
Endocrine manifestations inLangerhans cell histiocytosisPolyzois Makras1, Krystallenia I. Alexandraki2, George P. Chrousos3,Ashley B. Grossman4 and Gregory A. Kaltsas2,4
1 Department of Endocrinology and Diabetes, 251 Hellenic Air-Force and VA General Hospital, Athens, Greece2 Division of Endocrinology, Department of Pathophysiology, Laiko University Hospital, Athens, Greece3 First Department of Pediatrics, National and Kapodistrian University of Athens, Athens, Greece4 Department of Endocrinology, St Bartholomew’s Hospital, London, UK
Review TRENDS in Endocrinology and Metabolism Vol.18 No.6
Langerhans cell histiocytosis is a rare, multisystemdisease that shows a particular predilection for hypotha-lamo–pituitary axis involvement. Diabetes insipidus isthe most frequent permanent consequence of Langer-hans cell histiocytosis, developing in around a quarter ofpatients. Although the exact prevalence of anterior pitu-itary hormone deficiencies is not known, it is probablyhigh and is almost always associated with diabetesinsipidus. Established pituitary hormone deficienciesare mostly permanent and require prompt diagnosisand treatment, whereas continuous follow-up is neededto detect deficiencies that might evolve later during thecourse of the disease. Involvement of endocrine tissuesother than the pituitary has also been described but isrelatively rare. Further studies are needed to evaluate theeffect that endocrine deficiencies exert on the overallprognosis of patients with Langerhans cell histiocytosis.
IntroductionLangerhans cell histiocytosis (LCH) is a rare diseasecharacterized by the clonal accumulation and/or prolifer-ation of specific dendritic cells resembling normal epider-mal Langerhans cells (LCs) [1]. These cells can infiltratevirtually any organ, albeit without necessarily inducingdysfunction [2]. LCH is also regarded as an inflammatorydisease because an altered expression of cytokines andcellular adhesion molecules important for the migrationand homing of LCs has been demonstrated [1,2]. Thecourse of the disease is fairly unpredictable because itcan resolve spontaneously or progress to a disseminatedform, compromising vital functions with occasionally fatalconsequences [3]. LCH is more often encountered in chil-dren, with a peak age range of 1–3 years and an incidenceof 3–5 cases per million per year, with a male to femaleratio of 2:1 [4]. LCH in adults is even rarer, and is regardedas an ‘orphan disease’, with an estimated prevalence of 1–2cases per million; although LCH can develop at any age,themean age at diagnosis is 33 years [5,6].Making an earlyand accurate diagnosis is important because multisystemLCH is associated with a 20% mortality rate, and 50%of those who survive develop at least one permanentconsequence [7,8].
Corresponding author: Kaltsas, G.A. ([email protected]).Available online 27 June 2007.
www.sciencedirect.com 1043-2760/$ – see front matter � 2007 Elsevier Ltd. All rights reserve
Although LCH is a rare disorder in adults, it shows aparticular predilection for involvement of the hypotha-lamo–pituitary axis (HPA), leading to diabetes insipidus(DI) and/or anterior pituitary dysfunction [9,10]. In arecent multicenter analysis, DI was the most commonpermanent consequence of LCH, occurring in 24% ofpatients [8]. Other endocrine deficiencies can also developin up to 20% of patients [9,11,12]. It is, therefore, importantthat endocrinologists should consider LCH in the evalu-ation of sellar and/or parasellar lesions, particularly whenassociated with pituitary hormone deficiencies. Here,we describe current knowledge regarding endocrinedeficiencies in adult patients with LCH, and their potentialimplications, and discuss the effect that established andevolving therapies could exert on endocrine deficiencies.
Diagnostic criteriaAccording to the recently developed classification scheme,‘definitive diagnosis’ of LCH requires the expression of theCD1a antigen on the lesional cell surface (Figure 1); ‘diag-nosis’ requires the characteristic lesional appearance bylight microscopy and two or more features from a positivelesional cell stain for ATPase, S-100 protein (Figure 1),a-D-mannosidase or characteristic binding of peanut lectin[13]. ‘Presumptive diagnosis’ is suggested when findingsaremerely consistent with those described in the literature[14]. In addition, and according to the criteria of the LCH-A1 therapeutic protocol of the Histiocyte Society for adults(http://www.histio.org/site), the state of the disease isdefined as ‘active’ in those patients with persistence orprogression of disease-related signs or symptoms, with orwithout the appearance of new lesions.
Common non-endocrine manifestations of LCHThere is a wide spectrum of clinical manifestations of LCH,and almost every tissue can be involved with or withoutassociated dysfunction [15]. Organ infiltration commonlycauses distress and disability but is of prognostic import-ance only if vital functions are affected [16]. Two differentphenotypes are usually seen in adults and children withLCH; involvement of bone, lung, skin and DI usuallypredominates in adults, whereas involvement of liver,spleen, lymph nodes and bone marrow is more commonin children [17].
d. doi:10.1016/j.tem.2007.06.003

Figure 1. Expression of the CD1a antigen on the lesional cell surface is mandatory
for the ‘definitive diagnosis’, whereas a positive lesional cell stain for S-100 protein
is a characteristic pathological finding. (a) LCs demonstrating indistinct
cytoplasmic borders and oval nuclei with an irregular outline and occasional
grooves. Many cells are immunoreactive for S-100 protein (�20 magnification). (b)
Immunoreactivity for CD1a shows cytoplasmic immunoreactivity with enhanced
membrane distribution (�20 magnification). Reproduced, with permission, from
Ref. [31].
