Embed Size (px)
Citation preview

Fast comparison of microbial genomes using the ChaosGames Representation for metagenomic applications
Jhon Trujillo
Seminario GebixJohn Trujillo - Universidad Del Valle - Cali
24 de abril de 2014
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 1 / 54

Overview
1 Abstract
2 Introduction
3 MetodologıaDatos genomicos usadosUsando los CGR para comprar genomasUsando los CGR para comprar genomas
4 ResultadosTamanos de los fragmentos de las secuenciasComparacion con BlastComparacion con BlastComparacion con Blast
5 Discussion and future work
6 Conclusiones
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 2 / 54

Abstract
Genome sequencing technology.
Secuenciamiento de genomas genera grandes bases de datos.
El Hardware existente no es suficiente y es muy costo.
Se requiere de algoritmos eficientes.
Se propone un metodo eficiente : Teorıa del Caos y analisisMultifractral.
Realizar busquedas y filtrar datos.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 3 / 54

Ejemplo
Conjunto de 1500 genomas.
Blast puede tomar hasta 25 horas en buscar esta secuencia confragmentos grandes, por ejemplo de 100k.
Usando el metodo propuesto reduce el conjunto de busqueda en un95 %. (1500 a 50) en solo 15 minutos.
Predice una secuencia con un 67 % de exactitud.
La idea es demostrar que este metodo es mas rapido para comprarsecuencias en grandes conjuntos de datos.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 4 / 54

Introduction
El Secuenciamiento de genomas genera grandes datos.
La secuenciacion por nanoporos genera grandes “reads” de 100.000pb en longitudes de 100kb.
El poder computacional no es suficiente : Una opcion es usar Blast.
Segunda opcion es usar: la teorıa del caos y el analisismultifractral.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 5 / 54

Introduction
Cada secuencia tiene una representacion fractal. Una combinacion denucleotidos tiene una escala independiente.
Un CGR puede ser usado para visualizar estas representaciones.
El CGR puede mostrar la frecuencia de una “word” del codigogenetico usando una imagen simple donde un conjunto de pixelspuede representar la frecuencia de una”word”. Mostrando lafrecuencia de cada palabra con un color determinado.
Luego, un analisis multifractral puede ser aplicado al CGR.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 6 / 54
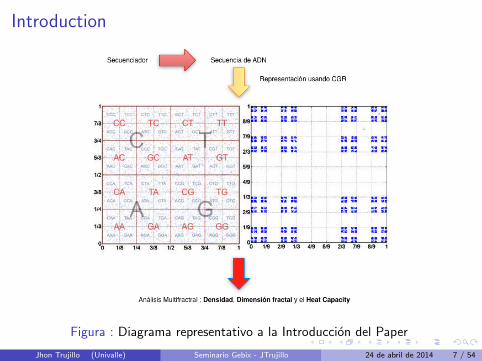
Introduction
Figura : Diagrama representativo a la Introduccion del Paper
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 7 / 54

Introduction
Demostrar [Z.-G.Yu,V.Anh,K.-S.Lau] donde usaron esta metodologıa paraclasificar 33 genomas bacterianos.
Los autores mostraron la similaridad filogenetica de las bacteriasagrupadas en 2 y 3 dimensiones ( Estas dimensiones fueron derivadasa partir de la dimension fractal ).
Usaron conceptos como el Heat Capacity para distinguir exones eintrones en el genoma bacteriano.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 8 / 54

Introduction
La dimension fractal era mas alta en secuencias no codificantes queen secuencias codificantes.
Similares aproximaciones fueron realizadas para el genoma humano ydistinguir los genomas del HIV-1 [A.Pandit,A.Dasanna] .
No es claro como escalar este tipo de analisis a los cientos degenomas presentes en los repositorios publicos.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 9 / 54

