
UNIVERSIDAD AUTÓNOMA DE PUEBLA FACULTAD DE CIENCIAS QUÍMICAS
CURSO DE FISICOQUÍMICA II
(TERMODINÁMICA DE SOLUCIONES, EQUILIBRIO HETEROGÉNEO Y CINÉTICA QUÍMICA)
PERIODO OTOÑO 2015
DR. ROBERTO PORTILLO Y REYES

PA
RTE I
TERM
OD
INÁ
MIC
A DE
SOLU
CIO
NES
2

Soluciones
Ideales
SOLU
CIO
NES
Una solución es un sistem
a homogéneo constituido por m
ás de un componente,
usualmente líquido.
El componente que se encuentra en m
ayor proporción se denomina solvente y
los que están en menor proporción se llam
an solutos.
Infinitamente diluidas: 1-10 m
M
(se aproximan a la idealidad)
Reales
0.1-10 M
Las interacciones moleculares soluto-solvente son de igual
magnitud a las solvente-solvente y las soluto-soluto
3

Solución (SC)
Soluto (ST) Solvente (SV
)
electrolito no electrolito
En
solución
se disocia
total o parcialm
ente en
ion
es
En
solución
especies
molecu
lares
Solución (SC)
Soluto (ST) Solvente (SV
)
GA
SE
S , L
IQU
IDO
S O
SÓ
LID
OS
LIQ
UID
OS
O S
ÓL
IDO
S
LIQ
UID
OS
M
ÁS
US
UA
LE
S
4

¾L
a disolu
ción se d
efine com
o el proceso
en el q
ue u
na su
stancia
q
uím
ica (S
Q) se d
isuelve en
un
disolven
te (SV
), generalm
ente agu
a.
¾ L
as formu
laciones acu
osas pu
eden
conten
er otros líqu
idos, com
o glicerin
a, etan
ol y
prop
ilenglicol,
qu
e se
den
omin
an
codisolven
tes p
orqu
e increm
entan
la solub
ilidad
del p
rincip
io activo.
¾E
n
med
ios b
iológicos la
disolu
ción
se realiza
siemp
re en
m
edio
acuoso y su
ele ser un
paso p
revio a la absorción
sistémica
¾L
a d
isolución
está
cond
icionad
a
por
distin
tos factores
fisicoqu
ímicos y d
e formu
lación q
ue p
ued
en m
odificar la can
tidad
d
isuelta, i. e., la solu
bilid
ad.
INT
RO
DU
CC
IÓN
:
5

¾L
a solu
bilid
ad
es la
concen
tración
de
soluto
en
un
a d
isolución
saturad
a.
¾L
a Tem
peratu
ra, natu
raleza , polarid
ad y p
H d
el med
io, son
algun
os de los factores q
ue m
odifican
la solub
ilidad
.
¾M
olaridad
(M), m
olalidad
(m) y fracción
molar (X
) son las
un
idad
es mas frecu
entes p
ara expresar solu
bilid
ad d
e un
solu
to o la concen
tración d
e un
a solución
.
¾E
s fun
dam
ental en
la solub
ilidad
el rol de las fu
erzas in
termolecu
lares entre solu
to y solvente: cu
and
o son d
el m
ismo tip
o y magn
itud
, mayor será la solu
bilid
ad, i. e., lo
mism
o disu
elve a lo mism
o.
6

PR
OC
ES
O D
E D
ISO
LU
CIÓ
N
SE
RIG
E P
OR
LO
S P
RIN
CIP
IOS
DE
LA
TE
RM
OD
INÁ
MIC
A
EN
ER
GÉ
TIC
O
EN
TR
ÓP
ICO
EX
OT
ÉR
MIC
O E
ND
OT
ÉR
MIC
O
DIS
OL
UC
IÓN
AU
ME
NT
A E
L D
ES
OR
DE
N
FA
VO
RE
CE
L
A
SO
LU
BIL
IDA
D
DE
U
NA
S
US
TA
NC
IA
INC
LU
SO
S
I E
L
PR
OC
ES
O E
S E
ND
OT
ÉR
MIC
O
FA
CT
OR
ES
7

PR
OC
ES
O D
E D
ISO
LU
CIO
N
8

PR
OC
ES
O D
E D
ISO
LU
CIO
N
9

SIS
TE
MA
M
UL
TIC
OM
PO
NE
NT
E
ME
ZC
LA
S S
IMP
LE
S EN
ER
GÍA
LIB
RE
MO
LA
R P
AR
CIA
L
Ó P
OT
EN
CIA
L Q
UÍM
ICO
i
MA
GN
ITU
DE
S
MO
LA
RE
S P
AR
CIA
LE
S
VO
LU
ME
N M
OL
AR
P
AR
CIA
L
Gi =
(dG
/dn
i )T,P
,nj =
Vi =
(dV
/dn
i )T,P
,nj
V = n
i Vi + n
j Vj
G = n
i i + nj j
10
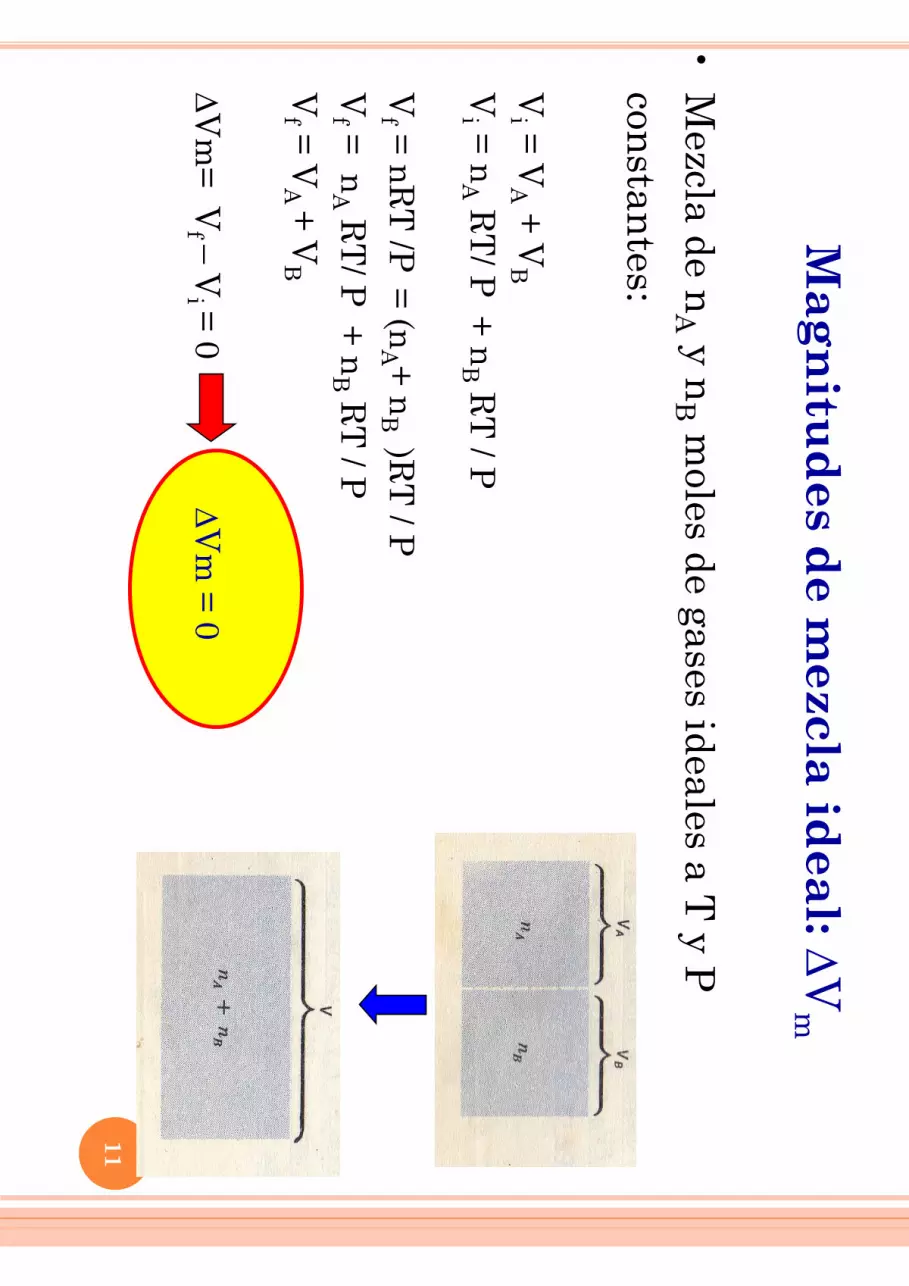
Mag
nitu
des d
e mezcla id
eal: DV
m
•M
ezcla de nA y n
B moles de gases ideales a T
y P
constan
tes:
Vi =
VA +
VB
V
i = n
A RT
/ P +
nB R
T / P
Vf =
nR
T /P
= (n
A + n
B )R
T / P
Vf =
nA R
T/ P
+ n
B RT
/ P
V
f = V
A +
VB
D
Vm
= V
f – Vi =
0 DV
m =
0
11

