
www.elsevier.com/locate/ynbdi
Neurobiology of Disease 23 (2006) 23 – 35
Prostaglandin E2 and BDNF levels in rat hippocampus are negatively
correlated with status epilepticus severity: No impact on survival of
seizure-generated neurons
Maria Antonietta Ajmone-Cat,a,b,d,* Robert E. Iosif,b,d Christine T. Ekdahl,b,d Zaal Kokaia,c,d
Luisa Minghetti,a and Olle Lindvall b,d
aDepartment of Cell Biology and Neuroscience, Istituto Superiore di Sanita, Viale Regina Elena 299, 00161 Rome, ItalybLaboratory of Neurogenesis and Cell Therapy, Section of Restorative Neurology, Wallenberg Neuroscience Center,
University Hospital, SE-221 84 Lund, SwedencLaboratory of Neural Stem Cell Biology, Section of Restorative Neurology, Stem Cell Institute, University Hospital, SE-221 84 Lund, SwedendLund Strategic Research Center for Stem Cell Biology and Cell Therapy, Lund, Sweden
Received 10 September 2005; revised 24 January 2006; accepted 27 January 2006
Available online 13 March 2006
Partial and generalized status epilepticus (pSE and gSE) trigger the
same level of progenitor cell proliferation in adult dentate gyrus, but
survival of new neurons is poor after gSE. Here, we show markedly
elevated levels of prostaglandin E2 (PGE2) and brain-derived neuro-
trophic factor (BDNF) in rat hippocampal formation at 7 days
following pSE but not gSE. Administration of the cyclooxygenase
(COX) inhibitor flurbiprofen for 1 week, starting at day 8 post-SE,
abated PGE2 and decreased BDNF levels, but did not affect survival of
new neurons 4 weeks later. Thus, high PGE2 and BDNF levels induced
by pSE are probably not of major importance for survival of new
neurons during the first days after formation. We propose that they
modulate other aspects of synaptic and cellular plasticity, and thereby
may influence epileptogenesis.
D 2006 Elsevier Inc. All rights reserved.
Keywords: Prostaglandin E2 (PGE2); Cyclooxygenase-2 (COX-2); Brain-
derived neurotrophic factor (BDNF); EP2; EP3; Isoprostanes; Flurbiprofen;
Status epilepticus; Neurogenesis
Introduction
The restorative potential of the adult brain following injury and
its plasticity to environmental and behavioral cues arise partly from
the ability of its own neural stem cells to react to physio-
pathological changes in their ‘‘niche’’ with a complex neurogenic
response (Gage, 2002). The process of neurogenesis comprises at
0969-9961/$ - see front matter D 2006 Elsevier Inc. All rights reserved.
doi:10.1016/j.nbd.2006.01.010
* Corresponding author. Department of Cell Biology and Neuroscience,
Istituto Superiore di Sanita, Viale Regina Elena 299, 00161 Rome, Italy.
Fax: +39 06 4957821.
E-mail address: [email protected] (M.A. Ajmone-Cat).
Available online on ScienceDirect (www.sciencedirect.com).
least four distinct steps: proliferation, survival, migration, and
differentiation, with each step having a specific regulatory
machinery. In the adult dentate gyrus (DG), multipotent progen-
itors located in the subgranular zone (SGZ) continuously generate
neuroblasts, which migrate into the granule cell layer (GCL), adopt
the morphological characteristics of granule cells, and extend
axonal projections to their appropriate target, the CA3 region (Lie
et al., 2004). The new neurons develop into functional granule cells
(van Praag et al., 2002) but have also been reported to differentiate
into inhibitory interneurons (Liu et al., 2003). The formation of
new neurons in the adult SGZ is modulated by different
physiological stimuli, and circumstantial evidence suggests a link
between level of hippocampal neurogenesis and cognitive function
(Lie et al., 2004). Insults to the adult brain, such as epileptic
seizures and cerebral ischemia, trigger increased formation of
neurons in the SGZ (Bengzon et al., 1997; Parent et al., 1997; Jin et
al., 2001; Arvidsson et al., 2001; Liu et al., 1998). Whether the
new DG neurons generated after brain insults contribute to
functional recovery or impairment is not known. For example, it
has been proposed that the new neurons formed after epileptic
seizures participate in the neural circuits which underlie the
pathological excitability in chronic epilepsy (Scharfman, 2004).
Following status epilepticus (SE), SGZ cell proliferation is high
during about 2 weeks after the insult but then returns to baseline
(Parent et al., 1997). We have previously demonstrated that increase
of cell proliferation in the SGZ following electrically induced SE,
peaking at 7 days post-SE, is independent of the severity of SE
(Mohapel et al., 2004). In contrast, the degree of survival of the
newborn neurons is clearly influenced by the severity of the initial
epileptic insult. In rats exhibiting partial SE (pSE), defined by the
predominance of non-clonic convulsions, the new neurons formed at
1 week showed no significant decrease over the subsequent 4 weeks.
In contrast, there was a 65% loss of new neurons during the same

M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–3524
time period in rats with generalized SE (gSE), defined by the
predominance of clonic convulsions (Mohapel et al., 2004). The
severity of the initial injury and the associated local inflammation,
sustained by microglial cells, were probably main causes of poor
survival (Ekdahl et al., 2003a,b; Monje et al., 2003, Mohapel et al.,
2004). Better knowledge of mechanisms involved in the marked loss
of the newborn neurons, which is observed also during striatal
neurogenesis after stroke (Arvidsson et al., 2002), is crucial to
provide opportunities of manipulating endogenous neurogenesis
and exploit its possible therapeutic potential.
Here, we have induced SE by electrical stimulation in the
hippocampus, and compared pSE and gSE rats in order to identify
factors in the microenvironment which are differentially regulated
by SE severity and may underlie differences in neurogenesis. We
particularly addressed the question whether SE severity differen-
tially affects the synthesis of prostaglandin E2 (PGE2) and
cyclooxygenase (COX)-2, the enzyme catalyzing the first com-
mitted step in PGE2 synthesis. COX-2 is expressed by neurons in
an activity-dependent way (Yamagata et al., 1993; Kaufmann et al.,
1996), and increases dramatically after seizures (Marcheselli and
Bazan, 1996; Tu and Bazan, 2003). In addition, PGE2 has recently
emerged as a putative neuroprotective factor in several paradigms
of neurodegeneration, depending on the extent of induction,
cellular source, and subset of specific receptors dominantly
expressed in a given area (Minghetti, 2004). Finally, there is
experimental evidence indicating that COX-2 and PGE2 can
influence hippocampal neurogenesis by promoting proliferation
of SGZ progenitors (Uchida et al., 2002; Sasaki et al., 2003).
Recent evidence suggests a functional link between PGE2 and
the neurotrophin brain-derived neurotrophic factor (BDNF), in
which BDNF expression in the rat hippocampus appears to be under
the control of COX-2 activity (Shaw et al., 2003). In accordancewith
this idea, also BDNF protein levels are markedly increased in
hippocampal subregions after recurring seizures (Elmer et al., 1998).
BDNF has been shown to promote differentiation and survival of
neuronal progenitors in rat hippocampus and cortex (Lee et al.,
2002; Barnabe-Heider and Miller, 2003), and functional BDNF
signaling is required for long-term survival of newborn DG neurons
in themouse (Sairanen et al., 2005). However, depending on the type
of the insult, and the level of BDNF and its mode of delivery, this
neurotrophic factor may also counteract neurogenesis (Larsson et
al., 2002; Gustafsson et al., 2003).
The specific objectives of the present study were threefold: first,
to quantify PGE2 and BDNF levels and determine the distribution
and magnitude of COX-2 expression in hippocampal subregions at
different time points after pSE and gSE; second, to analyze the
expression of PGE2 receptors on different DG cell types, especially
on the newly formed neuroblasts, to assess if these cells can be
directly influenced by PGE2 in the early phases of neurogenesis;
finally, to explore whether manipulation of PGE2, and possibly
BDNF synthesis by the non-selective COX inhibitor flurbiprofen,
could affect the survival of the newborn neurons generated
following SE.
Materials and methods
Animals and surgery
125 male Sprague–Dawley rats (Mollegaard’s Breeding Center,
Copenhagen, Denmark), weighing 250 g at the time of surgery,
were housed separately under 12 h light/12 h dark conditions with
ad libitum access to food and water. 116 rats were anesthetized
with halothane and implanted unilaterally with a twisted insulated
stainless-steel stimulating/recording electrode (Plastics One, Roa-
noke, VA) into the right ventral hippocampal CA1–CA3 region
(coordinates: 4.8 mm caudal, 5.2 mm lateral to bregma, 6.3 mm
ventral from dura, toothbar at �3.3 mm; Paxinos and Watson,
1997). Rats were then either subjected to electrically induced SE
(n = 76) or used as non-stimulated controls and referred as to sham
(n = 40). Nine rats not subjected to electrode implantation were
used as intact controls and referred as to controls. Experiments
followed guidelines set by the Malmo-Lund Ethical Committee for
use and care of laboratory animals.
