Embed Size (px)
Citation preview

Dr.Abdallah Rabba
Shereen Rawajbeh
Sondos Hassouneh
Areej Abu Hanieh1

Valproic acid (VPA) is a broad-spectrum,
carboxylic acid-derived anticonvulsant that has
been used in the treatment of first line therapy of
mania ,epilepsy, bipolar disease, schizophrenia,
and migraine headache.
2
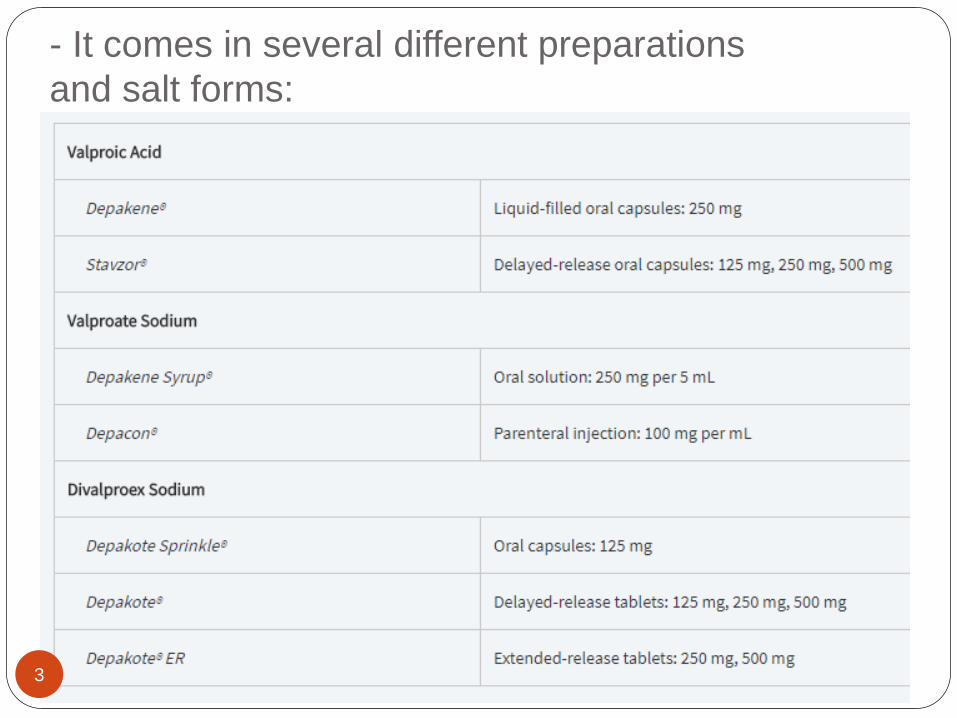
- It comes in several different preparations
and salt forms:
3

For example :
Divalproex sodium is a mixture of equal parts of
the acid and sodium salts of valproic acid.
The delayed-release (Depakote) and extended-
release (Depakote ER) formulations are not
bioequivalent.1 A 20 percent increase in the daily
dose is recommended when switching from
Depakote to Depakote ER to account for the
differences in rate and extent of absorption.
4

5

Dosing initial oral dosing for patients 10 years of age and
older is 10–15 mg/kg/day. Dosing intervals for the
oral and parenteral preparations are typically
every 8–12 hours (although dosing every 6 hours
may be needed in some patients), Who are those
patients ?
with the exception of the extended-release
formulation, which can be administered once
daily.
6

Dosing : The daily dose can be titrated weekly by 5–10
mg/kg/day to a maximum recommended dose of
60 mg/kg/day.
Loading doses are not recommended for the oral
VPA formulations due to intolerable
gastrointestinal side effects.
Intravenous loading doses of valproate sodium 25
mg/kg are commonly given for patients in status
epilepticus.
Intravenous loading doses can be given at a rate
of 1.5–3 mg/kg/min, but faster rates of up to 6
mg/kg/min appear to be safe.
7

