Embed Size (px)
Citation preview

FIBRO-OSSEOUS LESIONS“The term fibro-osseous lesion (FOL) is a
generic designation of a group of jaw
disorders” characterized by the
replacement of bone by a benign
connective tissue matrix.
This matrix displays varying degrees of
mineralization in the form of woven bone
or of cementum-like round acellular
intensely basophilic structures.

ClassificationCharles Waldron Classification Of The Fibro-Osseous Lesions Of
The Jaws (1985)
1. Fibrous Dysplasia
a. Monostotic
b. Polyostotic
2. Fibro-Osseous (Cemental) Lesions Presumably Arising In The Periodontal Ligament
a. Periapical Cemental Dysplasia
b. Localized Fibro-Osseous-Cemental Lesions (Probably Reactive In Nature)
c. Florid Cement-Osseous Dysplasia (Gigantiform Cementoma)
d. Ossifying & Cemenifying Fibroma
3. Fibro-Osseous Neoplasms Of Uncertain Or Detectable Relationship To Those Arising In The Periodontal Ligament (Category II)
a. Cemetoblastoma, Osteoblastoma & Osteoid Osteoma
b. Juvenile Active Ossifying Fibroma & Other So Called Aggressive, Active Ossifying /Cementifying Fibromas.

Eversole Classification, 2008

By definition, all benign fibro-osseous
lesions possess an osseous and fibrous
tissue component.

The ossifications in BFOL can be quiteheterogeneous even within a specificdisease entity.
Newly formed bone - woven pattern ofcollagen fiber orientation.
Mature bone - lamellar pattern.
Many have both irregular trabeculaeas well as spheroidal cementiclecalcifications, so called “ Cemento-ossifying” lesions.
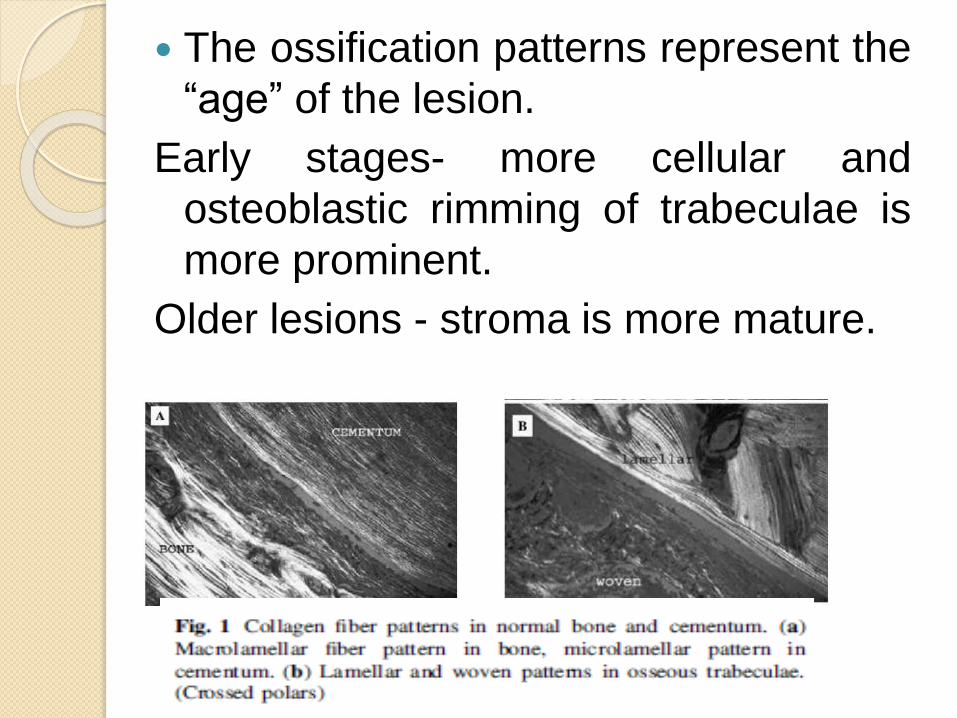
The ossification patterns represent the
“age” of the lesion.
Early stages- more cellular and
osteoblastic rimming of trabeculae is
more prominent.
Older lesions - stroma is more mature.

FIBROUS DYSPLASIA Lichtenstein,1938
It is a disease of bone maturation andremodeling in which the normal medullarybone and cortices are replaced by adisorganized fibrous woven bone.
The resultant fibro-osseous bone is moreelastic and structurally weaker than theoriginal bone.
Caused by postzygotic mutation in theGNAS1 gene.
Mutations of the GSa gene
No hereditary influence.

Investigation of the GSa gene in
the diagnosis of fibrous
dysplasia Done by direct sequencing of the Gsa
gene.
High prevalence of GSa gene
mutations in fibrous dysplasia.
Int. J. Oral Maxillofac. Surg. 2004; 33: 498–501
Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2011;111:618-
626

