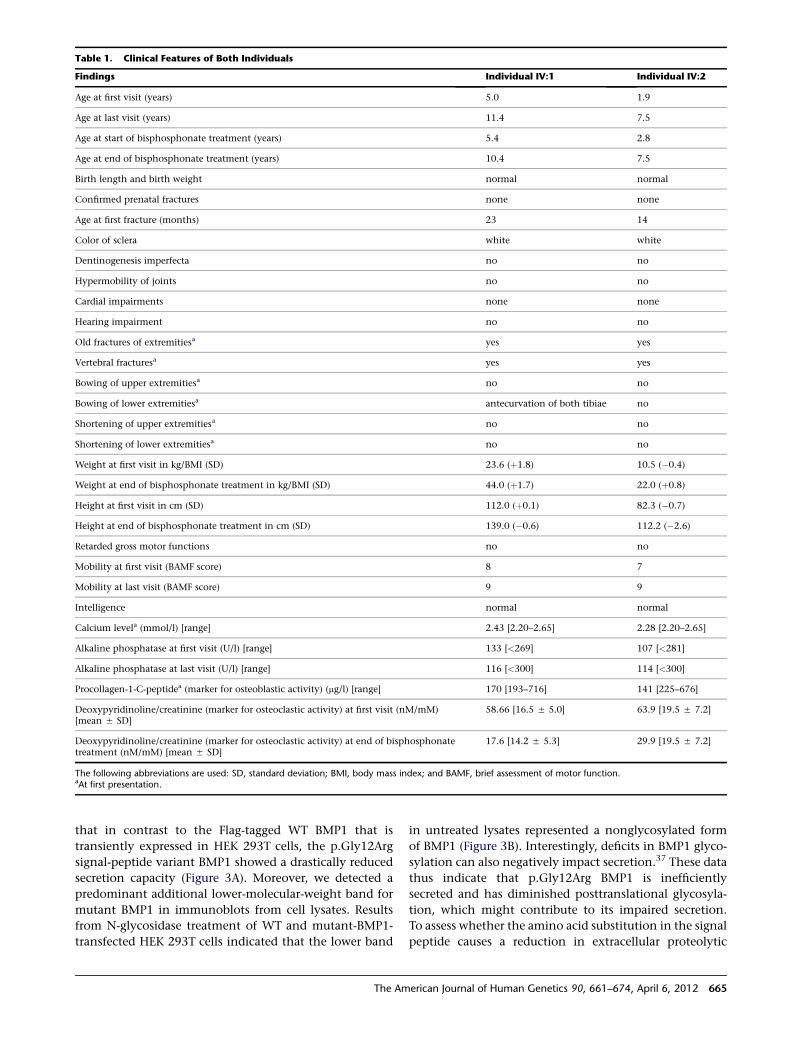
that in contrast to the Flag-tagged WT BMP1 that is
transiently expressed in HEK 293T cells, the p.Gly12Arg
signal-peptide variant BMP1 showed a drastically reduced
secretion capacity (Figure 3A). Moreover, we detected a
predominant additional lower-molecular-weight band for
mutant BMP1 in immunoblots from cell lysates. Results
from N-glycosidase treatment of WT and mutant-BMP1-
transfected HEK 293T cells indicated that the lower band
in untreated lysates represented a nonglycosylated form
of BMP1 (Figure 3B). Interestingly, deficits in BMP1 glyco-
sylation can also negatively impact secretion.37 These data
thus indicate that p.Gly12Arg BMP1 is inefficiently
secreted and has diminished posttranslational glycosyla-
tion, which might contribute to its impaired secretion.
To assess whether the amino acid substitution in the signal
peptide causes a reduction in extracellular proteolytic
Table 1. Clinical Features of Both Individuals
Findings Individual IV:1 Individual IV:2
Age at first visit (years) 5.0 1.9
Age at last visit (years) 11.4 7.5
Age at start of bisphosphonate treatment (years) 5.4 2.8
Age at end of bisphosphonate treatment (years) 10.4 7.5
Birth length and birth weight normal normal
Confirmed prenatal fractures none none
Age at first fracture (months) 23 14
Color of sclera white white
Dentinogenesis imperfecta no no
Hypermobility of joints no no
Cardial impairments none none
Hearing impairment no no
Old fractures of extremitiesa yes yes
Vertebral fracturesa yes yes
Bowing of upper extremitiesa no no
Bowing of lower extremitiesa antecurvation of both tibiae no
Shortening of upper extremitiesa no no
Shortening of lower extremitiesa no no
Weight at first visit in kg/BMI (SD) 23.6 (þ1.8) 10.5 (�0.4)
Weight at end of bisphosphonate treatment in kg/BMI (SD) 44.0 (þ1.7) 22.0 (þ0.8)
Height at first visit in cm (SD) 112.0 (þ0.1) 82.3 (�0.7)
Height at end of bisphosphonate treatment in cm (SD) 139.0 (�0.6) 112.2 (�2.6)
Retarded gross motor functions no no
Mobility at first visit (BAMF score) 8 7
Mobility at last visit (BAMF score) 9 9
Intelligence normal normal
Calcium levela (mmol/l) [range] 2.43 [2.20–2.65] 2.28 [2.20–2.65]
Alkaline phosphatase at first visit (U/l) [range] 133 [<269] 107 [<281]
Alkaline phosphatase at last visit (U/l) [range] 116 [<300] 114 [<300]
Procollagen-1-C-peptidea (marker for osteoblastic activity) (mg/l) [range] 170 [193–716] 141 [225–676]
Deoxypyridinoline/creatinine (marker for osteoclastic activity) at first visit (nM/mM)[mean 5 SD]
58.66 [16.5 5 5.0] 63.9 [19.5 5 7.2]
Deoxypyridinoline/creatinine (marker for osteoclastic activity) at end of bisphosphonatetreatment (nM/mM) [mean 5 SD]
17.6 [14.2 5 5.3] 29.9 [19.5 5 7.2]
The following abbreviations are used: SD, standard deviation; BMI, body mass index; and BAMF, brief assessment of motor function.aAt first presentation.
The American Journal of Human Genetics 90, 661–674, April 6, 2012 665
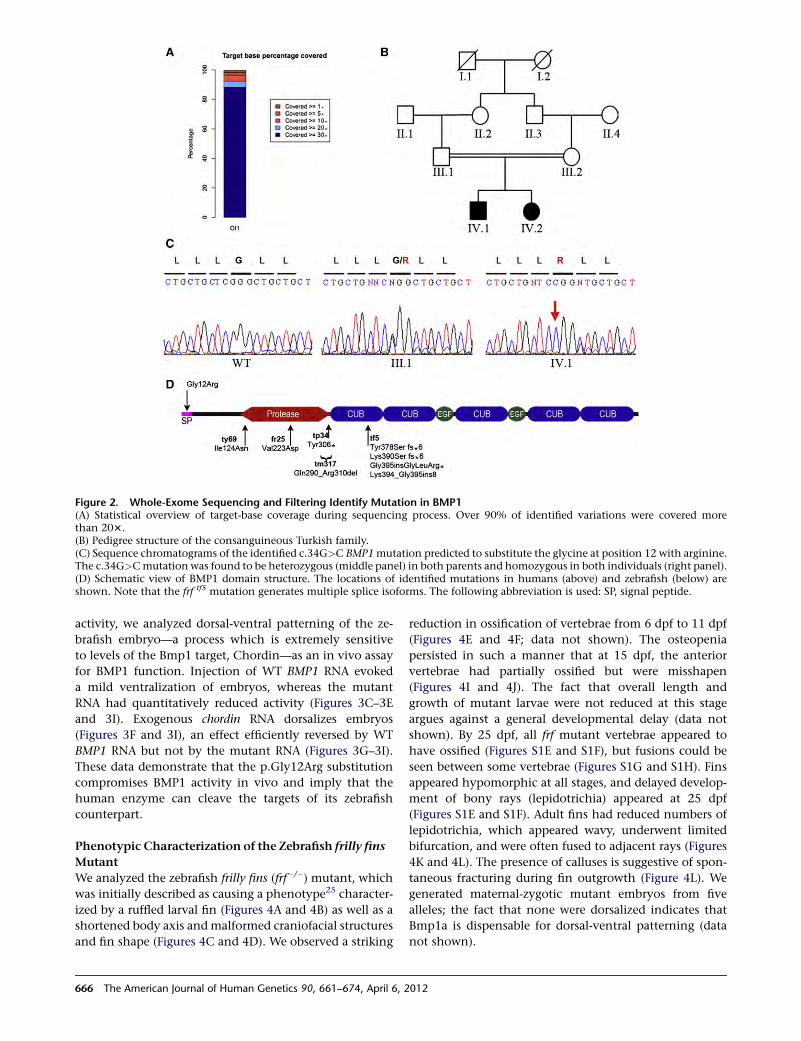
activity, we analyzed dorsal-ventral patterning of the ze-
brafish embryo—a process which is extremely sensitive
to levels of the Bmp1 target, Chordin—as an in vivo assay
for BMP1 function. Injection of WT BMP1 RNA evoked
a mild ventralization of embryos, whereas the mutant
RNA had quantitatively reduced activity (Figures 3C–3E
and 3I). Exogenous chordin RNA dorsalizes embryos
(Figures 3F and 3I), an effect efficiently reversed by WT
BMP1 RNA but not by the mutant RNA (Figures 3G–3I).
These data demonstrate that the p.Gly12Arg substitution
compromises BMP1 activity in vivo and imply that the
human enzyme can cleave the targets of its zebrafish
counterpart.
Phenotypic Characterization of the Zebrafish frilly fins
Mutant
We analyzed the zebrafish frilly fins (frf�/�) mutant, which
was initially described as causing a phenotype25 character-
ized by a ruffled larval fin (Figures 4A and 4B) as well as a
shortened body axis andmalformed craniofacial structures
and fin shape (Figures 4C and 4D). We observed a striking
reduction in ossification of vertebrae from 6 dpf to 11 dpf
(Figures 4E and 4F; data not shown). The osteopenia
persisted in such a manner that at 15 dpf, the anterior
vertebrae had partially ossified but were misshapen
(Figures 4I and 4J). The fact that overall length and
growth of mutant larvae were not reduced at this stage
argues against a general developmental delay (data not
shown). By 25 dpf, all frf mutant vertebrae appeared to
have ossified (Figures S1E and S1F), but fusions could be
seen between some vertebrae (Figures S1G and S1H). Fins
appeared hypomorphic at all stages, and delayed develop-
ment of bony rays (lepidotrichia) appeared at 25 dpf
(Figures S1E and S1F). Adult fins had reduced numbers of
lepidotrichia, which appeared wavy, underwent limited
bifurcation, and were often fused to adjacent rays (Figures
4K and 4L). The presence of calluses is suggestive of spon-
taneous fracturing during fin outgrowth (Figure 4L). We
generated maternal-zygotic mutant embryos from five
alleles; the fact that none were dorsalized indicates that
Bmp1a is dispensable for dorsal-ventral patterning (data
not shown).
Figure 2. Whole-Exome Sequencing and Filtering Identify Mutation in BMP1(A) Statistical overview of target-base coverage during sequencing process. Over 90% of identified variations were covered morethan 203.(B) Pedigree structure of the consanguineous Turkish family.(C) Sequence chromatograms of the identified c.34G>C BMP1mutation predicted to substitute the glycine at position 12 with arginine.The c.34G>Cmutation was found to be heterozygous (middle panel) in both parents and homozygous in both individuals (right panel).(D) Schematic view of BMP1 domain structure. The locations of identified mutations in humans (above) and zebrafish (below) areshown. Note that the frf tf5 mutation generates multiple splice isoforms. The following abbreviation is used: SP, signal peptide.
666 The American Journal of Human Genetics 90, 661–674, April 6, 2012
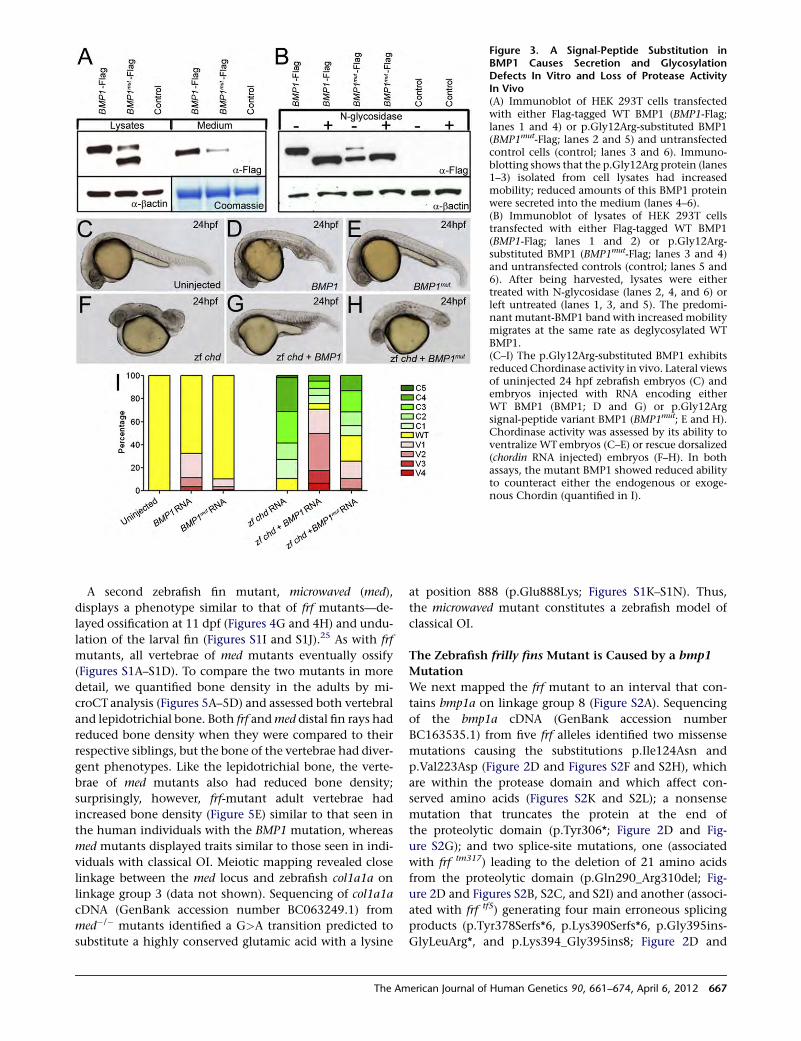
A second zebrafish fin mutant, microwaved (med),
displays a phenotype similar to that of frf mutants—de-
layed ossification at 11 dpf (Figures 4G and 4H) and undu-
lation of the larval fin (Figures S1I and S1J).25 As with frf
mutants, all vertebrae of med mutants eventually ossify
(Figures S1A–S1D). To compare the two mutants in more
detail, we quantified bone density in the adults by mi-
croCT analysis (Figures 5A–5D) and assessed both vertebral
and lepidotrichial bone. Both frf andmed distal fin rays had
reduced bone density when they were compared to their
respective siblings, but the bone of the vertebrae had diver-
gent phenotypes. Like the lepidotrichial bone, the verte-
brae of med mutants also had reduced bone density;
surprisingly, however, frf-mutant adult vertebrae had
increased bone density (Figure 5E) similar to that seen in
the human individuals with the BMP1 mutation, whereas
med mutants displayed traits similar to those seen in indi-
viduals with classical OI. Meiotic mapping revealed close
linkage between the med locus and zebrafish col1a1a on
linkage group 3 (data not shown). Sequencing of col1a1a
cDNA (GenBank accession number BC063249.1) from
med�/� mutants identified a G>A transition predicted to
substitute a highly conserved glutamic acid with a lysine
at position 888 (p.Glu888Lys; Figures S1K–S1N). Thus,
the microwaved mutant constitutes a zebrafish model of
classical OI.
The Zebrafish frilly fins Mutant is Caused by a bmp1
Mutation
We next mapped the frf mutant to an interval that con-
tains bmp1a on linkage group 8 (Figure S2A). Sequencing
of the bmp1a cDNA (GenBank accession number
BC163535.1) from five frf alleles identified two missense
mutations causing the substitutions p.Ile124Asn and
p.Val223Asp (Figure 2D and Figures S2F and S2H), which
are within the protease domain and which affect con-
served amino acids (Figures S2K and S2L); a nonsense
mutation that truncates the protein at the end of
the proteolytic domain (p.Tyr306*; Figure 2D and Fig-
ure S2G); and two splice-site mutations, one (associated
with frf tm317) leading to the deletion of 21 amino acids
from the proteolytic domain (p.Gln290_Arg310del; Fig-
ure 2D and Figures S2B, S2C, and S2I) and another (associ-
ated with frf tf5) generating four main erroneous splicing
products (p.Tyr378Serfs*6, p.Lys390Serfs*6, p.Gly395ins-
GlyLeuArg*, and p.Lys394_Gly395ins8; Figure 2D and
Figure 3. A Signal-Peptide Substitution inBMP1 Causes Secretion and GlycosylationDefects In Vitro and Loss of Protease ActivityIn Vivo(A) Immunoblot of HEK 293T cells transfectedwith either Flag-tagged WT BMP1 (BMP1-Flag;lanes 1 and 4) or p.Gly12Arg-substituted BMP1(BMP1mut-Flag; lanes 2 and 5) and untransfectedcontrol cells (control; lanes 3 and 6). Immuno-blotting shows that the p.Gly12Arg protein (lanes1–3) isolated from cell lysates had increasedmobility; reduced amounts of this BMP1 proteinwere secreted into the medium (lanes 4–6).(B) Immunoblot of lysates of HEK 293T cellstransfected with either Flag-tagged WT BMP1(BMP1-Flag; lanes 1 and 2) or p.Gly12Arg-substituted BMP1 (BMP1mut-Flag; lanes 3 and 4)and untransfected controls (control; lanes 5 and6). After being harvested, lysates were eithertreated with N-glycosidase (lanes 2, 4, and 6) orleft untreated (lanes 1, 3, and 5). The predomi-nant mutant-BMP1 band with increased mobilitymigrates at the same rate as deglycosylated WTBMP1.(C–I) The p.Gly12Arg-substituted BMP1 exhibitsreduced Chordinase activity in vivo. Lateral viewsof uninjected 24 hpf zebrafish embryos (C) andembryos injected with RNA encoding eitherWT BMP1 (BMP1; D and G) or p.Gly12Argsignal-peptide variant BMP1 (BMP1mut; E and H).Chordinase activity was assessed by its ability toventralize WT embryos (C–E) or rescue dorsalized(chordin RNA injected) embryos (F–H). In bothassays, the mutant BMP1 showed reduced abilityto counteract either the endogenous or exoge-nous Chordin (quantified in I).
The American Journal of Human Genetics 90, 661–674, April 6, 2012 667

Figures S2D, S2E, and S2J). We were able to rescue the frf
larval fin phenotype by injecting a DNA construct driving
zebrafish Bmp1a expression from a heat-shock promoter,
further supporting the conclusion that frilly fins represents
a bmp1a mutant (Figures S3A–S3C).
Bmp1 Function in Bone Formation and Development
To understand the function of Bmp1 in bone formation,
we characterized zebrafish bmp1a gene expression and
the frf mutant phenotype in more detail. Zebrafish
bmp1a is expressed in osteoblasts. During larval stages,
we observed strong expression in fin mesenchyme cells
within the fin fold at a stage contemporaneous with the
appearance of the fin-fold defect in frf mutants (Figures
6A and 6B). In addition, we noted expression in the floor
plate and hypochord, branchial arches and operculum
(Figures 6A and 6C), and sites of bone formation in the
head. We confirmed expression of bmp1a in osteoblasts
by double-fluorescent in situ hybridization with the zebra-
fish osteoblast maker col10a1. We found consistent overlap
of both mRNAs in osteoblasts on the operculum and
cleithrum (Figures 6E and 6E00; data not shown). We also
noted expression in clusters of cells arranged metameri-
cally adjacent to the notochord, a location and arrange-
ment consistent with osteoblasts of the vertebral column
(Figure 6D).
To determine whether the compromised ossification in
frilly fins mutant larvae is due to insufficient generation
of osteoblasts, we analyzed expression of sp7, osteopontin,
and collagen10a1 by in situ hybridization in frf mutants.
Osteoblasts were found to be normal in number and
location, consistent with the interpretation that loss of
bmp1a does not disrupt osteoblast generation, number,
localization, or differentiation (Figures 6F–6K). We con-
firmed the presence of normal osteoblast numbers by first
crossing frf into the sp7:mcherry transgenic line,26 which
labels osteoblasts on skeletal structures. Compared to WT
cells, mCherry-positive cells did not decrease in number
in the frfmutant vertebrae or fins, even at the earliest times
that such cells are visible (Figures 6L and 6Mand Figure S4A
and S4B). We obtained a similar result by immunostaining
with the osteoblast-specific zns5 antibody, although we did
observe altered cellular morphology—osteoblasts appeared
more cuboidal in the mutant than did the flattened cells in
the WT bone (Figures 7A–7D). The latter effect was also
seen in the lepidotrichia when imaged by transmission
electron microscopy (Figures 7E and 7F). However, no
significant change in osteoblast number was observed
in fin rays (Figure S4C). To test whether this altered-
morphology phenotype is concomitant with a loss of oste-
oblast activity, we exposed frf mutants to RA, which was
previously described as stimulating osteoblast activity
and causing precocious hyperossification of the entire
vertebral column.26,33 Unlike in WT (Figures 7G and 7H),
ossification in frf mutants was almost completely refrac-
tory to RA treatment (Figures 7I and 7J), consistent with
a defect in the ability of osteoblasts to effectively generate
osteoid, downstream of osteoblast differentiation and
activity.
Bmp1 could potentially affect the ossification process
through proteolytic cleavage processes involving a number
of targets. Among these, its ability to cleave and inactivate
the BMP2/4 inhibitor Chordin is well documented and has
the potential to affect the ossification process.38,39 To test
whether the frf phenotypes could be due to an excess of
uncleaved Chordin, we generated frf�/� chordin�/�
double-mutant adults (in which the early ventralized
phenotype was rescued by chordin mRNA injection). As
previously described,40 Chordin function is dispensable
after gastrulation for axial skeleton generation and
patterning (Figures S5A and S5C), yet it remains possible
that elevated levels might perturb osteogenesis. However,
Chordin loss at late larval stages failed to rescue the frf
Figure 4. The Zebrafish frilly finsMutant Displays Larval Fin-FoldRuffling and Osteogenesis Defects(A and B) Ventral view of the posterior medial fin fold at 3 dpf ina frilly fins (frf) (B) larva showing undulations in the fin fold, whichnormally has a linear morphology (A).(C and D) Compared to the siblings, 4-month-old frf �/� adults (D)are short and display axis defects, body curvature, and fin andcraniofacial dysmorphogenesis.(E–L) Alizarin-red staining of frf �/� (F, J, and L),microwaved (med�/�)(H), andWTsiblings (E, G, I, and K) at 11 dpf (E–H), 15 dpf (I and J),and 4 months (K and L). Both frf andmed display reduced ossifica-tion of the vertebrae (F and H), whereas nascent vertebrae areosteopenic and dysmorphic (J). Tail fins in frf �/� have lost theWT fan shape (K) and display fracture calluses, reduced bifurca-tions (L), and crinkled lepidotrichia, which often fuse to eachother (L; inset).
668 The American Journal of Human Genetics 90, 661–674, April 6, 2012

defect (Figures S5B and S5D). In addition to Chordin
cleavage, Bmp1 also plays a role in generating mature
Collagen I through the cleavage of the C-terminal propep-
tide domain (Figure 8A). Indeed, the inability of RA to
rescue the ossification process in frf mutants and the simi-
larity between the frf and med vertebrae phenotypes
suggest that the major requirement for Bmp1 in bone
formation is the generation of mature Collagen I. Using
Figure 5. CT Analysis of frilly fins and microwaved Bone Reveals Altered Adult Bone Densities(A–D) microCT analysis of bone density in a 7-month-old frf�/� mutant (B) and its WT sibling (A) as well as a med�/� mutant (D) and itsWT sibling (C). Note that although themedmutant looks overtly normal (D), the frfmutant skeleton displays axial curvature and defectsin the head skeleton (B).(E) Box plots of density measurements derived from microCT analysis of mutants and sibling vertebrae and fin lepidotrichia. medmutants have osteopenia of both lepidotrichia in the fins and vertebrae (blue boxes). Although frf mutants also display osteopenia ofthe fins, they show an increased bone density in the vertebrae (green boxes). The Mann-Whitney U test was performed for comparingdensities between mutants and siblings for each bone type (*** denotes p < 0.001; ** denotes p < 0.01; and n ¼ 8 for all data sets). Boxesindicate the median and the 25th and 75th percentiles, whereas the whiskers display the largest and smallest values.
Figure 6. bmp1a Is Expressed in Osteoblasts, which Appear Normally Differentiated in frilly fins(A–D) In situ hybridization of bmp1a at 4 dpf (A), 2 dpf (B), 3 dpf (C) and 8 dpf (D). Expression is seen in fin mesenchyme cells of thefin fold (A and B), floor plate and hypochord (A, open and filled arrowheads, respectively), branchial arches (C, asterisk), and operculum(C, arrowhead) and perichordal cells of the anterior notochord (D, arrowheads).(E–E00) Confocal images of double-fluorescent in situ hybridizations showing coexpression of bmp1a (E and E00; red) and the osteoblastmarker collagen10a1 (E0 and E00; green) in osteoblasts on the operculum at 4 dpf. Most cells express both markers (three cases highlightedby arrowheads), and central, more mature osteoblasts express slightly higher levels of col10a1.(F–K) Perichordal expression of osteoblast markers is not disrupted in frf mutants. Lateral images of anterior notochord of 8 dpf WT(F, H, and J) and frf�/� (G, I, and K) larvae hybridized with probes for sp7 (F and G), osteopontin (H and I), and collagen10a1 (J and K).(L and M) Confocal images of sp7:mCherry-expressing osteoblasts on vertebrae of WT (L) and frfmutant (M) larvae at 20 dpf. There is noreduction in osteoblast numbers in the mutant.
The American Journal of Human Genetics 90, 661–674, April 6, 2012 669
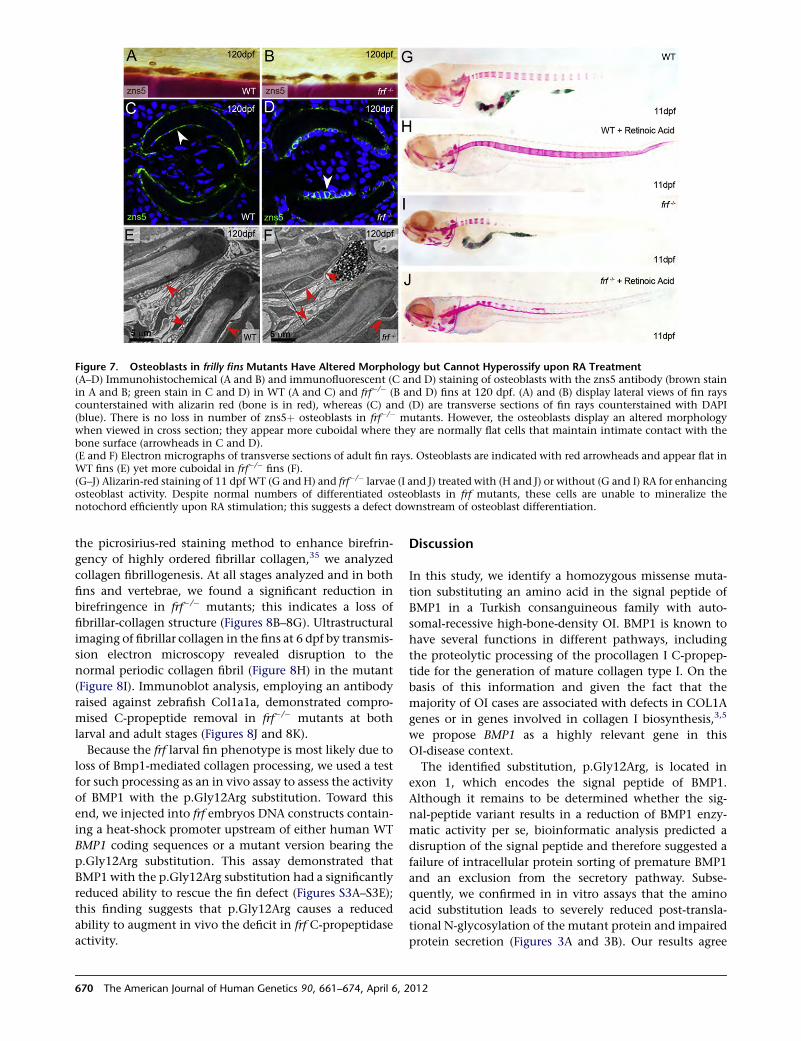
the picrosirius-red staining method to enhance birefrin-
gency of highly ordered fibrillar collagen,35 we analyzed
collagen fibrillogenesis. At all stages analyzed and in both
fins and vertebrae, we found a significant reduction in
birefringence in frf�/� mutants; this indicates a loss of
fibrillar-collagen structure (Figures 8B–8G). Ultrastructural
imaging of fibrillar collagen in the fins at 6 dpf by transmis-
sion electron microscopy revealed disruption to the
normal periodic collagen fibril (Figure 8H) in the mutant
(Figure 8I). Immunoblot analysis, employing an antibody
raised against zebrafish Col1a1a, demonstrated compro-
mised C-propeptide removal in frf�/� mutants at both
larval and adult stages (Figures 8J and 8K).
Because the frf larval fin phenotype is most likely due to
loss of Bmp1-mediated collagen processing, we used a test
for such processing as an in vivo assay to assess the activity
of BMP1 with the p.Gly12Arg substitution. Toward this
end, we injected into frf embryos DNA constructs contain-
ing a heat-shock promoter upstream of either human WT
BMP1 coding sequences or a mutant version bearing the
p.Gly12Arg substitution. This assay demonstrated that
BMP1 with the p.Gly12Arg substitution had a significantly
reduced ability to rescue the fin defect (Figures S3A–S3E);
this finding suggests that p.Gly12Arg causes a reduced
ability to augment in vivo the deficit in frf C-propeptidase
activity.
Discussion
In this study, we identify a homozygous missense muta-
tion substituting an amino acid in the signal peptide of
BMP1 in a Turkish consanguineous family with auto-
somal-recessive high-bone-density OI. BMP1 is known to
have several functions in different pathways, including
the proteolytic processing of the procollagen I C-propep-
tide for the generation of mature collagen type I. On the
basis of this information and given the fact that the
majority of OI cases are associated with defects in COL1A
genes or in genes involved in collagen I biosynthesis,3,5
we propose BMP1 as a highly relevant gene in this
OI-disease context.
The identified substitution, p.Gly12Arg, is located in
exon 1, which encodes the signal peptide of BMP1.
Although it remains to be determined whether the sig-
nal-peptide variant results in a reduction of BMP1 enzy-
matic activity per se, bioinformatic analysis predicted a
disruption of the signal peptide and therefore suggested a
failure of intracellular protein sorting of premature BMP1
and an exclusion from the secretory pathway. Subse-
quently, we confirmed in in vitro assays that the amino
acid substitution leads to severely reduced post-transla-
tional N-glycosylation of the mutant protein and impaired
protein secretion (Figures 3A and 3B). Our results agree
Figure 7. Osteoblasts in frilly fins Mutants Have Altered Morphology but Cannot Hyperossify upon RA Treatment(A–D) Immunohistochemical (A and B) and immunofluorescent (C and D) staining of osteoblasts with the zns5 antibody (brown stainin A and B; green stain in C and D) in WT (A and C) and frf�/� (B and D) fins at 120 dpf. (A) and (B) display lateral views of fin rayscounterstained with alizarin red (bone is in red), whereas (C) and (D) are transverse sections of fin rays counterstained with DAPI(blue). There is no loss in number of zns5þ osteoblasts in frf�/� mutants. However, the osteoblasts display an altered morphologywhen viewed in cross section; they appear more cuboidal where they are normally flat cells that maintain intimate contact with thebone surface (arrowheads in C and D).(E and F) Electron micrographs of transverse sections of adult fin rays. Osteoblasts are indicated with red arrowheads and appear flat inWT fins (E) yet more cuboidal in frf�/� fins (F).(G–J) Alizarin-red staining of 11 dpfWT (G and H) and frf�/� larvae (I and J) treated with (H and J) or without (G and I) RA for enhancingosteoblast activity. Despite normal numbers of differentiated osteoblasts in frf mutants, these cells are unable to mineralize thenotochord efficiently upon RA stimulation; this suggests a defect downstream of osteoblast differentiation.
670 The American Journal of Human Genetics 90, 661–674, April 6, 2012

with those published in a previous study (Garrigue-Antar
et al.37), which showed the importance of N-glycosylation
for secretion and stability of BMP1. Compromised BMP1
secretion thus reduces the availability of BMP1 in the
extracellular matrix, potentially leading to the insufficient
processing of substrates, including the procollagen I C-pro-
peptide. Indeed, we were able to demonstrate measurably
reduced processing of two substrates by p.Gly12Arg-
substituted BMP1 in two in vivo assays in zebrafish. Exog-
enous Bmp1 has been described as cleaving the dorsal
determinant, Chordin, and leading to ventralization of
zebrafish embryos.41 We exploited this finding to show
that the p.Gly12Arg-substituted version of BMP1 was less
efficient at ventralizing the embryo than the WT BMP1
(Figure 3). In addition, unlike the WT BMP1, the
p.Gly12Arg-substituted BMP1 was unable to measurably
rescue the larval fin ruffling of the bmp1a zebrafish mutant
(Figure S3); larval fin ruffling is associated with defective
collagen-rod formation. Thus, we have shown that
p.Gly12Arg leads to both reduced secretion and subse-
quent reduced processing of the substrates Chordin and
Collagen I.
Such reduced C-propeptide cleavage predicts the
assembly of procollagen instead of mature collagen into
collagen I fibrils. Immunoblot analysis of both larval and
adult zebrafish protein samples demonstrated that forms
retaining the C-propeptide predominate upon reduction
of Bmp1 function (Figure 8). Unfortunately, no material
was available to show this effect in the affected individuals.
We hypothesize that this results in an impairment of the
collagen matrix within the bone structure and could be
the major cause in the underlying pathomechanism.
Supporting this hypothesis, use of the birefringent
collagen stain, picrosirius red, demonstrated less ordered
collagen-fiber structure in zebrafish bmp1a mutants (Fig-
ure 8). Interestingly, recent studies have assumed that
collagen C-propeptides assembled within collagen fibrils
would either increase the intrafibrillar spacing or directly
serve as nucleators of mineralization.15 This might explain
the high bone mineral density we noted in our individuals
(Figure 1 and Table 1). Our findings provide evidence that
insufficient collagen processing caused by p.Gly12Arg
most likely leads to ectopic accumulation of minerals in
the bone. However, this bone is nonetheless structurally
compromised, leading to fragility. The precise pathophysi-
ology of increased bone mineralization upon defective
collagen processing remains unclear and needs to be inves-
tigated further.
We note similar defects in bone formation in the zebra-
fish frilly fins mutant, and we show that these defects
Figure 8. frf Displays Defects in Fibrillar Collagen Order and Col1a1a Processing(A) Major proteolytic roles of Bmp1 include removing the C-propeptide (orange ovals) of pro-Collagen I and cleaving the BMP2/4 inhib-itor, Chordin (red hexagon), to release free BMP2/4 (green oval).(B–G) Picrosirius-red stained sagittal (B–E) and transverse (F and G) sections of WT (B, D, and F) and frf�/� (C, E, and G) larvae at 11 dpf(B and C), 20 dpf (D and E), and 4 months (F and G). Sections viewed under polarized light reveal the reduced collagen-fiber-associatedbirefringency in the Centra region (B–E) and fin rays (F and G) in frf�/� mutant (C, E, and G) and WT (B, D, and F) larvae.(H and I) Transmission electron micrographs of longitudinal sections of WT (H) and frf�/� (I) larval medial fins at 6 dpf show loss ofstructured collagen fibers in the mutant.(J and K) Immunoblots of protein extracted fromWT (lane 1) and frf�/� mutant (lane 2) larvae probed with an antibody directed againstzebrafish Collagen1a1a (upper panels in both J and K) or an antibody against b-actin as a loading control (lower panels). The fourpossible Collagen1a1 forms are indicated on the right; these forms include Procollagen1a1 retaining both C- and N- terminal propetides(Pro a1[I]), mature collagen a1(I) retaining neither propeptide (a1[I]), a form retaining only the N-propeptide (pN a1[I]), and a formretaining only the C-propeptide (pC a1[I]). In 6 dpf (J) and 4-month-old (K) frf�/� mutants, the two forms retaining the C-propeptidepredominate.
The American Journal of Human Genetics 90, 661–674, April 6, 2012 671

correspond to mutations in bmp1a. Alizarin-red staining
demonstrated delayed ossification at larval stages and mal-
formation of adult skeletal structures with evidence of
fractures (Figure 4). Quantification of mineral content by
microCT analysis demonstrated a higher mineral content
of mature bone, as seen in the individuals. The reason for
the divergent mineral-content phenotypes between the
larval-stage frilly fins mutant and the adult-stage mutant
is currently unclear. We hypothesize that there is a differ-
ence in mineralization rate between the two stages and
that mature collagen is rate limiting during the initial
deposition in larval stages, whereas in adult stages, the
retained telopeptide gradually induces increased minerali-
zation through a mechanism that has not been deter-
mined. Accordingly, the only location found to have
reduced mineralization in adult frilly fins is the distal fin,
a site of bone deposition.
As the first described animal model with reduced
Bmp1 function in an adult, frilly fins was used for the
investigation of the role of this protease in bone forma-
tion. We showed that although generation and patterning
of osteoblasts is unaffected (Figure 6), there was an
intriguing alteration in morphology of the osteoblasts—
they adopted a cuboidal shape (Figure 7). Although this
might indicate a defect in the osteoblasts themselves, we
favor the interpretation that this is a result of reduced
adhesion to the compromised bone matrix. Supporting
this is the in vitro observation that osteoblasts cultured
on bone matrix lacking collagen appeared rounded com-
pared to flattened cells cultured on a purified mineralized
collagen matrix.42
The role of Bmp1 (and other Tolloid-related proteins) in
dorsal-ventral patterning through Chordin cleavage is
well documented. We could, however, conclusively
show that this was not the relevant substrate underlying
the frilly fins bone phenotype (Figure S5). In fact, multiple
lines of evidence from our analysis support the interpreta-
tion that the major role of Bmp1 in ossification is
removal of the C-propeptide from Collagen I. First, we
note similarity of frf to the microwaved mutant, which
we identified as a collagen1a1a mutant (Figure 4). Second,
frf larvae are not able to hyperossify the vertebral column
upon retinoic-acid stimulation of the osteoblasts (Fig-
ure 7), most consistent with a defect that is considerably
downstream in the process of osteoid formation. Finally,
we show compromised Collagen-I processing and
higher-order structure in frf biochemically and histologi-
cally (Figure 8).
While we were preparing this manuscript, Martinez-Glez
et al.43 described a homozygous missense mutation
causing an alteration in the protease domain of BMP1 in
two affected individuals from an Egyptian family affected
by severe autosomal-recessive OI. In line with our findings,
concomitant abnormal procollagen I C-propeptide pro-
cessing was described. Comparison of the individuals’
phenotypes in both studies showed the following differ-
ences: affected individuals in the Egyptian family pre-
sented with classical autosomal-recessive OI, whereas our
individuals, as well as the zebrafish model, presented
with bone fragility associated with an increase in bone
mineral density. Interestingly, Lindahl K. et al.15 very
recently described COL1A1 mutations affecting the BMP1
C-propeptide cleavage site; also, the individuals in this
study presented with an increased-mineralization OI
phenotype very similar to our individuals, suggesting
that impaired BMP1-related collagen C-propeptide cleav-
age (either by mutations in BMP1 or mutations affecting
the BMP1 cleavage site in COL1A1) causes a distinct
form of OI. In contrast, the missense mutation described
by Martinez-Glez et al.43 might have different functional
consequences and thereby cause phenotypic variability
and differences in the severity of the disease.
All together, our combined data in humans and
zebrafish define the molecular and cellular bases of
BMP1-dependent osteogenesis and show the importance
of this protein for bone formation and stability. These
data, in both humans and zebrafish, support the finding
that deficits in removal of the C-propeptide from Collagen
I result in autosomal-recessive OI with high bone mineral
density.15
Supplemental Data
Supplemental Data include five figures and one table and can be
found with this article online at http://www.cell.com/AJHG.
Acknowledgments
We are grateful to all family members that participated in this
study, Esther Milz for excellent technical assistance, Karin Boss
for critically reading the manuscript, and Kaicheng Liang from
the Singapore Bioimaging Consortium for microCT imaging.
This work was supported by the German Federal Ministry of
Education and Research by grant 01GM0880 (SKELNET) to B.W.
The authors would like to thank the National Heart, Lung, and
Blood Institute Grand Opportunity (GO) Exome Sequencing
Project and the following ongoing studies that produced and
provided exome-variant calls for comparison: the Lung GO
Sequencing Project (HL-102923), the Women’s Health Initiative
Sequencing Project (HL-102924), the Broad GO Sequencing
Project (HL-102925), the Seattle GO Sequencing Project (HL-
102926), and the Heart GO Sequencing Project (HL-103010).
Received: November 17, 2011
Revised: January 23, 2012
Accepted: February 24, 2012
Published online: April 5, 2012
Web Resources
The URLs for data presented herein are as follows:
ENSEMBL, http://www.ensembl.org
Exome Variant Server, http://snp.gs.washington.edu/EVS/
OMIM, http://www.ncbi.nlm.nih.gov/omim
PolyPhen, http://coot.embl.de/PolyPhen
UCSC Genome Browser, http://www.genome.ucsc.edu
672 The American Journal of Human Genetics 90, 661–674, April 6, 2012

References
1. Byers, P.H., and Cole, W.G. (2002). Osteogenesis Imperfecta. In
Connective Tissue and its Heritable Disorders: Molecular,
Genetic, and Medical Aspects, Second Edition, P. Royce and B.
Steinmann,eds. (Hoboken,NJ: JohnWiley&Sons),pp.385–430.
2. Sillence, D.O., and Rimoin, D.L. (1978). Classification of
osteogenesis imperfect. Lancet 1, 1041–1042.
3. Basel, D., and Steiner, R.D. (2009). Osteogenesis imperfecta:
Recent findings shed new light on this once well-understood
condition. Genet. Med. 11, 375–385.
4. Rauch, F., and Glorieux, F.H. (2004). Osteogenesis imperfecta.
Lancet 363, 1377–1385.
5. Marini, J.C., Forlino,A.,Cabral,W.A.,Barnes,A.M., SanAntonio,
J.D., Milgrom, S., Hyland, J.C., Korkko, J., Prockop, D.J.,
De Paepe, A., et al. (2007). Consortium for osteogenesis imper-
fectamutations in the helical domain of type I collagen: Regions
rich in lethal mutations align with collagen binding sites for
integrins and proteoglycans. Hum. Mutat. 28, 209–221.
6. Pollitt, R., McMahon, R., Nunn, J., Bamford, R., Afifi, A.,
Bishop, N., and Dalton, A. (2006). Mutation analysis of
COL1A1 andCOL1A2 in patients diagnosed with osteogenesis
imperfecta type I-IV. Hum. Mutat. 27, 716.
7. Morello, R., Bertin, T.K., Chen, Y., Hicks, J., Tonachini, L.,
Monticone, M., Castagnola, P., Rauch, F., Glorieux, F.H.,
Vranka, J., et al. (2006). CRTAP is required for prolyl 3- hydrox-
ylation and mutations cause recessive osteogenesis imper-
fecta. Cell 127, 291–304.
8. Cabral, W.A., Chang, W., Barnes, A.M., Weis, M., Scott, M.A.,
Leikin, S., Makareeva, E., Kuznetsova, N.V., Rosenbaum,
K.N., Tifft, C.J., et al. (2007). Prolyl 3-hydroxylase 1 deficiency
causes a recessive metabolic bone disorder resembling lethal/
severe osteogenesis imperfecta. Nat. Genet. 39, 359–365.
9. Christiansen, H.E., Schwarze, U., Pyott, S.M., AlSwaid, A., Al
Balwi, M., Alrasheed, S., Pepin, M.G., Weis, M.A., Eyre, D.R.,
and Byers, P.H. (2010). Homozygosity for a missense mutation
in SERPINH1, which encodes the collagen chaperone protein
HSP47, results in severe recessive osteogenesis imperfecta. Am.
J. Hum. Genet. 86, 389–398.
10. van Dijk, F.S., Nesbitt, I.M., Zwikstra, E.H., Nikkels, P.G.,
Piersma, S.R., Fratantoni, S.A., Jimenez, C.R., Huizer, M.,
Morsman, A.C., Cobben, J.M., et al. (2009). PPIB mutations
cause severe osteogenesis imperfecta. Am. J. Hum. Genet.
85, 521–527.
11. Lapunzina, P., Aglan, M., Temtamy, S., Caparros-Martın, J.A.,
Valencia, M., Leton, R., Martınez-Glez, V., Elhossini, R., Amr,
K., Vilaboa, N., and Ruiz-Perez, V.L. (2010). Identification of
a frameshift mutation in Osterix in a patient with recessive
osteogenesis imperfecta. Am. J. Hum. Genet. 87, 110–114.
12. Becker, J., Semler, O., Gilissen, C., Li, Y., Bolz, H.J., Giunta, C.,
Bergmann, C., Rohrbach, M., Koerber, F., Zimmermann, K.,
et al. (2011). Exome sequencing identifies truncating muta-
tions in human SERPINF1 in autosomal-recessive osteogenesis
imperfecta. Am. J. Hum. Genet. 88, 362–371.
13. Alanay, Y., Avaygan, H., Camacho, N., Utine, G.E., Boduroglu,
K., Aktas, D., Alikasifoglu, M., Tuncbilek, E., Orhan, D., Bakar,
F.T., et al. (2010). Mutations in the gene encoding the RER
protein FKBP65 cause autosomal-recessive osteogenesis im-
perfecta. Am. J. Hum. Genet. 86, 551–559.
14. Mann, V., and Ralston, S.H. (2003). Meta-analysis of COL1A1
Sp1 polymorphism in relation to bone mineral density and
osteoporotic fracture. Bone 32, 711–717.
15. Lindahl, K., Barnes, A.M., Fratzl-Zelman, N., Whyte, M.P.,
Hefferan, T.E., Makareeva, E., Brusel, M., Yaszemski, M.J., Ru-
bin, C.J., Kindmark, A., et al. (2011). COL1 C-propeptide
cleavage site mutations cause high bone mass osteogenesis
imperfecta. Hum. Mutat. 32, 598–609.
16. Myllyharju, J., and Kivirikko, K.I. (2004). Collagens, modi-
fying enzymes and their mutations in humans, flies and
worms. Trends Genet. 20, 33–43.
17. Bond, J.S., and Beynon, R.J. (1995). The astacin family of met-
alloendopeptidases. Protein Sci. 4, 1247–1261.
18. Sterchi, E.E., Stocker, W., and Bond, J.S. (2008). Meprins,
membrane-bound and secreted astacin metalloproteinases.
Mol. Aspects Med. 29, 309–328.
19. Ge, G., and Greenspan, D.S. (2006). Developmental roles of
the BMP1/TLD metalloproteinases. Birth Defects Res. C
Embryo Today 78, 47–68.
20. Ge, G., and Greenspan, D.S. (2006). BMP1 controls TGFbeta1
activation via cleavage of latent TGFbeta-binding protein.
J. Cell Biol. 175, 111–120.
21. Kessler, E., Takahara, K., Biniaminov, L., Brusel, M., and
Greenspan, D.S. (1996). Bone morphogenetic protein-1: The
type I procollagen C-proteinase. Science 271, 360–362.
22. Canty, E.G., and Kadler, K.E. (2005). Procollagen trafficking,
processing and fibrillogenesis. J. Cell Sci. 118, 1341–1353.
23. Suzuki, N., Labosky, P.A., Furuta, Y., Hargett, L., Dunn, R.,
Fogo, A.B., Takahara, K., Peters, D.M., Greenspan, D.S., and
Hogan, B.L. (1996). Failure of ventral body wall closure in
mouse embryos lacking a procollagen C-proteinase encoded
by Bmp1, a mammalian gene related to Drosophila tolloid.
Development 122, 3587–3595.
24. Parichy, D.M., Elizondo, M.R., Mills, M.G., Gordon, T.N., and
Engeszer, R.E. (2009). Normal table of postembryonic zebra-
fish development: Staging by externally visible anatomy of
the living fish. Dev. Dyn. 238, 2975–3015.
25. van Eeden, F.J., Granato, M., Schach, U., Brand, M., Furutani-
Seiki, M., Haffter, P., Hammerschmidt, M., Heisenberg, C.P.,
Jiang,Y.J., Kane,D.A., et al. (1996).Genetic analysis of fin forma-
tion in the zebrafish, Danio rerio. Development 123, 255–262.
26. Spoorendonk, K.M., Peterson-Maduro, J., Renn, J., Trowe, T.,
Kranenbarg, S., Winkler, C., and Schulte-Merker, S. (2008).
Retinoic acid and Cyp26b1 are critical regulators of osteogen-
esis in the axial skeleton. Development 135, 3765–3774.
27. Geisler, R. (2002). Mapping and cloning. In Zebrafish: A
practical approach, C. Nusslein-Volhard and R. Dahm, eds.
(Oxford: Oxford University Press), pp. 175–212.
28. Kwan, K.M., Fujimoto, E., Grabher, C., Mangum, B.D., Hardy,
M.E., Campbell, D.S., Parant, J.M., Yost, H.J., Kanki, J.P., and
Chien, C.B. (2007). The Tol2kit: A multisite gateway-based
construction kit for Tol2 transposon transgenesis constructs.
Dev. Dyn. 236, 3088–3099.
29. Rentzsch, F., Zhang, J., Kramer, C., Sebald, W., and Ham-
merschmidt, M. (2006). Crossveinless 2 is an essential positive
feedback regulator of Bmp signaling during zebrafish gastrula-
tion. Development 133, 801–811.
30. Balciunas, D.,Wangensteen, K.J.,Wilber, A., Bell, J., Geurts, A.,
Sivasubbu, S., Wang, X., Hackett, P.B., Largaespada, D.A.,
McIvor, R.S., and Ekker, S.C. (2006). Harnessing a high
cargo-capacity transposon for genetic applications in verte-
brates. PLoS Genet. 2, e169.
31. Thisse, C., and Thisse, B. (2008). High-resolution in situ
hybridization to whole-mount zebrafish embryos. Nat. Pro-
toc. 3, 59–69.
The American Journal of Human Genetics 90, 661–674, April 6, 2012 673

32. Brend, T., and Holley, S.A. (2009). Zebrafish whole mount
high-resolution double fluorescent in situ hybridization. J.
Vis. Exp. 25, 1229.
33. Laue, K., Janicke, M., Plaster, N., Sonntag, C., and Ham-
merschmidt, M. (2008). Restriction of retinoic acid activity
by Cyp26b1 is required for proper timing and patterning of
osteogenesis during zebrafish development. Development
135, 3775–3787.
34. Walker, M.B., and Kimmel, C.B. (2007). A two-color acid-free
cartilage and bone stain for zebrafish larvae. Biotech. Histo-
chem. 82, 23–28.
35. Borges, L.F., Gutierrez, P.S., Marana, H.R., and Taboga, S.R.
(2007). Picrosirius-polarization staining method as an effi-
cient histopathological tool for collagenolysis detection in
vesical prolapse lesions. Micron 38, 580–583.
36. Brown, A.M., Fisher, S., and Iovine, M.K. (2009). Osteoblast
maturation occurs in overlapping proximal-distal compart-
ments during fin regeneration in zebrafish. Dev. Dyn. 238,
2922–2928.
37. Garrigue-Antar, L., Hartigan, N., and Kadler, K.E. (2002). Post-
translational modification of bone morphogenetic protein-1
is required for secretion and stability of the protein. J. Biol.
Chem. 277, 43327–43334.
38. Scott, I.C., Blitz, I.L., Pappano, W.N., Imamura, Y., Clark, T.G.,
Steiglitz, B.M., Thomas, C.L., Maas, S.A., Takahara, K., Cho,
K.W., and Greenspan, D.S. (1999). Mammalian BMP-1/
Tolloid-related metalloproteinases, including novel family
member mammalian Tolloid-like 2, have differential enzy-
matic activities and distributions of expression relevant to
patterning and skeletogenesis. Dev. Biol. 213, 283–300.
39. Lee, K.S., Kim, H.J., Li, Q.L., Chi, X.Z., Ueta, C., Komori, T.,
Wozney, J.M., Kim, E.G., Choi, J.Y., Ryoo, H.M., and Bae,
S.C. (2000). Runx2 is a common target of transforming
growth factor beta1 and bone morphogenetic protein 2, and
cooperation between Runx2 and Smad5 induces osteoblast-
specific gene expression in the pluripotent mesenchymal
precursor cell line C2C12. Mol. Cell. Biol. 20, 8783–8792.
40. Fisher, S., and Halpern, M.E. (1999). Patterning the zebrafish
axial skeleton requires early chordin function. Nat. Genet.
23, 442–446.
41. Muraoka, O., Shimizu, T., Yabe, T., Nojima, H., Bae, Y.K.,
Hashimoto, H., and Hibi, M. (2006). Sizzled controls dorso-
ventral polarity by repressing cleavage of the Chordin protein.
Nat. Cell Biol. 8, 329–338.
42. Basle, M.F., Grizon, F., Pascaretti, C., Lesourd, M., and Chap-
pard, D. (1998). Shape and orientation of osteoblast-like cells
(Saos-2) are influenced by collagen fibers in xenogenic bone
biomaterial. J. Biomed. Mater. Res. 40, 350–357.
43. Martınez-Glez, V., Valencia, M., Caparros-Martın, J.A., Aglan,
M., Temtamy, S., Tenorio, J., Pulido, V., Lindert, U., Rohrbach,
M., Eyre, D., et al. (2012). Identification of a mutation
causing deficient BMP1/mTLD proteolytic activity in auto-
somal recessive osteogenesis imperfecta. Hum. Mutat. 33,
343–350.
674 The American Journal of Human Genetics 90, 661–674, April 6, 2012

ARTICLE
A ‘‘Copernican’’ Reassessment of the HumanMitochondrial DNA Tree from its Root
Doron M. Behar,1,2,* Mannis van Oven,3,* Saharon Rosset,4 Mait Metspalu,1 Eva-Liis Loogvali,1
Nuno M. Silva,5 Toomas Kivisild,1,6 Antonio Torroni,7 and Richard Villems1,8
Mutational events along the human mtDNA phylogeny are traditionally identified relative to the revised Cambridge Reference
Sequence, a contemporary European sequence published in 1981. This historical choice is a continuous source of inconsistencies,
misinterpretations, and errors in medical, forensic, and population genetic studies. Here, after having refined the human mtDNA
phylogeny to an unprecedented level by adding information from 8,216 modern mitogenomes, we propose switching the reference
to a Reconstructed Sapiens Reference Sequence, which was identified by considering all available mitogenomes from Homo neandertha-
lensis. This ‘‘Copernican’’ reassessment of the human mtDNA tree from its deepest root should resolve previous problems and will
have a substantial practical and educational influence on the scientific and public perception of human evolution by clarifying the
core principles of common ancestry for extant descendants.
Introduction
Nested hierarchy of species, resulting from the descent
with modification process,1 is fundamental to our under-
standing of the evolution of biological diversity and
life in general. In molecular genealogy, the sequential
accumulation of mutations since the time of the most
recent common ancestor (MRCA) is reflected within the
ever-evolving phylogeny of any genetic locus. Accordingly,
the reconstructed ancestral sequence of a locus should
optimally serve as the reference point for its derived
alleles.2 The human mtDNA phylogeny3–7 is an almost
perfect molecular prototype for a nonrecombining locus,
and knowledge on its variation has been and is extensively
used in medical, genealogical, forensic, and popula-
tion genetic studies.8–11 Boosted by rapid advances in
sequencing and genotyping technology, its mode of inher-
itance, high mutation rate, lack of recombination, and
high cellular copy number have proved critical in making
this locus the primary choice in the field of archaeoge-
netics and ancient DNA.12–14 Although its early synthesis
was based on restriction-fragment-length polymor-
phisms,15–18 control-region variation,19,20 or a combina-
tion of both,21 the human mtDNA phylogeny is now
reconstructed from complete mtDNA sequences,4,6,7,22
thus stretching the phylogenetic resolution to its maxi-
mum. mtDNA also became the main target of ancient-
DNA studies because it is much more abundant than
nuclear DNA.13 The recently published Homo neandertha-
lensis mitogenomes23,24 represent the best available out-
group source for rooting the human mtDNA phylogeny
known to lay inside the contemporary African varia-
tion.22,25,26 Despite these major advances, the extinct
human mtDNA complete root sequence was never
precisely determined, and mtDNA nomenclature remains
cumbersome because it refers to the first completely
sequenced mtDNA,27,28 labeled rCRS, which is now
known to belong to the recently coalescing European
haplogroup H2a2a1.7 The use of the rCRS as a reference
resulted in a number of practical problems such as (1)
the misidentification of derived versus ancestral states
of alleles and (2) the count of nonsynonymous muta-
tions that map to the path between the rCRS and
the case sequences.29 For instance, clinical and func-
tional studies frequently include among the putative
nonsynonymous candidate mutations the haplogroup-
HV-defining transition at position 14766 (CYTB) simply
because the revised Cambridge Reference Sequence
(rCRS) belongs to its derived haplogroup H.30
In this study, to definitively address these issues,
we propose a ‘‘Copernican’’ reassessment of the human
mtDNA phylogeny by switching to a Reconstructed
Sapiens Reference Sequence (RSRS) as the phylogenetically
valid reference point. To this end, the previously suggested
root7,22,25 was updated tomost parsimoniously incorporate
the available mitogenomes from H. neanderthalensis.23,24
Moreover, we further refined the human mtDNA
phylogeny to an unprecedented level by adding informa-
tion from 8,216 mitogenomes and evaluated the ranges
of nucleotide substitutions from the root RSRS rather
than the rCRS28 as a reference point (Figure 1 and Figure S1,
available online).
1Estonian Biocentre and Department of Evolutionary Biology, University of Tartu, Tartu 51010, Estonia; 2Molecular Medicine Laboratory, Rambam Health
Care Campus, Haifa 31096, Israel; 3Department of Forensic Molecular Biology, Erasmus MC, University Medical Center Rotterdam, 3000 CA Rotterdam,
The Netherlands; 4Department of Statistics and Operations Research, School of Mathematical Sciences, Tel Aviv University, Tel Aviv 69978, Israel; 5Instituto
de Patologia e Imunologia Molecular da Universidade do Porto, Porto 4200-465, Portugal; 6Department of Biological Anthropology, University of
Cambridge, Cambridge CB2 1QH, UK; 7Dipartimento di Biologia e Biotecnologie ‘‘L. Spallanzani,’’ Universita di Pavia, Pavia 27100, Italy; 8Estonian
Academy of Sciences, 6 Kohtu Street, Tallinn 10130, Estonia
*Correspondence: [email protected] (D.M.B.), [email protected] (M.v.O.)
DOI 10.1016/j.ajhg.2012.03.002. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 675–684, April 6, 2012 675
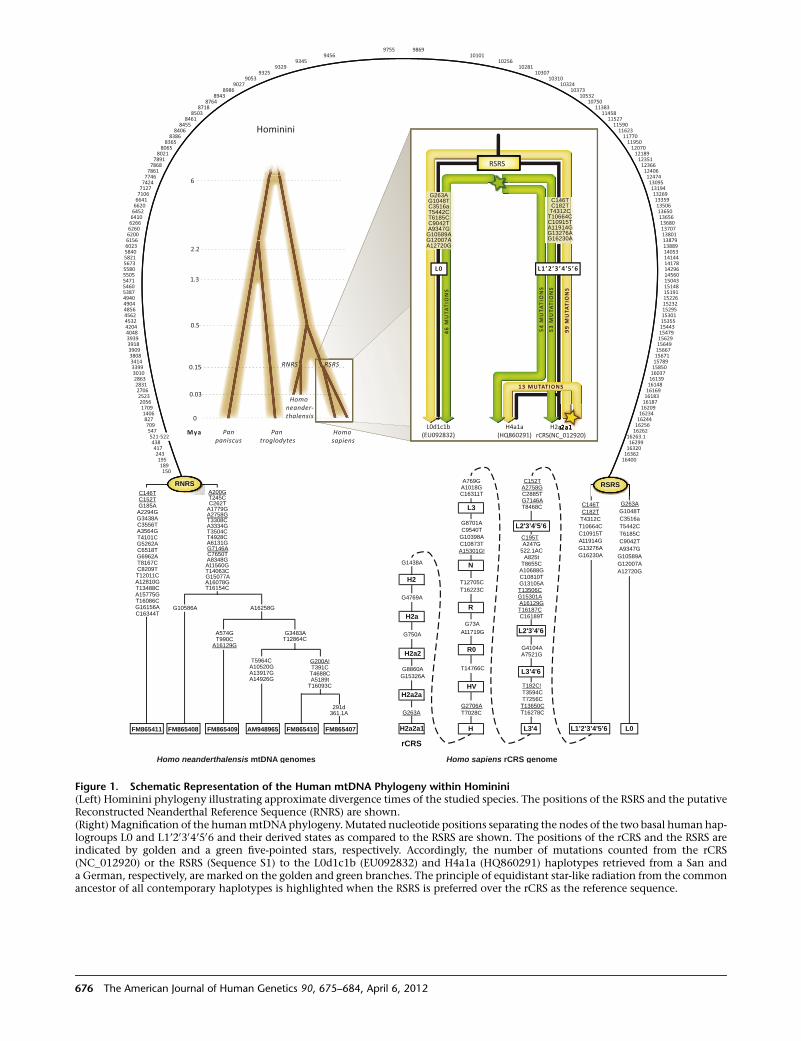
6
1.3
2.2
0.5
0.15
0.03
0L0d1c1b
(EU092832)H2a2a1
rCRS(NC_012920)H4a1a
(HQ860291)
53 M
UTA
TIO
NS
54 M
UTA
TIO
NS
46 M
UTA
TIO
NS
99 M
UTA
TIO
NS
13 MUTATIONS
G10589A
A12720GG12007A
T5442C
C9042TA9347G
G263AG1048TC3516a
T6185C
L0 L1’2’3’4’5’6
A11914GG13276A
C10915T
G16230A
C182TT4312C
C146T
T10664C
Pan paniscus
Pan troglodytes
Homo neander-thalensis
Homo sapiens
RSRSRNRS
Mya
Hominini
RSRS
222a1a1111111a222a1a111111a1a1aa aaa 1122
C8209T
A8348G
T12011C
A11560G
G5262AT4928C
C6518TA6131G
G6962AG7146A
A3564GA3334G
T4101CT3504C
G3438A
T6185C
T245CG263A
C152TG185A C262T
A2294G A1779G
C146T A200G
C146T
T13488C
G15077A
G1048TC182T
T8167C
C7650T
C10915TC9042TA11914G
A15775G
A16078G
C3516aT4312C
T16086C
T16154C
T5442CT10664C
A12810G
T14063C
A2758G
C3556TT3308C
A12720G
A574G G3483AT990C T12864C
C16344T
A9347GG13276AG10589AG16230A
G10586A A16258G
G12007A
G16156A
A14926G A5189tT16093C
291d361.1A
A16129G
T5964C G200A!A10520G T391CA13917G T4688C
L0L1’2’3’4'5’6FM865411 FM865408 FM865409 AM948965 FM865410 FM865407 H2a2a1
H2
H2a2a
H2a
H2a2
C152TA2758GC2885TG7146A
A825tT8655C
A10688GC10810TG13105AT13506C
T8468C
L2'3’4’5’6
C195TA247G
522.1AC
A7521G
L3’4'6
T182C!T3594CT7256CT13650C
G15301AA16129GT16187CC16189T
L2'3’4’6
G4104A
G8701AC9540T
G10398AC10873TA15301G!
N
T16278C
L3'4
A769GA1018GC16311T
L3
T14766C
HV
G2706AT7028C
H
G1438A
T12705CT16223C
R
G73AA11719G
R0
G8860AG15326A
rCRS
G4769A
G750A
G263A
RSRS
97559456
93459329
93259053
90278986
89438764
87188503
84618455
84068386
83658065
80217891
78687861774674247127710666416620645264106266626062006156602358405821567355805505547154605387494049044856456245324204404839393918390938083414339930102863283127062523205617091406827709547521-522438417243195189150
986910101
1025610281
1030710310
1032410373
1053210750
1138311458
11527115901162311770119501207012189123511236612406124741309513194132691335913506136501365613680137071380113879138891405314144141781429614560150431514815191152261523215295153011535515443154791562915649156671567115789158501603716139161481616916183161871620916234162441625616262
16263.116299163201636216400
Homo neanderthalensis mtDNA genomes Homo sapiens rCRS genome
RNRS
Figure 1. Schematic Representation of the Human mtDNA Phylogeny within Hominini(Left) Hominini phylogeny illustrating approximate divergence times of the studied species. The positions of the RSRS and the putativeReconstructed Neanderthal Reference Sequence (RNRS) are shown.(Right)Magnification of the humanmtDNA phylogeny. Mutated nucleotide positions separating the nodes of the two basal human hap-logroups L0 and L1’20304’506 and their derived states as compared to the RSRS are shown. The positions of the rCRS and the RSRS areindicated by golden and a green five-pointed stars, respectively. Accordingly, the number of mutations counted from the rCRS(NC_012920) or the RSRS (Sequence S1) to the L0d1c1b (EU092832) and H4a1a (HQ860291) haplotypes retrieved from a San anda German, respectively, are marked on the golden and green branches. The principle of equidistant star-like radiation from the commonancestor of all contemporary haplotypes is highlighted when the RSRS is preferred over the rCRS as the reference sequence.
676 The American Journal of Human Genetics 90, 675–684, April 6, 2012