Induction of status epilepticus
Seven days after electrode implantation, SE was induced as
originally described by Lothman et al. (1989). Afterdischarge (AD)
threshold was determined for each rat through a 1 s 50 Hz electrical
current, starting at 10 AA and increasing in 10 AA increments at 1
min intervals until an AD lasting 5 s or more was registered (Chart
3.6.3, PowerLab/MacLab; AD Systems, Hastings, UK). Thirty
minutes later, rats received 1 h supra-threshold stimulation with 10
s trains of 1 ms biphasic square wave pulses, at a frequency of 50
Hz. Every 10 min, stimulations were interrupted for 1 min of
electroencephalogram (EEG) recordings and AD measurements.
After 1 h of stimulation, all rats exhibited continuous self-sustained
ictal EEG activity. Based on the severity of behavioral convulsions,
two different SE profiles were distinguished (Mohapel et al.,
2004): partial SE (pSE, including grade 1–2 according to Racine’s
scoring system for kindled seizures; Racine, 1972) and generalized
SE (gSE, including grade 3–4). Examples of typical EEG
recordings from sham, pSE, and gSE rats are given in Figs. 1A–
C. Behavioral convulsions and ictal EEG activity were arrested
with pentobarbital (65 mg/kg, i.p.) 2 h after cessation of
stimulation.
Immunocytochemistry
Thirty-five rats with partial and 13 rats with generalized SE,
and 26 electrode-implanted, non-stimulated rats were used for
immunocytochemistry. Rats received an overdose of sodium
penthobarbital (200 mg/kg, i.p.) and were transcardially perfused
with 250 ml of saline followed by 250 ml of ice-cold formaldehyde
solution (4% paraformaldehyde in 0.1 M phosphate-buffered saline
(PBS), pH 7.4). Brains were removed, post-fixed overnight in the
same fixative, and then placed in 20% sucrose/0.1M phosphate
buffer for at least 24 h. Coronal sections (30 Am) were cut on a
freezing microtome and stored in cryoprotective solution at
�20-C. For COX-2/neuron-specific nuclear protein (NeuN)
double-labeling, free-floating sections were first microwaved in
citrate buffer (0.01M, pH 6) for 2 min for COX-2 retrieval, then
rinsed in potassium phosphate-buffered saline (KPBS) before pre-
incubation with 5% donkey and horse serum in 0.25% Triton X-
100 for 1 h at room temperature. The sections were incubated with
a goat polyclonal anti-COX-2 antibody (1:1000, M19, Santa Cruz
Biotechnology Inc., Santa Cruz, CA) and a mouse anti-NeuN
antibody (1:100, Chemicon, Temecula, CA) overnight at +4-C,followed by rinsing and incubation in the dark for 2 h with Cy3-
conjugated donkey anti-goat IgG antibody (1:200, Jackson
ImmunoResearch, West Grove, PA) and biotinylated horse anti-

Fig. 1. Typical EEG recordings from a sham-operated rat (A) and, during the period of self-sustained seizures, from rats exhibiting pSE (B) and gSE (C).
Behavioral convulsions were scored into different grades, indicated below the recordings, according to Racine (1972). Levels of PGE2 (D, E) and 8-epi-PGF2a(F, G) concentrations in hippocampal subregions at 7 (D, F) and 35 days (E, G) following pSE and gSE, and in sham-operated and control animals, n = 4–6.
Values are means T SEM. *P < 0.05.
M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–35 25
mouse IgG antibody (1:200, Vector Laboratories, Burlingame,
CA). After rinsing, sections were incubated in streptavidin Alexa
Fluor 488 (1:200, Molecular Probes, The Netherlands) in the dark
for 2 h, mounted on glass slides and cover-slipped with glycerol-
based mounting medium.
The staining protocol was similar for COX-2/glial fibrillary
acidic protein (GFAP) and COX-2/activated microglia marker
(ED1) double-labeling, for which mouse anti-GFAP (1:1000
Sigma) and mouse anti-ED1 (1:200, Serotec, Oslo, Norway)
primary antibodies were used for labeling of glial cells. For COX-
2/Doublecortin (Dcx) double-labeling, performed with primary
antibodies both made in goat, the protocol was modified as
follows: in the first pre-incubation step, horse serum was omitted.
After incubation with Cy3-conjugated donkey anti-goat IgG,
sections were rinsed, incubated 1 h in the dark with 5% donkey
and rabbit serum in 0.25% Triton X-100, then incubated with 17.5
Ag/ml of F(abV)2 fragment of donkey anti-goat IgG (H + L)
(Jackson ImmunoResearch) in T-KPBS for 6 h at +4-C, followedby rinses and incubation with goat anti-Dcx (1:400, C-18 Santa
Cruz) overnight at +4-C. The anti-Dcx antibody was then revealed
by using a biotinylated rabbit anti-goat IgG antibody (1:200,
Vector Laboratories) and streptavidin Alexa Fluor 488 as
described below.
The procedure for BrdU/NeuN double-labeling has been
described in detail elsewhere (Ekdahl et al., 2001). A monoclonal
rat anti-BrdU antibody (Oxford Biotechnology, UK) was used. For
BrdU/EP2 and BrdU/EP3 double-labeling, rabbit anti-EP2 (1:500,
Cayman Chemical, Ann Arbor, MI) or rabbit anti-EP3 (1:250,
Cayman Chemical) antibodies were used, preceded by an overnight
incubation in 0.25% Triton X-100, and revealed by a biotinylated
horse anti-rabbit IgG antibody (1:200, Vector Laboratories)
followed by Alexa Fluor 488 (1:200, Molecular Probes). The
same secondary antibody was used to reveal EP2 and EP3 in
double-staining with ED1 and GFAP that were instead shown by
Cy3-conjugated horse anti-mouse IgG. For EP2/Dcx and EP3/Dcx
double-labeling, secondary antibodies were Cy3-conjugated don-
key anti-rabbit IgG for EP2 and EP3, and biotinylated horse anti-
goat IgG for Dcx.
Brain dissection and PGE2, 8-epi-PGF2a, and BDNF extraction
Nineteen rats with partial and 9 rats with generalized SE, 14
electrode-implanted non-stimulated rats, and 9 intact rats were
used for PGE2, 8-epi-PGF2a, and BDNF measurements. After
decapitation, brains were immediately removed from the skull
and transferred into ice-cold saline. The hippocampal formation

M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–3526
was then subdissected as described by Elmer et al. (1998). In
brief, DG was carefully separated from the rest of the
hippocampus, which was divided in two parts: the CA1 region
and subiculum, and the CA3 region. Dissected tissues from both
hemispheres were placed in Eppendorf tubes, immediately frozen
on dry ice and stored at �80-C until metabolite extraction. A
detailed procedure for PGE2 and 8-epi-PGF2a extraction has been
described elsewhere (Minghetti et al., 2000). In brief, 200 Al ofice-cold Tris–HCl buffer pH 7.5 containing 10 Ag/ml of the COX
inhibitor indomethacin (stock solution 100� in ethanol) to avoid
ex vivo PGE2 synthesis, and 10 AM of the radical scavenger BHT
(stock solution 100� in ethanol) to avoid auto-oxidation, were
added to each frozen sample, which was quickly thawed,
homogenized with a Teflon pestle (Sigma) �20 cycles in an
ice bath-vigorously vortexed, and centrifuged at 14,000 rpm for
45 min at +4-C. The supernatants were collected and stored at
�80-C until analysis. Pellets were resuspended in 200 Al of 0.1M NaOH for protein determination (see above). For BDNF
extraction, tissue samples were rapidly thawed in ice-cold PBS
containing a protease inhibitor cocktail (1:10 of a stock solution
in H20, prepared following the manufacturer’s instructions.
Sigma), and processed as before.
PGE2 and 8-epi-PGF2a measurement
PGE2 and 8-epi-PGF2a were measured in tissue extracts by
high sensitivity colorimetric enzyme immunoassays (EIA kits,
detection limit for PGE2: 7.8 pg/ml, Assay Designs, Inc. Ann
Arbor, MI; detection limit for 8-epi-PGF2a: 2 pg/ml; Cayman
Chemical, Ann Arbor, MI). According to the manufacturers, the
cross-reactivity of the anti-PGE2 antibody with 8-epi-PGF2a was
less than 0.25% and that of anti-8-epi-PGF2a antibody for other
prostaglandins was less than 1% (0.02% for PGE2). All measure-
ments were run at least in duplicate for each sample. No differences
in metabolite concentrations were observed between the side
ipsilateral and contralateral to electrode implantation. Results were
expressed as pg/mg of total proteins measured in the pellets
obtained after the extraction procedure.
BDNF measurement
The amounts of free mature BDNF in tissue extracts were
measured (duplicate or triplicate) by a high sensitivity colorimetric
enzyme immunoassay (EIA kit, detection limit: 7.8 pg/ml,
Promega Corporation, Madison, WI), following the manufacturer’s
instructions. According to the manufacturer, the cross-reactivity of
the anti-BDNF antibody with other related neurotrophic factors
(NGF, NT-3, and NT-4) was less than 3%. No differences in BDNF
concentrations were observed between the side ipsilateral and
controlateral to electrode implantation. Results were expressed as
pg/mg of total proteins.
