Embed Size (px)
Citation preview

Approaches to analysing 1000s of bacterial isolates
A/Prof Torsten Seemann
Victorian Life Sciences Computation Initiative (VLSCI)Microbiological Diagnostic Unit Public Health Laboratory (MDU PHL)
Doherty Centre for Applied Microbial Genomics (DCAMG)The University of Melbourne
ICEID 2015 - Atlanta, USA - Mon 24 Aug 2015

Introduction
Melbourne, Australia

Microbial genomics
Doherty Centre for Applied Microbial Genomics

Microbiological Diagnostic Unit
∷ Oldest public health lab in Australia: established 1897 in Melbourne: large historical isolate collection back to 1950s
∷ National reference laboratory: Salmonella, Listeria, EHEC
∷ WHO regional reference lab: vaccine preventable invasive bacterial pathogens

The shift to WGS
∷ New director: Prof Ben Howden - clinician, microbiologist, pathologist
∷ New building: Doherty Institute for Immunity and Infection
∷ Mandate: modernise service delivery: nationally lead the conversion to WGS: enhance research output and collaboration

The one true assay?
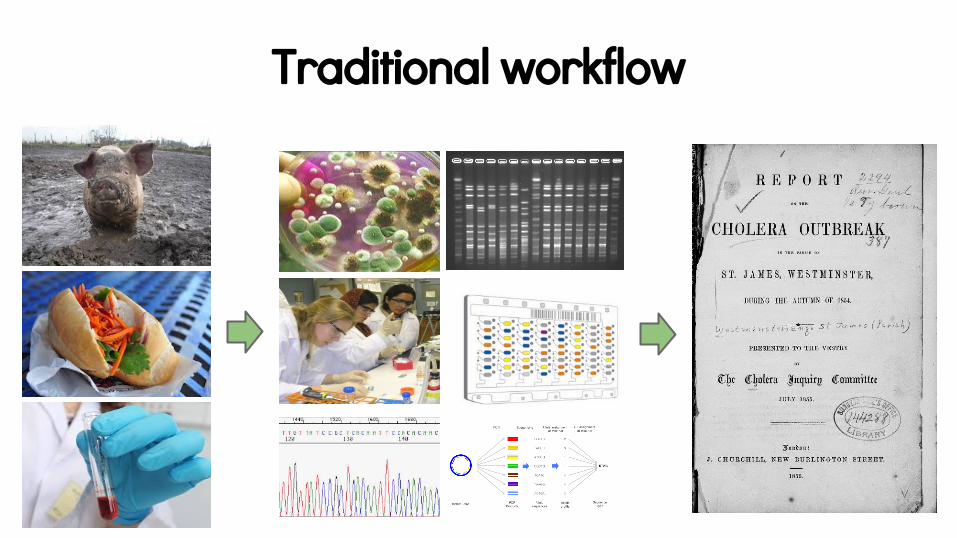
Traditional workflow
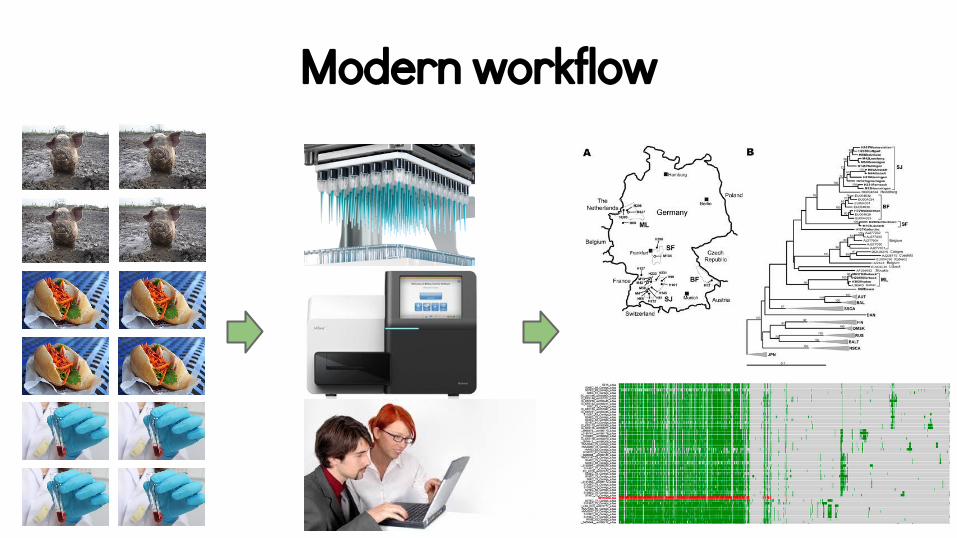
Modern workflow
Does it deliver?
Yes!Bioinformatics Epidemiology
Technology
Microbiology
This meansscientists
not just software
Domain expertise
Always changing...