Subjects and Methods
Updating the Human mtDNA Phylogeny and
Inference of the Ancestral Root HaplotypeMtDNA Genomes Comprising the Phylogeny
A total of 18,843 complete mtDNA sequences were used to refine
the human mtDNA phylogeny of which 10,627 were previously
reported and used for the mtDNA tree Build 13 (28 Dec 2011)
as posted by PhyloTree.7 The remaining 8,216 sequences are
mainly from the large complete mtDNA database available at
FamilyTreeDNA and in part from data sets maintained by the
authors. The large database available at FamilyTreeDNA was
privately obtained by the sample donors, usually for genealogical
purposes. Most donors were of western Eurasian ancestry, but
donors with matrilineal ancestry from other geographical regions
have also contributed. Once the mtDNA sequences were obtained,
donors had several options: keep them confidential, share them
with peer genealogists, submit them to the National Center for
Biotechnology Information (NCBI) GenBank, and/or consent to
contribute them anonymously to a research database maintained
by FamilyTreeDNA to improve the mtDNA phylogeny. In turn,
this contribution rewards and enriches the genealogical experi-
ence as well as benefits the scientific community. All the proce-
dures followed in this study were in accordance with the ethical
standards of the responsible committee on human experimenta-
tion of the participating research centers.
Likewise, it is important to clarify that because the complete
sequences were obtained privately, some donors have indepen-
dently uploaded their sequence to NCBI. Currently (as of February
28, 2012), a total of 1,220 complete mtDNA sequences that were
generated at FamilyTreeDNA were privately deposited in NCBI
GenBank. Most of these sequences were already considered in
the previous PhyloTree Builds.7 Because we have no way to
know which of the sequences were autonomously uploaded to
NCBI, all duplicate sequences that matched precisely between
NCBI and our database were excluded from our analysis. There-
fore, even if multiple samples were excluded, no topological infor-
mation was lost. Accordingly, out of the 8,216 sequences used
to verify the phylogeny, a total of 4,265 sequences are released
and deposited in NCBI GenBank under accession numbers
JQ701803–JQ706067. The complete mtDNA sequences of the
Neanderthals were retrieved from the literature.23,24
Complete mtDNA Sequencing
DNAwas extracted from buccal swabs. MtDNAwas amplified with
18 primers to yield nine overlapping fragments as previously
reported.22 PCR products were cleaned with magnetic-particle
technology (BioSprint 96; QIAGEN). After purification, the nine
fragments were sequenced by means of 92 internal primers to
obtain the complete mtDNA genome. Sequencing was performed
on a 3730xl DNA Analyzer (Applied Biosystems), and the resulting
sequences were analyzed with the Sequencher software (Gene
Codes Corporation). Mutations were scored relative to the rCRS
and the suggested RSRS. Sample quality control was assured as
follows:
(1) After the PCR amplification of the nine fragments, DNA
handling and distribution to the 96 sequencing reactions
was aided by the Beckman Coulter Biomek FX liquid
handler to minimize the chance for human pipetting
errors.
(2) All 96 sequencing reactions of each sample were performed
simultaneously in the same sequencing run. Most observed
mutations were determined by at least two sequence reads.
However, in a minority of the cases only one sequence read
was available because of various technical reasons, usually
related to the amount and quality of the DNA available.
(3) Any fragment that failed the first sequencing attempt or
any ambiguous base call was tested by additional and
independent PCR and sequencing reactions. In these cases,
the first hypervariable segment (HVS-I) of the control
region was resequenced too to assure that the correct
sample was retrieved.
(4) Genotyping history for each sample was recorded to help
in the search for DNA handling errors and artificial recom-
bination events.
(5) All sequences were aligned with the software Sequencher
(Gene Codes Corporation), and all positions with a Phred
score less than 30 were manually evaluated by an operator.
Two independent operators read each sequence. All posi-
tions that differed from the reference sequences were
recorded electronically to minimize typographic errors.
(6) Any sequence that did not comfortably fit within the estab-
lished human mtDNA phylogeny was highlighted and
resequenced to exclude potential lab errors.
(7) Any comments and remarks raised by external investiga-
tors after release of the data will be addressed by reassessing
the original sequences for accuracy. After that, any unre-
solved result will be further examined by resequencing
and, if necessary, immediately corrected.
Tree Reconstruction and Notation of MutationsThe phylogeny was reconstructed by evaluating both all previ-
ously available published and the herein released complete
mtDNA sequences aiming at the most parsimonious solution
and aided by the software mtPhyl. Polymorphic positions are
shown on the branches and reticulations were resolved by consid-
ering the degree of mutability of individual positions as counted
by their number of occurrences in the overall phylogeny. Both
the ancestral and derived base status for each mutation appearing
in the phylogeny according to the International Union Of Pure
And Applied Chemistry (IUPAC) nucleotide code are reported.
We use capital letters for transitions (e.g., G73A) and lowercase
letters for transversions (e.g., A73t). Although heteroplasmies are
not noted in the phylogeny, we recommend labeling them by
using IUPAC code and capital letters (e.g., G73R). Throughout
the phylogeny indels are given with respect to the RSRS andmain-
tain the traditional nucleotide position numbering as in the rCRS.
Sequencing alignment prefers 30 placement for indels, except in
cases where the phylogeny suggests otherwise.31 Deletions are
indicated by a ‘‘d’’ after the deleted nucleotide position (e.g.,
T15944d). Insertions are indicated by a dot followed by the posi-
tion number and type of inserted nucleotide(s) (e.g., 5899.1C for
a C insertion at the first inserted nucleotide position after position
5899 and 5899.2C for a subsequent C insertion, and these are
abbreviated as 5899.1CC when occurring on the same branch).
We label polynucleotide stretches of unknown length as follows:
573.XC. In cases where an insertion occurred at an ancestral
branch but a reversion of this insertion (¼ deletion) took place
at a descendant branch, we noted the latter as follows:
5899.1Cd. An exclamationmark (!) at the end of a labeled position
denotes a reversion to the ancestral state. The number of exclama-
tion marks stands for the number of sequential reversions in
the given position from the RSRS (e.g., C152T, T152C!, and
The American Journal of Human Genetics 90, 675–684, April 6, 2012 677

C152T!!). Some indel positions have been a source of confusion
because multiple alignment solutions enable alternative scoring.
Notably, the dinucleotide repeat in hypervariable segment II
(HVS-II) of the control region can be viewed either as a CA repeat
starting at position 514 or as an AC repeat starting at position 515,
leading to two different notations being in use for a repeat loss:
522–523d versus 523–524d. We adhered to the guidelines for
consistent treatment of mtDNA-length variants that were estab-
lished by the forensic genetic community31 and favor the AC
interpretation. As the RSRS has one AC unit less compared to
the rCRS, we filled positions 523 and 524 of the RSRS with "NN,"
thereby preserving the historical genome annotation numbering.
Consequently, an AC insertion compared to the RSRS is scored as
522.1AC, whereas an AC deletion is scored as 521–522d. Table S2
presents all common indel positions throughout the complete
mtDNA sequence and the way we labeled them. Transitions at
the hypervariable position 16519, insertions of one or two Cs at
positions 309, 315, and 16193, A to C transversions at 16182
and 16183, as well as length variation of the AC dinucleotide
repeat spanning 515–522, were excluded from the phylogeny.
Haplogroup labels were re-evaluated and the following sugges-
tions were made:
(1) Monophyletic clades that are composed of two or more
previously named haplogroups are labeled by concate-
nating their names and separating them by apostrophe
(e.g., L0a’b). This is not applied in the case of capital-
letter-only labeled haplogroups (e.g., JT);
(2) We suggest labeling an extant sample that matches
a haplogroup root with the superscript case letter n for
‘‘nodal’’ (e.g., Hn);
(3) We note that when completemtDNA sequences are consid-
ered, the inability to differentiate a nodal haplotype from
an unresolved paraphyletic clade is eliminated. Accord-
ingly, the haplogroup label of each observed complete
mtDNA sequences can: (1) mark it in a nodal position; (2)
affiliate it with a previously labeled haplogroup; (3) suggest
a, so far, unlabeled haplogroup; or (4) in the absence of
two additional samples to justify the labeling of a, so far,
unidentified haplogroup, affiliate it with the ancestral
haplogroup. So, the label of a given sample as ‘‘H’’ means
that it is an unlabeled descendent of haplogroup H that
cannot be affiliated to any known H haplogroup clade
at the time of report and based on complete mtDNA
sequence. We suggest restricting the use of label ‘‘H*’’ to
cases where the haplogroup labeling is based on partial
mtDNA sequence;
(4) To aid the nonexpert in understanding the mtDNA hap-
logroup nomenclature system, we summarize in Table S3
the cases where haplogroup labels do not logically follow
from the hierarchy and hence could lead to confusion.
Changing these haplogroup labels to make them more
logical is undesirable at this stage because they are already
used extensively in the literature and therefore changing
them would probably cause even more confusion. In addi-
tion, we note that for the most basal nodes of the
phylogeny, historically the following shorthand names
have been in use: L1’5 ¼ L1’20304’506; L205 ¼ L20304’506;L206 ¼ L20304’6; and L4’6 ¼ L304’6, which we will herein
refer to by their full name. One shorthand haplogroup
name, M4’’67, is maintained because writing it in full
(M4’18’30’37’38’43’45’63’64’65’66’67) seems impractical.
It is important to note that the aim of this study is to publish the
most up-to-date human mtDNA phylogeny, and it cannot be
regarded by any means as a population-level survey exploring
the frequencies and distributions of the various haplogroups.
Therefore, although all sequences were used to establish the tree
topology, the subset of sequences actually presented in the
phylogeny is lower because for each branch up to two representa-
tive example sequences are provided. In most cases, we labeled
haplogroups only when supported by at least three distinct haplo-
types to maximize the accuracy of the haplogroup defining array
of mutations and to avoid the establishment of haplogroups
resulting from sequencing errors. Exceptions included previously
established haplogroups or haplogroups supported by a particu-
larly long array of mutations. Accordingly, the tips of the herein
released phylogeny are in fact internal haplogroup nodes, thus
private mutations (if any) of individual haplotypes were not
included.
Evaluation of the mtDNA Clock and Age EstimatesSubstitution Counts and Molecular Clock
To calculate the substitution counts from the RSRS to every extant
mitogenome (which is a tip in the mtDNA phylogeny), we
summed up the number of mutations on the path leading to
each noted haplogroup in the phylogeny and added to this the
number of positions that differed between the tip and the root
of the haplogroup. Thus, we are guaranteed to correctly count
all parallel and back mutations, except for the case where two
mutations affecting the same position occurred on a branch in
the tree (in which case we either count zero instead of two, if
the second is a back mutation, or one instead of two, if the second
mutation is not back to the initial state). As has been argued in the
past, such repeatedmutations within a single branch in the highly
resolved human mtDNA tree are highly unlikely,32 and are even
more so if the fastest mutating sites (16519 and the A to C trans-
versions and poly-C insertions around the HVS-I position 16189)
are eliminated, as was done in our analysis.
To test the validity of molecular clock assumption on human
mtDNA substitutions, we used PAML 4.4 with the HKY85 substitu-
tion model to generate maximum likelihood estimates of branch
lengths with and without the molecular clock assumption. We
chose to sample around 200–300 sequences and analyze their
coalescent tree (a subtree of the complete tree) in each PAML
run, to accommodate PAML’s computational limitations, and
also to sample mostly deep branches (such as M44), rather than
the recent and very short branches (such as D4a1b1) of the over-
sampled haplogroups such as H and D. Thus, we preferentially
sampled haplogroups whose coalescence with other samples in
the tree was more ancient. This ensured that even in such
a sample, the deeper clades such as the basal M clades would
be represented with high probability, whereas more recently
coalescing haplogroups such as the ones of haplogroup D would
be rarely sampled.
The generalized likelihood ratio (GLR) test for validity of the
clock assumption then uses the test statistic 2 3 (log-likelihood
of non-clock model � log-likelihood of clock model), which,
under the null hypothesis of molecular clock, has a c2 distribution
with degrees of freedom equal to the number of parameters under
no clock (¼ number of branches in the tree) minus number of
parameters under clock (¼ number of internal nodes in the tree).
We performed the analyses on two sets of the mtDNA
sequences: once by using the coding region alone and once on
the entire molecule. This was done as another sanity check for
678 The American Journal of Human Genetics 90, 675–684, April 6, 2012

the validity and generality of our results. All obtained p values are
presented in Table S4.
Age Calculations Assuming a Molecular Clock
In spite of thediscovered clockviolations,wewere still interested in
applying the best available tools for estimating the ages of ancestral
nodes in the tree assuming a molecular clock. We adopted the
calculation approach andmutation rate estimate of,32 who suggest
to estimate ages in substitutions and then transform them to years
in a nonlinear manner accounting for the selection effect on non-
synonymous mutations. We used PAML 4.433 with the HKY85
substitution model to generate maximum likelihood estimates of
internal node ages under a molecular clock assumption. Because
PAML is computationally limited in the size of trees it can analyze,
weperformed estimation for thewhole tree in several separate runs.
We divided the tree into seven collections of haplogroups:
d All L haplogroups (i.e., the entire phylogeny excluding M
and N)
d All of M excluding D
d D and JT
d H excluding H1 and H5
d B4’5 and HV excluding H but including H1 and H5
d U
d N excluding HV, U, JT and B4’5
For each PAML run, we selected all sequences belonging to one
of these sets, and added a small random sample of other samples
from the rest of the phylogeny to maintain ‘‘calibration.’’ Putting
together the estimates from all seven runs provided us with age
estimates for all nodes in our tree. Estimates are given in Table S5.
Data TransitionWe are aware that the suggested change can raise difficulties and
even antagonism from the scientific community. On the other
hand, a scenario in which a reference sequence of a genetic locus
does not represent its ancestral sequence should, indisputably, be
corrected. The realization of the superiority of complete mtDNA
sequence analysis compared to other approaches, combined
with the emergence of deep sequencing technologies, will possibly
shift the entire field into the use of only complete mtDNA
sequences in the near future.34–36 Therefore, the sooner the
change is made the less ‘‘painful’’ it will be. As the common
practice for reporting complete mtDNA sequences is by posting
the sequences as FASTA files to NCBI, rather than reporting the
substitutions with respect to a reference sequence (as in the case
of many data sets restricted to control-region variation), no major
change is needed. When a FASTA file is available or created, the
only change needed is to switch the reference sequence to the
RSRS. For control-region-based data sets, the conversion might
be more problematic as the common practice to report the
sequences in literature did not involve FASTA files but recorded
mutations as compared to the rCRS. Table S6 compares the classic
diagnostic mutations for the major haplogroups relative to the
rCRS or the RSRS.
To facilitate data transition we release the tools ‘‘FASTmtDNA,’’
which allows transformation of Excel list-type reports of mtDNA
haplotypes into FASTA files, and ‘‘mtDNAble,’’ which labels
haplogroups, performs a phylogeny-based quality check and
identifies private substitutions. These noted features are fully
supported in a web interface or as standalone versions, which
can be freely downloaded from thewebsite including theirmanual
and example files. In addition, the web interface allows the
benefit of comparing private substitutions between submitted
and previously stored mitogenomes to suggest the labeling of
additional haplogroups. Following quality check and consent, the
web interface enables the storing of complete mtDNA sequences
by members of the mtDNA community to enrich a growing
database. This in turn is expected to strengthen the data set used
by the website to label haplogroups, perform quality control and
refine the phylogeny. Additional tools will be periodically added
and updated.
Results
The RSRS
Since the sub-Saharan haplogroup L0 was defined,37 it
became clear that the root of the extant variation
of human mitochondrial genomes is allocated between
haplogroups L0 and L1’20304’506, which are separated
from each other by 14 coding and four control-region
mutations22 (Figure 1). Until now, our understanding of
the root of the human mtDNA tree was incomplete
because of the absence of reliable closely related outgroup
mitogenomes, and the exact placement of the 18 muta-
tions separating the L0 and L1’20304’506 nodes remained
vague. In principle, ancient mtDNA from early human
fossils might be informative but unreachable because of
considerable technical problems inherent to the analysis
process.13 However, as the split between H. sapiens and
H. neanderthalensis certainly predates the appearance of
the RSRS,38 a resolution of the deepest node might
be achieved by rooting the human phylogeny with
H. neanderthalensis complete mtDNA sequences23,24
(Figure 1). Table S1 shows all substitutions separating hap-
logroup L0 from L1’20304’506, their status in the six
H. neanderthalensis mitogenomes and their most parsimo-
nious allocation around the human root. Accordingly,
the ancestral mtDNA sequence of extant humans should
correspond to the bifurcation of L0 and L1’20304’506.Although it cannot be excluded that further sampling of
the African mtDNA variation might reveal yet another
more basal clade of the human mtDNA tree, it is at least
equally valid to indicate that, in spite of the many
thousands of reported complete mtDNA sequences,7 such
a clade has not been found so far. Operating under this
assumption we established the reference point, RSRS,
which is made available as Sequence S1.
We present the most resolved human mtDNA
phylogeny by compiling the information from 18,843
mitochondrial genomes of which 10,627 were previously
summarized in PhyloTree Build 13 (28 Dec 2011).7 We fol-
lowed the established cladistic notation for haplogroup
labeling adjusted for complete mtDNA genomes.7,39 Yet,
in contrast with the previously reported phylogeny, all
mutational changes noted on the branches of the tree indi-
cate the actual descendant nucleotide state relative to the
state in the RSRS. Although this has no effect on the tree
topology per se, it is critical to emphasize its major conse-
quences in the way of reporting the list of mutations
The American Journal of Human Genetics 90, 675–684, April 6, 2012 679
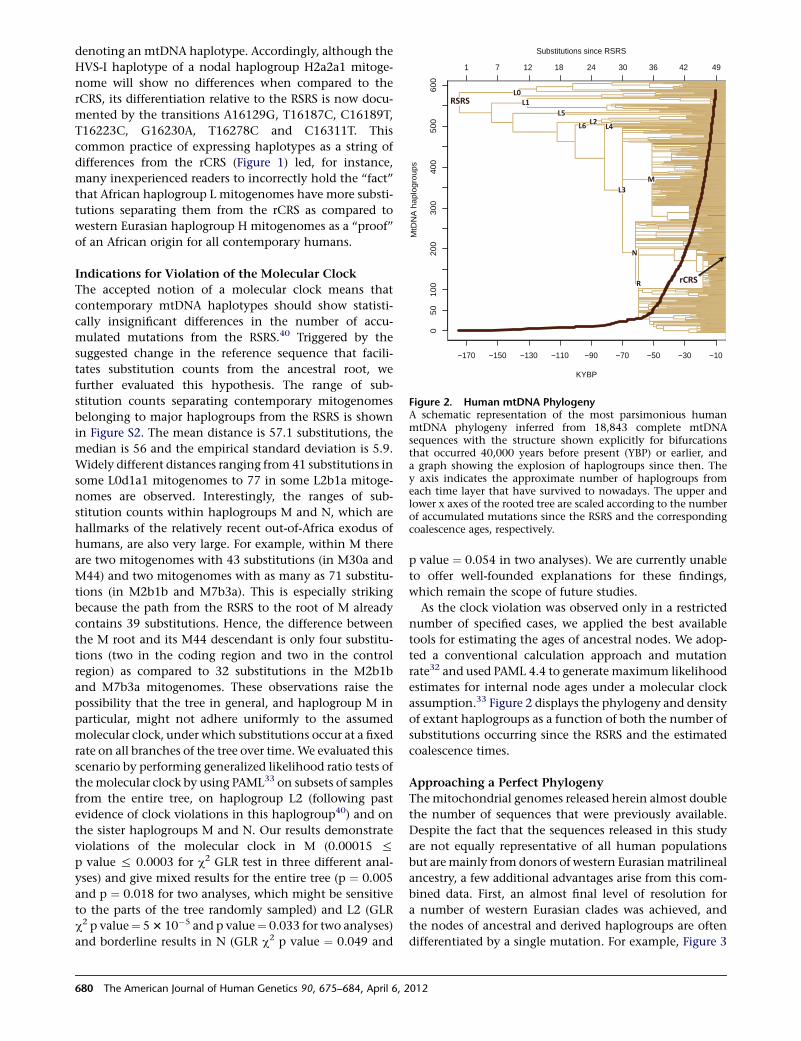
denoting an mtDNA haplotype. Accordingly, although the
HVS-I haplotype of a nodal haplogroup H2a2a1 mitoge-
nome will show no differences when compared to the
rCRS, its differentiation relative to the RSRS is now docu-
mented by the transitions A16129G, T16187C, C16189T,
T16223C, G16230A, T16278C and C16311T. This
common practice of expressing haplotypes as a string of
differences from the rCRS (Figure 1) led, for instance,
many inexperienced readers to incorrectly hold the ‘‘fact’’
that African haplogroup L mitogenomes have more substi-
tutions separating them from the rCRS as compared to
western Eurasian haplogroup H mitogenomes as a ‘‘proof’’
of an African origin for all contemporary humans.
Indications for Violation of the Molecular Clock
The accepted notion of a molecular clock means that
contemporary mtDNA haplotypes should show statisti-
cally insignificant differences in the number of accu-
mulated mutations from the RSRS.40 Triggered by the
suggested change in the reference sequence that facili-
tates substitution counts from the ancestral root, we
further evaluated this hypothesis. The range of sub-
stitution counts separating contemporary mitogenomes
belonging to major haplogroups from the RSRS is shown
in Figure S2. The mean distance is 57.1 substitutions, the
median is 56 and the empirical standard deviation is 5.9.
Widely different distances ranging from 41 substitutions in
some L0d1a1 mitogenomes to 77 in some L2b1a mitoge-
nomes are observed. Interestingly, the ranges of sub-
stitution counts within haplogroups M and N, which are
hallmarks of the relatively recent out-of-Africa exodus of
humans, are also very large. For example, within M there
are two mitogenomes with 43 substitutions (in M30a and
M44) and two mitogenomes with as many as 71 substitu-
tions (in M2b1b and M7b3a). This is especially striking
because the path from the RSRS to the root of M already
contains 39 substitutions. Hence, the difference between
the M root and its M44 descendant is only four substitu-
tions (two in the coding region and two in the control
region) as compared to 32 substitutions in the M2b1b
and M7b3a mitogenomes. These observations raise the
possibility that the tree in general, and haplogroup M in
particular, might not adhere uniformly to the assumed
molecular clock, under which substitutions occur at a fixed
rate on all branches of the tree over time.We evaluated this
scenario by performing generalized likelihood ratio tests of
the molecular clock by using PAML33 on subsets of samples
from the entire tree, on haplogroup L2 (following past
evidence of clock violations in this haplogroup40) and on
the sister haplogroups M and N. Our results demonstrate
violations of the molecular clock in M (0.00015 %
p value % 0.0003 for c2 GLR test in three different anal-
yses) and give mixed results for the entire tree (p ¼ 0.005
and p ¼ 0.018 for two analyses, which might be sensitive
to the parts of the tree randomly sampled) and L2 (GLR
c2 p value¼ 53 10�5 and p value¼ 0.033 for two analyses)
and borderline results in N (GLR c2 p value ¼ 0.049 and
p value ¼ 0.054 in two analyses). We are currently unable
to offer well-founded explanations for these findings,
which remain the scope of future studies.
As the clock violation was observed only in a restricted
number of specified cases, we applied the best available
tools for estimating the ages of ancestral nodes. We adop-
ted a conventional calculation approach and mutation
rate32 and used PAML 4.4 to generate maximum likelihood
estimates for internal node ages under a molecular clock
assumption.33 Figure 2 displays the phylogeny and density
of extant haplogroups as a function of both the number of
substitutions occurring since the RSRS and the estimated
coalescence times.
Approaching a Perfect Phylogeny
Themitochondrial genomes released herein almost double
the number of sequences that were previously available.
Despite the fact that the sequences released in this study
are not equally representative of all human populations
but aremainly from donors of western Eurasianmatrilineal
ancestry, a few additional advantages arise from this com-
bined data. First, an almost final level of resolution for
a number of western Eurasian clades was achieved, and
the nodes of ancestral and derived haplogroups are often
differentiated by a single mutation. For example, Figure 3
−170 −150 −130 −110 −90 −70 −50 −30 −10
050
100
200
300
400
500
600
KYBP
MtD
NA
hap
logr
oups
1 7 12 18 24 30 36 42 49
Substitutions since RSRS
L0L1
L5L2L6 L4
L3M
N
R rCRS
RSRS
Figure 2. Human mtDNA PhylogenyA schematic representation of the most parsimonious humanmtDNA phylogeny inferred from 18,843 complete mtDNAsequences with the structure shown explicitly for bifurcationsthat occurred 40,000 years before present (YBP) or earlier, anda graph showing the explosion of haplogroups since then. They axis indicates the approximate number of haplogroups fromeach time layer that have survived to nowadays. The upper andlower x axes of the rooted tree are scaled according to the numberof accumulated mutations since the RSRS and the correspondingcoalescence ages, respectively.
680 The American Journal of Human Genetics 90, 675–684, April 6, 2012
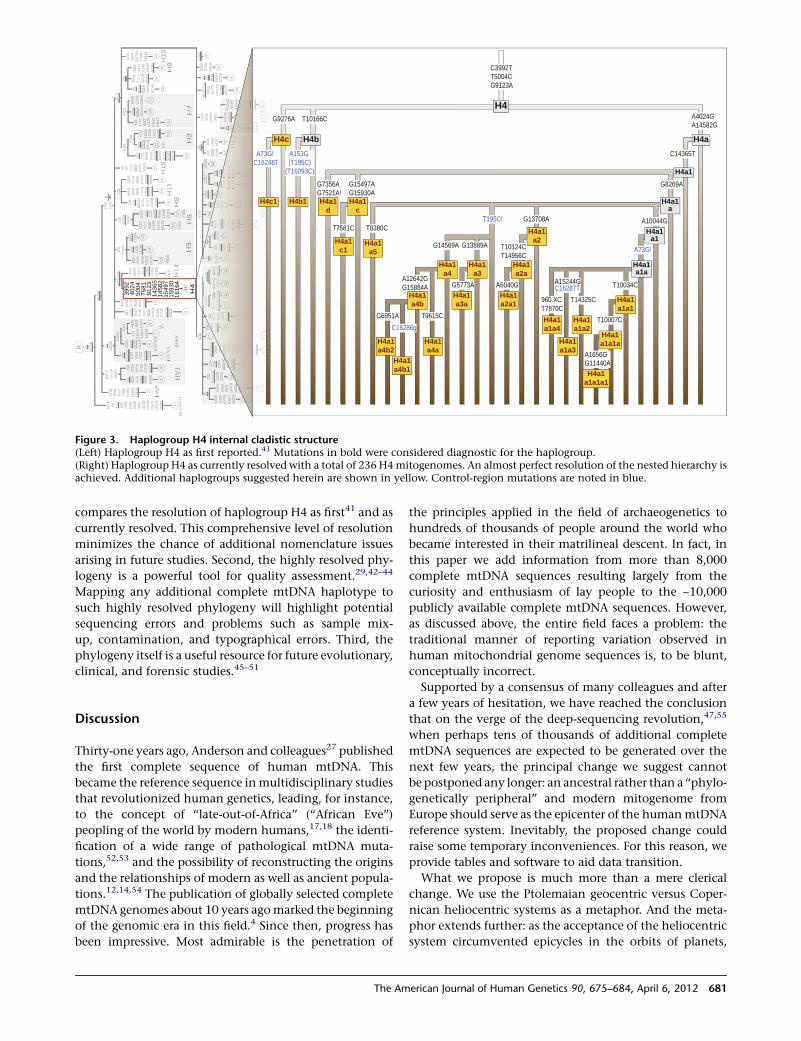
compares the resolution of haplogroup H4 as first41 and as
currently resolved. This comprehensive level of resolution
minimizes the chance of additional nomenclature issues
arising in future studies. Second, the highly resolved phy-
logeny is a powerful tool for quality assessment.29,42–44
Mapping any additional complete mtDNA haplotype to
such highly resolved phylogeny will highlight potential
sequencing errors and problems such as sample mix-
up, contamination, and typographical errors. Third, the
phylogeny itself is a useful resource for future evolutionary,
clinical, and forensic studies.45–51
Discussion
Thirty-one years ago, Anderson and colleagues27 published
the first complete sequence of human mtDNA. This
became the reference sequence inmultidisciplinary studies
that revolutionized human genetics, leading, for instance,
to the concept of ‘‘late-out-of-Africa’’ (‘‘African Eve’’)
peopling of the world by modern humans,17,18 the identi-
fication of a wide range of pathological mtDNA muta-
tions,52,53 and the possibility of reconstructing the origins
and the relationships of modern as well as ancient popula-
tions.12,14,54 The publication of globally selected complete
mtDNA genomes about 10 years agomarked the beginning
of the genomic era in this field.4 Since then, progress has
been impressive. Most admirable is the penetration of
the principles applied in the field of archaeogenetics to
hundreds of thousands of people around the world who
became interested in their matrilineal descent. In fact, in
this paper we add information from more than 8,000
complete mtDNA sequences resulting largely from the
curiosity and enthusiasm of lay people to the ~10,000
publicly available complete mtDNA sequences. However,
as discussed above, the entire field faces a problem: the
traditional manner of reporting variation observed in
human mitochondrial genome sequences is, to be blunt,
conceptually incorrect.
Supported by a consensus of many colleagues and after
a few years of hesitation, we have reached the conclusion
that on the verge of the deep-sequencing revolution,47,55
when perhaps tens of thousands of additional complete
mtDNA sequences are expected to be generated over the
next few years, the principal change we suggest cannot
be postponed any longer: an ancestral rather than a ‘‘phylo-
genetically peripheral’’ and modern mitogenome from
Europe should serve as the epicenter of the humanmtDNA
reference system. Inevitably, the proposed change could
raise some temporary inconveniences. For this reason, we
provide tables and software to aid data transition.
What we propose is much more than a mere clerical
change. We use the Ptolemaian geocentric versus Coper-
nican heliocentric systems as a metaphor. And the meta-
phor extends further: as the acceptance of the heliocentric
system circumvented epicycles in the orbits of planets,
7311
719
R
1476
6
d522
-523 1276
4510
217
1137
712
879
1476
616
256
1635
2
3992
4024
5004
7581
9123
1436
514
582
1549
715
930
1616
411 H4
d522
-523
9033
1077
513
513h
1620
916
215T
59
H1
4
456
1630
4
200
4336
5839
1552
116
093
5471
1286
4
13 H5
a
H5
15
709
1608
1618
9
14
239
1636
216
482 44
+ C15
221
462
6386
6814
040
1630
0
3915
4727
9380
1058
916
129
1624
9
16
H6
aH
6b
H617
55 57 1117
3847
6253
1099
3
21
H1
5
1651
9
152 72 183
1598
1606
616
239
60
3460
3786
1153
6
61
1636
2
62
73 8557
9368
1235
816
145
28
6908
7711
1551
916
291
29
3591
4310
9148
1302
016
168
30 H9
3010
6776
73
6320
8468
9921
1497
816
051
1616
216
259
H1
a
33
1808
5460
1378
215
817
1631
8
32
d522
-523
2483
3796
5899
+2C
7870
8348
9022
1256
116
189
1635
616
362
H1
b
36
236
709
1900
5899
+C60
4016
294
35
228
523+
CA
1129
916
233
34
368
1000
316
291
38
723
7271
8952
1154
916
311
39
1428
7
3666
1171
940
6216
294
4041
1623
4
42
573+
3C13
943
43
1504
716
189
37
4769
152
1081
016
274
1842
1123
313
708
1432
316
291
23
H224
H2c
1438
152
319
8598
1328
113
928
1626
616
311
1636
216
519
22
93
95C
1555
8258
1590
2
45
5471
1479
8
46
152
4679
1287
913
404
1415
216
239G
1631
1
47 H3
a
73 761
1432
5
44
183
709
2581
3387
G59
11
49
1295
7
72 150
1536
1066
714
467
195
1555
1420
016
176
1651
9
5251
1555
1623
4
50
1629
0
53
4793
185
1719
8573
1310
514
560
1621
3
1598
6296
A16
265
26 H7
25
48
195
961G
8448
8898
1375
916
278
1631
1
2392
6719
9530
1263
316
209
1639
9
252
2308
1036
1
19
54
H1
1
146
709
1310
1C16
111
1616
716
288
1636
2
3936
1455
216
287
18
55
H8
H1
2
20
195
4216
5378
1447
0A14
548
1611
4
H1
031
2259
4745
1368
014
872
93 7337
1304
213
326
573+
C16
519
7471
+C94
4911
563
1354
215
712
1627
816
311
H1
3
56
57
H3
H1
58
H1
3a
2706
7028
*
275348
1235
113
266C
60+T 64 152
153
2355
2442
3438
3847
1072
813
188
1567
416
126
1636
2
150
3290
5134
6263
9585
1269
6
2758
3834
6317
7094
1035
611
252
1616
843
711
674
1480
016
320
(pre
-HV
)1H
V1
HV
*V
2
3
1
7
195
523+
CA
5093
6059
7762
1171
913
933
5
7216
298
pre
*V1
1590
4
5581
8557
1522
116
222
6
pre
* V2
pre
-V
8014
T15
218
1606
7 750
7569
8376
9755
1353
516
519
4
4919
6285
1273
214
299
1624
116
311
237
1555
3531
4715
5201
8838
1045
412
362
1273
013
928
1633
5
10
9
4639
8869
1037
9
8
4580
737311
719
1171
9
R
1476
614
766
d522
d522
-523523 1276
4510
217
1137
712
879
1287
914
766
1476
616
256
1635
2
d522
d522
-52352
390
3310
775
1351
3h16
209
1620
916
215T
59
H1
4
456
1630
4
200
4336
5839
1552
116
093
5471
5471
1286
4
13
aH
5a
5H
5
15
709
709
1608
1618
916
189
14
239
1636
216
362
1648
2 44+ C
152
152
214
6263
6263
8668
1404
016
300
3915
4727
9380
1058
916
129
1624
9
16
aH
6a
bH
6b
6H
617
55 57 1117
3847
6253
1099
3
21
H1
5
1651
916
519
152
152 7272 183
183
1598
1598
1606
616
239
60
3460
3786
1153
6
61
1636
216
362
62
7373 8557
8557
9368
1235
816
145
28
6908
7711
1551
916
291
1629
1
29
3591
4310
9148
1302
016
168
1616
8
30 H9
3010
6776
7373
6320
8468
9921
1497
816
051
161
1808
5460
1378
215
817
1631
8
32
d522
d522
-523523
2483
3796
5
236
709
709
1900
5899
+C60
4
228
523+
CA
523
CA
1129
916
233
34
368
1037
4769
152
152
1081
016
274
1842
1123
313
708
1432
316
291
1629
123
2H
224
H2c
1438
152
152
319
8598
1328
113
928
1392
816
266
1631
116
311
1636
216
362
1651
916
519
22
93
218
318
370
9
1295
7
7272 5019
519
515
55555
1555
1555
1623
416
234
1629
0
53
4793
185
1719
8573
1310
514
560
1621
3
1598
1598
6296
A16
265
26
7H
7
25
48
195
961G
8448
8898
1375
916
278
1627
816
311
1631
1
2392
6719
9530
1263
316
209
1620
916
399
252
2308
1036
1
19
54
H1
1
146
709
709
1310
1C16
111
1616
716
288
1636
216
362
3936
1455
216
287
18
55
H8
20
195
195
4216
5378
1447
0A14
548
1611
4
H1
031
2259
4745
1368
014
872
9393 7337
573+
C16
519
1651
974
71+C
9449
1156
3 2
2706
7028
*
275348
1235
113
266C
60+T 64 152
152
153
2355
2442
3438
3847
1072
813
188
1567
416
126
1636
216
362
150
150
3290
5134
6263
6263
9585
1269
6
2758
3834
6317
7094
1035
611
252
1616
816
168
437
1167
414
800
1632
0
(pre
-HV
--)1
HV
1
HV
*VV
V
2
3
1
7
195
195
523+
CA
523
CA
5093
6059
7762
1171
911
719
1393
3
5
727216
298
pre
*V1
**
1590
4
5581
8557
8557
1522
116
222
6
pre
*2
V2
**
pre
-V
8014
T15
218
1606
7 750
7569
8376
9755
1353
516
519
1651
9
4
4919
6285
1273
214
299
1624
116
311
237
1555
3531
4715
5201
8838
1045
412
362
1273
013
928
1633
5
10
9
4639
8869
1037
9
8
4580
aH
1a
3316
362
1636
2
H1
b
36
aH
3a
1631
116
311
3H
13
3H
3H
158
H1
3a
1635
6
162
1616
216
259
1625
932
796
3796
5899
+2C
7870
8348
9022
9022
9022
1256
116
189
1618
916
356
1635
6
C60
4016
294
1629
4
3535
8810
003
1629
116
291
38
723
723
7271
7271
8952
1154
916
311
1631
1
393939
1428
78
3666
3666
1171
911
719
4062
4062
1629
416
294
4041
1623
416
234
63
42
573+
573+
3C3C13
943
43
1504
715
047
1618
916
189
939393
95C
95C
1555
1555
1555
8258
1590
2
454545
5471
5471
5471
5471
1479
8
46
152
152
4679
4679
1287
912
879
1287
913
404
1415
216
239G
1631
116
311
1631
116
311
1631
1
4747
73737373 761
1432
5
44
709
709
709
709
2581
3387
G59
11
494949
150
150
150
1536
1066
714
467
115 1420
016
176
1651
9
52525151
50
5
H1
273 13
042
1304
213
326
1332
61 13
542
1571
215
712
1627
816
278
1627
85656
575757
1635
6
C3992T T5004CG9123A
A4024GA14582G
C14365T
G8269A
A10044G
T10034C
T10007C
A1656GG11440A
T14325C
A15244G
960.XC T7870C
G13708A
T10124CT14956C
A6040G
G13889A
G5773A
G14569A
T9615C
A12642GG15884A
G6951A
T8380C
G15497AG15930A
T7581C
G7356A G7521A!
T10166CG9276A
A73G!
C16287T
T195C!
C16286g
A153G (T195C)
(T16093C)
A73G! C16248T
H4a1
c
H4a1
c1
H4a1
d
H4b1
H4c
H4c1
H4a1
a3
H4a1
a3a
H4a1
a4
H4a1
a4a
H4a1
a4b
H4a1
a4b1
H4a1
a4b2
H4a1
a5
H4a1
a1a1
H4a1
a1a1a
H4a1
a1a1a1
H4a1
a1a2
H4a1
a1a3
H4a1
a1a4
H4a1
a2
H4a1
a2a
H4a1
a2a1
H4a1
c
H4a1
c1
H4a1
d
H4b1
H4c
H4c1
H4a1
a3
H4a1
a3a
H4a1
a4
H4a1
a4a
H4a1
a4b
H4a1
a4b1
H4a1
a4b2
H4a1
a5
H4a1
a1a1
H4a1
a1a1a
H4a1
a1a1a1
H4a1
a1a2
H4a1
a1a3
H4a1
a1a4
H4a1
a2
H4a1
a2a
H4a1
a2a1
H4b
H4
H4a
H4a1
H4a1
a
H4a1
a1
H4a1
a1a
H4b
H4
H4a
H4a1
H4a1
a
H4a1
a1
H4a1
a1a
Figure 3. Haplogroup H4 internal cladistic structure(Left) Haplogroup H4 as first reported.41 Mutations in bold were considered diagnostic for the haplogroup.(Right) Haplogroup H4 as currently resolved with a total of 236 H4mitogenomes. An almost perfect resolution of the nested hierarchy isachieved. Additional haplogroups suggested herein are shown in yellow. Control-region mutations are noted in blue.
The American Journal of Human Genetics 90, 675–684, April 6, 2012 681

switching the mtDNA reference to an ancestral RSRS will
end an academically inadmissible conjuncture where
virtually all mitochondrial genome sequences are scored
in part from derived-to-ancestral states and in part from
ancestral-to-derived states. We aim to trigger the radical
but necessary change in the way mtDNA mutations are
reported relative to their ancestral versus derived status,
thus establishing an intellectual cohesiveness with the
current consensus of shared common ancestry of all con-
temporary human mitochondrial genomes.
Note that the problem is not restricted to mtDNA.
Indeed, in themuch larger perspective of complete nuclear
genomes in which comparisons are often currently made
relative to modern human reference sequences, often of
European origin, it seems worthwhile to begin consid-
ering, as valuable alternatives, public reference sequences
of ancestral alleles (common in all primates) whereby
derived alleles (common to some human populations)
would be distinguished.
Supplemental Data
Supplemental Data include two figures, six tables, and one
sequence and can be found with this article online at http://
www.cell.com/AJHG/.
Acknowledgments
We thank the genealogical community for donating their
privately obtained complete mtDNA sequences for scientific
studies and FamilyTreeDNA for compiling the data. We thank
FamilyTreeDNA for supporting the establishment of the herein
released website. We thank Eileen Krauss-Murphy of Family-
TreeDNA for help with assembly of the database. We thank
Rebekah Canada and William R. Hurst for help with the assembly
of haplogroup H and K samples, respectively. R.V. and D.M.B.
thank the European Commission, Directorate-General for
Research for FP7 Ecogene grant 205419. D.M.B. is a shareholder
of FamilyTreeDNA and a member of its scientific advisory board.
R.V. and M.M. thank the European Union, Regional Development
Fund for a Centre of Excellence in Genomics grant, and R.V.
thanks the Swedish Collegium for Advanced Studies for support
during the initial stage of this study. M.M. thanks Estonian Science
Foundation for grant 8973. A.T. received support from Fondazione
Alma Mater Ticinensis and the Italian Ministry of Education,
University and Research: Progetti Ricerca Interesse Nazionale
2009. S.R. thanks the Israeli Science Foundation for grant 1227/
09 and IBM for an Open Collaborative Research grant. FCT, the
Portuguese Foundation for Science and Technology, partially sup-
ported this work through the personal grant N.M.S. (SFRH/BD/
69119/2010). Instituto de Patologia e Imunologia Molecular da
Universidade do Porto is an Associate Laboratory of the Portuguese
Ministry of Science, Technology and Higher Education and is
partially supported by the Portuguese Foundation for Science
and Technology.
Received: January 9, 2012
Revised: February 22, 2012
Accepted: March 2, 2012
Published online: April 5, 2012
Web Resources
The URLs for data presented herein are as follows:
FASTmtDNA, http://www.mtdnacommunity.org
mtDNAble, http://www.mtdnacommunity.org
mtPhyl, http://eltsov.org/mtphyl.aspx
PhyloTree, http://www.phylotree.org
Accession Numbers
The 4,265 complete mtDNA sequences reported herein have been
submitted to GenBank (accession numbers JQ701803–JQ706067).
References
1. Darwin, C. (1859). Natural Selection. On the Origin of
Species by Means of Natural Selection, or, The Preservation
of Favoured Races in the Struggle for Life, Chapter 4 (London:
John Murray).
2. Delsuc, F., Brinkmann, H., and Philippe, H. (2005). Phyloge-
nomics and the reconstruction of the tree of life. Nat. Rev.
Genet. 6, 361–375.
3. Kivisild, T., Metspalu, E., Bandelt, H.J., Richards, M., and
Villems, R. (2006). The world mtDNA phylogeny. In Human
mitochondrial DNA and the evolution of Homo sapiens, H.J.
Bandelt, V. Macaulay, and M. Richards, eds. (Berlin: Springer-
Verlag), pp. 149–179.
4. Ingman, M., Kaessmann, H., Paabo, S., and Gyllensten, U.
(2000). Mitochondrial genome variation and the origin of
modern humans. Nature 408, 708–713.
5. Richards, M., and Macaulay, V. (2001). The mitochondrial
gene tree comes of age. Am. J. Hum. Genet. 68, 1315–1320.
6. Torroni, A., Achilli, A., Macaulay, V., Richards, M., and
Bandelt, H.J. (2006). Harvesting the fruit of the human
mtDNA tree. Trends Genet. 22, 339–345.
7. van Oven, M., and Kayser, M. (2009). Updated comprehensive
phylogenetic tree of global human mitochondrial DNA
variation. Hum. Mutat. 30, E386–E394.
8. Underhill, P.A., and Kivisild, T. (2007). Use of y chromosome
and mitochondrial DNA population structure in tracing
human migrations. Annu. Rev. Genet. 41, 539–564.
9. Salas, A., Bandelt, H.J., Macaulay, V., and Richards, M.B.
(2007). Phylogeographic investigations: The role of trees in
forensic genetics. Forensic Sci. Int. 168, 1–13.
10. Shriver, M.D., and Kittles, R.A. (2004). Genetic ancestry and
the search for personalized genetic histories. Nat. Rev. Genet.
5, 611–618.
11. Taylor, R.W., and Turnbull, D.M. (2005). Mitochondrial DNA
mutations in human disease. Nat. Rev. Genet. 6, 389–402.
12. Gilbert,M.T.,Kivisild,T.,Grønnow,B.,Andersen, P.K.,Metspalu,
E., Reidla,M., Tamm, E., Axelsson, E., Gotherstrom,A., Campos,
P.F., et al. (2008). Paleo-Eskimo mtDNA genome reveals matri-
lineal discontinuity in Greenland. Science 320, 1787–1789.
13. Gilbert, M.T., Hansen, A.J., Willerslev, E., Rudbeck, L., Barnes,
I., Lynnerup, N., and Cooper, A. (2003). Characterization of
genetic miscoding lesions caused by postmortem damage.
Am. J. Hum. Genet. 72, 48–61.
14. Haak, W., Forster, P., Bramanti, B., Matsumura, S., Brandt, G.,
Tanzer, M., Villems, R., Renfrew, C., Gronenborn, D., Alt,
K.W., and Burger, J. (2005). Ancient DNA from the first Euro-
pean farmers in 7500-year-old Neolithic sites. Science 310,
1016–1018.
682 The American Journal of Human Genetics 90, 675–684, April 6, 2012

15. Denaro, M., Blanc, H., Johnson, M.J., Chen, K.H., Wilmsen,
E., Cavalli-Sforza, L.L., and Wallace, D.C. (1981). Ethnic vari-
ation in Hpa 1 endonuclease cleavage patterns of human
mitochondrial DNA. Proc. Natl. Acad. Sci. USA 78, 5768–5772.
16. Brown,W.M. (1980). Polymorphism inmitochondrial DNA of
humans as revealed by restriction endonuclease analysis. Proc.
Natl. Acad. Sci. USA 77, 3605–3609.
17. Cann, R.L., Stoneking, M., and Wilson, A.C. (1987). Mito-
chondrial DNA and human evolution. Nature 325, 31–36.
18. Vigilant, L., Stoneking, M., Harpending, H., Hawkes, K., and
Wilson, A.C. (1991). African populations and the evolution
of human mitochondrial DNA. Science 253, 1503–1507.
19. Richards, M., Corte-Real, H., Forster, P., Macaulay, V.,
Wilkinson-Herbots, H., Demaine, A., Papiha, S., Hedges, R.,
Bandelt, H.J., and Sykes, B. (1996). Paleolithic and neolithic
lineages in the European mitochondrial gene pool. Am. J.
Hum. Genet. 59, 185–203.
20. Torroni, A., Bandelt, H.J., D’Urbano, L., Lahermo, P., Moral, P.,
Sellitto, D., Rengo, C., Forster, P., Savontaus, M.L., Bonne-
Tamir, B., and Scozzari, R. (1998). mtDNA analysis reveals
a major late Paleolithic population expansion from south-
western to northeastern Europe. Am. J. Hum. Genet. 62,
1137–1152.
21. Torroni, A., Schurr, T.G., Cabell, M.F., Brown, M.D., Neel, J.V.,
Larsen, M., Smith, D.G., Vullo, C.M., and Wallace, D.C.
(1993). Asian affinities and continental radiation of the four
founding Native American mtDNAs. Am. J. Hum. Genet. 53,
563–590.
22. Behar, D.M., Villems, R., Soodyall, H., Blue-Smith, J., Pereira,
L., Metspalu, E., Scozzari, R., Makkan, H., Tzur, S., Comas,
D., et al; Genographic Consortium. (2008). The dawn of
human matrilineal diversity. Am. J. Hum. Genet. 82, 1130–
1140.
23. Briggs, A.W., Good, J.M., Green, R.E., Krause, J., Maricic, T.,
Stenzel, U., Lalueza-Fox, C., Rudan, P., Brajkovic, D., Kucan,
Z., et al. (2009). Targeted retrieval and analysis of five Nean-
dertal mtDNA genomes. Science 325, 318–321.
24. Green, R.E., Malaspinas, A.S., Krause, J., Briggs, A.W., Johnson,
P.L., Uhler, C., Meyer, M., Good, J.M., Maricic, T., Stenzel, U.,
et al. (2008). A complete Neandertal mitochondrial genome
sequence determined by high-throughput sequencing. Cell
134, 416–426.
25. Kivisild, T., Shen, P., Wall, D.P., Do, B., Sung, R., Davis, K.,
Passarino, G., Underhill, P.A., Scharfe, C., Torroni, A., et al.
(2006). The role of selection in the evolution of human mito-
chondrial genomes. Genetics 172, 373–387.
26. Kivisild, T., Reidla, M., Metspalu, E., Rosa, A., Brehm, A.,
Pennarun, E., Parik, J., Geberhiwot, T., Usanga, E., and
Villems, R. (2004). Ethiopian mitochondrial DNA heritage:
Tracking gene flow across and around the gate of tears. Am.
J. Hum. Genet. 75, 752–770.
27. Anderson, S., Bankier, A.T., Barrell, B.G., de Bruijn, M.H.,
Coulson, A.R., Drouin, J., Eperon, I.C., Nierlich, D.P., Roe,
B.A., Sanger, F., et al. (1981). Sequence and organization of
the human mitochondrial genome. Nature 290, 457–465.
28. Andrews, R.M., Kubacka, I., Chinnery, P.F., Lightowlers, R.N.,
Turnbull, D.M., and Howell, N. (1999). Reanalysis and
revision of the Cambridge reference sequence for human
mitochondrial DNA. Nat. Genet. 23, 147.
29. Yao, Y.G., Salas, A., Bravi, C.M., and Bandelt, H.J. (2006).
A reappraisal of completemtDNAvariation in East Asian fami-
lies with hearing impairment. Hum. Genet. 119, 505–515.
30. Pello, R., Martın, M.A., Carelli, V., Nijtmans, L.G., Achilli, A.,
Pala, M., Torroni, A., Gomez-Duran, A., Ruiz-Pesini, E., Marti-
nuzzi, A., et al. (2008). Mitochondrial DNA background
modulates the assembly kinetics of OXPHOS complexes in
a cellular model of mitochondrial disease. Hum. Mol. Genet.
17, 4001–4011.
31. Bandelt, H.J., and Parson, W. (2008). Consistent treatment
of length variants in the human mtDNA control region:
A reappraisal. Int. J. Legal Med. 122, 11–21.
32. Soares, P., Ermini, L., Thomson, N., Mormina, M., Rito, T.,
Rohl, A., Salas, A., Oppenheimer, S., Macaulay, V., and Ri-
chards, M.B. (2009). Correcting for purifying selection: An
improved human mitochondrial molecular clock. Am. J.
Hum. Genet. 84, 740–759.
33. Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum
likelihood. Mol. Biol. Evol. 24, 1586–1591.
34. Tang, S., and Huang, T. (2010). Characterization of mitochon-
drial DNA heteroplasmy using a parallel sequencing system.
Biotechniques 48, 287–296.
35. Li, M., Schonberg, A., Schaefer, M., Schroeder, R., Nasidze, I.,
and Stoneking, M. (2010). Detecting heteroplasmy from
high-throughput sequencing of complete human mitochon-
drial DNA genomes. Am. J. Hum. Genet. 87, 237–249.
36. Zaragoza, M.V., Fass, J., Diegoli, M., Lin, D., and Arbustini, E.
(2010). Mitochondrial DNA variant discovery and evaluation
in human Cardiomyopathies through next-generation
sequencing. PLoS ONE 5, e12295.
37. Mishmar, D., Ruiz-Pesini, E., Golik, P., Macaulay, V., Clark,
A.G., Hosseini, S., Brandon, M., Easley, K., Chen, E., Brown,
M.D., et al. (2003). Natural selection shaped regional mtDNA
variation in humans. Proc. Natl. Acad. Sci. USA 100, 171–176.
38. Green, R.E., Krause, J., Briggs, A.W., Maricic, T., Stenzel, U.,
Kircher, M., Patterson, N., Li, H., Zhai, W., Fritz, M.H., et al.
(2010). A draft sequence of the Neandertal genome. Science
328, 710–722.
39. Richards, M.B., Macaulay, V.A., Bandelt, H.J., and Sykes, B.C.
(1998). Phylogeography of mitochondrial DNA in western
Europe. Ann. Hum. Genet. 62, 241–260.
40. Torroni, A., Rengo, C., Guida, V., Cruciani, F., Sellitto, D.,
Coppa, A., Calderon, F.L., Simionati, B., Valle, G., Richards,
M., et al. (2001). Do the four clades of the mtDNA haplogroup
L2evolve at different rates?Am. J.Hum.Genet.69, 1348–1356.
41. Achilli, A., Rengo, C., Magri, C., Battaglia, V., Olivieri, A., Scoz-
zari, R., Cruciani, F., Zeviani, M., Briem, E., Carelli, V., et al.
(2004). The molecular dissection of mtDNA haplogroup H
confirms that the Franco-Cantabrian glacial refugewas amajor
source for the European gene pool. Am. J. Hum. Genet. 75,
910–918.
42. Parson, W., and Bandelt, H.J. (2007). Extended guidelines for
mtDNA typing of population data in forensic science. Forensic
Sci. Int. Genet. 1, 13–19.
43. Salas, A., Carracedo, A., Macaulay, V., Richards, M., and
Bandelt, H.J. (2005). A practical guide to mitochondrial DNA
error prevention in clinical, forensic, and population genetics.
Biochem. Biophys. Res. Commun. 335, 891–899.
44. Bandelt, H.J., Lahermo, P., Richards, M., and Macaulay, V.
(2001). Detecting errors in mtDNA data by phylogenetic
analysis. Int. J. Legal Med. 115, 64–69.
45. Ballantyne, K.N., vanOven,M., Ralf, A., Stoneking,M., Mitch-
ell, R.J., van Oorschot, R.A., and Kayser, M. (2011). MtDNA
SNP multiplexes for efficient inference of matrilineal genetic
ancestry within Oceania. Forensic Sci. Int. Genet., in press.
The American Journal of Human Genetics 90, 675–684, April 6, 2012 683

Published online September 20, 2011. 10.1016/j.fsigen.2011.
08.010.
46. Pereira, L., Soares, P., Radivojac, P., Li, B., and Samuels, D.C.
(2011).Comparing phylogeny and thepredictedpathogenicity
of protein variations reveals equal purifying selection across
the global human mtDNA diversity. Am. J. Hum. Genet. 88,
433–439.
47. Behar, D.M., Harmant, C., Manry, J., van Oven, M., Haak, W.,
Martinez-Cruz, B., Salaberria, J., Oyharcabal, B., Bauduer, F.,
Comas, D., and Quintana-Murci, L.; Consortium. TG.
(2012). The Basque paradigm: Genetic evidence of a maternal
continuity in the Franco-Cantabrian Region since pre-
Neolithic times. Am. J. Hum. Genet. 90, 486–493.
48. Zeviani, M., and Carelli, V. (2007). Mitochondrial disorders.
Curr. Opin. Neurol. 20, 564–571.
49. Gunnarsdottir, E.D., Nandineni, M.R., Li, M., Myles, S., Gil,
D., Pakendorf, B., and Stoneking, M. (2011). Larger mitochon-
drial DNA than Y-chromosome differences betweenmatrilocal
and patrilocal groups from Sumatra. Nat. Commun. 2, 228.
50. Baum, D.A., Smith, S.D., and Donovan, S.S. (2005). Evolution.
The tree-thinking challenge. Science 310, 979–980.
51. Behar, D.M., Metspalu, E., Kivisild, T., Rosset, S., Tzur, S.,
Hadid, Y., Yudkovsky, G., Rosengarten, D., Pereira, L.,
Amorim, A., et al. (2008). Counting the founders: The matri-
lineal genetic ancestry of the Jewish Diaspora. PLoS ONE 3,
e2062.
52. Wallace, D.C., Singh, G., Lott, M.T., Hodge, J.A., Schurr, T.G.,
Lezza, A.M., Elsas, L.J., 2nd, and Nikoskelainen, E.K. (1988).
Mitochondrial DNA mutation associated with Leber’s heredi-
tary optic neuropathy. Science 242, 1427–1430.
53. MITOMAP. (2011) A Human Mitochondrial Genome Data-
base. http://www.mitomap.org.
54. Quintana-Murci, L., Harmant, C., Quach, H., Balanovsky, O.,
Zaporozhchenko, V., Bormans, C., van Helden, P.D., Hoal,
E.G., and Behar, D.M. (2010). Strongmaternal Khoisan contri-
bution to the South African coloured population: A case of
gender-biased admixture. Am. J. Hum. Genet. 86, 611–620.
55. Schonberg, A., Theunert, C., Li, M., Stoneking, M., and
Nasidze, I. (2011). High-throughput sequencing of complete
human mtDNA genomes from the Caucasus and West Asia:
High diversity and demographic inferences. Eur. J. Hum.
Genet. 19, 988–994.
684 The American Journal of Human Genetics 90, 675–684, April 6, 2012

