Embed Size (px)
Citation preview

Modelling Assignment
Submitted to :- Submitted by:-
Dr. Durg Vijay Singh Swati KumariRoll No-22M.Sc. Bioinformatics2Nd semester
I-TASSER● I-TASSER (Iterative Threading ASSEmbly Refinement)
● It is web appliocation for protein structure & function prediction.
● Model are built based on Multiple threading alignment by LOMETS and iterative TASSER simulation.
● It was ranked as the No 1 server in recent CASP7(Critical Assessment of technique for protein Structure Prediction) and CASP8 experiment.
I-TASSER● I-TASSER (Iterative Threading ASSEmbly Refinement)
● It is web appliocation for protein structure & function prediction.
● Model are built based on Multiple threading alignment by LOMETS and iterative TASSER simulation.
● It was ranked as the No 1 server in recent CASP7(Critical Assessment of technique for protein Structure Prediction) and CASP8 experiment.

CONTENTS● Objective
● Protein threading
● Ab initio method
● I-TASSER
● MUSTER
● DALI server
● Robetta
● Validation
● Result and discussion
● Conclusion
● References

●We have to geerate a model of given sequence Ecdl [Emeicella rugulosa] have 604 residue.
●This protein have not shown significant alignment with any solved structure .
●On other hand, it can be modeled with either Fold recognition method or Ab intio method.
OBJECTIVE

Sequence is...
>gi|407259499|gb|AFT91383.1| EcdL [Emericella rugulosa]MDDSPWPQCDIRVQDTFGPQVSGCYEDFDFTLLFEESILYLPPLLIAASVALLRIWQLRSTENLLKRSGLLSILKPTSTTRLSNAAIAIGFVASPIFAWLSFWEHARSLRPSTILNVYLLGTIPMDAARARTLFRMPGNSAIASIFATIVVCKVVLLVVEAMEKQRLLLDRGWAPEETAGILNRSFLWWFNPLLLSGYKQALTVDKLLAVDEDIGVEKSKDEIRRRWAQAVKQNASSLQDVLLAVYRTELWGGFLPRLCLIGVNYAQPFLVNRVVTFLGQPDTSTSRGVASGLIAAYAIVYMGIAVATAAFHHRSYRMVMMVRGGLILLIYDHTLTLNALSPSKNDSYTLITADIERIVSGLRSLHETWASLIEIALSLWLLETKIRVSAVAAAMVVLVCLLVSGALSGLLGVHQNLWLEAMQKRLNATLATIGSIKGIKATGRTNTLYETILQLRRTEIQKSLKFRELLVALVTLSYLSTTMAPTFAFGTYSILAKIRNMTPLLAAPAFSSLTIMTLLGQAVSGFVESLMGLRQAMASLERIRQYLVGKEAPEPSPNKPGVASTEGLVAWSASLDEPGLDPRVEMRRMSSLQHRFYNLGELQD

Protein Threading
●Also known as Fold recognition method.
●It is a Template Based Model.
●It is a method of protein modeling which is used to model those proteins which have the same fold as protein of known structures, but don't have homologous proteins with known structure.
●There are many popular software for Fold Recognotion-- I-TASSER- MUSTER- PHYRE2- RaptorX server

Ab intio method
●It is a Template Free Model.
● It done when we have not any information about structure of our protein of intrest.
●The most popular software for ab initio are -- Robetta

I-TASSER
●Iterative - Threading ASSEmbly Refinement
●It is web appliocation for protein structure & function prediction.
●Model are built based on Multiple threading alignment by LOMETS (Local Meta Threading Server) and iterative TASSER simulation.
●It was ranked as the No 1 server in recent CASP7(Critical Assessment of technique for protein Structure Prediction)
and CASP8 experiment.