Types of Fibrous DysplasiaForms of presentation of fibrous dysplasia
Bone Involvement
Single Multiple Cafἐ au
Lait Spots
Endocrine
disorders
Soft-
tissues
masses
Monostotic X
Polyostotic X
McCune
Albright
Syndrome
X X X
Mazabrau
d disease
X X
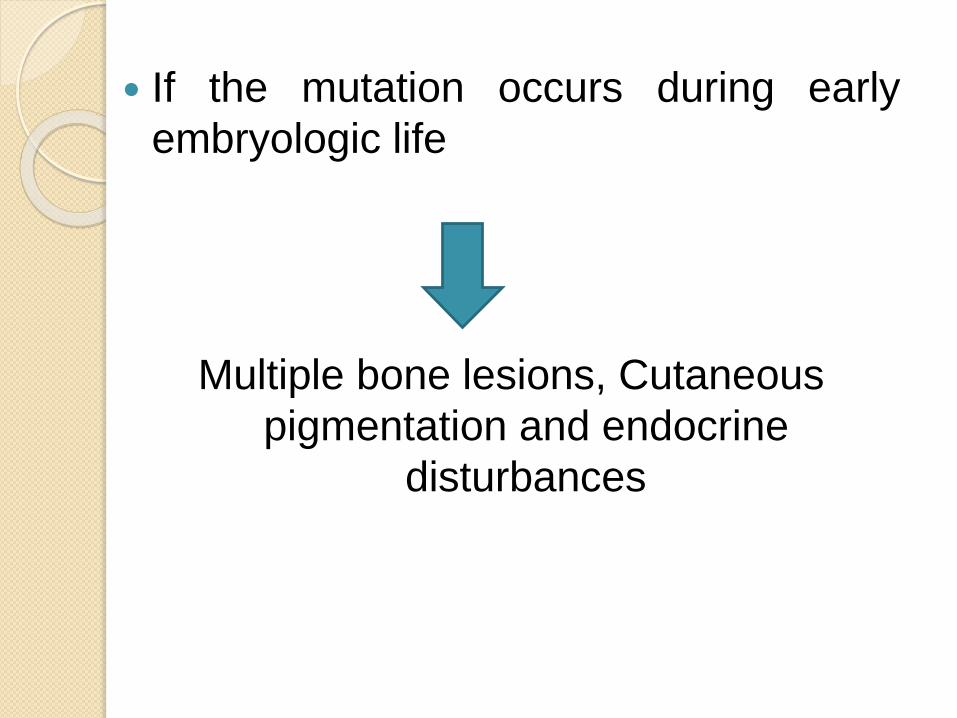
If the mutation occurs during early
embryologic life
Multiple bone lesions, Cutaneous
pigmentation and endocrine
disturbances
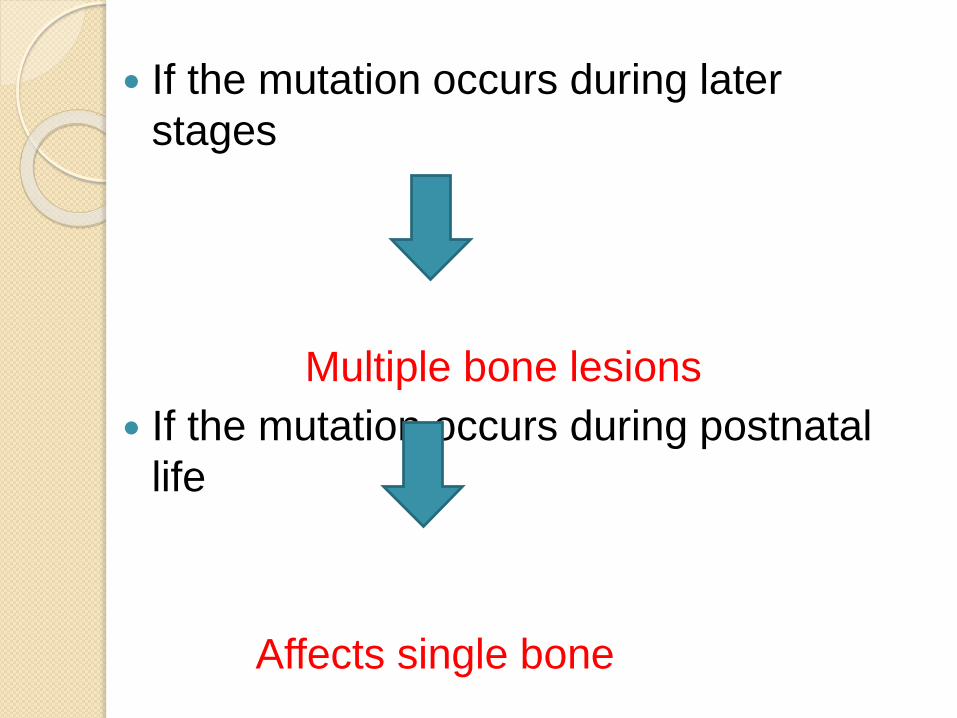
If the mutation occurs during later
stages
Multiple bone lesions
If the mutation occurs during postnatal
life
Affects single bone

Monostotic Fibrous Dysplasia
Limited to a single bone. Accounts for 80% to 85% of all cases M:F = 1:1 Painless swelling. Growth is generally slow but occasionally
rapid. Maxilla > Mandible. 60% of cases causes displacement of
mandibular canal. Often detected during the first two
decades of life.
Fibro-osseous Iesions of the jaws. J Oral MaxilIofac Surg 43:249, 1985

Mandibular lesions are truly
monostotic.
Maxillary lesions often involve
adjacent bones(e.g., zygoma,
sphenoid, occipital)
Craniofacial Fibrous Dysplasia.

Polyostotic Fibrous dysplasia;
Jaffe-Lichtenstein Syndrome;
McCune – Albright Syndrome
Involvement of two or more bones.
When seen with cafἐ au lait
pigmentation Jaffe- Lichtenstein
syndrome.
Polyostotic fibrous dysplasia + cafἐ au
lait pigmentation + multiple
endocrinopathies McCune Albright
Syndrome.

Causes facial asymmetry.
Pathologic fractures with pain and
deformity.
Leg length discrepency is common
due to involvement of upper portion of
the femur ( Hockey Stick deformity).