Review TRENDS in Endocrinology and Metabolism Vol.18 No.6 253
LCH can be stratified according to system involvementas a single-system disease, and is then subdivided accord-ing to whether there is single- or multisite involvement.With a single site, this is usually a single bone lesion,isolated skin disease, or solitary lymph node involvement.Formultiple sites, this would includemultiple bone lesionsor multiple lymph node involvement. Multisystem diseaseincorporates multiple organ disease, with or without dys-function [18]. Single-system disease is usually associatedwith a good prognosis and might require no treatment[19,20]. By contrast, multisystem disease requires severaldifferent therapeutic modalities, including surgery,administration of cytotoxic drugs with or without gluco-corticoids and/or local radiotherapy, and can lead to a pooroutcome [19–21]. Recent multicenter analyses revealedthat patients with multisystem disease have the majorityof permanent consequences, whereas patients with lunginvolvement exhibit the highest mortality [8,16]. LCHfrequently affects the bones; the majority of the lesionsare asymptomatic but can also present as a painful swel-ling [2,4]. Skin rashes are common in infants but adults canalso develop red or brown papular lesions [4,6]. Commonskin manifestations of LCH include crusted dermal areas
www.sciencedirect.com
scattered over the trunk, often healing with depigmenta-tion; and lesions in the perianal region, genitalia, scalp orbehind the ears that often ulcerate [4,6]. LCH can alsopresent as gingival hypertrophy or ulcerative lesions onthe soft or hard palate, buccal mucosa, tongue and lips, andcan infiltrate the cervical, mediastinal and abdominallymph nodes [4,6]. Pulmonary LCH runs an unpredictableand variable course in adults, and could be related to,and/or exacerbated by, cigarette smoking [22]. The clinicalpresentation varies, and patients can be asymptomatic, ordevelop pneumothoraces and/or a restrictive pattern oflung disease that can lead to respiratory failure [6,22].Aural involvement can present as external otitis and infil-tration of the mastoids [4]. Fever and weight loss can alsobe presenting symptoms [16].
Endocrine manifestations of LCHOnly a few studies have investigated the prevalence ofendocrine abnormalities in patients with LCH, usingadequate endocrine testing in a prospective manner[9–12,17,23]. Based on these studies and on epidemi-ological information, endocrine manifestations seem tobe common and mostly develop in the context of multi-system disease [8–12,16,17,23,24] (Table 1).
DI
The HPA is involved in 5–50% of children with LCH, and�17–25% develop DI, which is the most common disease-related endocrine manifestation [9,16,17]. In two largeseries including 1741 children and 274 adults with LCH,a 12% and 30% overall prevalence of DI was found, respect-ively [16,24].When only patients withmultisystem diseaseare included, the prevalence of DI can be as high as 40%and is considered to be the most common disease-relatedpermanent consequence [8]. This high prevalence is notsurprising because LCH and germinoma are