Embed Size (px)
Citation preview

UNIVERSIDAD NACIONAL DE
CAJAMARCA
FACULTAD DE MEDICINA
Dr. Juan C. Salazar PajaresDEPARTAMENTO DE MEDICINA
HOSPITAL II ESSALUD CAJAMARCA
13. ESCLEROSIS MÚLTIPLE
Enfoque Diagnóstico y Manejo

I. ESCLEROSIS MULTIPLE
A. Forma encefalomielopática crónica recurrente
B. Esclerosis múltiple aguda o variante de Marburgo.
C. Esclerosis cerebral difusa o Enfermedad de Shilder
D. Esclerosis concéntrica o Enfermedad de Baló.
II. NEUROMIELITIS ÓPTICA DE DEVIC.
III. ENCEFALOMIELITIS DISEMINADA AGUDA
A. Postinfecciosa: post sarampión ,varicela, viruela, parotiditis , rubeola,
influenza y otras virales y bacterias (mycoplasma, rickettssia )
B. Postvacunal: rabia o viruela y raramente otros tipos de vacuna.
IV. ENCEFALITIS HEMORRÁGICA NECROTIZANTE AGUDA Y SUBAGUDA
A. Encefalopatía aguda ( leucoencefalitis hemorrágica de Hurst )
B. Mielopatía necrótica subaguda.
Dr. Juan C. Salazar Pajares 2ROPPER A. , SAMUELS ,M.: Adams and Victor´s Principles of Neurology . 9va Ed., Edit. Mc Graw Hill. México. 2011

Autoinmune Esclerosis múltiple Probablemente autoinmune Encefalomielitis post infecciosa y post vacunal Virus
Virus JC Leucoencefalopatía multifocal progresiva Virus del sarampión Panencefalitis esclerosante subaguda Virus de la rubeola Síndrome de rubeola progresivo VIH Leucoencefalopatía
Metabólica adquirida Mielinolisis póntica central Enfermedad de Marchiafava Bignami Deficiencia de B12 Degeneración combinada de la médula Metabólica hereditaria Leucodistrofia Vascular Encefalopatía arterioesclerótica subcortical (
Binswanger ) Irradiación Leucoencefalopatía Toxinas:
Solventes: tolueno Leucoencefalopatía espongiforme Monóxido de carbono Leucoencefalopatía
Drogas: Anfotericina B Leucoencefalopatía multifocal Metrotexate Leucoencefalopatía necrotizante
Edema crónico Leucoencefalopatía Condiciones de Inmunosupresión Leucoencefalopatía multifocal necrotizante.
Dr. Juan C. Salazar Pajares 3HABERLAND, C.: Clinical Neuropathology : Text and Color Atlas. Demos Medical Publishing, New York, 2007..

La mielina es una membrana proteo lipídica, producida por los oligodendrocitos , que envuelve a los axones nerviosos , para mejora la capacidad de conducción nerviosa, formando hendiduras denominados Nódulos de Ranvier , donde se concentran los canales iónicos y por tanto se generan potenciales de acción.
Cuando se separa la mielina del axón, la membrana no tiene los canales de Na , K y otros para permitir el flujo suficiente y generar despolarización y potenciales de acción.
La pérdida de mielina hace imposible la despolarización de membrana nerviosa para conducir el potencial de acción ,así que el nervio queda inservible.
Dr. Juan C. Salazar Pajares 4ROLAK, L: Neurología, secretos. 5ta. Ed. Edt. Elsevier Mosby. Barcelona España, 2011.

I. DEFINICIÓNII. EPIDEMOLOGÍAIII. ETIOPATOGENIAIV. ANATOMOPATOLOGÍAV. APROXIMACIÓN CLÍNICA
1. Cuadro clínico
2. Formas evolutivas
3. Criterios diagnósticos
VI. DIAGNÓSTICO DIFERENCIALVII. EXAMENES AUXILIARESVIII. TRATAMIENTO
Dr. Juan C. Salazar Pajares 5

Enfermedad desmielinizante más frecuente y antigua, que cursa con lesiones destructivas de mielina, focales y múltiples en el SNC , que se expresa por alteración en de los diversos sistemas motor, sensitivo, cerebeloso, visual y otros, teniendo una evolución intermitente de recidivas y remisiones , con una alteración neurológica progresiva generalmente.
Esclerosis diseminada ( Inglaterra)
Esclerosis en placas (Francia )
Esclerosis múltiple ( USA)
Dr. Juan C. Salazar Pajares 6
Diagrama muestra sitios comunes de
lesiones en placas en la Esclerosis
Múltiple
cerebelo

USA: incidencia 350 000 y 450 000 /año Enfermedad esporádica, pero hay caso familiares ( 21-31% de
gemelos); Gemelos: Homocigotos 30%, heterocigotos 5%. Más frecuente blancos. Menos frecuente en japoneses, nativos americanos. Riesgo de adquirir la enfermedad ,si la migración es alrededor de
los 15 años Sexo: afecta H/M: 1 / 4 .
Dr. Juan C. Salazar Pajares 7
LATITUD (° N) PREVALENCIA
ESCOCIA 60 309 / 100,000 hb
CANADA, EEUU, EUROPA 40-60 35-68/ 100,000 hb
INGLATERRA 51 63/ 100,000 hb
ITALIA 41 13/ 100,000 hb
PAÍSES DEL CARIBE Y ZONA ECUATORIAL 0-20 < 5/ 100,000 hb

