Embed Size (px)
Citation preview

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency (PID)?Eczema or Secondary Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal abnormalities “Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions

The term masquerade syndrome encompasses a group of malignant diseases that mimic clinical presentations of ocular inflammation.
The following differential diagnoses ought to be considered:
❃ Intraocular:
❃ In the area of the conjunctiva and eyelids:
Masquerade syndromes’ definiton
lymphoma, choroid melanoma, intraocular metastases, retinoblastoma, para-neoplastic retinopathy
melanoma of the conjunctiva, squamous cell carcinoma, conjunctival lymphoma, basal cell carcinoma, sebaceous gland carcinoma of the eyelid,chalazion.

The term masquerade syndrome encompasses a group of malignant diseases that mimic clinical presentations of ocular inflammation.
The following differential diagnoses ought to be considered:
❃ Intraocular:
❃ In the area of the conjunctiva and eyelids:
Masquerade syndromes’ definiton
lymphoma, choroid melanoma, intraocular metastases, retinoblastoma, para-neoplastic retinopathy
melanoma of the conjunctiva, squamous cell carcinoma, conjunctival lymphoma, basal cell carcinoma, sebaceous gland carcinoma of the eyelid,chalazion.
They are usually poorly, if not at all, responsive to corticosteroid.
One must be suspicious when the apparent intraocular inflammation :
is unilateral
occurs either in very young children or in the elderly

The term masquerade syndrome encompasses a group of malignant diseases that mimic clinical presentations of ocular inflammation.
The following differential diagnoses ought to be considered:
❃ Intraocular:
❃ In the area of the conjunctiva and eyelids:
Masquerade syndromes’ definiton
lymphoma, choroid melanoma, intraocular metastases, retinoblastoma, para-neoplastic retinopathy
melanoma of the conjunctiva, squamous cell carcinoma, conjunctival lymphoma, basal cell carcinoma, sebaceous gland carcinoma of the eyelid,chalazion.
………………….and masquerading as allergic diseases?

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency
(PID)?Eczema or Secondary Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal abnormalities “Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions

Primary Immunodeficiency Masquerading as Allergic Disease
Primary immune deficiencies (PIDs)
have an incidence and prevalence estimated between:
1:10,000 to 1:2000
from registries in > 40 countries.
This is likely an underestimation as many cases remain undiagnosed; a random telephone survey in the United States estimated the prevalence of all PIDs at 1:2000 to 1:800.Boyle J, J Clin Immunol 2007;27(5):497–502.
The hallmark clinical pattern is an increased susceptibility to infection

Jeffrey Modell Foundation (JMF): 10 warning signs of primary immunodeficiency
(PI)
Hernandez-Trujillo VP. Immunol Allergy Clin N Am 2015;35:625–636
Pediatrics
1. ≥ 4 new ear infections within 1 year2. ≥ 2 serious sinus infections within 1 year3. ≥ 2 months on antibiotics with little effect4. ≥ 2 pneumonias within 1 year5. Failure of an infant to gain weight or grow normally6. Recurrent deep skin or organ abscesses7. Persistent thrush in mouth or fungal infection on skin8. Need for intravenous antibiotics to clear infections9. ≥ 2 deep-seated infections including septicemia10. A family history of PI
≥ 2 months
≥ 2/yr
≥ 2/yr≥ 4/yr
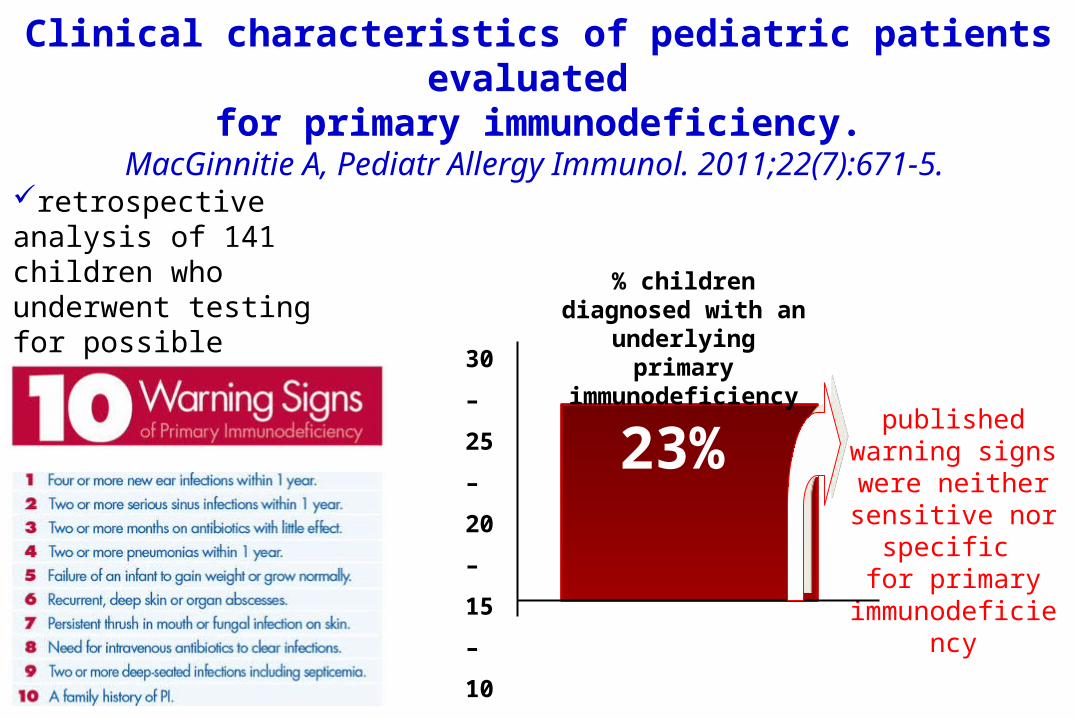
Clinical characteristics of pediatric patients evaluated
for primary immunodeficiency.MacGinnitie A, Pediatr Allergy Immunol. 2011;22(7):671-5.
retrospective analysis of 141 children who underwent testing for possible primary immunodeficiency 30
–25 –20 –15 –10 –05 –00
% children diagnosed with an
underlying primary
immunodeficiency
23% published warning signs were neither sensitive nor
specific for primary
immunodeficiency
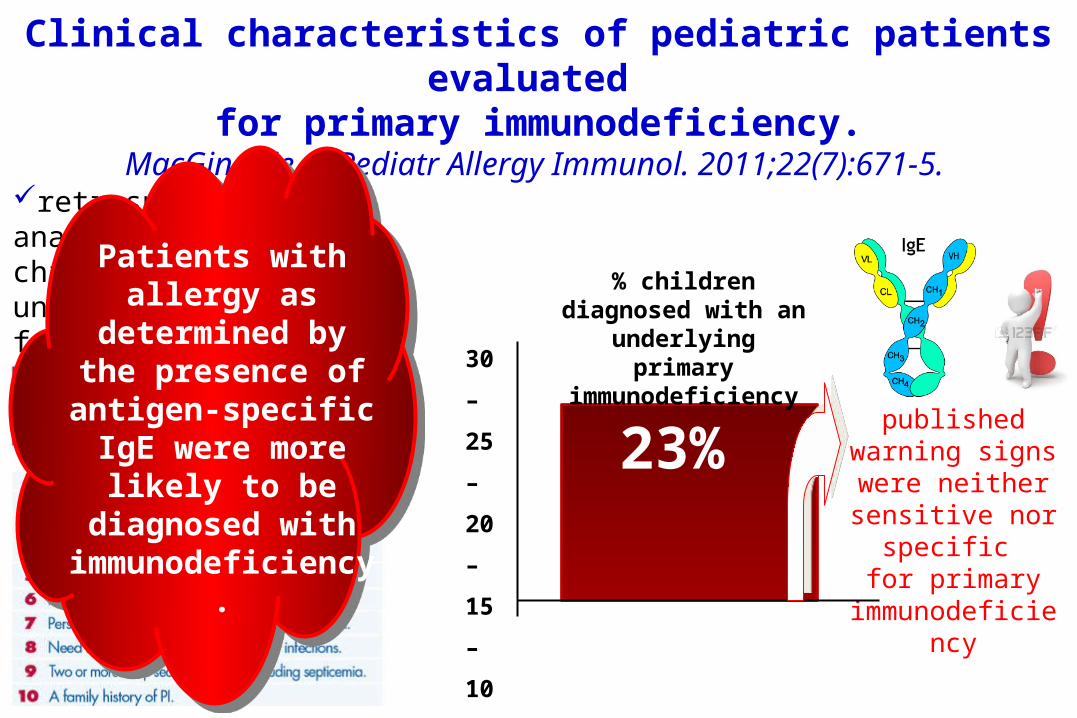
Clinical characteristics of pediatric patients evaluated
for primary immunodeficiency.MacGinnitie A, Pediatr Allergy Immunol. 2011;22(7):671-5.
retrospective analysis of 141 children who underwent testing for possible primary immunodeficiency 30
–25 –20 –15 –10 –05 –00
% children diagnosed with an
underlying primary
immunodeficiency
23% published warning signs were neither sensitive nor
specific for primary
immunodeficiency
Patients with allergy as
determined by the presence of antigen-specific IgE were more
likely to be diagnosed with
immunodeficiency.
Features of primary immunodeficiencies
Raje N, Immunol Allergy Clin N Am 2015;35:599–623

Common allergic symptoms can also be manifestations
of an underlying and severe Immune Deficiencythe allergic triad
in immunodeficienciesAllergic diseases share common factors with primary immunodeficiencies including:
1) skin rashes such as eczema,
2) increased IgE,
3) eosinophilia.

Primary immune deficiencies with aberrant IgE production Ozcan E, J Allergy Clin Immunol
2008;122(6):1054–62
Omenn syndrome; Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX); Wiskott-Aldrich syndrome; Hyper-IgE syndrome; Atypical complete DiGeorge syndrome.
PID with elevated serum IgE
levels
Increased IgE levels in IPEX, Wiskott-Aldrich syndrome and Omenn syndrome are likely related to increased T(H)2 cytokine production caused by decreased a number or function of CD4(+)CD25(+)forkhead box protein P3(+) regulatory T cells.
The link between signal transducer and activator of transcription 3 mutations and elevated serum IgE levels in hyper-IgE syndrome is unclear.
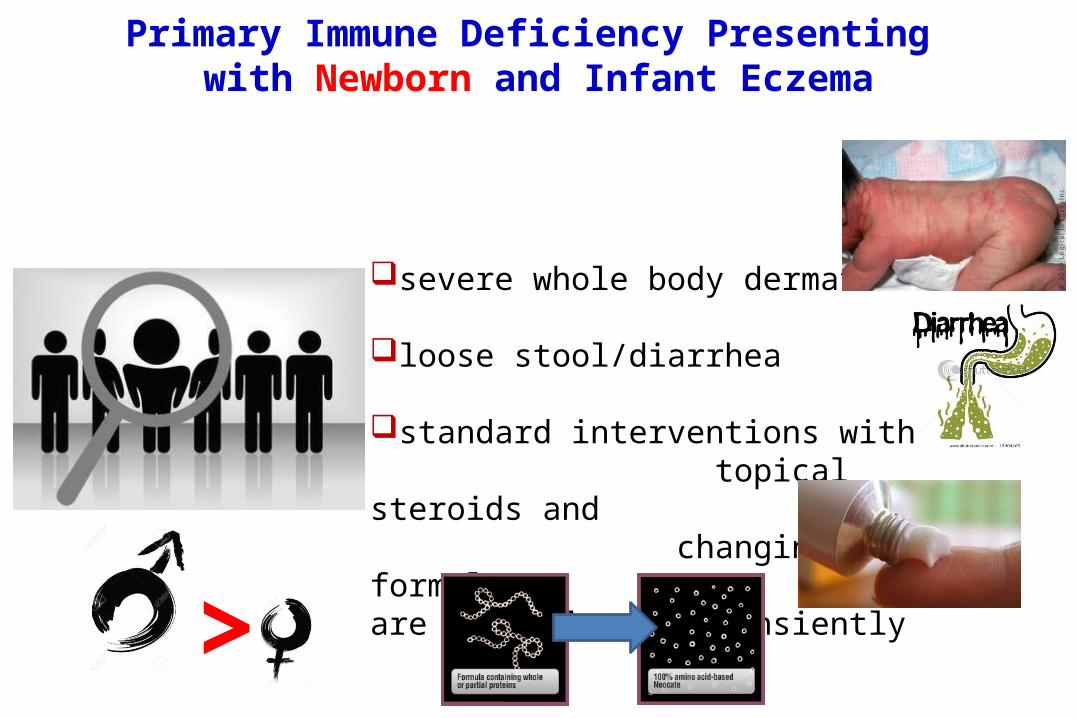
Primary Immune Deficiency Presenting with Newborn and Infant Eczema
severe whole body dermatitis
loose stool/diarrhea
standard interventions with topical steroids and changing formulas are helpful only transiently
>

Originally described in 1965 with reticuloendotheliosis and eosinophilia. Omenn G. N Engl J Med 1965;273:427–32.
The prevalence is estimated at less than 1/1,000,000.
It is linked to Severe Combined Immune Deficiency (SCID) and presents in infancy.
Omenn Syndrome
erythroderma, desquamatous alopecia, chronic diarrhea, failure to thrive, lymphedema, hepatomegaly.
Omenn syndrome is an
inflammatory condition
characterized by: Aleman K, Eur J Pediatr 2001;160(12):718–25

Reviewing Omenn syndrome. Aleman K, Eur J Pediatr 2001;160(12):718–25.
literature search in the period 1965-1999
68 patients
100 –090 –080 –070 –060 –050 –040 –030 –020 –010 –000 erythematous
rash
98%
HepatoSplenomegaly
88%
lymphadenopathy
80%
alopecia
57%
% infants with

Reviewing Omenn syndrome. Aleman K, Eur J Pediatr 2001;160(12):718–25.
literature search in the period 1965-1999
68 patients
100 –090 –080 –070 –060 –050 –040 –030 –020 –010 –000 erythematous
rash
98%
HepatoSplenomegaly
88%
lymphadenopathy
80%
alopecia
57%
% infants with
recurrent infections in 72%

It is an inflammatory phenotype of SCID that has been linked to mutations in the recombination-activating gene 1 (RAG1) or RAG2, RNA component of mitochondrial RNA processing endoribonuclease, adenosine deaminase, interleukin (IL)-2R, IL-7R, artemis-nuclease-DCLRE1C DNA ligase 4, and 22q11 microdeletion.
It usually presents in an autosomal-recessive pattern.
This is a “leaky” T/B-SCID phenotype where some T or B cells are present and suggestive of mutations that lead to impairment of theV(D)J DNA recombination involved in generating immunoglobulins as well as T-cell receptors.
Omen Syndrome Pathogenesis
Chan SK, Immunol Allergy Clin North Am. 2015 Nov;35(4):767-78
XX

In contrast to classic SCID, patients have low, normal or even markedly increased circulating activated T-lymphocyte levels; however, they are oligoclonal and associated with poor antigenic responses.
Typically, B cells are absent with loss of IgG, IgA, IgM, but increased serum IgE.
Peripheral blood eosinophilia resulting from Th2 skewing.
Skin biopsy of lesions show acanthosis (thickening of the skin) and parakeratosis (retention of nuclei in the stratum corneum).
Omen Syndrome Diagnosis
Chan SK, Immunol Allergy Clin North Am. 2015 Nov;35(4):767-78

Immunosuppressive treatment for their marked lymphoproliferation and inflammation targeting the janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway has been used to control inflammation. Borie DC, Curr Opin Investig Drugs. 2003;4(11):1297-303
Suppression can result in almost complete resolution of the dermatitis, alopecia, hepatosplenomegaly, and lymphadenopathy with significant weight gain. Ege M, Blood 2005;105(11):4179–86.
Care should be taken to treat underlying infections and screening for opportunistic infections should be initiated before immune suppression, given the underlying SCID phenotype.
To date, curative treatment and ultimate resolution requires Hematopoietic stem cell transplantation HSCT. Gomez L. J Pediatr 1995;127(1):76–81.
Omen Syndrome Management

An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy.
Powell BR, J Pediatr 1982;100(5):731–7.
Features of 8 patients from a family in which 17 male infants died in the first years of life.
The features included:•diarrhea, •diabetes mellitus, •hemolytic anemia, •eczematoid rashes, •exaggerated responses to viral illnesses, •pathologic evidence of autodestruction of endocrine glands, insulitis, and thyroiditis with thyroid autoantibodies.
(IPEX)
X-linked recessive disease
Immune dysregulation,
Polyendocrinopathy,
Enteropathy, X-linked (IPEX)
syndrome.
X-linked Immunodysregulation, Polyendocrinopathy,
and Enteropathy (IPEX)
Most commonly, these patients present with: early onset severe and watery diarrhea,type 1 diabetes mellitus, and failure to thrive
Patients with (IPEX) frequently present
with:
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
eosinophilia
severe atopy. (+) the classic triad of: enteropathy, (severe darrhea) endocrinopathy, dermatitis.
•usually type 1 diabetes mellitus, •sometimes thyroiditis
Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78

X-linked Immunodysregulation, Polyendocrinopathy,
and Enteropathy (IPEX): PathogenesisIt is a X-linked recessive disease caused by a mutation in the FoxP3 gene on chromosome Xp11 resulting in absent or dysfunctional Tregs
This transcription factor plays an important role in the developmentand function of T regulatory cells that are actively involved in suppressing the immune system and in self-tolerance.Ricciardelli I, Immunology 2008;125(2):178–83.
These T regulatory cells (CD41251) control T- and B-cell inflammatory reactions.
FoxP3 binds to the FOX-binding site within the IL-2 promoter and suppresses its activity.

X-linked Immunodysregulation, Polyendocrinopathy,
and Enteropathy (IPEX): Diagnosis
Elevated IgE (possible sIgE to foods), IgA, and blood eosinophilia.
The diagnosis is often made after endoscopy.
Evaluation for IPEX should follow the diagnosis of male infant-onset insulin-dependent diabetes mellitus.
Enteric biopsy may reveal villous atrophy reminiscent of severe gastrointestinal graft versus host disease or similar to that described in celiac disease or autoimmune enteropathy (with the presence of anti-enterocyte, harmonin, and villin autoantibodies).
normal villous atrophy

X-linked Immunodysregulation, Polyendocrinopathy,
and Enteropathy (IPEX): Differential DiagnosisThe triad of endocrine dysfunction, enteropathy, and eczema in infant males is suggestive of IPEX, although other diseases should be considered, including:IPEX-like syndrome in CD25 mutations that affect both genders; STAT5b mutation; ITCH syndrome, Nedd4 family of HECT domain E3 ubiquitin ligase mutation;Schmidt syndrome/autoimmune polyendocrinopathy syndrome type 1; autoimmune polyendocrinopathy, candidiasis, ectodermal dystrophy; Wiskott–Aldrich; STAT1 (gain of function); X-linked autoimmune enteropathy; X-linked thrombocytopenia.
(the IL-2 receptor)
No mutations in FoxP3 gene

X-linked Immunodysregulation, Polyendocrinopathy,
and Enteropathy (IPEX): Management
high-dose corticosteroids, cyclosporine, tacrolimus,methotrexate, infliximab, rituximab, Sirolimus has been effective.
the mainstays of treatment
is supportive care
parenteral nutrition,
blood transfusions,
controlling diabetes
have been used:
Bindl L, J Pediatr 2005;147(2):256–9.
stop
arrests lymphocytes in the late G1 or S-phase leaving the regulatory T cells
relatively unphased and fully functional.

A challenging undertaking: Stem cell transplantation
for immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome Kucuk ZY, J Allergy Clin Immunol 2016;137:953-954
Our results suggest a higher risk of graft failure
compared with a previous report
or compared with other immune disorders:
6 of 7 patients experienceddecreasing engraftment
Retrospective analysis of 7 patients with IPEX syndrome who underwent hematopoietic stem cell transplantation (SCT)
Median age at diagnosis was 4.5 years
Median age at SCT was 6.7 years

Pedigree demonstrating a sex-linked recessive condition characterized by draining ears,
eczematoid dermatitis and bloody diarrhea. Aldrich R, Pediatrics 1954;13(2):133–9.
Wiskott–Aldrich Syndrome
†
††
††
†† †
†
††
† †
†
††
†
†X-linked recessive disease

Wiskott–Aldrich Syndrome
Wiskott–Aldrich syndrome
Infant males
severe diarrhea,
eczema, and
thrombocytopenia
Because of severe thrombocytopenia
bloody diarrheahemorrhagic manifestations
(small platelets and a reduced number of platelets)

Wiskott–Aldrich Syndrome
Wiskott–Aldrich syndrome
Infant males
severe diarrhea,
eczema, and
thrombocytopenia
Because of severe thrombocytopenia
bloody diarrheahemorrhagic manifestations
(small platelets and a reduced number of platelets)
skin, nose, intestine,brain. spontaneous or as the result of an injury
(approximately 36,000/mm3)

Wiskott–Aldrich Syndrome
Wiskott–Aldrich syndrome
Infant males
severe diarrhea,
eczema, and
thrombocytopenia
Because of severe thrombocytopenia
bloody diarrheahemorrhagic manifestations
(small platelets and a reduced number of platelets)
skin, nose, intestine,brain. spontaneous or as the result of an injury
(approximately 36,000/mm3)Symptoms of WAS vary significantly between patients and even between patients in the same
family. Some patients show symptoms soon
after birth whereas in others, symptoms are only seen in late infancy.