Mag
nitu
des d
e mezcla id
eal: DG
m
•M
ezcla de
nA
y n
B m
oles de
gases ideales
a T
y
P
constan
tes.
�
Estad
o inicial (i): A
,B pu
ros = º + RT
lnP
�E
stado fin
al (f): A,B
mezcla = º + R
Tln
Pp
Gi = n
A A
+ nB
B = n
A (ºA + RT
lnP
)+nB (ºB + R
Tln
P)
Gf = n
A A
+ nB
B = n
A (ºA + RT
lnP
A )+nB (ºB + R
Tln
PB )
+
=
12

Magn
itud
es de m
ezcla ideal: D
Gm
�D
Gm
= Gf - G
i D
Gm
= n
A RT
ln P
A /P +
nB R
T ln
PB /P
sien
do n =
nA +
nB , X
A = n
A /n, n
A = X
A n y P
A = X
A P
DG
m = n
XA
RT
ln x
A + n X
B RT
ln x
B
DG
m = n
RT
(xA ln
xA + x
B ln x
B )
D
ado que x<
1 D
Gm
< 0 M
ezclado:
Proceso esp
ontán
eo
13
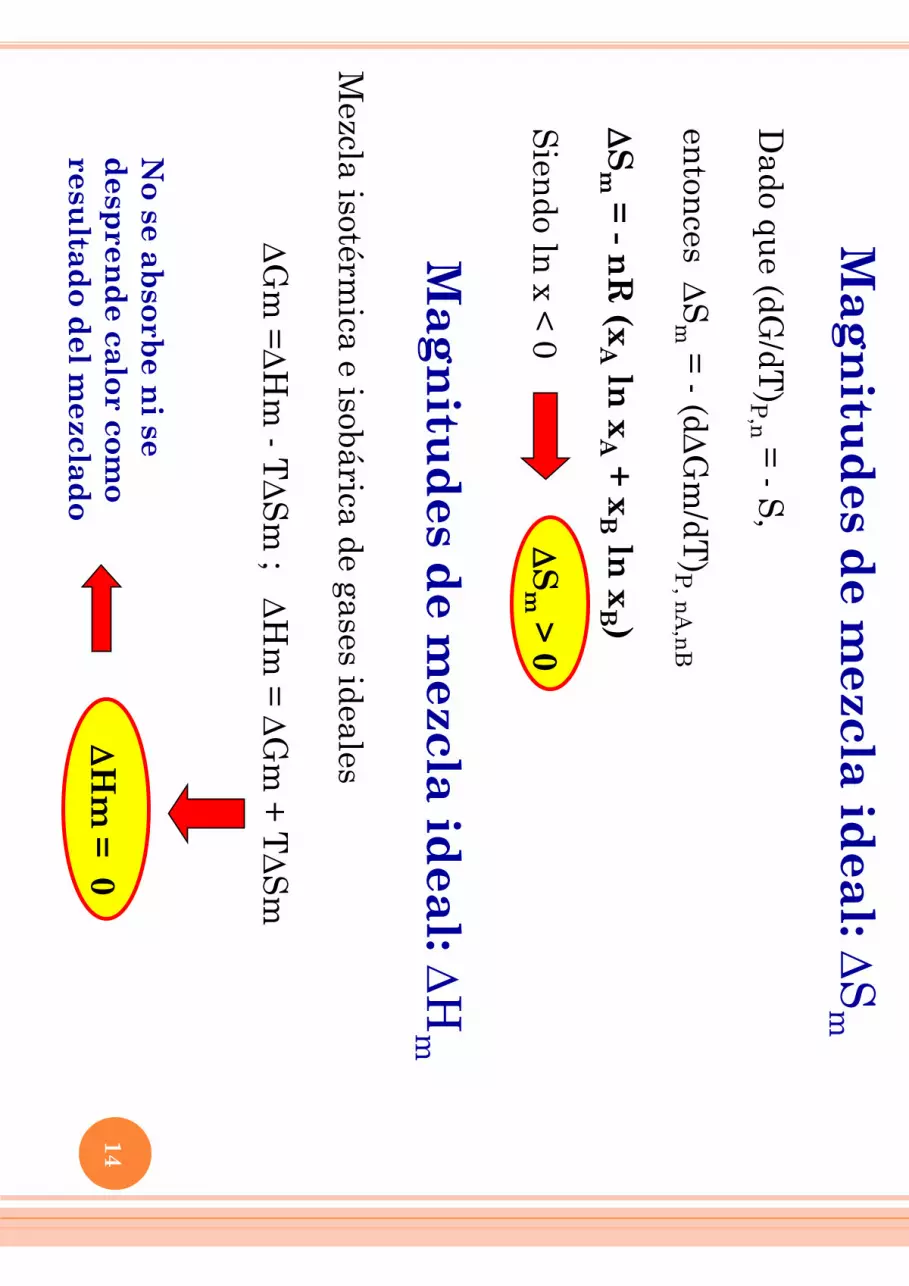
Mag
nitu
des d
e mezcla id
eal: DS
m
Dado qu
e (dG/dT
)P,n =
- S,
enton
ces DS
m =
- (dDG
m/dT
)P, n
A,n
B D
Sm
= - nR
(xA ln
xA + x
B ln x
B ) S
iendo ln
x < 0
DS
m > 0
Mag
nitu
des d
e mezcla id
eal: DH
m
Mezcla isotérm
ica e isobárica de gases ideales
D
Gm
=DH
m - T
DS
m ; D
Hm
= D
Gm
+ T
DS
m
D
Hm
= 0 N
o se absorb
e ni se
desp
rend
e calor como
resultad
o del m
ezclado
14

Variación
de las magn
itudes term
odinám
icas de mezclado
para un
a solución
ideal:
15

Soluciones ideales: m
ezcla de dos líquidos volátiles
CO
MP
OS
ICIO
N D
EL
LIQ
UID
O
XA
,L XB
,L
LE
Y D
E R
AO
UL
T
CO
MP
OS
ICIO
N D
EL
GA
S
YA
,G YB
,G
LE
Y D
E D
AL
TO
N
16

Soluciones ideales: m
ezcla de dos líquidos volátiles
A, l =
A ,g
A
pu
ro A
,l * = A ,g =
A º + RT
ln P
A * A
mezcla
A,l =
A ,g =
A º + RT
ln P
A
A
,l = A
,l * + RT
ln P
A
PA *
P
A = XA
,L
PA *
A =
A * + RT
ln X
A
B =
B * + RT
ln X
B
LE
Y D
E R
AO
UL
T 17

Mezcla de dos líquidos volátiles: Ley de R
aoult diagram
a presión-composición
•Cad
a comp
onen
te de la solu
ción
ejerce un
a presión
de vap
or qu
e es p
roporcion
al a su fracción
molar
en el líq
uid
o y a la presión
de
vapor d
el comp
onen
te pu
ro (a T
dad
a).
i
ii
PX
P
Ley de Raoult
18

En una solución ideal ambos com
ponentes siguen la ley de Raoult:
Mezcla de dos líquidos volátiles
**
**
*1
21
12
12
12
11
PP
PP
XP
XP
PP
X
Suponiendo comportam
iento ideal de la fase gas sobre la solución:
11
P
yP
*1
1
XP
*1
11
1*
**
21
21
PP
Xy
PP
PP
X
Com
o 1
1*1
y
PX
Pentonces
**
*1
22
12
1
PP
PP
PP
X19