Sexo/ Edad: H : 35-40 a. ; M: 20-35 años
Edad: adolescentes – 35 años ( El 66% inicia enfermedad : 20-40
años)
La afección se asocia a los alelos HLA-DRB1, HLA-DQB1.
El locus del CMH II(complejo mayor de histocompatibilidad) en
cromosoma 6 p.
Existe un elemento de respuesta e interacción a la Vit. D en la
región promotora del HLA-DRB1 y HLA-DQB1, que reduce la
sensibilidad del portador para desarrollar la enfermedad.
El riesgo de adquirir la enfermedad es 20-40(10-20) veces mayor en
los familiares en primer grado de un enfermo, con respecto a la
población general .
Dr. Juan C. Salazar Pajares 8

Dr. Juan C. Salazar Pajares 9
> 100 20-60
60-100
5-20
0-5
0-5
309

No se conoce exactamente la causa. Se estima que las causas son multifactoriales.
Infección viral persistente Proceso autoinmune (pérdida de tolerancia hacia antígenos de
la mielina) Errores biológicos: Fenómenos de mímica molecular entre
antígenos virales y proteínas de la mielina
Dr. Juan C. Salazar Pajares 10
RESPUESTA
AUTOINMUNE
DESORDENADA
EDAD DE EXPOSICION
DEL INDIVIDUO
FACTORES
AMBIENTALES
DESARROLLO DEL
ESCLEROSIS MULTIPLE
PREDISPOSICION
GENETICA

Dr. Juan C. Salazar Pajares 11
Factores genéticos y ambientales: virus,
polisacáridos bacterianos, superantígenos,
metabolitos, stress metabólico
Factores locales:
Virus, Estrés metabólico
citosinasCell T a
Proteína básica de mielina,
glicoproteína oligodendrocítica
de la mielina (MOG),
proteína proteolipídica, α β-
cristalina, fosfodiesterasas, y
la proteína S-100

Dr. Juan C. Salazar Pajares 12

Posible mecanismo de lesión y reparación de la Esclerosis Múltiple. Factores genéticos y ambientales (incluyendo la infección viral, lipopolisacáridos bacterianos, superantígenos, metabolitos reactivos, y el estrés metabólico) puede facilitar la penetración o movimiento de las células T autoreactivas y los anticuerpos desmielinizantes desde la circulación sistémica al sistema nervioso central a través de la interrupción de la barrera hematoencefálica. En el sistema nervioso central, los factores locales (incluyendo infección viral y el estrés metabólico) puede regular al máximo la expresión de moléculas de adhesión endoteliales, como la molécula 1 de adhesión intercelular (ICAM-1), molécula 1 de adhesión vascular – celular (VCAM-1), y E-selectina, facilitando aún más la entrada de células T en el sistema nervioso central. Proteasas, incluyendo metaloproteinasas de la matriz, pueden mejorar aún más la migración de células inmunes autorreactivas ,por degradación de macromoléculas de la matriz extracelular .
Citosinas proinflamatorias liberadas por las células T activadas, tales como interferón γ ,factor de necrosis tumoral β (TNF β ), puede hasta regular la expresión de moléculas de la superficie celular de los linfocitos vecinos y células presentadoras de antígeno. La unión de antígenos supuestos de la esclerosis múltiple (MS), tales como la proteína básica de mielina, glicoproteína asociada a la mielina , glicoproteína oligodendrocítica de la mielina (MOG), proteína proteolipídica, α β-cristalina, fosfodiesterasas, y la proteína S-100 ; con el complejo trimolecular - el receptor de células T (TCR) y las moléculas del complejo de histocompatibilidad principal clase II (MHC) en las células presentadoras de antígeno, pueden desencadenar o incrementar la respuesta inmune contra el antígeno unido o anergia, en función del tipo de señal que resulta de la interacción con las moléculas coestimuladoras de la superficie (por ejemplo, CD28 y CTLA-4) y sus ligandos ( B7-1 y B7-2). Una disminución de la regulación de la respuesta inmune (anergia) puede resultar en la liberación de citosinas antiinflamatorias (Interleucina-1, la interleucina-4 e interleucina-10) a partir de células T CD4 +, lo que conlleva a la proliferación de las células T ayudadoras antiinflamatorias tipo 2(Th2) CD4 + .
Dr. Juan C. Salazar Pajares 13