Steps of I-TASSER
1. Threading
2. Structral assembly
3. Model Selection and Refinment
4. Structral based Functional Annotation
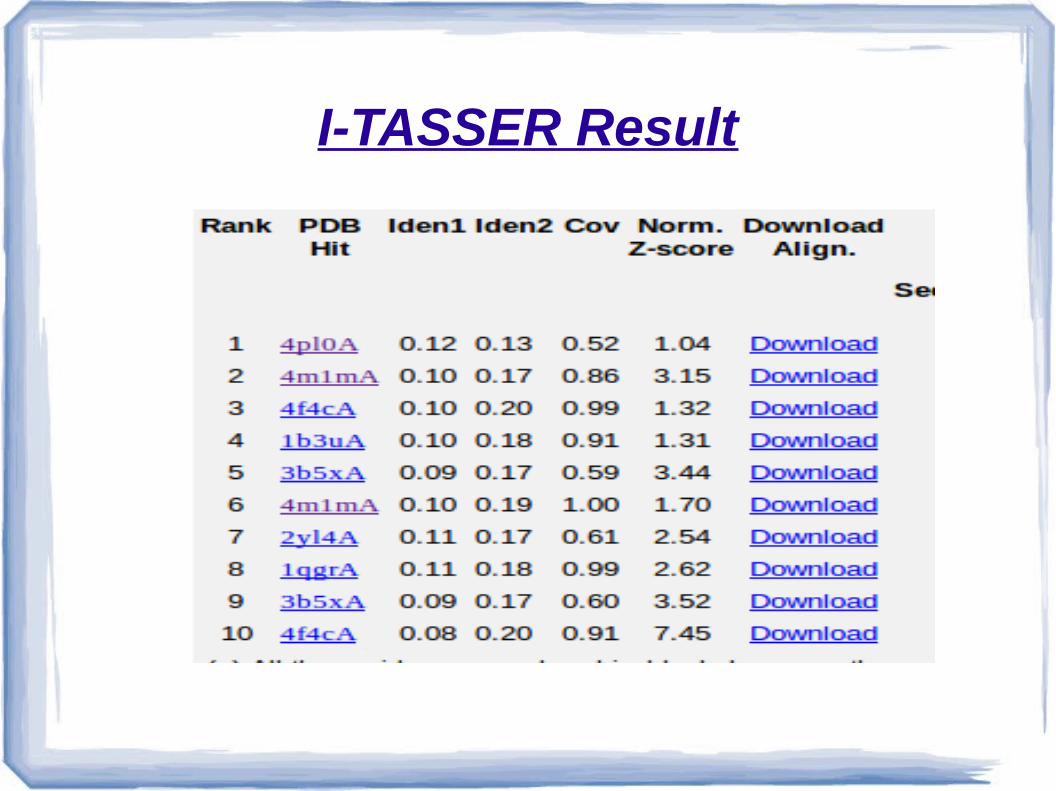
I-TASSER Result

MUSTER
●Muti-Source ThreER
●It is also one of best software for new protein to identify the templates structure from the PDB library.
●It generate sequence template alignment by comninig sequence profile-profile alignment (PPA) with multiple structural information.
●It was successful for TBM in CASP experiment.

Steps of MUSTER
1. Scoring Function2. Sequence Profile3. Secondary Stucture Match4. Stucture Profile5. Solvent Accesiability6. Backbon of Dihyral Angle7. Hydrophobic Scoring Matrix8. Dynamic Programming9. Template taking scheme

MUSTER Result

Result of I-TASSER and MUSTER
●The result of I-TASSER and MUSTER both give the model of our protein based on template – 4PL0A.
●“I consider 4PL0A this model for our protein of intrest”.

MUSTER - Target-template alignments

MUSTER best Model

● Structure of an antibacterial peptide ATP-binding cassette transporter.
●Molecular Description :- Classification: Transport ProteinMolecule: Microcin-J25 export ATP-
binding/permease protein McjDType: protein Length: 580Chains: A, BOrganism: Escherichia coli
4Pl0A

DALI Server●Distance mAtrix aLIgnment
●The Dali server is a network service for comparing protein structures in 3D.
●Be used to imply evolutionary relationship between protein that share very little common sequence.
●The output of sructural Alignment are : - 1.Z score- Structural similarity is measured by Doli z-score.