the mostcommon diagnoses in patients presenting with centralDI and pituitary stalk enlargement, accounting for 15%and 8% of cases, respectively [25]. When other pituitaryhormone deficiencies are present, the prevalence of DI ismuch higher, reaching 94% [9,10] (Table 1). Although DIusually develops within a year following the diagnosis ofLCH, it can occur at any time, extending to many years[9,12]. DI can also be the presenting feature predating thediagnosis of LCH [25]; 51% of patients presenting with DIwill develop other LCH manifestations within a year[26].The pathogenesis of DI has been attributed to eitherinfiltration and/or scarring of the HPA, or to an auto-immune process involving reacting antibodies againstvasopressin [24]. Established DI is generally permanentand does not respond to any available treatment, exceptsymptomatically [16]. In an attempt to identify risk factorsfor DI, 1741 children with LCH were analyzed [24];patients with multisystem disease and craniofacial invol-vement, particularly of the ear, eye and oral region at thetime of diagnosis, had a 4.6-fold relative risk for developingDI [24]. This risk was attenuated if the disease remainedactive for a long period or became reactivated after a periodof inactivity [24]. DI associated with structural abnormal-ities of the HPA often heralds the development of anterior
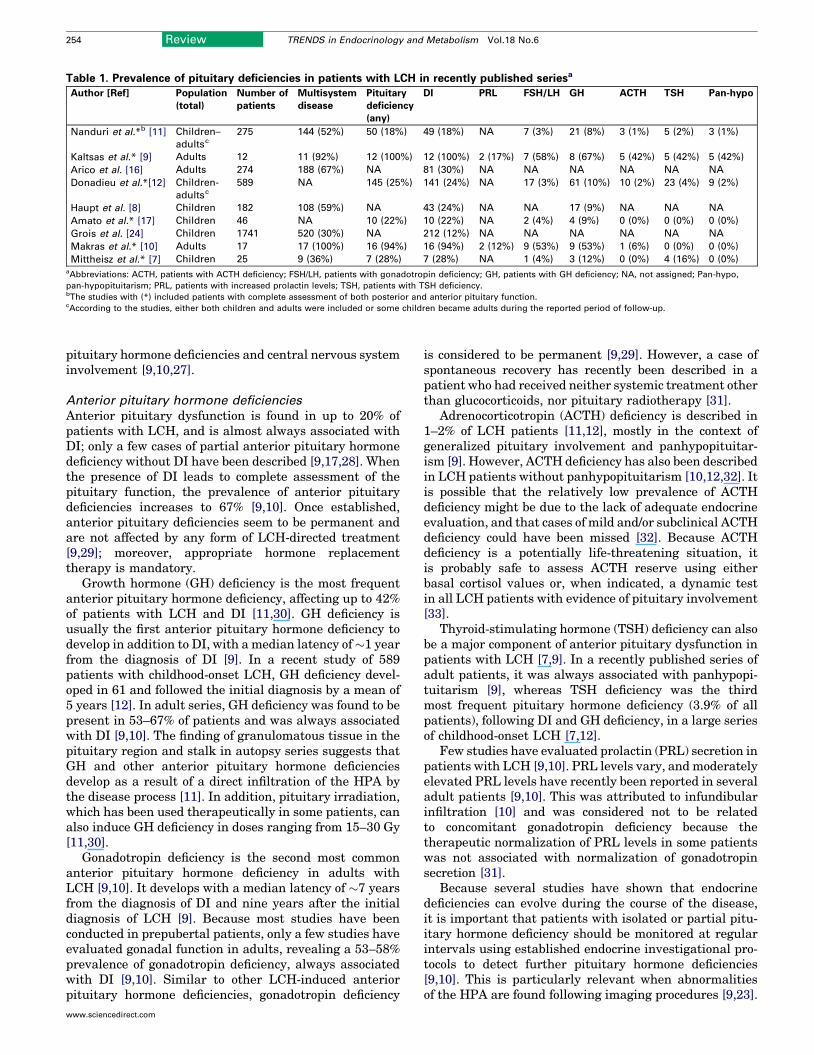
Table 1. Prevalence of pituitary deficiencies in patients with LCH in recently published seriesa
Author [Ref] Population
(total)
Number of
patients
Multisystem
disease
Pituitary
deficiency
(any)
DI PRL FSH/LH GH ACTH TSH Pan-hypo
Nanduri et al.*b [11] Children–
adultsc
275 144 (52%) 50 (18%) 49 (18%) NA 7 (3%) 21 (8%) 3 (1%) 5 (2%) 3 (1%)
Kaltsas et al.* [9] Adults 12 11 (92%) 12 (100%) 12 (100%) 2 (17%) 7 (58%) 8 (67%) 5 (42%) 5 (42%) 5 (42%)
Arico et al. [16] Adults 274 188 (67%) NA 81 (30%) NA NA NA NA NA NA
Donadieu et al.*[12] Children-
adultsc
589 NA 145 (25%) 141 (24%) NA 17 (3%) 61 (10%) 10 (2%) 23 (4%) 9 (2%)
Haupt et al. [8] Children 182 108 (59%) NA 43 (24%) NA NA 17 (9%) NA NA NA
Amato et al.* [17] Children 46 NA 10 (22%) 10 (22%) NA 2 (4%) 4 (9%) 0 (0%) 0 (0%) 0 (0%)
Grois et al. [24] Children 1741 520 (30%) NA 212 (12%) NA NA NA NA NA NA
Makras et al.* [10] Adults 17 17 (100%) 16 (94%) 16 (94%) 2 (12%) 9 (53%) 9 (53%) 1 (6%) 0 (0%) 0 (0%)
Mittheisz et al.* [7] Children 25 9 (36%) 7 (28%) 7 (28%) NA 1 (4%) 3 (12%) 0 (0%) 4 (16%) 0 (0%)aAbbreviations: ACTH, patients with ACTH deficiency; FSH/LH, patients with gonadotropin deficiency; GH, patients with GH deficiency; NA, not assigned; Pan-hypo,
pan-hypopituitarism; PRL, patients with increased prolactin levels; TSH, patients with TSH deficiency.bThe studies with (*) included patients with complete assessment of both posterior and anterior pituitary function.cAccording to the studies, either both children and adults were included or some children became adults during the reported period of follow-up.
254 Review TRENDS in Endocrinology and Metabolism Vol.18 No.6
pituitary hormone deficiencies and central nervous systeminvolvement [9,10,27].