Therapeutic drug monitoring
(TDM)
Is used in conjunction with the clinical exam to
optimize seizure control and minimize toxicity.
Measuring serum concentrations is routinely
performed via immunoassay ( what is
immunoassay ?), and the therapeutic range is
reported as 50–100 mcg/mL, although individual
patients may have optimal responses outside
these ranges.
8

Toxicity : Concentrations higher than 150 mcg/mL are
associated with a high incidence of CNS side effects. Free (unbound) concentrations are also available with a therapeutic range reported to be 6–22 mcg/mL with toxicity occurring above 50 mcg/mL.
Diurnal variations in the serum concentration of VPA have been reported, so it is important to be consistent when sampling in order to properly compare levels and adjust dosing regimens. Clearance tends to be higher in the evening compared to the morning times in both young adults and elderly subjects , Why ? , so trough levels are recommended before the morning dose.
9
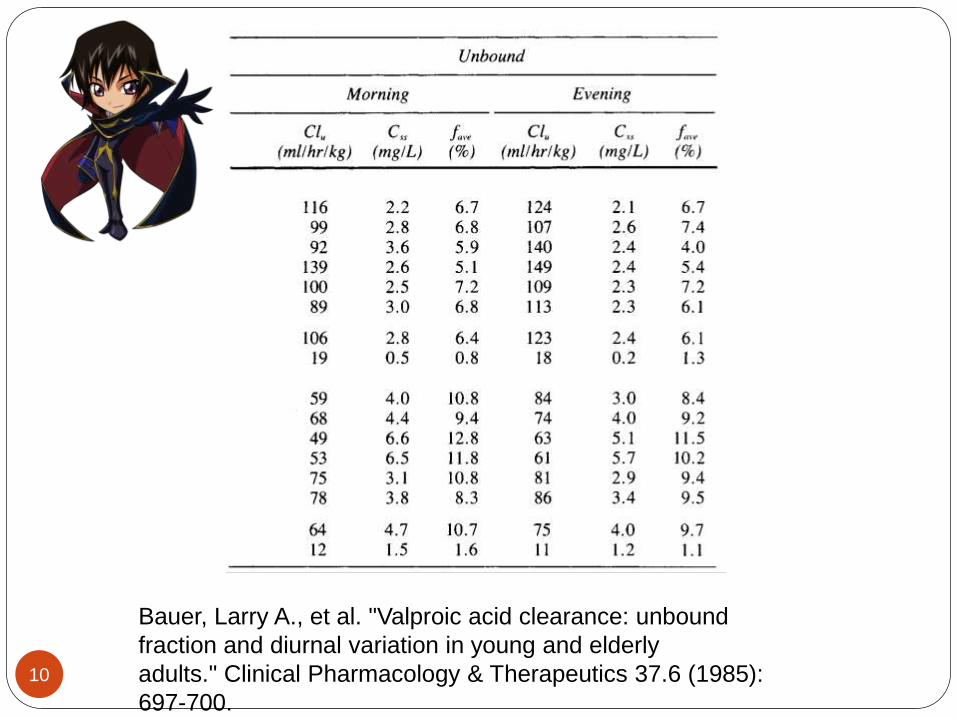
Bauer, Larry A., et al. "Valproic acid clearance: unbound
fraction and diurnal variation in young and elderly
adults." Clinical Pharmacology & Therapeutics 37.6 (1985):
697-700.
10

BIOAVAILABILITY:
All of the formulations of valproic acid have
bioavailabilities near 100 percent (F = 1),
reflecting the absence of a first-pass effect
in the liver.
The enteric-coated formulation has a
bioavailability of approximately 90 percent
(F = 0.9)
Valproate sodium is rapidly converted to
valproic acid in the stomach and then
readily absorbed via the gastrointestinal
tract. 11

