Embed Size (px)
Citation preview

Research Signpost
37/661 (2), Fort P.O.
Trivandrum-695 023
Kerala, India
Evaluation of Electrochemical Reactors as a New Way to Environmental Protection, 2014: 59-78 ISBN: 978-81-308-0549-8 Editors: Juan M. Peralta-Hernández, Manuel A. Rodrigo-Rodrigo and
Carlos A. Martínez-Huitle
4. BDD electrochemical reactors
Ignasi Sirés and Enric Brillas Laboratori d’Electroquímica dels Materials i del Medi Ambient
Departament de Química Física, Facultat de Química, Universitat de Barcelona c/ Martí i Franquès 1-11, 08028 Barcelona, Spain
Abstract. The extraordinary performance of BDD electrodes at
laboratory scale for a wide range of applications, particularly the
environmental protection, has fostered in recent years their
integration in electrochemical reactors as high power, large
O2-evolution overpotential anodes. A large variety of both,
commercial and purpose-made systems, have been used for the
decontamination and disinfection of synthetic and real aqueous
solutions. This chapter pays a look to the fundamentals of direct and
mediated electro-oxidation with BDD, and then describes the
influence of key operation parameters on the degradation rate,
current efficiency and energy consumption. Special focus is put on
the remediation of waters containing organic pollutants, since the
vast majority of studies in the literature refer to the degradation of
industrial chemicals, pesticides, pharmaceuticals and dyes.
Introduction
Over the last 15 years, the use of boron-doped diamond (BDD) thin-film
electrodes for the electrochemical treatment (primordially via
electrochemical oxidation (EO)) of organic contaminants contained in waters
Correspondence/Reprint request: Prof. Enric Brillas, Departament de Química Física, Facultat de Química
Universitat de Barcelona, c/ Martí i Franquès 1-11, 08028 Barcelona, Spain. E-mail: [email protected]

Ignasi Sirés & Enric Brillas 60
has received increasing attention. BDD is synthesized by chemical reduction
of CH4(g) with H2(g) in the presence of B2O3(g) at near 825 ºC and deposited onto
a conductive substrate such as Si, Ti, Nb or a carbonaceous material [1-3].
Thin films thus obtained, usually having between 1 and 10 m thickness,
possess technologically relevant characteristics including an inert surface with
low adsorption properties, remarkable corrosion stability even in strongly
acidic media and extremely large O2-evolution overpotential. Thanks to these
properties, they are excellent anodic materials for EO, with much higher
oxidation ability than other common materials like Pt and PbO2 [2,3]. This
chapter is devoted to describe the main applications of BDD electrochemical
reactors. Firstly, the fundamentals of the oxidation process with BDD anodes
are explained. The treatment of polluted waters by different kinds of either
bench-scale or pilot plants is further discussed, giving details of the most
typical parameters for assessing the current efficiency and energy
consumption. Finally, the effects of relevant operating variables on the
performance of EO with a BDD anode are examined.
1. Fundamentals
EO is the most popular electrochemical method for removing organic
pollutants from wastewaters [2]. This technique has been recently applied to
degrade industrial chemicals, pesticides, pharmaceuticals and dyes from
aqueous solutions. It is based on the oxidation of pollutants and their
reaction intermediates in an electrolytic cell by:
(i) Direct anodic oxidation (or direct electron transfer to the anode), which
yields very poor decontamination.
(ii) Chemical reaction with species that are electrogenerated from water
discharge at the anode M, such as physically adsorbed active oxygen
(physisorbed hydroxyl radical M( OH)) or chemisorbed active oxygen
(oxygen in the lattice of a metal oxide anode (MO)). These oxidizing
species can yield total or partial decontamination, respectively.
Two main approaches have been proposed for the abatement of
wastewater pollution by EO based on the indirect or mediated oxidation with
different heterogeneous species formed from water discharge [2,4,5]:
(i) The electrochemical conversion method, in which organics are
selectively converted into biodegradable compounds, usually carboxylic
acids, by chemisorbed active oxygen.
(ii) The electrochemical combustion (or electrochemical incineration)
method, in which organics become totally mineralized, i.e., oxidized to
CO2, water and inorganic ions, by physisorbed OH. This radical is the

BDD electrochemical reactors 61
second strongest oxidant known after fluorine, with a high standard
reduction potential (Eº = 2.80 V/SHE) that ensures its fast reaction with
most organics giving dehydrogenated or hydroxylated derivatives up to
their mineralization.
In both cases, high potential differences are required between the
electrodes of the electrolytic cell to simultaneously oxidize pollutants and
water, thus maintaining the anode activity. In contrast, low potential
differences hinder the O2 evolution and frequently cause the loss of the
anode surface activity due to the adsorption of by-products involved in the
direct anodic oxidation of the initial organics and, consequently, EO cannot
actually be utilized for wastewater treatment. The nature of the anode
material has a strong influence on both the selectivity and efficiency of the
EO process. This was explained by Comninellis [4] through a
comprehensive model for organics destruction in acidic medium including
the competition with the O2-evolution reaction (OER). Its predictions fit
quite well with results obtained with BDD anodes, which present the largest
O2-overpotential known [6].
According to the Comninellis’ model, the anodes can be classified
according to two extreme cases: the so-called active and non-active anodes.
Typical examples are Pt, IrO2 and RuO2 for the former and PbO2, SnO2 and
BDD for the latter. The proposed model assumes that the initial reaction in
both kind of anodes (denoted as M) is the oxidation of water molecules by
reaction (1) to give the physisorbed M( OH):
M + H2O M( OH) + H+ + e (1)
Both the electrochemical and chemical reactivity of M( OH) depend on
the electrode material. The surface of active anodes interacts strongly with
OH and, as a result, a higher oxide or superoxide MO can be formed from
reaction (2). This occurs when higher oxidation states are available for a
metal oxide anode above the standard potential for O2 evolution (Eº = 1.23
V/SHE).
M( OH) MO + H+ + e (2)
The redox couple MO/M acts as a mediator in organics oxidation via
reaction (3), which competes with the side OER via chemical decomposition
of the higher oxide species from reaction (4).
MO + R M + RO (3)
MO M + ½ O2(g) (4)

