Embed Size (px)
Citation preview


Important cause of death in young children
Malignancies Need to distinguish from benign lesions
Heterotopia / Choristoma
Hamartoma
Early diagnosis is the aim
Treatment is better tolerated
NEOPLASTIC LESIONS:

Hemangioma
Lymphangioma
Sacrococcygeal Teratoma
BENIGN TUMORS

Most common Benign Tumor of Childhood
1. Capillary
2. Cavernous
? Malformations.. ? Hamartoma
Majority are superficial lesions
Malignant Transformation is very Rare
Hemangioma

Site - Skin , Subcutaneous tissue , Mucosa of oral
cavity / lips
Morphology:: Bright red - Blue , Few mm - few cms.
Strawberry Hemangioma Present since Birth.
Capillary Hemangioma
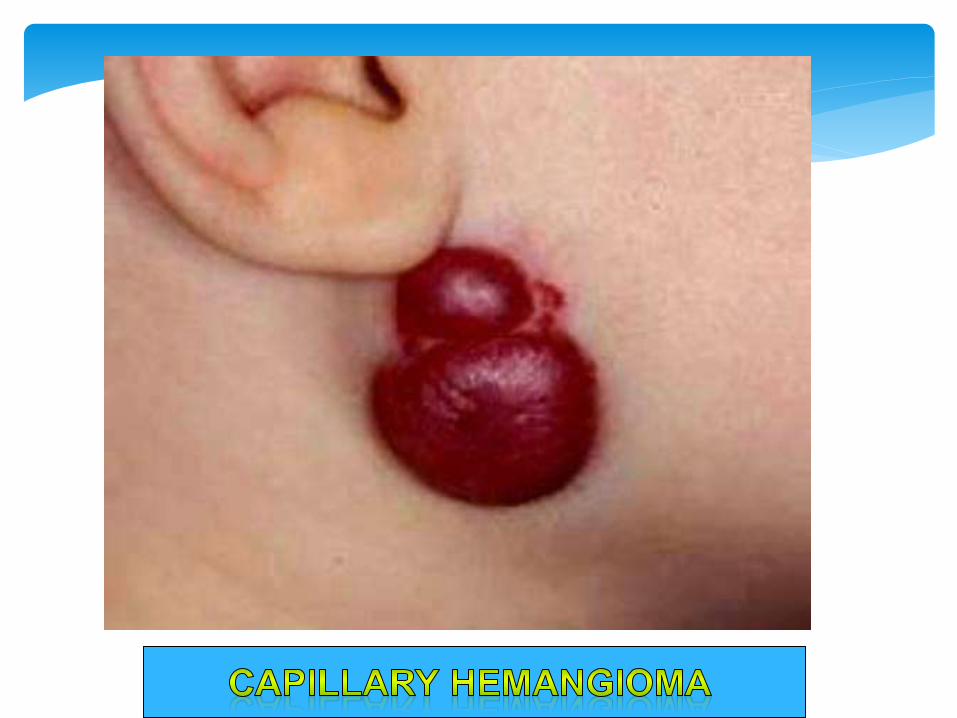
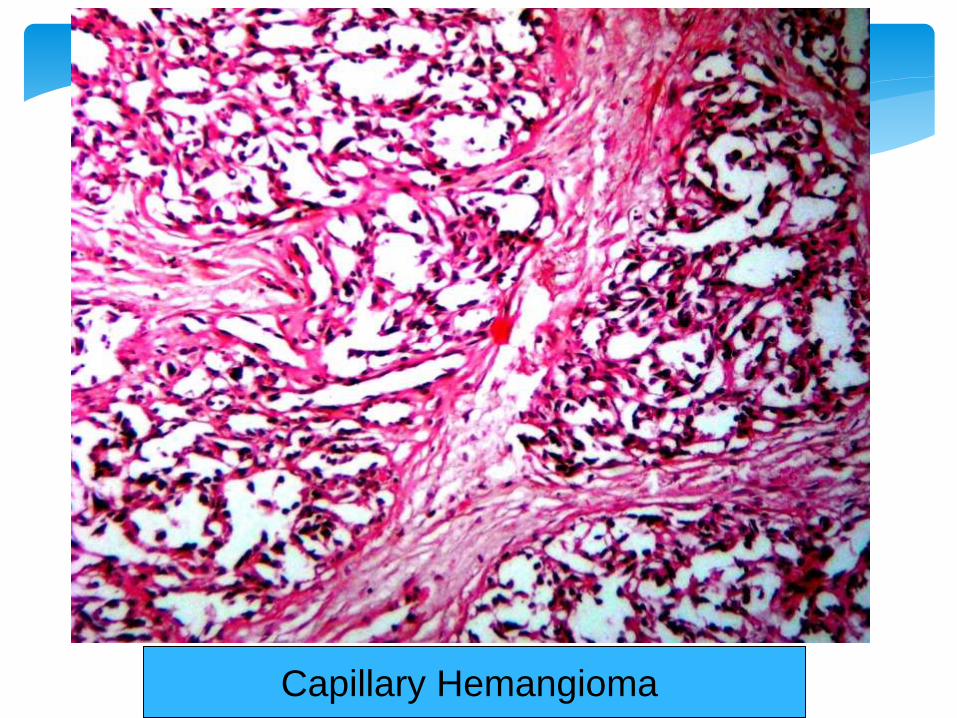
Capillary Hemangioma