Radiographic Features
Depends upon the stage of the
disease.
Early onset lesions are radiolucent
and later progressively calcify,
culminating in a “Ground Glass” or
Mottled Mixed radiolucent/ radiopaque
pattern.
Critical feature to diagnosis- FD fails
to manifest any discreate margins;
rather, the lesional bone subtly blends
into the surrounding normal appearing
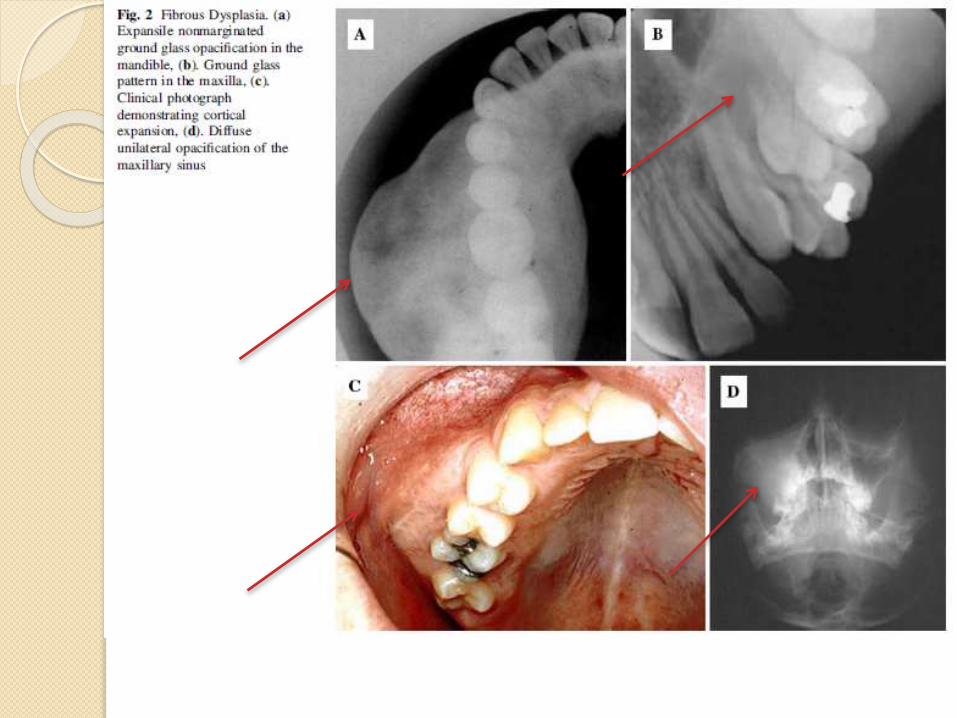
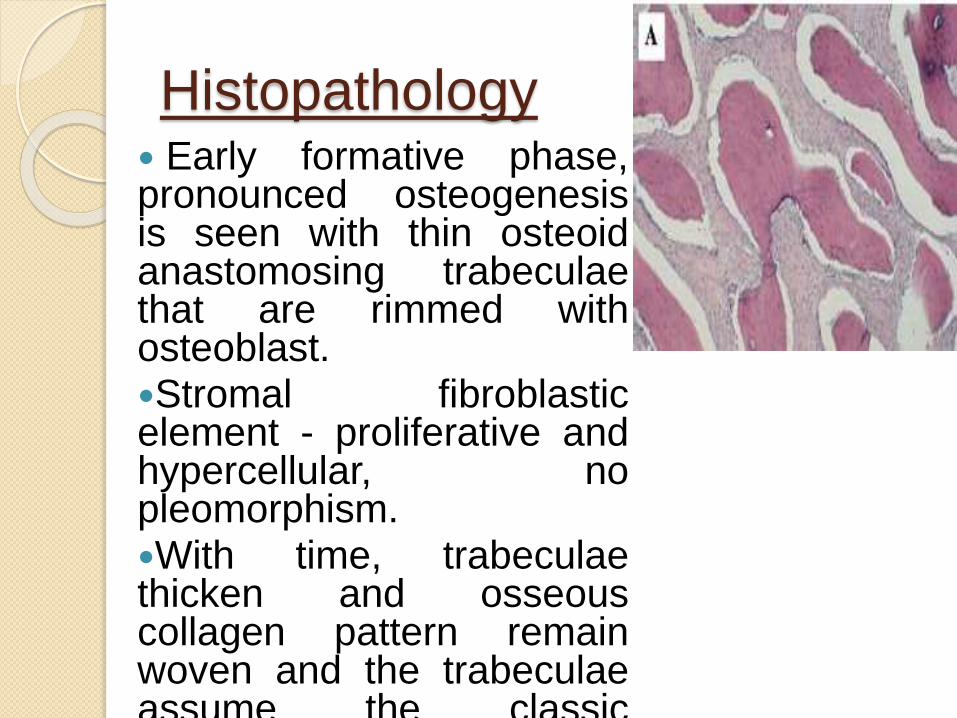
Histopathology Early formative phase,pronounced osteogenesisis seen with thin osteoidanastomosing trabeculaethat are rimmed withosteoblast.Stromal fibroblasticelement - proliferative andhypercellular, nopleomorphism.With time, trabeculaethicken and osseouscollagen pattern remainwoven and the trabeculaeassume the classic“Chinese figure” pattern.

Long-standing polyostotic FD is
sarcomatous , which can occur in
absence of radiation therapy.
Most frequent site - craniofacial
skeleton.
Differentiating features on radiograph
between sarcoma and FD are:
◦ Permeative ill-defined borders,
◦ Destroyed cortical outline and/or spiculated
periosteal new bone formation
◦ Periodontal ligament space widening.

Malignant Transformation of
FD: Occur in <1% of the cases.
Osteosarcoma is the most common
histologic type, followed by
fibrosarcoma, chondrosarcoma, and
malignant fibrohistiocytoma.
Most common in the maxilla and
mandible
Calvarium – rare involvement.
Spontaneous Conversion of Fibrous Dysplasia
Into Osteosarcoma(J Craniofac Surg 2011;22: 959 -961)

Treatment and Prognosis
Timing of intervention is based on the
symptoms manifesting as a result of the
disease.
Recommended treatment options can be
divided into 4 categories:
1. Observation
2. Medical therapy
3. Surgical remodelling
4. Radical excision and reconstruction

1. Observation:
Monitoring is through serial radiographs,CT scans, and clinical examinations. Inlesions that present in childhood,monitoring until skeletal maturity mayallow for ultimately less radical treatmentand less overall morbidity.
Special attention to cranial nervefunction during monitoring of theselesions should be exercised
Decreased nerve function may be anindication for surgical therapy.