Secuenciacion
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 10 / 54

Secuenciacion
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 11 / 54

Secuenciacion
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 12 / 54

Secuenciacion
El Secuenciamiento moderno solo hace lecturas muy pequenas.
Unir cada palabra es un juego de rompecabezas.
¿Como saber cual es el mejor metodo de ensamblaje?.
¿Como saber que tan bueno es el ensamblaje y su correctaorganizacion?.
Se compararon ambos genomas relacionados por medio de sus atributostaxonomicos :
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 13 / 54
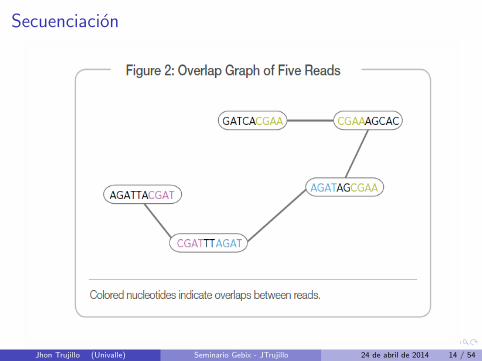
Secuenciacion
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 14 / 54
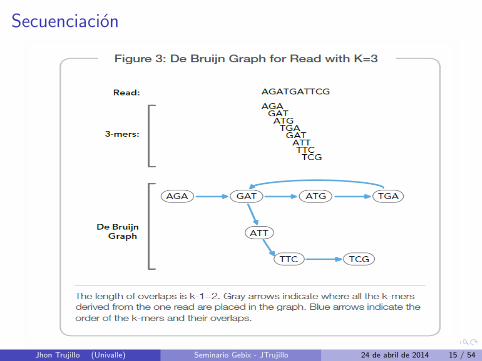
Secuenciacion
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 15 / 54

Secuenciacion
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 16 / 54
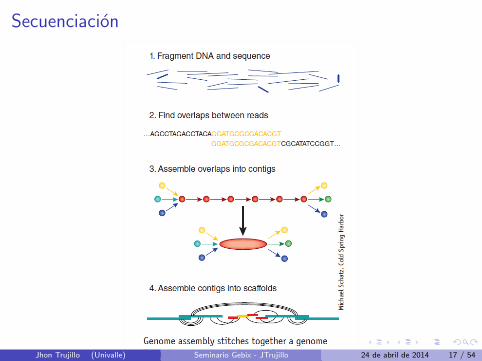
Secuenciacion
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 17 / 54

Introduction
Objetivo
Investigar la utilidad de los CGR para comparar y estudiar metagenomasdonde los fragmentos de cientos de genomas necesitan ser clasificadostaxonomicamente e identificados.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 18 / 54

MetodologıaSe presenta un breve resumen de la metodologıa.
Un grafico CGR en dos dimensiones se representa con un cuadrado dedos dimensiones y cada vertice del cuadrado representa uno de los 4nucleotidos base(g).
A=(0,0)T=(1,0)C=(0,1)G=(1,1)
Ejemplo : ATGCGAGTGT.
Figura : Representacion CGRJhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 19 / 54

Metodologıa
Una secuencia puede ser graficada dentro del area del cuadrado, de talmanera que la posicion de un nucleotido i corresponde a una posicion Ri .
ri = 0,5 ∗ (r(i − 1) + gi )
Donde el primer nucleotido dibujado en el cuadrado es graficado en elcentro del cuadrado, en la posicion (0.5 , 0.5).
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 20 / 54
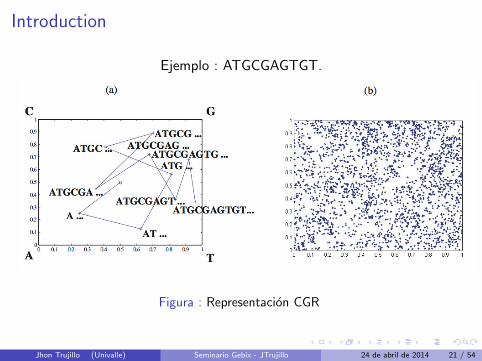
Introduction
Ejemplo : ATGCGAGTGT.
Figura : Representacion CGR
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 21 / 54

Metodologıa
Cada sub-cuadrado tiene definido un tamano r .
Por ejemplo:Un cuadrado con 100sub-cuadrados tieneun r = 1/10
Ejemplo : ATGCGAGTGT.
Figura : Resolucion de la 8-esima coordenada en lasecuencia. Cada coordenada tiene una precision de 4 bits.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 22 / 54