Site - Skin , Mucosa , Viscera (Liver)
Morphology Red-Blue , compressible , Spongy , well
defined lesions , 2 - 3 cms
Cavernous Lymphangioma - Cystic Hygroma
Cavernous Hemangioma
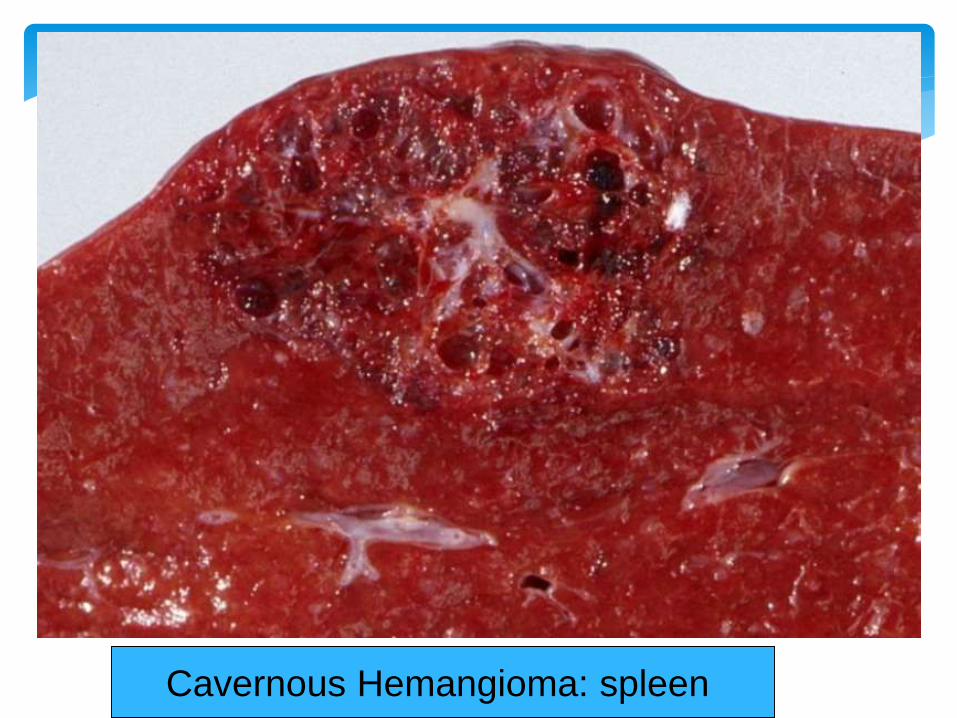