Ignasi Sirés & Enric Brillas 62
In contrast, the surface of a non-active anode interacts so weakly with
OH that organics react with M( OH) to yield fully-oxidized reaction
products such as CO2 via the overall reaction (5):
a M( OH) + R M + m CO2 + n H2O + x H+ + y e (5)
where R is an organic compound with m carbon atoms and without any
heteroatom, which needs a = (2m + n) oxygen atoms to be totally
mineralized to CO2. The oxidative reaction (3) with chemisorbed MO is
much more selective than the mineralization reaction (5) with physisorbed
hydroxyl radical. The latter reaction also competes with waste reactions of
M( OH) such as its direct oxidation to O2 by reaction (6) or its indirect
consumption through dimerization to hydrogen peroxide by reaction (7):
M( OH) M + ½ O2 + H+ + e (6)
2 M( OH) 2 M + H2O2 (7)
According to the aforementioned model, it is expected that a non-active
anode acts as an inert substrate and as a sink for the removal of electrons,
only allowing outer-sphere reactions and water oxidation. Thus, hydroxyl
radical produced from water discharge by reaction (1) is subsequently
involved in the oxidation process of organics. Furthermore, the
electrochemical activity (related to the overvoltage for O2 evolution) and
chemical reactivity (related to the rate of organics oxidation) of physisorbed
M( OH) are closely related to the strength of the M- OH interaction. In
general, the weaker the interaction, the faster the chemical reaction of
organics with M( OH). Since BDD thin-films are the best non-active
electrodes verifying this behavior, they have been proposed as the most
suitable anodes for EO [6].
On the other hand, other weaker oxidants such as ozone can be produced
from water oxidation at the anode by reaction (8) with Eº = 1.51 V/SHE [5]:
3 H2O O3 + 6 H+ + 6 e (8)
Although in EO, reactive oxygen species (ROS) such as M( OH), H2O2
and O3 are formed by reaction (1), (7) and (8), respectively, pollutants are
primordially oxidized by the strongest oxidant M( OH). In fact, this radical
has such a short lifetime that only acts while direct current is supplied to the
anode.

BDD electrochemical reactors 63
When a BDD anode is utilized, weaker oxidizing species like
peroxodisulfate, peroxodicarbonate and peroxodiphosphate ions can be
competitively produced along with ROS from the anodic oxidation of
sulfate, bicarbonate and phosphate ions present in the electrolyte as follows
[1]:
2 HSO4 S2O82
+ 2 H+ + 2 e (9)
2 HCO3 C2O62
+ 2 H+ + 2 e (10)
2 PO43
P2O84
+ 2 e (11)
A different behavior is found when solutions to be treated by EO contain
chloride ions. Active chlorine species (mainly Cl2, HClO and/or ClO )
formed from Cl oxidation at the anode can effectively degrade some
pollutants in competition with ROS. It is known [2,7] that the electrolysis of
Cl aqueous solutions involves its direct oxidation on the anode to yield
soluble chlorine by reaction (12):
2 Cl Cl2(aq) + 2 e (12)
If the local concentration of dissolved chlorine exceeds its solubility,
supersaturation drives the formation of chlorine gas bubbles. When
electrogenerated chlorine diffuses away from the anode, it can react with
chloride ion to form trichloride ion by reaction (13) or it can be
disproportionated through hydrolysis to yield hypochlorous acid and Cl ion
by reaction (14).
Cl2(aq) + Cl Cl3 (13)
Cl2(aq) + H2O HClO + Cl + H+ (14)
In the solution bulk, hypochlorous acid is in equilibrium with
hypochlorite ion (pKa = 7.55) according to reaction (15):
HClO ClO + H+ (15)
Usually, Cl3 ion is formed in very low concentration up to pH near 4,
whereas the predominant species is Cl2(aq) up to pH close to 3, HClO in the
pH range 3-8 and ClO for pH > 8.0. The mediated oxidation of organics
with these species is then expected to be faster in acidic than in alkaline
media, due to the higher standard potential of Cl2(aq) (Eº = 1.36 V/SHE) and
HClO (Eº = 1.49 V/SHE) compared to that of ClO ion (Eº = 0.89 V/SHE).

Ignasi Sirés & Enric Brillas 64
The concentration of electrogenerated ClO can be limited by its anodic
oxidation to chlorite ion from reaction (16) and consecutive oxidation to
chlorate and perchlorate ions from reactions (17) and (18), respectively
[2,7]:
ClO + H2O ClO2 + 2 H+ + 2 e (16)
ClO2 + H2O ClO3 + 2 H+ + 2 e (17)
ClO3 + H2O ClO4 + 2 H+ + 2 e (18)
The loss of ClO ion is also possible from waste reactions (19)-(21) in
the solution bulk as well as by reduction to Cl ion on the cathode of an
undivided cell from reaction (22):
2 HClO + ClO ClO3 + 2 Cl + 2 H+ (19)
2 ClO 2 Cl + O2 (20)
ClO + H2 Cl + H2O (21)
ClO + H2O + 2 e Cl + 2 OH (22)
The rate of electrode reactions for generation of M( OH) and other ROS
as well as for Cl2(aq), ClO2 , ClO3 and ClO4 production at the BDD anode
depends mainly on current density, Cl concentration, flow rate and
temperature. In contrast, the rate of homogeneous chemical reactions is a
function of the diffusion rate of organic pollutants through the solution,
which is influenced by the flow rate, the concentration of such pollutants,
temperature and pH. In sections below, the role of these variables during the
degradation of organics using BDD electrochemical reactors will be
discussed.
2. Treatment of organics using BDD electrochemical reactors
A large variety of electrochemical plants and reactors has been tested for
the removal of organic pollutants from waters by EO with a BDD anode.
Figure 1 exemplifies some bench-scale systems containing different
electrochemical reactors operating in batch or in continuous mode [8-14].
Most reactors in that figure are undivided flow cells equipped with planar
(rectangular or circular) and monopolar electrodes in parallel-plate
configuration, like reactors (a), (b) and (d). The bipolar trickle tower reactor
shown in Figure 1c contained Raschig rings coated with BDD, thus giving