Anterior pituitary hormone deficiencies
Anterior pituitary dysfunction is found in up to 20% ofpatients with LCH, and is almost always associated withDI; only a few cases of partial anterior pituitary hormonedeficiency without DI have been described [9,17,28]. Whenthe presence of DI leads to complete assessment of thepituitary function, the prevalence of anterior pituitarydeficiencies increases to 67% [9,10]. Once established,anterior pituitary deficiencies seem to be permanent andare not affected by any form of LCH-directed treatment[9,29]; moreover, appropriate hormone replacementtherapy is mandatory.
Growth hormone (GH) deficiency is the most frequentanterior pituitary hormone deficiency, affecting up to 42%of patients with LCH and DI [11,30]. GH deficiency isusually the first anterior pituitary hormone deficiency todevelop in addition to DI, with a median latency of�1 yearfrom the diagnosis of DI [9]. In a recent study of 589patients with childhood-onset LCH, GH deficiency devel-oped in 61 and followed the initial diagnosis by a mean of5 years [12]. In adult series, GH deficiency was found to bepresent in 53–67% of patients and was always associatedwith DI [9,10]. The finding of granulomatous tissue in thepituitary region and stalk in autopsy series suggests thatGH and other anterior pituitary hormone deficienciesdevelop as a result of a direct infiltration of the HPA bythe disease process [11]. In addition, pituitary irradiation,which has been used therapeutically in some patients, canalso induce GH deficiency in doses ranging from 15–30 Gy[11,30].
Gonadotropin deficiency is the second most commonanterior pituitary hormone deficiency in adults withLCH [9,10]. It develops with a median latency of �7 yearsfrom the diagnosis of DI and nine years after the initialdiagnosis of LCH [9]. Because most studies have beenconducted in prepubertal patients, only a few studies haveevaluated gonadal function in adults, revealing a 53–58%prevalence of gonadotropin deficiency, always associatedwith DI [9,10]. Similar to other LCH-induced anteriorpituitary hormone deficiencies, gonadotropin deficiency
www.sciencedirect.com
is considered to be permanent [9,29]. However, a case ofspontaneous recovery has recently been described in apatient who had received neither systemic treatment otherthan glucocorticoids, nor pituitary radiotherapy [31].
Adrenocorticotropin (ACTH) deficiency is described in1–2% of LCH patients [11,12], mostly in the context ofgeneralized pituitary involvement and panhypopituitar-ism [9]. However, ACTH deficiency has also been describedin LCH patients without panhypopituitarism [10,12,32]. Itis possible that the relatively low prevalence of ACTHdeficiency might be due to the lack of adequate endocrineevaluation, and that cases of mild and/or subclinical ACTHdeficiency could have been missed [32]. Because ACTHdeficiency is a potentially life-threatening situation, itis probably safe to assess ACTH reserve using eitherbasal cortisol values or, when indicated, a dynamic testin all LCH patients with evidence of pituitary involvement[33].
Thyroid-stimulating hormone (TSH) deficiency can alsobe a major component of anterior pituitary dysfunction inpatients with LCH [7,9]. In a recently published series ofadult patients, it was always associated with panhypopi-tuitarism [9], whereas TSH deficiency was the thirdmost frequent pituitary hormone deficiency (3.9% of allpatients), following DI and GH deficiency, in a large seriesof childhood-onset LCH [7,12].
Few studies have evaluated prolactin (PRL) secretion inpatients with LCH [9,10]. PRL levels vary, and moderatelyelevated PRL levels have recently been reported in severaladult patients [9,10]. This was attributed to infundibularinfiltration [10] and was considered not to be relatedto concomitant gonadotropin deficiency because thetherapeutic normalization of PRL levels in some patientswas not associated with normalization of gonadotropinsecretion [31].
Because several studies have shown that endocrinedeficiencies can evolve during the course of the disease,it is important that patients with isolated or partial pitu-itary hormone deficiency should be monitored at regularintervals using established endocrine investigational pro-tocols to detect further pituitary hormone deficiencies[9,10]. This is particularly relevant when abnormalitiesof the HPA are found following imaging procedures [9,23].

Figure 2. Infundibular infiltration, empty sella and hypothalamic lesions are
among the most common radiological findings in LCH patients and HPA
involvement. Coronal T1-weighted MRI scan without (a) and after (b) contrast
enhancement, indicating enlargement of the infundibulum. (c) Coronal
T1-weighted MRI scan, postcontrast, showing atrophy of the infundibular
stalk and a partially empty sella. (d) Sagittal T1-weighted contrast-enhanced
image showing hypothalamic involvement. Reproduced, with permission, from
Ref. [10].
Review TRENDS in Endocrinology and Metabolism Vol.18 No.6 255
Hypothalamic involvement
Hypothalamic involvement can also occur in patients withLCH, leading to pituitary dysfunction, neuropsychiatricand behavioral disorders, and autonomic and metabolicabnormalities [34]. Neurological and neuropsychologicalinvolvement leading to disturbances of appetite, bodytemperature regulation, sleeping pattern and behavioralskills has been reported, depending on the affected hypo-thalamic region [35]. Increased appetite, resulting inhyperphagia and secondary obesity, is the most frequentlyreported hypothalamic abnormality [9,17]. Furtherabnormalities, such as memory impairment, can resultin problems with treatment compliance, whereas hypo-thalamic-related adipsia might complicate the manage-ment of DI [9]. The combination of DI, adipsia andmemory impairment is a particularly complex manage-ment situation.