Las células Th2 pueden enviar señales antiinflamatorias de células presentadoras de antígenos
activadas y estimular a las células B productoras de anticuerpos patológicos o reparadores
(remielinizantes ).
Alternativamente, si el resultado del procesamiento de antígenos en una respuesta inmune mejorada, las citoquinas proinflamatorias ( la interleucina-12 y interferón γ ) pueden
desencadenar una cascada de eventos, lo que resulta en la proliferación de células T ayudadoras
tipo 1 CD4+ pro-inflamatorias tipo 1 (Th1) y finalmente una injuria inmune a la mielina y
oligodendrocitos.
Los múltiple los mecanismos de la lesión de la mielina mediada por inmunidad : 1)
citoquinas lesionan los oligodendrocitos y la mielina; 2) los macrófagos digieren los
antígenos de superficie de la mielina , incluyendo anticuerpos unidos a la mielina y los
oligodendrocitos ( citotoxicidad dependiente de anticuerpos ), 3) lesión mediada por el
complemento y 4) lesión directa de los oligodendrocitos por los linfocitos CD4 + y CD8 + T.
La lesión de la mielina ,resulta en los axones desnudos, incapaces de transmitir los
potenciales de acción de manera eficiente dentro de el sistema nervioso central (pérdida de la
conducción saltatoria). Esta disminución o bloqueo de los potenciales de acción produce
síntomas neurológicos. Los segmentos del axón expuesto puede ser susceptible a mayor daño
por mediadores solubles de la lesión ( citosinas, quimiocinas, complemento y proteasas),
resultando en una lesión axonal irreversible ( transección axonal y terminaciones ovoides del
axón ).
Hay varios posibles mecanismos de reparación de la membrana de mielina, incluyendo la
resolución de la respuesta inflamatoria ,seguida de remielinización espontánea ; la propagación de
los canales de sodio de los nodos de Ranvier para cubrir los segmentos desnudos de los axones y
restaurar la conducción ; la remielinización mediada por anticuerpos ; y la remielinización derivada
de la proliferación, migración y diferenciación de células precursoras de oligodendrocitos
residentes. Adaptado de esquema- Fundación Clínica Mayo.
Dr. Juan C. Salazar Pajares 14

Lesión característica : Inflamación Placa de desmielinización.Los linfocitos, macrófagos, anticuerpos, se acumulan alrededor de las vénulas del sistema nerviosos central y destruyen la mielina en un proceso autoinmune.
Localización en regiones: Peri ventriculares Yuxtacorticales Tronco cerebral Cerebelo Médula espinal
Dr. Juan C. Salazar Pajares 15
Dedo de Dawson
Las lesiones desmielinizadas
de forma más o menos ovoide,
perpendiculares al borde de
los ventrículos cerebrales son
una de las características de la
esclerosis múltiple. Estas
lesiones suelen ser visibles por
resonancia magnética.
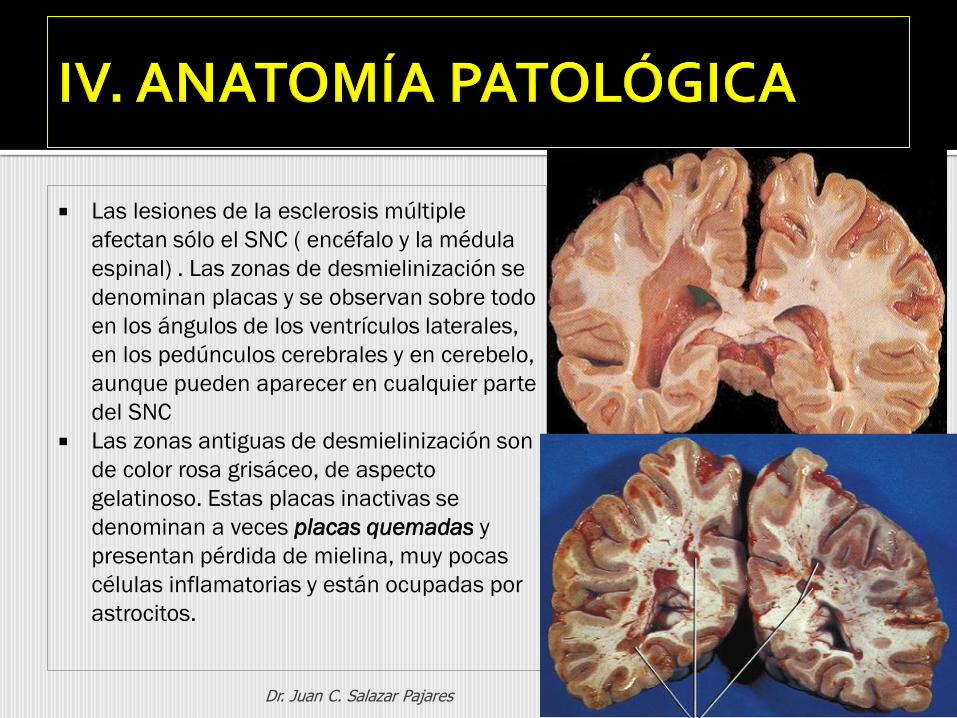
Las lesiones de la esclerosis múltiple
afectan sólo el SNC ( encéfalo y la médula
espinal) . Las zonas de desmielinización se
denominan placas y se observan sobre todo
en los ángulos de los ventrículos laterales,
en los pedúnculos cerebrales y en cerebelo,
aunque pueden aparecer en cualquier parte
del SNC
Las zonas antiguas de desmielinización son
de color rosa grisáceo, de aspecto
gelatinoso. Estas placas inactivas se
denominan a veces placas quemadas y
presentan pérdida de mielina, muy pocas
células inflamatorias y están ocupadas por
astrocitos.
Dr. Juan C. Salazar Pajares 16

Dr. Juan C. Salazar Pajares 17

(A). FISIOPATOLOGÍA DEL PATRÓN VASCULAR. La imagen muestra lesión microvascular en la
pared y / o Intraluminal que induce la lesión isquémica del parénquima. Se nota la hialinización
mural causada por Arteriopatía hipertensiva (flecha) y la vacuolización isquémica adyacente (*);
(B) FISIOPATOLOGÍA DEL PATRÓN PERIVASCULAR. La imagen muestra una proceso patológico
perivascular y la lesión del parénquima adyacente. Capa desmielinizante causada por la inflamación
perivenular en un paciente esclerosis múltiple.Dr. Juan C. Salazar Pajares 18