Protein measurement
The BCA Protein Assay kit (Pierce, Rockford, IL), based on
bicinchoninic acid for the colorimetric detection and quantification
of total proteins, was used to determine the amount of proteins in
the pellets obtained after extraction of PGE2, 8-epi-PGF2a, and
BDNF. Protein concentrations were reported with reference to
standards curves of bovine serum albumin (working range: 5 Ag/ml–400 Ag/ml).
Bromodeoxyuridine (BrdU) and flurbiprofen administration
Seven days after SE, 24 rats which had exhibited partial SE
and 24 electrode-implanted, non-stimulated rats (sham) received 4
injections (every 2 h during a 6 h-period) of the thymidine
analogue BrdU (50 mg/kg, i.p.; Sigma) dissolved in KPBS (pH
7–7.4) for labeling of mitotic cells. Starting from day 8 post-SE,
animals were treated with the non-selective COX inhibitor
flurbiprofen (10 mg/kg, s.c.; Sigma) or vehicle (DMSO/EtOH/
Castor Oil in a ratio of 20:5:75). A first group of 12 rats (6 partial
SE rats, and 6 sham rats) were sacrificed 24 h after the first
injection of flurbiprofen or vehicle, and the hippocampi sub-
dissected and homogenized as described above. A second group
of 36 rats (18 partial SE rats, and 18 sham rats) received daily
injections of flurbiprofen or vehicle for 1 days, and were
sacrificed at 35 days post-SE. Brains were then processed for
immunocytochemistry.
Cell counting
All analyses were conducted by an observer blind to treatment
conditions. BrdU-stained sections were examined in an Olympus
AX-70 fluorescence microscope with 40� objective (Olympus,
Albertslund, Denmark). Three to five coronal sections per rat,
located between 3.3 and 4.3 mm posterior to bregma (encompass-
ing the dorsal hippocampal region), were counted for BrdU-
positive cells. Counts were conducted in the SGZ/GCL in both
hemispheres and no hemispheric differences were detected. Only
cells located in the GCL or within two cell diameters from this
region in the SGZ were counted. In order to reduce counting bias,
only central cell profiles (Coggeshall and Lekan, 1996) exceeding
4 Am were included. All counts are reported as mean number of
cells per section. Co-localization of BrdU with NeuN was assessed
using a confocal scanning light microscope (Leica) with the Kr/Ar
488 and 568 excitation filters. The percentage of BrdU/NeuN
double-labeled cells in each rat was obtained by analyzing 50
BrdU-positive cells with respect to NeuN double-labeling from the
same hippocampal region used for the BrdU counts.
Statistical analysis
Comparisons were performed using one way analysis of
variance (ANOVA) followed by post hoc Fisher’s test. Data are
presented as mean T SEM, and differences are considered
significant at P < 0.05.
Results
Status epilepticus severity differentially regulates prostaglandin E2
synthesis and 8-epi-PGF2a generation in rat hippocampus
Partial and generalized SE induce the same magnitude of neural
progenitor proliferation in SGZ at 7 days post-insult, but there is a
marked loss of the new neurons at 35 days only following the more
severe gSE (Mohapel et al., 2004). In order to explore which
factors could underlie this difference in survival of newborn
neurons, 7 and 35 days post-SE were chosen as critical time points.
We first assessed the levels of PGE2 in hippocampal subregions
(DG, CA3, and CA1) after electrically evoked pSE and gSE. The
PGE2 content in hippocampal formation of intact (control) and

M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–35 27
electrode-implanted, non-stimulated (sham) rats was similar at both
7 and 35 days (Figs. 1D, E). In pSE rats, PGE2 levels were
significantly increased at 7 days in all hippocampal subregions as
compared to both control and sham rats. The pSE-induced PGE2
amounted to about 510, 707, and 289% of the levels in DG, CA3,
and CA1 of sham rats, respectively. The highest PGE2 levels were
detected in DG (Fig. 1D). In contrast, in gSE rats, PGE2 levels at 7
days did not differ from those in control and sham rats (Fig. 1D).
At 35 days after SE, PGE2 levels were low and comparable in the
four animal groups (Fig. 1E).
The prostaglandin biosynthetic cascade is accompanied by
generation of free radicals, which arise from the peroxidase
activity of COX, and can contribute to neuronal damage
following excitotoxic insults (Pepicelli et al., 2002, 2005) and
seizures (Patel et al., 2001). In parallel to PGE2 measurement, we
therefore determined the levels of the F2-isoprostane 8-epi-PGF2a,
a well recognized marker of free radical-dependent lipid
peroxidation, in the same samples. pSE gave rise to a significant
increase of 8-epi-PGF2a levels in the DG and CA3 at 7 days
compared to sham (Fig. 1F), which returned to control levels at
35 days (Fig. 1G). In contrast, 8-epi-PGF2a levels were lower
after gSE as compared to sham animals at 7 days (Fig. 1F), but
moderately enhanced in all subregions at 35 days post-SE (Fig.
1G). The 8-epi-PGF2a levels were significantly elevated in sham
rats at 7 days as compared to intact controls, most likely due to
the electrode implantation per se (Fig. 1F). This effect was no
longer detectable at 35 days (Fig. 1G).
Status epilepticus severity differentially regulates COX-2
expression in rat hippocampus
To gain information about the cellular source of PGE2, in
particular in neurons vs. glia, and describe the regulation of its
biosynthetic enzyme after SE of different severities, we analyzed
the expression of COX-2 by immunocytochemistry at 1–2, 3,
7–8, and 35 days after pSE or gSE. We found that COX-2 was
markedly up-regulated at days 1–2 post-SE. Both pSE and gSE
profiles led to comparable and strong COX-2 expression in
dentate GCL (Figs. 2B, F) and hilus, in CA3 and CA1
pyramidal layers, and in thalamus, amygdala, piriform cortex,
and neocortex (data not shown). Interestingly, the expression of
COX-2 differed between the two SE profiles at the later time
points (Fig. 2 and Table 1). Following pSE, strong COX-2
immunoreactivity was detected at 3 and 7 days but declined
thereafter, reaching low levels after 35 days (Figs. 2C–E and
Table 1). This decrement in COX-2 immunoreactivity was
observed already at 7 days after gSE (Fig. 2H and Table 1),
in accordance with the low level of PGE2 at this time point in
the hippocampal homogenates.
Expression of COX-2 was restricted to mature neuronal
populations, e.g., dentate granule cells and hilar neurons, and
CA1 and CA3 pyramidal neurons, at all time points after both
pSE and gSE, as assessed by double staining with NeuN, a
marker for mature neurons (Figs. 3A–I). The newly formed
neuroblasts, identified by Dcx expression, showed no COX-2
immunoreactivity (Figs. 3L–N), in accordance with the devel-
opment-dependent expression of this enzyme (Kaufmann et al.,
1997). No co-localization of COX-2 was observed in cells
expressing astrocytic (GFAP) or macrophagic-microglial markers
(ED1) (Figs. 4A–C and D–F, respectively) at any time point
after SE.
PGE2 receptors are not expressed on newly formed hippocampal
neuroblasts
We then explored whether the Dcx-immunoreactive neuroblasts
generated after SE expressed PGE2 receptors, which could mediate
a direct action of the ligand. We focused on the two most
abundantly expressed PGE2 receptor subtypes in the hippocampus,
EP2 and EP3, whose activation has been shown to be neuro-
protective in paradigms of excitoxicity (McCullough et al., 2004;
Bilak et al., 2004). Strong EP2 immunoreactivity was observed in
the GCL (Fig. 5A), as previously reported by McCullough et al.
(2004), and in ED1+ and GFAP+ cells (Figs. 5E–L). In contrast,
we detected no EP2 immunoreactivity in Dcx+ neuroblasts (Figs.
5A–C). The new neurons developed EP2 immunoreactivity at later
stages of maturation, between the fourth and fifth week after their
birth, as indicated by double-label immunocytochemistry and
confocal microscopical analysis performed on SE-rats injected
with BrdU on day 7 post-SE and sacrificed on day 35. At this time
point, virtually all (95%) BrdU+ cells in the SGZ/GCL co-
expressed EP2 (Fig. 5D) and NeuN (Fig. 7C). Weak EP3
immunoreactivity was found in mature GCL neurons, as reported
in previous studies (Nakamura et al., 2000). Similarly to EP2, EP3
expression was absent in Dcx+ neuroblasts but was acquired at
later stages, being detected in some BrdU+ cells at 35 days from
SE (not shown).
Status epilepticus severity differentially regulates BDNF protein
levels in rat hippocampus
Given the proposed action of COX-2 on BDNF expression
(Shaw et al., 2003), we also explored the possibility that SE
severity differentially regulates, besides PGE2, BDNF synthesis in
rat hippocampal formation. Levels of BDNF were measured at 7
and 35 days after pSE or gSE. As shown in Fig. 6A, BDNF levels
were significantly elevated in DG and CA3 at 7 days following
pSE (263 and 246% of sham, respectively), but were unchanged in
gSE rats. After 35 days, all groups showed low levels of BDNF
protein (Fig. 6B).