Cavernous Hemangioma: spleen
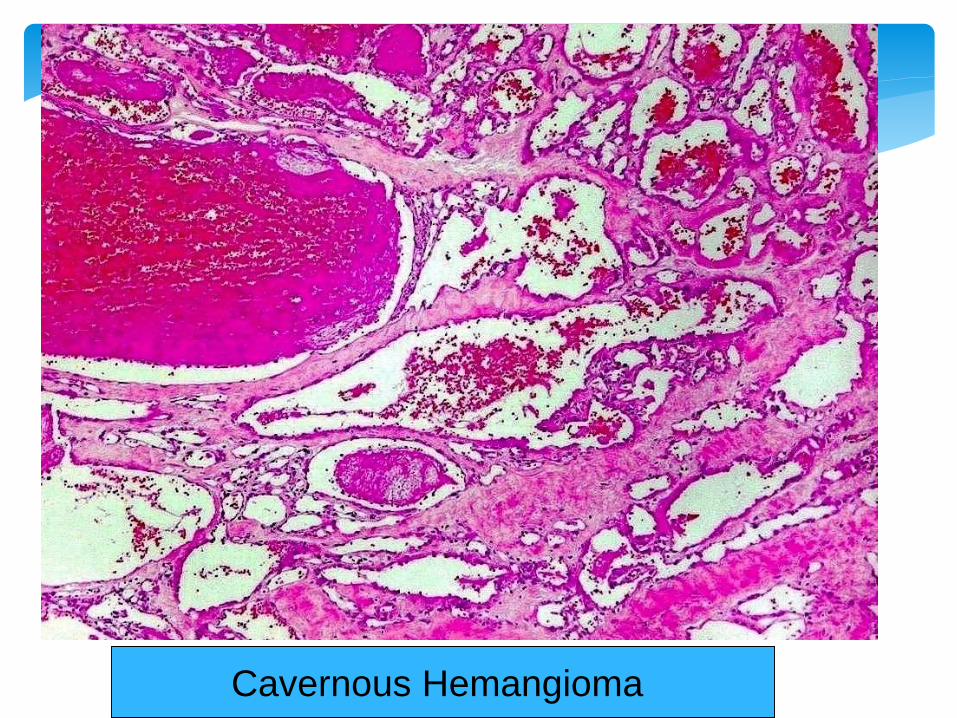
Cavernous Hemangioma
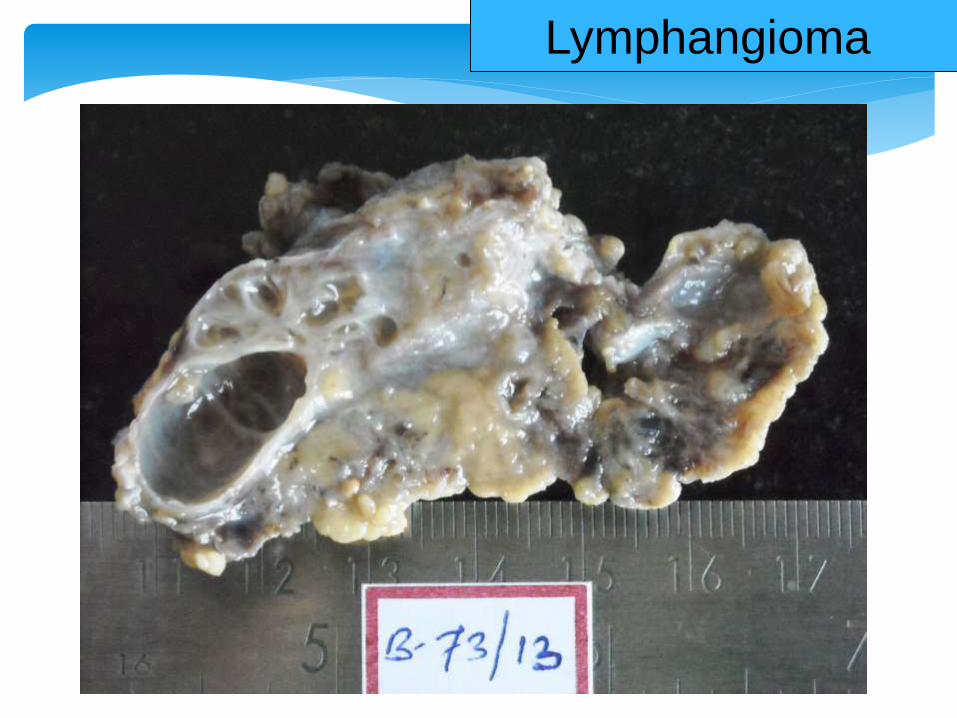
Lymphangioma
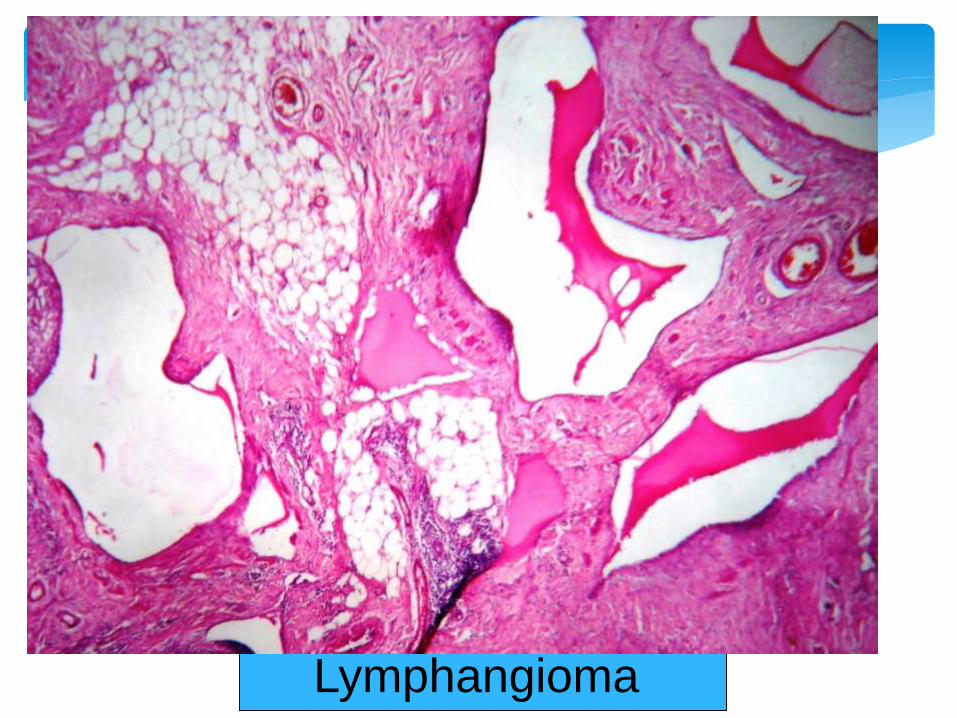
Lymphangioma

