
Corresponding author:
Petra Nytrova, MD
Dpt. of Neurology and Center for Clinical Neuroscience, First Medical Faculty, Charles
University in Prague,
Katerinska 30, 120 00 Praha 2, Czech Republic
Tel. + 420 2 2496 6422
Fax: + 420 2 2491 7907
e-mail:[email protected]
Complement activation in patients with Neuromyelitis optica.
Petra Nytrova1,2, Eliska Potlukova3, David Kemlink1, Mark Woodhall2, Dana Horakova1,
Patrick Waters2, Eva Havrdova1, Dana Zivorova4, Angela Vincent2, Marten Trendelenburg5
1Department of Neurology and Center of Clinical Neuroscience First Faculty of Medicine,
General University Hospital and First Faculty of Medicine, Charles University in Prague,
Czech Republic
2Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, UK
3Third Department of Medicine, General University Hospital and First Faculty of Medicine,
Charles University in Prague, Czech Republic
4Laboratory of Clinical Immunology, Institute of Clinical Biochemistry and Laboratory
Diagnostics, General University in Prague, Czech Republic
5Laboratory of Clinical Immunology, Department of Biomedicine, University Hospital
Basel, Switzerland

Abstract
The role of complement has been demonstrated in experimental models of neuromyeltis
optica (NMO), however, only few studies have analysed complement components
longitudinally in NMO patients. We measured serum or plasma concentrations of anti-C1q
antibodies and complement split products C3a, C4a and soluble C5b-9 in patients with NMO,
multiple sclerosis and healthy controls. NMO patients had higher levels of C3a and anti-C1q
antibodies than healthy controls. C3a levels correlated with disease activity, neurological
disability and aquaporin-4 IgG in NMO patients suggesting a role of the alternative pathway
of complement in the pathogenesis of NMO and supporting the strategy of therapeutic
complement inhibition.
Keywords: neuromyelitis optica, complement, aquaporin-4 IgG, C1q antibodies

1. Introduction
Neuromyelitis optica (NMO), also referred to as Devic´s disease, is a rare, severely disabling
inflammatory disorder of the central nervous system (CNS), usually with a relapsing-
remitting course (Wingerchuk et al., 1999). This autoimmune disease predominantly affects
the optic nerves and spinal cord. NMO is associated with serum antibodies against aquaporin-
4 (also known as NMO-IgG or AQP4-IgG) in up to 80% of cases (Lennon et al., 2004;
Waters et al., 2008).
The pathogenesis of NMO seems to be closely linked to the activation of the complement
system. In experimental models, AQP4-IgG binding to AQP4 causes cytotoxicity only in the
presence of complement (Hinson et al., 2007; Saadoun et al., 2010) suggesting that this is
crucial for pathogenicity in NMO. In addition, recent studies reported that exacerbations of
NMO were reflected by changes in concentrations of complement activation products in sera
(C4d) as well as in the cerebrospinal fluid (C5a) (Kuroda et al., 2012; Tuzun et al., 2011).
Furthermore, although Veszeli et al. did not find substantial systemic complement activation
in NMO patients without disease activity, the complement system was found to be
abnormally affected even during remission (Veszeli et al., 2014). Eculizumab, a therapeutic
monoclonal IgG that neutralises the complement protein C5, has effectively reduced the
relapse frequency of NMO in open label trial (Pittock et al., 2013).
AQP4 is a water channel that is most abundantly expressed in the processes of astrocytes. Its
monomers assemble as tetramers. These AQP4 tetramers aggregate in cell plasma membranes
to form supramolecular clusters which are targeted by AQP4-IgG. These clustered AQP4-IgG
are crucial for complement-dependent cytotoxicity through multivalent C1q binding
(Papadopoulos et al., 2013). AQP4-IgG are mainly of the IgG1 subclass that usually

activates the classical pathway of complement (Waters et al., 2008). Formation of
AQP4:AQP4-IgG immune complexes, with subsequent complement-mediated destruction of
astrocytes, is paralleled by loss of AQP4 and GFAP staining in perivascular lesions in
histological sections. This loss is localised in the same areas that exhibit accumulation of
eosinophils and plasma cells as well as vasculocentric deposits of immunoglobulins and
products of complement activation (Jarius et al., 2008; Lucchinetti et al., 2002; Roemer et al.,
2007). These characteristics also resemble the histopathological features of murine brain
lesions induced by injection of human AQP4-IgG with human complement into the brain
(Saadoun et al. 2010).
Autoantibodies against complement C1q (anti-C1q) have been found in a number of
autoimmune diseases (Potlukova et al., 2008; Siegert et al., 1999; Trendelenburg, 2005;
Wisnieski et al., 1992). Most interestingly, anti-C1q were found in more than 97% of patients
with biopsy-proven active lupus nephritis supporting the hypothesis of a pathogenic role of
anti-C1q in systemic lupus erythematosus (Trendelenburg et al., 2006). In other autoimmune
diseases, the role of anti-C1q remains unclear. Interestingly, C1q-targeting monoclonal
antibodies prevented complement-mediated damage in animal models of NMO (Phuan et al.,
2013), may have a possible role in regulation of the immune response, and providing a proof-
of-concept for C1q-targeted monoclonal antibody therapies in NMO.
In contrast to NMO, studies on complement activation in multiple sclerosis (MS) led to more
conflicting results. As a consequence, complement activation is generally not considered to
be of crucial pathogenic relevance in MS (Brink et al., 2005; Sellebjerg et al., 1998; Urich et
al., 2006). The role of anti-C1q in MS remains to be determined.