Acquiring genome data

Sequencing a sample
∷ Simple preparation∷ Multiplexing∷ Robotics∷ Reliable instruments
Isolation is a bottleneck→ Direct sequencing?

Where is the genome data?
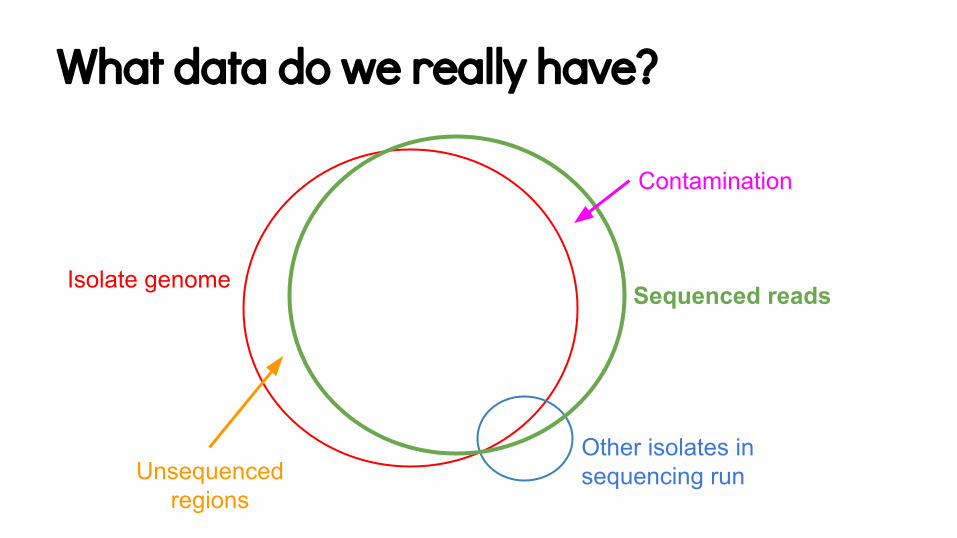
What data do we really have?
Isolate genome Sequenced reads
Other isolates in sequencing run
Contamination
Unsequenced regions
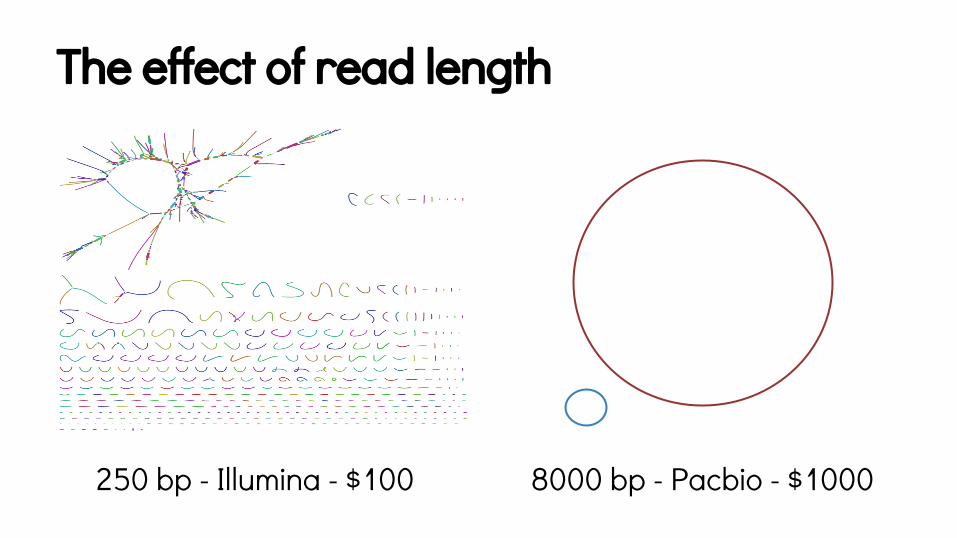
The effect of read length
250 bp - Illumina - $100 8000 bp - Pacbio - $1000
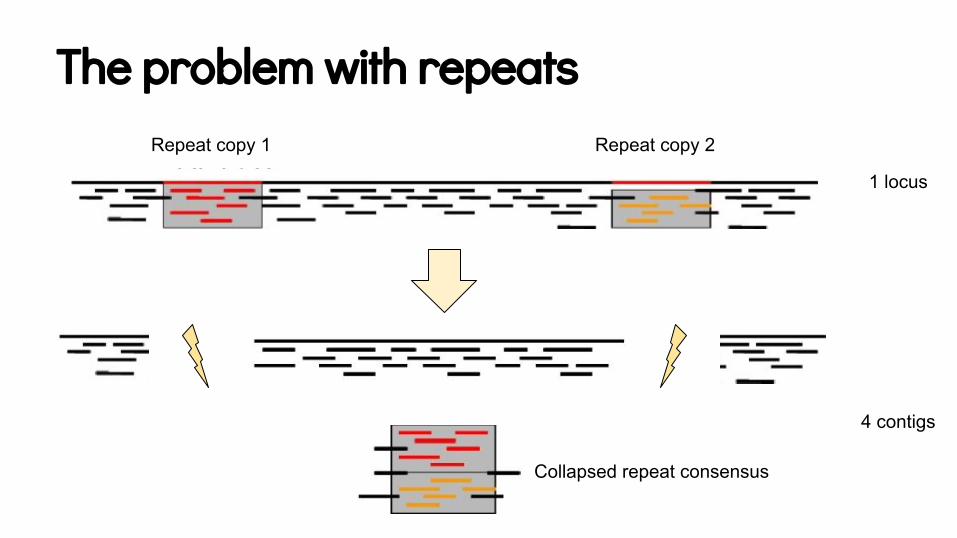
The problem with repeatsRepeat copy 1 Repeat copy 2
Collapsed repeat consensus
1 locus
4 contigs