La relación entre la P total y la fracción m
olar en la fase gas (y1 )
vendrá dado por:
*
*1
2*
**
11
21
PP
PP
PP
y
Mientras que la relación entre la P total y
la fracción molar en la fase líquida (X
1 ) vendrá dada por:
**
*2
12
1P
PP
PX
El conocimiento de la P del sistem
a en función de las fracciones m
olares en el líquido y en el gas permite dibujar el diagram
a de Presión de vapor frente a la C
omposición de am
bas fases a T constante.
20
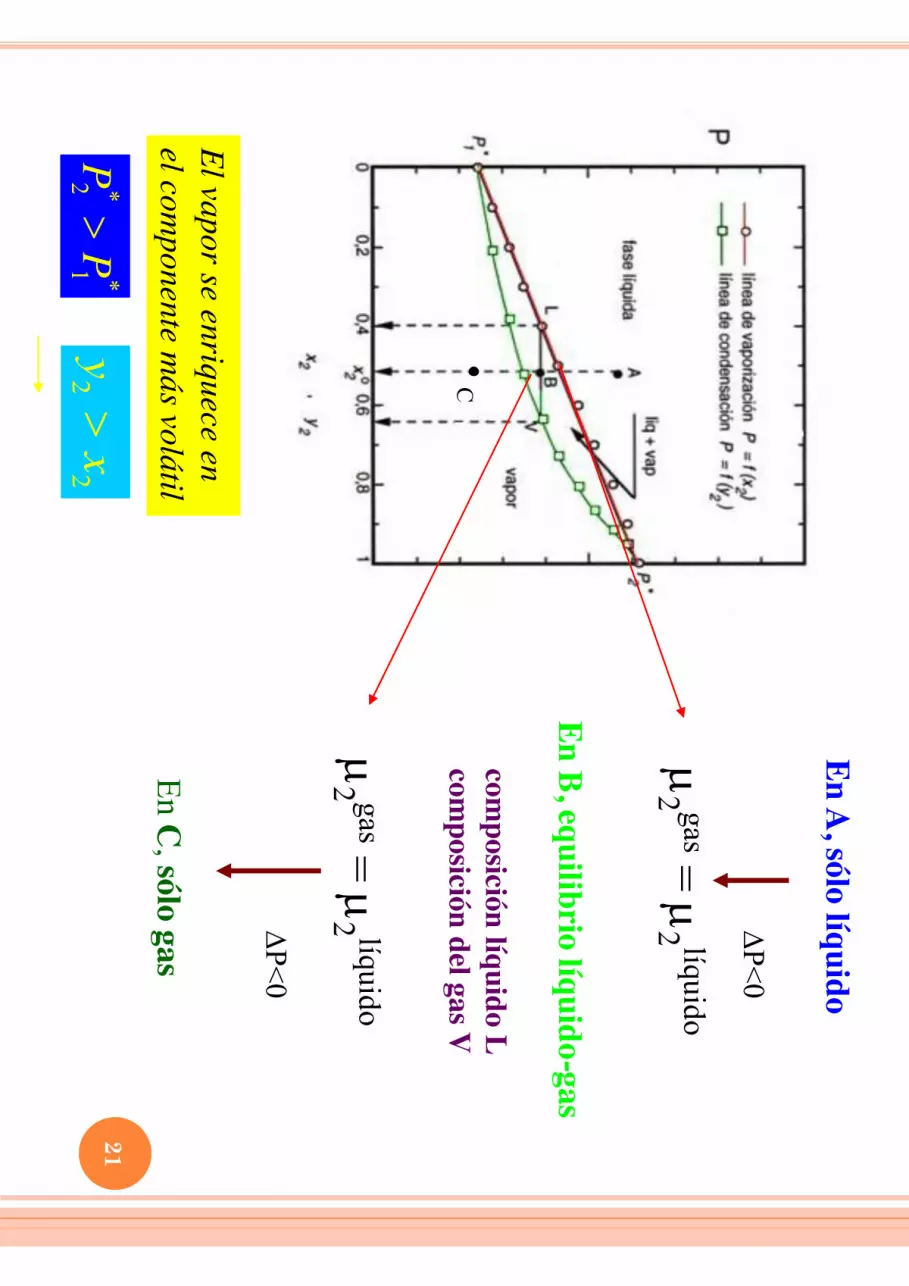
En A, sólo líquido
DP<0
2 gas =
2 líquido
En B, equilibrio líquido-gas
composición líquido L
composición del gas V
2 gas =
2 líquido
C
DP<0
En C, sólo gas
El vapor se enriquece en el com
ponente más volátil
**
21
P
P2
2
yx
21

Soluciones Reales: D
esviaciones de la ley de Raoult
Desviaciones positivas
Desviaciones negativas
Interacciones intermoleculares < que
interacciones en el líquido puro
ideali
iP
P
Interacciones intermoleculares > que
Interacciones en líquido puro
ideali
iP
P
Proceso en
dotérm
ico, DH
> 0 P
roceso exotérmico, D
H < 0 22

23
Pvap soluciones reales P
vap B Ley de Henry B
Ley de Henry A Raoult B Raoult A
(100%A, 0%
B) A
XB B
(100%B, 0%
A)
XA
Considerando el estado de concentración del solvente y los solutos, una solución
altamente diluida puede presentar dos com
portamientos extrem
os:
C
omportam
iento de Raoult (solvente)
Existen 2 tipos de soluciones ideales
muy diluidas (no consideran
interacción molecular)
C
omportam
iento de Henry (soluto)
Solu
ciones R
eales: Infin
itamen
te dilu
idas

24
(A)Com
portamiento de Raoult
En soluciones ideales m
uy diluidas (Xsoluto
0), el solvente cumple con la Ley de
Raoult en ese rango de composición:
P
solv = Xsolv P
solv o donde
Psolv = presión de vapor del solvente en la solución
X
solv = fracción molar del solvente en la solución
P
solv o = presión de vapor del solvente puro en las condiciones dadas
(B) Comportam
iento de Henry E
n soluciones ideales muy diluidas (X
soluto 0), el soluto cum
ple con la Ley de Henry:
Psolut = X
solut KH
donde
Psolut = presión de vapor del soluto en la solución
X
solut = fracción molar del soluto en la solución
K
H = constante de Henry para el soluto
En una solución ideal diluida:
• el solvente se aproxim
a a un comportam
iento descrito por la Ley de Raoult
• el soluto se aproxima a un com
portamiento descrito por la Ley de Henry