REPORT
Mutations in the GlycosylphosphatidylinositolGene PIGL Cause CHIME Syndrome
Bobby G. Ng,1 Karl Hackmann,2 Melanie A. Jones,3 Alexey M. Eroshkin,1 Ping He,1 Roy Wiliams,1
Shruti Bhide,3 Vincent Cantagrel,4 Joseph G. Gleeson,4 Amy S. Paller,5 Rhonda E. Schnur,6
Sigrid Tinschert,2 Janice Zunich,7 Madhuri R. Hegde,3 and Hudson H. Freeze1,*
CHIME syndrome is characterized by colobomas, heart defects, ichthyosiform dermatosis, mental retardation (intellectual disability),
and ear anomalies, including conductive hearing loss. Whole-exome sequencing on five previously reported cases identified PIGL,
the de-N-acetylase required for glycosylphosphatidylinositol (GPI) anchor formation, as a strong candidate. Furthermore, cell lines
derived from these cases had significantly reduced levels of the two GPI anchor markers, CD59 and a GPI-binding toxin, aerolysin
(FLAER), confirming the pathogenicity of the mutations.
CHIME syndrome (MIM 280000), also known as Zunich
neuroectodermal syndrome, is an extremely rare auto-
somal recessive multisystemic disorder clinically character-
ized by colobomas, congenital heart defects, early onset
migratory ichthyosiform dermatosis, mental retardation
(intellectual disability), and ear anomalies, including
conductive hearing loss. Other clinical manifestations
include distinctive facial features, abnormal growth, geni-
tourinary abnormalities, seizures, and feeding difficul-
ties.1 To date, eight cases have been reported, all having
nearly identical phenotypes.
In 2010, Cantagrel et al.2 described a congenital disorder
of glycosylation (CDG) in which individuals with patho-
logical mutations in SRD5A3 (MIM 611715) presented
with CHIME-like features (MIM 612379). Yet individuals
with Classical CHIME as described by Zunich1 lacked
mutations in SRD5A3. We hypothesized that CHIME
syndrome could be a glycosylation disorder on the basis
of the clinical similarity to those individuals identified by
Cantagrel.2
To test this, we obtained DNA samples from six of the
eight previously described cases from five unrelated fami-
lies for the purpose of whole-exome sequencing (WES).
All clinical samples were obtained with proper informed
consent in accordance with the Sanford-BurnhamMedical
Research Institute’s institutional review board consent
guidelines.
To identify the genetic cause of CHIME syndrome,
we performed WES on five of six previously described
cases.3,4 Exome sequences were enriched with the Roche
Nimblegen Seqcap EZ whole-exome Ver 2.0 on an Illumina
HiSeq platform, and the raw data were aligned to hg18.
Analysis employed Agilent’s AVADIS NGS software.
The five sequenced exomes had an average of 12,549
total variants and an average of 3,774 novel variants with
87% of the exome targets having at least 103 coverage
(Table 1).
A sixth case previously described by Tinschert et al.5 was
the only sample analyzed by comparative genomic hybrid-
ization (CGH) array; it showed a 1MBmaternally inherited
deletion on chromosome 17. DNA was isolated from
whole blood with QIAGEN’s DNA blood kit according to
the manufacturer’s protocol (QIAGEN, Hilden, Germany).
Array CGH was performed on Agilent’s SurePrint G3
Human CGH Microarray Kit 2x400K (Design ID 021850,
Agilent, Santa Clara, CA, USA) according to the manufac-
turer’s protocol, except that dyes were used inversely on
sample and reference. An Agilent microarray scanner
provided the raw data that were processed by Feature
Extraction 9.5. Deleted and amplified regions were identi-
fied on Agilent’s Genomic Workbench Standard Edition
5.0.14. Customized CGH array confirmed copy number
variants and familial segregation. Agilent’s eArray platform
had a general probe density of 1 per 200 bp to 1 per 2.5 kb
depending on the size of the variant. The coordinates of
the array result were mapped to hg18.
We focused exome analysis on the 17 genes in this
region (chromosome 17: 15,620,754–16,698,489 hg18) as
likely candidates. We excluded synonymous changes, vari-
ants in dbSNP v133, and variants present in a limited (30)
in-house exome library. All cases had compound heterozy-
gous mutations in only PIGL [RefSeq NM_004278.3]
within that region (Figure 1). Sanger sequencing confirmed
all mutations in PIGL, and carrier status in each available
parent or sibling (family 665 was not available) excluded
de novo events.
PIGL (NP_004269 [MIM 605947]) is an endoplasmic
reticulum (ER)-localized enzyme that catalyzes the second
1Genetic Disease Program, Sanford Children’s Health Research Center, Sanford-BurnhamMedical Research Institute, La Jolla, CA 92037, USA; 2Institut fuer
Klinische Genetik, Medizinische Fakultaet Carl Gustav Carus, Technische Universitaet Dresden, 01307 Dresden, Germany; 3Department of Human
Genetics, Emory University School of Medicine, Atlanta, GA 30322, USA; 4Neurogenetics Laboratory, Institute for Genomic Medicine, Howard Hughes
Medical Institute, Department of Neurosciences and Pediatrics, University of California, San Diego, La Jolla, CA 92093, USA; 5Department of Dermatology,
Northwestern University, Feinberg School of Medicine, Chicago, IL 60611, USA; 6Division of Genetics, Department of Pediatrics, Cooper Medical School of
Rowan University, Camden, NJ 08103, USA; 7Genetics Center, Indiana University School of Medicine–Northwest, Gary, IN 46408, USA
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.010. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 685–688, April 6, 2012 685
step of glycosylphosphatidylinositol (GPI) biosynthesis,
the de-N-acetylation of N-acetylglucosaminyl-phosphati-
dylinositol (GlcNAc-PI / GlcN-PI) that occurs on the
cytoplasmic side of the ER.6 Following de-N-acetylation,
glucosaminyl-phosphatidylinositol (GlcN-PI) flips to the
luminal side of the ER where GlcN-PI undergoes further
extensions prior to its transfer to acceptor proteins.7
Aside from a possible founder missense mutation, the
other mutations identified in our six CHIME cases are
predicted to be highly damaging (frameshift, nonsense,
essential splice site, and entire gene deletion) (Table 2).
The c.500T>C (p.Leu167Pro) mutation found in all six
cases is at a highly conserved residue (Table 3) located in
the catalytic domain and predicted by both PolyPhen
and SIFT to be damaging.
Utilizing two large public databases, we found the
heterozygous missense mutation c.500T>C in eight out
of nearly 13,000 alleles. In the National Heart, Lung, and
Blood Institute (NHLBI) Exome Sequencing Project, the
c.500T>C mutation appears at a frequency of 6:10,752
alleles (5,376 genomes; with all six heterozygotes being
of European origin). In the 1000 Genomes database, it
was present at 2:2,188 alleles (1,094 genomes). Given the
relatively rare frequency (<0.1%) and the fact that all six
CHIME cases are of European ancestry, we hypothesized
that c.500T>C was due to a founder mutation.
We compared twomicrosatellite markers, 17xATT (trinu-
cleotide marker) and 19xGT (dinucleotide marker), on
chromosome 17: 15,620,754–16,698,489 (hg18) flanking
PIGL. The c.500T>C missense mutation was tightly linked
with the 17xATT in all CHIME cases with an ATT repeat
size of 22, whereas European controls (16) had a repeat
size of 17 repeats. Furthermore, all CHIME cases had a
19xGT repeat size of 13, whereas the European controls
(15) had a repeat size of 19. These results support linkage
disequilibrium between the c.500T>C allele and allele 22
of the 17xATT and allele 13 of the 19xGT marker (data
not shown).
To confirm that the mutations in PIGL are pathological,
we utilized a primary fibroblast cell line from individual
3988 and an Epstein-Barr virus (EBV)-transformed
lymphoblast cell line from individual 33300 to measure
two separate cell surface GPI-anchor-containing markers,
CD59 and a GPI-binding toxin, aerolysin (FLAER). In
agreement with other proven GPI deficiencies, cells
available from CHIME syndrome cases are also deficient
for both GPI anchor markers (Figure 2). It is important
to note that individual 33300 carries the chromo-
some 17 deletion and c.500T>C mutation, making her
hemizygous for the mutation and proving that it is
pathogenic.
Three inherited genetic disorders were previously identi-
fied in GPI biosynthetic genes: PIGM (MIM 610273), PIGN
(MIM 606097), and PIGV (MIM 610274).8–10 Somatic
mutations in PIGA (MIM 311770) cause paroxysmal
nocturnal hemoglobinuria (MIM 300818), a hematologic
disorder.11 Interestingly, epidermal-specific knockout
of mouse PigA recreates features of human Harlequin
Table 1. Summary of WES Statistics for Five CHIME Syndrome Cases
Individual
Average680-2-2 680-2-3 682-2-1 665-3-1 3988
Total number of sequenced reads 53,341,240 54,687,243 47,134,146 117,769,811 121,450,190 78,876,526
Total number of unmapped reads (%) 1,257,149 (2.4%) 765,214 (1.4%) 738,306 (1.6%) 934,246 (0.8%) 988,578 (0.8%) 936,699 (1.2%)
Total number of mapped reads 52,084,091 53,922,029 46,395,840 116,835,565 120,461,612 77,939,827
Percentage Targets with 103 coverage 78 88 85 88 95 87
Percentage Targets with 203 coverage 43 65 59 79 89 67
Total NS-SS-Indels 10,022 12,211 11,921 15,614 12,978 12,549
Novel NS-SS-Indels 2,338 3,211 3,181 7,173 2,966 3,774
Figure 1. Organization of Human PIGL andAssociated MutationsSchematic representation showing both genomicand protein organization of human PIGL withcorresponding mutations as well as functionalprotein domains including a 20aa transmem-brane domain (TMD) and the core de-N-acetylasedomain.
686 The American Journal of Human Genetics 90, 685–688, April 6, 2012
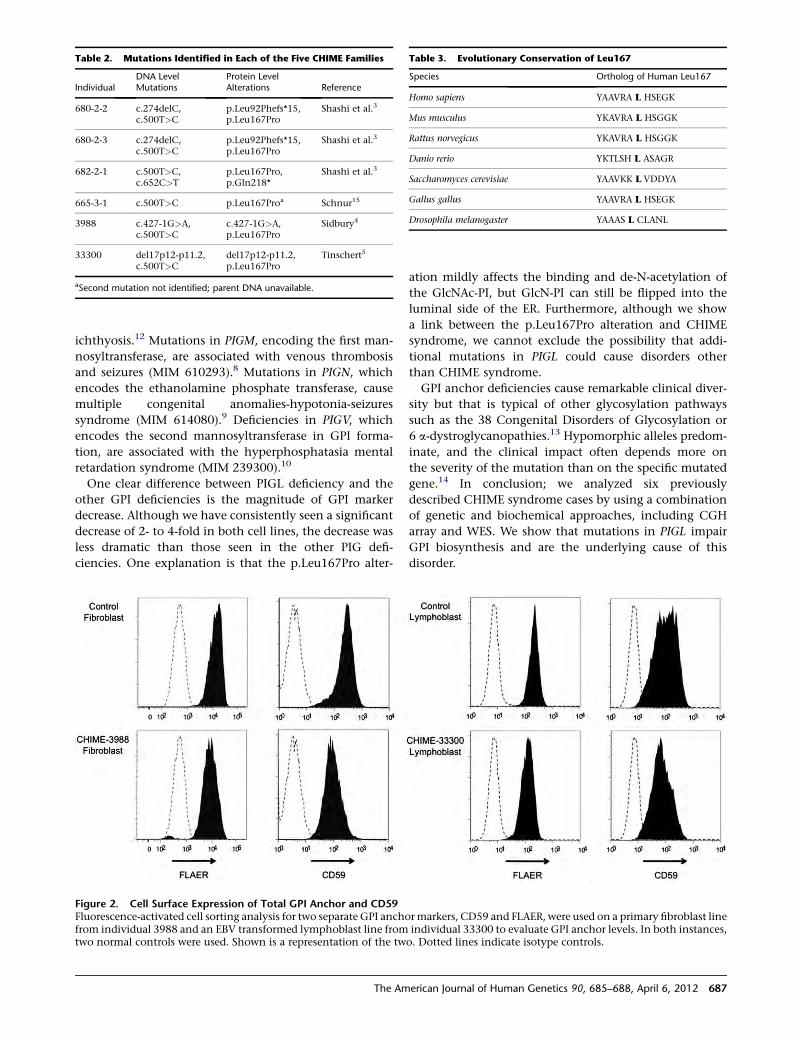
ichthyosis.12 Mutations in PIGM, encoding the first man-
nosyltransferase, are associated with venous thrombosis
and seizures (MIM 610293).8 Mutations in PIGN, which
encodes the ethanolamine phosphate transferase, cause
multiple congenital anomalies-hypotonia-seizures
syndrome (MIM 614080).9 Deficiencies in PIGV, which
encodes the second mannosyltransferase in GPI forma-
tion, are associated with the hyperphosphatasia mental
retardation syndrome (MIM 239300).10
One clear difference between PIGL deficiency and the
other GPI deficiencies is the magnitude of GPI marker
decrease. Although we have consistently seen a significant
decrease of 2- to 4-fold in both cell lines, the decrease was
less dramatic than those seen in the other PIG defi-
ciencies. One explanation is that the p.Leu167Pro alter-
ation mildly affects the binding and de-N-acetylation of
the GlcNAc-PI, but GlcN-PI can still be flipped into the
luminal side of the ER. Furthermore, although we show
a link between the p.Leu167Pro alteration and CHIME
syndrome, we cannot exclude the possibility that addi-
tional mutations in PIGL could cause disorders other
than CHIME syndrome.
GPI anchor deficiencies cause remarkable clinical diver-
sity but that is typical of other glycosylation pathways
such as the 38 Congenital Disorders of Glycosylation or
6 a-dystroglycanopathies.13 Hypomorphic alleles predom-
inate, and the clinical impact often depends more on
the severity of the mutation than on the specific mutated
gene.14 In conclusion; we analyzed six previously
described CHIME syndrome cases by using a combination
of genetic and biochemical approaches, including CGH
array and WES. We show that mutations in PIGL impair
GPI biosynthesis and are the underlying cause of this
disorder.
Table 3. Evolutionary Conservation of Leu167
Species Ortholog of Human Leu167
Homo sapiens YAAVRA L HSEGK
Mus musculus YKAVRA L HSGGK
Rattus norvegicus YKAVRA L HSGGK
Danio rerio YKTLSH L ASAGR
Saccharomyces cerevisiae YAAVKK L VDDYA
Gallus gallus YAAVRA L HSEGK
Drosophila melanogaster YAAAS L CLANL
Table 2. Mutations Identified in Each of the Five CHIME Families
IndividualDNA LevelMutations
Protein LevelAlterations Reference
680-2-2 c.274delC,c.500T>C
p.Leu92Phefs*15,p.Leu167Pro
Shashi et al.3
680-2-3 c.274delC,c.500T>C
p.Leu92Phefs*15,p.Leu167Pro
Shashi et al.3
682-2-1 c.500T>C,c.652C>T
p.Leu167Pro,p.Gln218*
Shashi et al.3
665-3-1 c.500T>C p.Leu167Proa Schnur15
3988 c.427-1G>A,c.500T>C
c.427-1G>A,p.Leu167Pro
Sidbury4
33300 del17p12-p11.2,c.500T>C
del17p12-p11.2,p.Leu167Pro
Tinschert5
aSecond mutation not identified; parent DNA unavailable.
Figure 2. Cell Surface Expression of Total GPI Anchor and CD59Fluorescence-activated cell sorting analysis for two separate GPI anchormarkers, CD59 and FLAER, were used on a primary fibroblast linefrom individual 3988 and an EBV transformed lymphoblast line from individual 33300 to evaluate GPI anchor levels. In both instances,two normal controls were used. Shown is a representation of the two. Dotted lines indicate isotype controls.
The American Journal of Human Genetics 90, 685–688, April 6, 2012 687

Acknowledgments
Supported by The Rocket Fund, a Sanford Professorship (H.H.F.)
and R01 DK55615. K.H. was supported by the Bundesministerium
fur Bildung und Forschung network grant MR-NET 01GS08166.
Received: January 5, 2012
Revised: January 30, 2012
Accepted: February 9, 2012
Published online: March 22, 2012
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
Agilent eARRAY, https://earray.chem.agilent.com/earray/
NHLBI Exome Sequencing Project (ESP), http://evs.gs.
washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
UCSC Genome Browser, www.genome.ucsc.edu
References
1. Zunich, J., and Esterly, N. (2008). In CHIME syndrome
(Zunich syndrome) Neurocutaneous Disorders Phakomatoses
and Hamartoneoplastic Syndromes, M. Ruggieri, I. Pascual-
Castroviejo, C. Di Rocco, and G. Weinheim, eds. (Wien:
Springer-Verlag), pp. 949–955.
2. Cantagrel, V., Lefeber, D.J., Ng, B.G., Guan, Z., Silhavy, J.L.,
Bielas, S.L., Lehle, L., Hombauer, H., Adamowicz, M., Swiezew-
ska, E., et al. (2010). SRD5A3 is required for converting
polyprenol to dolichol and is mutated in a congenital glyco-
sylation disorder. Cell 142, 203–217.
3. Shashi, V., Zunich, J., Kelly, T.E., and Fryburg, J.S. (1995).
Neuroectodermal (CHIME) syndrome: an additional case
with long term follow up of all reported cases. J. Med. Genet.
32, 465–469.
4. Sidbury, R., and Paller, A.S. (2001). What syndrome is this?
CHIME syndrome. Pediatr. Dermatol. 18, 252–254.
5. Tinschert, S., Anton-Lamprecht, I., Albrecht-Nebe, H., and
Audring, H. (1996). Zunich neuroectodermal syndrome:
migratory ichthyosiform dermatosis, colobomas, and other
abnormalities. Pediatr. Dermatol. 13, 363–371.
6. Watanabe, R., Ohishi, K., Maeda, Y., Nakamura, N., and
Kinoshita, T. (1999). Mammalian PIG-L and its yeast homo-
logue Gpi12p are N-acetylglucosaminylphosphatidylinositol
de-N-acetylases essential in glycosylphosphatidylinositol
biosynthesis. Biochem. J. 339, 185–192.
7. Ferguson, M.A.J., Kinoshita, T., and Hart, G.W. (2009).
Glycosylphosphatidylinositol Anchors. In Essentials of Glyco-
biology, A. Varki, R.D. Cummings, J.D. Esko, H. Freeze, G.
Hart, and J. Marth, eds. (Cold Spring Harbor, NY: Cold Spring
Harbor Laboratory Press), pp. 143–161.
8. Almeida, A.M., Murakami, Y., Layton, D.M., Hillmen, P., Sell-
ick, G.S., Maeda, Y., Richards, S., Patterson, S., Kotsianidis, I.,
Mollica, L., et al. (2006). Hypomorphic promoter mutation
in PIGM causes inherited glycosylphosphatidylinositol defi-
ciency. Nat. Med. 12, 846–851.
9. Maydan, G., Noyman, I., Har-Zahav, A., Neriah, Z.B., Pasma-
nik-Chor, M., Yeheskel, A., Albin-Kaplanski, A., Maya, I.,
Magal, N., Birk, E., et al. (2011). Multiple congenital anoma-
lies-hypotonia-seizures syndrome is caused by a mutation in
PIGN. J. Med. Genet. 48, 383–389.
10. Krawitz, P.M., Schweiger, M.R., Rodelsperger, C., Marcelis, C.,
Kolsch, U., Meisel, C., Stephani, F., Kinoshita, T., Murakami,
Y., Bauer, S., et al. (2010). Identity-by-descentfilteringof exome
sequence data identifies PIGV mutations in hyperphosphata-
sia mental retardation syndrome. Nat. Genet. 42, 827–829.
11. Takeda, J., Miyata, T., Kawagoe, K., Iida, Y., Endo, Y., Fujita, T.,
Takahashi, M., Kitani, T., and Kinoshita, T. (1993). Deficiency
of the GPI anchor caused by a somatic mutation of the PIG-
A gene in paroxysmal nocturnal hemoglobinuria. Cell 73,
703–711.
12. Hara-Chikuma, M., Takeda, J., Tarutani, M., Uchida, Y., Hol-
leran, W.M., Endo, Y., Elias, P.M., and Inoue, S. (2004).
Epidermal-specific defect of GPI anchor in Pig-a null mice
results in Harlequin ichthyosis-like features. J. Invest. Derma-
tol. 123, 464–469.
13. Freeze, H.H., and Ng, B.G. (2011). Golgi glycosylation and
human inherited diseases. In Cold Spring Harb Perspect
Biol, G. Warren and J. Rothman, eds. (Cold Spring Harbor,
NY: Cold Spring Harbor Laboratory Press), pp. 35–56.
14. Godfrey, C., Foley, A.R., Clement, E., and Muntoni, F. (2011).
Dystroglycanopathies: coming into focus. Curr. Opin. Genet.
Dev. 21, 278–285.
15. Schnur, R.E., Greenbaum, B.H., Heymann, W.R., Christensen,
K., Buck, A.S., and Reid, C.S. (1997). Acute lymphoblastic
leukemia in a child with the CHIME neuroectodermal
dysplasia syndrome. Am. J. Med. Genet. 72, 24–29.
688 The American Journal of Human Genetics 90, 685–688, April 6, 2012

REPORT
SKIV2L Mutations Cause Syndromic Diarrhea,or Trichohepatoenteric Syndrome
Alexandre Fabre,1,2 Bernard Charroux,3 Christine Martinez-Vinson,4 Bertrand Roquelaure,2
Egritas Odul,5 Ersin Sayar,6 Hilary Smith,7 Virginie Colomb,8 Nicolas Andre,9 Jean-Pierre Hugot,4
Olivier Goulet,8 Caroline Lacoste,10 Jacques Sarles,2 Julien Royet,3 Nicolas Levy,1,10
and Catherine Badens1,10,*
Syndromic diarrhea (or trichohepatoenteric syndrome) is a rare congenital bowel disorder characterized by intractable diarrhea and
woolly hair, and it has recently been associated with mutations in TTC37. Although databases report TTC37 as being the human ortho-
log of Ski3p, one of the yeast Ski-complex cofactors, this lead was not investigated in initial studies. The Ski complex is a multiprotein
complex required for exosome-mediated RNA surveillance, including the regulation of normal mRNA and the decay of nonfunctional
mRNA. Considering the fact that TTC37 is homologous to Ski3p, we explored a gene encoding another Ski-complex cofactor, SKIV2L, in
six individuals presenting with typical syndromic diarrhea without variation in TTC37. We identified mutations in all six individuals.
Our results show that mutations in genes encoding cofactors of the human Ski complex cause syndromic diarrhea, establishing a link
between defects of the human exosome complex and a Mendelian disease.
Syndromic diarrhea (SD) is a rare and severe disease charac-
terized by intractable diarrhea, facial dysmorphism, intra-
uterine growth retardation, immunodeficiency, and hair
abnormalities.1 Trichohepatoenteric (THE [MIM 222470])
syndrome, described initially by Verloes et al. in 19972 as
a different syndrome, has since been grouped with syn-
dromic diarrhea because the main clinical features in
both syndromes are identical3 (for simplicity, we now
refer to both syndromes as a singular disorder, SD/THE
syndrome). After a homozygosity-mapping analysis, we
and others recently identified TTC37 mutations as being
responsible for this syndrome in 21 individuals.4,5 The
precise function of TTC37 (also called Thespin) was not
elucidated even after the description of its involvement
in SD/THE. This protein shares no sequence homology
with other human proteins and shows no known func-
tional domains except several tetratrico-peptide-repeat
(TPR) domains that are structural motifs found in over
300 human proteins.6 In some databases, TTC37 is re-
ported as being the ortholog of yeast SKI3, which encodes
a key component of the Ski complex, a multiprotein
complex required for exosome-mediated RNA surveil-
lance.7 However, this lead was not explored in the initial
studies. In our series, 6 out of 15 individuals did not carry
a mutation in TTC37 but presented with typical character-
istics of SD/THE syndrome. Considering the fact that
TTC37 is homologous to Ski3p, we assumed that other
genes encoding Ski-complex proteins might be responsible
for SD/THE syndrome in these individuals; we confronted
this hypothesis with the results of the linkage analysis of
one of the consanguineous families.
Out of the six individuals with typical SD/THE syndrome
and no mutation in TTC37, only one was previously re-
ported.8 All procedures followed for clinical and genetic
analyses in this study were in accordance with the ethical
standards of the institutional and national committees on
human experimentation, and proper informed consent
was obtained from the parents of the affected children. All
individuals presented with severe and intractable diarrhea
that occurred between 1 and 12 weeks after birth, hair ab-
normalities (sparse, fragile, and uncombable hair and tri-
chorrhexis nodosa), and facial dysmorphism characterized
byhypertelorism, a broadflatnasal bridge, and aprominent
forehead (Figure 1). All children received parenteral nutri-
tion, but the amount of time varied between individuals
and ranged from a few weeks to several years. Immunodefi-
ciencywasmostly due to low immunoglobulin levels and to
the absence of an immune response to vaccines. One indi-
vidual out of the three with immunodeficiency died from
a measles infection. These six individuals harbor no muta-
tion in TTC37, but their clinical presentation is undistin-
guishable from those whohaveTTC37mutations (Table 1).
Searching for a candidate gene in this group, we investi-
gated the possible homology (reported in the Ensembl
database) between TTC37 and yeast SKI3. Interspecies
sequence-alignment analysis (with the bioinformatics
prediction software BLAST) revealed that TTC37 shares
significant amino-acid-sequence similarity with yeast
1UMR_S 910, Inserm-Faculte de Medecine, Aix-Marseille Universite, 13385 Marseille, France; 2AP-HM, Service de Pediatrie Multidisciplinaire, Hopital d’En-
fants de la Timone, 13385 Marseille, France; 3Institut de Biologie du Developpement de Marseille-Luminy, Aix-Marseille Universite, 13288 Marseille,
France; 4AP-HP, Service de Gastroenterologie, Hopital Robert Debre, 75019 Paris, France; 5Department of Pediatric Gastroenterology, Gazi University School
of Medicine, 06500 Ankara, Turkey; 6Department of Pediatric Gastroenterology, Akdeniz University, 07058 Antalya, Turkey; 7Department of Pediatrics,
Children’s Memorial Hospital, Chicago, IL 60614, USA; 8AP-HP, Service de Gastroenterologie, Hopital Necker-Enfants Malades, 75004 Paris, France;9AP-HM, Service d’Oncologie Pediatrique, Hopital d’Enfants de la Timone, 13385 Marseille, France; 10AP-HM, Laboratoire de Genetique Moleculaire,
Hopital d’Enfants de la Timone, 13385 Marseille, France
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.009. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 689–692, April 6, 2012 689

Ski3p. Between TTC37 residues 5 and 84, TTC37 and Ski3p
were 35% identical and 58% similar, and between residues
488 and 1223, they were 20% identical and 38% similar,
indicating that TTC37 is the human ortholog of SKI3.
Because Ski3p ispart of amultiprotein complex, thisfinding
prompted us to test whether mutations in other human or-
thologs of the Ski proteins could be associatedwith SD/THE
syndrome. For one consanguineous family, we performed
a linkage analysis in which the TTC37 critical interval
was excluded, and this analysis revealed that there was a
region of homozygosity spanning nucleotides 9,138,488–
40,453,008 and containing 856 genes in chromosomal
region 6p21.2–6p24.3. When looking for functional candi-
date genes in this region, we noticed that SKIV2L (RefSeq
accessionnumberNM_006929.4), a genedescribed asbeing
the human ortholog of the SKI2,9,10 was present in region
6p21.3. Thus, we performed direct sequencing of SKIV2L
in our cohort of six individuals affected by typical SD/THE
syndrome and found the presence of mutations predicted
to be deleterious in all six. The identified mutations
and their mode of inheritance are described in Table 2.
All of the unaffected parents available for analysis were
heterozygous for one of the mutations identified in
their child. We identified eight different variations dis-
tributed throughout the protein (Figure 1). Seven of these
variations correspond to nonsense or frameshift mutations
that introduce apremature terminationcodon, and theyare
Figure 1. Clinical Presentation of Individualswith SD/THE Syndrome and SKIV2L Mutations(A) Clinical presentation of four individuals withSD/THE syndrome. One individual has muta-tions in TTC37 (first picture on the left), andthree others have mutations in SKIV2L (rightpanel).(B) A schematic overview of the predicted SKIV2Lstructure with a helicase ATP-binding domainand a helicase C-terminal domain (UniProt).The location of the domains is annotated byamino acid position. The eight amino acid substi-tutions are indicated by arrows.(C) Nucleotide sequences are shown for thecontrols (upper sequence) and for each SKIV2Lvariant (lower sequence). Arrows indicate muta-tion positions on the reference sequences.
c.848G>A (p.Trp283*), c.1434del (p.Ser479-
Alafs*3), c.1635_1636insA (p.Gly546Argfs*
35), c.226C>T (p.Arg756*), c.2442G>A
(p.Trp814*), c.2572del (p.Val858*), and
c.2662_2663del (p.Arg888Glyfs*12). The
mutation types suggest that the disease
mechanism is loss of function.We identified
one missense mutation, c.1022T>G
(p.Val341Gly), that is located in the helicase
ATP-binding domain (Figure 1) and con-
cerns a highly conserved residue. PolyPhen
predicted that this specificmutation is prob-
ably damaging (the prediction score was 2.8
[mutations predicted to be pathological have an index
above 0.5]).
It isnowclear that themajorityof genomic information is
transcribed into RNAmolecules; this process generates very
abundant and complex pools of RNAs that the cells have to
control.While performing numerous RNA-processing reac-
tions, the cell must, at the same time, eradicate surplus and
aberrantmaterial.11 This task is, at least partially, performed
by the RNA exosome multiprotein complex that contains
both 30-to-50 exonuclease and 30-to-50 endonuclease activityand acts in both the nucleus and the cytoplasm.12 Initially
discovered in yeast but also present in higher eukaryotes,
including humans, this exosome complex is involved in
the decay pathways of normal mRNA but is also required
to maintain the fidelity of gene expression through RNA
surveillance processes, such as nonsense-mediated decay,
nonstop decay, and no-go decay.13–16 Genetic screens in
yeast have identified three proteins (Ski2p, Ski3p, and
Ski8p) that form the Ski (Superkiller) complex and act as
specific cofactors required for all the functions of the cyto-
plasmic exosome but not the nuclear exosome.17 The puta-
tive DExH-box RNA helicase activity of Ski2 suggests that it
is the only Ski protein with a catalytic function, whereas
Ski3p and Ski8p contain repeated domains thought to be
needed for protein-protein interactions.18 In humans, the
cytoplasmic exosome is involved in various mRNA decay
pathways and is required for normal cell growth.19
690 The American Journal of Human Genetics 90, 689–692, April 6, 2012
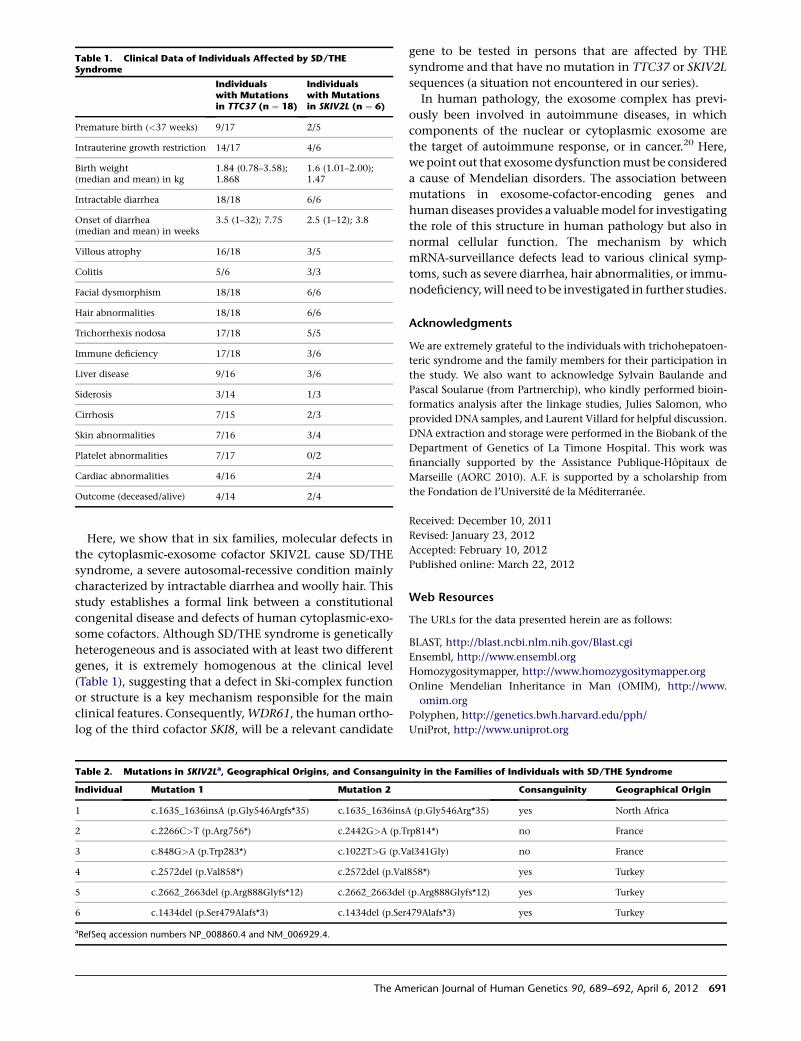
Here, we show that in six families, molecular defects in
the cytoplasmic-exosome cofactor SKIV2L cause SD/THE
syndrome, a severe autosomal-recessive condition mainly
characterized by intractable diarrhea and woolly hair. This
study establishes a formal link between a constitutional
congenital disease and defects of human cytoplasmic-exo-
some cofactors. Although SD/THE syndrome is genetically
heterogeneous and is associated with at least two different
genes, it is extremely homogenous at the clinical level
(Table 1), suggesting that a defect in Ski-complex function
or structure is a key mechanism responsible for the main
clinical features. Consequently,WDR61, the human ortho-
log of the third cofactor SKI8, will be a relevant candidate
gene to be tested in persons that are affected by THE
syndrome and that have no mutation in TTC37 or SKIV2L
sequences (a situation not encountered in our series).
In human pathology, the exosome complex has previ-
ously been involved in autoimmune diseases, in which
components of the nuclear or cytoplasmic exosome are
the target of autoimmune response, or in cancer.20 Here,
we point out that exosome dysfunctionmust be considered
a cause of Mendelian disorders. The association between
mutations in exosome-cofactor-encoding genes and
human diseases provides a valuablemodel for investigating
the role of this structure in human pathology but also in
normal cellular function. The mechanism by which
mRNA-surveillance defects lead to various clinical symp-
toms, such as severe diarrhea, hair abnormalities, or immu-
nodeficiency, will need to be investigated in further studies.
Acknowledgments
We are extremely grateful to the individuals with trichohepatoen-
teric syndrome and the family members for their participation in
the study. We also want to acknowledge Sylvain Baulande and
Pascal Soularue (from Partnerchip), who kindly performed bioin-
formatics analysis after the linkage studies, Julies Salomon, who
provided DNA samples, and Laurent Villard for helpful discussion.
DNA extraction and storage were performed in the Biobank of the
Department of Genetics of La Timone Hospital. This work was
financially supported by the Assistance Publique-Hopitaux de
Marseille (AORC 2010). A.F. is supported by a scholarship from
the Fondation de l’Universite de la Mediterranee.
Received: December 10, 2011
Revised: January 23, 2012
Accepted: February 10, 2012
Published online: March 22, 2012
Web Resources
The URLs for the data presented herein are as follows:
BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi
Ensembl, http://www.ensembl.org
Homozygositymapper, http://www.homozygositymapper.org
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
Polyphen, http://genetics.bwh.harvard.edu/pph/
UniProt, http://www.uniprot.org
Table 1. Clinical Data of Individuals Affected by SD/THESyndrome
Individualswith Mutationsin TTC37 (n ¼ 18)
Individualswith Mutationsin SKIV2L (n ¼ 6)
Premature birth (<37 weeks) 9/17 2/5
Intrauterine growth restriction 14/17 4/6
Birth weight(median and mean) in kg
1.84 (0.78–3.58);1.868
1.6 (1.01–2.00);1.47
Intractable diarrhea 18/18 6/6
Onset of diarrhea(median and mean) in weeks
3.5 (1–32); 7.75 2.5 (1–12); 3.8
Villous atrophy 16/18 3/5
Colitis 5/6 3/3
Facial dysmorphism 18/18 6/6
Hair abnormalities 18/18 6/6
Trichorrhexis nodosa 17/18 5/5
Immune deficiency 17/18 3/6
Liver disease 9/16 3/6
Siderosis 3/14 1/3
Cirrhosis 7/15 2/3
Skin abnormalities 7/16 3/4
Platelet abnormalities 7/17 0/2
Cardiac abnormalities 4/16 2/4
Outcome (deceased/alive) 4/14 2/4
Table 2. Mutations in SKIV2La, Geographical Origins, and Consanguinity in the Families of Individuals with SD/THE Syndrome
Individual Mutation 1 Mutation 2 Consanguinity Geographical Origin
1 c.1635_1636insA (p.Gly546Argfs*35) c.1635_1636insA (p.Gly546Arg*35) yes North Africa
2 c.2266C>T (p.Arg756*) c.2442G>A (p.Trp814*) no France
3 c.848G>A (p.Trp283*) c.1022T>G (p.Val341Gly) no France
4 c.2572del (p.Val858*) c.2572del (p.Val858*) yes Turkey
5 c.2662_2663del (p.Arg888Glyfs*12) c.2662_2663del (p.Arg888Glyfs*12) yes Turkey
6 c.1434del (p.Ser479Alafs*3) c.1434del (p.Ser479Alafs*3) yes Turkey
aRefSeq accession numbers NP_008860.4 and NM_006929.4.
The American Journal of Human Genetics 90, 689–692, April 6, 2012 691

References
1. Girault, D., Goulet, O., Le Deist, F., Brousse, N., Colomb, V.,
Cesarini, J.P., de Potter, S., Canioni, D., Griscelli, C., Fischer,
A., et al. (1994). Intractable infant diarrhea associated with
phenotypic abnormalities and immunodeficiency. J. Pediatr.
125, 36–42.
2. Verloes, A., Lombet, J., Lambert, Y., Hubert, A.F., Deprez, M.,
Fridman, V., Gosseye, S., Rigo, J., and Sokal, E. (1997). Tri-
cho-hepato-enteric syndrome: Further delineation of a distinct
syndrome with neonatal hemochromatosis phenotype,
intractable diarrhea, and hair anomalies. Am. J. Med. Genet.
68, 391–395.
3. Fabre, A., Andre, N., Breton, A., Broue, P., Badens, C., and
Roquelaure, B. (2007). Intractable diarrhea with ‘‘phenotypic
anomalies’’ and tricho-hepato-enteric syndrome: Two names
for the same disorder. Am. J. Med. Genet. A. 143, 584–588.
4. Hartley, J.L., Zachos, N.C., Dawood, B., Donowitz, M., For-
man, J., Pollitt, R.J., Morgan, N.V., Tee, L., Gissen, P., Kahr,
W.H., et al. (2010). Mutations in TTC37 cause trichohepatoen-
teric syndrome (phenotypic diarrhea of infancy). Gastroenter-
ology 138, 2388–2398, 2398, e1–e2.
5. Fabre, A., Martinez-Vinson, C., Roquelaure, B., Missirian, C.,
Andre, N., Breton, A., Lachaux, A., Odul, E., Colomb, V.,
Lemale, J., et al. (2011). Novel mutations in TTC37 associated
with tricho-hepato-enteric syndrome. Hum. Mutat. 32,
277–281.
6. Blatch, G.L., and Lassle, M. (1999). The tetratricopeptide
repeat: A structural motif mediating protein-protein interac-
tions. Bioessays 21, 932–939.
7. Brown, J.T., Bai, X., and Johnson, A.W. (2000). The yeast
antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex
in vivo. RNA 6, 449–457.
8. Egritas, O., Dalgic, B., and Onder, M. (2009). Tricho-hepato-
enteric syndrome presenting with mild colitis. Eur. J. Pediatr.
168, 933–935.
9. Lee, S.-G., Lee, I., Park, S.H., Kang, C., and Song, K. (1995).
Identification and characterization of a human cDNA homol-
ogous to yeast SKI2. Genomics 25, 660–666.
10. Dangel, A.W., Shen, L., Mendoza, A.R., Wu, L.-C., and Yu, C.Y.
(1995). Human helicase gene SKI2W in the HLA class III
region exhibits striking structural similarities to the yeast anti-
viral gene SKI2 and to the human gene KIAA0052: Emergence
of a new gene family. Nucleic Acids Res. 23, 2120–2126.
11. Garneau, N.L., Wilusz, J., and Wilusz, C.J. (2007). The high-
ways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol.
8, 113–126.
12. Houseley, J., and Tollervey, D. (2009). The many pathways of
RNA degradation. Cell 136, 763–776.
13. Schmid,M., and Jensen, T.H. (2008). The exosome: Amultipur-
pose RNA-decay machine. Trends Biochem. Sci. 33, 501–510.
14. Parker, R., and Song,H. (2004). The enzymes and control of eu-
karyotic mRNA turnover. Nat. Struct. Mol. Biol. 11, 121–127.
15. Zhu, B., Mandal, S.S., Pham, A.D., Zheng, Y., Erdjument-Brom-
age, H., Batra, S.K., Tempst, P., and Reinberg, D. (2005). The
human PAF complex coordinates transcription with events
downstream of RNA synthesis. Genes Dev. 19, 1668–1673.
16. Toh-E, A., and Wickner, R.B. (1980). ‘‘Superkiller’’ mutations
suppress chromosomal mutations affecting double-stranded
RNA killer plasmid replication in saccharomyces cerevisiae.
Proc. Natl. Acad. Sci. USA 77, 527–530.
17. Schaeffer, D., Clark, A., Klauer, A.A., Tsanova, B., and van
Hoof, A. (2010). Functions of the cytoplasmic exosome. Adv.
Exp. Med. Biol. 702, 79–90.
18. Wang, L., Lewis, M.S., and Johnson, A.W. (2005). Domain
interactions within the Ski2/3/8 complex and between the
Ski complex and Ski7p. RNA 11, 1291–1302.
19. vanDijk, E.L., Schilders, G., and Pruijn, G.J. (2007). Human cell
growth requires a functional cytoplasmic exosome, which is
involved invariousmRNAdecaypathways.RNA13, 1027–1035.
20. Staals, R.H., and Pruijn, G.J. (2010). The human exosome and
disease. Adv. Exp. Med. Biol. 702, 132–142.
692 The American Journal of Human Genetics 90, 689–692, April 6, 2012

REPORT
Mutations in C5ORF42 Cause Joubert Syndromein the French Canadian Population
Myriam Srour,1,11 Jeremy Schwartzentruber,2,11 Fadi F. Hamdan,1 Luis H. Ospina,3 Lysanne Patry,1
Damian Labuda,4 Christine Massicotte,4 Sylvia Dobrzeniecka,1 Jose-Mario Capo-Chichi,1
Simon Papillon-Cavanagh,4 Mark E. Samuels,4 Kym M. Boycott,5 Michael I. Shevell,6
Rachel Laframboise,7 Valerie Desilets,4 FORGE Canada Consortium,12 Bruno Maranda,8
Guy A. Rouleau,9 Jacek Majewski,10 and Jacques L. Michaud1,*
Joubert syndrome ( JBTS) is an autosomal-recessive disorder characterized by a distinctive mid-hindbrain malformation, developmental
delay with hypotonia, ocular-motor apraxia, and breathing abnormalities. Although JBTS was first described more than 40 years ago in
French Canadian siblings, the causal mutations have not yet been identified in this family nor in most French Canadian individuals
subsequently described.We ascertained a cluster of 16 JBTS-affected individuals from 11 families living in the Lower St. Lawrence region.
SNP genotyping excluded the presence of a common homozygous mutation that would explain the clustering of these individuals.
Exome sequencing performed on 15 subjects showed that nine affected individuals from seven families (including the original JBTS
family) carried rare compound-heterozygous mutations in C5ORF42. Two missense variants (c.4006C>T [p.Arg1336Trp] and
c.4690G>A [p.Ala1564Thr]) and a splicing mutation (c.7400þ1G>A), which causes exon skipping, were found in multiple subjects
that were not known to be related, whereas three other truncating mutations (c.6407del [p.Pro2136Hisfs*31], c.4804C>T
[p.Arg1602*], and c.7477C>T [p.Arg2493*]) were identified in single individuals. None of the unaffected first-degree relatives were
compound heterozygous for these mutations. Moreover, none of the six putative mutations were detected among 477 French Canadian
controls. Our data suggest that mutations in C5ORF42 explain a large portion of French Canadian individuals with JBTS.
Joubert syndrome (JBTS [MIM 213300]) is an autosomal-
recessive disorder characterized by the presence of hypo-
tonia, apnea or hyperpnea in infancy, oculomotor apraxia,
and variable developmental delay or intellectual impair-
ment (reviewed in Sattar et al.1). The diagnostic hallmark
of JBTS is the presence of a complex malformation of the
midbrain-hindbrain junction that comprises cerebellar
vermis hypoplasia or aplasia, deepened interpeduncular
fossa, and elongated superior cerebellar peduncles. This
malformation appears like a molar tooth on an axial brain
MRI (magnetic resonance imaging). In a subset of individ-
uals, JBTS also involves other organs and results in cystic
kidneys, retinopathy, or polydactyly. JBTS is a genetically
heterogeneous condition for which 15 genes have been
described to date.2–19 All of these genes appear to play a
role in the development and/or function of nonmotile
cilia. Although JBTS was first described in French Canadian
siblings more than 40 years ago by Marie Joubert and
colleagues, until now, the causal mutations have not yet
been identified in the original family nor in most French
Canadians subjects.20,21
There is a highprevalence of JBTS in the FrenchCanadian
population living in the Lower St. Lawrence (‘‘Bas-du-
Fleuve’’ in French) region of the province of Quebec
(Figure 1). In total, we identified 16 living affected individ-
uals (from 11 unrelated families) who have at least one
grandparent originating from that region. Informed con-
sent was obtained from all individuals or their legal guard-
ians. This project was approved by our institutional ethics
committee. We were initially able to collect blood-derived
DNA from 15 of these individuals, including an affected
individual (II-1 in family 394; individual BD in Joubert
et al.20) from the original JBTS family described by Marie
Joubert and colleagues in 1969. There was a striking cluster
of seven families from the east end of the region (Matapedia
region); one family is fromMont-Joli (population of 6,568),
three families are from Amqui (population of 6,261), and
three other families are from Sayabec (population of
1,877). Individual II-1 from family 394 did not undergo
brain-imaging studies, but an MRI scan performed on her
brother (II-2) showed the molar-tooth sign (MTS) (Fig-
ure 2B).21 All the other affected individuals showed the
MTS and variable expression of the classical JBTS features.
The cohort included three families with two affected
siblings, and the parents were not affected in any family
(consistent with a recessive mode of transmission).
1Centre of Excellence in Neurosciences, Universite de Montreal and Sainte-Justine Hospital Research Center, Montreal H3T 1C5, Canada; 2McGill Univer-
sity and Genome Quebec Innovation Centre, Montreal H3A 1A4, Canada; 3Department of Ophthalmology, Sainte-Justine Hospital Research Center,
Montreal H3T 1C5, Canada; 4Sainte-Justine Hospital Research Center, Montreal H3T 1C5, Canada; 5Children’s Hospital of Eastern Ontario Research
Institute, Ottawa K1H 8L1, Canada; 6Division of Pediatric Neurology, Montreal Children’s Hospital-McGill University Health Center, Montreal H3H
1P3, Canada; 7Department of Medical Genetics, Centre Hospitalier Universitaire Laval, Quebec G1V 4G2, Canada; 8Division of Genetics, Centre Hospitalier
Universitaire de Sherbrooke, Sherbrooke J1H 5N4, Canada; 9Centre of Excellence in Neurosciences of Universite de Montreal, Centre Hospitalier de
l’Universite de Montreal Research Center and Department of Medicine, Montreal H2L 2W5, Canada; 10Department of Human Genetics, McGill University,
Montreal H3A 1A4, Canada11These authors contributed equally to this work12FORGE Steering Committee is listed in Acknowledgements
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.011. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 693–700, April 6, 2012 693

It was initially established that the population of the
Lower St. Lawrence region was a result of both the immi-
gration of a limited number of settlers (6,000 individuals)
from Quebec City and its surrounding areas in the late
17th century and beginning of the 18th century and a rapid
increase in settlers resulting from a high fertility rate.22
The establishment of settlers in the region followed a
west-to-east pattern, and settlers later migrated to regions
farther east. A small number of Acadians also contributed
to the early population of the Matapedia region.23 The
demographic growth of this population thus appears to
be characterized by a series of bottlenecks that might
have resulted in regional founder effects. We hypothesized
that a founder effect could underlie the clustering of
individuals with JBTS in the Lower St. Lawrence region,
raising the possibility that a common homozygous muta-
tion explains a large portion of them. We performed
whole-genome SNP genotyping in all 15 individuals with
JBTS by using the Illumina Human 610 Genotyping
BeadChip panel, which interrogates 620,901 SNPs, and
we used PLINK24 to search for homozygosity regions con-
taining >30 consecutive SNPs and extending over >1Mb.
We identified several overlapping regions of shared
homozygosity, but these regions were not found in more
than five families, were small (1 megabase or less), and
contained genes that are unlikely to play a role in cilia
development and/or function (Table S1, available online).
Altogether, the genotyping data suggest the presence of
allelic and/or genetic heterogeneity within our cohort.
Given the lack of hints from genotype-based mapping,
we decided to sequence the protein-coding exomes of all
our JBTS-affected subjects in the hopes of identifying a
unique candidate gene harboring private pathogenic vari-
ants in a large fraction of the samples. Genomic DNA from
each sample was captured with the Agilent SureSelect
Figure 1. Distribution of Individuals with JBTSin the Lower St. Lawrence RegionNumbers refer to families (pedigrees in Figure 2).Note the cluster of families along Route 132,which follows the Matapedia River.
50 Mb oligonucleotide library, and the
captured DNA was sequenced with paired-
end 100 bp reads on Illumina HiSeq2000.
The result was an average of 14.7 Gb of
raw sequence for each sample. Data were
analyzed as previously described.25 After
we used Picard (v. 1.48) to remove putative
PCR-generated duplicate reads, we aligned
the reads to human genome assembly
hg19 by using a Burroughs-Wheeler algo-
rithm (BWA v. 0.5.9). The median read
depth of the bases in CCDS (consensus
coding sequence) exons was 115 (deter-
mined with Broad Institute Genome
Analysis Toolkit v. 1.0.4418).26 On average,
88% (52.9%) of the bases in CCDS exons were covered
by at least 20 reads. We called sequence variants by
using custom scripts for Samtools (v. 0.1.17), Pileup, and
varFilter, and we required at least three variant reads as
well as >20% variant reads for each called position.
Single-nucleotide variants (SNVs) had Phred-like quality
scores of at least 20, and small insertions or deletions
(indels) had scores of at least 50. We used Annovar to
annotate variants according to the type of mutation,
occurrence in dbSNP, SIFT score, and 1,000 Genomes allele
frequency.27 To identify potentially pathogenic variants,
we filtered out (1) synonymous variants or intronic vari-
ants other than those affecting the consensus splice sites,
(2) variants seen in more than one of 261 exomes from
individuals with rare, monogenic diseases unrelated to
JBTS (these individuals were sequenced at the McGill
University and Genome Quebec Innovation Centre), and
(3) variants with a frequency greater than 0.5% in the
1,000 Genomes Browser (Tables 1 and 2).
We first examined the exome datasets to look for rare
variants in the 15 genes already associated with JBTS (these
genes are INPP5E [MIM 613037], TMEM216 [MIM 613277],
AHI1 [MIM 608894], NPHP1 [MIM 607100], CEP290
[MIM 610142], TMEM67 [MIM 609884], RPGRIP1L
[MIM 610937], ARL13B [MIM 608922], CC2D2A [MIM
612013], CXORF5 [MIM 300170], KIF7 [MIM 611254],
TCTN1 [MIM 609863], TCTN2 [MIM 613885], TMEM237
[MIM 614424], and CEP41)2–19 as well as in the JBTS
candidate gene, TTC21B (MIM 612014).28 Two individuals
(II-1 from family 484 and II-2 from family 473, Figure S2)
that are not known to be related were each found to be
carrying two heterozygous missense variants (c.4667A>T
[p.Asp1556Val] and c.3376G>A [p.Glu1126Lys]) in
CC2D2A (RefSeq accession number NM_001080522.2).13,14
These amino acids are highly conserved, and both
694 The American Journal of Human Genetics 90, 693–700, April 6, 2012

mutations are predicted to be deleterious according to
SIFT (scores < 0.05)29 and Polyphen-2 (scores > 0.90)30
(Figure S1). The c.4667A>T (p.Asp1556Val) mutation has
already been reported in individuals with JBTS.31 Segrega-
tion studies have indicated that the affected individuals
but none of their unaffected first-degree relatives were
compound heterozygous for these mutations (Figure S2).
We conclude that these mutations are probably patho-
genic. Both individuals have a mild phenotype. They
have oculomotor apraxia and only mild motor delay
(they walked at 18 [II-1 from family 484] and 19 [II-2 from
family 473] months of age and do not have gait ataxia).
The individual who is of school age performs well in
a regular classroom. Four additional individuals were sin-
gly heterozygous for rare variants in the other known
B Family 394
WTc.4006C>T
C Family 474
WT6407del
WTc.4006C>T
c.6407delc.4006C>T
WTWT
D Family 480 E Family 479
A Family 406/301
c.7400+1G>Ac.4006C>T
WTc.7400+1G>A
WTc.4006C>T
WTc.4690G>A
WTc.7400+1G>A
c.7400+1G>Ac.4690G>A
c.7400+1G>Ac.4690G>A
I
II
III
IV
1 2 3 4
1 2 3 4
1 2 3 4
1 2 3
G Family 468
WTWT
c.4006C>Tc.4690G>A
I
II
WTc.4006C>T
WTc.4690G>A
1 2
1 2
F Family 489
I
II
I
II
WTc.4006C>T
c.4006C>Tc.7400+1G>A
WTc.7400+1G>A
WTc.7400+1G>A
1 2
1 2
WTc.4006C>T
WTc.4804C>T
c.4006C>Tc.4804C>T
I
II
1 2
1
1 2
1 2
c.7400+1G>Ac.4006C>T
WTc.4006C>T
1 2
1 2 3 4
c.7400+1G>Ac.4006C>T
I
II
I
II
WTc.7477C>T
WTc.4690G>A
c.4690G>Ac.7477C>T
WTc.7477C>T
WTWT
1 2
1 2 3
Figure 2. Segregation of C5ORF42Muta-tions in Families Affected by JBTS
JBTS-associated genes; such variants
are c.265C>T (p.Leu89Phe) in
TMEM216 (M_001173991.2),
c.3257A>G (p.Glu1086Gly) in
AHI1 (NM_001134831), c.1600G>A
(p.Glu534Lys) in CEP290 (NM_
025114.3), and c.3032T>C
(p.Met1011Thr) in TTC21B (NM_
024753.4). Because each of these
genes has previously been associated
with recessive JBTS, these heterozy-
gous variants are unlikely to fully
explain the disorder that these indi-
viduals have.
We next looked at the whole-exome
data for the other protein-coding
genes containing homozygous or
multiple heterozygous variants in the
13 affected individuals who did not
have mutations in CC2D2A (Tables 1
and 2). Strikingly, five subjects,
including a member of the initial
JBTS family, carried two different
heterozygous variants in an un-
studied anonymous gene, C5ORF42
(NM_023073.3). Mutations in six
other genes were found in affected
individuals among sets of three
families (Tables 1 and2). Because these
latter genes (MUC5B, PLEC, FAT3, FLG,
TTN, and LAMA5) are known to accu-
mulate mutations at a high rate, they
are unlikely to be linked to the disease
(Table S2). All five affected individuals
with changes in C5ORF42 carried the
same missense mutation, c.4006C>T
(p.Arg1336Trp) (NM_023073.3), as
well as one of three different mutations: one mutation
that affects a consensus donor splice site, c.7400þ1G>A
(NM_023073.3), and two truncating mutations, c.6407del
(p.Pro2136Hisfs*31) and c.4804C>T (p.Arg1602*) (NM_
023073.3) (Figures 2 and 3 and Table 3). Sanger sequencing
in the five affected individuals confirmed the presence of
these variants. Segregation studies indicated that the
affected individuals, but not their unaffected first-degree
relatives, were compound heterozygotes for these variants
(Figure 2 and Table 3). Subsequently, wewere able to collect
DNA from individual II-2 (individual M.D.19-20), the
affected brother of II-1 in the initial JBTS family (family
394), and we found that he was compound heterozygous
for the same C5ORF42 mutations identified in his affected
sister (Figure 2B and Table 3). None of these four variants
The American Journal of Human Genetics 90, 693–700, April 6, 2012 695

was detected in 261 in-house control exomes, which were
derived from other projects including some French Cana-
dian subjects, and in the 1,000 Genomes Browser. RT-PCR
performed on RNA extracted from the blood of individuals
II-2 (from family 394) and III-4 (from family 406/301), who
both carry the c.7400þ1G>A splicing mutation, showed
that this mutation causes skipping of exon 35 in C5ORF42
(NM_023073.3) and results in the creation of a premature
stop codon (Figure S3). The p.Arg1336Trp amino acid
substitution is predicted to be damaging (SIFT¼ 0.00; Poly-
phen-2 ¼ 0.99) and to affect a residue that is conserved
across vertebrate species (Figure 3B).
On the basis of the exome-sequencing data, four
additional JBTS-affected individuals from three families
(301, 468, and 489) were each carrying a single heterozy-
gous C5ORF42 mutation, including the already described
c.4006C>T (p.Arg1336Trp) and c.7400þ1G>A mutations
and the truncating mutation c.7477C>T (p.Arg2493*)
(NM_023073.3) (Figure 2 and Table 3). The c.7477C>T
(p.Arg2493*) mutation was absent from our 261 control
exomes and the 1,000 Genomes Browser. Our SNP geno-
typing data suggest that these four individuals—but not
the other individuals with JBTS in our cohort—are hetero-
zygous for a unique 5 Mb haplotype that encompasses
C5ORF42 (Figure S4). It seemed unlikely that this haplo-
type would be carrying three different rare mutations;
therefore, this observation suggests that the four individ-
uals might carry a second mutation linked to this haplo-
type. Upon further inspection of the exome data, we
discovered that all four individuals are also heterozygous
for another missense variant, c.4690G>A (p.Ala1564Thr)
(based on the ENST00000388739 transcript annotated by
the Ensemble Genome Browser). This allele was not
included in our original filtered dataset because it is located
in an internal coding exon (chr5: 37,157,522–37,157,415)
not annotated by RefSeq for the longest isoform of the
gene (NM_023073.3). Sanger sequencing confirmed the
presence of the various mutations in the four affected
individuals. Segregation studies showed that the four
affected individuals but none of their unaffected first-
degree relatives were compound heterozygous for
c.4690G>A (p.Ala1564Thr) and for one of the three other
mutations (c.4006C>T [p.Arg1336Trp], c.7400þ1G>A,
and c.7477C>T [p.Arg2493*]) (Figure 2). The additional,
alternative exon (which we designate exon 40a) with the
c.4690G>A (p.Ala1564Thr) mutation occurs between
RefSeq annotated exons 40 and 41 (NM_023073.3), is
present in brain expressed sequence tag (EST) clones with
GenBank accession numbers AK096581 and BC144070,
and retains the large open reading frame of the gene. Using
RNA-sequencing data made publicly available by Illumi-
na’s Body Map 2.0 (see Web Resources), we were able to
confirm the expression of the exon. The assembly of raw
data from 16 different tissues identified a large number of
reads that mapped to that exon in both brain and testes
samples; significantly fewer reads mapped to other tissues
(Figure S5). Reads that covered both ends of the exon and
spliced correctly to neighboring exons were found in either
brain or testes samples. The c.4690G>A (p.Ala1564Thr)
mutation was also absent from our 261 control exomes
and from the 1,000 Genomes Browser. It was not possible
to get accurate SIFT or Polyphen-2 predictions for this
mutation because the corresponding exon was not anno-
tated across species.
We further addressed the frequency of the six putative
C5ORF42 mutations identified in our JBTS individuals in
the French Canadian population. Genotyping 477 French
Canadian controls, including 96 Acadians subjects and 96
subjects from the Gaspesie region located immediately east
of Matapedia, did not identify a carrier of any of the six
C5ORF42 mutations. However, some of these mutations
are reported in the heterozygous state at very low frequen-
cies in the National Heart, Lung, and Blood Institute
(NHLBI) Go Exome Sequencing Project (ESP) dataset; these
mutations are c.4006C>T (p.Arg1336Trp) (2/10,754;
minor allele frequency [MAF] ¼ 0.0186%), c.7477C>T
(p.Arg2493*) (1/10,755; MAF ¼ 0.009%; rs139675596),
and c.4690G>A (p.Ala1564Thr) (12/4,574; MAF ¼0.262%; rs111294855). It should be noted that
c.4006C>T and c.7477C>T correspond to CpG sites,
Table 2. Genes with Rare Homozygous or Multiple HeterozygousVariants from the Combined Exome Sequences from 13 Individualswith JBTS
Number of Familieswith Mutationsin the Same Gene
Numberof Genes Gene Identity
1 family 528 C5ORF42, .
2 families 16 C5ORF42, ACAN, ADAMTS18,C10orf68, FSIP2, LRP1B,MUC12, MUC16, MUC4,MYO16, PKD1L2, PKHD1L1,RGPD4, SHROOM4,TMEM231, ZNF717
3 families 7 C5ORF42, MUC5B, PLEC,FAT3, FLG, TTN, LAMA5
4 families 1 C5ORF42
5 families 1 C5ORF42
>5 families 0 -
Table 1. Variant Prioritization Steps in the Analysis of CombinedExome Sequences from 13 Individuals with JBTS
Filters Applied (Sequentially)Number ofVariants Retained
Nonsynonymous, splicing, and coding indelvariants
34,157a
After excluding variants present in >1in-house exome
7,075
After excluding variants reported in 1,000Genomes Browser (frequency > 0.5%)
6,911
aTotal number of variants identified in the combined 13 exomes; redundantvariants were counted only once.
696 The American Journal of Human Genetics 90, 693–700, April 6, 2012