Processing the data
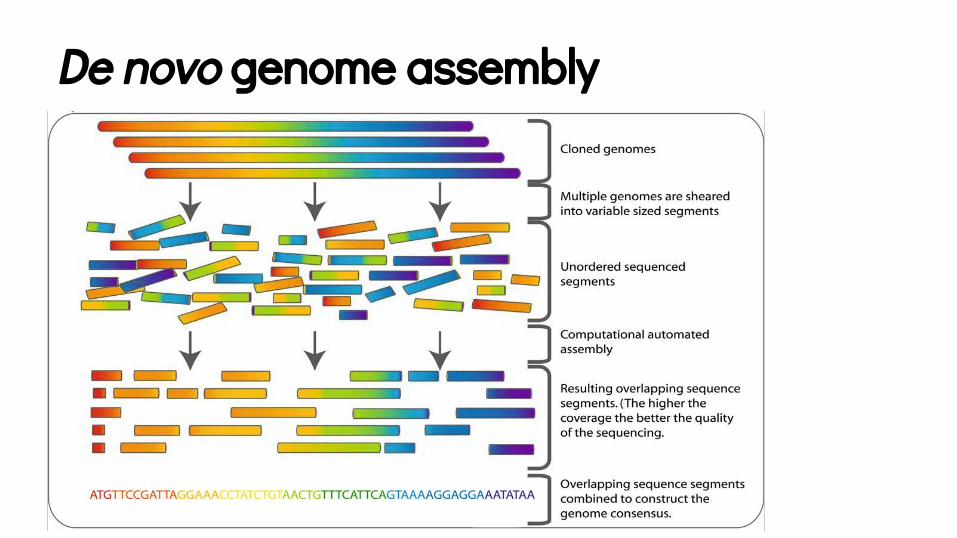
De novo genome assembly

Compare to already assembled genomes
AGTCTGATTAGCTTAGCTTGTAGCGCTATATTATAGTCTGATTAGCTTAGAT
ATTAGCTTAGATTGTAG
CTTAGATTGTAGC-C
TGATTAGCTTAGATTGTAGC-CTATAT
TAGCTTAGATTGTAGC-CTATATT
TAGATTGTAGC-CTATATTA
TAGATTGTAGC-CTATATTAT
SNP Deletion
Reference
Reads

Best practice
■ Use both approaches□ reference-based + de novo
■ Best of both worlds□ and worst of both worlds - interpretation is non-trivial
■ Still need□ good epidemiology, metadata and domain knowledge!