25
Podemos visualizar la diferencia entre am
bos comportam
ientos si hacemos la
siguiente abstracción. Supongam
os una
solución com
puesta por
agua (solvente),
y acetona
infinitamente diluida (soluto). Si vam
os a escala microscópica, verem
os lo siguiente: • Si som
os una molécula de agua (solvente), verem
os a nuestro alrededor casi solam
ente moléculas de agua, y harem
os caso omiso de las poquísim
as m
oléculas de acetona presentes. C
omo solvente, nuestra presión de vapor va a depender solam
ente de nuestra fracción m
olar en todo el rango composicional.
• Si
somos
una m
olécula de
acetona (soluto),
veremos
solamente
las poquísim
as moléculas de acetona a nuestro alrededor, sin interesar com
o interactúan con las de agua. C
omo soluto, a dilución infinita, nuestra presión de vapor dependerá sólo de
nuestra fracción molar.
26
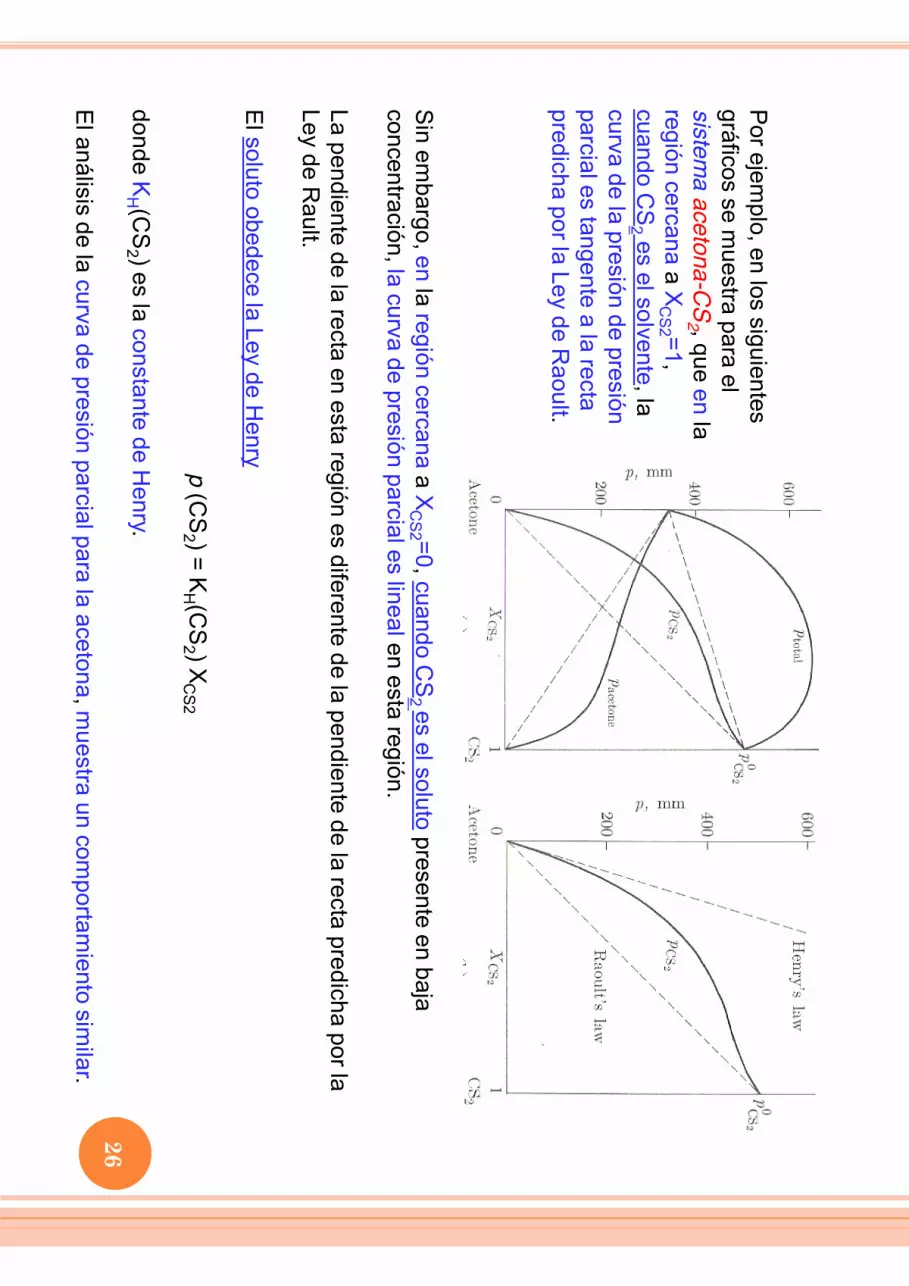
Por ejem
plo, en los siguientes gráficos se m
uestra para el sistem
a acetona-CS
2 , que en la región cercana a X
CS2 =1,
cuando CS
2 es el solvente, la curva de la presión de presión parcial es tangente a la recta predicha por la Ley de R
aoult.
Sin em
bargo, en la región cercana a XC
S2 =0, cuando CS
2 es el soluto presente en baja concentración, la curva de presión parcial es lineal en esta región. La pendiente de la recta en esta región es diferente de la pendiente de la recta predicha por la Ley de R
ault. E
l soluto obedece la Ley de Henry
p (C
S2 ) = K
H (CS
2 ) XC
S2 donde K
H (CS
2 ) es la constante de Henry.
El análisis de la curva de presión parcial para la acetona, m
uestra un comportam
iento similar.

Soluciones Reales: Infinitam
ente diluidas
XB = 0
XB = 1 P
B
PA = P
A . X
A
Solvente Ley de R
aoult
PB = k
H . XB
PB = k’H . m
Soluto Ley de H
enry
Soluciones infinitamente diluidas
kH
PA
XA = 1
XA = 0
Solu
ción d
iluid
a id
eal:
Ley d
e Hen
ry
Solu
ción d
iluid
a id
eal:
Ley d
e Rao
ult
27

28
El
modelo
de Solución Ideal no
considera las
interacciones entre
partículas, pero el caso más general es aquél en el que existe algún tipo
de interacción entre los componentes, llam
ado solución real, en la que cabe
esperar una
desviación del
comportam
iento ideal
como
consecuencia de la interacción de las moléculas.
Para expresar la desviación, introducim
os un “factor de corrección” para la fracción m
olar que da una idea del “grado de idealidad” de la solución, denom
inado coeficiente de actividad (), aplicable a soluciones líquidas y sólidas:
μ i, real = μºi + RT ln[i X
i ] por lo que la expresión de concentración “real” de un com
ponente “i” en una m
ezcla real ahora se denomina actividad (a
i ):
ai = i X
i
Soluciones Reales
Soluciones Líquidas y Sólidas: Actividad

29
La actividad corresponde a la “concentración” para soluciones reales, pero ni las actividades ni los coeficientes de actividad tienen unidades. P
ara el caso ideal, tenem
os que los coeficientes de actividad i son iguales a la unidad y:
a
i = Xi
El coeficiente de actividad puede m
edirse o calcularse teóricamente, o
puede ignorarse, haciendo que i = 1.0. Podem
os expresar ahora en forma
general el potencial químico de una especie “i” en una m
ezcla real como:
μ i, real = μºi + R
Tln ai
Para un com
puesto puro “i”, ai = 1.0, puesto que X
i = 1.0 y i = 1.0, y de la expresión
μi – G
ºi = RTlnX
i i , se tiene que μ
i = Gºi para una sustancia pura.
_
_

30
La mezcla de gases tam
bién es una solución y de la mism
a forma com
o las soluciones líquidas y sólidas se desvían del com
portamiento ideal,
las soluciones gaseosas también se desvían de la idealidad.
Para describir la com
posición de los componentes de una solución
gaseosa se utiliza la Presión Parcial (término análogo a la fracción
molar). S
i una solución gaseosa tiene una presión total (Ptotal ) y los
componentes gaseosos tienen fracciones m
olares X1 , X
2 , X3 , ..., las
presiones parciales de los componentes se definen com
o
P1 = X
1 ・P
total
P2 = X
2 ・P
total
Pn = X
n ・P
total R
ecordando el potencial químico para los gases:
μ p = μ p º + R
Tln(P/P
o) donde “P” es la presión y “Pº ” es una presión de referencia.
Soluciones Gaseosas: Fugacidad

31
Se define la fugacidad (f) de un com
ponente gaseoso en una mezcla com
o una m
edida de la “presión parcial real” de ese componente, y se define
como:
fi = λi P
i es decir, com
o una desviación de la idealidad (representada por la presión parcial), relacionada a través de un coeficiente de fugacidad (λ
i ). La “actividad” de una m
ezcla de gases es igual a la fugacidad. La fugacidad representa una “tendencia de escape” del com
ponente en la solución (tiene unidades de presión), cuyo potencial quím
ico será ahora:
μ i, real = μºi + RTln fi
En
condiciones ideales,
el coeficiente
de fugacidad
es unitario,
y la
fugacidad es igual a la presión parcial:
fi = P
i

32
Soluciones acuosas La unidad
de concentración
más
comúnm
ente utilizada para
las soluciones acuosas es la m
olalidad, por lo que la actividad para las soluciones acuosas es
ai = m
i Hi donde m
i es la molalidad de i y Hi otro tipo de coeficiente de actividad,
en esta
ocasión, basado
en un
tipo de
solución ideal
de tipo
Henryano.
De este m
odo, vemos cóm
o la actividad es un término que nos
permite
relacionar la
concentración de
cualquier com
puesto en
solución con su energía libre. «La actividad tiene diferentes form
as, en función del tipo de solución. Podem
os pensar en la actividad como en un tipo de concentración, ya
que, de hecho, en todos los casos es una concentración multiplicada
por un factor que nos indica las diferencias existentes entre una solución ideal y una solución real».

Soluciones Reales (no ideales)
En general, las soluciones no se comportan com
o soluciones ideales. Sólo en el caso en que la fracción m
olar del disolvente tienda a uno, solución diluida ideal, su com
portamiento se puede asem
ejar al de una solución ideal.
En el caso de soluciones no ideales el potencial químico es dado por:
0ln
ii
iRT
a
Actividad = concentración efectiva o real
ai = P
i real/Pi o
el coeficiente de actividad, γi , es una m
edida de la discrepancia del com
portamiento de la sustancia i respecto a la idealidad.
33
a = γi χ
i

34
Determinación de la actividad y del coeficiente de actividad
La actividad de constituyentes volátiles en una mezcla líquida se
determina a partir de la m
edición de la presión parcial del constituyente en la fase de vapor en equilibrio con el líquido. Ya que en el equilibrio para cada constituyente
i (liq) =
i (gas) entonces, suponiendo gas ideal,
μ oi (liq) + R
T ln ai = μ
oi (gas)+ + RT ln p
i . P
ara el líquido puro:
μ oi (liq) = μ
oi (gas)+ + RT ln p
oi .