2. Medical treatment
Currently, no medical therapy exists for the
permanent cure of fibrous dysplasia.
1. Biphosphonates.
2. Systemic steroids
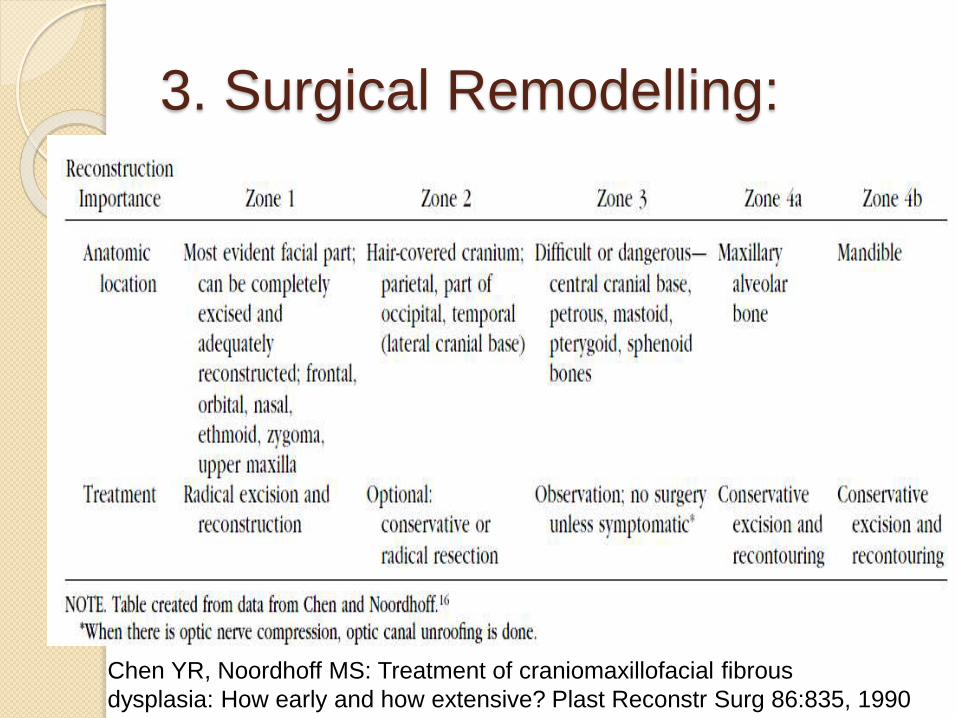
3. Surgical Remodelling:
Chen YR, Noordhoff MS: Treatment of craniomaxillofacial fibrous
dysplasia: How early and how extensive? Plast Reconstr Surg 86:835, 1990

4. Surgical Treatment
Indications:Functional concernFacial recontouringCompression of the optic canal, recommended
decompression
Other indications for surgical therapy:Cosmetic DeformityPainPathologic FractureHearing LossSinus Or Nasal ObstructionEpistaxisMalocclusion and Impeded mastication.

Osteitis Deformans( Paget
Disease) Rapid turnover remodeling of bone
throughout the skeleton.
Disease of the elderly.
Although two Paget-like bone
dysplasias that arise during childhood.

Etiology
Defective function of the
osteoprotegerin/TNFRSF11A or
B/RANKL/RANK pathway, a molecular
regulator of osteoclastogenesis.

Classic Paget Disease of Bone
( CPDB) Late adult onset.
Characterized by rapid turnover of boneremodeling and osseous expansion withprogressive skeleton deformities.
Tubular bones show bowing and spinalcurvature with vertebral collapse.
Elevated serum alkaline phosphatase,
Normal calcium and phosphorus levels.
Cranial nerve neuropathies can developas a consequence of foraminanarrowing.
Deafness

Radiographic Features
In early stages, radiolucent “ Coin
shaped” lesions appear in the flat
bones of the skull, a condition called
as “ Osteitis Circumscripta”
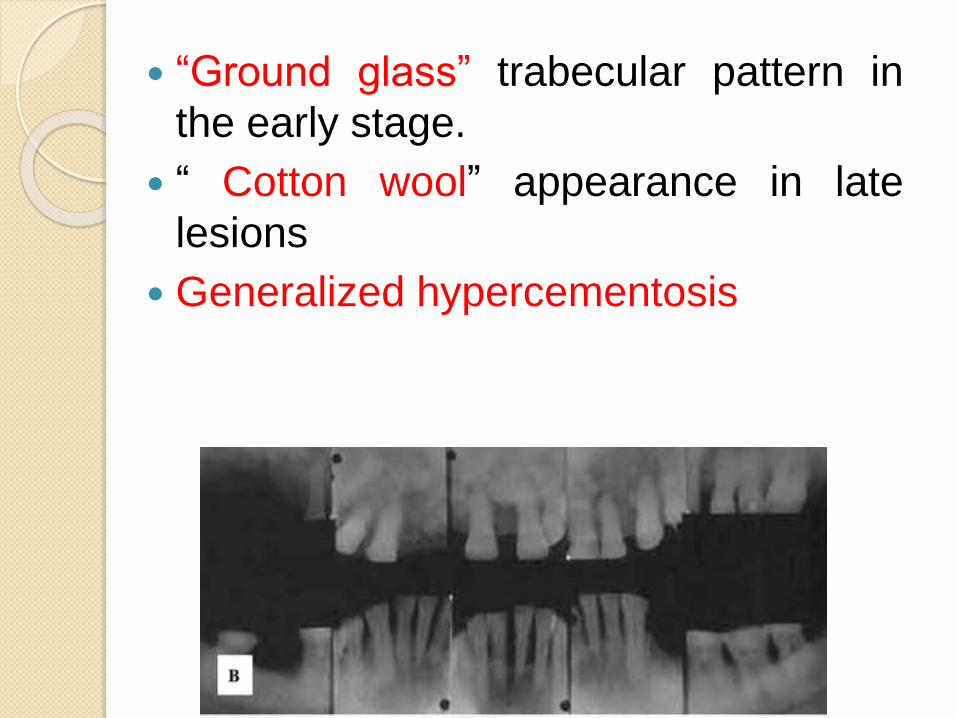
“Ground glass” trabecular pattern in
the early stage.
“ Cotton wool” appearance in late
lesions
Generalized hypercementosis
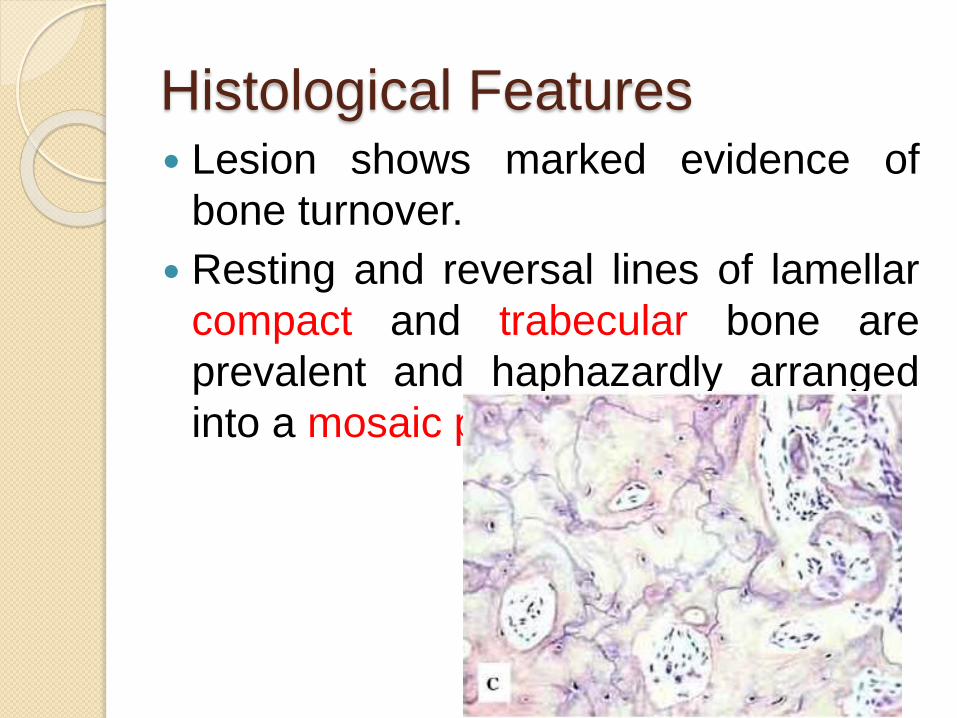
Histological Features Lesion shows marked evidence of
bone turnover.
Resting and reversal lines of lamellar
compact and trabecular bone are
prevalent and haphazardly arranged
into a mosaic pattern.