Contains the tissues derived from all ectoderm,
endoderm and mesoderm.
Skin is most commonly seen: hence the name dermoid
cyst.
Any tissues can be seen: cartilage, bone, tooth, hair,
sebaceous glands, brain tissues…..etc.
Most commonly seen in the midline structures.
Congenital Teratoma:
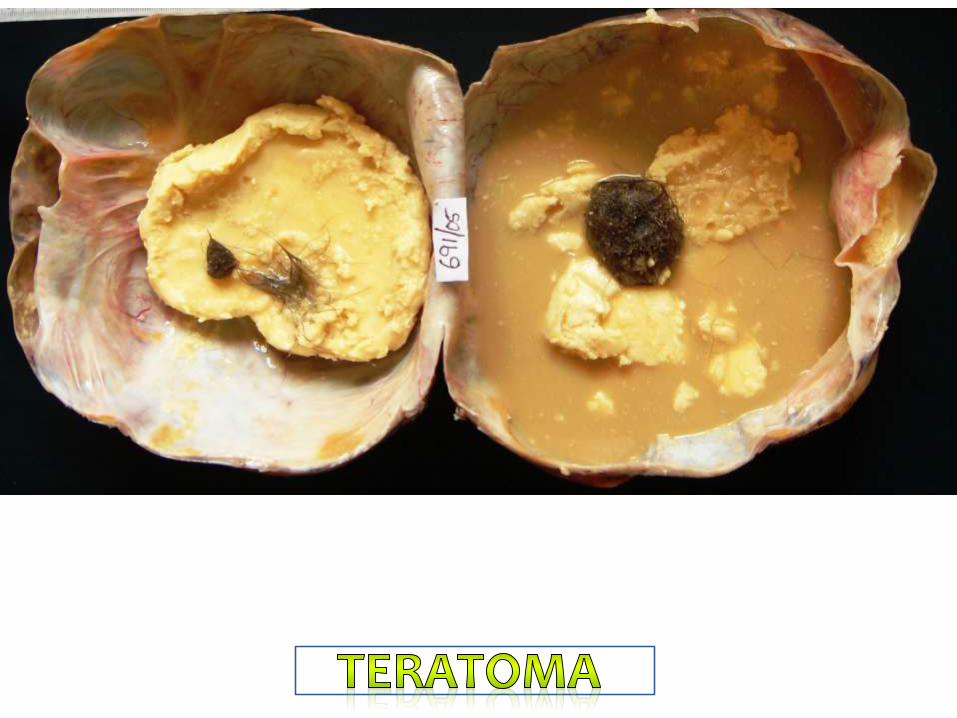