DALI Server result
2. Super positionof atomic co ordinate set - Its use to compare multiple conformation of same protein and to evaluate the quality of alignment produced using only two or more known sequences .
3. Minimal RMSD between structure - RMSD of two align stucture shows the divergance from one to another.
4. Nresu - its show the number of align residue.
5. Lali - its the number of align position.

DALI result “Low rmsd and high nres shows the better alignment.”
●If both rmsd and nres is high or low, not possible to establish an order between the alignment.
●Rmsd- It is the measure of the average deviation in distance between aligned alpha carbons (i.e, calculate the diversance from one to another b/w two sequences)
●Note:- DALI package is based on Fartran programming and perl script.
“The best alignment shows with low rmsd 0.6 and high lali score 403.”
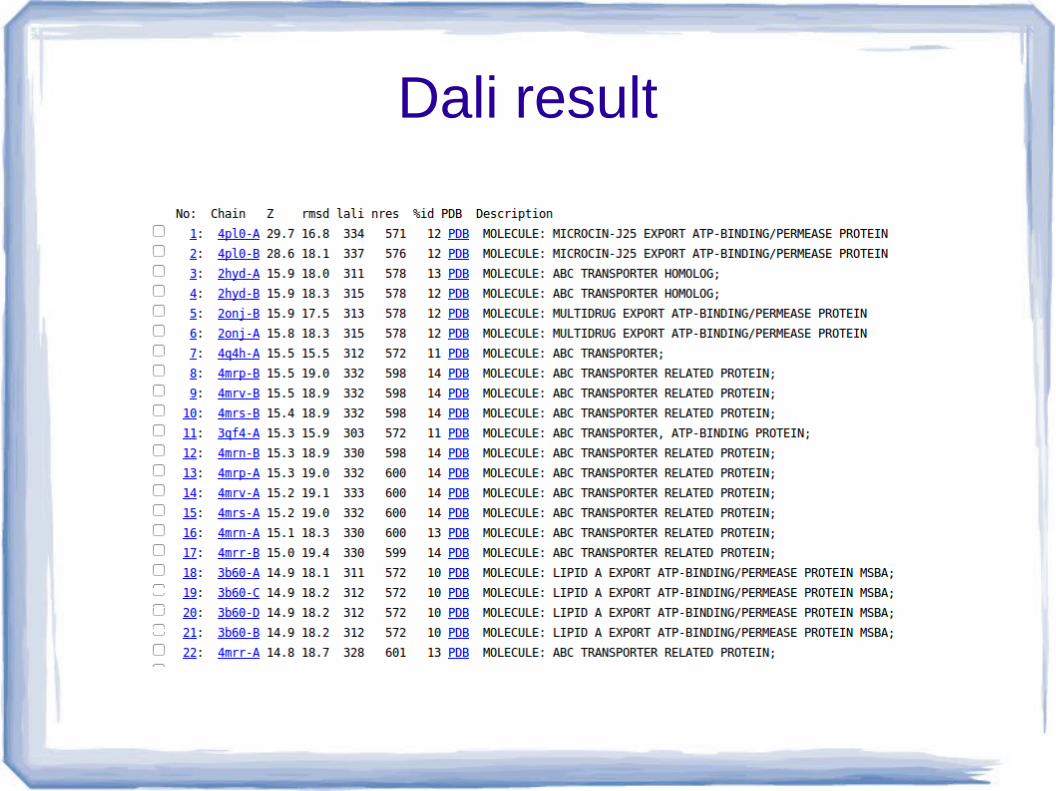
Dali result

STRUCTURE-STRUCTURE ALIGNMENT BY PYMOL

Ab initio by Robetta
● I am going to used Robetta server for Ab initio structure prediction.
● At first, we go on robetta server.
● Register on Robetta home server.
● Open id with username or email.
● Paste protein sequence.
● Filling the all field which is neccessaey .
● Submit.
● After submiting, we received a job id 54467 on date 24 march 2015.
● Check result by clicking Queue on Robetta page.