Neoplastic Transformation
Two neoplastic processes may occur
1. Giant cell tumors
2. Sarcomas( Osteogenic sarcoma,
fibrosarcoma, chondrosarcoma and
undifferentiated sarcoma)

Treatment
Biphosphonates
Histological appearance also appear
more normal after drug therapy.

Juvenile Paget Disease
(Idiopathic Hyperphosphatasia) Inherited as an autosomal recessive
trait
Deformities in the long bones,
kyphosis, acetabular protrusion and
pathophysiologically by rapid turnover.
Long bone widening with pathologic
fracture and thickening of skull.
Elevated serum alkaline phosphatase
Osteopenia and skeletal deformity
with bowed limbs

Familial Expansile Osteolysis(
FEO) Autosomal dominant trait
Manifest in the second decade
Osteoclastic resorption with cancellousbone expansion and elevated serumalkaline phosphatase.
Early tooth loss by external resorption
Deafness
Histologically - focal collection ofmultinucleated giant cells and viral-likeinclusions.

Expansile Skeletal
Hyperphosphatasia(ESH) Autosomal dominant trait
Accelerated bone turnover withhyperostotic expansion of the long bones
Pain in phalanges
Premature tooth exfoliation and deafness
Episodic hypercalcemia
Absence of large osteolytic lesions withcortical thinning.
Elevated alkaline phosphatase.
No viral like inclusions.

Segmental Odontomaxillary
Dysplasia Lesion confined to a single segment of
the maxilla, usually in premolar and
molar.
Clinical Features:
◦ Teeth fail to erupt
◦ Dental anomalies like- malformed, mis
shapened and/ or teeth of anomalous
size.
Expansion of alveolar bone

Radiographic Features
Falling Sheet pattern
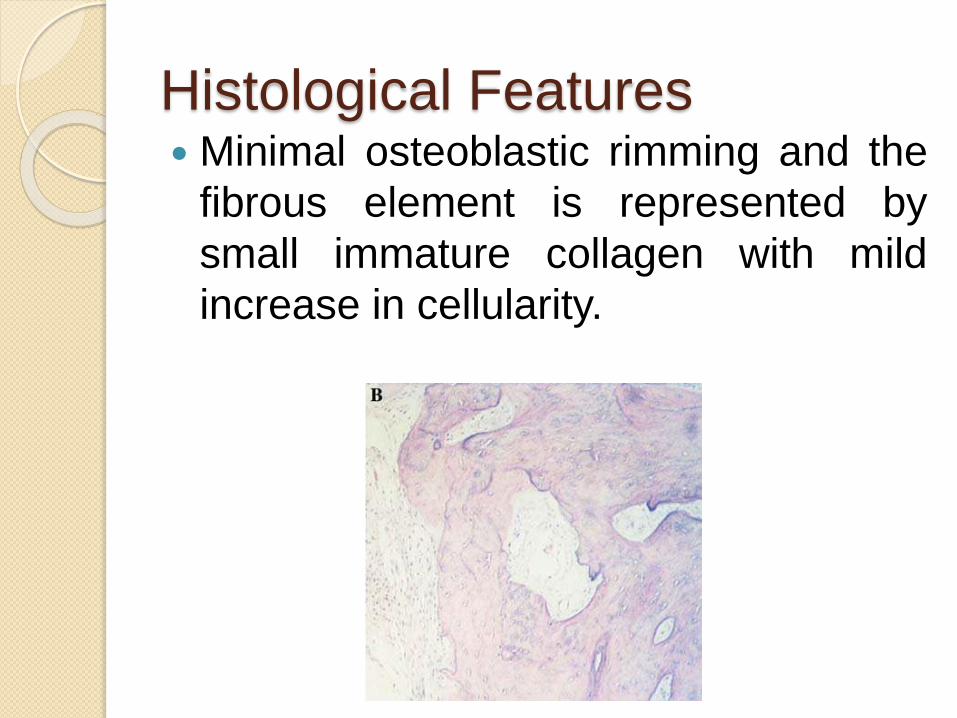
Histological Features Minimal osteoblastic rimming and the
fibrous element is represented by
small immature collagen with mild
increase in cellularity.

Cemento-Osseus Dysplasia
These conditions are defined by
specific clinical and pathological
features and have been classified as
Cemento-Osseus Dysplasia
Periapical Cemental Dysplasia
Focal Cemento-Osseus Dysplasia
Florid Osseous Dysplasia

Precise etiology is unknown.
Disorders of metabolism of cells
normally involved in production of
bone and cementum matrices.
The aberrant activity of the tissues
may be the result of an unusual
response to undefined local factors.