35
Substrayendo las dos últim
as ecuaciones y dividiendo por RT, se obtiene
ln a
i = ln (pi / p
oi ) de donde
ai = p
i / poi
que es análoga a la Ley de Raoult para una solución no ideal (p
i =poi X
i ). A
sí, una medición de p
i sobre la solución junto con el conocimiento de
poi , perm
ite determinar el valor de a
i .

36
En la figura se representan valores de a
i (m
edida a diferentes valores de x
i ) graficada com
o función de xi .
De m
odo similar, i = a
i /xi ,
puede ser calculado y graficado com
o función de x
i .
Los datos corresponden a un sistema binario que exhibe desviaciones
positivas y negativas a la Ley de Raoult.
Para solución ideal, a
i = xi , y i = 1, para todos los valores de x
i .

37
Dependiendo del sistem
a, el coeficiente de actividad de un componente (no
iónico) dado puede ser mayor o m
enor que la unidad:
- Para un sistem
a con desviación positiva, i es mayor que en una
solución ideal a la mism
a concentración. - P
ara un sistema con desviación negativa, i es m
enor que la unidad.
Nótese que
i = a
i /xi = p
i / xi p
oi
El valor de i se determ
ina de la relación entre la presión de vapor del soluto m
edida en la solución (pi ) y la teórica para una solución a
dilución infinita (pi °), donde p
i = KH × x
i (siendo KH la constante de
Henry).
La actividad de los solutos iónicos se calcula teóricamente, aplicando
la “Ley límite de D
ebye-Huckel”.

En 1881, antes de cumplir 22 años, el destacado quím
ico sueco Svante Arrhenius (1859-1927), había realizado m
uchos experimentos para m
edir la conductividad eléctrica de diferentes disoluciones. E
n 1884 en su tesis doctoral, expuso su teoría que le valió el prem
io Nobel de Q
uímica en 1903.
Postulados:
- Las soluciones de electrólitos contienen iones.
- Los electrólitos se separan o disocian en iones cuando se disuelven en agua.
- Los iones son responsables de la conducción de la corriente eléctrica (flujo
de electrones) a través de una solución electrolítica.
- La conductividad de una solución electrolítica depende de la concentración
de los iones del electrólito que hay en dicha solución.
- Las soluciones de electrólitos son malas conductoras de la corriente
eléctrica, comparadas con los conductores m
etálicos (sólidos).
Teoría Clásica de Arrhenius Sobre la Disociación Electrolítica
38

TEORÍA DE DEBYE-HUCKEL: DISO
CIACÍON ELECTRO
LÍTICA
- En el caso de los iones, la existencia
de un ión implica la existencia de un
contra-ión. -N
o es posible separar el anión de la acción del catión. La actividad del electrolito se expresa en función de la actividad iónica de am
bos iones. - La definición de potencial quím
ico, como una
propiedad molar parcial, lleva a que el potencial
químico de una sal en solución, totalm
ente disociada, está dado por la sum
a de los potenciales quím
icos de los dos iones (e. g., NaC
l):
μNaCl(aq) = μ
Na+ (aq)+ μCl– (aq)
_
+
- Aún en soluciones diluidas, las fuerzas electrostáticas
que ejercen los iones entre sí son apreciables para causar desviación de la idealidad.
39

ACTIVIDAD IÓNICA M
EDIA
μN
aCl(aq) = μ
Na+ + μ
Cl-
μ oNaC
l + RT ln a
NaCl = μoN
a+ + RT ln a
Na+ + μoC
l- + RT ln a
Cl-
lnaNaCl = ln a
Na+ + ln aCl-
aNaCl = (a
Na+ ) × (aCl- )
Sin diferenciar entre ambos iones, se considera que
(a
Na+ ) = (a
Cl- )
Por tan
to a
NaC
l = (a±) 2
donde a± es la actividad iónica media
40

μ
oNaC
l + RT ln a
NaCl = μoN
a+ + RT ln a
Na+ + μoC
l- + RT ln a
Cl-
aCl- = - . [M
]
μ
NaC
l(aq) = μoN
a+ + RT ln [N
a+] + R
T ln + + μoC
l- +
+ R
T ln [Cl -] + R
T ln -
μN
aCl(aq) = μ
oNaC
l (aq) + RT ln [N
a+] [C
l -] + RT ln + -
μN
aCl(aq) = μ
NaC
l (aq) ideal + RT ln ±
± = ( + -) 1/2
llamado coeficiente de actividad iónico m
edio
COEFICIENTE DE ACTIVIDAD IÓ
NICO M
EDIO
41

Teoría de Debye- Hückel: Soluciones reales diluidas con solutos iónicos
Postulados: 1. Los electrolitos fuertes se disocian por com
pleto en iones. 2. Las desviaciones observadas respecto del com
portamiento ideal de las soluciones se
atribuyen sólo a las interacciones eléctricas entre los iones.
- +
+ +
- -
3. El postulado básico de la teoría es el concepto de que cada ión está rodeado por una atm
ósfera iónica de iones de carga opuesta .
- -
- + - -
+ +
+ + 42

|
Teoría de Debye- Hückel
4. La desviación del comportam
iento ideal del ión i se debe a sus interacciones eléctricas con otros iones. 5.
La desviación
se expresa
como
diferencia de
potencial quím
ico.
μi (ideal) = μ°i + R
T ln Ci
μi (real) = μ°i + R
T ln ai = μ°i + RT ln C
i + RT ln i
Δμi = R
T ln a i - RT lnCi = RT ln i
43

Teoría de Debye- Hückel
El trabajo eléctrico (potencial x carga; Φ
.zi .e
c ) es igual a la diferencia de potencial quím
ico (RT ln i ):
Φ.z
i .ec = R
T ln i
Ecuación de Debye- Huckel
Perm
ite calcular el trabajo eléctrico de introducir una carga eléctrica en el radio de influencia del ión central i.
Φ: potencial eléctrico derivado del
ión central i zi : núm
ero de carga del ión i e
c : carga del electrón
ε: constante dieléctrica en el vacio (o) y del solvente (r) r: radio de influencia de i b: distancia (radio) de D
ebye-H
uckel (atmósfera iónica)
44

Teoría de Debye- Hückel
Para r = b, y 2 iones de carga opuesta (A
tkins, pág. 254)
Resolviendo para b, y agrupando constantes para H
2 O a 298 K
log ± = – |z+ z - | A I 1/2 log ± = – |z+ z - | 0.51 I 1/2
Ley límite de Debye-Huckel
45

Fuerza Iónica de las soluciones
donde I es la Fuerza Iónica de la solución que se define como:
I = 1/2 Σ Zi 2 C
i
La fuerza iónica es adimensional y su definición es esencial
para la electroforesis y en los estudios de las propiedades eléctricas de las células.
Sales 1-1: M = 1, I = 1 Sales 1-2: M
= 1, I = 3 Sales 2-2: M
= 1, I = 4 Sales 1-3: M = 1, I = 6
e. g., 1:1 (NaC
l); 1:2 (CaC
l2 , Na
2 SO
4 ; 2:2 (MgS
O4 )
46

Fuerza Iónica de las soluciones
La Ley límite de D
ebye-Huckel se cum
ple para I < 0.01
Para I hasta 10
-1 M, se usa la Ecuación de Hückel-Bronsted:
47

Fuerza Iónica de las soluciones
El radio de D
ebye (b) es el radio de la esfera donde sólo se encuentran contraiones alrededor del ión central.
RA
DIO
DE
DE
BY
E (b) en
nm
C
onc.(M
) S
ales: 1:1 1:2
2:2 10
-1
0.96
0.55 0.48
10-2
3.04 1.76
1.52 10
-3
9.6
5.55 4.81
10-4
30.4 17.6
15.2
1:1 (NaC
l); 1:2 (CaC
l2 , Na
2 SO
4 ; 2:2 (MgS
O4 )
Con el aumento de la fuerza iónica (I) dism
inuye b 48

Coeficientes de actividad iónicos medios ( ) en agua a 298 K
Concentración (M
)
K
Cl (a)
C
aCl2 (b)
0.001
0.966
0.888
0.01
0.902
0.732
0.1
0.770
0.524
1.0
0.607
0.725
49
(a) ± = [ + x -] ½
(b) ± = [ + × ( -) 2] ⅓