Involvement of other endocrine tissue
Thyroid involvement is rare and can develop either as anisolated LCH lesion or in the context of multisystem dis-ease [9,10,36]. In such cases, infiltration can extend beyondthe capsule, causing adherence of the gland to the sur-rounding soft tissues or muscles [37]. Occasionally, invol-vement of the thyroid is found incidentally followingsurgical excision for unrelated conditions [38]. Histologi-cally, these can be mistaken for poorly differentiated car-cinoma [38] or papillary carcinoma on fine needleaspiration [39]. Hypocalcemia in the absence of compensa-tory parathyroid hormone elevation has been reported inan adult patient with diffuse thyroidal involvement andinfiltration of all excised parathyroid glands [37]. AlthoughLCH can frequently affect the lower genital tract [4],involvement of the ovaries is rare; only one of 42 patientswith LCH genital lesions had ovarian involvement in thecontext of disseminated disease [40]. Infiltration of theadrenals glands has been documented from autopsy stu-dies [41] but no significant clinical consequences have beendescribed [4,6]. Direct involvement of the pancreas by LCHis extremely rare; it almost always occurs as a part ofmultisystem disease, and histological documentation iseven more rarely obtained [42].
Radiological findings of the HPAMagnetic resonance imaging (MRI) is currently consideredto be the method of choice for the evaluation of the HPA,following the administration of gadolinium-dimegluminegadopentetate [43]. Although there is no specific MRIappearance of LCH related HPA lesions [9,27], almostall patients with LCH-induced DI demonstrate loss ofthe physiological intense signal (‘bright spot’) of theposterior pituitary [9,10,26,43,44]. Another common MRIfinding is infundibular enlargement [10,43], describedin up to 71% of patients at the time of diagnosis of DI[43], and/or the presence of hypothalamic mass lesions,described in 8–18% of patients exhibiting one or morepituitary hormone deficiencies [9,10] (Figure 2). Pituitaryenlargement can also be found in up to 16% of adultpatients, always associated with DI [10]; partially or com-pletely empty sella and infundibular atrophy are lesscommon radiological abnormalities of HPA involvement
www.sciencedirect.com
[9,10] (Figure 2). The majority of these appearances havebeen attributed to either direct infiltration or cytokine-induced gliosis and/or fibrosis [45]. Anterior pituitary hor-mone deficiency can also be found in the absence of anystructural HPA abnormalities [10,46]. In such cases,microinjury leading to vascular impairment and scarring,or cytokine modulation from adjacent osseous lesionsand/or an autoimmune effect, have been implicated [47,48].
Bone and calcium metabolism in LCHSkeletal involvement is one of the most frequentpresenting features of adults with LCH [6]. The classicalappearance is that of a lytic lesion, although osteoblasticlesions can also develop [49,50]. In addition, LCs cansecrete bone-resorbing cytokines, such as interleukin-1and prostaglandin E2, associatedwith increased bone turn-over [51,52]. Hypercalcemia has been described in fourpatients [50,53]; this was attributed to increased 1,25dihydroxyvitamin D levels in an adult patient [53] andadministration of vinblastine in two children [50].Although there is no information regarding the exactprevalence of osteoporosis in adults with LCH, it is likelyto be high owing to the presence of several predisposingfactors, such as inflammatory cytokines, anterior pituitaryhormone deficiencies and treatment, particularly withglucocorticoids [54,55].
Metabolic alterations in LCH and cardiovascular riskfactorsAlthough LCH is a clonal disorder [56], it also exhibitsfeatures of an inflammatory disease [51,57]. The ongoinginflammatory process, presence of hypopituitarism andvarious therapies used for the treatment of LCH, particu-larly glucocorticoids, are considered to be independent risk

256 Review TRENDS in Endocrinology and Metabolism Vol.18 No.6
factors for cardiovascular diseases, probably through theinduction of insulin resistance (IR) [58–60]. It is thereforepossible that patients with LCH represent a group athigher risk for cardiovascular diseases through the addi-tive effect of several different mechanisms known to induceIR. Only a few previous case reports have describedabnormalities of carbohydrate metabolism and/or diabetesmellitus (DM) in patients with LCH [61,62]. Althoughthese patients were not studied in detail, it was claimedthat hepatic and/or pancreatic involvement by the diseaseprocess, with subsequent insulin deficiency, was the prob-able underlying pathogenic mechanism [61,62]. However,patients with DI and DM without hepatic and/or pancrea-tic involvement and evidence of IR have also beendescribed [15]. A prospective trial looking at the prevalenceof IR, abnormalities of carbohydrate metabolism and othercardiovascular risk factors in patients with LCH is neededto clarify this issue.