which are associated with a higher mutation rate, possibly
explaining the recurrence of these nonetheless rare muta-
tions in different populations.
The presence of five potentially deleterious C5ORF42
mutations that segregate with the disease in seven presum-
ably unrelated (though all French Canadian) families
strongly suggests that disruption of this gene causes JBTS
in our subjects. It remains uncertain whether c.4690G>A
(p.Ala1564Thr) is pathogenic, considering that it is not
clearly deleterious and that it is found at a higher
frequency (0.26%) in the ESP dataset than are the other
mutations. It is possible that this variant is linked to
another mutation—not identified by our exome-
sequencing approach—on the same haplotype.
Very little is known about C5ORF42 function. The Ref-
Seq version of the full-length transcript (NM_023073.3;
Ensemble accession number ENST00000425232) appar-
ently derives from virtual assembly of overlapping mRNA
and EST clones. The predicted major mRNA isoform
comprises 11,199 bp and contains 52 exons; the putative
encoded protein is similarly large and comprises 3,198
amino acids. With the exception of c.4690G>A
(p.Ala1564Thr), all mutations reported herein are common
to all annotated protein-coding transcripts (Figure 3A). The
predicted protein sequence is well conserved across much
of the gene length in other vertebrates. It does not appear
to contain any specific known functional domains,
although the Gene Ontology project suggests that it might
be a transmembrane protein and ProtoNet predicts a
coiled-coil structure within the protein. Proteomic studies
have reported interactions among C5ORF42, the p21-
activating kinase 1 (PAK1), and the small ubiquitin-like
modifier 1 (SUMO1).32,33 Although the significance of
these interactions remains to be validated and further
investigated, it is noteworthy that these latter genes play
a role in neural development.34,35 EST-expression (Unig-
ene data), microarray profiling (Allen Brain Atlas), and
BioGPS indicate that C5ORF42 is widely expressed in
a variety of tissues, including the brain.
In terms of genotype-phenotype correlation, all JBTS
individuals with mutations in C5ORF42 showed global
developmental delay, and the onset of independent
Figure 3. C5ORF42 Mutations Identified in Individuals with JBTS(A) Scheme showing the positions of the mutations with respect to the different C5ORF42 Ensembl-annotated transcripts that are pre-dicted to produce proteins. The numbering on top is based on the cDNA positions of ENST00000425232 (identical to RefSeq accessionnumber NM_023073.3). Mutation c.7957þ288G>A is annotated as part of a coding exon in ENST00000388739 and causes a missensechange (p.Ala1564Thr).(B) NCBI HomoloGene-generated amino acid alignment of C5ORF42. Its predicted orthologs show the conservation of the Arg1336residue.
The American Journal of Human Genetics 90, 693–700, April 6, 2012 697
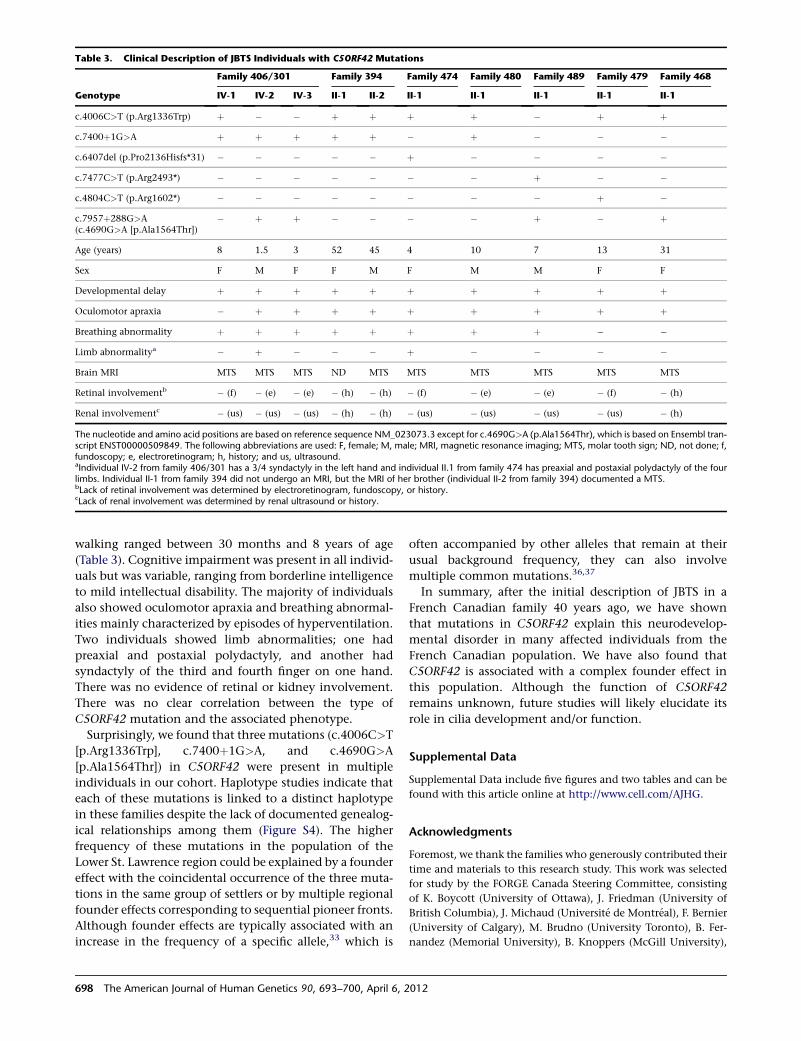
walking ranged between 30 months and 8 years of age
(Table 3). Cognitive impairment was present in all individ-
uals but was variable, ranging from borderline intelligence
to mild intellectual disability. The majority of individuals
also showed oculomotor apraxia and breathing abnormal-
ities mainly characterized by episodes of hyperventilation.
Two individuals showed limb abnormalities; one had
preaxial and postaxial polydactyly, and another had
syndactyly of the third and fourth finger on one hand.
There was no evidence of retinal or kidney involvement.
There was no clear correlation between the type of
C5ORF42 mutation and the associated phenotype.
Surprisingly, we found that three mutations (c.4006C>T
[p.Arg1336Trp], c.7400þ1G>A, and c.4690G>A
[p.Ala1564Thr]) in C5ORF42 were present in multiple
individuals in our cohort. Haplotype studies indicate that
each of these mutations is linked to a distinct haplotype
in these families despite the lack of documented genealog-
ical relationships among them (Figure S4). The higher
frequency of these mutations in the population of the
Lower St. Lawrence region could be explained by a founder
effect with the coincidental occurrence of the three muta-
tions in the same group of settlers or by multiple regional
founder effects corresponding to sequential pioneer fronts.
Although founder effects are typically associated with an
increase in the frequency of a specific allele,33 which is
often accompanied by other alleles that remain at their
usual background frequency, they can also involve
multiple common mutations.36,37
In summary, after the initial description of JBTS in a
French Canadian family 40 years ago, we have shown
that mutations in C5ORF42 explain this neurodevelop-
mental disorder in many affected individuals from the
French Canadian population. We have also found that
C5ORF42 is associated with a complex founder effect in
this population. Although the function of C5ORF42
remains unknown, future studies will likely elucidate its
role in cilia development and/or function.
Supplemental Data
Supplemental Data include five figures and two tables and can be
found with this article online at http://www.cell.com/AJHG.
Acknowledgments
Foremost, we thank the families who generously contributed their
time and materials to this research study. This work was selected
for study by the FORGE Canada Steering Committee, consisting
of K. Boycott (University of Ottawa), J. Friedman (University of
British Columbia), J. Michaud (Universite de Montreal), F. Bernier
(University of Calgary), M. Brudno (University Toronto), B. Fer-
nandez (Memorial University), B. Knoppers (McGill University),
Table 3. Clinical Description of JBTS Individuals with C5ORF42 Mutations
Genotype
Family 406/301 Family 394 Family 474 Family 480 Family 489 Family 479 Family 468
IV-1 IV-2 IV-3 II-1 II-2 II-1 II-1 II-1 II-1 II-1
c.4006C>T (p.Arg1336Trp) þ � � þ þ þ þ � þ þ
c.7400þ1G>A þ þ þ þ þ � þ � � �
c.6407del (p.Pro2136Hisfs*31) � � � � � þ � � � �
c.7477C>T (p.Arg2493*) � � � � � � � þ � �
c.4804C>T (p.Arg1602*) � � � � � � � � þ �
c.7957þ288G>A(c.4690G>A [p.Ala1564Thr])
� þ þ � � � � þ � þ
Age (years) 8 1.5 3 52 45 4 10 7 13 31
Sex F M F F M F M M F F
Developmental delay þ þ þ þ þ þ þ þ þ þ
Oculomotor apraxia � þ þ þ þ þ þ þ þ þ
Breathing abnormality þ þ þ þ þ þ þ þ � �
Limb abnormalitya � þ � � � þ � � � �
Brain MRI MTS MTS MTS ND MTS MTS MTS MTS MTS MTS
Retinal involvementb � (f) � (e) � (e) � (h) � (h) � (f) � (e) � (e) � (f) � (h)
Renal involvementc � (us) � (us) � (us) � (h) � (h) � (us) � (us) � (us) � (us) � (h)
The nucleotide and amino acid positions are based on reference sequence NM_023073.3 except for c.4690G>A (p.Ala1564Thr), which is based on Ensembl tran-script ENST00000509849. The following abbreviations are used: F, female; M, male; MRI, magnetic resonance imaging; MTS, molar tooth sign; ND, not done; f,fundoscopy; e, electroretinogram; h, history; and us, ultrasound.aIndividual IV-2 from family 406/301 has a 3/4 syndactyly in the left hand and individual II.1 from family 474 has preaxial and postaxial polydactyly of the fourlimbs. Individual II-1 from family 394 did not undergo an MRI, but the MRI of her brother (individual II-2 from family 394) documented a MTS.bLack of retinal involvement was determined by electroretinogram, fundoscopy, or history.cLack of renal involvement was determined by renal ultrasound or history.
698 The American Journal of Human Genetics 90, 693–700, April 6, 2012

M. Samuels (Universite de Montreal), and S. Scherer (University of
Toronto). We would like to thank Janet Marcadier (clinical
coordinator) and Chandree Beaulieu (project manager) for their
contribution to the infrastructure of the FORGE Canada Consor-
tium. The authors also wish to acknowledge the contribution of
the high-throughput sequencing platform of the McGill
University and Genome Quebec Innovation Centre (Montreal,
Canada). This work was funded by the Government of Canada
through Genome Canada, the Canadian Institutes of Health
Research (CIHR), and the Ontario Genomics Institute (OGI-049).
Additional funding was provided by Genome Quebec and
Genome British Columbia. K. Boycott is supported by a Clinical
Investigatorship Award from the CIHR Institute of Genetics.
J.L. Michaud is a National Scholar from the Fonds de la Recherche
en Sante du Quebec (FRSQ). M. Srour holds a training award from
the FRSQ.
Received: December 15, 2011
Revised: January 23, 2012
Accepted: February 13, 2012
Published online: March 15, 2012
Web Resources
The URLs for data presented herein are as follows:
1,000 Genomes Browser, http://browser.1000genomes.org/index.
html
Allen Brain Atlas, http://www.brain-map.org/
BioGPS, http://biogps.org
dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
Ensemble Genome Browser, http://www.ensembl.org
ESP Exome Variant Server, http://evs.gs.washington.edu/EVS/
Gene Ontology, http://www.geneontology.org/
Illumina’s Body Map 2.0 transcriptome, http://www.ebi.ac.uk/
arrayexpress/browse.html?keywords ¼ E-MTAB-513
NCBI HomoloGene, http://www.ncbi.nlm.nih.gov/homologene
NCBI Nucleotide Database, http://www.ncbi.nlm.nih.gov/nuccore
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
Polyphen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
Unigene, http://www.ncbi.nlm.nih.gov/unigene
References
1. Sattar, S., and Gleeson, J.G. (2011). The ciliopathies in
neuronal development: a clinical approach to investigation
of Joubert syndrome and Joubert syndrome-related disorders.
Dev. Med. Child Neurol. 53, 793–798.
2. Bielas, S.L., Silhavy, J.L., Brancati, F., Kisseleva, M.V., Al-Gazali,
L., Sztriha, L., Bayoumi, R.A., Zaki, M.S., Abdel-Aleem, A.,
Rosti, R.O., et al. (2009). Mutations in INPP5E, encoding
inositol polyphosphate-5-phosphatase E, link phosphatidyl
inositol signaling to the ciliopathies. Nat. Genet. 41, 1032–
1036.
3. Edvardson, S., Shaag, A., Zenvirt, S., Erlich, Y., Hannon, G.J.,
Shanske, A.L., Gomori, J.M., Ekstein, J., and Elpeleg, O.
(2010). Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is
associated with a TMEM216 mutation. Am. J. Hum. Genet.
86, 93–97.
4. Valente, E.M., Logan, C.V., Mougou-Zerelli, S., Lee, J.H.,
Silhavy, J.L., Brancati, F., Iannicelli, M., Travaglini, L., Romani,
S., Illi, B., et al. (2010). Mutations in TMEM216 perturb cilio-
genesis and cause Joubert, Meckel and related syndromes.
Nat. Genet. 42, 619–625.
5. Dixon-Salazar, T., Silhavy, J.L., Marsh, S.E., Louie, C.M., Scott,
L.C., Gururaj, A., Al-Gazali, L., Al-Tawari, A.A., Kayserili, H.,
Sztriha, L., and Gleeson, J.G. (2004). Mutations in the AHI1
gene, encoding jouberin, cause Joubert syndrome with
cortical polymicrogyria. Am. J. Hum. Genet. 75, 979–987.
6. Parisi, M.A., Bennett, C.L., Eckert, M.L., Dobyns, W.B., Glee-
son, J.G., Shaw, D.W., McDonald, R., Eddy, A., Chance, P.F.,
and Glass, I.A. (2004). The NPHP1 gene deletion associated
with juvenile nephronophthisis is present in a subset of indi-
viduals with Joubert syndrome. Am. J. Hum. Genet. 75, 82–91.
7. Valente, E.M., Silhavy, J.L., Brancati, F., Barrano, G., Krishnas-
wami, S.R., Castori, M., Lancaster, M.A., Boltshauser, E., Boc-
cone, L., Al-Gazali, L., et al; International Joubert Syndrome
Related Disorders Study Group. (2006). Mutations in
CEP290, which encodes a centrosomal protein, cause pleio-
tropic forms of Joubert syndrome. Nat. Genet. 38, 623–625.
8. Sayer, J.A., Otto, E.A., O’Toole, J.F., Nurnberg, G., Kennedy,
M.A., Becker, C., Hennies, H.C., Helou, J., Attanasio, M.,
Fausett, B.V., et al. (2006). The centrosomal protein nephro-
cystin-6 is mutated in Joubert syndrome and activates
transcription factor ATF4. Nat. Genet. 38, 674–681.
9. Baala, L., Romano, S., Khaddour, R., Saunier, S., Smith,U.M., Au-
dollent, S., Ozilou, C., Faivre, L., Laurent, N., Foliguet, B., et al.
(2007). The Meckel-Gruber syndrome gene, MKS3, is mutated
in Joubert syndrome. Am. J. Hum. Genet. 80, 186–194.
10. Arts, H.H., Doherty, D., van Beersum, S.E., Parisi, M.A.,
Letteboer, S.J., Gorden, N.T., Peters, T.A., Marker, T., Voesenek,
K., Kartono, A., et al. (2007). Mutations in the gene encoding
the basal body protein RPGRIP1L, a nephrocystin-4 interactor,
cause Joubert syndrome. Nat. Genet. 39, 882–888.
11. Delous, M., Baala, L., Salomon, R., Laclef, C., Vierkotten, J.,
Tory, K., Golzio, C., Lacoste, T., Besse, L., Ozilou, C., et al.
(2007). The ciliary gene RPGRIP1L is mutated in cerebello-
oculo-renal syndrome (Joubert syndrome type B) and Meckel
syndrome. Nat. Genet. 39, 875–881.
12. Cantagrel, V., Silhavy, J.L., Bielas, S.L., Swistun, D., Marsh,
S.E., Bertrand, J.Y., Audollent, S., Attie-Bitach, T., Holden,
K.R., Dobyns, W.B., et al; International Joubert Syndrome
Related Disorders Study Group. (2008). Mutations in the cilia
gene ARL13B lead to the classical form of Joubert syndrome.
Am. J. Hum. Genet. 83, 170–179.
13. Noor, A.,Windpassinger, C., Patel, M., Stachowiak, B., Mikhai-
lov, A., Azam, M., Irfan, M., Siddiqui, Z.K., Naeem, F., Pater-
son, A.D., et al. (2008). CC2D2A, encoding a coiled-coil and
C2 domain protein, causes autosomal-recessive mental retar-
dation with retinitis pigmentosa. Am. J. Hum. Genet. 82,
1011–1018.
14. Gorden, N.T., Arts, H.H., Parisi, M.A., Coene, K.L., Letteboer,
S.J., van Beersum, S.E., Mans, D.A., Hikida, A., Eckert, M.,
Knutzen, D., et al. (2008). CC2D2A is mutated in Joubert
syndrome and interacts with the ciliopathy-associated basal
body protein CEP290. Am. J. Hum. Genet. 83, 559–571.
15. Dafinger, C., Liebau, M.C., Elsayed, S.M., Hellenbroich, Y.,
Boltshauser, E., Korenke, G.C., Fabretti, F., Janecke, A.R., Eber-
mann, I., Nurnberg, G., et al. (2011). Mutations in KIF7 link
Joubert syndrome with Sonic Hedgehog signaling and micro-
tubule dynamics. J. Clin. Invest. 121, 2662–2667.
The American Journal of Human Genetics 90, 693–700, April 6, 2012 699

16. Garcia-Gonzalo, F.R., Corbit, K.C., Sirerol-Piquer, M.S., Ramas-
wami, G., Otto, E.A., Noriega, T.R., Seol, A.D., Robinson, J.F.,
Bennett, C.L., Josifova, D.J., et al. (2011). A transition zone
complex regulates mammalian ciliogenesis and ciliary
membrane composition. Nat. Genet. 43, 776–784.
17. Sang, L., Miller, J.J., Corbit, K.C., Giles, R.H., Brauer, M.J.,
Otto, E.A., Baye, L.M., Wen, X., Scales, S.J., Kwong, M., et al.
(2011). Mapping the NPHP-JBTS-MKS protein network
reveals ciliopathy disease genes and pathways. Cell 145,
513–528.
18. Huang, L.J., Szymanska, K., Jensen, V.L., Janecke, A.R., Innes,
A.M., Davis, E.E., Frosk, P., Li, C.M., Willer, J.R., Chodirker,
B.N., et al. (2011). TMEM237 is mutated in individuals with
a Joubert syndrome related disorder and expands the role of
the TMEM family at the ciliary transition zone. Am. J. Hum.
Genet. 89, 713–730.
19. Lee, J.E., Silhavy, J.L., Zaki, M.S., Schroth, J., Bielas, S.L.,
Marsh, S.E., Olvera, J., Brancati, F., Iannicelli, M., Ikegami,
K., et al. (2012). CEP41 is mutated in Joubert syndrome and
is required for tubulin glutamylation at the cilium. Nat.
Genet. 44, 193–199.
20. Joubert, M., Eisenring, J.J., Robb, J.P., and Andermann, F.
(1969). Familial agenesis of the cerebellar vermis. A syndrome
of episodic hyperpnea, abnormal eye movements, ataxia, and
retardation. Neurology 19, 813–825.
21. Andermann, F., Andermann, E., Ptito, A., Fontaine, S., and
Joubert, M. (1999). History of Joubert syndrome and a 30-
year follow-up of the original proband. J. Child Neurol. 14,
565–569.
22. Fortin, J.C., and Lechasseur, A. (1999). Le Bas-Saint-Laurent
(Quebec: Presses de l’Universite Laval).
23. Hebert, P.M. (1994). Les Acadiens du Quebec (Montreal:
Editions de l0echo).24. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira,
M.A., Bender, D., Maller, J., Sklar, P., de Bakker, P.I., Daly,
M.J., and Sham, P.C. (2007). PLINK: A tool set for whole-
genome association and population-based linkage analyses.
Am. J. Hum. Genet. 81, 559–575.
25. Majewski, J., Schwartzentruber, J.A., Caqueret, A., Patry, L.,
Marcadier, J., Fryns, J.P., Boycott, K.M., Ste-Marie, L.G.,
McKiernan, F.E., Marik, I., et al; FORGE Canada Consortium.
(2011). Mutations in NOTCH2 in families with Hajdu-Cheney
syndrome. Hum. Mutat. 32, 1114–1117.
26. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis,
K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly,
M., and DePristo, M.A. (2010). The Genome Analysis Toolkit:
A MapReduce framework for analyzing next-generation DNA
sequencing data. Genome Res. 20, 1297–1303.
27. Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR:
Functional annotation of genetic variants from high-
throughput sequencing data. Nucleic Acids Res. 38, e164.
28. Davis, E.E., Zhang, Q., Liu, Q., Diplas, B.H., Davey, L.M., Hart-
ley, J., Stoetzel, C., Szymanska, K., Ramaswami, G., Logan,
C.V., et al; NISC Comparative Sequencing Program. (2011).
TTC21B contributes both causal and modifying alleles across
the ciliopathy spectrum. Nat. Genet. 43, 189–196.
29. Kumar, P., Henikoff, S., and Ng, P.C. (2009). Predicting the
effects of coding non-synonymous variants on protein func-
tion using the SIFT algorithm. Nat. Protoc. 4, 1073–1081.
30. Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gera-
simova, A., Bork, P., Kondrashov, A.S., and Sunyaev, S.R.
(2010). A method and server for predicting damaging
missense mutations. Nat. Methods 7, 248–249.
31. Mougou-Zerelli, S., Thomas, S., Szenker, E., Audollent, S.,
Elkhartoufi, N., Babarit, C., Romano, S., Salomon, R., Amiel,
J., Esculpavit, C., et al. (2009). CC2D2A mutations in Meckel
and Joubert syndromes indicate a genotype-phenotype corre-
lation. Hum. Mutat. 30, 1574–1582.
32. Bandyopadhyay, S., Chiang, C.Y., Srivastava, J., Gersten, M.,
White, S., Bell, R., Kurschner, C., Martin, C.H., Smoot, M.,
Sahasrabudhe, S., et al. (2010). A human MAP kinase interac-
tome. Nat. Methods 7, 801–805.
33. Ganesan, A.K., Kho, Y., Kim, S.C., Chen, Y., Zhao, Y., and
White, M.A. (2007). Broad spectrum identification of SUMO
substrates in melanoma cells. Proteomics 7, 2216–2221.
34. Huang, W., Zhou, Z., Asrar, S., Henkelman, M., Xie, W., and
Jia, Z. (2011). p21-Activated kinases 1 and 3 control brain
size through coordinating neuronal complexity and synaptic
properties. Mol. Cell. Biol. 31, 388–403.
35. Wilkinson, K.A., Nakamura, Y., and Henley, J.M. (2010).
Targets and consequences of protein SUMOylation in
neurons. Brain Res. Brain Res. Rev. 64, 195–212.
36. Yotova, V., Labuda, D., Zietkiewicz, E., Gehl, D., Lovell, A.,
Lefebvre, J.F., Bourgeois, S., Lemieux-Blanchard, E., Labuda,
M., Vezina, H., et al. (2005). Anatomy of a founder effect:
Myotonic dystrophy in Northeastern Quebec. Hum. Genet.
117, 177–187.
37. Roddier, K., Thomas, T., Marleau, G., Gagnon, A.M., Dicaire,
M.J., St-Denis, A., Gosselin, I., Sarrazin, A.M., Larbrisseau, A.,
Lambert, M., et al. (2005). Two mutations in the HSN2 gene
explain the high prevalence of HSAN2 in French Canadians.
Neurology 64, 1762–1767.
700 The American Journal of Human Genetics 90, 693–700, April 6, 2012

REPORT
Mutations in ROGDI Cause Kohlschutter-Tonz Syndrome
Anna Schossig,1,3,14 Nicole I. Wolf,2,4,14 Christine Fischer,3 Maria Fischer,5 Gernot Stocker,5
Stephan Pabinger,5 Andreas Dander,5 Bernhard Steiner,6 Otmar Tonz,6 Dieter Kotzot,1 Edda Haberlandt,7
Albert Amberger,1 Barbara Burwinkel,8,9 Katharina Wimmer,1 Christine Fauth,1
Caspar Grond-Ginsbach,10 Martin J. Koch,11 Annette Deichmann,12 Christof von Kalle,12
Claus R. Bartram,3 Alfried Kohlschutter,13 Zlatko Trajanoski,5 and Johannes Zschocke1,3,*
Kohlschutter-Tonz syndrome (KTS) is an autosomal-recessive disease characterized by the combination of epilepsy, psychomotor regres-
sion, and amelogenesis imperfecta. The molecular basis has not yet been elucidated. Here, we report that KTS is caused by mutations in
ROGDI. Using a combination of autozygosity mapping and exome sequencing, we identified a homozygous frameshift deletion,
c.229_230del (p.Leu77Alafs*64), in ROGDI in two affected individuals from a consanguineous family. Molecular studies in two addi-
tional KTS-affected individuals from two unrelated Austrian and Swiss families revealed homozygosity for nonsense mutation
c.286C>T (p.Gln96*) and compound heterozygosity for the splice-site mutations c.531þ5G>C and c.532-2A>T in ROGDI, respectively.
The latter mutation was also found to be heterozygous in the mother of the Swiss affected individual in whom KTS was reported for
the first time in 1974. ROGDI is highly expressed throughout the brain and other organs, but its function is largely unknown. Possible
interactions with DISC1, a protein involved in diverse cytoskeletal functions, have been suggested. Our finding that ROGDI mutations
cause KTS indicates that the protein product of this gene plays an important role in neuronal development as well as amelogenesis.
Kohlschutter-Tonz syndrome (KTS, MIM 226750) is a rare
genetic disorder characterized by the combination of
epilepsy, psychomotor delay and regression, and amelogen-
esis imperfecta. So far, 24 individuals with the clinical diag-
nosis of KTS have been reported.1–9 Pedigrees suggest an
autosomal-recessive mode of inheritance, but genetic
heterogeneity cannot be excluded. The molecular basis of
KTS has not yet been elucidated. The most striking feature
is global enamel deficiency (amelogenesis imperfecta) of
the hypoplastic or hypocalcified type; this deficiency affects
primary as well as permanent teeth right from the moment
of eruption. The enamel is very thin, rough, prone todisinte-
gration, and stained in various shades of brown. Onset of
epilepsy usually occurs in the first year of life; seizures are
difficult to treat or might be refractory to therapy. Affected
children show severe psychomotor delay or regression,
which might be present after birth but more frequently
develops after the onset of seizures. Both gross and fine
motor skills are usually impaired, and intellectual disability
might be severe. The natural course is variable; several
affected individuals developed spastic tetraplegia, and
somedied in childhood.There arenoconsistentdysmorphic
features or metabolic abnormalities, although nonspecific
facial anomalieshavebeenreported insomeaffected individ-
uals. Cranial imaging frequently shows mild brain atrophy.
In order to identify the genetic basis of KTS, we investi-
gated four affected children from three families as well as
healthy members of the index family reported in 1974.1
Clinical features of the affected individuals are summa-
rized in Table 1. Family A is a consanguineous Moroccan
family with two affected children (A-IV:3 and A-IV:4;
Figure 1);9 the parents are first cousins. Initial development
of the affected boy (A-IV:3) appeared normal, but treat-
ment-resistant epilepsy started when he was 4 months
old and led to loss of fixation and global developmental
delay. The affected younger sister (A-IV:4) showed psycho-
motor delay from birth onward. Epileptic seizures, which
were difficult to treat, started when she was 12 months
old. The first teeth in both children erupted when they
were 13 and 14 months old, respectively; from the begin-
ning, their teeth were lusterless and had a brownish dis-
coloration. Family B has been reported previously;8 the
parents of the affected boy (B-II:1) are not knowingly
related but come from neighboring villages in East Tyrol
(Austria). Epilepsy started when the boy was 5 months
old but later improved; there were no seizures after 7 years
of age, and medication was discontinued when he was
15 years old. Primary and permanent teeth were yellow,
hypoplastic, and crowded. Family C has one affected girl
(C-XI:2) who has not yet been reported. Left-sided hemi-
convulsive seizures started when she was 6 months old
and were initially difficult to treat, but when she was
6 years old, anticonvulsive treatment could be discontin-
ued. Primary and secondary dentition showed enamel
1Division of Human Genetics, Medical University Innsbruck, 6020 Innsbruck, Austria; 2Department of Child Neurology, VU University Medical Center,
1007 MB Amsterdam, The Netherlands; 3Institute of Human Genetics, Heidelberg University, 69120 Heidelberg, Germany; 4Department of Child
Neurology, Heidelberg University, 69120 Heidelberg, Germany; 5Division of Bioinformatics, Medical University Innsbruck, 6020 Innsbruck, Austria; 6Chil-
dren’s Hospital, 6000 Lucerne, Switzerland; 7Department of Pediatrics, Medical University Innsbruck, 6020 Innsbruck, Austria; 8German Cancer Research
Center, 69120 Heidelberg, Germany; 9Department of Obstetrics and Gynecology, Heidelberg University, 69120 Heidelberg, Germany; 10Department of
Neurology, Heidelberg University, 69120 Heidelberg, Germany; 11Department of Oral, Dental, and Maxillofacial Diseases, Heidelberg University, 69120
Heidelberg, Germany; 12National Center for Tumor Diseases and German Cancer Research Center, 69120 Heidelberg, Germany; 13University Hospital
for Child and Adolescent Medicine, 20246 Hamburg, Germany14These authors contributed equally to this work
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.012. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 701–707, April 6, 2012 701
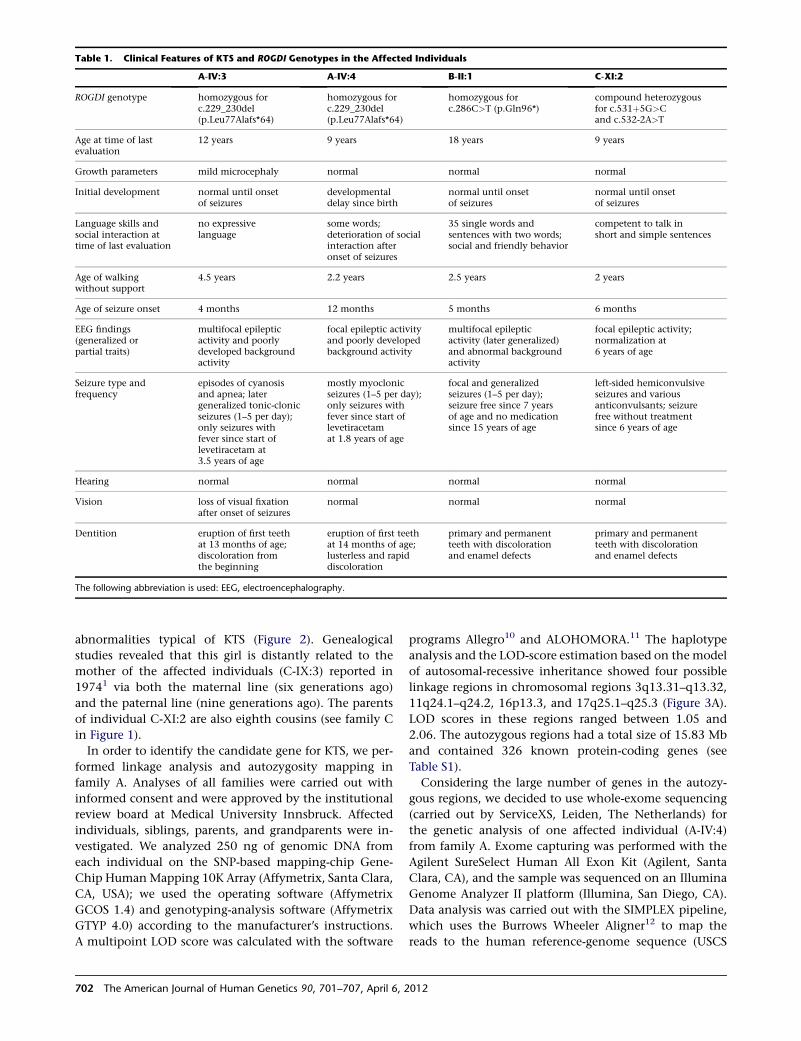
abnormalities typical of KTS (Figure 2). Genealogical
studies revealed that this girl is distantly related to the
mother of the affected individuals (C-IX:3) reported in
19741 via both the maternal line (six generations ago)
and the paternal line (nine generations ago). The parents
of individual C-XI:2 are also eighth cousins (see family C
in Figure 1).
In order to identify the candidate gene for KTS, we per-
formed linkage analysis and autozygosity mapping in
family A. Analyses of all families were carried out with
informed consent and were approved by the institutional
review board at Medical University Innsbruck. Affected
individuals, siblings, parents, and grandparents were in-
vestigated. We analyzed 250 ng of genomic DNA from
each individual on the SNP-based mapping-chip Gene-
Chip HumanMapping 10K Array (Affymetrix, Santa Clara,
CA, USA); we used the operating software (Affymetrix
GCOS 1.4) and genotyping-analysis software (Affymetrix
GTYP 4.0) according to the manufacturer’s instructions.
A multipoint LOD score was calculated with the software
programs Allegro10 and ALOHOMORA.11 The haplotype
analysis and the LOD-score estimation based on the model
of autosomal-recessive inheritance showed four possible
linkage regions in chromosomal regions 3q13.31–q13.32,
11q24.1–q24.2, 16p13.3, and 17q25.1–q25.3 (Figure 3A).
LOD scores in these regions ranged between 1.05 and
2.06. The autozygous regions had a total size of 15.83 Mb
and contained 326 known protein-coding genes (see
Table S1).
Considering the large number of genes in the autozy-
gous regions, we decided to use whole-exome sequencing
(carried out by ServiceXS, Leiden, The Netherlands) for
the genetic analysis of one affected individual (A-IV:4)
from family A. Exome capturing was performed with the
Agilent SureSelect Human All Exon Kit (Agilent, Santa
Clara, CA), and the sample was sequenced on an Illumina
Genome Analyzer II platform (Illumina, San Diego, CA).
Data analysis was carried out with the SIMPLEX pipeline,
which uses the Burrows Wheeler Aligner12 to map the
reads to the human reference-genome sequence (USCS
Table 1. Clinical Features of KTS and ROGDI Genotypes in the Affected Individuals
A-IV:3 A-IV:4 B-II:1 C-XI:2
ROGDI genotype homozygous forc.229_230del(p.Leu77Alafs*64)
homozygous forc.229_230del(p.Leu77Alafs*64)
homozygous forc.286C>T (p.Gln96*)
compound heterozygousfor c.531þ5G>Cand c.532-2A>T
Age at time of lastevaluation
12 years 9 years 18 years 9 years
Growth parameters mild microcephaly normal normal normal
Initial development normal until onsetof seizures
developmentaldelay since birth
normal until onsetof seizures
normal until onsetof seizures
Language skills andsocial interaction attime of last evaluation
no expressivelanguage
some words;deterioration of socialinteraction afteronset of seizures
35 single words andsentences with two words;social and friendly behavior
competent to talk inshort and simple sentences
Age of walkingwithout support
4.5 years 2.2 years 2.5 years 2 years
Age of seizure onset 4 months 12 months 5 months 6 months
EEG findings(generalized orpartial traits)
multifocal epilepticactivity and poorlydeveloped backgroundactivity
focal epileptic activityand poorly developedbackground activity
multifocal epilepticactivity (later generalized)and abnormal backgroundactivity
focal epileptic activity;normalization at6 years of age
Seizure type andfrequency
episodes of cyanosisand apnea; latergeneralized tonic-clonicseizures (1–5 per day);only seizures withfever since start oflevetiracetam at3.5 years of age
mostly myoclonicseizures (1–5 per day);only seizures withfever since start oflevetiracetamat 1.8 years of age
focal and generalizedseizures (1–5 per day);seizure free since 7 yearsof age and no medicationsince 15 years of age
left-sided hemiconvulsiveseizures and variousanticonvulsants; seizurefree without treatmentsince 6 years of age
Hearing normal normal normal normal
Vision loss of visual fixationafter onset of seizures
normal normal normal
Dentition eruption of first teethat 13 months of age;discoloration fromthe beginning
eruption of first teethat 14 months of age;lusterless and rapiddiscoloration
primary and permanentteeth with discolorationand enamel defects
primary and permanentteeth with discolorationand enamel defects
The following abbreviation is used: EEG, electroencephalography.
702 The American Journal of Human Genetics 90, 701–707, April 6, 2012

hg19, February 2009, Genome Reference Consortium
GRCh37). For SNP and DIP (deletion-insertion polymor-
phism) calling, as well as for realignment around indels,
we applied the Genome Analysis Toolkit (GATK).13 Exon
boundaries were specified by the Consensus Coding
Sequence (CCDS).10 An exome coverage depth of 233
was achieved: 46% of exons showed high coverage
(R203), and around 10% of exons showed low coverage
(%53). Variant detection identified 20,454 SNPs as well
as 1,208 DIPs. We annotated all variants with additional
information by using GATK and ANNOVAR14 to facilitate
the identification of disease-causing mutations. Subse-
quently, we applied the auto_annovar functionality to
filter variants against dbSNP (build 132), the 1,000
Genomes Project (Nov 2010), and previously assigned
conservation scores (for filtering details, see Table S2). After
all filtering steps, only a single strong candidate gene,
ROGDI (rogdi homolog [Drosophila], RefSeq accession
number NM_024589.1) in chromosomal region 16p13.3,
remained in the autozygous regions of interest. In exon 4
of this gene, we found a homozygous frameshift deletion,
c.229_230del (p.Leu77Alafs*64), which is predicted to
disrupt the amino acid structure and cause a premature
stop codon (Figure 3B). The filtering algorithm also called
a missense variant, c.2273G>C (p.Cys758Ser), in EVPL
(envoplakin [MIM 601590]) in the linkage region of
17q25.1; this variant (rs142251448) was included in build
134/135 of dbSNP and had a heterozygote frequency of
0.3% in the North American population.
After completing exome sequencing, we found a PhD
thesis that reports the results of autozygosity mapping in
five families affected by KTS.15 That study identified 30
candidate genes, including ROGDI, but did not find any
linkage to chromosomal region 17q25.1. There is no
other report on a possible link between KTS and ROGDI
or EVPL. Also considering the expected severity of the
frameshift deletion found in family A, we focused our
subsequent studies on ROGDI (Ensembl accession number
ENSG00000067836). This gene stretches over 5.98 kb in
chromosomal region 16p13.3 and contains 11 exons, all
of which are coding. Bioinformatics analysis showed that
the transcript of ROGDI codes for 287 amino acids and
results in a molecular weight of 32 kDa (RefSeq accession
number NP_078865.1). There is only one known func-
tional transcript. Dye-terminator sequencing of all exons
and adjacent intron sequences of ROGDI (NM_024589.1)
(ABI Prism 7000 sequence detection system, Applied Bio-
systems, Carlsbad, CA; primer sequences are available in
Table S3) confirmed the homozygous presence of the
mutation c.229_230del in both affected siblings of family
A (Figure 3C). As expected, both parents were found to
be heterozygous. Sequence analysis in family B revealed
Figure 1. Pedigrees of Investigated FamiliesThe pedigree for family A shows a consanguineous Moroccan family9 in which linkage analysis and exome sequencing were performed.The pedigree for family B shows a Tyrolean family affected by KTS,8 and the parents are not knowingly related. The pedigree for familyC shows the newly diagnosed Swiss family (the parents are X:1 and X:2) and its relationship with the distantly related index familyreported in 1974 (parents IX-3 und IX-4).1 Note that the parents in the newly identified family are distantly related to each other,but the affected child is compound heterozygous for two different mutations.
The American Journal of Human Genetics 90, 701–707, April 6, 2012 703
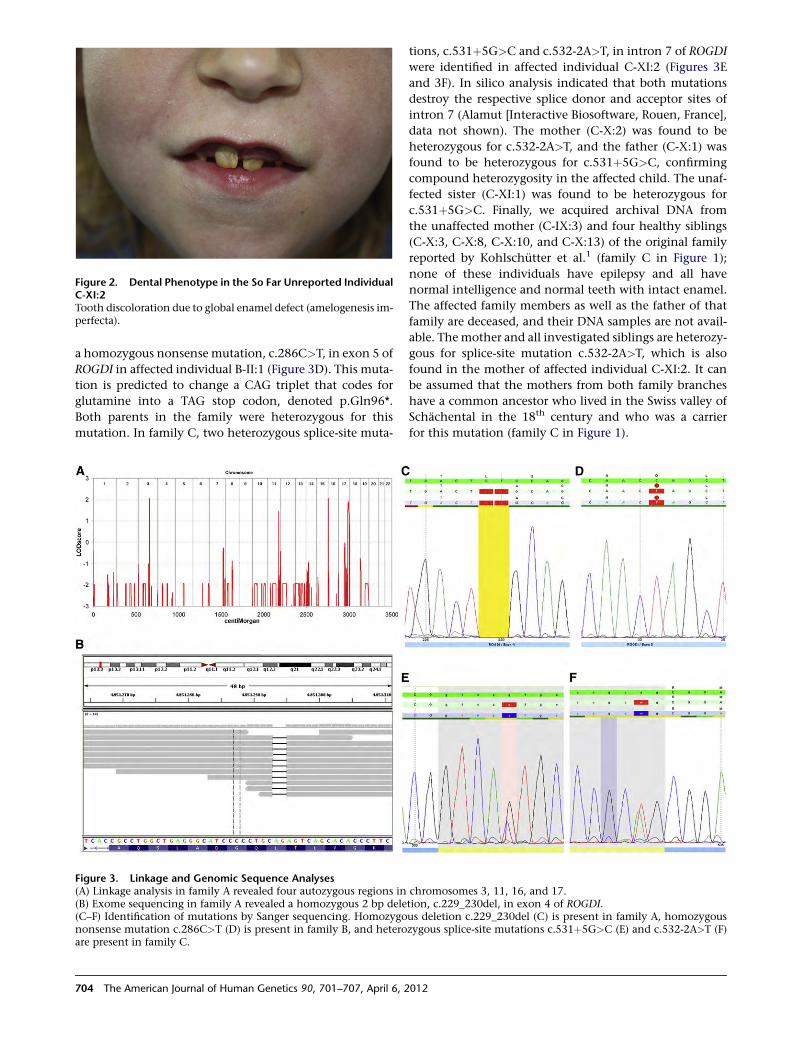
a homozygous nonsense mutation, c.286C>T, in exon 5 of
ROGDI in affected individual B-II:1 (Figure 3D). This muta-
tion is predicted to change a CAG triplet that codes for
glutamine into a TAG stop codon, denoted p.Gln96*.
Both parents in the family were heterozygous for this
mutation. In family C, two heterozygous splice-site muta-
tions, c.531þ5G>C and c.532-2A>T, in intron 7 of ROGDI
were identified in affected individual C-XI:2 (Figures 3E
and 3F). In silico analysis indicated that both mutations
destroy the respective splice donor and acceptor sites of
intron 7 (Alamut [Interactive Biosoftware, Rouen, France],
data not shown). The mother (C-X:2) was found to be
heterozygous for c.532-2A>T, and the father (C-X:1) was
found to be heterozygous for c.531þ5G>C, confirming
compound heterozygosity in the affected child. The unaf-
fected sister (C-XI:1) was found to be heterozygous for
c.531þ5G>C. Finally, we acquired archival DNA from
the unaffected mother (C-IX:3) and four healthy siblings
(C-X:3, C-X:8, C-X:10, and C-X:13) of the original family
reported by Kohlschutter et al.1 (family C in Figure 1);
none of these individuals have epilepsy and all have
normal intelligence and normal teeth with intact enamel.
The affected family members as well as the father of that
family are deceased, and their DNA samples are not avail-
able. The mother and all investigated siblings are heterozy-
gous for splice-site mutation c.532-2A>T, which is also
found in the mother of affected individual C-XI:2. It can
be assumed that the mothers from both family branches
have a common ancestor who lived in the Swiss valley of
Schachental in the 18th century and who was a carrier
for this mutation (family C in Figure 1).
Figure 2. Dental Phenotype in the So Far Unreported IndividualC-XI:2Tooth discoloration due to global enamel defect (amelogenesis im-perfecta).
Figure 3. Linkage and Genomic Sequence Analyses(A) Linkage analysis in family A revealed four autozygous regions in chromosomes 3, 11, 16, and 17.(B) Exome sequencing in family A revealed a homozygous 2 bp deletion, c.229_230del, in exon 4 of ROGDI.(C–F) Identification of mutations by Sanger sequencing. Homozygous deletion c.229_230del (C) is present in family A, homozygousnonsense mutation c.286C>T (D) is present in family B, and heterozygous splice-site mutations c.531þ5G>C (E) and c.532-2A>T (F)are present in family C.
704 The American Journal of Human Genetics 90, 701–707, April 6, 2012
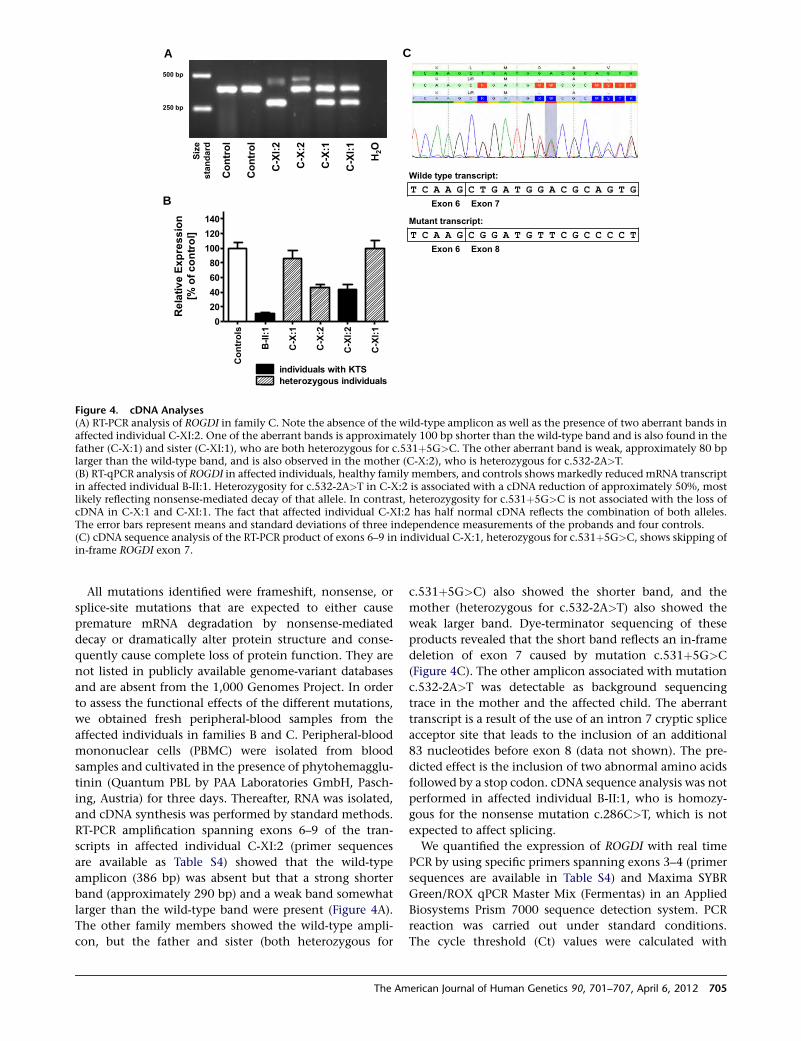
All mutations identified were frameshift, nonsense, or
splice-site mutations that are expected to either cause
premature mRNA degradation by nonsense-mediated
decay or dramatically alter protein structure and conse-
quently cause complete loss of protein function. They are
not listed in publicly available genome-variant databases
and are absent from the 1,000 Genomes Project. In order
to assess the functional effects of the different mutations,
we obtained fresh peripheral-blood samples from the
affected individuals in families B and C. Peripheral-blood
mononuclear cells (PBMC) were isolated from blood
samples and cultivated in the presence of phytohemagglu-
tinin (Quantum PBL by PAA Laboratories GmbH, Pasch-
ing, Austria) for three days. Thereafter, RNA was isolated,
and cDNA synthesis was performed by standard methods.
RT-PCR amplification spanning exons 6–9 of the tran-
scripts in affected individual C-XI:2 (primer sequences
are available as Table S4) showed that the wild-type
amplicon (386 bp) was absent but that a strong shorter
band (approximately 290 bp) and a weak band somewhat
larger than the wild-type band were present (Figure 4A).
The other family members showed the wild-type ampli-
con, but the father and sister (both heterozygous for
c.531þ5G>C) also showed the shorter band, and the
mother (heterozygous for c.532-2A>T) also showed the
weak larger band. Dye-terminator sequencing of these
products revealed that the short band reflects an in-frame
deletion of exon 7 caused by mutation c.531þ5G>C
(Figure 4C). The other amplicon associated with mutation
c.532-2A>T was detectable as background sequencing
trace in the mother and the affected child. The aberrant
transcript is a result of the use of an intron 7 cryptic splice
acceptor site that leads to the inclusion of an additional
83 nucleotides before exon 8 (data not shown). The pre-
dicted effect is the inclusion of two abnormal amino acids
followed by a stop codon. cDNA sequence analysis was not
performed in affected individual B-II:1, who is homozy-
gous for the nonsense mutation c.286C>T, which is not
expected to affect splicing.
We quantified the expression of ROGDI with real time
PCR by using specific primers spanning exons 3–4 (primer
sequences are available in Table S4) and Maxima SYBR
Green/ROX qPCR Master Mix (Fermentas) in an Applied
Biosystems Prism 7000 sequence detection system. PCR
reaction was carried out under standard conditions.
The cycle threshold (Ct) values were calculated with
A
B
C
Figure 4. cDNA Analyses(A) RT-PCR analysis of ROGDI in family C. Note the absence of the wild-type amplicon as well as the presence of two aberrant bands inaffected individual C-XI:2. One of the aberrant bands is approximately 100 bp shorter than the wild-type band and is also found in thefather (C-X:1) and sister (C-XI:1), who are both heterozygous for c.531þ5G>C. The other aberrant band is weak, approximately 80 bplarger than the wild-type band, and is also observed in the mother (C-X:2), who is heterozygous for c.532-2A>T.(B) RT-qPCR analysis of ROGDI in affected individuals, healthy family members, and controls shows markedly reduced mRNA transcriptin affected individual B-II:1. Heterozygosity for c.532-2A>T in C-X:2 is associated with a cDNA reduction of approximately 50%, mostlikely reflecting nonsense-mediated decay of that allele. In contrast, heterozygosity for c.531þ5G>C is not associated with the loss ofcDNA in C-X:1 and C-XI:1. The fact that affected individual C-XI:2 has half normal cDNA reflects the combination of both alleles.The error bars represent means and standard deviations of three independence measurements of the probands and four controls.(C) cDNA sequence analysis of the RT-PCR product of exons 6–9 in individual C-X:1, heterozygous for c.531þ5G>C, shows skipping ofin-frame ROGDI exon 7.
The American Journal of Human Genetics 90, 701–707, April 6, 2012 705

sequence-detection system (SDS) software v1.2 (Applied
Biosystems). We quantified relative gene expression with
the comparative DDCt method by using HPRT1 (RefSeq
accession number NM_000194.2) as a reference gene.
These analyses showed that the amount of ROGDI cDNA
was markedly reduced to 10.6% (much lower than the
mean of the four controls) in affected individual B-II:1
(Figure 4B). The amount of cDNA in affected individual
C-XI:2 was 43.6%, similar to the value of 46.9% in her
mother (C-X:2). The amount of ROGDI cDNA in the father
and sister was in the normal range (86.6% and 100.2%,
respectively).
In summary, the cDNA analyses confirm that the muta-
tions in affected individuals B-II:1 and C-XI:2 severely
disrupt the normal ROGDI transcript. Mutation
c.531þ5G>C causes skipping of in-frame exon 7 but
does not lead to a translational frameshift and is not asso-
ciated with nonsense-mediated decay. In contrast, muta-
tion c.532-2A>T triggers the use of a cryptic intronic splice
acceptor site, explaining both a larger size of the cDNA
amplicon and nonsense-mediated decay. The latter effect
was also observed for nonsense mutation c.286C>T.
Thus, the mutations in all three KTS-affected families are
expected to be severe (null) mutations that are likely to
cause complete loss of ROGDI function.
The exact function of the protein encoded by ROGDI
is unknown. Using ANNIE,16 sequence-structure analysis
showed neither relevant features (e.g., transmembrane
regions or signal peptides) nor relevant protein domains.
Protein prediction methods17 indicate that ROGDI is a
globular protein and that the secondary structure consists
of 45% helixmotifs, 37% loop structures, and 17% strands.
The gene is highly conserved and has orthologs in many
species, including Drosophila melanogaster. It shows partic-
ularly high expression levels in various human brain
regions,18 in line with the CNS phenotype of KTS. A
Drosophila mutant of this gene showed a possible defi-
ciency in olfactory memory.19 Yeast two-hybrid screens20
suggested a possible interaction between ROGDI and
DISC1 (MIM 605210), a protein implicated in the develop-
ment of schizophrenia and involved in processes of cyto-
skeletal stability and organization, neuronal migration,
intracellular transport, and cell division.21 There are no
published studies that examined the role of ROGDI in
tooth development and amelogenesis. Our own data
provide robust information on the clinical effects of the
loss of ROGDI function in humans and provide interesting
perspectives for research into the molecular causes of
epilepsy and other conditions.
In conclusion, we report that KTS is caused by putative
loss-of-function mutations in ROGDI. All mutations
identified are predicted to be severe (null) mutations that
are likely to cause complete loss of protein function.
Heterozygosity for ROGDI-null mutations does not appear
to have any adverse effects. It is possible that individuals
with homozygosity or compound heterozygosity for
hypomorphic missense mutations in ROGDI could present
with isolated epilepsy independently from minor enamel
defects or vice versa. Assessing potential genotype-pheno-
type correlations will require molecular studies on addi-
tional affected individuals. Although we found ROGDI
mutations in all KTS-affected individuals investigated so
far, we cannot rule out genetic heterogeneity. Future
work will hopefully elucidate the exact function of ROGDI
in neuronal development and amelogenesis.
Supplemental Data
Supplemental Data include four tables and can be found with this
article online at http://www.cell.com/AJHG.
Acknowledgments
This work was supported by a grant from the Standortagentur
Tirol. We wish to thank Josef Muheim (Greppen, Switzerland)
for considerable help with the genealogical studies that allowed
us to link the two Swiss nuclear families into a single pedigree.
Long-term medical care to the affected individuals in the study
was provided by Thomas Schmitt-Mechelke and Petra Kolditz,
(both from Children’s Hospital, Lucerne, Switzerland). Bart
Janssen and Thomas Chin-A-Woeng (both from ServiceXS,
Leiden, The Netherlands) assisted with the exome sequencing.
We gratefully acknowledge expert technical assistance by Brunhild
Schagen (Department of Oral, Dental, and Maxillofacial Diseases,
Heidelberg University, Germany) as well as by Pia Traunfellner,
Sandra Unterkirchner, and Ramona Berberich (all from the Divi-
sion of Human Genetics, Medical University Innsbruck, Austria).
Received: December 23, 2011
Revised: January 31, 2012
Accepted: February 15, 2012
Published online: March 15, 2012
Web Resources
The URLs for data presented herein are as follows:
dbSNP, http://www.ncbi.nlm.nih.gov/snp/
Ensembl, http://www.ensembl.org
GenBank, http://www.ncbi.nlm.nih.gov/genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
References
1. Kohlschutter, A., Chappuis, D., Meier, C., Tonz, O., Vassella, F.,
and Herschkowitz, N. (1974). Familial epilepsy and yellow
teeth—a disease of the CNS associated with enamel hypo-
plasia. Helv. Paediatr. Acta 29, 283–294.
2. Christodoulou, J., Hall, R.K., Menahem, S., Hopkins, I.J., and
Rogers, J.G. (1988). A syndrome of epilepsy, dementia, and
amelogenesis imperfecta: Genetic and clinical features. J.
Med. Genet. 25, 827–830.
3. Petermoller, M., Kunze, J., and Gross-Selbeck, G. (1993).
Kohlschutter syndrome: Syndrome of epilepsy—dementia—
amelogenesis imperfecta. Neuropediatrics 24, 337–338.
706 The American Journal of Human Genetics 90, 701–707, April 6, 2012

4. Zlotogora, J., Fuks, A., Borochowitz, Z., and Tal, Y. (1993).
Kohlschutter-Tonz syndrome: Epilepsy, dementia, and amelo-
genesis imperfecta. Am. J. Med. Genet. 46, 453–454.
5. Musumeci, S.A., Elia, M., Ferri, R., Romano, C., Scuderi, C.,
and Del Gracco, S. (1995). A further family with epilepsy,
dementia and yellow teeth: The Kohlschutter syndrome. Brain
Dev. 17, 133–138, discussion 142–133.
6. Wygold, T., Kurlemann, G., and Schuierer, G. (1996). Kohl-
schutter syndrome—an example of a rare progressive neuroec-
todermal disease. Case report and review of the literature. Klin.
Padiatr. 208, 271–275.
7. Donnai, D., Tomlin, P.I., and Winter, R.M. (2005). Kohlschut-
ter syndrome in siblings. Clin. Dysmorphol. 14, 123–126.
8. Haberlandt, E., Svejda, C., Felber, S., Baumgartner, S., Gunther,
B., Utermann, G., and Kotzot, D. (2006). Yellow teeth,
seizures, and mental retardation: A less severe case of Kohl-
schutter-Tonz syndrome. Am. J. Med. Genet. A. 140, 281–283.
9. Schossig, A., Wolf, N., Grond-Ginsbach, C., Schagen, B.,
Koch, M., Rating, D., and Zschocke, J. (2007). Epileptische
Enzephalopathie und Zahnschmelzdefekt (Kohlschutter-
Tonz-Syndrom): Drei Fallberichte und Literaturubersicht.
Med. Genetik 19, 422–426.
10. Pruitt, K.D., Tatusova, T., andMaglott, D.R. (2007). NCBI refer-
ence sequences (RefSeq): A curated non-redundant sequence
database of genomes, transcripts and proteins. Nucleic Acids
Res. 35 (Database issue), D61–D65.
11. Ruschendorf, F., and Nurnberg, P. (2005). ALOHOMORA:
A tool for linkage analysis using 10K SNP array data. Bioinfor-
matics 21, 2123–2125.
12. Li, H., and Durbin, R. (2009). Fast and accurate short read
alignment with Burrows-Wheeler transform. Bioinformatics
25, 1754–1760.
13. DePristo, M.A., Banks, E., Poplin, R., Garimella, K.V., Maguire,
J.R., Hartl, C., Philippakis, A.A., del Angel, G., Rivas, M.A.,
Hanna, M., et al. (2011). A framework for variation discovery
and genotyping using next-generation DNA sequencing data.
Nat. Genet. 43, 491–498.
14. Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR:
Functional annotation of genetic variants from high-
throughput sequencing data. Nucleic Acids Res. 38, e164.
15. Lo, C. (2009). Genetics in Epilepsy. PhD thesis, University
College London, London, UK.
16. Ooi, H.S., Kwo, C.Y.,Wildpaner, M., Sirota, F.L., Eisenhaber, B.,
Maurer-Stroh, S., Wong, W.C., Schleiffer, A., Eisenhaber, F.,
and Schneider, G. (2009). ANNIE: Integrated de novo protein
sequence annotation. Nucleic Acids Res. 37 (Web Server issue),
W435–W440.
17. Rost, B., Yachdav, G., and Liu, J. (2004). The PredictProtein
server. Nucleic Acids Res. 32 (Web Server issue), W321–W326.
18. Wu, C., Orozco, C., Boyer, J., Leglise, M., Goodale, J., Batalov,
S., Hodge, C.L., Haase, J., Janes, J., Huss, J.W., 3rd, and Su, A.I.
(2009). BioGPS: An extensible and customizable portal for
querying and organizing gene annotation resources. Genome
Biol. 10, R130.
19. Dubnau, J., Chiang, A.S., Grady, L., Barditch, J., Gossweiler, S.,
McNeil, J., Smith, P., Buldoc, F., Scott, R., Certa, U., et al.
(2003). The staufen/pumilio pathway is involved in
Drosophila long-term memory. Curr. Biol. 13, 286–296.
20. Camargo, L.M., Collura, V., Rain, J.C., Mizuguchi, K., Hermja-
kob, H., Kerrien, S., Bonnert, T.P., Whiting, P.J., and Brandon,
N.J. (2007). Disrupted in Schizophrenia 1 Interactome:
Evidence for the close connectivity of risk genes andapotential
synaptic basis for schizophrenia. Mol. Psychiatry 12, 74–86.
21. Brandon, N.J., and Sawa, A. (2011). Linking neurodevelop-
mental and synaptic theories of mental illness through
DISC1. Nat. Rev. Neurosci. 12, 707–722.
The American Journal of Human Genetics 90, 701–707, April 6, 2012 707