The aims of our study were to elucidate the role of complement activation and anti-C1q in
patients with NMO by means of serological assessments during the relapse and remission of
disease and to evaluate the potential use of plasma complement parameters as biomarkers of
disease activity NMO. To support our observations we assessed complement binding to
AQP4-IgG on frozen primate optic nerve.
2. Materials and Methods
2.1. Patients
The study was designed as a prospective cohort study in patients with demyelinating
disorders at the Multiple Sclerosis Centre of the Department of Neurology, General
University Hospital in Prague in 2010-2011. NMO and MS patients were included to the
study. At inclusion, time of relapse and at 6-months follow-up, clinical data were recorded
and serum as well as plasma samples were collected.
The diagnosis of NMO was based on the Wingerchuk’s diagnostic criteria (Wingerchuk,
2007). Plasma (n=43) and serum samples (n=43) were analysed from 19 AQP4-IgG positive
patients. In eight patients with NMO, we obtained three or more samples at different time
points (range 2 weeks to 6 months). For the comparison of all groups, only first sample value
of individual patient was included for statistical analysis. All forty-three AQP4-IgG positive
samples were used for intragroup NMO analysis and correlations. The plasma samples were
used for complement breakdown product assessment and the serum samples for C1q and
AQP4 antibody measurement.

Thirty five MS patients were included in the study for measurement of complement
activation products. Furthermore, for the analysis of anti-C1q, we used additional 41 serum
samples of different MS patients (in total, 76 serum samples were frozen in 2010). All MS
patients fulfilled the McDonald's criteria for MS relapse remitting (n=64), secondary (n=9) or
primary progressive form (n=3) (Polman et al., 2005).
Patient-reported symptoms or objectively observed signs typical of an acute inflammatory
demyelinating event in the CNS with duration of at least 24 hours, in the absence of fever or
infection, were considered as a relapse (Polman et al., 2005). Samples were taken during both
remission and relapse. Samples of the NMO or MS patients in relapse were obtained prior to
initiation of treatment by high-dose methylprednisolone, or plasma exchange. Samples of
patients treated with intravenous immunoglobulins (IVIG), cyclophosphamide, natalizumab
and rituximab were drawn before regular medicament infusion.
All patients were of Caucasian origin except two NMO female patients from Asia. Patients
were observed by a specialist in demyelinating disorders of the CNS. Conventional brain and
spinal cord MRI were used for establishment of diagnosis and evaluation of spinal cord
involvement. Neurological disability was evaluated by Kurtzke expanded disability status
scale (EDSS) (Kurtzke, 1983). As controls, 40 volunteers without signs of an autoimmune
disorder or infection at the time of blood sampling were included; most of them worked as
health care professionals.
Blood samples were collected by venepuncture in the collection tubes with EDTA. The serum
and plasma samples were centrifuged at 3000 rpm for 10 minutes and frozen within forty five

minutes after centrifugation and aliquoting. The samples were stored at -80oC and not thawed
until assessment for complement split products and antibodies against AQP4 and C1q.
All subjects gave an informed consent to the procedures, which were approved by the Ethical
Committee of the General University Hospital and the First Faculty of Medicine, Charles
University in Prague.
2.2. Serological analysis
2.2.1. Serum antibodies against aquaporin-4 and complement C1q
All samples were tested in a blinded manner by a commercially available
immunofluorescence cell-based assay (CBA) using recombinant human M1-AQP4
(Euroimmun, Lübeck, Germany) as antigen following the manufacturer’s instructions. Sera
of 19 NMO AQP4-IgG positive patients were retested by an in-house CBA using human
M23-AQP4 expressed on live cells, and serial dilutions to determine the end-point titres. Sera
of all MS and healthy controls were AQP4-IgG negative. Anti-C1q were measured in serum
using a commercially available ELISA kit (Bühlmann Laboratories, Schönenbuch,
Switzerland). In this assay, undigested purified human C1q served as the antigen, and sera
were diluted and incubated in a high-salt buffer (1M NaCl). The optical densities were
measured at 450 nm and converted into international units per ml (IU/ml) using the standards
provided by the manufacturer. The manufacturer’s cut-off of 15 IU/ml was used to
determined positivity.
2.2.2. Assays for complement split products