TreatmentEstablished complete or partial anterior and/or posteriorpituitary hormone deficiencies should be treated withstandard replacement regimens [33]. Administration ofGH has always been a matter of concern, owing to thepossibility of a GH-induced promoting effect in diseaseactivity [30]. However, the use of GH, particularly inchildren with short stature, was found not to be associatedwith any alterations in disease activity and/or recurrences[11,30]. There are no data regarding the administration ofGH in adults, although its use could be potentiallybeneficial, owing to its effects on metabolic parameters,general well being and quality of life [63].
The standard therapeutic approach to adult LCH is yetto be defined [16]. The combination of vinblastine withsteroids is the most frequently used initial therapy formultisystem disease in adults [16] and is the currentstandard treatment of multisystem disease in children[64]. Clinical trials carried out by the Histiocyte Societyno longer support the use of etoposide because of itsassociated high risk for the development of leukemia[16], whereas reports have shown that the purine analogcladribine (2-chlorodeoxyadenosine) can be effective foradults with recurrent and/or disseminated disease [65].Radiotherapy, at doses up to 25 Gy, has been used inpatients with endocrine deficiencies and radiological evi-dence of HPA involvement, and has been associated withpartial or temporary radiological improvement only [9].Established pituitary hormone deficiencies are consideredto be permanent and do not respond to any form of treat-ment [8,9,29]. This information is mainly derived fromretrospective studies involving subjects who were treatedseveral years ago, using non-homogeneous criteria, andbefore the international protocols proposed by the Histio-cyte Society were implemented [64,66]. It is thereforepossible that the endocrine sequelae described in long-term survivors with LCH might be different in subjectswho received more ‘modern’ treatment. Two large coopera-tive studies in patients with LCH, one including mainlychildren and the other using a different and more ‘aggres-sive’ therapeutic protocol, found a 14% prevalence of DI[35,64]. Although this prevalence of DI was lower than
www.sciencedirect.com
previously reported, this might well be related to therelatively short follow-up of these patients; it could alsobe related to the administered treatment [64]. In light ofmultisystem involvement, the different number of bio-chemical, radiological and histopathological investigationsrequired to evaluate the extent and activity of the disease,and the different therapeutic modalities that can beapplied, patients should be treated using a multidisciplin-ary approach.
ConclusionsLCH should be included in the differential diagnosis ofadults presenting with DI, anterior pituitary hormonedeficiencies and sellar pathology. The possibility of disse-minated disease should always be considered, and patientsshould undergo a careful multidisciplinary evaluation andfollow-up to screen for possible asymptomatic localizationsthat could warrant prompt treatment. In the case of partialpituitary hormone deficiency, continuous monitoring isrequired because further deficiencies can develop duringthe course of the disease. Aside from hormonal replace-ment therapy, the optimal therapeutic approach to adultpatients with LCH has not yet been established. Furtherpopulation-based incidence, quality-of-life and late-effectstudies are urgently required.
References1 Arceci, R.J. (1999) The histiocytoses: the fall of the Tower of Babel.Eur.
J. Cancer 35, 747–7672 Arico, M. and Egeler, R.M. (1998) Clinical aspects of Langerhans cell
histiocytosis. Hematol. Oncol. Clin. North Am. 12, 247–2583 Schmitz, L. and Favara, B.E. (1998) Nosology and pathology of
Langerhans cell histiocytosis. Hematol. Oncol. Clin. North Am. 12,221–246
4 Broadbent, V. et al. (1994) Langerhans cell histiocytosis – clinical andepidemiological aspects. Br. J. Cancer Suppl. 23, S11–S16
5 Baumgartner, I. et al. (1997) Langerhans’-cell histiocytosis in adults.Med. Pediatr. Oncol. 28, 9–14
6 Malpas, J.S. (1998) Langerhans cell histiocytosis in adults. Hematol.Oncol. Clin. North Am. 12, 259–268
7 Mittheisz, E. et al. (2007) Central nervous system-related permanentconsequences in patients with Langerhans cell histiocytosis. Pediatr.Blood Cancer 48, 50–56
8 Haupt, R. et al. (2004) Permanent consequences in Langerhans cellhistiocytosis patients: a pilot study from the Histiocyte Society–LateEffects Study Group. Pediatr. Blood Cancer 42, 438–444