Peak concentrations are achieved within
0.5–2 hours after oral administration. The
presence of food will delay the peak
concentration but not the extent of
absorption.
The volume of distribution in adults is
reported to range from 0.1 to 0.4
L/kg, indicating that VPA remains primarily
within the intravascular and extracellular
space. The volume of distribution tends to
increase with higher doses due to
saturable protein binding.
12
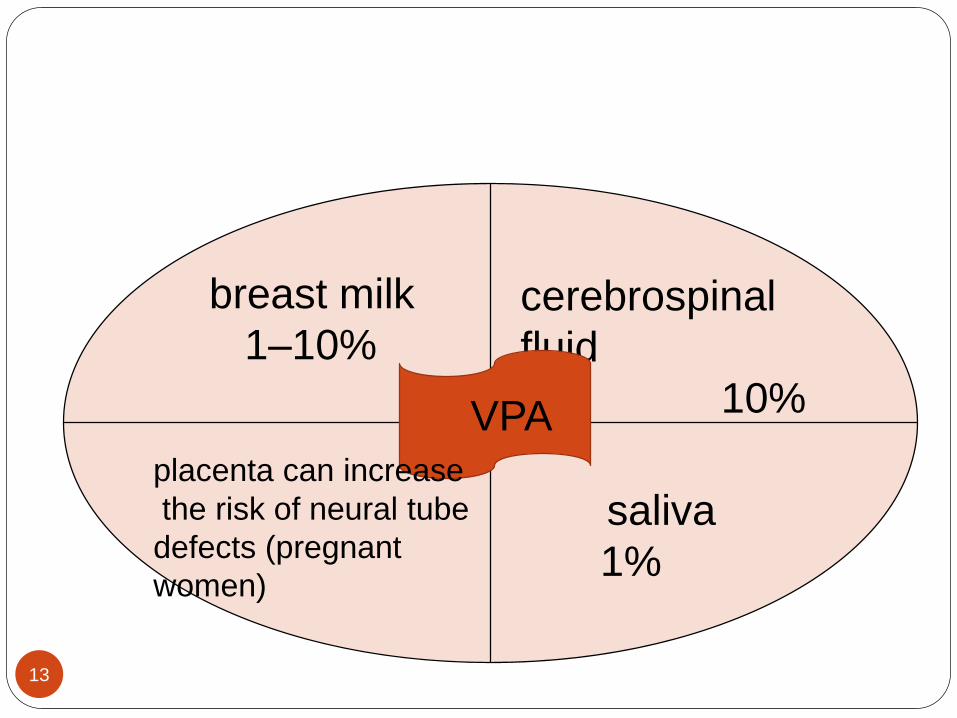
cerebrospinal
fluid
10%
breast milk
1–10%
VPA
saliva
1%
placenta can increase
the risk of neural tube
defects (pregnant
women)
13
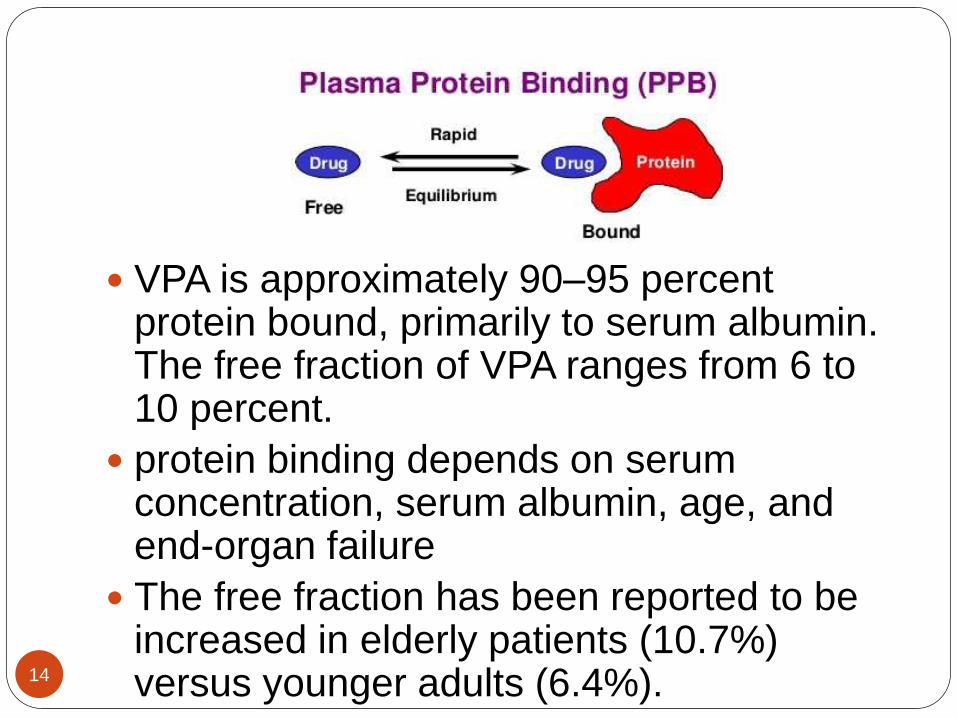
VPA is approximately 90–95 percent protein bound, primarily to serum albumin. The free fraction of VPA ranges from 6 to 10 percent.
protein binding depends on serum concentration, serum albumin, age, and end-organ failure
The free fraction has been reported to be increased in elderly patients (10.7%) versus younger adults (6.4%).14