Short-term treatment with COX inhibitor flurbiprofen attenuates
hippocampal PGE2 and BDNF levels after partial status
epilepticus but does not impair long-term survival of new neurons
The high levels of PGE2 and BDNF at 7 days after pSE raised
the possibility that the increased synthesis of these two factors
could be neuroprotective and underlie the much better survival of
newborn neurons following pSE as compared to gSE. To explore
this hypothesis, rats that had exhibited pSE were first given BrdU
on day 7 and then daily injections of the non-selective COX
inhibitor flurbiprofen or vehicle, from day 8 post-SE and for 7 days
thereafter. Although COX-2 is the major isoform in paradigms of
excitotoxicity, including epilepsy, a contribution of COX-1 activity
to PGE2 levels cannot be ruled out (Candelario-Jalil et al., 2003;
Pepicelli et al., 2005). Thus, we decided to use flurbiprofen rather
than a specific COX-2 inhibitor to achieve a complete inhibition of
PGE2 synthesis, regardless of enzyme isoform. Flurbiprofen was
administered starting from 8 days post-SE to evaluate the effect of
PGE2 suppression specifically on the survival of the newly formed,
BrdU-labeled neurons without interfering with the proliferative
phase of neurogenesis. Given the natural decline of COX-2
expression after the initial induction, the flurbiprofen treatment

Fig. 2. Photomicrographs showing COX-2 immunoreactivity in the dentate GCL in sham-operated rats (A) or in rats at days 1–2, 3, 7–8, and 35 following
pSE (B–E) or gSE (F– I). Scale bar in panel I = 50 Am.
M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–3528
was limited to 1 week to minimize gastrointestinal adverse effects
related to prolonged administration.
A first group of animals was sacrificed already on day 9 post-
SE, i.e., 24 h after the first flurbiprofen injection, with the dual aim
to verify the efficacy of COX inhibition and evaluate the
consequences of PGE2 suppression on BDNF levels. As shown
in Fig. 7A, PGE2 levels in the DG were completely abated to
baseline and the flurbiprofen treatment also markedly reduced
BDNF levels (Fig. 7B).
A second group of animals was sacrificed on day 35 post-SE,
and the number of BrdU+ and BrdU+/NeuN+ cells was counted in
the SGZ/GCL (Figs. 7C, D).
The total number of BrdU+ cells was unaffected by the
flurbiprofen treatment both in sham and pSE rats (Fig. 7D).
Confocal microscopy revealed that the percentage of BrdU cells
double-labeled with NeuN was virtually identical in vehicle- and
flurbiprofen-treated pSE rats (95% and 93% of total BrdU+
number, respectively) at 35 days post-SE. Thus, 1 week of
flurbiprofen treatment administered directly after the birth of the
neuroblasts did not alter the number of surviving mature BrdU/
NeuN immunoreactive neurons 4 weeks later. Given the crucial
role of inflammation in neurogenesis, we finally evaluated the
number of ED1+ macrophages-microglia in the DG of vehicle- and
flurbiprofen-injected pSE rats. No significant differences were
found between the groups (data not shown).
Discussion
The present data show that the microenvironment encountering
the new hippocampal neurons generated at 1 week after SE differs
markedly depending on the severity of the insult. Our previous study
demonstrated that neuronal damage and associated inflammation are
more pronounced after gSE than following pSE, which contributes
to the marked loss of the new neurons over the subsequent weeks in
animals with gSE (Mohapel et al., 2004). Here we show that the
milder pSE profile gives rise to higher and more sustained increase
of PGE2 and BDNF in the hippocampal formation as compared to
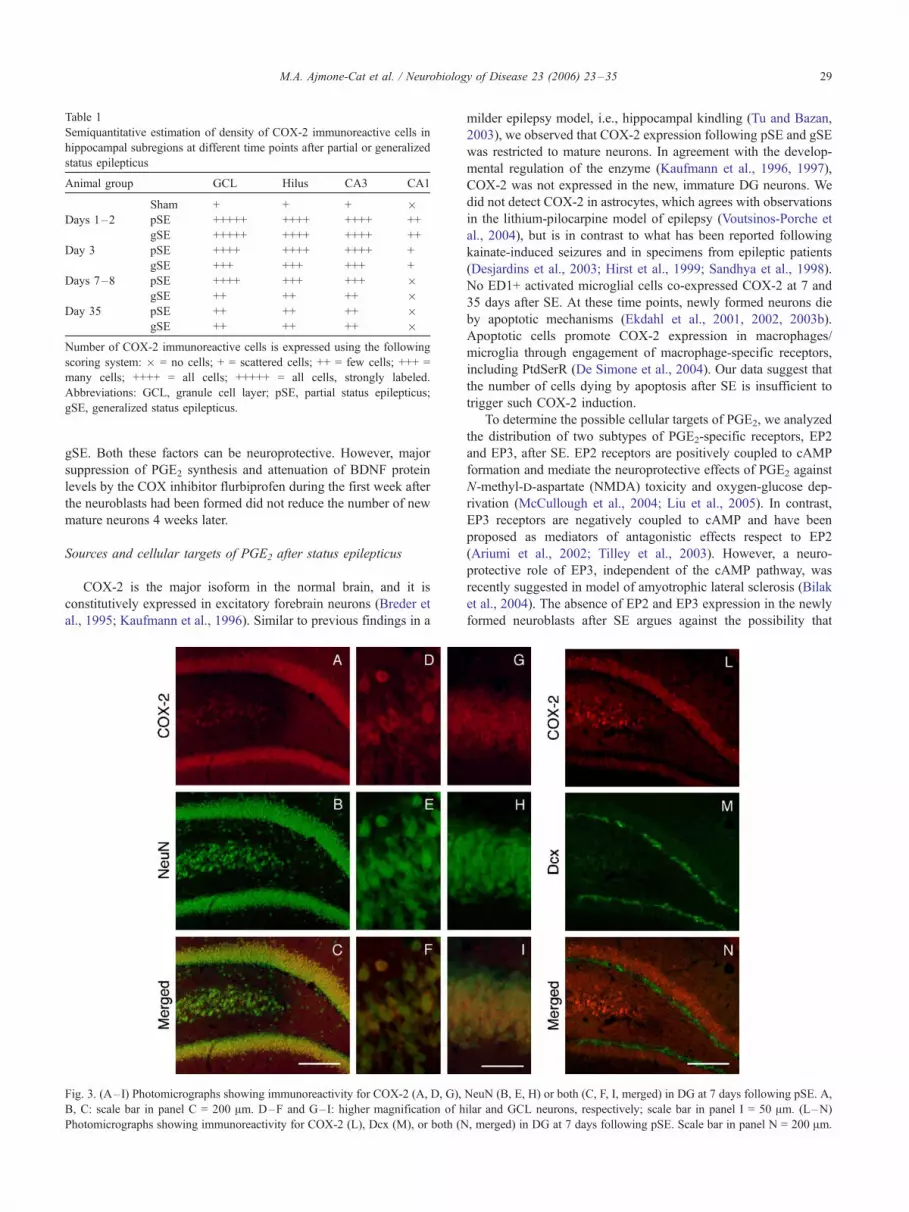
Table 1
Semiquantitative estimation of density of COX-2 immunoreactive cells in
hippocampal subregions at different time points after partial or generalized
status epilepticus
Animal group GCL Hilus CA3 CA1
Sham + + + �Days 1–2 pSE +++++ ++++ ++++ ++
gSE +++++ ++++ ++++ ++
Day 3 pSE ++++ ++++ ++++ +
gSE +++ +++ +++ +
Days 7–8 pSE ++++ +++ +++ �gSE ++ ++ ++ �
Day 35 pSE ++ ++ ++ �gSE ++ ++ ++ �
Number of COX-2 immunoreactive cells is expressed using the following
scoring system: � = no cells; + = scattered cells; ++ = few cells; +++ =
many cells; ++++ = all cells; +++++ = all cells, strongly labeled.
Abbreviations: GCL, granule cell layer; pSE, partial status epilepticus;
gSE, generalized status epilepticus.
M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–35 29
gSE. Both these factors can be neuroprotective. However, major
suppression of PGE2 synthesis and attenuation of BDNF protein
levels by the COX inhibitor flurbiprofen during the first week after
the neuroblasts had been formed did not reduce the number of new
mature neurons 4 weeks later.
Sources and cellular targets of PGE2 after status epilepticus
COX-2 is the major isoform in the normal brain, and it is
constitutively expressed in excitatory forebrain neurons (Breder et
al., 1995; Kaufmann et al., 1996). Similar to previous findings in a
Fig. 3. (A–I) Photomicrographs showing immunoreactivity for COX-2 (A, D, G),
B, C: scale bar in panel C = 200 Am. D–F and G–I: higher magnification of h
Photomicrographs showing immunoreactivity for COX-2 (L), Dcx (M), or both (N
milder epilepsy model, i.e., hippocampal kindling (Tu and Bazan,
2003), we observed that COX-2 expression following pSE and gSE
was restricted to mature neurons. In agreement with the develop-
mental regulation of the enzyme (Kaufmann et al., 1996, 1997),
COX-2 was not expressed in the new, immature DG neurons. We
did not detect COX-2 in astrocytes, which agrees with observations
in the lithium-pilocarpine model of epilepsy (Voutsinos-Porche et
al., 2004), but is in contrast to what has been reported following
kainate-induced seizures and in specimens from epileptic patients
(Desjardins et al., 2003; Hirst et al., 1999; Sandhya et al., 1998).