Periapical Cemental Dysplasia Seen at the apices of the teeth with vital , non-
inflamed pulps.
Predominantly involves mandibular incisors.
Usually detected incidentally on routine
radiographic examination.
Radiographic Examination
Consist of multiple round to ovoid , radiolucent
lesions at the apex of the vital teeth.
May mimic periapical pathology of pulpal origin.
Individual lesions are seldom more than 1.0 cm in
diameter and most are less than 0.5 cm in size.
The radiolucences maybe discrete with well
defined borders or they maybe large enough to
appear confluent as they overlap and merge.

Histopathology
On Biopsy, they consist of multiplefragments of moderately cellular,collagenous tissue.
Amount and degree of mineralizationcomponent are variable, dependent on thelength of time the lesions present andtherefore the stage of the process.
The calcified tissue is associated withosteoblasts and the cementoblasts alongthe surface and is deposited in a variety ofconfigurations.
Inflammatory cells are present

Focal Cemento-Osseus
Dysplasia
Clinical Features :
Female predominance.
4th -5th decades of life.
Solitary lesions , in posterior mandible.

Radiographic Features Diagnosed in routine radiographs,
characteristically, asymptomatic.
Most lesions appear as radiolucent – radio-opaque areas.
In edentulous areas development ofidiopathic bone cavities, result in bonyexpansion of affected area.

Histopathology
Consistency of tissueremoved for biopsies isimportant .
Difficult to curette formthe sockets, is removedas multiple fragments ofgritty tissues
Distinction betweenossifying fibroma, whichtypically can beremoved as largefragmented that can beseparated cavity formbone.
“Ginger root” Pattern

Treatment
Lesions exhibit only limited potential
for progressive growth, more lesion
require no additional treatment
following biopsy.
Periodic observation

Florid Cemento-Osseous
Dysplasia Middle-aged females
Painless non-expansile lesion often
involving two or more jaw quadrants.
Radiographically - multiple confluent
lobular radiopaque masses in tooth-
bearing areas
Tendency toward bilateral,
symmetrical involvement
Asymptomatic and detected incidentally

Jaw expansion - large lesions.
Dull pain or drainage are always
associated with exposure of the
sclerotic calcified masses to the oral
cavity as the result of progressive
alveolar atrophy under a denture or
after extraction of teeth in the involved
area.


Treatment
Often difficult, not very satisfactory.
In the asymptomatic patient
observation
Antibiotics
Sequestration of the cementum-like
masses will occur slowly and followed
by healing.
Saucerization or surgical excision
Inflammatory/ Reactive
Processes This include:
◦ Focal Sclerosing Osteomyelitis(
Condensing Osteitis)
◦ Diffuse Sclerosing Osteomyelitis
◦ Proliferative Periostitis

Focal Sclerosing Osteomyelitis
( Condensing Osteitis) Mildest and most self-limiting form
Posterior mandible at apices of molar
teeth
Bacterial origin
Pathogenic bacteria are of low
virulence
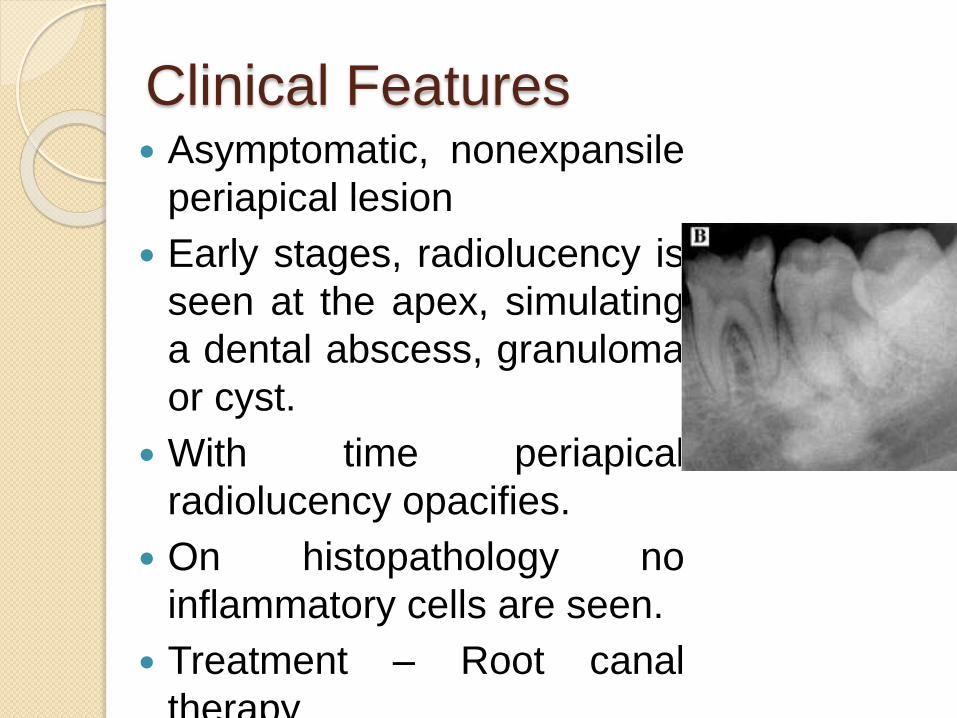
Clinical Features Asymptomatic, nonexpansile
periapical lesion
Early stages, radiolucency is
seen at the apex, simulating
a dental abscess, granuloma
or cyst.
With time periapical
radiolucency opacifies.
On histopathology no
inflammatory cells are seen.
Treatment – Root canal
therapy
Differential Diagnosis
Focal osteosclerosis
Focal cemento osseous dysplasia

Diffuse Sclerosing
Osteomyelitis Unilateral diffuse ground glass
opacification without defined boundariesof the mandibular body.
Cortical expansion H/O dull episodic pain that last for weeks
to subside and later becomesymptomatic again.
Caused by gram negative anaerobicbacteria of low virulence.
Definitive anaerobic culture Source of infection - Odontogenic


Microscopically, bone exhibits a fibro
osseous pattern with intervening foci
of dense sclerotic bone.
Osseous elements are trabecular
Cementifying areas - absent.