BDD electrochemical reactors 65
Figure 1. Sketches of electrochemical plants and cells used in EO. Bench-scale flow
plants with a: (a) One-compartment flow reactor with a turbulence promoter (C)
operating either in batch (A) or in continuous (B) mode (adapted from ref. [8,9]), (b)
one-compartment flow reactor with parallel-plate electrodes working in batch mode
(adapted from ref. [10]) and (c) bipolar trickle tower reactor in batch mode (adapted
from ref. [11,12]). (d) FM01-LC filter-press flow reactor (adapted from ref. [13,14]).
rise to a large total surface of this material. Scialdone et al. [15,16] have
utilized microreactors with a micro-gap between electrodes that are even able
to destroy pollutants in pure water without the addition of any electrolyte.
The high oxidation ability of BDD( OH) formed from water discharge by
reaction (1) on the BDD surface in the above reactors ensures the large
effectiveness of EO, although slower reactions with other ROS (H2O2 and O3)
and weaker electrogenerated oxidants (peroxodisulfate, peroxodicarbonate or
peroxodiphosphate) are also feasible. This behavior has been corroborated for
synthetic solutions of several industrial chemicals, pesticides, dyes and

Ignasi Sirés & Enric Brillas 66
pharmaceuticals [12,13,15-35], as well as for urban and industrial wastewaters
[9-11,36-44]. The decontamination process of organic pollutants in the
effluents is monitored from the abatement of their chemical oxygen demand
(COD) and/or total organic carbon (TOC). From COD data, for example, the
percentage of COD removal is calculated as follows:
(23)
where COD is the experimental COD decay (in mg L-1
) at electrolysis
time t and COD0 is the corresponding initial value before treatment. A
similar equation can be defined for the percentage of TOC removal.
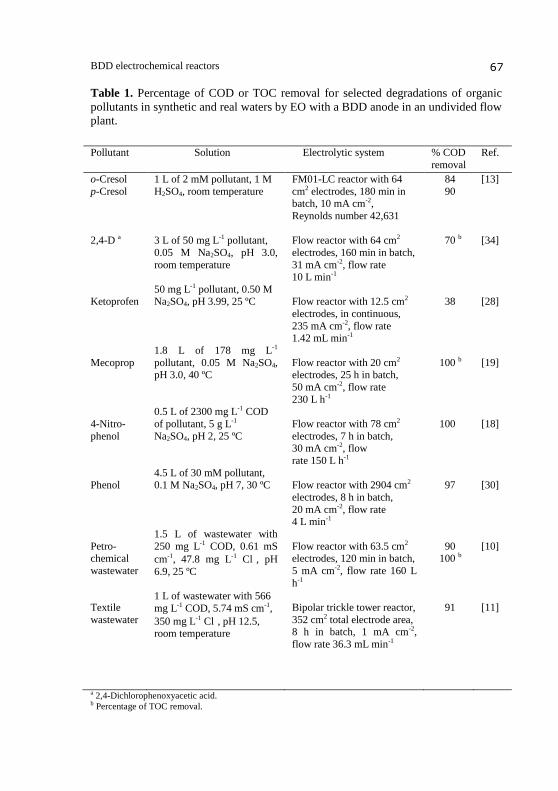
Table 1 collects the percentage of COD or TOC removal for selected EO
experiments. As can be seen, large percentages of organic matter removal
were found in most cases operating in batch mode during prolonged
electrolysis time. In contrast, a poor mineralization (only 38%) was obtained
for 50 mg L-1
ketoprofen under continuous conditions working at a high
current density of 235 mA cm-2
and a very low liquid flow rate of 1.42 mL
min-1
[28]. The presence of Cl ion in the effluent leads to the additional
production of active chlorine species (Cl2/HClO/ClO ) from reactions (12)-
(15), which can also destroy organics in competence with BDD( OH), other
ROS and weaker oxidants produced from other anions. Although the
degradation is usually enhanced in the presence of Cl ion, several works
have demonstrated the generation of hazardous by-products such as
organochlorides, chloramines, trihalomethanes and inorganic ions like ClO3
and ClO4 [9,10,21,27], even in drinking water [45]. Determination of all
these species during the EO processes with a BDD anode is then necessary
to confirm whether their viability is possible, especially when actual urban
and industrial wastewaters with high Cl contents are treated. Several coupled and integrated processes have been proposed and
applied to the treatment of wastewaters in order to enhance the degradation
power of EO with a BDD anode [46-58]. Coupled processes involve solar
photoelectro-Fenton [46,49,51,53], EO/Microwave [48], EO with reverse
osmosis/nanofiltration [52], EO with generated H2O2/UV-C photolysis [58]
and EO/Microfiltration [57]. Examples of electrochemical plants used for the
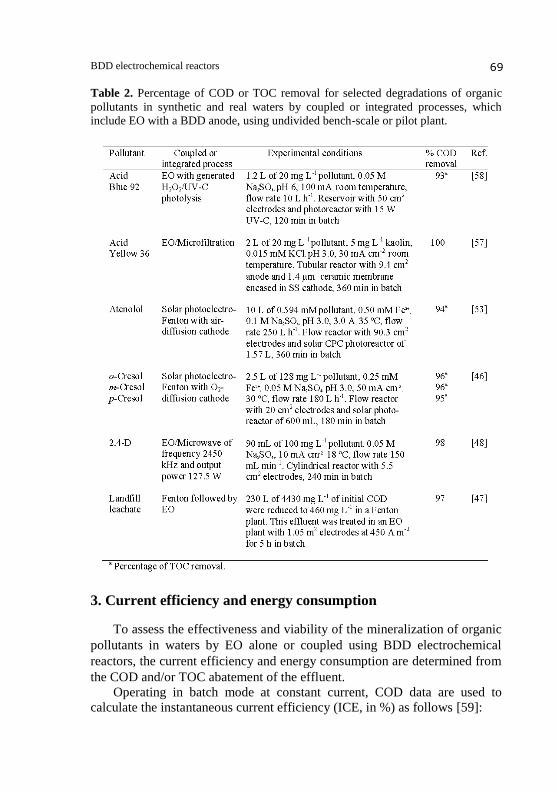
two first techniques are schematized in Figure 2. Table 2 shows that ≥ 93%
mineralization was achieved for different synthetic solutions under treatment
with the above coupled processes, which were more effective than the
corresponding EO ones. For example, the use of EO/Microwave for the
degradation of 100 mg L-1
of the pesticide 2,4-D (2,4-dichlorophenoxyacetic
acid) under the conditions given in Table 2 yielded 98% COD removal after
240 min of electrolysis in batch, whereas the comparative EO process only
BDD electrochemical reactors 67
Table 1. Percentage of COD or TOC removal for selected degradations of organic
pollutants in synthetic and real waters by EO with a BDD anode in an undivided flow
plant.
Pollutant Solution Electrolytic system % COD
removal
Ref.
o-Cresol
p-Cresol
2,4-D a
Ketoprofen
Mecoprop
4-Nitro-
phenol
Phenol
Petro- chemical
wastewater
Textile
wastewater
1 L of 2 mM pollutant, 1 M
H2SO4, room temperature
3 L of 50 mg L-1 pollutant,
0.05 M Na2SO4, pH 3.0,
room temperature
50 mg L-1 pollutant, 0.50 M Na2SO4, pH 3.99, 25 ºC
1.8 L of 178 mg L-1
pollutant, 0.05 M Na2SO4, pH 3.0, 40 ºC
0.5 L of 2300 mg L-1 COD
of pollutant, 5 g L-1
Na2SO4, pH 2, 25 ºC
4.5 L of 30 mM pollutant, 0.1 M Na2SO4, pH 7, 30 ºC
1.5 L of wastewater with
250 mg L-1 COD, 0.61 mS
cm-1, 47.8 mg L-1 Cl , pH
6.9, 25 ºC
1 L of wastewater with 566
mg L-1 COD, 5.74 mS cm-1,
350 mg L-1 Cl , pH 12.5, room temperature
FM01-LC reactor with 64
cm2 electrodes, 180 min in batch, 10 mA cm-2,
Reynolds number 42,631
Flow reactor with 64 cm2
electrodes, 160 min in batch,
31 mA cm-2, flow rate 10 L min-1
Flow reactor with 12.5 cm2
electrodes, in continuous,
235 mA cm-2, flow rate 1.42 mL min-1
Flow reactor with 20 cm2 electrodes, 25 h in batch,
50 mA cm-2, flow rate
230 L h-1
Flow reactor with 78 cm2
electrodes, 7 h in batch, 30 mA cm-2, flow
rate 150 L h-1
Flow reactor with 2904 cm2
electrodes, 8 h in batch,
20 mA cm-2, flow rate 4 L min-1
Flow reactor with 63.5 cm2 electrodes, 120 min in batch,
5 mA cm-2, flow rate 160 L
h-1
Bipolar trickle tower reactor,
352 cm2 total electrode area, 8 h in batch, 1 mA cm-2,
flow rate 36.3 mL min-1
84
90
70 b
38
100 b
100
97
90 100 b
91
[13]
[34]
[28]
[19]
[18]
[30]
[10]
[11]
a 2,4-Dichlorophenoxyacetic acid. b Percentage of TOC removal.