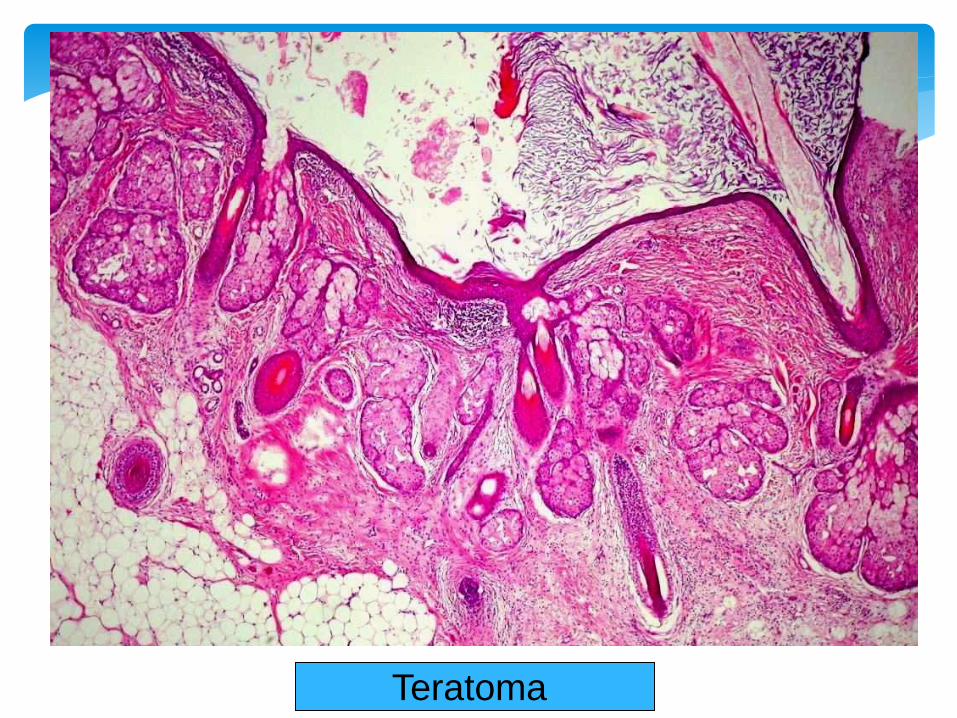
Teratoma
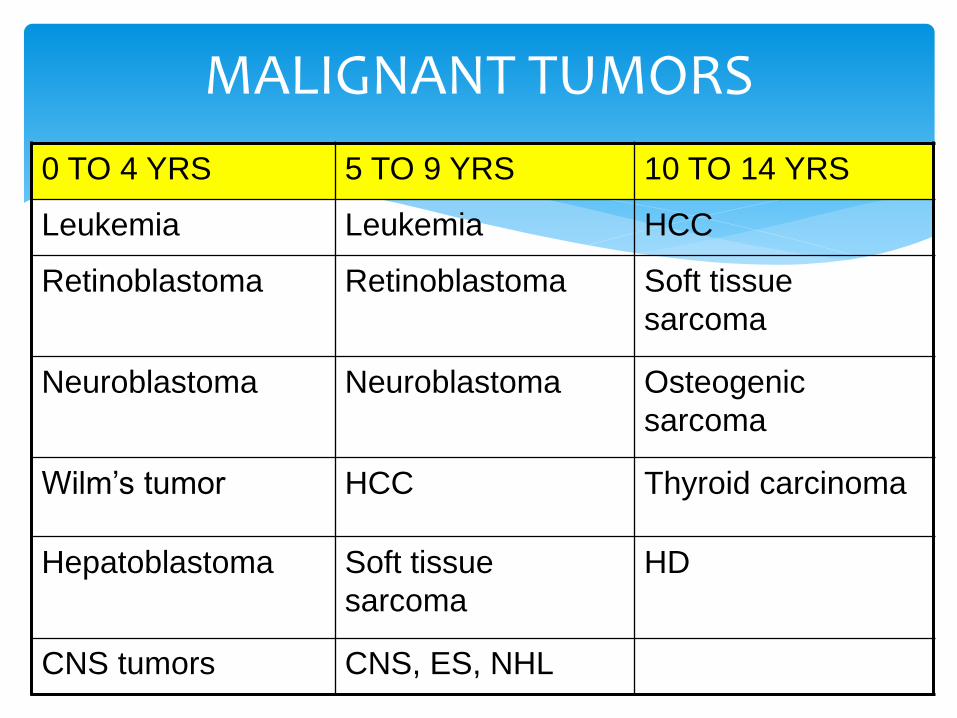
MALIGNANT TUMORS
0 TO 4 YRS 5 TO 9 YRS 10 TO 14 YRS
Leukemia Leukemia HCC
Retinoblastoma Retinoblastoma Soft tissue
sarcoma
Neuroblastoma Neuroblastoma Osteogenic
sarcoma
Wilm’s tumor HCC Thyroid carcinoma
Hepatoblastoma Soft tissue
sarcoma
HD
CNS tumors CNS, ES, NHL