No ED1+ activated microglial cells co-expressed COX-2 at 7 and
35 days after SE. At these time points, newly formed neurons die
by apoptotic mechanisms (Ekdahl et al., 2001, 2002, 2003b).
Apoptotic cells promote COX-2 expression in macrophages/
microglia through engagement of macrophage-specific receptors,
including PtdSerR (De Simone et al., 2004). Our data suggest that
the number of cells dying by apoptosis after SE is insufficient to
trigger such COX-2 induction.
To determine the possible cellular targets of PGE2, we analyzed
the distribution of two subtypes of PGE2-specific receptors, EP2
and EP3, after SE. EP2 receptors are positively coupled to cAMP
formation and mediate the neuroprotective effects of PGE2 against
N-methyl-d-aspartate (NMDA) toxicity and oxygen-glucose dep-
rivation (McCullough et al., 2004; Liu et al., 2005). In contrast,
EP3 receptors are negatively coupled to cAMP and have been
proposed as mediators of antagonistic effects respect to EP2
(Ariumi et al., 2002; Tilley et al., 2003). However, a neuro-
protective role of EP3, independent of the cAMP pathway, was
recently suggested in model of amyotrophic lateral sclerosis (Bilak
et al., 2004). The absence of EP2 and EP3 expression in the newly
formed neuroblasts after SE argues against the possibility that
NeuN (B, E, H) or both (C, F, I, merged) in DG at 7 days following pSE. A,
ilar and GCL neurons, respectively; scale bar in panel I = 50 Am. (L–N)
, merged) in DG at 7 days following pSE. Scale bar in panel N = 200 Am.

Fig. 4. (A–F) Photomicrographs showing immunoreactivity for COX-2 (A), GFAP (B), or both (C, merged), and for COX-2 (D), ED1 (E), or both (F, merged),
in DG at 7 days following pSE. Scale bar in panel F = 200 Am.
M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–3530
PGE2 acts directly on these cells. It is also inconceivable that the
previously reported enhancement of cell proliferation in the SGZ
exerted by PGE2 under normal conditions and following cerebral
ischemia (Uchida et al., 2002; Sasaki et al., 2003) is exerted
through direct stimulation of the progenitor cells. The presence of
other EP receptor subtypes on the neuroblasts seems unlikely,
given the very low expression of their mRNA in DG (Batshake et
al., 1995; Zhang and Rivest, 1999; Zhu et al., 2005).
The expression of EP2 receptors in macrophages/microglial cells
and astrocytes after SE is of particular interest since PGE2 exerts
immunomodulatory functions on these cells. Through stimulation of
this receptor subtype, PGE2 reduces expression of tumor necrosis
factor-a, major histocompatibility complex II, and inducible nitric
oxide synthase, and induces synthesis of anti-inflammatory cyto-
kines, such as transforming growth factor-h (Levi et al., 1998;
Zhang and Rivest, 2001). We have previously shown that microglial
cells become activated following SE, their number being signifi-
cantly lower in pSE than in gSE, and correlating negatively with the
survival of newly formed neurons (Ekdahl et al., 2003a). It is
tempting to speculate that the reduced inflammatory reaction
observed in the partial profile is a consequence of a down-regulation
of microglia by the sustained levels of PGE2.
Elevated PGE2 and COX-2 expression following status epilepticus:
consequences for survival of mature and newly generated
hippocampal neurons
The consequences for neuronal viability of the enhancement of
COX-2 activity and PGE2 synthesis in response to pathological
conditions are controversial (Minghetti, 2004). It has been
proposed that COX-2 activity exerts a protective or toxic role
depending on the specific stage of the pathological process. As an
example, post-treatment, but not pre-treatment, with a selective
COX-2 inhibitor has been shown to enhance functional recovery
from kainic acid-induced neurodegeneration (Gobbo and O’Mara,
2004). It remains unclear whether the neuronal damage associated
with these conditions arises from COX-derived prostanoids per se
or from the bystander generation of free radicals during COX
peroxidase activity (Andreasson et al., 2001; Patel et al., 2001;
Pepicelli et al., 2002; Jiang et al., 2004; Manabe et al., 2004;
Turrin and Rivest, 2004). The sustained, markedly elevated levels
of COX-2, PGE2, and isoprostanes here demonstrated at 7 days
after pSE, characterized by the highest survival of newborn and
mature neurons as compared to the gSE profile, suggest that
activation of the prostanoid synthetic pathway is not necessarily
coupled with neuronal toxicity. Our findings favor a beneficial
role of a protracted PGE2 production for the final outcome of
tissue repair and neurogenesis after SE. On the other hand, the
shorter time course of COX-2 expression after gSE, demonstrated
by immunocytochemistry, may be associated with an initial, more
intense burst of PGE2 and free radical production that can
compromise tissue integrity and make the microenvironment
surrounding newly formed neurons non-permissive for their
survival and maturation. The increased levels of isoprostanes,
found in the gSE group at 35 days after the insult, indicate that, in
this profile, oxidative injury is still occurring in the hippocampus
at delayed time points after SE, independently from PGE2
synthesis, which is already at baseline level at 7 days. The fact

Fig. 5. (A–C) Photomicrographs showing expression of EP2 (A) and Dcx (B), but lack of co-expression (C), in the GCL/SGZ at 7 days following pSE. Scale
bar in C = 50 Am. (D) Confocal image with orthogonal projection of a BrdU (red)/EP2 (green) double-labeled cell in the SGZ/GCL. (E–L) Photomicrographs
showing immunoreactivity for EP2 (E), ED1 (F), or both (G, merged), and for EP2 (H), GFAP (I), or both (L, merged) in DG at 7 days after pSE. Scale bar in
panel L = 25 Am.
Fig. 6. Levels of BDNF protein in hippocampal subregions at 7 (A) and 35 days (B) following pSE and gSE, and in sham-operated and controls rats. n = 4–6
for 7 days, n = 3–5 for 35 days. Values are means T SEM. *P < 0.05.
M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–35 31

Fig. 7. Levels of PGE2 (A) and BDNF protein (B) in hippocampal subregions at 24 h following one injection of flurbiprofen (10 mg/kg, s.c.) or vehicle,
administered 8 days after pSE. n = 3. Values are means T SEM. (C) Confocal image with orthogonal projection of a BrdU (red)/NeuN (green) double-labeled
neuron in the SGZ/GCL. (D) Number of BrdU-labeled cells in SGZ/GCL at 35 days after pSE in rats that received BrdU injections at 7 days, and daily
flurbiprofen or vehicle injections from day 8, and for 1 week thereafter. n = 8–10.
M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–3532
that administration of the COX inhibitor flurbiprofen, beginning
just after the formation of the BrdU-labeled neuroblasts at 8 days
after pSE and continuing for 1 week thereafter, did not influence
the number of mature BrdU/NeuN-double-labeled neurons at 35
days may have several hypothetical explanations: first, the COX-2
activation and elevated levels of PGE2 in the hippocampal
subregions in the pSE animals do not contribute significantly to
the good survival of the new neurons after this epileptic insult.
Second, the treatment period was too short to reveal a presumed
neuroprotective effect. Finally, it may be possible that already
prior to flurbiprofen administration, the COX-2 activation and
high PGE2 levels had promoted the survival of newly formed
neurons by establishing a favorable niche through modulation of
important functions in mature neurons or glial cells.
COX-2 synthesized prostaglandins are involved in long-term
potentiation (LTP) (Chen et al., 2002), and PGE2 influences
membrane excitability, synaptic transmission, and plasticity in
hippocampal pyramidal neurons (Chen and Bazan, 2005). COX-2
expression and enzymatic activity are regulated by neuronal
activity, and increased during excitotoxic insults, such as global
ischemia and electrically and chemically induced seizures (Mar-
cheselli and Bazan, 1996; Nakayama et al., 1998; Sandhya et al.,
1998), with mechanisms involving the overactivation of glutama-
tergic NMDA receptors (Yamagata et al., 1993; Adams et al., 1996;
Pepicelli et al., 2002, 2005). As a reflection of the complex
interplay between neuronal activity and COX-2, both pro-
convulsant or anti-convulsant effects have been claimed for
PGE2, depending on seizure model and type and timing of COX
inhibitors used (Steinhauer and Hertting, 1981; Forstermann et al.,
1984; Seregi et al., 1984; Paoletti et al., 1998; Baran et al., 1994;
Baik et al., 1999; Kunz and Oliw, 2001; Tu and Bazan, 2003). The
effects of PGE2 inhibition by flurbiprofen on epileptogenesis were
not specifically addressed in our study. Although the occurrence of
spontaneous behavioral seizures, occasionally observed in both SE
profiles, was not macroscopically altered by flurbiprofen treatment,
modifications of the frequency of EEG discharges cannot be
excluded. Further investigations are needed to address this issue.