Ignasi Sirés & Enric Brillas 68
Figure 2. Schemes of (a) a 2.5 L bench-scale flow plant and (b) one-compartment
filter-press electrochemical reactor with a BDD anode and a gas (O2 or air) diffusion
cathode, both of 20 cm2 area, used for solar photoelectro-Fenton (SPEF) treatment.
Adapted from ref. [46]. (c) Experimental setup for EO degradation with microwave
activation of the BDD anode at frequency of 2450 MHz and output power of 127.5 W
using a one-compartment cylindrical reactor. Adapted from ref. [48].
reduced the COD by 67% [48]. This increase in mineralization degree was
associated to the enhancement of the mass transport of organics toward the
BDD anode, which was induced by the high frequency of the microwave
radiation applied. On the other hand, interesting integrated pilot plants have
been used by the Ortiz group for the decontamination of landfill leachate by
Fenton followed by EO [47] and municipal wastewaters by ultrafiltration/
reverse osmosis followed by EO [55]. For the former integrated method, for
example, 97% COD removal was found starting from a landfill leachate with
4430 mg L-1
COD, as shown in Table 2.
BDD electrochemical reactors 69
Table 2. Percentage of COD or TOC removal for selected degradations of organic
pollutants in synthetic and real waters by coupled or integrated processes, which
include EO with a BDD anode, using undivided bench-scale or pilot plant.
3. Current efficiency and energy consumption
To assess the effectiveness and viability of the mineralization of organic
pollutants in waters by EO alone or coupled using BDD electrochemical
reactors, the current efficiency and energy consumption are determined from
the COD and/or TOC abatement of the effluent.
Operating in batch mode at constant current, COD data are used to
calculate the instantaneous current efficiency (ICE, in %) as follows [59]:

Ignasi Sirés & Enric Brillas 70
(24)
where F is the Faraday constant (96,485 C mol-1
), Vs is the solution volume
(in L), COD is the experimental COD decay (in mg L-1
) at the time
interval t (in s) and I is the current (in A).
The Comninellis’ group [60] has proposed a kinetic model to explain the
change in COD and ICE when a pure organic of formula CxHyOz undergoes
electrochemical incineration in a recirculation plant at constant current. This
model is based on the variation of the limiting current density during the
electrolysis, which is determined from Eq. (25):
(25)
where ilim(t) is the limiting current density (in A m-2
) at a given time t, km is
the average mass-transport coefficient in the electrochemical reactor (in m s-1
)
and COD(t) (in mol m-3
) is the chemical oxygen demand at the same time t.
Figures 3a and b show the two regimes (A and B) identified depending
on the ratio = iappl/ilimo, where iappl is the applied current density and ilim
o is
the initial limiting current density:
(i) Regime A (iappl < ilim(t)): the electrolysis is under current control, COD
decreases linearly with time and ICE is 100%.
(ii) Regime B (iappl > ilim(t)): the electrolysis is under mass transport control
and secondary reactions (such as O2 evolution) start. Both COD removal
and ICE follow an exponential trend due to mass-transport limitation.
A good agreement between experimental and predicted values from the
above kinetic model has been found for both COD and ICE evolutions
during the EO with a BDD anode of different organic compounds like acetic
acid, isopropanol, phenol, 4-chlorophenol and 2-naphthol in 1 M H2SO4
[61].
When TOC removal of a solution containing a pure organic is
monitored, the mineralization current efficiency (MCE, in %) at constant I
(in A) and electrolysis time t (in h) can be estimated from the expression
[62]:
(26)
where n is the number of electrons exchanged in the mineralization process
of the organic compound, 4.32×107 is a conversion factor (= 3,600 s h
-1
12,000 mg carbon mol-1
) and m is the number of carbon atoms of the
molecule under study.

BDD electrochemical reactors 71
Energy parameters are essential figures-of-merit to assess the viability of
the electrochemical treatment for industrial application. Operating at
constant I, the energy consumptions per unit volume (EC) and unit TOC
mass (ECTOC) are obtained from Eqs. (27) and (28), respectively [51]:
(27)
(28)
where Ecell is the average potential difference of the cell (in V). A similar
equation to (28) is applied to determine the energy consumption per unit
COD mass (ECCOD, in kWh (g COD)-1
).
Figure 3. Evolution of (a) COD and (b) instantaneous current efficiency with
electrolysis time. (A) represents the charge transfer control and (B) represents the
mass transport control. Symbols: COD0 = initial chemical oxygen demand (in mol
O2 m-3), = iappl/ilim
o, VR = reservoir volume (in m3), km
= mass-transport coefficient
in the reactor (in m s-1) and A= electrode area (m2). Adapted from ref. [60].
4. Effect of operating parameters
It has been reported that the mineralization rate, current efficiency and
energy consumption for the EO treatment of organic pollutants in a BDD
electrochemical reactor depend on several operating parameters that include
primordially the applied current (or current density), temperature, flow rate,
Ignasi Sirés & Enric Brillas 72
pollutant concentration, pH and the electrolyte type and its concentration.
For each electrolysis, a careful study of these variables is necessary to find
the best experimental conditions to be applied.
The current density is a key parameter since it regulates the amounts of
ROS and other weaker electrogenerated oxidants that are able to destroy the
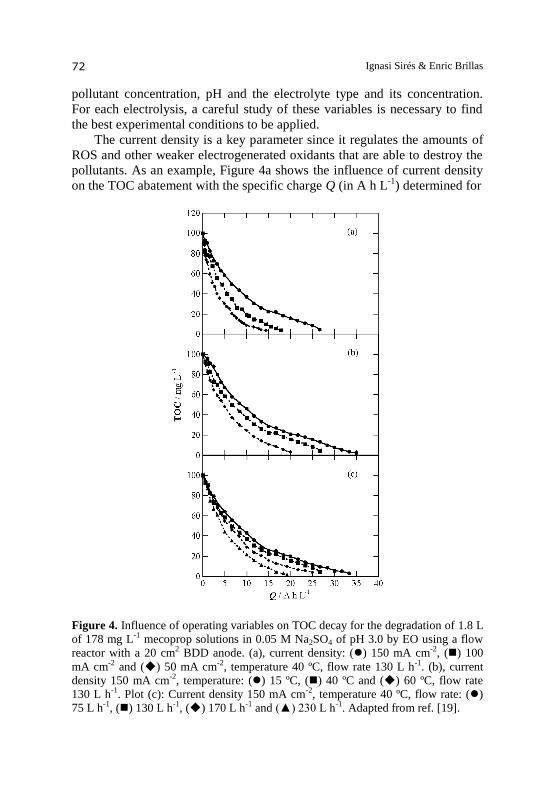
pollutants. As an example, Figure 4a shows the influence of current density
on the TOC abatement with the specific charge Q (in A h L-1
) determined for
Figure 4. Influence of operating variables on TOC decay for the degradation of 1.8 L
of 178 mg L-1 mecoprop solutions in 0.05 M Na2SO4 of pH 3.0 by EO using a flow
reactor with a 20 cm2 BDD anode. (a), current density: () 150 mA cm-2, () 100
mA cm-2 and () 50 mA cm-2, temperature 40 ºC, flow rate 130 L h-1. (b), current
density 150 mA cm-2, temperature: () 15 ºC, () 40 ºC and () 60 ºC, flow rate
130 L h-1. Plot (c): Current density 150 mA cm-2, temperature 40 ºC, flow rate: ()
75 L h-1, () 130 L h-1, () 170 L h-1 and (▲) 230 L h-1. Adapted from ref. [19].