RobettaSteps of Robetta -
1. After giving target sequence it will start doing Profile - profile alignment based on fragments library.
2. phase 1 : Monte Carlo fragment assembly
Low resolution model ( predict of strc with about near accuracy )
3. Phase 2 : physics base atomic refinement
High resolution model (absolute accuracy)
4. Final atomic model
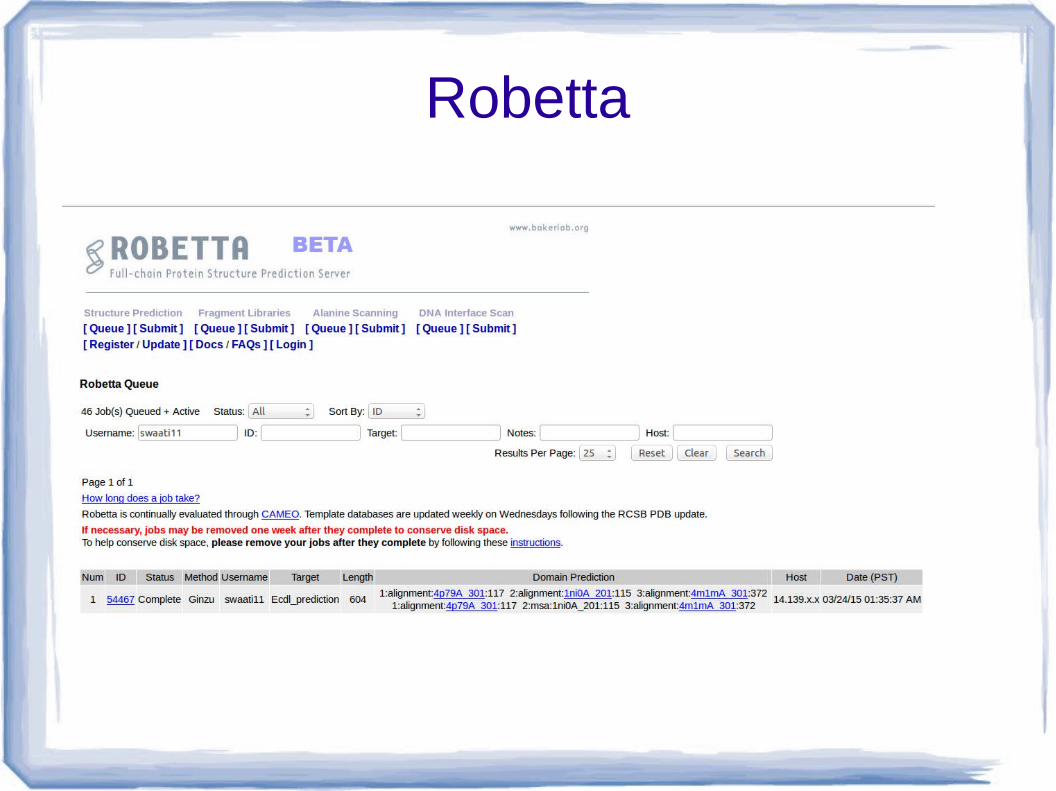
Robetta
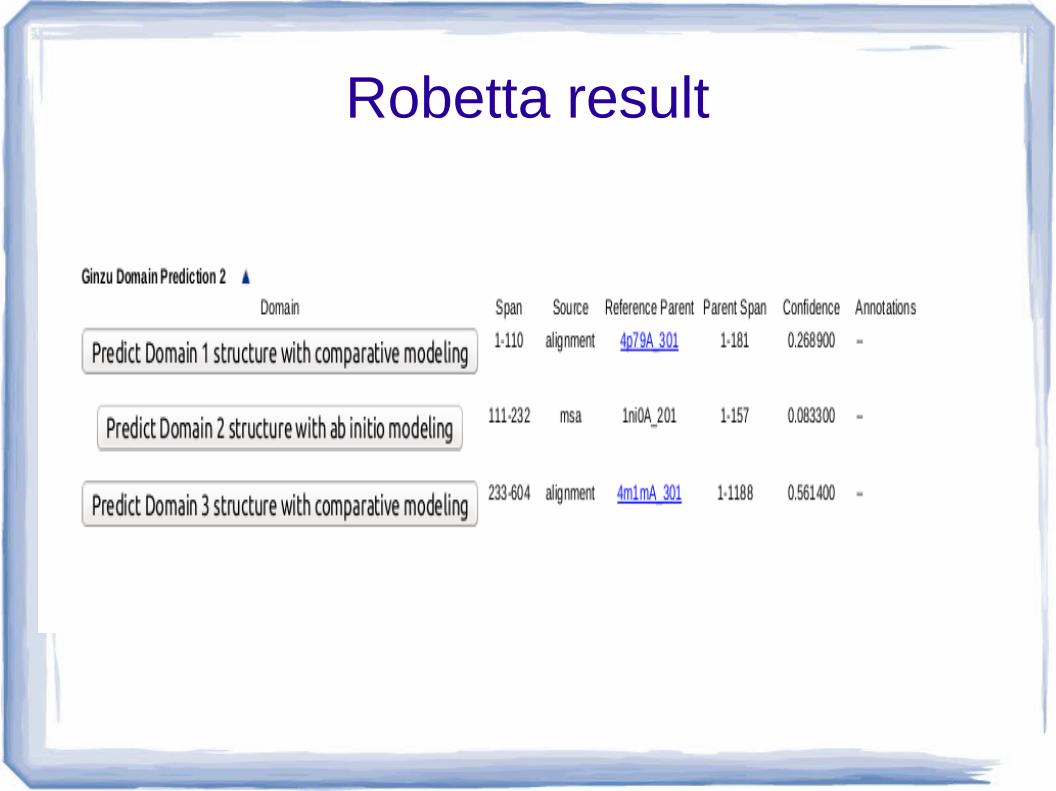
Robetta result