Applications

Backward compatibility
∷ MLST∷ NG-MAST∷ Resistome∷ Virulome
∷ MLVA∷ VNTR∷ Serotyping
∷ PFGE∷ Phage typing

New assays
∷ Species identification: build a “signature” from k-mer/oligos in the reads: compare to database of known signatures: strain level accuracy
∷ Features
: very fast screening, < 1 minute per isolate: identify contamination, mixed samples: discover wrongly labelled samples!
Phylogenomics

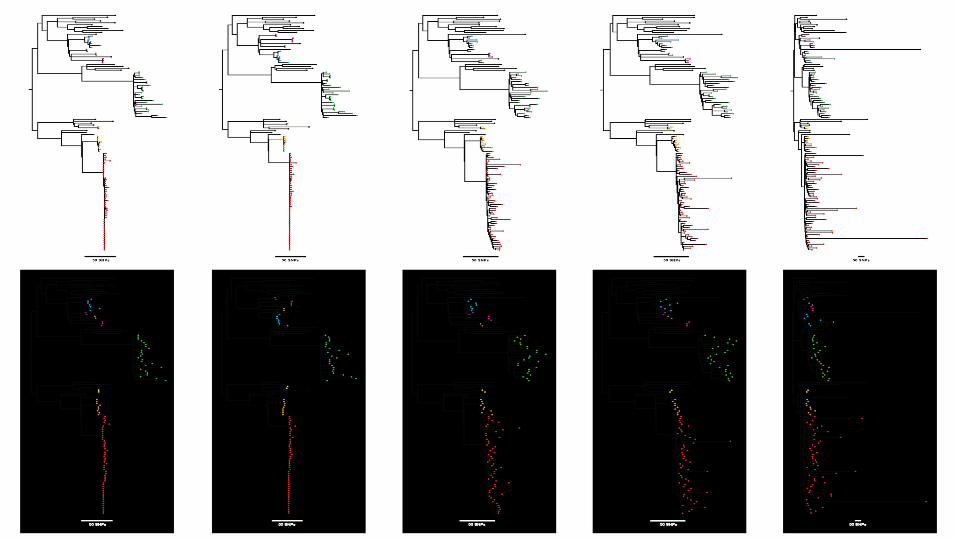
Reference based analysis
∷ Implies you have a “close” reference: need to be careful with draft genomes
∷ Very sensitive: single mutation precision
∷ May not be complete: ignores novel DNA in your isolate

The pan genome

The core genome
Core is common to all & has similar sequence.
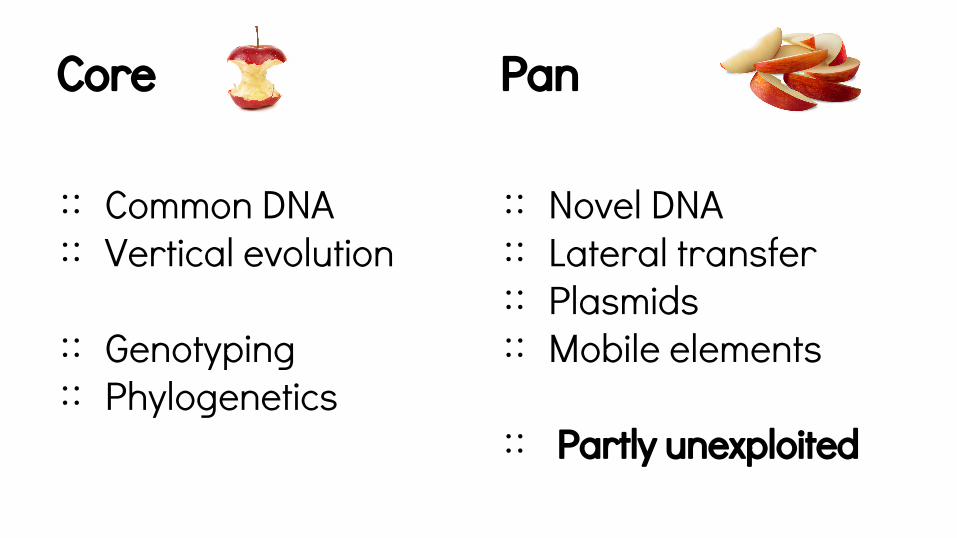
Core
∷ Common DNA∷ Vertical evolution
∷ Genotyping∷ Phylogenetics
∷ Novel DNA∷ Lateral transfer∷ Plasmids∷ Mobile elements
∷ Partly unexploited
Pan

Pan genome analysis
Rows are genomes, columns are genes.

Conclusion

Bioinformatics challenges
∷ Metagenomics: avoiding the isolation bottleneck
∷ Incremental update of data analyses: both core and pan genome: phylogenetic trees
∷ Data distribution: finding and getting appropriate comparator isolates
Open science
∷ Crowd-sourcing provably works: EHEC outbreak 2011: Ebola: MERS
∷ But only if people share: sequencing data: metadata: software source code for analysis


The EndThank you for listening.