The soluble terminal complement complex (sC5b-9) as well as C3a and C4a fragments of
complement proteins were measured by commercially available enzyme immunoassays
(MicroVue™SC5b-9 Plus EIA Kit, MicroVue™ C3a Plus EIA Kit, MicroVue™ C4a Plus
EIA Kit; Quidel, San Diego, USA) following the manufacturer’s instructions. Briefly, test
specimens were added to microassay wells precoated with specific monoclonal antibodies to
each of the complement split products used for their capturing. Horseradish peroxidase
conjugated antibodies to different epitopes of the same antigen were added to each test well.
Following addition of a chromogen, the plates were measured spectrophotometrically. The
reference ranges for the complement split products for healthy individuals stated by the
manufacturer were 33.8 – 268.1 ng/ml (mean 129.6 ng/ml) for C3a and 383.5 – 8168.2 ng/ml
(mean 1694.7 ng/ml) for C4a. The reference values for sC5b-9 assay were not provided by
the manufacturer.
Total C3 and C4 serum levels in NMO patients were measured by routine nephelometry
(Dade Behring, Vienna Austria).
2.3. Immunohistochemistry - indirect immunofluorescence
Fourteen serum samples from NMO patients and 20 healthy blood donors were tested on
multiplex biochip based slides (Euroimmun). Each field, that is screened with an individual
serum, consisted of four individual chips with fixed frozen tissue (rat hippocampus, primate
optic nerve, primate and rat cerebellum) and four individual chips with HEK cells transfected
with human M1-AQP4, LGI1, CASRP2 and GlyR-alpha1, giving a total of 8 biochips per
field. Phosphate-buffered saline (PBS) containing 5% Tween-20 was used for samples
dilution. For each serum (diluted 1:10), indirect immunofluorescence was carried out on
individual chips: A) diluted sample / FITC-conjugated anti-human IgG (Euroimmun), B)
diluted sample / serum of blood donors as complement source (Euroimmun)/FITC-

conjugated anti-complement antibodies [polyclonal rabbit anti-human C1q (Dako, Glostrup,
Denmark), polyclonal rabbit anti-human C3c (Euroimmun), polyclonal rabbit anti-human
C4c (Dako)]. Since C3b and C4b were progressively degraded, we used anti-C3c and
anti-C4c antibodies for immunohistochemistry analysis. The lyophilized complement source
(Euroimmun) from 10 healthy donors was resuspended in sterile water prior using. The
patterns were visually interpreted by fluorescence microscopy (Zeiss LSM 700 Confocal,
Carl Zeiss Microscopy GmbH, Jena, Germany).
2.4. Statistical analysis
Statistical intergroup analyses were conducted by Kruskal Wallis non-parametric ANOVA
and its post hoc multiple comparison z-test. For correlation statistics we used the Spearman’s
test. For NMO group comparisons we used Mann-Whitney non-parametric tests. Tests were
performed with the help of StatSoft, Inc. (2011) STATISTICA (Statsoft, Tulsa, OK, USA),
version 10 (www.statsoft.com) and GraphPad PRISM 5. For the construction of receiver
operating characteristics (ROC) and its statistics we used the program JROCFIT1.0.2
(www.jrocfit.org). Differences were considered as being statistically significant in case of p <
0.05.
3. Results
3.1. Clinical features and treatment
Nineteen patients with NMO were included in our study. Ten NMO patients had active
disease (defined as ≥1 relapse during the last 6 months prior to sample collecting) and nine
were inactive (relapse-free during the period of 6 months). In eight patients with NMO, we

obtained three or more plasma samples at different time points ranging from 2 weeks to 6
months from the first.
The majority of patients had long histories and had been treated with variable regimens. Six
NMO patients were treated by a combination of drugs, i.e. low dose of prednisone with
azathioprine. Monotherapy was used in 10 patients (low dose of prednisone, mycophenolate
mofetil, cyclophosphamide, rituximab). One female patient underwent autologous stem cell
transplantation three months before samples could be collected. Two patients were without
treatment.
MS patients were treated by a monotherapy (natalizumab, interferon beta, prednisone, IVIG,
mycophenolate mofetil, cyclophosphamide) or received a combination of drugs (interferon
beta and prednisone or azathioprine or methotrexate). Eleven MS patients were without
treatment.
Demographic data of NMO patients, MS patients, and healthy controls are summarised in
Table 1.
3.2. Complement activation products analysis
Plasma levels of complement activation products differed substantially between groups (Fig.
1A-C). Both NMO and MS patient had significantly higher levels of C3a and sC5b-9 and
lower levels of C4a compared with controls. However, patients with NMO had lower levels
of C4a than the MS patients (p < 0.05). Serum levels of anti-C1q were higher in the NMO
group compared to both MS and controls (Fig. 1D). Surprisingly, the MS patients had lower
anti-C1q levels than controls.

3.3. Correlation of complement activation products with disease activity
In NMO patients (Table 2) there was a significant correlation between EDSS score and C3a
or the ratio C3a:C3 (Fig. 2A). This correlation was as strong as the correlation between
EDSS score and titres of AQP4-IgG (Fig. 2B). In addition, plasma levels of C3a in NMO
patients with clinically active disease were significantly higher than in NMO patients with
inactive disease (Fig. 3A), and C3a levels were higher during relapses than during remission
periods (Fig. 3B). Using ROC curves, the best cut-off for the distinction between patients
with active disease compared to those who were inactive was 500 ng/ml with a sensitivity of
94.7% and a specificity of 90.3% (Fig. 4). However, we did not find any correlation between
C3a levels and the time to the next relapse after plasma sampling.
In contrast to C3a, the other complement split products and anti-C1q did not correlate with
disease activity. We also did not find any correlation between complement split products or
C1q antibodies and the clinical activity or severity of disease including EDSS score in MS
patients.
3.4. Immunohistochemical studies
Using diluted patients´ sera and FITC-conjugated anti-human IgG, all patients with NMO
were found to be AQP4-IgG positive, showing weak reactivity with optic nerve and
cerebellum, but strong membrane reactions to the AQP4-expressing recombinant cells which
have high AQP4 expression (Fig.5 row I-III: B). All control sera remained negative (Fig.5
row I-III: A).