Wiskott–Aldrich Syndrome: Pathogenesis
Wiskott-Aldrich syndrome (WAS) protein (WASp) belongs to a family of proteins that relay signals from the surface of the cell to the actin cytoskeleton.
Mutations in the WAS gene have various effects on the level of WASp, which, in turn, correlates with the severity of the disease.
The absence of functional WASp leads to a severe clinical phenotype that can result in death if not diagnosed and treated early in life.
In addition to WAS, mutations in the WAS gene can result in the mild variant X-linked thrombocytopenia, or in X-linked neutropenia, characterized by neutropenia with myelodysplasia.
Massaad MJ, Ann N Y Acad Sci. 2013;1285:26-43
The three fibers of the cytoskeleton–microtubules in blue, intermediate filaments in red, and actin in green
XXXXX
X X X
XX

Wiskott–Aldrich Syndrome: Pathogenesis
Wiskott-Aldrich syndrome (WAS) protein (WASp) belongs to a family of proteins that relay signals from the surface of the cell to the actin cytoskeleton.
Mutations in the WAS gene have various effects on the level of WASp, which, in turn, correlates with the severity of the disease.
The absence of functional WASp leads to a severe clinical phenotype that can result in death if not diagnosed and treated early in life.
In addition to WAS, mutations in the WAS gene can result in the mild variant X-linked thrombocytopenia, or in X-linked neutropenia, characterized by neutropenia with myelodysplasia.
Massaad MJ, Ann N Y Acad Sci. 2013;1285:26-43
The three fibers of the cytoskeleton–microtubules in blue, intermediate filaments in red, and actin in green
XXXXX
X X X
XX
All immune cells, B, NK, and T cells are affected, resulting in a combined
phenotype.
Wiskott–Aldrich Syndrome: Diagnosis
It has a typical triad:
thrombocytopenia,
eczema,
recurrent infections.
eosinophiliaelevated IgE, IgA,impaired antibody production, (mainly antipolysaccharide antibodies),decreased serum IgM levels
with reduced platelet size and a normal number of
megakaryocytes

The bleeding in the skin is in the form of purpura which are red or purple discolorations measuring 0.3-1cm, under the skin or inside the mouth.
Petechiae are purpura which appear as pin-head sized red or blue dots and measure less than 3 mm.
Ecchymoses are those discolorations from bleeding that measure greater than 1 cm.
These boys can have prolonged bleeding after circumcision, bleeding from the gum especially after the loss of a tooth and prolonged bloody noses.
Copious hemorrhage, during infancy is often the presenting symptom.
Internal bleeding, as in the brain, liver or spleen can be life threatening.
Wiskott–Aldrich Syndrome: Clinical ManifestationsMicrothrombocytopenia

It has a typical triad:
thrombocytopenia,
eczema,
Patients with WAS tend to have allergies. These can be in the form of food allergies, asthma, hay fever.
Patients with Classic WAS tend to have severe eczema (80% of cases) which can be persistent and difficult to control.
Eczema usually starts in infancy, and it can manifest as "cradle cap" on the scalp or a diaper rash.
Wiskott–Aldrich Syndrome: Clinical Manifestations

It has a typical triad:
thrombocytopenia,
eczema,
recurrent infections.
Frequent and severe bacterial infections ranging from ear infection and sinusitis to more severe infections such as pneumonia, meningitis and sepsis. Those patients are particularly susceptible to infections with bacteria that have a capsule covering them (encapsulated) such as the Pneumococcus and Hemophilus influenzae. The infections usually start between 3-6 months of age when the defense acquired from the mother prior to birth begins to wane. The immune function tends to decline over the years with infections becoming more serious and frequent.
impaired antibody production
Wiskott–Aldrich Syndrome: Clinical Manifestations

It has a typical triad:
thrombocytopenia,
eczema,
recurrent infections. T-lymphocytes defects
Frequent and recurrent viral infections such as with the herpes virus, disseminated varicella
Encephalitis and hepatitis with cytomegalovirus,
Patients are unable to clear the EBV (mononucleosis) completely from their immune system, and thus they are more susceptible to lymphoma's associated with this virus,
Fungal and opportunistic infections, especially those caused by Candida and Aspergillus and Pneumocystis jirovecii.
Wiskott–Aldrich Syndrome: Clinical Manifestations
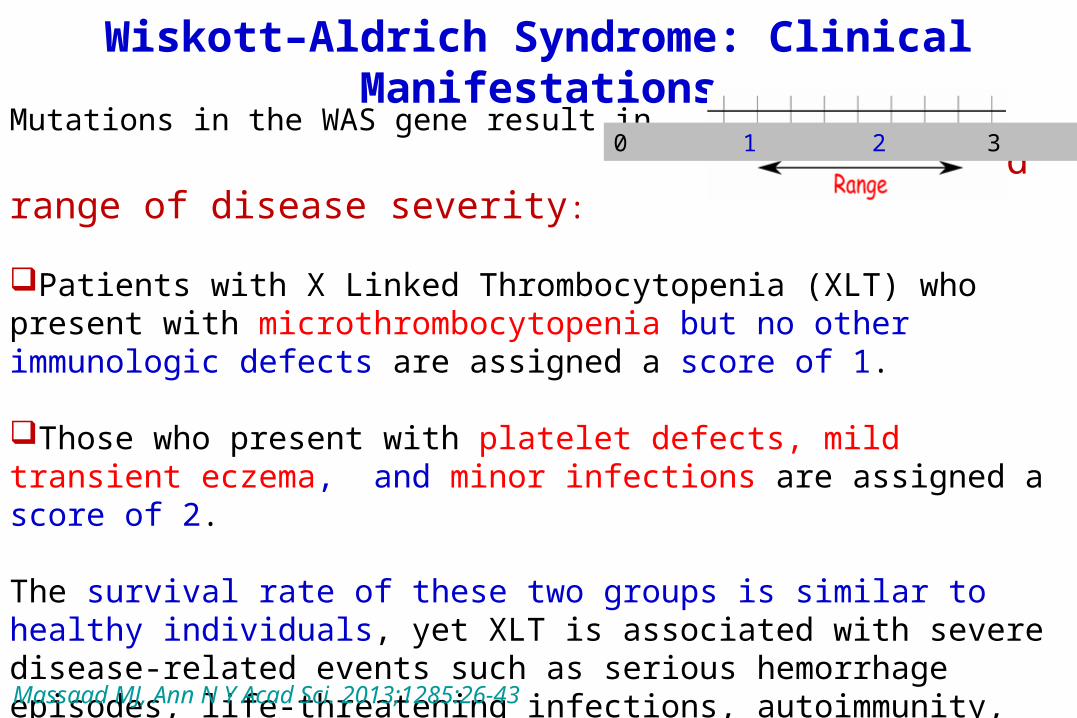
Wiskott–Aldrich Syndrome: Clinical Manifestations
Mutations in the WAS gene result in a broad range of disease severity:
Patients with X Linked Thrombocytopenia (XLT) who present with microthrombocytopenia but no other immunologic defects are assigned a score of 1.
Those who present with platelet defects, mild transient eczema, and minor infections are assigned a score of 2.
The survival rate of these two groups is similar to healthy individuals, yet XLT is associated with severe disease-related events such as serious hemorrhage episodes, life-threatening infections, autoimmunity, and cancer.
Massaad MJ, Ann N Y Acad Sci. 2013;1285:26-43
0 1 2 3 4

Wiskott–Aldrich Syndrome: Clinical Manifestations
Mutations in the WAS gene result in a broad range of disease severity:
Massaad MJ, Ann N Y Acad Sci. 2013;1285:26-43
Patients who present with thrombocytopenia and small platelets, persistent but manageable eczema, and/or recurrent infections are assigned a score of 3.
Those who present with difficult-to-treat eczema and multiple severe infections are assigned a score of 4.
Because diagnosis could be before the full manifestation of the disease, patients with XLT should be reevaluated at a laterstage in life and their clinical scores updated accordingly.
0 1 2 3 4

A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP
Lanzi G. J Exp Med. 2012; 209(1):29–34A female offspring of consanguineous parents, showed features of Wiskott-Aldrich syndrome (WAS), including recurrent infections, eczema, thrombocytopenia, defective T cell proliferation and chemotaxis, and impaired natural killer cell function.
Cells from this patient had undetectable WAS protein (WASP), but normal WAS sequence and messenger RNA levels.
WASP interacting protein (WIP), which stabilizes WASP, was also undetectable. A homozygous c.1301C>G stop codon mutation was found in the WIPF1 gene, which encodes WIP. Introduction of WIP into the patient’s T cells restored WASP expression.
These findings indicate that WIP deficiency should be suspected in patients with features of WAS in whom WAS sequence and mRNA levels are normal.
WIP
X
OKOK

Wiskott–Aldrich Syndrome: Differential Diagnosis
Wiskott-Aldrich syndrome-2 (WAS2) caused by mutations in the WAS/WASL-interacting protein family member 1 (WIPF1) gene on chromosome 2q31.1 coding for a protein that stabilizes and prevents the degradation of WASp, (both male and female can be affected)
Acute or chronic idiopathic thrombocytopenia,
Thrombocytopenia absent radius syndrome,
X-linked thrombocytopenia (a mild form of WAS with no other immunologic defects).
radius
ulna

Wiskott–Aldrich Syndrome: Management
The goal is to control bleeding disorder and the autoimmune manifestations (autoimmune arthritis and anemia), without further suppressing the immune system.
Patients who are on preventative medication for Pneumocystis (Bactrim), Aspergillus, Candida (oral anti-fungal agent such as fluconazole) and herpes virus (Acyclovir) were noticed to have a reduced rate of infection.
This underscores the importance of starting preventative medication early on in infancy in patients with severe WAS.

Wiskott–Aldrich Syndrome: Management
Patients with Wiskott–Aldrich syndrome may require
intensive support with parenteral nutrition, blood transfusions, platelet transfusions, intravenous immunoglobulin.s
plenectomy
•increase level and volume of platelets in patients with X Linked Thrombocytopenia,
•does not decrease the risks of autoimmunity and lymphoproliferative disorders,
•T and B cell function are reduced in patients who undergo HSCT following splenectomy, making it a less desirable intervention if HSCT is considered.
•Nevertheless, because of the introduction of efficient vaccines and antibiotics, splenectomy remains a reasonable treatment for XLT patients with mild disease.

Wiskott–Aldrich Syndrome: Management
The only curative treatment to date is Hematopoietic stem cell transplantation (HSCT).
The use of HSCT for XLT is still controversial, considering the excellent long-term survival with conventional treatment.
However, in light of possible severe infections, autoimmunity, and lymphoproliferative disorders in some XLT patients, HSCT can be considered, especially if a human leukocyte antigen (HLA)–identical related sibling donor is available.
The best transplantation outcome has been achieved with HLA-identical sibling donors and matched unrelated donors when the age of the recipient is < 5 years at the time of the transplant.Massaad MJ, Ann N Y Acad Sci. 2013;1285:26-43

Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Davis SD, Lancet
1966;1(7445):1013–5.
Job’s syndrome was first described in 1966 with 2 patients who had recurrent staphylococcal abscesses, similar to the boils borne by the prophet Job in the Bible.
This clinical syndrome, which was first characterized as a triad of:
recurrent staphylococcal abscesses,
pulmonary infections,
eczematous dermatitis

Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Davis SD, Lancet
1966;1(7445):1013–5.
Job’s syndrome was first described in 1966 with 2 patients who had recurrent staphylococcal abscesses, similar to the boils borne by the prophet Job in the Bible.
This clinical syndrome, which was first characterized as a triad of:
recurrent staphylococcal abscesses,
pulmonary infections,
eczematous dermatitis
was later found to be associated with
increased serum IgE levels, leading to the name autosomal dominant hyper IgE syndrome
(AD-HIES).Buckley RH. Pediatrics 1972;49:59-70.
Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome)
it may not just be atopy
eosinophilia, increased IgE, eczema
the prevalence is estimated between 1 and 9 in 100,000.
≈ 1 in 10,000
most of cases are de novo mutations
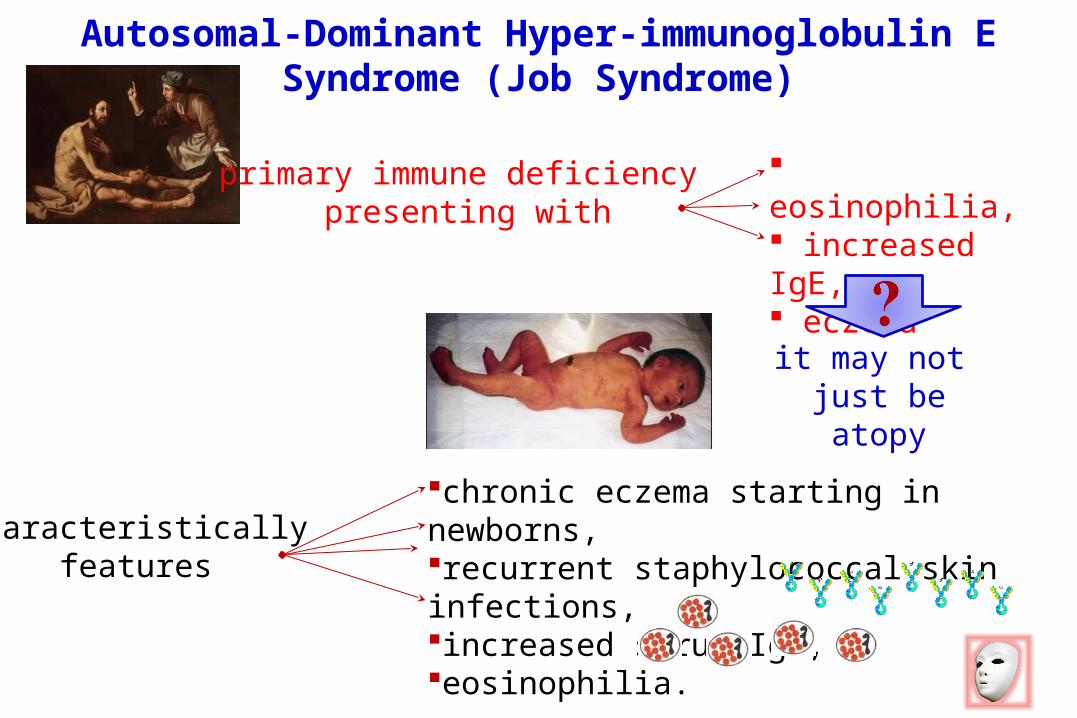
Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome)
it may not just be atopy
primary immune deficiency presenting with
eosinophilia, increased IgE, eczema
chronic eczema starting in newborns, recurrent staphylococcal skin infections,increased serum IgE, eosinophilia.
characteristically features
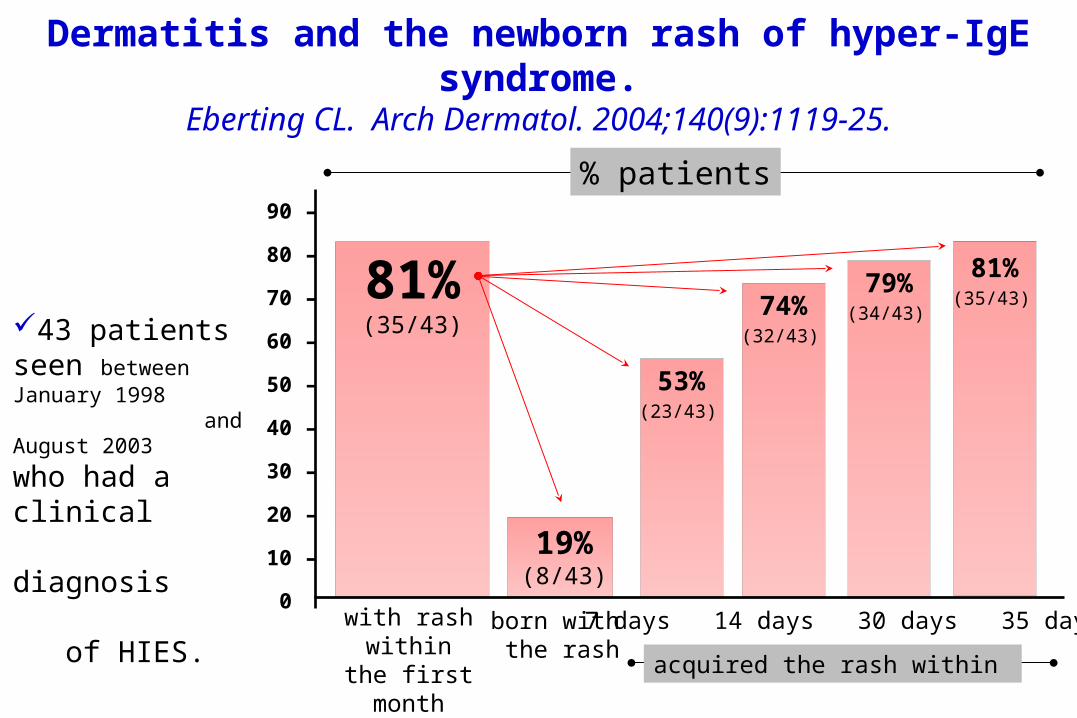
Dermatitis and the newborn rash of hyper-IgE syndrome.
Eberting CL. Arch Dermatol. 2004;140(9):1119-25.
90 –
80 –
70 –
60 –
50 –
40 –
30 –
20 –
10 –
00
% patients
81%(35/43)
born with the rash
7 days 14 days 30 days 35 daysacquired the rash within
19% (8/43)
53%(23/43)
74%(32/43)
79%(34/43)
81%(35/43)
with rashwithin the
first month
43 patients seen between January 1998 and August 2003 who had a clinical diagnosis of HIES.
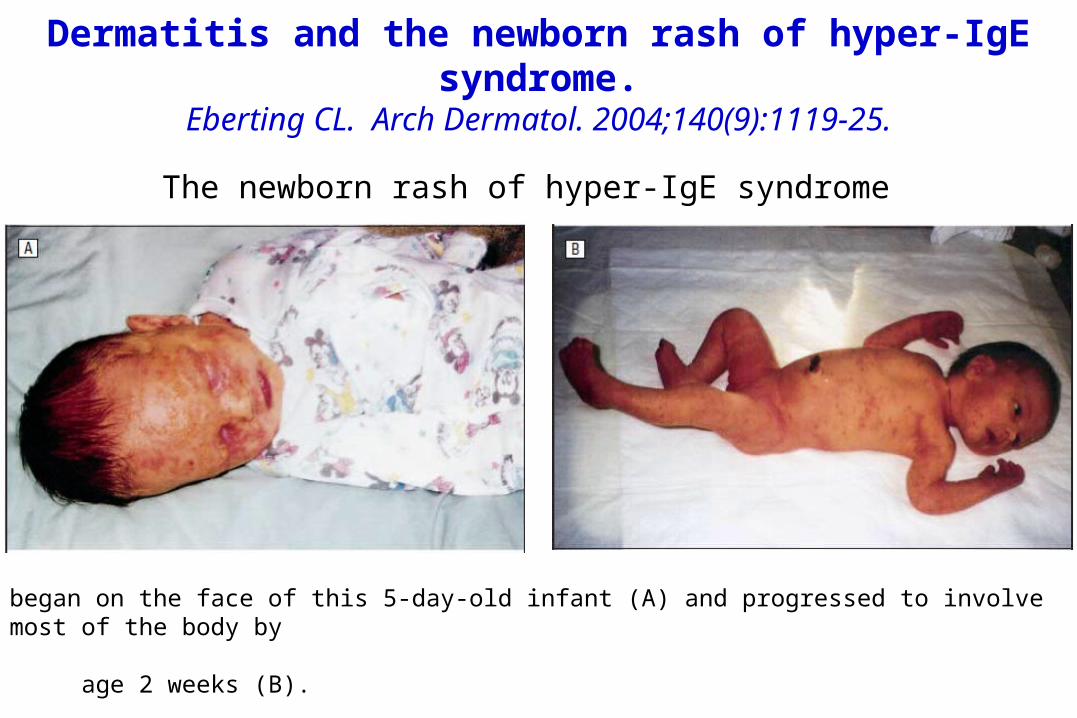
Dermatitis and the newborn rash of hyper-IgE syndrome.
Eberting CL. Arch Dermatol. 2004;140(9):1119-25.
began on the face of this 5-day-old infant (A) and progressed to involve most of the body by age 2 weeks (B).
The newborn rash of hyper-IgE syndrome
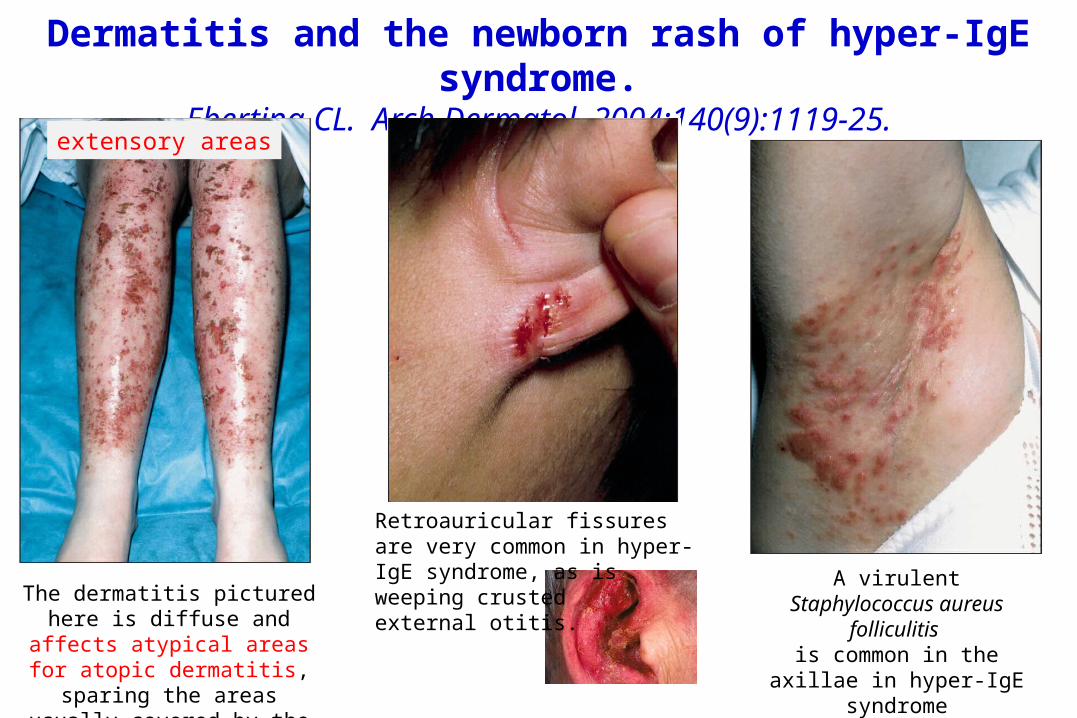
Dermatitis and the newborn rash of hyper-IgE syndrome.
Eberting CL. Arch Dermatol. 2004;140(9):1119-25.
The dermatitis pictured here is diffuse and affects atypical areas for atopic dermatitis, sparing the areas usually covered by the patient’s
socks.
Retroauricular fissures are very common in hyper-IgE syndrome, as isweeping crusted external otitis.
A virulent Staphylococcus aureus folliculitis is common in the
axillae in hyper-IgE syndrome
extensory areas
Dermatitis and the newborn rash of hyper-IgE syndrome.
Eberting CL. Arch Dermatol. 2004;140(9):1119-25.
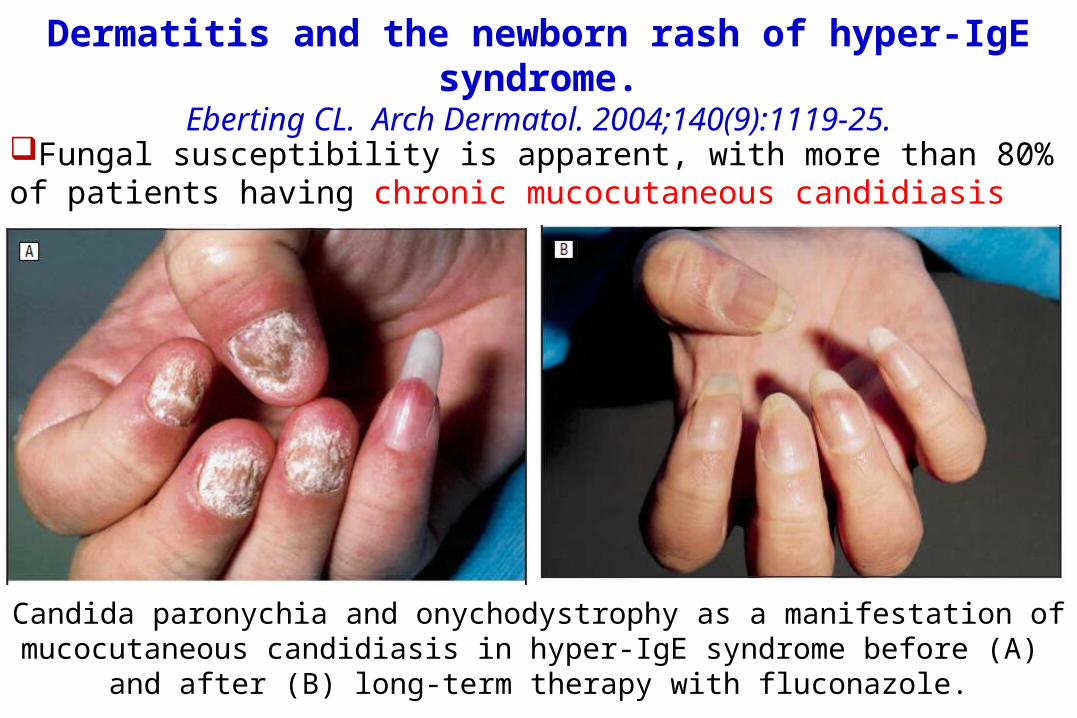
Candida paronychia and onychodystrophy as a manifestation ofmucocutaneous candidiasis in hyper-IgE syndrome before (A)
and after (B) long-term therapy with fluconazole.
Fungal susceptibility is apparent, with more than 80% of patients having chronic mucocutaneous candidiasis
Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome)
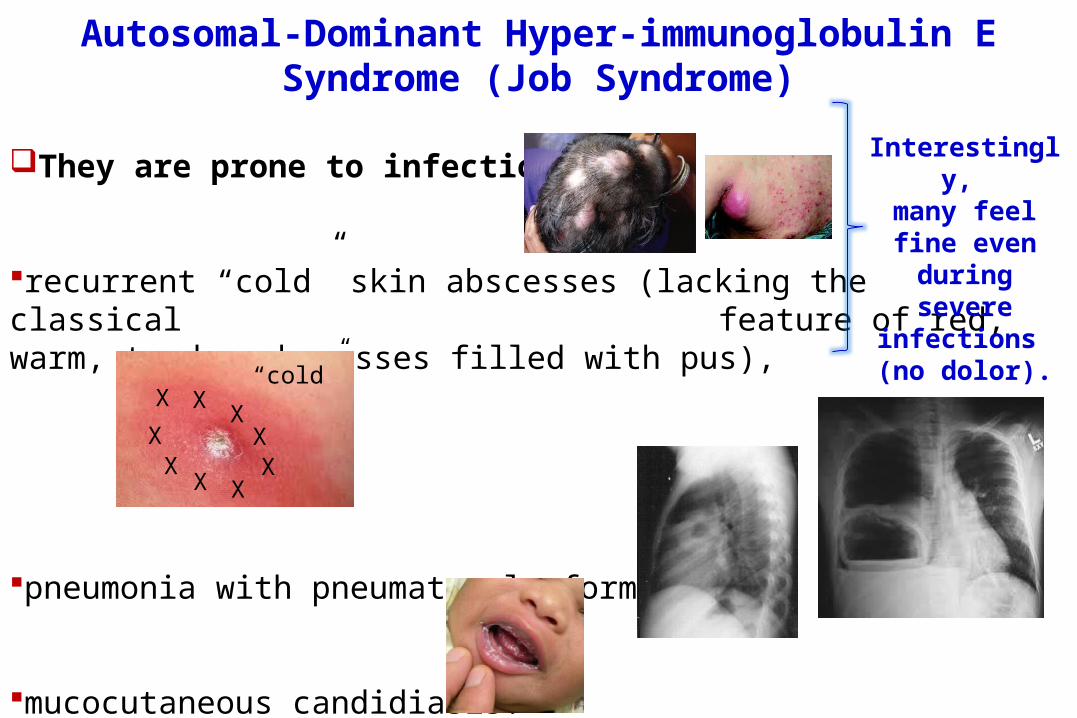
They are prone to infections:
recurrent “cold” skin abscesses (lacking the classical feature of red, warm, tender abscesses filled with pus),
pneumonia with pneumatocele formation,
mucocutaneous candidiasis.
Interestingly,
many feel fine even
during severe
infections (no dolor).
XX
X X XX
XXX
“cold”

Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome)
Recurrent sinopulmonary infections generally start in the first several years of life, with S. aureus being the most common pathogen implicated in the pneumonias.
Streptococcus pneumoniae and Haemophilus influenzae also occur frequently, and the first presentation of pneumonia in infancy may be caused by Pneumocystis jirovecii.
As with the cold abscesses, patients with AD-HIES with pneumonia lack systemic signs of inflammation, including fever, frequently delaying diagnosis leading to parenchymal lung damage
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544multiple pneumatoceles, and an evident aspergilloma (arrows).

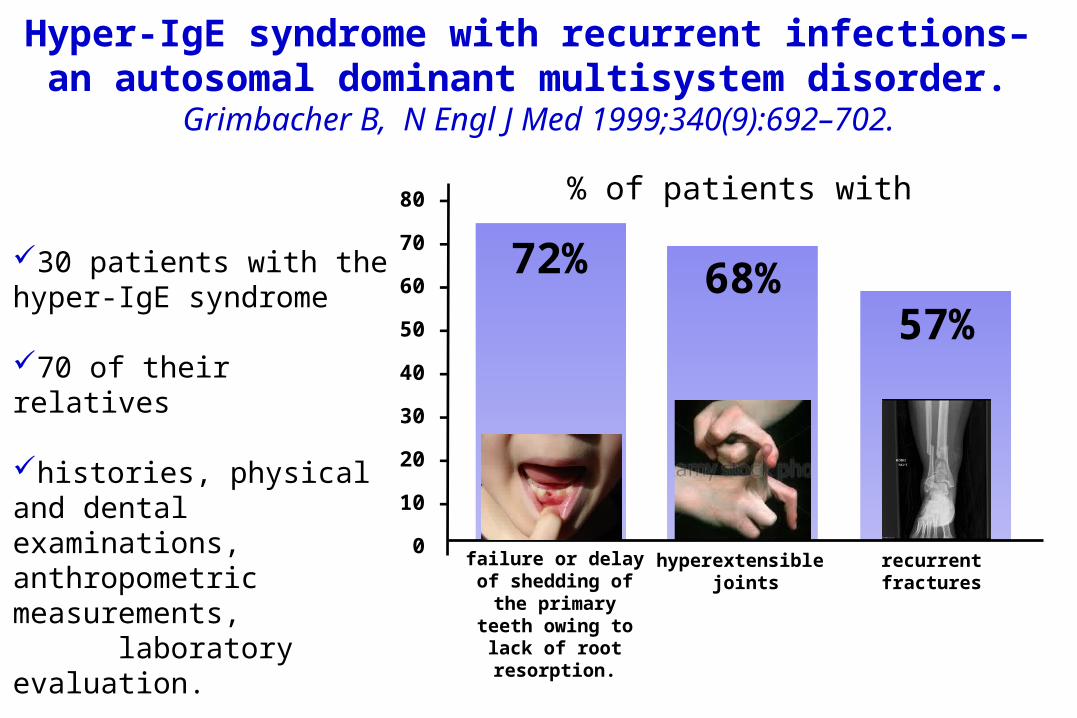
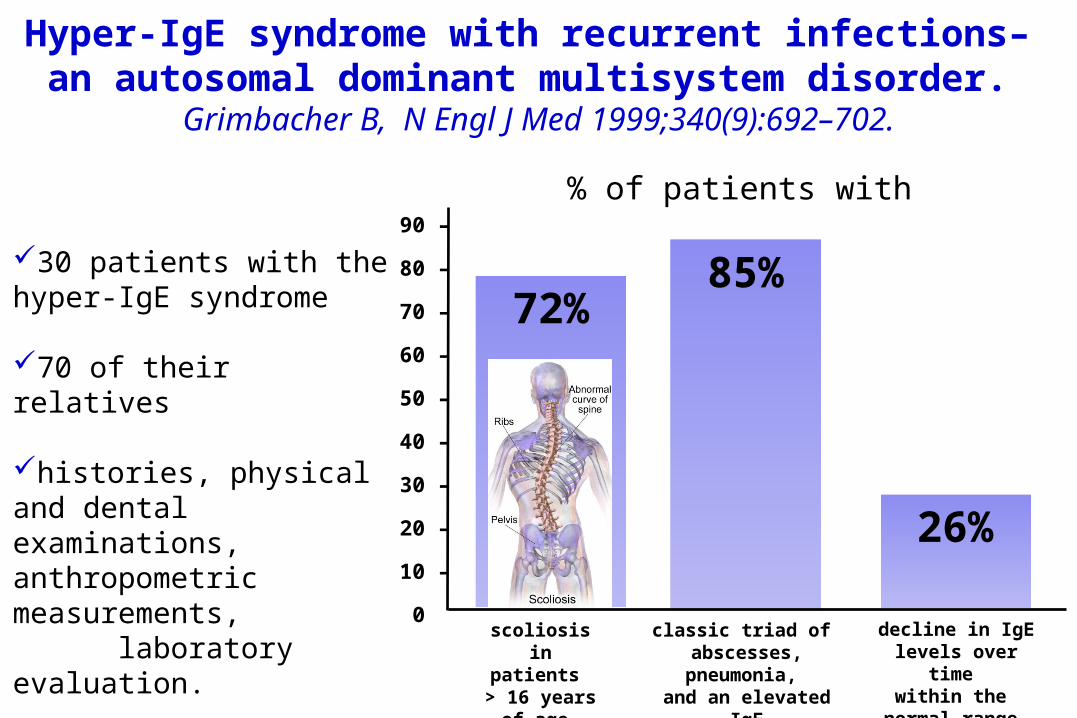
Hyper-IgE syndrome with recurrent infections– an autosomal dominant multisystem disorder.
Grimbacher B, N Engl J Med 1999;340(9):692–702.
30 patients with the hyper-IgE syndrome
70 of their relatives
histories, physical and dental examinations, anthropometric measurements, laboratory evaluation.
Nonimmunologic features of the hyper-IgE syndrome were present in all patients > 8 years.

Hyper-IgE syndrome with recurrent infections– an autosomal dominant multisystem disorder.
Grimbacher B, N Engl J Med 1999;340(9):692–702.Characteristic Facial Appearance of Men and Women
of Different Races with the Hyper-IgE Syndrome. Patients had: facial asymmetry with a suggestion of hemihypertrophy; facial skin is rough, with prominent poresprominent forehead;prominent inferior lip deep-set eyes; wide, fleshy nasal tip; mild prognathism.broad nasal bridge with increased distance between alae.
(carnosa)
Hyper-IgE syndrome with recurrent infections– an autosomal dominant multisystem disorder.
Grimbacher B, N Engl J Med 1999;340(9):692–702.
30 patients with the hyper-IgE syndrome
70 of their relatives
histories, physical and dental examinations, anthropometric measurements, laboratory evaluation.
80 –
70 –
60 –
50 –
40 –
30 –
20 –
10 –
00 failure or delay of shedding of
the primary teeth owing to
lack of root resorption.
72%
recurrent fractures
57%
hyperextensible joints
68%
% of patients with
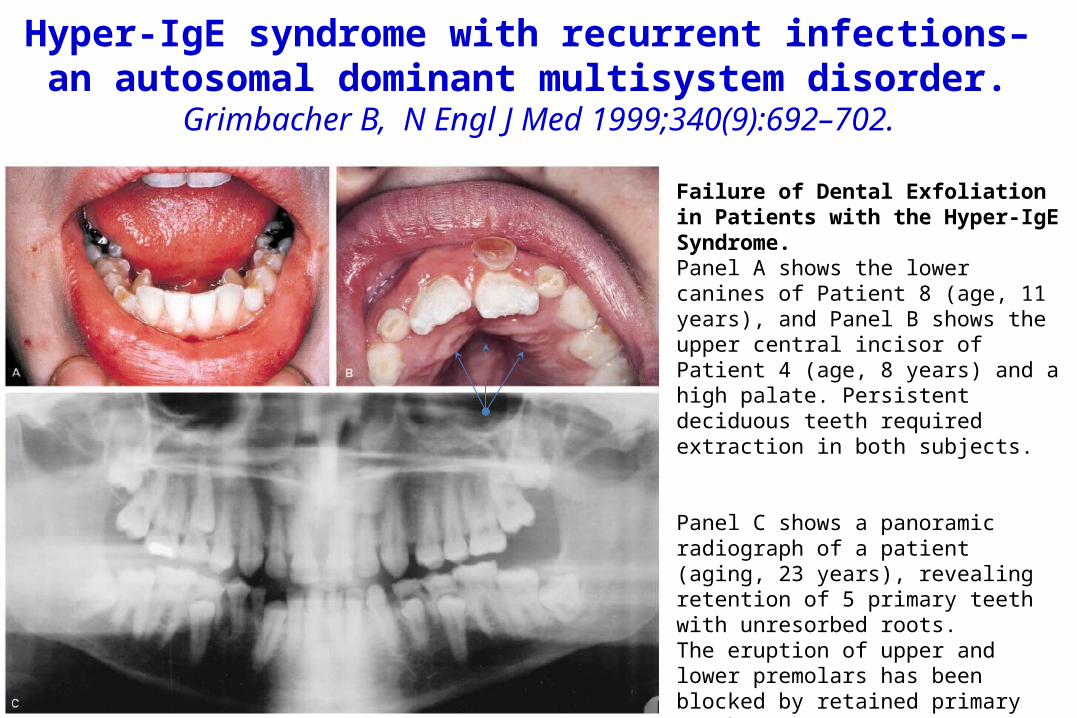
Hyper-IgE syndrome with recurrent infections– an autosomal dominant multisystem disorder.
Grimbacher B, N Engl J Med 1999;340(9):692–702.
Failure of Dental Exfoliation in Patients with the Hyper-IgE Syndrome.Panel A shows the lower canines of Patient 8 (age, 11 years), and Panel B shows the upper central incisor of Patient 4 (age, 8 years) and a high palate. Persistent deciduous teeth required extraction in both subjects.
Panel C shows a panoramic radiograph of a patient (aging, 23 years), revealing retention of 5 primary teeth with unresorbed roots. The eruption of upper and lower premolars has been blocked by retained primary teeth,
Hyper-IgE syndrome with recurrent infections– an autosomal dominant multisystem disorder.
Grimbacher B, N Engl J Med 1999;340(9):692–702.
30 patients with the hyper-IgE syndrome
70 of their relatives
histories, physical and dental examinations, anthropometric measurements, laboratory evaluation.
90 –
80 –
70 –
60 –
50 –
40 –
30 –
20 –
10 –
00 scoliosis in patients
> 16 years of age
72%
classic triad of abscesses,
pneumonia, and an elevated
IgEin pts > 8 yrs of
age
85%
% of patients with
decline in IgE levels over time
within the normal range.
26%

Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome): Pathogenesis
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Dominant-negative mutations in STAT3 cause AD-HIES.
These mutations are primarily missense or single-codon in-frame deletions that result in single amino acid changes.
STAT3 is a transcription factor that plays an essential role in the signal transduction of many cytokines, including IL-6, IL-10, IL-21, IL-22, IL-23 and generation of Th
17.
Because of its involvement with many cytokines, STAT3 plays a crucial role in immunity, inflammation, wound healing, cell
survival, embryogenesis, and oncogenesis.
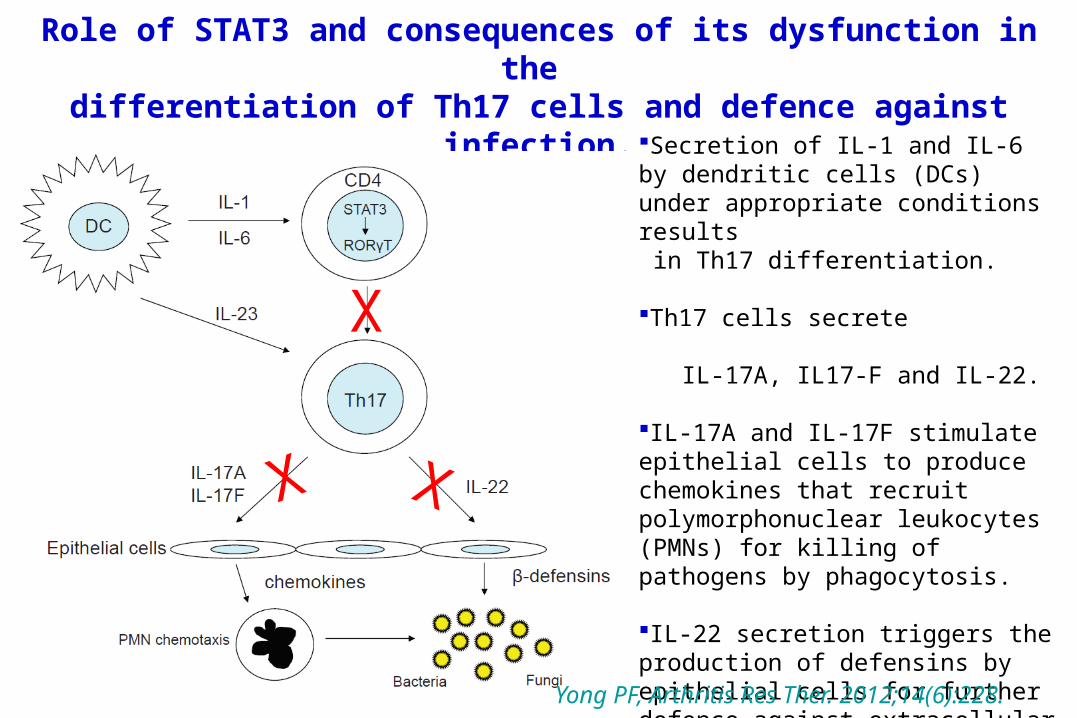
Role of STAT3 and consequences of its dysfunction in the differentiation of Th17 cells and defence against
infection.Secretion of IL-1 and IL-6 by dendritic cells (DCs) under appropriate conditions results in Th17 differentiation.
Th17 cells secrete IL-17A, IL17-F and IL-22.
IL-17A and IL-17F stimulate epithelial cells to produce chemokines that recruit polymorphonuclear leukocytes (PMNs) for killing of pathogens by phagocytosis.
IL-22 secretion triggers the production of defensins by epithelial cells for further defence against extracellular pathogens.Yong PF, Arthritis Res Ther.
2012;14(6):228.
X
X X

Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome): Diagnosis
Serum IgE levels are often > 2000 IU/mL,
Eosinophilia often > 700 cells/µL,
Almost absent Th 17
but
patients usually lack any symptomatic allergic disease such as allergic rhinitis, food allergy, or anaphylaxis.Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78
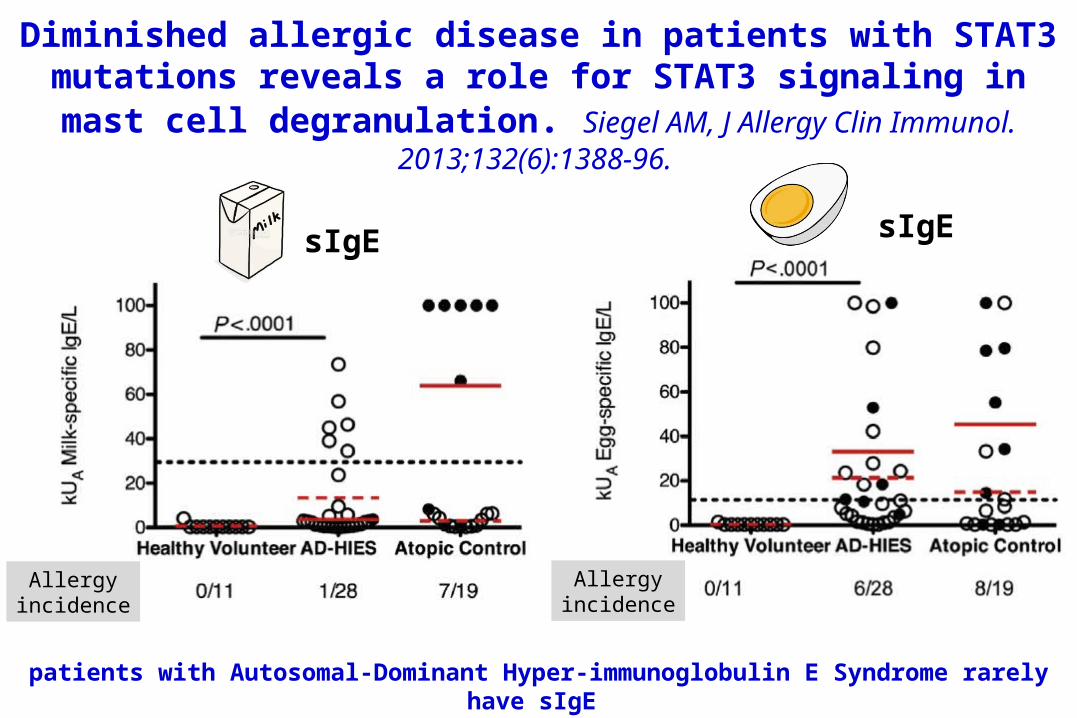
Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast
cell degranulation. Siegel AM, J Allergy Clin Immunol. 2013;132(6):1388-96.
Allergyincidence
Allergyincidence
sIgE sIgE
patients with Autosomal-Dominant Hyper-immunoglobulin E Syndrome rarely have sIgE

Grimbacher B, Am J Hum Genet 1999;65:735-44.
A score > 15 indicated that ADHIES is likely owing to
STAT3 deficiency.
Clinical Scoring Criteria to assess the probability Autosomal-Dominant Hyper-IgE Syndrome (AD-
HIES),

Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome): Diagnosis
Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78
AD-HIES as a result of STAT3 dominant-negative mutations lead to eosinophilia and increased serum IgE.
Often, the baseline increased total white blood cell count does not increase with acute infection.
Neutropenia is rare, but has been reported, as have normal Ig levels.
Numbers of T helper 17 cells assessed by flow cytometry or STAT3 phosphorylation are low.
The gold standard is molecular diagnosis with sequencing of STAT3.
X

Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome): Prognosis
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Parenchymal lung disease and subsequent chronic infection with molds, such as Aspergillus, and gram-negative bacteria, such as Pseudomonas, contribute to most cases of death in AD-HIES.
Because of this situation, pyogenic pneumonias should be aggressively diagnosed and treated to minimize parenchymal damage.
Lung abscesses may require operative intervention and aggressivemanagement of possible complications.
Although there is significant mortality, life expectancy can reach 50 years.

Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome): Management
Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78
Prevention and management of infections with long-term systemic antibiotics and antifungals is important.
The dermatitis in HIES is largely driven by ongoing infection, particularly S. aureus. Consequently, treatment of the skin includes bleach baths (1 mL/ L of water) or chlorhexidine washes as well as prophylactic antibiotics (cotrimoxazole, which targets S. aureus) frequently leads to minimal dermatitis.
The development of skin abscesses has reduced following the introduction of prophylactic antibiotics, although these sometimes require surgical intervention.Yong PF, Arthritis Res Ther. 2012;14(6):228.

Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome): Management
Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78Yong PF, Arthritis Res Ther. 2012;14(6):228.
A further point of note is that the aberrant tissue healing following pulmonary infections can result in parenchymal abnormalities that allow colonisation with P. aeruginosa, fungal infections and non-tuberculous mycobacteria.
Superinfection with these organisms represents the most challenging aspect of long-term management. Eradication of these organisms is difficult and the role of surgery for areas of parenchymal abnormality is uncertain.
Pulmonary surgery appears to be associated with a greater risk of complications and should be carefully considered and only undertaken in a centre with particular experience in the disease.

Autosomal-Dominant Hyper-immunoglobulin E Syndrome (Job Syndrome): Management
Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78
Because some patients fail to sustain protective vaccination titers, Ig replacement should be initiated.
Immunoglobulin replacement should be considered in those patients with impaired specific antibody responses, bronchiectasis, and breakthrough infections while on prophylaxis.
HSCT is under investigation, but has been used in severe disease.
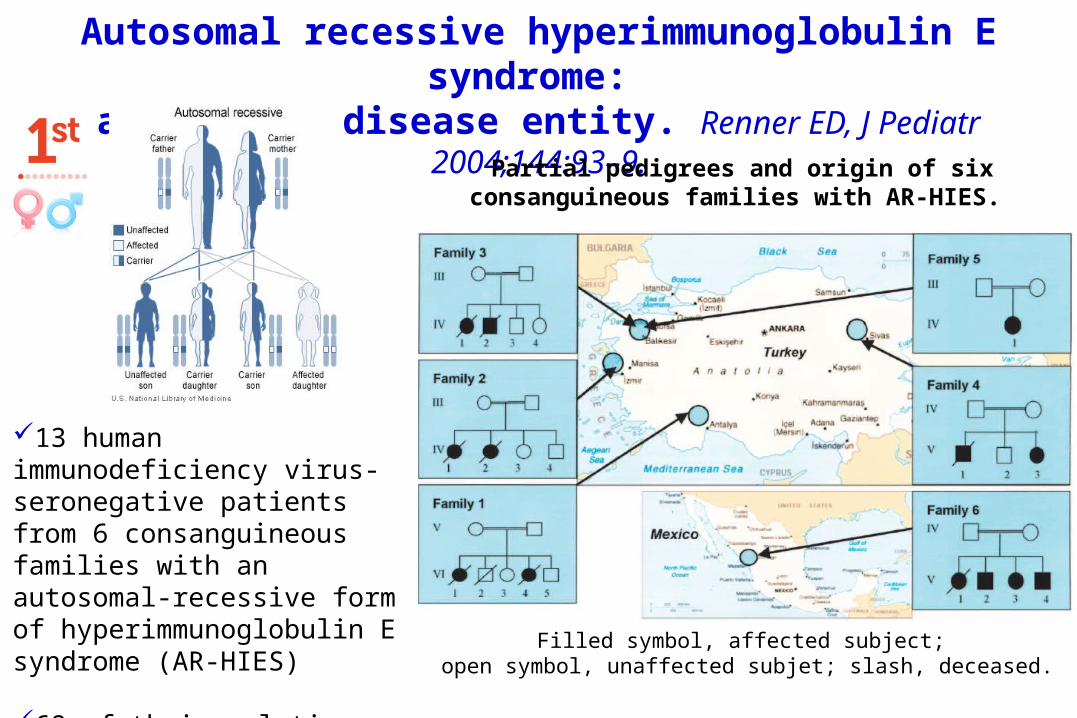
Autosomal recessive hyperimmunoglobulin E syndrome:
a distinct disease entity. Renner ED, J Pediatr 2004;144:93–9.
13 human immunodeficiency virus-seronegative patients from 6 consanguineous families with an autosomal-recessive form of hyperimmunoglobulin E syndrome (AR-HIES)
68 of their relatives.
Partial pedigrees and origin of six consanguineous families with AR-HIES.
Filled symbol, affected subject; open symbol, unaffected subjet; slash, deceased.

Autosomal recessive hyperimmunoglobulin E syndrome:
a distinct disease entity. Renner ED, J Pediatr 2004;144:93–9.
13 human immunodeficiency virus-seronegative patients from 6 consanguineous families with an autosomal-recessive form of hyperimmunoglobulin E syndrome (AR-HIES)
68 of their relatives.
Partial pedigrees and origin of six consanguineous families with AR-HIES.
Filled symbol, affected subject; open symbol, unaffected subjet; slash, deceased.
Also known as
Dedicator of Cytokinesis 8 DeficiencyWilliams KW,
Immunol Allergy Clin N Am 2015;35:523–544
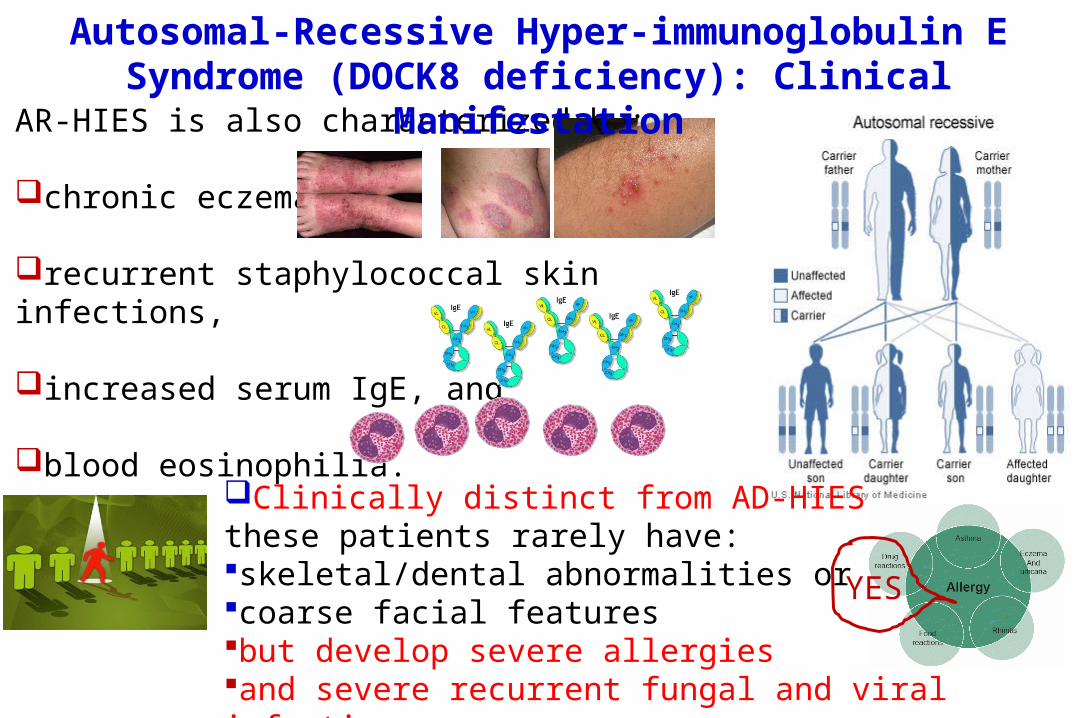
AR-HIES is also characterized by:
chronic eczema,
recurrent staphylococcal skin infections,
increased serum IgE, and
blood eosinophilia. Clinically distinct from AD-HIES, these patients rarely have: skeletal/dental abnormalities or coarse facial featuresbut develop severe allergies and severe recurrent fungal and viral infections
Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Clinical
Manifestation
YES
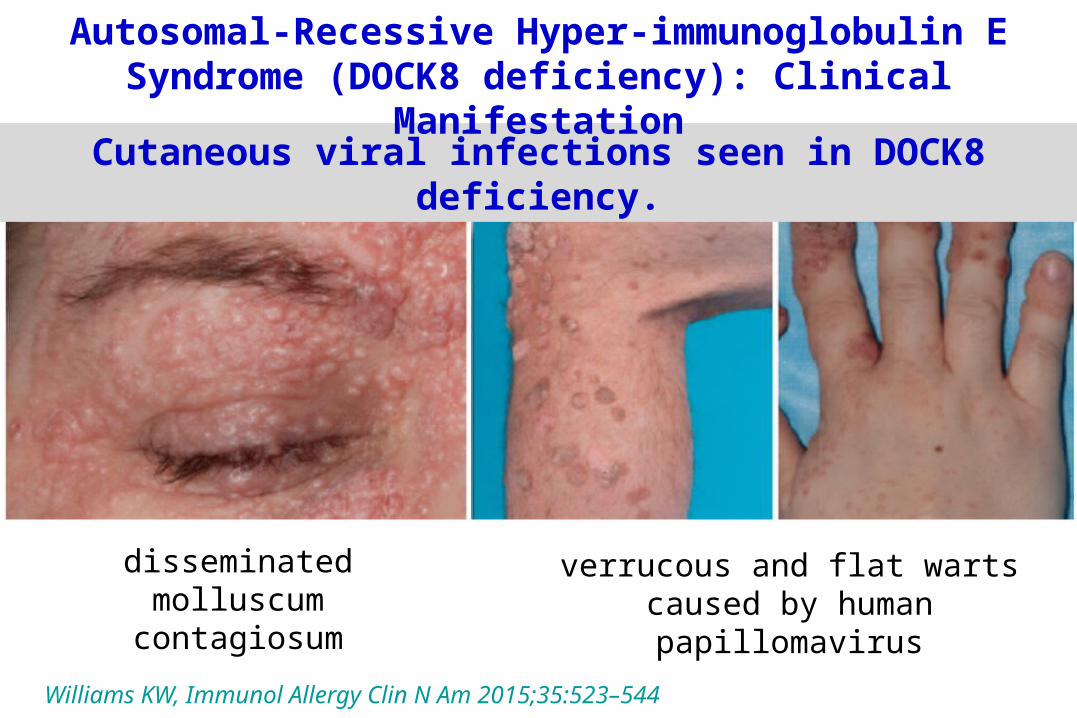
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
disseminatedmolluscum contagiosum
verrucous and flat warts caused by human papillomavirus
Cutaneous viral infections seen in DOCK8 deficiency.
Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Clinical
Manifestation
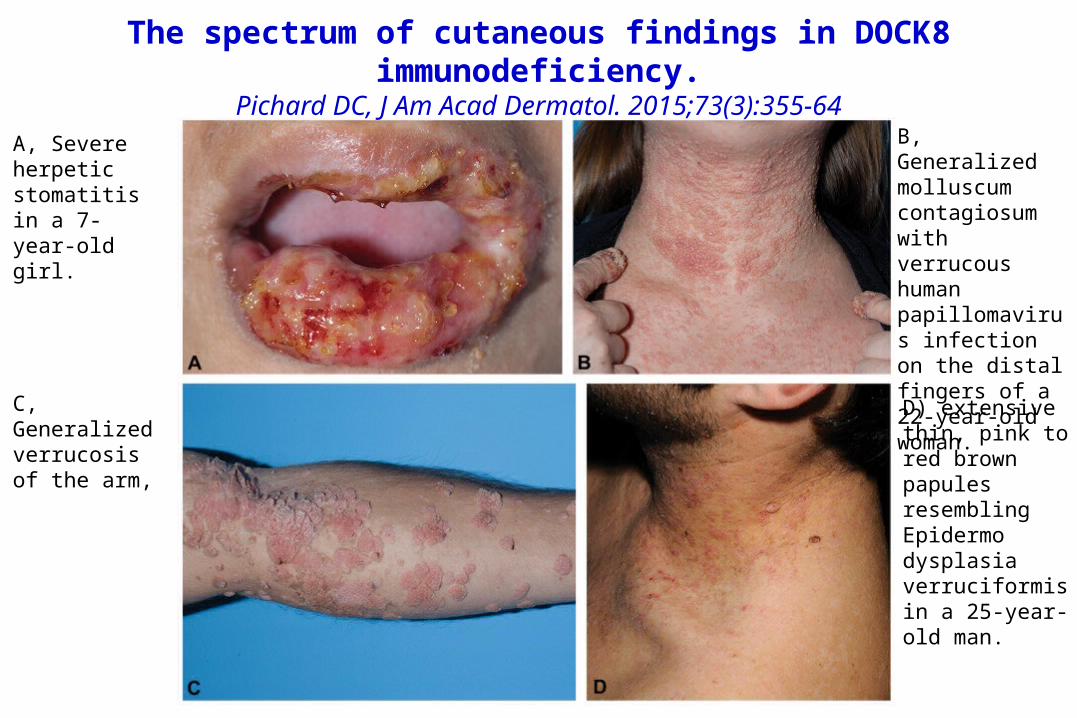
The spectrum of cutaneous findings in DOCK8 immunodeficiency.
Pichard DC, J Am Acad Dermatol. 2015;73(3):355-64A, Severe herpeticstomatitis in a 7-year-old girl.
B, Generalized molluscum contagiosum with verrucous humanpapillomavirus infection on the distal fingers of a 22-year-old woman.
C, Generalizedverrucosis of the arm,
D) extensive thin, pink to red brown papules resemblingEpidermodysplasia verruciformis in a 25-year-old man.

Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78
2) Central nervous system manifestations (facial paralysis, hemiplegia, ischemic infarction, and subarachnoid hemorrhage),
3) Autoimmune diseases
4) Vascular disorders (aneurysms) are variably associated.
5) Poor growth and failure to thrive is common,
6) Malignancy.
autoimmune hemolytic anemia, hypothyroidism, vasculitis
They are susceptible to:
Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Clinical
Manifestation
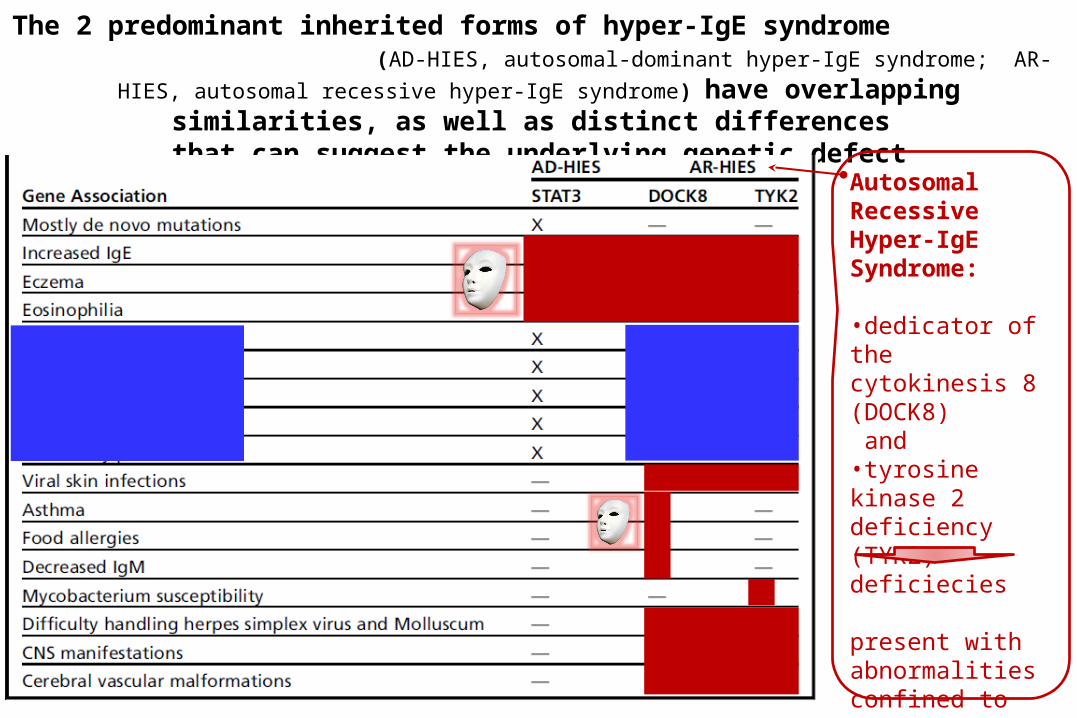
The 2 predominant inherited forms of hyper-IgE syndrome (AD-HIES, autosomal-dominant hyper-IgE syndrome; AR-HIES, autosomal recessive
hyper-IgE syndrome) have overlapping similarities, as well as distinct differences
that can suggest the underlying genetic defectAutosomal Recessive Hyper-IgE Syndrome:
•dedicator of the cytokinesis 8 (DOCK8) and •tyrosine kinase 2 deficiency (TYK2) deficiecies
present with abnormalities confined to the immune system.
Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78
Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Pathogenesis
AR-HIES is a combined immune deficiency where dedicator of the cytokinesis 8 (DOCK8) gene or the signaling protein tyrosine kinase 2 (TYK2) mutations affect CD4 and CD8 proliferation, B cells, and memory cells.
Homozygous mutations of DOCK8 (9p24.3) result in its loss of function.
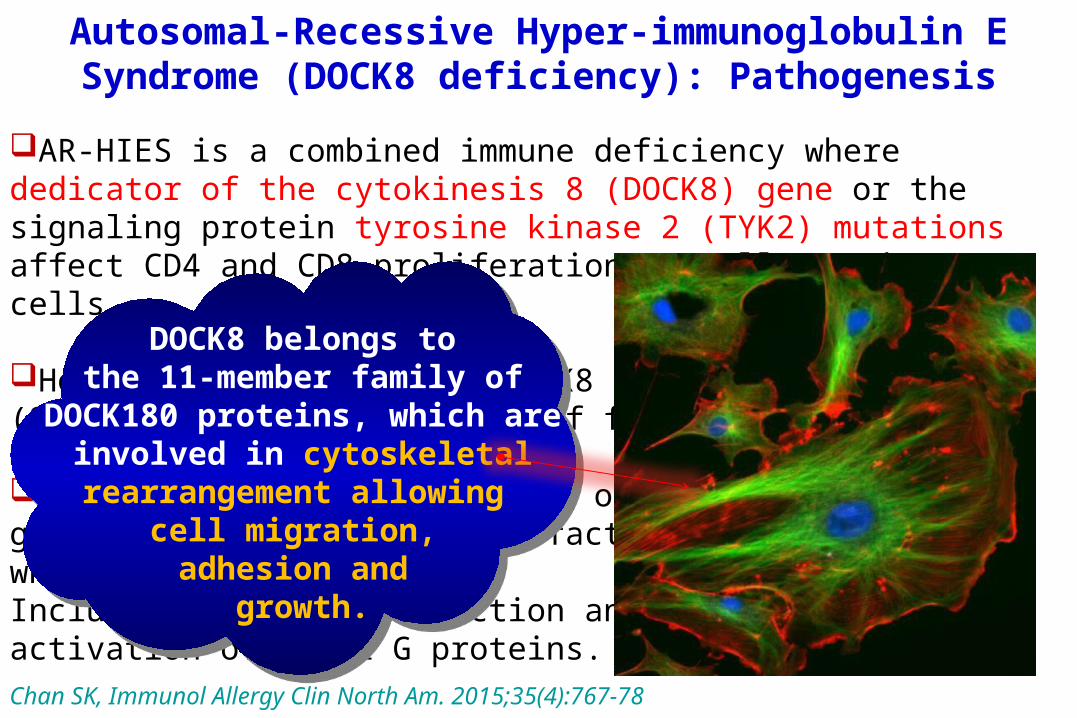
DOCK8 belongs to a subfamily of guanine nucleotide exchange factors, which have multiple roles, Including signal transduction and activation of small G proteins.
DOCK8 belongs tothe 11-member family of DOCK180 proteins, which
areinvolved in cytoskeletal rearrangement allowing
cell migration, adhesion and
growth.

Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78
Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Diagnosis
low T-cell numbers (CD4+and CD8+), low serum IgM levels BUT poor specific responses, (failure to sustain protective titers against vaccination).
Flow cytometry for T helper 17 cells may be low.
T-lymphocyte activation and proliferation are diminished, particularly in the CD8+ population, thus contributing their increased viral susceptibility.
elevated total white blood cell counts that do not increase with acute infection, Eosinophilia typically > 1000 µL, high IgE, Serum IgG levels are normal or increased
Similar to AD-HIES, AR-HIES patients have:
Diagnosis is confirmed with DOCK8 sequencing and can be strongly
suggested by absence of expression on flow cytometry.
Zhang Q. N Engl J Med 2009;361(21):2046–55.

Chan SK, Immunol Allergy Clin North Am. 2015;35(4):767-78
Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Prognosis
There is higher mortality at a younger age in DOCK8 deficiency, with death often occurring before the age of 20.
The prognosis is poor worse than AD-HIES, with most failing to reach adulthood as a result of sepsis, neurologic disorders, and malignancies.
Vascular complications (aneurysms) have been documented,including those secondary to vaccine-strain varicella zoster. Sabry A, J Allergy Clin Immunol 2014;133(4):1225–7.

Specific antibody production in AR-HIES can be variable (despite normal IgG levels) and replacement immunoglobulin therapy because of poor specific antibody production has been used with anecdotal improvement in respiratory tract infections.
Viral skin infections have unfortunately not improved with replacement immunoglobulin therapy.
Widespread molluscum and human papilloma virus
infection has been difficult to treat - standard therapies with salicylic acid, cryotherapy and imiquimod have had limited success; interferon alpha has been used anecdotally with mixed results.
Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Management
Yong PF. Arthritis Research & Therapy 2012;14:228

Specific antibody production in AR-HIES can be variable (despite normal IgG levels) and replacement immunoglobulin therapy because of poor specific antibody production has been used with anecdotal improvement in respiratory tract infections.
Viral skin infections have unfortunately not improved with replacement immunoglobulin therapy.
Widespread molluscum and human papilloma virus
infection has been difficult to treat - standard therapies with salicylic acid, cryotherapy and imiquimod have had limited success; interferon alpha has been used anecdotally with mixed results.
Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Management
Yong PF. Arthritis Research & Therapy 2012;14:228
Acyclovir or valacyclovir should be considered for prophylaxis to prevent
recurrent HSV andVZV infections.
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544

Autosomal-Recessive Hyper-immunoglobulin E Syndrome (DOCK8 deficiency): Management
Yong PF. Arthritis Research & Therapy 2012;14:228
Haematopoeitic stem cell transplantation has been reported in DOCK8 deficiency patients.
In all individuals, resolution of recurrent infections (particularly viral skin infections with molluscum) and eczema occurred, although one individual continued to suffer from food allergies.
Improvement in IgE levels and resolution of vasculitis were reported
These initial results suggest that stem cell transplantationin AR-HIES may represent an excellent curative option given the high morbidity and mortality seen in the disease.

Autosomal Recessive Hyper-IgE syndrome with Phosphoglucomutase 3 Deficiency
Mutations in phosphoglucomutase 3 (PGM3) were recently defined, leading to a multisystem disease sharing some clinical features with AD-HIES and DOCK8 deficiency.
cutaneous leukocytoclastic vasculitis, eczema-like rash, cognitive impairment, myoclonus, ataxia, dysarthria (motor speech disorder) delayed visual and sensory hearing evoked potentials, severe bronchiectasis.
Are all present
Phosphoglucomutase catalyzes the reversible transfer of a phosphate group between the I- and 6-positions of glucose and mediates both glycogen formation and utilization

Autosomal Recessive Hyper-IgE syndrome with Phosphoglucomutase 3 Deficiency
Mutations in phosphoglucomutase 3 (PGM3) were recently defined, leading to a multisystem disease sharing some clinical features with AD-HIES and DOCK8 deficiency.
cutaneous leukocytoclastic vasculitis, eczema-like rash, cognitive impairment, myoclonus, ataxia, dysarthria (motor speech disorder) delayed visual and sensory hearing evoked potentials, severe bronchiectasis.
Are all present
Phosphoglucomutase catalyzes the reversible transfer of a phosphate group between the I- and 6-positions of glucose and mediates both glycogen formation and utilization
PGM3 deficiency should be suspected in an individual with an exaggerated allergic
phenotype, recurrent infections, and neurologic deficits.

The Diverse Clinical Features of Chromosome 22q11.2 Deletion Syndrome (DiGeorge Syndrome)
Maggadottir SM,J Allergy Clin Immunol Pract 2013;1:589-94
The estimated prevalence for chromosome 22q11.2 deletion syndrome is 1 in 4000 births.
The inheritance pattern is autosomal dominant. The prevalence of chromosome 22q11.2 deletion syndrome will rise because this is a haplosufficiency genetic disorder*, and, therefore, the chance of an affected parent having an affected child is approximately 50%.
Spontaneous or de novo mutations account for approximately 90%, of 22q11.2 deletions; only approximately 10% are inherited from an affected parent. *Any gene that allows the production of viable adults even when one copy of the gene in diploids is mutant or deleted from one of the homologous chromosomes.

Presenting phenotype in 100 children with the 22q11 deletion syndrome. Oskarsdottir S, Eur J Pediatr
2005;164:146-53.
most common clinical features that lead to diagnosis:
•Cardiac defects, •Absent or hypoplastic thymus, •Hypocalcemia, and•Characteristic dysmorphic features.
Cardiac defects, Recurrent infections, Speech delay, Developmental delay, or learning difficulties Characteristic dysmorphic features
in children younger than
2 years of age
in children older than 2 years of age

Presenting phenotype in 100 children with the 22q11 deletion syndrome. Oskarsdottir S, Eur J Pediatr
2005;164:146-53.
most common clinical features that lead to diagnosis:
•Cardiac defects, •Absent or hypoplastic thymus, •Hypocalcemia, and•Characteristic dysmorphic features.
Cardiac defects, Recurrent infections, Speech delay, developmental delay, or learning difficulties Characteristic dysmorphic features
in children younger than
2 years of age
in children older than 2 years of age
In very few patients, <1% with chromosome 22q11.2 deletion syndrome, the thymus and the T cells are absent,
and this is referred to as complete DiGeorge syndrome.
In 75% of patients, the immune system is affected to some degree, which leaves approximately 20% of patients with normal
T-cell counts.
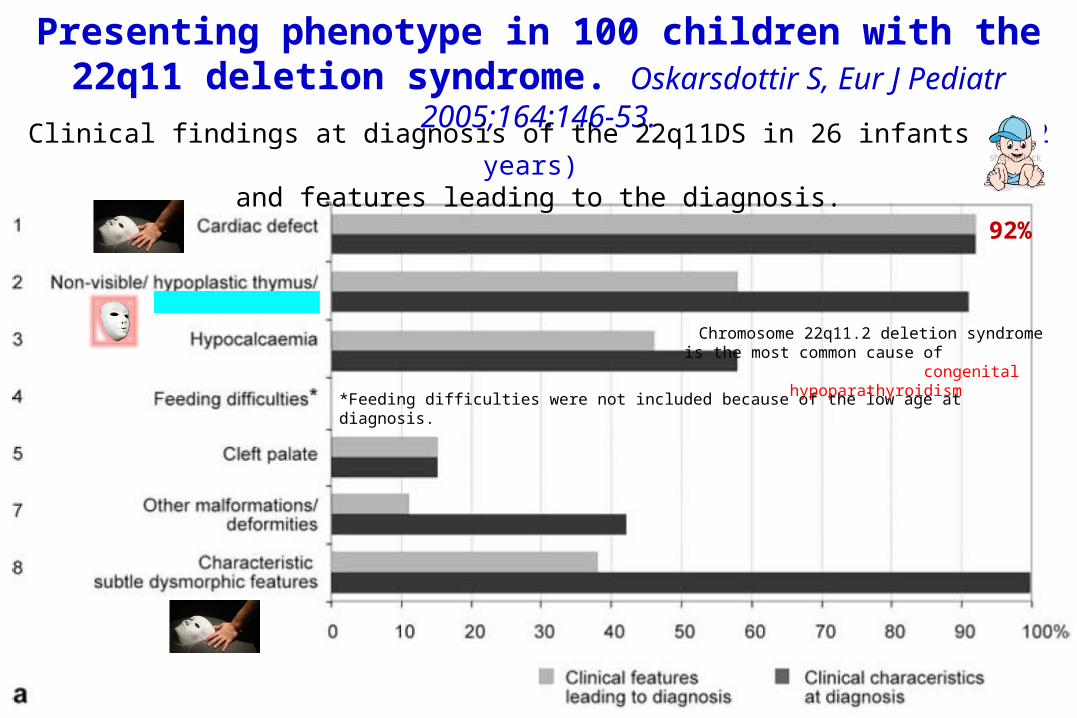
Presenting phenotype in 100 children with the 22q11 deletion syndrome. Oskarsdottir S, Eur J Pediatr
2005;164:146-53.Clinical findings at diagnosis of the 22q11DS in 26 infants (< 2 years) and features leading to the diagnosis.
92%
*Feeding difficulties were not included because of the low age at diagnosis.
Chromosome 22q11.2 deletion syndrome is the most common cause of
congenital hypoparathyroidism
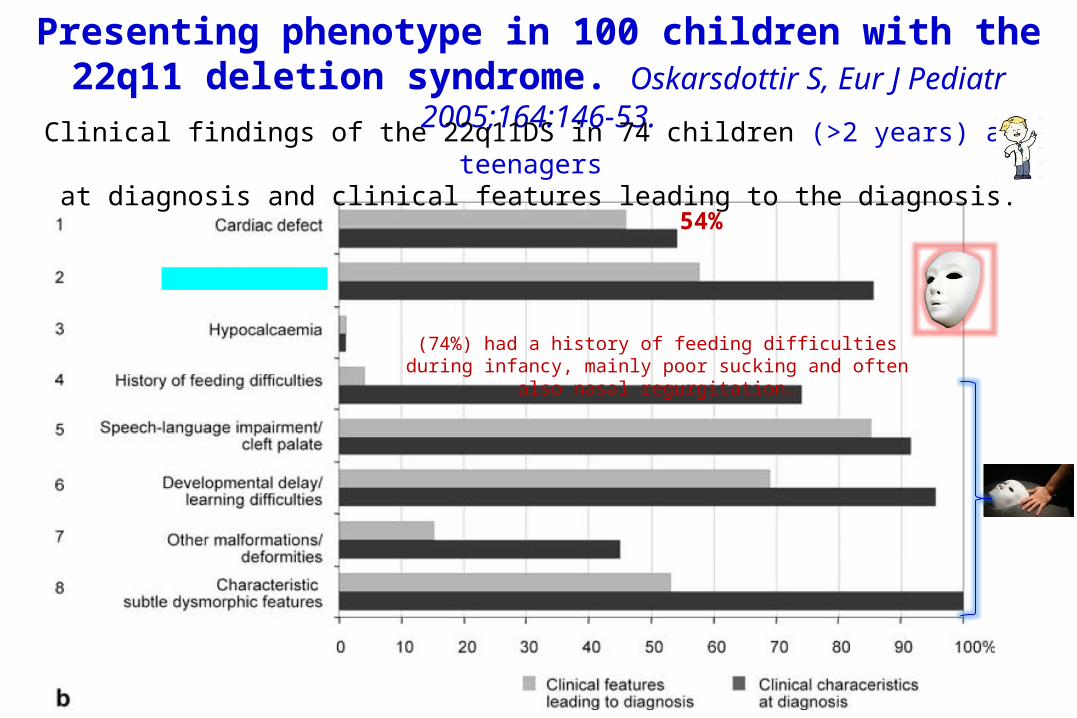
Presenting phenotype in 100 children with the 22q11 deletion syndrome. Oskarsdottir S, Eur J Pediatr
2005;164:146-53.Clinical findings of the 22q11DS in 74 children (>2 years) and teenagers at diagnosis and clinical features leading to the diagnosis.
54%
(74%) had a history of feeding difficulties during infancy, mainly poor sucking and often also nasal
regurgitation.

All patients had mild dysmorphic features considered to be typical for 22q11 deletion, such as:
•minor auricular anomalies (small round ears, posterior rotation of the ears, simple helices, or low-set ears),
•typically shaped nose (thin alae nasae, broad nose tip, tubular form of the nose),
•small, arch-shaped mouth,
•hooded eyelids,
•malar flatness (cheekbones may be flat),
•myotonic-looking face,
•long, slender and/or tapering fingers.
Presenting phenotype in 100 children with the 22q11 deletion syndrome. Oskarsdottir S, Eur J Pediatr
2005;164:146-53.
(lunghe e snelle)
OK

All patients had mild dysmorphic features considered to be typical for 22q11 deletion, such as:
•minor auricular anomalies (small round ears, posterior rotation of the ears, simple helices, or low-set ears),
•typically shaped nose (thin alae nasae, broad nose tip, tubular form of the nose),
•small, arch-shaped mouth,
•hooded eyelids,
•malar flatness (cheekbones may be flat),
•myotonic-looking face,
•long, slender and/or tapering fingers.
Presenting phenotype in 100 children with the 22q11 deletion syndrome. Oskarsdottir S, Eur J Pediatr
2005;164:146-53.
(lunghe e snelle)
OK
Images A show normal ear position. Low set ears are positioned below the
horizontal line as illustrated in B.
Low-set ears are often posteriorly rotated, reflecting an arrest in the
normal anterior rotation that occurred during fetal ear development. Low set
ears
Normal set
ears

Chromosome 22q11.2 Deletion Syndrome (DiGeorge Syndrome): Clinical Manifestations
Maggadottir SM,J Allergy Clin Immunol Pract 2013;1:589-94
contributions to recurrent infections in these children:1) palatal dysfunction (majority of patients will have palatal weakness and a minority will have submucous clefting, a cleft lip is extraordinarily uncommon in the syndrome.) with dysfunctional swallowing.
2) Nasal regurgitation is a major problem and together with Eustachian tube dysfunction and poor drainage contributes to recurrent ear infections.
3) Decreased T-cell responses and proliferation. (avoid live viral vaccine if CD4+ T-cell count is <400 cells). However, even if T-cell counts may be low-for-age in infancy but in most patients typically improves in the first year of life. Despite poor thymic output of T cells, antiviral defenses and B-cell help by existing T cells is not greatly impaired,

What is a submucous cleft palate? A submucous cleft of the soft palate is characterized by a midline deficiency or lack of muscular tissue and incorrect positioning of the muscles.
A submucous cleft of the hard palate is defined as a bony defect in the midline or center of the bony palate. This can sometimes be felt as a notch or depression in the bony palate when the palate is palpated with a finger.
Often a submucous cleft palate is associated with a bifid or cleft uvula.
What are the effects of submucous cleft palate?When a submucous cleft is present, the muscles of the soft palate may not function properly and the individual is at risk for speech problems, middle ear disease, and swallowing difficulties.
How can a submucous cleft palate be identified?The most common reason that a child is evaluated for a submucous cleft palate is abnormal nasal speech. Other symptoms may include persistent middle ear disease and feeding/swallowing difficulties. A submucous cleft palate may be identified by the presence of a bifid uvula; a very thin translucent strip of lining (mucosa) in the middle of the roof of the mouth; and, a notch at the back edge of the hard palate that can be felt by the fingertip.
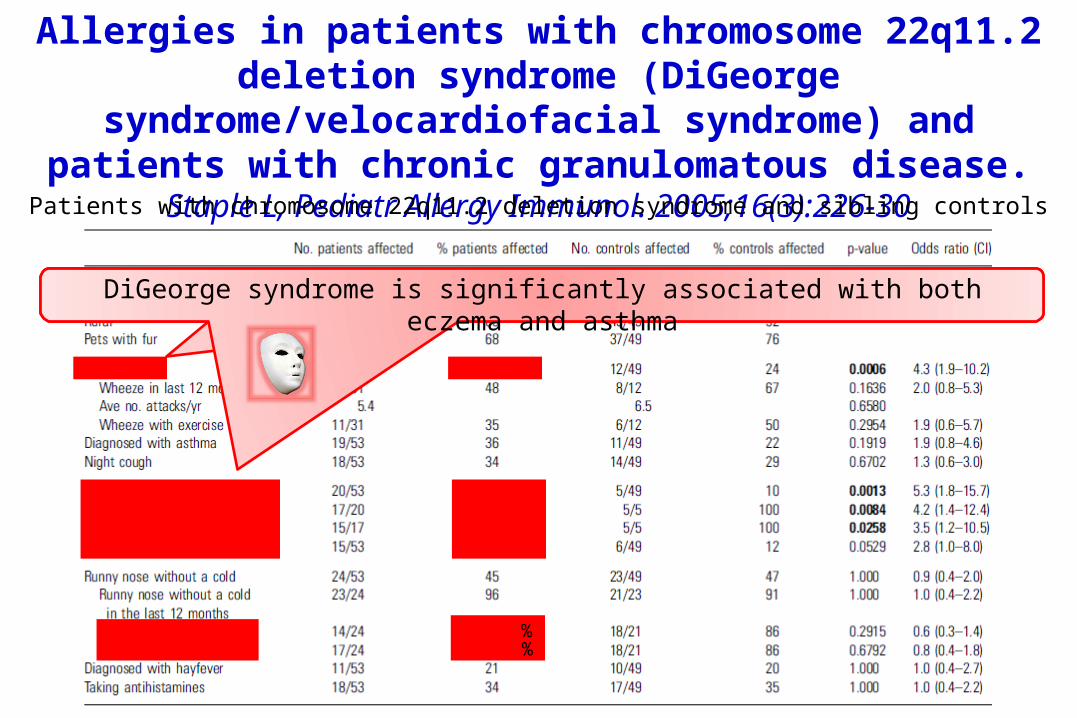
Allergies in patients with chromosome 22q11.2 deletion syndrome (DiGeorge
syndrome/velocardiofacial syndrome) and patients with chronic granulomatous disease.
Staple L, Pediatr Allergy Immunol. 2005;16(3):226-30Patients with chromosome 22q11.2 deletion syndrome and sibling controls
%
%%%%
DiGeorge syndrome is significantly associated with both eczema and asthma
%%

IMMUNE DEFICIENCY in Complete DiGeorge Syndrome
Patients with rare complete DiGeorge syndrome have true thymus aplasia and absence of T cells and severe immune compromise.
This group of patients can be defined by <50 T cells/mm3 and/or <50 naive(CD45RA+/CD62L+ T cells/mm3). Markert ML, J Allergy Clin Immunol 2004;113:734-41.
These patients need protection from infection and blood products. Antipneumocystis and antifungal prophylaxis should be promptly initiated and immunoglobulin replacement therapy in those with hypogammaglobulinemia.
Live viral vaccines should be avoided, and respiratory syncytial virus (RSV) prophylaxis is provided.
If cardiac surgery is needed these patients should receive cytomegalovirus negative and irradiated blood products.