BDD electrochemical reactors 73
the EO degradation of 1.8 L of 178 mg L-1
mecoprop solutions in 0.05 M
Na2SO4 at pH 3.0 and 40 ºC using a plant like that of Figure 2a, but only
equipped with a flow reactor with a 20 cm2 BDD anode, at flow rate of
130 L h-1
[19]. An increase in current density from 50 to 150 mA cm-2
caused a rise in specific charge from 14 to 27 A h L-1
for reaching overall
decontamination, although TOC was more rapidly reduced since the time
needed for total mineralization dropped from 25 to 17 h. The faster TOC
removal with time at higher current density can then be ascribed to the
concomitant greater production of BDD( OH) from reaction (1), thus
accelerating the oxidation rate of all organics. However, the higher specific
charge required for total decontamination indicates a gradually slower
accumulation of BDD( OH) due to the progressive acceleration of its
parasitic non-oxidizing reactions. These wasting reactions involve the
primary oxidation of this radical to O2 by reaction (6) or its reaction with
H2O2 (generated via reaction (7)) to produce hydroperoxyl radical (HO2 ) by
reaction (29) [2,19,34,51]. Moreover, the parallel increase in rate of O3
(reaction (8)) and peroxodisulfate (reaction (9)) formation can also reduce
the accumulation of BDD( OH) at the anode.
(29)
The above behavior is also reflected in the concomitant decay in current
efficiency and the higher energy consumption. At 3 h of electrolysis, for
example, MCE dropped from 23% at 50 mA cm-2
(Q = 1.67 A h L-1
) to 8.9%
at 150 mA cm-2
(Q = 5 A h L-1
). In contrast, the final EC was raised from
149 kWh m-3
at 50 mA cm-2
in 25 h to 700 kWh m-3
at 150 mA cm-2
in 17 h.
The increase in temperature is expected to cause a greater mass transport
toward the BDD anode due to the decrease of medium viscosity. This
phenomenon can explain the tendency found when the temperature of the
above 178 mg L-1
mecoprop solution varied from 15 to 60 ºC operating at
150 mA cm-2
and flow rate of 130 L h-1
. Under these conditions, Figure 4b
shows a more rapid TOC destruction at higher temperature, decreasing the
required specific charge for total decontamination from 35 A h L-1
at 15 ºC
to 20 A h L-1
at 60 ºC. The mecoprop mineralization was then accelerated
because of the faster reaction of pollutants with BDD(●OH) as a result of the
inhibition of its wasting reactions since more amounts of organics could be
transported toward the BDD surface. This mass-transport limitation
undergone by organics to arrive to the anode was confirmed by increasing
the flow rate of the solution, as shown in Figure 4c for the same solution
electrolyzed at 150 mA cm-2
and 40 ºC. A progressively faster TOC decay
was observed when the flow rate was raised, then dropping the specific
Ignasi Sirés & Enric Brillas 74
charge for total mineralization from 34 A h L-1
(20 h) at 75 L h-1
to 18 A h L-1
(11 h) at 230 L h-1
.
The aforementioned results indicate an enhancement of the EO process
with a BDD anode upon increase of temperature and flow rate, yielding
higher current efficiency and lower energy consumption. The same behavior
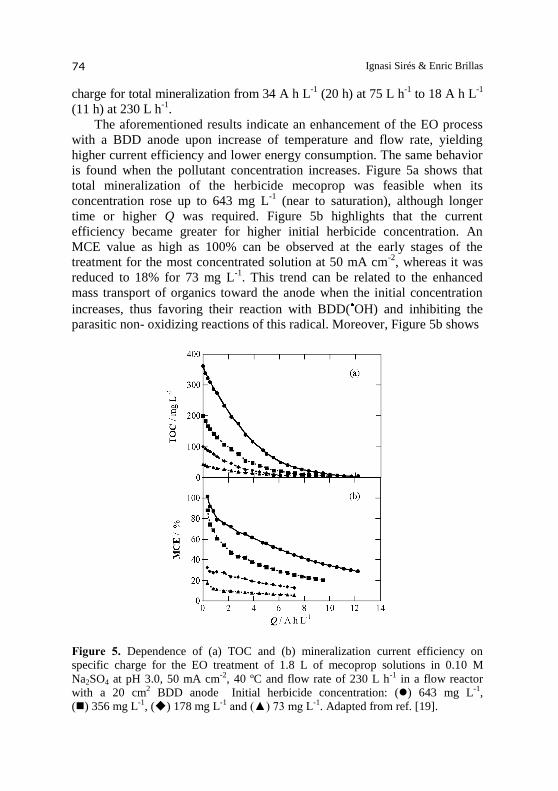
is found when the pollutant concentration increases. Figure 5a shows that
total mineralization of the herbicide mecoprop was feasible when its
concentration rose up to 643 mg L-1
(near to saturation), although longer
time or higher Q was required. Figure 5b highlights that the current
efficiency became greater for higher initial herbicide concentration. An
MCE value as high as 100% can be observed at the early stages of the
treatment for the most concentrated solution at 50 mA cm-2
, whereas it was
reduced to 18% for 73 mg L-1
. This trend can be related to the enhanced
mass transport of organics toward the anode when the initial concentration
increases, thus favoring their reaction with BDD( OH) and inhibiting the
parasitic non- oxidizing reactions of this radical. Moreover, Figure 5b shows
Figure 5. Dependence of (a) TOC and (b) mineralization current efficiency on
specific charge for the EO treatment of 1.8 L of mecoprop solutions in 0.10 M
Na2SO4 at pH 3.0, 50 mA cm-2, 40 ºC and flow rate of 230 L h-1 in a flow reactor
with a 20 cm2 BDD anode Initial herbicide concentration: () 643 mg L-1,
() 356 mg L-1, () 178 mg L-1 and (▲) 73 mg L-1. Adapted from ref. [19].