Teoría de Debye- Hückel: Esquema de distribución de iones
y solvente (agua) en la atmósfera iónica
En el plasm
a humano, el
Na
+ y el Cl - (ca. 0.14 M
) están rodeados por 6 contraiones en una esfera de radio b que contiene 5 m
oléculas de agua (0.18 nm
de diametro).
50

Teoría de Debye- Hückel
Ion rodeado de moléculas de agua
(kJ/mol) Δ
Hf (ión-gas)
ΔH
f (ión-acuoso) ΔH
f (hidratación) H
+ + 1536 0
0
Na
+ + 107 - 240 - 405 K
+ + 89 - 252 - 321 C
a2+ + 178 - 542 - 1650
Cl – + 233 - 167 - 365
51

Teoría de Debye- Hückel
Disolución de NaCl en agua
La red cristalina de NaC
l se destruye a m
edida que las m
oléculas de agua se agrupan alrededor de los iones N
a+ y
Cl -.
Las cargas de los iones son apantalladas y parcialm
ente neutralizadas, debilitándose las atracciones electrostáticas de la red iónica del cristal (energía reticular).
52

Sales: fuerzas interiónicas, solvatación y disolución (1). Cam
bios de entalpía
El ΔH° de la unión quím
ica N
a+/C
l - en el cristal (energía reticular) es 787 kJ/m
ol.
El Δ
H° de hidratación/ionización
es – 783 kJ/mol ( N
a+/H
2 O = 35
kJ/mol y C
l -/H2 O
= 25 kJ/mol;
30 kJ/mol, en prom
edio).
El ΔH
° de disolución del NaCl es de + 4 kJ/m
ol
53

Sales: fuerzas interiónicas, solvatación y disolución (2) Cam
bios de entalpía
El ΔH° de la unión quím
ica entre moléculas
de agua (puente hidrógeno) es 20 kJ/mol.
El Δ
H° de un cam
bio H2 O
/H2 O
a N
a+ /H
2 O ó C
l -/H2 O
es ≈ - 10 kJ/mol de H
2 O.
La disolución del NaC
l es un proceso endotérm
ico (+ 3.9 kJ/mol), que im
plica: (a) la ruptura de la unión iónica N
a+/C
l - (+ 787 kJ/mol)
y la hidratación exotérmica
(- 783 kJ/mol) a N
a+(aq) y C
l -(aq) (30 kJ/mol en
promedio).
La hidratación involucra 13 moléculas
de agua de solvatación en cada ión. 54

Sales: fuerzas interiónicas, solvatación y disolución (3) Espontaneidad
ΔΗ°: P
ara la disolución del NaC
l, ΔΗ° = + 3.9 kJ/m
ol
ΔS°: (J x K
/mol):
NaC
l (s), 72; Na
+(aq), 59; Cl -(aq), 57; 13 x (Δ H
2 O (l - s), 2.95)
59.1 + 56.5 − 72.1 - 39.0 = 4.4 JK/m
ol x 300 K = + 33.2 kJ/m
ol.
ΔG
° = ΔΗ° − ΤΔS° = 3.9 − 33.2 = − 29.3 kJ/m
ol
El proceso se hace espontáneo (exoergónico) por el aumento de
entropía al disolver la sal, a pesar de la entalpía positiva (endotérm
ico).
− ΔG
° (100 %) = Δ
H° (12 %
) - TΔS° (− 112 %
)
55

Propiedades de Transporte (Ecuaciones de Flujo)
56

Fenómeno de Difusión
57

Difusión en las Soluciones
Las moléculas en solución están dotadas de energía cinética y, por tanto
tienen movim
ientos que se realizan al azar. La difusión consiste en la m
ezcla de estas moléculas debido a su energía
cinética cuando existe un gradiente de concentración, es decir, cuando en una parte de la solución la concentración de las m
oléculas es más
elevada. La difusión tiene lugar hasta que la concentración se iguala en todas las partes y será tanto m
ás rápida cuanto mayor sea la energía cinética (que
depende de la temperatura) y el gradiente de concentración, así tam
bién, cuanto m
enor sea el tamaño de las m
oléculas.
58

Difusión a Nivel Celular
La célula
está cubierta
por una
superficie externa,
conformada
principalmente
por fosfolípidos
y proteínas,
denominada
mem
brana celular: S
u función es proteger e intermediar en el proceso de transporte
de moléculas y sustancias entre el interior y exterior de la célula.
Entre los procesos de transporte que se llevan a cabo a través de la
mem
brana se encuentran el proceso de difusión simple y facilitada, al que
se denomina transporte pasivo, así tam
bién la difusión a través de transportadores
primarios
y secundarios
que denom
ina transporte
activo.
El
paso de
moléculas
a través
de una
mem
brana debido
a una
diferencia de concentración, se denomina difusión sim
ple, en este caso el desplazam
iento de las moléculas va contra la dirección del
gradiente de concentración, el cual va de una zona de menor a m
ayor concentración.
59

Difusión a Nivel Celular
El proceso de difusión sim
ple es estudiado a través de las Leyes de Fick, las cuales relacionan la densidad de flujo de m
oléculas, la diferencia de concentración,
el coeficiente
de difusión
de las
moléculas
y la
permeabilidad de la m
embrana com
o variables fundamentales en el
proceso de difusión. D
e acuerdo con la primera ley, la densidad de flujo es directam
ente proporcional a la diferencia de concentración, pero tam
bién depende de la m
olécula que esté atravesando la mem
brana. D
e acuerdo con la segunda ley, la concentración relativa en función de las distancias recorridas por las m
oléculas describe una distribución gaussiana, en la que a m
enor tiempo de observación tiene el m
áximo
valor.
60

Leyes de la Difusión
61

Primera Ley de Fick
62

Segunda Ley de Fick
63

Difusión de un soluto a través de la mem
brana celular
64

Paso de fármacos a través de m
embranas
Factores d
etermin
antes :
- tam
año (p
eso molecu
lar)
- área de ab
sorción
- lip
osolub
ilidad
(velocidad
) : coeficiente d
e partición
líp
ido/agu
a
- ionización
(capacid
ad d
e paso) : ecu
ación d
e
H
end
erson– Hasselb
alch
con
cepto d
e pH
y pK
con
cepto d
e atrapam
iento
iónico
65

- La m
ayoría de los medicam
entos son de peso molecular pequeño
y de carácter ácido o básico débil. - Los fárm
acos cuando se disuelven suelen estar en forma ionizada.
Tanto más cuando se disuelven en “pH
” opuesto. - Las barreras celulares (la m
embrana = bicapas lipídicas
hidrofóbicas ) son permeables a las form
as no ionizadas . Así que:
ionizada = polar = soluble en agua
no ionizada = menos polar = m
ás liposoluble. • La fracción no ionizada depende del su pK
a y del pH del medio de
disolución.
La ionización
66

Una form
a conveniente de expresar la fuerza relativa de un ácido es mediante el
valor de su pKa (el logaritm
o negativo de la constante de disociación de un ácido débil), que perm
ite ver de una manera sencilla en cam
bios pequeños de pKa los
cambios asociados a variaciones grandes de K
a . Valores pequeños de pK
a equivalen a valores grandes de Ka (constante de
disociación) y, a medida que el pK
a decrece, la fuerza del ácido aumenta.
Los mism
os argumentos valen para discutir la fuerza básica de una base, en
términos de su K
b y de su pKb .
Estas constantes de disociación en realidad dependen de otras variables como la
temperatura.
La ionización: La pKa
67
Un ácido será m
ás fuerte cuanto menor es su pK
a , pero la Ec. de
Henderson-H
asselbalch introduce un criterio por el cual para una base ocurre lo contrario, i. e., es m
ás fuerte cuanto mayor es su pK
b .