To assess complement activation by the antibodies, patients’ sera together with a complement
source and FITC-conjugated anti-human complement (anti-human C1q, anti-human C3c,
anti-human C4c, in separate assays) were applied to the cells and the tissue sections. First, as
a positive control, the recombinant AQP4 expressing cells were tested in this three-step
staining procedure (Fig.5, row I: C-E). All NMO sera but none of the controls produced
fluorescent signals on the AQP4-expressing cells indicating complement activation by the
bound human antibodies. The primate optic nerve transverse sections fluoresced in a
mesh-like staining pattern (Fig. 5, row II: C-E) corresponding to the glial supporting
meshwork that separates the optic nerve fibers. The cerebellum exhibited mainly membrane
staining, particularly in the granule cell layers of cerebellum (Fig. 5, row III: C-E).
4. Discussion
Previous studies have implicated complement-activating IgG1 AQP4 antibodies in the
pathogenesis of NMO, but these have concentrated on the classical pathway. Here we provide
clinical, serological and immunohistochemical data suggesting that activation of C3 is
implicated in the pathogenesis of NMO, and might provide a biomarker of disease activity.
Moreover, by immunofluorescence staining we show that patients´ AQP4-IgG form immune
complexes, including C1q, C3c and C4c, not only on AQP4 transfected cells but also on
native neuronal tissues when normal human serum is added as a source of complement. The
staining pattern was similar to that described by Waters et al. on mouse brain tissue sections
(Waters et al., 2008). Based on the similar staining patterns observed for AQP4 and
AQP4:AQP4-IgG complexes and complement deposits, particularly C3c, we conclude that

AQP4-IgG binds to brain tissue AQP4 with subsequent complement activation of the
alternative pathway as well as the better classical pathway.
We did not find any correlation between C3a and sC5b-9 levels, which might suggest a more
complex mechanism of astrocyte damage including the possibility of C3a-mediated
recruitment of eosinophils and neutrophils into NMO lesions. C3a is proinflammatory
mediator and anaphylotoxin with immune and non-immune biological functions. For
instance, C3a is involved in excitotoxicity-mediated neuronal death through astrocytes
stimulation (van Beek et al., 2001). Receptor for C3aR is expresses by
monocytes/macrophages, microglia, astrocytes, neurons and endothelial cell too (Davoust et
al., 1999; Ischenko et al., 1998; Klos et al., 1992). Interestingly, C3a has been shown to
induce an increase in interleukin 6 (IL-6) mRNA expression by astrocyte cell lines (Jauneau
et al., 2003; Sayah et al., 1999), which is in line with the observation that cerebrospinal fluid
concentrations of IL-6 are increased during the initial attack of NMO (Uzawa et al., 2013).
Low C4a levels as observed in our NMO patients might be the result of complement
dysregulation and/or the more complex interplay between complement-activating AQP4-IgG
and anti-C1q. The biological function of C4a in NMO remains more speculative but might be
linked to regulatory properties of the fragment. Recombinant C4a has been demonstrated to
inhibit C3a and C5a-stimulated degranulation of mast cells (Xie et al., 2012). In addition, an
anti-inflammatory role of C4a in glomerulonephritis has been described (Welch et al., 2001).
Considering that the lack of early components of the classical pathway of complement (C1q
and C4) are associated with systemic autoimmunity (Pickering et al., 2000), these
components might also have a protective effects in NMO.

To date, only a few analyses of complement split products in plasma/serum or cerebrospinal
fluid have been reported and led to conflicting results. Tuzun et al. described an increased
activity of the classical pathway during relapses of NMO by measuring the levels of
breakdown products for the classical (C4d), alternative (fragment Bb) and terminal
complement (sC5b-9) pathways (Tuzun et al., 2011). Moreover, they described a negative
correlation between EDSS and levels of C4d, fragment Bb and sC5b-9. The differing results
of our study could partially be explained by the use of different assays, by different sampling
time points, differences in the accompanying treatment of the patients and the assessment in
sera of AQP4-IgG negative NMO patients. More in line with our findings is the report
demonstrating significantly elevated C5a levels in the cerebrospinal fluid of NMO patients, in
particular in patients with multiple enhanced lesions on MRI (Kuroda et al., 2013).
Independent of the complement activation products, we found that anti-C1q Abs levels were
significantly higher in NMO patients as compared to the MS patients and healthy controls,
respectively. This might be of particular interest with regard to the recent study by Phuan et
al. who demonstrated that monoclonal neutralising antibodies targeting C1q significantly
improved the course of NMO in an experimental model of NMO (Phuan et al.,2013).
However, the effects of anti-C1q on C1q could be different from neutralizing monoclonal
antibodies as mentioned before. Again, data derived from studies in systemic autoimmunity
point towards a disease exacerbating effect of autoantibodies against C1q (Trendelenburg et
al., 2006), e.g. by the inhibition of protective effects of the C1q molecule.
Our study has several limitations: the descriptive character of our data does not allow final
statements on the role of complement in NMO. In addition, the relatively small and locally
recruited cohort of NMO patients, due to the fact that NMO is a rare disease in Caucasians,