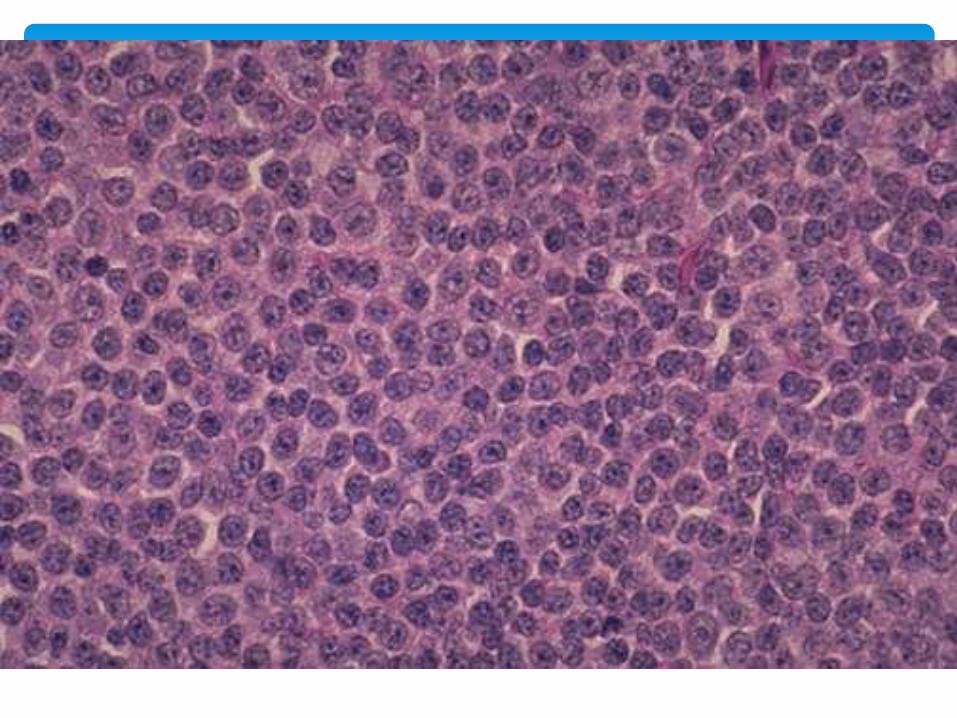
Histologically malignant pediatric tumors are alike
Primitive embryonal morphology
Sheets of small cells, scant cytoplasm, dense round nucleus
SBRCT
Neuroblastoma, Lymphoma, RMS, ES, Wilm’s tumor