9 Kaltsas,G.A. et al. (2000)Hypothalamo–pituitary abnormalities inadultpatients with Langerhans cell histiocytosis: clinical, endocrinological,and radiological features and response to treatment.J.Clin. Endocrinol.Metab. 85, 1370–1376
10 Makras, P. et al. (2006) Evolving radiological features of hypothalamo–pituitary lesions in adult patients with Langerhans cell histiocytosis(LCH). Neuroradiology 48, 37–44
11 Nanduri, V.R. et al. (2000) Growth and endocrine disorders inmultisystem Langerhans’ cell histiocytosis. Clin. Endocrinol. (Oxf.)53, 509–515
12 Donadieu, J. et al. (2004) Endocrine involvement in pediatric-onsetLangerhans’ cell histiocytosis: a population-based study. J. Pediatr.144, 344–350
13 Favara, B.E. et al. (1997) Contemporary classification of histiocyticdisorders. The WHO Committee on Histiocytic/Reticulum CellProliferations. Reclassification Working Group of the HistiocyteSociety. Med. Pediatr. Oncol. 29, 157–166
14 Chu, T. et al. (1987) Histiocytosis syndromes in children. Lancet 2,41–42
15 Dunger, D.B. et al. (1989) The frequency and natural history of diabetesinsipidus in children with Langerhans-cell histiocytosis. N. Engl.J. Med. 321, 1157–1162

Review TRENDS in Endocrinology and Metabolism Vol.18 No.6 257
16 Arico, M. et al. (2003) Langerhans cell histiocytosis in adults. Reportfrom the International Registry of the Histiocyte Society. Eur. J.Cancer 39, 2341–2348
17 Amato, M.C. et al. (2006) Endocrine disorders in pediatric-onsetLangerhans cell histiocytosis. Horm. Metab. Res. 38, 746–751
18 Broadbent, V. and Gadner, H. (1998) Current therapy for Langerhanscell histiocytosis. Hematol. Oncol. Clin. North Am. 12, 327–338
19 Ceci, A. et al. (1993) Langerhans cell histiocytosis in childhood: resultsfrom the Italian Cooperative AIEOP–CNR–HX ’83 study.Med. Pediatr.Oncol. 21, 259–264
20 Gadner, H. et al. (1994) VP-16 and the treatment of histiocytosis. Eur.J. Pediatr. 153, 389
21 Komp, D.M. et al. (1981) A staging system for histiocytosis X: aSouthwest Oncology Group Study. Cancer 47, 798–800
22 Vassallo, R. et al. (2002) Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N. Engl. J. Med. 346, 484–490
23 Broadbent, V. et al. (1993) Anterior pituitary function and computedtomography/magnetic resonance imaging in patients with Langerhanscell histiocytosis and diabetes insipidus. Med. Pediatr. Oncol. 21,649–654
24 Grois, N. et al. (2006) Risk factors for diabetes insipidus in Langerhanscell histiocytosis. Pediatr. Blood Cancer 46, 228–233
25 Maghnie, M. et al. (2000) Central diabetes insipidus in children andyoung adults. N. Engl. J. Med. 343, 998–1007
26 Prosch, H. et al. (2004) Central diabetes insipidus as presentingsymptom of Langerhans cell histiocytosis. Pediatr. Blood Cancer 43,594–599
27 Grois, N.G. et al. (1998) Central nervous system disease in Langerhanscell histiocytosis. Hematol. Oncol. Clin. North Am. 12, 287–305
28 Tabarin, A. et al. (1991) Histiocytosis X of the hypothalamus.J. Endocrinol. Invest. 14, 139–145
29 Kilpatrick, S.E. et al. (1995) Langerhans’ cell histiocytosis(histiocytosis X) of bone. A clinicopathologic analysis of 263 pediatricand adult cases. Cancer 76, 2471–2484
30 Donadieu, J. et al. (2004) Incidence of growth hormone deficiencyin pediatric-onset Langerhans cell histiocytosis: efficacy andsafety of growth hormone treatment. J. Clin. Endocrinol. Metab. 89,604–609
31 Makras, P. et al. (2005) Spontaneous gonadotrophin deficiency recoveryin an adult patient with Langerhans cell histiocytosis (LCH). Pituitary8, 169–174
32 Modan–Moses, D. et al. (2001) Hypopituitarism in Langerhans cellhistiocytosis: seven cases and literature review. J. Endocrinol. Invest.24, 612–617
33 Stewart, P.M. (2003) The adrenal cortex. In Williams Textbook ofEndocrinology (10th edn) (Larsen, R. et al., eds), pp. 491–551,Saunders
34 Cone, R. et al. (2003) Neuroendocrinology. In Williams Textbook ofEndocrinology (10th edn) (Larsen, R. et al., eds), pp. 81–176, Saunders
35 Grois, N. et al. (1995) Diabetes insipidus in Langerhans cellhistiocytosis: results from the DAL-HX 83 study. Med. Pediatr.Oncol. 24, 248–256
36 Thompson, L.D. et al. (1996) Langerhans cell histiocytosis of thethyroid: a series of seven cases and a review of the literature. Mod.Pathol. 9, 145–149
37 Yap, W.M. et al. (2001) Langerhans cell histiocytosis involving thethyroid and parathyroid glands. Mod. Pathol. 14, 111–115
38 Coode, P.E. and Shaikh, M.U. (1988) Histiocytosis X of the thyroidmasquerading as thyroid carcinoma. Hum. Pathol. 19, 239–241