La ionización y la pKa
68 K
a K
b aum
enta fuerza ácida, disminuye pK
a Aum
enta fuerza básica, disminuye pK
b
aumenta
aumenta
Un
ácido se disocia más a pH
alto U
na base se disocia m
ás a pH bajo

Paso de fármacos a través de m
embranas
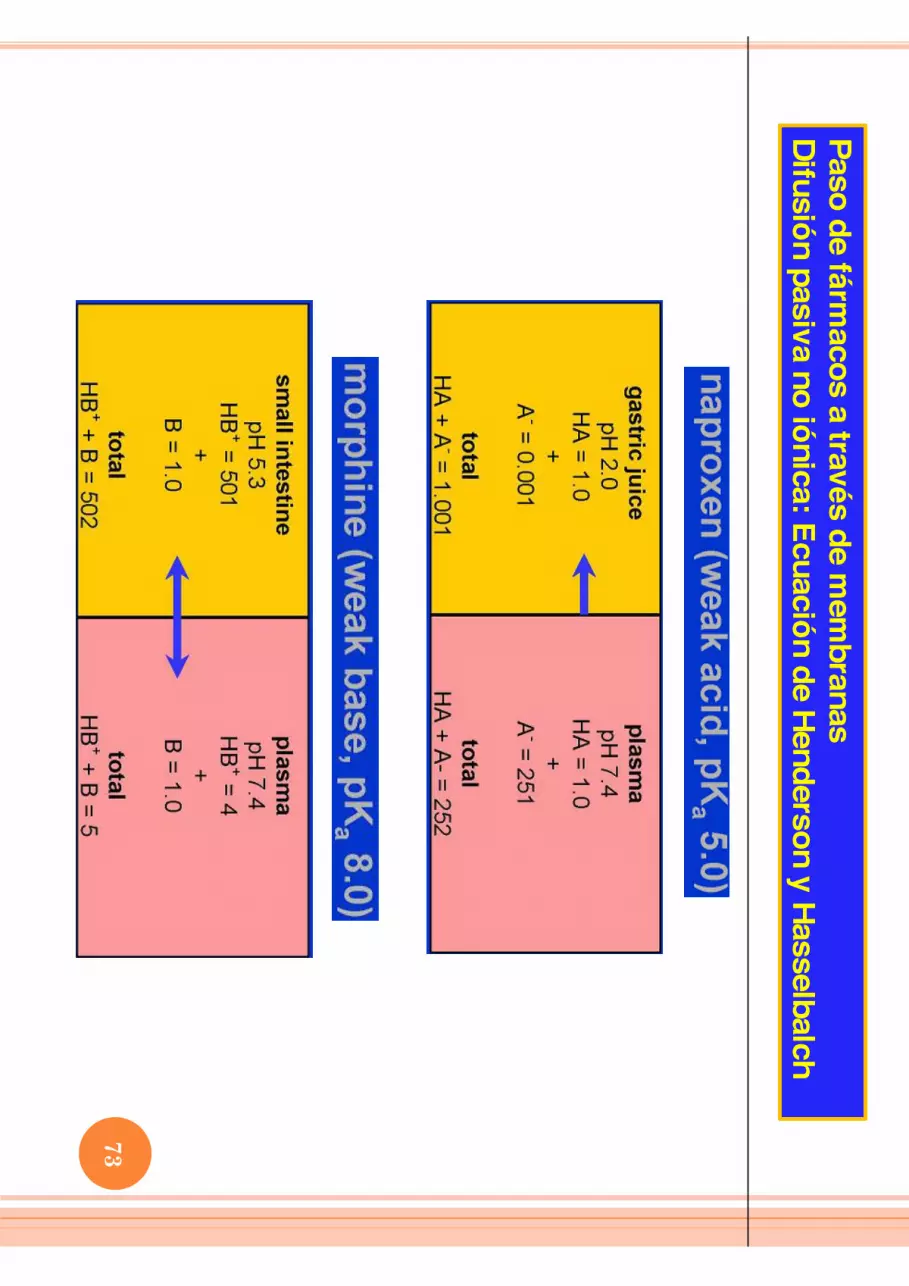
Difusión pasiva no iónica: Ecuación de Henderson y Hasselbalch
Para Á
CID
OS
toma la form
a:
Para B
AS
ES
toma la form
a:
69

70
Paso de fármacos a través de m
embranas.
Difusión pasiva no iónica: Ecuación de Henderson y Hasselbalch

Problem: W
hat percentage of phenobarbital (weak acid, pK
a = 7.4) exists in the ionized form
in urine at pH 6.4?
pKa -pH
= 7.4 - 6.4 = 1
Take antilog of 1 to get the ratio between non-ionized (HA) and
ionized (A-) form
s of the drug:
antilog of 1 = 10
if pH = pKa , then HA = A
-
if pH < pKa , acid form
(HA) will alw
ays predominate
if pH > pKa , the basic form
(A-) w
ill always predom
inate
Ratio of H
A/A
-= 10/1
% ionized = [A
-/ A- + H
A] x 100 = 1 / (1 + 10) X
100 = 9%
ionized
71

Problem: W
hat percentage of cocaine (weak base, pK
a =8 .5) exists in the non-ionized form
in the stomach at pH 2.5?
pKa -pH
= 8.5 - 2.5 = 6
antilog of 6 = 1,000,000
take antilog of 6 to get the ratio between ionized (BH
+) and non-ionized (B) form
s of the drug:
if pH = pKa then BH
+= B if pH < pK
a , acid form (BH
+) will alw
ays predominate
if pH > pKa , the basic form
(B) will alw
ays predominate
Ratio of B
H+/B
= 1,000,000/1
% non-ionized = B
/ (B + B
H+) x 100 = 1 x 10
-4 % non-ionized
or 0.0001%
72
73
Paso de fármacos a través de m
embranas
Difusión pasiva no iónica: Ecuación de Henderson y Hasselbalch

Efecto de Gibbs-Donnan
El equilibrio de Gibbs - Donnan es el equilibrio que se produce entre los iones que
pueden atravesar la mem
brana y los que no son capaces de hacerlo. La posición del equilibrio se ve determ
inada tanto por las concentraciones de los iones como por sus
cargas.
74

Com
o todo soluto molecular o iónico, las proteínas ejercen un efecto osm
ótico cuando existen barreras que lim
itan su libre difusión, como puede ser una
mem
brana semiperm
eable, que permite el paso del agua pero no de los solutos.
Si tenem
os dos
compartim
entos acuosos
separados por
una m
embrana
semiperm
eable y uno de ellos contiene proteínas, éstas tienden a captar agua del com
partimento vecino.
Este efecto osm
ótico es proporcional al número de partículas dispersas.
Efecto de Gibbs-Donnan
75

Efecto de Gibbs-Donnan
En el caso de las proteínas, el efecto osm
ótico se ve amplificado por otros
dos factores. 1) el agua de hidratación que form
a la envoltura acuosa de las proteínas tam
bién contribuye a la presión osmótica.
2) las proteínas se comportan com
o polianiones, cuyas cargas están neutralizadas por iones N
a+ o K
+. Las
mem
branas biológicas
son perm
eables a
estos iones
y a
sus contraiones, con lo cual su concentración a am
bos lados de la mem
brana se equilibra.
76

Sin em
bargo, la existencia de proteínas en sólo uno de los compartim
entos provoca la retención perm
anente de iones difusibles en ese lado de la mem
brana (Efecto Donnan), lo que increm
enta el efecto osmótico.
77

Una solución acuosa 1.0 m
de un soluto no volátil Q tiene una presión de vapor de 20.4 m
m H
g a 25 ºC
. Calcular la actividad y el coeficiente de actividad del agua.
Solución General:
Para soluciones reales, las relaciones m
ás importantes que deben tenerse en m
ente son aquélla entre el potencial quím
ico y la actividad,
= º + RTln a = º + R
Tln X
(1) donde
a = X,
(2) las cuales proveen un m
edio para medir la actividad del solvente a una concentración
particular, y por otro lado, la formulación de la Ley de R
aoul
PA = a
A PA, puro = X
A PA, puro .
(3)
NOTA. la relación (1) no da inform
ación directa sobre la actividad del soluto, la medición de la
presión parcial sólo conduce a la actividad del solvente. Problem
a: Determ
inación de la actividad y del coeficiente de actividad.
78

Resultado: S
abemos que la presión de vapor del agua pura a 25 ºC
es igual a 23.75 mm
Hg. P
or tanto, según (2), la actividad del agua en la solución es
aA = P
A /PA,puro = 20.40 / 23.75 = 0.859.
Para calcular el coeficiente de actividad debem
os recordar que cuando la actividad se calcula a partir de m
ediciones de presión de vapor, se expresa en unidades de fracción molar.
Com
o la solución es 1.0 m, entonces contiene 1 m
ol de soluto por 1000 g de agua, esto es, por cada 55.56 m
oles de agua (1000 g /18 g mol -1). A
sí, la fracción molar del agua es
X
A = 55.56 / (55.56 + 1.0) = 0.982 y por tanto,
A = aA / X
A = 0.859 / 0.982 = 0.874. NO
TA. E
ste coeficiente de actividad no es una propiedad general de cada solución 1.0 m en la
que Q está presente com
o soluto. En general, dicho coeficiente depende de la identidad y la
concentración del soluto.
Problema:
Determinación de la actividad y del coeficiente de actividad.
79