At concentrations above 70 mcg/mL, VPA displays a higher free fraction with minimal changes in total concentration.
Renal failure may increase the free fraction to 18 percent while cirrhosis can increase it to 29 percent. Caution must be used when interpreting total VPA serum levels in these patients because they can be falsely low while their free levels may be therapeutic or higher.
In addition, VPA may displace other highly protein-bound drugs, such as phenytoin; therefore, free phenytoin levels should be monitored when these drugs are given concomitantly
15

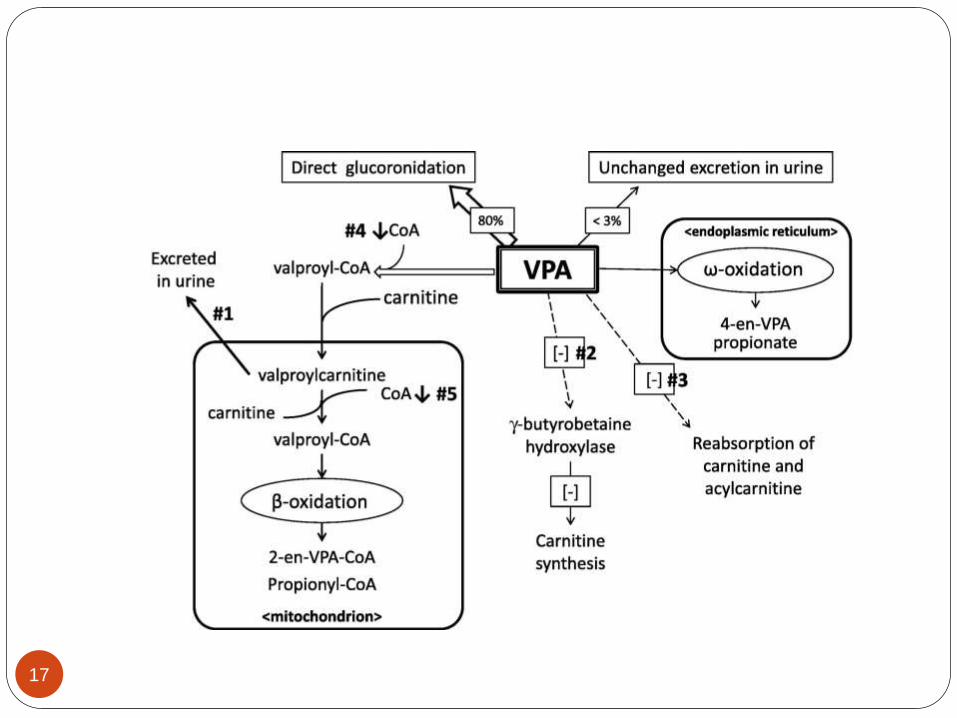
METABOLISM
VPA is primarily metabolized in the liver via
glucuronidation (40%), β-oxidation, and ω-
oxidation (20%) and then eliminated via the
kidneys, with less than 3 percent of the drug
excreted in the urine unchanged.
VPA is characterized as a low-extraction drug with
its clearance being independent of hepatic blood
flow. Many of the metabolites are thought to be
responsible for anticonvulsant activity as well as
toxicity
16
17