Metodologıa
Suma de Particiones
Zr (q) =∑i
(Pi )q (1)
Donde q pertenece a los Reales y se usa para indicar las regiones conmucha y poca densidad. En este paper : −15 <= q <= 15
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 23 / 54

Metodologıa
Exponente de escalamiento del espectrum
τq = lımr→0
log(Zr (q))
log(r)(2)
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 24 / 54

Metodologıa
Dimension fractal en funcion del espectrum
Para q diferente de 1.
Dq =τ
q − 1(3)
Para q igual a 1.
Dq = lımr→0
∑(Pi ) ∗ log(Zr (q))
log(r)(4)
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 25 / 54

Metodologıa
Exponente de escalamiento del espectrum
Calculado la segunda derivada diferencial de τ con respecto a q.
Cq =δ2 ∗ τqδ ∗ q2
= 2 ∗ τq − τ(q − 1) − τ(q + 1) (5)
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 26 / 54
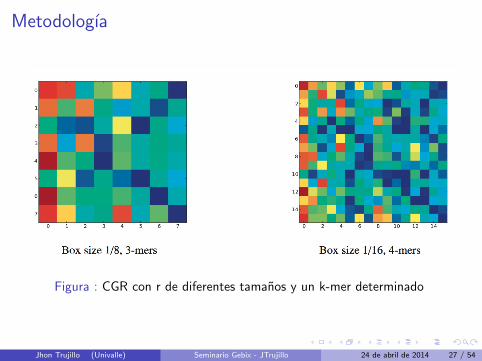
Metodologıa
Figura : CGR con r de diferentes tamanos y un k-mer determinado
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 27 / 54
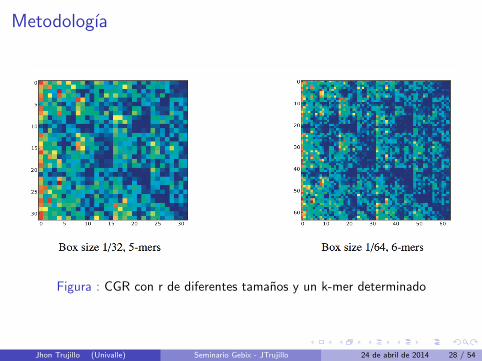
Metodologıa
Figura : CGR con r de diferentes tamanos y un k-mer determinado
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 28 / 54
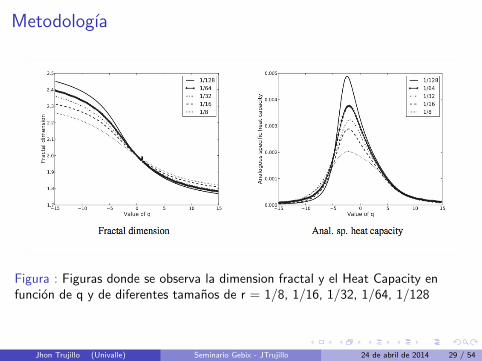
Metodologıa
Figura : Figuras donde se observa la dimension fractal y el Heat Capacity enfuncion de q y de diferentes tamanos de r = 1/8, 1/16, 1/32, 1/64, 1/128
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 29 / 54

Fuente de los Datos
Se obtuvieron los datos del programa GENOMETA.
de 2550 genomas se sacaron 1551 genomas donde cada uno de ellosson de una especie diferente (species-target).
Los genomas restantes (999 genomas) se usaron como datos deconsulta.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 30 / 54

Usando los CGR
Se usaron varios tamanos para los r y las siguientes propiedades de losCGRs:
1 La densidad de la caja Pi .
2 2. La dimension Fractal Di .
3 El heat Capacity Cq.
Por cada una de estas medidas se uso un RMSD de la siguiente forma:
√√√√ Nb∑i ,j
(di − dj)2
Nb(6)
Para este caso di y dj son las densidades de los cuadrados i y j en cadauno de los dos CGRs.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 31 / 54