does not allow definite conclusions on the correlation between plasma/serum parameters and
disease activity. The limited relapse samples and the time frame of the study may preclude
the finding of associations between these products and disease activity. Finally, different
treatment profiles in MS and NMO patients might have strongly affected our comparative
analyses. Thus, further studies on the role of complement in NMO, in particular with a focus
on outcome measures such as disability, will be necessary.
In conclusion, our data strongly support the hypothesis that complement activation is
involved in the pathogenesis of NMO in vivo. Independently, complement C3a as a
biomarker might play an important role not only for the diagnosis of NMO but also for the
evaluation of disease activity at follow-up. Moreover, our data support strategies of
therapeutic complement inhibition in NMO patients in particular during relapses of disease.
Abbreviations
AQP4 aquaporin-4; AQP4-IgG antibodies against aquaporin-4; Anti-C1q antibodies against
C1q; AUC Area Under Curve; CASPR2 contactin-associated protein 2; CNS central nervous
system; EDSS Expanded Disability Status Scale; FPF False Positive Fraction; GlyR glycine
receptor; HSD Honestly Significant Difference; IVIG intravenous immunoglobulins; LGI1
leucine-rich, glioma inactivated 1; MS multiple sclerosis; NMO neuromyelitis optica; ROC
Receiver Operating Characteristic; sC5b-9 soluble terminal complement complex; TPF True
Positive Fraction
Competing interests

Nytrova Petra, her research stay at Dpt. of Clinical Neurosciences at University of Oxford
was supported by Euroimmun and a John Newsom-Davis Fellowship from the Guarantors of
Brain.
Potlukova Eliska has nothing to disclose.
Kemlink David has nothing to disclose.
Woodhall Mark has nothing to disclose.
Waters Patrick is a named inventor on patents for antibody assays and has received
royalties, and he has received a speaker honorarium from Biogen-Idec Japan.
Horakova Dana received speaker honoraria and consultant fees from Biogen Idec, Novartis,
Merck Serono, Teva and Bayer Healthcare and financial support for research activities from
Biogen Idec.
Havrdova Eva received speaker and consulting honoraria from Biogen Idec, Novartis,
Sanofi Genzyme, Roche, Merck Serono, Teva and Bayer Healthcare.
Zivorova Dana has nothing to disclose.
Vincent Angela has received funding from Euroimmun AG and is a consultant for Athena
Diagnostics. The University of Oxford holds patents and receives royalties and payments for
antibody tests.
Trendelenburg Marten receives financial support for research activities from Roche Pharma
and is member of the Swiss advisory board of GSK.
Acknowledgements
Supported by Grant Agency of the Charles University (grant GAUK 132010), the Czech
Ministries of Education and Health (PRVOUK-P26/LF1/4, NT13237-4/2012) and by the
National Health Service National Specialised Commissioning Group for Neuromyelitis
Optica and the National Institute for Health Research Oxford Biomedical Research Centre for

funding. The research stay of main author PN at Dpt.of Clinical Neuroscience at University
of Oxford was supported by a John Newsom-Davis Fellowship from the Guarantors of Brain,
Fond Mobility of the Charles University and Euroimmun. MT is a recipient of a grant from
the Swiss National Foundation (310030_134900/1).
References:
Brink, B.P., Veerhuis, R., Breij, E.C., van der Valk, P., Dijkstra, C.D., Bo, L., 2005. The
pathology of multiple sclerosis is location-dependent: no significant complement
activation is detected in purely cortical lesions. J. Neuropathol. Exp. Neurol. 64, 147-
155.
Davoust, N., Jones, J., Stahel, P.F., Ames, R.S., Barnum, S.R., 1999. Receptor for the C3a
anaphylatoxin is expressed by neurons and glial cells. Glia 26, 201-211.
Hinson, S.R., Pittock, S.J, Lucchinetti, C.F., Roemer, S.F., Fryer, J.P., Kryzer, T.J., Lennon,
V.A., 2007. Pathogenic potential of IgG binding to water channel extracellular
domain in neuromyelitis optica. Neurology 69, 2221-2231.
Ischenko, A., Sayah, S., Patte, C., Andreev, S., Gasque, P., Schouft, M.T., Vaudry, H.,
Fontaine, M., 1998. Expression of a functional anaphylatoxin C3a receptor by
astrocytes. J. Neurochem. 71, 2487-2496.
Jarius, S., Paul, F., Franciotta, D., Waters, P., Zipp, F., Hohlfeld, R., Vincent, A., Wildemann,
B., 2008. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat.
Clin. Pract. Neurol. 4, 202-214.
Jauneau, A.C., Ischenko, A., Chan, P., Fontaine, M., 2003. Complement component
anaphylatoxins upregulate chemokine expression by human astrocytes. FEBS Lett.
537, 17-22.