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency (PID)?Eczema or Secondary
Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal abnormalities “Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions

Human immunodeficiency virus infection is characterized by a progressive depletion of helper T-lymphocytes and, like allergic diseases, is associated with altered T cell regulation. It is one of the more common immunodeficiencies and should always be excluded when considering a Primary Immune Deficiency. Virologic testing with HIV DNA polymerase or HIV RNA assays should be used.
Human Immunodeficiency Virus (HIV)
Infants with HIV infection (a secondary immunodeficiency)can present with:
1) eczematous rash, 2) high IgE, and 3) failure to thrive. Mankahla A,
Am J Clin Dermatol. 2012;13(3):153-66
in about 25% of children with
HIV infection
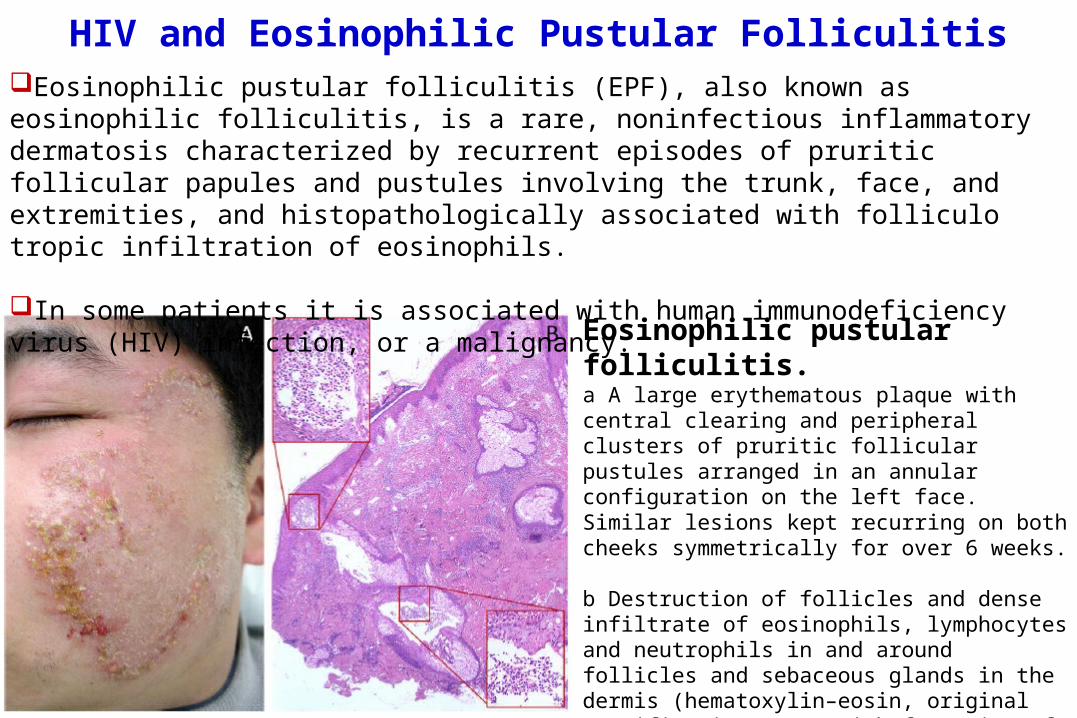
HIV and Eosinophilic Pustular Folliculitis
Eosinophilic pustular folliculitis. a A large erythematous plaque with central clearing and peripheral clusters of pruritic follicular pustules arranged in an annular configuration on the left face. Similar lesions kept recurring on both cheeks symmetrically for over 6 weeks.
b Destruction of follicles and dense infiltrate of eosinophils, lymphocytes and neutrophils in and around follicles and sebaceous glands in the dermis (hematoxylin–eosin, original magnification ×40), with formation of eosinophilic microabscess (red boxes original magnification ×400)
Eosinophilic pustular folliculitis (EPF), also known as eosinophilic folliculitis, is a rare, noninfectious inflammatory dermatosis characterized by recurrent episodes of pruritic follicular papules and pustules involving the trunk, face, and extremities, and histopathologically associated with folliculo tropic infiltration of eosinophils.
In some patients it is associated with human immunodeficiency virus (HIV) infection, or a malignancy.

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency (PID)?Eczema or Secondary Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal abnormalities “Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions
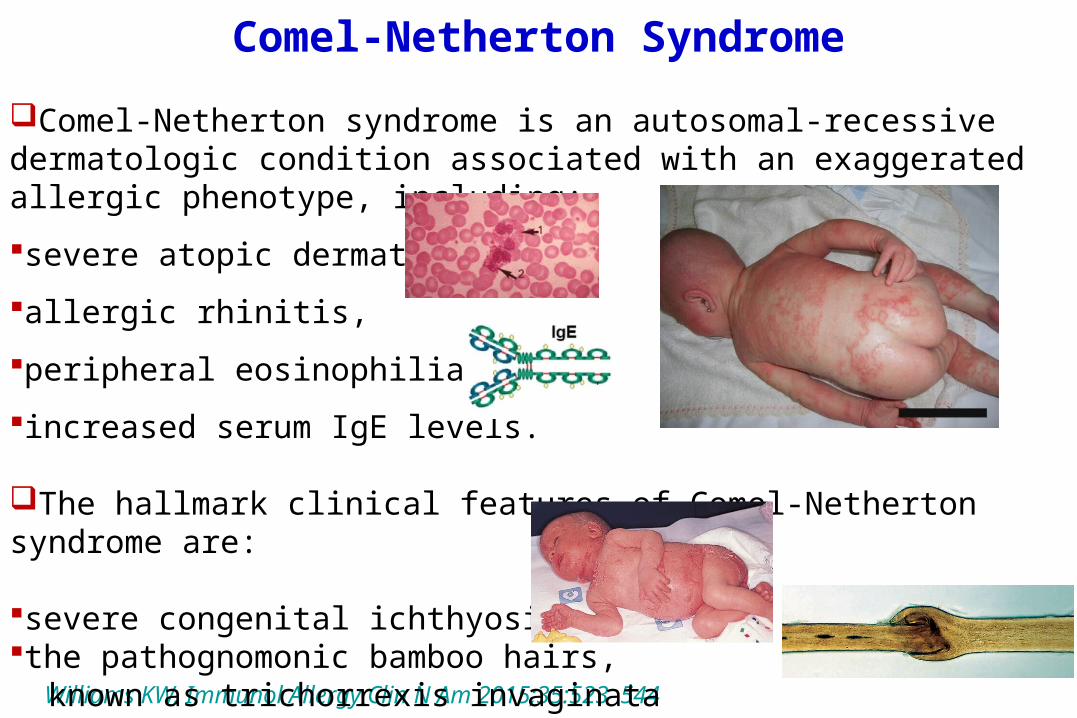
Comel-Netherton Syndrome
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Comel-Netherton syndrome is an autosomal-recessive dermatologic condition associated with an exaggerated allergic phenotype, including:severe atopic dermatitis, allergic rhinitis, peripheral eosinophilia, increased serum IgE levels.
The hallmark clinical features of Comel-Netherton syndrome are:
severe congenital ichthyosis and the pathognomonic bamboo hairs, known as trichorrexis invaginata

Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
The extent of skin involvement varies and may include: erythroderma or migrating and scaly plaques.
The skin lesions are usually pruritic secondary to the profound skin barrier defect and enhanced inflammation.
S aureus skin infections are frequent.
Additional findings include: failure to thrive, sparse or absent hair, eyebrows and eye lashes at birth, chronic diarrhea
Comel-Netherton Syndrome

Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
The extent of skin involvement varies and may include: erythroderma or migrating and scaly plaques.
The skin lesions are usually pruritic secondary to the profound skin barrier defect and enhanced inflammation.
S aureus skin infections are frequent.
Additional findings include: failure to thrive, sparse or absent hair, eyebrows and eye lashes at birth, chronic diarrhea
Comel-Netherton Syndrome
Mortality is highest in the neonatal period from sepsis, dehydration due to
transepidermal water loss, or malnutrition.
Hovnanian A. Cell Tissue Res 2013;351(2):289–300.
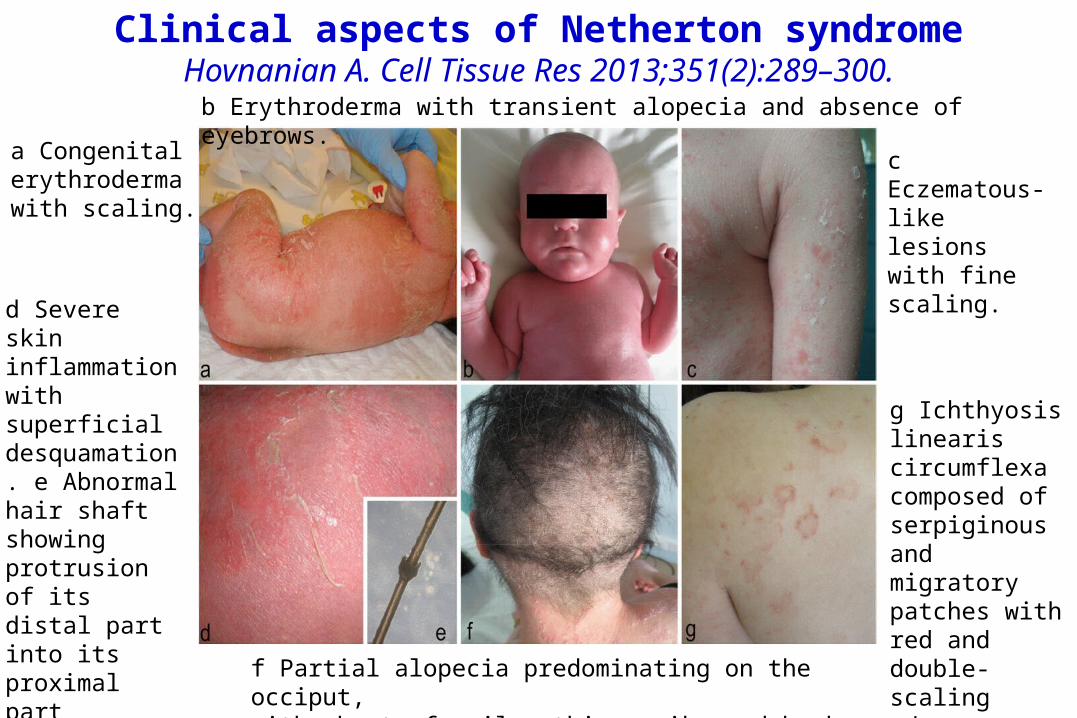
f Partial alopecia predominating on the occiput, with short, fragile, thin, spiky and broken hair.
Clinical aspects of Netherton syndromeHovnanian A. Cell Tissue Res 2013;351(2):289–300.
a Congenitalerythroderma with scaling.
b Erythroderma with transient alopecia and absence of eyebrows.
c Eczematous-like lesions with fine scaling.
d Severe skin inflammation with superficial desquamation. e Abnormalhair shaft showing protrusion of its distal part into its proximal part(trichorrhexis invaginata or bamboo hair).
g Ichthyosis linearis circumflexa composed of serpiginous and migratorypatches with red and double-scaling edges

Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Comel-Netherton Syndrome: Genetics and Pathogenesis
Comel-Netherton syndrome stems from loss-of-function mutations in SPINK5 (serine protease inhibitor Kazal-type 5) , which encodes the serine protease inhibitor LEKTI (lympho-epithelial kazal type related inhibitor type 5) that inhibits enzymes having a trypsin or chymotrypsin-like activity in the skin. LEKTI is expressed in both the epithelia and the thymus.
Murine models have shown that LEKTI deficiency leads to unopposed peptidase activity, causing impaired epidermal differentiation, defective cornification, and poor skin barrier formation.
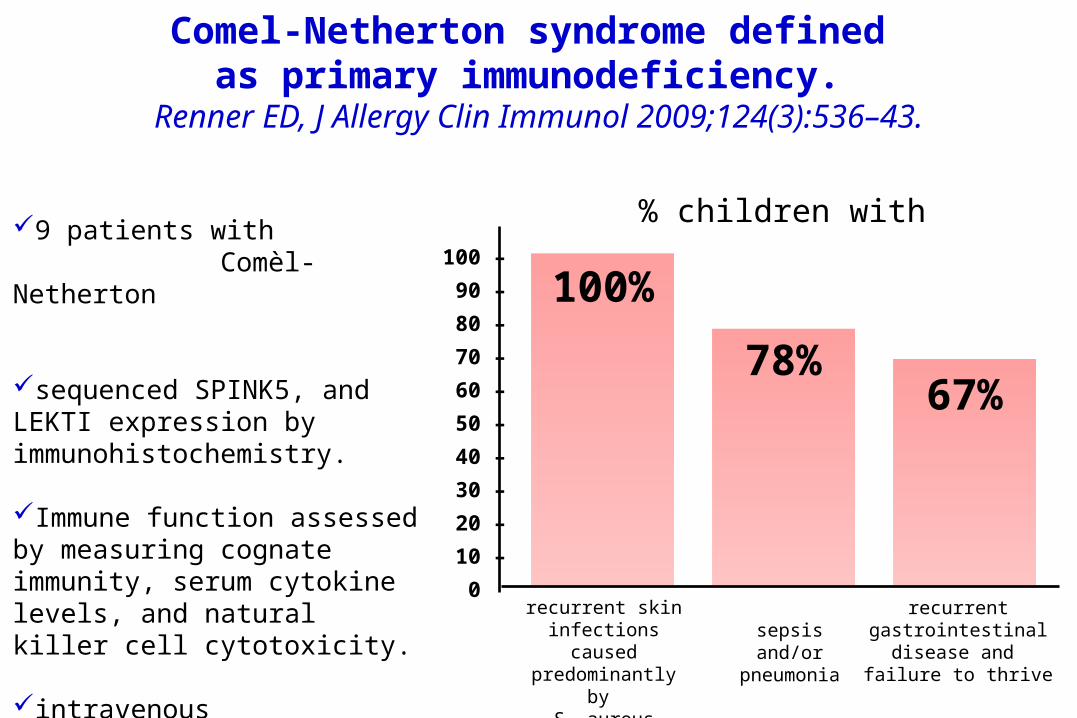
Comel-Netherton syndrome defined as primary immunodeficiency.
Renner ED, J Allergy Clin Immunol 2009;124(3):536–43.
9 patients with Comèl-Netherton
sequenced SPINK5, and LEKTI expression by immunohistochemistry.
Immune function assessed by measuring cognate immunity, serum cytokine levels, and natural killer cell cytotoxicity.
intravenous immunoglobulin replacement therapy.
100 –090 –080 –070 –060 –050 –040 –030 –020 –010 –000
recurrent skin infections caused predominantly by
S. aureus
sepsis and/or pneumonia
recurrent gastrointestinal
disease and failure to thrive
100% 78%
67%
% children with

Comel-Netherton syndrome defined as primary immunodeficiency.
Renner ED, J Allergy Clin Immunol 2009;124(3):536–43.
Treatment with intravenous immunoglobulin resulted in remarkable clinical improvement and temporarily
increased natural killer cell cytotoxicity.
9 patients with Comèl-Netherton
sequenced SPINK5, and LEKTI expression by immunohistochemistry.
Immune function assessed by measuring cognate immunity, serum cytokine levels, and natural killer cell cytotoxicity.
intravenous immunoglobulin replacement therapy.

Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Comel-Netherton Syndrome: Management
Management is aimed at systemic and cutaneous symptom management. Emollients are essential for their skin, and adequate nutrition and hydration is critical. •Renner ED, Comel-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol 2009;124(3):536–43.
Intravenous immunoglobulin and anti-TNF-a monoclonal antibodies have been effective in reducing skin inflammation.•Fontao L, Infliximab infusions for Netherton syndrome:sustained clinical improvement correlates with a reduction of thymic stromal lymphopoietin levels in the skin. J Invest Dermatol 2011;131(9):1947–50.

Severe Dermatitis, Multiple Allergies, and Metabolic Wasting Syndrome (SAM)
clinical features
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome closely resembles Comel-Netherton syndrome,
Unique to SAM syndrome is:
early and severe development of food allergies
prominent metabolic wasting.
malabsorption
failure to thrive.are also common

Severe Dermatitis, Multiple Allergies, and Metabolic Wasting Syndrome (SAM)
clinical features
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome closely resembles Comel-Netherton syndrome,
Unique to SAM syndrome is:
early and severe development of food allergies
prominent metabolic wasting.
malabsorption
failure to thrive.are also common
Recurrent infections, developmental delay, and minor cardiac defects,
such as ventricular septal defects, have also been described

Severe Dermatitis, Multiple Allergies, and Metabolic Wasting Syndrome (SAM)
clinical laboratory findings
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Increased serum IgE levels and absolute eosinophil counts.
Keratinocytes show upregulation of proinflammatory cytokine genes, such as IL5, which likely contributes to the eosinophilia.
Skin biopsy shows abnormally formed desmosomes and loss of cell-cell adhesion. Samuelov L, Nat Genet 2013; 45(10):1244–8.
+IL-5

Severe Dermatitis, Multiple Allergies, and Metabolic Wasting Syndrome (SAM)
genetics and pathogenesis
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Homozygous loss-of-function mutations in desmoglein-1 (DSG-1) were identified in 2 consanguineous families with SAM syndrome.
DSG-1 plays a crucial role in cell-cell adhesion in keratinocytes, as well as in myocardial cells. Amagai M. J Invest Dermatol 2012;132(3 Pt 2):776–84.
DSG-1 deficiency leads to absent cell-cell adhesion and abnormally formed epidermal desmosomes with consequent epidermal barrier dysfunction.

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency (PID)?Eczema or Secondary Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal
abnormalities “Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions

Loeys-Dietz Syndrome: Clinical features
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Loeys-Dietz syndrome (LDS) is a connective tissue disorder with a great clinical overlap with Autosomal Dominant Hyper IgE Syndrome.
Musculoskeletal abnormalitiesare variable and can include:
craniosynostosis, retained primary dentition, facial asymmetry, pectus deformity, scoliosis, flat feet, joint hyperextensibility, high arched or cleft palate, abnormal uvula, hypertelorism.

Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Their skin is classically thin and translucent, and easy bruising and poor wound healing are not uncommon.
Vascular anomalies are a prominent feature of LDS.
Diffuse arterial abnormalities, such as aneurysms and tortuosity, put patients at great risk for dissection or hemorrhages.
Patients with LDS have an exaggerated allergic phenotype, which can include: asthma, food allergy, atopic dermatitis, allergic rhinitis
Loeys-Dietz Syndrome: Clinical features

Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Their skin is classically thin and translucent, and easy bruising and poor wound healing are not uncommon.
Vascular anomalies are a prominent feature of LDS.
Diffuse arterial abnormalities, such as aneurysms and tortuosity, put patients at great risk for dissection or hemorrhages.
Patients with LDS have an exaggerated allergic phenotype, which can include: asthma, food allergy, atopic dermatitis, allergic rhinitis
Loeys-Dietz Syndrome: Clinical features
Not present in theAutosomal Dominant Hyper IgE SyndromeBut present in the Autosomal Recessive Hyper IgE Syndrome

Gastrointestinal complaints, such as:
chronic abdominal pain, poor growth, constipation, vomiting,
Eosinophilic gastrointestinal disease (eosinophilic esophagitis, colitis, or gastritis)
Elimination diets seem to be beneficial in reducing clinical symptoms.
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
are common
is often diagnosed pathologically.
Loeys-Dietz Syndrome: Clinical features

Loeys-Dietz Syndrome: Genetics and pathogenesis
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Four genetic mutations have been identified in LDS.
These mutations include heterozygous mutations in TGFBR1, TGFBR2, SMAD3, and TGFB2.
Each mutation results in altered TGF-β signaling, yielding abnormal collagen and connective tissue growth, and effects on lymphocyte differentiation.