METABOLISM
VPA has an average elimination half-life of 11
hours and follows first-order kinetics. The mean
plasma clearance for total VPA is reported to be
approximately 6–7 mL/hr/kg in adults (range 5–10
mL/hr/kg), which may be decreased in patients
with renal or hepatic failure, and increased in
patients taking concomitant hepatic enzyme-
inducing agents.
Total clearance increases with higher doses due
to saturable protein binding, which leads to a
higher free fraction and more unbound drug
available for metabolism (assuming normal
hepatic function). 18

VPA acts as a substrate and inhibitor of various
cytochrome P450 enzymes, reflecting a high
potential for drug interactions
Enzymes That Metabolize VPA
Enzymes That VPAInhibits
Enzymes That VPAInduces
CYP2A6, 2B6, 2C9, 2C19, 2E1
CYP2C9 (weak), 2C19 (weak), 2D6 (weak), 3A4
(weak)
CYP2A6 (weak)
19

Drug interaction Several drugs significantly influence the clearance of VPA
including aspirin, caffeine, and various antibiotics. Aspirin and caffeine act by displacing VPA binding to serum proteins, inhibiting its clearance. Though this may be used to reduce the daily dose of VPA, it can also increase the risk of toxicity of VPA .
Carbapenem antibiotics increase the glucuronidation of VPA and increase clearance .The risk for hepatotoxicity with VPA is greatest in infants under 2 years old treated with additional drugs
Drugs that activate cytochrome P450 enzymes increase the clearance of VPA, increase the amount of N-acetylcysteineconjugates of VPA and may have the greatest risk for hepatoxicity.
VPA can also influence the clearance and metabolism of other drugs. A significant interaction can occur with coadministration of lamotrigine, where VPA doubles its half- life and increases risk of the ADRs Stevens-Johnson syndrome and toxic epidermal necrolysis. Interaction with topiramate, another AED, has been shown to increase risk for hyperammonia and encephalopathy .
20

VPA also interferes with HIV medications
zidovudine lopinavir and ritonavir ,patients
receiving valproic acid may require a zidovudine
dosage reduction to maintain unchanged serum
zidovudine concentrations
21

Drugs That DECREASE VPA Concentration
Drugs That May INCREASE VPA Concentration
Bile acid–binding resins –decreased absorptionCarbamazepine – hepatic metabolism inducedCarbapenem antibiotics –multiple mechanismsLamotrigine – hepatic metabolism inducedPhenytoin – hepatic metabolism inducedPhenobarbital – hepatic metabolism inducedRifampin – hepatic metabolism inducedRitonavir – hepatic metabolism inducedSevelamer – decreased absorption
Aspirin (high dose) – protein binding displacement/decreased intrinsic clearanceCimetidine – hepatic metabolism inhibitedMacrolides – unknown mechanismFelbamate – hepatic metabolism inhibited
22

QUESTION
JJ is a 54-year-old woman admitted with acute
kidney injury from interstitial nephritis. Her
baseline serum creatinine is 0.9 mg/dL, and
currently it is 2.3 mg/dL. She has a history of
seizures for which she takes Depakote 250 mg
PO TID.
What is the best recommendation for dose
adjustments in a patient with renal insufficiency?
23
24





