BDD electrochemical reactors 75
a dramatic decay of the current efficiency when all electrolyses were
prolonged up to achieve total decontamination. This behavior has been
explained by the progressive decrease of organic matter with formation of
more hardly oxidizable by-products [2]. At the end of the degradation of the
643 mg L-1
mecoprop solution after consumption of 12.2 A h L-1
(22 h) by
applying 50 mA cm-2
at 40 ºC and flow rate of 230 L h-1
, MCE = 29% and
EC = 143 kWh m-3
(ECTOC = 0.401 kWh (g TOC)-1
) were obtained.
While high temperature, flow rate and organic concentration and low
current density are desirable for improving the performance of the EO
treatment with a BDD anode, the effect of pH seems related to the effluent
composition. In the presence of Na2SO4, several authors [19,28] have
reported a slight influence of pH in the range 2-12 on the degradation of
organics, even when varying the electrolyte concentration from 0.05 to 0.50 M
[19]. In contrast, Weiss et al. [22] have described that solutions of sodium
dodecylbenzenesulfonate, a common surfactant, treated in a system like that
of Figure 1b exhibited lower TOC removal rates in alkaline media of pH 12
than in acidic or neutral solutions, due to concurrent oxidation of dissolved
carbonates from reaction (10) at potentials less positive than that required for
water oxidation. When Cl ion is present in the medium, the active chlorine
species formed via reactions (12)-(15) also attack the organic matter and the
degradation is enhanced in acidic media compared to the alkaline ones
because of the higher standard potential of Cl2/HClO than of ClO ion,
which predominates in the latter effluent. This behavior has been confirmed,
for example, by Scialdone et al. [27] for the electrochemical incineration of
250 mL of 100 mg L-1
oxalic acid in Na2SO4 (pH 2) or NaOH (pH 12)
solutions using a flow plant and an electrochemical reactor like that of
Figure 1b after injection of 7,000 C at 39 mA cm-2
, 25 ºC and flow rate of
0.2 L min-1
. In the absence of NaCl, 81-83% mineralization with 56-57%
current efficiency was found in both media. In contrast, by adding 10 g L-1
NaCl, the mineralization increased up to 93% with 65% current efficiency at
pH 2, but it was only reduced by 72% with 48% current efficiency at pH 12
as a result of the greater oxidation ability of active chlorine species
generated in the former medium, as stated above.
5. Conclusion
A variety of BDD electrochemical reactors are available nowadays for
water treatment. Robust equipments based on the commercial DiaCell® have
been successfully integrated in swimming pools and spas for ensuring an
effective disinfection. The conventional FM01-LC reactor, as well as
innovative microfluidic cells based on the ElectroCell AB, have also been well

Ignasi Sirés & Enric Brillas 76
proven for batch or continuous decontamination. However, the preponderant
systems for the EO treatment of organic pollutants involve purpose-made
filter-press reactors, which have shown a high ability for the complete
degradation of pesticides, dyes, pharmaceuticals and common industrial
contaminants contained in synthetic solutions and real wastewaters. BDD,
when used as an anode, is able to produce a very oxidizing mixture, which
depending on the composition of the electrolyte can contain different ROS
( OH, H2O2, O3), active chlorine and peroxosalts. In such media, the organic
pollutants and their reaction by-products can be even completely mineralized.
Furthermore, if current density and flow rate, which tend to become the two
key operating parameters, are accurately chosen, a 100% TOC removal can be
attained along with a high current efficiency and moderate energy
consumption when highly concentrated solutions are treated. The integration
of EO with BDD anodes in existing water treatment facilities, as well as
further development of coupled processes recently proposed can definitely
enhance the economic viability of the BDD technology, finally allowing its
real implementation for treating wastewaters at real scale.
References
1. Kraft, A. 2007, Int. J. Electrochem. Soc., 2, 355.
2. Panizza, M., Cerisola, G. 2009, Chem. Rev., 109, 6541.
3. Brillas, E., Martínez-Huitle, C. A., 2011, Synthetic Diamond Films:
Preparation, Electrochemistry, Characterization and Applications, John Wiley
& Sons, Hoboken, NJ, USA.
4. Comninellis, Ch. 1994, Electrochim. Acta, 39, 1857.
5. Martínez-Huitle, C. A., Ferro, S. 2006, Chem. Soc. Rev., 35, 1324.
6. Marselli, B., Garcia-Gomez, J., Michaud, P. A., Rodrigo, M. A., Comninellis,
Ch. 2003, J. Electrochem. Soc., 150, D79.
7. Rajkumar, D., Kim, J. G. 2006, J. Hazard. Mater., B136, 203.
8. Vacca, A., Mascia, M., Palmas, S., Mais, L., Rizzardini, S. 2013, J. Chem.
Technol. Biotechnol., 88, 2244.
9. Mascia, M., Vacca, A., Palmas, S. 2013, Chem. Eng. J., 219, 512.
10. Cabral da Silva, A. J., Vieira dos Santos, E., Morais, C. C. O., Martínez-Huitle,
C. A., Castro, S. S. L. 2013, Chem. Eng. J., 233, 47.
11. Koparal, A. S., Yavuz, Y., Gürel, C., Öğütveren, U. B. 2007, J. Hazard. Mater.,
145, 100.
12. Yavuz, Y., Koparal, A. S., Öğütveren, U. B. 2011, J. Chem. Technol.
Biotechnol., 86, 261.
13. Nava, J. L., Núñez, F., González, I. 2007, Electrochim. Acta, 52, 3229.
14. Pérez, T., León, M. I., Nava, J. L. 2013, J. Electroanal. Chem., 707, 1.
15. Scialdone, O., Guarisco, C., Galia, A., Filardo, G., Silvestri, G., Amatore, C.,
Sella, C., Thouin, L. 2010, J. Electroanal. Chem., 638, 293.