Differential diagnosis
Lesion Clinical Features Histological
Features
Fibrous Dysplasia Painless lesion Chinese figure
pattern
Florid cememto
osseous dysplasia
Multiquadrant
opaque lesions with
associated
radiolucencies
Show both
trabecular and
cementifying areas
along with hollow
bone cavities
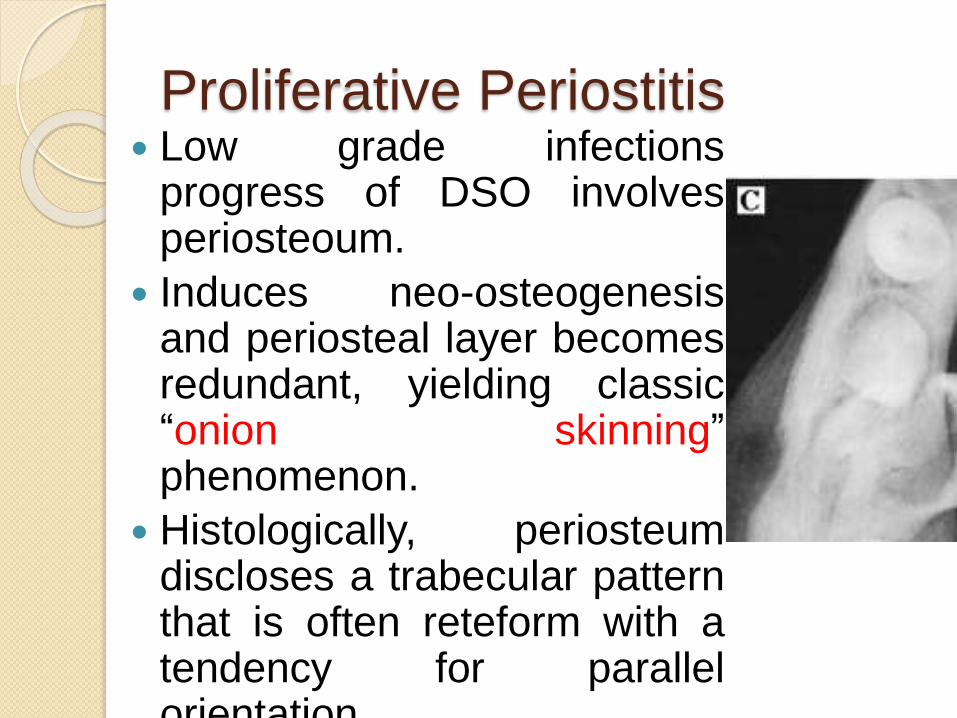
Proliferative Periostitis Low grade infections
progress of DSO involvesperiosteoum.
Induces neo-osteogenesisand periosteal layer becomesredundant, yielding classic“onion skinning”phenomenon.
Histologically, periosteumdiscloses a trabecular patternthat is often reteform with atendency for parallelorientation

Treatment
Removing odontogenic infection
source by extraction or RCT.
Long term antibiotic therapy.

Hyperparathyroidism
The “brown tumor” of
hyperparathyroidism, a giant cell
lesion, may be encountered anywhere
in the skeleton in both primary and
secondary HPT.
Reported among patients with renal
osteodystrophy.

Clinical Features
Marked facial deformity
Thickening of dipole and massive
maxillomandibular enlargement with
protrusion of anterior teeth and wide
diastemas.
Also known as – Sagliker Syndrome
and the Uglifying Human Face
Syndrome.
Histologically, trabecular pattern is
seen

Enlarged regions of facial bones, jaws
and skull manifest a dense ground
glass opacification.
Parathyroidectomy does not result in
resolution of the bony enlargements.

Ossifying Fibromas
Neoplasms with a fibro-osseous
histology represented by the ossifying
fibroma group of lesions.
Neoplasms in the true sense
exhibiting progressive proliferative
capabilities with bony expansion and
well defined margins radiologically.

Types
Ossifying/ Cementifying Fibroma
Juvenile Ossifying Fibroma
Trabecular Juvenile Ossifying Fibroma
Psammomatoid Juvenile Ossifying
Fibroma
Gigantiform Cementoma

Ossifying/Cementifying
Fibroma Most common form of OF occurs in
maxilla and mandible.
Mutation in HRPT2 gene that encodesparafibromin protein.
Painless with expansion of both cortices.
Larger lesions may expand the inferioraspect of mandible.
Teeth are displaced superiorly (mandibular lesion) and inferiorly(maxillary lesion) and expand into theantrum.

Radiographic Features
Early lesions appears radiolucent
With time it appears radiopaque, as
more matrix calcifies.

Histological Features
Shows three
histological forms
◦ Ossifying
◦ Cementifying
◦ Storiform

Juvenile Ossifying Fibroma
Also known as Juvenile Active
Ossifying Fibroma and Juvenile
Aggressive Ossifying Fibroma.
2 clinicopathologic entities
1. Trabecular Juvenile Ossifying Fibroma
( TrJOF)
2. Psammomatoid Juvenile Ossifying
Fibroma (PsJOF)

Trabecular Juvenile Ossifying
Fibroma(TrJOF) Also known as trabecular desmo-
osteoblastoma.
Majority of patients are children and
adolescents.
Only 20% are over 15 years of age.
M:F = 1:1
Maxilla and mandible are dominant
sites.
Maxilla is slightly more affected.