Klos, A., Bank, S., Gietz, C., Bautsch, W., Kohl, J., Burg, M., Kretzschmar, T., 1992. C3a
receptor on dibutyryl-cAMP-differentiated U937 cells and human neutrophils: the
human C3a receptor characterized by functional responses and 125I-C3a binding.
Biochemistry 31, 11274-11282.
Kuroda, H., Fujihara, K., Takano, R., Takai, Y., Takahashi, T., Misu, T., Nakashima, I., Sato,
S., Itoyama, Y., Aoki, M., 2013. Increase of complement fragment C5a in
cerebrospinal fluid during exacerbation of neuromyelitis optica. J. Neuroimmunol.
254, 178-182
Kurtzke, J.F., 1983. Rating neurologic impairment in multiple sclerosis: an expanded
disability status scale (EDSS). Neurology 33, 1444-1452.
Lennon, V.A., Wingerchuk, D.M., Kryzer ,T.J., Pittock, S.J., Lucchinetti, C.F., Fujihara, K.,
Nakashima, I., Weinshenker, B.G., 2004. A serum autoantibody marker of
neuromyelitis optica: distinction from multiple sclerosis. Lancet 364, 2106-2112.
Lucchinetti, C.F., Mandler, R.N., McGavern, D., Bruck, W., Gleich, G., Ransohoff, R.M.,
Trebst, C., Weinshenker, B., Wingerchuk, D.M., Parisi, J.E., Lassmann, H., 2002. A
role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica.
Brain 125, 1450-1461.
Papadopoulos, M.C., Verkman, A.S., 2012. Aquaporin 4 and neuromyelitis optica. Lancet.
Neurol. 11, 535-544.
Phuan, P.W., Zhang, H., Asavapanumas, N., Leviten, M., Rosenthal, A., Tradtrantip, L.,
Verkman, A.S., 2013. C1q-targeted monoclonal antibody prevents complement-
dependent cytotoxicity and neuropathology in in vitro and mouse models of
neuromyelitis optica. Acta. Neuropathol. 125, 829-840.

Pickering, M.C., Botto, M., Taylor, P.R., Lachmann, P.J., Walport, M.J., 2000. Systemic
lupus erythematosus, complement deficiency, and apoptosis. Advances in
immunology 76, 227-324.
Pittock, S.J., Lennon, V.A., McKeon, A., Mandrekar, J., Weinshenker, B.G., Lucchinetti,
C.F., O'Toole, O., Wingerchuk, D.M., 2013. Eculizumab in AQP4-IgG-positive
relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet
Neurol. 12, 554-562.
Polman, C.H., Reingold, S.C., Edan, G., Filippi, M., Hartung, H.P., Kappos, L., Lublin, F.D.,
Metz, L.M., McFarland, H.F., O'Connor, P.W., Sandberg-Wollheim, M., Thompson,
A.J., Weinshenker, B.G., Wolinsky, J.S., 2005. Diagnostic criteria for multiple
sclerosis: 2005 revisions to the "McDonald Criteria". Ann. Neurol. 58, 840-846.
Potlukova, E., Kralikova, P., 2008. Complement component c1q and anti-c1q antibodies in
theory and in clinical practice. Scand. J. Immunol. 67, 423-430.
Roemer, S.F., Parisi, J.E., Lennon, V.A., Benarroch, E.E., Lassmann, H., Bruck, W.,
Mandler, R.N., Weinshenker, B.G., Pittock, S.J., Wingerchuk, D.M., Lucchinetti,
C.F., 2007. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes
neuromyelitis optica from multiple sclerosis. Brain 130, 1194-1205.
Saadoun, S., Waters, P., Bell, B.A., Vincent, A., Verkman, A.S., Papadopoulos, M.C., 2010.
Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human
complement produces neuromyelitis optica lesions in mice. Brain 133, 349-361.
Sayah, S., Ischenko, A.M., Zhakhov, A., Bonnard, A.S., Fontaine, M., 1999. Expression of
cytokines by human astrocytomas following stimulation by C3a and C5a
anaphylatoxins: specific increase in interleukin-6 mRNA expression. J. Neurochem.
72, 2426-2436.

Sellebjerg, F., Jaliashvili, I., Christiansen, M., Garred, P., 1998. Intrathecal activation of the
complement system and disability in multiple sclerosis. J. Neurol. Sci. 157, 168-174.
Siegert, C.E., Kazatchkine, M.D., Sjoholm, A., Wurzner, R., Loos, M., Daha, M.R., 1999.
Autoantibodies against C1q: view on clinical relevance and pathogenic role. Clin.
Exp. Immunol. 116, 4-8.
Trendelenburg, M., 2005. Antibodies against C1q in patients with systemic lupus
erythematosus. Springer Semin. Immunopathol. 27, 276-285.
Trendelenburg, M., Lopez-Trascasa, M., Potlukova, E., Moll, S., Regenass, S., Fremeaux-
Bacchi, V., Martinez-Ara, J., Jancova, E., Picazo, M.L., Honsova, E., Tesar, V.,
Sadallah, S., Schifferli, J., 2006. High prevalence of anti-C1q antibodies in biopsy-
proven active lupus nephritis. Nephrol. Dial. Transplant. 21, 3115-3121.
Tuzun, E., Kurtuncu, M., Turkoglu, R., Icoz, S., Pehlivan, M., Birisik, O., Eraksoy, M.,
Akman-Demir, G., 2011. Enhanced complement consumption in neuromyelitis optica
and Behcet's disease patients. J. Neuroimmunol. 233, 211-215.
Urich, E., Gutcher, I., Prinz, M., Becher, B., 2006. Autoantibody-mediated demyelination
depends on complement activation but not activatory Fc-receptors. Proc. Natl. Acad.
Sci. U S A 103, 18697-18702.
Uzawa, A., Mori, M., Sawai, S., Masuda, S., Muto, M., Uchida, T., Ito, S., Nomura, F.,
Kuwabara, S., 2013. Cerebrospinal fluid interleukin-6 and glial fibrillary acidic
protein levels are increased during initial neuromyelitis optica attacks. Clin. Chim.
Acta 421, 181-183.
van Beek, J., Nicole, O., Ali, C., Ischenko, A., MacKenzie, E.T., Buisson, A., Fontaine, M.,
2001. Complement anaphylatoxin C3a is selectively protective against NMDA-
induced neuronal cell death. Neuroreport 12, 289-293.