REPORT
A Nonsense Mutation in the Human Homologof Drosophila rogdi Causes Kohlschutter–Tonz Syndrome
Adi Mory,1,2 Efrat Dagan,2,3 Barbara Illi,4 Philippe Duquesnoy,5 Shikma Mordechai,2 Ishai Shahor,6
Sveva Romani,4 Nivin Hawash-Moustafa,2 Hanna Mandel,1,6 Enza M. Valente,4 Serge Amselem,5
and Ruth Gershoni-Baruch1,2,*
Kohlschutter–Tonz syndrome (KTS) is a rare autosomal-recessive disorder of childhood onset, and it is characterized by global develop-
mental delay, spasticity, epilepsy, and amelogenesis imperfecta. In 12 KTS-affected individuals from a Druze village in northern Israel,
homozygosity mapping localized the gene linked to the disease to a 586,513 bp region (with a LOD score of 6.4) in chromosomal region
16p13.3. Sequencing of genes (from genomic DNA of an affected individual) in the linked region revealed chr16: 4,848,632 G>A, which
corresponds to ROGDI c.469C>T (p.Arg157*). The nonsensemutation was homozygous in all affected individuals, heterozygous in 10 of
100 unaffected individuals from the same Druze community, and absent from Druze controls from elsewhere. Wild-type ROGDI local-
izes to the nuclear envelope; ROGDI was not detectable in cells of affected individuals. All affected individuals suffered seizures, were
unable to speak, and had amelogenesis imperfecta. However, age of onset and the severity of mental and motor handicaps and that
of convulsions varied among affected individuals homozygous for the same nonsense allele.
Kohlschutter–Tonz syndrome (KTS) (MIM 226750) is
described as a rare autosomal-recessive neurodegenerative
disorder characterized by progressive dementia, spasticity,
and epilepsy.1,2 A clinical marker of KTS is a generalized
enamel defect, amelogenesis imperfecta, which is most
obvious as yellowed teeth. KTS was first identified in fami-
lies from Switzerland,1,2 Sicily,3 the Druze community of
northern Israel,4 and, subsequently, other locations in
western Europe.5–9 To date, only 21 affected individuals
have been reported. Seizures, intellectual impairment,
and amelogenesis imperfecta were reported in all families,
and other clinical features varied among reports. The goal
of this project was to identify the gene and the mutation
responsible for KTS in the highly consanguineous Druze
community.
We have compiled 14 new KTS cases pertaining to five
families, all of which originate from the same small Druze
village in northern Israel. The index case (II-1 in family 1),
who was referred to us at the age of 13 months for the eval-
uation of seizures, was noted to have amelogenesis imper-
fecta. Individual II-1 and her parents and siblings, as well
as informative relatives of four other families with affected
children, were enrolled (Figure S1, available online). A
consanguineous liaison is evident for families 3, 4, and 5,
although the parents of the affected children in family 4
are not known to be related. Family 2, unrelated to families
3–5, is consanguineous too (Figure S1). The study was
approved by the institutional review board at Rambam
Health Care Campus, Haifa, and after signed informed
consent (self and parental), a blood sample was drawn
for DNA extraction from all available family members
(both affected and healthy individuals). All affected indi-
viduals were clinically evaluated by a pediatric clinical
geneticist, medical records were reviewed, and parents
were interviewed.
The clinical characteristics of 14 KTS-affected individuals
(seven males and seven females; ages 2–24 years) are de-
picted in Table 1. Born at term after normal gestation,
they all appeared normal at birth and had no apparent dys-
morphology.AlthoughallKTScasesultimatelydisplayedan
unequivocal phenotype heralded by seizures and ‘‘yellow
teeth,’’ they varied widely with regard to the severity of
the manifestations, even within the same nuclear family.
Family 4 has five affected children (V-4 to V-8) who all
had epileptic episodes that varied in age of onset, intensity,
frequency, and response to treatment. The firstborn child
(V-4), who died at the age of 2 years, was vegetative, failed
to thrive, and suffered from intractable convulsions and
microcephaly. Their second-born affected child (V-6) dis-
played impaired psychomotor development from the
first months of life and convulsive episodes, starting at
9 months of age, that were refractory to treatment. She
was nonverbal and nonambulant. It was noted that her
brother (V-5) lagged developmentally starting at 6 months
of age, and he is regularly maintained on anticonvulsants
(he has had a partial response). At 16.5 years of age, he is
awkwardly ambulant and nonverbal, performs mostly by
shouting and yelling, and is irritable and self-mutilating.
His 15-year-old sister (V-7) suffers from a convulsive
disorder that responds well to treatment, and, although
intellectually disabled, she manages to communicate
with her mother, utters a few words, and is ambulant.
The youngest sibling (V-8), currently 3.5 years old, has
a convulsive disorder that is only partially controlled,
1The Ruth and Bruce Rappaport Faculty of Medicine, Technion-Israel Institute of Technology, 31096 Haifa, Israel; 2Institute of Human Genetics, Rambam
Health Care Campus, 31096 Haifa, Israel; 3Department of Nursing, Faculty of Social Welfare and Health Sciences, University of Haifa, 31905 Haifa, Israel;4CSS-Mendel Institute, viale Regina Margherita 261, 00198 Rome, Italy; 5Institut National de la Sante et de la Recherche Medicale U.933 and Universite
Pierre et Marie Curie, Hopital Armand-Trousseau, 75012 Paris, France; 6Department of Pediatrics, Rambam Health Care Campus, 31096 Haifa, Israel
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.03.005. �2012 by The American Society of Human Genetics. All rights reserved.
708 The American Journal of Human Genetics 90, 708–714, April 6, 2012
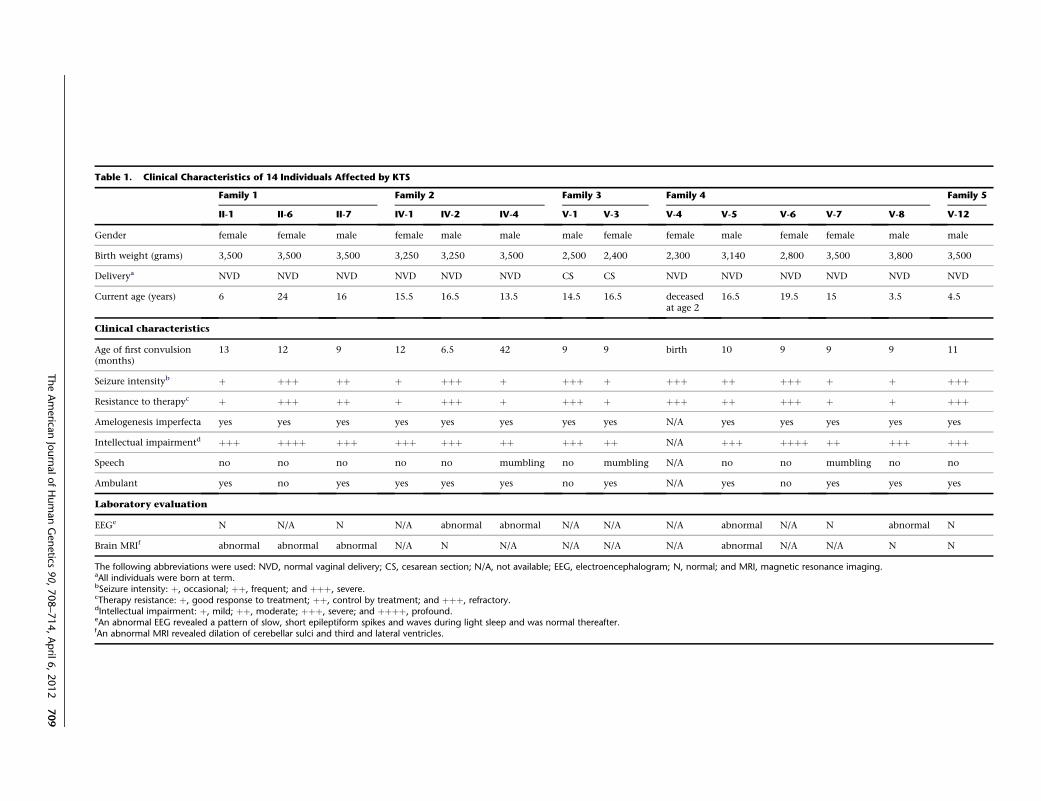
Table 1. Clinical Characteristics of 14 Individuals Affected by KTS
Family 1 Family 2 Family 3 Family 4 Family 5
II-1 II-6 II-7 IV-1 IV-2 IV-4 V-1 V-3 V-4 V-5 V-6 V-7 V-8 V-12
Gender female female male female male male male female female male female female male male
Birth weight (grams) 3,500 3,500 3,500 3,250 3,250 3,500 2,500 2,400 2,300 3,140 2,800 3,500 3,800 3,500
Deliverya NVD NVD NVD NVD NVD NVD CS CS NVD NVD NVD NVD NVD NVD
Current age (years) 6 24 16 15.5 16.5 13.5 14.5 16.5 deceasedat age 2
16.5 19.5 15 3.5 4.5
Clinical characteristics
Age of first convulsion(months)
13 12 9 12 6.5 42 9 9 birth 10 9 9 9 11
Seizure intensityb þ þþþ þþ þ þþþ þ þþþ þ þþþ þþ þþþ þ þ þþþ
Resistance to therapyc þ þþþ þþ þ þþþ þ þþþ þ þþþ þþ þþþ þ þ þþþ
Amelogenesis imperfecta yes yes yes yes yes yes yes yes N/A yes yes yes yes yes
Intellectual impairmentd þþþ þþþþ þþþ þþþ þþþ þþ þþþ þþ N/A þþþ þþþþ þþ þþþ þþþ
Speech no no no no no mumbling no mumbling N/A no no mumbling no no
Ambulant yes no yes yes yes yes no yes N/A yes no yes yes yes
Laboratory evaluation
EEGe N N/A N N/A abnormal abnormal N/A N/A N/A abnormal N/A N abnormal N
Brain MRIf abnormal abnormal abnormal N/A N N/A N/A N/A N/A abnormal N/A N/A N N
The following abbreviations were used: NVD, normal vaginal delivery; CS, cesarean section; N/A, not available; EEG, electroencephalogram; N, normal; and MRI, magnetic resonance imaging.aAll individuals were born at term.bSeizure intensity: þ, occasional; þþ, frequent; and þþþ, severe.cTherapy resistance: þ, good response to treatment; þþ, control by treatment; and þþþ, refractory.dIntellectual impairment: þ, mild; þþ, moderate; þþþ, severe; and þþþþ, profound.eAn abnormal EEG revealed a pattern of slow, short epileptiform spikes and waves during light sleep and was normal thereafter.fAn abnormal MRI revealed dilation of cerebellar sulci and third and lateral ventricles.
TheAmerica
nJournalofHumanGenetics
90,708–714,April
6,2012
709

and he is hyperactive, nonverbal, relatively ambulant, and
mentally disabled.
A wide interfamilial variability was again noted in
families 1, 2, and 3. Family 2 has three affected children
(IV-1, IV-2, and IV-4). The first affected boy (IV-2), who
was hypotonic and suckedpoorly as a neonate, experienced
intractable seizures beginning at 6.5 months of age. The
youngest boy (IV-4) was considered normal until, at the
age of three years, he experienced a convulsive episode.
Accordingly, IV-4 performs better than his older siblings.
Affected children from family 1 (II-1, II-6, and II-7) and
family 3 (V-1 and V-3) all presented with seizures at around
one year of age (between 9 and 13 months). The severity of
the epileptic events ranged from short convulsive episodes
that resolved under antiepileptic treatment (such as with
individuals II-1 and V-3) to generalized convulsions
partially controlled by anticonvulsants (II-7) to frequent
convulsive episodes resistant to therapy (II-6 and V-1).
Intellectual disability was noted to correlate with the
severity of the epileptic events. The phenotype displayed
by our KTS cases is consistent but variable. All cases dis-
played amelogenesis imperfecta, seizures, severe develop-
mental delay, and lack of speech. The magnitude of the
intellectual disability, although severe to profound, is still
directly related to the severity of the convulsive disorder
as manifested by age of onset and response to treatment.
Affected individuals lost their gross motor skills in either
early or late adolescence because they became spastic. The
most afflicted individualswerebedriddenearly in life.Meta-
bolicworkupwasnegative as a rule. Electroencephalograms
(EEGs) undertaken shortly after a convulsive episode were
interpreted, in most cases, as normal. Magnetic resonance
images (MRIs) were available for seven individuals. In four
of these individuals, dilatation of cerebellar sulci and third
and lateral ventricles was evident and should probably be
regarded as a correlate of cerebral atrophy.
With this in mind, the term ‘‘dementia’’ previously used
for defining a major characteristic of KTS is not valid for
the cases described here. KTS-affected children have global
developmental delay that is cortical in nature (intellectual
disability, spasticity, and seizures), and the progressive
nature of their neurological decline might be attributed
to, among other things, the intractability of their seizures
and, as such, epileptic encephalopathy.
Homozygosity mapping was carried out with the
assumptions that the disease follows a recessive model
and that a single responsible allele is shared by all affected
individuals. First, genomic DNA from ten affected individ-
uals, one healthy sibling, and one obligate carrier parent
was genotyped with the Illumina 6000 SNP array (BioRap
Technologies at the Rappaport Institute); thereafter, five
affected individuals were genotyped with the Affimetrix
GeneChip Human Mapping 250K Nsp microarray (Biolog-
ical Services Unit at the Weizmann Institute). Homozy-
gosity-by-descent analysis was carried out manually and
explored identical homozygous intervals in all affected
individuals. Candidate homozygous loci were genotyped
with microsatellite markers derived from Marshfield
maps or with markers designed by us (our markers were
based on the Tandem Repeats Finder program and the
UCSC Human Genome Database). Haplotypes of family
members were manually constructed and analyzed with
SUPERLINK online. In the 6000 SNP array, the analysis
identified a candidate segment, in chromosomal region
16p13.3, spanning 2,670,304 bp between rs2075852
and rs1012259; only one homozygous SNP (rs85930)
shared homozygosity in all ten affected individuals,
whereas the healthy mother and sibling were heterozy-
gous. Evaluating this interval in the 250K SNP array,
we identified a homozygous segment spanning only
873,963 bp between rs11865087 and rs3760030; this
segment was shared by four of the five genotyped individ-
uals. The fifth affected individual was clinically reevaluated
and excluded from the research as a non-KTS case (data
not shown). We developed and genotyped the following
three microsatellite markers in this interval: GT32 at
chr16: 4,533,109–4,533,282 (marker 1); TG19 at chr16:
4,854,835–4,855,072 (marker 2); and GT31 at chr16:
5,105,840–5,106,041 (marker 3). We depict haplotypes in
Figure 1 to show the segregation of markers and SNPs in
affected individuals and healthy family members; there
are three affected and seven healthy individuals in family
1, three affected and two healthy individuals in family 2,
and six affected and eight healthy individuals in families
3–5 (Figure 1A). Individual V-4 (family 4), who died at
2 years of age, and individual V-8 (family 4), who has not
yet been diagnosed with KTS at this stage, were excluded
from the analysis. All affected individuals shared the
same homozygous haplotype for markers 2 and 3, and
a 586,513 bp segment between microsatellite marker 1
(chr16: 4,533,109) and rs3760030 (chr16: 5,119,872) was
defined as the linkage locus for KTS (Figure 1A). Under
a recessive model of full penetrance, the LOD score for
linkage of this region to KTS was 6.4.
Chromosomal region 16p13.3 harbors 17 genes. We
performed Sanger sequencing of coding regions and flank-
ing intron-exon boundaries on genomic DNA from one
affected individual (II-1 in family 1) by using the primers
that we designed. The genomic sequences were retrieved
from the UCSC Genome Browser (GRCh37/hg19 assembly
[Feb. 2009]). In the first ten genes, no potentially
damaging variants were found. In contrast, sequencing
of theDrosophila rogdi homolog, ROGDI (FLJ22386) (RefSeq
accession number NM_024589.1), yielded a homozygous
nonsensemutation (c.469C>T in exon 7) causing a prema-
ture stop codon, p.Arg157* (Figure 1B). This nonsense
mutation cosegregated with KTS in all five families; all
affected individuals were homozygous for the mutation,
all parents were heterozygous, and unaffected siblings
were either heterozygous or homozygous for the wild-
type allele. Of 100 unaffected adults from the same Druze
community, ten were heterozygous carriers of the muta-
tion; none were homozygous for the mutation. The muta-
tion was not observed in 100 Druze individuals from other
710 The American Journal of Human Genetics 90, 708–714, April 6, 2012
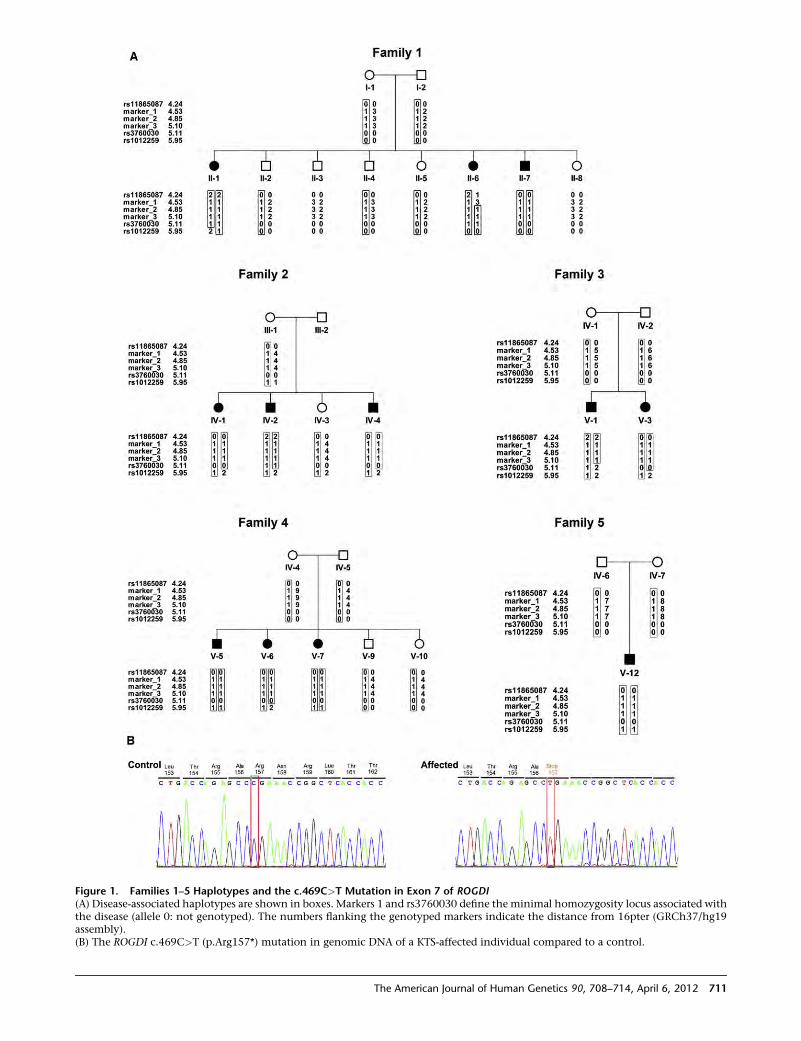
Figure 1. Families 1–5 Haplotypes and the c.469C>T Mutation in Exon 7 of ROGDI(A) Disease-associated haplotypes are shown in boxes. Markers 1 and rs3760030 define the minimal homozygosity locus associated withthe disease (allele 0: not genotyped). The numbers flanking the genotyped markers indicate the distance from 16pter (GRCh37/hg19assembly).(B) The ROGDI c.469C>T (p.Arg157*) mutation in genomic DNA of a KTS-affected individual compared to a control.
The American Journal of Human Genetics 90, 708–714, April 6, 2012 711

parts of Israel. Primer sequences and PCR conditions are
available upon request. The c.469C>T mutation was not
reported in the Exome Variant Server. No clinical links
were reported for SNPs in the gene.
ROGDI (FLJ22386) is the human homolog of Drosophila
rogdi. The human gene encodes a 287 amino acid leucine
zipper protein of unknown function. Drosophila rogdi
encodes 343 and 268 amino acid isoforms. The stop at
Figure 2. Subcellular Localization of ROGDI in Transfected HEK 293 Cells and Blood Mononuclear Cells(A–F) The immunostaining of ROGDI-transfected HEK 293 cells was performedwith a ROGDI polyclonal antibody incubated in the pres-ence of a permeabilizing reagent (0.2% saponin). ROGDI labeling (A) was revealed with an Alexa Fluor-488 goat anti-rabbit secondaryantibody (green). Nuclei (B) were stained with DAPI (blue). The merged picture (C) shows the ROGDI-antibody staining (green) togetherwith nuclei staining (blue). As a control, the same experiment was performed with the secondary antibody in the absence of ROGDIantibody (D, E, and F).(G–R) The same ROGDI antibody was used for immunolocalization of native ROGDI in blood mononuclear cells (green signal in G, K,and O). Colabeling was performed with a LAMIN A monoclonal antibody (red signal in H, L, and P). Nuclei (I, M, and Q) were stainedwith DAPI (blue). The partial colocalization of ROGDI with LAMIN A is shown in (J), (N), and (R). Cells were observed by confocalmicroscopy. Three cell sections from the middle to the top of cells are shown (G–J, K–N, and O–R, respectively). White scale bars repre-sent 10mm.
712 The American Journal of Human Genetics 90, 708–714, April 6, 2012
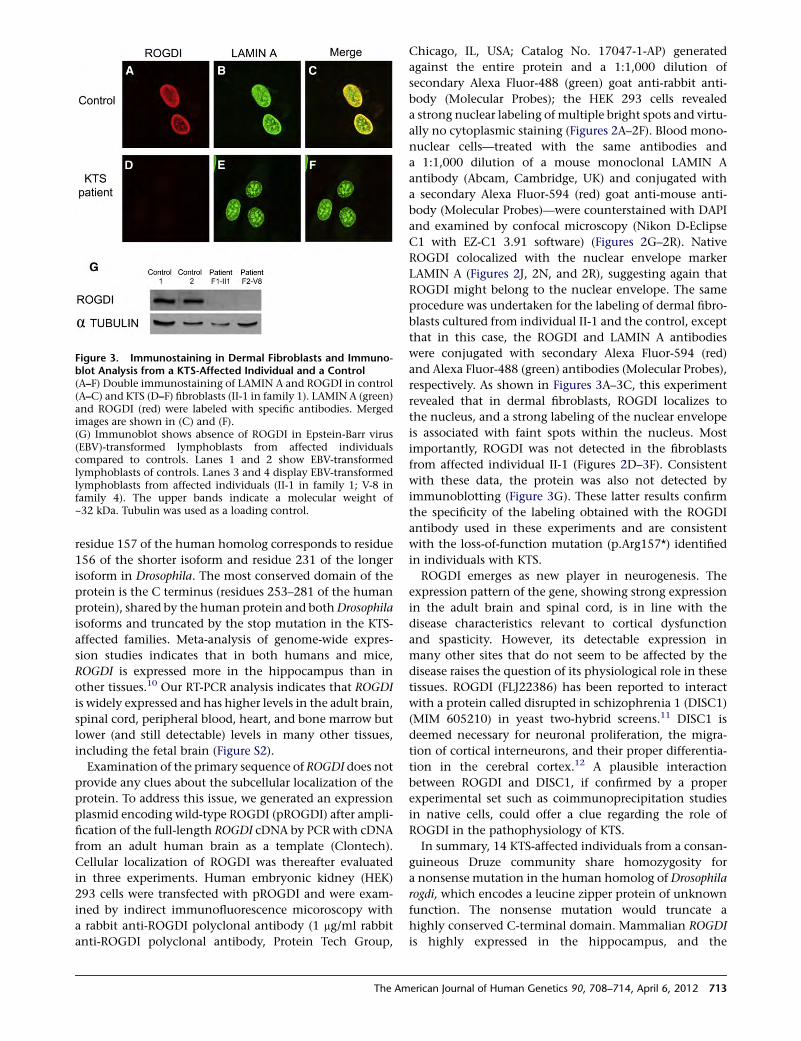
residue 157 of the human homolog corresponds to residue
156 of the shorter isoform and residue 231 of the longer
isoform in Drosophila. The most conserved domain of the
protein is the C terminus (residues 253–281 of the human
protein), shared by the human protein and bothDrosophila
isoforms and truncated by the stop mutation in the KTS-
affected families. Meta-analysis of genome-wide expres-
sion studies indicates that in both humans and mice,
ROGDI is expressed more in the hippocampus than in
other tissues.10 Our RT-PCR analysis indicates that ROGDI
is widely expressed and has higher levels in the adult brain,
spinal cord, peripheral blood, heart, and bone marrow but
lower (and still detectable) levels in many other tissues,
including the fetal brain (Figure S2).
Examination of the primary sequence of ROGDI does not
provide any clues about the subcellular localization of the
protein. To address this issue, we generated an expression
plasmid encoding wild-type ROGDI (pROGDI) after ampli-
fication of the full-length ROGDI cDNA by PCR with cDNA
from an adult human brain as a template (Clontech).
Cellular localization of ROGDI was thereafter evaluated
in three experiments. Human embryonic kidney (HEK)
293 cells were transfected with pROGDI and were exam-
ined by indirect immunofluorescence micoroscopy with
a rabbit anti-ROGDI polyclonal antibody (1 mg/ml rabbit
anti-ROGDI polyclonal antibody, Protein Tech Group,
Chicago, IL, USA; Catalog No. 17047-1-AP) generated
against the entire protein and a 1:1,000 dilution of
secondary Alexa Fluor-488 (green) goat anti-rabbit anti-
body (Molecular Probes); the HEK 293 cells revealed
a strong nuclear labeling of multiple bright spots and virtu-
ally no cytoplasmic staining (Figures 2A–2F). Blood mono-
nuclear cells—treated with the same antibodies and
a 1:1,000 dilution of a mouse monoclonal LAMIN A
antibody (Abcam, Cambridge, UK) and conjugated with
a secondary Alexa Fluor-594 (red) goat anti-mouse anti-
body (Molecular Probes)—were counterstained with DAPI
and examined by confocal microscopy (Nikon D-Eclipse
C1 with EZ-C1 3.91 software) (Figures 2G–2R). Native
ROGDI colocalized with the nuclear envelope marker
LAMIN A (Figures 2J, 2N, and 2R), suggesting again that
ROGDI might belong to the nuclear envelope. The same
procedure was undertaken for the labeling of dermal fibro-
blasts cultured from individual II-1 and the control, except
that in this case, the ROGDI and LAMIN A antibodies
were conjugated with secondary Alexa Fluor-594 (red)
and Alexa Fluor-488 (green) antibodies (Molecular Probes),
respectively. As shown in Figures 3A–3C, this experiment
revealed that in dermal fibroblasts, ROGDI localizes to
the nucleus, and a strong labeling of the nuclear envelope
is associated with faint spots within the nucleus. Most
importantly, ROGDI was not detected in the fibroblasts
from affected individual II-1 (Figures 2D–3F). Consistent
with these data, the protein was also not detected by
immunoblotting (Figure 3G). These latter results confirm
the specificity of the labeling obtained with the ROGDI
antibody used in these experiments and are consistent
with the loss-of-function mutation (p.Arg157*) identified
in individuals with KTS.
ROGDI emerges as new player in neurogenesis. The
expression pattern of the gene, showing strong expression
in the adult brain and spinal cord, is in line with the
disease characteristics relevant to cortical dysfunction
and spasticity. However, its detectable expression in
many other sites that do not seem to be affected by the
disease raises the question of its physiological role in these
tissues. ROGDI (FLJ22386) has been reported to interact
with a protein called disrupted in schizophrenia 1 (DISC1)
(MIM 605210) in yeast two-hybrid screens.11 DISC1 is
deemed necessary for neuronal proliferation, the migra-
tion of cortical interneurons, and their proper differentia-
tion in the cerebral cortex.12 A plausible interaction
between ROGDI and DISC1, if confirmed by a proper
experimental set such as coimmunoprecipitation studies
in native cells, could offer a clue regarding the role of
ROGDI in the pathophysiology of KTS.
In summary, 14 KTS-affected individuals from a consan-
guineous Druze community share homozygosity for
a nonsense mutation in the human homolog of Drosophila
rogdi, which encodes a leucine zipper protein of unknown
function. The nonsense mutation would truncate a
highly conserved C-terminal domain. Mammalian ROGDI
is highly expressed in the hippocampus, and the
Figure 3. Immunostaining in Dermal Fibroblasts and Immuno-blot Analysis from a KTS-Affected Individual and a Control(A–F) Double immunostaining of LAMIN A and ROGDI in control(A–C) and KTS (D–F) fibroblasts (II-1 in family 1). LAMIN A (green)and ROGDI (red) were labeled with specific antibodies. Mergedimages are shown in (C) and (F).(G) Immunoblot shows absence of ROGDI in Epstein-Barr virus(EBV)-transformed lymphoblasts from affected individualscompared to controls. Lanes 1 and 2 show EBV-transformedlymphoblasts of controls. Lanes 3 and 4 display EBV-transformedlymphoblasts from affected individuals (II-1 in family 1; V-8 infamily 4). The upper bands indicate a molecular weight of~32 kDa. Tubulin was used as a loading control.
The American Journal of Human Genetics 90, 708–714, April 6, 2012 713

corresponding protein localizes to the nuclear envelope.
Age of onset and the severity of the degenerative KTS
phenotype vary considerably among individuals who are
homozygous for the same disease allele and who are
from the same small community.
Supplemental Data
Supplemental Data include two figures and can be found with this
article online at http://www.cell.com/AJHG.
Received: February 24, 2012
Revised: March 13, 2012
Accepted: March 15, 2012
Published online: April 5, 2012
Web Resources
The URLs for data presented herein are as follows:
BLAST, http://blast.ncbi.nlm.nih.gov
GeneBank, http://www.ncbi.nlm.nih.gov/nuccore/nm_024589.1
GeneCards, http://www.genecards.org/
Mutalyzer, http://www.mutalyzer.nl/2.0
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
PRIMER3, http://frodo.wi.mit.edu/primer3/
SNP Database, http://www.ncbi.nlm.nih.gov/snp/
SuperLink, http://bioinfo.cs.technion.ac.il/superlink-online/
Tandem Repeat Finder, http://tandem.bu.edu/trf/trf.html
UCSC, http://genome.ucsc.edu
References
1. Kohlschutter, A., Chappuis, D., Meier, C., Tonz, O., Vassella, F.,
and Herschkowitz, N. (1974). Familial epilepsy and yellow
teeth—a disease of the CNS associated with enamel hypo-
plasia. Helv. Paediatr. Acta 29, 283–294.
2. Witkop, C.J., Jr., and Sauk, J.J., Jr. (1976). Heritable defects
of enamel. In Oral Facial Genetics, R.E. Stewart and G.H.
Prescott, eds. (St. Louis: C.V. Mosby), pp. 200–202.
3. Christodoulou, J., Hall, R.K., Menahem, S., Hopkins, I.J., and
Rogers, J.G. (1988). A syndrome of epilepsy, dementia, and
amelogenesis imperfecta: Genetic and clinical features. J.
Med. Genet. 25, 827–830.
4. Zlotogora, J., Fuks, A., Borochowitz, Z., and Tal, Y. (1993).
Kohlschutter-Tonz syndrome: epilepsy, dementia, and amelo-
genesis imperfecta. Am. J. Med. Genet. 46, 453–454.
5. Petermoller, M., Kunze, J., and Gross-Selbeck, G. (1993). Kohl-
schutter syndrome: Syndrome of epilepsy—dementia—ame-
logenesis imperfecta. Neuropediatrics 24, 337–338.
6. Musumeci, S.A., Elia, M., Ferri, R., Romano, C., Scuderi, C.,
and Del Gracco, S. (1995). A further family with epilepsy,
dementia and yellow teeth: The Kohlschutter syndrome. Brain
Dev. 17, 133–138, discussion 142–143.
7. Wygold, T., Kurlemann, G., and Schuierer, G. (1996). Kohl-
schutter syndrome—an example of a rare progressive neuroec-
todermal disease. Case report and review of the literature. Klin.
Padiatr. 208, 271–275.
8. Donnai, D., Tomlin, P.I., and Winter, R.M. (2005). Kohlschut-
ter syndrome in siblings. Clin. Dysmorphol. 14, 123–126.
9. Haberlandt, E., Svejda, C., Felber, S., Baumgartner, S., Gunther,
B., Utermann, G., and Kotzot, D. (2006). Yellow teeth,
seizures, and mental retardation: A less severe case of Kohl-
schutter-Tonz syndrome. Am. J. Med. Genet. A. 140, 281–283.
10. Kapushesky, M., Adamusiak, T., Burdett, T., Culhane, A.,
Farne, A., Filippov, A., Holloway, E., Klebanov, A., Kryvych,
N., Kurbatova, N., et al. (2012). Gene Expression Atlas
update—a value-added database of microarray and
sequencing-based functional genomics experiments. Nucleic
Acids Res. 40 (Database issue), D1077–D1081.
11. Camargo, L.M., Collura, V., Rain, J.C., Mizuguchi, K., Hermja-
kob, H., Kerrien, S., Bonnert, T.P., Whiting, P.J., and Brandon,
N.J. (2007). Disrupted in Schizophrenia 1 Interactome:
Evidence for the close connectivity of risk genes and a
potential synaptic basis for schizophrenia. Mol. Psychiatry
12, 74–86.
12. Kamiya, A., Kubo, K.I., Tomoda, T., Takaki, M., Youn, R.,
Ozeki, Y., Sawamura, N., Park, U., Kudo, C., Okawa, M.,
et al. (2005). A schizophrenia-associated mutation of DISC1
perturbs cerebral cortex development. Nat. Cell Biol. 7,
1167–1178.
714 The American Journal of Human Genetics 90, 708–714, April 6, 2012

REPORT
Maternal Inheritance of a PromoterVariant in the Imprinted PHLDA2 GeneSignificantly Increases Birth Weight
Miho Ishida,1 David Monk,1,5 Andrew J. Duncan,1 Sayeda Abu-Amero,1 Jiehan Chong,1,6
Susan M. Ring,2 Marcus E. Pembrey,1,2 Peter C. Hindmarsh,1 John C. Whittaker,3 Philip Stanier,4
and Gudrun E. Moore1,*
Birth weight is an important indicator of both perinatal and adult health, but little is known about the genetic factors contributing to its
variability. Intrauterine growth restriction is a leading cause of perinatal morbidity and mortality and is also associated with adult
disease. A significant correlation has been reported between lower birth weight and increased expression of the maternal PHLDA2 allele
in term placenta (the normal imprinting pattern wasmaintained). However, a mechanism that explains the transcriptional regulation of
PHLDA2 on in utero growth has yet to be described. In this study, we sequenced the PHLDA2 promoter region in 263 fetal DNA samples
to identify polymorphic variants. We used a luciferase reporter assay to identify in the PHLDA2 promoter a 15 bp repeat sequence (RS1)
variant that significantly reduces PHLDA2-promoter efficiency. RS1 genotyping was then performed in three independent white Euro-
pean normal birth cohorts. Meta-analysis of all three (total n ¼ 9,433) showed that maternal inheritance of RS1 resulted in a significant
93 g increase in birth weight (p ¼ 0.01; 95% confidence interval [CI] ¼ 22–163). Moreover, when the mother was homozygous for RS1,
the influence on birthweight was 155 g (p¼ 0.04; 95%CI¼ 9–300), which is a similarmagnitude to the reduction in birth weight caused
by maternal smoking.
Very low birth weight shows a strong association with
perinatal mortality and morbidity and is linked to an
increased risk of developing adulthood diseases, such as
obesity and type 2 diabetes (MIM 125853).1,2 Fetal growth
relies on an effective nutrient supply from the mother to
the fetus via the placenta; this nutrient supply is in-
fluenced by a complex interrelationship between the
environment and genetics. Of particular interest are
imprinted genes, which show expression from only one
allele in a parent-of-origin dependent manner. Genomic
imprinting is found almost exclusively in placental
mammals. Its evolution is probably best explained by
the ‘‘conflict hypothesis,’’ which suggests that paternally
expressed imprinted genes promote fetal growth and
ensure inheritance of the paternal genome to successive
generations, whereas maternally expressed imprinted
genes limit growth in order for the mother to survive
and reproduce again.3
PHLDA2 (MIM 602131) encodes the pleckstrin
homology-like domain, family A, member 2 protein and
is a maternally expressed imprinted gene found in one of
the most extensively studied imprinting clusters in human
chromosomal region 11p15.5. Consistent with the
‘‘conflict hypothesis,’’ Phlda2-null mice exhibit placenta
overgrowth, whereas doubling the Phlda2 expression in
transgenic mice results in placental stunting accompanied
by a 13% reduction in fetal weight; both of these findings
suggest that Phlda2 has a growth-suppressing role.4,5 In hu-
mans, PHLDA2 is expressed in a variety of tissues but is
predominantly expressed in the villous cytotrophoblast
of the placenta throughout gestation,6,7 and upregulation
has been observed in intrauterine growth restriction
(IUGR) placentas.8–10 This complements our previous
finding that PHLDA2 expression is significantly higher in
the term placenta of lower-birth-weight babies.11 However,
sequence analysis of all informative samples in the ‘‘Moore
cohort’’ of white European normal births confirmed that
only maternal, monoallelic PHLDA2 expression was
present.11 This indicates that loss of imprinting (LOI) was
not responsible for the increased PHLDA2 expression and
suggests that additional regulatory mechanisms, including
the PHLDA2 promoter, other than imprinting must be
involved.
In this study, we examined the PHLDA2 promoter region
for genetic polymorphisms that might affect PHLDA2 tran-
scriptional activity and therefore could affect birth weight.
From the Moore cohort (n ¼ 263), recruited from Queen
Charlotte and Chelsea Hospital,11 we sequenced a ~2 kb
upstream region beginning at the transcription start site
and overlapping the promoter CpG island. The UCSC
Genome Browser (build GRCh37/hg19) listed 20 SNPs,
encompassing rs12798267 to rs412300, in this region.
1Clinical andMolecular Genetics Unit, Institute of Child Health, University College London, LondonWC1N 1EH, UK; 2Avon Longitudinal Study of Parents
and Children, Department of Social Medicine, Oakfield House, Oakfield Grove, University of Bristol, Bristol BS8 2BN, UK; 3Noncommunicable Disease
Epidemiology Unit, London School of Hygiene and Tropical Medicine, University of London, LondonWC1E 7HT, UK; 4Neural Development Unit, Institute
of Child Health, University College London, London WC1N 1EH, UK5Present address: Imprinting and Cancer Group, Epigenetics and Cancer Biology Program, Bellvitge Institute for Biomedical Research, L’Hospitalet de Llo-
bregat, Barcelona 08907, Spain6Present address: Ipswich Hospital NHS Trust, Ipswich, IP4 5PD, UK
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.021. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 715–719, April 6, 2012 715

However, none of these SNPs were identified in this cohort,
suggesting that they either are rare in the white European
population or have not been accurately validated. We only
detected one variable sequence: a tandem 15 bp (50-GGGG
CGGGGAGGGGC- 30; bp 4,934–4,967 of NG_009266.1)
repeat sequence (RS) variant present 48 bp upstream of
the PHLDA2 transcription start site (Figure S1, available
online). The tandem repeat (RS2) is most common (it is
present in 87% of chromosomes), and the minor allele is
a single copy (RS1) that is found in the remaining 13% of
chromosomes (Figure 1). In addition, RS1 was not found
to be in linkage disequilibrium (LD) with nearby SNPs
rs3847646 (located ~3 kb upstream) or rs13390 or
rs1056819 (present within PHLDA2 exons 1 and 2,
respectively).
We investigated the effect of the PHLDA2 RS on the
gene’s promoter activity by transiently transfecting lucif-
erase reporter constructs into the transformed human
embryonic kidney (HEK) 293T cell line and the human
trophoblast cell line 1 (TCL-1). We made the promoter
constructs by cloning 300 bp (with the use of HindIII
and Xho1) and 600 bp (with the use of HindIII and Sac1)
DNA fragments upstream of the PHLDA2 start site and in-
serting them into the pGL3.1-Basic vector (Promega, UK).
These sequences contained either RS1 or RS2. RS1 showed
significantly lower PHLDA2 promoter activity for both the
300 bp (74% decrease; t test, p ¼ 0.004) and 600 bp (42%
decrease; t test, p ¼ 0.001) constructs (Figure 2). The exper-
iments were performed in duplicate for HEK293T cells and
in triplicate for TCL-1 cells (Figure S2), and each assay
included six replicates. TFSEARCH (Transcriptional Factor
Search) shows that the RS2 allele potentially harbors four
SP1 and two MZF1 binding sites. However, losing a 15 bp
copy (RS1) removes three of these sites, suggesting the
possibility that the number of available transcription-
factor binding sites might be important for promoter
efficiency.
Given that high expression of PHLDA2 is associated with
lower birth weight11 and that RS1 reduces the PHLDA2
Figure 1. Sequence ElectropherogramsShowing the 15 bp RS in the PHLDA2 PromoterRegionRS1/RS1 homozygous, RS2/RS2 homozygous,and RS1/RS2 heterozygous sequences are shown.Each black bar represents the location of a single15 bp copy. The start of the overlapping sequencein the heterozygous sample is indicated by theblack arrow and dotted bars.
promoter efficiency in vitro, we investi-
gated whether RS1 could be associated
with increased birth weight. Because
PHLDA2 is maternally expressed and pater-
nally silenced, RS1 homozygotes and
heterozygotes with a maternally inherited
RS1 were grouped and deemed the ‘‘RS1
effect group.’’ RS2 homozygotes and hetero-
zygotes with a paternally inherited RS1 were named the
‘‘unaffected group’’ (Table S1). We assessed the parental
origin of the RS1 allele in the heterozygous babies by
genotyping their corresponding parental DNA samples.
Uninformative cases were those in which both parents
were heterozygotes.
We first genotyped the parental DNA samples corre-
sponding to the heterozygous babies in the Moore cohort,
and 28 babies were revealed to have maternally inherited
RS1 (Table 1). To investigate the effect of maternally in-
herited RS1 on birth weight, we applied a linear-regression
model and corrected for the following covariates: the
gender of the baby,maternal weight, gestational age, parity
and maternal diabetes, hypertension, and smoking habits
(Table S2). A two-tailed test was used throughout; p values
were based on Wald tests, and standard residual plots were
examined and showed no evidence of departure from
model assumptions. This analysis showed that babies in
the RS1 effect group tended to have an average birth
weight 122 g higher than that of the babies in the unaf-
fected group (p ¼ 0.15; 95% confidence interval [CI] ¼�43–286) (Table 1). We then carried out the same analysis
on the UCL-FGS cohort (baby-parent trios, n ¼ 385) from
the University College London Fetal Growth Study.12
This produced a similar trend to the Moore cohort: babies
in the RS1 effect group (n ¼ 16) were on average 68 g
heavier (p ¼ 0.61; 95% CI ¼ �196–332) (Table 1). The
reproducibility of this trend suggests a potentially valid
finding despite the fact that statistical confidence could
not be achieved because too few individuals (approxi-
mately 13%) had maternal RS1 (Table 1).
To address this, we then introduced a third and larger
collection, the ALSPAC cohort (n ¼ 8,785) from the Avon
Longitudinal Study of Parents and Children study.13
Because this cohort only includes samples from the
mother and child and because PHLDA2 is a maternally ex-
pressed transcript, the RS1 effect group (n¼ 179) consisted
of homozygous RS1/RS1 babies and heterozygous babies
with homozygous RS1/RS1 mothers (Table S3). Using the
716 The American Journal of Human Genetics 90, 715–719, April 6, 2012
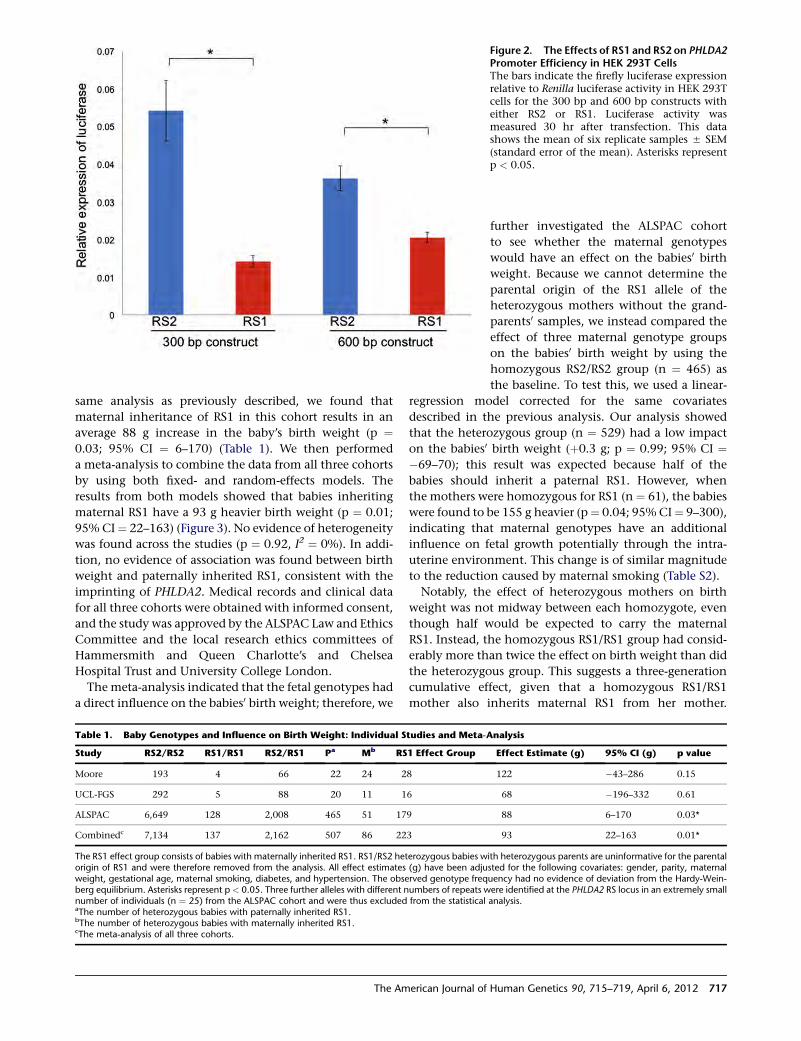
same analysis as previously described, we found that
maternal inheritance of RS1 in this cohort results in an
average 88 g increase in the baby’s birth weight (p ¼0.03; 95% CI ¼ 6–170) (Table 1). We then performed
a meta-analysis to combine the data from all three cohorts
by using both fixed- and random-effects models. The
results from both models showed that babies inheriting
maternal RS1 have a 93 g heavier birth weight (p ¼ 0.01;
95% CI¼ 22–163) (Figure 3). No evidence of heterogeneity
was found across the studies (p ¼ 0.92, I2 ¼ 0%). In addi-
tion, no evidence of association was found between birth
weight and paternally inherited RS1, consistent with the
imprinting of PHLDA2. Medical records and clinical data
for all three cohorts were obtained with informed consent,
and the study was approved by the ALSPAC Law and Ethics
Committee and the local research ethics committees of
Hammersmith and Queen Charlotte’s and Chelsea
Hospital Trust and University College London.
The meta-analysis indicated that the fetal genotypes had
a direct influence on the babies0 birth weight; therefore, we
Figure 2. The Effects of RS1 and RS2 on PHLDA2Promoter Efficiency in HEK 293T CellsThe bars indicate the firefly luciferase expressionrelative to Renilla luciferase activity in HEK 293Tcells for the 300 bp and 600 bp constructs witheither RS2 or RS1. Luciferase activity wasmeasured 30 hr after transfection. This datashows the mean of six replicate samples 5 SEM(standard error of the mean). Asterisks representp < 0.05.
Table 1. Baby Genotypes and Influence on Birth Weight: Individual Studies and Meta-Analysis
Study RS2/RS2 RS1/RS1 RS2/RS1 Pa Mb RS1 Effect Group Effect Estimate (g) 95% CI (g) p value
Moore 193 4 66 22 24 28 122 �43–286 0.15
UCL-FGS 292 5 88 20 11 16 68 �196–332 0.61
ALSPAC 6,649 128 2,008 465 51 179 88 6–170 0.03*
Combinedc 7,134 137 2,162 507 86 223 93 22–163 0.01*
The RS1 effect group consists of babies with maternally inherited RS1. RS1/RS2 heterozygous babies with heterozygous parents are uninformative for the parentalorigin of RS1 and were therefore removed from the analysis. All effect estimates (g) have been adjusted for the following covariates: gender, parity, maternalweight, gestational age, maternal smoking, diabetes, and hypertension. The observed genotype frequency had no evidence of deviation from the Hardy-Wein-berg equilibrium. Asterisks represent p< 0.05. Three further alleles with different numbers of repeats were identified at the PHLDA2 RS locus in an extremely smallnumber of individuals (n ¼ 25) from the ALSPAC cohort and were thus excluded from the statistical analysis.aThe number of heterozygous babies with paternally inherited RS1.bThe number of heterozygous babies with maternally inherited RS1.cThe meta-analysis of all three cohorts.
further investigated the ALSPAC cohort
to see whether the maternal genotypes
would have an effect on the babies0 birthweight. Because we cannot determine the
parental origin of the RS1 allele of the
heterozygous mothers without the grand-
parents0 samples, we instead compared the
effect of three maternal genotype groups
on the babies0 birth weight by using the
homozygous RS2/RS2 group (n ¼ 465) as
the baseline. To test this, we used a linear-
regression model corrected for the same covariates
described in the previous analysis. Our analysis showed
that the heterozygous group (n ¼ 529) had a low impact
on the babies0 birth weight (þ0.3 g; p ¼ 0.99; 95% CI ¼�69–70); this result was expected because half of the
babies should inherit a paternal RS1. However, when
the mothers were homozygous for RS1 (n ¼ 61), the babies
were found to be 155 g heavier (p¼ 0.04; 95% CI¼ 9–300),
indicating that maternal genotypes have an additional
influence on fetal growth potentially through the intra-
uterine environment. This change is of similar magnitude
to the reduction caused by maternal smoking (Table S2).
Notably, the effect of heterozygous mothers on birth
weight was not midway between each homozygote, even
though half would be expected to carry the maternal
RS1. Instead, the homozygous RS1/RS1 group had consid-
erably more than twice the effect on birth weight than did
the heterozygous group. This suggests a three-generation
cumulative effect, given that a homozygous RS1/RS1
mother also inherits maternal RS1 from her mother.
The American Journal of Human Genetics 90, 715–719, April 6, 2012 717

Alternatively, homozygous RS1/RS1 mothers could also
affect babies0 birth weight by influencing the circulating
PHLDA2 protein/mRNA levels in the maternal blood,
given that PHLDA2 shows biallelic expression in adult
blood.14
Maternal inheritance of RS1 did not affect the placental
weight (þ2.5 g; p ¼ 0.93; 95% CI ¼ �62–67) but did
have a small and statistically-significant influence on
head circumference (þ0.23 cm; p ¼ 0.04; 95% CI ¼ 0.01–
0.45). Interestingly, although the RS1 sequence is con-
served in monkeys, the duplicated RS2 allele seems to be
exclusive to humans (Figure S1). This implies an evolu-
tionary role in human reproductive success. Consistent
with the conflict hypothesis, maternal PHLDA2 RS1 is asso-
ciated with both increased growth of the baby and head
circumference. Conversely, the net effect of the common
(RS2/RS2) allele in humans is limited birth weight and
head circumference, an effect which might provide an
evolutionary advantage—protecting the mother and her
birth canal.
Given the perinatal and life-long health complications
associated with very low birth weight,1,2,15 a number of
studies have investigated the genetic contribution of puta-
tive growth-regulating genes, including the imprinted
genes IGF2 (MIM 147470) and H19 (MIM 103280).16–19
Genome-wide linkage or association studies have located
several loci associated with birth weight,20–23 although
none have yet been directly associated with actual gene
function. PHLDA2 has not previously been detected in
these screens, perhaps as a result of the complexity intro-
duced by the parent-of-origin effect but also because
PHLDA2 RS1 is not in linkage disequilibrium with nearby
SNPs and is therefore not well-represented on the genotyp-
ing platforms used. In addition, our study maximizes the
information content for this allele because we specifically
genotyped all informative individuals for what is essen-
tially the functional and presumably causal variant. The
biochemical function of PHLDA2 remains unknown. It is
a small cytoplasmic protein that binds to phosphoinosi-
tide lipids via its PH domain.24 A recent study showed a
relationship between PHLDA2 expression and lower
growth velocity of the fetal femur; this relationship
suggests that PHLDA2 possibly plays a role in bone devel-
opment.25 Although increased Phlda2 expression in
transgenic mice resulted in a smaller placenta and a corre-
sponding reduction in birth weight,5 we could not
replicate this finding on the human placenta either in
a comparative study with PHLDA2 expression11 or indi-
rectly via association with the promoter RS genotype.
Nevertheless, a profound effect on the babies0 birth weight
was still detected, suggesting that placental weight was not
the predominant regulatory factor. It also appears that
this effect is controlled through the maternal expression
of the gene, which is consistent with the conflict hypoth-
esis and is mediated by the maternal genetic inheritance at
the DNA level in the promoter. This provides the first
example of a maternal genetic effect working together
with a maternally driven epigenetic effect. We suspect it
will be the first of many examples once further details of
the interactions of the genome with the epigenome are
unraveled. The PHLDA2 promoter RS and its expression
might serve as a useful genetic biomarker that can be
used to predict birth size. Further insight into the function
of PHLDA2 along with other imprinted genes will help us
understand the genetic basis of fetal growth as well as
the common and serious complications—such as IUGR—
of pregnancy.
Supplemental Data
Supplemental Data include three figures and three tables and can
be found with this article online at http://www.cell.com/AJHG.
Acknowledgments
Wewould like to thankM. Sweeney for her help at the sequencing
facility at the Institute of Neurology and all the members of
Professor Moore’s Development and Growth research group for
valuable suggestions and help. This research was funded by the
Child Health Research Appeal Trust (the Institute of Child Health
and theGreat Ormond Street Hospital for Children [GOSH]), Over-
seas Research Studentship (M.I.), the Medical Research Council,
Wellbeing of Women, March of Dimes, PARKS, and the GOSH
Charity. P.S. is supported by the GOSH Charity. We are extremely
grateful to all the familieswho took part in this study, themidwives
for their help in recruiting the families, and the whole ALSPAC
(Avon Longitudinal Study of Parents and Children) team, which
includes interviewers, computer and laboratory technicians, cler-
ical workers, research scientists, volunteers, managers, reception-
ists, and nurses. The UK Medical Research Council, the Wellcome
Trust, and the University of Bristol provide core support for
ALSPAC. We would also like to thank the University College Lon-
don Fetal Growth Study cohort team for their collaborative work.
Received: December 21, 2011
Revised: February 17, 2012
Accepted: February 22, 2012
Published online: March 22, 2012
Figure 3. Meta-Analysis Showing the Relationship betweenBirth Weight and PHLDA2 Promoter RS1 EffectThe data is depicted in a Forest plot; the 95% CI for each study isrepresented by a horizontal line, and the estimated effect sizes areshown as gray squares. The weight of the study in the meta-anal-ysis is represented by the size of the squares. The scale used is ingrams (g). The diamond shape indicates the mean and 95% CIfor the total estimate of the effect. Both random- and fixed-effectmodels produced the same results, and the plot represents theresults from the fixed-effect model.
718 The American Journal of Human Genetics 90, 715–719, April 6, 2012

Web Resources
The URLs for data presented herein are as follows:
ALSPAC, http://www.bristol.ac.uk/alspac
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
TFSEARCH, http://www.cbrc.jp/research/db/TFSEARCH
UCSC Genome Browser, http://genome.ucsc.edu
References
1. Barker, D.J. (2004). The developmental origins of adult disease.
J. Am. Coll. Nutr. 23 (6, Suppl), 588S–595S.
2. McIntire, D.D., Bloom, S.L., Casey, B.M., and Leveno, K.J.
(1999). Birth weight in relation to morbidity and mortality
among newborn infants. N. Engl. J. Med. 340, 1234–1238.
3. Moore, T., and Haig, D. (1991). Genomic imprinting in
mammalian development: A parental tug-of-war. Trends
Genet. 7, 45–49.
4. Frank, D., Fortino,W., Clark, L., Musalo, R., Wang,W., Saxena,
A., Li, C.M., Reik, W., Ludwig, T., and Tycko, B. (2002).
Placental overgrowth in mice lacking the imprinted gene Ipl.
Proc. Natl. Acad. Sci. USA 99, 7490–7495.
5. Tunster, S.J., Tycko, B., and John, R.M. (2010). The imprinted
Phlda2 gene regulates extraembryonic energy stores. Mol.
Cell. Biol. 30, 295–306.
6. Saxena, A., Frank, D., Panichkul, P., Van den Veyver, I.B.,
Tycko, B., and Thaker, H. (2003). The product of the imprinted
gene IPL marks human villous cytotrophoblast and is lost in
complete hydatidiform mole. Placenta 24, 835–842.
7. Qian, N., Frank, D., O’Keefe, D., Dao, D., Zhao, L., Yuan, L.,
Wang, Q., Keating, M., Walsh, C., and Tycko, B. (1997). The
IPL gene on chromosome 11p15.5 is imprinted in humans
and mice and is similar to TDAG51, implicated in Fas expres-
sion and apoptosis. Hum. Mol. Genet. 6, 2021–2029.
8. McMinn, J., Wei, M., Schupf, N., Cusmai, J., Johnson, E.B.,
Smith, A.C., Weksberg, R., Thaker, H.M., and Tycko, B.
(2006). Unbalanced placental expression of imprinted
genes in human intrauterine growth restriction. Placenta 27,
540–549.
9. Diplas, A.I., Lambertini, L., Lee, M.-J., Sperling, R., Lee, Y.L.,
Wetmur, J., and Chen, J. (2009). Differential expression of
imprinted genes in normal and IUGR human placentas.
Epigenetics 4, 235–240.
10. Kumar, N., Leverence, J., Bick, D., and Sampath, V. (2012).
Ontogeny of growth-regulating genes in the placenta.
Placenta 33, 94–99.
11. Apostolidou, S., Abu-Amero, S., O’Donoghue, K., Frost, J.,
Olafsdottir, O., Chavele, K.M., Whittaker, J.C., Loughna, P.,
Stanier, P., and Moore, G.E. (2007). Elevated placental expres-
sion of the imprinted PHLDA2 gene is associated with low
birth weight. J. Mol. Med. 85, 379–387.
12. Hindmarsh, P.C., Geary, M.P., Rodeck, C.H., Kingdom, J.C.,
and Cole, T.J. (2002). Intrauterine growth and its relationship
to size and shape at birth. Pediatr. Res. 52, 263–268.
13. Jones, R.W., Ring, S., Tyfield, L., Hamvas, R., Simmons, H.,
Pembrey, M., and Golding, J.; ALSPAC Study Team. (2000).
A new human genetic resource: A DNA bank established as
part of the Avon longitudinal study of pregnancy and child-
hood (ALSPAC). Eur. J. Hum. Genet. 8, 653–660.
14. Muller, S., van den Boom, D., Zirkel, D., Koster, H., Berthold,
F., Schwab, M., Westphal, M., and Zumkeller, W. (2000).
Retention of imprinting of the human apoptosis-related
gene TSSC3 in human brain tumors. Hum. Mol. Genet. 9,
757–763.
15. Simmons, R.A. (2009). Developmental origins of adult disease.
Pediatr. Clin. North Am. 56, 449–466.
16. Adkins, R.M., Somes, G., Morrison, J.C., Hill, J.B., Watson,
E.M., Magann, E.F., and Krushkal, J. (2010). Association of
birth weight with polymorphisms in the IGF2, H19, and
IGF2R genes. Pediatr. Res. 68, 429–434.
17. Gomes, M.V., Soares, M.R., Pasqualim-Neto, A., Marcondes,
C.R., Lobo, R.B., and Ramos, E.S. (2005). Association between
birth weight, body mass index and IGF2/ApaI polymorphism.
Growth Horm. IGF Res. 15, 360–362.
18. Petry, C.J., Ong, K.K., Barratt, B.J., Wingate, D., Cordell, H.J.,
Ring, S.M., Pembrey, M.E., Reik, W., Todd, J.A., and Dunger,
D.B.; ALSPAC Study Team. (2005). Common polymorphism
in H19 associated with birthweight and cord blood IGF-II
levels in humans. BMC Genet. 6, 22.
19. Petry, C.J., Seear, R.V., Wingate, D.L., Acerini, C.L., Ong, K.K.,
Hughes, I.A., and Dunger, D.B. (2011). Maternally transmitted
foetal H19 variants and associations with birth weight. Hum.
Genet. 130, 663–670.
20. Andersson, E.A., Pilgaard, K., Pisinger, C., Harder, M.N.,
Grarup, N., Faerch, K., Poulsen, P., Witte, D.R., Jørgensen, T.,
Vaag, A., et al. (2010). Type 2 diabetes risk alleles near
ADCY5, CDKAL1 and HHEX-IDE are associated with reduced
birthweight. Diabetologia 53, 1908–1916.
21. Arya, R., Demerath, E., Jenkinson, C.P., Goring, H.H., Puppala,
S., Farook, V., Fowler, S., Schneider, J., Granato, R., Resendez,
R.G., et al. (2006). A quantitative trait locus (QTL) on chromo-
some 6q influences birth weight in two independent family
studies. Hum. Mol. Genet. 15, 1569–1579.
22. Fradin, D., Heath, S., Lepercq, J., Lathrop, M., and Bougneres,
P. (2006). Identification of distinct quantitative trait Loci
affecting length or weight variability at birth in humans. J.
Clin. Endocrinol. Metab. 91, 4164–4170.
23. Freathy, R.M., Mook-Kanamori, D.O., Sovio, U., Prokopenko,
I., Timpson, N.J., Berry, D.J., Warrington, N.M., Widen, E.,
Hottenga, J.J., Kaakinen, M., et al; Genetic Investigation of
ANthropometric Traits (GIANT) Consortium; Meta-Analyses
of Glucose and Insulin-related traits Consortium; Wellcome
Trust Case Control Consortium; Early Growth Genetics
(EGG) Consortium. (2010). Variants in ADCY5 and near
CCNL1 are associated with fetal growth and birth weight.
Nat. Genet. 42, 430–435.
24. Saxena, A., Morozov, P., Frank, D., Musalo, R., Lemmon, M.A.,
Skolnik, E.Y., and Tycko, B. (2002). Phosphoinositide binding
by the pleckstrin homology domains of Ipl and Tih1. J. Biol.
Chem. 277, 49935–49944.
25. Lewis, R.M., Cleal, J.K., Ntani, G., Crozier, S.R., Mahon, P.A.,
Robinson, S.M., Harvey, N.C., Cooper, C., Inskip, H.M., God-
frey, K.M., et al; Southampton Women’s Survey Study Group.
(2012). Relationship between placental expression of the
imprinted PHLDA2 gene, intrauterine skeletal growth and
childhood bone mass. Bone 50, 337–342.
The American Journal of Human Genetics 90, 715–719, April 6, 2012 719