BDNF synthesis following status epilepticus: possible link with
PGE2 and consequences for neuronal survival
PGE2 has been shown to stimulate the production of neuro-
trophins, such as NGF and BDNF, in embryonic rat hippocampal
cultures containing both neurons and glial cells and in mouse
astrocyte cultures (Friedman et al., 1990; Toyomoto et al., 2004). A
further link between the expression of BDNF and COX-2 activity
has been proposed by recent studies, showing that specific and
non-specific COX inhibitors blocked the increases of both PGE2
and BDNF following spatial learning and LTP (Shaw et al., 2003,
2005). Finally, it has been reported that cAMP elevating agents
enhance TrkB signaling and trafficking in hippocampal neurons (Ji
et al., 2005). The concomitant, high levels of PGE2 and BDNF
following pSE may therefore result in potentiated BDNF signaling,
due to stimulation of EP2 and cAMP elevation by PGE2.

M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–35 33
The pSE profile was characterized by elevated levels of BDNF
in the DG and CA3, paralleling the levels of PGE2, at 7 days post-
pSE but not gSE, where a more transient induction probably
occurred. Moreover, COX-2 inhibition by flurbiprofen at 8 days
post-SE attenuated the levels of BDNF. These observations support
the possibility that COX activity promotes BDNF synthesis.
However, a COX-2 independent effect of flurbiprofen on BDNF
synthesis, or a modification of neuronal activity due to COX-2
inhibition, which may in turn affect BDNF expression (Castren et
al., 1998; Fukuchi et al., 2005), cannot be excluded.
Studies in epilepsy models and transgenic mice with altered
BDNF expression or signaling suggest both facilitatory and
hampering roles for BDNF in seizure generation, neuronal damage,
and repair (Binder et al., 2001; Binder, 2004; Barton and Shannon,
2005). BDNF plays a central role during nervous system
development and adult neurogenesis as a necessary factor for the
differentiation and/or survival of neuronal progenitors (Lee et al.,
2002; Barnabe-Heider and Miller, 2003; Sairanen et al., 2005).
Because the flurbiprofen treatment did not completely suppress
BDNF levels, it still remains a possibility that, in our system, the
prolonged synthesis of BDNF in the pSE but not gSE profile may
contribute to the long-term survival of newborn neurons observed
in the partial profile. Both direct and indirect effects of BDNF on
survival are possible, since hippocampal neuroblasts very likely
express the BDNF receptor TrkB (Linnarsson et al., 2000), as
demonstrated for cortical precursors in vivo and in vitro and for
subventricular zone-derived interneurons (Fukumitsu et al., 1998;
Barnabe-Heider and Miller, 2003; Gascon et al., 2005). It should
be pointed out, though, that the effects of BDNF on neurogenesis
are complex and dependent on, e.g., tissue level, mode of delivery,
and type of insult. Long-term viral vector-mediated delivery of
BDNF and infusion of TrkB-Fc fusion protein, which scavenges
endogenous BDNF, have been shown to counteract and promote,
respectively, hippocampal neurogenesis after global cerebral
ischemia by influencing neuronal differentiation (Larsson et al.,
2002; Gustafsson et al., 2003). As discussed for PGE2, such an
action of BDNF could result from the modulation of important
functions in mature neurons or glial cells in the early phases after
SE. The reduction of BDNF levels as a consequence of
flurbiprofen treatment starting from 8 days post-SE may then
occur too late to have any effect on neuronal survival.
Conclusions
The dramatic differences between pSE and gSE rats with
respect to hippocampal PGE2 and BDNF levels at 7 days after the
initial epileptic insult could have several functional implications.
The fact that cell proliferation in pSE and gSE animals is similar at
this time point, when PGE2 and BDNF levels are widely different,
argues against a major role of these factors for the proliferation step
in SE-induced hippocampal neurogenesis. Our data also provide no
evidence that the high PGE2 and BDNF levels during the first days
after the formation of the new neurons are of major importance for
their long-term survival. Based on previous findings, we propose
that the sustained, high PGE2 and BDNF levels in the pSE but not
in the gSE animals could modulate hippocampal cellular and
synaptic plasticity, be neuroprotective, and influence epilepto-
genesis. Further investigations will be necessary to address these
issues and deepen our knowledge of how PGE2 and BDNF may
affect the destiny of the newly generated neurons following SE.
Acknowledgments
This work was supported by the Swedish Research Council, EU
project LSHBCT-2003-503005 (EUROSTEMCELL), The Soder-
berg, Crafoord, and Kock Foundations, and by the Italian Ministry
of Education, University and Research (FIRB-MIUR, Grant no.
H91/1: ‘‘Synaptic plasticity and brain repair’’). The Lund Stem Cell
Center is supported by a Center of Excellence grant in Life
Sciences from the Swedish Foundation for Strategic Research.
References
Adams, J., Collaco-Moraes, Y., de Belleroche, J., 1996. Cyclooxygenase-2
induction in cerebral cortex: an intracellular response to synaptic
excitation. J. Neurochem. 66, 6–13.
Andreasson, K.I., Savonenko, A., Vidensky, S., Goellner, J.J., Zhang, Y.,
Shaffer, A., Kaufmann, W.E., Worley, P.F., Isakson, P., Markowska,
A.L., 2001. Age-dependent cognitive deficits and neuronal apoptosis in
cyclooxygenase-2 transgenic mice. J. Neurosci. 21, 8198–8209.
Ariumi, H., Takano, Y., Masumi, A., Takahashi, S., Hirabara, Y., Honda,
K., Saito, R., Kamiya, H.O., 2002. Roles of the central prostaglandin
EP3 receptors in cardiovascular regulation in rats. Neurosci. Lett. 324,
61–64.
Arvidsson, A., Kokaia, Z., Lindvall, O., 2001. N-methyl-d-aspartate
receptor-mediated increase of neurogenesis in adult rat dentate gyrus
following stroke. Eur. J. Neurosci. 14, 10–18.
Arvidsson, A., Collin, T., Kirik, D., Kokaia, Z., Lindvall, O., 2002.
Neuronal replacement from endogenous precursors in the adult brain
after stroke. Nat. Med. 8, 963–970 (Epub 2002 Aug 5).
Baik, E.J., Kim, E.J., Lee, S.H., Moon, C., 1999. Cyclooxygenase-2
selective inhibitors aggravate kainic acid induced seizure and neuronal
cell death in the hippocampus. Brain Res. 843, 118–129.
Baran, H., Vass, K., Lassmann, H., Hornykiewicz, O., 1994. The
cyclooxygenase and lipoxygenase inhibitor BW755C protects rats
against kainic acid-induced seizures and neurotoxicity. Brain Res.
646, 201–206.
Barnabe-Heider, F., Miller, F.D., 2003. Endogenously produced neuro-
trophins regulate survival and differentiation of cortical progenitors via
distinct signaling pathways. J. Neurosci. 23, 5149–5160.
Barton, M.E., Shannon, H.E., 2005. The seizure-related phenotype of
brain-derived neurotrophic factor knockdown mice. Neuroscience 136,
563–569 (Electronic publication 2005 Sep 28).
Batshake, B., Nilsson, C., Sundelin, J., 1995. Molecular characterization
of the mouse prostanoid EP1 receptor gene. Eur. J. Biochem. 231,
809–814.
Bengzon, J., Kokaia, Z., Elmer, E., Nanobashvili, A., Kokaia, M., Lindvall,
O., 1997. Apoptosis and proliferation of dentate gyrus neurons after
single and intermittent limbic seizures. Proc. Natl. Acad. Sci. U. S. A.
94, 10432–10437.
Bilak, M., Wu, L., Wang, Q., Haughey, N., Conant, K., St Hillaire, C.,
Andreasson, K., 2004. PGE2 receptors rescue motor neurons in a model
of amyotrophic lateral sclerosis. Ann. Neurol. 56, 240–248.
Binder, D.K., 2004. The role of BDNF in epilepsy and other diseases of the
mature nervous system. Adv. Exp. Med. Biol. 548, 34–56.
Binder, D.K., Croll, S.D., Gall, C.M., Scharfman, H.E., 2001. BDNF
and epilepsy: too much of a good thing? Trends Neurosci. 24,
47–53.
Breder, C.D., Dewitt, D., Kraig, R.P., 1995. Characterization of inducible
cyclooxygenase in rat brain. J. Comp. Neurol. 355, 296–315.
Candelario-Jalil, E., Gonzalez-Falcon, A., Garcia-Cabrera, M., Alvarez, D.,
Al-Dalain, S., Martinez, G., Leon, O.S., Springer, J.E., 2003.
Assessment of the relative contribution of COX-1 and COX-2 isoforms
to ischemia-induced oxidative damage and neurodegeneration following
transient global cerebral ischemia. J. Neurochem. 86, 545–555.
Castren, E., Berninger, B., Leingartner, A., Lindholm, D., 1998. Regulation

M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–3534
of brain-derived neurotrophic factor mRNA levels in hippocampus by
neuronal activity. Prog. Brain Res. 117, 57–64.
Chen, C., Bazan, N.G., 2005. Endogenous PGE2 regulates membrane
excitability and synaptic transmission in hippocampal CA1 pyramidal
neurons. J. Neurophysiol. 93, 929–941.
Chen, C., Magee, J.C., Bazan, N.G., 2002. Cyclooxygenase-2 regulates
prostaglandin E2 signaling in hippocampal long-term synaptic plastic-
ity. J. Neurophysiol. 87, 2851–2857.