39 Kitahama, S. et al. (1996) Thyroid involvement by malignanthistiocytosis of Langerhans’ cell type. Clin. Endocrinol. (Oxf.) 45,357–363
40 Axiotis, C.A. et al. (1991) Langerhans cell histiocytosis of the femalegenital tract. Cancer 67, 1650–1660
41 Miller, A.A. and Ramsden, F. (1965) Neural and visceralxanthomatosis in adults. J. Clin. Pathol. 18, 622–635
42 Goyal, R. et al. (2006) Langerhans cell histiocytosis infiltration intopancreas and kidney. Pediatr. Blood Cancer DOI: 10.1002/pbc.20746(http://www3.interscience.wiley.com/cgi-bin/jhome/106561790)
www.sciencedirect.com
43 Grois, N. et al. (2004) Course and clinical impact of magnetic resonanceimaging findings in diabetes insipidus associated with Langerhans cellhistiocytosis. Pediatr. Blood Cancer 43, 59–65
44 Maghnie, M. et al. (1993) Role of MR imaging in the evaluation of thefunctional status of the posterior pituitary gland: the view of a pediatricendocrinologist. AJNR Am. J. Neuroradiol. 14, 1443–1445
45 Barthez,M.A. et al. (2000) Langerhans cell histiocytosis and the centralnervous system in childhood: evolution and prognostic factors. Resultsof a collaborative study. J. Child Neurol. 15, 150–156
46 Howarth, D.M. et al. (1999) Langerhans cell histiocytosis: diagnosis,natural history, management, and outcome. Cancer 85, 2278–2290
47 Maghnie, M. et al. (1994) Evolving pituitary hormone deficiency isassociated with pituitary vasculopathy: dynamic MR study in childrenwith hypopituitarism, diabetes insipidus, and Langerhans cellhistiocytosis. Radiology 193, 493–499
48 Scherbaum, W.A. et al. (1986) Autoimmune cranial diabetes insipidus:its association with other endocrine diseases and with histiocytosis X.Clin. Endocrinol. (Oxf.) 25, 411–420
49 Meyer, J.S. and De Camargo, B. (1998) The role of radiology in thediagnosis and follow-up of Langerhans cell histiocytosis. Hematol.Oncol. Clin. North Am. 12, 307–326
50 Jubinsky, P.T. (2003) Hypercalcemia in Langerhans cell histiocytosis:is it therapy-related? J. Pediatr. Hematol. Oncol. 25, 176–179
51 Egeler, R.M. et al. (1999) Differential in situ cytokine profiles ofLangerhans-like cells and T cells in Langerhans cell histiocytosis:abundant expression of cytokines relevant to disease and treatment.Blood 94, 4195–4201
52 Arenzana–Seisdedos, F. et al. (1986) Histiocytosis X. Purified (T6+)cells from bone granuloma produce interleukin 1 and prostaglandin E2in culture. J. Clin. Invest. 77, 326–329
53 Al Ali, H. et al. (2002) Hypercalcemia in Langerhans’ cellgranulomatosis with elevated 1,25 dihydroxyvitamin D (calcitriol)level. Bone 30, 331–334
54 Dalle, C.L. et al. (2001) Comparison of trabecular bonemicroarchitecture and remodeling in glucocorticoid-induced andpostmenopausal osteoporosis. J. Bone Miner. Res. 16, 97–103
55 Wuster, C. et al. (2001) The influence of growth hormone deficiency,growth hormone replacement therapy, and other aspects ofhypopituitarism on fracture rate and bone mineral density. J. BoneMiner. Res. 16, 398–405
56 Willman, C.L. et al. (1994) Langerhans’-cell histiocytosis (histiocytosisX)-a clonal proliferative disease. N. Engl. J. Med. 331, 154–160
57 de Graaf, J.H. et al. (1996) The presence of cytokines in Langerhans’cell histiocytosis. J. Pathol. 180, 400–406
58 Wei, L. et al. (2004) Taking glucocorticoids by prescription is associatedwith subsequent cardiovascular disease. Ann. Intern. Med. 141,764–770
59 Alexander, R.W. (1994) Inflammation and coronary artery disease. N.Engl. J. Med. 331, 468–469
60 Rosen, T. and Bengtsson, B.A. (1990) Premature mortality due tocardiovascular disease in hypopituitarism. Lancet 336, 285–288
61 Ben, G. et al. (2001) A disseminated form of Langerhans histiocytosisassociated with diabetes insipidus and diabetes mellitus. Rev. Med.Interne 22, 469–474 [In French]
62 Trochtenberg, D.S. and Dessypris, E.N. (1990) Reversible hepatomegalyand diabetesmellitus in an adultwith disseminated histiocytosis X.Am.J. Med. Sci. 299, 179–184
63 Papadogias, D.S. et al. (2003) GH deficiency in adults. Hormones(Athens) 2, 217–218
64 Gadner, H. et al. (2001) A randomized trial of treatmentfor multisystem Langerhans’ cell histiocytosis. J. Pediatr. 138,728–734
65 Pardanani, A. et al. (2003) 2-Chlorodeoxyadenosine therapy fordisseminated Langerhans cell histiocytosis. Mayo Clin. Proc. 78,301–306
66 Gadner, H. et al. (1994) Treatment strategy for disseminatedLangerhans cell histiocytosis. DAL HX–83 Study Group. Med.Pediatr. Oncol. 23, 72–80
Recommended

