Usando los CGR
De igual manera se usa para medir las otras dos propiedades:
Para la dimension fractal di y dj hacen referencia a la dimension.
Para el Heat Capacity di y dj hacen referencia al hear capacity en undeterminado q.
La Sumatoria se da en funcion de promedio de todos los valores de q.
Yo propongo las siguientes ecuaciones ( el paper no las presenta ):
√√√√√ Nq∑i ,j
(deni − denj)2
Nq(7)
√√√√√ Nq∑i ,j
(Ci − Cj)2
Nq(8)
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 32 / 54

¡ Recordar !
Que el string-query no se encuentra en la secuencia target.
Un emparejamiento exacto entre dos secuencias no es posible
Por lo tanto :El menor valor medido por el RMSD (menor diferencia) deberıa identificarel genoma desde el conjunto Target.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 33 / 54

¿Como validar la presicion del proceso ?
Usandoinformacion taxonomica descargada desde el NCBI(ftp://ftp.ncbi.nih.gov/pub/taxonomy/) de cada genoma.
Y usando un algoritmo para la construccion del arbol taxonomico.
Se compararon ambos genomas relacionados por medio de sus atributostaxonomicos :
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 34 / 54

¿Como validar la presicion del proceso ?
Especies.
Genus
family
order
Phylum
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 35 / 54

Resultados
Para calibrar y la evaluacion del metodo se evaluaron los siguientesexperimentos se realizaron:
Tamanos de los fragmentos de las secuencias.
Comparacion con Blast.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 36 / 54

Resultados
Tener en cuenta que :
Un CGR generado a partir de una fraccion de una secuencia de ungenoma es similar la secuencia completa.
Se sabe que una dimension fractal difiere significativamente paraCGRs generados para regiones codificantes y no codificantes.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 37 / 54