REPORT
Alzheimer Disease Susceptibility Loci:Evidence for a Protein Network under Natural Selection
Towfique Raj,1,2,3,4 Joshua M. Shulman,1,3,4 Brendan T. Keenan,1,4 Lori B. Chibnik,1,3,4
Denis A. Evans,5,6 David A. Bennett,5,6 Barbara E. Stranger,2,3,4 and Philip L. De Jager1,3,4,*
Recent genome-wide association studies have identified a number of susceptibility loci for Alzheimer disease (AD). To understand the
functional consequences and potential interactions of the associated loci, we explored large-scale data sets interrogating the human
genome for evidence of positive natural selection. Our findings provide significant evidence for signatures of recent positive selection
acting on several haplotypes carrying AD susceptibility alleles; interestingly, the genes found in these selected haplotypes can be assem-
bled, independently, into a molecular complex via a protein-protein interaction (PPI) network approach. These results suggest a possible
coevolution of genes encoding physically-interacting proteins that underlie AD susceptibility and are coexpressed in different tissues. In
particular, PICALM, BIN1, CD2AP, and EPHA1 are interconnected through multiple interacting proteins and appear to have coordinated
evidence of selection in the same human population, suggesting that they may be involved in the execution of a shared molecular func-
tion. This observationmay be AD-specific, as the 12 loci associated with Parkinson disease do not demonstrate excess evidence of natural
selection. The context for selection is probably unrelated to AD itself; it is likely that these genes interact in another context, such as in
immune cells, where we observe cis-regulatory effects at several of the selected AD loci.
Alzheimer disease (AD [MIM 104300]) is themost common
neurodegenerative disease and is a leading cause of
dementia.1 Sporadic late-onset AD has a genetic compo-
nent that includes the well-known ε4 haplotype of APOE
(MIM 107741) and other loci that harbor susceptibility
alleles.2–6 However, the functional consequences and
evolutionary history of these loci and their possible
interactions remain largely unknown. Previous studies
reporting evidence of selection at the APOE locus7,8 sug-
gested the hypothesis that AD-associated pathways may
have experienced selection in human populations, quite
possibly due to selective pressure on a phenotype un-
related to AD, given that AD has little impact on an
individual’s reproductive fitness. To further explore this
hypothesis, we integrated evidence for natural selection
within validated AD loci with a pathway-based analysis
of these loci and an examination of transcriptional
patterns in immune cells. We identified several genes in
AD susceptibility loci that appear to physically interact
and that show evidence of having undergone natural
selection. Additionally, several of these loci exhibit a cis-
regulatory effect on the transcription levels of the selected
genes in immune cells, suggesting one plausible mecha-
nism by which an AD-related molecular pathway may
have evolved in response to environmental pressures in
early human history.
Given reports that the APOE ε4 haplotype exhibits
evidence for natural selection,7,8 we assessed all validated
and well-replicated AD susceptibility loci3,4 for evidence
of recent (<60,000 years ago) positive selection, using
linkage disequilibrium (LD)-based methods to detect
genomic regions harboring genetic variants inferred to
have recently and rapidly increased in frequency within
human populations. We applied the integrated haplotype
score (iHS)9 statistic, which measures the lengths of
the haplotypes around a given SNP, to identify evidence
of positive selection in human populations of African
(Yoruba from Ibadan, Nigeria [YRI]), European (Centre
d0Etude du Polymorphisme Humain samples from Utah
residents of European descent [CEU]), and East Asian
(Japanese subjects from Tokyo and Han Chinese sub-
jects from Beijing representing Asian populations [ASI])
ancestry from phase II of the International HapMap
Project.10 The iHS statistic was implemented with a mean
of 0 and a variance of 1. To discover signatures of selec-
tion on the haplotype bearing AD susceptibility alleles,
we searched for SNPs meeting a stringent threshold of
jiHSj > 2 (corresponding to the most extreme 5% of iHS
values across the genome among HapMap II SNPs with
minor allele frequency > 0.05) within the LD block con-
taining the index SNP of each locus from two large-scale
AD genome-wide association studies (GWAS).3,4 Further-
more, we restricted our analysis to SNPs with r2 > 0.5
and/or D0 ¼ 1, the published index SNP being associated
with AD in each locus. Although we tested only 11 loci,
we applied a stringent correction for genome-wide testing
to identify only the most robust signals of selection. Using
this approach, we found evidence for selection acting on
haplotypes carrying AD-associated alleles in 3 of 11 loci,
including PICALM (MIM 603025; rs561655), BIN1 (MIM
1Program in Translational NeuroPsychiatric Genomics, Institute for the Neurosciences Department of Neurology, Brigham and Women’s Hospital, 77
Avenue Louis Pasteur, Boston, MA 02115, USA; 2Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, 77 Avenue Louis Pasteur,
Boston, MA 02115, USA; 3Harvard Medical School, Boston, MA 02115, USA; 4Program in Medical and Population Genetics, The Broad Institute, 7 Cam-
bridge Center, Cambridge, MA 02142, USA; 5Departments of Internal Medicine and Neurological Science, Rush University Medical Center, 600 S Paulina
Street, Chicago, IL 60612, USA; 6Rush Alzheimer’s Disease Center, Rush University Medical Center, 600 S Paulina Street, Chicago, IL 60612, USA
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.022. �2012 by The American Society of Human Genetics. All rights reserved.
720 The American Journal of Human Genetics 90, 720–726, April 6, 2012
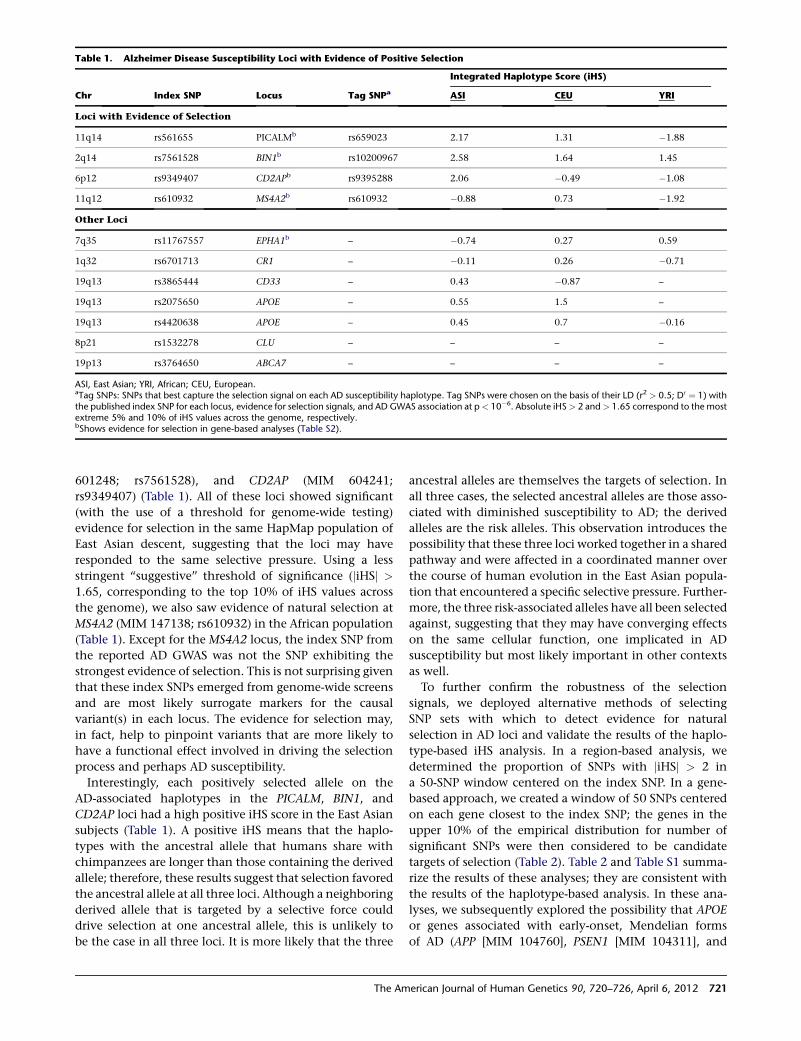
601248; rs7561528), and CD2AP (MIM 604241;
rs9349407) (Table 1). All of these loci showed significant
(with the use of a threshold for genome-wide testing)
evidence for selection in the same HapMap population of
East Asian descent, suggesting that the loci may have
responded to the same selective pressure. Using a less
stringent ‘‘suggestive’’ threshold of significance (jiHSj >
1.65, corresponding to the top 10% of iHS values across
the genome), we also saw evidence of natural selection at
MS4A2 (MIM 147138; rs610932) in the African population
(Table 1). Except for the MS4A2 locus, the index SNP from
the reported AD GWAS was not the SNP exhibiting the
strongest evidence of selection. This is not surprising given
that these index SNPs emerged from genome-wide screens
and are most likely surrogate markers for the causal
variant(s) in each locus. The evidence for selection may,
in fact, help to pinpoint variants that are more likely to
have a functional effect involved in driving the selection
process and perhaps AD susceptibility.
Interestingly, each positively selected allele on the
AD-associated haplotypes in the PICALM, BIN1, and
CD2AP loci had a high positive iHS score in the East Asian
subjects (Table 1). A positive iHS means that the haplo-
types with the ancestral allele that humans share with
chimpanzees are longer than those containing the derived
allele; therefore, these results suggest that selection favored
the ancestral allele at all three loci. Although a neighboring
derived allele that is targeted by a selective force could
drive selection at one ancestral allele, this is unlikely to
be the case in all three loci. It is more likely that the three
ancestral alleles are themselves the targets of selection. In
all three cases, the selected ancestral alleles are those asso-
ciated with diminished susceptibility to AD; the derived
alleles are the risk alleles. This observation introduces the
possibility that these three loci worked together in a shared
pathway and were affected in a coordinated manner over
the course of human evolution in the East Asian popula-
tion that encountered a specific selective pressure. Further-
more, the three risk-associated alleles have all been selected
against, suggesting that they may have converging effects
on the same cellular function, one implicated in AD
susceptibility but most likely important in other contexts
as well.
To further confirm the robustness of the selection
signals, we deployed alternative methods of selecting
SNP sets with which to detect evidence for natural
selection in AD loci and validate the results of the haplo-
type-based iHS analysis. In a region-based analysis, we
determined the proportion of SNPs with jiHSj > 2 in
a 50-SNP window centered on the index SNP. In a gene-
based approach, we created a window of 50 SNPs centered
on each gene closest to the index SNP; the genes in the
upper 10% of the empirical distribution for number of
significant SNPs were then considered to be candidate
targets of selection (Table 2). Table 2 and Table S1 summa-
rize the results of these analyses; they are consistent with
the results of the haplotype-based analysis. In these ana-
lyses, we subsequently explored the possibility that APOE
or genes associated with early-onset, Mendelian forms
of AD (APP [MIM 104760], PSEN1 [MIM 104311], and
Table 1. Alzheimer Disease Susceptibility Loci with Evidence of Positive Selection
Chr Index SNP Locus Tag SNPa
Integrated Haplotype Score (iHS)
ASI CEU YRI
Loci with Evidence of Selection
11q14 rs561655 PICALMb rs659023 2.17 1.31 �1.88
2q14 rs7561528 BIN1b rs10200967 2.58 1.64 1.45
6p12 rs9349407 CD2APb rs9395288 2.06 �0.49 �1.08
11q12 rs610932 MS4A2b rs610932 �0.88 0.73 �1.92
Other Loci
7q35 rs11767557 EPHA1b – �0.74 0.27 0.59
1q32 rs6701713 CR1 – �0.11 0.26 �0.71
19q13 rs3865444 CD33 – 0.43 �0.87 –
19q13 rs2075650 APOE – 0.55 1.5 –
19q13 rs4420638 APOE – 0.45 0.7 �0.16
8p21 rs1532278 CLU – – – –
19p13 rs3764650 ABCA7 – – – –
ASI, East Asian; YRI, African; CEU, European.aTag SNPs: SNPs that best capture the selection signal on each AD susceptibility haplotype. Tag SNPs were chosen on the basis of their LD (r2 > 0.5; D0 ¼ 1) withthe published index SNP for each locus, evidence for selection signals, and AD GWAS association at p< 10�6. Absolute iHS> 2 and> 1.65 correspond to the mostextreme 5% and 10% of iHS values across the genome, respectively.bShows evidence for selection in gene-based analyses (Table S2).
The American Journal of Human Genetics 90, 720–726, April 6, 2012 721
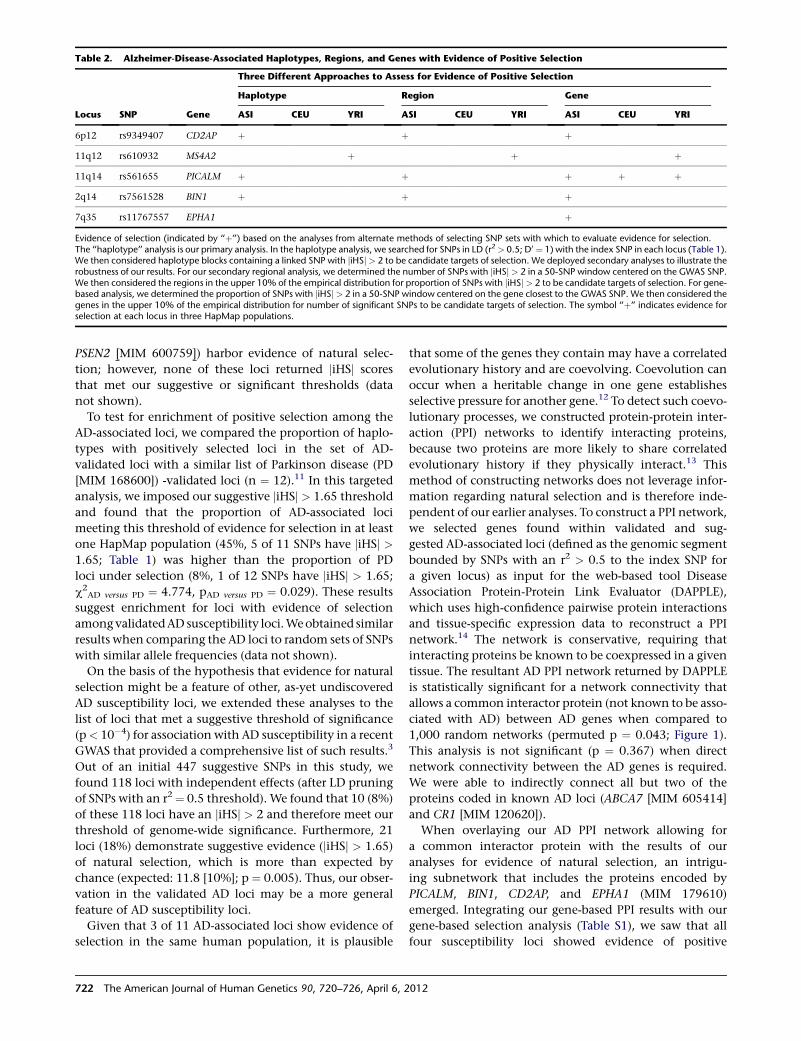
PSEN2 [MIM 600759]) harbor evidence of natural selec-
tion; however, none of these loci returned jiHSj scores
that met our suggestive or significant thresholds (data
not shown).
To test for enrichment of positive selection among the
AD-associated loci, we compared the proportion of haplo-
types with positively selected loci in the set of AD-
validated loci with a similar list of Parkinson disease (PD
[MIM 168600]) -validated loci (n ¼ 12).11 In this targeted
analysis, we imposed our suggestive jiHSj > 1.65 threshold
and found that the proportion of AD-associated loci
meeting this threshold of evidence for selection in at least
one HapMap population (45%, 5 of 11 SNPs have jiHSj >1.65; Table 1) was higher than the proportion of PD
loci under selection (8%, 1 of 12 SNPs have jiHSj > 1.65;
c2AD versus PD ¼ 4.774, pAD versus PD ¼ 0.029). These results
suggest enrichment for loci with evidence of selection
amongvalidatedAD susceptibility loci.Weobtained similar
results when comparing the AD loci to random sets of SNPs
with similar allele frequencies (data not shown).
On the basis of the hypothesis that evidence for natural
selection might be a feature of other, as-yet undiscovered
AD susceptibility loci, we extended these analyses to the
list of loci that met a suggestive threshold of significance
(p< 10�4) for association with AD susceptibility in a recent
GWAS that provided a comprehensive list of such results.3
Out of an initial 447 suggestive SNPs in this study, we
found 118 loci with independent effects (after LD pruning
of SNPs with an r2 ¼ 0.5 threshold). We found that 10 (8%)
of these 118 loci have an jiHSj > 2 and therefore meet our
threshold of genome-wide significance. Furthermore, 21
loci (18%) demonstrate suggestive evidence (jiHSj > 1.65)
of natural selection, which is more than expected by
chance (expected: 11.8 [10%]; p ¼ 0.005). Thus, our obser-
vation in the validated AD loci may be a more general
feature of AD susceptibility loci.
Given that 3 of 11 AD-associated loci show evidence of
selection in the same human population, it is plausible
that some of the genes they contain may have a correlated
evolutionary history and are coevolving. Coevolution can
occur when a heritable change in one gene establishes
selective pressure for another gene.12 To detect such coevo-
lutionary processes, we constructed protein-protein inter-
action (PPI) networks to identify interacting proteins,
because two proteins are more likely to share correlated
evolutionary history if they physically interact.13 This
method of constructing networks does not leverage infor-
mation regarding natural selection and is therefore inde-
pendent of our earlier analyses. To construct a PPI network,
we selected genes found within validated and sug-
gested AD-associated loci (defined as the genomic segment
bounded by SNPs with an r2 > 0.5 to the index SNP for
a given locus) as input for the web-based tool Disease
Association Protein-Protein Link Evaluator (DAPPLE),
which uses high-confidence pairwise protein interactions
and tissue-specific expression data to reconstruct a PPI
network.14 The network is conservative, requiring that
interacting proteins be known to be coexpressed in a given
tissue. The resultant AD PPI network returned by DAPPLE
is statistically significant for a network connectivity that
allows a common interactor protein (not known to be asso-
ciated with AD) between AD genes when compared to
1,000 random networks (permuted p ¼ 0.043; Figure 1).
This analysis is not significant (p ¼ 0.367) when direct
network connectivity between the AD genes is required.
We were able to indirectly connect all but two of the
proteins coded in known AD loci (ABCA7 [MIM 605414]
and CR1 [MIM 120620]).
When overlaying our AD PPI network allowing for
a common interactor protein with the results of our
analyses for evidence of natural selection, an intrigu-
ing subnetwork that includes the proteins encoded by
PICALM, BIN1, CD2AP, and EPHA1 (MIM 179610)
emerged. Integrating our gene-based PPI results with our
gene-based selection analysis (Table S1), we saw that all
four susceptibility loci showed evidence of positive
Table 2. Alzheimer-Disease-Associated Haplotypes, Regions, and Genes with Evidence of Positive Selection
Locus SNP Gene
Three Different Approaches to Assess for Evidence of Positive Selection
Haplotype Region Gene
ASI CEU YRI ASI CEU YRI ASI CEU YRI
6p12 rs9349407 CD2AP þ þ þ
11q12 rs610932 MS4A2 þ þ þ
11q14 rs561655 PICALM þ þ þ þ þ
2q14 rs7561528 BIN1 þ þ þ
7q35 rs11767557 EPHA1 þ
Evidence of selection (indicated by ‘‘þ’’) based on the analyses from alternate methods of selecting SNP sets with which to evaluate evidence for selection.The ‘‘haplotype’’ analysis is our primary analysis. In the haplotype analysis, we searched for SNPs in LD (r2> 0.5; D0 ¼ 1) with the index SNP in each locus (Table 1).We then considered haplotype blocks containing a linked SNP with jiHSj> 2 to be candidate targets of selection. We deployed secondary analyses to illustrate therobustness of our results. For our secondary regional analysis, we determined the number of SNPs with jiHSj> 2 in a 50-SNP window centered on the GWAS SNP.We then considered the regions in the upper 10% of the empirical distribution for proportion of SNPs with jiHSj> 2 to be candidate targets of selection. For gene-based analysis, we determined the proportion of SNPs with jiHSj> 2 in a 50-SNP window centered on the gene closest to the GWAS SNP. We then considered thegenes in the upper 10% of the empirical distribution for number of significant SNPs to be candidate targets of selection. The symbol ‘‘þ’’ indicates evidence forselection at each locus in three HapMap populations.
722 The American Journal of Human Genetics 90, 720–726, April 6, 2012

selection in East Asians (Table 2 and Table S1). This
evidence suggests a possible coevolution of these four
genes, which, on the basis of the PPI analysis, may interact
in a macromolecular complex (Figures 1 and 2). To assess
the statistical significance of this subnetwork, we con-
structed a new PPI network by using these four genes as
the seed regions, and found that the subnetwork was statis-
tically significant for indirect connectivity (permuted p ¼0.033; Figure 2). We observed that many of the interacting
proteins connecting the AD-associated proteins also
showed significant evidence for selection (Figure 2), and,
interestingly, one of these proteins is encoded by a gene
(GAB2 [MIM 606203]) that has been previously suggested
to be associated with AD.15,16 Thus, these AD-associated
and ‘‘connector’’ genes may have been under selection
because of their participation in a single functional module
over evolutionary time, and common function may also
explain their individual associations with AD suscepti-
bility. Given the late age-at-onset of AD, it is unlikely
that this phenotype itself has been the target of selec-
tion over evolutionary time; rather, it is likely that these
four genes, along with their interaction partners, com-
pose a functional module that is important in other biolog-
ical contexts.
Some of the strongest selective pressures acting during
human evolution have been attributed to human interac-
tions with pathogens; we thus investigated the functional
consequences of AD-associated variants with evidence of
selection on gene expression levels in immune cells. This
approach was additionally motivated by the observation
Figure 1. Protein-Protein Interaction NetworkGenerated from Proteins Encoding for AD-Associated and -Suggested GenesThe 118 LD-pruned SNPs (p < 10-4) from the Alz-heimer Disease Genetics Consortium (ADGC)GWAS3, which provided a complete list of sugges-tive results, were included as an input to DAPPLE.DAPPLE selects genes from a SNP list based on theregion containing SNPs with an r2 > 0.5 to eachindex SNP; this region is then extended to thenearest recombination hot spot. AD-associatedproteins are represented as nodes connected byan edge if there is in vitro evidence for high-confi-dence interaction. The large colored circles repre-sent AD-associated and -suggestive proteins, andsmall circles in grey represent the connectedproteins (not known to be associated with AD).Most of the AD-associated proteins are connectedvia common interactor proteins (gray) withwhich the associated proteins each share anedge. The red points indicate genes (CD2AP,PICALM, BIN1, EPHA1, and MS4A2) under posi-tive selection in our gene-based analyses ofHapMap II populations (Table S1). The PPInetwork is statistically significant for indirectconnectivity (PPI network permuted p ¼ 0.043).
that some AD-associated variants map close
to genes with putative immune function
(i.e., CD33 [MIM 159590], CR1, and
MS4A2) and others, such as PICALM, BIN1, CD2AP, and
EPHA1, are coexpressed in immune cells, particularly in
the myeloid lineage.17 Thus, we assessed each of the 11
validated and well-replicated AD loci for evidence of an
effect on RNA expression in cis; that is, we performed an
expression quantitative trait locus (eQTL) analysis in
each locus for genes in the vicinity of the index SNP (see
Supplemental Material andMethods). We first investigated
genetic variation and mRNA expression in an available
data set derived from peripheral blood mononuclear cells
(PBMCs) of 228 individuals of European ancestry with
demyelinating disease,18 representing a set of subjects
with an activated immune system. PBMCs are purified
from peripheral blood and contain both myeloid and
lymphoid cells. After gene-based permutation assessing
significance of SNP-gene association p values, we identified
five cis-regulatory effects in the 11 tested loci (Table S2).
Specifically, we found three AD-associated index vari-
ants—rs610932, rs7561528, and rs3752246—with cis-
regulatory effects in PBMCs: rs610932 influences the
expression of neighboring MS4A2 (nominal p ¼ 5.04 3
10�8), whereas rs7561528 and rs3752246 affect the ex-
pression of BIN1 (p ¼ 4.76 3 10�4) and ABCA7 (p ¼6.87 3 10�5), respectively. These cis-regulatory effects are
all significant at a p < 0.05 permutation threshold; the
same MS4A2 and BIN1 haplotypes harbor evidence of
natural selection (Table 1).
In the PICALM locus, which has strong evidence of selec-
tion, the current best ADmarker does not have a strong cis-
regulatory effect on gene expression, but other SNPs that
The American Journal of Human Genetics 90, 720–726, April 6, 2012 723

have evidence of association with AD (p < 5x10�8 in pub-
lished studies) 3,4 and are in strong LD with the index SNP
have replicated evidence of having a cis-regulatory effect
on gene expression. Specifically, the AD risk allele
rs659023G (pAD ¼ 2.78 3 10�10)3 (r2 ¼ 0.8 with index
PICALM SNP rs561655) is significantly associated with
decreased expression of PICALM in PBMC (p ¼ 4.76 3
10�4; Figure S1). We replicate this cis-regulatory effect in
CD4þ T lymphocytes (which are constituents of the
PBMC cell mixture) of 40 healthy individuals of European
ancestry (p ¼ 2.48 3 10�4).
In the case of PICALM, the evidence for selection and the
effect on gene expression converge (Figure 3; Table S3): the
rs659023 SNP discussed above exhibits a strong correlation
with PICALM RNA expression and has a high iHS (eQTL
p ¼ 4.76 3 10�4, jiHSj ¼ 2.17). These results suggest that
one or more potential functional variants that influence
PICALM expression may have been selected for over the
course of human history in this well-validated AD locus.
This information can be leveraged to focus fine-mapping
efforts onto a short list of candidate causal variants that
can be targeted in well-powered AD susceptibility analyses.
In summary, we observe significant evidence for signa-
tures of positive selection on haplotypes associated with
late-onset Alzheimer disease. The most intriguing finding
of our study is that multiple AD-associated genes (i.e.,
PICALM, BIN1, CD2AP, and EPHA1) with evidence for posi-
tive selection encode proteins that physically interact
within an independently defined PPI network. Further-
more, some of the linking proteins in the network also
Figure 2. Protein-Protein Interaction Subnet-work of AD-Associated Genes under PositiveSelectionThe subnetwork is simply a highly intercon-nected subset of the larger network that emergesfrom DAPPLE and is illustrated in Figure 1.The subnetwork is statistically significant forindirect connectivity (PPI subnetwork permutedp¼ 0.033). As in Figure 1, the red dotsmark geneswith evidence for natural selection.
show evidence of positive selection, and
one of these,GAB2, has previously been sug-
gested to be associated with AD.15,16 The
convergence of results in these two comple-
mentary analyses suggests that these four
interacting proteins encoding for AD
susceptibility genes may have had a corre-
lated evolutionary history, perhaps in
response to a single evolutionary pressure
that affected East Asian populations and,
to a lesser degree, European populations
(Table 2). Moreover, we found that several
of these loci exhibit a cis-regulatory effect
on the transcription of the selected genes
in immune cells, suggesting that changes
in gene expression may be one mechanism
bywhich anAD-relatedmolecular pathway evolved in early
human history, most likely in a non-AD context.
The resultspresentedhereprovide convincing support for
recent positive selection of loci underlying late-onset AD,
but what are the selective pressures underlying the signa-
tures? It is difficult to identify the precise selective pressure
that led to our observations. Selection may result from an
array of forces including pathogen resistance aswell as envi-
ronmental and dietary changes as modern humans
migrated out of Africa and spread throughout the world.
For thegeneswithputative immune functions,we speculate
that pathogens may have exerted selective pressures on
populations and consequently influenced the frequency
of AD-associated alleles. Regardless of what the selection
pressure might have been, our results suggest that several
different loci implicated in AD susceptibility may have
worked together in another context to enhance survival
over the course of recent human evolutionary history.
Evidence for natural selection at a given variant suggests
that such a variant may have functional consequences,
given that the selection process was mediated by an alter-
ation of a biological process. One can think of selective pres-
sures asnatural, invivohumanexperiments inwhichwecan
measure the response of human populations to unknown
perturbations, and these alterations can inform the function
of geneswithin a given locus.However, by itself, evidence of
selection is not sufficient for identifying causal variants in
susceptibility loci. Rather, it offers another key dimension
of functional information to integrative analyses that
include disease association, protein-protein interaction,
724 The American Journal of Human Genetics 90, 720–726, April 6, 2012
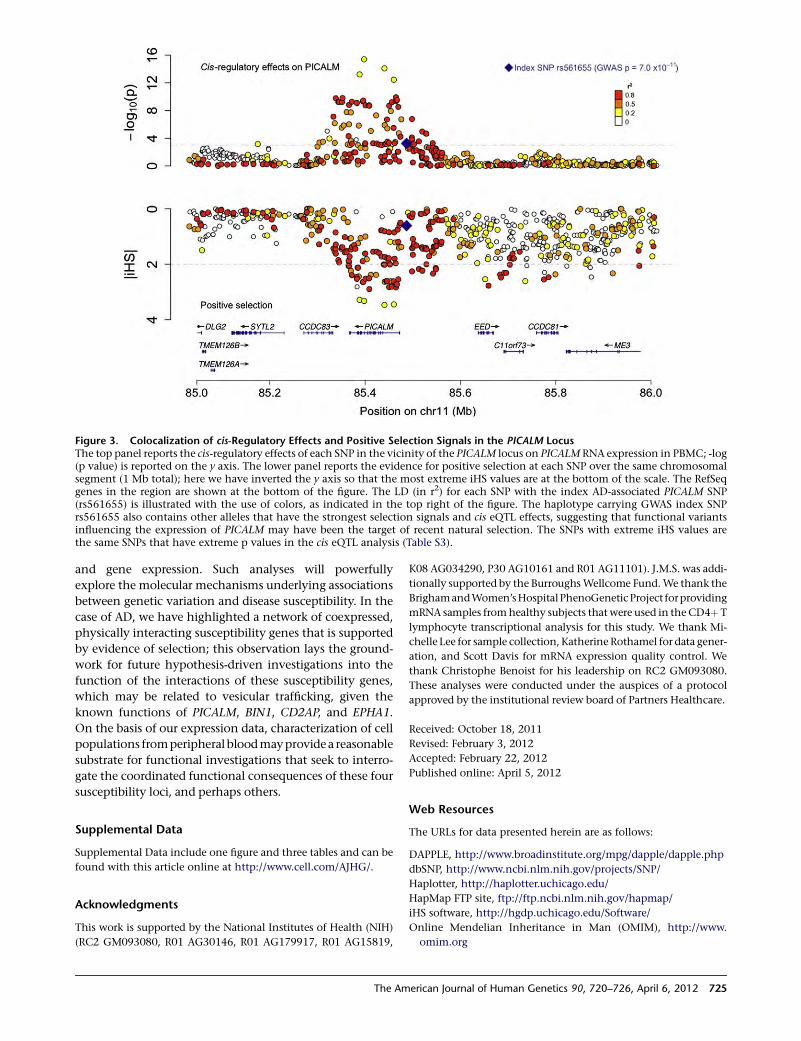
and gene expression. Such analyses will powerfully
explore the molecular mechanisms underlying associations
between genetic variation and disease susceptibility. In the
case of AD, we have highlighted a network of coexpressed,
physically interacting susceptibility genes that is supported
by evidence of selection; this observation lays the ground-
work for future hypothesis-driven investigations into the
function of the interactions of these susceptibility genes,
which may be related to vesicular trafficking, given the
known functions of PICALM, BIN1, CD2AP, and EPHA1.
On the basis of our expression data, characterization of cell
populations fromperipheral bloodmayprovidea reasonable
substrate for functional investigations that seek to interro-
gate the coordinated functional consequences of these four
susceptibility loci, and perhaps others.
Supplemental Data
Supplemental Data include one figure and three tables and can be
found with this article online at http://www.cell.com/AJHG/.
Acknowledgments
This work is supported by the National Institutes of Health (NIH)
(RC2 GM093080, R01 AG30146, R01 AG179917, R01 AG15819,
K08 AG034290, P30 AG10161 and R01 AG11101). J.M.S. was addi-
tionally supported by the BurroughsWellcome Fund.We thank the
BrighamandWomen’sHospitalPhenoGeneticProject forproviding
mRNA samples fromhealthy subjects that were used in theCD4þ T
lymphocyte transcriptional analysis for this study. We thank Mi-
chelle Lee for sample collection, Katherine Rothamel for data gener-
ation, and Scott Davis for mRNA expression quality control. We
thank Christophe Benoist for his leadership on RC2 GM093080.
These analyses were conducted under the auspices of a protocol
approved by the institutional review board of Partners Healthcare.
Received: October 18, 2011
Revised: February 3, 2012
Accepted: February 22, 2012
Published online: April 5, 2012
Web Resources
The URLs for data presented herein are as follows:
DAPPLE, http://www.broadinstitute.org/mpg/dapple/dapple.php
dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
Haplotter, http://haplotter.uchicago.edu/
HapMap FTP site, ftp://ftp.ncbi.nlm.nih.gov/hapmap/
iHS software, http://hgdp.uchicago.edu/Software/
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
Figure 3. Colocalization of cis-Regulatory Effects and Positive Selection Signals in the PICALM LocusThe top panel reports the cis-regulatory effects of each SNP in the vicinity of the PICALM locus on PICALM RNA expression in PBMC; -log(p value) is reported on the y axis. The lower panel reports the evidence for positive selection at each SNP over the same chromosomalsegment (1 Mb total); here we have inverted the y axis so that the most extreme iHS values are at the bottom of the scale. The RefSeqgenes in the region are shown at the bottom of the figure. The LD (in r2) for each SNP with the index AD-associated PICALM SNP(rs561655) is illustrated with the use of colors, as indicated in the top right of the figure. The haplotype carrying GWAS index SNPrs561655 also contains other alleles that have the strongest selection signals and cis eQTL effects, suggesting that functional variantsinfluencing the expression of PICALM may have been the target of recent natural selection. The SNPs with extreme iHS values arethe same SNPs that have extreme p values in the cis eQTL analysis (Table S3).
The American Journal of Human Genetics 90, 720–726, April 6, 2012 725

References
1. Avramopoulos, D. (2009). Genetics of Alzheimer’s disease:
recent advances. Genome Med 1, 34.
2. Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish,
A., Hamshere, M.L., Pahwa, J.S., Moskvina, V., Dowzell, K.,
Williams, A., et al. (2009). Genome-wide association study
identifies variants at CLU and PICALM associated with
Alzheimer’s disease. Nat. Genet. 41, 1088–1093.
3. Naj, A.C., Jun, G., Beecham, G.W., Wang, L.S., Vardarajan,
B.N., Buros, J., Gallins, P.J., Buxbaum, J.D., Jarvik, G.P., Crane,
P.K., et al. (2011). Common variants at MS4A4/MS4A6E,
CD2AP, CD33 and EPHA1 are associated with late-onset
Alzheimer’s disease. Nat. Genet. 43, 436–441.
4. Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert,
J.C., Carrasquillo, M.M., Abraham, R., Hamshere, M.L.,
Pahwa, J.S., Moskvina, V., et al. (2011). Common variants at
ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are
associated with Alzheimer’s disease. Nat. Genet. 43, 429–435.
5. Lambert, J.C., Heath, S., Even, G., Campion, D., Sleegers, K.,
Hiltunen, M., Combarros, O., Zelenika, D., Bullido, M.J.,
Tavernier, B., et al; European Alzheimer’s Disease Initiative
Investigators. (2009). Genome-wide association study iden-
tifies variants at CLU and CR1 associated with Alzheimer’s
disease. Nat. Genet. 41, 1094–1099.
6. Seshadri, S., Fitzpatrick, A.L., Ikram, M.A., DeStefano, A.L.,
Gudnason, V., Boada, M., Bis, J.C., Smith, A.V., Carassquillo,
M.M., Lambert, J.C., et al; CHARGE Consortium; GERAD1
Consortium; EADI1 Consortium. (2010). Genome-wide anal-
ysis of genetic loci associated with Alzheimer disease. JAMA
303, 1832–1840.
7. Drenos, F., and Kirkwood, T.B. (2010). Selection on alleles
affecting human longevity and late-life disease: the example
of apolipoprotein E. PLoS ONE 5, e10022.
8. Vamathevan, J.J., Hasan, S., Emes, R.D., Amrine-Madsen, H.,
Rajagopalan, D., Topp, S.D., Kumar, V., Word, M., Simmons,
M.D., Foord, S.M., et al. (2008). The role of positive selection
in determining the molecular cause of species differences in
disease. BMC Evol. Biol. 8, 273.
9. Voight, B.F., Kudaravalli, S., Wen, X., and Pritchard, J.K.
(2006). A map of recent positive selection in the human
genome. PLoS Biol. 4, e72.
10. Frazer, K.A., Ballinger, D.G., Cox, D.R., Hinds, D.A., Stuve,
L.L., Gibbs, R.A., Belmont, J.W., Boudreau, A., Hardenbol, P.,
Leal, S.M., et al; International HapMap Consortium. (2007).
A second generation human haplotype map of over 3.1
million SNPs. Nature 449, 851–861.
11. Nalls, M.A., Plagnol, V., Hernandez, D.G., Sharma, M.,
Sheerin, U.M., Saad, M., Simon-Sanchez, J., Schulte, C.,
Lesage, S., Sveinbjornsdottir, S., et al; International Parkinson
Disease Genomics Consortium. (2011). Imputation of
sequence variants for identification of genetic risks for Parkin-
son’s disease: a meta-analysis of genome-wide association
studies. Lancet 377, 641–649.
12. Fraser, H.B., Hirsh, A.E., Wall, D.P., and Eisen, M.B. (2004).
Coevolution of gene expression among interacting proteins.
Proc. Natl. Acad. Sci. USA 101, 9033–9038.
13. Tillier, E.R., and Charlebois, R.L. (2009). The human protein
coevolution network. Genome Res. 19, 1861–1871.
14. Rossin, E.J., Lage, K., Raychaudhuri, S., Xavier, R.J., Tatar, D.,
Benita, Y., Cotsapas, C., and Daly, M.J.; International Inflam-
matory Bowel Disease Genetics Constortium. (2011). Proteins
encoded in genomic regions associated with immune-
mediated disease physically interact and suggest underlying
biology. PLoS Genet. 7, e1001273.
15. Reiman, E.M., Webster, J.A., Myers, A.J., Hardy, J., Dunckley, T.,
Zismann, V.L., Joshipura, K.D., Pearson, J.V., Hu-Lince, D.,
Huentelman, M.J., et al. (2007). GAB2 alleles modify
Alzheimer’s risk inAPOEepsilon4 carriers.Neuron54, 713–720.
16. Ikram, M.A., Liu, F., Oostra, B.A., Hofman, A., van Duijn,
C.M., and Breteler, M.M. (2009). The GAB2 gene and the
risk of Alzheimer’s disease: replication and meta-analysis.
Biol. Psychiatry 65, 995–999.
17. Wu, C., Orozco, C., Boyer, J., Leglise, M., Goodale, J., Batalov,
S., Hodge, C.L., Haase, J., Janes, J., Huss, J.W., 3rd, and Su, A.I.
(2009). BioGPS: an extensible and customizable portal for
querying and organizing gene annotation resources. Genome
Biol. 10, R130.
18. De Jager, P., Jia, X., Wang, J., de Bakker, P., Ottoboni, L., Aggar-
wal, N., Piccio, L., Raychaudhuri, S., Tran, D., Aubin, C., et al.
(2009). Meta-analysis of genome scans and replication
identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis
susceptibility loci. Nat. Genet. 41, 776–782.
726 The American Journal of Human Genetics 90, 720–726, April 6, 2012

REPORT
Linkage-Disequilibrium-Based BinningAffects the Interpretation of GWASs
Andrea Christoforou,1,2,16,* Michael Dondrup,3,16 Morten Mattingsdal,4,5,16 Manuel Mattheisen,6,7,8,9,16
Sudheer Giddaluru,1,2 Markus M. Nothen,6,7,10 Marcella Rietschel,11 Sven Cichon,1,6,7,12
Srdjan Djurovic,4,13,14 Ole A. Andreassen,4,14 Inge Jonassen,3,15 Vidar M. Steen,1,2 Pal Puntervoll,3
and Stephanie Le Hellard1,2
Genome-wide association studies (GWASs) are critically dependent on detailed knowledge of the pattern of linkage disequilibrium (LD)
in the human genome. GWASs generate lists of variants, usually SNPs, ranked according to the significance of their association to a trait.
Downstream analyses generally focus on the gene or genes that are physically closest to these SNPs and ignore their LD profile with other
SNPs. We have developed a flexible R package (LDsnpR) that efficiently assigns SNPs to genes on the basis of both their physical position
and their pairwise LD with other SNPs. We used the positional-binning and LD-based-binning approaches to investigate whether
including these ‘‘LD-based’’ SNPs would affect the interpretation of three published GWASs on bipolar affective disorder (BP) and of
the imputed versions of two of these GWASs. We show how including LD can be important for interpreting and comparing GWASs.
In the published, unimputed GWASs, LD-based binning effectively ‘‘recovered’’ 6.1%–8.3% of Ensembl-defined genes. It altered the
ranks of the genes and resulted in nonnegligible differences between the lists of the top 2,000 genes emerging from the two binning
approaches. It also improved the overall gene-based concordance between independent BP studies. In the imputed datasets, although
the increases in coverage (>0.4%) and rank changes were more modest, even greater concordance between the studies was observed,
attesting to the potential of LD-based binning on imputed data as well. Thus, ignoring LD can result in the misinterpretation of the
GWAS findings and have an impact on subsequent genetic and functional studies.
Over the past decade, genome-wide association studies
(GWASs) have revolutionized the analysis of human
complex genetic traits. By scanning hundreds of thou-
sands of genetic variants, typically SNPs, in hundreds or
thousands of individuals, they search for the variant(s)
that associate with a particular disease or trait. Critical to
the development and evolution of GWASs has been the
creation of the International HapMap Project,1 which
has cataloged the common patterns of human genetic vari-
ation, including the linkage disequilibrium (LD) between
SNPs. Knowledge of this LD, or nonrandom association
of alleles at multiple loci, has made it possible to identify
informative subsets of SNPs (i.e., ‘‘tagging SNPs’’) that
capture the bulk of genome-wide variation and has re-
sulted in affordable genome-wide genotyping. To date,
almost 1,000 GWASs have been published and have tested
hundreds of human traits and reported thousands of
significant associations (Catalog of Published Genome-
Wide Association Studies2). Previously known associations
have been confirmed, and new candidates have been
implicated.3 However, a general sense of disappointment
lingers because GWASs have fallen short of the initial
expectation that they would unravel the genetic basis of
complex traits.4,5 Recent analyses reveal that a large
proportion of the ‘‘missing heritability’’5,6 can be ex-
plained by a polygenic model that considers all GWAS
SNPs simultaneously,7–9 but these studies provide no clues
about the identity of the susceptibility variants or the
underlying biology of the trait.6 Thus, much attention
has been given to uncovering and characterizing this
‘‘missing’’ or ‘‘hidden’’ heritability.6,10
In a conventional GWAS, each SNP is considered sepa-
rately (the ‘‘single-marker’’ approach), resulting in a list
of variants ranked according to the statistical significance
of their association to the trait (i.e., their p value).11 The
‘‘top hits’’ are typically reported, and the relevance of
each finding, as well as the focus of future work, is
primarily based on the functional unit(s), namely gene(s),
implicated by the associated SNP. Furthermore, gene-based
methods are increasingly being applied as complementary
approaches to the analysis of GWAS data. These methods
take the gene instead of the individual SNP as the basic
unit of association and thus allow aggregation of SNPs of
smaller effect, potentially increasing power and reducing
1Dr. Einar Martens Research Group for Biological Psychiatry, Department of Clinical Medicine, University of Bergen, 5021 Bergen, Norway; 2Center for
Medical Genetics and Molecular Medicine, Haukeland University Hospital, 5021 Bergen, Norway; 3Computational Biology Unit, Uni Computing, Uni
Research, 5008 Bergen, Norway; 4Institute of Clinical Medicine, University of Oslo, 0318 Oslo, Norway; 5Research Unit, Sørlandet Hospital HF, 4604
Kristiansand, Norway; 6Department of Genomics, Life and Brain Center, University of Bonn, 53127 Bonn, Germany; 7Institute of Human Genetics,
University of Bonn, 53127 Bonn, Germany; 8Institute for Genomic Mathematics, University of Bonn, 53127 Bonn, Germany; 9Department of Biostatistics,
Harvard School of Public Health, Boston, MA 02115, USA; 10German Centre for Neurodegenerative Disorders, 53175 Bonn, Germany; 11Department of
Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, University of Mannheim, 68159 Mannheim, Germany; 12Structural and
Functional Organization of the Brain, Institute of Neuroscience and Medicine, Research Center Julich, 52425 Julich, Germany; 13Department of
Medical Genetics, Oslo University Hospital, 0424 Oslo, Norway; 14Division of Mental Health and Addiction, Oslo University Hospital, 0424 Oslo, Norway;15Department of Informatics, University of Bergen, 5008 Bergen, Norway16These authors contributed equally to this work
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.025. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 727–733, April 6, 2012 727
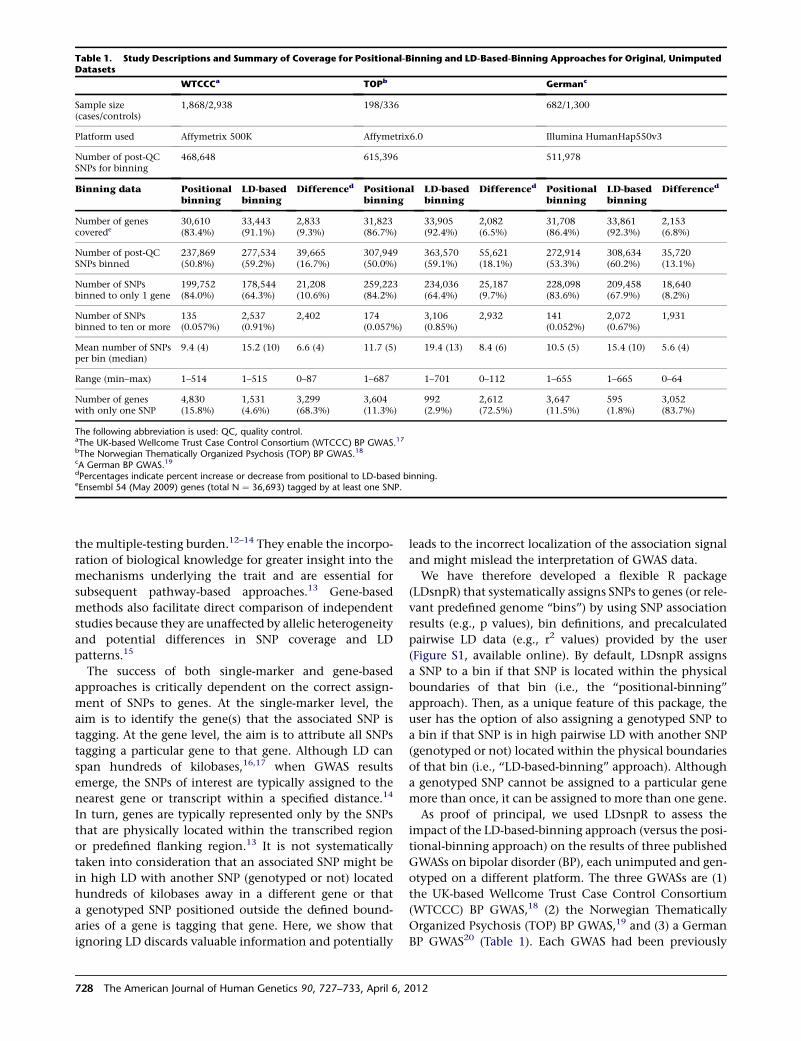
the multiple-testing burden.12–14 They enable the incorpo-
ration of biological knowledge for greater insight into the
mechanisms underlying the trait and are essential for
subsequent pathway-based approaches.13 Gene-based
methods also facilitate direct comparison of independent
studies because they are unaffected by allelic heterogeneity
and potential differences in SNP coverage and LD
patterns.15
The success of both single-marker and gene-based
approaches is critically dependent on the correct assign-
ment of SNPs to genes. At the single-marker level, the
aim is to identify the gene(s) that the associated SNP is
tagging. At the gene level, the aim is to attribute all SNPs
tagging a particular gene to that gene. Although LD can
span hundreds of kilobases,16,17 when GWAS results
emerge, the SNPs of interest are typically assigned to the
nearest gene or transcript within a specified distance.14
In turn, genes are typically represented only by the SNPs
that are physically located within the transcribed region
or predefined flanking region.13 It is not systematically
taken into consideration that an associated SNP might be
in high LD with another SNP (genotyped or not) located
hundreds of kilobases away in a different gene or that
a genotyped SNP positioned outside the defined bound-
aries of a gene is tagging that gene. Here, we show that
ignoring LD discards valuable information and potentially
leads to the incorrect localization of the association signal
and might mislead the interpretation of GWAS data.
We have therefore developed a flexible R package
(LDsnpR) that systematically assigns SNPs to genes (or rele-
vant predefined genome ‘‘bins’’) by using SNP association
results (e.g., p values), bin definitions, and precalculated
pairwise LD data (e.g., r2 values) provided by the user
(Figure S1, available online). By default, LDsnpR assigns
a SNP to a bin if that SNP is located within the physical
boundaries of that bin (i.e., the ‘‘positional-binning’’
approach). Then, as a unique feature of this package, the
user has the option of also assigning a genotyped SNP to
a bin if that SNP is in high pairwise LD with another SNP
(genotyped or not) located within the physical boundaries
of that bin (i.e., ‘‘LD-based-binning’’ approach). Although
a genotyped SNP cannot be assigned to a particular gene
more than once, it can be assigned to more than one gene.
As proof of principal, we used LDsnpR to assess the
impact of the LD-based-binning approach (versus the posi-
tional-binning approach) on the results of three published
GWASs on bipolar disorder (BP), each unimputed and gen-
otyped on a different platform. The three GWASs are (1)
the UK-based Wellcome Trust Case Control Consortium
(WTCCC) BP GWAS,18 (2) the Norwegian Thematically
Organized Psychosis (TOP) BP GWAS,19 and (3) a German
BP GWAS20 (Table 1). Each GWAS had been previously
Table 1. Study Descriptions and Summary of Coverage for Positional-Binning and LD-Based-Binning Approaches for Original, UnimputedDatasets
WTCCCa TOPb Germanc
Sample size(cases/controls)
1,868/2,938 198/336 682/1,300
Platform used Affymetrix 500K Affymetrix6.0 Illumina HumanHap550v3
Number of post-QCSNPs for binning
468,648 615,396 511,978
Binning data Positionalbinning
LD-basedbinning
Differenced Positionalbinning
LD-basedbinning
Differenced Positionalbinning
LD-basedbinning
Differenced
Number of genescoverede
30,610(83.4%)
33,443(91.1%)
2,833(9.3%)
31,823(86.7%)
33,905(92.4%)
2,082(6.5%)
31,708(86.4%)
33,861(92.3%)
2,153(6.8%)
Number of post-QCSNPs binned
237,869(50.8%)
277,534(59.2%)
39,665(16.7%)
307,949(50.0%)
363,570(59.1%)
55,621(18.1%)
272,914(53.3%)
308,634(60.2%)
35,720(13.1%)
Number of SNPsbinned to only 1 gene
199,752(84.0%)
178,544(64.3%)
21,208(10.6%)
259,223(84.2%)
234,036(64.4%)
25,187(9.7%)
228,098(83.6%)
209,458(67.9%)
18,640(8.2%)
Number of SNPsbinned to ten or more
135(0.057%)
2,537(0.91%)
2,402 174(0.057%)
3,106(0.85%)
2,932 141(0.052%)
2,072(0.67%)
1,931
Mean number of SNPsper bin (median)
9.4 (4) 15.2 (10) 6.6 (4) 11.7 (5) 19.4 (13) 8.4 (6) 10.5 (5) 15.4 (10) 5.6 (4)
Range (min–max) 1–514 1–515 0–87 1–687 1–701 0–112 1–655 1–665 0–64
Number of geneswith only one SNP
4,830(15.8%)
1,531(4.6%)
3,299(68.3%)
3,604(11.3%)
992(2.9%)
2,612(72.5%)
3,647(11.5%)
595(1.8%)
3,052(83.7%)
The following abbreviation is used: QC, quality control.aThe UK-based Wellcome Trust Case Control Consortium (WTCCC) BP GWAS.17bThe Norwegian Thematically Organized Psychosis (TOP) BP GWAS.18cA German BP GWAS.19dPercentages indicate percent increase or decrease from positional to LD-based binning.eEnsembl 54 (May 2009) genes (total N ¼ 36,693) tagged by at least one SNP.
728 The American Journal of Human Genetics 90, 727–733, April 6, 2012
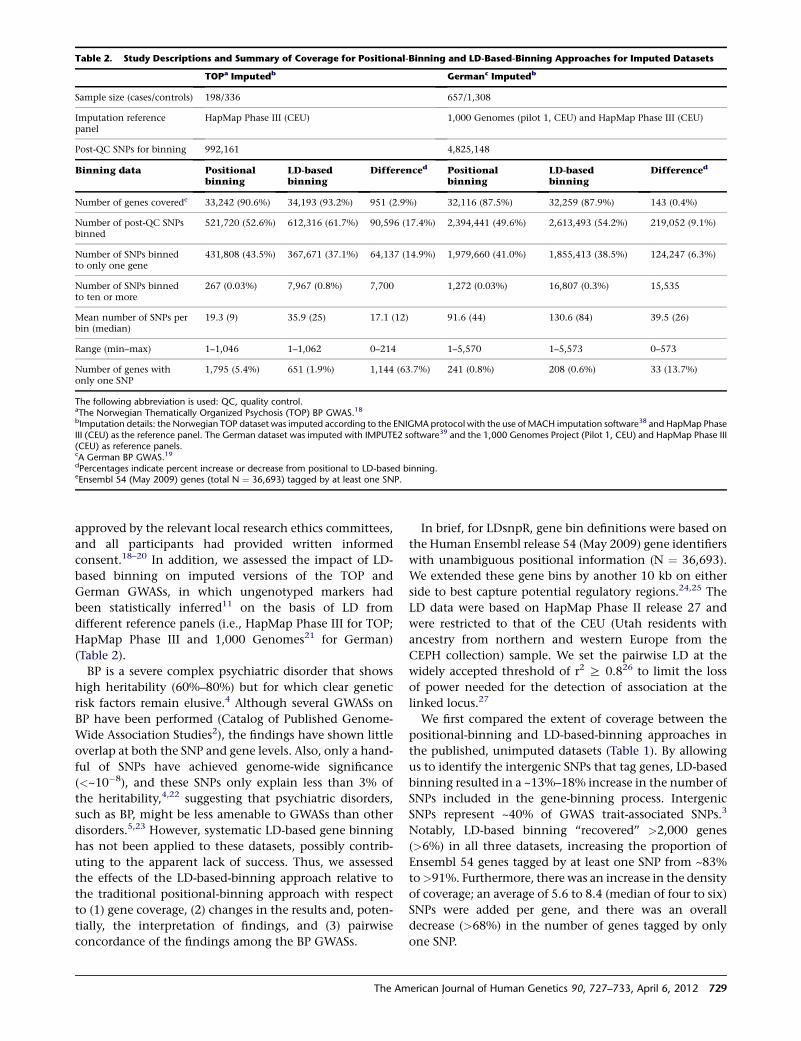
approved by the relevant local research ethics committees,
and all participants had provided written informed
consent.18–20 In addition, we assessed the impact of LD-
based binning on imputed versions of the TOP and
German GWASs, in which ungenotyped markers had
been statistically inferred11 on the basis of LD from
different reference panels (i.e., HapMap Phase III for TOP;
HapMap Phase III and 1,000 Genomes21 for German)
(Table 2).
BP is a severe complex psychiatric disorder that shows
high heritability (60%–80%) but for which clear genetic
risk factors remain elusive.4 Although several GWASs on
BP have been performed (Catalog of Published Genome-
Wide Association Studies2), the findings have shown little
overlap at both the SNP and gene levels. Also, only a hand-
ful of SNPs have achieved genome-wide significance
(<~10�8), and these SNPs only explain less than 3% of
the heritability,4,22 suggesting that psychiatric disorders,
such as BP, might be less amenable to GWASs than other
disorders.5,23 However, systematic LD-based gene binning
has not been applied to these datasets, possibly contrib-
uting to the apparent lack of success. Thus, we assessed
the effects of the LD-based-binning approach relative to
the traditional positional-binning approach with respect
to (1) gene coverage, (2) changes in the results and, poten-
tially, the interpretation of findings, and (3) pairwise
concordance of the findings among the BP GWASs.
In brief, for LDsnpR, gene bin definitions were based on
the Human Ensembl release 54 (May 2009) gene identifiers
with unambiguous positional information (N ¼ 36,693).
We extended these gene bins by another 10 kb on either
side to best capture potential regulatory regions.24,25 The
LD data were based on HapMap Phase II release 27 and
were restricted to that of the CEU (Utah residents with
ancestry from northern and western Europe from the
CEPH collection) sample. We set the pairwise LD at the
widely accepted threshold of r2 R 0.826 to limit the loss
of power needed for the detection of association at the
linked locus.27
We first compared the extent of coverage between the
positional-binning and LD-based-binning approaches in
the published, unimputed datasets (Table 1). By allowing
us to identify the intergenic SNPs that tag genes, LD-based
binning resulted in a ~13%–18% increase in the number of
SNPs included in the gene-binning process. Intergenic
SNPs represent ~40% of GWAS trait-associated SNPs.3
Notably, LD-based binning ‘‘recovered’’ >2,000 genes
(>6%) in all three datasets, increasing the proportion of
Ensembl 54 genes tagged by at least one SNP from ~83%
to>91%. Furthermore, there was an increase in the density
of coverage; an average of 5.6 to 8.4 (median of four to six)
SNPs were added per gene, and there was an overall
decrease (>68%) in the number of genes tagged by only
one SNP.
Table 2. Study Descriptions and Summary of Coverage for Positional-Binning and LD-Based-Binning Approaches for Imputed Datasets
TOPa Imputedb Germanc Imputedb
Sample size (cases/controls) 198/336 657/1,308
Imputation referencepanel
HapMap Phase III (CEU) 1,000 Genomes (pilot 1, CEU) and HapMap Phase III (CEU)
Post-QC SNPs for binning 992,161 4,825,148
Binning data Positionalbinning
LD-basedbinning
Differenced Positionalbinning
LD-basedbinning
Differenced
Number of genes coverede 33,242 (90.6%) 34,193 (93.2%) 951 (2.9%) 32,116 (87.5%) 32,259 (87.9%) 143 (0.4%)
Number of post-QC SNPsbinned
521,720 (52.6%) 612,316 (61.7%) 90,596 (17.4%) 2,394,441 (49.6%) 2,613,493 (54.2%) 219,052 (9.1%)
Number of SNPs binnedto only one gene
431,808 (43.5%) 367,671 (37.1%) 64,137 (14.9%) 1,979,660 (41.0%) 1,855,413 (38.5%) 124,247 (6.3%)
Number of SNPs binnedto ten or more
267 (0.03%) 7,967 (0.8%) 7,700 1,272 (0.03%) 16,807 (0.3%) 15,535
Mean number of SNPs perbin (median)
19.3 (9) 35.9 (25) 17.1 (12) 91.6 (44) 130.6 (84) 39.5 (26)
Range (min–max) 1–1,046 1–1,062 0–214 1–5,570 1–5,573 0–573
Number of genes withonly one SNP
1,795 (5.4%) 651 (1.9%) 1,144 (63.7%) 241 (0.8%) 208 (0.6%) 33 (13.7%)
The following abbreviation is used: QC, quality control.aThe Norwegian Thematically Organized Psychosis (TOP) BP GWAS.18bImputation details: the Norwegian TOP dataset was imputed according to the ENIGMA protocol with the use of MACH imputation software38 and HapMap PhaseIII (CEU) as the reference panel. The German dataset was imputed with IMPUTE2 software39 and the 1,000 Genomes Project (Pilot 1, CEU) and HapMap Phase III(CEU) as reference panels.cA German BP GWAS.19dPercentages indicate percent increase or decrease from positional to LD-based binning.eEnsembl 54 (May 2009) genes (total N ¼ 36,693) tagged by at least one SNP.
The American Journal of Human Genetics 90, 727–733, April 6, 2012 729