Figura 3. Las fotomicrografías de una placa crónica de EM. A) región hipocelular bien delimitada de
pérdida de la mielina , evidente en la sustancia blanca periventricular (luxol azul y ácido periódico de
Schiff-tinción de mielina, x15). B) la tinción de neurofilamentos para los axones en la misma lesión
demuestra una reducción en la densidad axonal (x15). N Engl J Med 2000;343(13)26:938-52.
Dr. Juan C. Salazar Pajares 19

La Figura 4. Fotomicrografías de lesión desmielinizante activa de EM (inmuno tinción de mielina
glicoproteína de mielina [ marrón] con tinción de hematoxilina de los núcleos [Azul]).
A) borde activo de lesión de la esclerosis múltiple (indicado por el asterisco), los productos de la
degradación de la mielina están presentes en numerosos macrófagos (puntas de flecha) (x100).
B )Los macrófagos que contienen restos de mielina (puntas de flecha) están entrelazados con las vainas
de mielina en degeneración. N Engl J Med 2000;343(13)26:938-52.
Dr. Juan C. Salazar Pajares 20

Figura 5. La remielinización de lesión crónica de EM. El área manchada de azul pálido (indicado por el
asterisco) representa una región de la remielinización parcial (una placa de sombra) a lo largo
el borde periventricular de una lesión crónica en un paciente EM (luxol azul y ácido periódico-Schiff
mielina mancha, x15). NAWM denota cuestión de apariencia blanco normal.
N Engl J Med 2000; 343 (13) 26:938-52.
Dr. Juan C. Salazar Pajares 21
Placas agudas de EM, en la sustancia blanca del hemiferio
cerebral mal definidos, de color amarillento y ligeramente
granular.

Dr. Juan C. Salazar Pajares 22

El diagnóstico depende de reconocimiento de variantes clínicas de la enfermedad.
Generalmente en mujeres de 20 a 35 años ( 90 %), varones (35-40 años)
Menores de 40 años, debut con déficit neurológico subaguda que tiende aparecer y desaparecer.
Mayores de 50 años, con desarrollo de déficit neurológico permanente, con remisiones y recaídas; y compromiso medular progresivo.
Síntoma de inicio : 50% de los enfermos manifiesta parestesias o pérdida de fuerza en uno o más miembros: TORPEZA MOTRIZ.
Dr. Juan C. Salazar Pajares 23

Manifestaciones clínicas mas frecuentes:
1. Déficit motor piramidal 45%
2. Pérdida de agudeza visual 40%
3. Perdida sensitiva 35%
4. Disfunción de tronco encéfalo 30%
5. Ataxia y temblor cerebeloso 25%
6. Trastornos esfinterianos 20%
- EM es una enfermedad de mielina (sustancia blanca), rara
vez produce afasias, convulsiones, dolor y movimientos
involuntarios.
Dr. Juan C. Salazar Pajares 24ROLAK, L: Neurología, secretos. 5ta. Ed. Edt. Elsevier Mosby. Barcelona España, 2011.

1. Síntomas iniciales: alteraciones de la sensibilidad, fuerza muscular,
agudeza visual, diplopía, marcha atáxica , torpeza, vértigo , alteraciones
esfinterianas. Otras más inespecíficas: malestar, fatiga, cefalea previas.
2. Síndrome sensitivo (1ra) (40%) parestesias, disestesias, hipostesias,
síntoma de Lhermitte , neuralgia del trigémino.
3. Neuritis óptica (2da) ( 17%): pérdida de la visión monocular parcial o
total, alteración de la visión a colores , escotomas, dolor periorbitario al
movimiento. FO: palidez relativa del nervio óptico, papilitis de la cabeza
del nervio, envainamiento de los vasos venosos retinianos.
4. Síndrome motor: (12%)Mono , para o hemiparesia o plejia, alteración de
la destreza motora , espasticidad, hiperreflexia, con signos de liberación ,
5. Diplopía : (11%) a veces sutil , alteración de movimientos conjugados .
Oftalmología internuclear (OI) , síndrome del 1 y medio.
Dr. Juan C. Salazar Pajares 25

6. Síndrome cerebeloso : (20%)ataxia de la marcha y temblor de actitud ,
dismetría, adiadococinesia, asinergia, Signo de Romberg, nistagmos ,
reflejos osteotendinosos pendulares, prueba de Stewart Holmes positiva.
7. Síndrome neuropsiquiátrico: deterioro cognitivo, amnesia, alteraciones
de la atención, dificultad en la resolución de problemas, labilidad
emocional, depresión (60%), parálisis pseudobulbar con risa y llanto
inmotivado, en ocasiones euforia, anosognosia, anosodiaforia “la belle
indifference”.
8. Síndrome neurovegetativo: (20%)disfunción vesical e intestinal. Vejiga
espástica o atónica, estreñimiento crónico.
9. Síndrome vertiginoso: (20%) inicial, pasajero, con signos de tronco
cerebral, nistagmos, parálisis facial, diplopía, hipoacusia unilateral, con
pruebas de Dix Hall Pike (-)
Dr. Juan C. Salazar Pajares 26