Resultados
El objetivo de esta prueba consiste en :
Determinar cuanto varia la identificacion de una secuencia de ungenoma dependiendo del tamano del fragmento usado a partir de lasecuencia.
En otras palabras determinar el mınimo tamano del fragmento sin quehaya un cambio significativo.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 38 / 54
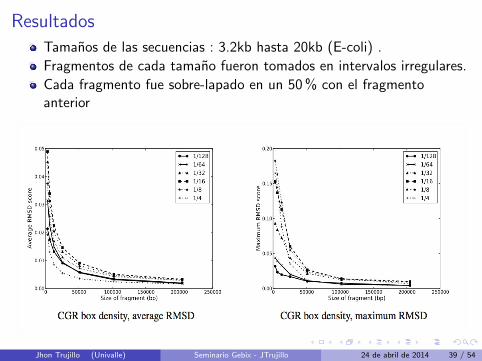
Resultados
Tamanos de las secuencias : 3.2kb hasta 20kb (E-coli) .
Fragmentos de cada tamano fueron tomados en intervalos irregulares.
Cada fragmento fue sobre-lapado en un 50 % con el fragmentoanterior
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 39 / 54
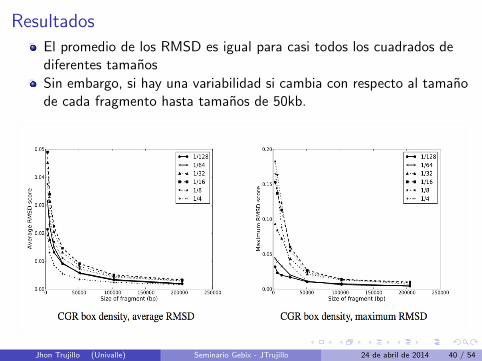
Resultados
El promedio de los RMSD es igual para casi todos los cuadrados dediferentes tamanos
Sin embargo, si hay una variabilidad si cambia con respecto al tamanode cada fragmento hasta tamanos de 50kb.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 40 / 54
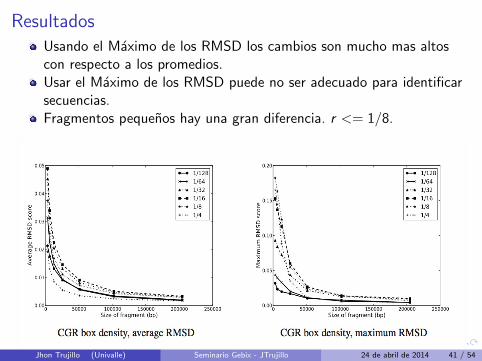
ResultadosUsando el Maximo de los RMSD los cambios son mucho mas altoscon respecto a los promedios.Usar el Maximo de los RMSD puede no ser adecuado para identificarsecuencias.Fragmentos pequenos hay una gran diferencia. r <= 1/8.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 41 / 54
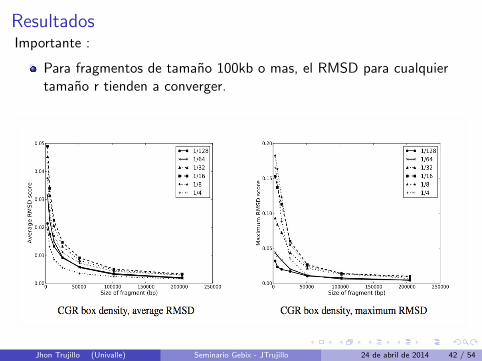
ResultadosImportante :
Para fragmentos de tamano 100kb o mas, el RMSD para cualquiertamano r tienden a converger.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 42 / 54
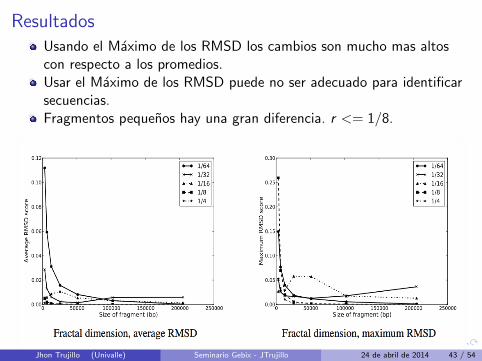
ResultadosUsando el Maximo de los RMSD los cambios son mucho mas altoscon respecto a los promedios.Usar el Maximo de los RMSD puede no ser adecuado para identificarsecuencias.Fragmentos pequenos hay una gran diferencia. r <= 1/8.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 43 / 54
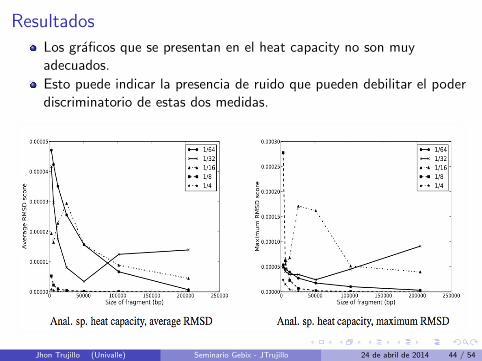
Resultados
Los graficos que se presentan en el heat capacity no son muyadecuados.
Esto puede indicar la presencia de ruido que pueden debilitar el poderdiscriminatorio de estas dos medidas.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 44 / 54
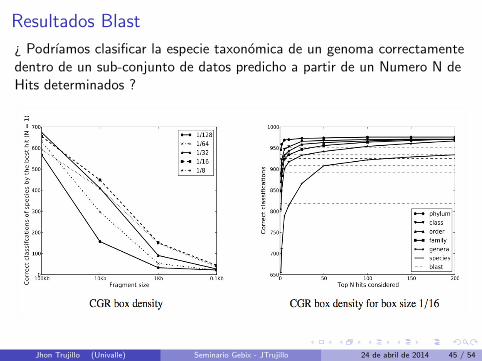
Resultados Blast
¿ Podrıamos clasificar la especie taxonomica de un genoma correctamentedentro de un sub-conjunto de datos predicho a partir de un Numero N deHits determinados ?
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 45 / 54
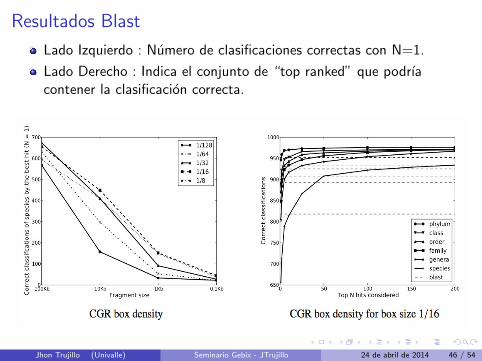
Resultados Blast
Lado Izquierdo : Numero de clasificaciones correctas con N=1.
Lado Derecho : Indica el conjunto de “top ranked” que podrıacontener la clasificacion correcta.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 46 / 54
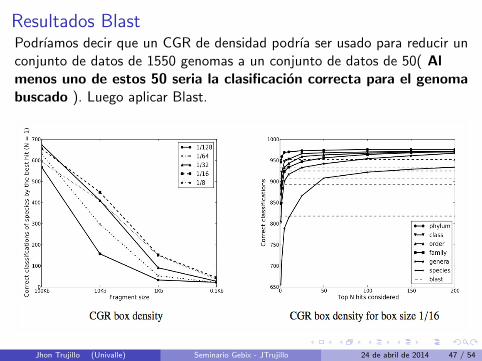
Resultados BlastPodrıamos decir que un CGR de densidad podrıa ser usado para reducir unconjunto de datos de 1550 genomas a un conjunto de datos de 50( Almenos uno de estos 50 seria la clasificacion correcta para el genomabuscado ). Luego aplicar Blast.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 47 / 54
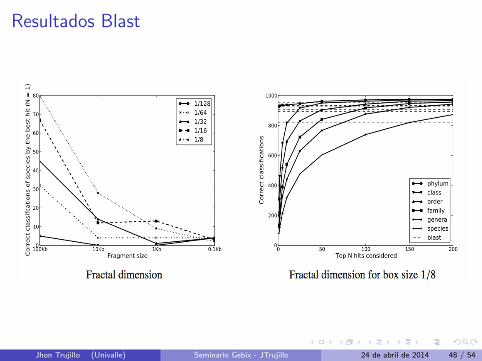
Resultados Blast
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 48 / 54
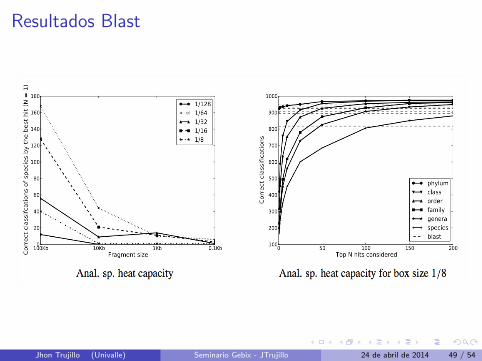
Resultados Blast
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 49 / 54