BDD electrochemical reactors 77
16. Scialdone, O., Galia, A., Guarisco, C., La Mantia, S. 2012, Chem. Eng. J.,
189-190, 229.
17. Panizza, M., Cerisola, G. 2004, Electrochim. Acta, 49, 3221.
18. Cañizares, P., Lobato, J., Paz, R., Rodrigo, M. A., Sáez, C. 2005, Water Res.,
39, 2687.
19. Flox, C., Cabot, P. L., Centellas, F., Garrido, J. A., Rodríguez, R. M., Arias, C.,
Brillas, E. 2006, Chemosphere, 64, 892.
20. Waterston, K., Wang, J. W., Bejan, D., Bunce, N. J. 2006, J. Appl. Electrochem.,
36, 227.
21. Mascia, M., Vacca, A., Palmas, S., Polcaro, A. M. 2007, J. Appl. Electrochem.,
37, 71.
22. Weiss, E., Groenen-Serrano, K., Savall, A. 2007, J. Appl. Electrochem.,
37, 1337.
23. Weiss, E., Groenen-Serrano, K., Savall, A. 2008, J. Appl. Electrochem., 38, 329.
24. Carter, K. E., Farrell, J. 2008, Environ. Sci. Technol., 42, 6111.
25. Sirés, I., Brillas, E., Cerisola, G., Panizza, M. 2008, J. Electroanal. Chem.,
613, 151.
26. Scialdone, O., Randazzo, S., Galia, A., Silvestri, G. 2009, Water Res., 43, 2260.
27. Mascia, M., Vacca, A., Polcaro, A. M., Palmas, S., Rodriguez Ruiz, J., Da
Pozzo, A. 2010, J. Hazard. Mater., 174, 314.
28. Domínguez, J. R., González, T., Palo, P., Sánchez-Martín, J. 2010, Chem. Eng.
J., 162, 1012.
29. Anglada, A., Urtiaga, A. M., Ortiz, I. 2010, J. Hazard. Mater., 181, 729.
30. Dominguez-Ramos, A., Aldaco, R., Irabien, A. 2010, J. Chem. Technol.
Biotechnol., 85, 821.
31. Zhu, X., Ni, J., Wei, J., Chen, P. 2011, Electrochim. Acta, 56, 9439.
32. Brinzila, C. I., Pacheco, M. J., Ciríaco, L., Ciobanu, R. C., Lopes, A. 2012,
Chem. Eng. J., 209, 54.
33. Reis, R. M., Baio, J. A. F., Migliorini, F. L., Rocha, T. S., Baldan, M. R.,
Ferreira, N. G., Lanza, M. R. V. 2013, J. Electroanal. Chem., 690, 89.
34. García, O., Isarain-Chávez, E., Garcia-Segura, S., Brillas, E., Peralta-Hernández,
J. M. 2013, Electrocatalysis, 4, 224.
35. Isarain-Chávez, E., de la Rosa, C., Martínez-Huitle, C. A., Peralta-Hernández, J.
M. 2013, Int. J. Electrochem. Sci., 8, 3084.
36. Cañizares, P., Martinez, L., Paz, R., Sáez, C., Lobato, J., Rodrigo, M. A. 2006,
J. Chem. Technol. Biotechnol., 81, 1331.
37. Cañizares, P., Louhichi, B., Gadri, A., Nasr, B., Paz, R., Rodrigo, M. A., Saez,
C. 2007, J. Hazard. Mater., 146, 552.
38. Menapace, H. M., Diaz, N., Weiss, S. 2008, J. Environ. Sci. Health Part A,
43, 961.
39. Zhang, C., Wang, J., Zhou, H., Fu, D., Gu, Z. 2010, Chem. Eng. J., 161, 93.
40. Panizza, M., Cerisola, G. 2010, J. Electroanal. Chem., 638, 28.
41. Alvarez-Guerra, E., Dominguez-Ramos, A., Irabien, A. 2011, Chem. Eng. J., 170, 7.
42. Panizza, M., Michaud, P. A., Cerisola, G., Comninellis, Ch. 2001, Electrochem.
Commun. 3, 336.

Ignasi Sirés & Enric Brillas 78
43. Aquino, J. M., Pereira, G. F., Rocha-Filho, R. C., Bocchi, N., Biaggio, S. R.
2011, J. Hazard. Mater., 192, 1275.
44. Dominguez-Ramos, A., Irabien, A. 2013, Ind. Eng. Chem. Res., 52, 7534.
45. Bergmann, M. E. H., Rollin, J., Iourtchouk, T. 2009, Electrochim. Acta,
54, 2102.
46. Flox, C., Cabot, P. L., Centellas, F., Garrido, J. A., Rodríguez, R. M., Arias, C.,
Brillas, E. 2007, Appl. Catal. B: Environ., 75, 17.
47. Urtiaga, A., Rueda, A., Anglada, A., Ortiz, I. 2009, J. Hazard. Mater.,
166, 1530.
48. Gao, J., Zhao, G., Liu, M., Li, D. 2009, J. Phys. Chem. A, 113, 10466.
49. Guinea, E., Centellas, F., Garrido, J. A., Rodríguez, R. M., Arias, C., Cabot, P.
L., Brillas, E. 2009, Appl. Catal. B: Environ., 89, 459.
50. Grafias, P., Xekoukoulotakis, N. P., Mantzavinos, D., Diamadopoulos, E. 2010,
Water Res., 44, 2773.
51. Ruiz, E. J., Hernández-Ramírez, A., Peralta-Hernández, J. M., Arias, C., Brillas,
E. 2011, Chem. Eng. J., 171, 385.
52. Durante, C., Cuscov, M., Isse, A. A., Sandonà, G., Gennaro, A. 2011, Water
Res., 45, 2122.
53. Isarain-Chávez, E., Rodríguez, R. M., Cabot, P. L., Centellas, F., Arias, C.,
Garrido, J. A., Brillas, E. 2011, Water Res., 45, 4119.
54. Diogo, J. C., Morao, A., Lopes, A. 2011, Environ. Progress Sust. Energy,
30, 399.
55. Urtiaga, A. M., Perez, G., Ibañez, R., Ortiz, I. 2013, Desalination, 331, 26.
56. Cotillas, S., Llanos, J., Cañizares, P., Mateo, S., Rodrigo, M. A. 2013, Water
Res., 47, 1741.
57. Juang, Y., Nurhayati, E., Huang, C., Pan, J. R., Huang, S. 2013, Sep. Purif.
Technol., 120, 289.
58. Vahid, B., Khataee, A. 2013, Electrochim. Acta, 88, 614.
59. Comninellis, Ch., Pulgarin, C. 1991, J. Appl. Electrochem., 21, 703.
60. Panizza, M., Michaud, P. A., Cerisola G., Comninellis, Ch. 2001, J. Electroanal.
Chem. 507, 206.
61. Panizza, M., Brillas, E., Comninellis, Ch. 2008, J. Environ. Eng. Manage.,
18, 139.
62. Skoumal, M., Arias, C., Cabot, P. L., Centellas, F., Garrido, J. A., Rodríguez, R.
M., Brillas, E. 2008, Chemosphere, 71, 1718.
















![Electrochemical study of biologically relevant molecules at ......petitive with boron-doped diamond electrodes [2,4]. Finally, GUITAR was found to be less susceptible to air oxidation](https://img.pdfslide.net/doc/110x75/60f57e069c558d717c62d00a/electrochemical-study-of-biologically-relevant-molecules-at-petitive-with.jpg)