The imputed datasets also yielded increased coverage
(Table 2) but, as expected, to a lesser extent depending
on the reference panel used for imputation. Although
HapMap II (i.e., LDsnpR reference panel) is denser than
HapMap III28 (i.e., reference panel for the TOP and
German studies), imputation on the 1,000 Genomes data
(i.e., reference panel for the German study) potentially
gives the densest coverage. For the TOP and German
imputed datasets, LD-based binning resulted in an increase
of 17.4% and 9.1%, respectively, in the number of SNPs
included in the gene-binning process and the recovery of
951 (2.9%) and 143 (0.4%) genes, respectively. Although
this is only a small proportion of the total gene coverage,
the recovery of these genes enables them to be considered
as candidates for BP association and might lead to a better
understanding of the biology should the true association
stem from them. Also of note, in the German GWAS, LD-
based binning alone achieved an overall gene coverage of
92.3% (imputation achieved 87.5% coverage, and imputa-
tion combined with LD-based binning achieved 87.9%
coverage), suggesting that under some scenarios, LD-based
binning alone can offer the most coverage. As with the
original GWASs, there was an increase in the density of
coverage; an average of 17.1 and 39.5 (median 12 and
26) SNPs were added per gene for the TOP and German
imputed datasets, respectively. There was also a decrease
in the number of genes tagged by only one SNP (63.7%);
the decrease was not as notable for the German imputed
dataset (13.7%).
We next assessed the effects of the LD-based-binning
approach on the results of the three GWASs at both the
single-marker and gene levels. At the single-marker level,
we used the positional-binning and LD-based-binning
approaches to compare the genes tagged by the most
significant SNPs reported in the original publications18–20
(Table S1). Although LD-based binningmade no difference
to the results of the TOP BP study, three of the 14 SNPs in
the WTCCC BP study and three of the eight SNPs in the
German BP study implicated additional or alternative
genes. Interpreting GWAS single-marker results demands
fastidious consideration because when given only the
p value, it is not immediately clear where the true source
of the association originates17 and thus which is the true
candidate gene. The overall potential for mislocalizing the
association signal was underscored by the reduced number
of SNPs tagging only one gene and the increased number of
SNPs tagging ten or more genes after LD-based binning
(Tables 1 and 2). Further investigations, such as expression
studies,20 are therefore warranted before attributing puta-
tive causality to a gene and, as a result, nominating it as
the focus of future fine-mapping, functional, and other
expensive and time-consuming follow-up studies.29
As previously stated, gene-based analyses are ideal for
pathway approaches, which aid in the interpretation of
GWAS results by exploiting prior biological annotation to
determine whether certain biological functions are en-
riched (i.e., overrepresented) among the more significant
genes in a dataset. These methods require one measure of
association (or score) for each gene on the basis of the indi-
vidual SNP association signals. Here, we used a function in
LDsnpR to score each gene with the most significant
p value (i.e., the minimum p value approach), which was
adjusted for the number of SNPs tagging that gene by
a modification of Sidak’s correction.30 The minimum
p value approach is the most widely used gene-scoring
approach31 and assumes an underlying genetic architec-
ture in which a single SNP, or locus, within the gene
contributes to the disorder. The modification performs at
least as well as a powerful regression-based method in cor-
recting for the bias due to SNP number.32 In this study, the
correlation between the gene score and the number
of SNPs in the bin was reduced from Pearson r2 > 0.30 to
r2 < 0.020 in all three datasets after the modified Sidak
correction was applied. Also, permutation-based gene-set
analysis, as implemented in PLINK,33 on the German
GWAS confirmed the high correlation between modified
Sidak-corrected p values and permutation-based p values
(r2 > 0.95). The genes were scored for both the posi-
tional-binning and LD-based-binning approaches and
were compared.
The overall correlation in the ranks of the genes between
the two approaches was <0.83 in the three original data-
sets and the TOP imputed dataset, indicating that LD-
based binning altered the scores and the subsequent ranks
of the genes. Although not as large, changes in rank were
also observed in the German imputed dataset (Table 3).
When a resampling analysis was performed on the unim-
puted WTCCC dataset (it randomly excluded 5% of the
samples [20 repetitions]), the average overall correlation
in ranks due to LD-based binning (0.80) was lower than
that resulting from random fluctuations in the datasets
(>0.87), indicating greater changes due to LD (Table S2).
Such changes in rank are likely to impact threshold-free,
rank-based pathway approaches, such as gene-set-enrich-
ment analysis,34 which aims to determine whether a prede-
fined set of genes is enriched at the top of a ranked list. By
inspecting the top 2,000 genes emerging from the two
binning approaches, we found a 27%–34% difference
between the two gene lists in the three unimputed and
the TOP imputed datasets and a 15.5% difference in the
German imputed dataset. Here, the resampling analysis
in the WTCCC GWAS found that random fluctuations in
Table 3. Effect of LD-Based Binning on Ranks of Genes within EachGWAS
WTCCC TOP GermanTOPImputed
GermanImputed
Correlationa ofgene ranks
0.79 0.83 0.83 0.83 0.92
Number of genesmoving into top 2,000with LD-based binning
681(34.0%)
601(30.0%)
538(26.9%)
558(27.9%)
309(15.5%)
aSpearman rank correlation (i.e., rho).
730 The American Journal of Human Genetics 90, 727–733, April 6, 2012

the dataset led to a 25.6% change in the top 2,000 genes,
whereas LD-based binning resulted in a 30.7% difference
(Table S2). For threshold-based approaches, such as Inge-
nuity Pathway Analysis and ALIGATOR,35 in which a list
of genes meeting a specified threshold is tested for overrep-
resentation of a particular biological function, LD-based
binning could result in the submission of a substantially
different list. Changes in the ranks of the genes are thus
likely to impact the outcome of these analyses and possibly
the overall biological interpretation of the findings. The
extent to which these LD-based changes are meaningful
will also depend on the study design and resulting power,
given that the resampling analysis shows that substantial
changes in results can also occur as a result of slight
changes in the dataset.
Finally, we assessed whether LD-based binning
improved the concordance of results across studies, espe-
cially in light of the aforementioned changes in the ranks
of the genes. We compared the positional-binning and LD-
based-binning approaches by performing pairwise rank-
correlation analyses of the three GWAS datasets at both
the SNP level and the gene level (Table 4). When the posi-
tional-binning approach was used, little to no correlation
was observed at both the SNP and gene levels. However,
with LD-based binning, the overall rank correlation
increased by ~3% and was more significant for all pairwise
comparisons, including the imputed datasets. Interest-
ingly, the greatest concordance was observed when LD-
based binning was combined with imputation, high-
lighting the complementary nature of the two methods.
Although there was no obvious increase in overlap in the
top gene hits (data not shown), this increase in overall
concordance warrants the use of the LD-based-binning
approach for the reanalysis of these and other datasets in
the search for common functional gene sets and pathways.
The observed increase in correlation persisted even when
regions of high LD, such as the MHC (major histocompat-
ibility complex) region on chromosome 6, were excluded
(data not shown).
Our study illustrates the importance of systematically
accounting for LD in the interpretation of GWAS results.
To the best of our knowledge, our study is the first to quan-
tify the added value of LD-based binning; in particular, it
shows an increase in the concordance of results across
independent GWASs of a trait as complex as BP. Excluding
LD defies the basic premise of the GWAS approach by dis-
carding valuable genetic information and risking the
incorrect localization of the association signal and the
misinterpretation of the biology of the findings. Our find-
ings call for a reanalysis of previously published GWAS
data via the LD-based-binning approach and for future
GWASs to adopt this method automatically. LDsnpR facil-
itates this process by efficiently assigning SNPs to genes
and provides the option of scoring the genes for direct
entry into pathway-analysis tools. LDsnpR’s flexible frame-
work allows the application of different gene-scoring
methods; the application of such methods is necessary
for detecting gene-based associations under different
genetic architectures for the traits.31 The user-definable r2
parameter enables the scanning of a greater range of allele
frequencies at the linked locus.27 Bin definitions and pre-
calculated pairwise LD information can be updated on
the basis of the user’s interests and the information avail-
able. LD-based binning might also serve as a complemen-
tary and/or alternative approach to imputation. In partic-
ular, as high-quality LD data from the 1,000 Genomes
Project21 emerges, all GWASs, including those previously
subjected to imputation, might benefit from simple and
efficient LD-based binning at no extra cost. As we show
here, LD-based binning can further enhance imputed
GWASs, albeit to a lesser extent than unimputed datasets.
More tools that allow for incorporation of LD into the
interpretation of GWAS data are emerging,36,37 further
testifying to the importance of this approach. Also, for
studies genotyped on different platforms and/or imputed
with the use of different reference panels, LD-based
binning enables uniform comparison at both the gene
and pathway levels.
It is crucial to note that our study, as well as LDsnpR,
only addresses SNP-to-gene assignment. Issues involving
the derivation of the most accurate gene score (which
accounts for gene size and LD between SNPs), the handling
of SNPs that are assigned tomultiple, possibly overlapping,
genes, and the correlation between genes are unresolved
obstacles for pathway-analysis approaches13 and are
beyond the scope of this paper. Furthermore, the benefits
of LD-based binning will be unique to each GWAS depend-
ing on the trait and its true underlying genetic architec-
ture, the study design, and the extent of SNP coverage.
Supplemental Data
Supplemental Data include one figure and two tables and can be
found with this article online at http://www.cell.com/AJHG.
Table 4. Pairwise Concordance between GWASs at SNP and Gene Levels
WTCCC vs. TOP WTCCC vs. German TOP vs. GermanTOP Imputed vs.German Imputed
SNP level 0.0066 (0.00018) 0.0037 (0.31) �0.0018 (0.51) �0.00023 (0.83)
Gene level (positional binning) 0.030 (1.78 3 10�7) �0.0017 (0.78) 0.023 (4.78 3 10�5) 0.068 (<2.2 3 10�16)
Gene level (LD-based binning) 0.077 (<2.2 3 10�16) 0.027 (7.24 3 10�7) 0.053 (<2.2 3 10�16) 0.098 (<2.2 3 10�16)
The Spearman rank correlation and p value (in parentheses) are shown for each pairwise comparison.
The American Journal of Human Genetics 90, 727–733, April 6, 2012 731

Acknowledgments
We acknowledge Isabel Hanson Scientific Writing for critical help
with the manuscript preparation. This work was supported by
grants from the Bergen Research Foundation, the University of
Bergen, the Research Council of Norway (FUGE, Psyksik Helse,
and eVita), UNI Computing, Western Norway Regional Health
Authority (Helse Vest), the Dr. Einar Martens Fund, South-Eastern
Norway Regional Health Authority (Helse Sør-Øst), the National
Institutes of Health and the National Heart, Lung, and Blood Insti-
tute (U01 HL089856, RO1 MH087590 and R01 MH081862), and
the German Federal Ministry of Education and Research (National
Genome Research Network 2, the National Genome Research
Network plus, and the Integrated Genome Research Network
MooDS [grant 01GS08144 to S.C.]). LDsnpR was developed within
the eSysbio project. We acknowledge Hakon Sagehaug for contrib-
uting Java code and members of the BioStar QA community for
their help and interesting discussions.
Received: September 6, 2011
Revised: February 16, 2012
Accepted: February 27, 2012
Published online: March 22, 2012
Web Resources
The URLs for data presented herein are as follows:
1,000 Genomes Project, http://www.1000genomes.org/
Catalog of Published Genome-Wide Association Studies, www.
genome.gov/gwastudies/
ENIGMAprotocol, http://enigma.loni.ucla.edu/protocols/genetics-
protocols/
HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Human Ensembl Release 54, http://may2009.archive.ensembl.
org/biomart/martview/11839bb5ec82fb10bf0333540fa09c46
IMPUTE2 Software, http://mathgen.stats.ox.ac.uk/impute/
impute_v2.html
Ingenuity Pathway Analysis, http://www.ingenuity.com/
LDsnpR, http://services.cbu.uib.no/software/ldsnpr
PLINK, http://pngu.mgh.harvard.edu/~purcell/plink/
R Archive Network, http://cran.r-project.org
References
1. International HapMap Consortium. (2005). A haplotype map
of the human genome. Nature 437, 1299–1320.
2. Hindorff, L.A., Sethupathy, P., Junkins, H.A., Ramos, E.M.,
Mehta, J.P., Collins, F.S., and Manolio, T.A. (2009). Potential
etiologic and functional implications of genome-wide associa-
tion loci for human diseases and traits. Proc. Natl. Acad. Sci.
USA 106, 9362–9367.
3. Manolio, T.A. (2010). Genomewide association studies and
assessment of the risk of disease. N. Engl. J. Med. 363, 166–176.
4. Bondy, B. (2011). Genetics in psychiatry: Are the promises
met? World J. Biol. Psychiatry 12, 81–88.
5. Gershon, E.S., Alliey-Rodriguez, N., and Liu, C. (2011). After
GWAS: Searching for genetic risk for schizophrenia and
bipolar disorder. Am. J. Psychiatry 168, 253–256.
6. Stranger, B.E., Stahl, E.A., and Raj, T. (2011). Progress and
promise of genome-wide association studies for human
complex trait genetics. Genetics 187, 367–383.
7. Gibson, G. (2010). Hints of hidden heritability in GWAS. Nat.
Genet. 42, 558–560.
8. Davies, G., Tenesa, A., Payton, A., Yang, J., Harris, S.E., Lie-
wald, D., Ke, X., Le Hellard, S., Christoforou, A., Luciano,
M., et al. (2011). Genome-wide association studies establish
that human intelligence is highly heritable and polygenic.
Mol. Psychiatry 16, 996–1005.
9. Lee, S.H., Wray, N.R., Goddard, M.E., and Visscher, P.M.
(2011). Estimating missing heritability for disease from
genome-wide association studies. Am. J. Hum. Genet. 88,
294–305.
10. Cantor, R.M., Lange, K., and Sinsheimer, J.S. (2010). Priori-
tizing GWAS results: A review of statistical methods and
recommendations for their application. Am. J. Hum. Genet.
86, 6–22.
11. McCarthy, M.I., Abecasis, G.R., Cardon, L.R., Goldstein, D.B.,
Little, J., Ioannidis, J.P., and Hirschhorn, J.N. (2008). Genome-
wide association studies for complex traits: Consensus, uncer-
tainty and challenges. Nat. Rev. Genet. 9, 356–369.
12. Bergen, S.E., Balhara, Y.P., Christoforou, A., Cole, J., Degen-
hardt, F., Dempster, E., Fatjo-Vilas, M., Khedr, Y., Lopez,
L.M., Lysenko, L., et al. (2011). Summaries from the XVIII
World Congress of Psychiatric Genetics, Athens, Greece, 3-7
October 2010. Psychiatr. Genet. 21, 136–172.
13. Wang, K., Li, M., and Hakonarson, H. (2010). Analysing bio-
logical pathways in genome-wide association studies. Nat.
Rev. Genet. 11, 843–854.
14. Wang, K., Li, M., and Bucan, M. (2007). Pathway-based
approaches for analysis of genomewide association studies.
Am. J. Hum. Genet. 81, 1278–1283.
15. Neale, B.M., and Sham, P.C. (2004). The future of association
studies: Gene-based analysis and replication. Am. J. Hum.
Genet. 75, 353–362.
16. Hinds, D.A., Stuve, L.L., Nilsen, G.B., Halperin, E., Eskin, E.,
Ballinger, D.G., Frazer, K.A., and Cox, D.R. (2005). Whole-
genome patterns of common DNA variation in three human
populations. Science 307, 1072–1079.
17. Lawrence, R., Evans, D.M., Morris, A.P., Ke, X., Hunt, S., Pao-
lucci, M., Ragoussis, J., Deloukas, P., Bentley, D., and Cardon,
L.R. (2005). Genetically indistinguishable SNPs and their
influence on inferring the location of disease-associated vari-
ants. Genome Res. 15, 1503–1510.
18. Wellcome Trust Case Control Consortium. (2007). Genome-
wide association study of 14,000 cases of seven common
diseases and 3,000 shared controls. Nature 447, 661–678.
19. Djurovic, S., Gustafsson, O., Mattingsdal, M., Athanasiu, L.,
Bjella, T., Tesli, M., Agartz, I., Lorentzen, S., Melle, I., Morken,
G., and Andreassen, O.A. (2010). A genome-wide association
study of bipolar disorder in Norwegian individuals, followed
by replication in Icelandic sample. J. Affect. Disord. 126,
312–316.
20. Cichon, S., Muhleisen, T.W., Degenhardt, F.A., Mattheisen,
M., Miro, X., Strohmaier, J., Steffens, M., Meesters, C., Herms,
S., Weingarten, M., et al; Bipolar Disorder Genome Study
(BiGS) Consortium. (2011). Genome-wide association study
identifies genetic variation in neurocan as a susceptibility
factor for bipolar disorder. Am. J. Hum. Genet. 88, 372–381.
21. 1000 Genomes Consortium. (2010). A map of human genome
variation from population-scale sequencing. Nature 467,
1061–1073.
22. So, H.C., Gui, A.H., Cherny, S.S., and Sham, P.C. (2011). Eval-
uating the heritability explained by known susceptibility vari-
ants: A survey of ten complex diseases. Genet. Epidemiol. 35,
310–317.
732 The American Journal of Human Genetics 90, 727–733, April 6, 2012

23. Neale, B.M., and Purcell, S. (2008). The positives, protocols,
and perils of genome-wide association. Am. J. Med. Genet.
B. Neuropsychiatr. Genet. 147B, 1288–1294.
24. Blow, M.J., McCulley, D.J., Li, Z., Zhang, T., Akiyama, J.A.,
Holt, A., Plajzer-Frick, I., Shoukry, M., Wright, C., Chen, F.,
et al. (2010). ChIP-Seq identification of weakly conserved
heart enhancers. Nat. Genet. 42, 806–810.
25. Vandiedonck, C., Taylor, M.S., Lockstone, H.E., Plant, K., Tay-
lor, J.M., Durrant, C., Broxholme, J., Fairfax, B.P., and Knight,
J.C. (2011). Pervasive haplotypic variation in the spliceo-tran-
scriptome of the human major histocompatibility complex.
Genome Res. 21, 1042–1054.
26. Spencer, C.C., Su, Z., Donnelly, P., and Marchini, J. (2009).
Designing genome-wide association studies: Sample size,
power, imputation, and the choice of genotyping chip. PLoS
Genet. 5, e1000477.
27. Wray, N.R. (2005). Allele frequencies and the r2 measure of
linkage disequilibrium: Impact on design and interpretation
of association studies. Twin Res. Hum. Genet. 8, 87–94.
28. Santos, P.S., Hohne, J., Poerner, F., da Graca Bicalho, M.,
Uchanska-Ziegler, B., and Ziegler, A. (2011). Does the new
HapMap throw the baby out with the bath water? Eur. J.
Hum. Genet. 19, 733–734.
29. Ioannidis, J.P., Thomas, G., and Daly, M.J. (2009). Validating,
augmenting and refining genome-wide association signals.
Nat. Rev. Genet. 10, 318–329.
30. Saccone, S.F., Hinrichs, A.L., Saccone, N.L., Chase, G.A., Kon-
vicka, K., Madden, P.A., Breslau, N., Johnson, E.O., Hatsukami,
D., Pomerleau, O., et al. (2007). Cholinergic nicotinic receptor
genes implicated in a nicotine dependence association study
targeting 348 candidate genes with 3713 SNPs. Hum. Mol.
Genet. 16, 36–49.
31. Lehne, B., Lewis, C.M., and Schlitt, T. (2011). From SNPs to
genes: disease association at the gene level. PLoS ONE 6,
e20133.
32. Segre, A.V., Groop, L., Mootha, V.K., Daly, M.J., and Altshuler,
D.; DIAGRAM Consortium; MAGIC investigators. (2010).
Common inherited variation in mitochondrial genes is not
enriched for associations with type 2 diabetes or related glyce-
mic traits. PLoS Genet. 6, e1001058.
33. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira,
M.A., Bender, D., Maller, J., Sklar, P., de Bakker, P.I., Daly,
M.J., and Sham, P.C. (2007). PLINK: A tool set for whole-
genome association and population-based linkage analyses.
Am. J. Hum. Genet. 81, 559–575.
34. Subramanian, A., Tamayo, P., Mootha, V.K., Mukherjee, S.,
Ebert, B.L., Gillette, M.A., Paulovich, A., Pomeroy, S.L., Golub,
T.R., Lander, E.S., and Mesirov, J.P. (2005). Gene set enrich-
ment analysis: A knowledge-based approach for interpreting
genome-wide expression profiles. Proc. Natl. Acad. Sci. USA
102, 15545–15550.
35. Holmans, P., Green, E.K., Pahwa, J.S., Ferreira, M.A., Purcell,
S.M., Sklar, P., Owen, M.J., O’Donovan, M.C., and Craddock,
N.; Wellcome Trust Case-Control Consortium. (2009). Gene
ontology analysis of GWA study data sets provides insights
into the biology of bipolar disorder. Am. J. Hum. Genet. 85,
13–24.
36. Hong, M.G., Pawitan, Y., Magnusson, P.K., and Prince, J.A.
(2009). Strategies and issues in the detection of pathway
enrichment in genome-wide association studies. Hum. Genet.
126, 289–301.
37. Zhang, K., Chang, S., Cui, S., Guo, L., Zhang, L., and Wang, J.
(2011). ICSNPathway: Identify candidate causal SNPs and
pathways from genome-wide association study by one analyt-
ical framework. Nucleic Acids Res. 39 (Web Server issue),
W437–443.
38. Li, Y., Willer, C.J., Ding, J., Scheet, P., and Abecasis, G.R.
(2010). MaCH: Using sequence and genotype data to estimate
haplotypes and unobserved genotypes. Genet. Epidemiol. 34,
816–834.
39. Howie, B.N., Donnelly, P., andMarchini, J.A. (2009). A flexible
and accurate genotype imputationmethod for the next gener-
ation of genome-wide association studies. PLoS Genet. 5,
e1000529.
The American Journal of Human Genetics 90, 727–733, April 6, 2012 733

REPORT
Rare Mutations in XRCC2 Increasethe Risk of Breast Cancer
D.J. Park,1,20 F. Lesueur,2,20 T. Nguyen-Dumont,1 M. Pertesi,2 F. Odefrey,1 F. Hammet,1 S.L. Neuhausen,3
E.M. John,4,5 I.L. Andrulis,6 M.B. Terry,7 M. Daly,8 S. Buys,9 F. Le Calvez-Kelm,2 A. Lonie,10 B.J. Pope,10
H. Tsimiklis,1 C. Voegele,2 F.M. Hilbers,11 N. Hoogerbrugge,12 A. Barroso,13 A. Osorio,13,14 the BreastCancer Family Registry, the Kathleen Cuningham Foundation Consortium for Research into FamilialBreast Cancer, G.G. Giles,15 P. Devilee,11,16 J. Benitez,13,14 J.L. Hopper,17 S.V. Tavtigian,18 D.E. Goldgar,19
and M.C. Southey1,*
An exome-sequencing study of families with multiple breast-cancer-affected individuals identified two families with XRCC2mutations,
one with a protein-truncatingmutation and one with a probably deleterious missensemutation.We performed a population-based case-
control mutation-screening study that identified six probably pathogenic coding variants in 1,308 cases with early-onset breast cancer
and no variants in 1,120 controls (the severity grading was p< 0.02). We also performed additional mutation screening in 689 multiple-
case families. We identified ten breast-cancer-affected families with protein-truncating or probably deleterious rare missense variants in
XRCC2. Our identification of XRCC2 as a breast cancer susceptibility gene thus increases the proportion of breast cancers that are asso-
ciated with homologous recombination-DNA-repair dysfunction and Fanconi anemia and could therefore benefit from specific targeted
treatments such as PARP (poly ADP ribose polymerase) inhibitors. This study demonstrates the power of massively parallel sequencing
for discovering susceptibility genes for common, complex diseases.
Currently, only approximately 30% of the familial risk for
breast cancer has been explained, leaving the substantial
majority unaccounted for.1 Recently, exome sequencing
has been demonstrated to be a powerful tool for identi-
fying the underlying cause of rare Mendelian disorders.
However, diseases such as breast cancer present substan-
tially increased complexity in terms of locus, allelic and
phenotypic heterogeneity, and relationships between
genotype and phenotype.
As part of a collaborative (Leiden University Medical
Centre, the Spanish National Cancer Center, and The
University of Melbourne) project involving the exome
capture and massively parallel sequencing of multiple-
case breast-cancer-affected families, we applied whole-
exome sequencing to DNA frommultiple affected relatives
from 13 families (family structure and sample availability
were considered before the affected relatives were chosen).
Bioinformatic analysis of the resulting exome sequences
identified a protein-truncating mutation, c.651_652del
(p.Cys217*), in X-ray repair cross complementing gene-2
(XRCC2 [MIM 600375; NM_005431.1]) in the peripheral-
blood DNA of a man participating in the Australian Breast
Cancer Family Registry2 (ABCFR; Figure 1A); this man (III-4
in Figure 1A) had been diagnosed with breast cancer at
29 years of age, and his mother (II-3), sister (III-5), and
cousin (III-1) had been diagnosed with breast cancer at
37, 41, and 34 years of age, respectively. The cousin
(III-1), who had also been selected for exome sequencing,
did not carry this mutation, the sister’s DNA was Sanger
sequenced and was found to carry the mutation, and there
was no DNA available for testing of the mother. Exome
sequencing of three individuals from a family participating
in a Dutch research study of multiple-case breast-cancer-
affected families identified a probably deleterious missense
mutation (c.271C>T [p.Arg91Trp] in XRCC2) (Figure 2) in
two sisters (II-6 and II-8 in Figure 1B) diagnosed with breast
cancer at 40 and 48 years of age, respectively, but not in
their cousin (II-1), who was diagnosed at 47 years of age.
Genotyping of XRCC2 mutations c.651_652del
(p.Cys217*) and c.271C>T (p.Arg91Trp) in 1,344 cases
1Genetic Epidemiology Laboratory, The University of Melbourne, Victoria 3010, Australia; 2Genetic Cancer Susceptibility Group, International Agency for
Research on Cancer, 69372 Lyon, France; 3Department of Population Sciences, Beckman Research Institute of City of Hope, Duarte, CA 91010, USA;4Cancer Prevention Institute of California, Fremont, CA 94538, USA; 5Department of Health Research and Policy, Stanford Cancer Center Institute, Stan-
ford, CA 94305, USA; 6Department of Molecular Genetics, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, ON M5G 1X5, Canada;7Department of Epidemiology, Mailman School of Public Health, Columbia University, New York, NY 10032, USA; 8Fox Chase Cancer Center, Philadelphia,
PA 19111, USA; 9Huntsman Cancer Institute, University of Utah Health Sciences Center, Salt Lake City, UT 84112, USA; 10Victorian Life Sciences Compu-
tation Initiative, Carlton, Victoria 3010, Australia; 11Department of Human Genetics, Leiden University Medical Center, Leiden, 2300 RC Leiden, The
Netherlands; 12Department of Human Genetics, Radboud University Nijmegen Medical Center, 6525 GA Nijmegen, The Netherlands; 13Human Genetics
Group, Human Cancer Genetics Program, Spanish National Cancer Center, 28029 Madrid, Spain; 14Spanish Network on Rare Diseases, 46010 Valencia,
Spain; 15Centre for Cancer Epidemiology, The Cancer Council Victoria, Carlton, Victoria 3052, Australia; 16Department of Pathology, Leiden University
Medical Center, Leiden, 2300 RC Leiden, The Netherlands; 17Centre for Molecular, Environmental, Genetic, and Analytical Epidemiology, School of Pop-
ulation Health, The University of Melbourne, Victoria 3010, Australia; 18Department of Oncological Sciences, Huntsman Cancer Institute, University of
Utah School of Medicine, Salt Lake City, UT 84112, USA; 19Department of Dermatology, University of Utah School of Medicine, Salt Lake City, UT
84132, USA20These authors contributed equally to this work
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.02.027. �2012 by The American Society of Human Genetics. All rights reserved.
734 The American Journal of Human Genetics 90, 734–739, April 6, 2012

and 1,436 controls from the Melbourne Collaborative
Cohort Study3 (MCCS) and the ABCFR revealed one
control (II-2, Figure 1C) who carried c.651_652del
(p.Cys217*). Intriguingly, this control individual’s sister
(II-1) was diagnosed with breast cancer at 63 years of age,
and her mother (I-2) was diagnosed with melanoma at
69 years of age (Figure 1C, Tables 1 and 2).
XRCC2, a RAD51 paralog, was cloned because of its
ability to complement the DNA-damage sensitivity of the
irs1 hamster cell line.4 Cells derived from Xrcc2-knockout
mice exhibit profound genetic instability as a result of
homologous recombination (HR) deficiency.5 XRCC2 is
highly conserved, and most truncations of the protein
destroy its ability to protect cells from the effects of the
DNA cross-linking agent mitomycin C.6 The involvement
of the HR DNA repair genes BRCA1 (MIM 113705),
BRCA2 (MIM 600185), ATM (MIM 607585), CHEK2 (MIM
604373), BRIP1 (MIM 605882), PALB2 (MIM 610355),
and RAD51C (MIM 602774) in breast cancer risk empha-
sizes the importance of this mechanism in the etiology
of breast cancer.7–9 Biallelic mutations in three of these
genes are associated with Fanconi anemia (FA), and, most
interestingly, Shamseldin et al.10 have recently reported
a homozygous frameshift mutation in XRCC2 as being
associated with a previously unrecognized form of FA.
XRCC2 binds directly to the C-terminal portion of the
product of the breast cancer susceptibility pathway gene
RAD51 (MIM 179617), which is central to HR.6,11 XRCC2
also complexes in vivo with RAD51B (RAD51L1 [MIM
602948]), the product of the breast and ovarian cancer
susceptibility gene RAD51C9 and the product of the
ovarian cancer risk gene RAD51D (MIM 602954),12,13 and
localizes to sites of DNA damage.6 Cells deficient in
XRCC2 also show centrosome disruption, a key compo-
nent of mitotic-apparatus dysfunction, which is often
linked to the onset of mitotic catastrophe. XRCC2 is
important in preventing chromosome missegregation
leading to aneuploidy.14 Studies of common genetic varia-
tion in XRCC2 have reported some evidence of association
with breast cancer risk (e.g., rs3218408),15 subtle effects on
DNA-repair capacity,16 and poor survival associated with
rs3218536 (XRCC2, Arg188His).15
On the basis of the exome-sequencing results, the subse-
quent genotyping of the two probably pathogenic variants
*
*
** *
*
A B
C D
EF
G H IJ
Figure 1. Pedigrees of Families Found to Carry XRCC2 MutationsMutation status is indicated for all family members for whom a DNA sample was available. Cancer diagnosis and age of onset are indi-cated for affected members. Asterisks indicate that DNA underwent exome sequencing (libraries for 50 bp fragment reads were preparedaccording to the SOLiD Baylor protocol 2.1 and the Nimblegen exome-capture protocol v.1.2 with some variations). The followingabbreviations are used: BC, breast cancer (black filled symbols); PC, pancreatic cancer; BwC, bowel cancer; UC, uterine cancer; MM,malignant melanoma; UK, unknown age; BlC, bladder cancer; OC, ovarian cancer; BCC, basal cell carcinoma; L, lung cancer; (allgray-filled symbols); V, verified cancer (via cancer registry or pathology report); and wt, wild-type. Some symbols represent more thanone person as indicated by a numeral.
The American Journal of Human Genetics 90, 734–739, April 6, 2012 735
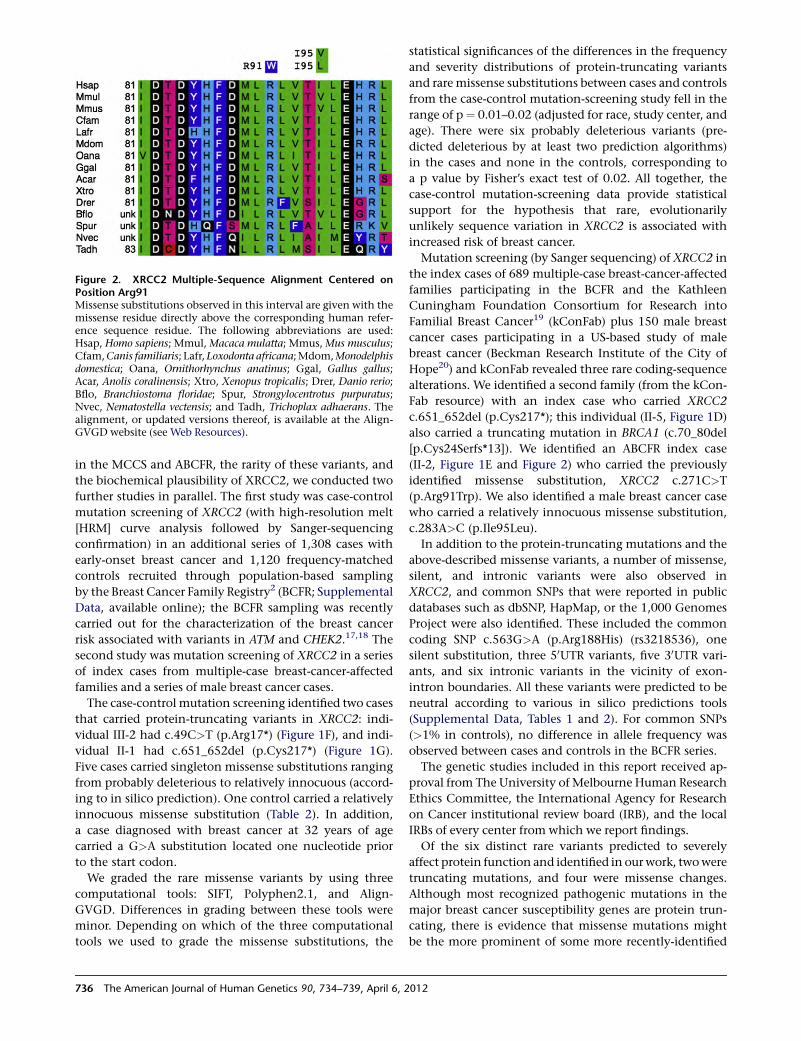
in the MCCS and ABCFR, the rarity of these variants, and
the biochemical plausibility of XRCC2, we conducted two
further studies in parallel. The first study was case-control
mutation screening of XRCC2 (with high-resolution melt
[HRM] curve analysis followed by Sanger-sequencing
confirmation) in an additional series of 1,308 cases with
early-onset breast cancer and 1,120 frequency-matched
controls recruited through population-based sampling
by the Breast Cancer Family Registry2 (BCFR; Supplemental
Data, available online); the BCFR sampling was recently
carried out for the characterization of the breast cancer
risk associated with variants in ATM and CHEK2.17,18 The
second study was mutation screening of XRCC2 in a series
of index cases from multiple-case breast-cancer-affected
families and a series of male breast cancer cases.
The case-control mutation screening identified two cases
that carried protein-truncating variants in XRCC2: indi-
vidual III-2 had c.49C>T (p.Arg17*) (Figure 1F), and indi-
vidual II-1 had c.651_652del (p.Cys217*) (Figure 1G).
Five cases carried singleton missense substitutions ranging
from probably deleterious to relatively innocuous (accord-
ing to in silico prediction). One control carried a relatively
innocuous missense substitution (Table 2). In addition,
a case diagnosed with breast cancer at 32 years of age
carried a G>A substitution located one nucleotide prior
to the start codon.
We graded the rare missense variants by using three
computational tools: SIFT, Polyphen2.1, and Align-
GVGD. Differences in grading between these tools were
minor. Depending on which of the three computational
tools we used to grade the missense substitutions, the
statistical significances of the differences in the frequency
and severity distributions of protein-truncating variants
and rare missense substitutions between cases and controls
from the case-control mutation-screening study fell in the
range of p¼ 0.01–0.02 (adjusted for race, study center, and
age). There were six probably deleterious variants (pre-
dicted deleterious by at least two prediction algorithms)
in the cases and none in the controls, corresponding to
a p value by Fisher’s exact test of 0.02. All together, the
case-control mutation-screening data provide statistical
support for the hypothesis that rare, evolutionarily
unlikely sequence variation in XRCC2 is associated with
increased risk of breast cancer.
Mutation screening (by Sanger sequencing) of XRCC2 in
the index cases of 689 multiple-case breast-cancer-affected
families participating in the BCFR and the Kathleen
Cuningham Foundation Consortium for Research into
Familial Breast Cancer19 (kConFab) plus 150 male breast
cancer cases participating in a US-based study of male
breast cancer (Beckman Research Institute of the City of
Hope20) and kConFab revealed three rare coding-sequence
alterations. We identified a second family (from the kCon-
Fab resource) with an index case who carried XRCC2
c.651_652del (p.Cys217*); this individual (II-5, Figure 1D)
also carried a truncating mutation in BRCA1 (c.70_80del
[p.Cys24Serfs*13]). We identified an ABCFR index case
(II-2, Figure 1E and Figure 2) who carried the previously
identified missense substitution, XRCC2 c.271C>T
(p.Arg91Trp). We also identified a male breast cancer case
who carried a relatively innocuous missense substitution,
c.283A>C (p.Ile95Leu).
In addition to the protein-truncating mutations and the
above-described missense variants, a number of missense,
silent, and intronic variants were also observed in
XRCC2, and common SNPs that were reported in public
databases such as dbSNP, HapMap, or the 1,000 Genomes
Project were also identified. These included the common
coding SNP c.563G>A (p.Arg188His) (rs3218536), one
silent substitution, three 50UTR variants, five 30UTR vari-
ants, and six intronic variants in the vicinity of exon-
intron boundaries. All these variants were predicted to be
neutral according to various in silico predictions tools
(Supplemental Data, Tables 1 and 2). For common SNPs
(>1% in controls), no difference in allele frequency was
observed between cases and controls in the BCFR series.
The genetic studies included in this report received ap-
proval from The University of Melbourne Human Research
Ethics Committee, the International Agency for Research
on Cancer institutional review board (IRB), and the local
IRBs of every center from which we report findings.
Of the six distinct rare variants predicted to severely
affect protein function and identified in ourwork, twowere
truncating mutations, and four were missense changes.
Although most recognized pathogenic mutations in the
major breast cancer susceptibility genes are protein trun-
cating, there is evidence that missense mutations might
be the more prominent of some more recently-identified
Figure 2. XRCC2 Multiple-Sequence Alignment Centered onPosition Arg91Missense substitutions observed in this interval are given with themissense residue directly above the corresponding human refer-ence sequence residue. The following abbreviations are used:Hsap, Homo sapiens; Mmul, Macaca mulatta; Mmus,Mus musculus;Cfam,Canis familiaris; Lafr,Loxodonta africana;Mdom,Monodelphisdomestica; Oana, Ornithorhynchus anatinus; Ggal, Gallus gallus;Acar, Anolis coralinensis; Xtro, Xenopus tropicalis; Drer, Danio rerio;Bflo, Branchiostoma floridae; Spur, Strongylocentrotus purpuratus;Nvec, Nematostella vectensis; and Tadh, Trichoplax adhaerans. Thealignment, or updated versions thereof, is available at the Align-GVGD website (see Web Resources).
736 The American Journal of Human Genetics 90, 734–739, April 6, 2012
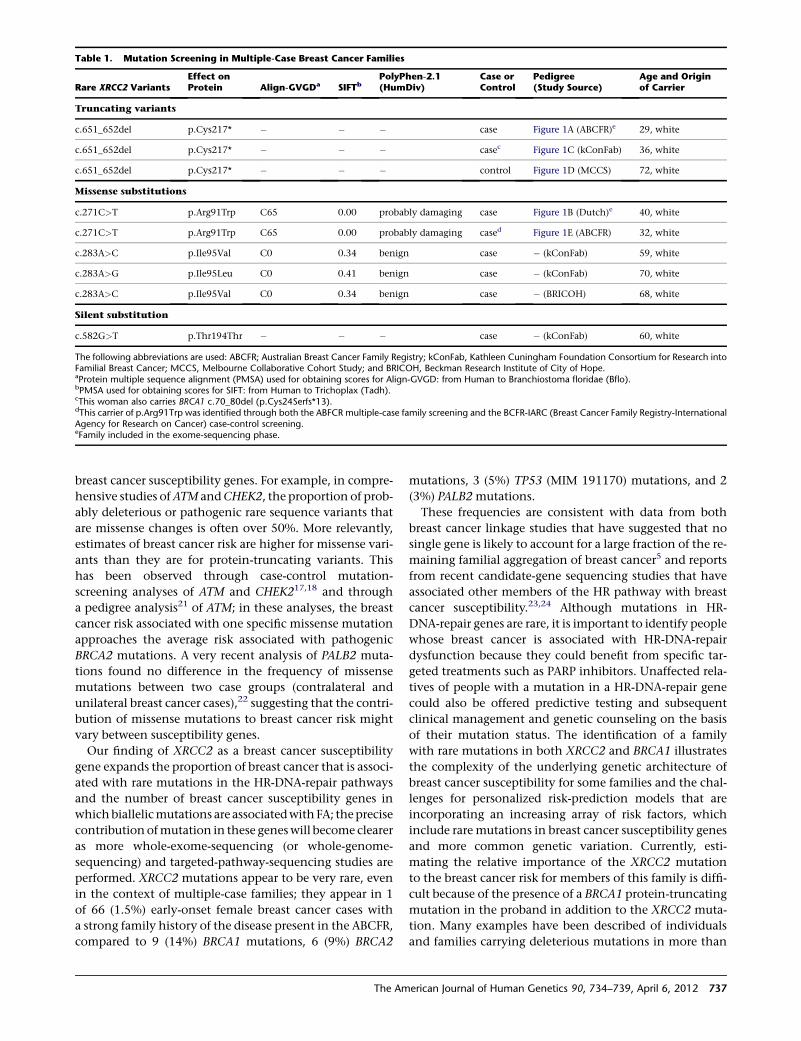
breast cancer susceptibility genes. For example, in compre-
hensive studies ofATM andCHEK2, the proportion of prob-
ably deleterious or pathogenic rare sequence variants that
are missense changes is often over 50%. More relevantly,
estimates of breast cancer risk are higher for missense vari-
ants than they are for protein-truncating variants. This
has been observed through case-control mutation-
screening analyses of ATM and CHEK217,18 and through
a pedigree analysis21 of ATM; in these analyses, the breast
cancer risk associated with one specific missense mutation
approaches the average risk associated with pathogenic
BRCA2 mutations. A very recent analysis of PALB2 muta-
tions found no difference in the frequency of missense
mutations between two case groups (contralateral and
unilateral breast cancer cases),22 suggesting that the contri-
bution of missense mutations to breast cancer risk might
vary between susceptibility genes.
Our finding of XRCC2 as a breast cancer susceptibility
gene expands the proportion of breast cancer that is associ-
ated with rare mutations in the HR-DNA-repair pathways
and the number of breast cancer susceptibility genes in
whichbiallelicmutations are associatedwith FA; theprecise
contribution ofmutation in these geneswill become clearer
as more whole-exome-sequencing (or whole-genome-
sequencing) and targeted-pathway-sequencing studies are
performed. XRCC2 mutations appear to be very rare, even
in the context of multiple-case families; they appear in 1
of 66 (1.5%) early-onset female breast cancer cases with
a strong family history of the disease present in the ABCFR,
compared to 9 (14%) BRCA1 mutations, 6 (9%) BRCA2
mutations, 3 (5%) TP53 (MIM 191170) mutations, and 2
(3%) PALB2mutations.
These frequencies are consistent with data from both
breast cancer linkage studies that have suggested that no
single gene is likely to account for a large fraction of the re-
maining familial aggregation of breast cancer5 and reports
from recent candidate-gene sequencing studies that have
associated other members of the HR pathway with breast
cancer susceptibility.23,24 Although mutations in HR-
DNA-repair genes are rare, it is important to identify people
whose breast cancer is associated with HR-DNA-repair
dysfunction because they could benefit from specific tar-
geted treatments such as PARP inhibitors. Unaffected rela-
tives of people with a mutation in a HR-DNA-repair gene
could also be offered predictive testing and subsequent
clinical management and genetic counseling on the basis
of their mutation status. The identification of a family
with rare mutations in both XRCC2 and BRCA1 illustrates
the complexity of the underlying genetic architecture of
breast cancer susceptibility for some families and the chal-
lenges for personalized risk-prediction models that are
incorporating an increasing array of risk factors, which
include rare mutations in breast cancer susceptibility genes
and more common genetic variation. Currently, esti-
mating the relative importance of the XRCC2 mutation
to the breast cancer risk for members of this family is diffi-
cult because of the presence of a BRCA1 protein-truncating
mutation in the proband in addition to the XRCC2 muta-
tion. Many examples have been described of individuals
and families carrying deleterious mutations in more than
Table 1. Mutation Screening in Multiple-Case Breast Cancer Families
Rare XRCC2 VariantsEffect onProtein Align-GVGDa SIFTb
PolyPhen-2.1(HumDiv)
Case orControl
Pedigree(Study Source)
Age and Originof Carrier
Truncating variants
c.651_652del p.Cys217* � � � case Figure 1A (ABCFR)e 29, white
c.651_652del p.Cys217* � � � casec Figure 1C (kConFab) 36, white
c.651_652del p.Cys217* � � � control Figure 1D (MCCS) 72, white
Missense substitutions
c.271C>T p.Arg91Trp C65 0.00 probably damaging case Figure 1B (Dutch)e 40, white
c.271C>T p.Arg91Trp C65 0.00 probably damaging cased Figure 1E (ABCFR) 32, white
c.283A>C p.Ile95Val C0 0.34 benign case � (kConFab) 59, white
c.283A>G p.Ile95Leu C0 0.41 benign case � (kConFab) 70, white
c.283A>C p.Ile95Val C0 0.34 benign case � (BRICOH) 68, white
Silent substitution
c.582G>T p.Thr194Thr � � � case � (kConFab) 60, white
The following abbreviations are used: ABCFR; Australian Breast Cancer Family Registry; kConFab, Kathleen Cuningham Foundation Consortium for Research intoFamilial Breast Cancer; MCCS, Melbourne Collaborative Cohort Study; and BRICOH, Beckman Research Institute of City of Hope.aProtein multiple sequence alignment (PMSA) used for obtaining scores for Align-GVGD: from Human to Branchiostoma floridae (Bflo).bPMSA used for obtaining scores for SIFT: from Human to Trichoplax (Tadh).cThis woman also carries BRCA1 c.70_80del (p.Cys24Serfs*13).dThis carrier of p.Arg91Trp was identified through both the ABFCR multiple-case family screening and the BCFR-IARC (Breast Cancer Family Registry-InternationalAgency for Research on Cancer) case-control screening.eFamily included in the exome-sequencing phase.
The American Journal of Human Genetics 90, 734–739, April 6, 2012 737
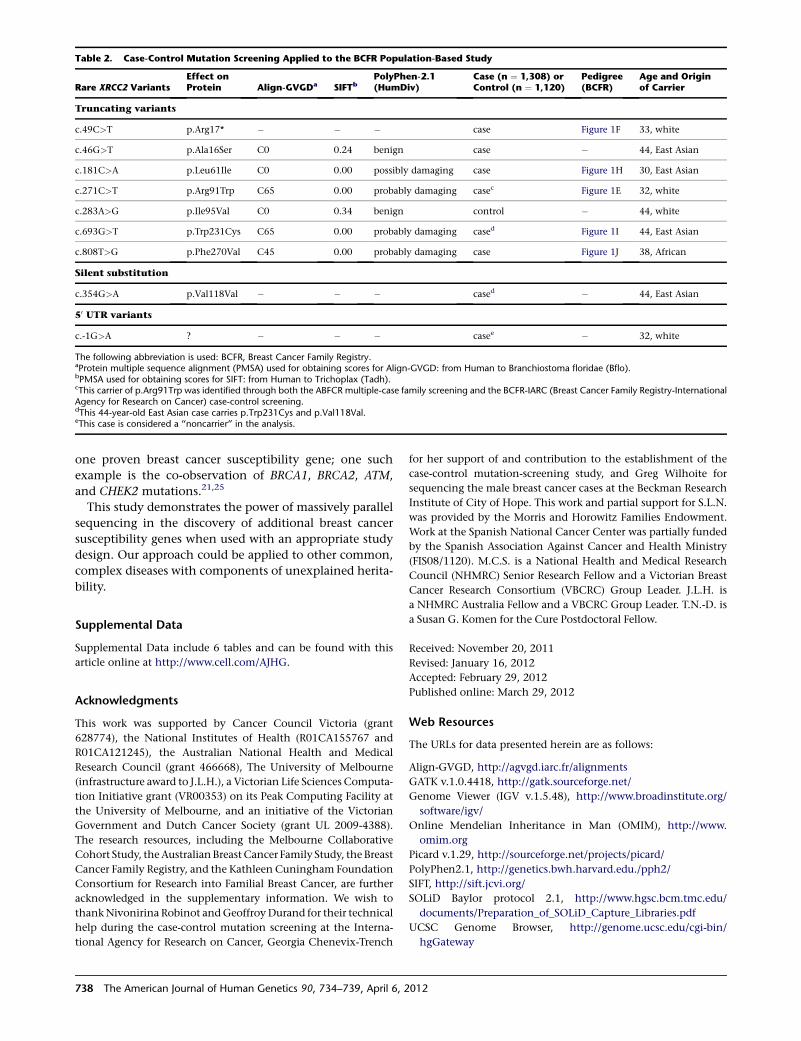
one proven breast cancer susceptibility gene; one such
example is the co-observation of BRCA1, BRCA2, ATM,
and CHEK2 mutations.21,25
This study demonstrates the power of massively parallel
sequencing in the discovery of additional breast cancer
susceptibility genes when used with an appropriate study
design. Our approach could be applied to other common,
complex diseases with components of unexplained herita-
bility.
Supplemental Data
Supplemental Data include 6 tables and can be found with this
article online at http://www.cell.com/AJHG.
Acknowledgments
This work was supported by Cancer Council Victoria (grant
628774), the National Institutes of Health (R01CA155767 and
R01CA121245), the Australian National Health and Medical
Research Council (grant 466668), The University of Melbourne
(infrastructure award to J.L.H.), a Victorian Life Sciences Computa-
tion Initiative grant (VR00353) on its Peak Computing Facility at
the University of Melbourne, and an initiative of the Victorian
Government and Dutch Cancer Society (grant UL 2009-4388).
The research resources, including the Melbourne Collaborative
Cohort Study, theAustralianBreast Cancer Family Study, the Breast
Cancer Family Registry, and the Kathleen Cuningham Foundation
Consortium for Research into Familial Breast Cancer, are further
acknowledged in the supplementary information. We wish to
thankNivonirina Robinot andGeoffroyDurand for their technical
help during the case-control mutation screening at the Interna-
tional Agency for Research on Cancer, Georgia Chenevix-Trench
for her support of and contribution to the establishment of the
case-control mutation-screening study, and Greg Wilhoite for
sequencing the male breast cancer cases at the Beckman Research
Institute of City of Hope. This work and partial support for S.L.N.
was provided by the Morris and Horowitz Families Endowment.
Work at the Spanish National Cancer Center was partially funded
by the Spanish Association Against Cancer and Health Ministry
(FIS08/1120). M.C.S. is a National Health and Medical Research
Council (NHMRC) Senior Research Fellow and a Victorian Breast
Cancer Research Consortium (VBCRC) Group Leader. J.L.H. is
a NHMRC Australia Fellow and a VBCRC Group Leader. T.N.-D. is
a Susan G. Komen for the Cure Postdoctoral Fellow.
Received: November 20, 2011
Revised: January 16, 2012
Accepted: February 29, 2012
Published online: March 29, 2012
Web Resources
The URLs for data presented herein are as follows:
Align-GVGD, http://agvgd.iarc.fr/alignments
GATK v.1.0.4418, http://gatk.sourceforge.net/
Genome Viewer (IGV v.1.5.48), http://www.broadinstitute.org/
software/igv/
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
Picard v.1.29, http://sourceforge.net/projects/picard/
PolyPhen2.1, http://genetics.bwh.harvard.edu./pph2/
SIFT, http://sift.jcvi.org/
SOLiD Baylor protocol 2.1, http://www.hgsc.bcm.tmc.edu/
documents/Preparation_of_SOLiD_Capture_Libraries.pdf
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/
hgGateway
Table 2. Case-Control Mutation Screening Applied to the BCFR Population-Based Study
Rare XRCC2 VariantsEffect onProtein Align-GVGDa SIFTb
PolyPhen-2.1(HumDiv)
Case (n ¼ 1,308) orControl (n ¼ 1,120)
Pedigree(BCFR)
Age and Originof Carrier
Truncating variants
c.49C>T p.Arg17* � � � case Figure 1F 33, white
c.46G>T p.Ala16Ser C0 0.24 benign case � 44, East Asian
c.181C>A p.Leu61Ile C0 0.00 possibly damaging case Figure 1H 30, East Asian
c.271C>T p.Arg91Trp C65 0.00 probably damaging casec Figure 1E 32, white
c.283A>G p.Ile95Val C0 0.34 benign control � 44, white
c.693G>T p.Trp231Cys C65 0.00 probably damaging cased Figure 1I 44, East Asian
c.808T>G p.Phe270Val C45 0.00 probably damaging case Figure 1J 38, African
Silent substitution
c.354G>A p.Val118Val � � � cased � 44, East Asian
50 UTR variants
c.-1G>A ? � � � casee � 32, white
The following abbreviation is used: BCFR, Breast Cancer Family Registry.aProtein multiple sequence alignment (PMSA) used for obtaining scores for Align-GVGD: from Human to Branchiostoma floridae (Bflo).bPMSA used for obtaining scores for SIFT: from Human to Trichoplax (Tadh).cThis carrier of p.Arg91Trp was identified through both the ABFCR multiple-case family screening and the BCFR-IARC (Breast Cancer Family Registry-InternationalAgency for Research on Cancer) case-control screening.dThis 44-year-old East Asian case carries p.Trp231Cys and p.Val118Val.eThis case is considered a ‘‘noncarrier’’ in the analysis.
738 The American Journal of Human Genetics 90, 734–739, April 6, 2012

References
1. Turnbull, C., and Rahman, N. (2008). Genetic predisposition
to breast cancer: Past, present, and future. Annu. Rev. Geno-
mics Hum. Genet. 9, 321–345.
2. John, E.M., Hopper, J.L., Beck, J.C., Knight, J.A., Neuhausen,
S.L., Senie, R.T., Ziogas, A., Andrulis, I.L., Anton-Culver, H.,
Boyd, N., et al; Breast Cancer Family Registry. (2004). The
Breast Cancer Family Registry: An infrastructure for coopera-
tive multinational, interdisciplinary and translational studies
of the genetic epidemiology of breast cancer. Breast Cancer
Res. 6, R375–R389.
3. Giles, G.G., and R, E.D. (2002). The Melbourne Collaborative
Cohort Study. IARC Sci Publ 156, 2.
4. Cartwright, R., Tambini, C.E., Simpson, P.J., and Thacker, J.
(1998). The XRCC2 DNA repair gene from human and mouse
encodes a novel member of the recA/RAD51 family. Nucleic
Acids Res. 26, 3084–3089.
5. Deans, B., Griffin, C.S., O’Regan, P., Jasin, M., and Thacker, J.
(2003). Homologous recombination deficiency leads to
profound genetic instability in cells derived from Xrcc2-
knockout mice. Cancer Res. 63, 8181–8187.
6. Tambini, C.E., Spink, K.G., Ross, C.J., Hill, M.A., and Thacker,
J. (2010). The importance of XRCC2 in RAD51-related DNA
damage repair. DNA Repair (Amst.) 9, 517–525.
7. Moynahan,M.E., Chiu, J.W., Koller, B.H., and Jasin,M. (1999).
Brca1 controls homology-directed DNA repair. Mol. Cell 4,
511–518.
8. Moynahan, M.E., Pierce, A.J., and Jasin, M. (2001). BRCA2 is
required for homology-directed repair of chromosomal breaks.
Mol. Cell 7, 263–272.
9. Meindl, A., Hellebrand, H., Wiek, C., Erven, V., Wappensch-
midt, B., Niederacher, D., Freund, M., Lichtner, P., Hartmann,
L., Schaal, H., et al. (2010). Germline mutations in breast and
ovarian cancer pedigrees establish RAD51C as a human cancer
susceptibility gene. Nat. Genet. 42, 410–414.
10. Shamseldin, H.E., Elfaki, M., and Alkuraya, F.S. (2012). Exome
sequencing reveals a novel Fanconi group defined by XRCC2
mutation. J. Med. Genet. 49, 184–186.
11. Gao, L.-B., Pan, X.-M., Li, L.-J., Liang, W.-B., Zhu, Y., Zhang,
L.-S., Wei, Y.-G., Tang, M., and Zhang, L. (2011). RAD51
135G/C polymorphism and breast cancer risk: Ameta-analysis
from 21 studies. Breast Cancer Res. Treat. 125, 827–835.
12. Loveday, C., Turnbull, C., Ramsay, E., Hughes, D., Ruark, E.,
Frankum, J.R., Bowden, G., Kalmyrzaev, B., Warren-Perry,
M., Snape, K., et al; Breast Cancer Susceptibility Collaboration
(UK). (2011). Germlinemutations in RAD51D confer suscepti-
bility to ovarian cancer. Nat. Genet. 43, 879–882.
13. Liu, N., Schild, D., Thelen, M.P., and Thompson, L.H. (2002).
Involvement of Rad51C in two distinct protein complexes
of Rad51 paralogs in human cells. Nucleic Acids Res. 30,
1009–1015.
14. Griffin, C.S., Simpson, P.J., Wilson, C.R., and Thacker, J.
(2000). Mammalian recombination-repair genes XRCC2 and
XRCC3 promote correct chromosome segregation. Nat. Cell
Biol. 2, 757–761.
15. Lin,W.-Y., Camp, N.J., Cannon-Albright, L.A., Allen-Brady, K.,
Balasubramanian, S., Reed, M.W.R., Hopper, J.L., Apicella, C.,
Giles, G.G., Southey, M.C., et al. (2011). A role for XRCC2
gene polymorphisms in breast cancer risk and survival. J.
Med. Genet. 48, 477–484.
16. Rafii, S., O’Regan, P., Xinarianos, G., Azmy, I., Stephenson, T.,
Reed, M., Meuth, M., Thacker, J., and Cox, A. (2002). A poten-
tial role for the XRCC2 R188H polymorphic site in DNA-
damage repair and breast cancer. Hum. Mol. Genet. 11,
1433–1438.
17. Le Calvez-Kelm, F., Lesueur, F., Damiola, F., Vallee, M.,
Voegele, C., Babikyan, D., Durand, G., Forey, N., McKay-
Chopin, S., Robinot, N., et al; Breast Cancer Family Registry.
(2011). Rare, evolutionarily unlikely missense substitutions
in CHEK2 contribute to breast cancer susceptibility: results
from a breast cancer family registry case-control mutation-
screening study. Breast Cancer Res. 13, R6.
18. Tavtigian, S.V., Oefner, P.J., Babikyan, D., Hartmann, A.,
Healey, S., Le Calvez-Kelm, F., Lesueur, F., Byrnes, G.B.,
Chuang, S.-C., Forey, N., et al; Australian Cancer Study; Breast
Cancer Family Registries (BCFR); Kathleen Cuningham
Foundation Consortium for Research into Familial Aspects
of Breast Cancer (kConFab). (2009). Rare, evolutionarily
unlikely missense substitutions in ATM confer increased risk
of breast cancer. Am. J. Hum. Genet. 85, 427–446.
19. Mann, G.J., Thorne, H., Balleine, R.L., Butow, P.N., Clarke,
C.L., Edkins, E., Evans, G.M., Fereday, S., Haan, E., Gattas,
M., et al; Kathleen Cuningham Consortium for Research in
Familial Breast Cancer. (2006). Analysis of cancer risk and
BRCA1 and BRCA2 mutation prevalence in the kConFab
familial breast cancer resource. Breast Cancer Res. 8, R12.
20. Ding, Y.C., Steele, L., Chu, L.-H., Kelley, K., Davis, H., John,
E.M., Tomlinson, G.E., and Neuhausen, S.L. (2011). Germline
mutations in PALB2 in African-American breast cancer cases.
Breast Cancer Res. Treat. 126, 227–230.
21. Goldgar, D.E., Healey, S., Dowty, J.G., Da Silva, L., Chen, X.,
Spurdle, A.B., Terry, M.B., Daly, M.J., Buys, S.M., Southey,
M.C., et al; BCFR; kConFab. (2011). Rare variants in the
ATM gene and risk of breast cancer. Breast Cancer Res. 13, R73.
22. Tischkowitz, M., Capanu, M., Sabbaghian, N., Li, L., Liang, X.,
Vallee, M.P., Tavtigian, S.V., Concannon, P., Foulkes, W.D.,
Bernstein, L., et al; The WECARE Study Collaborative Group.
(2012). Rare germline mutations in PALB2 and breast cancer
risk: A population-based study. Hum Mutat 33, 674–680.
23. Rahman, N., Seal, S., Thompson, D., Kelly, P., Renwick, A.,
Elliott, A., Reid, S., Spanova, K., Barfoot, R., Chagtai, T., et al;
Breast Cancer Susceptibility Collaboration (UK). (2007).
PALB2, which encodes a BRCA2-interacting protein, is a breast
cancer susceptibility gene. Nat. Genet. 39, 165–167.
24. Seal, S., Thompson, D., Renwick, A., Elliott, A., Kelly, P.,
Barfoot, R., Chagtai, T., Jayatilake, H., Ahmed, M., Spanova,
K., et al; Breast Cancer Susceptibility Collaboration (UK).
(2006). Truncating mutations in the Fanconi anemia J gene
BRIP1 are low-penetrance breast cancer susceptibility alleles.
Nat. Genet. 38, 1239–1241.
25. Turnbull, C., Seal, S., Renwick, A., Warren-Perry, M., Hughes,
D., Elliott, A., Pernet, D., Peock, S., Adlard, J.W., Barwell, J.,
et al; Breast Cancer Susceptibility Collaboration (UK),
EMBRACE. (2012). Gene-gene interactions in breast cancer
susceptibility. Hum. Mol. Genet. 21, 958–962.
The American Journal of Human Genetics 90, 734–739, April 6, 2012 739