10. Disartria: flácida o espástica, escandida.
11. Disfagia: a líquidos
12. Paresia facial , similar a parálisis de Bell ( No hipoageusia,
hiperacusia, dolor retroauricular,)
13. Fatiga: alteración del rendimiento laboral , falta de fuerza-
depresión , insomnio,
14. Espasmos flexores: espasmo flexor de la extremidad precedidos
de parestesias, disestesias, angustia.
15. Mioquimia facial:
Dr. Juan C. Salazar Pajares 27

Dr. Juan C. Salazar Pajares 28

Dr. Juan C. Salazar Pajares 29
MUMENTHALER,M.; MATTLE, : Fundamentals of Neurology . 4th. Ed. Thieme, Stuttgart · New York,2006

Dr. Juan C. Salazar Pajares 30

Dr. Juan C. Salazar Pajares 31
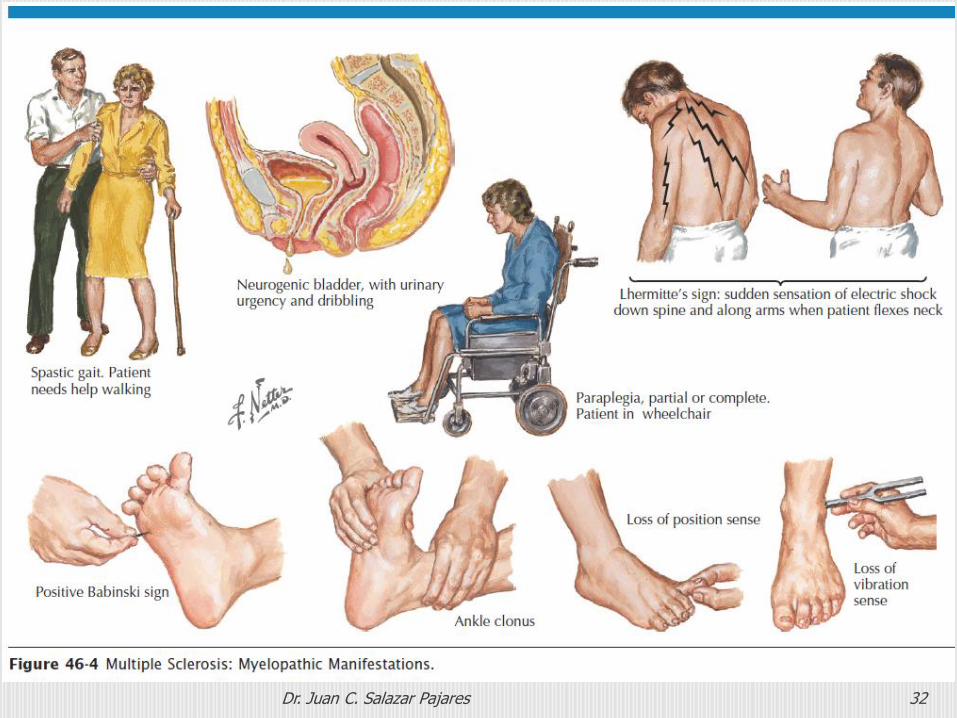
Dr. Juan C. Salazar Pajares 32
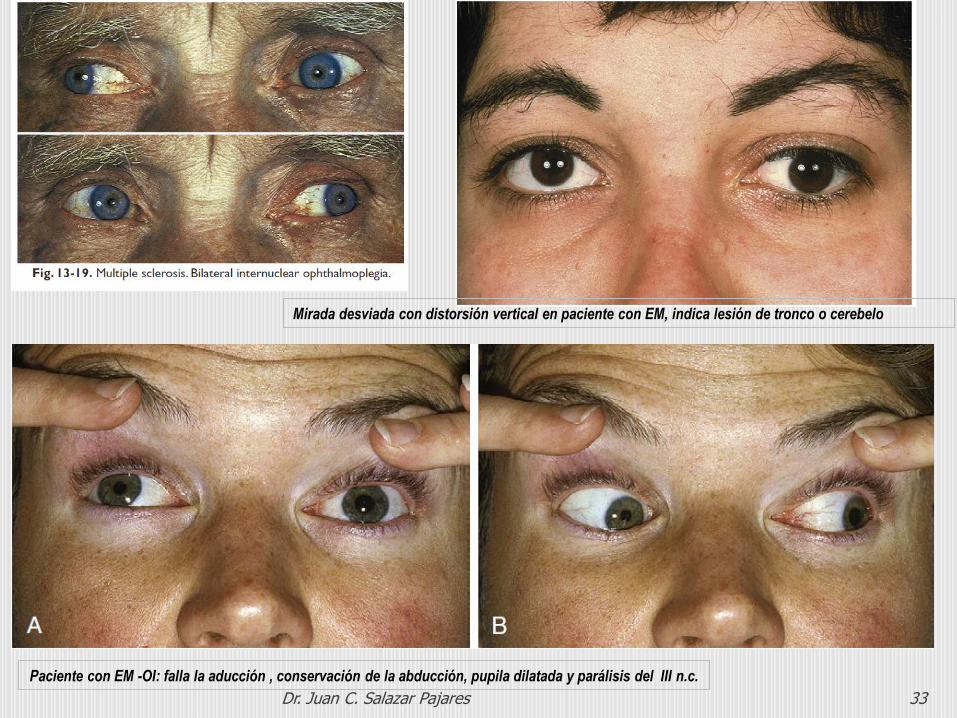
Dr. Juan C. Salazar Pajares 33
Paciente con EM -OI: falla la aducción , conservación de la abducción, pupila dilatada y parálisis del III n.c.
Mirada desviada con distorsión vertical en paciente con EM, indica lesión de tronco o cerebelo