Most common solid tumor of childhood next to CNS
tumors
Most occur below 5 yrs of age
Tendency to regress spontaneously
Head to toe Sympathetic chain
75% arise within abdomen Adrenals
Can arise in brain, Head & neck

Range in size
Advanced tumors may invade the renal vein
Grey-White, Soft, Friable, Large areas of hemorrhage,
Cystic, Calcification
Tendency to metastasize early and invade adjacent
structures
MORPHOLOGY- NEUROBLASTOMA

SBRCT
Some areas of differentiation seen
Homer-Wright pseudorosettes
Some ganglion like cells Maturation
Ganglioneuroblastoma
Ganglioneuroma Better differentiated
Presence of Schwann cell stroma
MICROSCOPY- NEUROBLASTOMA

Protuberant abdomen, fever, weight loss
Hepatomegaly, metastasis, bone pain
Elevated blood levels of catecholamines: VMA
Staging of the tumors & age of patient decides the prognosis
Del (1p36) & amplification of N-myc oncogene Bad
prognosis
CLINICAL FEATURES
Mc Malignant tumor of eye in childhood
Multifocal or Bilateral
Spontaneous regression is noted, secondary primary
tumors possible
Familial & Sporadic
Rb gene mutation
RETINOBLASTOMA

RETINOBLASTOMA
1 in 20000 infants and children
60% Familial; 40% Sporadic
Autosomal dominant trait
Gene is present on chromosome 13q14,
Loss of both alleles leads to Retinoblastoma
(deletions)

Arise from neuroepithelial cell
SBRCT
Some differentiated structures may be present
Flexner- Wintersteiner rosettes
Unlike pseudorosettes
Dissemination by Optic nerve CNS. Skull bones, LN.
MORPHOLOGY

Median age at presentation is 2 yrs
Poor vision, strabismus, Leukocoria (Cat’s eye reflex)
Rx: Enucleation
Spontaneous regression, with or without Rx is also noted
Secondary tumors like OS & Soft tissue sarcomas.
CLINICAL FEATURES


Most common renal tumour in children
10 / million, under 15 yrs,
Common age – 2 to 5 yrs,
5 to 10% bilateral – synchronous or metachronous
Wilms tumour

PATHOGENESIS AND GENETICS:
1. WAGR syndrome: Aniridia, Genital anomalies, Mental
retardation .
2. Denys- Drash syndrome: Gonadal dysgenesis (male
Psuedohermaproditism)
3. Beckwith-Wiedmann syndrome: Organomegaly,
Macroglossia, Hemihypertrophy, Omphalocele, Adrenal
cytomegaly.
Wilms tumour

Tissue features showing an attempt to recapitulate nephrogenesis
Triphasic Tumor:: Blastemal, Stromal, Epithelial components +
Sheets of small blue cells without differentiation-Blastemal
Fibromyxoid stromal component, Smooth muscle, cartilage
Abortive tubules & Glomeruli
MICROSCOPY

2 to 5 yrs common
Palpable abdominal mass
Hematuria,
Pain,
Intestinal obstruction,
Hypertension
Pulmonary metastases
Wilms tumour – Clinical Course

PROGNOSIS
Very Good,
Nephrectomy + chemotherapy,
2 yrs survival – 90%
Prone for second primary tumours.
Wilms tumour – Clinical Course






