REPORT
Exome Sequencing Identifies PDE4DMutations as Another Cause of Acrodysostosis
Caroline Michot,1,10 Carine Le Goff,1,10 Alice Goldenberg,2 Avinash Abhyankar,3 Celine Klein,1
Esther Kinning,4 Anne-Marie Guerrot,2 Philippe Flahaut,5 Alice Duncombe,6 Genevieve Baujat,1
Stanislas Lyonnet,1 Caroline Thalassinos,7 Patrick Nitschke,8 Jean-Laurent Casanova,3,9
Martine Le Merrer,1 Arnold Munnich,1 and Valerie Cormier-Daire1,*
Acrodysostosis is a rare autosomal-dominant condition characterized by facial dysostosis, severe brachydactyly with cone-shaped epiph-
yses, and short stature. Moderate intellectual disability and resistance to multiple hormones might also be present. Recently, a recurrent
mutation (c.1102C>T [p.Arg368*]) in PRKAR1A has been identified in three individuals with acrodysostosis and resistance to multiple
hormones. After studying ten unrelated acrodysostosis cases, we report here de novo PRKAR1A mutations in five out of the ten individ-
uals (we found c.1102C>T [p.Arg368*] in four of the ten and c.1117T>C [p.Tyr373His] in one of the ten). We performed exome
sequencing in two of the five remaining individuals and selected phosphodiesterase 4D (PDE4D) as a candidate gene. PDE4D encodes
a class IV cyclic AMP (cAMP)-specific phosphodiesterase that regulates cAMP concentration. Exome analysis detected heterozygous
PDE4D mutations (c.673C>A [p.Pro225Thr] and c.677T>C [p.Phe226Ser]) in these two individuals. Screening of PDE4D identified
heterozygous mutations (c.568T>G [p.Ser190Ala] and c.1759A>C [p.Thr587Pro]) in two additional acrodysostosis cases. These
mutations occurred de novo in all four cases. The four individuals with PDE4D mutations shared common clinical features, namely
characteristic midface and nasal hypoplasia and moderate intellectual disability. Metabolic screening was normal in three of these
four individuals. However, resistance to parathyroid hormone and thyrotropin was consistently observed in the five cases with PRKAR1A
mutations. Finally, our study further supports the key role of the cAMP signaling pathway in skeletogenesis.
Acrodysostosis (MIM 101800) is a dominantly inherited
condition consisting of (1) skeletal dysplasia characterized
by facial dysostosis with nasal hypoplasia (a depressed
nasal bridge and prominent mandible), severe brachydac-
tyly with short broad metatarsals, metacarpals, and
phalanges, cone-shaped epiphyses, advanced bone matu-
ration, spinal stenosis, and short stature; (2) resistance to
multiple hormones, including parathyroid hormone and
thyrotropin; and (3) possible neurological involvement
(moderate to mild intellectual disability).1,2 Differential
diagnoses include Albright hereditary osteodystrophy
(MIM 103580) and pseudopseudohypoparathyroidism
(MIM 612463), which are both due to loss-of-function
mutations in GNAS (a-stimulary subunit of the G protein)
(MIM 139320) and are characterized by less severe hand
and foot involvement.3
A recurrent c.1102C>T mutation in PRKAR1A (MIM
188830) has been recently identified in three cases of acro-
dysostosis with resistance to multiple hormones.4 This
gene encodes the cyclic AMP (cAMP)-dependent regula-
tory subunit of protein kinase A. The mutated subunit
impairs the protein-kinase-A response to cAMP and
accounts for hormone resistance and skeletal abnormali-
ties resembling those observed in Albright hereditary
osteodystrophy.
After studying ten unrelated individuals with acrodysos-
tosis, we found PRKAR1A mutations in five out of the
ten, and we show that most of the remaining cases were
accounted for by mutations in phosphodiesterase, 4D
(PDE4D [MIM 600129]), which is also involved in cAMP
metabolism.
Ten unrelated cases were included in this study. There
was no family history, and each individual was the only
affected member in his family. Inclusion criteria were the
following: (1) the presence of severe generalized brachy-
dactyly affecting metacarpals and phalanges and associ-
ated with cone-shaped epiphyses and (2) the exclusion of
Albright hereditary osteodystrophy on the basis of normal
bioactivity of the Gs alpha subunit and normal GNAS
sequencing.
We performed a complete screening of phosphocalcic
metabolism and blood levels of creatinine, calcium,
phosphorus, thyroxin, thyrotropin, 25-hydroxyvitaminD,
1,25-dihydroxyvitaminD, parathyroid hormone (PTH),
and fibroblast growth factor 23, as well as urinary levels
of creatinine, calcium, and phosphorus. The clinical
1Unite Institut National de la Sante et de la Recherche Medicale U781, Departement de Genetique, Universite Paris Descartes, Sorbonne Paris Cite, Hopital
Necker Enfants Malades, Paris 75015, France; 2Service de Genetique Medicale, Centre Hospitalier Universitaire-Hopitaux de Rouen, Rouen 76100, France;3St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065, USA; 4The Ferguson-
Smith Centre for Clinical Genetics, Royal Hospital for Sick Children-Yorkhill, Dalnair Street, Glasgow G3 8SJ, Scotland; 5Service de Pediatrie, Centre Hos-
pitalier Universitaire-Hopitaux de Rouen, Rouen 76100, France; 6Service d’Ophtalmologie, Centre Hospitalier Universitaire-Hopitaux de Rouen, Rouen
76100, France; 7Endocrinologie Gynecologie Diabetologie Pediatrique, Assistance Publique-Hopitaux de Paris, Paris 75015, France; 8Plateforme de Bioin-
formatique, Universite Paris Descartes, Paris 75015, France; 9Unite Institut National de la Sante et de la Recherche Medicale U980, Laboratory of Human
Genetics of Infectious Diseases, Necker Medical School, University Paris Descartes, Paris 75015, France10These authors contributed equally to this work
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.03.003. �2012 by The American Society of Human Genetics. All rights reserved.
740 The American Journal of Human Genetics 90, 740–745, April 6, 2012

radiological and biochemical details are summarized in
Table 1 and Figure 1.
Informed consent for participation, sample collection,
and photograph publication was obtained via protocols
approved by the Necker Hospital ethics committee.
We sequenced PRKAR1A (RefSeq accession number
NM_002734.3) by using specific primers (available upon
request) in the ten individuals. De novo PRKAR1A muta-
tions, including the recurrent mutation,4 were identified
in five out of the ten individuals (c.1102C>T [p.Arg368*]
was found in four of the five, and c.1117T>C [p.Tyr373His]
was found in one of the five) (Table 1). This missense
mutation was predicted to be damaging by PolyPhen, was
found to alter a conserved amino acid located in the cata-
lytic domain, and was not identified in alleles from 200
ethnically matched controls.
The exclusion of PRKAR1A in five acrodysostosis cases
prompted us to perform exome sequencing in two of these
five individuals. Exome capture was performed with the
SureSelect Human All Exon kit (Agilent Technologies).5
Single-end sequencing was performed on an Illumina
Genome Analyzer IIx (Illumina) and generated 72 bp
reads. For sequence alignment, variant calling, and anno-
tation, we aligned the sequences to the human genome
reference sequence (hg18 build) by using the Burrows-
Wheeler Aligner.6 Downstream processing was carried out
with the Genome Analysis Toolkit (GATK7), SAMtools,8
and Picard tools. Substitution calls were made with
GATK Unified Genotyper, whereas indel calls were made
with a GATK IndelGenotyperV2. All calls with a read
coverage %23 and a Phred-scaled SNP quality of %20
were filtered out. All the variants were annotated with an
in-house -developed annotation software system. We first
focused our analyses on nonsynonymous variants, splice-
acceptor and donor-site mutations, and coding indels
because we anticipated that synonymous variants would
be far less likely to cause disease (Table S1, available
online). We also defined variants as previously unidenti-
fied if they were absent from control populations and
from all datasets, including dbSNP129, the 1000 Genomes
Project, and in-house exome data.
On the basis of the dominant mode of inheritance of
acrodysostosis, we selected eight candidate genes that all
harbor heterozygous mutations (Table S2). Given the
involvement of PRKAR1A, a cAMP-activated protein kinase
A, in some acrodysostosis cases,4 we then only considered
gene(s) that encode proteins involved in the cAMP
signaling pathway. Therefore, we regarded PDE4D (RefSeq
accession number NM_001104631) as the best candidate
gene. Indeed,PDE4D encodes a class IVcAMP-specificphos-
phodiesterase that regulates cAMP concentration. Exome
analysis detected two PDE4D mutations (c.673C>A
[p.Pro225Thr] and c.677T>C [p.Phe226Ser]) in the two
individuals. These results were confirmed by Sanger
sequencing. Subsequent screening of the 15 PDE4D coding
exons in the three remaining cases led to the identifica-
tion of two distinct heterozygous missense mutations
(c.568T>G [p.Ser190Ala] and c.1759A>C [p.Thr587Pro])
in two additional cases. Thesemutations were not observed
in the parents of acrodysostosis-affected individuals, con-
firming that they occurred de novo.
We identified a total of four distinct heterozygous
PDE4D mutations in four individuals (Table 1). Among
them, the p.Ser190Ala substitution affected a serine
residue predicted to be phosphorylated (Uniprot database),
and the p.Thr587Pro substitution disturbed the conserved
catalytic PDEase_I domain (pfam database), which confers
the 3050-cyclic nucleotide phosphodiesterase activity. The
two remaining alterations (p.Pro225Thr and p.Phe226Ser)
affected conserved residues across species. All four muta-
tions were considered to be pathogenic by PolyPhen
and were absent from alleles in 200 ethnically matched
controls.
Here, we report PDE4D mutations in four unrelated
cases of acrodysostosis and PRKAR1A mutations in five
cases. All mutations occurred de novo, providing further
evidence that acrodysostosis has a dominant mode of
inheritance.
After we divided up the acrodysostosis-affected individ-
uals and grouped them according to the mutations they
had, our study revealed interesting genotype-phenotype
correlations. Indeed, the four individuals with PDE4D
mutations shared characteristic facial features, namely
midface hypoplasia with the canonical nasal hypoplasia
initially reported in acrodysostosis and moderate intellec-
tual disability with speech delay.1,2 The characteristic
facial dysostosis and intellectual disability were neither
observed in our individuals with PRKAR1A mutations nor
mentioned in the three previously reported cases.4 Along
the same lines, hormone resistance was observed in only
one person with PDE4D mutations—case 6 had increased
PTH levels and normal serum-phosphate levels—whereas
hormone resistance was consistently observed in individ-
uals carrying PRKAR1A mutations (all five suffered from
chronic resistance to parathyroid hormone, and four of
the five had peripheral hypothyroidism). Although our
study does not allow generalized conclusions, our find-
ings might suggest that individuals with facial dysostosis
and moderate intellectual disability should be screened
for PDE4D mutations, whereas individuals with less char-
acteristic facial features, no intellectual disability, and
hormone resistance should be screened for mutations in
PRKAR1A.
The five individuals harboring PRKAR1A mutations pre-
sented with growth retardation (<�2 standard deviations
[SDs]) and decreased growth speed in late childhood
(between 7 and 13 years of age). The adult individuals
had a final height <�3 SDs. Alternatively, the cases
harboring PDE4D mutations presently have normal
growth charts, but they are only 3–7 years old, and predict-
ing final adult height is therefore impossible. It is worth
noting that two out of the four PDE4D cases presented
with an acute intracranial hypertension due to sinus
thrombosis; both of these individuals required derivation
The American Journal of Human Genetics 90, 740–745, April 6, 2012 741
Table 1. Clinical, Radiological, and Biochemical Data of the Ten Acrodysostosis Individuals Reported
Patient 1 Patient 2 Patient 3 Patient 4 Patient 5 Patient 6 Patient 7 Patient 8 Patient 9 Patient 10
Sex female male female female female male male male male female
PRKAR1A mutation c.1102C>T c.1102C>T c.1102C>T c.1117T>C c.1102C>T � � � � �
PDE4D mutation � � � � � c.673C>A c.677T>C c.568T>G c.1759A>C �
IUGR no no no yes no yes no no no no
Postnatal growthretardation (<�2 SDs)
yes(26 years old)
no(8 years old)
yes(13 years old)
yes(22 years old)
yes(34 years old)
no(7 years old)
no(4 years old)
no(4 years old)
no(3 years old)
yes(38 years old)
Advanced bone age � yes yes � � yes yes yes yes �
Facial dysostosis
Nasal hypoplasia no no no no no yes yes yes yes no
Depressed nasal bridge no no yes no yes yes yes yes yes no
Prominent mandible no no no no yes no no yes no yes
Peripheral dysostosis
Severe brachydactyly yes yes yes yes yes yes yes yes yes yes
Short metatarsals,metacarpals, andphalanges
yes yes yes yes yes yes yes yes yes yes
Cone-shaped epiphyses yes(childhood)
yes yes yes(childhood)
yes(childhood)
yes yes yes yes nd
Hormonal screening
PTH (n ¼ 10–46 ng/l) 95 79 116 84 142 76 39 24 19 normal
Calcemia(n ¼ 2.2–2.7 mmol/l)
2.35 2.47 2.4 2.57 2.37 2.18 2.47 2.4 2.5 2.38
Phosphoremia(n ¼ 1.3–1.85 mmol/l)
1.23 1.56 1.76 nd 1.3 1.54 1.68 1.81 1.7 1.8
25-OHvitD(n ¼ 30–80 ng/ml)
nd 26 16 nd 22 26 25 45 30 nd
1,25-diOHvitD (pg/ml) nd 39 106 65 53 nd nd nd
FGF23 (n ¼ 1–120 UI/ml) nd nd nd nd 145 90 112 171.2 60.9 nd
Free T4(n ¼ 7.5–15 pmol/l)
8.65 hypothyroidism hypothyroidism hypothyroidism 16.71 treatment 10.4 10 14.3 17 17
TSH (n ¼ 0.34–5.6 mUI/l) 2.67 13.41 15.42 increased 0.16 2.59 2.51 3.58 2.77 1.8l
Calciuria(n ¼ 1.5–6 mmol/l)
nd <0.2 nd nd nd 0.86 1.28 1.44 2.27 nd
742
TheAmerica
nJournalofHumanGenetics
90,740–745,April
6,2012

surgery and medical treatment. This observation should
prompt the careful investigation of headache complaints
in such cases. This feature, hitherto unreported in
PRKAR1A-mutation-positive individuals, might be another
distinctive characteristic specific to the clinical spectrum of
symptoms associated with PDE4D mutations. Finally,
neither PDE4D nor PRKAR1A mutations were found in
one adult individual who had characteristic skeletal
features but no hormone resistance or facial dysostosis.
One cannot exclude a molecular defect not detectable by
Sanger sequencing, but it is also conceivable that other
genes might account for acrodysostosis.
Considering PRKAR1A mutations, we confirm that
c.1102C>T is a recurrent mutation observed in seven of
the eight patients reported so far, whereas only one
missense mutation that changes a conserved amino acid
located in the cAMP binding domain has been identified.
Interestingly, the Arg388* substitution is considered
a gain-of-function mutation because it decreases protein-
kinase-A sensitivity to cAMP.4 In contrast, germ-line loss-
of-function mutations resulting in constitutive activation
of protein kinase A are responsible for Carney complex
(MIM 160980), an autosomal-dominant multiple-
neoplasia syndrome characterized by cardiac, endocrine,
cutaneous, and neural myxomatous tumors and pig-
mented lesions of the skin and mucosae.9
All mutations that we have identified in PDE4D are
heterozygous missense mutations and are presumably
responsible for impaired phosphodiesterase activity.
PDE4D belongs to the cAMP-hydrolyzing phosphodies-
terase family, which is directly involved in the rate of
cAMP degradation. Considering the crucial role of cAMP
in intracellular signaling in response to a number of
membrane-impermeable hormones, a dysregulation of
cAMP levels could be the underlying mechanism of the
acrodysostosis that results from PDE4D mutations.
The cAMP-specific PDE4 family is widely expressed, and
PDE4 isoforms have similar catalytic functions, but they
have distinct cellular functions because of differences in
specific intracellular trafficking and signaling-complex
formation.10,11 PDE4D uses different promoters to
generate multiple alternatively spliced transcript variants
(at least nine) that encode functional proteins; this might
explain the phenotype variability observed in the four re-
ported cases.
Of note, mice deficient in PDE4D exhibited delayed
growth and female infertility due to impaired ovula-
tion;12 these two symptoms have also been described in
acrodysostosis cases.13 Mouse models have also revealed
that PDE4D plays a critical role in the memory and hippo-
campal neurogenesis mediated by cAMP signaling.14
Flies deficient in the PDE4D homolog, dunce, also display
impaired central-nervous-system and reproductive func-
tion.15 All together, these data support the involvement
of PDE4D impairment in the regulation of cAMP signal-
ing, especially in growth and central-nervous-system
development.Table
1.
Continued
Patient1
Patient2
Patient3
Patient4
Patient5
Patient6
Patient7
Patient8
Patient9
Patient10
Phosp
haturia
(n¼
10–5
0mmol/l)
nd
14.8
nd
nd
nd
13.76
7.12
36.7
4.70
nd
Creatininuria(m
mol/l)
nd
4.5
nd
nd
nd
6.3
2.08
11.7
4.15
nd
Neurolo
gy
Intellectual
disab
ility
no
no
no
no
no
yes;sp
eech
delay
requiring
orthophony;
fine-motor-skill
impairm
ent
yes;sp
eech
delay
requiring
orthophony;
fine-motor-skill
impairm
ent
yes;sp
eech
delay
;psych
omotor
delay
(walked
at17month
sofag
e)
yes;sp
eech
delay
;psych
omotor
delay
requiring
physioth
erap
y
no
Oth
ersp
inal
sten
osis;
carpal
tunnel
intracranial
hypertension
withjugular
sten
osisrequiring
derivation
intracranial
hypertensionwith
thrombophlebitis
ofth
etran
sverse
sinus
andjugular(treated
byacetazolamide,
antico
agulant,
andderivation)
spinal
sten
osis
Thefollo
wingabbreviationsare
used:IUGR,intrauterinegrowth
retardation;SDs,standard
deviations;nd,notdone;PTH,parathyroid
horm
one;25-O
HvitD
,25-hydroxyvitaminD;1,25-diO
HvitD
,1,25-dihydroxyvitaminD;
FGF2
3,fibroblast
growth
factor23;T4,thyroxin;andTSH,thyrotropin.
The American Journal of Human Genetics 90, 740–745, April 6, 2012 743

Finally, our findings further support the key role of the
cAMP signaling pathway in skeletogenesis, as previously
shown for Albright hereditary osteodystrophy due to
GNAS mutations. Ongoing studies will highlight the
specific link between PRKAR1A and PDE4D, which are
both involved in cAMP signaling and responsible for acro-
dysostosis.
Supplemental Data
Supplemental Data include two tables and can be found with this
article online at http://www.cell.com/AJHG.
Acknowledgments
We thank all individuals and their families for their contribution
to this work.
Received: December 9, 2011
Revised: January 11, 2012
Accepted: March 6, 2012
Published online: March 29, 2012
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
Pfam, http://www.sanger.ac.uk/resources/databases/pfam.html
Picard Tools, http://picard.sourceforge.net
Polyphen, http://genetics.bwh.harvard.edu/pph/
Uniprot, http://www.uniprot.org/
References
1. Maroteaux, P., and Malamut, G. (1968). Acrodysostosis. Presse
Med. 76, 2189–2192.
2. Robinow, M., Pfeiffer, R.A., Gorlin, R.J., McKusick, V.A.,
Renuart, A.W., Johnson, G.F., and Summitt, R.L. (1971).
Acrodysostosis. A syndrome of peripheral dysostosis, nasal
hypoplasia, and mental retardation. Am. J. Dis. Child. 121,
195–203.
3. Bastepe, M., and Juppner, H. (2005). GNAS locus and pseudo-
hypoparathyroidism. Horm. Res. 63, 65–74.
4. Linglart, A., Menguy, C., Couvineau, A., Auzan, C., Gunes,
Y., Cancel, M., Motte, E., Pinto, G., Chanson, P., Bougneres,
P., et al. (2011). Recurrent PRKAR1A mutation in acrodysos-
tosis with hormone resistance. N. Engl. J. Med. 364, 2218–
2226.
5. Byun, M., Abhyankar, A., Lelarge, V., Plancoulaine, S., Palan-
duz, A., Telhan, L., Boisson, B., Picard, C., Dewell, S., Zhao,
C., et al. (2010). Whole-exome sequencing-based discovery
of STIM1 deficiency in a child with fatal classic Kaposi
sarcoma. J. Exp. Med. 207, 2307–2312.
6. Li, H., and Durbin, R. (2009). Fast and accurate short read
alignment with Burrows-Wheeler transform. Bioinformatics
25, 1754–1760.
7. McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis,
K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly,
M., and DePristo, M.A. (2010). The Genome Analysis Toolkit:
a MapReduce framework for analyzing next-generation DNA
sequencing data. Genome Res. 20, 1297–1303.
8. Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer,
N., Marth, G., Abecasis, G., and Durbin, R.; 1000 Genome
Project Data Processing Subgroup. (2009). The Sequence
Alignment/Map format and SAMtools. Bioinformatics 25,
2078–2079.
9. Kirschner, L.S., Carney, J.A., Pack, S.D., Taymans, S.E., Giatza-
kis, C., Cho, Y.S., Cho-Chung, Y.S., and Stratakis, C.A. (2000).
Mutations of the gene encoding the protein kinase A type I-
alpha regulatory subunit in patients with the Carney complex.
Nat. Genet. 26, 89–92.
10. Rall, T.W., and Sutherland, E.W. (1958). Formation of a cyclic
adenine ribonucleotide by tissue particles. J. Biol. Chem. 232,
1065–1076.
Figure 1. Pictures and X-Rays of Individuals 6and 8 with PDE4D Mutations(A1 and B1) Full-face pictures of individuals 6 (A)and 8 (B) showing facial dysostosis with a flatnasal bridge and nasal hypoplasia.(A2 and B2) Profile pictures show malar hypo-plasia.(A3) Palmar face of right hand.(A4 and B3) Dorsal face of hands, which are broadand shortened.(A5 and B4) Standard X-rays of both hands showsevere brachydactyly with short, broad meta-carpals and phalanges, cone-shaped epiphyses(arrows), and advanced carpal maturation.
744 The American Journal of Human Genetics 90, 740–745, April 6, 2012

11. Sutherland, E.W., and Rall, T.W. (1958). Fractionation and
characterization of a cyclic adenine ribonucleotide formed
by tissue particles. J. Biol. Chem. 232, 1077–1091.
12. Jin, S.L., Richard, F.J., Kuo, W.P., D’Ercole, A.J., and Conti, M.
(1999). Impaired growth and fertility of cAMP-specific phos-
phodiesterase PDE4D-deficient mice. Proc. Natl. Acad. Sci.
USA 96, 11998–12003.
13. Graham, J.M., Jr., Krakow, D., Tolo, V.T., Smith, A.K., and
Lachman, R.S. (2001). Radiographic findings and Gs-alpha
bioactivity studies and mutation screening in acrodysostosis
indicate a different etiology from pseudohypoparathyroidism.
Pediatr. Radiol. 31, 2–9.
14. Li, Y.F., Cheng, Y.F., Huang, Y., Conti, M., Wilson, S.P.,
O’Donnell, J.M., and Zhang, H.T. (2011). Phosphodiesterase-
4D knock-out and RNA interference-mediated knock-down
enhance memory and increase hippocampal neurogenesis
via increased cAMP signaling. J. Neurosci. 31, 172–183.
15. Dudai, Y., Jan, Y.N., Byers, D., Quinn, W.G., and Benzer, S.
(1976). dunce, a mutant of Drosophila deficient in learning.
Proc. Natl. Acad. Sci. USA 73, 1684–1688.
The American Journal of Human Genetics 90, 740–745, April 6, 2012 745

REPORT
Exome Sequencing IdentifiesPDE4D Mutations in Acrodysostosis
Hane Lee,1,2 John M. Graham, Jr.,3,9 David L. Rimoin,1,3,4,9 Ralph S. Lachman,5,9 Pavel Krejci,9,10
Stuart W. Tompson,8 Stanley F. Nelson,1,2 Deborah Krakow,1,6,7 and Daniel H. Cohn7,8,*
Acrodysostosis is a dominantly-inherited, multisystem disorder characterized by skeletal, endocrine, and neurological abnormalities. To
identify the molecular basis of acrodysostosis, we performed exome sequencing on five genetically independent cases. Three different
missense mutations in PDE4D, which encodes cyclic AMP (cAMP)-specific phosphodiesterase 4D, were found to be heterozygous in
three of the cases. Two of the mutations were demonstrated to have occurred de novo, providing strong genetic evidence of causation.
Two additional cases were heterozygous for de novo missense mutations in PRKAR1A, which encodes the cAMP-dependent regulatory
subunit of protein kinase A and which has been recently reported to be the cause of a form of acrodysostosis resistant to multiple
hormones. These findings demonstrate that acrodysostosis is genetically heterogeneous and underscore the exquisite sensitivity of
many tissues to alterations in cAMP homeostasis.
Acrodysostosis (MIM 101800), also known as Arkless-
Graham syndrome or Maroteaux-Malamut syndrome, is
a pleiotropic disorder characterized by skeletal, endocrine,
and neurological abnormalities.1,2 Skeletal features include
brachycephaly, midface hypoplasia with a small upturned
nose, brachydactyly, and lumbar spinal stenosis. Endo-
crine abnormalities have been reported and include hypo-
thyroidism and hypogonadism in males and irregular
menses in females (summarized in Butler et al.). Develop-
mental disability is a common finding but is variable in
severity and can be associated with significant behavioral
problems. Most cases are sporadic, and there is evidence
of a paternal age effect,3 suggesting that the phenotype
might result from de novo point mutations. Perhaps as
a result of the developmental disability and/or endocrine
abnormalities, there are only a few examples of dominant
transmission.4,5 Recently, dominant mutations in
PRKAR1A (MIM 188830), which encodes the cyclic AMP
(cAMP)-dependent regulatory subunit of protein kinase A
(PKA), were found in a subset of acrodysostosis cases
resistant to multiple hormones.6 The mutations resulted
in reduced PKA activation by cAMP and led to a reduced
hormone response in multiple tissues.
We studied five sporadic cases, four males and one
female, who had clinical and radiographic phenotypes
(Figure 1, Table 1) consistent with the diagnosis of acrody-
sostosis. A prior publication7 contains additional clinical
details on three of the five cases (International Skeletal
Dysplasia Registry reference numbers R99-101A [case 1 in
Graham et al.7], R99-514A [case 2], and 95-141A [case
R1]). All studies were carried out with informed consent
under a protocol approved by the institutional review
board at Cedars-Sinai Medical Center. To determine the
molecular basis of the phenotype in each case, we per-
formed exome capture and sequence analysis. In three
cases (R02-309A, R06-434A, and R95-141A), DNA from
the unaffected parents was also available at the outset
of the study, and we determined the exome sequences for
the six parents to facilitate the identification of de novo
mutations in these trios. High-molecular-weight genomic
DNA was extracted from either blood or lymphoblastoid
cell lines; the quality of the DNA samples was determined
by a Qubit Fluorometer (Invitrogen) and a Bioanalyzer
(Agilent). For each sample, we prepared the sequencing
library with 3 mg of genomic DNA and used the Agilent
SureSelect Target Enrichment System to construct an
Illumina Paired-End Sequencing library (protocol version
2.0.1). The Agilent SureSelect Human All Exon 50Mb kit
was used for the exome capture. Sequences for each sample
(50 bp paired end) were determined on a single lane of an
Illumina HiSeq2000 instrument, and a total of 82–123
million paired-end reads per sample were generated. We
performed base-calling by using the real-time analysis
(RTA) software provided by Illumina.
We aligned the sequence reads to human reference
genome human_g1k_v37.fasta (downloaded from the
Genome Analysis Toolkit [GATK] resource bundle in
November 2010) by using Novoalign from the Novocraft
Short Read Alignment Package; the adaptor-stripping
and base-quality-calibration options were on. We used
SAMtools version 0.1.15 to sort the aligned BAM files,
and we removed potential PCR duplicates (rmdup) by
1Department of Human Genetics, University of California, Los Angeles, Los Angeles, CA 90095, USA; 2Department of Pathology, University of California,
Los Angeles, Los Angeles, CA 90095, USA; 3Department of Pediatrics, University of California, Los Angeles, Los Angeles, CA 90095, USA; 4Department of
Medicine, University of California, Los Angeles, Los Angeles, CA 90095, USA; 5Department of Radiological Sciences, University of California, Los Angeles,
Los Angeles, CA 90095, USA; 6Department of Obstetrics and Gynecology, University of California, Los Angeles, Los Angeles, CA 90095, USA; 7Department
of Orthopedic Surgery, University of California, Los Angeles, Los Angeles, CA 90095, USA; 8Department of Molecular, Cell, and Developmental Biology,
University of California, Los Angeles, Los Angeles, CA 90095, USA; 9Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, CA 90048,
USA; 10Institute of Experimental Biology, Masaryk University and Department of Cytokinetics, Institute of Biophysics AS CR, 61265 Brno, Czech Republic
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.03.004. �2012 by The American Society of Human Genetics. All rights reserved.
746 The American Journal of Human Genetics 90, 746–751, April 6, 2012
using Picard. On average, 88.2% of the reads were uniquely
aligned to the reference genome. The PCR duplication rate
varied between 5.1% and 8.2%, and there was an average
estimated library size of 704 million unique fragments.
The on-target rate, or capture specificity, varied from 60%
to 63.8%. The mean coverage across the captured regions
was 973, and approximately 92% of the targeted bases
were covered by R10 reads for each exome.
We performed local realignment for each sample by
using the GATK ‘‘IndelRealigner’’ tool, and we recalibrated
base qualities by using the GATK ‘‘TableRecalibration’’ tool
according to GATK’s recommendation (Best Practice
Variant Detection with the GATK version 2). Variants
were simultaneously called with the GATK ‘‘Unified
Genotyper’’ tool for all 11 samples (the five cases and six
unaffected parents). Small indels were called with the
‘‘-glm DINDEL’’ option. The dbSNP132 file downloaded
from the GATK resource bundle was used so that the
known SNP positions were annotated in the output
VCF (variant call format) file. Only the variants found
within the protein coding regions of the captured exons
were reported with the –L option. The interval file that
we used is available upon request. Using the GATK
‘‘VariantFiltrationWalker’’ tool, we hard filtered both the
SNPs and INDELs to remove low-quality variants. As sug-
gested by GATK, we used the following parameters for
standard filtration: (1) the clusterWindowSize was 10, (2)
mapping quality of zero was >40, (3) quality by depth
was <5.0, and (4) strand bias was >�0.10.
We annotated the ‘‘PASS’’-ed variants that were not
found at dbSNP132 positions by using SeattleSeqAnnota-
tion version 6.16 (SNPs and INDELs were annotated sepa-
rately). Both NCBI (National Center for Biotechnology
Information) full genes and CCDS (consensus coding
sequence) 2010 gene models were used for the annotation.
Variants present in the 1,000 Genomes Database (March
2010 release) or dbSNP131 as well as those resulting in
synonymous coding changes or found outside the coding
region were removed from further analysis.
The annotated variants were first examined in the trios
and were further filtered under a rare dominant model.
Because acrodysostosis is dominantly inherited and was
sporadic in the cases studied, we prioritized the variants
to examine the de novo variants. We identified potential
de novo variants by selecting the heterozygous variants
found only in the case but not in the parents, and we
Figure 1. Radiographic Phenotype in Acrodysostosis CasesFor four of the cases, anteroposterior hand (A–D), lateral-skull (E–H), and lumbar-spine (I–L) radiographs are shown. Individuals R02-309and R99-101 have mutations in PRKAR1A, and individuals R06-434 and R95-141 have mutations in PDE4D. Arrows on the lateral-skullfilms identify midface hypoplasia. Arrows on the lumbar-spine films indicate absence of normal interpedicular widening in the lumbarvertebrae; the absence of such widening predisposes the affected individuals to spinal stenosis. The case numbers are indicated acrossthe top.
The American Journal of Human Genetics 90, 746–751, April 6, 2012 747
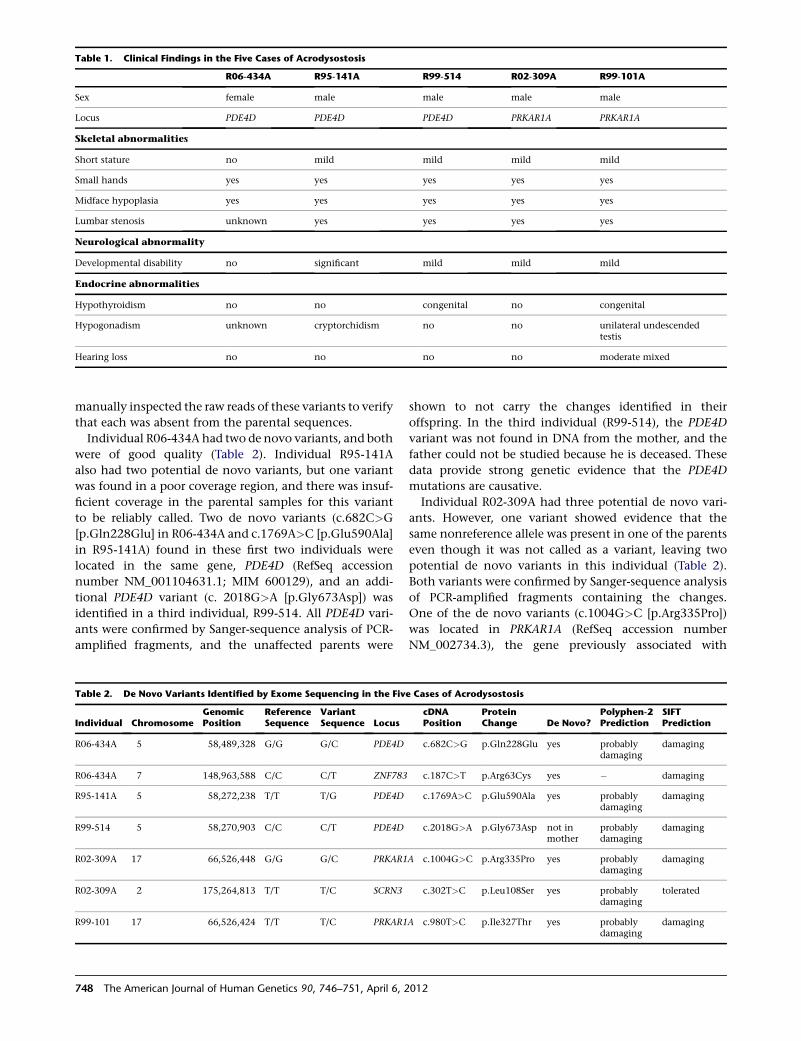
manually inspected the raw reads of these variants to verify
that each was absent from the parental sequences.
Individual R06-434A had two de novo variants, and both
were of good quality (Table 2). Individual R95-141A
also had two potential de novo variants, but one variant
was found in a poor coverage region, and there was insuf-
ficient coverage in the parental samples for this variant
to be reliably called. Two de novo variants (c.682C>G
[p.Gln228Glu] in R06-434A and c.1769A>C [p.Glu590Ala]
in R95-141A) found in these first two individuals were
located in the same gene, PDE4D (RefSeq accession
number NM_001104631.1; MIM 600129), and an addi-
tional PDE4D variant (c. 2018G>A [p.Gly673Asp]) was
identified in a third individual, R99-514. All PDE4D vari-
ants were confirmed by Sanger-sequence analysis of PCR-
amplified fragments, and the unaffected parents were
shown to not carry the changes identified in their
offspring. In the third individual (R99-514), the PDE4D
variant was not found in DNA from the mother, and the
father could not be studied because he is deceased. These
data provide strong genetic evidence that the PDE4D
mutations are causative.
Individual R02-309A had three potential de novo vari-
ants. However, one variant showed evidence that the
same nonreference allele was present in one of the parents
even though it was not called as a variant, leaving two
potential de novo variants in this individual (Table 2).
Both variants were confirmed by Sanger-sequence analysis
of PCR-amplified fragments containing the changes.
One of the de novo variants (c.1004G>C [p.Arg335Pro])
was located in PRKAR1A (RefSeq accession number
NM_002734.3), the gene previously associated with
Table 1. Clinical Findings in the Five Cases of Acrodysostosis
R06-434A R95-141A R99-514 R02-309A R99-101A
Sex female male male male male
Locus PDE4D PDE4D PDE4D PRKAR1A PRKAR1A
Skeletal abnormalities
Short stature no mild mild mild mild
Small hands yes yes yes yes yes
Midface hypoplasia yes yes yes yes yes
Lumbar stenosis unknown yes yes yes yes
Neurological abnormality
Developmental disability no significant mild mild mild
Endocrine abnormalities
Hypothyroidism no no congenital no congenital
Hypogonadism unknown cryptorchidism no no unilateral undescendedtestis
Hearing loss no no no no moderate mixed
Table 2. De Novo Variants Identified by Exome Sequencing in the Five Cases of Acrodysostosis
Individual ChromosomeGenomicPosition
ReferenceSequence
VariantSequence Locus
cDNAPosition
ProteinChange De Novo?
Polyphen-2Prediction
SIFTPrediction
R06-434A 5 58,489,328 G/G G/C PDE4D c.682C>G p.Gln228Glu yes probablydamaging
damaging
R06-434A 7 148,963,588 C/C C/T ZNF783 c.187C>T p.Arg63Cys yes � damaging
R95-141A 5 58,272,238 T/T T/G PDE4D c.1769A>C p.Glu590Ala yes probablydamaging
damaging
R99-514 5 58,270,903 C/C C/T PDE4D c.2018G>A p.Gly673Asp not inmother
probablydamaging
damaging
R02-309A 17 66,526,448 G/G G/C PRKAR1A c.1004G>C p.Arg335Pro yes probablydamaging
damaging
R02-309A 2 175,264,813 T/T T/C SCRN3 c.302T>C p.Leu108Ser yes probablydamaging
tolerated
R99-101 17 66,526,424 T/T T/C PRKAR1A c.980T>C p.Ile327Thr yes probablydamaging
damaging
748 The American Journal of Human Genetics 90, 746–751, April 6, 2012

acrodysostosis with hormone resistance.6 Individual R99-
101 was also found to have a variant (c.980T>C
[p.Ile327Thr]) in PRKAR1A, and subsequent Sanger-
sequence analysis of a PCR-amplified fragment confirmed
the mutation and demonstrated its absence from DNA
derived from the parents; this analysis indicated that the
variant resulted from a de novo event. Therefore, acrody-
sostosis in these latter two individuals appears to have re-
sulted from PRKAR1A mutations.
All five missense variants (three in PDE4D and two in
PRKAR1A) were predicted to be damaging by PolyPhen-2
(Polymorphism Phenotyping version 2) and/or SIFT, two
commonly used tools that predict the functional conse-
quences of amino acid changes on the basis of sequence
homology and the physical properties of the amino acids.
None of these variants were observed in an internal exome
dataset of 48 individuals affected by different medical
conditions, in a group of 250 published exome data-
sets,8,9 or among the 5,379 exomes available from the
National Heart, Lung, and Blood Institute (NHLBI) Exome
Sequencing Project Exome Variant Server (ESP5400).
The findings described here thus demonstrate that acro-
dysostosis can result from missense mutations in PDE4D,
the gene encoding cAMP-dependent phosphodiesterase
4D. PDE4D encodes at least five isoforms that differ at their
amino-terminal ends as a result of alternate transcription
start sites or alternative splicing.10 The encoded proteins
range in size from 508 to 810 amino acids, and the three
longer isoforms contain two highly evolutionarily con-
served upstream regions (UCR1 and UCR2) and the large
catalytic domain. The two shorter isoforms lack the
amino-terminal UCR1 domain, which regulates catalytic
activity along with UCR2.11 The p.Gln228Glu substitution
alters a conserved residue in the UCR1 region, indicating
that disruption of the longer isoforms alone is enough to
cause a phenotypic effect in the target tissues and result
in acrodysostosis. The p.Glu590Ala and p.Gly673Asp sub-
stitutions alter conserved catalytic-domain amino acids,
indicating that these residues are essential for normal
PDE4D activity.
The results of this study also confirm that mutations in
PRKAR1A, which encodes the cyclic AMP-dependent regu-
latory subunit of PKA, can also lead to acrodysostosis.6 The
two substitutions, p.Arg335Pro and p.Ile327Thr, found in
PRKAR1A were different than the recurrent mutation
(p.R368*) previously reported,6 but all three mutations
were in exon 11, which encodes part of the highly
conserved cAMP-binding domain B. Binding of cAMP by
PRKAR1A is required for the release and activation of
PKA (Figure 2), which then phosphorylates and activates
CREB; this process then leads to the expression of down-
stream targets. This suggests that these mutations could
cause reduced cAMP binding and result in reduced PKA
activation and, consequently, reduced downstream signal-
ing. This mechanism would distinguish the acrodysostosis
mutations from the PRKAR1Amutations that cause Carney
Figure 2. cAMP Signaling CascadeLigand binding (represented in this example by PTH, but other ligands and receptors can stimulate cAMP synthesis), activates Gs-a andstimulates cAMP synthesis by adenylate cyclase. The binding of cAMP by PRKAR1A, the cAMP-dependent regulatory subunit, leads tothe dissociation and activation of PKA and the subsequent phosphorylation of cAMP response element binding (CREB), nuclear trans-location, and expression of downstream genes. PDE4D phosphodiesterase activitymodulates cAMP levels.Mutations (indicated by aster-isks) in the genes encoding these three components of the pathway result in a spectrum of clinically related disorders— acrodysostosisfor mutations in PDE4D or PRKAR1A or Albright hereditary osteodystrophy for mutations in GNAS, the gene encoding Gs-a.
The American Journal of Human Genetics 90, 746–751, April 6, 2012 749

complex (the mutations that cause Carney complex
primarily lead to reduced PRKAR1A synthesis, lack of regu-
latory control of PKA activation, and derepression of
CREB-mediated targets).12
The clinical and radiographic phenotypes (summarized
in Table 1) facilitated comparing the acrodysostosis cases
with the typical symptoms associated with either PDE4D
or PRKAR1A mutations. Mild short stature with small
hands was present in all of the cases, including those
with PRKAR1Amutations previously described,6 regardless
of the locus involved. Similarly, stenosis of the lumbar
spine and midface hypoplasia with a small nose were
consistent findings both clinically and radiographically
(Figure 1). However, endocrine abnormalities were vari-
able; hypothyroidism was documented in just two of the
individuals, R99-514 (who had a PDE4D mutation) and
R99-101 (who had a PRKAR1Amutation). Hypothyroidism
persisted in individual R99-101 but spontaneously
resolved in individual R99-514 when he reached three
years of age. However, firm conclusions cannot be made
from these observations because the number of cases
studied thus far is too small. One of the four male individ-
uals with a PDE4D mutation (R95-141A) had cryptorchi-
dism. One of the PRKAR1A individuals described here,
R99-101, exhibited a unilateral undescended testis, and
both of the males previously described6 had cryptorchi-
dism, indicating that hypogonadism can be found in cases
with defects in either gene. From a neurological viewpoint,
four of the five individuals studied had some degree of
developmental disability, and one individual (R95-141A)
displayed significant behavioral problems. Thus, it is diffi-
cult to distinguish acrodysostosis cases with PDE4D muta-
tions from those with PRKAR1A mutations by clinical
observation only.
The acrodysostosis phenotype is similar to that of Pde4d-
knockout mice.13 As in humans with acrodysostosis,
Pde4d-nullmice exhibit reduced growth andmidface hypo-
plasia. Females with acrodysostosis have been reported to
have irregular menses, and knockout mice have reduced
fertility associated with decreased ovulation and oocyte
degeneration. These observations suggest that the human
mutations lead to reduced PDE4D activity. Because the
heterozygous knockout mice were phenotypically normal
and had essentially normal phosphodiesterase activity,13
it appears that haploinsufficiency for PDE4D activity has
no phenotypic consequence. Because PDE4D is a dimer,
the data suggest the possibility that the missense alleles
identified in the acrodysostosis cases might cause the
phenotype via a dominant-negative effect on the protein.
Albright hereditary osteodystrophy (MIM 103580)
shares phenotypic features, including short stature, bra-
chydactyly, hormone resistance, and varying degrees of
developmental disability, with acrodysostosis and results
frommutations in GNAS14 (MIM 139320), the gene encod-
ing the adenylate cyclase activating protein Gs-a. Gs-a,
PDE4D, and PRKAR1A are all components of the cAMP
signaling pathway (Figure 2). The disruption of PRKAR1A
and GNAS causes downregulation of the cAMP signaling
cascade in response to an external signal, such as parathy-
roid hormone (PTH). Although decreased PDE4D activity
might be predicted to increase cAMP levels, it has been sug-
gested13 that inactivation of PDE4D-mediated negative
feedback would cause a permanent desensitization state
of the cAMP signaling pathway; this desensitization would
paradoxically lead to a significant reduction in the cAMP
response. Consequently, the phenotypic effects resulting
from PDE4Dmutations would be similar to those resulting
from PRKAR1A and GNAS defects.
PDE4D is orthologous to Drosophila dunce, which has
been shown to play a role in learning and memory in
flies.15 Flies deficient in dunce have reduced cAMP
phosphodiesterase activity,16 a reduction which results in
defects in both associative and nonassociative memory.17
Although increased branching of terminal neuronal
processes has been observed in dunce larvae (implicating
abnormal brain morphology as an element of the pheno-
type18), alterations that occur in the biochemical process
of memory as a result of altered cAMP levels in the
mushroom body of the Drosophila brain appear to be the
predominant effect of dunce mutations.19 Because most
acrodysostosis cases exhibit significant developmental
disabilities, the data presented here raise the possibility
that PDE4D deficiency disrupts a highly evolutionarily
conserved neurological pathway.
Thus, a variety of genetic defects that alter cAMP metab-
olism produce disorders with a related constellation of
findings, which include short stature with brachydactyly,
endocrine abnormalities, and developmental disability.
However, the precise role of PDE4D in the skeleton, partic-
ularly in growth-plate cartilage, is not well understood.
Loss of cAMP activity as a result of a chondrocyte-specific
knockout of Gs-a revealed severe growth-plate abnormali-
ties, accelerated hypertrophic chondrocyte differentiation
with ectopic cartilage formation, and increased parathy-
roid hormone-related peptide expression in periarticular
chondrocytes.20 Individuals with acrodysostosis have
been reported to exhibit accelerated bone maturation
as well as ectopic bone formation,21,22 supporting the
hypothesis that a component of the cartilage phenotype
might be reduced activity of the cAMP signaling cascade.
It remains to be determined whether modulation of cAMP
levels could ameliorate the phenotypic consequences of
mutations in the pathway in any meaningful way, espe-
cially in the primary target tissues of the skeleton, brain,
and endocrine organs. Understanding the complexity of
cAMP regulation among the affected tissues would be an
important step in achieving this goal.
Acknowledgments
We would like to thank Traci Toy and Bret Harry at the University
of California, Los Angeles (UCLA) DNA Microarray Core for their
assistance with constructing the sequencing libraries and compu-
tational support, Suhua Feng at the UCLA Broad Stem Cell
750 The American Journal of Human Genetics 90, 746–751, April 6, 2012

Research Center for his assistance in running the HiSeq2000
instrument, Lisette Nevarez for assistance with Sanger-sequence
analysis, and both Nancy Kramer and Daniel Gruskin for assis-
tance with the clinical information. This study was supported in
part by the Steven Spielberg Pediatric Research Center at Cedars-
Sinai Medical Center and by National Institutes of Health grant
HD22657.
Received: December 2, 2011
Revised: March 1, 2012
Accepted: March 6, 2012
Published online: March 29, 2012
Web Resources
The URLs for data presented herein are as follows:
Exome Variant Server, http://evs.gs.washington.edu/EVS/
Genome Analysis Toolkit, ftp://gsapubftp-anonymous@ftp.
broadinstitute.org
Novocraft Short Read Alignment Package, http://www.novocraft.
com
Online Mendelian Inheritance in Man (OMIM), http://www.
omim.org
Picard, http://picard.sourceforge.net/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/bgi.shtml
SAMtools, http://samtools.sourceforge.net/
SeattleSeqAnnotation, http://snp.gs.washington.edu/
SeattleSeqAnnotation131/
SIFT, http://sift.jcvi.org/
References
1. Maroteaux, P., and Malamut, G. (1968). Acrodysostosis. Presse
Med. 76, 2189–2192.
2. Robinow, M., Pfeiffer, R.A., Gorlin, R.J., McKusick, V.A.,
Renuart, A.W., Johnson, G.F., and Summitt, R.L. (1971). Acro-
dysostosis. A syndrome of peripheral dysostosis, nasal hypo-
plasia, andmental retardation.Am. J. Dis. Child. 121, 195–203.
3. Jones, K.L., Smith, D.W., Harvey, M.A.S., Hall, B.D., and Quan,
L. (1975). Older paternal age and fresh genemutation: Data on
additional disorders. J. Pediatr. 86, 84–88.
4. Steiner, R.D., and Pagon, R.A. (1992). Autosomal dominant
transmission of acrodysostosis. Clin. Dysmorphol. 1, 201–206.
5. Sheela, S.R., Perti, A., and Thomas, G. (2005). Acrodysostosis:
Autosomal dominant transmission. Indian Pediatr. 42,
822–826.
6. Linglart, A., Menguy, C., Couvineau, A., Auzan, C., Gunes, Y.,
Cancel, M., Motte, E., Pinto, G., Chanson, P., Bougneres, P.,
et al. (2011). Recurrent PRKAR1A mutation in acrodysostosis
with hormone resistance. N. Engl. J. Med. 364, 2218–2226.
7. Graham, J.M., Jr., Krakow, D., Tolo, V.T., Smith, A.K., and
Lachman, R.S. (2001). Radiographic findings and Gs-alpha
bioactivity studies and mutation screening in acrodysostosis
indicate a different etiology from pseudohypoparathyroidism.
Pediatr. Radiol. 31, 2–9.
8. Yi, X., Liang, Y., Huerta-Sanchez, E., Jin, X., Cuo, Z.X., Pool,
J.E., Xu, X., Jiang, H., Vinckenbosch, N., Korneliussen, T.S.,
et al. (2010). Sequencing of 50 human exomes reveals adapta-
tion to high altitude. Science 329, 75–78.
9. Li, Y., Vinckenbosch, N., Tian, G., Huerta-Sanchez, E., Jiang,
T., Jiang, H., Albrechtsen, A., Andersen, G., Cao, H., Kornelius-
sen, T., et al. (2010). Resequencing of 200 human exomes
identifies an excess of low-frequency non-synonymous
coding variants. Nat. Genet. 42, 969–972.
10. Bolger, G.B., Erdogan, S., Jones, R.E., Loughney, K., Scotland,
G., Hoffmann, R., Wilkinson, I., Farrell, C., and Houslay,
M.D. (1997). Characterization of five different proteins
produced by alternatively spliced mRNAs from the human
cAMP-specific phosphodiesterase PDE4D gene. Biochem. J.
328, 539–548.
11. Houslay, M.D., and Adams, D.R. (2003). PDE4 cAMP phospho-
diesterases: Modular enzymes that orchestrate signalling
cross-talk, desensitization and compartmentalization. Bio-
chem. J. 370, 1–18.
12. Bertherat, J., Horvath, A., Groussin, L., Grabar, S., Boikos, S.,
Cazabat, L., Libe, R., Rene-Corail, F., Stergiopoulos, S., Bour-
deau, I., et al. (2009). Mutations in regulatory subunit type
1A of cyclic adenosine 50-monophosphate-dependent protein
kinase (PRKAR1A): Phenotype analysis in 353 patients and 80
different genotypes. J. Clin. Endocrinol. Metab. 94, 2085–
2091.
13. Jin, S.-L.C., Richard, F.J., Kuo, W.-P., D’Ercole, A.J., and Conti,
M. (1999). Impaired growth and fertility of cAMP-specific
phosphodiesterase PDE4D-deficient mice. Proc. Natl. Acad.
Sci. USA 96, 11998–12003.
14. Patten, J.L., Johns, D.R., Valle, D., Eil, C., Gruppuso, P.A.,
Steele, G., Smallwood, P.M., and Levine, M.A. (1990). Muta-
tion in the gene encoding the stimulatory G protein of adeny-
late cyclase in Albright’s hereditary osteodystrophy. N. Engl.
J. Med. 322, 1412–1419.
15. Dudai, Y., Jan, Y.N., Byers, D., Quinn, W.G., and Benzer, S.
(1976). dunce, a mutant of Drosophila deficient in learning.
Proc. Natl. Acad. Sci. USA 73, 1684–1688.
16. Byers, D., Davis, R.L., and Kiger, J.A., Jr. (1981). Defect in cyclic
AMP phosphodiesterase due to the dunce mutation of
learning in Drosophila melanogaster. Nature 289, 79–81.
17. Gong, Z., Xia, S., Liu, L., Feng, C., and Guo, A. (1998). Operant
visual learning and memory in Drosophila mutants dunce,
amnesiac and radish. J. Insect Physiol. 44, 1149–1158.
18. Zhong, Y., Budnik, V., and Wu, C.F. (1992). Synaptic plasticity
in Drosophila memory and hyperexcitable mutants: Role of
cAMP cascade. J. Neurosci. 12, 644–651.
19. Davis, R.L. (1996). Physiology and biochemistry of Drosophila
learning mutants. Physiol. Rev. 76, 299–317.
20. Sakamoto, A., Chen,M., Kobayashi, T., Kronenberg, H.M., and
Weinstein, L.S. (2005). Chondrocyte-specific knockout of the
G protein G(s)alpha leads to epiphyseal and growth plate
abnormalities and ectopic chondrocyte formation. J. Bone
Miner. Res. 20, 663–671.
21. Butler, M.G., Rames, L.J., and Wadlington, W.B. (1988). Acro-
dysostosis: Report of a 13-year-old boy with review of litera-
ture and metacarpophalangeal pattern profile analysis. Am.
J. Med. Genet. 30, 971–980.
22. Becker, S., Mausolf, A., and Laszig, R. (1989). Acrodysostosis:
an autosomal inherited form of peripheral dysostosis. HNO
37, 165–168.
The American Journal of Human Genetics 90, 746–751, April 6, 2012 751

CORRECTION
This Month in Genetics
Kathryn B. Garber*
(The American Journal of Human Genetics 90, 383–384; March 9, 2012)
In the summary titledNew Tools for Interpretation of Newborn-Screening Results, the first author of the paper being discussed
should have been Marquardt,’’ not ‘‘Marquard.’’
Marquardt et al. (2012) Genet Med. Published online February 16, 2012. 10.138/gim.2012.2.
*Correspondence: [email protected]
DOI 10.1016/j.ajhg.2012.03.010. �2012 by The American Society of Human Genetics. All rights reserved.
752 The American Journal of Human Genetics 90, 752, April 6, 2012

ERRATUM
Large-Scale Gene-Centric Meta-Analysis across39 Studies Identifies Type 2 Diabetes Loci
Richa Saxena,* Clara C. Elbers, Yiran Guo, Inga Peter, Tom R. Gaunt, Jessica L. Mega,Matthew B. Lanktree, Archana Tare, Berta Almoguera Castillo, Yun R. Li, Toby Johnson,Marcel Bruinenberg, Diane Gilbert-Diamond, Ramakrishnan Rajagopalan, Benjamin F. Voight,Ashok Balasubramanyam, John Barnard, Florianne Bauer, Jens Baumert, Tushar Bhangale,Bernhard O. Bohm, Peter S. Braund, Paul R. Burton, Hareesh R. Chandrupatla, Robert Clarke,Rhonda M. Cooper-DeHoff, Errol D. Crook, George Davey-Smith, Ian N. Day, Anthonius de Boer,Mark C.H. de Groot, Fotios Drenos, Jane Ferguson, Caroline S. Fox, Clement E. Furlong,Quince Gibson, Christian Gieger, Lisa A. Gilhuijs-Pederson, Joseph T. Glessner, Anuj Goel,Yan Gong, Struan F.A. Grant, Diederick E. Grobbee, Claire Hastie, Steve E. Humphries,Cecilia E. Kim, Mika Kivimaki, Marcus Kleber, Christa Meisinger, Meena Kumari, Taimour Y. Langaee,Debbie A. Lawlor, Mingyao Li, Maximilian T. Lobmeyer, Anke-Hilse Maitland-van der Zee,Matthijs F.L. Meijs, Cliona M. Molony, David A. Morrow, Gurunathan Murugesan, Solomon K. Musani,Christopher P. Nelson, Stephen J. Newhouse, Jeffery R. O’Connell, Sandosh Padmanabhan,Jutta Palmen, Sanjey R. Patel, Carl J. Pepine, Mary Pettinger, Thomas S. Price, Suzanne Rafelt,Jane Ranchalis, Asif Rasheed, Elisabeth Rosenthal, Ingo Ruczinski, Sonia Shah, Haiqing Shen,Gunther Silbernagel, Erin N. Smith, Annemieke W.M. Spijkerman, Alice Stanton,Michael W. Steffes, Barbara Thorand, Mieke Trip, Pim van der Harst, Daphne L. van der A,Erik P.A. van Iperen, Jessica van Setten, Jana V. van Vliet-Ostaptchouk, Niek Verweij,Bruce H.R. Wolffenbuttel, Taylor Young, M. Hadi Zafarmand, Joseph M. Zmuda,the Look AHEAD Research Group, DIAGRAM consortium, Michael Boehnke, David Altshuler,Mark McCarthy, W.H. Linda Kao, James S. Pankow, Thomas P. Cappola, Peter Sever, Neil Poulter,Mark Caulfield, Anna Dominiczak, Denis C. Shields, Deepak L. Bhatt, Li Zhang, Sean P. Curtis,John Danesh, Juan P. Casas, Yvonne T. van der Schouw, N. Charlotte Onland-Moret,Pieter A. Doevendans, Gerald W. Dorn II, Martin Farrall, Garret A. FitzGerald,Anders Hamsten Robert Hegele, Aroon D. Hingorani, Marten H. Hofker, Gordon S. Huggins,Thomas Illig, Gail P. Jarvik, Julie A. Johnson, Olaf H. Klungel, William C. Knowler, Wolfgang Koenig,Winfried Marz, James B. Meigs, Olle Melander, Patricia B. Munroe, Braxton D. Mitchell,Susan J. Bielinski, Daniel J. Rader, Muredach P. Reilly, Stephen S. Rich, Jerome I. Rotter,Danish Saleheen, Nilesh J. Samani, Eric E. Schadt, Alan R. Shuldiner, Roy Silverstein,Kandice Kottke-Marchant, Philippa J. Talmud, Hugh Watkins, Folkert W. Asselbergs,Paul I.W. de Bakker, Jeanne McCaffery, Cisca Wijmenga, Marc S. Sabatine, James G. Wilson,Alex Reiner, Donald W. Bowden, Hakon Hakonarson, David S. Siscovick, and Brendan J. Keating*
The American Journal of Human Genetics, 90, 410–425; March 2012
The originally published online version of this paper omitted two authors, Peter Sever and Neil Poulter, who have now
been added. Middle initials have also been added for Deepak L. Bhatt and Folkert W. Asselbergs. In addition, the ASCOT
and INVEST portions of the Supplemental Acknowledgments have been updated. The authors regret the errors.
*Correspondence: [email protected] (R.S.), [email protected] (B.J.K.)
DOI 10.1016/j.ajhg.2012.03.001. �2012 by The American Society of Human Genetics. All rights reserved.
The American Journal of Human Genetics 90, 753, April 6, 2012 753





