Robetta result ● Robetta generate 3 model -
Sl. no. Protein ID Discription
1 4p79 ● Crystal str of cloudin provides insight into the architecture of tight junction
● Ion channel regulator, alpha helical● Membrane protein
2 1ni0 ● Hydrolase● Restriction endonuclease PuvII from proteus vulgaris,
class alpha/beta protein● EC 3.1.21.4
3 4m1m ● Multidrug resistant protein ● ATP binding cassate transpoter● Pgp

Validation of Model
Validation of generated model is validate by these server -
1. ANOLEA
2. PROCHECK
3. PROSA

ANOLEA
● Atomic Non-Local Environment Assessment
● It check the non local environment of protein itself with their different amino acid on the basis of energy with Euclidean distance (7°A) with 11 residue Amino acid.
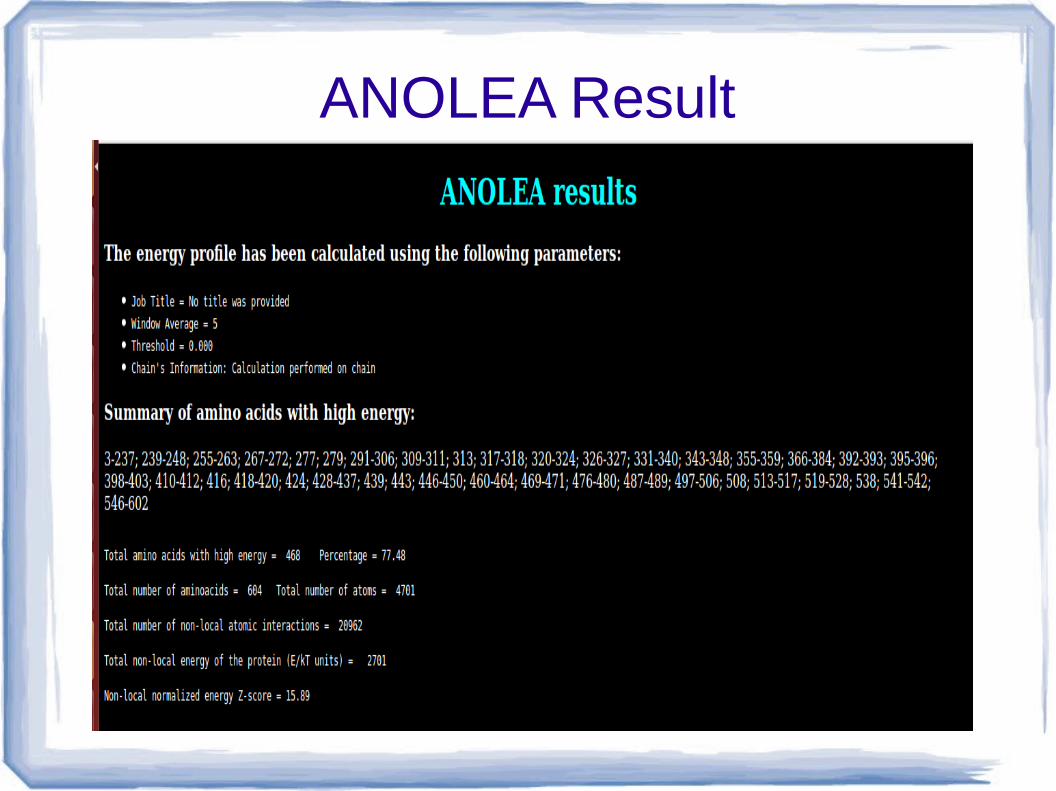
ANOLEA Result