Resultados Blast
Comparison of timings using CGR box densities and BLAST.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 50 / 54

Discusion y trabajo futuro
Las comunidades cientıficas trabajan fragmentos pequenos de lecturas(100pb).
El desafıo esta en poder usar fragmentos pequenos por que seria maspreciso: Identificar secuencias novel y raras.
Los procesos de secuenciacion aun generan “reads” muy pequenos.
En este estudio se uso una funcion RMSD muy sencilla.
Se deberıa aplicar un metodo que pretermita identificar puntos claveen los CGR.
Aplicar conceptos estadısticos mas fuertes como los propuestos enalgunos papers de referencia que ellos recomiendan.
Desarrollar algoritmos de computer vision que permitan extraerinformacion importante del CGR.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 51 / 54

Discusion y trabajo futuro
La teorıa del caos para representar genomas fue introducida hace masde dos decadas. Es una forma conveniente para visualizar y comprargenomas de secuencias.
Estos resultados pueden ser usados por otros metodos para elalineamiento de las secuencias.
Aun falta muchas pruebas con los nuevos metodos de secuenciacionexistentes.
Se mostro que la propiedad CGR Density Box puede ser usada comoun metodo para filtrar espacios de busqueda.
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 52 / 54

References
Z.-G.Yu,V.Anh,K.-S.Lau (2001)
Measure representation and multifractal analysis of complete genomes
Phys.Rev.E64 (2001) 031903.
A.Pandit,A.Dasanna,S.Sinha, (2012)
Multifractal analysis of hiv-1 genomes.
MolPhylogenet Evol 62 (2) (2012) 756–63..
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 53 / 54

¡Gracias!
Jhon Trujillo (Univalle) Seminario Gebix - JTrujillo 24 de abril de 2014 54 / 54





