Loeys-Dietz Syndrome: Management
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Despite the genetic and phenotypic variation seen in the LDS types, medical management is similar and very complex:
Cardiovascular care
Orthopedic care
Allergies care
Gastroenterology and nutrition care

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency (PID)?Eczema or Secondary Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal abnormalities“Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions

Food Protein-Induced Enterocolitis Syndrome.Leonard SA, Pediatr Clin North Am. 2015;62(6):1463-77
Food protein-induced enterocolitis syndrome (FPIES) is a rare, non-IgE-mediated gastrointestinal food allergy primarily diagnosed in infancy, but has also been reported in older children and adults.
Acute FPIES reactions typically present within 1 to 4 hours of food ingestion with:
Chronic FPIES typically presents with:
delayed, repetitive vomiting, lethargy, pallorcollapse
protracted vomiting and/or diarrhea, weight loss or poor growth.

Food Protein-Induced Enterocolitis Syndrome.Leonard SA, Pediatr Clin North Am. 2015;62(6):1463-77
Food protein-induced enterocolitis syndrome (FPIES) is a rare, non-IgE-mediated gastrointestinal food allergy primarily diagnosed in infancy, but has also been reported in older children and adults.
Acute FPIES reactions typically present within 1 to 4 hours of food ingestion with:
Chronic FPIES typically presents with:
delayed, repetitive vomiting, lethargy, pallorcollapse
protracted vomiting and/or diarrhea, weight loss or poor growth.
Common foods triggering FPIES include: •cow's milk, •soy, •rice, •oats, •fish, •egg.

Food Protein-Induced Enterocolitis Syndrome.Leonard SA, Pediatr Clin North Am. 2015;62(6):1463-77
Food protein-induced enterocolitis syndrome (FPIES) is a rare, non-IgE-mediated gastrointestinal food allergy primarily diagnosed in infancy, but has also been reported in older children and adults.
Acute FPIES reactions typically present within 1 to 4 hours of food ingestion with:
Chronic FPIES typically presents with:
delayed, repetitive vomiting, lethargy, pallorcollapse
protracted vomiting and/or diarrhea, weight loss or poor growth.
most Cow Milk or Soy FPIES presented with chronic symptoms, such as diarrhea,
colitis, reflux, or failure to thrive, occurring shortly after CM or soy formula introduction,
which resolved with avoidance.

Food Protein-Induced Enterocolitis Syndrome.Leonard SA, Pediatr Clin North Am. 2015;62(6):1463-77
Food protein-induced enterocolitis syndrome (FPIES) is a rare, non-IgE-mediated gastrointestinal food allergy primarily diagnosed in infancy, but has also been reported in older children and adults.
Acute FPIES reactions typically present within 1 to 4 hours of food ingestion with:
Chronic FPIES typically presents with:
delayed, repetitive vomiting, lethargy, pallorcollapse
protracted vomiting and/or diarrhea, weight loss or poor growth.
Owing to nonspecific symptoms and the lack of diagnostic testing, FPIES is often
initially misdiagnosed, leading
to a delay in diagnosis and increased morbidity from extensive workups and
hospitalizations.

Food Protein-Induced Enterocolitis Syndrome.Leonard SA, Pediatr Clin North Am. 2015;62(6):1463-77
Many FPIES patients have an atopic background.
Eczema was reported in 9% to 57%,
Wheezing or asthma was reported in 3% to 25%,
Allergic rhinitis was reported in 38%,
IgE-mediated food allergy to other foods in 11% to 30%,
A family history of allergic disease was reported in 20% to 77%,
A family history of FPIES in 6% and of food allergy in 34%.
less than 10% of subjects had a positive SPT to trigger foods, and between 11% and 24% had detectable sIgE levels.

The pendulum between food protein-induced enterocolitis syndrome and IgE-mediated milk
allergy.Kessel A, Acta Paediatr. 2011;100(10):e183-5.The transition from milk protein-induced enterocolitis syndrome
to IgE-mediated milk allergy is uncommon.
Herein, we describe three infants that suffered from recurrent vomiting and restlessness in response to cow's milk formula with negative skin prick to milk and therefore diagnosed as milk protein-induced enterocolitis syndrome.
After recovering and reintroducing cow's milk formula, they developed disseminated urticaria and positive skin prick test to cow milk compatible with IgE-mediated milk allergy.
CONCLUSION: An infant that recovers from cow milk food-induced enterocolitis syndrome might develop afterward IgE-mediated cow milk allergy.
IgEFPIES

Unusual shift from IgE-mediated milk allergy to food protein-induced enterocolitis syndrome.
Banzato C, Eur Ann Allergy Clin Immunol. 2013;45(6):209-11
In children with FPIES, the presence of sIgE to the causative food, either at presentation or during follow-up, defines an "atypical form" of FPIES characterized by a lesser probability of developing tolerance and a potential progression to typical IgE-mediated hypersensitivity.
Although it is uncommon, the shift from non-IgE-mediated milk-protein induced enterocolitis syndrome to IgE-mediated milk allergy has recently been described.
We report the first case, to our knowledge, of a shift from IgE-mediated cow's milk allergy to pure non-IgE-mediated FPIES, in a 4-month-old male infant.
IgE
FPIES

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency (PID)?Eczema or Secondary Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal abnormalities “Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions

Pulmonary Manifestations of Primary Immunodeficiency Disorders
Nonas S. Immunol Allergy Clin North Am. 2015;35(4):753-66.
Pulmonary complications of primary immunodeficiency disorders (PIDDs) are common and contribute significantly to morbidity and mortality in these patients.
Recurrent pulmonary infections are often the first warning sign of PIDD and remain a leading cause of death from infectious causes in adults with PIDD.
Recurrent pulmonary infections, including pneumonia, lung abscess, and/or empyema formation, account for significant morbidity and mortality in PIDD, accounting for 29% to 44% of deaths.

Pulmonary Manifestations of Primary Immunodeficiency Disorders
Nonas S. Immunol Allergy Clin North Am. 2015;35(4):753-66.
The mortality from infectious complications of PIDD has significantly decreased in the past several decades, following the widespread standard use of immunoglobulin replacement therapy starting in the 1980s.
With this increased longevity came an increasing prevalence in, and mortality from, noninfectious complications in PIDD.
In short, patients now live long enough to develop chronic noninfectious complications, particularly in the lungs including bronchiectasis, interstitial lung disease (ILD) , pulmonary malignancy, and autoimmunity.

Nonas S. Immunol Allergy Clin North Am. 2015;35(4):753-66.
Obstructive Airway Disease and BronchiectasisIn CVID the prevalence of bronchiectasis is greater than 70%. Thickett KM, QJM 2002;95:655–62.
classic tram-trackappearance.
Obstructive airway disease, including asthma, bronchiolitis, and bronchiectasis, is extremely common in primary immunodeficiency.
Obstructive spirometry is noted in 50% to 94% of patients with primary immunodeficiency disease (PIDDs). Touw CM, Pediatr Allergy Immunol 2009;21:793–805.
Asthma is particularly common in patients with antibody deficiency, with rates between 15% and 42% of patients with common variable immune deficiency (CVID), selective immunoglobulin (Ig) A deficiency, and X-linked agammaglobulinemia (XLA). Agondi RC, Allergy 2010;65:510–5. Ozcan C, Allergol Immunopathol (Madr) 2015;43:57–61.
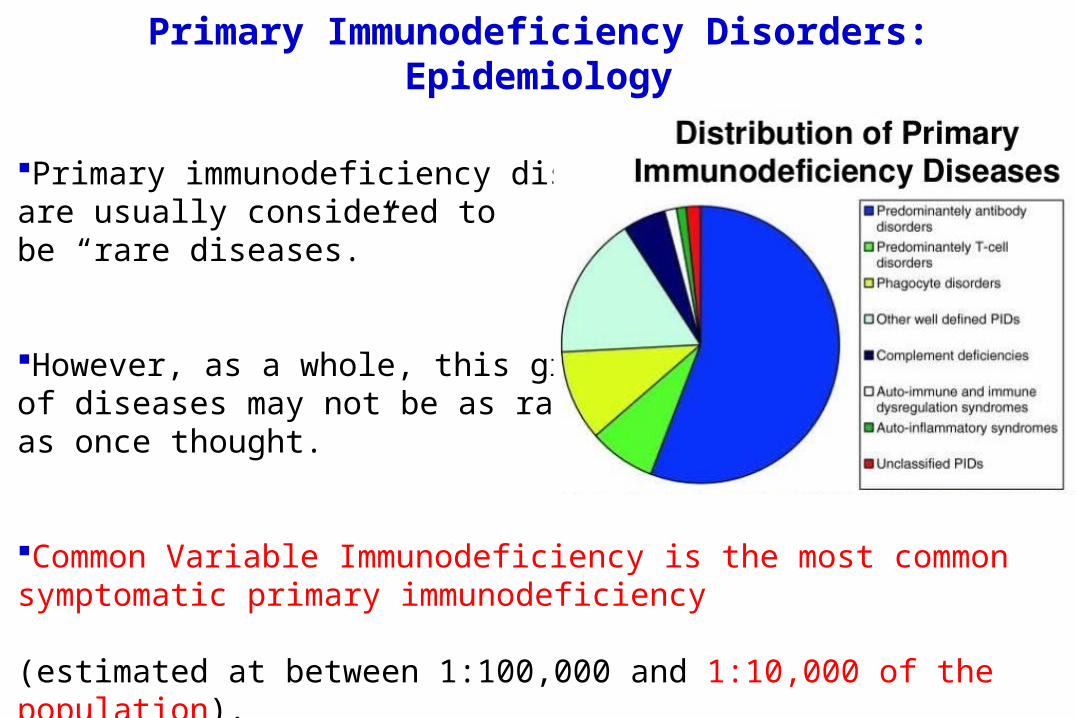
Primary Immunodeficiency Disorders: Epidemiology
Primary immunodeficiency disordersare usually considered tobe “rare diseases.”
However, as a whole, this group of diseases may not be as rare as once thought.
Common Variable Immunodeficiency is the most common symptomatic primary immunodeficiency (estimated at between 1:100,000 and 1:10,000 of the population).

Common Variable Immunodeficiency: Diagnosis, Management, and Treatment.
Abbott JK, Immunol Allergy Clin North Am. 2015;35(4):637-58
CVID occurs equally in males and females, although, among children, boys predominate.
CVID age of onset is variable, but it peaks in early childhood (34% < 10 years of age) and around the third decade of life.
Several noninfectious complications are commonly reported, including: splenomegaly, chronic gastrointestinal (GI) disease, bronchiectasis, chronic lung disease, autoimmune cytopenia, andother autoimmune, malignant, and granulomatous diseases.
0 5 10 20 30age years

Common Variable Immunodeficiency: Diagnosis, Management, and Treatment.
Abbott JK, Immunol Allergy Clin North Am. 2015;35(4):637-58
Common variable immunodeficiency (CVID) is a grouping of heterogeneous diseases with the common finding of impaired antibody production.
the following criteria for probable CVID were proposed: 1) > 2 years of age, 2) IgG, IgA, IgM < 2 standard deviations from the mean for age, 3) either absent isohemagglutinin* or absent vaccine responses, and 4) no other defined causes of hypogammaglobulinemia.* Isohemagglutinin is the naturally occurring anti-A and anti-B antibodies against the antigens present on red blood cells.
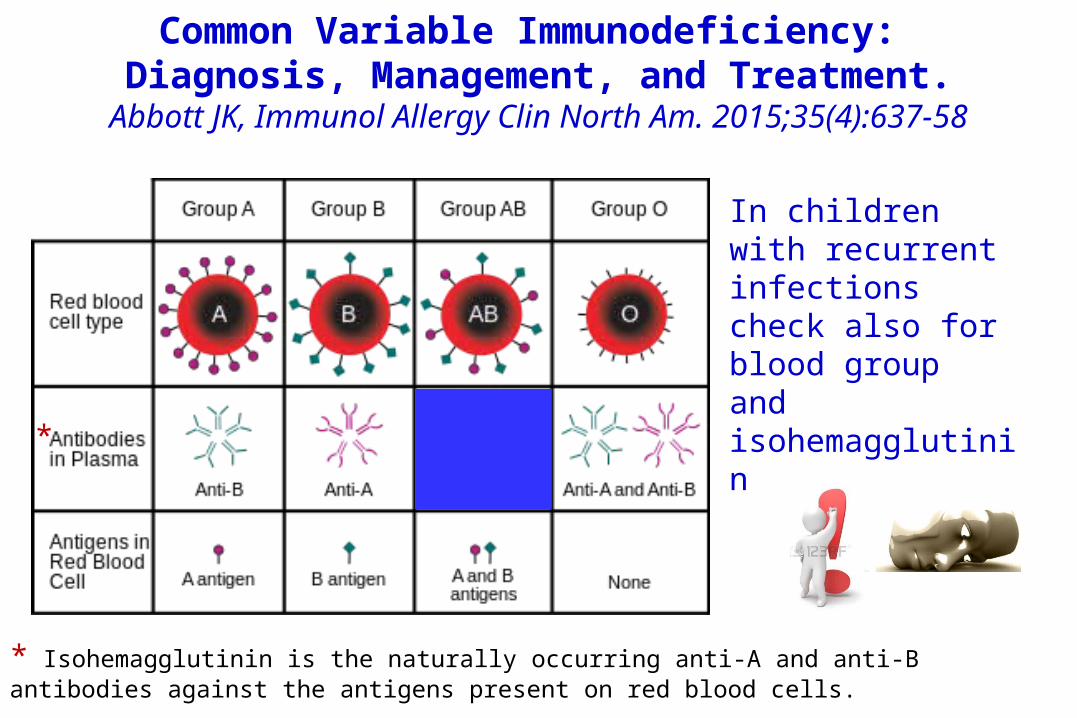
Common Variable Immunodeficiency: Diagnosis, Management, and Treatment.
Abbott JK, Immunol Allergy Clin North Am. 2015;35(4):637-58
* Isohemagglutinin is the naturally occurring anti-A and anti-B antibodies against the antigens present on red blood cells.
*
In children with recurrent infections check also for blood group and isohemagglutinin
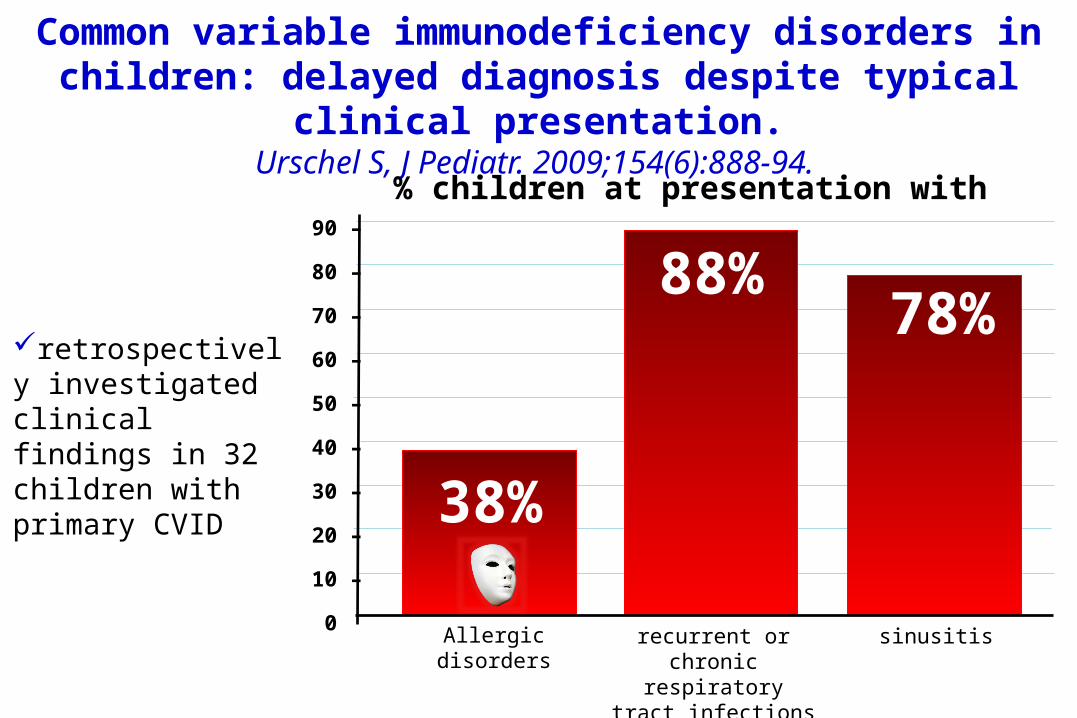
Common variable immunodeficiency disorders in children: delayed diagnosis despite typical
clinical presentation.Urschel S, J Pediatr. 2009;154(6):888-94.
retrospectively investigated clinical findings in 32 children with primary CVID
90 –
80 –
70 –
60 –
50 –
40 –
30 –
20 –
10 –
00
% children at presentation with
88%
recurrent or chronic respiratory
tract infections
sinusitis
78%
Allergic disorders
38%
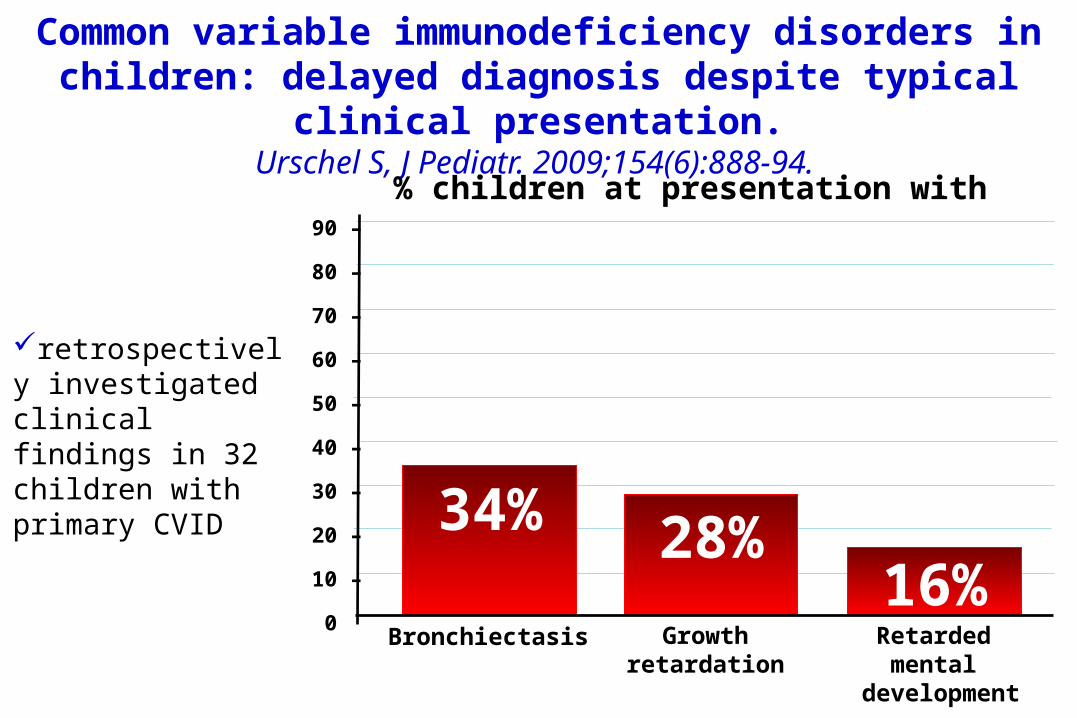
Common variable immunodeficiency disorders in children: delayed diagnosis despite typical
clinical presentation.Urschel S, J Pediatr. 2009;154(6):888-94.
retrospectively investigated clinical findings in 32 children with primary CVID
90 –
80 –
70 –
60 –
50 –
40 –
30 –
20 –
10 –
00
% children at presentation with
28%16%
34%Bronchiectasis Growth
retardation Retarded mental
development
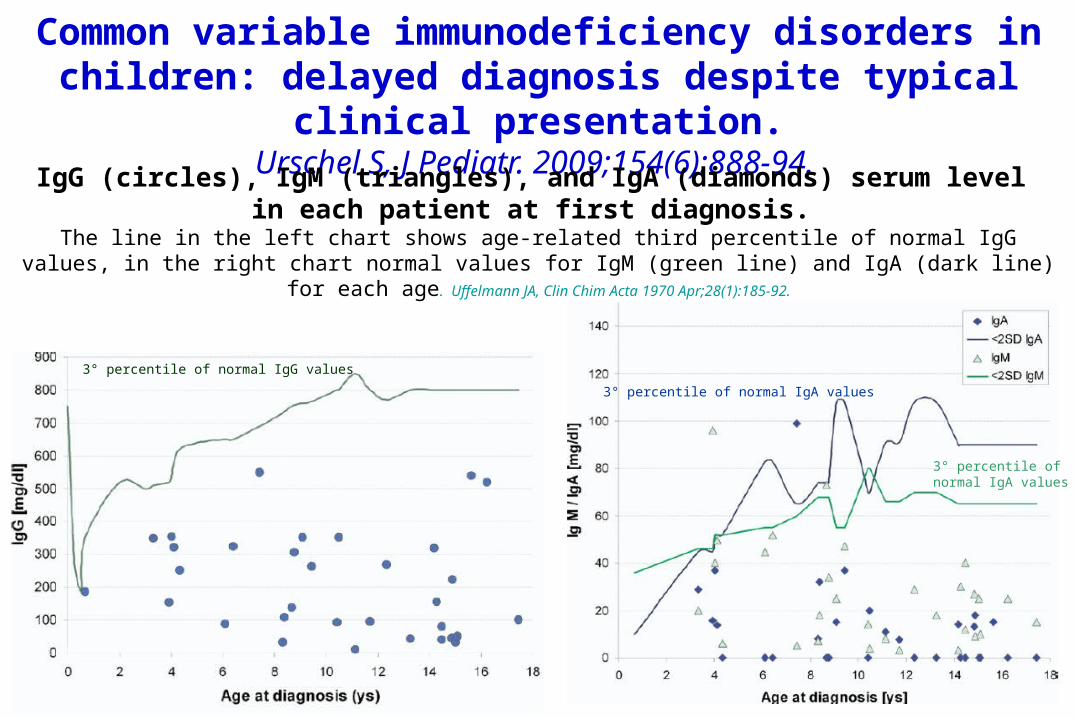
Common variable immunodeficiency disorders in children: delayed diagnosis despite typical
clinical presentation.Urschel S, J Pediatr. 2009;154(6):888-94. IgG (circles), IgM (triangles), and IgA (diamonds) serum level
in each patient at first diagnosis. The line in the left chart shows age-related third percentile of normal IgG values, in the right chart
normal values for IgM (green line) and IgA (dark line) for each age. Uffelmann JA, Clin Chim Acta 1970 Apr;28(1):185-92.
3° percentile of normal IgG values3° percentile of normal IgA values
3° percentile of normal IgA values
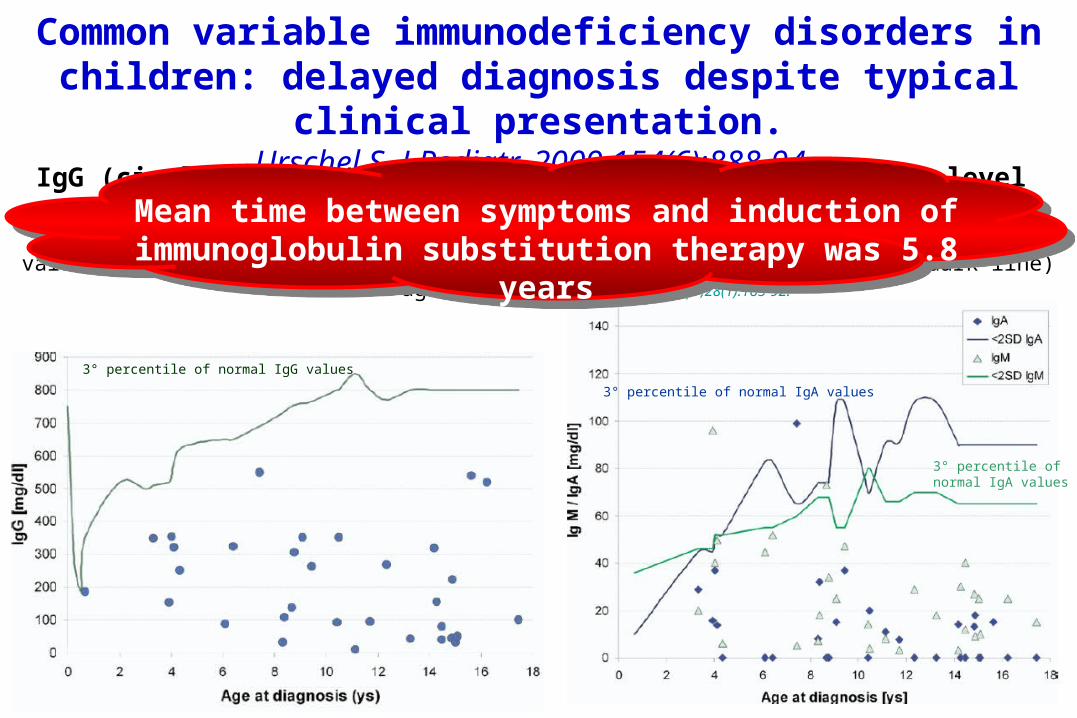
Common variable immunodeficiency disorders in children: delayed diagnosis despite typical
clinical presentation.Urschel S, J Pediatr. 2009;154(6):888-94. IgG (circles), IgM (triangles), and IgA (diamonds) serum level
in each patient at first diagnosis. The line in the left chart shows age-related third percentile of normal IgG values, in the right chart
normal values for IgM (green line) and IgA (dark line) for each age. Uffelmann JA, Clin Chim Acta 1970 Apr;28(1):185-92.
3° percentile of normal IgG values3° percentile of normal IgA values
3° percentile of normal IgA values
Mean time between symptoms and induction of immunoglobulin substitution therapy was 5.8
years