Dr. Juan C. Salazar Pajares 34
Lesion pontina: combinación de lesión del VI nc y VII craneal derechos
Parálisis Facial periférica izquierda similar a la PFP de Belll, puede definirse en una RM

Como complicaciones de los primarios: Contracturas Atrofias musculares Infección Urinaria Inmovilidad - Osteoporosis - Fracturas
Dr. Juan C. Salazar Pajares 35

Se deben al impacto psicológico:
Alteraciones psicológicas
Repercusión Social
Problemas laborales
Conflictos de parejas (divorcios)
Dr. Juan C. Salazar Pajares 36

1. EM recaídas y remisiones (EMRR) – Trastorno netamente inflamatorio con desmielinización y remielinización
2. EM primaria progresiva (EMPP) – Trastorno con trasección axonal y no remielinización
3. EM secundaria progresiva (EMSP) – Trastorno que comienza con inflamación y termina sin regeneración
4. EM progresiva con recaídas (EMPR)
Dr. Juan C. Salazar Pajares 37

Dr. Juan C. Salazar Pajares 38

Dr. Juan C. Salazar Pajares 39

A- EM recaidas y remisiones
(EMRR)
B- EM primaria progresiva
(EMPP)
C- EM secundaria progresiva
(EMSP)
D- EM progresiva con
recaidas (EMPR)
Dr. Juan C. Salazar Pajares 40

A. EM recaidas y
remisiones (EMRR)
B. EM primaria
progresiva (EMPP)
C. EM secundaria
progresiva (EMSP)
D. EM progresiva con
recaidas (EMPR)
Dr. Juan C. Salazar Pajares 41

A. EM recaídas y
remisiones (EMRR)
B. EM primaria
progresiva (EMPP)
C. EM secundaria
progresiva (EMSP)
D. EM progresiva con
recaídas (EMPR)
Dr. Juan C. Salazar Pajares 42

A. EM recaídas y
remisiones (EMRR)
B. EM primaria
progresiva (EMPP)
C. EM secundaria
progresiva (EMSP)
D. EM progresiva con
recaídas (EMPR)
Dr. Juan C. Salazar Pajares 43

1. Examen neurológico: anormalidades objetivas de función SNC
2. La historia indica compromiso de 2 o mas partes del
SNC.(Diseminación Espacial)
3. Enfermedad del SNC, revela predominantemente afección de las
sustancia blanca.
4. Compromiso del SNC, siguiendo 1 de 2 modelos:
a) Diseminación en el tiempo: 2 o más episodios que duran más
de 24 horas y separados por más de 30 días.
b) Progresión lenta de síntomas y signos, durante al menos 6m.
5. Paciente entre 10 y 50 años al inicio de los síntomas
6. Signos y Síntomas que no pueden explicarse por otras
afecciones
Dr. Juan C. Salazar Pajares 44

1. EM definida clínicamente a) 2 ataques + evidencia clínica de 2 lesiones separadasb) 2 ataques + evidencia clínica 1 lesión + evidencia paraclínica otra lesión
separada. 2. EM definida, fundamentada con laboratorio
a) 2 ataques + evidencia clínica o paraclínica de 1 lesión + anormalidades inmunológicas en el LCR
b) 1 ataque + evidencia clínica de 2 lesiones separadas + anormalidades del LCR
c) 1 ataque + evidencia clínica de 1 lesión + evidencia paraclínica de otra lesión separada + anormalidades del LCR.
3. EM probable clínicamente a) 2 ataques + evidencia clínica de 1 lesiónb) 1 ataque + evidencia clínica de 2 lesiones separadac) 1 ataque + evidencia clínica de 1 lesión + evidencia paraclínica de otra
lesión separada4. EM probable ,fundamentada por laboratorio :
a) 2 ataques y anormalidades en el LCR
Dr. Juan C. Salazar Pajares 45

1. EM definida clínicamente a) 2 ataques + evidencia clínica de 2 lesiones separadasb) 2 ataques + evidencia clínica 1 lesión + evidencia paraclínica otra lesión
separada. 2. EM definida, fundamentada con laboratorio
a) 2 ataques + evidencia clínica o paraclínica de 1 lesión + anormalidades inmunológicas en el LCR
b) 1 ataque + evidencia clínica de 2 lesiones separadas + anormalidades del LCR
c) 1 ataque + evidencia clínica de 1 lesión + evidencia paraclínica de otra lesión separada + anormalidades del LCR.
3. EM probable clínicamente a) 2 ataques + evidencia clínica de 1 lesiónb) 1 ataque + evidencia clínica de 2 lesiones separadac) 1 ataque + evidencia clínica de 1 lesión + evidencia paraclínica de otra
lesión separada4. EM probable ,fundamentada por laboratorio :
a) 2 ataques y anormalidades en el LCR
Dr. Juan C. Salazar Pajares 46