Veszeli, N., Fust, G., Csuka, D., Trauninger, A., Bors, L., Rozsa, C., Nagy, Z., Jobbagy, Z.,
Eizler, K., Prohaszka, Z., Varga, L., Illes, Z., 2014. A systematic analysis of the
complement pathways in patients with neuromyelitis optica indicates alteration but no
activation during remission. Mol. Immunol. 57, 200-209.
Waters, P., Jarius, S., Littleton, E., Leite, M.I., Jacob, S., Gray, B., Geraldes, R., Vale, T.,
Jacob, A., Palace, J., Maxwell, S., Beeson, D., Vincent, A., 2008. Aquaporin-4
antibodies in neuromyelitis optica and longitudinally extensive transverse myelitis.
Arch. Neurol. 65, 913-919.
Welch, T.R., Frenzke, M., Carroll, M.C., Witte, D.P., 2001. Evidence of a role for C4 in
modulating interstitial inflammation in experimental glomerulonephritis. Clin.
Immunol. 101, 366-370.
Wingerchuk, D.M., 2007. Diagnosis and treatment of neuromyelitis optica. Neurologist 13, 2-
11.
Wingerchuk, D.M., Hogancamp, W.F., O'Brien, P.C., Weinshenker, B.G., 1999. The clinical
course of neuromyelitis optica (Devic's syndrome). Neurology 53, 1107-1114.
Wisnieski, J.J., Jones, S.M., 1992. IgG autoantibody to the collagen-like region of Clq in
hypocomplementemic urticarial vasculitis syndrome, systemic lupus erythematosus,
and 6 other musculoskeletal or rheumatic diseases. J. Rheumatol. 19, 884-888.
Xie, P., Nishiura, H., Semba, U., Chen, J., Zhao, R., Kuniyasu, A., Yamamoto, T., 2012.
Inhibitory effects of C4a on chemoattractant and secretagogue functions of the other
anaphylatoxins via Gi protein-adenylyl cyclase inhibition pathway in mast cells. Int.
Immunopharmacol. 12, 158-168.