Coggeshall, R.E., Lekan, H.A., 1996. Methods for determining numbers
of cells and synapses: a case for more uniform standards of review.
J. Comp. Neurol. 364, 6–15 (Erratum in: J. Comp. Neurol. 1996.
369, 162).
De Simone, R., Ajmone-Cat, M.A., Minghetti, L., 2004. Atypical
antiinflammatory activation of microglia induced by apoptotic neurons:
possible role of phosphatidylserine–phosphatidylserine receptor inter-
action. Mol. Neurobiol. 29, 197–212.
Desjardins, P., Sauvageau, A., Bouthillier, A., Navarro, D., Hazell, A.S.,
Rose, C., Butterworth, R.F., 2003. Induction of astrocytic cyclo-
oxygenase-2 in epileptic patients with hippocampal sclerosis. Neuro-
chem. Int. 42, 299–303.
Ekdahl, C.T., Mohapel, P., Elmer, E., Lindvall, O., 2001. Caspase
inhibitors increase short-term survival of progenitor-cell progeny in
the adult rat dentate gyrus following status epilepticus. Eur. J.
Neurosci. 14, 937–945.
Ekdahl, C.T., Mohapel, P., Weber, E., Bahr, B., Blomgren, K., Lindvall,
O., 2002. Caspase-mediated death of newly formed neurons in the
adult rat dentate gyrus following status epilepticus. Eur. J. Neurosci.
16, 1463–1471.
Ekdahl, C.T., Claasen, J.H., Bonde, S., Kokaia, Z., Lindvall, O., 2003a.
Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl.
Acad. Sci. U. S. A. 100, 13632–13637.
Ekdahl, C.T., Zhu, C., Bonde, S., Bahr, B.A., Blomgren, K., Lindvall, O.,
2003b. Death mechanisms in status epilepticus-generated neurons and
effects of additional seizures on their survival. Neurobiol. Dis. 14,
513–523.
Elmer, E., Kokaia, Z., Kokaia, M., Carnahan, J., Nawa, H., Lindvall, O.,
1998. Dynamic changes of brain-derived neurotrophic factor protein
levels in the rat forebrain after single and recurring kindling-induced
seizures. Neuroscience 83, 351–362.
Forstermann, U., Seregi, A., Hertting, G., 1984. Anticonvulsive effects of
endogenous prostaglandins formed in brain of spontaneously convuls-
ing gerbils. Prostaglandins 27, 913–923.
Friedman, W.J., Larkfors, L., Ayer-LeLievre, C., Ebendal, T., Olson, L.,
Persson, H., 1990. Regulation of beta-nerve growth factor expression by
inflammatory mediators in hippocampal cultures. J. Neurosci. Res. 27,
374–382.
Fukuchi, M., Tabuchi, A., Tsuda, M., 2005. Transcriptional regulation
of neuronal genes and its effect on neural functions: cumulative
mRNA expression of PACAP and BDNF genes controlled by
calcium and cAMP signals in neurons. J. Pharmacol. Sci. 98,
212–218.
Fukumitsu, H., Furukawa, Y., Tsusaka, M., Kinukawa, H., Nitta, A.,
Nomoto, H., Mima, T., Furukawa, S., 1998. Simultaneous expression of
brain-derived neurotrophic factor and neurotrophin-3 in Cajal-Retzius,
subplate and ventricular progenitor cells during early development
stages of the rat cerebral cortex. Neuroscience 84, 115–127.
Gage, F.H., 2002. Neurogenesis in the adult brain. J. Neurosci. 22,
612–613.
Gascon, E., Vutskits, L., Zhang, H., Barral-Moran, M.J., Kiss, P.J., Mas, C.,
Kiss, J.Z., 2005. Sequential activation of p75 and TrkB is involved in
dendritic development of subventricular zone-derived neuronal progen-
itors in vitro. Eur. J. Neurosci. 21, 69–80.
Gobbo, O.L., O’Mara, S.M., 2004. Post-treatment, but not pre-treatment,
with the selective cyclooxygenase-2 inhibitor celecoxib markedly
enhances functional recovery from kainic acid-induced neurodegenera-
tion. Neuroscience 125, 317–327.
Gustafsson, E., Lindvall, O., Kokaia, Z., 2003. Intraventricular infusion of
TrkB-Fc fusion protein promotes ischemia-induced neurogenesis in
adult rat dentate gyrus. Stroke 34, 2710–2715.
Hirst, W.D., Young, K.A., Newton, R., Allport, V.C., Marriott, D.R.,
Wilkin, G.P., 1999. Expression of COX-2 by normal and reactive
astrocytes in the adult rat central nervous system. Mol. Cell. Neurosci.
13, 57–68.
Ji, Y., Pang, P.T., Feng, L., Lu, B., 2005. Cyclic AMP controls BDNF-
induced TrkB phosphorylation and dendritic spine formation in mature
hippocampal neurons. Nat. Neurosci. 8, 164–172.
Jiang, J., Borisenko, G.G., Osipov, A., Martin, I., Chen, R., Shvedova,
A.A., Sorokin, A., Tyurina, Y.Y., Potapovich, A., Tyurin, V.A., Graham,
S.H., Kagan, V.E., 2004. Arachidonic acid-induced carbon-centered
radicals and phospholipid peroxidation in cyclo-oxygenase-2-trans-
fected PC12 cells. J. Neurochem. 90, 1036–1049.
Jin, K., Minami, M., Lan, J.Q., Mao, X.O., Batteur, S., Simon, R.P.,
Greenberg, D.A., 2001. Neurogenesis in dentate subgranular zone and
rostral subventricular zone after focal cerebral ischemia in the rat. Proc.
Natl. Acad. Sci. U. S. A. 98, 4710–4715.
Kaufmann, W.E., Worley, P.F., Pegg, J., Bremer, M., Isakson, P., 1996.
COX-2, a synaptically induced enzyme, is expressed by excitatory
neurons as postsynaptic sites in rat cerebral cortex. Proc. Natl. Acad.
Sci. U. S. A. 93, 2317–2321.
Kaufmann, W.E., Worley, P.F., Taylor, C.V., Bremer, M., Isakson, P.C.,
1997. Cyclooxygenase-2 expression during rat neocortical development
and in Rett syndrome. Brain Dev. 19, 25–34.
Kunz, T., Oliw, E.H., 2001. The selective cyclooxygenase-2 inhibitor
rofecoxib reduces kainate-induced cell death in the rat hippocampus.
Eur. J. Neurosci. 13, 569–575.
Larsson, E., Mandel, R.J., Klein, R.L., Muzyczka, N., Lindvall, O., Kokaia,
Z., 2002. Suppression of insult-induced neurogenesis in adult rat brain
by brain-derived neurotrophic factor. Exp. Neurol. 177, 1–8.
Lee, J., Duan, W., Mattson, M.P., 2002. Evidence that brain-derived
neurotrophic factor is required for basal neurogenesis and mediates, in
part, the enhancement of neurogenesis by dietary restriction in the
hippocampus of adult mice. J. Neurochem. 82, 1367–1375.
Levi, G., Minghetti, L., Aloisi, F., 1998. Regulation of prostanoid synthesis
in microglial cells and effects of prostaglandin E2 on microglial
functions. Biochimie 80, 899–904.
Lie, D.C., Song, H., Colamarino, S.A., Ming, G.L., Gage, F.H., 2004.
Neurogenesis in the adult brain: new strategies for central nervous
system diseases. Annu. Rev. Pharmacol. Toxicol. 44, 399–421.
Linnarsson, S., Willson, C.A., Ernfors, P., 2000. Cell death in regenerating
populations of neurons in BDNF mutant mice. Brain Res. Mol. Brain
Res. 75, 61–69.
Liu, J., Solway, K., Messing, R.O., Sharp, F.R., 1998. Increased neuro-
genesis in the dentate gyrus after transient global ischemia in gerbils.
J. Neurosci. 18, 7768–7778.
Liu, H., Kaur, J., Dashtipour, K., Kinyamu, R., Ribak, C.E., Friedman,
L.K., 2003. Suppression of hippocampal neurogenesis is associated
with developmental stage, number of perinatal seizure episodes, and
glucocorticosteroid level. Exp. Neurol. 184, 196–213.
Liu, D., Wu, L., Breyer, R., Mattson, M.P., Andreasson, K., 2005.
Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral
ischemia. Ann. Neurol. 57, 758–761.
Lothman, E.W., Bertram, E.H., Bekenstein, J.W., Perlin, J.B., 1989. Self-
sustaining limbic status epilepticus induced by Fcontinuous_ hippocam-
pal stimulation: electrographic and behavioral characteristics. Epilepsy
Res. 3, 107–119.
Manabe, Y., Anrather, J., Kawano, T., Niwa, K., Zhou, P., Ross, M.E.,
Iadecola, C., 2004. Prostanoids, not reactive oxygen species, mediate
COX-2-dependent neurotoxicity. Ann. Neurol. 55, 668–675.
Marcheselli, V.L., Bazan, N.G., 1996. Sustained induction of prostaglandin
endoperoxide synthase-2 by seizures in hippocampus. Inhibition by a
platelet-activating factor antagonist. J. Biol. Chem. 271, 24794–24799.