Common Variable Immunodeficiency: Treatment.Abbott JK, Immunol Allergy Clin North Am. 2015;35(4):637-58
Effective treatment of CVID depends on adequate treatment of both infectious and noninfectious medical issues.
IgG replacement is generally effective in preventing further infections, although, for patients who have bronchiectasis, aggressive pulmonary physiotherapy should also be implemented.
Antibiotic prophylaxis is frequently used in patients with persistent infections.
Treatment of noninfectious complications requires specificmanagement approaches tailored to the specific problem. Medications can include immunosuppressives and even cytotoxic therapies.

Adenosine Deaminase–Severe Combined Immunodeficiency
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Peripheral eosinophilia is a commonly encountered clinical manifestation seen in adenosine deaminase (ADA)-SCID.
Because of the profound T-cell, B-cell, and NK-cell lymphopenia seen in ADA-SCID, affected individuals typically present in infancy with severe opportunistic infections, such as with Pneumocysti jirovecii pneumonia.
Recurrent or severe respiratory infections typically occur within the first few months of life, along with failure to thrive, frequent thrush, and chronic diarrhea.
Cysts of Pneumocystis jiroveci in smear from bronchoalveolar lavage. Methenamine silver
stain

Adenosine Deaminase–Severe Combined Immunodeficiency
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Atopy is a common feature in ADA-SCID, found in about half of those with early onset disease.
Of the atopic manifestations:allergic rhinitis, wheezy bronchitis, asthma, food allergy,mild atopic dermatitis, urticaria.
were the most common identified in this population
(+) sIgE

Adenosine Deaminase–Severe Combined Immunodeficiency
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Atopy is a common feature in ADA-SCID, found in about half of those with early onset disease.
Of the atopic manifestations:allergic rhinitis, wheezy bronchitis, asthma, food allergy,mild atopic dermatitis, urticaria.
were the most common identified in this population
(+) sIgE
Without early recognition and
treatment, ADA-SCID is often fatal within the
first year of life.

Adenosine Deaminase–Severe Combined Immunodeficiency
Diagnosis
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
Profound deficiency of circulating T lymphocytes, B lymphocytes, and NK cells.
Serum IgG levels are usually normal at presentation as a result of maternal transfer of IgG but decrease soon after.
Serum IgA and IgM levels are variable, although more commonly IgM levels are low.
Serum eosinophilia and IgE levels are frequently increased among both those with early and delayed onset, presumably because of their increased CD4+ Th2 cytokine production and their clinical features of atopy.
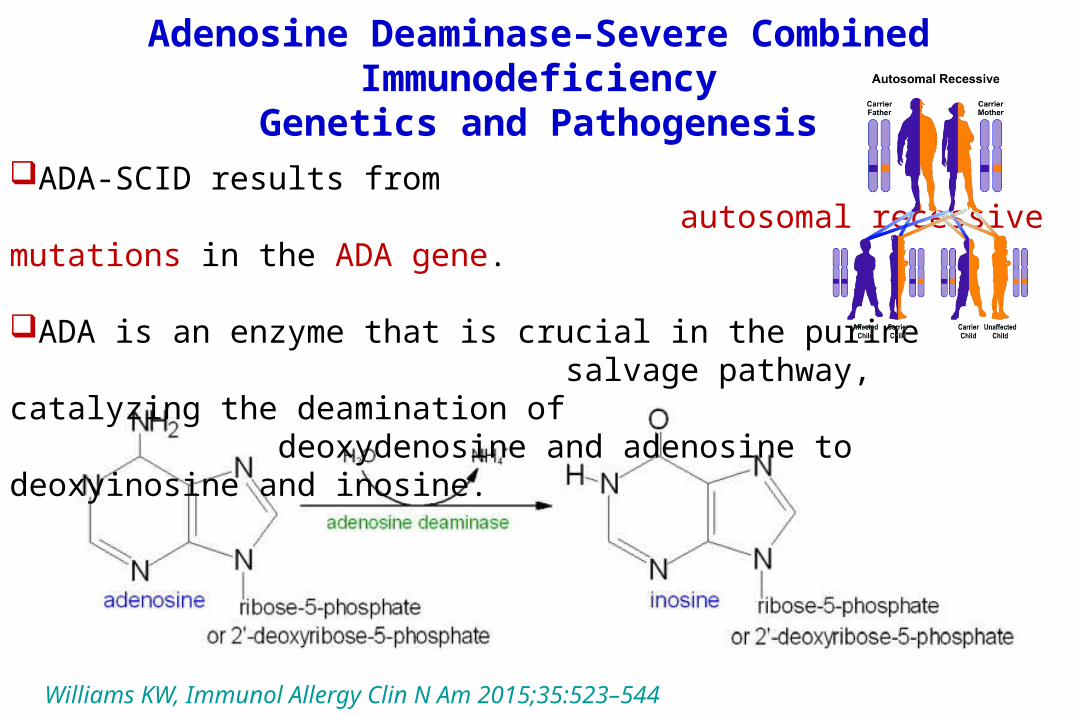
Adenosine Deaminase–Severe Combined Immunodeficiency
Genetics and Pathogenesis
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
ADA-SCID results from autosomal recessive mutations in the ADA gene.
ADA is an enzyme that is crucial in the purine salvage pathway, catalyzing the deamination of deoxydenosine and adenosine to deoxyinosine and inosine.
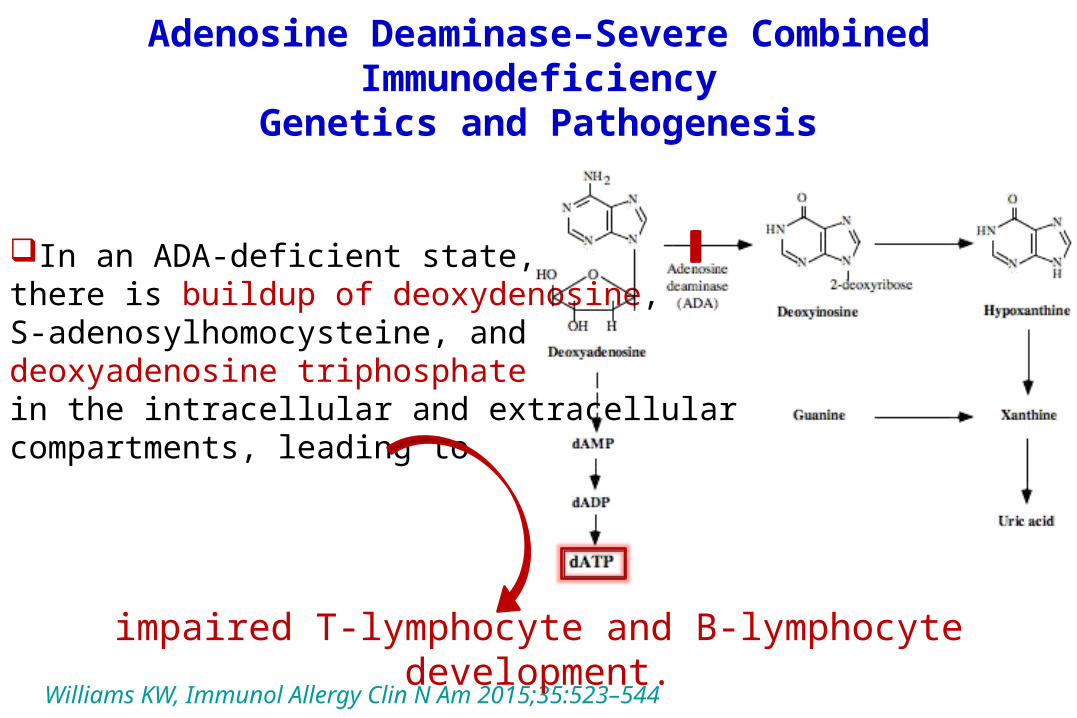
Adenosine Deaminase–Severe Combined Immunodeficiency
Genetics and Pathogenesis
Williams KW, Immunol Allergy Clin N Am 2015;35:523–544
In an ADA-deficient state, there is buildup of deoxydenosine,S-adenosylhomocysteine, and deoxyadenosine triphosphate in the intracellular and extracellularcompartments, leading to
impaired T-lymphocyte and B-lymphocyte development.
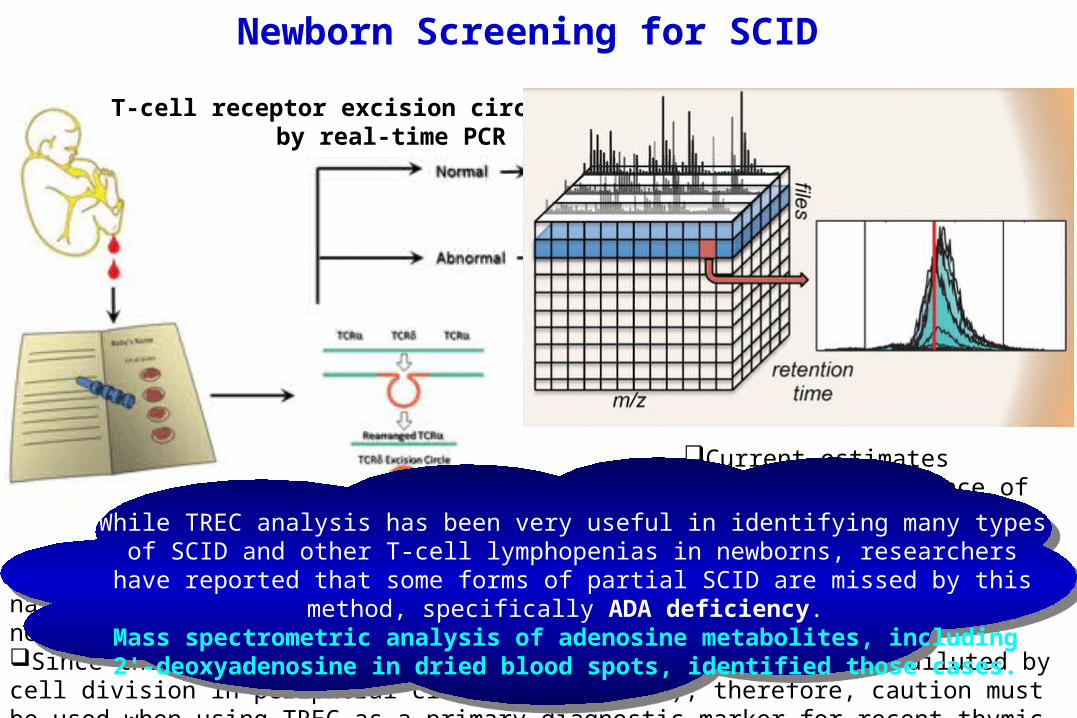
Newborn Screening for SCID
Newborn screening for SCID begins with analysis of a dried blood spot collected at birth.
Babies with a positive result should have additional confirmatory tests.
Current estimates suggest the incidence of this group of immunodeficiency disorders is as high as 1 in 40,000 births.
T-cell receptor excision circle (TREC) serves as a helpful marker of naive T-cell development and production in the thymus, especially in the neonate. Since these excision circles are extra-chromosomal, they are diluted by cell division in peripheral circulation (blood); therefore, caution must be used when using TREC as a primary diagnostic marker for recent thymic emigration.
T-cell receptor excision circle (TREC) by real-time PCR
While TREC analysis has been very useful in identifying many types of SCID and other T-cell lymphopenias in newborns, researchers have reported that
some forms of partial SCID are missed by this method, specifically ADA deficiency.
Mass spectrometric analysis of adenosine metabolites, including 2'-deoxyadenosine in dried blood spots, identified those cases.

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency (PID)?Eczema or Secondary Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal abnormalities “Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions

that can be helped with some type of corticosteroid therapy.
The majority of patients with
eczema, eosinophilia, elevated IgE
are likely to be atopic individuals
oral corticosteroidshave long been the mainstay of
therapy to treat severe flares of allergic
diseases.
inhibit production of proinflammatory cytokines, chemokines, arachidonic acid
metabolites, and adhesion molecules while concurrently upregulating anti-inflammatory
mediators.

immune suppression by administered corticosteroids can lead to detrimental effects if
an underlying PID goes unrecognized
patients with the PIDs rely on the remaining functioning parts of their immune system to control latent infections and combat opportunistic diseases Pneumocystis jirovecii,
Mycobacterium avium complex, fungi

immune suppression by administered corticosteroids can lead to detrimental effects if
an underlying PID goes unrecognized
patients with the PIDs rely on the remaining functioning parts of their immune system to control latent infections and combat opportunistic diseases Pneumocystis jirovecii,
Mycobacterium avium complex, fungi
practitioners should exercise prudence before using elective systemic
corticosteroids or other immune suppressants
if there is a history of recurrent or opportunistic infection(s).

Masquerade syndromes in allergic diseases
Attilio BonerUniversity ofVerona, Italy
IntroductionEczema or Primary Immunodeficiency (PID)?Eczema or Secondary Immunodeficiency?Eczema with an exagerated skin barrier
defect Allergy with musculoskeletal abnormalities “Food allergy” without sIgE Respiratory Tract Manifestations in PIDCaution is importantSummary & Conclusions

PIDs are rare diseases that can be masked or not considered because of the predominant clinical features of atopy.
The differential diagnosis of eosinophilia and Hyper IgE is broad and includes disorders of immune deficiency or dysfunction.
Since there is a great deal of overlap in many of these disorders, and thus, clinical and laboratory evaluation is warranted.
Summary & Conclusions
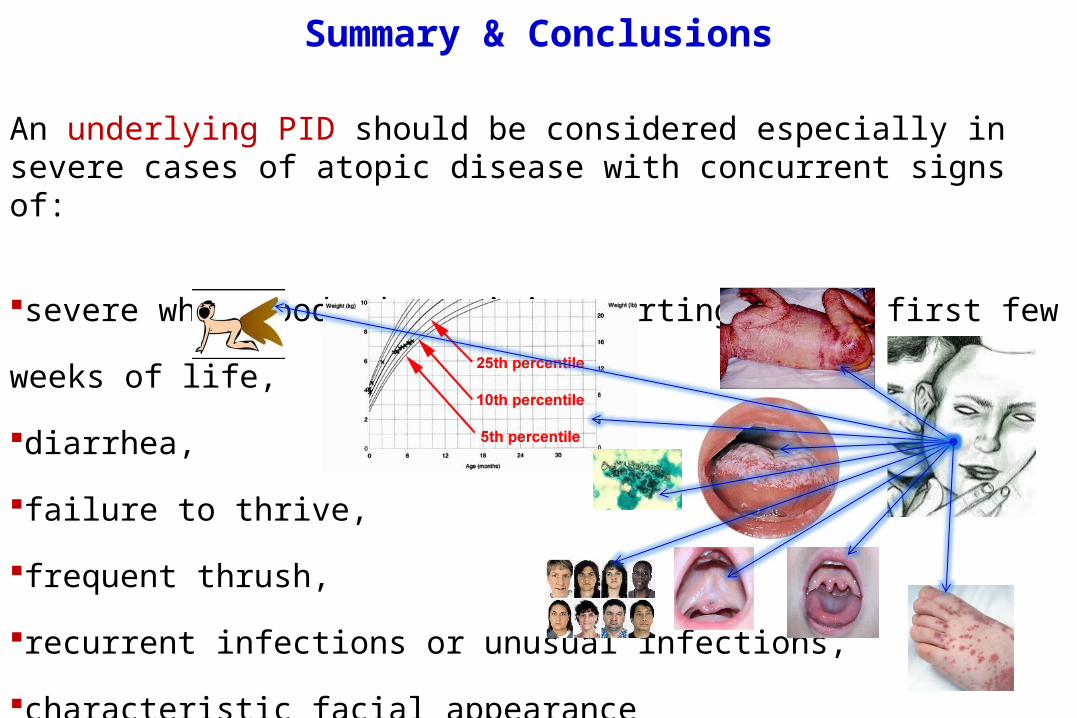
An underlying PID should be considered especially in severe cases of atopic disease with concurrent signs of:
severe whole body dermatitis starting in the first few weeks of life,diarrhea, failure to thrive,frequent thrush, recurrent infections or unusual infections,characteristic facial appearanceautoimmunity (early onset diabetes, thrombocytopenia)
Summary & Conclusions

An underlying PID should be considered especially in severe cases of atopic disease with concurrent signs of:
severe whole body dermatitis starting in the first few weeks of life,diarrhea, failure to thrive,frequent thrush, recurrent infections or unusual infections,characteristic facial appearanceautoimmunity (early onset diabetes, thrombocytopenia)
Summary & Conclusions
Without considering an underlying PID, some individuals will remain
undiagnosed and untreated, and this risk impacts
their morbidity and mortality, especially when exposed to agents that
further reduce immune competence.

19° FORMAT Verona 12-13/05/2017

Grazie per la vostra attenzione alla storia che
vi ha raccontato il mio nonno.
Ciao a tutti.Mia Charlize Powell





