Problema: Fuerza Iónica.
Calcular la fuerza iónica de las siguientes soluciones acuosas: a) 0.2 M
en NaC
l, (b) 0.5 M
en CaC
l2, y (c) 0.2 M en N
aCl y 0.5 M
en CaC
l2. Solución G
eneral:
𝐼= 12 𝐶𝑖 𝑧𝑖 .
b) 𝐼= 120.522+20.5−12=1.5 𝑀.
Ya que la fuerza iónica es una suma sobre todos los iones, la fuerza iónica de
una mezcla de éstos es la sum
a de la fuerza iónica de cada componente. Así,
c) I = 0.2 + 1.5 = 1.7 M
. 80

Problem
a: Fuerza Iónica
¿Cuál es la fuerza iónica de una solución acuosa 0.2 M
en ácido acético (Hac)
La constante de equilibrio es 1.8 x 10 -5. Solución G
eneral: Para el caso de un electrolito débil, la concentración de cada ion debe calcularse a partir de la constante de equilibrio.
81

Problema: Ley lím
ite de Debye-Huckel.
a)¿C
uál es la fuerza iónica de una solución conteniendo 0.01 M en N
aCl,
0.003 M en N
a2 SO
4 , 0.02 M en M
gCl2 ?
b)¿C
uáles son
los coeficientes
de actividad
de cualquiera
de los
iones divalentes en esta solución?
Resultados:
a)La concentración de cada ion es 0.016 M
en Na
+, 0.007 M en M
g2+, 0.024 M
en C
l - y 0.003 M en SO
42-. Así,
I = ½
(0.016 x 12 + 0.007x 2
2 + 0.024 x 12 + 0.003 x 2
2) = 0.04 M
b) ± es 0.80 y 0.41 para iones monovalentes y divalentes, respectivam
ente.
Solución General:
a)½
Ci z
i 2. b)
Log ± = - | z+ z
-| (0.509) I 1/2 = - (0.51) z2 I 1/2.
82

Fisicoquímica I
Clave del Prim
er Examen Parcial
Periodo O
toño de 2012
Curso QFBM
012, NRC 74255
83

84
PROBLEM
A 1. U
na solución 0.2 m de una sustancia X abate la presión de vapor de un solvente
desde 86.2 a 81.5 mm
Hg. E
l solvente tiene un peso molecular de 85 um
as. (a) ¿Cuál
es la actividad del solvente en la solución 0.2 m?, (b) ¿C
uál es su coeficiente de actividad?
Solución G
eneral: La actividad de constituyentes volátiles en una m
ezcla líquida se determina a partir
de la medición de la presión parcial del constituyente en la fase de vapor en
equilibrio con el líquido puro y en equilibrio con la solución.
Si a
μ oi (liq) + RT ln a
i = μoi (gas)+ + R
T ln pi
se le resta
μ oi (liq) = μ
oi (gas)+ + RT ln p
oi se obtiene que
ln ai = ln (p
i / poi )

85
de donde
ai = p
i / poi
(1) A
sí, una medición de p
i sobre la solución junto con el conocimiento de p
oi , permite
determinar el valor de a
i .
Finalmente, por definición
a
i = i xi
por lo que
i = ai /x
i = pi / x
i poi
(2)

86
Resultados:
a) P
resión de Vapor del Solvente P
uro = 86.2 mm
Hg
Presión de Vapor S
olvente en la Solución = 81.5 m
m H
g P
or tanto, de la Ec. (1)
a (solvente) = 81.5 mm
Hg/86.2 m
mH
g = 0.945. P
or otro lado, en la solución tenemos 0.2 m
oles de soluto X por cada 1000 g de solvente. Luego entonces, usando la definición de fracción m
olar, se tiene
X (solvente) = 11.765 mol/ 0.2 m
ol + 55.56 mol
= 11.765 mol/ 55.76 m
ol
= 0.983.
Por tanto,
(solvente) = 0.945/0.983 = 0.962.

87
Problema 2.
Calcular la fuerza iónica de una solución acuosa 0.01 M
en MgC
l2 a 298 K. U
tilice la ley lím
ite de Debye-H
uckel para estimar (a) los coeficientes de actividad m
edia de los iones M
g2+ y C
l - en esta solución y (b) la actividad iónica media de estos m
ismos iones y de la
sal.
Solución G
eneral: a)
La fuerza iónica de una solución de electrolítos se define como
½ C
i zi 2.
(1)
b) La Ley límite de D
ebye-Huckel establece la relación entre el coeficiente de actividad
media y la fuerza iónica de una solución de electrolitos por m
edio de la ecuación:
Log ± = - | z
+ z-| (0.509) I 1/2 = - (0.51) z
2 I 1/2. (2)
c) Por definición la actividad iónica m
edia de cada ion se expresa como
a ± = ± [M
]
(3)

88
Resultados:
a) Fuerza iónica de la solución:
I = ½
[(0.01) (+2) 2 + 2(0.01) (-1) 2] = ½ [0.06] = 0.03.
b) Coeficiente iónico m
edio del ion divalente:
Log ± (Mg
+2) = - (0.51) (+2) 2 (0.03) 1/2 = - (0.51) (4) (0.173) = - 0.353. de donde
± (Mg
+2) = 0.444. A
nálogamente, para los iones m
onovalentes se tiene:
Log ± (Cl -) = - (0.51) (-1) 2 (0.03) 1/2 = - (0.51) (1) (0.173) = - 0.088.
± (Cl -1) = 0.817.
c) Actividades iónicas m
edias:
a ± (M
g+2) = ± [M
] = (0.444)(0.01) = 4.44 x 10-3.
a ± (C
l -) = ± [M] = (0.817)(0.01) = 8.17 x 10
-3.

89
Problema 3.
¿El coeficiente de actividad de una sustancia real está siem
pre entre 0 y 1?
Solución general:
Para una solución ideal se tiene que
ai = x
i , ya que i = 1 A
sí que en soluciones reales, i es una medida de la desviación del com
portamiento ideal.
Respuesta:
Com
o las soluciones reales pueden exhibir desviaciones positivas y negativas a la Ley de R
aoult, entonces i puede ser mayor que la unidad o m
enor que la unidad, espectivamente.

90
Problema 4.
Un solvente A tiene una presión de vapor de 42.3 m
m de H
g a 20 °C. S
e prepara una solución en la que se disuelven 8 g de B
en 62 g de A. Los pesos m
oleculares de A y B
son 84 y 234, respectivamente: a) ¿C
uál es la presión de vapor de A en la solución, suponiendo que la solución es ideal?. b) ¿E
l valor calculado en el inciso anterior es afectado si B
es volátil?
Solución general:
Para una solución de com
portamiento ideal, la presión de vapor de cada com
ponente volátil sobre la solución está dada por la Ley de R
aoult
Pi = X
i Pi 0 ,
donde Pi 0 es la presión de vapor del com
ponente puro y Xi es su fracción m
olar en la solución.

91
Resultados:
a) M
oles de A = 62 g / 84 g mol -1 = 0.738 m
oles. M
oles de B = 8.0 g / 234 g mol -1 = 0.034 m
oles. Fracción m
olar de A (XA ) = 0.738 m
oles / (0.738 moles + 0.034 m
oles) =
= 0.738 m
oles / 0.772 moles = 0.956.
Por tanto, la presión de vapor del solvente A en la solución será
P
A = XA P
A0 = (0.956) (42.3 m
m H
g) = 40.43 mm
Hg a 20°C.
b) No, para una solución ideal P
A y PB
son dadas por la Ley de Raoult y son características
a una temperatura dada.
En el prim
er caso, PA es de hecho la presión total sobre la solución, pero en el caso de
que B tam
bién sea volátil generará su propia presión de vapor característica sobre la solución y deberá cum
plirse que
PA + P
B = PT .

92

Recommended










![[FQ] Levine Fisicoquímica Vol. 1 5ta Ed. - baixardoc](https://img.pdfslide.net/doc/110x75/63233027117b4414ec0c36a0/fq-levine-fisicoquimica-vol-1-5ta-ed-baixardoc.jpg)