PROCHECK
● Checks the stereochemical quality of a protein structure by analyzing residue-by-residue geometry and overall structure geometry.
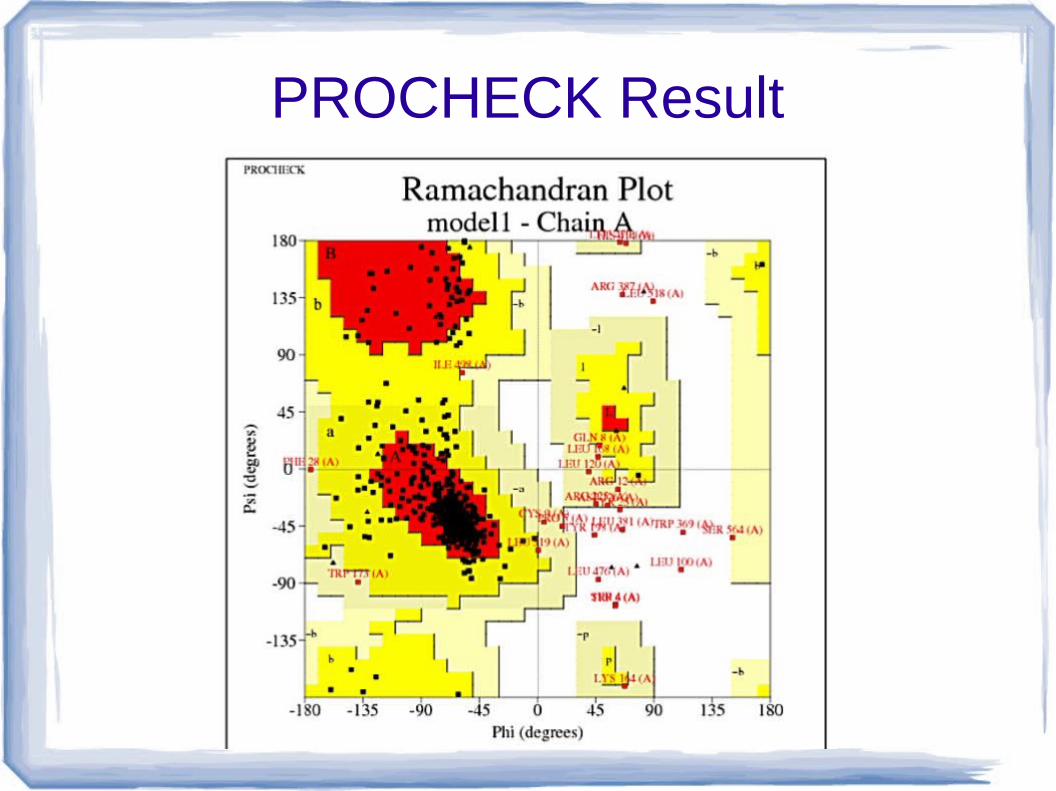
PROCHECK Result