Dr. Juan C. Salazar Pajares 47
PRESENTACIO N CLÍNICA DATOS ADICIONALES NECESARIOS PARA EL DX DE EM
2 o + ataques
Evidencia clínica objetiva de 2 o + lesiones
Ninguno
2 o mas ataques
Evidencia clínica objetiva de 1 lesión
Diseminación espacial, demostrada por :
RM, o
2 o más lesiones consistentes detectadas por RM +
LCR (+): detección de bandas oligoclonales o ↑ Ig G. / o
Ataque clínico adicional, implique otro sitio diferente.
Diseminación temporal, demostrada por
RM ,o
2do ataque clínico.
1 ataque
Evidencia clínica objetiva de 2 o + lesiones
Diseminación espacial, demostrada por :
RM, o
2 o más lesiones consistentes detectadas por RM +
LCR: detección de bandas oligoclonales o ↑ Ig G. / y
Diseminación temporal, demostrada por:
RM , o
2do ataque clínico.
1 ataque
Evidencia clínica objetiva de 1 lesión (monosintomática, síndrome clínico aislado )
ídem
Progresión insidiosa progresiva sugestiva de esclerosis múltiple. LCR (+) , y
Diseminación espacial, demostrada por :
9 o más lesiones cerebrales en T2 , o
2 o más lesiones en médula espinal , o
4 a 8 lesiones cerebrales + 1 lesión en médula espinal , o
PEV anormales , con 4 a 8 lesiones cerebrales, o
PEV anormales , con menos de 4 lesiones cerebrales + 1 lesión medular/ y
Diseminación temporal, demostrada por :
RM ,o
Progresión continua durante un 1 año.

1. Debe excluirse todas las enfermedades infecciosas o sistémicas con sintomatología similar.
2. Trastornos inflamatorios: LES, poliarteritis nodosa, Enfermedad de Becet´s
3. Enfermedades Granulomatosas; Sarcoidosis, Granulomatosis de Wegerner.
4. Alteraciones de la medula espinal / foramen magno: Tumores extrínsecos e intrínsecos, Esclerosis combinada por deficiencia de B12
5. Alteraciones cerebrales aisladas: MAV, meningioma 6. Otros: Degeneración espinocerebelosa, enfermedades
mitocondriales, adrenoleucodistrofia, enfermedad de Lyme, Encefalomielitis Aguda Diseminada.
Dr. Juan C. Salazar Pajares 48

Dr. Juan C. Salazar Pajares 49

1. FORMA ENCEFALOMIELOPÁTICA CRÓNICA RECURRENTE2. ENFERMEDAD DE DEVIC 3. MIELITIS TRANSVERSA AGUDA 4. ESCLEROSIS MÚLTIPLE AGUDA O VARIANTE DE MARBURG: rara,
fulminante, con lesiones pseudotumorales, mortalidad al 1 año.5. ESCLEROSIS CEREBRAL DIFUSA O ENFERMEDAD DE SHILDER
:enfermedad progresiva, con exacerbaciones progresivas de los síntomas. Demencia, hemianopsias, ceguera, sordera, hemi o cuadriplejia, parálisis pseudobulbar. En la RM grandes lesiones que comprometen un hemisferio o un lóbulo, invade cuerpo calloso y hemisferio contralateral. LCR: aumento de proteínas, sin bandas oligoclonales.
6. ESCLEROSIS CONCÉNTRICA O ENFERMEDAD DE BALÓ: clínicamente y distribución de las lesiones son similares a la anterior. Característica histopatológica: bandas alternantes de destrucción de mielina en anillos concéntricos.
Dr. Juan C. Salazar Pajares 50

No existe prueba o marcador único para
realizar el diagnóstico de EM. Puede ser
avalado por :
RMN en T2 – ESPECTROSCOPIA.
PEV – PESS - PEA
LCR: bandas oligoclonales.
Dr. Juan C. Salazar Pajares 51

Dr. Juan C. Salazar Pajares 52

Dr. Juan C. Salazar Pajares 53

Dr. Juan C. Salazar Pajares 54

Dr. Juan C. Salazar Pajares 55

Dr. Juan C. Salazar Pajares 56

a A (Axial TSE T2):
múltiples lesiones
hiperintensas. Predominio
de afectación periférica
y en la región ventral de
la protuberancia.
A
B
B (Axial TSE T2):
Lesión hiperintensa
en el brazo
posterior de la
cápsula interna
derecha.
D
C
C (Axial FLAIR):
lesión hiperintensa
periventricular en el
pedúnculo
cerebeloso superior
izquierdo.
D (Axial TSE T2):
lesión hiperintensa
situada en el núcleo
rojo izquierdo.
Aunque la localización de las lesiones sería sugestiva de Enfermedad de
Behçet, no se podría excluir una enfermedad desmielinizante (EM)
En este caso la clínica del paciente (uveítis y aftas orales) es fundamental para establecer el diagnóstico.
La localización peri ventricular es menos
frecuente en la EB que en la EM. La lesión del
núcleo rojo excluye la enfermedad de Behçet
Dr. Juan C. Salazar Pajares 57