Figure legends
Figure 1. Comparison of plasma levels of complement activation products and serum levels
of anti-C1q antibodies in patients with demyelinating disorders versus normal controls. A:
C3a; B: C4a; C: sC5b-9; D: anti-C1q. For this analysis, only the first samples of each patient
(before escalation of treatment) were used. Horizontal lines depict the medians of the
measurements, the dotted horizontal lines represent the mean of normal controls (C3a and
C4a, not available for sC5b-9) or the upper limit of normal values (anti-C1q) as stated by the
manufacturer. (n.s.: not significant).
Figure 2. Linear regression between neurological disability as expressed by EDSS score and
plasma levels of C3a (A) and AQP4-IgG (B). The analysis was performed using only the
first samples available.
Figure 3. Plasma C3a levels in NMO patients with 1 ≥ relapse within the last 6 months prior
to sample collecting (Group I) versus patients > 6 months relapse-free (Group II) (A); and
during the relapse as compared to remission periods (B). Analysis in Fig. 3A was performed
using only the first samples available whereas in Fig. 3B all available samples from 19
patients were used (i.e. including those at follow-up).
Figure 4. ROC curve of complement C3a levels for the distinction between patients with ≥ 1
relapse during the last 6 months prior to sampling and patients that remained free of relapse
during the last 6 months. AUC: Area Under Curve, TPF: True Positive Fraction, FPF: False
Positive Fraction
Figure 5.
Row I: Cell-based assay for AQP4-IgG. AQP4 transfected HEK cells incubated with serum
of a healthy control and a patient’s serum and FITC-conjugated anti-hIgG (A,B); AQP4
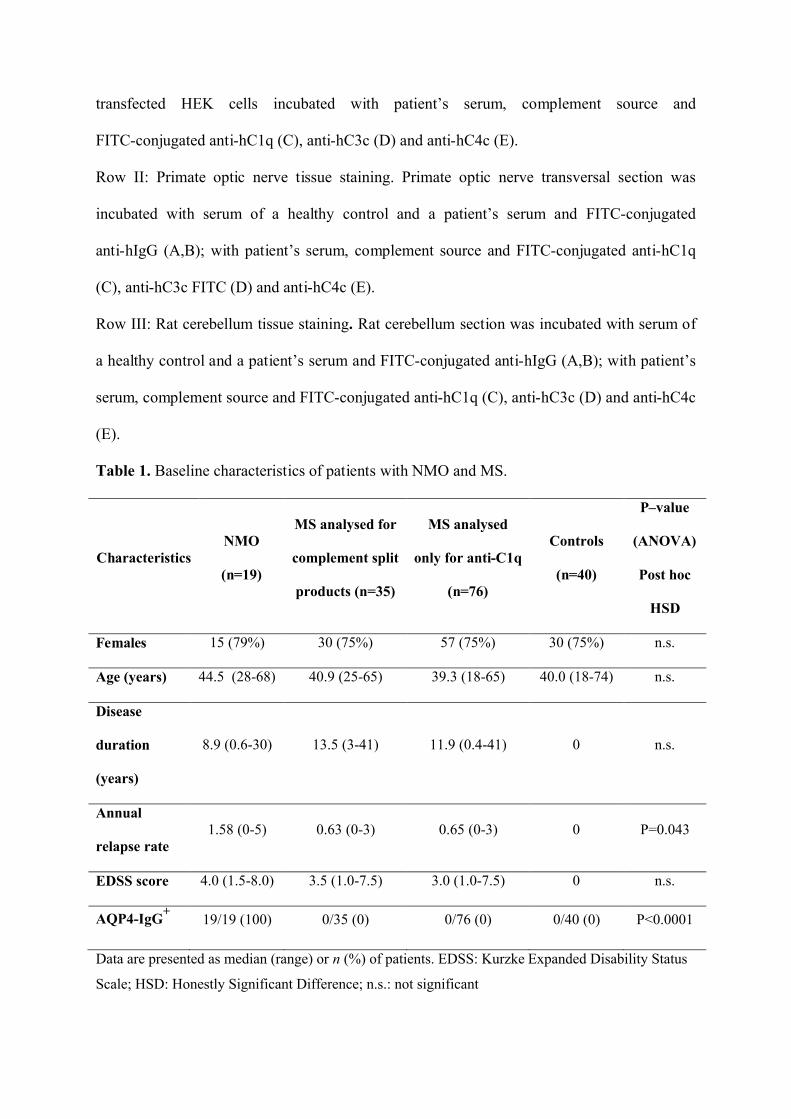
transfected HEK cells incubated with patient’s serum, complement source and
FITC-conjugated anti-hC1q (C), anti-hC3c (D) and anti-hC4c (E).
Row II: Primate optic nerve tissue staining. Primate optic nerve transversal section was
incubated with serum of a healthy control and a patient’s serum and FITC-conjugated
anti-hIgG (A,B); with patient’s serum, complement source and FITC-conjugated anti-hC1q
(C), anti-hC3c FITC (D) and anti-hC4c (E).
Row III: Rat cerebellum tissue staining. Rat cerebellum section was incubated with serum of
a healthy control and a patient’s serum and FITC-conjugated anti-hIgG (A,B); with patient’s
serum, complement source and FITC-conjugated anti-hC1q (C), anti-hC3c (D) and anti-hC4c
(E).
Table 1. Baseline characteristics of patients with NMO and MS.
CharacteristicsNMO
(n=19)
MS analysed for
complement split
products (n=35)
MS analysed
only for anti-C1q
(n=76)
Controls
(n=40)
P–value
(ANOVA)
Post hoc
HSD
Females 15 (79%) 30 (75%) 57 (75%) 30 (75%) n.s.
Age (years) 44.5 (28-68) 40.9 (25-65) 39.3 (18-65) 40.0 (18-74) n.s.
Disease
duration
(years)
8.9 (0.6-30) 13.5 (3-41) 11.9 (0.4-41) 0 n.s.
Annual
relapse rate1.58 (0-5) 0.63 (0-3) 0.65 (0-3) 0 P=0.043
EDSS score 4.0 (1.5-8.0) 3.5 (1.0-7.5) 3.0 (1.0-7.5) 0 n.s.
AQP4-IgG+
19/19 (100) 0/35 (0) 0/76 (0) 0/40 (0) P<0.0001
Data are presented as median (range) or n (%) of patients. EDSS: Kurzke Expanded Disability Status
Scale; HSD: Honestly Significant Difference; n.s.: not significant

Table 2. Spearman rank order coefficients of correlations between complement split
products, AQP4-IgG, anti-C1q and EDSS in NMO.
EDSS
scoreAnti-C1q C3a C4a sC5b-9
AQP4-IgG
titresC3a/C3 C4a/C4
EDSS
score- n.s.
0.683
p<0.05n.s. n.s.
0.590
p<0.05
0.705
p<0.05n.s.
AQP4-IgG
titres
0.590
p<0.05n.s.
0.447
p<0.05n.s. n.s. -
0.482
p<0.05n.s.
Anti-C1q n.s. - n.s. n.s. n.s. n.s. n.s. n.s.
For this analysis, all available samples were used, including those from follow-up (n=43). EDSS:
Kurzke expanded disability status scale; n.s. not significant
Figures


Figure 5.

Recommended

