PROCHECK
● Plot Statics

PROSA
● Protein Structure Analysis● It check the quality of c alpha carbon.
● Output of prosa shows-
1. Z score- it shows the overall quality of model value display of all experimentally determined protein chain in PDB.
"more negative z score- best structure.
more positive z score not well structure."
2. Plot of residue score- shows local quality of model by plotting energy as sum of AA sequence position i (take window size 40)

PROSA
– Positive value correspond problematic part of structure.
3. Prosa web visualizer picture 3D model (J mol c-alpha trace)- the 3D structure of protein of interest is visualised by the use molecular viewer Jmol.
– Residue are colored from blue to red in order of increasing residue energy.
● The Z score of this model is -6.41

PROSA Result
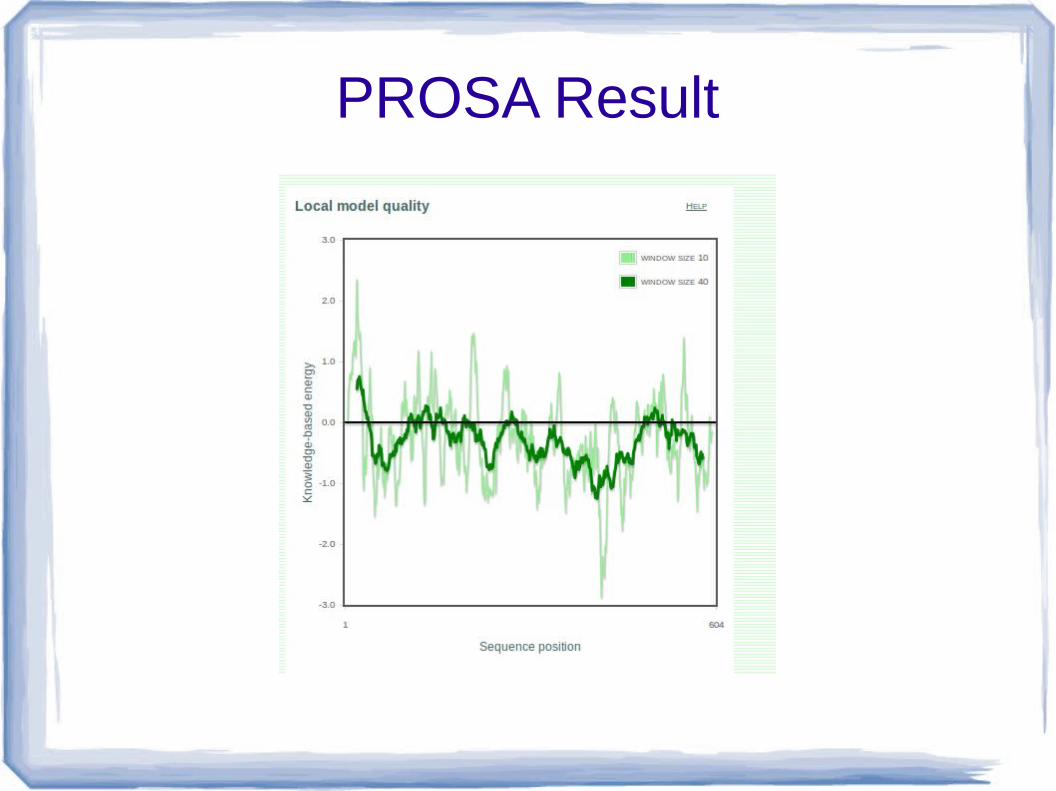
PROSA Result
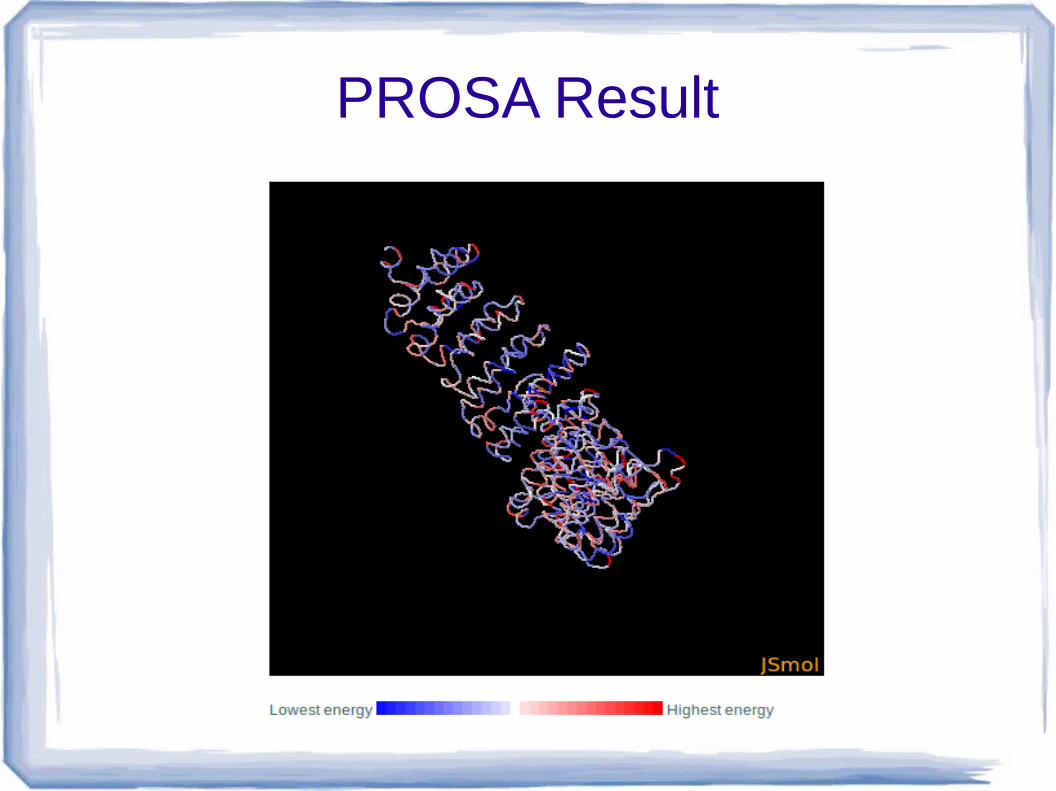
PROSA Result