McCullough, L., Wu, L., Haughey, N., Liang, X., Hand, T., Wang, Q.,
Breyer, R.M., Andreasson, K., 2004. Neuroprotective function of the
PGE2 EP2 receptor in cerebral ischemia. J. Neurosci. 24, 257–268.

M.A. Ajmone-Cat et al. / Neurobiology of Disease 23 (2006) 23–35 35
Minghetti, L., 2004. Cyclooxygenase-2 (COX-2) in inflammatory and
degenerative brain diseases. J. Neuropathol. Exp. Neurol. 63, 901–910.
Minghetti, L., Greco, A., Cardone, F., Puopolo, M., Ladogana, A., Almonti,
S., Cunningham, C., Perry, V.H., Pocchiari, M., Levi, G., 2000.
Increased brain synthesis of prostaglandin E2 and F2-isoprostane in
human and experimental transmissible spongiform encephalopathies.
J. Neuropathol. Exp. Neurol. 59, 866–871.
Mohapel, P., Ekdahl, C.T., Lindvall, O., 2004. Status epilepticus severity
influences the long-term outcome of neurogenesis in the adult dentate
gyrus. Neurobiol. Dis. 15, 196–205.
Monje, M.L., Toda, H., Palmer, T.D., 2003. Inflammatory blockade restores
adult hippocampal neurogenesis. Science 302, 1760–1765.
Nakamura, K., Kaneko, T., Yamashita, Y., Hasegawa, H., Katoh, H.,
Negishi, M., 2000. Immunohistochemical localization of prostaglan-
din EP3 receptor in the rat nervous system. J. Comp. Neurol. 421,
543–569.
Nakayama, M., Uchimura, K., Zhu, R.L., Nagayama, T., Rose, M.E.,
Stetler, R.A., Isakson, P.C., Chen, J., Graham, S.H., 1998. Cyclo-
oxygenase-2 inhibition prevents delayed death of CA1 hippocampal
neurons following global ischemia. Proc. Natl. Acad. Sci. U. S. A. 95,
10954–10959.
Paoletti, A.M., Piccirilli, S., Costa, N., Rotiroti, D., Bagetta, G., Nistico, G.,
1998. Systemic administration of N omega-nitro-l-arginine methyl
ester and indomethacin reduces the elevation of brain PGE2 content and
prevents seizures and hippocampal damage evoked by LiCl and tacrine
in rat. Exp. Neurol. 149, 349–355.
Parent, J.M., Yu, T.W., Leibowitz, R.T., Geschwind, D.H., Sloviter, R.S.,
Lowenstein, D.H., 1997. Dentate granule cell neurogenesis is increased
by seizures and contributes to aberrant network reorganization in the
adult rat hippocampus. J. Neurosci. 17, 3727–3738.
Patel, M., Liang, L.P., Roberts II, L.J., 2001. Enhanced hippocampal F2-
isoprostane formation following kainate-induced seizures. J. Neuro-
chem. 79, 1065–1069.
Paxinos, G., Watson, C., 1997. The Rat Brain in Stereotaxic Coordinates.
Academic Press, Paper Back, New York.
Pepicelli, O., Fedele, E., Bonanno, G., Raiteri, M., Ajmone-Cat, M.A.,
Greco, A., Levi, G., Minghetti, L., 2002. In vivo activation of N-
methyl-d-aspartate receptors in the rat hippocampus increases prosta-
glandin E(2) extracellular levels and triggers lipid peroxidation through
cyclooxygenase-mediated mechanisms. J. Neurochem. 81, 1028–1034.
Pepicelli, O., Fedele, E., Berardi, M., Raiteri, M., Levi, G., Greco, A.,
Ajmone-Cat, M.A., Minghetti, L., 2005. Cyclo-oxygenase-1 and -2
differently contribute to prostaglandin E2 synthesis and lipid perox-
idation after in vivo activation of N-methyl-d-aspartate receptors in rat
hippocampus. J. Neurochem. 93, 1561–1567.
Racine, R.J., 1972. Modification of seizure activity by electrical stimula-
tion: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32,
281–294.
Sairanen, M., Lucas, G., Ernfors, P., Castren, M., Castren, E., 2005. Brain-
derived neurotrophic factor and antidepressant drugs have different but
coordinated effects on neuronal turnover, proliferation, and survival in
the adult dentate gyrus. J. Neurosci. 25, 1089–1094.
Sandhya, T.L., Ong, W.Y., Horrocks, L.A., Farooqui, A.A., 1998. A light
and electron microscopic study of cytoplasmic phospholipase A2 and
cyclooxygenase-2 in the hippocampus after kainate lesions. Brain Res.
788, 223–231.
Sasaki, T., Kitagawa, K., Sugiura, S., Omura-Matsuoka, E., Tanaka, S.,
Yagita, Y., Okano, H., Matsumoto, M., Hori, M., 2003. Implication of
cyclooxygenase-2 on enhanced proliferation of neural progenitor cells
in the adult mouse hippocampus after ischemia. J. Neurosci. Res. 72,
461–471.
Scharfman, H.E., 2004. Functional implications of seizure-induced neuro-
genesis. Adv. Exp. Med. Biol. 548, 192–212.
Seregi, A., Forstermann, U., Hertting, G., 1984. Decreased levels of
brain cyclo-oxygenase products as a possible cause of increased
seizure susceptibility in convulsion-prone gerbils. Brain Res. 305,
393–395.
Shaw, K.N., Commins, S., O’Mara, S.M., 2003. Deficits in spatial learning
and synaptic plasticity induced by the rapid and competitive broad-
spectrum cyclooxygenase inhibitor ibuprofen are reversed by increasing
endogenous brain-derived neurotrophic factor. Eur. J. Neurosci. 17,
2438–2446.
Shaw, K.N., Commins, S., O’Mara, S.M., 2005. Cyclooxygenase inhibition
attenuates endotoxin-induced spatial learning deficits, but not an
endotoxin-induced blockade of long-term potentiation. Brain Res.
1038, 231–237.
Steinhauer, H.B., Hertting, G., 1981. Lowering of the convulsive
threshold by non-steroidal anti-inflammatory drugs. Eur. J. Pharma-
col. 69, 199–203.
Tilley, S.L., Hartney, J.M., Erikson, C.J., Jania, C., Nguyen, M., Stock, J.,
McNeisch, J., Valancius, C., Panettieri Jr., R.A., Penn, R.B., Koller,
B.H., 2003. Receptors and pathways mediating the effects of prosta-
glandin E2 on airway tone. Am. J. Physiol.: Lung Cell. Mol. Physiol.
284, 599–606.
Toyomoto, M., Ohta, M., Okumura, K., Yano, H., Matsumoto, K., Inoue,
S., Hayashi, K., Ikeda, K., 2004. Prostaglandins are powerful inducers
of NGF and BDNF production in mouse astrocyte cultures. FEBS Lett.
562, 211–215.
Tu, B., Bazan, N.G., 2003. Hippocampal kindling epileptogenesis upregu-
lates neuronal cyclooxygenase-2 expression in neocortex. Exp. Neurol.
179, 167–175.
Turrin, N.P., Rivest, S., 2004. Innate immune reaction in response to
seizures: implications for the neuropathology associated with epilepsy.
Neurobiol. Dis. 16, 321–334.
Uchida, K., Kumihashi, K., Kurosawa, S., Kobayashi, T., Itoi, K., Machida,
T., 2002. Stimulatory effects of prostaglandin E2 on neurogenesis in the
dentate gyrus of the adult rat. Zool. Sci. 19, 1211–1216.
van Praag, H., Schinder, A.F., Christie, B.R., Toni, N., Palmer, T.D., Gage,
F.H., 2002. Functional neurogenesis in the adult hippocampus. Nature
415, 1030–1034.
Voutsinos-Porche, B., Koning, E., Kaplan, H., Ferrandon, A., Guenounou,
M., Nehlig, A., Motte, J., 2004. Temporal patterns of the cerebral
inflammatory response in the rat lithium-pilocarpine model of temporal
lobe epilepsy. Neurobiol. Dis. 17, 385–402.
Yamagata, K.I., Andreasson, W.E., Kaufmann, C.A., Worley, P.F., 1993.
Expression of a mitogen-inducible cyclooxygenase in brain neurons:
regulation by synaptic activity and glucocorticoids. Neuron 11,
371–386.
Zhang, J., Rivest, S., 1999. Distribution, regulation and colocalization of
the genes encoding the EP2- and EP4-PGE2 receptors in the rat brain
and neuronal responses to systemic inflammation. Eur. J. Neurosci. 11,
2651–2668.
Zhang, J., Rivest, S., 2001. Anti-inflammatory effects of prostaglandin E2
in the central nervous system in response to brain injury and circulating
lipopolysaccharide. J. Neurochem. 76, 855–864.
Zhu, P., Genc, A., Zhang, X., Zhang, J., Bazan, N.G., Chen, C., 2005.
Heterogeneous expression and regulation of hippocampal prostaglandin
E2 receptors. J. Neurosci. Res. 81, 817–826.
Recommended

