Clinical Features
Progressive and sometimes rapid
expansion of bone
In maxilla, obstruction of nasal
passages and epistaxis may be
present.
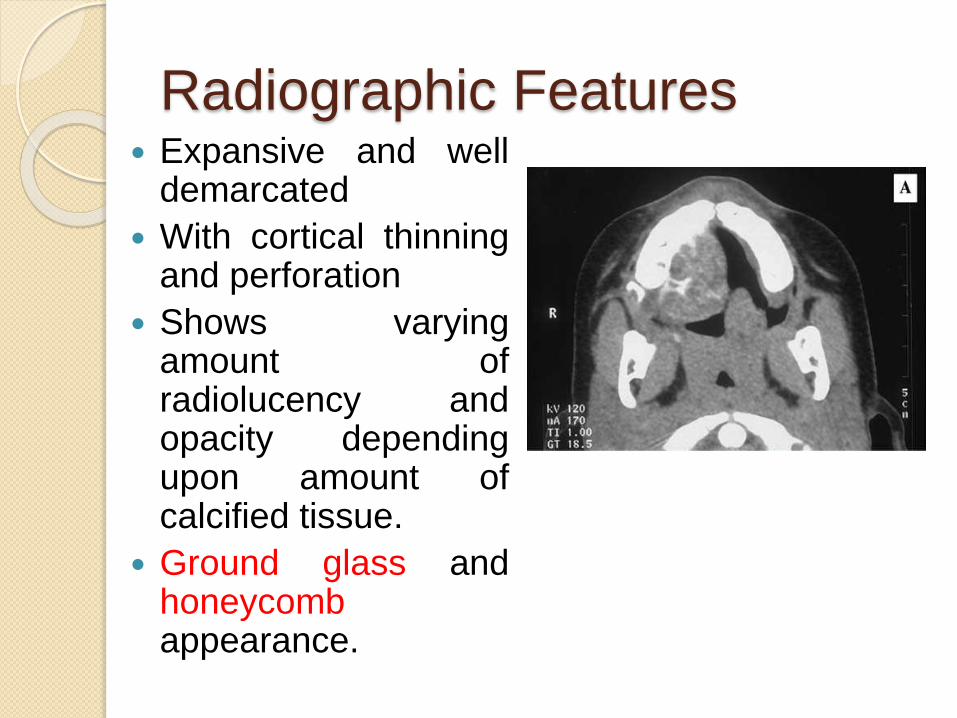
Radiographic Features Expansive and well
demarcated
With cortical thinningand perforation
Shows varyingamount ofradiolucency andopacity dependingupon amount ofcalcified tissue.
Ground glass andhoneycombappearance.

Histological Features Uncapsulated and shows
infiltration of surroundingbone
Characteristic loosestructure
Stroma is cell rich, withspindle or polyhedralcells, produce littlecollagen
Cellular, immatureosteoid forms strands.
Irregular mineralizationtakes place at the centreof the strands
Local aggregates ofosetoclastic giant cellsare invariably present inthe stroma.

Clinical course is characterized by
infrequent recurrence following
conservative excision.
Complete cure could be achieved in
those cases without resorting to
radical surgical intervention.
Malignant transformation not reported

Psammomatoid Juvenile
Ossifying Fibroma Affects extragnathic craniofacial
bones, particularly periorbital, frontal
and ethmoid bones.
Gogl,1949, as psammomatoid fibroma
of the nose and paranasal sinuses.
Margo,1985 described as a distinctive
solitary fibro-osseous lesion that
affects the orbit and shows distinctive
histologic features.

16 to 33 years.
Range of 3 months to 72 years.
Majority originates in paranasal
sinuses ( frontal and ethmoid).
10 % in calvarium
7% in mandible( Makek in 1983)

Clinical Features
Bone expansion involves orbital or
nasal bones and sinuses.
Orbital extension- proptosis and visual
complaints including blindness, nasal
obstruction, ptosis, papilledema and
disturbances in ocular mobility.

Radiographic Features Round, well defined,
corticated osteolyticlesion with a cysticappearance.
In CT scans, appear lessdense than normal bone,appear multiloculated
Size range from 2 to 8cm
In facial skeleton a wellcircumscribed expansivemass with a thick wall ofbone density on CT scanis strongly suggestive ofpsammomatoid juvenileossifying fibroma.
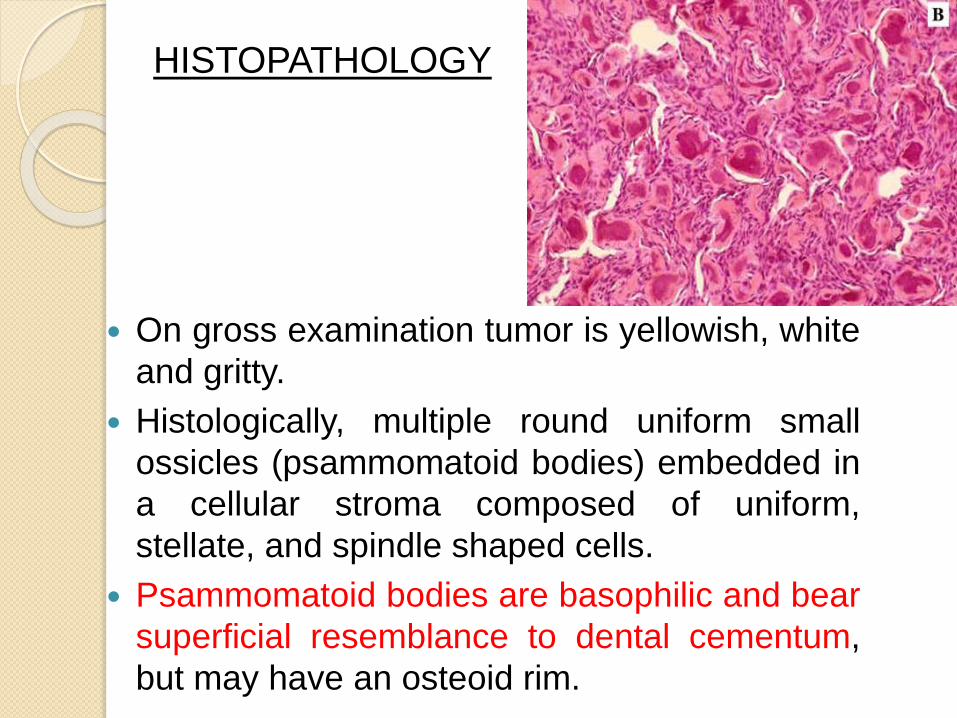
On gross examination tumor is yellowish, white
and gritty.
Histologically, multiple round uniform small
ossicles (psammomatoid bodies) embedded in
a cellular stroma composed of uniform,
stellate, and spindle shaped cells.
Psammomatoid bodies are basophilic and bear
superficial resemblance to dental cementum,
but may have an osteoid rim.
HISTOPATHOLOGY

Surgical excision is treatment of
choice.
Recurrence rate of >30% reported.
No malignant change observed.

Familial Gigantiform
Cementoma An autosomal dominant variant
usually involving multiple quadrants
with variably expansile lesions.
Anterior mandible.
No racial predilection.
Often evolve during childhood and
can grow rapidly.

Radiographically,expansion with aradiolucent masscontainingfloccularcalcifications.
Microscopically,benignhypercellularstroma withmonomorphicappearingfibroblasts andmature collagenfibers.
Ovoid, laminated,psammomatoidcalcifications arepresent.

Treatment is resection with immediate
or staged reconstruction.






