Dr. Juan C. Salazar Pajares 58
NEUROSARCOIDOSIS

Mujer de 25 años, con EM - recaída-remisión. A) FLAIR axial (recuperación de la
inversión de líquido atenuado) imágenes hiperintensas múltiples ovoides y lesiones
confluentes en la sustancia blanca periventricular . B). Nueve meses después ,
aumento del número y tamaño de las lesiones C)Con Gadolinio muchas de las
lesiones demuestran anillo o realce periférico, lo que indica la ruptura de la barrera
hemato - encefálica. D) RM parasagital en T1, muestra múltiples regiones en las
que está disminuida la señal (en adelante, "los agujeros negros") en la sustancia
blanca periventricular y el cuerpo calloso. Estas regiones corresponden a las lesiones
crónicas de la esclerosis múltiple
N Engl J Med 2000;343(13)26:938-52.
Dr. Juan C. Salazar Pajares 59
A
D
CB
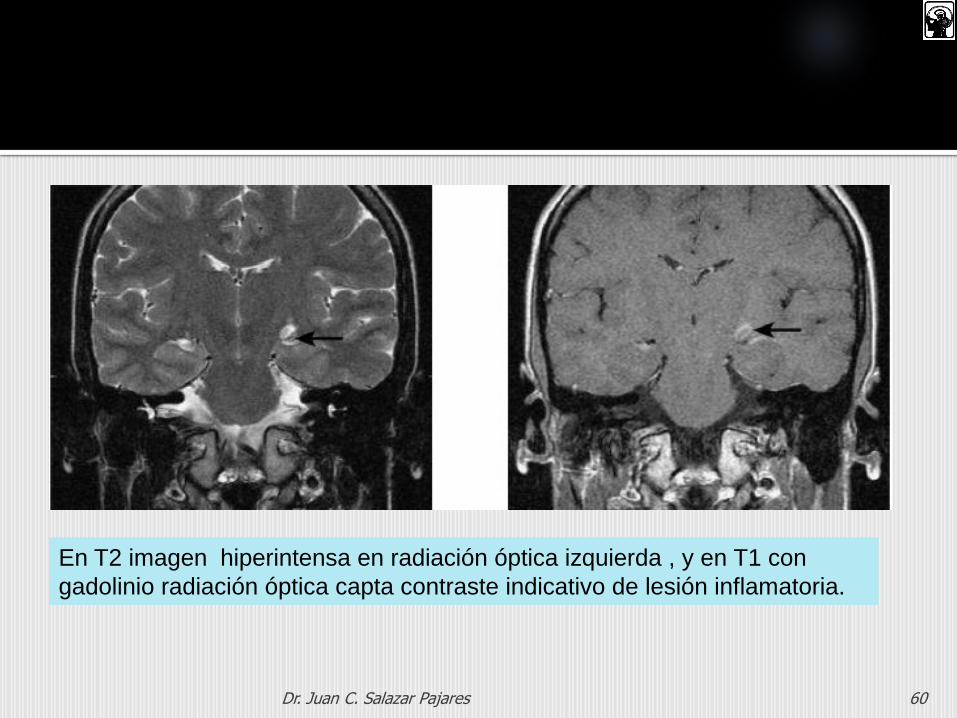
Dr. Juan C. Salazar Pajares 60
En T2 imagen hiperintensa en radiación óptica izquierda , y en T1 con
gadolinio radiación óptica capta contraste indicativo de lesión inflamatoria.

Dr. Juan C. Salazar Pajares 61
RM muestra múltiples áreas de
hiperseñal en la sustancia
blanca debido a EM.
Diagrama- hallazgos
anormales inespecíficos
de la EM.
La RM es útil.
Anormalidades en
globulinas del LCR.
Los PEA VCMC,
PESS, PEV
retrasados o
enlentecidos.
Bandas oligoclonales
en el LCR.

Dr. Juan C. Salazar Pajares 62

Dr. Juan C. Salazar Pajares 63
ESCELROSIS MULTIPLE AGUDA O ENFERMEDAD DE MARBUG

Dr. Juan C. Salazar Pajares 64

SINTOMÁTICO
ESPECÍFICO
DE LAS RECAÍDAS
DE REHABILITACIÓN
Dr. Juan C. Salazar Pajares 65

Dr. Juan C. Salazar Pajares 66N Engl J Med 2000;343(13)26:938-52.

Dr. Juan C. Salazar Pajares 67N Engl J Med 2000;343(13)26:938-52.

1. DEL BROTE:
a) Corticoides a dosis altas, i.v., durante cortos períodos de tiempo (3-5 días)
Metilprednisolona 1 gr IV / día /3- 5 días.
Seguido de Prednisona 60 mg SD/ 3 días- reducir 10mg c/3- VO
O ACTH 25-60 UI IM o EV (8hs), por 2 – 4 s.
b) Plasmaféresis
2. A LARGO PLAZO:a) El objetivo es intentar de disminuir el número de brotes, las secuelas y la
progresión de la incapacidad.
b) Básicamente se emplean inmunosupresores (azatioprina, ciclofosfamida, metotrexate) e interferones (alfa)
Dr. Juan C. Salazar Pajares 68

Forma remitente recidivante:
Interferón β 1a y 1b.
Acetato de Glatiramer
Natalizumab
Fingolimod
Forma progresiva
Azotioprina, ciclofosfamida, ciclosporina
Interferón B-1 b
Mitoxantrona
Dr. Juan C. Salazar Pajares 69

1. CON RESPECTO AL TRATAMIENTO SINTOMÁTICO,
se puede resumir de la siguiente manera:
Espasticidad (baclofeno, diazepam, dantrolene sódico)
Fatiga (amantadita)
Dolor (incluyendo la neuralgia del trigémino) (carbamacepina,
fenitoína, gabapentina, pregabaliana, amitriptilina)
Hiperreflexia vesical (oxibutinia, betanecol)
Retención urinaria por hiporreflexia del detrusor
Infecciones intercurrentes, úlceras de decúbito, dolor por
hipertonía muscular.
Gran importancia tiene el tratamiento rehabilitador.
Dr. Juan C. Salazar Pajares 70