Result and Discussion●As the given instructions the given sequence Ecdl having 604 aa long can't modeling by homology modeling.
●Thus, we have to move towards on the other method of prediction either Fold recognition methid or Ab initio method.
●Threading method was done by MUSTER and I-TASSER server which gives common reslutt .
●And the Ab initio method was done by Robetta server,as the result we get information about domain and generate structure on the basis of founded domain.
●With the help of prosa/ Procheck , analysed the ramachandran plot of the model.and validate our model by the using of other tools ti check quality of model.
●We found the many protein of ABC transporter and p glyco protein as result.

Conclusion
Result of I-TASSER and MUSTER which for fold recognition and Robetta for ab initio structure prediction shows the given protein is higher similar to those protein which are -
●ABC transporter super family protein (ATP binding Cassette )●Transmembrane protein

REFERENCE● http://zhanglab.ccmb.med.umich.edu/I-TASSER/● http://zhanglab.ccmb.med.umich.edu/MUSTER/● http://robetta.bakerlab.org/● http://melolab.org/anolea/● https://prosa.services.came.sbg.ac.at/prosa.php● http://ekhidna.biocenter.helsinki.fi/dali_server/resu
lts/20150324-0049-69ef51112579617192cac4dcad7075f2/index.html

THANK YOU.....




























